(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Hematologia
acp-medicine Hematologia
Última revisão: 23/10/2012
Comentários de assinantes: 0
Elizabeth A. Price, MD, MPH
Clinical Instructor, Department of Medicine, Division of Hematology, Stanford University School of Medicine.
Stanley L. Schrier, MD, FACP
Professor of Medicine, Division of Hematology, Stanford University School of Medicine, and Staff Physician, Medicine-Hematology, Stanford Hospital and Clinics
Artigo original: Price EA, Schrier SL. Hemoglobinopathies and hemolytic anemias. ACP Medicine. 2008;1-34.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
A alteração de membrana geralmente sinaliza aos macrófagos do sistema reticuloendotelial para que removam o eritrócito danificado da circulação. Em circunstâncias extraordinárias, todavia, o dano à membrana é tão grande que o conteúdo intracelular, incluindo a hemoglobina, é liberado no plasma. Este capítulo descreve os aspectos estruturais e funcionais dos eritrócitos normais e das doenças que envolvem a arquitetura da membrana, proteínas eritrocitárias e fatores extracorpusculares que levam ao encurtamento da sobrevida das hemácias.
As células precursoras dos eritrócitos sofrem 4 ou 5 divisões na medula óssea e, em seguida, expelem seus núcleos e transformam-se em reticulócitos. À medida que estas células enucleadas amadurecem, a síntese de hemoglobina diminui. As células perdem a maioria de seus receptores de transferrina e entram na circulação sanguínea periférica, onde sobrevivem por cerca de 4 meses.
Conforme se movem pela circulação, os eritrócitos precisam enfrentar intensos estresses mecânicos e metabólicos, sofrem deformação durante a passagem por capilares com diâmetro equivalente à metade do próprio diâmetro, resistem a altas forças de cisalhamento ao se moverem através das válvulas cardíacas, sobrevivem a episódios repetitivos de depleção de substrato e acidemia estase-induzida e têm de evitar a remoção pelos macrófagos do sistema reticuloendotelial. Os eritrócitos também precisam manter um ambiente interno que proteja a hemoglobina do ataque oxidativo, bem como uma concentração ideal de 2,3-difosfoglicerato (2,3-DPG) necessário à função da hemoglobina.
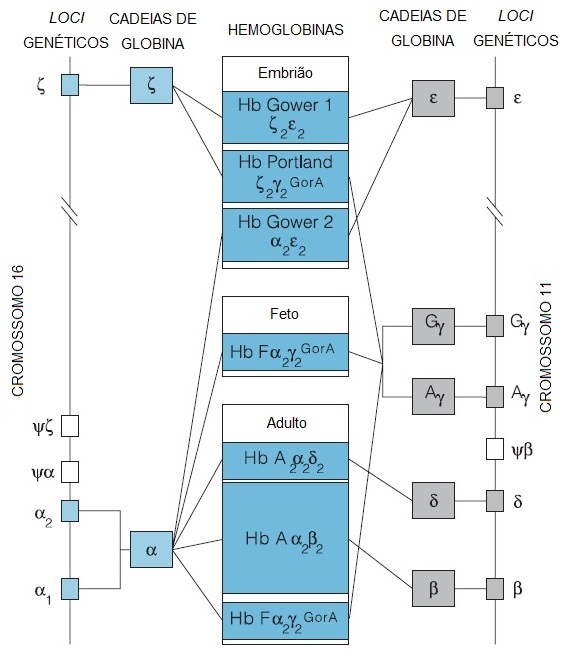
O eritrócito de um adulto normal contém 3 formas de hemoglobina: HbA (96%), HbA2 (2 a 3%) e HbF (< 2%). A HbA normal (alfa-2-beta-2) é composta por 2 cadeias alfa (codificadas por 4 genes localizados no cromossomo 16) e 2 cadeias beta (codificadas no cromossomo 11). A HbA2 é constituída por 2 cadeias alfa e 2 cadeias delta (alfa-2-delta-2), enquanto a hemoglobina fetal (HbF) é composta por 2 cadeias alfa e 2 cadeias gama (alfa-2-gama-2). Os genes codificadores das cadeias beta, delta e gama estão estreitamente ligados entre si no cromossomo 11. A concentração extraordinariamente alta de hemoglobina existente no eritrócito – 33 a 35 g/dL (concentração de hemoglobina corpuscular média [CHCM]) – produz uma solução intracelular viscosa.
Os eritrócitos utilizam principalmente a glicose para manter o potencial de redução que protege a célula contra o ataque oxidativo, gerar 2,3-DPG necessário à modulação da função da hemoglobina e controlar o conteúdo de sais e, consequentemente, de água do eritrócito por ação do trifosfato de adenosina (ATP) e das adenosina trifosfatases transportadoras (ATPases) [Tabela 1]. O conteúdo de água e hemoglobina do eritrócito determina o volume corpuscular médio (VCM) e a CHCM.
Tabela 1. Metabolismo do eritrócito
|
Via |
Produto |
Funções dos produtos metabólicos |
|
Glicólise pela via de Embden-Meyerhof |
ATP |
Serve de substrato para todas as reações envolvendo quinases, bomba de sódio/potássio acoplada à ATPase, bomba de efluxo de cálcio acoplada à ATPase e outras ATPases existentes na membrana eritrocitária, entre as quais a aminofosfolipídio translocase Mantém o estado deformável da membrana do eritrócito |
|
2,3-DPG |
Interage com a desoxiemoglobina, deslocando o equilíbrio em favor do descarregamento de O2 a partir da oxiemoglobina Atua como ânion intracelular incapaz de cruzar a membrana eritrocitária | |
|
NADH |
Atua como substrato da metemoglobina redutase, capacitando-a a reduzir a metemoglobina (Fe3+) em hemoglobina (Fe2+) | |
|
Via da pentose-fosfato (desvio da hexose monofosfato) |
NADPH |
Serve de substrato para outra metemoglobina redutase na redução da metemoglobina (mecanismo contra falhas) Serve de coenzima para a glutationa redutase na redução da glutationa oxidada; a glutationa reduzida protege a hemácia contra a desnaturação oxidativa |
ATP = trifosfato de adenosina; ATPase = adenosina trifosfatase; 2,3-DPG = 2,3-difosfoglicerato; NADH = nicotinamida adenina dinucleotídeo reduzido; NADPH = fosfato de nicotinamida adenina dinucleotídeo reduzido.
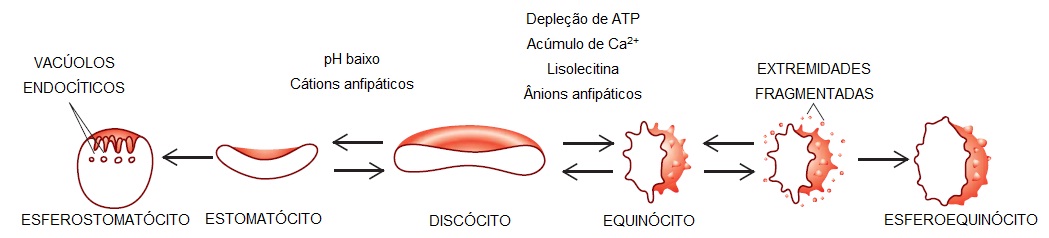
A hemácia normalmente apresenta um formato discoide, com diâmetro de 7 a 8 mcm, VCM da ordem de 85 a 90 fL (1 fL = 10-15 L) e uma área de superfície igual a 140 mcm2 [Figura 1]. O formato exclusivo permite que a hemácia se esprema entre capilares com diâmetros da ordem de 3 mcm.
Lipídios (fosfolipídios e colesterol) são responsáveis por 50% do peso da membrana superficial. Os fosfolípidos são distribuídos assimetricamente na bicamada da membrana; os carregados com carga positiva ficam na parte externa e aqueles carregados com carga negativa ficam predominantemente na parte interna. Esta assimetria permite a passagem seletiva de pequenas moléculas carregadas, seja para o exterior ou para o interior da bicamada, produzindo equinócitos ou estomatócitos [Figura 1].

Figura 1. O eritrócito normal (ou discócito) sofre alterações de formato em resposta às condições criadas pelo tratamento com certos agentes. A maioria das alterações é reversível, desde que os agentes sejam removidos antes da perda permanente de material de membrana.
ATP = trifosfato de adenosina.
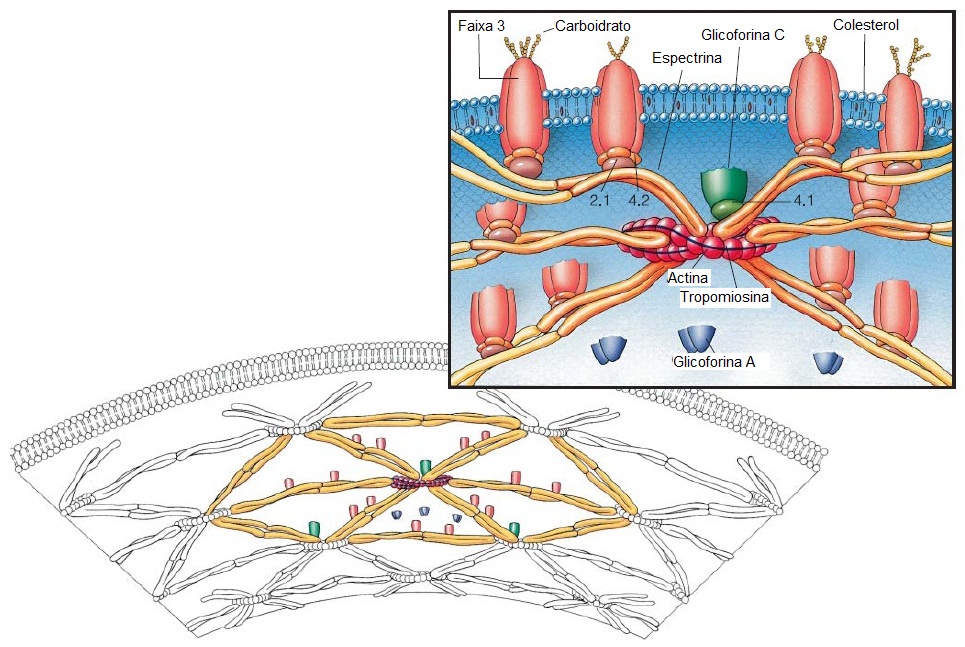
As proteínas da membrana das hemácias incluem proteínas integrais e periféricas. As proteínas integrais interagem e estendem-se sobre a bicamada fosfolipídica hidrofóbica [Figura 2]. As principais proteínas integrais da membrana eritrocitária são as glicoforinas (que contêm a maior parte do ácido siálico e carregam os antígenos do grupo sanguíneo MNS) e a faixa 3, que é um transportador de ânion e bicarbonato.
As proteínas periféricas são todas encontradas na face citosólica da membrana. A interação destas proteínas periféricas, que incluem a espectrina e a actina, resulta no citoesqueleto resistente – e, contudo, elástico – das hemácias. O citoesqueleto periférico, por sua vez, está conectado às proteínas integrais [Figura 2].1,2
Figura 2. A faixa 3, um canal de transporte de ânions (laranja), e as outras proteínas integrais, glicoforina A (não mostrada), glicoforina B (não mostrada) e glicoforina C (verde), estendem-se sobre a membrana eritrocitária. As cadeias laterais de carboidrato externas ramificadas estão fixas a estas proteínas. As cabeças polares hidrofílicas das moléculas de fosfolipídio que compõem a bicamada estão orientadas na direção da superfície celular, enquanto as cadeias laterais de ácidos graxos hidrofóbicos estão direcionadas para o interior da bicamada. O colesterol está intercalado entre as cadeias de ácidos graxos. A faixa 3 está ligada à hemoglobina e à gliceraldeído-3-fosfato desidrogenase em sua superfície citosólica. A espectrina (amarelo), actina (vermelho), tropomiosina (azul) e faixa 4,1 (verde-claro) formam uma rede entrelaçada sobre a superfície interna da membrana. Os heterodímeros de espectrina associam-se para formar heterotetrâmeros. A figura inferior representa a rede entrelaçada do citoesqueleto hexagonal junto à superfície interna da membrana. A faixa 2,1 (ancirina) liga a proteína integral faixa 3 ao citoesqueleto periférico através da cadeia beta da espectrina. Ligações extras são fornecidas pela glicoforina C e pela faixa 4,1.
Os carboidratos da membrana contribuem para a carga negativa externa da membrana e atuam parcialmente como antígenos de grupo sanguíneo. Alguns destes glicolipídios se associam ao fosfatidilinositol para formar uma âncora glicolipídica, denominada âncora de glicosilfosfatidilinositol (GPI). Estas âncoras de GPI fornecem um sítio de ancoragem de membrana para várias classes de proteínas que exercem funções biológicas importantes junto às superfícies de membrana, entre as quais as diversas proteínas controladoras da ação do complemento [ver Hemoglobinúria paroxística noturna (HPN), adiante].3
O controle do volume das hemácias possui uma considerável importância patofisiológica, porque o conteúdo de água e cátions destas células determinam a viscosidade intracelular e a proporção da área de superfície em relação ao volume. O conteúdo de Na+ e K+ é determinado pela difusão passiva e pelo transporte ativo, primariamente via Na+/K+ ATPase. O principal ânion intracelular é o Cl-, que entra na hemácia com alta permeabilidade através da faixa 3. O cotransportador de K+/Cl- dirige o gradiente de K+/Cl- e é ativado pelo inchaço da hemácia e pelo baixo pH intracelular, causando uma perda líquida de K+ e Cl-. A ATPase de Ca2+ bombeia ativamente o Ca2+ para fora da hemácia, tornando o conteúdo de Ca2+ citosólico livre inferior a 0,1 mcM – 4 ordens de magnitude abaixo da concentração plasmática de 1 mM. O canal de Gardos, que é um canal de efluxo de K+ ativado por Ca2+, exerce papel importante na regulação do volume. A água entra e sai através de um canal de água denominado CHIP28 (proteína de membrana integral formadora de canal de 28 kDa) ou aquaporina. Outros ânions intracelulares importantes são o 2,3-DPG e a hemoglobina, que são incapazes de penetrar a membrana celular. Quando a concentração de Ca2+ citosólico livre sobe e atinge níveis da ordem de 0,3 mcM, o canal é ativado, e isto resulta em uma perda líquida de K+. Se esta perda não for corrigida, a hemácia afetada se torna desidratada.4
A depleção de ATP, o acúmulo de íons de cálcio ou o tratamento com lisolecitina ou compostos anfipáticos aniônicos transformam o eritrócito normal (ou discócito) em equinócito – uma célula espiculada, crenulada, por vezes chamada de hemácia crenada [Figura 1]. O cálcio, atuando sozinho ou aliado à proteína ligadora de cálcio (calmodulina), pode afetar a alteração de formato equinocítica. Quando o processo equinocítico persiste, a fragmentação ou brotamento das extremidades do equinócito acarretam perda de componentes de membrana, particularmente de faixa 3 e fosfolipídios. Isto resulta em perda de área de superfície, diminuição da proporção da área de superfície em relação ao volume e formação de esferoequinócitos pouco derformáveis.
Os principais determinantes do fluxo sanguíneo são: o hematócrito; a concentração plasmática de proteínas (p. ex., fibrinogênio e imunoglobulinas) que influenciam no grau de formação de rouleau ou agregação; a deformabilidade das hemácias; o calibre dos vasos sanguíneos; e a taxa de cisalhamento (proporção velocidade de fluxo/raio do tubo). Diante das taxas de cisalhamento menores encontradas nas vênulas pós-capilares, as hemácias tendem a se aglomerar como massas assimétricas, com consequente aumento da viscosidade do sangue e resistência ao fluxo.
Na medula óssea, os reticulócitos em desenvolvimento perdem progressivamente seu RNA residual no decorrer de um período de 4 dias após a extrusão nuclear. Ao final deste estágio, o reticulócito torna-se incapaz de sintetizar proteínas. O cotransporte ativo de K+/Cl- atua para diminuir o volume celular. Com a montagem de proteínas de membrana completa, a célula madura resultante entra na circulação e sobrevive durante um período de 100 a 120 dias.5 A morte do eritrócito é um fenômeno dependente da idade, podendo estar relacionada aos estresses mecânicos e químicos com que a célula se depara na circulação. Conforme envelhece, o eritrócito perde água, e sua área de superfície diminui. A proporção entre área de superfície e volume diminui, e a CHCM aumenta, comprometendo a deformabilidade celular. Além disso, a atividade enzimática reduzida diminui a capacidade celular de resistência ao estresse metabólico. A manifestação do envelhecimento pode ocorrer via alterações na superfície do eritrócito, como uma diminuição da densidade ou do tipo de carga de superfície ou o aparecimento de um neoantígeno de senescência (talvez, um agrupamento oxidativo de faixa 3 [Figura 2]) que se liga a imunoglobulinas específicas e componentes do complemento.6 Por meio destas alterações, o eritrócito senil sinaliza sua incapacidade ao sistema reticuloendotelial, deflagrando sua própria remoção pelos macrófagos.
Sob condições fisiológicas, pouco menos de 1% das hemácias são destruídas diariamente e substituídas por um número quase idêntico de células novas. Em um homem pesando 70 kg e cujo volume de sangue aproximado seja de 5 L, cerca de 50 mL de sangue total (contendo cerca de 22 mL de eritrócitos concentrados) são destruídos e repostos todos os dias. Considerando que 1/3 de cada eritrócito é hemoglobina, a substituição destas células requer a síntese de cerca de 7 g de hemoglobina por dia. A medula óssea de um adulto normal consegue quintuplicar prontamente seu débito eritroide. Após um extensivo e prolongado estresse anêmico, é possível aumentar a produção em até 7 a 8 vezes. O suprimento de ferro, contudo, impõe uma limitação significativa à reposição de hemácias: 3/4 do ferro utilizado na síntese celular em um dia provém das células destruídas no dia anterior.
A severidade da anemia é determinada pela taxa de destruição de hemácias e pela capacidade da medula de aumentar a produção eritroide. Quando um indivíduo conta com uma medula sadia, o tempo de sobrevida do eritrócito pode ser reduzido de 120 para 20 dias sem indução de anemia nem icterícia. Entretanto, nestes casos, pode haver uma reticulocitose substancial.
A maioria das formas de hemólise é extravascular. Através da membrana, a célula danificada sinaliza sua condição alterada ao sistema reticuloendotelial e é removida. Sob circunstâncias inusitadas, quando o dano ao eritrócito é devastador – como ocorre em algumas formas de lise mediada pelo complemento – ou em situações em que o sistema reticuloendotelial não consegue lidar com a carga de células danificadas, há desenvolvimento de uma lise intravascular que leva à hemoglobinemia.
A hemoglobina liberada no plasma é degradada em dímeros alfa-beta que, por sua vez, ligam-se à haptoglobina. Os complexos hemoglobina-haptoglobina são removidos pelo sistema reticuloendotelial. Quando a capacidade de ligação da haptoglobina é excedida, os dímeros alfa-beta passam para dentro do filtrado glomerular. Alguns dímeros alfa-beta são excretados diretamente na urina e produzem hemoglobinúria, enquanto outros são captados pelas células dos túbulos renais. As células dos túbulos renais que contêm ferro podem ser excretadas durante vários dias após um episódio de hemólise intravascular. A hemossidenúria pode ser identificada pela coloração com azul da Prússia. A hemoglobina plasmática livre pode se dissociar em globina e hemina. A hemina pode se ligar à hemopexina e, nesta forma ligada, alcançar as células tubulares renais, ou pode se ligar à albumina plasmática e produzir metemalbuminemia.
A hemólise intravascular pode produzir uma anemia severa, de maneira aguda. Além disso, as partículas da membrana eritrocítica liberadas no plasma podem atuar como poderosos estímulos de coagulação intravascular disseminada. A hemólise severa aguda também causa insuficiência renal aguda [ver 10:VI Insuficiência renal aguda]. Quando um paciente em estado de compensação de um aumento acentuado da hemólise adquire uma infecção que compromete agudamente a atividade eritroide da medula,7 os níveis de hemoglobina podem sofrer uma queda dramática – uma condição denominada crise aplásica. Com a hemólise crônica, frequentemente há desenvolvimento de cálculos de pigmento junto à vesícula biliar.
As causas de hemólise podem ser classificadas como extra ou intracorpusculares. As causas intracorpusculares, que são essencialmente defeitos eritrocitários, abrangem as anomalias de membrana, distúrbios metabólicos e distúrbios relacionados à estrutura ou biossíntese da hemoglobina. As causas extracorpusculares representam os elementos anormais encontrados junto ao leito vascular, que atacam e destroem os eritrócitos normais. Como os eritrócitos com defeitos intracorpusculares causadores de hemólise são intrinsecamente anormais, ao serem transfundidos em receptores normais, apresentam um tempo de sobrevida curto característico. Dentre os defeitos intracorpusculares, somente um distúrbio – a hemoglobinúria paroxística noturna (HPN) – não é hereditário.
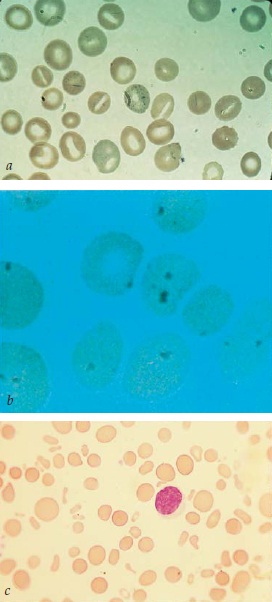
A hidrocitose é um distúrbio hereditário que costuma se manifestar nas primeiras fases da vida, sob a forma de uma anemia hemolítica compensada. Ocasionalmente, o baço é apalpável. O VCM em geral é alto. O esfregaço de sangue periférico contém estomatócitos [Figura 3]. Há aumento significativo do fluxo passivo tanto de Na+ como de K+. A Na+/K+ ATPase está inibida. A concentração de cátions e, portanto, o conteúdo de água das hemácias aumentam, provocando aumento do VCM e diminuição da proporção entre área de superfície e volume. Os estomatócitos parecem aderir com maior avidez do que as hemácias normais, e este achado pode ser responsável pelo aumento descrito do número de eventos tromboembólicos.8 Talvez de modo mais significativo, o número de hemácias com fosfatidilserina exposta na superfície da membrana está aumentado. A fosfatidilserina – um fosfolipídio com carga relativamente negativa e que normalmente predomina na camada interna da membrana – proporciona um foco de formação de trombina e, assim, também pode contribuir para a tendência ao desenvolvimento de trombose.9 A esplenectomia pode conduzir à melhora da anemia, porém os pacientes devem ser seguidos atentamente, dada a possibilidade de trombose pós-operatória.10 Outras terapias eventualmente podem se mostrar úteis. Em um paciente, os eventos vaso-oclusivos foram controlados com transfusão prolongada de hemácias, e, em outro, com terapia à base de pentoxifilina.8
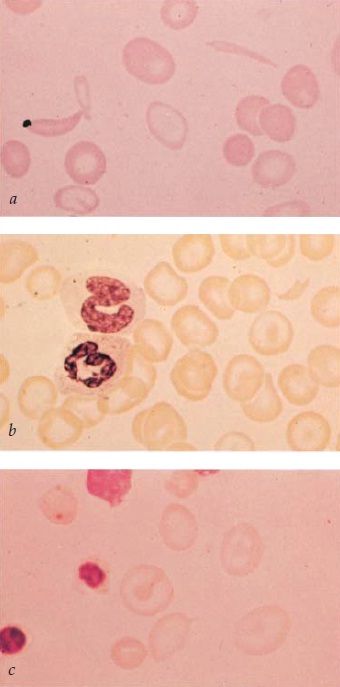
Figura 3. Os estomatócitos são identificados pela observação de áreas claras, semelhantes a fendas (a); o esfregaço também mostra microesferócitos, que correspondem a um estágio mais avançado da estomatocitose. Na varredura por microscopia eletrônica ou no exame de preparações a fresco, os microesferócitos são demonstrados como sendo estomatócitos. Os microesferócitos são observados na esferocitose hereditária e na anemia hemolítica autoimune, bem como em outras condições caracterizadas por uma perda relativamente seletiva de material de membrana ou por um aumento do volume celular. A coloração supravital dos eritrócitos (b) revela a presença de corpúsculos de Heinz corados em azul, isolados ou múltiplos, junto aos eritrócitos contracorados. A microscopia de fase pode ser utilizada para demonstrar os corpúsculos de Heinz. Os eliptócitos são visualizados em um esfregaço obtido de um paciente com eliptocitose (c).
A xenocitose, outro distúrbio hemolítico hereditário, é caracterizada por um defeito de membrana que acarreta perda de cátions, em particular de K+. Os eritrócitos sofrem desidratação porque o vazamento de K+ excede o influxo de Na+, possivelmente como resultado da ação de um cotransportador de K+/Cl- hiperativo. Os pacientes desenvolvem uma hemólise com graus variáveis de compensação. A esplenomegalia não constitui um aspecto proeminente. O esfregaço de sangue periférico é variável, apresentando células-alvo, estomatócitos, equinócitos ou as conhecidas “poças de hemoglobina” (isto é, hemoglobina acumulada em torno da circunferência da célula). A CHCM está aumentada. Como estas células rígidas são removidas em muitas partes do sistema reticuloendotelial, a esplenectomia tem pouca utilidade.11 Em raros casos, a xerocitose pode causar hidropsia fetal não imune.12
Em uma população de 1 milhão de indivíduos, talvez existam 250 a 500 casos de eliptocitose hereditária.11 Três variantes morfológicas foram observadas nesta condição: (1) eliptocitose hereditária comum; (2) eliptocitose hereditária esferocítica; e (3) eliptocitose hereditária estomatocítica.13 A maioria dos pacientes com eliptocitose hereditária comum são heterozigotos para este distúrbio autossômico dominante e apresentam apenas eritrócitos elípticos ou, na pior das hipóteses, uma hemólise compensada. Os indivíduos homozigotos para o distúrbio podem apresentar anemia hemolítica descompensada.
Diante da aplicação de um estresse de cisalhamento, os eritrócitos assumem uma conformação elíptica. Quando este estresse é retirado, a célula normalmente retrocede a seu formato discoide. Foi hipotetizado que os defeitos de membrana observados na eliptocitose hereditária interferem no retrocesso ao formato normal. O defeito de membrana parece ser uma lesão afetando o citoesqueleto da membrana. As membranas eritrocitárias de pacientes com eliptocitose hereditária são quase sempre mecanicamente frágeis.
O diagnóstico é estabelecido em casos de pacientes com hemólise intracorpuscular extravascular, que apresentam eliptócitos no esfregaço de sangue periférico. A eliptocitose também pode ser observada em casos de deficiência de ferro severa, distúrbios mieloproliferativos e mielodisplásicos e, às vezes, deficiências de cobalamina e folato.13 Os resultados do teste de fragilidade osmótica geralmente são normais. A esplenectomia tem sido útil em casos de pacientes com eliptocitose hereditária comum severa.
A síndrome hereditária (autossômica recessiva) da piropoiquilocitose, uma variante da eliptocitose hereditária, causa hemólise severa em crianças pequenas. A doença é produzida por uma espectrina alfa anormal ou por uma mutação envolvendo a espectrina beta. O esfregaço sanguíneo mostra a ocorrência de uma extrema microcitose e de uma extraordinária variação de tamanho e formato dos eritrócitos [Figura 3]. A esplenectomia pode diminuir a taxa de hemólise.
Em geral, a esferocitose hereditária é herdada como um traço autossômico dominante e afeta cerca de 220 indivíduos a cada 1 milhão de pessoas em todo o mundo. Foi descrita uma rara variante autossômica recessiva da esferocitose hereditária.14
Devido à perda de membrana de superfície, os eritrócitos assumem um formato microesferocítico e, assim, não conseguem se deformar o suficiente para passar pela vasculatura esplênica. Como consequência, há captura esplênica dos eritrócitos, hemólise e aumento compensatório da produção de hemácias. Os defeitos de membrana subjacentes levam ao brotamento de vesículas de membrana sob condições de depleção metabólica. Estas vesículas de membrana são enriquecidas com fosfolipídios oriundos da bicamada, assim como as proteínas transmembrânicas associadas [Figura 2]. As lesões moleculares subjacentes parecem consistir em deficiências de espectrina, espectrina-anquirina, faixa 3 e faixa 4,2 (paladina).13,15
Cerca de 25% dos pacientes com esferocitose hereditária apresentam hemólise totalmente compensada e sem anemia. Nestes pacientes, o distúrbio é diagnosticado somente diante da existência de uma condição concomitante (p. ex., infecção ou gestação) que aumenta a taxa de hemólise ou diminui a capacidade compensatória da medula. Em outros pacientes, pode haver desenvolvimento de uma anemia branda, cálculos pigmentados, úlceras na perna e ruptura esplênica. As crises aplásicas podem ser precipitadas por infecções comuns no trato respiratório, especialmente a infecção pelo parvovírus.7 É importante lembrar que esta doença pode se tornar evidente durante o 1º ano de vida, quando uma aumentada maturação esplênica com consequente remoção de eritrócitos e combinada a uma lenta resposta eritropoética pode produzir uma anemia severa o bastante para fazer o paciente necessitar de transfusão de hemácias.16
Este diagnóstico é sugerido pela predominância de microesferócitos no esfregaço periférico [Figura 3b], uma CHCM = 35 g/dL, reticulocitose, icterícia leve, esplenomegalia e história familiar positiva, ainda que pelo menos a metade dos pacientes recém-diagnosticados não possua história familiar. A confirmação do diagnóstico é feita por meio do teste de fragilidade osmótica com incubação de 24 horas. Um teste de Coombs negativo e história familiar positiva para esferocitose hereditária contam contra um diagnóstico de anemia hemolítica autoimune adquirida. Em casos de hemólise moderada a severa, deve ser feita a administração de folato (1 mg/dia). A esplenectomia erradica as manifestações clínicas do distúrbio, incluindo as crises aplásicas. Deve-se realizar uma colecistectomia concomitante, se forem observados sintomas de doença da vesícula biliar.
A HPN consiste em um distúrbio clonal somático de células-tronco hematopoéticas. A HPN envolve o gene PIG-A, que foi mapeado junto ao braço curto do cromossomo X.17 A mutação deste gene resulta na deficiência de uma proteína ancoradora de membrana, a fosfatidilinosil glicana de classe A. A maioria das células hematopoéticas maduras resultantes é quimérica. Os eritrócitos humanos normais – e provavelmente as plaquetas e os neutrófilos – modulam o ataque do complemento por pelo menos 3 proteínas GPI ligadas à membrana: DAF (CD55), proteína ligadora de C8 (C8BP) e MIRL (CD59). Na ausência da âncora de GPI, todas as proteínas que usam esta âncora de membrana tornam-se invariavelmente deficientes nas hemácias dos indivíduos com HPN.18 Como a síntese defeituosa de GPI afeta todas as células hematopoéticas, os pacientes com HPN podem apresentar graus variáveis de anemia, neutropenia ou trombocitopenia, ou podem desenvolver uma falência total de medula óssea.19
Classicamente, os episódios agudos de hemólise intravascular se sobrepõem a um fundo de hemólise crônica. O paciente tipicamente nota uma hemoglobinúria durante a micção, após ter dormido.20,21 As obstruções venosas recorrentes acarretam embolia, além de trombose hepática e mesentérica, possivelmente como resultado da liberação de micropartículas pró-coagulantes derivadas das plaquetas.22 Uma revisão da literatura demonstrou que os eventos trombóticos foram responsáveis por 22% dos casos de morte de pacientes com HPN.23 Ocasionalmente, a condição dos pacientes com HPN que sofrem trombose é confundida com distúrbios psicossomáticos, porque estes indivíduos se queixam de dores severas recorrentes no abdome e nas costas, sem causa evidente ou acompanhadas de impotência.24 Nestes casos, a anemia e a hemólise associadas podem ser bastante leves, sendo que os episódios de hemólise não necessariamente estão correlacionados aos sintomas.
Um diagnóstico de HPN deve ser considerado para qualquer paciente que apresente hemólise crônica ou episódica. O diagnóstico também deve ser considerado em casos de pacientes que apresentam tromboembolismo venoso recorrente, sobretudo quando o trombo se forma em um local como a veia cava inferior ou o sistema mesentérico portal, ou quando se produz a síndrome de Budd-Chiari. O diagnóstico é sugerido por evidências de hemólise intravascular, como hemoglobinemia, níveis séricos de haptoglobina diminuídos, níveis séricos de metemalbumina aumentados, hemoglobinúria ou hemossidenúria. A combinação de hipoplasia medular e hemólise constitui um indício importante. A HPN pode ocorrer associada à anemia aplásica, síndrome mielodisplásica ou outros distúrbios primários da medula óssea. A morfologia eritrocitária geralmente é normal. O diagnóstico é estabelecido por meio da realização de exames específicos baseados na análise de separação de células ativadas por fluorescência utilizando anticorpos que avaliam quantitativamente a DAF (CD55) e, particularmente, a MIRL (CD59), além de outras proteínas ligadas ao GPI nas hemácias ou na superfície de granulócitos.25 É importante testar mais de uma linhagem celular, especialmente no contexto das transfusões. As células totalmente deficientes de proteínas ligadas ao GPI são classificadas como células de HPN de tipo III, enquanto aquelas com deficiência parcial são classificadas como células de HPN de tipo II, e aquelas que apresentam expressão normal são as células de HPN de tipo I.24 As células de HPN de tipo III são mais suscetíveis à lise mediada pelo complemento do que as células de HPN de tipo II.26 O risco de trombose está correlacionado ao tamanho do clone de HPN entre os granulócitos.27
Na HPN, a anemia ocasionalmente é tão severa (níveis de hemoglobina < 8 g/dL) que o paciente necessita de transfusões regulares.21 Por isso, a escolha do componente a ser transfundido é decisiva. Acredita-se que a infusão de produtos do sangue contendo complemento pode intensificar a hemólise. A infusão de leucócitos doadores, que comumente estão presentes em uma unidade de hemácias concentradas, em um receptor imunizado contra o antígeno leucocitário humano (HLA – em inglês, human leukocyte antigen), pode provocar uma reação antígeno-anticorpo que ativa a via clássica do complemento. Neste caso, pode ser benéfico usar unidades especiais contendo poucos leucócitos [ver Medicina transfusional].
Um teste com prednisona (p. ex., 60 mg/dia com desmame rápido; ou 20 a 60 mg em dias intercalados) pode diminuir os requerimentos da transfusão, além de ser útil para aliviar a anemia de uma exacerbação aguda. Os benefícios proporcionados pela esplenectomia são bastante questionáveis. A cirurgia constitui um procedimento arriscado em casos de pacientes com HPN, porque a estase e o trauma acentuam a hemólise e a obstrução venosa. Se for necessário realizar uma cirurgia, deve ser considerada a instituição de uma anticoagulação profilática à base de varfarina durante o perioperatório.
Os pacientes com HPN frequentemente são deficientes de ferro. A simples administração de ferro para corrigir este defeito, todavia, muitas vezes agrava a hemólise, pois a terapia à base de ferro produz uma coorte de células novas, das quais uma ampla parte é de células suscetíveis à lise mediada pelo complemento. A realização da transfusão antes da instituição da terapia à base de ferro pode ajudar a contornar este problema, porque diminuirá a estimulação eritropoética sobre a medula. A administração de 5 mg/dia de folato é igualmente recomendada.24
O eculizumab é um anticorpo monoclonal que se liga ao componente C5 do complemento e inibe a ativação terminal deste sistema. Em um estudo de fase III, duplo-cego, randomizado e placebo-controlado, envolvendo pacientes com HPN que haviam necessitado de transfusões nos últimos 12 meses, os indivíduos tratados com eculizumab durante 26 semanas apresentaram requerimentos bem menores de transfusão de hemácias (em média, 0 unidades vs. 10 unidades); níveis diminuídos de lactato desidrogenase (DHL – de uma média de 2.199 U/L no momento basal a 327 U/L após 26 semanas); e melhora da qualidade de vida, em comparação ao observado no grupo tratado com placebo.28 O eculizumab também pode ter ação protetora contra a trombose.29 Os pacientes tratados com eculizumab devem ser vacinados contra Neisseria meningitidis.
A trombocitopenia resultante de uma produção precária de plaquetas pode requerer transfusões de plaquetas [ver Medicina transfusional].20 A síndrome de Budd-Chiari e a trombose da veia cava inferior devem ser diagnosticadas e tratadas rapidamente com heparina, seguida da administração de varfarina por tempo prolongado. Se a heparinização for ineficaz, pode ser utilizada uma terapia trombolítica (p. ex., estreptoquinase).38 Crianças e adolescentes com HPN complicada por anemia aplásica devem ser considerados para um possível transplante de medula óssea alogênica.21,31 Em relatos de caso, a anemia associada à HPN respondeu à eritropoetina,32 sendo que 4 pacientes com trombocitopenia e neutropenia severa responderam ao uso combinado de fator estimulador de colônias de granulócitos (G-CSF – em inglês, granulocyte colony-stimulating factor) e ciclosporina.33
Em um estudo envolvendo 80 pacientes com HPN, a média do tempo de sobrevida foi 10 anos.20 As causas das mortes associadas à HPN foram: trombocitopenia, hemólise da HPN, tromboses ou anemia aplásica associada à HPN [ver Anemia: defeitos de produção]. É interessante notar que 15% dos pacientes apresentaram remissão espontânea.20 Em casos raros, pode haver uma perda de ferro severa e prolongada, decorrente da hemossidenúria crônica, com consequente desenvolvimento de deficiência de ferro. Alguns pacientes desenvolvem hemocromatose associada à transfusão.21
A leucemia mieloide aguda pode se desenvolver ao longo do curso da HPN. Em uma série, foi isto que aconteceu em 3 dos 80 pacientes estudados. Em outra série, que envolveu 220 pacientes, a incidência de síndromes mielodisplásicas foi de 5%, e a incidência de leucemia aguda foi igual a 1%.21
O poder redutor do eritrócito é dado pela glutationa reduzida (GSH) e pelas coenzimas reduzidas nicotinamida adenina dinucleotídeo (NADH – em inglês, nicotinamide adenine dinucleotide) e fosfato de nicotinamida adenina dinucleotídeo (NADPH – em inglês, nicotinamide adenine dinucleotide phosphate) [Tabela 1]. Quando as reservas eritrocitárias destes materiais são inadequadas, a hemoglobina e as proteínas associadas à membrana podem ser oxidadas, com consequente produção de corpúsculos de Heinz, que, por sua vez, são predominantemente constituídos de produtos da degradação oxidativa da hemoglobina [Figura 3b]. Os eritrócitos que contêm corpúsculos de Heinz são rígidos e, portanto, seletivamente removidos pelo sistema reticuloendotelial.
As deficiências de certas enzimas envolvidas na síntese de GSH acarretam ataques oxidativos aos eritrócitos e hemólise. Vários relatos descreveram famílias cujos membros apresentavam uma síntese de GSH quase desprezível e hemólise associada à produção de corpúsculos de Heinz. A deficiência de glutationa peroxidase parece contribuir para a hemólise em recém-nascidos.
A glicose-6-fosfato desidrogenase (G6PD – em inglês, glucose-6-phosphate dehydrogenase) é a primeira enzima atuante na via da pentose fosfato, ou desvio da hexose monofosfato. Esta enzima catalisa a conversão de NADP+ em NADPH, que é um poderoso agente redutor. O NADPH é um cofator da glutationa redutase e, como tal, atua na proteção da célula contra o ataque oxidativo, reduzindo a glutationa oxidada formada na reação catalisada pela glutationa peroxidase. As hemácias deficientes de G6PD são, portanto, suscetíveis à oxidação e hemólise.34,35
A deficiência de G6PD constitui um dos distúrbios mais frequentes em todo o mundo. Cerca de 10% dos homens afrodescendentes que vivem nos Estados Unidos são afetados, assim como números significativos de africanos e alguns habitantes do litoral Mediterrâneo. Este distúrbio confere uma vantagem seletiva contra a malária endêmica. Exemplificando, em um estudo realizado em Gana envolvendo gestantes (que eram altamente suscetíveis à malária falcípara e suas consequências), a prevalência da infecção foi igual a 66% entre as mulheres normais, 58% entre as mulheres heterozigotas para G6PD, e 50% entre as mulheres homozigotas.36
O gene codificador da G6PD está localizado no cromossomo X, banda q28. Indivíduos do sexo masculino carregam apenas 1 gene para esta enzima, de modo que os homens afetados pelo distúrbio são hemizigotos. As mulheres são afetadas bem menos frequentemente, porque têm de carregar 2 genes de G6PD defeituosos para apresentarem uma doença clínica com a mesma severidade da doença que se manifesta nos homens. Entretanto, a expressão de um gene de G6PD defeituoso não é totalmente mascarada nas mulheres heterozigotas. De fato, estas mulheres exibem uma atividade enzimática de G6PD altamente variável. De acordo com a hipótese de inativação do X, ou hipótese de Lyon-Beutler, indivíduos do sexo feminino heterozigotos para G6PD apresentam 2 linhagens celulares: uma que contém um cromossomo X ativo com um gene codificador de G6PD normal e outra contendo um cromossomo X ativo com um gene determinante de deficiência de G6PD.34 O acaso determina parcialmente as proporções relativas destas 2 linhagens celulares que, por sua vez, controlam a severidade clínica do defeito.
A deficiência de G6PD é causa comum de icterícia neonatal no sexo masculino e pode acarretar uma complicação devastadora conhecida como kernicterus.37 Isto parece ser devido nem tanto ao grau de hemólise, mas principalmente à reduzida capacidade do fígado neonatal de conjugar a bilirrubina. O risco é maior diante da co-herança do polimorfismo do promotor do gene da UDP glicuronosil transferase 1, que leva ao desenvolvimento da síndrome de Gilbert.38
Existem 3 classes de deficiência de G6PD: classe I, que consiste em uma anemia hemolítica não esferocítica congênita crônica incomum; classe II, em que a deficiência enzimática é severa e a hemólise tende a ser episódica; e classe III, a variante mais comum, em que a deficiência enzimática é moderada e a hemólise é causada pelo ataque oxidativo. A severidade da hemólise e a anemia estão diretamente relacionadas à magnitude da deficiência enzimática. Esta, por sua vez, é determinada pela meia-vida da enzima. A meia-vida normal da G6PD é 62 dias. Na deficiência de G6PD de classe III, a enzima possui meia-vida de 13 dias, enquanto na deficiência de classe II, a meia-vida da G6PD é de várias horas. A clonagem e o sequenciamento do gene da G6PD esclareceram a classificação da deficiência de G6PD. Antes do sequenciamento deste gene, mais de 300 variantes de deficiência de G6PD haviam sido descritas.34
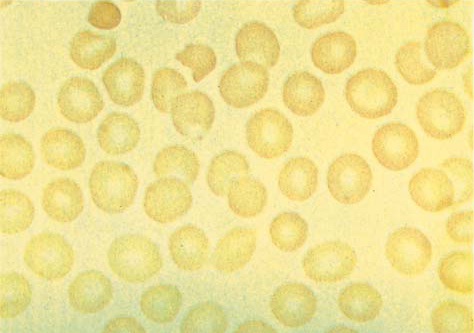
A hemólise ocorre em indivíduos com deficiência de G6PD classe III após a exposição a fármacos ou substâncias que produzem estresse oxidativo. A ingesta ou exposição aos feijões da fava pode causar uma hemólise intravascular devastadora (conhecida como favismo) em pacientes com deficiência de G6PD, no entanto esta reação geralmente ocorre apenas em indivíduos com a variante Mediterrânea da deficiência de classe II. A fava contém isouramil e divicina, que são 2 agentes fortemente redutores, cujas ações resultam na oxidação das proteínas de membrana. Como consequência, a célula torna-se rígida, e a hemoglobina fica confinada em uma porção do citosol. A outra parte do citosol exibe uma aparência de sombra clara (isto é, a clássica célula mordida, hemibolha ou de ligação cruzada) [Figura 4]. Estes defeitos de membrana produzem hemólise extra e intravascular.35 Também há relatos de casos de hemólise deflagrada por infecções severas, cetoacidose diabética e insuficiência renal.
Figura 4. Células mordidas, hemibolhas ou células de ligação cruzada são indicativas de ataque oxidativo, com consequente hemólise oxidativa.
A anemia hemolítica caracterizada pelo aparecimento de “células mordidas” e corpúsculos de Heinz após a administração de certos fármacos sugere a possibilidade de uma deficiência de G6PD [Tabela 2]. A dapsona, que é capaz de induzir hemólise do tipo oxidante, tem sido cada vez mais utilizada na profilaxia contra pneumonia por Pneumocystis carinii em pacientes infectados pelo HIV [ver HIV e Aids]. Desta forma, é importante submeter os potenciais usuários de dapsona a um rastreamento para detecção de deficiência de G6PD empregando os testes enzimáticos padrão. Outros agentes dotados de potencial oxidante, como o amil nitrito (“poppers”), podem causar hemólise.39
Tabela 2. Fármacos produtores de hemólise em pacientes com deficiência de G6PD
|
Classe |
Exemplo |
|
Antimaláricos |
Primaquina |
|
Cloroquina | |
|
Sulfonamidas |
Sulfametoxazol |
|
Sulfapiridina | |
|
Sulfonas |
Dapsona |
|
Analgésicos |
Acetanilida |
|
Fenacetina | |
|
Ácido acetilsalicílico (10 g/dia) | |
|
Nitrofuranas |
Nitrofurantoína |
|
Furazolidona | |
|
Derivados de vitamina K hidrossolúveis |
Menadiol |
G6PD = glucose-6-fosfato desidrogenase.
Outros distúrbios a serem considerados no diagnóstico diferencial da hemólise oxidativa são a hemoglobinopatia instável, a doença da hemoglobina M e deficiências de outras enzimas essenciais ao metabolismo da glutationa. Um teste para G6PD ou um ensaio enzimático geralmente resolvem esta questão. Pacientes com deficiência de G6PD de tipo A (deficiência de tipo III) e reticulocitose ativa, contudo, podem apresentar níveis de G6PD quase normais, porque suas hemácias jovens contêm níveis de G6PD relativamente altos. Nesses casos, é melhor repetir os testes quando a contagem de reticulócitos voltar ao normal. Informações sobre o teste genético para deficiência de G6PD podem ser encontradas na internet (www.geneclinics.org).
Evitar o uso de fármacos que possam produzir hemólise é uma medida decisiva no tratamento. O favismo agudo requer suporte circulatório, manutenção de um fluxo sanguíneo renal adequado e realização de transfusões de eritrócitos que não sejam deficientes de G6PD. O médico também deve estar alerta para o possível aparecimento de coagulação intravascular disseminada.
As reações em série que constituem a via glicolítica geram vários produtos, como o ATP, que exercem diversas funções essenciais no metabolismo eritrocitário [Tabela 1]. Os defeitos envolvem a principal via glicolítica (via de Embden-Meyerhof) e geralmente interferem na produção de ATP.
A piruvato quinase (PK) catalisa a formação de piruvato em uma reação associada à síntese de ATP. Depois da deficiência de G6PD, a deficiência de PK (autossômica recessiva) é a 2ª enzimopatia hereditária mais frequente. A hemólise, icterícia branda e, ocasionalmente, uma esplenomegalia palpável são os problemas apresentados pelo paciente. O exame do esfregaço de sangue periférico geralmente revela a presença de hemácias normais, porém há alguns casos em que as hemácias são extremamente espiculadas. Pode haver crises aplásicas.40
A ocorrência de hemólise não esferocítica congênita levanta a possibilidade de deficiência de PK. Um ensaio enzimático estabelece o diagnóstico. A esplenectomia deve ser considerada para pacientes que necessitam de transfusões.
A deficiência de glicose-6-fosfato isomerase constitui a 3ª enzimopatia mais comum a resultar em hemólise. Outras enzimopatias são bastante raras. Existem testes e ensaios específicos disponíveis para a detecção de deficiências de enzimas como hexoquinase, fosfofrutoquinase, triose fosfato isomerase, fosfoglicerato quinase e aldolase.
Na anemia hemolítica associada à deficiência de pirimidina 5’-nucleotidase, observa-se a persistência de um pontilhado basofílico grosseiro nos eritrócitos maduros, provavelmente porque a deficiência enzimática impede a degradação do RNA do reticulócito. Este acúmulo resulta na expansão do pool de nucleotídeos eritrocitários total, que atinge níveis 5 vezes maiores do que o nível normal. Os nucleotídeos pirimidínicos acumulam-se, enquanto a concentração de nucleotídeos de adenina diminui. A glicólise é comprometida por um mecanismo desconhecido.
As hemoglobinopatias de importância clínica são classificadas em 5 categorias, com base no defeito subjacente. Estes defeitos são:
1. Tendência da hemoglobina a virar gel ou sofrer cristalização (p. ex., anemia falciforme ou doença da hemoglobina C).
2. Instabilidade da hemoglobina (p. ex., anemias por corpúsculos de Heinz congênitas).
3. Hemoglobina com propriedades anormais de ligação ao oxigênio (p. ex., distúrbio causado pela hemoglobina de Chesapeake).
4. Pronta oxidação da hemoglobina em metemoglobina (p. ex., metemoglobinemia).
5. Diferentes taxas de síntese das cadeias da hemoglobina (p. ex., talassemias).
Definição. A anemia falciforme é uma doença autossômica recessiva causada pela substituição do aminoácido valina pela glutamina na 6ª posição da cadeia beta-hemoglobina, resultando na produção de HbS.
Epidemiologia. De 8 a 10% dos afro-americanos e um percentual menor de indivíduos descendentes de povos do leste do Mediterrâneo, Índia ou Arábia Saudita possuem o gene falciforme (HbS). A doença desenvolve-se em indivíduos homozigotos para este gene (HbSS), nos quais 70 a 98% da hemoglobina é do tipo S. Cerca de 0,2% dos afro-americanos têm anemia falciforme. O fato de o gene falciforme estar presente nas populações que vivem em regiões endêmicas de malária falcípara sugere que a heterozigose falciforme confere uma vantagem protetora contra a malária.41
As análises com endonuclease de restrição indicam que a mutação no gene falciforme provavelmente ocorre de maneira espontânea, pelo menos em 5 regiões geográficas. Estas variações são denominadas como sendo do Senegal, Benin, República da África Central (ou Bantu), Saudita-Asiática, Camarões e Índia (que pode ser idêntica à variante Saudita-Asiática). Estas variantes possuem importância clínica, porque algumas estão associadas a uma produção aumentada de cadeias gamaglobina (e, assim, a níveis maiores de HbF), enquanto outras estão mais frequentemente associadas à expressão de um gene determinante de alfatalassemia-2 [ver As talassemias, adiante]. Qualquer uma destas associações pode suavizar alguns aspectos do processo de falcização.41
Patofisiologia. Existem 2 aspectos clínicos principais que caracterizam a anemia falciforme: (1) hemólise crônica; e (2) crises vaso-oclusivas episódicas agudas, que causam insuficiência de órgãos e são responsáveis pela maior parte da morbidade e mortalidade associadas a esta doença.
A HbS ligada ao oxigênio ou monóxido de carbono apresenta solubilidade quase normal. Quando a molécula libera seu oxigênio e se transforma em desoxiemoglobina S, contudo, sua solubilidade diminui. Em um ambiente com níveis de oxigênio reduzidos, a HbS sofre polimerização e origina longas fibras semelhantes a tubos que induzem a falcização eritrocítica.42
O polímero da desoxi-hemoglobina S está em equilíbrio com as moléculas solúveis circundantes de desoxi-hemoglobina S. Um aumento da concentração de HbS, uma diminuição do pH ou a elevação da concentração de 2,3-DPG tendem a estabilizar a forma de desoxi-hemoglobina S e intensificar a gelificação.42 Além disso, os eritrócitos falciformes retêm a função de cotransporte de K+/Cl- e contêm cálcio intracelular em quantidade suficiente para ativar o canal de efluxo de Gardos43 [ver Controle da hidratação e volume, anteriormente]. Estes 2 mecanismos atuam juntos para produzir uma população de eritrócitos falciformes bastante densos, com CHCM que chegam a 50 g/dL.43 A HbF inibe a polimerização,43 de modo que os pacientes com valores altos de HbF (p. ex., indivíduos com a variante Saudita-Asiática da anemia falciforme) desenvolvem uma doença mais branda.41 Quando a hipoxemia e a CHCM atingem níveis críticos, a polimerização passa a ocorrer após um período variável de atraso.43 Este atraso representa o período em que os tetrâmeros de desoxi-hemoglobina S se associam lentamente para formar um núcleo. Quando o núcleo atinge um tamanho crítico, ocorre uma gelificação rápida, quase explosiva. Os tetrâmeros de desoxi-hemoglobina S livres rapidamente se fixam ao núcleo para produzir as fibras longas semelhantes a tubos que, por sua vez, alinham-se para formar estruturas semelhantes a tubos que torcem a célula e lhe conferem um formato de foice [Figura 5].
Figura 5. A anemia falciforme é caracterizada pela presença de células falciformes acentuadamente torcidas, entre as quais são encontradas formas alongadas (a). As células-alvo (b) são observadas em diversas condições, como hipocromia decorrente da deficiência de ferro, hemoglobinopatias (p. ex., variantes de HbC) e talassemias, além da doença hepática. A anemia de Cooley (c), ou betatalassemia major, é indicada por uma profunda hipocromia, alvejamento, variação de tamanho e forma dos eritrócitos, bem como presença de hemácias nucleadas.
A maioria das células encontradas na circulação venosa não é falciforme. Entretanto, a falcização ocorre no momento em que a polimerização é abreviada para menos de 1 segundo ou quando as hemácias são capturadas junto à microcirculação. Algumas hemácias contêm hemoglobina falciforme polimerizada mesmo estando na circulação arterial. Outra manifestação do dano à membrana nas células falciformes consiste na célula irreversivelmente falciforme, que retém o formato de foice mesmo quando é reoxigenada.44 Algumas destas hemácias pouco deformáveis derivam diretamente de uma subpopulação de reticulócitos pobres em HbF44 e são removidas de maneira predominante no sistema reticuloendotelial. A rápida remoção destas células jovens, assim como das células rígidas, densas e mais velhas que são incapazes de atravessar o sistema de monócitos-macrófagos, resulta em hemólise extravascular crônica.
Devido à extrema sensibilidade da falcização ao ambiente local, têm sido enfocados os fatores celulares. A extrema hiperosmolaridade da medula renal (1.200 mOsm) desidrata as hemácias e eleva a CHCM. Em consequência, uma falcização suficiente para anular a capacidade de concentração da medula renal pode ser observada até mesmo em pacientes que apresentam apenas o traço falciforme.
Foi proposto que a hemólise intravascular resulta na presença de quantidades aumentadas de hemoglobina livre no plasma. Esta, por sua vez, captura e remove óxido nítrico (NO). A redução do NO leva à diminuição da vasodilatação, podendo ampliar ainda mais os problemas de hipertensão pulmonar, priapismo e, talvez, acidente vascular cerebral (AVC).45
Crise falciforme e infarto isquêmico. A crise falciforme constitui uma complicação vaso-oclusiva da anemia falciforme, que é potencialmente prejudicial à vida. O evento iniciador da crise falciforme é desconhecido, e também não se sabe por que apenas alguns pacientes apresentam crises severas.
Aglomerados de células falciformes cada vez mais rígidas acabam obstruindo a microvasculatura, nas seguintes circunstâncias: (1) queda de pH, aumento da desoxigenação ou elevação da CHCM; (2) diminuição da produção de NO ou captura e remoção do NO pela hemoglobina livre existente no plasma;46 (3) existência de doença microvascular; ou (4) prolongamento do tempo de trânsito capilar. A trombose também pode exercer um papel na oclusão falciforme. Ocorre certo grau de desorganização da bicamada fosfolipídica da membrana, com a fosfatidilserina se movendo para o folheto externo e possivelmente intensificando as manifestações tromboembólicas da anemia falciforme.47 Na anemia falciforme, também parece haver um aumento do número de células endoteliais circulantes, que apresentam expressão anormal de fator tecidual e podem fornecer uma base adicional ao tromboembolismo.48
O bloqueio acarreta infarto isquêmico, liberação de citocinas inflamatórias e uma sequência de amplificação da obstrução estase-induzida, que pode progredir para crise falciforme. As circulações portais onde a tensão de oxigênio é baixa, como no fígado ou nos rins, estão particularmente expostas ao risco de sofrer obstrução.
Os fatores de risco predisponentes ao desenvolvimento de crises dolorosas incluem níveis de hemoglobina acima de 8,5 g/dL, gestação, tempo frio e uma contagem de reticulócitos elevada. A hipoxemia noturna representa um fator risco importante em crianças.49 Contudo, o hematócrito baixo observado na anemia falciforme diminui a viscosidade do sangue e tem ação protetora. Os pacientes com anemia falciforme também apresentam níveis plasmáticos de fibrinogênio caracteristicamente altos. Isto intensifica a agregação dos eritrócitos já rígidos e aumenta a viscosidade do sangue, sobretudo diante das baixas velocidades de cisalhamento encontradas junto à microcirculação.50 Quando comparadas às hemácias normais, as hemácias falciformes também tendem mais a aderir às células endoteliais.51 O papel dos leucócitos neste processo de adesão está se tornando mais nítido. A administração de G-CSF provocou crises falciformes e até a morte de pacientes.52,53 O fator estimulador de colônias de granulócitos-macrófagos (GM-CSF – em inglês, granulocyte-macrophage colony-stimulating factor) tem causado crises similares. A severidade da anemia falciforme parece ser paralela ao nível de contagem de leucócitos, enquanto as moléculas de adesão celular leucocitárias parecem ser essenciais à ocorrência da vaso-oclusão falciforme.54,55
Diagnóstico da anemia falciforme. Antigamente, o diagnóstico de anemia falciforme costumava ser estabelecido com base nas manifestações clínicas ocorridas durante a infância. A criança afetada apresentava limitações de tolerância ao exercício, falta de ar, taquicardia, infecções severas frequentes e episódios de dactilia bastante dolorosos. Atualmente, muitos casos são identificados por meio de testes de rastreamento, que podem ser prontamente indicados pelo diagnóstico da condição em um familiar ou realizados como procedimento de rotina neonatal. Na Califórnia, assim como em muitos estados norte-americanos, todas as amostras de sangue de cordão umbilical fetal são examinadas por cromatografia líquida de alto desempenho (HPLC – em inglês, high-performance liquid chromatography). Em raros casos, o distúrbio é diagnosticado na fase adulta, às vezes durante a 1ª gestação, quando os exames de pré-natal revelam a existência de anemia. Os sintomas gerais são: tolerância limitada ao exercício, dispneia por esforço, crises dolorosas, ataques de icterícia e até mesmo cólica biliar.
O aspecto clínico do paciente e um esfregaço sanguíneo mostrando células falciformes, células em formato de folha de azevinho e eritrócitos contendo corpúsculo de Howell-Jolly são bastante sugestivos de anemia falciforme. Os corpúsculos de Jowell-Jolly representam os resquícios de cromatina nuclear que normalmente são removidos pelo baço. As contagens de plaquetas e leucócitos em geral estão altas. Exceto diante de uma crise aplásica em progressão que esteja levando praticamente à ausência de normoblastos, a medula apresenta hiperplasia eritroide. O diagnóstico é confirmado por meio do exame de uma preparação falciforme: uma gota de sangue é incubada com metabissulfito de sódio a 2% e a proporção de células falciformes é medida imediatamente e também após 1 hora. Kits de teste comerciais, como o Sickledex, baseiam-se na relativa insolubilidade da HbS em tampões fosfato 1 M para fornecer o diagnóstico. Os testes mais definitivos para anemia falciforme, todavia, são a eletroforese de hemoglobina ou o HPLC, que indicam os percentuais relativos de HbS e HbF. Todos estes testes também são úteis na avaliação de familiares para detecção do traço da célula falciforme. Pacientes heterozigotos para os genes da HbS e da betatalassemia podem parecer serem homozigotos para HbS. Outras variedades de hemoglobina falciforme são observadas muito raramente. Os métodos baseados em análise de DNA também podem ser utilizados para apontar a anomalia genética específica e identificar as subpopulações das quais o paciente é descendente.41 Descrições e informações adicionais sobre exames diagnósticos são disponibilizadas no website www.geneclinics.org. Indivíduos com anemia falciforme e alfatalassemia apresentam níveis maiores de hemoglobina, contagens de reticulócitos mais baixas, CHCM menor, VCM menor e hemácias menos densas do que os indivíduos que sofrem apenas de anemia falciforme. Tais pacientes podem apresentar expectativa de vida maior e, talvez, um padrão diferente de manifestações de complicações veno-oclusivas.56 A combinação de deficiência de G6PD e anemia falciforme produz resultados que não são benéficos nem prejudiciais.57,58
Crise falciforme. O tratamento conservador padrão da crise falciforme concentra-se em repouso, hidratação e analgesia. Em pacientes com acidose demonstrável, deve-se induzir uma leve alcalinização por meio da administração de uma solução de bicarbonato. Esta solução é preparada pela adição de uma ampola de bicarbonato de sódio a 1 L de solução de dextrose a 5% em água ou solução salina hipotônica (NaCl 0,45%). A solução de bicarbonato deve ser infundida a uma velocidade de 5 a 7 mL/kg/h durante as primeiras 4 horas, e a 4 mL/kg/h durante as próximas 20 horas. Ainda não foi avaliado o papel do oxigênio suplementar em casos de pacientes com tensão de oxigênio arterial (PaO2) normal e sem problemas cardiopulmonares.
Tratamento da dor. A dor [ver Dor] é a principal preocupação de 10 a 20% dos pacientes que sofrem de anemia falciforme. A necrose avascular da medula óssea produz uma dor aflitiva que pode durar 8 a 10 dias. A necessidade de aliviar a dor às vezes resulta em habituação ou vício, embora isto seja relativamente incomum.
O paciente com anemia falciforme deve receber analgésicos orais para usar em casa, na tentativa de eliminar a crise de dor no momento em que surgir. Os fármacos anti-inflamatórios não hormonais (AINH), como o naproxeno (500 mg) e o cetorolaco (10 mg), podem ser utilizados no início. Se o uso isolado dos AINH não for suficiente, é possível instituir o uso subsequente de uma combinação narcótica-analgésica, como a hidrocodona e o acetaminofeno ou a oxicodona e a aspirina. O cetorolaco pode precipitar a insuficiência renal59 e deve ser utilizado com cautela em casos de pacientes com doença renal preexistente ou em estado de desidratação. Adjuvantes, como a difenidramina oral (50 mg) ou o lorazepam (1 a 2 mg) podem acalmar o paciente e, talvez, antagonizar as ações da histamina liberada.60 Quando tais ações (talvez repetidas a cada 6 horas) não controlam a dor, é comum o paciente acabar necessitando de tratamento parenteral. O fornecimento de tratamento pelo médico habitual do paciente é certamente preferível a ter que contar com prestadores de assistência pouco familiares em departamentos de emergência.60 O paciente precisa passar por uma rápida avaliação para detecção de uma possível infecção, síndrome torácica aguda, infarto ósseo e outras complicações. A dor severa deve ser considerada uma emergência médica,61 sendo essencial a rápida instituição de uma terapia à base de opiáceos. É importante observar o regime de opiáceos crônico do paciente, bem como as doses de opiáceos que foram previamente efetivas no tratamento de dores moderadas a severas. Uma dose de carga inicial de 5 a 10 mg de morfina endovenosa ou uma dose equivalente de um opiáceo alternativo podem ser utilizadas no tratamento de dores moderadas a severas de pacientes opiáceos-naive com peso corporal mínimo de 50 kg.61,62 Caso não haja alívio da dor ou o alívio da dor seja inadequado decorridos 30 minutos da administração da 1ª dose, podem ser administrados 50% da dose inicial de opiáceos. Outras doses adicionais, administradas a cada 15 a 30 minutos, devem ser tituladas para obtenção de uma analgesia adequada. A frequência respiratória deve ser monitorada atentamente, em particular se estiver próxima de 10 respirações/minuto. Algumas unidades que têm utilizado a analgesia controlada pelo paciente vêm obtendo bons resultados. É importante continuar a administrar a analgesia parenteral a intervalos regulares e fornecer doses maiores em caso de dores adicionais. O paciente provavelmente precisará de um laxante e poderá necessitar de um antiemético, como a proclorperazina (10 mg, por via oral ou intramuscular). Quando o paciente responde, a terapia realizada em casa com morfina oral de liberação controlada costuma ser efetiva. Se a dor persistir por mais de 8 a 12 horas, o paciente provavelmente terá de ser internado para receber tratamento prolongado com doses maiores de analgesia e líquidos parenterais, aliado à observação.60 O uso do opiáceo meperidina é contraindicado para pacientes com insuficiência renal ou história de convulsões, devido ao acúmulo de seu metabólito tóxico – a normeperidina63 – e, em geral, não é adotado de forma rotineira no tratamento da dor associada à anemia falciforme.
Alteração da patofisiologia da célula falciforme. Uma compreensão mais nítida acerca da cinética da falcização sugere algumas perspectivas futuras para a terapia da anemia falciforme. A diminuição da CHCM deve reduzir a gelificação. Uma abordagem que tenta bloquear o efluxo de K+ dependente de Ca2+ (canal de Gardos) [ver Controle da hidratação e volume, anteriormente] foi testada em um modelo experimental murino de doença falciforme e mostrou-se promissora em termos de prevenção da desidratação das hemácias.64,65 Em um estudo de fase II de um bloqueador de canal de Gardos, o Senicapoc, envolvendo indivíduos adultos com doença da HbSS, aqueles que foram tratados com o fármaco apresentaram aumento significativo dos níveis de Hb (de 0,68 g/dL), bem como diminuição da contagem de reticulócitos e dos níveis de DHL e bilirrubina indireta, embora os grupos de tratamento não tenham diferido quanto ao número de crises de dor apresentadas pelos pacientes.66
Terapias destinadas a interferir na falcização estão sendo ativamente buscadas. A presença de 20 a 30% de HbF nas hemácias falciformes retarda acentuadamente a gelificação, do mesmo modo como agiria um mecanismo que ligasse os genes controladores da síntese de hemoglobina fetal. Isso faz a amenização da severidade da anemia falciforme parecer viável.67,68 A hidroxiureia produz um aumento dos níveis de reticulócito F e HbF. Em um estudo de fase III, os pacientes tratados com hidroxiureia (dose inicial de 15 mg/kg/dia) apresentaram menos crises dolorosas, menos admissões hospitalares em decorrência das crises e menos episódios de síndrome torácica aguda, além de terem necessitado de menos transfusões do que os pacientes que receberam placebo.69 O tratamento não teve efeitos sobre a ocorrência de AVC. No entanto, após 8 anos de seguimento, a mortalidade foi reduzida em 40%.70 O efeito benéfico da hidroxiureia adveio depois de cerca de 8 semanas de terapia e foi acompanhado de aumento do VCM e da proporção de células F. Além disso, houve uma diminuição do número de neutrófilos e da adesão das hemácias falciformes às células endoteliais.71 A hidroxiureia causa uma diminuição dose-associada e reversível das contagens sanguíneas, que deve ser monitorada regularmente ao longo do tratamento. Embora existam relatos de caso de desenvolvimento de leucemia em pacientes com anemia falciforme tratados com hidroxiureia,72,73 o seguimento prolongado de adultos70 e crianças74 não demonstrou a existência de um risco aumentado de leucemia. Diante da existência de aspectos preocupantes quanto a uma possível teratogenicidade, recomenda-se que homens e mulheres sob tratamento com hidroxiureia adotem métodos anticoncepcionais.75
Também estão sendo realizados estudos com butirato, que é capaz de aumentar a produção de cadeias gama e, assim, elevar os níveis de HbF e interferir na gelificação.76,77 Os agentes hipometiladores, como 5-azacitidina e decitabina, também podem aumentar os níveis de HbF a valores terapeuticamente úteis. Como as células falciformes aderem de maneira anormal ao endotélio, foram realizadas tentativas de bloquear a adesão. Até agora, estes esforços ainda não se mostraram produtivos.
As citocinas inflamatórias parecem exercer um papel importante na crise falciforme, e isto é evidenciado pelo fato de um preditor de sucesso da terapia com hidroxiureia ser a ocorrência de diminuição da contagem de leucócitos.70,71 Outros pesquisadores estão estudando o possível papel vasodilatador do NO.
O transplante de medula óssea alogênica de doador irmão pode resultar em cura ou levar à substituição do traço falciforme pela anemia falciforme. Costuma-se reservar o transplante para casos de doença severa, e sua indicação inclui as crises vaso-oclusivas frequentes e persistentes e/ou a síndrome torácica aguda, mesmo com a terapia à base de hidroxiureia, bem como resultados anormais de exames de Doppler transcranianos, mesmo com terapia de transfusão crônica. Na maior série de transplantes realizada até hoje, envolvendo 87 pacientes com HbSS ou HbS/beta-0 com idades entre 2 e 22 anos, a maioria dos quais recebeu enxertos de doador irmão de HLA idêntico, a sobrevida de 5 anos livre de eventos foi de 86%,78 de modo similar às taxas observadas em outros estudos.79 Houve 6 casos de morte associada ao transplante primariamente atribuíveis à doença do enxerto vs. hospedeiro (DEVH), além de 6 casos de rejeição, dos quais 4 ocorreram antes da adição de globulina antitimócito ao regime de condicionamento. Convulsões associadas ao transplante foram observadas em 16 (24%) pacientes, sendo que 7 pacientes desenvolveram leucoencefalopatia posterior reversível atribuível à toxicidade da ciclosporina; 20% dos pacientes desenvolveram DEVH aguda de grau II ou maior; e a incidência cumulativa da DEVH crônica foi igual a 13%. O uso do transplante é limitado em função da disponibilidade de doadores irmãos compatíveis. A limitada experiência com transfusão de sangue de cordão existente até o momento é encorajadora.78
Terapia transfusional prolongada. Foi demonstrado que esta terapia previne o AVC.80 Alguns pesquisadores demonstraram que as transfusões preventivas diminuem ou eliminam as crises de dor, episódios de síndrome torácica aguda, infecção bacteriana e internação.81 Outros autores, contudo, alertam quanto aos perigos da sobrecarga de ferro,82,83 hepatite associada ao transplante, problemas com acesso venoso e aloimunização eritrocitária.84 Estudos adicionais podem esclarecer o papel da terapia transfusional prolongada.
Problemas esqueléticos. A necrose asséptica (osteonecrose) da cabeça do femur ocorre em cerca de 10% dos pacientes, particularmente naqueles que também sofrem de alfatalassemia. A artroplastia tem sido um procedimento relativamente inefetivo, em parte devido à existência de osso rígido adjacente, que interfere na colocação da prótese, e também por causa do risco aumentado de infecção.85
Problemas cardíacos. As complicações cardíacas associadas à anemia resultam do aumento significativo do débito cardíaco. Estas complicações incluem o aumento das câmaras, cardiomegalia, hipertrofia ventricular esquerda e murmúrios de fluxo.86 O infarto do miocárdio agudo ocorre em indivíduos adultos relativamente jovens e sem doença coronariana.87
Problemas pulmonares. As complicações pulmonares agudas constituem a principal causa de morbidade e mortalidade. Estas complicações incluem infecção local, obstruções vasculares em vasos pulmonares (trombose in situ e embolia) e embolia gordurosa pulmonar a partir da necrose gordurosa da medula isquêmica.88 Um amplo estudo sobre a síndrome torácica aguda constatou que os pacientes adultos eram afebris, mas apresentavam falta de ar, calafrios e dor no tórax, bem como dor em pelo um dos membros.89 Os infartos das vértebras torácicas contribuem de maneira significativa para a manifestação da dor.90 O exame físico frequentemente não revela achados torácicos anormais. Em um estudo, constatou-se que a PaO2 estava baixa, atingindo em média 71 mmHg, porém caindo a níveis abaixo de 60 mmHg em 25% dos pacientes.89 Neste estudo, a taxa de morte entre adultos foi de 4,3%. A morte foi precedida de valores mais baixos de hemoglobina, contagens mais altas de leucócitos e envolvimento multilobar. A autópsia de 16 casos mostrou que 9 pacientes tiveram embolia pulmonar e formação de êmbolos fatais, sendo que possivelmente 20% apresentavam infecções bacterianas. Em pacientes com síndrome torácica aguda e infecção pulmonar, o organismo infeccioso mais comumente encontrado foi Chlamydia pneumoniae (30%), seguida por Mycoplasma pneumoniae (21%), vírus sincicial respiratório (10%), Staphylococcus aureus (4%) e Streptococcus pneumoniae (3%).91
Geralmente, a terapia para casos de síndrome torácica aguda deve incluir a espirometria de incentivo,90 terapia antimicrobiana para pacientes com evidências de infecção, analgesia, reposição de líquidos para euvolemia e consideração da possibilidade de realizar um lavado broncoalveolar para identificar infecções microbianas ou detectar macrófagos repletos de gordura em êmbolos gordurosos. É necessário realizar um monitoramento meticuloso. Devem ser realizadas medições repetidas da oxigenação e, quando clinicamente necessário, transfusões. Se uma transfusão simples não proporcionar benefícios, a exsanguíneo transfusão deve ser realizada para diminuir o percentual de HbS para menos de 30% e manter os níveis de Hb total < 10 g/dL.92 Um dos benefícios mais importantes da terapia com hidroxiureia reside em sua habilidade de diminuir a frequência da síndrome torácica aguda.69,93 As crianças também podem precisar de suplementação profilática de penicilina.94
A hipertensão pulmonar, definida como uma velocidade de jato regurgitante tricúspide = 2,5 m/s ou uma pressão arterial pulmonar sistólica = 35 mmHg, ocorre em 30 a 40% dos pacientes adultos com HbSS ou HbS/beta-0.95,96 A hipertensão pulmonar está associada a uma mortalidade aumentada.95,97 Em um estudo, a hipertensão pulmonar foi a causa da morte de 1/4 dos pacientes com anemia falciforme avaliados.98 Dada a alta prevalência e mortalidade associada, alguns especialistas recomendam que todos os pacientes sejam submetidos a um rastreamento com ecocardiografia.99 Os níveis plasmáticos de pró-peptídeo natriurético cerebral N-terminal também podem ser úteis como ferramenta de estudo. Em um estudo, níveis acima de 160 pg/mL apresentaram um valor preditivo de 78% para a hipertensão pulmonar.100 A avaliação para detecção de hipertensão pulmonar deve ser realizada quando o paciente estiver clinicamente estável, uma vez que as pressões pulmonares podem subir durante as crises de dor.101 A avaliação da hipertensão pulmonar em pacientes com anemia falciforme deve seguir a avaliação padrão para hipertensão pulmonar em pacientes sem anemia falciforme,99 e devem ser investigadas as causas adicionais de hipertensão pulmonar. Estão sendo conduzidos estudos para determinar o tratamento ideal da hipertensão pulmonar na anemia falciforme.
Doença hepatobiliar. A colelitíase ocorre em 30 a 70% dos pacientes, alguns dos quais exibem sinais e sintomas de colecistite.102 Existem dados conflitantes sobre a frequência da colecistite ou obstrução do ducto biliar comum.102,103 Se for necessário realizar uma colecistectomia, será preciso esperar a crise dolorosa terminar. Antes da cirurgia, devem ser realizadas transfusões para elevar os níveis de Hb para 10 g/dL, quando necessário, e o procedimento deve ser realizado por laparoscopia.102
As complicações hepáticas incluem hepatopatia congestiva secundária à insuficiência cardíaca e hepatite viral decorrente de transfusões frequentes. A ocorrência falcização no fígado também pode produzir hepatopatia. Os níveis séricos de bilirrubina frequentemente excedem 30 mg/dL em pacientes com colestase intra-hepática, sendo que as anomalias de coagulação podem levar ao desenvolvimento de complicações hemorrágicas e morte.
Complicações renais e urológicas. A perda de água como resultado de uma incapacidade de concentrar a urina pode intensificar o processo de falcização. O milieu extremamente hipertônico da medula renal induz um severo processo de falcização e destruição dos vasos retos. Têm lugar a hematúria e a necrose papilar. Estas complicações também são observadas em pacientes com traço falciforme e naqueles com anemia falciforme/doença da hemoglobina C. O defeito que afeta a capacidade de concentração renal parece ser dependente da quantidade de polímero HbS contidos nas células e, portanto, é menos severo em pacientes que também apresentam variantes de alfatalassemia.104
As complicações incluem acidose tubular renal, hipercalemia e proteinúria. Em um estudo, 42% dos pacientes com HbSS apresentaram albuminúria microscópica (definida como níveis na faixa de 30 a 299 mg/g de creatinina), e 26% tinham albuminúria macroscópica (definida como níveis de albuminúria = 300 mg/g de creatinina).105 O tratamento com enalapril diminui a proteinúria, sugerindo a existência de um componente de hipertensão capilar glomerular.106 A insuficiência renal, que está associada à piora da anemia, contribui para a morte de cerca de 1/5 dos pacientes com mais de 40 anos de idade homozigotos para anemia falciforme.
O priapismo constitui uma complicação extraordinariamente dolorosa da anemia falciforme e pode resultar em impotência.107 Um estudo realizado no Reino Unido relatou uma resposta satisfatória em 13 dentre 18 pacientes tratados com etilefrina (um agonista alfa-adrenérgico) para priapismo. No entanto, este agente não é disponibilizado para uso nos Estados Unidos.108
Distúrbios neurológicos. As complicações neurológicas da anemia falciforme são o AVC, a hemorragia subaracnóidea e perdas funcionais isoladas sugestivas de uma obstrução focal. A patogênese da obstrução de grandes artérias cerebrais provavelmente é diferente daquela associada aos eventos oclusivos microvasculares que ocorrem nos leitos capilares hipóxicos. As causas subjacentes mais prováveis são o dano ao endotélio vascular, seguido de uma extensiva proliferação da íntima e, então, trombose junto ao leito vascular danificado.56 Em um estudo multi-institucional envolvendo 4.082 pacientes, a prevalência dos AVC foi de 4 a 5%, com uma incidência de 0,61 a cada 100 pacientes-anos.109 Dentre os AVC, 54% foram infartos, 34% tiveram natureza hemorrágica, 11% foram ataques isquêmicos transitórios e 1% apresentaram aspectos de infarto e hemorragia. Dentre os pacientes sobreviventes, a taxa de recidivas de AVC foi igual a 14%. A mortalidade foi de 11%. Quase todos os pacientes que morreram tiveram AVC hemorrágicos.
Em um estudo prospectivo, que utilizou ultrassonografia com Doppler transcraniana para apontar crianças que apresentavam risco de sofrer AVC, o tratamento com terapia-padrão ou terapia transfusional (para diminuir a concentração de HbS < 30%) resultou na ocorrência de apenas 1 caso de AVC, em comparação aos 10 casos de AVC e 1 caso de hematoma intracerebral observados no grupo de controle (p < 0,002).80 O estudo foi encerrado precocemente. Em outro estudo de seguimento, crianças cujos exames de Doppler transcraniano reverteram para a normalidade foram randomizadas para continuar ou parar de receber transfusões. No grupo de 41 crianças randomizadas para interromper as transfusões, 14 apresentaram sonogramas de Doppler anormais, e 2 desenvolveram AVC, enquanto aquelas que continuaram recebendo transfusão não apresentaram nenhum evento.110 Este estudo também foi encerrado precocemente. É importante notar que os pacientes randomizados para prosseguirem com as transfusões apresentaram níveis médios de ferritina da ordem de 3.562 ng/mL, apesar da quelação com deferoxamina. Estes resultados levantaram muitas questões sérias acerca da frequência ideal de avaliações com Doppler transcraniano; duração ideal da terapia transfusional; consequências inevitáveis da hemocromatose transfusional [ver Betatalassemia major (anemia de Cooley), adiante] e a necessidade de sangue eticamente compatível para minimizar a reação de alotransfusão; disposição do paciente e seus familiares em aceitar a terapia transfusional; e o papel do transplante de medula óssea alogênica como possível alternativa.80,111 O risco de eventos cerebrovasculares é maior em pacientes submetidos à terapia transfusional prolongada que têm múltiplos vasos colaterais cerebrais resultantes da doença moyamoya (taxa de risco = 2,4).112
Complicações oculares. Os principais problemas oculares associados à anemia falciforme são a retinopatia, a hemorragia vítrea e a neovascularização. Recomenda-se a realização de avaliações oftalmológicas anuais. Atualmente, está sendo investigada a eficácia da fotocoagulação a laser no tratamento das alterações oculares induzidas pelas células falciformes.
Complicações dermatológicas. Úlceras mal cicatrizadas na perna podem ser causa importante de morbidade entre pacientes com anemia falciforme. O grau de anemia não parece estar correlacionado à presença ou severidade destas úlceras, mas a incompetência das válvulas venosas e a resultante insuficiência venosa foram associadas à ulceração.113 O tratamento-padrão inclui desbridamento, controle da infecção local, uso de curativos do tipo wet-dry (umedecidos em salina e deixados secar para serem removidos) e, possivelmente, transfusão de hemácias. O tratamento local com GM-CSF intensifica a cicatrização, talvez por estimular o desenvolvimento local de macrófagos.114 O GM-CSF pode ser tanto injetado perilesionalmente como aplicado topicamente na ferida, contudo o método de aplicação mais bem-sucedido envolve a injeção subcutânea de 100 mcg de GM-CSF em 4 sítios estabelecidos circunferencialmente em torno da úlcera, a uma distância de 5 mm de sua borda (resultando na aplicação de uma dose total de 400 mcg na ferida). Em alguns casos 1 tratamento foi suficiente, enquanto em outros foi necessário instituir tratamentos semanais por um período de 4 a 12 semanas. Esta terapia não teve o uso aprovado pelo Food and Drug Administration.
Crise aplásica. A crise aplásica diminui rapidamente os níveis de hemoglobina e hematócrito, além de produzir reticulocitopenia, do mesmo modo como atua em qualquer estado hemolítico crônico. Constatou-se que a infecção pelo parvovírus, assim como a necrose de medula óssea, causa crise aplásica.115
Suscetibilidade a infecções. Pacientes com anemia falciforme são hipoesplênicos e exibem anormalidades de sistema complemento. Uma atividade opsonizadora sérica deficiente para micro-organismos de Salmonella pode conferir maior suscetibilidade a infecções, inclusive ao desenvolvimento de osteomielite.
Complicações da anestesia. A hipoxemia e a estase vascular que podem ocorrer durante a anestesia geral intensificam a falcização e podem acarretar crise falciforme durante o período pós-operatório. Em uma análise de quase 4.000 pacientes, 12 mortes estavam associadas à realização de 1.079 procedimentos, sendo que houve mais complicações após a aplicação de anestesia regional do que subsequentemente à anestesia geral.116 Um programa simples de transfusão para elevar os níveis de hemoglobina para 10 g/dL foi tão eficaz quanto os programas pré-operatórios mais agressivos, em termos de redução da taxa de complicações.117
Gestação e contracepção. Os riscos da gestação para mulheres com anemia falciforme incluem problemas pulmonares e uma incidência aumentada de infecção no trato urinário, hematúria, pré-eclâmpsia e morte materna. Provavelmente, a hipoxemia pélvica e a sobrecarga vascular associadas à gestação levam à intensificação da falcização, com as consequentes complicações associadas. A vaso-oclusão placentária pode ser responsável pela morte fetal e pelo baixo peso ao nascimento.
Os clínicos experientes diferem quanto ao modo de abordar as pacientes gestantes com anemia falciforme. Alguns defendem apenas a instituição de um meticuloso tratamento conservador, enquanto outros recomendam a realização de transfusões profiláticas. Um estudo controlado indicou que as transfusões profiláticas não proporcionam nenhuma vantagem.118
A amostragem de vilosidades coriônicas (que podem fornecer DNA para análise durante o 1º trimestre da gestação), técnicas de amplificação de DNA e sondas que identificam a alteração nucleotídica específica da anemia falciforme podem fornecer um diagnóstico pré-natal relativamente seguro e bastante confiável.119
Os anticoncepcionais orais podem impor um risco especial às mulheres com anemia falciforme, porque foram associados a um discreto aumento da incidência de AVC, tromboembolia venosa e infarto do miocárdio. No entanto, as evidências emergentes de que o uso diário de anticoncepcionais orais contendo menos de 50 mg de estrogênios sintéticos é relativamente seguro sugerem que as pacientes com anemia falciforme podem tomar esta medicação com um grau razoável de confiança. O uso de alguns dispositivos anticoncepcionais implantáveis constitui outra alternativa para algumas pacientes. Em qualquer dos casos, tanto a gestação como o aborto estão associados a um alto risco na anemia falciforme.118
Aconselhamento genético. Um elemento essencial a ser considerado na prestação de aconselhamento genético a pacientes com traço falciforme ou anemia falciforme é a morbidade significativa entre crianças e adultos afetados. Casais com anemia falciforme ou traço falciforme podem desejar ter filhos, mesmo com os riscos maternos e fetais implicados. Ocorrem cerca de 4.000 a 5.000 gestações desse tipo nos Estados Unidos, a cada ano.129 Em um estudo, 286 das 445 gestações (64%) em mães com anemia falciforme prosseguiram até o parto; 21% dos bebês eram pequenos, e, assim, já se esperava que necessitassem de cuidados adicionais, os quais as mães poderiam ter dificuldade para fornecer. Neste estudo, houve apenas uma morte materna causada pela anemia falciforme120 [ver Aconselhamento genético e diagnóstico pré-natal, adiante].
Prognóstico da anemia falciforme. Embora no passado se afirmasse que a maioria dos pacientes com anemia falciforme morreria ao redor dos 20 anos de idade, atualmente se considera que a média da idade em que a morte ocorre é 42 anos entre os homens e 48 anos entre as mulheres.67 Esta expectativa de vida é 25 a 30 anos menor do que a expectativa de vida para a população afro-americana em geral. Dentre as causas de morte identificadas, somente 18% envolveram insuficiência orgânica – principalmente doença renal, insuficiência cardíaca, doença pulmonar ou as consequências de AVC crônicos; 33% dos pacientes morreram durante as crises de dor aguda, as quais frequentemente estavam associadas à síndrome torácica aguda e, com menos frequência, ao AVC. A existência de alfatalassemia não produziu nenhum efeito mensurável. Os fatores preditores de um resultado precário foram uma contagem de leucócitos > 15.000/mL, níveis baixos de HbF e envolvimento orgânico manifestado por doença renal, síndrome torácica aguda e eventos neurológicos. Tomar hidroxiureia afetou significativamente o prognóstico, produzindo uma diminuição de 40% na mortalidade e uma redução dos episódios de crise dolorosa.70 É significativo que existam muitas barreiras à prestação de um tratamento efetivo para os pacientes com anemia falciforme, tais como o acesso limitado a centros de tratamento da anemia falciforme, dependência dos departamentos de emergência para tratamento das crises álgicas, além dos desafios inerentes à mudança de um prestador de assistência pediátrico para um prestador de atendimento para adultos.75
Traço falciforme. A heterozigose para o gene determinante da célula falciforme resulta na expressão do traço falciforme (HbAS). As hemácias de indivíduos com traço falciforme possuem concentração de HbS inferior a 50% (com frequência, níveis da ordem de 30%).
Em geral, os indivíduos com traço falciforme levam vidas normais e sadias. Algumas complicações são observadas: hipostenúria, hematúria renal e, durante a gestação, bacteriúria e pielonefrite. O infarto esplênico ocorre sob condições de hipóxia, bem como em altitudes elevadas, predominantemente em indivíduos não afrodescendentes que têm anemia falciforme.
O traço falciforme foi identificado como um dos principais fatores de risco de ocorrência de morte súbita durante o treinamento básico do exército.121 Nestes casos, a morte resultou de uma parada cardíaca inexplicável, termoplegia, estresse do calor ou rabdomiólise. A idade crescente foi correlacionada ao risco aumentado de morte súbita. Entretanto, estes eventos ocorreram sob condições extremas: atividade física bastante extenuante, geralmente envolvendo indivíduos despreparados e, às vezes, associada a altas altitudes ou calor extremo. A maioria dos indivíduos com traço falciforme acostumados à atividade física não apresenta risco aumentado de morte súbita. Exemplificando, a incidência de morte súbita entre jogadores de futebol afro-americanos com traço falciforme não é maior do que entre os demais jogadores.122
As opções terapêuticas para hematúria renal incluem a administração de diuréticos, bicarbonato por via parenteral, transfusões ou ácido épsilon-aminocaproico.
Célula falciforme/betatalassemia. Quando combinado ao traço falciforme, um defeito no gene determinante da betatalassemia produz uma doença bastante semelhante à anemia falciforme. O gene determinante da betatalassemia diminui a taxa de síntese da cadeia beta-A, resultando na predominância de beta-S em pacientes com traço falciforme. Dependendo de o paciente ter beta-0 ou beta+-talassemia, as hemácias contêm quantidades variáveis de HbS, HbA, HbA2 e HbF. Pacientes com beta-0-talassemia não possuem HbA, mas apenas HbS, HbF e HbA2. Por isso, estes pacientes desenvolvem uma doença severa. O diagnóstico é baseado na elevação dos níveis de HbA2, HbF ou ambas detectada por eletroforese de hemoglobina, bem como pela história familiar positiva de talassemia e presença de gene falciforme. Em um estudo envolvendo 55 pacientes gregos, o tratamento com hidroxiureia resultou em uma distinta melhora clínica.123,124 Descrições e informações adicionais sobre testes diagnósticos são disponibilizadas on-line, no website www.geneclinics.org.
Doença da célula falciforme/hemoglobina C. Na doença da célula falciforme/hemoglobina C (HbSC), quantidades quase iguais de HbS e HbC são formadas. Entre 1 e 2% da hemoglobina é HbF, e também há pequenas quantidades de HbA2. Entretanto, a HbA está ausente. A falcização aumentada observada nestes pacientes resulta do efeito patológico da HbC [ver Doença da hemoglobina C, adiante].125 Até 30 a 50% dos pacientes com este distúrbio não são anêmicos e apresentam apenas uma modesta reticulocitose. Os pacientes podem ser identificados somente quando o próprio distúrbio se manifesta sob a forma de crise vaso-oclusiva durante uma cirurgia, gestação ou emergência médica.126 Também pode haver esplenomegalia, retinopatia proliferativa, necrose asséptica em ossos longos e síndrome torácica aguda.125 O paciente pode sofrer AVC, embora esta ocorrência seja menos frequente do que na doença da HbSS.109 Além disso, os benefícios proporcionados pelo rastreamento com Doppler transcraniano e pela terapia transfusional, em termos de prevenção de AVC, ainda não foram comprovados junto a esta população.127 O esfregaço de sangue periférico [Figura 5] mostra a presença de células irreversivelmente falciformes, além de células-alvo, estomatócitos e eritrócitos com deposições excêntricas de hemoglobina, provavelmente representando agregados de HbC ou cristais. O diagnóstico é confirmado por eletroforese de hemoglobina ou HPLC.126 Informações adicionais sobre testes diagnósticos são disponibilizadas on-line, no website http://www.geneclinics.org.
O tratamento é realizado do mesmo modo como na anemia falciforme. Em um estudo envolvendo 6 pacientes com doença da HbSC, o tratamento com hidroxiureia (1.000 mg/dia) resultou em aumento do VCM, diminuição do número dos reticulócitos do estresse, aumento dos níveis de hemoglobina e, provavelmente, diminuição da densidade celular. Ainda que não seja definitivo, este pequeno estudo sugere que a hidroxiureia beneficia pacientes com doença da HbSC.128 A expectativa de vida para os pacientes com doença da HbSC é quase 20 anos maior do que para aqueles com Doença da HbSS.67
Doença da hemoglobina C. A molécula HbC é a alfa-2-beta-26gli-lis. A mutação genética provavelmente teve origem em um único lugar, em Burkina Faso, na África Ocidental.126 A presença desta hemoglobina praticamente não produz doença no estado heterozigoto, mas causa uma hemólise compensada branda e esplenomegalia palpável no estado homozigoto.
A relativa insolubilidade da HbC é responsável pelas alterações patológicas associadas a sua presença. É provável que a HbC interaja com o cotransportador de K+/Cl-, mantendo-o ativo. Este cotransportador normalmente é desligado nas hemácias após o estágio de reticulócito. O resultado é a perda de K+, desidratação celular com elevada CHCM e, então, agregação e cristalização da HbC pouco solúvel.126 A relativa insolubilidade da HbC faz com que os eritrócitos se tornem rígidos e, assim, fiquem sujeitos à fragmentação e à perda de material de membrana, com consequente observação de microesferócitos no esfregaço de sangue periférico [Figura 3].
As células-alvo, que representam um importante achado morfológico, constituem cerca de 80% dos eritrócitos. Os cristais de HbC estão presentes no estado de oxiemoglobina e são dissolvidos quando as hemácias são desoxigenadas, sendo provavelmente responsáveis pela ausência de episódios vaso-oclusivos.
O diagnóstico da doença da HbC baseia-se nos achados do esfregaço sanguíneo e na ausência de evidências de deficiência de ferro ou de talassemia. A confirmação do diagnóstico é feita por eletroforese de hemoglobina. Nenhuma terapia é necessária.
Doença da hemoglobina E. Na doença da HbE, a lisina é substituída por ácido glutâmico na posição 26 da cadeia de betaglobina, resultando em uma molécula instável em termos oxidativos. O traço HbE é encontrado de maneira predominante no Sudeste Asiático e na Índia subcontinental – a frequência da HbE pode chegar a 60% em certas regiões do Sudeste Asiático.129 Nos Estados Unidos, esta condição despertou a atenção clínica em decorrência do influxo de indivíduos oriundos daquela região.
Pacientes heterozigotos para HbE apresentam valores normais de hemoglobina, microcitose e ausência de esplenomegalia. A eletroforese revela que 70% da hemoglobina é HbA, 25% é HbE, e o restante é HbA2 ou HbF. Laboratórios inexperientes podem confundir a HbE com a HbA2. O indício deste erro está no fato de a HbA2 jamais corresponder a mais de 8% do conteúdo total de hemoglobina. Um relatório laboratorial de níveis de HbA2 da ordem de 25% requer a imediata revisão dos dados.
Os pacientes homozigotos para HbE apresentam anemia branda, com níveis de hemoglobina aproximados de 12 a 13 g/dL, VCM baixo e contagens de hemácias elevadas, contudo sem reticulocitose. Estes indivíduos exibem micrócitos e células-alvo. A eletroforese mostra apenas a HbE. Não ocorre hemólise crônica. Fármacos oxidantes, como a dapsona, devem ser evitados por indivíduos hetero e homozigotos.
Um sério problema clínico tem lugar diante da heterozigosidade duvidosa para HbE e betatalassemia. Pacientes deste tipo apresentam uma ampla variabilidade clínica de manifestações. Os pacientes mais gravemente afetados se parecem com os pacientes com betatalassemia major e correm risco de apresentar complicações relacionadas à sobrecarga de ferro, doença cardiopulmonar e hipercoagulabilidade (ver adiante).129 Os níveis de HbF constituem um poderoso fator prognóstico da morbidade e da resposta à hidroxiureia.129 Informações adicionais sobre testes diagnósticos são disponibilizadas on-line, no website www.geneclinics.org.
Muitas variantes individuais caracterizam as hemoglobinopatias instáveis. As instabilidades da hemoglobina decorrem de substituições de aminoácidos que deprivam a molécula de seu grupo heme, alteram a bolsa do heme, afrouxam a ligação existente entre suas cadeias alfa e beta, ou enfraquecem a estrutura da subunidade [ver Abordagem de distúrbios hematológicos]. O resultado é a ruptura e precipitação da hemoglobina, particularmente quando sujeita ao ataque oxidativo. A hemoglobina precipitada forma os corpúsculos de Heinz, que são observados inclusive em indivíduos heterozigotos para a variante instável da hemoglobina. Devido aos efeitos deletérios produzidos pelos corpúsculos de Heinz sobre o eritrócito e sua membrana, pode haver uma hemólise significativa até mesmo no estado de heterozigose.
O diagnóstico de uma hemoglobinopatia instável é sugerido pela ocorrência de uma hemólise não esferocítica crônica parcialmente compensada. Os corpúsculos de Heinz são observados nos eritrócitos de pacientes submetidos à esplenectomia. Os eritrócitos de pacientes que não se submeteram à esplenectomia apresentam corpúsculos de Heinz ao serem incubados com corante azul de cresil brilhante. O diagnóstico diferencial de uma hemoglobinopatia inclui a deficiência de G6PD. Este distúrbio geralmente pode ser excluído por meio do ensaio direto para detecção da enzima.
O tratamento implica em evitar o uso de fármacos oxidantes. A esplenectomia pode ser considerada em casos de hemólise severa e inadequadamente compensada.
A presença de hemoglobina com afinidade aumentada pelo oxigênio é uma hipótese a ser considerada no diagnóstico diferencial da eritrocitose de causa inexplicada, em especial se houver associação familiar [ver As policitemias]. A eletroforese de hemoglobina pode revelar o distúrbio, mas a determinação da curva de dissociação da oxiemoglobina [ver Abordagem de distúrbios hematológicos] é preferível como base do diagnóstico, em casos de suspeita. A hemoglobina de Chesapeake e a hemoglobina de Rainier exemplificam algumas formas que apresentam afinidade pelo oxigênio particularmente aumentada.
Os raros casos em que a hemoglobina apresenta baixa afinidade pelo oxigênio, como a hemoglobina de Kansas, resultam de mutações. Os pacientes com hemoglobinopatia de baixa afinidade pelo oxigênio às vezes são cianóticos devido ao maior descarregamento de oxigênio.
A metemoglobina consiste no produto da oxidação da hemoglobina, em que o ferro é encontrado na forma férrica. Assim, a molécula é incapaz de se ligar ao oxigênio de maneira reversível. Comumente, 1% da hemoglobina é encontrada no estado férrico. Diariamente, entre 0,5 e 3% da desoxiemoglobina é normalmente oxidada de modo espontâneo e transforma-se em metemoglobina. O poder redutor normal dos eritrócitos [Tabela 1] mantém o equilíbrio entre oxidação e redução. O sistema enzimático que reduz 95% da metemoglobina a hemoglobina envolve 2 proteínas, NADH-citocromo b5 redutase e citocromo b5, além de também requerer NADH. Conforme o nome sugere, a NADH-citocromo b5 redutase usa NADH para reduzir o citocromo b5 e, então, reduz a metemoglobina.130,131 Foram descritas novas mutações nos genes afetados.132
Mais frequentemente, a metemoglobinemia é adquirida pela ingesta ou exposição a agentes oxidantes que oxidam o Fe2+ tão rápido que os sistemas redutores acabam sendo vencidos [ver Mecanismo do ataque oxidativo, adiante].
Existem 2 formas congênitas de metemoglobinemia. Na forma enzimopênica hereditária de metemoglobinemia, os pacientes são homozigotos ou duplo-heterozigotos para uma deficiência de NADH-citocromo b5 redutase.133 Estes pacientes exibem uma cor azulada mesmo quando apenas cerca de 10% do conteúdo de hemoglobina está na forma de metemoglobina. No entanto, estes indivíduos não estão doentes e toleram facilmente níveis de metemoglobina iguais ou superiores a 25%. Em contraste, a presença de cerca de 5 g/dL de hemoglobina reduzida, desoxigenada, produz cianose. Pacientes com esta forma de metemoglobinemia não apresentam hemólise e geralmente dispensam tratamento. O diagnóstico pode ser estabelecido por ensaio de NADH-citocromo b5 redutase, realizado em laboratório especial. Havendo interesse, pode ser feita a administração oral de azul de metileno (100 a 300 mg/dia), porém este composto causa desconforto urinário.130 O azul de metileno transfere elétrons do NADPH para a metemoglobina.
A outra forma hereditária de metemoglobinemia é causada pela HbM, que possui 5 variantes raras. Cada uma dessas variantes contém uma substituição de aminoácido junto à bolsa do heme, que permite a formação de ligações estáveis entre o ferro do heme e as cadeias laterais de aminoácidos. Estas ligações mantêm a hemoglobina na forma de Fe2+ – que é incapaz de se ligar ao oxigênio e é inacessível às enzimas redutoras. O distúrbio é observado apenas em indivíduos heterozigotos. Cerca de 30% da hemoglobina é anormal, conforme detectado por eletroforese. A cianose é observada ao nascimento. A hemólise é mínima, e não há necessidade de terapia.
As talassemias apresentam uma distribuição mundial. Em muitas regiões, são responsáveis por perturbações médicas, sociais e econômicas significativas. Em todo o mundo, as regiões em que as talassemias ocorrem são consíguas às regiões endêmicas de malária, indicando que as formas heterozigotas de talassemia conferem proteção contra a malária.134 As técnicas de biologia molecular têm ajudado a elucidar a patofisiologia destas síndromes,134 o que, por sua vez, tem permitido aos pesquisadores estabelecer diagnósticos pré-natais sem ambiguidade. Utilizando estes dados, os futuros pais podem fazer escolhas pensadas e bem informadas acerca do resultado de gestações nas quais os fetos serão gravemente afetados.
As talassemias resultam de deleção genética, de anormalidades de transcrição e translação [ver Genética básica para clínicos] e de instabilidade do RNA mensageiro direcionador da síntese de globina ou da própria globina. Os genes que controlam a síntese das cadeias alfa e não alfa da hemoglobina estão localizados nos cromossomos 11 e 16 [Figura 6].
Figura 6. Os genes codificadores de cadeias alfa e não alfa que se unem para formar o tetrâmero da hemoglobina estão localizados nos cromossomos 16 e 11, respectivamente. Os genes da cadeia alfa estão presentes como loci duplicados. Foram descritas 6 espécies distintas de hemoglobina normal, das quais 3 são sintetizadas apenas durante os estágios embrionários do desenvolvimento (Hb Gower 1, Hb Portland e Hb Gower 2). A HbF predomina durante o desenvolvimento fetal, sendo que uma pequena quantidade continua sendo sintetizada durante a vida adulta. A HbA e a HbA2 constituem as principais formas de hemoglobina no adulto. Diferentes genes de hemoglobina são ativados nos vários estágios do desenvolvimento. No embrião, as cadeias zeta se combinam às cadeias épsilon para produzir a Hb Gower 1, bem como às cadeias gama para forma a Hb Portland. As cadeias alfa e épsilon ligam-se para formar a Hb Gower 2. Duas variedades de cadeias gama derivam de loci separados e diferem quanto a um único aminoácido. G-gama contém glicina na posição 136, enquanto A-gama contém alanina nesta posição. Os genes codificadores de outras 2 cadeias não alfa – delta e beta – que são requeridos para a síntese das hemoglobinas do adulto, são ativados em fases posteriores do desenvolvimento fetal. Ainda são desconhecidos os fatores que regulam este processo e coordenam com precisão a sequência de alterações na produção de hemoglobina. Algumas evidências sugerem que segmentos de DNA interpostos entre os vários genes da hemoglobina podem controlar as taxas relativas de síntese dos produtos dos genes adjacentes.
Em um indivíduo sadio, a síntese das cadeias alfa e beta é meticulosamente coordenada para produzir a HbA do adulto (alfa-2-beta-2). Em contraste, pacientes com talassemia geralmente apresentam uma síntese desequilibrada de cadeias de globina normais. Em alguns casos, todavia, síndromes do tipo talassemia podem resultar da produção diminuída de uma cadeia estruturalmente anormal.135 Como uma das cadeias de globina está presente em quantidade reduzida, a cadeia não pareada acumula-se na célula precursora eritroide em desenvolvimento, com consequente toxicidade [Figura 6]. Em consequência, as células eritroides morrem na medula e dão origem à forma clássica de eritropoese inefetiva [ver Anemia: defeitos de produção]. Os eritrócitos afetados sofrem hemólise no sangue periférico.
As betatalassemias são caracterizadas pela produção diminuída das cadeias de betaglobina, que causa acúmulo e agregação das cadeias de alfaglobina sem par. Estes agregados de cadeias alfa precipitam, causando diminuição da síntese de ATP, vazamento de potássio e redução das quantidades de ácido siálico de superfície. Os eritrócitos afetados são deformados e relativamente rígidos. A barreira de membrana à entrada do Ca2+ é rompida, permitindo a entrada do cátion. Esses agregados de alfaglobina também parecem manter o cotransportador de K+/Cl- funcionando. Como resultado, nas formas severas de betatalassemia, observam-se graus variáveis de desidratação.136 As membranas das hemácias são instáveis e fragmentam-se com facilidade. Há evidências de oxidação das proteínas 4,1 e espectrina, localizadas na membrana das hemácias [Figura 2]. Além disso, a fosfatidilserina migra para a camada externa da membrana, talvez formando um ninho para eventos tromboembólicos.137,138 Estas alterações destrutivas da membrana, que podem ser detectadas pelos macrófagos, podem ser parcialmente causadas pela oxidação local.139,140 O acúmulo anormal de cadeias alfa provavelmente é responsável pela apoptose acelerada e eritropoese inefetiva observadas nos precursores eritroides medulares.140 A diminuição geral da síntese de hemoglobina por célula produz a hipocromia e a formação de célula-alvo observadas.
Os pacientes com alfatalassemia apresentam acúmulos de cadeias beta em excesso que, quando estão presentes em quantidade suficiente, formam tetrâmeros beta-4 (HbH) [Figura 6]. Estes tetrâmeros possuem alta afinidade pelo oxigênio e são instáveis, agregando-se diante de estresses oxidativos (p. ex., infecção). Os agregados de betaglobina também se fixam ao esqueleto da membrana do eritrócito, contudo produzem lesões diferentes daquelas produzidas pelos agregados de alfaglobina. Nas alfatalassemias severas, a eritropoese inefetiva é menos proeminente. Em vez disso, a principal característica é a destruição periférica de hemácias. Nas alfatalassemias severas, as hemácias são rígidas e, em contraste com o observado na betatalassemia, suas membranas são mais estáveis do que o normal.137,138 Do mesmo modo, em contraste com a betatalassemia, as hemácias são uniformemente super-hidratadas na alfatalassemia.136
Ambas as talassemias, alfa e beta, caracterizam-se por graus variáveis de anemia. Esta variação é atribuível a graus variáveis de eritropoese inefetiva e hemólise.140 Quando a anemia é severa, a hipóxia associada induz uma vigorosa eritropoese compensatória, levando à expansão da cavidade medular, osteopenia e ampliação dos órgãos reticuloendoteliais. Pode haver aparecimento de tumores nos sítios com atividade eritropoética extramedular. A destruição dos eritroblastos e eritrócitos pode predispor ao desenvolvimento de colelitíase e icterícia obstrutiva. Os pacientes que apresentam as formas mais severas de talassemia necessitam de transfusões regulares, que eventualmente podem gerar uma sobrecarga de ferro significativa do ponto de vista clínico [ver Função das hemácias e distúrbios do metabolismo do ferro].
As quantificações de HbF e HbA2 auxiliares no diagnóstico das betatalassemias são prontamente disponibilizadas nos laboratórios clínicos que empregam eletroforese de hemoglobina e, mais recentemente, HPLC. Em contraste, os exames requeridos para diagnosticar as alfatalassemias são bastante sofisticados; no passado, eram realizados somente em instituições especificamente dedicadas à pesquisa das talassemias. Na época atual, os laboratórios especializados podem detectar o número e a posição dos genes de alfaglobina deletados. Informações adicionais sobre testes diagnósticos são disponibilizadas on-line, no website www.geneclinics.org.
As ferramentas de diagnóstico clínico utilizadas para avaliar pacientes com suspeita de terem alfatalassemia incluem a história clínica, avaliação de esfregaço, cálculo de índices, coloração com azul de cresil brilhante e estudos familiares. Na prática, o traço alfatalassêmico é diagnosticado com base no achado de microcitose em um paciente com repleção de ferro, cujos níveis de HbA2 e HbF são normais.
O diagnóstico da alfatalassemia e da betatalassemia deve ser suspeitado diante VCM < 75 fL e contagem de hemácias > 5 milhões de células/mcL. Um paciente que apresente estes 2 achados apresenta 85% de chances de ter uma síndrome talassêmica.141 Em um estudo, o diagnóstico da talassemia não foi considerado em cerca da metade dos casos de pacientes que tinham a doença.141
A síntese deficiente de betaglobina característica da betatalassemia leva ao acúmulo de cadeias alfa sem par. Na betatalassemia, um desenvolvimento significativo em termos de diagnóstico consiste no aumento compensatório parcial das cadeias delta e gama, que resulta em níveis elevados de HbA2 (alfa-2-delta-2) e HbF (alfa-2-gama-2), respectivamente [Figura 5]. As variantes da betatalassemia produzem 3 síndromes clínicas: betatalassemia major, betatalassemia minor e talassemia intermediária.
A betatalassemia major geralmente é uma condição homozigota ou duplamente heterozigota. Ambos os pais do indivíduo afetado carregam o traço betatalassêmico. Na beta-0-talassemia, a variante mais severa, nenhuma cadeia beta é sintetizada. Somente HbF e HbA2 são encontradas. A beta+-talassemia é um pouco menos severa e caracteriza-se por pequenas quantidades de cadeias beta e de HbA, além de HbF e HbA2. A delta-betatalassemia é ainda mais branda; é causada pela deleção dos genes codificadores de deltaglobina e betaglobina. Esta mutação impede a produção de HbA2 e HbA e permite a síntese isolada de hemoglobina fetal.
A betatalassemia major caracteriza-se por uma severa anemia que surge ainda no 1º ano de vida. Os pacientes também apresentam icterícia, hepatoesplenomegalia, expansão da medula eritroide com alterações corporais secundárias (incluindo retardo do crescimento) e maior suscetibilidade a infecções.
O diagnóstico é simples. Não existe outra condição que seja semelhante à anemia de Cooley. O exame do esfregaço de sangue periférico revela a presença de hemácias nucleadas, eritrócitos hipocrômicos distorcidos e pontilhado basofílico, que representa agregados de RNA ribossômico [Figura 5]. A coloração supravital mostra a existência de acúmulos de cadeias alfa em excesso não pareadas.
Suporte geral. Terapia transfusional. A estratégia envolve a realização de transfusões para atingir níveis de hemoglobina de aproximadamente 12 g/dL e, então, permitir que estes níveis caiam para cerca de 9 g/dL, pouco antes de realizar a próxima transfusão. Este procedimento evita complicações, como insuficiência cardíaca, sobrecarga de líquidos e deformidade esquelética. Geralmente, é necessário realizar uma esplenectomia para aumentar a sobrevida das hemácias do próprio paciente, bem como das hemácias transfundidas.142 A vacinação com vacina pneumocócica é indicada, devido ao risco de sepse pneumocócica após a esplenectomia. A terapia transfusional está associada ao risco de aloimunização.
Quelação do ferro. As transfusões prolongadas eventualmente geram sobrecarga de ferro que, quando não tratadas, causam a morte por hemocromatose cardíaca durante a adolescência. A sobrecarga de ferro leva ao acúmulo de ferro ligado à proteína não transferrina e à ampliação do pool de ferro lábil intracelular. Estas espécies reativas do ferro estimulam a formação de radicais livres que, por sua vez, danificam os lipídios de membrana e causam lesão celular.143 Para prevenir esta lesão, é necessário tratar profilaticamente a sobrecarga de ferro utilizando quelantes de ferro. Atualmente, existem 2 quelantes de ferro disponibilizados para uso nos Estados Unidos: deferoxamina (administrada por via subcutânea ou endovenosa) e deferasirox (um agente de uso oral). Outro quelante de ferro adicional, a deferiprona, é disponibilizado para uso fora dos Estados Unidos. A deferoxamina subcutânea (50 mg/kg/dia) pode promover perdas de ferro da ordem de 50 a 200 mg/dia, mas apenas ao ser infundida de modo contínuo, ao longo de 8 a 12 horas, durante 5 dias/semana.144 Esta terapia não só previne a disfunção ventricular esquerda, como também reverte as anormalidades já estabelecidas.145 Os efeitos benéficos da quelação do ferro têm melhorado o prognóstico de pacientes com anemia de Cooley:145 a morte dos pacientes em torno dos 20 anos de idade em decorrência de arritmia ou insuficiência ventricular esquerda deixou de ser inevitável. Graças à atual terapia com deferoxamina, 61% dos pacientes nascidos antes de 1976 não apresentam doença cardiovascular. Os pacientes complacentes, cujos níveis de ferritina estão principalmente abaixo de 2.500 ng/mL, apresentam uma taxa de sobrevida de 91% após 15 anos.146 Entretanto, a complacência com a terapia à base de deferoxamina constitui um problema, ao passo que o custo do fármaco, aliado ao custo da bomba e das tubulações necessárias à administração, tornam o tratamento inviável para a maioria dos pacientes que vivem nos países em desenvolvimento. A deferiprona, cuja molécula é menor e mais lipofílica, pode ser mais efetiva do que a deferoxamina em termos de quelação do ferro intracelular. Desta forma, a deferiprona também pode ser mais efetiva na remoção do ferro cardíaco, em comparação à deferoxamina.147,148 Foi proposto que o tratamento combinado com deferoxamina-deferiprona permite que o ferro seja deslocado para fora das células pela deferiprona e, então, transferido para a deferoxamina, propiciando uma quelação mais efetiva do ferro miocárdico, mesmo diante de uma sobrecarga de ferro severa.149 Em um estudo envolvendo pacientes com sobrecarga leve a moderada de ferro miocárdico, aqueles que foram randomizados para receber tratamento com combinação deferoxamina-deferiprona, vs. aqueles que receberam apenas deferoxamina, apresentaram uma redução relativa significativa da sobrecarga de ferro miocárdico detectada por imagem de T2* (tempo de relaxamento, transversal, ou spin-spin).150 Os pacientes tratados com deferoxamina devem ser monitorados quanto à ototoxidade e toxicidade oftalmológica, enquanto os pacientes tratados com deferiprona devem ser monitorados quanto à neutropenia ou agranulocitose e hepatotoxicidade.
Os métodos utilizados para diagnosticar a sobrecarga de ferro incluem técnicas invasivas (biópsia de fígado) e não invasivas (quantificação dos níveis séricos de ferritina, dispositivo supercondutor de interferência quântica [SQUID – em inglês, superconducting quantum interference device] e imagem de ressonância magnética [RM]). Embora o exame de biópsia hepática seja considerado o padrão-ouro para a determinação do grau de sobrecarga de ferro, está sujeito a erros de amostragem e traz um risco associado de 0,5% de sangramento severo.151 Os níveis séricos de ferritina são rapidamente disponibilizados, e sua determinação é de execução simples, além de ser um teste útil para acompanhar as tendências ao longo do tempo. Entretanto, esses valores estarão falsamente elevados nos estados de inflamação. O SQUID, uma das primeiras técnicas não invasivas empregadas para medir a carga de ferro, possui disponibilidade limitada e não pode ser utilizado para detecção de ferro cardíaco.151 O uso de T2*, uma variável da ressonância magnética inversamente relacionada à concentração tecidual de ferro, tem sido diretamente validado contra as biópsias hepáticas,152 apresenta alta reprodutibilidade no fígado e no coração153 e recentemente emergiu como principal ferramenta não invasiva para quantificação do ferro em órgãos cardíacos, hepáticos e endócrinos.151,154 É importante avaliar os órgãos-alvo de maneira independente, uma vez que a deposição de ferro em um órgão pode não refletir o grau de deposição em outros órgãos.155
Problemas médicos específicos. Como o tratamento e, consequentemente, a sobrevida têm melhorado entre os pacientes com betatalassemia, os indivíduos se tornaram suscetíveis a doenças crônicas e complicações associadas à terapia.
Doença óssea. A osteoporose e a osteopenia afetam 52 a 96% dos pacientes com talassemia.156 As causas destas condições são multifatoriais e incluem endocrinopatias associadas, expansão da medula óssea e sobrecarga de ferro.157 As sequelas incluem baixa estatura e dor óssea, que pode ser severa. Em um estudo envolvendo pacientes com talassemia e osteoporose, 59% dos quais tinham história de fraturas patológicas, o tratamento com ácido zoledrônico (4 mg/3 meses) promoveu aumento significativo da densidade mineral óssea da espinha lombar, diminuição dos marcadores de reabsorção e formação óssea e melhora da dor óssea.158
Doença endócrina. As endocrinopatias são comuns na talassemia e em grande parte devidas à deposição de ferro em órgãos endócrinos, principalmente a hipófise anterior. Um estudo descreveu a ocorrência de baixa estatura em 39% dos casos, além de hipogonadismo em 22,9% dos meninos e 12,2% das meninas, hipotireoidismo primário em 7,7% dos casos e hipoparatireoidismo em 7,6% da população.159
Doença cardiopulmonar. A doença cardíaca é a principal causa de morte de pacientes com talassemia major dependente de transfusão.143 Os principais fatores contribuidores para o desenvolvimento de doença cardíaca são a sobrecarga de ferro e o débito cardíaco aumentado, secundário à anemia e à expansão da medula. Além do acompanhamento da condição do ferro por RM T2*, recomenda-se a realização de uma ecocardiografia anual. Uma diminuição de 10% na fração de ejeção ventricular esquerda ou um decréscimo para menos de 45% requerem uma quelação de ferro imediata e agressiva com o intuito de melhorar a sobreviva.157,160 Assim como na anemia falciforme, a hipertensão pulmonar é uma complicação cada vez mais reconhecida em pacientes com talassemia.161 A patofisiologia subjacente não é totalmente compreendida, embora a esplenectomia pareça ser um fator de risco.162 Relatos anedóticos sugerem que o sidenafil ou epoprostenal podem ser benéficos para a redução da hipertensão pulmonar.163,164
Vírus da hepatite C (HCV). Os pacientes com talassemia que dependem de transfusões correm o risco de contrair patógenos transmissíveis pelo sangue. O rastreamento de produtos do sangue para detecção do vírus da hepatite C (HCV) foi implantada em 1990, e os adultos que receberam transfusões antes desta época apresentam risco de exposição ao HCV. Em uma pesquisa recente sobre pacientes adultos talassêmicos, realizada na América do Norte, 70% dos indivíduos avaliados eram soropositivos ou positivos para RNA de HCV,165 sendo que nos países industrializados até mesmo a exposição de pacientes jovens ainda pode ser alta.166 Os indivíduos com infecção por HCV crônica, particularmente aqueles que sofriam de sobrecarga de ferro crônica, correm risco de desenvolvimento de cirrose e carcinoma hepatocelular. O tratamento da infecção pelo HCV em pacientes com talassemia é complicado pelo risco de hemólise induzida por ribavirina.155
Hipercoagulabilidade. Tem sido cada vez mais reconhecido que a talassemia, particularmente a betatalassemia, pode aumentar a ocorrência de eventos tromboembólicos. Em um estudo envolvendo 8.860 pacientes com betatalassemia, a prevalência de eventos tromboembólicos foi de 1,65% entre aqueles com talassemia major e de 4% entre aqueles com talassemia intermediária.167 A patofisiologia pode incluir trombocitose em pacientes esplenectomizados, ativação plaquetária aumentada, intensificação da expressão de fosfatidilserina na membrana celular externa e níveis diminuídos de proteínas C e S.157,168,169 Atualmente, não há um consenso quanto às recomendações referentes à profilaxia anticoagulante de rotina para pacientes com betatalassemia.169
Tratamento. Agentes modificadores de doença. A meta da terapia com estes agentes é aumentar a síntese das cadeias de gamaglobina (consequentemente, os níveis de HbF) e diminuir o excesso de cadeias alfa.157 O mecanismo pelo qual a hidroxiureia aumenta os níveis de HbF não é totalmente compreendido.157 Entretanto, os relatos recentes sobre o uso de hidroxiureia por esta população foram encorajadores, uma vez que os pacientes dependentes de transfusão se tornaram independentes.170,171 Os agentes hipometiladores de DNA podem sobrepujar a inibição da síntese de cadeias gama,157 sendo que tanto a 5-azacitidina como a 5-aza-2’-desoxicitidina (decitabina) foram empregadas no tratamento de pacientes com talassemia, promovendo respostas variáveis em termos de aumento dos níveis de HbF e/ou hemoglobina.157 A variabilidade da resposta clínica é pouco compreendida. Além disso, as preocupações com a toxicidade têm limitado as pesquisas investigativas em grande escala sobre a 5-azacitidina.172 Há evidências de que os inibidores de histona desacetilase (HDAC) também podem aumentar a expressão da transcrição da cadeia de gamaglobina.173 Os inibidores de HDAC incluem o butirato de arginina administrado por via endovenosa e o fenilbutirato de sódio administrado por via oral. Os pacientes tratados com estes agentes também apresentaram respostas variáveis. Não foi possível predizer o sucesso alcançado com estas terapias, tendo como base os parâmetros conhecidos dos pacientes.174 A eritropoetina também tem sido utilizada com certo benefício, apesar das limitações de seu uso envolverem o custo, a inconveniência da administração por via subcutânea e o efeito passageiro.174
Terapia genética. Embora a transferência de um gene de globina para células-tronco hematopoéticas autólogas seja um objetivo promissor, ainda existem numerosos desafios a serem enfrentados para se tentar trazer essa modalidade terapêutica para a prática clínica.174 As dificuldades incluem o potencial de mutagênese associado ao uso de vetores virais, particularmente após o desenvolvimento de um distúrbio linfoproliferativo T observado em 2 crianças com imunodeficiência ligada ao X tratadas com terapia genética,175 além das atuais restrições impostas à pesquisa com células-tronco embrionárias.174
Transplante. O transplante da medula óssea tem sido realizado com doadores irmãos HLA-compatíveis. Até agora, mais de 1.000 pacientes foram submetidos ao transplante alogênico de medula óssea fornecida por doadores irmãos normais ou com traço betatalassêmico.157 Alguns pacientes com betatalassemia por HbE exibem um fenótipo tão severo quanto a betatalassemia major e requer a mesma terapia, incluindo o transplante de medula óssea alogênica.176 A experiência com transfusão de sangue de cordão é mais limitada.177 Dependendo da condição do paciente no momento do transplante, a taxa de mortalidade associada ao transplante foi de 5 a 19%. A taxa de cura foi de 54 a 90%.178,179
Tratamento do pós-transplante ex-talassêmico. Embora o transplante de células-tronco proporcione a possibilidade de cura da talassemia, as complicações pré-transplante ainda persistem. As sequelas importantes são: sobrecarga de ferro, infecção pelo HCV, disfunção endócrina e complicações relacionadas ao transplante.157 Um regime de flebotomia em geral é iniciado 18 meses após o transplante, com o objetivo de diminuir os níveis séricos de ferritina para menos de 100 mcg/L.180 A infecção pelo HCV pode ser tratada com interferon.181
Pacientes com betatalassemia minor geralmente são heterozigotos para uma mutação envolvendo a betaglobina e podem ter ou não uma anemia leve. O esfregaço periférico mostra uma hipocromia distinta e microcitose com pontilhado basofílico. A esplenomegalia é um achado ocasional.
Os níveis de HbA2 sobem para mais de 5% em 90% dos pacientes, enquanto os níveis de HbF estão acima de 2% em 50% dos pacientes. Este aumento dos níveis de HbF é proporcionalmente variável para cada hemácia (um fenômeno conhecido como distribuição heterocelular), conforme demonstra a coloração de Kleihauer-Betke. Os pacientes com níveis de HbF mais altos apresentam uma anemia menos severa. Os heterozigotos para deltabetatalassemia produzem quantidades aumentadas de HbF, mas apenas quantidades normais de HbA2.
A possibilidade de anemia ferropriva (por deficiência de ferro) deve ser excluída do diagnóstico diferencial de traço betatalassêmico [ver Função da hemácia e distúrbios do metabolismo do ferro]. Em geral, é fácil distinguir os 2 distúrbios. Ambos estão associados à hipocromia e microcitose, mas a deficiência de ferro produz hipoproliferação de hemácias, enquanto a betatalassemia minor causa apenas uma redução mínima do número de eritrócitos. A um nível de hemoglobina de 9 g/dL, o paciente com deficiência de ferro apresenta uma contagem de hemácias aproximada de 3 milhões de células/mcL, enquanto um paciente com traço betatalassêmico apresenta uma contagem de hemácias de cerca 5 milhões de células/mcL. Se o diagnóstico continuar duvidoso, a quantificação dos níveis séricos de ferro e a determinação da capacidade de ligação de ferro ou dos níveis séricos de ferritina podem ser utilizadas para distinguir estes distúrbios. É importante lembrar, todavia, que um paciente com traço talassêmico também pode ter deficiência de ferro em consequência de sangramento vaginal, gastrintestinal ou ambos.
Talassemia intermediária. Conforme implica a denominação, a talassemia intermediária é caracterizada por manifestações clínicas de severidade moderada. Os pacientes com esta síndrome apresentam uma anemia distinta, com níveis de hemoglobina mínimos de 6 a 7 g/dL. Esses indivíduos exibem graus variáveis de hepatoesplenomegalia, mas geralmente dispensam a realização de transfusões regulares. Durante as infecções ou outras agressões eritropoéticas, contudo, pode haver uma necessidade passageira de realizar transfusões. Em 2 estudos clínicos pequenos, constatou-se que a isobutiramida proporciona benefícios.182,183 Apesar da ausência de requerimentos vigentes para as transfusões, os pacientes com talassemia intermediária podem desenvolver uma severa sobrecarga de ferro. Isto se deve em parte à ação da hepcidina, um peptídeo modulador do ferro produzido no fígado. A hepcidina atua prevenindo a absorção do ferro pelo enterócito duodenal e a liberação de ferro pelos macrófagos na medula óssea.184 A síntese de hepcidina é negativamente regulada em resposta ao impulso eritropoético aumentado, sendo que os níveis de hepcidina são indevidamente suprimidos em pacientes com talassemia intermediária, apesar da sobrecarga de ferro hepático.184
A hemoglobinopatia associada à hemoglobina Lepore constitui outra variante betatalassêmica. Os pacientes homozigotos para este distúrbio apresentam anemia de Cooley ou talassemia intermediária, uma vez que suas hemácias contêm apenas hemoglobina Lepore e HbF.135
As hemácias dos pacientes heterozigotos para a persistência da hemoglobina fetal (PHHF) contêm cerca de 50% de HbF, enquanto as hemácias dos indivíduos homozigotos possuem 100% de HbF. No passado, acreditava-se que os pacientes com PHHF viviam bem e tinham anemia mínima ou inexistente. Contudo, foram descritas algumas variantes clínicas da PHHF associadas a um tipo de anemia distinto.
Os genes da alfaglobina e da betaglobina diferem quanto a 2 aspectos principais. Primeiro, não existem substituições fetais, neonatais ou adultas para os genes da alfaglobina. Segundo, existem apenas 2 genes codificadores de betaglobina, porém 4 genes codificadores de alfaglobina – 2 genes de alfaglobina em cada cromossomo 16 [Figura 6]. O genótipo da alfaglobina normal é denominado alfa-alfa/alfa-alfa. Os pacientes portadores da variante alfatalassemia-1 exibem uma deleção de 2 genes de cadeia alfa junto ao mesmo cromossomo e, assim, apresentam o haplótipo —/ ou alfa-0. Esta deleção é comum entre pacientes asiáticos. Os pacientes que exibem a variante alfatalassemia-2 perderam 1 gene alfa junto a 1 cromossomo e apresentam haplótipo -alfa/ ou alfa+. Apesar de esta mutação ser particularmente frequente entre afrodescendentes, também é observada nas populações asiática e mediterrânea. Cinco síndromes clinicamente distintas foram reconhecidas entre pacientes com genótipos diferentes para o gene da alfaglobina: hemoglobina de Barts ou hidropsia fetal (—/—); doença da hemoglobina H (—/-alfa); alfatalassemia-1 heterozigota (—/alfa-alfa); alfatalassemia-2 homozigota (-alfa/-alfa); e síndrome do portador silencioso (-alfa/alfa-alfa).
Hemoglobina de Barts (hidropsia fetal). Crianças com síndrome da hemoglobina de Barts são homozigotas para alfatalassemia-1 (—/—) e, portanto, não produzem cadeias alfa. As cadeias gama não pareadas formam tetrâmeros gama-4 (hemoglobina de Barts). Todos os bebês com esta condição nascem hidrópicos, e a maioria morre, exceto quando são salvos pelo transplante intrauterino de células-tronco. Os pais geralmente são heterozigotos para a condição (—/alfa-alfa).
Doença da hemoglobina H. O quadro clínico da doença da HbH é o de anemia hemolítica variável, que ocorre em pacientes originários da Ásia, do Oriente Médio ou do Mediterrâneo. A HbH, que precipita mediante coloração com azul de cresil brilhante, geralmente pode ser detectada nas hemácias recém-coletadas do paciente. Os mecanismos moleculares podem envolver a deleção de 3 genes alfa, como seria o caso de um paciente duplamente heterozigoto para alfatalassemia-1 (—/) e alfatalassemia-2 (-alfa/), rendendo um genótipo —/-alfa. É comum haver esplenomegalia. Os pacientes em geral não necessitam de transfusões regulares, porém podem requerer um suporte transiente de hemácias diante de infecções ou estresses oxidativos que levem à precipitação da HH instável e à intensificação da hemólise. Durante a gestação, a anemia pode tornar-se clinicamente severa, e a paciente pode necessitar de transfusão de hemácias. Os parceiros de gestantes com doença da HbH devem ser submetidos a um rastreamento, pois se tiverem o traço alfatalassêmico, o feto pode apresentar hidropsia fetal alfatalassêmica homozigota. Ocasionalmente, ocorre retardo do crescimento associado à condição e até mesmo acúmulo de ferro na ausência de transfusões de hemácia.185
Alfatalassemia heterozigota. A alfatalassemia-1 heterozigota (—/alfa-alfa), que constitui um genótipo comumente encontrado em asiáticos, pode causar uma anemia branda. A condição produz hemácias distintamente hipocrômicas e microcíticas. Nos pacientes homozigotos para alfatalassemia-2 (-alfa/-alfa), um genótipo comum em afrodescendentes, faltam 2 genes alfa. As manifestações clínicas apresentadas pelos pacientes com este genótipo são semelhantes àquelas exibidas por pacientes heterozigotos para alfatalassemia-1. O estado de heterozigose para a alfatalassemia-2 (-alfa/alfa-alfa) é clinicamente indetectável e, assim, representa a síndrome do portador silencioso.
A hemoglobina de Constant Spring (HbCS) consiste em uma hemoglobina estruturalmente anormal, comumente encontrada em algumas populações asiáticas. O gene da alfaglobina apresenta uma mutação no códon de terminação que resulta na síntese de uma alfaglobina contendo 31 aminoácidos adicionais. Os pacientes heterozigotos para este defeito desenvolvem um quadro clínico semelhante ao quadro clínico dos pacientes homozigotos para alfatalassemia-2. Os pacientes homozigotos para HbCS tendem a apresentar manifestações clínicas discretamente mais severas do que aquelas observadas nos pacientes heterozigotos para alfatalassemia-1. Em pacientes duplamente heterozigotos para alfatalassemia-1 e alfa-CS (—/alfa-CS alfa), bem como em indivíduos com HbH/HbCS, a doença é levemente mais severa do que em pacientes com doença por HbH.140
É compreensível que pais de um bebê hidrópico natimorto ou de uma criança com anemia de Cooley relutem em repetir a experiência. Indivíduos adultos oriundos de famílias com casos de talassemia e que já sabem de sua própria condição heterozigota para talassemia muitas vezes anseiam por receber aconselhamento genético quando formam suas próprias famílias. O aconselhamento genético está vinculado a um estudo prospectivo sobre os pais, baseado na realização de exames diagnósticos de rotina e estudos familiares. Além disso, os avanços ocorridos na área de genética molecular podem atualmente fornecer diagnósticos pré-natais de talassemia, sem ambiguidades e com acurácia. Durante o 1º trimestre, a amostragem das vilosidades coriônicas aliada ao uso de marcadores de DNA polimórficos e sondas oligonucleotídicas sintéticas podem estabelecer o diagnóstico definitivo em cerca de 80% dos casos de betatalassemia [ver Genética básica para clínicos].186,187 De fato, a incidência de nascimentos de bebês com talassemia major sofreu uma queda em várias partes do mundo. Diferentes grupos étnicos respondem de maneiras diferentes ao aconselhamento genético.
Os eritrócitos podem ser danificados por traumatismo ou pela ação de anticorpos, fármacos, órgãos funcionalmente anormais e toxinas. Estas causas de defeitos extracorpusculares devem ser consideradas sempre que houver desenvolvimento de hemólise em um paciente sem história pessoal ou familiar de anemia.
A hemólise microangiopática caracteriza-se pelo aparecimento de eritrócitos fragmentados e bizarros (p. ex., esquistócitos ou células em capacete) em esfregaços de sangue periférico, bem como por sinais de hemólise intra e extravascular.
O eritrócito normal pode resistir a alongamentos e torsões consideráveis, porém se desintegra ao ser submetido a um estiramento forte ou a forças de cisalhamento. Estresses desta magnitude foram observados em jatos produzidos por válvas aórticas deformadas, shunts arteriovenosos, defeitos septais ventriculares ou com o uso de próteses valvares antigas.
Considera-se que a coagulação intravascular localizada, em que os filamentos de fibrina formam uma ponte junto ao lúmen arteriolar, ocorre nas arteríolas que suprem os tecidos inflamados ou neoplásicos. Os filamentos de fibrina “podam” os fragmentos de hemácias, cujas membranas são imediatamente vedadas de novo. Entretanto, uma parte do conteúdo dos eritrócitos vaza e produz vários graus de hemólise intravascular. As hemácias distorcidas são, então, removidas pelo sistema reticuloendotelial.
A hemólise, aliada aos achados típicos no esfregaço sanguíneo, é diagnóstica da hemólise microangiopática [Figura 5]. Quando a angiopatia é extensa, há desenvolvimento de trombocitopenia e coagulação intravascular disseminada. As causas incluem os jatos hemodinâmicos, vasculite,186 hemangiomas gigantes, púrpura trombocitopênica trombótica (PTT)/síndrome hemolítica-urêmica, câncer metastásico,189 certas infecções (em especial a meningocococemia, doenças causadas por riquétsias e infecção por hantavírus), a síndrome do anticorpo antifosfolípide catastrófica, coagulação intrasvascular disseminada, fármacos (cocaína, ciclosporina, mitomicina e tacrolimo) e até mesmo o uso de cateteres subclávios.189,190 A quinina foi identificada como sendo uma causa bastante comum de PTT trombótica fármaco-induzida e síndrome hemolítica-urêmica.191 Foi descrito um caso de hemólise microangiopática em um bebê com antraz cutâneo.192
No tratamento da hemólise microangiopática, os clínicos devem se concentrar principalmente na causa subjacente. Os pacientes podem desenvolver deficiência de ferro e necessitar de terapia à base de ferro. A suplementação das reservas de folato depletadas pode estimular a eritropoese. Em casos raros, a anemia causada por uma prótese de válvula aórtica antiga pode ser severa o bastante para justificar a troca da válvula. A PTT constitui uma emergência médica e requer uma plasmaférese urgente [ver Medicina transfusional; Distúrbios plaquetários e vasculares].
A hemoglobinúria de March, um distúrbio um tanto semelhante à hemólise microangiopática, geralmente acomete indivíduos jovens após um período prolongado de caminhada, corrida ou tocando bongô. Acredita-se que o traumatismo severo e repetitivo nos pés ou nas mãos destrói as hemácias circulantes no interior dos vasos das solas e palmas. O paciente percebe que sua urina está vermelha, a qual volta a ficar límpida em até 1 dia após a atividade. A hemoglobinemia transiente e a hemoglobinúria sem anemia, bem como as anormalidades observadas no esfregaço ou a reticulocitose confirmam o diagnóstico. Usar calçados acolchoados e evitar superfícies pavimentadas podem prevenir as recidivas em indivíduos que continuam a praticar corrida.
Um exemplo clássico e bem delineado de hemólise imune (não autoimune) envolve a incompatibilidade feto-materna ao nível do locus D do Rh. Neste caso, a mãe D negativa, após entrar em contato com os eritrócitos D positivos, pode passar a produzir anticorpos IgG anti-D. Estes anticorpos atravessam a placenta, atacando e destruindo os eritrócitos fetais. O feto torna-se ictérico e apresenta eritrócitos esferocíticos.
As hemácias fetais, agora recobertas com anticorpos maternos IgG anti-D, fixam-se aos macrófagos e monócitos fetais que contêm receptores para a porção Fc destas moléculas de IgG. A digestão macrofágica de porções da membrana eritrocitária acarreta uma considerável perda de área de superfície. Os esferócitos rígidos resultantes voltam para a circulação e são capturados pelo sistema reticuloendotelial, particularmente no baço. Como consequência, há hemólise. O anticorpo IgG é ativo em seu máximo a 37 °C. Em geral, a IgG não consegue promover uma ativação extensiva da via do complemento nem aglutinar as hemácias atacadas suspensas em salina.
O teste de antiglobulina de Coombs direto [Figura 7] é utilizado na prática clínica para detectar a IgG que recobre as hemácias. Este teste resulta negativo na mãe, cujos eritrócitos não possuem o antígeno D e, por isso, não são recobertos pelos anticorpos anti-D. O teste de Coombs indireto [Figura 7], que detecta no soro os anticorpos livres reativos contra as hemácias, resulta positivo para o soro materno, porque a mãe possui anticorpos anti-D circulantes. No feto, em contraste, o teste de Coombs direto resulta fortemente positivo, pois as hemácias fetais (que expressam o antígeno D) estão recobertas com anticorpos anti-D maternos. Os resultados fetais no teste de Coombs indireto podem ser positivos ou negativos, dependendo da quantidade de anticorpos anti-D transferida pela mãe, da avidez do anticorpo anti-D pelas hemácias D positivas fetais e da disponibilidade de sítios no antígeno D das hemácias do feto.
Estes anticorpos são descritos como quentes (atividade máxima a 37 °C [habitualmente, IgG1 ou IgG3]) ou frios (atividade máxima a 5 °C [habitualmente, IgM]). Os anticorpos também foram classificados como completos (isto é, capazes de aglutinar hemácias suspensas em salina) e incompletos (isto é, incapazes de aglutinar hemácias suspensas em salina). Sua detecção requer o uso de certas técnicas, como o teste de antiglobulina de Coombs direto [Figura 7] ou o tratamento enzimático das hemácias.193 Os autoanticorpos quentes costumam ser incompletos, enquanto as aglutininas frias (que são na maioria IgM) geralmente são completas.194
Figura 7. O teste de Coombs detecta a presença de anticorpos ou componentes do complemento humano em eritrócitos, ou a presença de anticorpos no soro. O teste é útil para o diagnóstico da doença hemolítica do recém-nascido associada ao Rh, hemólise autoimune ou potenciais reações hemolíticas transfusionais. A figura ilustra a doença hemolítica do recém-nascido associada ao Rh. No teste de Coombs direto (a), os eritrócitos fetais (hemácias) foram representados com o antígeno D fixo na superfície. Os anticorpos maternos anti-D ligam-se aos eritrócitos fetais nos sítios do antígeno D, in utero. O antissoro de Coombs, que contém anticorpos contra IgG humana, liga-se aos anticorpos anti-D existentes em uma amostra de eritrócitos fetais de lavado, fazendo-os se aglomerar (reação positiva). Os eritrócitos de lavado maternos, que não possuem antígeno D, não terão anticorpos anti-D fixos e, portanto, não serão aglutinados pelo soro de Coombs (reação negativa). No teste de Coombs indireto (b), o soro materno ou fetal é adicionado às hemácias de outra pessoa ou a painéis de eritrócitos de especificidade antigênica conhecida. Em seguida, o antissoro de Coombs é adicionado. Neste caso, ocorre aglutinamento somente quando o soro testado (p. ex., soro materno) contém anticorpos anti-D e se as hemácias selecionadas expressarem antígeno D. O teste de Coombs direto é utilizado para detectar moléculas de imunoglobulina já fixas nas hemácias, como se observa nos eritrócitos fetais em casos de doença hemolítica do recém-nascido associada ao Rh ou na anemia hemolítica autoimune. Desta forma, o teste utiliza os eritrócitos totalmente lavados do paciente. O teste de Coombs indireto é empregado para determinar se anticorpos específicos estão presentes em uma amostra de soro, sendo realizado com o soro do paciente.
A anemia hemolítica autoimune geralmente consiste em um distúrbio agudo caracterizado por hemólise extravascular. A hemólise intravascular raramente ocorre nesta condição; ela indica o andamento de uma destruição de hemácias a uma velocidade extremamente rápida ou que os mecanismos de remoção extravascular foram subjugados.
Patofisiologia. Na anemia hemolítica autoimune, por motivos ainda obscuros, há formação de autoanticorpos que são dirigidos contra componentes centrais dos eritrócitos (p. ex., antígeno Rh, antígeno de Kell,193 glicoforina A).195 Como alternativa, as hemácias do paciente são sensibilizadas com anticorpo IgG e com um componente do complemento (habitualmente, C3d). Em outras circunstâncias, todavia, parece haver fixação do complemento na superfície das hemácias por um anticorpo IgM, que subsequentemente é eliminado por lavagem. Ocasionalmente, as hemácias exibem apenas componentes do complemento, não sendo possível detectar nenhuma IgG pelo teste de Coombs. A fixação do complemento, nestes casos, pode ser explicada pela presença contínua de IgG em níveis abaixo do detectável pelo teste de antiglobulina direto usual. Como alternativa, um anticorpo IgG ou IgM fixador de complemento fixa-se à célula, mas é eluído durante os procedimentos de execução do teste.194
A severidade da hemólise está correlacionada ao número e à classe de IgG e, em raros casos, de moléculas de IgA fixas à superfície da hemácia. As hemácias recobertas com anticorpos fixam-se aos receptores dos macrófagos (FcRI, FcRII ou FcRIII) via porção Fc dos anticorpos. A ligação firme da hemácia a estes receptores de macrófagos, então, é sucedida pela remoção de uma parte da membrana da hemácia, resultando na produção de um esferócito, ou pela fagocitose de toda a hemácia.193 Níveis relativamente baixos de fixação de IgG1 às hemácias produzem um resultado positivo no teste de antiglobulina de Coombs direto, sem evidências de hemólise (cerca de 1.000 moléculas/hemácia). Contudo, níveis significativamente mais altos de autoanticorpos IgG1 por hemácia estão associados a uma franca hemólise.193 A presença combinada de IgG e componentes do complemento pode intensificar a severidade da hemólise.
Eritrócitos sensibilizados apenas contra a IgG costumam ser removidos no baço, enquanto as hemácias sensibilizadas contra IgG e complemento ou apenas contra o complemento geralmente são destruídas no fígado, uma vez que as células de Kupffer possuem receptores específicos para o componente do complemento C3b.
Diagnóstico diferencial. Foram descritas 2 variedades: uma anemia hemolítica autoimune idiopática e uma variedade secundária a outros distúrbios. Estes distúrbios primários incluem: lúpus eritematoso sistêmico, linfoma não Hodgkin (em especial, a leucemia linfocítica crônica), doença de Hodgkin, câncer, mieloma, cisto dermoide, infecção pelo HIV, linfadenopatia com disproteinemia, hepatite C196 e colite ulcerativa crônica.
Diagnóstico. As manifestações apresentadas pelo paciente variam notavelmente, de assintomáticas a severas. Pode ocorrer de um indivíduo apresentar resultado positivo no teste de Coombs ao passar pela avaliação do banco de sangue ou em doações de sangue. Em geral, é possível demonstrar que esses indivíduos possuem complemento ou a combinação de complemento e IgG (habitualmente, IgG1 ou IgG4) em suas hemácias, que, todavia, geralmente não sofrem hemólise. Em contraste, um episódio hemolítico agudo pode diminuir o hematócrito de 45% para 15% em 2 dias. Diante destas manifestações extremas, há desenvolvimento de fadiga severa e sintomas cardiorrespiratórios, aliados a icterícia, linfadenopatia e hepatoesplenomegalia.
Nos casos graves, o esfregaço sanguíneo revela a ocorrência de macrocitose, policromatofilia, esferocitose variável e autoaglutinação de hemácias. A contagem de plaquetas ocasionalmente também está deprimida (síndrome de Evans), e pode haver leucopenia. Cerca de 1/3 dos pacientes podem apresentar reticulocitopenia no momento da apresentação.197 O teste de Coombs direto resultará positivo. Qualquer um ou todos estes achados podem estar ausentes na doença branda.
A presença de complemento, IgG ou ambos nas hemácias deve ser determinada pelo uso dos reagentes de Coombs especificamente dirigidos contra IgG, IgA ou componentes do complemento. Em alguns casos, há suspeita de anemia hemolítica autoimune, porém o teste de Coombs direto resulta negativo repetidas vezes. Nestes casos, os níveis de autoanticorpos podem estar abaixo dos níveis de detectabilidade para autoanticorpos bastante ativos, como os autoanticorpos da subclasse IgG3, ou os autoanticorpos podem ser IgA ou IgM.193
Os pacientes com evidências de anemia hemolítica devem ser submetidos a um rastreamento para detecção de doenças autoimunes (p. ex., lúpus eritematoso sistêmico) e outras formas de hemólise, como HPN, doença da aglutinina fria e hemoglobinúria paroxística fria.
Tratamento. O tratamento de pacientes clinicamente afetados é voltado para a diminuição da produção de autoanticorpos e minimização do ataque macrofágico às hemácias. A terapia inicial geralmente consiste na administração de 60 a 100 mg/dia de prednisona, em doses divididas. Esta abordagem costuma produzir uma lenta diminuição da cobertura das hemácias com anticorpos, e parece interferir no ataque fagocítico aos eritrócitos recobertos. Uma resposta satisfatória ao uso de corticosteroide – indicada pela elevação da contagem de reticulócitos e pela melhora dos níveis de hemoglobina e hematócrito – pode ser evidente em 1 a 2 dias. Recomenda-se o uso da suplementação com 1 mg/dia de ácido fólico.
Após a resposta inicial à terapia, que costuma ser satisfatória, os níveis de hemoglobina e a contagem de reticulócitos podem voltar ao normal. O teste de Coombs, então, é repetido para determinar se a resposta enfraqueceu. Caso seja detectado um enfraquecimento, a dosagem de prednisona é desmamada com cuidado. Cerca de 20% dos pacientes permanecem bem por tempo indefinido, no entanto a maioria sofre de uma doença crônica traiçoeira que pode produzir recaídas súbitas com anemia abrupta. A prednisona deve ser titulada de acordo com os níveis de hemoglobina, a contagem de reticulócitos (cuja elevação indica a ocorrência de uma hemólise contínua) e o título de Coombs direto. A terapia em dias alternados deve ser considerada com o intuito de minimizar os efeitos colaterais do esteroide. Se os pacientes forem irresponsivos à terapia-padrão com prednisona, uma alternativa possivelmente efetiva consiste na administração de altas doses de dexametasona (p. ex., 40 mg/dia, por via oral, durante 4 dias consecutivos em ciclos de 28 dias).198
Se a dose de corticosteroide requerida para a terapia de longa duração produzir uma morbidade significativa, uma alternativa é agir de maneira empírica e proceder à esplenectomia ou ao uso de agentes imunossupressores. As medidas do sequestro esplênico de eritrócitos marcados com cromo 51 (51Cr-eritrócitos) não são indicadores confiáveis dos benefícios da esplenectomia. A esplenectomia raramente resulta em remissão estendida, porém é valiosa como medida poupadora de prednisona. Depois que este procedimento é realizado, a administração de uma baixa dose de prednisona (5 a 10 mg/dia) pode estabilizar a concentração de hemoglobina.
A azatioprina, que é um agente imunossupressor, ou a ciclofosfamida podem ser utilizadas como alternativas à esplenectomia. Não existem evidências confiáveis que sustentem a preferência pelo uso de um destes agentes em relação ao outro. Em casos de pacientes com doença agressiva severa, a ciclosporina tem sido utilizada com sucesso.199 O curso de azatioprina deve ser iniciado a uma dosagem de 100 a 200 mg/dia. A contagem sanguínea periférica deve ser monitorada com o intuito de prevenir uma reticulocitopenia ou neutropenia. O curso de ciclofosfamida é iniciado a uma dosagem de 100 a 200 mg/dia, com monitoramento das contagens sanguíneas e da urina. Entretanto, como a ciclofosfamida pode causar leucemia mieloide aguda ou síndrome mielodisplásica associadas à terapia, seu uso deve ser limitado [ver Leucemia mielógena crônica e outros distúrbios mieloproliferativos].
As doses de azatioprina ou ciclofosfamida precisam ser ajustadas para reduzir a contagem de leucócitos para aproximadamente 3.000/mm3. A melhora geralmente é observada em 3 a 4 semanas. Quando uma resposta é obtida, a dose de prednisona pode ser reduzida, e os níveis de hemoglobina, contagem de reticulócitos e título de Coombs podem ser monitorados para determinar a terapia mínima necessária. Em casos de pacientes com doença bastante refratária, pode ser experimentada a terapia com ciclofosfamida endovenosa nas doses utilizadas no transplante alogênico de medula óssea. Esta abordagem é nitidamente mielotóxica, e sua utilidade ainda precisa de confirmações adicionais. A administração endovenosa de uma dose alta de IgG foi utilizada no tratamento da anemia hemolítica autoimune. Em um relato, apenas 1/3 dos pacientes apresentaram uma resposta transiente, sendo necessárias doses maiores do que aquelas utilizadas em casos de PTT idiopática (isto é, 1 g/kg/dia, durante 5 dias).200,201
Existem relatos anedóticos sobre a utilidade do rituximabe para o tratamento de casos refratários e recidivantes.202,203 Entre estes relatos, há uma série retrospectiva envolvendo 11 indivíduos, dos quais 8 foram completamente responsivos à administração de 4 doses de rituximabe, com um tempo médio de seguimento de 604 dias.204 Entretanto, foi relatado 1 caso de anemia hemolítica autoimune subsequente à terapia com rituximabe para um distúrbio linfoproliferativo.205
Os pacientes com anemia sintomática necessitam de uma transfusão de hemácias, mas o banco de sangue frequentemente relata incompatilidade. Muitos bancos de sangue executam regularmente o teste de antiglobulina de Coombs direto nos receptores de hemácias. Um paciente que tenha anticorpos livres no soro exibirá uma reatividade bastante ampla e extensiva contra os painéis de doadores de hemácias e geralmente produzirá uma prova cruzada importante incompatível ao ser testado com a antiglobulina reagente. Se não forem necessária transfusões para sustentar as funções cardiorrespiratória e do sistema nervoso central, recomenda-se uma consulta imediata a um serviço de medicina transfusional.194 É inadmissível deixar que um paciente morra porque o banco de sangue não possui uma unidade de hemácias perfeitamente compatível. Se a transfusão for clinicamente indicada, o médico deve administrar as unidades menos incompatíveis de sangue disponíveis, pois foi demonstrado que estes pacientes conseguem tolerar bem até mesmo as hemácias de compatibilidade imperfeita.206
Esta condição é frequentemente indistinguível da anemia hemolítica autoimune. Existem 2 variantes: o tipo hapteno e a hemólise resultante da alteração de um antígeno de membrana.207
Tipo hapteno. Fármacos como as penicilinas e ciclosporinas ligam-se firmemente à membrana do eritrócito. Nas raras circunstâncias em que dosagens maciças de fármaco (p. ex., mais de 10 x 106 unidades/dia de penicilina) são necessárias, um fármaco acoplado a uma proteína pode atuar como hapteno e deflagrar uma resposta imune. Um anticorpo IgG que parece ser dirigido contra o complexo fármaco-hemácia é produzido.208,209 Isto leva à obtenção de um resultado positivo no teste de Coombs direto com reagente anti-IgG, bem como a um resultado negativo com o uso de um reagente anti-C3d. Quando o curso de um fármaco agressor é interrompido, a hemólise cessa em poucos dias. Em contraste, o fármaco pode ligar-se de modo frouxo e produzir um neoantígeno que gera a resposta imune.207 Nesta situação, o resultado do teste de Coombs direto com reagente anti-C3d geralmente é positivo, enquanto o resultado do teste realizado com reagente anti-IgG pode ser negativo.
Se o soro do paciente for testado contra células normais (ou seja, se o teste de Coombs indireto for usado), não ocorrerá nenhuma reação, a menos que o fármaco agressor e uma fonte de complemento sejam adicionados primeiro às hemácias normais. Suspender ou trocar o fármaco é uma ação efetiva para eliminar a hemólise, pois o anticorpo costuma ser bastante específico.
Alteração de um antígeno de membrana. Alguns fármacos podem alterar um antígeno de membrana e, deste modo, estimular a produção de anticorpos IgG que apresentam reação cruzada com o antígeno nativo. A metildopa constitui o exemplo clássico de um fármaco que causa anemia hemolítica autoimune. Outros exemplos são a levodopa, o ácido mefenâmico e a procainamida. A administração do fármaco leva à obtenção de resultado positivo no teste de Coombs direto com reagentes anti-IgG em 15 a 20% dos pacientes tratados. Contudo, a hemólise é observada em menos de 1% dos casos. O anticorpo eluído parece ser um autoanticorpo IgG clássico dirigido contra componentes Rh. O mecanismo da hemólise é idêntico ao da anemia hemolítica autoimune.210
Há relatos de que um AINH, o diclofenaco de sódio, causa uma anemia hemolítica aguda devastadora, com evidências de hemólise intra e extravascular acompanhada ocasionalmente de choque, insuficiência orgânica e até mesmo coagulação intravascular disseminada.211 Os pacientes desenvolvem autoanticorpos contra as hemácias e anticorpos fármaco-dependentes. Acredita-se que o diclofenaco de sódio se liga à superfície das hemácias e forma neoantígenos que levam à geração de autoanticorpos verdadeiros, bem como de anticorpos fármaco-dependentes. O teste de Coombs direto resulta positivo com o uso de reagentes contra IgG e contra C3d. Observa-se reatividade adicional de anticorpos com o acréscimo de metabólitos de diclofenaco de sódio obtidos a partir da urina de pacientes tratados com o fármaco. A terapia consiste na identificação da causa, suspensão do uso de diclofenaco de sódio e fornecimento de suporte ao paciente durante vários dias, até o processo cessar.211
O sangue geralmente é tipado apenas para os antígenos ABO e Rh-D. No entanto, outros antígenos também estão presentes nas hemácias. Portanto, um paciente que recebe transfusões extensivas ao longo de 1 a 2 semanas pode desenvolver uma resposta de anticorpos contra um ou mais destes outros antígenos. Os antígenos de Kell, Duffy, Kidd e Rh, além dos antígenos D, geralmente são os agentes agressores usuais. Quando o paciente com anticorpos recebe hemácias que expressam estes antígenos, pode haver uma hemólise autolimitada aguda, na maioria das vezes extravascular. Os indícios são uma história de transfusão, esferocitose em esfregaço periférico, resultado positivo no teste de Coombs direto e aparecimento recente de um anticorpo no soro do paciente (teste de Coombs indireto positivo). Em geral, não há necessidade de terapia, porém as transfusões adicionais devem ser submetidas à prova cruzada com o soro do paciente [ver Medicina transfusional]. Problemas similares são encontrados no transplante de medula óssea e outro tecido.212
A doença da aglutinina fria apresenta diversas variantes. Uma variante rara afeta adultos jovens e geralmente ocorre após a infecção por M. pneumoniae ou a mononucleose infecciosa, embora também haja relatos de vários casos associados à malária falcípara crônica. Uma variante mais comum acomete indivíduos com idade em torno de 60 anos e pode estar presente como doença da aglutinina fria idiopática, como um pródromo de um distúrbio linfoproliferativo ou imunoproliferativo, ou associada a um distúrbio linfoproliferativo já estabelecido.213
Patofisiologia. Em termos de sorologia, a doença da aglutinina fria é caracterizada por títulos elevados de aglutininas IgM (> 1:1.000 e geralmente > 1:10.000) no soro. Estes anticorpos são ativos em seu máximo a 4 °C, capazes de ativar a sequência do complemento e dirigidos contra antígenos polissacarídicos. Provavelmente, a IgM reage com os eritrócitos circulantes no sangue resfriado junto ao nariz, orelhas e queixo, onde fixa o complemento e, em seguida, se dissocia das hemácias quando estas atingem áreas mais quentes do corpo.
Na variedade pós-infecciosa deste distúrbio, a aglutinina fria IgM é oligoclonal e de curta duração. Ao contrário, a IgM é monoclonal na doença da aglutinina fria idiopática crônica ou nos casos associados à macroglobulinemia de Waldenström, leucemia linfocítica crônica ou outros linfomas. A IgM contém predominantemente cadeias leves lâmbda em pacientes com doença da aglutinina fria idiopática ou macroglobulinemia de Waldenström. Em pacientes com linfoma, todavia, a IgM contém sobretudo cadeias leves capa. Ocasionalmente, a aglutinina fria IgM é detectável como um pico de proteína M na eletroforese de proteínas séricas [ver Leucemia linfoide crônica e distúrbios de plasmócitos].
Na variante pós-Mycoplasma, os micoplasmas parecem ligar-se à superfície das hemácias no sítio de antígeno I. Esta interação receptor-ligante resulta na apresentação do antígeno I sob a forma imunogênica.214 Listeria monocytogenes contém antígeno I,213 e isto sustenta ainda mais a ideia de que alguns agentes infecciosos estimulam as aglutininas frias de ocorrência natural, assim como causam a doença da aglutinina fria pós-infecciosa.
Diagnóstico. A síndrome clínica da doença da aglutinina fria é bastante variável. Os pacientes ocasionalmente mostram apenas baixos títulos de aglutininas frias e não exibem outros sintomas nem apresentam história de pneumonia recente. Em pacientes com anemia hemolítica autoimune quente-e-fria, a hemólise associada tende a ser severa e crônica. As hemácias destes pacientes são recobertas com IgG e componentes do complemento, enquanto o soro contém títulos relativamente baixos de aglutininas frias que atuam a 30 °C e, talvez, inclusive, a temperaturas de até 37 °C.
O diagnóstico é sugerido pela detecção de anemia hemolítica acompanhada de sinais e sintomas acrais. Pode ser difícil extrair sangue, e as hemácias podem estar visivelmente aglutinadas dentro de uma seringa fria e no esfregaço sanguíneo. Os contadores de células automáticos podem contar as hemácias aglutinadas como células únicas e, assim, relatar valores absurdamente altos de VCM e CHCM. Geralmente, o laboratório detecta uma aglutinina fria amplamente ativa. O teste de Coombs direto resulta positivo com o uso de reagentes anticomplemento, porém menos frequentemente resulta positivo com anti-IgG.
Os achados que sustentam o diagnóstico de doença da aglutinina fria idiopática incluem um título elevado de aglutinina fria IgM acompanhado de ampla reatividade térmica193 e especificidade para I (reação com eritrócitos de adultos, mas ausência de reação com eritrócitos de cordão), composição puramente de cadeias leves capa, elevação ocasional dos níveis séricos de IgM absolutos e padrão de proteína M na eletroforese de proteínas séricas. A investigação deve ser voltada para a descoberta de um possível linfoma ou outro distúrbio subjacente nestes pacientes. Ao contrário, as aglutininas frias da mononucleose pós-infecciosa e pós-Mycoplasma são policlonais. O anticorpo associado à mononucleose pós-infecciosa costuma ser dirigido contra os antígenos I (hemácias de cordão).
Tratamento. As variantes pós-Mycoplasma ou mononucleose pós-infecciosa geralmente são brandas e autolimitadas e não requerem tratamento específico. Os pacientes com a variedade idiopática e que apresentam sintomas acrais devem mudar o estilo de vida, seja mudando-se para uma região de clima mais quente ou mantendo as orelhas, nariz, mãos e pés cobertos durante os períodos de frio. Tipicamente, os pacientes apresentam uma anemia crônica branda a moderada.215 Os pacientes severamente anêmicos podem necessitar de transfusões de hemácias concentradas. Nestes casos, o sangue precisa passar por uma cuidadosa prova cruzada e ser aquecido para minimizar a aglutinação a frio.
A esplenectomia e o uso de corticosteroides em geral não proporcionam benefícios em termos de controle da hemólise associada à doença da aglutinina fria. Provavelmente, as células recobertas com complemento são substancialmente removidas pelos macrófagos hepáticos, em vez dos macrófagos esplênicos, e as células produtoras de IgM são relativamente insensíveis aos efeitos dos corticosteroides. Em alguns casos, entretanto, altas doses de corticosteroides (p. ex., 100 mg/dia de prednisona) resultam em diminuição da taxa hemolítica em pacientes com títulos relativamente baixos de aglutininas frias. Na variante relativamente rara produzida pelas aglutininas frias IgG, o uso de corticosteroides e a esplenectomia podem ser benéficos. O uso de penicilamina ou outros agentes redutores contendo grupos sulfidrila não promove benefícios. Respostas satisfatórias ocasionalmente são obtidas com o uso de clorambucil em doses de 4 a 6 mg/dia. A transfusão de troca e a plasmaférese parecem ser as terapias lógicas para a doença aguda, porém estudos clínicos adicionais são necessários para avaliar estas técnicas. Foi relatado que a administração de interferon-alfa (3 x 106 U/m2, 3 vezes/semana) produz uma queda impressionante nos títulos de aglutinina fria, com diminuição dos níveis séricos de proteína monoclonal IgM e dos sintomas acrais ao longo de um período de 1 mês.193 O tratamento com rituximabe nas doses utilizadas para tratamento do linfoma não Hodgkin tem sido benéfico,216,217 sendo que a ciclofosfamida e a fludarabina também têm sido utilizadas.215
Os pacientes que sofrem do raro distúrbio de hemoglobinúria paroxística fria, mais comum em crianças, apresentam sinais e sintomas de hemólise intravascular. A hemólise está associada à presença de anticorpos IgG no soro, os quais são dirigidos contra o sistema P de hemácias. O anticorpo IgG é mais bem demonstrado por meio do teste de Donath-Landsteiner. O soro é misturado às células sanguíneas do próprio paciente ou de um indivíduo normal. A mistura é resfriada a 4 °C. Se o anticorpo IgG associado a este distúrbio estiver presente, ocorre hemólise após o aquecimento a 37 °C. Antigamente, a hemoglobinúria paroxística fria em geral era vista como uma complicação da sífilis. Contudo, recentemente, a condição foi observada associada a infecções virais e ao linfoma não Hodgkin.218 Embora a doença possa ser agudamente severa, é comum haver resolução espontânea em crianças.215 O tratamento inclui a evitação do frio. A terapia imunossupressora também é adotada.215
Os distúrbios hiperesplênicos constituem um grupo diverso de condições clínicas que compartilham aspectos comuns de esplenomegalia e hemólise. A ampliação esplênica e a hemólise ocorrem em muitos distúrbios, entre os quais: cirrose hepática com esplenomegalia congestiva, doença de Gaucher, linfoma, distúrbios do tecido conectivo, síndrome de Felty, sarcoidose, tuberculose e outras doenças infecciosas.
A estrutura única do baço é responsável por vários dos aspectos patofisiológicos do hiperesplenismo. As arteríolas esplênicas possuem alguns ramos diretos que levam aos sinusoides, porém a maioria das arteríolas terminais se abre nos cordões esplênicos. As células sanguíneas seguem dos cordões para a polpa através de fendas existentes nas paredes sinusais. As dimensões destas fendas são de aproximadamente 1 x 3 mcm.219 As células sanguíneas precisam se espremer através dos espaços longitudinais, que são revestidos por fibras reticulares, bem como entre as células adventícias localizadas fora do seio. Os macrófagos e as células endoteliais revestem o interior do seio. Contatos íntimos repetidos ocorrem entre as células sanguíneas e estes macrófagos.
No baço, o fluxo sanguíneo é lento. O pH e o nível de tensão de oxigênio nos eritrócitos diminuem, há consumo de glicose e o metabolismo celular é comprometido. O hematócrito pode aumentar, elevando ainda mais a viscosidade e a resistência ao fluxo. Em consequência, as células sanguíneas são expostas a estresses metabólicos e mecânicos na presença dos macrófagos e de outros leucócitos capazes de reconhecer danos à membrana celular. Conforme os eritrócitos envelhecem, os fagócitos removem as áreas defeituosas de suas superfícies, transformando os eritrócitos bicôncavos em esferócitos rígidos ou em fragmentos de hemácias. Estas partículas são posteriormente capturadas e removidas pelo sistema reticuloendotelial. Um baço grande possui um fluxo sanguíneo maior do que normal e expõe uma proporção inusitadamente ampla de células sanguíneas às suas atividades selecionadoras. Desta forma, no hiperesplenismo, o problema é essencialmente quantitativo. Pode haver um círculo vicioso em pacientes que sofrem de hemólise, porque a própria hemólise pode causar a esplenomegalia.
Se o baço não for apalpável, mas a situação clínica for fortemente sugestiva de esplenomegalia, pode ser útil realizar análises de ultrassonografia ou tomografia computadorizada. Como outras células sanguíneas, além dos eritrócitos, sofrem os efeitos de um baço grande, o paciente pode desenvolver pancitopenia. A menos que a doença subjacente envolva especificamente a medula óssea, a medula dos pacientes com hiperesplenismo geralmente é hiperplásica, devido à rápida regeneração de todas as linhagens celulares afetadas. O esfregaço de sangue periférico não é diagnóstico de hiperesplenismo.
Se o hiperesplenismo estiver produzindo complicações clinicamente significativas e a terapia para a doença primária do paciente não promover o colapso do baço, pode ser necessário realizar uma esplenectomia. Entretanto, a anemia não é necessariamente atribuível ao hiperesplenismo, seja qual for o tamanho do baço. A hemodiluição é outro mecanismo possível. Pacientes com esplenomegalia maciça, cujos valores de hematócrito e de hemoglobina sejam muito baixos, podem apresentar uma massa de hemácias normal, demonstrável via avaliação pela técnica do 51Cr. É comum a esplenomegalia maciça estar associada a um aumento do volume plasmático que resulta em uma notável hemodiluição. Além disso, baços significativamente ampliados podem conter um pool de eritrócitos que constitui até 25% da massa total de hemácias – em contraste com os baços normais, que não possuem este pool de hemácias. Em pacientes com esplenomegalia que apresentam uma diminuição real da massa de eritrócitos, a doença subjacente pode atuar reduzindo a produção de hemácias por meio da supressão da produção de eritropoetina, em vez da destruição acelerada. Sendo assim, é prudente determinar a massa de hemácias antes de estabelecer o diagnóstico de hiperesplenismo.
Patogênese. Dapsona, sulfassalazina, fenacetina, perclorato de sódio, nitroglicerina, fenazopiridina, primaquina,130 paraquate e análogos da vitamina K podem se inserir na fenda de ligação ao oxigênio existente na molécula de hemoglobina. Ao agirem deste modo, estes compostos conseguem gerar radicais livres oxidantes, como superóxido, radicais hidroxila livres e peróxido. Se os mecanismos redutores de proteção do eritrócitos forem vencidos [Tabela 1], a hemoglobina sofre oxidação e ocorre formação dos corpúsculos de Heinz e de metemoglobina. A sulfemoglobina também é produzida pelo ataque oxidativo. A molécula contém 1 átomo de enxofre no anel porfirínico, que confere a cor azul-esverdeada. A fonte de átomos de enxofre é obscura, mas a presença de enxofre no anel do heme o transforma em um transportador precário de oxigênio.220 A membrana eritrocitária também pode sofrer ataque oxidativo. As células danificadas são removidas junto ao sistema reticuloendotelial. A hemólise costuma ser (porém, não invarialvemente) extravascular e é possível observar os corpúsculos de Heinz em esfregaços sanguíneos tratados com corante especial. O esfregaço também pode mostrar as “células mordidas”, de hemibolha ou em ligação cruzada, que são tipicamente produzidas no ataque oxidativo aos eritrócitos [Figura 4]. Diversos danos oxidativos aparentemente fazem com que a hemoglobulina se acumule em um dos lados do eritrócito, deixando uma hemissombra encerrada junto à membrana plasmática como remanescente. Essas hemissombras podem ser detectadas no sangue periférico. Uma destruição oxidativa severa está associada a níveis aumentados de metemoglobina e à diminuição dos níveis eritrocitários de GSH. Os níveis de metemoglobina estão aumentados. Uma concentração da ordem 1,5 g/dL de metemoglobina ou de 0,5 g/dL de sulfemoglobina pode produzir o achado físico de cianose. Em contraste, 5 g/dL de desoxiemoglobina reduzida são necessários para produzir uma cianose comparável.130
Os nitritos podem oxidar a hemoglobina em metemoglobina. Consequentemente, o uso recreativo de nitritos de butil ou isobutil como estimulantes, psicodélicos e afrodisíacos tem causado problemas clínicos. Quando inalados nas quantidades habituais, estes agentes podem produzir um aumento leve a moderado dos níveis de metemoglobina, elevando os níveis normais de 1 a 2% para até 20%. Uma inalação ou ingesta mais extensiva destes agentes tem induzido uma severa metemoglobinemia, caracterizada por níveis de metemoglobina de aproximadamente 62%. Como a metemoglobina não transporta oxigênio, estes níveis elevados são acompanhados por manifestações de hipóxia tecidual, como cefaleia, falta de ar, letargia e estupor. O exame físico revela a ocorrência de taquicardia, hipotensão postural e cianose. O sangue venoso tem cor púrpura-acastanhada.221 Se a condição não for tratada, provavelmente será fatal.
Diagnóstico. O diagnóstico baseia-se na história de exposição a um fármaco ou outra toxina oxidante, aliada a achados característicos no esfregaço de sangue periférico e níveis elevados de metemoglobina.
Tratamento. O tratamento deve restaurar os níveis normais de metemoglobina. O tratamento tem início com a identificação e retirada do agente agressor. Os pacientes com metemoglobinemia severa devem ser tratados imediatamente com azul de metileno (1 a 2 mg/kg), cuja infusão é feita por via endovenosa em uma solução de 1 g/dL, durante 5 minutos. Na presença da enzima eritrocitária NAPDH-metemoglobina redutase e quantidades adequadas de NADPH doador de elétrons [Tabela 1], o azul de metileno é rapidamente reduzido a azul de leucometileno. Este produto, por sua vez, reduz rapidamente a metemoglobina a hemoglobina. A cianose, portanto, é revertida, e o paciente deve voltar a apresentar cor rosada imediatamente após a infusão. Decorridas várias horas, todavia, é possível que o paciente volte a ficar cianótico, provavelmente porque os nitratos liberados a partir dos tecidos terão reentrado no sangue periférico neste momento. A readministração de azul de metileno a uma dosagem de 1 mg/kg durante 5 minutos deve restaurar os níveis normais de hemoglobina.
O sucesso da terapia com azul de metileno requer o fornecimento adequado de NADPH. Os pacientes que apresentam anormalidades envolvendo a via da pentose fosfato, como deficiência de G6PD, são irresponsivos a esta abordagem e devem receber transfusões de troca emergenciais.221 Pacientes com níveis bastante altos de metemoglobina (pelo menos 60%) ou aqueles cujos esfregaços contêm muitas hemissombras devem ser submetidos à transfusão de troca, talvez com hemodiálise.130,221
A exposição ao chumbo resulta em encefalopatia hipertensiva, neuropatia e anemia hemolítica caracterizada pela presença de um pontilhado basofílico grosseiro nas hemácias. O mecanismo da hemólise induzida pelo chumbo é complexo, porque este metal exerce diversas ações: bloqueia a síntese de heme e, assim causa acúmulo de protoporfirina nas hemácias; produz deficiência de pirimidina 5’-nucleotidase;222 e ataca os fosfolipídios da membrana dos eritrócitos, produzindo vazamento de potássio e interferindo na atividade da ATPase de Na+/K+. As fontes de exposição do chumbo incluem tintas, tinturas de cabelo e coberturas esmaltadas de cerâmica.223
Diagnóstico. A avaliação para detecção de envenenamento com chumbo envolve a determinação dos níveis de protoporfirina eritrocitária livre (às vezes, denominados níveis de zinco protoporfirina), que estão aumentados em decorrência do bloqueio da última etapa da síntese do heme por ação do chumbo. O diagnóstico é confirmado pela medida dos níveis de chumbo no sangue e na urina.
Tratamento. Após a suspensão da exposição ao chumbo, pode-se considerar o uso de um agente quelante como o edetato de cálcio dissódico (CaNa2EDTA). O tratamento é iniciado com 0,5 a 1 g de CaNa2EDTA por via endovenosa, administrados ao longo de um período de 6 a 8 horas. O composto é administrado diariamente, por 5 dias.
Após este curso inicial, são administrados 0,5 g de CaNa2EDTA como bolo endovenoso ou injeção intramuscular, a cada 2 dias, durante 2 semanas. Neste período, os níveis de chumbo na urina devem ser monitorados. Como alternativa, o curso inicial de 5 dias de CaNa2EDTA pode ser sucedido pela administração oral de penicilamina: 1 g/dia durante os primeiros 7 dias; manutenção do fármaco durante os próximos 7 dias; e diminuição da dose de 1 g/dia nos últimos 7 dias do regime, quando os níveis de chumbo na urina são medidos ao final do dia. Outro estudo recomenda o fornecimento de 500 mg/dia de penicilamina e a continuidade desta dosagem por 60 dias após o paciente se tornar assintomático.224
Os exemplos clássicos de ataque enzimático são o veneno de cobra ou as lecitinases clostridiais (p. ex., fosfolipase C). Estas enzimas atacam os fosfolipídios da bicamada da membrana e produzem fragmentação das hemácias, esferocitose e hemólise intra e extravascular. Pode haver coagulação disseminada intravascular e choque. O pronto reconhecimento e tratamento do distúrbio primário são ações decisivas, bem como a instituição de uma terapia de suporte.
O veneno da aranha marrom, Loxosceles intermedia, libera esfingomielinases e metaloproteinases que clivam as glicoforinas da membrana dos eritrócitos. Isto, por sua vez, facilita a ativação do complemento e a lise das hemácias afetadas.225
O afogamento com água fresca ou a administração endovenosa acidental de água estéril podem causar hemólise intravascular por lise osmótica. Nestes casos, as hemácias incham e tornam-se esferoidais. O afogamento com água salgada pode induzir hemólise por desidratação de hemácias. As queimaduras causam desnaturação termoinduzida dos polipeptídeos da membrana eritrocitária, com consequente hemólise.
A malária é a causa infecciosa mais importante de hemólise. A anemia severa resultante causa a morte de um grande número de gestantes e de crianças com 2 a 5 anos de idade na África subsaariana. As espécies de Plasmodium, em particular P. falciparum, paralisam e destroem diretamente as hemácias, porém a anemia consiste em uma mistura complexa de produção comprometida de hemácias, eritropoese inefetiva e hemólise de hemácias parasitadas e não parasitadas.226,227 O diagnóstico é estabelecido com base em achados patognomônicos fornecidos pelo esfregaço sanguíneo. O tratamento é dirigido contra o parasita malárico, com fornecimento de suporte circulatório via transfusões de hemácias, quando necessário [ver Infecções por protozoários].
Outras causas infecciosas de hemólise. A infecção por M. pneumoniae e a mononucleose infecciosa podem causar hemólise por aglutininas frias. A infecção por Haemophilus influenzae do tipo b pode causar hemólise. O principal fator e virulência do H. influenzae, o polirribose ribosil fosfato [PRRP – em inglês, polyribose ribosyl phosphate], permite que o organismo escape da fagocitose. Ao ser liberado na circulação, o PRRP liga-se às hemácias. A ligação de anticorpos anti-PRRP, então, resulta na hemólise dependente do complemento.228 Os pacientes infectados por HIV ou citomegalovírus podem apresentar anemia hemolítica autoimune (ver anteriormente).229
A sepse clostridial pode ser devastadora. O aparecimento de hemoglobina livre no plasma ou a ocorrência de hemoglobinúria devem ser sugestivas da ocorrência desta infecção frequentemente letal. As espécies de Clostridium conseguem crescer de forma súbita e explosiva. Estes organismos liberam muitas enzimas, incluindo fosfolipases e proteases, que digerem as hemácias e produzem hemólise intravascular.
Algumas infecções podem causar esplenomegalia e hemólise hiperesplênica. A meningococcemia ou uma septicemia gram-negativa marcante com frequência produzem coagulação intravascular disseminada e hemólise microangiopática.
A babesiose é causada por um parasita que invade as hemácias e é transmitido a partir de um roedor-reservatório pelo mesmo carrapato causador da doença de Lyme e da erliquiose granulocítica humana. Esta doença vem sendo diagnosticada com maior frequência, particularmente em New England, Estados Unidos. Indivíduos imunocomprometidos, como aqueles infectados pelo HIV, são mais propensos a desenvolver infecções crônicas e severas. O diagnóstico é estabelecido com base no exame do esfregaço de sangue periférico, porém os métodos que empregam reação em cadeia da polimerase são mais sensíveis.230
Em pacientes com doença hepática, a anemia é mais frequentemente o resultado de um defeito de produção do que de hemólise, contudo os pacientes cirróticos podem apresentar esplenomegalia congestiva com hemólise hiperesplênica. Os macrófagos (com ou sem deficiência de vitamina B12 ou de folato) e as células-alvo (produzidas pela elevação do colesterol) também são achados comuns nestes casos.
A doença hepática severa, incluindo a cirrose alcoólica, pode resultar na formação de hemácias irregularmente espiculadas, conhecidas como acantócitos (células com esporão).231 Os acantócitos apresentam alterações de membrana (proporção diminuída de fosfolipídios em relação ao colesterol)232 que abreviam seu tempo de sobrevida e resultam no desenvolvimento de anemia hemolítica.
Em casos raros, a doença de Wilson – um distúrbio metabólico associado à deposição excessiva de cobre – é detectada pela primeira vez durante um episódio coincidente de uma dramática hemólise aguda. Um indício do diagnóstico subjacente é a existência de insuficiência hepática acompanhada de níveis baixos de fosfatase alcalina. A liberação de cobre livre no soro e sua entrada subsequente nas hemácias são consideradas o mecanismo hemolítico subjacente. Além dos níveis de hexoquinase dramaticamente afetados, o cobre intracelular parece promover a formação de radicais de oxigênio que reagem e oxidam os componentes da membrana. A penicilamina pode ser administrada a uma dosagem de 2 a 4 g/dia, por via oral, para reduzir os níveis de cobre livre, enquanto a troca de plasma pode ser utilizada nos casos severos.233 A administração de 1.000 a 2.000 UI/dia de vitamina E (alfatocoferol), durante vários dias, também pode ser útil, caso um ataque oxidativo represente um fator importante.
Os níveis plasmáticos de hemoglobina livre aumentam após a realização de um cirurgia de revascularização miocárdica com circulação extracorpórea. Acredita-se que este aumento seja causado pela ativação da via do complemento, que leva à deposição do complexo de ataque C5b-C9 na superfície das hemácias.234
Stanley L. Schrier, MD, FACP, atua como consultor junto às empresas Tularik, Inc., Amgen, Inc., Fibrogen, Inc., Apotex, Inc. e Receptron, Inc.
Elizabeth A. Price, MD, MPH, não possui relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
1. Liu SC, Derick LH. Molecular anatomy of the red blood cell membrane skeleton: structure-function relationships. Semin Hematol 1992;29:231–43.
2. Cohen CM, Gascard P. Regulation and post-translational modifi cation of erythrocyte membrane and membrane-skeletal proteins. Semin Hematol 1992;29:244–92.
3. Schwartz RS. PIG-A—the target gene in paroxysmal nocturnal hemoglobinuria. N Engl J Med 1994;330:283–4.
4. Canessa M. Red cell volume-related ion transport systems in hemoglobinopathies. Hematol Oncol Clin North Am 1991;5:495–516.
5. Hanspal M, Palek J. Biogenesis of normal and abnormal red blood cell membrane skeleton. Semin Hematol 1992;29:305–19.
6. Turrini F, Mannu F, Arese P, et al. Characterization of the autologous antibodies that opsonize erythrocytes with clustered integral membrane proteins. Blood 1993;81: 3146–52.
7. Potter CG, Potter AC, Hatton CS, et al. Variation of erythroid and myeloid precursors in the marrow and peripheral blood of volunteer subjects infected with human parvovirus (B19). J Clin Invest 1987;79:1486–92.
8. Smith BD, Segel GB. Abnormal erythrocyte endothelial adherence in hereditary stomatocytosis. Blood 1997;89: 3451–6.
9. Gallagher PG, Chang SH, Rettig MP, et al. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003;101: 4625–7.
10. Stewart GW, Amess JA, Eber SW, et al. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol 1996;93:303–10.
11. Vives Corrons JL, Besson I, Aymerich M, et al. Hereditary xerocytosis: a report of six unrelated Spanish families with leaky red cell syndrome and increased heat stability of the erythrocyte membrane. Br J Haematol 1995;90:817–22.
12. Ogburn PL Jr, Ramin KD, Danilenko-Dixon D, et al. In utero erythrocyte transfusion for fetal xerocytosis associated with severe anemia and non-immune hydrops fetalis. Am J Obstet Gynecol 2001;185:238–9.
13. Palek J, Jarolim P. Clinical expression and laboratory detection of red blood cell membrane protein mutations. Semin Hematol 1993;30:249–83.
14. Hassoun H, Vassiliadis JN, Murray J, et al. Characterization of the underlying molecular defect in spherocytosis associated with spectrin defi ciency. Blood 1997;90: 398–406.
15. Dhermy D, Galand C, Bournier O, et al. Heterogenous band 3 defi ciency in hereditary spherocytosis related to different band 3 gene defects. Br J Haematol 1997;98: 32–40.
16. Delhommeau F, Cynober T, Schischmanoff PO, et al. Natural history of hereditary spherocytosis during the fi rst year of life. Blood 2000;95:393–7.
17. Rosse WF. Hematopoiesis and the defect in paroxysmal nocturnal hemoglobinuria. J Clin Invest 1997;100:953–4.
18. Ohashi H, Hotta T, Ichikawa A, et al. Peripheral blood cells are predominantly chimeric of affected and normal cells in patients with paroxysmal nocturnal hemoglobinuria: simultaneous investigation on clonality and expression of glycophosphatidylinositol-anchored proteins. Blood 1994;83:853–9.
19. Rosti V. The molecular basis of paroxysmal nocturnal hemoglobinuria. Haematologica 2000;85:82–7.
20. Hillmen P, Lewis SM, Bessler M, et al. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med 1995;333:1253–8.
21. Socie G, Mary JY, de Gramont A, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet 1996;348: 573–7.
22. Hugel B, Socie G, Vu T, et al. Elevated levels of circulating procoagulant microparticles in patients with paroxysmal nocturnal hemoglobinuria and aplastic anemia. Blood 1999;93:3451–6.
23. Ray JG, Burows RF, Ginsberg JS, Burrows EA. Paroxysmal nocturnal hemoglobinuria and the risk of venous thrombosis: review and recommendations for management of the pregnant and nonpregnant patient. Haemostasis 2000; 30:103–17.
24. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005;106:3699–709.
25. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood 1996;87:5332–40.
26. Rosse WF, Hillmen P, Schreiber AD. Immune-mediated hemolytic anemia. Hematol Am Soc Hematol Educ Progr 2004;48–62.
27. Nishimura J, Kanakura Y, Ware RE, et al. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore) 2004;83:193–207.
28. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2006;355:1233–43.
29. Hillmen P, Muus P, Duhrsen U, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 2007;110:4123–8.
30. Sholar PW, Bell WR. Thrombolytic therapy for inferior vena cava thrombosis in paroxysmal nocturnal hemoglobinuria. Ann Intern Med 1985;103:539–41.
31. Saso R, Marsh J, Cevreska L, et al. Bone marrow transplants for paroxysmal nocturnal haemoglobinuria. Br J Haematol 1999;104:392–6.
32. Balleari E, Gatti AM, Mareni C, et al. Recombinant human erythropoietin for long-term treatment of anemia in paroxysmal nocturnal hemoglobinuria. Haematologica 1996;81: 143–7.
33. Schubert J, Scholz C, Geissler RG, et al. G-CSF and cyclosporin induce an increase of normal cells in hypoplastic paroxysmal nocturnal hemoglobinuria. Ann Hematol 1997;74:225–30.
34. Beutler E. Glucose-6-phosphate dehydrogenase def ciency: a historical perspective. Blood 2008;111:16–24.
35. Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase def ciency. Semin Hematol 1990;27:1–40.
36. Mockenhaupt FP, Mandelkow J, Till H, et al. Reduced prevalence of Plasmodium falciparum infection and of concomitant anaemia in pregnant women with heterozygous G6PD def ciency. Trop Med Int Health 2003;8: 118–24.
37. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase def ciency. Lancet 2008;371:64–74.
38. Kaplan M, Renbaum P, Levy-Lahad E, et al. Gilbert syndrome and glucose-6-phosphate dehydrogenase def ciency: a dose-dependent genetic interaction crucial to neonatal hyperbilirubinemia. Proc Natl Acad Sci U S A 1997;94:12128–32.
39. Neuberger A, Fishman S, Golik A. Hemolytic anemia in a G6PD-def cient man after inhalation of amyl nitrite (“poppers”). Isr Med Assoc J 2002;4:1085–6.
40. Duncan JR, Potter CB, Cappellini MD, et al. Aplastic crisis due to parvovirus infection in pyruvate kinase def ciency. Lancet 1983;2:14–6.
41. Nagel RL, Ranney HM. Genetic epidemiology of structural mutations of the beta-globin gene. Semin Hematol 1990; 27:342–59.
42. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
43. Steinberg MH, Rodgers GP. Pathophysiology of sickle cell disease: role of cellular and genetic modif ers. Semin Hematol 2001;38:299–306.
44. Hebbel RP. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood 1991;77:214–37.
45. Kato GJ, McGowan V, Machado RF, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 2006;107:2279–85.
46. Gladwin MT, Lancaster JR Jr, Freeman BA, Schechter AN. Nitric oxide’s reactions with hemoglobin: a view through the SNO-storm. Nat Med 2003;9:496–500.
47. Kuypers FA, Lewis RA, Hua M, et al. Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled annexin V. Blood 1996;87:1179–87.
48. Solovey A, Gui L, Key NS, Hebbel RP. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest 1998;101:1899–904.
49. Hargrave DR, Wade A, Evans JP, et al. Nocturnal oxygen saturation and painful sickle cell crises in children. Blood 2003;101:846–8.
50. Singhal A, Doherty JF, Raynes JG, et al. Is there an acute-phase response in steady-state sickle cell disease? Lancet 1993;341:651–3.
51. Hebbel RP. Perspectives series: cell adhesion in vascular biology. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest 1997;99:2561–4.
52. Grigg AP. Granulocyte colony-stimulating factor-induced sickle cell crisis and multiorgan dysfunction in a patient with compound heterozygous sickle cell/beta+ thalassemia. Blood 2001;97:3998–9.
53. Adler BK, Salzman DE, Carabasi MH, et al. Fatal sickle cell crisis after granulocyte colony-stimulating factor administration. Blood 2001;97:3313–4.
54. Okpala I, Daniel Y, Haynes R, et al. Relationship between the clinical manifestations of sickle cell disease and the expression of adhesion molecules on white blood cells. Eur J Haematol 2002;69:135–44.
55. Turhan A, Weiss LA, Mohandas N, et al. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A 2002;99:3047–51.
56. Francis RB Jr, Johnson CS. Vascular occlusion in sickle cell disease: current concepts and unanswered questions. Blood 1991;77:1405–14.
57. Steinberg MH, West MS, Gallagher D, Mentzer W. Effects of glucose-6-phosphate dehydrogenase def ciency upon sickle cell anemia. Blood 1988;71:748–52.
58. Nagel RL, Steinberg MH. Role of epistatic (modif er) genes in the modulation of the phenotypic diversity of sickle cell anemia. Pediatr Pathol Mol Med 2001;20:123–36.
59. Simckes AM, Chen SS, Osorio AV, et al. Ketorolac-induced irreversible renal failure in sickle cell disease: a case report. Pediatr Nephrol 1999;13:63–7.
60. Ballas SK. Neurobiology and treatment of pain. In: Embury S, Hebbel RP, Mohandas N, et al, editors. Sickle cell disease: basic principles and clinical practice. New York: Raven Press; 1994. p. 745.
61. Benjamin LJ, Dampier CD, Jacox AK, et al. Guideline for the management of acute and chronic pain in sickle-cell disease. Glenview (IL): American Pain Society; 1999.
62. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
63. Ballas SK. Meperidine for acute sickle cell pain in the emergency department: revisited controversy. Ann Emerg Med 2008;51:217.
64. Brugnara C, de Franceschi L, Alper SL. Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest 1993;92:520–6.
65. Bennekou P, de Franceschi L, Pedersen O, et al. Treatment with NS3623, a novel Cl- conductance blocker, ameliorates erythrocyte dehydration in transgenic SAD mice: a possible new therapeutic approach for sickle cell disease. Blood 2001;97:1451–7.
66. Ataga KI, Smith WR, De Castro LM, et al. Effi cacy and safety of the Gardos channel blocker, Senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 2008;111:3918–9.
67. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
68. Bunn HF. Induction of fetal hemoglobin in sickle cell disease. Blood 1999;93:1787–9.
69. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
70. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefi ts up to 9 years of treatment. JAMA 2003;289:1645–51.
71. Bridges KR, Barabino GD, Brugnara C, et al. A multi-parameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
72. Wilson S. Acute leukemia in a patient with sickle-cell anemia treated with hydroxyurea. Ann Intern Med 2000;133:925–6.
73. Ferster A, Sariban E, Meuleman N. Malignancies in sickle cell disease patients treated with hydroxyurea. Br J Haematol 2003;123:368–9.
74. Zimmerman SA, Schultz WH, Davis JS, et al. Sustained long-term hematologic effi cacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood 2004;103:2039–45.
75. Brawley OW, Cornelius LJ, Edwards LR, et al. NIH consensus development statement on hydroxyurea treatment for sickle cell disease: draft. NIH Consens State Sci Statements 2008;25.
76. Perrine SP, Ginder GD, Faller DV, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N Engl J Med 1993;328:81–6.
77. Atweh GF, Sutton M, Nassif I, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 1999;93:1790–7.
78. Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007;110:2749–56.
79. Bhatia M, Walters MC. Hematopoietic cell transplantation for thalassemia and sickle cell disease: past, present and future. Bone Marrow Transplant 2008;41:109–17.
80. Adams RJ, McKie VC, Hsu L, et al. Prevention of a fi rst stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 1998;339:5–11.
81. Ohene-Frempong K. Indications for red cell transfusion in sickle cell disease. Semin Hematol 2001;38(1 Suppl 1): 5–13.
82. Olivieri NF. Progression of iron overload in sickle cell disease. Semin Hematol 2001;38(1 Suppl 1):57–62.
83. Cohen AR, Martin MB. Iron chelation therapy in sickle cell disease. Semin Hematol 2001;38(1 Suppl 1):69–72.
84. Aygun B, Padmanabhan S, Paley C, Chandrasekaran V. Clinical signifi cance of RBC alloantibodies and autoantibodies in sickle cell patients who received transfusions. Transfusion 2002;42:37–43.
85. Milner PF, Kraus AP, Sebes JI, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med 1991;325:1476–81.
86. Koate P. [Cardiovascular pathology in sickle cell anemia]. Bull Acad Natl Med 1991;175:1055–62; discussion 62–3.
87. Norris S, Johnson CS, Haywood LJ. Sickle cell anemia: does myocardial ischemia occur during crisis? J Natl Med Assoc 1991;83:209–13.
88. Vichinsky E, Williams R, Das M, et al. Pulmonary fat embolism: a distinct cause of severe acute chest syndrome in sickle cell anemia. Blood 1994;83:3107–12.
89. Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 1997;89:1787–92.
90. Bellet PS, Kalinyak KA, Shukla R, et al. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N Engl J Med 1995;333:699–703.
91. Dean D, Neumayr L, Kelly DM, et al. Chlamydia pneumoniae and acute chest syndrome in patients with sickle cell disease. J Pediatr Hematol Oncol 2003;25:46–55.
92. Swerdlow PS. Red cell exchange in sickle cell disease. Hematol Am Soc Hematol Educ Progr 2006:48–53.
93. Styles LA, Schalkwijk CG, Aarsman AJ, et al. Phospholipase A2 levels in acute chest syndrome of sickle cell disease. Blood 1996;87:2573–8.
94. Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
95. Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886–95.
96. De Castro LM, Jonassaint JC, Graham FL, et al. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Am J Hematol 2008;83:19–25.
97. Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol 2006;134:109–15.
98. Darbari DS, Kple-Faget P, Kwagyan J, et al. Circumstances of death in adult sickle cell disease patients. Am J Hematol 2006;81:858–63.
99. Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and hemolytic anemia. Hematol Am Soc Hematol Educ Progr 2005:51–7.
100.Machado RF, Anthi A, Steinberg MH, et al. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA 2006;296:310–8.
101.Machado RF, Kyle Mack A, Martyr S, et al. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol 2007;136:319–25.
102.Haberkern CM, Neumayr LD, Orringer EP, et al. Cholecystectomy in sickle cell anemia patients: perioperative outcome of 364 cases from the National Preoperative Transfusion Study. Preoperative Transfusion in Sickle Cell Disease Study Group. Blood 1997;89:1533–42.
103.Serjeant GR. Sickle-cell disease. Lancet 1997;350:725–30.
104.Gupta AK, Kirchner KA, Nicholson R, et al. Effects of alpha-thalassemia and sickle polymerization tendency on the urine-concentrating defect of individuals with sickle cell trait. J Clin Invest 1991;88:1963–8.
105.Guasch A, Navarrete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol 2006;17:2228–35.
106.Falk RJ, Scheinman J, Phillips G, et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med 1992;326:910–5.
107.Bruno D, Wigfall DR, Zimmerman SA, et al. Genitourinary complications of sickle cell disease. J Urol 2001;166: 803–11.
108.Okpala I, Westerdale N, Jegede T, Cheung B. Etilefrine for the prevention of priapism in adult sickle cell disease. Br J Haematol 2002;118:918–21.
109.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998;91:288–94.
110.Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med 2005;353:2769–78.
111.Cohen AR. Sickle cell disease—new treatments, new questions. N Engl J Med 1998;339:42–4.
112.Dobson SR, Holden KR, Nietert PJ, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood 2002;99: 3144–50.
113.Mohan JS, Vigilance JE, Marshall JM, et al. Abnormal venous function in patients with homozygous sickle cell (SS) disease and chronic leg ulcers. Clin Sci (Lond) 2000;98:667–72.
114.Groves RW, Schmidt-Lucke JA. Recombinant human GM-CSF in the treatment of poorly healing wounds. Adv Skin Wound Care 2000;13(3 Pt 1):107–12.
115.Serjeant GR, Serjeant BE, Thomas PW, et al. Human parvovirus infection in homozygous sickle cell disease. Lancet 1993;341:1237–40.
116.Koshy M, Weiner SJ, Miller ST, et al. Surgery and anesthesia in sickle cell disease. Cooperative Study of Sickle Cell Diseases. Blood 1995;86:3676–84.
117.Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med 1995;333:206–13.
118.Koshy M, Burd L. Management of pregnancy in sickle cell syndromes. Hematol Oncol Clin North Am 1991;5: 585–96.
119.Embury SH. Prenatal diagnosis. In: Embury SH, Hebbel RP, Mohandas N, et al, editors. Sickle cell disease: basic principles and clinical practice. New York: Raven Press; 1994. p. 485.
120.Smith JA, Espeland M, Bellevue R, et al. Pregnancy in sickle cell disease: experience of the Cooperative Study of Sickle Cell Disease. Obstet Gynecol 1996;87:199–204.
121.Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
122.Sullivan LW. The risks of sickle-cell trait: caution and common sense. N Engl J Med 1987;317:830–1.
123.Loukopoulos D, Voskaridou E, Kalotychou V, et al. Reduction of the clinical severity of sickle cell/beta-thalassemia with hydroxyurea: the experience of a single center in Greece. Blood Cells Mol Dis 2000;26:453–66.
124.Rogers ZR. Hydroxyurea therapy for diverse pediatric populations with sickle cell disease. Semin Hematol 1997;34(3 Suppl 3):42–7.
125.Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
126.Nagel RL, Lawrence C. The distinct pathobiology of sickle cell-hemoglobin C disease. Therapeutic implications. Hematol Oncol Clin North Am 1991;5:433–51.
127.Debaun MR, Field JJ. Limitations of clinical trials in sickle cell disease: a case study of the Multi-center Study of Hydroxyurea (MSH) Trial and the Stroke Prevention (STOP) Trial. Hematol Am Soc Hematol Educ Progr 2007:482–8.
128.Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98: 838–44.
129.Vichinsky E. Hemoglobin E syndromes. Hematol Am Soc Hematol Educ Progr 2007:79–83.
130.Jaffe ER. Methemoglobinemia in the differential diagnosis of cyanosis. Hosp Pract (Off Ed) 1985;20:92–6, 101–3, 8– 10.
131.Charache S. Methemoglobinemia—sleuthing for a new cause. N Engl J Med 1986;314:776–8.
132.Manabe J, Arya R, Sumimoto H, et al. Two novel mutations in the reduced nicotinamide adenine dinucleotide (NADH)-cytochrome b5 reductase gene of a patient with generalized type, hereditary methemoglobinemia. Blood 1996;88:3208–15.
133.Percy MJ, Gillespie MJ, Savage G, et al. Familial idiopathic methemoglobinemia revisited: original cases reveal 2 novel mutations in NADH-cytochrome b5 reductase. Blood 2002;100:3447–9.
134.Weatherall DJ, Clegg JB. Thalassemia—a global public health problem. Nat Med 1996;2:847–9.
135.Adams JG 3rd, Coleman MB. Structural hemoglobin variants that produce the phenotype of thalassemia. Semin Hematol 1990;27:229–38
136.Schrier SL. Thalassemia: pathophysiology of red cell changes. Annu Rev Med 1994;45:211–8.
137.Schrier SL. Pathobiology of thalassemic erythrocytes. Curr Opin Hematol 1997;4:75–8.
138.Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol 2002;9:123–6.
139.Amer J, Goldfarb A, Fibach E. Flow cytometric measurement of reactive oxygen species production by normal and thalassaemic red blood cells. Eur J Haematol 2003;70: 84–90.
140.Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood 2000;96:2606–12.
141.Hansen RM, Hanson G, Anderson T. Failure to suspect and diagnose thalassemic syndromes. Interpretation of RBC indices by the nonhematologist. Arch Intern Med 1985;145:93–4.
142.Piomelli S. The management of patients with Cooley’s anemia: transfusions and splenectomy. Semin Hematol 1995;32:262–8.
143.Aessopos A, Berdoukas V, Tsironi M. The heart in transfusion dependent homozygous thalassaemia today—prediction, prevention and management. Eur J Haematol 2008; 80:93–106.
144.Hoffbrand AV. Oral iron chelation. Semin Hematol 1996;33:1–8.
145.Brittenham GM, Griffi th PM, Nienhuis AW, et al. Effi cacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med 1994;331:567–73.
146.Olivieri NF, Nathan DG, MacMillan JH, et al. Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med 1994;331:574–8.
147.Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood 2006;107:3733–7.
148.Pennell DJ, Berdoukas V, Karagiorga M, et al. Randomized controlled trial of deferiprone or deferoxamine in betathalassemia major patients with asymptomatic myocardial siderosis. Blood 2006;107:3738–44.
149.Tanner MA, Galanello R, Dessi C, et al. Combined chelation therapy in thalassemia major for the treatment of severe myocardial siderosis with left ventricular dysfunction. J Cardiovasc Magn Reson 2008;10:12.
150.Tanner MA, Galanello R, Dessi C, et al. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation 2007;115:1876–84.
151.Wood JC. Diagnosis and management of transfusion iron overload: the role of imaging. Am J Hematol 2007;82(12 Suppl):1132–5.
152.Wood JC, Enriquez C, Ghugre N, et al. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood 2005;106:1460–5.
153.Anderson LJ, Wonke B, Prescott E, et al. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet 2002;360:516–20.
154.Wood JC. Magnetic resonance imaging measurement of iron overload. Curr Opin Hematol 2007;14:183–90.
155.Cohen AR, Galanello R, Pennell DJ, et al. Thalassemia. Hematol Am Soc Hematol Educ Progr 2004:14–34.
156.Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Survival and complications in thalassemia. Ann N Y Acad Sci 2005;1054:40–7.
157.Schrier SL, Angelucci E. New strategies in the treatment of the thalassemias. Annu Rev Med 2005;56:157–71.
158.Voskaridou E, Anagnostopoulos A, Konstantopoulos K, et al. Zoledronic acid for the treatment of osteoporosis in patients with beta-thalassemia: results from a single-center, randomized, placebo-controlled trial. Haematologica 2006;91:1193–202.
159.Shamshirsaz AA, Bekheirnia MR, Kamgar M, et al. Metabolic and endocrinologic complications in beta-thalassemia major: a multicenter study in Tehran. BMC Endocr Disord 2003;3:4.
160.Davis BA, O’Sullivan C, Jarritt PH, Porter JB. Value of sequential monitoring of left ventricular ejection fraction in the management of thalassemia major. Blood 2004; 104:263–9.
161.Barnett CF, Hsue PY, Machado RF. Pulmonary hypertension: an increasingly recognized complication of hereditary hemolytic anemias and HIV infection. JAMA 2008; 299:324–31.
162.Phrommintikul A, Sukonthasarn A, Kanjanavanit R, Nawarawong W. Splenectomy: a strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart 2006;92:1467–72.
163.Tam DH, Farber HW. Pulmonary hypertension and betathalassemia major: report of a case, its treatment, and a review of the literature. Am J Hematol 2006;81:443–7.
164.Derchi G, Forni GL, Formisano F, et al. Effi cacy and safety of sildenafi l in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica 2005;90:452–8.
165.Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR. Complications of beta-thalassemia major in North America. Blood 2004;104:34–9.
166.Ansar MM, Kooloobandi A. Prevalence of hepatitis C virus infection in thalassemia and haemodialysis patients in north Iran-Rasht. J Viral Hepat 2002;9:390–2.
167.Cappellini MD, Grespi E, Cassinerio E, et al. Coagulation and splenectomy: an overview. Ann N Y Acad Sci 2005;1054:317–24.
168.Ataga KI, Cappellini MD, Rachmilewitz EA. Betathalassaemia and sickle cell anaemia as paradigms of hypercoagulability. Br J Haematol 2007;139:3–13.
169.Panigrahi I, Agarwal S. Thromboembolic complications in beta-thalassemia: beyond the horizon. Thromb Res 2007; 120:783–9.
170.Bradai M, Abad MT, Pissard S, et al. Hydroxyurea can eliminate transfusion requirements in children with severe beta-thalassemia. Blood 2003;102:1529–30.
171.Alebouyeh M, Moussavi F, Haddad-Deylami H, Vossough P. Hydroxyurea in the treatment of major beta-thalassemia and importance of genetic screening. Ann Hematol 2004; 83:430–3.
172.Lal A, Vichinsky E. The role of fetal hemoglobin-enhancing agents in thalassemia. Semin Hematol 2004;41(4 Suppl 6):17–22.
173.Fathallah H, Atweh GF. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematol Am Soc Hematol Educ Progr 2006:58–62.
174.Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. Br J Haematol 2007;138:291–304.
175.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2- associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003;302:415–9.
176.Rees DC, Styles L, Vichinsky EP, et al. The hemoglobin E syndromes. Ann N Y Acad Sci 1998;850:334–43.
177.Issaragrisil S, Visuthisakchai S, Suvatte V, et al. Brief report: transplantation of cord-blood stem cells into a patient with severe thalassemia. N Engl J Med 1995;332: 367–9.
178.Lucarelli G, Galimberti M, Giardini C, et al. Bone marrow transplantation in thalassemia. The experience of Pesaro. Ann N Y Acad Sci 1998;850:270–5.
179.Gaziev J, Lucarelli G. Stem cell transplantation for hemoglobinopathies. Curr Opin Pediatr 2003;15:24–31.
180.Angelucci E, Muretto P, Lucarelli G, et al. Phlebotomy to reduce iron overload in patients cured of thalassemia by bone marrow transplantation. Italian Cooperative Group for Phlebotomy Treatment of Transplanted Thalassemia Patients. Blood 1997;90:994–8.
181.Giardini C, Galimberti M, Lucarelli G, et al. Alpha-interferon treatment of chronic hepatitis C after bone marrow transplantation for homozygous beta-thalassemia. Bone Marrow Transplant 1997;20:767–72.
182.Domenica Cappellini M, Graziadei G, Ciceri L, et al. Oral isobutyramide therapy in patients with thalassemia intermedia: results of a phase II open study. Blood Cells Mol Dis 2000;26:105–11.
183.183 Reich S, Buhrer C, Henze G, et al. Oral isobutyramide reduces transfusion requirements in some patients with homozygous beta-thalassemia. Blood 2000;96:3357–63.
184.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica 2007;92:583–8.
185.Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood 2003;101: 791–800.
186.Kazazian HH Jr. The thalassemia syndromes: molecular basis and prenatal diagnosis in 1990. Semin Hematol 1990;27:209–28.
187.Dover GJ, Valle D. Therapy for beta-thalassemia—a paradigm for the treatment of genetic disorders. N Engl J Med 1994;331:609–10.
188.Ross CN, Reuter H, Scott D, Hamilton DV. Microangiopathic haemolytic anaemia and systemic vasculitis. Br J Rheumatol 1996;35:377–9.
189.Rytting M, Worth L, Jaffe N. Hemolytic disorders associated with cancer. Hematol Oncol Clin North Am 1996;10: 365–76.
190.Mach-Pascual S, Samii K, Beris P. Microangiopathic hemolytic anemia complicating FK506 (tacrolimus) therapy. Am J Hematol 1996;52:310–2.
191.Kojouri K, Vesely SK, George JN. Quinine-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: frequency, clinical features, and long-term outcomes. Ann Intern Med 2001;135:1047–51.
192.Freedman A, Afonja O, Chang MW, et al. Cutaneous anthrax associated with microangiopathic hemolytic anemia and coagulopathy in a 7-month-old infant. JAMA 2002;287:869–74.
193.Engelfriet CP, Overbeeke MA, von dem Borne AE. Autoimmune hemolytic anemia. Semin Hematol 1992;29:3–12.
194.Jefferies LC. Transfusion therapy in autoimmune hem olytic anemia. Hematol Oncol Clin North Am 1994;8: 1087–104.
195.Leddy JP, Falany JL, Kissel GE, et al. Erythrocyte membrane proteins reactive with human (warm-reacting) anti-red cell autoantibodies. J Clin Invest 1993;91:1672–80.
196.Ramos-Casals M, Garcia-Carrasco M, Lopez-Medrano F, et al. Severe autoimmune cytopenias in treatment-naive hepatitis C virus infection: clinical description of 35 cases. Medicine (Baltimore) 2003;82:87–96.
197.Liesveld JL, Rowe JM, Lichtman MA. Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood 1987;69:820–6.
198.Meyer O, Stahl D, Beckhove P, et al. Pulsed high-dose dexamethasone in chronic autoimmune haemolytic anaemia of warm type. Br J Haematol 1997;98:860–2.
199.Emilia G, Messora C, Longo G, Bertesi M. Long-term salvage treatment by cyclosporin in refractory autoimmune haematological disorders. Br J Haematol 1996;93:341–4.
200.Flores G, Cunningham-Rundles C, Newland AC, Bussel JB. Effi cacy of intravenous immunoglobulin in the treatment of autoimmune hemolytic anemia: results in 73 patients. Am J Hematol 1993;44:237–42.
201.Collins PW, Newland AC. Treatment modalities of auto-immune blood disorders. Semin Hematol 1992;29:64–74.
202.Gottardo NG, Baker DL, Willis FR. Successful induction and maintenance of long-term remission in a child with chronic relapsing autoimmune hemolytic anemia using rituximab. Pediatr Hematol Oncol 2003;20:557–61.
203.Trape G, Fianchi L, Lai M, et al. Rituximab chimeric anti-CD20 monoclonal antibody treatment for refractory hemolytic anemia in patients with lymphoproliferative disorders. Haematologica 2003;88:223–5.
204.D’Arena G, Califano C, Annunziata M, et al. Rituximab for warm-type idiopathic autoimmune hemolytic anemia: a retrospective study of 11 adult patients. Eur J Haematol 2007;79:53–8.
205.Jourdan E, Topart D, Richard B, et al. Severe autoimmune hemolytic anemia following rituximab therapy in a patient with a lymphoproliferative disorder. Leuk Lymphoma 2003;44:889–90.
206.Salama A, Berghofer H, Mueller-Eckhardt C. Red blood cell transfusion in warm-type autoimmune haemolytic anaemia. Lancet 1992;340:1515–7.
207.Salama A, Mueller-Eckhardt C. Immune-mediated blood cell dyscrasias related to drugs. Semin Hematol 1992;29: 54–63.
208.Garratty G. Immune cytopenia associated with antibiotics. Transfus Med Rev 1993;7:255–67.
209.Kopicky JA, Packman CH. The mechanisms of sulfonylurea-induced immune hemolysis: case report and review of the literature. Am J Hematol 1986;23:283–8.
210.Petz LD. Drug-induced autoimmune hemolytic anemia. Transfus Med Rev 1993;7:242–54.
211.Salama A, Kroll H, Wittmann G, et al. Diclofenac-induced immune haemolytic anaemia: simultaneous occurrence of red blood cell autoantibodies and drug-dependent antibodies. Br J Haematol 1996;95:640–4.
212.Ramsey G. Red cell antibodies arising from solid organ transplants. Transfusion 1991;31:76–86.
213.Silberstein LE. B-cell origin of cold agglutinins. Adv Exp Med Biol 1994;347:193–205.
214.Loomes LM, Uemura K, Childs RA, et al. Erythrocyte receptors for Mycoplasma pneumoniae are sialylated oligosaccharides of Ii antigen type. Nature 1984;307: 560–3.
215.Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev 2008;22:1–15.
216.Berentsen S, Tjonnfjord GE, Brudevold R, et al. Favourable response to therapy with the anti-CD20 monoclonal antibody rituximab in primary chronic cold agglutinin disease. Br J Haematol 2001;115:79–83.
217.Mori A, Tamaru J, Sumi H, Kondo H. Benefi cial effects of rituximab on primary cold agglutinin disease refractory to conventional therapy. Eur J Haematol 2002;68:243–6.
218.Sharara AI, Hillsley RE, Wax TD, Rosse WF. Paroxysmal cold hemoglobinuria associated with non-Hodgkin’s lymphoma. South Med J 1994;87:397–9.
219.Rosse WF. The spleen as a filter. N Engl J Med 1987;317: 704–6.
220.Lu HC, Shih RD, Marcus S, et al. Pseudomethemoglobinemia: a case report and review of sulfhemoglobinemia. Arch Pediatr Adolesc Med 1998;152:803–5.
221.Coleman MD, Coleman NA. Drug-induced methaemoglobinaemia. Treatment issues. Drug Saf 1996;14:394–405.
222.Valentine WN, Paglia DE, Fink K, Madokoro G. Lead poisoning: association with hemolytic anemia, basophilic stippling, erythrocyte pyrimidine 5’-nucleotidase defi - ciency, and intraerythrocytic accumulation of pyrimidines. J Clin Invest 1976;58:926–32.
223.Brown AC, Steensma DP, Tefferi A. The possibility of occult lead poisoning. Mayo Clin Proc 2008;83:368.
224.Carton JA, Maradona JA, Arribas JM. Acute-subacute lead poisoning. Clinical fi ndings and comparative study of diagnostic tests. Arch Intern Med 1987;147:697–703.
225.Tambourgi DV, De Sousa Da Silva M, Billington SJ, et al. Mechanism of induction of complement susceptibility of erythrocytes by spider and bacterial sphingomyelinases. Immunology 2002;107:93–101.
226.Casals-Pascual C, Roberts DJ. Severe malarial anaemia. Curr Mol Med 2006;6:155–68.
227.Nagel RL. Malarial anemia. Hemoglobin 2002;26:329–43.
228.Shurin SB, Anderson P, Zollinger J, Rathbun RK. Pathophysiology of hemolysis in infections with Hemophilus infl uenzae type b. J Clin Invest 1986;77:1340–8.
229.van Spronsen DJ, Breed WP. Cytomegalovirus-induced thrombocytopenia and haemolysis in an immunocompetent adult. Br J Haematol 1996;92:218–20.
230.Krause PJ, Spielman A, Telford SR 3rd, et al. Persistent parasitemia after acute babesiosis. N Engl J Med 1998; 339:160–5.
231.Owen JS, Brown DJ, Harry DS, et al. Erythrocyte echinocytosis in liver disease. Role of abnormal plasma high density lipoproteins. J Clin Invest 1985;76:2275–85.
232.Allen DW, Manning N. Cholesterol-loading of membranes of normal erythrocytes inhibits phospholipid repair and arachidonoyl-CoA:1-palmitoyl-sn-glycero-3-phosphocholine acyl transferase. A model of spur cell anemia. Blood 1996;87:3489–93.
233.Asfaha S, Almansori M, Qarni U, Gutfreund KS. Plasmapheresis for hemolytic crisis and impending acute liver failure in Wilson disease. J Clin Apher 2007;22:295–8.
234.Salama A, Hugo F, Heinrich D, et al. Deposition of terminal C5b-9 complement complexes on erythrocytes and leukocytes during cardiopulmonary bypass. N Engl J Med 1988;318:408–14.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.