(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Nefrologia
acp-medicine Nefrologia
Última revisão: 15/01/2014
Comentários de assinantes: 0
Assistant Professor, Department of Medicine, Division of Nephrology and Hypertension, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC
Vimal K. Derebail, MD, MPH
Assistant Professor, Department of Medicine, Division of Nephrology and Hypertension, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC
Abhijit V. Kshirsagar, MD, MPH
Associate Professor, Department of Medicine, Division of Nephrology and Hypertension, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC
Ronald J. Falk, MD, FACP
Allan Brewster Distinguished Professor of Medicine; Chief, Division of Nephrology and Hypertension; Director, UNC Kidney Center, Department of Medicine, Division of Nephrology and Hypertension, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC
Artigo original: McGregor JG, Derebail VK, Kshirsagar AV, Falk RJ. Vascular diseases of the kidney. ACP Medicine. 2007;1-16.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figura 1 – Dr. J. Charles Jennette, Department of Pathology, University of North Carolina at Chapel Hill University School of Medicine. Figura 2 – Dr. Romulo E. Colindres, Department of Medicine, Division of Nephrology and Hypertension, University of North Carolina at Chapel Hill University School of Medicine. Figuras 3, 5, 6 e 8 – Jerome Jacobs. Figura 4 – Gregory Bishop. Figura 7 – Cortesia de Dr. P. E. de Jong, Department of Medicine, State University, the Netherlands.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon
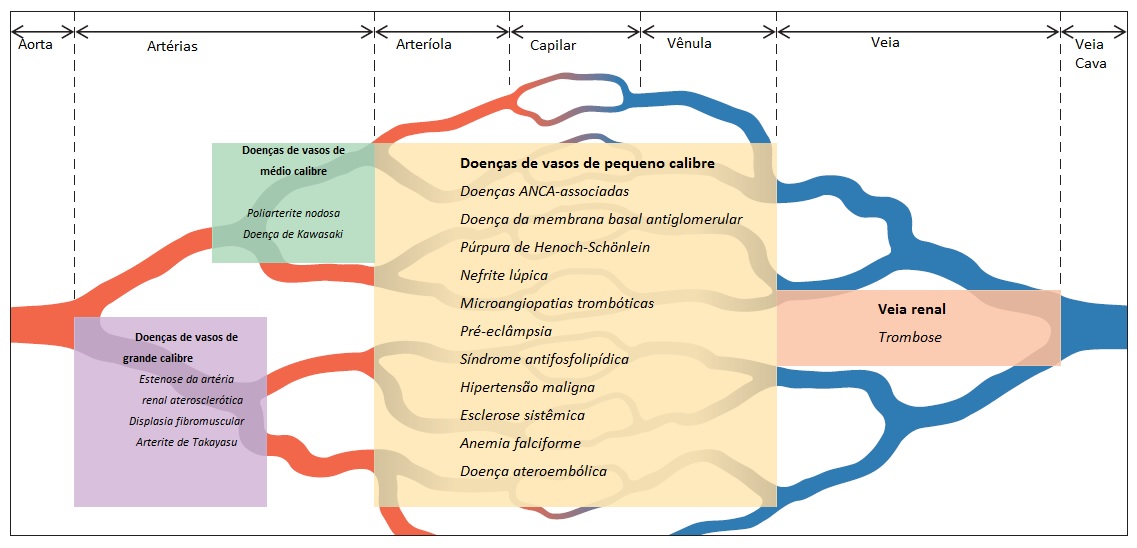
Existem vários processos patológicos locais e sistêmicos que afetam a árvore vascular renal [Figura 1]. Embora os mecanismos subjacentes possam ser distintos, as vasculopatias renais são todas caracteristicamente causadoras de graus variáveis de obstrução vascular, que levam ao eventual comprometimento do fluxo sanguíneo renal. A diminuição do fluxo sanguíneo renal frequentemente resulta em 2 adaptações fisiológicas importantes: uma elevação da pressão arterial e uma diminuição da taxa de filtração glomerular (TFG). A hipertensão e a insuficiência renal muitas vezes são indícios significativos do diagnóstico de vasculopatia renal. O rápido diagnóstico da vasculopatia subjacente é necessário à prevenção do desenvolvimento de uma doença renal irreversível, incluindo a doença renal em estágio terminal (DRET).

Figura 1. Visão geral da vasculopatia renal.
ANCA = anticorpos anticitoplasma de neutrófilo; DK = doença de Kawasaki; EAR = estenose arterial renal; DFM = displasia fibromuscular; MAT = microangiopatias trombóticas; PN = poliarterite nodosa SAF = síndrome antifosfolípide
Este capítulo discute as principais vasculopatias renais, que são classificadas de acordo com o tamanho do vaso afetado. Os vasos de grande calibre incluem as principais artérias renais e seus ramos principais, bem como a veia renal e seus ramos principais. Os vasos de médio calibre referem-se principalmente às artérias. Os vasos de pequeno calibre incluem as arteríolas, os capilares e as vênulas.
A estenose arterial renal (EAR) diz respeito a um grupo de doenças caracterizadas pelo estreitamento da artéria renal principal e de seus ramos. A aterosclerose e a displasia fibromuscular (DFM) são as 2 causas principais de EAR. Este capítulo enfoca principalmente a doença aterosclerótica, que é responsável por mais de 90% dos casos de EAR.
A EAR aterosclerótica foi definida como sendo um estreitamento de lúmen de pelo menos 50%, embora alguns defendam que o estreitamento deve ser de no mínimo 70% para ser considerado significativo do ponto de vista funcional. A aterosclerose da artéria renal é um achado relativamente comum tanto em homens como em mulheres, após a 5ª década da vida. Entretanto, a prevalência precisa da EAR aterosclerótica é desconhecida, pois não existem testes economicamente acessíveis para a população em geral. A avaliação da EAR em populações altamente selecionadas, como aquelas com alta carga de aterosclerose (ou seja, indivíduos com diabetes, episódio recente de acidente vascular cerebral [AVC] ou infarto agudo do miocárdio [IAM], vasculopatia periférica), revelou prevalência de 10 a 30%.1-4 Como seria esperado, os indivíduos com EAR aterosclerótica apresentam alto risco de doença cardiovascular.5,6
A EAR aterosclerótica é encontrada em indivíduos brancos e afrodescendentes.7,8 Não há relato de diferenças de prevalência entre os sexos. Os fatores de risco de desenvolvimento de EAR aterosclerótica incluem a hipercolesterolemia, idade avançada, tabagismo, hipertensão e diabetes melito.
As lesões ateroscleróticas ocorrem principalmente nas artérias de médio e grande calibre. Estas lesões são caracterizadas pela infiltração da camada íntima por macrófagos e monócitos, bem como pela deposição de lipoproteína de baixa densidade (LDL). A maioria dos casos de EAR aterosclerótica envolve a abertura da artéria renal a partir da aorta (óstio), o terço proximal da artéria renal principal ou a aorta perirrenal. A aterosclerose intrarrenal difusa e segmentar pode ser observada nos casos avançados de EAR, sobretudo em pacientes com diabetes melito.
A DFM constitui um grupo de doenças pouco comuns, que são causadoras comprovadas de estenose em vários leitos vasculares, notavelmente nas artérias renal e carótida. Estas doenças são caracterizadas por alterações displásicas não inflamatórias e não ateroscleróticas de qualquer uma das 3 camadas da artéria – íntima, média ou adventícia. A prevalência da DFM é pouco conhecida, porém a condição é provavelmente rara. A DFM é responsável por cerca de 10% de todos os casos de EAR, e esta, por sua vez, é considerada causadora de menos de 5% de todos os casos de hipertensão.
A vasculatura renal é mais frequentemente afetada pela DFM, sendo que a DFM renovascular corresponde a 60 a 75% de todos os casos de DFM. O envolvimento renal bilateral ocorre em 35% de todos os casos de envolvimento renal. A DFM dos vasos renais tende a afetar mulheres com menos de 50 anos – tipicamente, mulheres com menos de 30 anos de idade. A causa da DFM atualmente é desconhecida. A fibroplasia medial, que é o subtipo mais comum de DFM, caracteriza-se pela predominância de material fibrótico na camada média, com preservação da íntima e da adventícia. A condição afeta os 2/3 terços distais da artéria renal principal e de seus ramos. A natureza segmentar a fibroplasia medial, aliada à dilatação pós-estenótica localizada, resulta na clássica aparência de “colar de contas” à angiografia. As fibroplasias da íntima, que são o 2º tipo mais comum de DFM, caracterizam-se pela predominância de material fibrótico na íntima. A condição afeta a porção média da artéria renal principal e seus ramos. A avaliação angiográfica tipicamente revela um estreitamento uniforme e longo ou uma estenose concêntrica e focal. A hiperplasia de adventícia, que é um distúrbio extremamente raro, afeta as porções média a distal da artéria renal.
O estreitamento das artérias renais pode ter consequências clínicas significativas: elevação da pressão arterial e insuficiência renal. O mecanismo de elevação da pressão arterial é semelhante àquele demonstrado por Goldblatt, na década de 1930, empregando o modelo de clipagem de ligação da artéria renal. Dr. Goldblatt conseguiu induzir o desenvolvimento de hipertensão com a ligação de uma artéria renal. Posteriormente, foi entendido que a ligação causou ativação do sistema renina-angiotensina e reabsorção de sódio a partir da secreção da aldosterona.9
Os mecanismos por trás da insuficiência renal são obscuros. A insuficiência renal não é o mero resultado direto da diminuição do fluxo sanguíneo renal. Os modelos experimentais são imperfeitos como guia, pois empregam métodos que induzem estreitamento agudo, e não o estreitamento progressivo associado à aterosclerose e à DFM. Além disso, o agrupamento da aterosclerose a outros fatores de risco de insuficiência renal – notavelmente, o diabetes melito – dificulta a atribuição da insuficiência renal apenas à EAR aterosclerótica. Quando a insuficiência renal ocorre no contexto de EAR aterosclerótica, é comumente denominada nefropatia isquêmica.
Alguns achados clínicos e laboratoriais são sugestivos de EAR. O aparecimento abrupto ou piora da hipertensão em um indivíduo que apresenta ruídos abdominais e hipocalemia muitas vezes é considerado uma manifestação clássica da EAR. Outros indícios importantes incluem a presença de vasculopatia aterosclerótica em outros leitos, sobretudo nos vasos arteriais dos membros inferiores, aorta e carótidas; vasculopatia periférica; e doença aórtica. As anomalias renais são inespecíficas e incluem a azotemia inexplicável; azotemia induzida pelo uso de inibidor de enzima conversora de angiotensina (iECA) ou bloqueadores do receptor de angiotensina II (BRA); e uma diferença de tamanho dos rins, que é demonstrada por estudo radiológico. Além disso, os episódios recorrentes de edema pulmonar repentino no contexto de uma função sistólica normal do ventrículo esquerdo podem estar associados à ocorrência de EAR bilateral.
Em pacientes com quadro clínico compatível, a avaliação para EAR começa pela realização dos exames de radiografia. Muitos clínicos começam esta avaliação solicitando uma ultrassonografia renal, dada a natureza não invasiva do exame, bem como a ausência de efeitos colaterais e a facilidade de acesso. Entretanto, a ultrassonografia renal apresenta baixa sensibilidade e especificidade para EAR, e a observação de uma discrepância entre os tamanhos dos rins neste exame (isto é, > 2 cm no eixo longitudinal) sugere apenas uma história prévia de estenose arterial. Os exames que definem a anatomia ou funcionalidade são superiores à ultrassonografia renal para fins de avaliação. A escolha de um destes exames depende das características do paciente e das capacidades do centro médico (p. ex., alguns centros estão mais bem capacitados do que outros a realizar angiografia por ressonância magnética). No caso de pacientes com função renal relativamente preservada (isto é, concentração sérica de creatinina = 2 mg/dL), deve ser obtido um renograma com captopril. Este exame, com o emprego de uma variedade de radioisótopos, possui sensibilidade de 93% e especificidade de 70% para EAR.10 Uma perda da função renal, seja por depleção de volume ou doença renal intrínseca, acarreta diminuição da sensibilidade e especificidade do renograma com captopril. Desta forma, recomenda-se que os pacientes recebam hidratação adequada antes de realizarem o exame. A angiografia por ressonância magnética (ARM) é recomendada para os casos em que a função renal intrínseca está diminuída a ponto de a concentração sérica de creatinina estar acima de 2 mg/dL. A ARM intensificada por gadolínio possui sensibilidade de 88% e especificidade de 98% para EAR.11 Esta é uma técnica particularmente eficiente para a identificação de lesões de óstio. Todavia, em pacientes com insuficiência renal avançada ou DRET, o gadolínio deve ser usado com cautela, pois está associado ao desenvolvimento de uma condição cutânea incapacitante, conhecida como dermopatia fibrosante nefrogênica.12 A angiografia por tomografia com contraste foi proposta como teste radiológico alternativo, apresentando sensibilidade de 94% e especificidade de 63% para a EAR.13 Um ponto fraco deste teste reside no fato de os pacientes serem expostos ao meio de contraste iodado. A ultrassonografia com doppler, embora seja minimamente invasiva, depende de um operador, e os conhecimentos especializados necessários à sua utilização somente são disponibilizados em alguns centros acadêmicos.
O padrão-ouro do diagnóstico de EAR continua sendo a arteriografia renal. Devido aos riscos comprovados associados à exposição ao meio de contraste e à manipulação das placas ateroscleróticas, é importante limitar o uso da arteriografia renal aos pacientes que apresentam resultados positivos nos exames não invasivos. Alguns médicos recomendam a realização de uma avaliação funcional para excluir a hipótese de que EAR seja uma consequência fisiológica do estreitamento arterial, como ocorre na amostragem de renina a partir da veia renal.
Estenose arterial renal (EAR) aterosclerótica. Os exames radiográficos demonstraram que as lesões da EAR aterosclerótica evoluem, porém os exames funcionais que medem a TFG ou seus índices subordinados falharam em demonstrar de modo consistente a insuficiência renal progressiva. Desta forma, ainda não está claro se a intervenção altera o curso natural do processo patológico. Em consequência, a escolha entre tratamento médico e intervenção para EAR aterosclerótica ainda é controversa.14 O tratamento médico com vários agentes anti-hipertensivos continua sendo o método mais comum de tratamento da hipertensão e da insuficiência renal. Os agentes que preferencialmente diminuem o tônus arterial eferente – iECA e BRA – são contraindicados, devido ao risco de indução de insuficiência renal aguda. A angioplastia percutânea foi apresentada como uma alternativa favorável, porém numerosos estudos demonstraram que este exame não é superior à terapia farmacológica convencional.15 A angioplastia com colocação de stent foi elogiada como sendo tecnicamente superior à angioplastia isolada.16 Entretanto, uma revisão abrangente mostrou que este procedimento cura a hipertensão em menos de 20% dos pacientes e não promove benefícios evidentes em termos de função renal.17 De modo semelhante, os estudos de seguimento prolongado falharam em demonstrar que a revascularização cirúrgica da lesão estenótica é superior à terapia farmacológica.18 Assim, está sendo formado um consenso de que a terapia farmacológica é provavelmente equivalente às intervenções mais invasivas na vasta maioria dos pacientes com EAR aterosclerótica.
Não há agentes anti-hipertensivos conhecidos que sejam ideais. É preciso ter cautela ao usar iECA e BRA. Entretanto, o uso destes agentes não deve ser evitado, dada sua superioridade em relação aos outros agentes disponíveis na diminuição da proteinúria e retardo da progressão da doença renal crônica. Após o início de um curso de iECA ou de um regime de BRA, os exames laboratoriais de avaliação da função renal devem ser realizados em 1 a 2 semanas. Se os resultados destes exames mostrarem uma diminuição da função renal maior que 50%, o agente em uso deve ser descontinuado. Caso seja detectada uma redução da função renal da ordem de 25 a 50%, indica-se, então, que a dose do fármaco seja diminuída, sendo necessário realizar um teste de função renal dentro de 7 a 10 dias.19
Além das medicações anti-hipertensivas, o tratamento ideal da EAR envolve o tratamento das dislipidemias, controle do diabetes (quando apropriado) e aconselhamento para abandono do tabagismo.
Embora não se recomende de rotina a angioplastia para melhora da hipertensão ou da função renal, as preferências do paciente podem mudar a decisão em favor desta intervenção. Neste caso, existem certos fatores preditivos de falha da intervenção percutânea (p. ex., angioplastia ou colocação de stent) que podem ser úteis para guiar o processo de tomada de decisão. Os fatores correlacionados com a melhora incluem o envolvimento unilateral, estenose de menos de 50% do diâmetro luminal, tamanho basal do rim inferior a 8 a 9 cm no eixo longitudinal, nefropatia diabética e proteinúria severa. Recentemente, o índice resistivo passou a ser alvo de interesse como método preditivo do resultado. O índice resistivo é uma estimativa da resistência cumulativa das artérias segmentares e, possivelmente, dos vasos de menor calibre. A análise de variável única demonstrou que um índice resistivo alto é melhor como fator preditivo de falta de efeito da intervenção percutânea, em comparação ao nível de função renal, tamanho do rim e presença de proteinúria.20 O ultrassom com doppler é uma modalidade altamente dependente do operador, usada para prever o resultado da intervenção percutânea, e recomendada para uso de rotina por pouquíssimos centros devidamente especializados.
Displasia fibromuscular (DFM). A maioria das formas de DFM apresenta evolução radiográfica, mas ainda não foi esclarecido se há algum tipo de desenvolvimento associado de insuficiência renal. A intervenção percutânea é considerada o tratamento-padrão. Entretanto, a realização de estudos comparativos amplos é inviável, dada a relativa raridade destas condições. A angioplastia e a colocação de stent curam totalmente a hipertensão em cerca de 22% dos pacientes.21 A revascularização cirúrgica, embora esteja associada a um índice de cura maior (cerca de 79% em 1 ano),22 provavelmente causa mais morbidade a curto prazo do que a angioplastia. O reparo cirúrgico dos aneurismas (as “contas” observadas na arteriografia) torna-se necessário em casos de pacientes com hipertensão não controlada ou que estejam gestantes.
A arterite de Takayasu é uma arterite inflamatória que envolve a aorta e seus ramos. Afeta principalmente a aorta ascendente e seus ramos arteriais (as artérias carótidas, subclávia e axilar). O envolvimento dos ramos inferiores da aorta, incluindo a artéria renal principal e seus ramos, ocorre com a evolução da doença.
A arterite de Takayasu foi descrita pela 1ª vez no Japão, nos anos 1800. Os casos relatados envolvem sobretudo as mulheres, com uma proporção mulher:homem de até 9:1. A incidência varia drasticamente em diferentes partes do mundo. Na Ásia, o aparecimento de novos casos chega a ser 100 vezes mais frequente do que no Ocidente. Em média, a idade dos paciente no momento da manifestação da doença é entre 10 e 40 anos.
A patogênese da arterite de Takayasu permanece desconhecida, mas é provável que envolva uma combinação de fatores do hospedeiro e do meio-ambiente. Exemplificando, a arterite de Takayasu foi associada a certos antígenos leucocitários humanos (HLA), tais como HLA B52, DRBI*1502, DRB5*0102 e DQAI*0103. Além disso, as infecções crônicas, como a tuberculose, podem deflagrar uma resposta imune crônica que eventualmente resulta na arterite de Takayasu.
Em termos de histologia, a arterite de Takayasu apresenta uma fase inflamatória aguda e uma fase fibrótica crônica. Durante a fase aguda, ocorrem alterações nas 3 camadas da artéria: proliferação de células de músculo liso e deposição de mucopolissacarídeo na íntima; infiltração da camada média por linfócitos e células gigantes; e vasa vasoritis na adventícia, caracterizada pela infiltração de células T, células B e células dendríticas. A fase crônica é marcada pela fibrose e destruição da arquitetura vascular.
A suspeita clínica de arterite de Takayasu surge tipicamente diante de uma paciente que apresenta hipertensão não controlada. Os pulsos diminuídos ou ausentes são encontrados em 84 a 96% dos pacientes, aliados à claudicação de membro e discrepâncias de pressão arterial.23,24 Os ruídos vasculares, frequentemente múltiplos, são encontrados em 80 a 94% dos pacientes; e a regurgitação aórtica, que resulta da dilatação da aorta ascendente, pode ser detectada em até 25% dos pacientes.
Do ponto de vista histológico, a arterite de Takayasu pode ser indistinguível da arterite de células gigantes, que é outra vasculite sistêmica de artérias de grande calibre. A arterite de Takayasu afeta a aorta e seus ramos, enquanto a arterite de células gigantes afeta principalmente a carótida externa e seus ramos. Além disso, o envolvimento da artéria renal é extremamente raro na arterite de células gigantes. Outros fatores demográficos também ajudam a distinguir estes 2 distúrbios. O aparecimento da arterite de células gigantes costuma ocorrer após os 50 anos de idade, ao passo que o aparecimento da arterite de Takayasu acontece de forma característica antes dos 40 anos. A arterite de Takayasu é mais comum em asiáticos, enquanto a arterite de células gigantes é mais frequente entre os descendentes de europeus. A proporção mulher:homem, que chega a 9:1 na arterite de Takayasu, é de 3:2 na arterite de células gigantes.
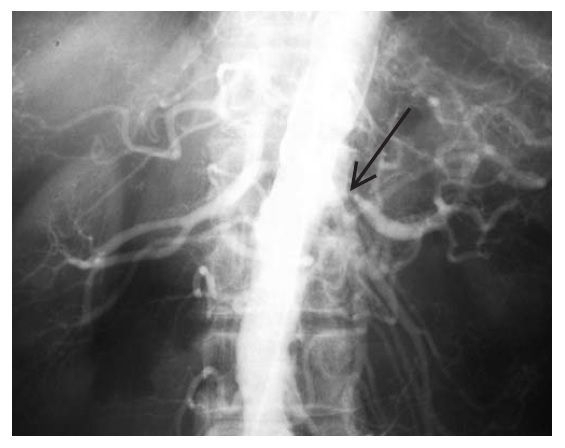
Em 1990, o American College of Rheumatology desenvolveu critérios diagnósticos específicos para a arterite de Takayasu [Tabela 1].25 Para estabelecer o diagnóstico, é necessário que pelo menos 3 dos 6 critérios sejam atendidos. A estratégia diagnóstica para estabelecer o envolvimento renal é similar ao discutido em termos de imagens que são feitas na EAR [Figura 2].
Tabela 1. Critérios de classificação do American College of Rheumatology para arterite de Takayasu*
|
Idade = 40 anos no momento do aparecimento da condição |
|
Claudicação dos membros† |
|
Pulsação braquial diminuída |
|
Diferença de pressão arterial > 10 mmHg‡ |
|
Ruído sobre as artérias subclávia ou aorta |
|
Anormalidades de arteriografia§ |
*Pelo menos 3 destes 6 critérios devem ser atendidos para estabelecer o diagnóstico.
†Fadiga e desconforto nos músculos de 1 ou mais membros, em especial nos membros superiores.
‡Tipicamente, a pressão arterial sistólica, entre os braços.
§Estreitamento ou obstrução da artéria aorta, de ramos primários ou de artérias de grande calibre na região proximal de membros superiores ou inferiores, que não sejam causados por outras condições (p. ex., aterosclerose, DFM).
DFM = displasia fibromuscular.
Figura 2. Arteriograma de estenose arterial renal (EAR) mostrando uma lesão de óstio na artéria renal esquerda (seta).
O tratamento da arterite de Takayasu consiste na terapia anti-hipertensiva, terapia imunossupressora e desvio cirúrgico das lesões arteriais severamente estenóticas. A maioria dos agentes anti-hipertensivos é apropriada para uso no tratamento da arterite de Takayasu, embora o uso dos iECA ou BRA deva ser estreitamente monitorado devido ao potencial de alterações abruptas na TFG. Os esteroides constituem a terapia imunossupressora de 1ª linha, com a qual cerca de 50% dos pacientes alcançam a remissão.26 Uma dose de 45 a 60 mg/dia de prednisona, por via oral (VO), ou uma dose equivalente de outro esteroide é administrada até que o paciente apresente melhora dos sintomas. Quando isto ocorre, a dose é diminuída e, se os sintomas voltarem, a dose é então restaurada à dose inicial. Dada a natureza crescente-decrescente da doença, a duração ideal da terapia com esteroide é variável. A terapia de 2ª linha, o metotrexato, geralmente é adotada por apresentar um perfil de efeitos colaterais reduzidos, em comparação ao observado com outros agentes citotóxicos, como a ciclofosfamida. Cerca de 50% dos pacientes respondem ao metotrexato VO administrado a uma dosagem de 5 a 20 mg/semana, sendo que o metotrexato tipicamente é combinado aos corticosteroides.27 A azatioprina tem sido usada com efeitos relatados similares àqueles produzidos pela ciclofosfamida.28 Agentes como o micofenolato de mofetil foram estudados, porém os dados obtidos são preliminares demais para sustentar recomendações formais.
A correção cirúrgica da estenose arterial é indicada para pacientes com EAR grave e hipertensão, claudicação de membro, doença cerebrovascular, isquemia esplâncnica, regurgitação aórtica ou doença coronariana. De modo significativo, a intervenção cirúrgica deve ser realizada durante uma fase quiescente de atividade da doença, em geral após a terapia imunossupressora.
Na arterite de Takayasu, o prognóstico varia conforme a extensão do envolvimento vascular. Embora a sobrevida de 5 anos para todos os pacientes com arterite de Takayasu seja de 80 a 90%,24,29 para os pacientes que apresentam envolvimento arterial múltiplo esta sobrevida é de 60%.
A trombose da veia renal (TVR) é uma complicação da síndrome nefrótica. A perda da seletividade da membrana basal glomerular (MBG) resulta na excreção de numerosas proteínas séricas, incluindo as proteínas anticoagulantes, como antitrombina III, proteína C e proteína S. Além disso, o sangue que flui pelas veias renais pode tornar-se relativamente hemoconcentrado após a ultrafiltração do plasma.
A prevalência exata da TVR é desconhecida. As estimativas variam amplamente. O distúrbio pode desenvolver-se em 5 a 60% dos pacientes com síndrome nefrótica. A TVR está mais frequentemente associada à nefropatia membranosa idiopática e secundária, sendo que 30% dos pacientes que apresentam estes distúrbios podem desenvolver TVR. É alarmante o fato de a vasta maioria destes casos ser assintomática.
A clássica manifestação clínica da TVR é a lombalgia aguda e a hematúria grosseira. Os pacientes tipicamente não apresentam insuficiência renal nem hipertensão. A TVR pode ser diagnosticada por tomografia computadorizada (TC), imagem de ressonância nuclear magnética (RNM) e venografia com contraste. O exame de imagem de ultrassonografia com Doppler é notoriamente dependente de operador e não deve ser usado para diagnosticar a TVR.
A anticoagulação com varfarina é indicada para pacientes com TVR. A meta terapêutica é uma um INR do tempo de protrombina entre 2 e 3. A duração apropriada da terapia provavelmente é a vida inteira. A terapia trombolítica ou trombectomia cirúrgica às vezes é considerada quando a TVR está associada à insuficiência renal aguda. A anticoagulação profilática foi defendida para os pacientes com síndrome nefrótica, devido ao risco aumentado de TVR, mas somente é recomendada diante da existência de evidências objetivas de doença tromboembólica. O prognóstico para os pacientes com TVR em geral é bom.
As doenças que afetam as artérias e veias de médio calibre existentes nos rins são relativamente raras. Existem 2 tipos de vasculites sistêmicas que afetam os rins de modo pouco frequente: a poliarterite nodosa (PN) e a doença de Kawasaki (DK).
A PN foi descrita pela 1ª vez por Kussmaul e Maier, em 1866, como periarterite nodosa. Trata-se de uma vasculite arterial sistêmica que pode envolver os rins. Durante muitos anos, as condições que hoje se sabe estarem comprovadamente associadas à presença de anticorpos anticitoplasma de neutrófilo (ANCA) podem ter sido diagnosticadas de maneira incorreta ou classificadas como PN. A Chapel Hill Consensus Conference estabeleceu critérios estritos para o diagnóstico das doenças associadas aos ANCA e da PN.30
Atualmente, a PN é definida com base em 3 critérios: (1) existência de uma vasculite necrotizante afetando pequenas artérias; (2) preservação dos vasos menores (isto é, arteríolas, capilares e vênulas); e (3) ausência de associação com uma glomerulopatia primária ou secundária. A condição é rara. A PN idiopática afeta anualmente 2 a 9 indivíduos a cada 1 milhão de pessoas,31,32 embora as taxas de incidência anuais cheguem a 77 casos por milhão nas áreas endêmicas de infecção pelo vírus da hepatite B.33 Nenhuma predileção racial, étnica ou sexual foi observada na PN idiopática.
A patogênese da PN é obscura. Parece haver uma associação com a infecção pelo vírus da hepatite B. No passado, a maioria dos casos relatados de PN idiopática podem, de fato, ter sido complicações da hepatite B resultantes da deposição de imunocomplexos. Os casos de PN infantil podem estar associados à infecção estreptocócica.
Em termos de histologia, a PN parece ser uma infiltração celular pleiomórfica da adventícia. A desgranulação das células polimorfonucleares resulta em leucocitoclasia e, eventualmente, em necrose transmural. Pode haver necrose segmentar arterial que, por sua vez, leva à formação de aneurismas. Nos estágios mais tardios do processo inflamatório, o dano endotelial e a trombose resultam na obstrução total do vaso afetado.
Do ponto de vista clínico, o desenvolvimento de PN é insidioso e está associado a sintomas constitutivos que surgem ao longo de várias semanas e até meses. Os sinais e sintomas inespecíficos iniciais são febre, perda de peso, mal-estar e anorexia. As artralgias de grandes articulações na ausência de artrite verdadeira podem ocorrer nos joelhos, tornozelos, cotovelos e punhos. Os achados mais sugestivos de PN incluem o livedo reticular (aparecimento de erupções arroxeadas sobre os membros inferiores e na parede abdominal), ulcerações ou nódulos dérmicos, e isquemia digital. A PN também pode afetar vários sistemas orgânicos, incluindo os intestinos, o coração e os rins. É comum o envolvimento renal acarretar hipertensão. Diante de uma envolvimento renal extensivo, também pode haver desenvolvimento de insuficiência renal.
O diagnóstico de PN é estabelecido pela demonstração da lesão característica em uma artéria. A biópsia de um nódulo cutâneo é preferida por sua relativa acessibilidade, contudo estes nódulos são notoriamente incomuns. A arteriografia pode representar uma alternativa conveniente. Embora os exames sorológicos não sejam diagnósticos na PN, títulos baixos de anticorpos antifator reumatoide (anti-FR) e anticorpos antinucleares (FAN) podem estar presentes. A PN secundária à hepatite B foi associada à hipocomplementemia. Enfim, a coloração por imunofluorescência com anticorpos para detecção de ANCA citoplasmáticos e perinucleares pode resultar positiva, contudo um teste mais específico, como o ensaio imunossorvente ligado à enzima (ELISA) para anticorpos dirigidos contra a protease sérica 3 (PR3) e contra a mieloperoxidase (MPO), resultará negativo.
Se a doença permanece sem tratamento, o prognóstico dos pacientes com PN é precário. Estes pacientes apresentam risco de desenvolver isquemia em numerosos órgãos. As principais causas de morbidade e mortalidade incluem insuficiência renal, isquemia mesentérica e AVC. Os corticosteroides e agentes citotóxicos constituem a base da terapia da PN idiopática, embora uma terapia ideal não tenha sido encontrada.
Cerca de 50% dos pacientes com PN idiopática alcançam a remissão com doses altas de esteroide (p. ex., 1 mg de prednisona/kg/dia) tomadas durante 3 a 6 meses.34 A ciclofosfamida, administrada por via intravenosa (IV, 0,6 g/m2/mês) ou VO (2 mg/kg/dia) durante 1 ano, é usada no tratamento de pacientes com doença irresponsiva aos esteroides e de pacientes que apresentam risco de desenvolvimento de complicações sérias. O escore Five Factor, um conjunto de critérios discretamente obsoleto, é usado em alguns casos para determinar quando há indicação para a terapia citotóxica: (1) sinais gastrintestinais, incluindo sangramento, perfuração, infarto ou pancreatite; (2) insuficiência renal; (3) proteinúria; (4) envolvimento do sistema nervoso central; e (5) envolvimento cardíaco.35 Segundo a nomenclatura de Chapel Hill, a ocorrência de proteinúria foi excluída dos critérios de indicação para terapia citotóxica em casos de PN. Os iECA e BRA devem ser usados com cautela, pois podem produzir um efeito renal que é o equivalente funcional da EAR clássica.
A DK é outra vasculite sistêmica que afeta as artérias de médio calibre. Na DK, o grau de envolvimento renal é variável. As lesões vasculíticas em vasos renais são raras e isto ainda é inexplicável. A piúria estéril, nefrite intersticial e proteinúria podem estar associadas à DK no rim. O diagnóstico e tratamento da DK são discutidos em detalhes em outro capítulo [ver 15:VIII Síndromes de vasculite sistêmica].
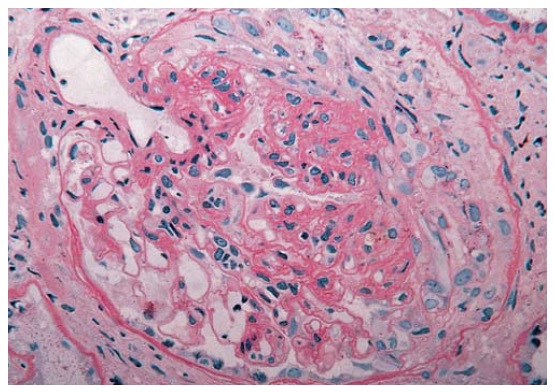
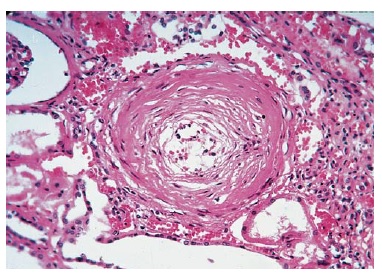
Muitas doenças dos vasos de pequeno calibre dos rins estão associadas à glomerulopatia. Embora estas doenças com frequência sejam consideradas formas de glomerulonefrite, a lesão-sentinela ocorre ao nível do capilar glomerular, onde os antígenos in situ ou exógenos induzem uma resposta inflamatória agressiva. Como consequência, pode haver uma degradação significativa da arquitetura glomerular, incluindo o rompimento da cápsula de Bowman (também conhecida como formação em crescente) e o vazamento de proteínas e hemácias para dentro do espaço tubular. Várias destas doenças vasculares foram descritas como sendo glomerulonefrite de progressão rápida (GNPR), devido à perda da função renal que ocorre em questão de dias a semanas [Figura 3].
Figura 3. Microscopia óptica de uma amostra de biópsia renal obtida de um paciente com glomerulonefrite em crescente. É possível visualizar a compressão dos tufos glomerulares por uma ampla formação em crescente. Embora uma lesão renal idêntica possa ser observada em diversos distúrbios renais, o achado de uma coloração de imunofluorescência negativa para complexos antígeno-anticorpo é altamente sugestivo de vasculite necrotizante sistêmica, como a granulomatose de Wegener ou a poliarterite nodosa (PN). A maioria das outras causas de glomerulonefrite em crescente é tipificada por padrões característicos de imunocoloração, como o padrão de imunofluorescência linear observado na síndrome de Goodpasture.
Estas doenças de vasos pequenos exibem manifestações clínicas semelhantes. A maioria dos pacientes apresenta evidências de doença sistêmica. Todos têm história de mal-estar, anorexia, perda de peso, febre, erupção, artralgia e alteração da cor da urina. Pacientes com síndrome pulmão-rim também podem apresentar hemoptise, epistaxe ou sinusite crônica. Os principais aspectos do exame físico para todas estas condições incluem a avaliação da pressão arterial, articulações, pele, nasofaringe, linfonodos, órgãos abdominais e olhos (retina). O exame de urina tipo 1 mostra hematúria ou proteinúria, enquanto a avaliação microscópica da urina revela a presença de cilindros de hemácias, corpúsculos adiposos ovais e hemácias dismórficas.
Vasculite associada aos anticorpos anticitoplasma de neutrófilo (ANCA). A vasculite associada à presença de ANCA é a forma mais comum de vasculite de vasos pequenos em adultos. A condição abrange 3 categorias principais: granulomatose de Wegener, poliangiite microscópica (PAM) e doença de Churg-Strauss. Estas 3 condições estão associadas à glomerulonefrite pauci-imune, ainda que tradicionalmente a glomerulopatia seja observada com menor frequência na doença de Churg-Strauss do que nos outros 2 distúrbios associados aos ANCA. A granulomatose de Wegener manifesta-se principalmente no rim, trato respiratório superior e pulmões, enquanto a PAM ocorre sobretudo no rim. A doença de Churg-Strauss é encontrada tipicamente nos pulmões e está associada à asma e eosinofilia [ver 15: VIII Síndromes de vasculite sistêmica]. De modo interessante, em um estudo recente sobre pacientes com doença de Churg-Strauss, foi demonstrado que a condição ANCA estava associada a diferentes manifestações clínicas.36 Exemplificando, os pacientes positivos para ANCA foram mais propensos a apresentar envolvimento renal (isto é, glomerulonefrite), enquanto aqueles negativos para ANCA tenderam mais a apresentar febre e envolvimento cardíaco.
A vasculite associada aos ANCA pode afetar indivíduos de todas as idades, contudo é mais comum entre os adultos de meia-idade e adultos de idade avançada (com mais de 50 anos). Apresenta a mesma distribuição entre homens e mulheres. Nos Estados Unidos, a condição é mais comum em brancos. A incidência comprovada da vasculite associada aos ANCA é de 1 a 2 casos em cada 100.000 indivíduos.37,38
A vasculite associada aos ANCA provavelmente resulta de uma interface entre fatores relacionados ao hospedeiro e meio-ambiente ou estímulos infecciosos, que atuam de modo isolado ou combinado. As infecções virais e bacterianas no trato respiratório superior frequentemente precedem a 1ª ocorrência da doença. A terapia de supressão das espécies estafilocócicas diminui a frequência das recorrências.
A presença de anticorpos antineutrófilo é central à patogênese tanto da doença renal como da doença sistêmica. Atualmente, existe um consenso de que estes anticorpos induzem os neutrófilos a liberarem seus grânulos citoplasmáticos em uma rápida explosão respiratória.39,40 Além disso, estes anticorpos também podem ativar a via alternativa do complemento.41
A aparência histológica da vasculite associada aos ANCA é a de uma infiltração leucocitoclástica de arteríolas, capilares e vênulas no corpo inteiro. Também pode haver uma necrose fibrinoide da camada muscular que em muitos casos se estende para dentro do tecido perivascular. De modo significativo, a lesão histológica (necrose fibrinoide) é indistinguível daquela observada em outras vasculites. O tipo e a localização do vaso envolvido ajudam a identificar o processo patológico específico.
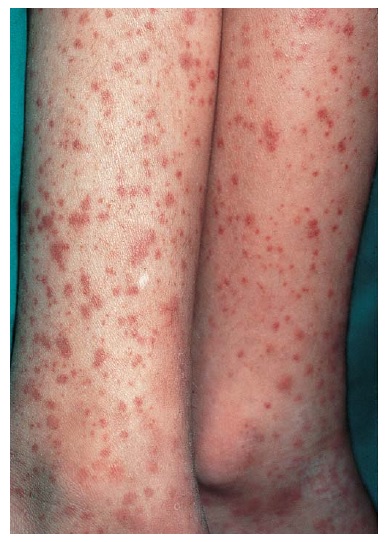
A manifestação clínica da vasculite de pequenos vasos frequentemente começa com o aparecimento de sinais e sintomas constitutivos, tais como febre, mal-estar, mialgias e artralgias. Os vasos sanguíneos em todas as partes do corpo – incluindo a musculatura esquelética, nervos periféricos, trato gastrintestinal, rins, trato respiratório e pele – podem ser afetados. Desta forma, os sinais e sintomas são variáveis. A angiite leucocitoclástica dos vasos dérmicos causa púrpura, que é o achado cutâneo mais comum [Figura 4]. Estas lesões de púrpura costumam ser salientes e, por motivos desconhecidos, tendem a se formar nos membros inferiores, em muitos casos em uma localização distal aos joelhos. A angiite dos vasos que suprem os nervos periféricos pode acarretar mononeurite múltipla, que é a manifestação neurológica mais comum.42,43 O envolvimento renal costuma resultar em glomerulopatia, com subsequente hematúria e proteinúria. O envolvimento do trato respiratório leva à epistaxe hemoptise. A asma é específica da doença de Churg-Strauss.
Figura 4. As erupções purpúricas podem ser observadas em diversas vasculites de pequenos vasos, na púrpura de Henoch-Schönlein, vasculite por hipersensibilidade e doença associada aos ANCA. As lesões são palpáveis, e a coloração de imunofluorescência dos vasos da derme em uma biópsia de pele pode ser diagnóstica. Uma deposição de IgA, por exemplo, é altamente sugestiva da púrpura de Henoch-Schönlein neste contexto.
O teste sorológico de ANCA é útil para estabelecer o diagnóstico da vasculite associada aos ANCA. A antiga nomenclatura dos ANCA foi substituída por uma terminologia mais específica, em que o C-ANCA foi sucedido pelo PR3-ANCA (serina protease 3) e o P-ANCA foi substituído pelo MPO-ANCA (mieloperoxidase). Cerca de 80% dos pacientes com granulomatose de Wegener apresentam altos títulos de anticorpos contra PR3-ANCA, enquanto 80% dos pacientes com PAM apresentam altos títulos de anticorpos contra MPO-ANCA. Significativamente, até 10% dos pacientes que exibem a manifestação clínica típica da vasculite associada aos ANCA podem apresentar sorologia negativa ou baixos títulos de anticorpos séricos contra a MPO ou a PR3.44
O prognóstico para os pacientes com vasculite associada aos ANCA não tratada é precário. A morte resulta da insuficiência renal ou de hemorragia pulmonar. Portanto, é importante intervir logo no início do curso da doença, de preferência antes que a concentração sérica de creatinina atinja 4 mg/dL.45 Os corticosteroides e a ciclofosfamida constituem a base da terapia de indução [Tabela 2]. Este regime produz remissão completa em cerca de 75% dos pacientes46 e melhora em até 90% dos casos.38 Os esteroides orais são iniciados a uma dose de 1 mg/kg/dia, a um máximo de 60 mg/kg/dia, durante as primeiras 4 a 6 semanas. Os esteroides são rapidamente afunilados ao longo das próximas 4 a 8 semanas subsequentes, de modo que ao final de 3 a 4 meses a maioria dos pacientes está livre de esteroides.
Tabela 2. Terapia de indução para a GNRP*
Pulso de corticosteroides:
Metilprednisolona, 7 mg/kg/dia, IV (dose máxima,500 mg) × 3 dias
Seguido de,
corticosteroides orais:
Prednisona, 1 mg/kg/dia, VO, 1 a 2 x/mês, com afunilamento rápido em 4 a 8 semanas
Mais
Ciclofosfamida:
VO: 2 a 3 mg/kg/dia, 6 x/mês; doses sucessivas até a mais próxima de 50 mg;
diminuir para 2 mg/kg/dia em casos de pacientes com idade > 55 anos
IV: 0,5 g/m2, mensalmente, 6 doses
Mais
Plasmaférese, transfusão de troca ou ambas, quando indicado
*Consultar no texto os detalhes referentes a cada uma das doenças.
GNRP = glomerulonefrite rapidamente progressiva; IV = via intravenosa; VO = via oral.
Em pacientes com hemorragia pulmonar concomitante, uma plasmaférese com ou sem troca de plasma deve ser iniciada imediatamente, aliada à terapia citotóxica. A plasmaférese refere-se à remoção do plasma (e do anticorpo patogênico) e infusão de salina e albumina para reposição do volume. Na troca de plasma, o plasma removido é substituído por plasma fresco congelado ou crioprecipitado. A dose ideal de plasmaférese é desconhecida. A plasmaférese é em geral realizada a cada 2 dias, com a realização de um total de 3 a 6 procedimentos, de modo que cada troca envolve 1 a 1,5 volume de plasma. Alguns centros monitoram os títulos de anticorpo para determinar a eficácia da troca. Recomenda-se usar a melhora clínica como fator primário para determinar o momento em que a plasmaférese deve ser concluída.
A terapia de manutenção ideal ainda precisa ser definida. Tanto a ciclofosfamida (VO e IV) como o micofenolato de mofetil têm sido usados. Recentemente, um agente antigo – a azatioprina – tem sido o alvo das atenções. A azatioprina VO mostrou-se equivalente à ciclofosfamida VO em termos de recidiva e efeitos colaterais.47 As recaídas da vasculite associada aos ANCA geralmente são tratadas como uma doença de novo, e o tratamento consiste na administração de doses altas de corticosteroides e ciclofosfamida.
Doença do anticorpo antimembrana basal glomerular (anti-MBG). Quase 20% dos casos de GNRP são causados pela doença do anticorpo antimembrana basal glomerular (anti-MBG). Esta condição é definida pela presença de anticorpos IgG circulantes dirigidos contra o colágeno de tipo IV – especificamente, contra o domínio NC1 da cadeia alfa-3.
A doença anti-MBG foi descrita como sendo uma condição que afeta indivíduos nos extremos de faixa etária. Nos casos relatados, os pacientes mais jovens tinham 4 anos e os pacientes de idade mais avançada tinha 81 anos. Classicamente, a doença ocorre em 2 grupos distintos: homens com menos de 30 anos e mulheres com mais de 60 anos.48 Os homens jovens com doença do anticorpo anti-MBG tendem a apresentar envolvimento renal e pulmonar. Esta condição recebe a denominação de síndrome de Goodpasture. A síndrome foi assim chamada em homenagem a Ernest Goodpasture, a quem é creditada a 1ª descrição da doença do anticorpo anti-MBG. O paciente por ele descrito era um jovem que tinha insuficiência renal e hemorragia pulmonar. Notavelmente, a manifestação observada foi atribuída à influenza pandêmica que ocorreu no início da década de 1900. As mulheres de idade mais avançada desenvolvem uma doença limitada aos rins. Tanto em homens como em mulheres, parece haver uma forte predileção racial por indivíduos brancos.
O(s) agente(s) indutor(es) do desenvolvimento de autoanticorpos na doença anti-MBG permanece(m) desconhecido(s). Relatos de casos isolados sugerem que a doença pode se desenvolver após infecções pulmonares ou exposição a aerossóis de hidrocarbonetos. Entretanto, estas observações ainda precisam ser sustentadas por estudos epidemiológicos amplos. O colágeno de tipo IV é encontrado de modo predominante na MBG e na membrana basal alveolar pulmonar. É interessante notar que os indivíduos com falta hereditária de colágeno de tipo IV (síndrome de Alport) podem desenvolver a doença do anticorpo anti-MBG, caso não recebam transplante renal de um doador sem grau de parentesco. O colágeno de tipo IV alveolar é relativamente pouco exposto ao soro, e isto talvez seja útil para explicar a frequência reduzida de envolvimento pulmonar. A lesão à barreira existente no pulmão, por ação de mecanismos como o tabagismo, pode expor o colágeno de tipo IV.
Ao exame sob microscópio óptico, o aspecto histológico associado à doença anti-MBG é semelhante àquele observado em outras formas de GNRP. As lesões apresentam alterações agressivas: expansão mesangial e celular difusa; cariorrexe; e formação em crescente (colapso da cápsula de Bowman). A doença do anticorpo anti-MBG é distinguida destas outras formas por meio da coloração imuno-histológica, que revela a presença de IgG linear ao longo da membrana basal.
A detecção de altos títulos séricos de anticorpos dirigidos contra a MBG é útil para confirmar o diagnóstico. Mais de 30% dos pacientes com doença anti-MBG também podem apresentar anticorpos ANCA.49 A presença de anticorpos ANCA é preditiva de uma resposta mais satisfatória à terapia imunossupressora.
O prognóstico dos pacientes com doença do anticorpo anti-MBG não tratada é precário. Quase todos os pacientes morrem por insuficiência renal ou hemorragia pulmonar. Com a terapia, o prognóstico é bom. A sobrevida de 1 ano é de aproximadamente 75 a 90%.48 Assim como na doença associada aos ANCA, a intervenção antecipada proporciona benefícios significativos em termos de sobrevida. Os corticosteroides, a ciclofosfamida e a troca de plasma representam a base da terapia [Tabela 2]. Estas 3 modalidades atuam removendo o anticorpo patogênico da circulação e, deste modo, limitando danos adicionais aos glomérulos e alvéolos. A troca de plasma consiste na troca de 4 litros de plasma por uma solução de albumina humana a 5%, realizada diariamente durante 14 dias. Como estas trocas podem depletar fatores de coagulação intrínsecos, o plasma fresco congelado deve ser fornecido dentro de 3 dias após a realização de qualquer tipo de procedimento invasivo planejado (p. ex., biópsia) ou no evento de uma hemorragia pulmonar. As recaídas são extremamente raras, e a terapia imunossupressora em geral pode ser descontinuada após o curso terapêutico inicial.
Glomerulonefrite por imunocomplexos. As glomerulonefrites por imunocomplexos constituem um grupo diverso de condições que contribuem para 30 a 40% dos casos de GNRP. Este tipo de glomerulonefrite desenvolve-se com frequência em pacientes com doença glomerular preexistente. Os exames histopatológicos mostram uma variação agressiva da glomerulonefrite subjacente. A nefropatia por IgA, nefrite lúpica, glomerulonefrite membranoproliferativa e glomerulonefrite pós-infecciosa são condições que podem se manifestar como uma GNRP por imunocomplexo. Os exames diagnósticos a serem realizados dependem da doença glomerular subjacente.
Na maioria das vezes, a nefrite lúpica manifesta-se como insuficiência renal com sedimento nefrítico. Este distúrbio afeta as mulheres de forma significativamente mais frequente do que os homens (9:1) e, em geral, surge entre 20 e 40 anos idade. Em termos de histologia, a nefrite lúpica é caracterizada de acordo com a extensão da proliferação celular e mesangial observada no glomérulo, sendo agrupada nas classes I a 5 da Organização Mundial da Saúde (OMS).50 A classe III (glomerulonefrite proliferativa focal) e a classe IV (glomerulonefrite proliferativa difusa) são, em particular, clinicamente importantes. Além disso, os pacientes com lúpus e anticorpos dirigidos contra o anticoagulante lúpico apresentam doença renal com aspectos de microangiopatia trombótica (MAT) (ver adiante).
Assim como no lúpus sistêmico, a causa da nefrite lúpica é desconhecida. O diagnóstico da nefrite lúpica é favorecido pelo fato de a condição geralmente ser acompanhada de outros aspectos do lúpus sistêmico, como a erupção ou a artrite. Os níveis séricos de complemento, de C3 e C4, estão tipicamente diminuídos. Outros exames sorológicos úteis incluem a quantificação dos títulos de FAN e de anticorpos anti-DNA de fita dupla (anti-dsDNA). Os testes para FAN possuem alta especificidade, e títulos acima de 1:160 são fortemente sugestivos da doença. Além disso, um resultado negativo para FAN confirma a ausência da doença. Um resultado positivo no ensaio para anticorpos anti-dsDNA ajuda a confirmar o diagnóstico de lúpus sistêmico [ver 15:IV Lúpus eritematoso sistêmico]. A nefrite lúpica é confirmada pelo exame de biópsia renal.
Sem a intervenção terapêutica, os pacientes com nefrite lúpica de classes III e IV, bem como os pacientes com MAT, apresentam um prognóstico ruim. A maioria dos pacientes evolui para DRET ou morre. Assim como em outros tipos de GNRP, a terapia consiste na administração de corticosteroides e agentes citotóxicos e, ocasionalmente, troca de plasma [Tabela 2]. A plasmaférese é indicada para os pacientes com MAT ou hemorragia pulmonar concomitante. Neste caso, o regime consiste na realização a cada 2 dias da troca de 1 a 1,5 volume de plasma, até serem realizados 3 a 6 procedimentos no total. Há um número crescente de dados sobre a eficácia do micofenolato de mofetil VO (500 a 2.000 mg/dia, por até 2 anos) como terapia primária51 ou como terapia de manutenção52 para a nefrite lúpica de classes III e IV.
A púrpura de Henoch-Schönlein (PHS) é uma vasculite sistêmica caracterizada pela deposição de imunocomplexos IgA-dominantes nas vênulas, capilares e arteríolas. O envolvimento renal que ocorre na PHS é indistinguível da nefropatia por IgA idiopática. O envolvimento de outros sistemas orgânicos, como a pele, nervos ou trato gastrintestinal, ajuda a distinguir entre PHS e nefropatia por IgA.
A PHS é uma doença da infância, cuja ocorrência atinge o pico aos 5 anos de idade. O envolvimento renal tende a ocorrer em crianças maiores e adultos. A PHS que ocorre em adultos costuma ser mais severa do que a forma infantil. Existe uma forte predileção da PHS por europeus e asiáticos, em comparação aos demais grupos étnicos, e a frequência da doença pode ser discretamente maior entre os homens do que nas mulheres.
Patogênese. O agente etiológico da PHS é desconhecido. A doença muitas vezes surge após uma infecção respiratória. A exposição aos vírus, como o adenovírus, pode deflagrar uma resposta de imunocomplexos que resulta na deposição de IgA (subtipo A1) em arteríolas, vênulas e capilares por todo o corpo.
Nos rins, a deposição de IgA é visualizada por coloração de imunofluorescência. Esta deposição frequentemente é acompanhada pela expansão da matriz mesangial e por hipercelularidade (ambas são achados inespecíficos para PHS). Na pele, a lesão característica é uma vasculite leucocitoclástica.
Diagnóstico. A manifestação clínica da PHS é variável. A clássica tétrade de sinais e sintomas consiste em erupções, artralgias, dor abdominal e doença renal. A erupção é purpúrica. As artralgias são simétricas e afetam as pequenas articulações. A dor abdominal é difusa, além de aumentar e diminuir. A doença renal costuma se manifestar com hematúria e proteinúria, sendo encontrada em mais de 50% dos pacientes com PHS. A insuficiência renal afeta apena 10 a 20% dos pacientes. Em casos raros, pode haver doença pulmonar e neuropatia periférica.
Tratamento. O prognóstico para a maioria dos pacientes com PHS é favorável. Quase 95% das crianças e 90% dos adultos se recuperam totalmente. A terapia de suporte, portanto, é suficiente para a maioria dos pacientes. A insuficiência renal progressiva e que leva à DRET ocorre em cerca de 5% dos pacientes. A terapia ideal para estes casos é desconhecida. Os agentes imunossupressores (p. ex., corticosteroides, ciclofosfamida, azatioprina) e os agentes anticoagulantes são empregados no tratamento da PHS, mas promovem resultados inconsistentes. Assim, a terapia geralmente é reservada para as formas agressivas de PHS, que são marcadas por insuficiência renal e envolvimento gastrintestinal difuso. Esta terapia inclui uma combinação de corticosteroides, ciclofosfamida e troca de plasma.
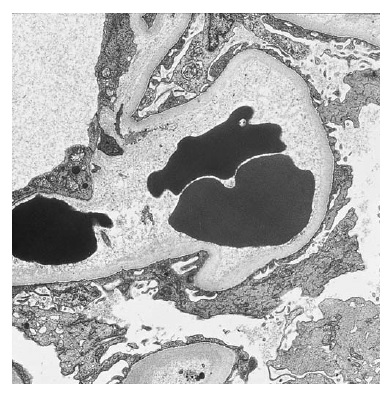
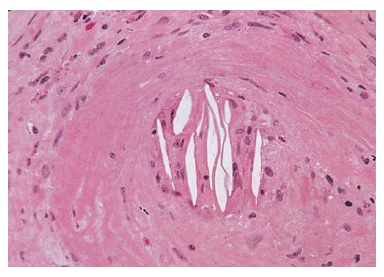
A MAT abrange um grupo de vasculopatias obstrutivas caracterizadas por ruptura endotelial, agregação plaquetária sistêmica ou intrarrenal, trombocitopenia e lesão mecânica dos eritrócitos. De forma significativa, os trombos de plaquetas contêm alta concentração de fator de von Willebrand, mas não contêm fibrina nem fibrinogênio. Além disso, o espessamento da íntima das artérias de pequeno calibre e arteríolas ocorre de modo difuso. Por sua aparência, a lesão é chamada “lesão em casca de cebola”.
As formas clássicas de MAT incluem a púrpura trombocitopênica trombótica (PTT) e a síndrome hemolítica-urêmica (SHU). A síndrome antifosfolípide (SAF), a pré-eclâmpsia, a esclerose sistêmica com crise renal, a hipertensão maligna e certos fármacos podem produzir um quadro clínico fenotipicamente similar ao de uma MAT clássica [Figuras 5 e 6]. Todas estas condições muitas vezes se manifestam como insuficiência renal aguda e podem causar necrose cortical aguda, que geralmente é considerada irreversível. Além disso, o paciente apresenta anemia hemolítica e trombocitopenia. Em todas estas condições, a lesão renal é notavelmente semelhante, de modo que a história frequentemente é decisiva para a determinação do tipo exato de MAT.
Figura 5. A microscopia óptica revela a característica lesão “em casca de cebola” observada na crise renal do escleroderma. Este achado é típico dos distúrbios que se manifestam como microangiopatia trombótica (MAT), tais como a hipertensão maligna, púrpura trombocitopênica trombótica (PTT), síndrome hemolítica-urêmica (SHU) e síndrome antifosfolípide (SAF) catastrófica.
Figura 6. Microscopia eletrônica mostrando um capilar glomerular de um paciente com púrpura trombocitopênica trombótica (PTT). O lúmen do capilar está no centro e contém 3 hemácias. O material de aspecto penuginoso observado entre as hemácias e a membrana basal glomerular (MBG) é característico deste distúrbio e pode representar fibrina incompletamente polimerizada. Este material está separando as células endoteliais da membrana basal. Nos casos mais crônicos, é possível observar uma lesão vascular similar àquela observada no escleroderma [Figura 5].
Eli Moschowitz foi quem descreveu a PTT pela 1ª vez, em 1924, em uma paciente de 14 anos de idade que apresentou febre e petéquias de aparecimento abrupto, seguidas de paralisia e morte. A autópsia da paciente revelou a presença de trombos hialinos difusos afetando as arteríolas terminais e capilares em numerosos órgãos. A PTT pode ocorrer em indivíduos de qualquer faixa etária. Não há diferenças evidentes em termos de ocorrência da doença associadas à raça ou sexo.
Patogênese. O papel decisivo da metaloproteinase ADAMTS-13 (uma desintegrina e metaloproteinase contendo domínios trombospondina-símile) na patogênese da PTT emergiu ao longo dos últimos 10 anos. A ADAMTS-13, que é encontrada na superfície das células endoteliais, normalmente cliva grandes multímeros de antígeno de von Willebrand, à medida que são secretados pela célula. Estes multímeros amplos se ligam às plaquetas (no componente Ib-alfa do receptor de glicoproteína Ib/IX/V vWA da plaqueta) de maneira mais eficiente do que clivam o antígeno de von Willebrand. A atividade da ADAMTS-13 plaquetária tem de ser severamente inibida para que haja desenvolvimento de PTT. Os níveis de atividade plasmática de ADAMTS-13 estão modestamente deprimidos em pacientes com hepatopatia, câncer disseminado, condições inflamatórias e metabólicas crônicas, na gestação e também em recém-nascidos.53 Quando a atividade plasmática da ADAMTS-13 cai para menos de 5 a 10% do normal, estes grandes antígenos de von Willebrand predominam, ligam-se às plaquetas e causam agregação plaquetária e formação de trombos nos vasos pequenos.
Foram postulados 2 mecanismos primários para a patogênese da PTT: o funcionamento anômalo da ADAMTS-13 e a não clivagem dos antígenos de von Willebrand. Na formas familiar ou crônica recidivante da PTT, os pacientes podem apresentar uma mutação no gene codificador da ADAMTS-13. Esta mutação resulta na ausência de ADAMTS nestes pacientes. Até o presente, mais de 55 mutações no gene da ADAMTS-13 foram descritas, bem como 26 polimorfismos genéticos.54,55 A falta de ADAMTS-13 ajuda a explicar por que a infusão de plasma fresco congelado, que comprovadamente contém a metaloproteinase, é a única terapia efetiva para estes indivíduos. Na PTT esporádica ou adquirida, pode haver formação de autoanticorpos dirigidos contra a ADAMTS-13 como resultado da exposição a vários antígenos, incluindo certos fármacos (p. ex., ticlopidina). A remoção dos autoanticorpos anti-ADAMTS-13 explicaria a eficácia da plasmaférese no tratamento da PTT, nestes pacientes.
Outro mecanismo recém-postulado para o desenvolvimento da forma familiar de PTT é uma deficiência de fator H plasmático. O fator H é uma proteína plasmática de 150 kDa, que inibe a ativação da via alternativa do complemento.56-58 Especificamente, o fator H expõe o componente C3b à clivagem pelo fator I. Uma deficiência de fator H, portanto, resultaria na desregulação de C3, potencializando a lesão endotelial glomerular mediada por autoanticorpos ou imunocomplexos. A exposição do subendotélio glomerular resulta em adesão e agregação plaquetária. Até agora, a maioria dos defeitos do fator H foi definida como sendo frameshifts, deleções e mutações pontuais.
Mais recentemente, foi sugerido que uma mutação envolvendo a proteína-cofator de membrana (MCP) causa uma forma de PTT fenotipicamente similar à deficiência de fator H.59 A MCP, um glicoproteína transmembrana amplamente expressa, atua como cofator essencial do fator I. Uma MCP anormal evitaria a inativação de C3b mediada pelo fator I, resultando na superativação da via alternativa do sistema complemento.
Diagnóstico. Classicamente, a PTT é definida por uma pêntade de achados clínicos: febre, trombocitopenia, anemia hemolítica microangiopática, anormalidades neurológicas e disfunção renal. Contudo, um diagnóstico provável pode ser estabelecido com base em uma tríade de observações laboratoriais: trombocitopenia, esquistócitos e elevação dos níveis séricos de lactato desidrogenase (oriunda dos eritrócitos fragmentados).
Diagnóstico diferencial. O diagnóstico diferencial inclui outras MATs. Uma história farmacológica que revele a exposição a agentes como ciclosporina, tacrolimos, mitomicina C, quinino, ticlopidina ou clopidogrel sugere a ocorrência de PTT induzida por fármacos. O quinino não é mais vendido sem receita médica, mas continua sendo encontrada na água tônica e em fitoterápicos contendo casca da árvore de cinchona. Uma história de ingesta de carne mal cozida ou laticínios/sucos não pasteurizados e possivelmente de uma doença diarreica anterior sugere um caso de SHU. A tríade de achados apresentados por uma paciente gestante levanta a possibilidade de pré-eclâmpsia e sua variante, a síndrome HELLP (hemolysis [hemólise], elevated liver enzymes [elevação de enzimas hepáticas], low platelets [baixa concentração de plaquetas]). Também pode ser difícil distinguir entre PTT e a fase de crise renal da esclerose sistêmica (escleroderma). Uma história de escleroderma, achados físicos compatíveis e a presença de anticorpos anti-Scl-70 ajudam a confirmar a ocorrência de crise renal do escleroderma. Por fim, uma história de hipertensão mal controlada e elevações severas da pressão arterial (em geral, com pressão diastólica > 130 mmHg), além de papiledema, é sugestiva de hipertensão maligna.
Além da MAT, também pode ser difícil distinguir a PTT de outras condições. A coagulação intravascular disseminada (CIVD), que parece ser deflagrada por sepse, choque ou complicações obstétricas, leva à ativação intravascular da cascata de coagulação e ao consumo de fatores de coagulação. A formação de trombos de fibrina intravasculares resulta em anemia hemolítica microangiopática. Talvez, seja possível distinguir presumivelmente entre CIVD e PTT por meio da determinação do tempo de protrombina (PT) e do tempo de tromboplastina parcial (PTT). Tanto o PT como o PTT estão elevados na CID.
Tratamento. O prognóstico para pacientes com PTT não tratada é ruim. A terapia é voltada para o suposto mecanismo subjacente. Como as formas familiar e crônica recidivante de PTT são consideradas resultantes da produção de ADAMTS-13 funcionalmente deficiente, estas condições são tratadas com a infusão de plasma fresco congelado pobre em plaquetas ou plasma pobre em crioprecipitado, em vez de plasmaférese. O plasma contém metaloproteinases ativas, incluindo a ADAMTS-13, e uma transfusão de plasma promoverá o aumento da atividade plasmática por até 3 semanas. A terapia genética poderá ser disponibilizada em breve, pois a sequência do gene codificador de ADAMTS-13 já foi determinada.
A PTT adquirida é tratada com troca de plasma. A troca de plasma possui 2 componentes: (1) plasmaférese para remoção dos multímeros de von Willebrand e autoanticorpos anti-ADAMTS-13; e (2) infusão de plasma fresco congelado para ajudar a reconstituir o volume e as proteínas do plasma, inclusive os fatores de coagulação. A troca de plasma é tipicamente realizada em dias alternados, em um total de 6 sessões. Os pacientes tratados com troca de plasma apresentam chances de 90% de sobreviverem a um episódio agudo de PTT.60 Nos casos de PTT irresponsiva à terapia médica, o paciente é submetido à esplenectomia.
A terapia antiplaquetária com aspirina deixou de ser recomendada para uso de rotina no tratamento da PTT, pois pode causar sangramento severo em pacientes com trombocitopenia grave.61 Um número crescente de relatos de caso tem demonstrado o tratamento bem-sucedido da PTT autoimune refratária com o rituximabe, um novo agente imunomodulador. Contudo, há dados limitados sobre esta forma de terapia.62,63 O rituximabe é um anticorpo monoclonal humano dirigido contra o CD20, que causa destruição das células CD20+ mediada pelo complemento e por outras células. Nos casos de PTT autoimune, acredita-se que o mecanismo de ação deste fármaco seja a destruição das células precursoras de células B CD20+, que produzem anticorpos anti-ADAMTS-13.
A SHU é definida como uma MAT subsequente à exposição à Escherichia coli 0157:H7 ou a outros sorotipos enteropatogênicos de E. coli, Shigella dysenteriae e, ocasionalmente, outros organismos enteropatogênicos. É uma doença que acomete predominantemente crianças, em especial com menos de 5 anos de idade. A SHU ocorre em 9 a 30% das crianças que apresentam diarreia sanguinolenta por Escherichia coli 0157:H7, manifestando-se em aproximadamente 1 semana após a infecção64-66 [ver 7:VIII Infecções causadas por Escherichia coli e outros bacilos entéricos gram-negativos]. O gado serve de reservatório para a Escherichia coli 0157:H7, que, por não conseguir penetrar a mucosa intestinal, não é prejudicial aos animais. As toxinas de Shiga produzidas pela bactéria podem ser transmitidas pelo leite e derivados não pasteurizados, e também por meio da ingesta de carnes mal cozidas.
Patogênese. Em seres humanos, a Escherichia coli 0157:H7 consegue invadir as células da mucosa do cólon e aderir a elas, onde então se replicam. A resposta inflamatória que se segue causa o colapso da arquitetura normal da mucosa e produção de diarreia sanguinolenta. Em seguida, há uma invasão bacteriana sistêmica. Ao nível sistêmico, as toxinas de Shiga exercem 2 papéis decisivos no desenvolvimento dos trombos. Primeiro, as toxinas impedem a secreção de ADAMST-13 atuando por um mecanismo não especificado, com consequente formação de amplos multímeros de von Willebrand. Em 2º lugar, as toxinas ativam a adesão plaquetária via componente Ib-alfa glicoproteína das glicoproteínas Ib/Ix/V1 e V2, que então podem promover agregação de plaquetas e do antígeno de von Willebrand.
Diagnóstico. A SHU caracteriza-se pelo aparecimento agudo de anemia hemolítica microangiopática, lesão renal e baixas contagens de plaquetas após um episódio agudo de diarreia sanguinolenta em uma criança até então saudável. A palidez, oligúria e petéquias ou contusões são manifestações comuns da condição. Os sintomas neurológicos, como letargia ou convulsões, também podem estar presentes. A ocorrência de uma condição diarreica é utilizada com frequência na clínica para distinguir entre SHU e PTT.
Tratamento. O prognóstico para pacientes com SHU é variável. As crianças que apresentam oligúria por menos de 24 horas em geral recuperam totalmente a função renal. Os adultos costumam apresentar um diagnóstico menos favorável em termos de recuperação da função renal. A terapia de suporte com diálise pode ser necessária.
Atualmente, não existe nenhuma terapia efetiva para SHU. A infusão e troca de plasma têm promovido resultados duvidosos.67,68 Entretanto, devido à sobreposição entre SHU e PTT, ambas as modalidades continuam sendo usadas com frequência. O tratamento ideal é a prevenção primária. Os produtos derivados de carne, em especial a carne moída e o cachorro-quente, devem ser consumidos sempre bem cozidos. Os laticínios não pasteurizados são contraindicados no tratamento dos pacientes com SHU, porque podem aumentar a liberação das toxinas de Shiga a partir da lise das bactérias.
A pré-eclâmpsia é uma vasculopatia multissistêmica de gestantes, que ocorre no 2º e 3º trimestres da gestação. Afeta 3 a 5% de todas as gestações.69 A pré-eclâmpsia continua sendo a principal causa de morbidade e mortalidade materno-fetal. Na pré-eclâmpsia, é comum haver envolvimento renal, que, por sua vez, pode ser usado para sustentar o diagnóstico.
Há muito tempo, os estudos têm sugerido que os fatores de risco de desenvolvimento de pré-eclâmpsia estão relacionados à uma perfusão placentária diminuída. Exemplificando, as mulheres em sua 1ª gestação, que tendem a ter uma placenta relativamente pequena, apresentam risco 2 vezes maior de desenvolver pré-eclâmpsia, em comparação às gestantes que passam pela 2ª ou 3ª gestação.70 Outros fatores de risco associados à diminuição da perfusão placentária incluem o diabetes melito, a hipertensão pré-existente, a trombofilia, a presença de mola hidatiforme e múltiplas gestações.
Patogênese. As hipóteses recentes acerca da fisiopatologia da pré-eclâmpsia enfocam a imunotolerância e os fatores angiogênicos circulantes. A placenta consiste em um conglomerado de 2 populações celulares genotipicamente distintas. A pré-eclâmpsia pode resultar de uma intolerância materna às células fetais. Repetidas exposições de baixo grau a estes antígenos podem induzir tolerância. Isto explicaria por que os estudos demonstraram que as múltiplas exposições aos espermatozoides de um mesmo parceiro, bem como as múltiplas gestações com um mesmo parceiro exercem efeito protetor contra o desenvolvimento de pré-eclâmpsia, enquanto a mudança de parceiro ou de doador de inseminação está associada ao risco aumentado de pré-eclâmpsia.71 A tirosina quinase fms-símile 1 solúvel (sFlt-1) também pode exercer papel importante na patogênese da pré-eclâmpsia. Esta molécula tipicamente se liga ao fator de crescimento endotelial vascular (VEGF), que é necessário à angiogênese normal, inclusive na placenta. Nas gestações normotensivas, os níveis de sFlt-1 começam a subir na 33ª a 36ª semana da gestação. Foi postulado que esta elevação ocorre precocemente na pré-eclâmpsia. A resultante diminuição dos níveis de VEGF pode, então, levar a uma angiogênese insuficiente na placenta. Um estudo de caso controle demonstrou que uma elevação dos níveis de sFlt-1 e a diminuição dos níveis de VEGF estão associadas ao desenvolvimento de pré-eclâmpsia.72
Diagnóstico. A manifestação clássica da pré-eclâmpsia é a tríade hipertensão, edema e proteinúria. A hipertensão (isto é, pressão arterial = 140/90 mmHg) é um achado relativamente sensível, apesar da baixa especificidade, que pode ocorrer em 12 a 22% de todas as gestações.73-75 O vazamento de proteínas dentro do espaço de Bowman e, subsequentemente, na urina é considerado resultante de endoteliose capilar glomerular (isto é, proliferação do endotélio). A excreção urinária de proteína ultrapassa 0,3 g em 24 horas e frequentemente chega a entrar na faixa nefrótica (> 3,5 g/24 horas). O edema é considerado resultante da perda de pressão oncótica junto aos vasos e parece afetar todos os órgãos. Entretanto, o edema muitas vezes só se torna evidente nos membros superiores ou na face. O edema hepático pode resultar no desenvolvimento de dor abdominal epigástrica e no quadrante superior direito. Sem dúvida, a obstrução capilar provoca diminuição do fluxo sanguíneo renal e da TFG. Entretanto, a insuficiência renal pode não ser detectada antes dos estágios tardios da pré-eclâmpsia, devido à elevação normal da TFG durante a gestação e à insensibilidade da concentração sérica de creatinina. Um aumento dos níveis séricos de ácido úrico, que é indicativo de diminuição da função secretória tubular, pode ser melhor como indicador da disfunção renal inicial em pacientes com pré-eclâmpsia. Nos países industrializados, a insuficiência renal aguda é rara em decorrência dos partos bem-sucedidos de fetos prematuros. A necrose tubular aguda isquêmica e a necrose cortical decorrente de pré-eclâmpsia atualmente contribuem para menos de 4% de todos os casos de insuficiência renal aguda associada à gestação.
Embora a pré-eclâmpsia possa ser facilmente diagnosticada, é frequentemente confundida com condições tão diversas quanto a pancreatite aguda, doença da vesícula biliar, apendicite, nefrolitíase e glomerulonefrite.76 Pode ser difícil distinguir entre pré-eclâmpsia e glomerulonefrite. Uma história de proteinúria e hematúria antes da gestação é fortemente sugestiva da existência de glomerulonefrite subjacente. No entanto, os registros antigos de uma paciente podem estar inacessíveis. Além disso, o nível de proteinúria e hematúria pode aumentar de forma acentuada durante a gestação. Desta forma, o exame de biópsia renal pode ser o único meio de distinguir entre pré-eclâmpsia e glomerulonefrite. Na biópsia, a alteração histológica considerada específica da pré-eclâmpsia – endoteliose – pode ajudar a distingui-la de outras formas de MAT. Uma biópsia renal é indicada para os casos em que há suspeita de pré-eclâmpsia antes da 28ª semana de gestação, quando a sobrevivência do feto é desejada.77 Em geral, o procedimento é bem tolerado tanto pela mãe como pelo feto.
Tratamento. A pré-eclâmpsia não tratada evolui rápido para eclâmpsia, que consiste na constelação de sinais resultantes do desenvolvimento de edema cerebral. Estes sinais incluem a encefalopatia, papiledema, perda visual central e convulsões. As sequelas neurológicas permanentes, necrose cortical aguda do rim e morte da paciente são todas possíveis. O parto do feto e a eliminação da placenta são considerados o tratamento de escolha para a pré-eclâmpsia. A prevenção desta condição constitui o aspecto-padrão do tratamento obstétrico. Os testes de fita urinária para proteinúria e a determinação da pressão arterial são realizados em todas as consultas de pré-natal. O desenvolvimento de hipertensão durante a gestação constitui uma indicação para tratamento mais intensivo, incluindo a realização mais frequente de avaliações para detecção de proteína na urina e a medida dos níveis séricos de ácido úrico. A terapia anti-hipertensiva é considerada quando a pressão arterial sobe para mais de 160/110 mmHg. A terapia anti-hipertensiva passa a ser obrigatória quando a pressão ultrapassa 170/110 mmHg.79 A meta da terapia é manter a pressão arterial média em 125 a 105 mmHg e a pressão arterial diastólica em 105 a 90 mmHg.76 Atualmente, a hidralazina, nifedipina, alfametildopa e labetalol são os agentes preferidos. Os iECA, que são usados com frequência no tratamento de outras doenças renais, são especificamente evitados por apresentarem uma forte ação teratogênica em potencial durante o 1º trimestre da gestação. Os corticosteroides são administrados para promover a produção de surfactante no feto. O sulfato de magnésio é usado para prevenir convulsões e parto prematuro. Várias intervenções adicionais, incluindo o uso oral de cálcio e aspirina, bem como a redução da ingesta de sal não promovem uma diminuição significativa comprovada da incidência de pré-eclâmpsia, em comparação ao placebo.
A SAF refere-se à hipercoagulabilidade em presença de autoanticorpos dirigidos contra fosfolipídios de carga negativa. Estes anticorpos são tradicionalmente considerados causadores da trombose de artérias e veias de grande calibre, bem como de abortos. O espectro total da doença já está elucidado. O envolvimento renal é comum na SAF. Além disso, quando o envolvimento renal ocorre, manifesta-se tipicamente como uma MAT.
O termo “anticorpos antifosfolípides” atualmente é considerado equivocado. Com exceção da cardiolipina, os antígenos-alvo são as proteínas plasmáticas ligadas aos fosfolipídios. Os anticorpos antifosfolipídio atualmente são agrupados em 4 categorias,80,81 ainda que 3 destas categorias possam apresentar sobreposição em decorrência do compartilhamento da mesma afinidade pelo antígeno entre os anticorpos. Os 4 subtipos são: (1) anticorpos anticardiolipina (ACA); (2) anticoagulante lúpico; (3) anticorpos antibeta-2-glicoproteína; e (4) anticorpos antiprotrombina. Os ACA foram isolados pela 1ª vez de pacientes com sífilis, há quase 100 anos. Posteriormente, descobriu-se que estes anticorpos eram dirigidos contra um fosfolipídio mitocondrial, a cardiolipina. O anticoagulante lúpico consiste em um grupo de imunoglobulinas originalmente encontradas em pacientes com lúpus eritematoso sistêmico, há cerca de 50 anos. Apesar do nome – que deriva da propriedade in vitro de retardamento da geração de trombina – o anticoagulante lúpico tem ação pró-coagulante in vivo. O anticoagulante lúpico é dirigido contra proteínas plasmáticas (em especial, a protrombina, mas também contra a anexina e a beta-2-glicoproteína) ligadas a fosfolipídios dotados de carga negativa. A beta-2-glicoproteína I é um importante inibidor da cascata de coagulação. Os anticorpos dirigidos contra esta proteína foram descritos há quase 10 anos. Os anticorpos dirigidos contra a protrombina, diferente do anticoagulante lúpico, foram descritos nos últimos 5 anos.
Patogênese. Ainda não foi esclarecido o que faz os anticorpos antifosfolípide causarem trombose. Estes anticorpos promovem efeitos pró e anticoagulantes. Do mesmo modo, os agentes que induzem a produção destes anticorpos são desconhecidos. Considerando que os anticorpos antibeta-2-glicoproteína podem apresentar reação cruzada com antígenos bacterianos, é possível especular que a exposição aos patógenos bacterianos (e, possivelmente, aos patógenos virais) induz a formação destes anticorpos.
Os anticorpos antifosfolipídio são bastante comuns e podem ser encontrados em 1 a 5% dos indivíduos sadios.82 Não está esclarecido se estes anticorpos podem causar SAF em indivíduos saudáveis, embora os estudos epidemiológicos tenham demonstrado a existência de uma associação entre tais anticorpos e a ocorrência de IM, AVC e trombose venosa. Os ACA estão presentes em 12 a 30% dos pacientes com lúpus sistêmico,83,84 enquanto 15 a 34% destes indivíduos têm anticoagulante lúpico.83,85 A SAF pode desenvolver-se em 50 a 70% dos pacientes com lúpus e anticorpos antifosfolipídio.82,86
Diagnóstico. A manifestação clínica da SAF mais frequentemente abrange um único evento trombótico, seja no sistema arterial ou venoso. A trombose venosa profunda nos membros inferiores é a ocorrência mais comum. A trombose arterial é menos frequente do que a trombose venosa, afetando principalmente o cérebro (AVC) ou os vasos coronarianos (IAM). Durante a gestação, pode haver aborto.
Um 2º tipo de manifestação é a SAF catastrófica, que é uma MAT.81 O rim é o órgão mais frequentemente afetado nestes casos, seguido pelo pulmão, sistema nervoso central, coração e pele. A maioria dos pacientes com envolvimento renal desenvolve hipertensão, e até 25% desenvolvem insuficiência renal progressiva com necessidade de diálise de suporte. Pode haver desenvolvimento de CID no contexto da SAF catastrófica.
Tratamento. O prognóstico da SAF varia de acordo com a severidade, sendo que as estratégias terapêuticas acompanham esta variação. Para os pacientes que possuem anticorpos antifosfolipídio e nunca tiveram SAF, ainda não está claramente definido se a anticoagulação é indicada para fins de prevenção primária da trombose. Uma revisão minuciosa deve ser realizada com o intuito de eliminar as condições ou situações que podem aumentar o risco de o paciente desenvolver trombose. Exemplificando, o uso de anticoncepcionais orais deve ser desestimulado, assim como o uso de cateteres venosos em pacientes sob hemodiálise. A anticoagulação profilática é indicada para os casos em que o uso de anticoncepcional oral ou de cateter venoso não puder ser eliminado.
Para os pacientes com anticorpos antifosfolipídio que sofreram um único episódio de trombose, a anticoagulação com varfarina é indicada para fins de prevenção secundária. A meta terapêutica é uma INR de 2 a 3. A terapia provavelmente deve ser mantida por toda a vida do paciente.
Atualmente, está em uso o tratamento com plasmaférese e troca de plasma aliado à terapia com altas doses de corticosteroide (similares às doses administradas no tratamento da síndrome pulmonar-renal). A terapia com imunoglobulina também pode ser tentada, para promover a ligação dos autoanticorpos agressores. Há poucos dados sobre a eficácia destas intervenções em casos de SAF catastrófica.
Prognóstico. A mortalidade entre os pacientes com SAF catastrófica é alta: chega a 50%. A morte geralmente resulta da falência de múltiplos órgãos.
A esclerose sistêmica é um subtipo dos distúrbios associados ao escleroderma. Esta, por sua vez, consiste em um grupo diverso de doenças que em geral compartilham o achado multiforme de lesões cutâneas escleróticas e espessas. Vários sistemas orgânicos são envolvidos, incluindo o coração, pulmões, trato gastrintestinal, músculos e rins.
A esclerose sistêmica é relativamente incomum. Nos Estados Unidos, a prevalência varia de 4 a 253 casos em 1 milhão de indivíduos.87 As mulheres são mais comumente afetadas do que os homens, em uma proporção de 3 a 8:1.88 Nos Estados Unidos, observa-se uma incidência discretamente maior entre os afrodescendentes do que entre os brancos.89
O envolvimento renal é comum na esclerose sistêmica. Cerca de 10 a 15% dos pacientes com esclerose sistêmica apresentam crise renal – uma síndrome marcada por insuficiência renal aguda, hipertensão grave, encefalopatia e, em alguns casos, insuficiência cardíaca. Mais frequentemente, os pacientes com esclerose sistêmica apresentam evidências menos dramáticas de envolvimento renal. Em quase todos os pacientes com esclerose sistêmica, o fluxo sanguíneo renal está anormal,90 sendo que 2/3 destes pacientes apresentam lesões vasculares renais.91 Metade de todos os pacientes com esclerose sistêmica pode ter hipertensão, concentração plasmática de creatinina elevada ou proteinúria.92,93 Na esclerose sistêmica, os fatores de risco significativos de desenvolvimento de doença renal incluem afrodescendência, uso de corticosteroides em doses altas (isto é, dosagens > 15 mg/dia) e envolvimento cutâneo difuso.
Etiologia e patogênese. A maioria dos casos de esclerose sistêmica é idiopática. Assim como a maioria das doenças do tecido conectivo, sem dúvida há envolvimento de uma complexa interface de fatores relacionados ao hospedeiro e fatores ambientais. Alguns casos de esclerose sistêmica apresentam sobreposição com outras condições reumatológicas, incluindo o lúpus eritematoso sistêmico, a artrite reumatoide e a doença mista do tecido conectivo indiferenciada. A exposição ambiental a solventes orgânicos (p. ex., benzeno, tolueno e tricloroetileno) foi implicada como causa secundária da esclerose sistêmica. No mecanismo proposto, estes solventes orgânicos podem ser metabolizados em compostos de epóxi que se ligam a proteínas e produzem autoantígenos,94 ou induzem lesões que resultam em anormalidades microvasculares e fibrose de órgãos-alvo.95 No entanto, uma revisão extensiva encontrou evidências insuficientes para sustentar a existência de uma ligação causal entre os agentes químicos e a esclerose sistêmica.96
Os achados patognomônicos encontrados na esclerose sistêmica são: a proliferação irreversível do tecido conectivo; o espessamento mucoide das paredes vasculares; e o estreitamento do lúmen vascular. Na maioria dos casos, as alterações mucoides são encontradas junto às arteríolas renais. Na crise renal, estas alterações histopatológicas ocorrem nas artérias arqueada e interlobular, bem como no glomérulo. Os trombos de fibrina são observados nas arteríolas e capilares, bem como nas áreas de necrose fibrinoide. A hipertrofia “em casca de cebola” concêntrica das artérias interlobulares resulta dos ciclos repetidos de doença ativa e quiescência.
Diagnóstico. O diagnóstico da doença renal pode ser simples de se estabelecer no contexto de esclerose sistêmica estabelecida. Os exames sorológicos são úteis para confirmar a esclerose sistêmica: os anticorpos anti-Scl-70 possuem alta especificidade, apesar da baixa sensibilidade. Um achado de perda capilar e ampliação do leito ungueal à capilaroscopia é sensível e específico.97 A biópsia renal não é indicada para uso de rotina, mas deve ser realizada em casos atípicos. Exemplificando, há um pequeno subgrupo de pacientes que apresentam esclerose sistêmica sem os típicos achados cutâneos (uma condição denominada escleroderma sem escleroderma). Em alguns pacientes que sofrem a crise renal do escleroderma, a pressão arterial pode não estar elevada. A biópsia da pele ou do rim pode ajudar a confirmar o diagnóstico, neste tipo de contexto.
Tratamento. A crise renal do escleroderma antigamente era considerada uma complicação uniformemente fatal da esclerose sistêmica. A introdução da terapia de diálise diminuiu a mortalidade, enquanto o uso da terapia farmacológica promoveu uma redução adicional de quase 50% nas taxas de morbidade e mortalidade.
O controle da pressão arterial sistêmica é a base da terapia da esclerose sistêmica com crise renal. O controle da pressão arterial é mais bem obtido de maneira gradual (isto é, diminuições de 10 a 15 mmHg/dia), até que a pressão arterial diastólica atinja 85 a 90 mmHg. Os iECA são os agentes de escolha. Foi demonstrado que estes inibidores diminuem o risco de DRET98 e a necessidade de terapia de substituição renal.99 Tais benefícios podem ser mediados por uma propriedade exclusiva desta classe de fármacos ou decorrentes da maior eficácia em termos de redução da pressão arterial. Uma observação importante é o fato de a maioria dos estudos ter usado captopril. Desta forma, o efeito benéfico pode não ocorrer com outros iECA ou com os BRA. Foi sugerido que um bloqueador de canal de cálcio di-hidropiridínico, a nifedipina, pode ser usado como agente adicional de controle da pressão arterial por pacientes que apresentam resposta inadequada a um iECA. As crises renais recorrentes são incomuns, quando é possível controlar a pressão arterial.
A hipertensão maligna é uma síndrome de dano vascular agudo aos rins, retina e cérebro, que ocorre no contexto de elevações graves da pressão arterial. Esta condição foi denominada “hipertensão maligna” porque está associada a uma expectativa de vida típica igual àquela associada a uma malignidade em estágio avançado.
Os fatores de risco tradicionais de desenvolvimento de hipertensão maligna são: sexo masculino, afrodescendência e falta de complacência dos pacientes com o uso da medicação. A prevalência da hipertensão maligna está em queda, provavelmente em consequência de um aumento da consciência aliado ao tratamento da hipertensão e ao maior acesso ao tratamento médico por parte das minorias.
Patogênese. O nível atual de conhecimento acerca da fisiopatologia da hipertensão maligna gira em torno do papel do endotélio.100 Durante os períodos de elevação severa da pressão arterial, o controle endotelial do tônus vascular é superado, e, em consequência, há desenvolvimento de hiperperfusão em órgão-alvo, necrose fibrinoide arteriolar e aumento da permeabilidade endotelial com formação de edema perivascular. Como resultado, a ação fibrinolítica do endotélio cessa e as plaquetas são expostas às moléculas de adesão celular.
Diagnóstico. Tipicamente, a pressão arterial sistólica excede 180 mmHg e a pressão arterial diastólica é superior a 120 mmHg. Em termos de histologia, a hipertensão maligna é caracterizada pela necrose fibrinoide difusa e pelo espessamento da íntima dos vasos de pequeno calibre (arteríolas). Nos rins, estas alterações são indistinguíveis daquelas observadas em outras MAT, esclerose sistêmica e pré-eclâmpsia. Na maioria dos casos, a função renal está diminuída e pode declinar rápido no decorrer de algumas semanas a meses, caso não haja tratamento.
Tratamento. O prognóstico para os pacientes com hipertensão maligna melhorou de modo importante ao longo dos últimos 40 anos. O controle agressivo da pressão arterial com o uso de vários agentes alivia a maior parte do dano aos órgãos-alvo [ver 1: III Hipertensão]. Inicialmente, existia a preocupação de que a diminuição da pressão arterial média pudesse causar uma azotemia progressiva, contudo estudos demonstraram que, em vez disto, a função renal é estabilizada ou melhorada a longo prazo, na maioria dos casos.
A anemia falciforme (AF) é uma condição autossômica recessiva, que resulta da substituição de uma valina por glutamina na 6ª posição da subunidade beta-hemoglobina. Os indivíduos homozigotos para a hemoglobina falciforme são suscetíveis à crise falciforme por obstrução capilar [ver 5:IV Hemoglobinopatias e anemias hemolíticas]. A heterozigosidade para hemoglobina falciforme pode conferir proteção contra a malária.
A AF ocorre essencialmente em indivíduos com ancestrais oriundos da região do Mediterrâneo Oriental e da África subsaariana. Nos Estados Unidos, a AF afeta predominantemente a população afro-americana. A AF é uma vasculopatia sistêmica, cujas taxas de morbidade e mortalidade derivam primariamente de lesões vaso-oclusivas nos pulmões, sistema nervoso central, baço e rins.101
Tradicionalmente, a hipoxemia é considerada central na fisiopatologia da hemoglobina falciforme. Sob condições de baixa pressão parcial de oxigênio, a molécula de hemoglobina sofre polimerização e, como consequência, ocorre o afoiçamento do eritrócito. Estas células falciformes produzem estase e obstrução nos capilares. Pesquisas recentes também sugeriram que a desregulação do volume do eritrócito e a interação entre a membrana celular eritrocitária e o endotélio vascular também podem contribuir para os eventos vaso-oclusivos.102 Por fim, a hipertrofia glomerular compensatória pode exercer algum papel no desenvolvimento da proteinúria e da insuficiência renal observada nos pacientes com AF.103-104
O envolvimento renal é relativamente comum na AF. A medula renal, que é uma área de hipóxia crônica, constitui a principal área de envolvimento. A estase dos eritrócitos nos vasos retos medulares pode levar ao aparecimento de áreas hemorrágicas e necróticas e, eventualmente, a inflamação intersticial, fibrose, atrofia tubular e infarto papilar (necrose). Com o passar do tempo, estes eventos vaso-oclusivos levam a uma perda quase total dos vasos retos [Figura 7].
Figura 7. Em um microangiograma por injeção de um rim normal (a), os vasos retos que irradiam para dentro da papila são visíveis. No microangiograma por injeção de um paciente com anemia falciforme (b), os vasos retos estão quase ausentes.
Existem 3 lesões renais patológicas descritas na AF. A necrose das papilas renais é uma complicação frequente, que ocorrem em 15 a 67% dos pacientes.105-107 Esta condição resulta do infarto papilar provocado por repetidos infartos vaso-oclusivos. Os pacientes com necrose papilar podem ser assintomáticos ou apresentar hematúria grosseira e insuficiência renal. A glomerulosclerose segmentar focal (GESF) é a glomerulopatia mais comumente encontrada em pacientes com AF e proteinúria.108-110 Foi proposto que a GESF resulta da hiperfiltração, exposição a espécies reativas do oxigênio e exposição às proteínas plasmáticas filtradas. Uma 3ª lesão patológica, a glomerulopatia membranoproliferativa de tipo I, foi descrita em apenas alguns pacientes com AF.103
Além dos achados patológicos, 26% dos pacientes com AF têm proteinúria e até 7% apresentam algum grau de insuficiência renal.110 Uma minoria significativa dos pacientes evolui para DRET. A reduzida capacidade de concentração da urina é quase universal na AF. Os episódios vaso-oclusivos junto aos vasos retos parecem interferir nas trocas concomitantes, produzindo uma osmolalidade urinária máxima de 400 a 450 mOsm/kg. Também pode haver desenvolvimento de uma acidose tubular renal distal incompleta, devido ao comprometimento da secreção de hidrogênio e potássio.
O diagnóstico de doença renal associada à AF é geralmente estabelecido com faciliade. As complicações renais tendem a ocorrer nas fases tardias do curso da doença, quando o diagnóstico primário já foi estabelecido [ver 5:IV Hemoglobinopatias e anemias hemolíticas].
O tratamento conservativo padrão da crise falciforme concentra-se em repouso, hidratação e analgesia [ver 5:IV Hemoglobinopatias e anemias hemolíticas]. Se o paciente apresentar evidências de hipertensão, envolvimento renal ou ambos, o tratamento com agentes antiproteinúricos (p. ex., iECA e BRA) é recomendado.
O prognóstico da AF melhorou tremendamente no decorrer dos últimos 30 anos. A AF foi inicialmente considerada uma doença da infância, pois a vasta maioria dos pacientes afetados não sobrevivia até a idade adulta. No entanto, a sobrevida tem melhorado,101,111 e uma proporção maior de pacientes afetados está conseguindo atingir a idade adulta. Um tratamento de suporte aprimorado, bem como o tratamento agressivo das infecções podem ser os responsáveis por esta melhora. A indução da produção de hemoglobina fetal é a base da terapia. A hidroxiureia, um fármaco antitumoral mielossupressor, comprovadamente diminui a ocorrência das dolorosas crises torácicas agudas, bem como a necessidade de transfusões.102 Entre as perspectivas futuras, estão o desenvolvimento de fármacos capazes de inibir a polimerização da molécula de hemoglobina falciforme; o transplante de medula óssea; e a introdução de beta-hemoglobina do tipo selvagem através de um vetor viral.
Ateroembolia é uma condição rara que afeta vários leitos vasculares e, mais notavelmente, os pequenos vasos do rim. É caracterizada pela deposição de cristais de colesterol nos capilares e arteríolas glomerulares. Outras denominações empregadas para esta condição incluem: doença da embolia por cristais de colesterol; doença ateroembólica do colesterol; e embolia por colesterol.
A epidemiologia da doença ateroembólica é obscura.112 É impossível estimar a incidência e prevalência da condição na população geral. A maior parte das informações é abstraída de grupos altamente seletos. Como regra geral, a doença ateroembólica tende a ocorrer em indivíduos que apresentam fatores de risco de aterosclerose e com aterosclerose generalizada extensiva. Portanto, os fatores de risco incluem a idade acima de 55 anos, hipercolerterolemia, diabetes melito e hipertensão, vasculopatia periférica, aterosclerose aórtica abdominal e EAR aterosclerótica. Os episódios de ateroembolia quase sempre sucedem a manipulação (ou traumatismo) de uma placa ateromatosa. Sendo assim, os episódios podem ocorrer após a realização de arteriografias, sobretudo de vasos grandes (como a aorta, as artérias renais e as artérias coronárias), ou após a terapia trombolítica. Em um estudo, a ateroembolia ocorreu em 12% dos pacientes submetidos à angiografia coronariana e cirurgia de revascularização.113
Do ponto de vista histológico, a lesão associada à doença ateroembólica é caracterizada pela obstrução das artérias arqueada e interlobular (150 a 200 mcm de diâmetro), bem como dos capilares glomerulares por cristais de colesterol embolizados. Estes cristais se dissolvem durante a preparação de rotina dos tecidos, mas deixam uma fenda destacada tipicamente biconvexa e com formato de agulha, que é referida como “fantasma” (ghost) [Figura 8]. Inicialmente, a infiltração de leucócitos polimorfonucleares e eosinófilos é observada em torno do vaso obstruído. Com o passar do tempo, os cristais podem atrair um infiltrado de células mononucleares inflamatórias, e pode haver desenvolvimento de hiperplasia da íntima e fibrose perivascular junto aos vasos afetados. É possível observar a recanalização do vaso obstruído.
Figura 8. A microscopia óptica revela um aterotrombo alojado em uma pequena artéria renal e a obstrução do lúmen vascular. As fendas paralelas bicôncavas, que representam os cristais de colesterol e foram dissolvidas durante a fixação em parafina, são características deste distúrbio.
A aterosclerose de vasos de grande calibre é sem dúvida um componente importante da patogênese. As placas ateromatosas encontradas na aorta e nas artérias renais são tipicamente a fonte dos êmbolos de colesterol que seguem para os rins. A ativação das vias do complemento (clássica e alternativa) pelos cristais de colesterol pode ajudar a explicar o componente inflamatório da doença observada ao exame de biópsia.
É difícil diagnosticar a doença ateroembólica. Os casos podem ocorrer de forma espontânea, mas a maioria está temporariamente relacionada aos procedimentos vasculares invasivos ou à terapia trombolítica. Contudo, a relação temporal com estes procedimentos vasculares invasivos é variável. A doença pode ocorrer imediatamente após o evento, ou pode se desenvolver de modo insidioso, no decorrer de algumas semanas ou meses. Além disso, os pacientes geralmente apresentam sinais e sintomas que envolvem múltiplos órgãos: rins (insuficiência renal, proteinúria, hematúria), pele (livedo reticular, dedos do pé arroxeados, gangrena), trato gastrintestinal (anorexia, náusea, isquemia intestinal ou infarto, pancreatite, enzimas hepáticas anormais), sistema nervoso (cefaleia, amaurose fugaz, mononeuropatia, doença cerebrovascular) e olhos (placas de Hollenhorst). Além disso, os sintomas constitutivos inespecíficos, como febre, mal-estar e perda de peso, frequentemente estão presentes e podem encobrir o diagnóstico ainda mais. Os achados laboratoriais incluem a hipocomplementemia (de C3 e C4) e a elevação dos níveis de marcadores inflamatórios (velocidade de sedimentação eritrocitária, níveis de proteína C reativa). Alguns pacientes apresentam elevados níveis séricos de creatinina, ureia, proteinúria e hematúria.
Uma abordagem prática para o diagnóstico da doença ateroembólica renal consiste em manter um alto índice de suspeita do distúrbio em pacientes com insuficiência renal aguda, sobretudo após a realização de procedimentos vasculares invasivos ou após a instituição da terapia trombolítica. Exemplificando, 80% dos pacientes com ateroembolia que apresentam envolvimento renal têm níveis séricos de creatinina acima de 2 mg/dL e uma hipertensão que se torna cada vez mais difícil de controlar.114 As manifestações cutâneas sugestivas de ateroembolia incluem o livedo reticular e os dedos (sobretudo do pé) arroxeados. Os baixos níveis de complemento fortalecem ainda mais a possibilidade de doença ateroembólica. O diagnóstico é estabelecido por biópsia da pele, músculo ou rim afetado.
O prognóstico para pacientes com doença ateroembólica, em especial aqueles com envolvimento renal, é precário. A mortalidade varia de 60 a 80%.114 Não há uma terapia específica para a condição. O tratamento geralmente consiste na terapia de suporte, que, quando necessário, inclui a diálise. A anticoagulação pode piorar o curso da doença ateroembólica e, portanto, é contraindicada.
Os autores não possuem relações comerciais com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
1. Harding MB, Smith LR, Himmelstein SI, et al: Renal artery stenosis: prevalence and associated risk factors in patients undergoing routine cardiac catheterization. J Am Soc Nephrol 2:1608, 1992
2. Missouris CG, Buckenham T, Cappuccio FP, et al: Renal artery stenosis: a common and important problem in patients with peripheral vascular disease. Am J Med 96:10, 1994
3. Uzu T, Inoue T, Fujii T, et al: Prevalence and predictors of renal artery stenosis in patients with myocardial infarction. Am J Kidney Dis 29:733, 1997
4. Kuroda S, Nishida N, Uzu T, et al: Prevalence of renal artery stenosis in autopsy patients with stroke. Stroke 31:61, 2000
5. Conlon PJ, Athirakul K, Kovalik E, et al: Survival in renal vascular disease. J Am Soc Nephrol 9:252, 1998
6. Edwards MS, Craven TE, Burke GL, et al: Renovascular disease and the risk of adverse coronary events in the elderly: a prospective, population-based study. Arch Intern Med 165:207, 2005.
7. Svetkey LP, Kadir S, Dunnick NR, et al: Similar prevalence of renovascular hypertension in selected blacks and whites. Hypertension 17:678, 1991
8. Emovon OE, Klotman PE, Dunnick NR, et al: Renovascular hypertension in blacks. Am J Hypertens 9:18, 1996
9. Goldblatt H, Lynch J, Hanzal RF, et al: Studies on experimental hypertension. 1. The production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med 59:347, 1934
10. McLean AG, Hilson AJ, Scoble JE, et al: Screening for renovascular disease with captopril-enhanced renography. Nephrol Dial Transplant 7:211, 1992
11. Thornton J, O’Callaghan J, Walshe J, et al: Comparison of digital subtraction angiography with gadolinium-enhanced magnetic resonance angiography in the diagnosis of renal artery stenosis. Eur Radiol 9:930, 1999
12. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium containing contrast agents—St. Louis, Missouri, 2002–2006. MMWR Morb Mortal Wkly Rep 56:137, 2007
13. Eklof H, Ahlstrom H, Magnusson A, et al: A prospective comparison of duplex ultrasonography, captopril renography, MRA, and CTA in assessing renal artery stenosis. Acta Radiol 47:764, 2006
14. Balk E, Raman G, Chung M, et al: Effectiveness of management strategies for renal artery stenosis: a systematic review. Ann Intern Med 145:901, 2006
15. Ramsay LE, Waller PC: Blood pressure response to percutaneous transluminal angioplasty for renovascular hypertension: an overview of published series. BMJ 300:569, 1990
16. Leertouwer TC, Gussenhoven EJ, Bosch JL, et al: Stent placement for renal arterial stenosis: where do we stand? A meta-analysis. Radiology 216:78, 2000
17. Isles CG, Robertson S, Hill D: Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 92:159, 1999
18. Alcazar JM, Rodicio JL: Ischemic nephropathy: clinical characteristics and treatment. Am J Kidney Dis 36:883, 2000
19. Bakris GL, Weir MR: Angiotensin-converting enzyme inhibitor-associated elevations in serum creatinine: is this a cause for concern? Arch Intern Med 160:685, 2000
20. Radermacher J, Chavan A, Bleck J, et al: Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Engl J Med 344:410, 2001
21. Bonelli FS, McKusick MA, Textor SC, et al: Renal artery angioplasty: technical results and clinical outcome in 320 patients. Mayo Clin Proc 70:1041, 1995
22. van Bockel JH, van Schilfgaarde R, Felthuis W, et al: Long-term results of in situ and extracorporeal surgery for renovascular hypertension caused by fibrodysplasia. J Vasc Surg 6:355, 1987
23. Lupi-Herrera E, Sanchez-Torres G, Marcushamer J, et al: Takayasu’s arteritis: clinical study of 107 cases. Am Heart J 93:94, 1977
24. Subramanyan R, Joy J, Balakrishnan KG: Natural history of aortoarteritis (Takayasu’s disease). Circulation 80:429, 1989
25. Arend WP, Michel BA, Bloch DA, et al: The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum 33:1129, 1990
26. Kerr GS, Hallahan CW, Giordano J, et al: Takayasu arteritis. Ann Intern Med 120:919, 1994
27. Hoffman GS, Leavitt RY, Kerr GS, et al: Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum 37:578, 1994
28. Valsakumar AK, Valappil UC, Jorapur V, et al: Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol 30:1793, 2003
29. Ishikawa K: Natural history and classification of occlusive thromboaortopathy (Takayasu’s disease). Circulation 57:27, 1978
30. Jennette JC, Falk RJ, Andrassy K, et al: Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 37:187, 1994
31. Sack M, Cassidy JT, Bole GG: Prognostic factors in polyarteritis. J Rheumatol 2:411, 1975
32. Scott DG, Bacon PA, Elliott PJ, et al: Systemic vasculitis in a district general hospital 1972–1980: clinical and laboratory features, classification and prognosis of 80 cases. QJM 51:292, 1982
33. McMahon BJ, Heyward WL, Templin DW, et al: Hepatitis B-associated polyarteritis nodosa in Alaskan Eskimos: clinical and epidemiologic features and long-term follow-up. Hepatology 9:97, 1989
34. Lam KC, Lai CL, Trepo C, et al: Deleterious effect of prednisolone in HBsAg-positive chronic active hepatitis. N Engl J Med 304:380, 1981
35. Guillevin L, Lhote F, Gayraud M, et al: Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome: a prospective study in 342 patients. Medicine (Baltimore) 75:17, 1996
36. Sable-Fourtassou R, Cohen P, Mahr A, et al: Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 143:632, 2005
37. Pettersson EE, Sundelin B, Heigl Z: Incidence and outcome of pauci-immune necrotizing and crescentic glomerulonephritis in adults. Clin Nephrol 43:141, 1995
38. Savage CO, Harper L, Adu D: Primary systemic vasculitis. Lancet 349:553, 1997
39. Falk RJ, Terrell R, Charles LA, et al: Anti-neutrophil cytoplasmic antibodies induce neutrophils to degranulate and produce oxygen radicals. Proc Natl Acad Sci USA 87:4115, 1990
40. Xiao H, Heeringa P, Hu P, et al: Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 110:955, 2002
41. Xiao H, Schreiber A, Heeringa P, et al: Alternative complement pathway in the pathogenesis of disease mediated by antineutrophil cytoplasmic autoantibodies. Am J Pathol 170:52, 2007.
42. Panegyres PK, Blumbergs PC, Leong AS, et al: Vasculitis of peripheral nerve and skeletal muscle: clinicopathological correlation and immunopathic mechanisms. J Neurol Sci 100:193, 1990
43. Kissel JT, Mendell JR: Vasculitic neuropathy. Neurol Clin 10:761, 1992
44. Jennette JC, Falk RJ: Small-vessel vasculitis. N Engl J Med 337:1512, 1997
45. Hogan SL, Nachman PH, Wilkman AS, et al: Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 7:23, 1996
46. Hogan SL, Falk RJ, Chin H, et al: Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann Intern Med 143:621, 2005
47. Jayne D, Rasmussen N, Andrassy K, et al: A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. European Vasculitis Study Group. N Engl J Med 349:36, 2003
48. Pusey CD: Anti-glomerular basement membrane disease. Kidney Int 64:1535, 2003
49. Hellmark T, Niles JL, Collins AB, et al: Comparison of anti-GBM antibodies in sera with or without ANCA. J Am Soc Nephrol 8:376, 1997
50. Weening JJ, D’Agati VD, Schwartz MM, et al: The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 15:241, 2004
51. Chan TM, Li FK, Tang CS, et al: Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong Kong-Guangzhou Nephrology Study Group. N Engl J Med 343:1156, 2000
52. Contreras G, Pardo V, Leclercq B, et al: Sequential therapies for proliferative lupus nephritis. N Engl J Med 350:971, 2004
53. Moake JL: Thrombotic microangiopathies. N Engl J Med 347:589, 2001
54. Tsai HM: The molecular biology of thrombotic microangiopathy. Kidney Int 70:16, 2006
55. Tsai HM: Current concepts in thrombotic thrombocytopenic purpura. Annu Rev Med 57:419, 2006
56. Warwicker P, Goodship TH, Donne RL, et al: Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 53:836, 1998
57. Caprioli J, Bettinaglio P, Zipfel PF, et al: The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. Italian Registry of Familial and Recurrent HUS/TTP. J Am Soc Nephrol 12:297, 2001
58. Sanchez-Corral P, Perez-Caballero D, Huarte O, et al: Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet 71:1285, 2002
59. Noris M, Brioschi S, Caprioli J, et al: Familial haemolytic uraemic syndrome and an MCP mutation. International Registry of Recurrent and Familial HUS/TTP. Lancet 362:1542, 2003
60. Bell WR, Braine HG, Ness PM, et al: Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: clinical experience in 108 patients. N Engl J Med 325:398, 1991
61. Rosove MH, Ho WG, Goldfinger D: Ineffectiveness of aspirin and dipyridamole in the treatment of thrombotic thrombocytopenic purpura. Ann Intern Med 96:27, 1982
62. Heidel F, Lipka DB, Huber C, et al: Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 97:228, 2007
63. Schleinitz N, Ebbo M, Mazodier K, et al: Rituximab as preventative therapy of a clinical relapse in TTP with ADAMTS 13 inhibitor. Am J Hematol 82:417, 2007
64. Karmali MA, Petric M, Lim C, et al: The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis 151:775, 1985
65. Cleary TG: Cytotoxin-producing Escherichia coli and the hemolytic uremic syndrome. Pediatr Clin North Am 35:485, 1988
66. Banatvala N, Griffin PM, Greene KD, et al: The United States National Prospective Hemolytic Uremic Syndrome Study: microbiologic, serologic, clinical, and epidemiologic findings. Hemolytic Uremic Syndrome Study Collaborators. J Infect Dis 183:1063, 2001
67. Misiani R, Appiani AC, Edefonti A, et al: Haemolytic uraemic syndrome: therapeutic effect of plasma infusion. Br Med J (Clin Res Ed) 285:1304, 1982
68. Sheth KJ, Gill JC, Hanna J, et al: Failure of fresh frozen plasma infusions to alter the course of hemolytic uremic syndrome. Child Nephrol Urol 9:38, 1988-89
69. Roberts JM, Cooper DW: Pathogenesis and genetics of pre-eclampsia. Lancet 357:53, 2001
70. Trupin LS, Simon LP, Eskenazi B: Change in paternity: a risk factor for preeclampsia in multiparas. Epidemiology 7:240, 1996
71. Dekker G, Sibai B: Primary, secondary, and tertiary prevention of pre-eclampsia. Lancet 357:209, 2001
72. Levine RJ, Maynard SE, Qian C, et al: Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350:672, 2004
73. Sibai BM, Caritis SN, Thom E, et al: Prevention of preeclampsia with low-dose aspirin in healthy, nulliparous pregnant women. The National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine Units. N Engl J Med 329:1213, 1993
74. Levine RJ, Hauth JC, Curet LB, et al: Trial of calcium to prevent preeclampsia. N Engl J Med 337:69, 1997
75. Knight M, Duley L, Henderson-Smart DJ, et al: Antiplatelet agents for preventing and treating pre-eclampsia. Cochrane Database Syst Rev (2):CD000492, 2000
76. Sibai BM: Treatment of hypertension in pregnant women. N Engl J Med 335:257, 1996
77. Lindheimer MD, Davison JM: Renal biopsy during pregnancy: ‘to b . . . or not to b . . .?’. Br J Obstet Gynaecol 94:932, 1987
78. Packham D, Fairley KF: Renal biopsy: indications and complications in pregnancy. Br J Obstet Gynaecol 94:935, 1987
79. Walker JJ: Pre-eclampsia. Lancet 356:1260, 2000
80. Greaves M: Antiphospholipid antibodies and thrombosis. Lancet 353:1348, 1999
81. Levine JS, Branch DW, Rauch J: The antiphospholipid syndrome. N Engl J Med 346:752, 2002
82. Petri M: Epidemiology of the antiphospholipid antibody syndrome. J Autoimmun 15:145, 2000
83. Cervera R, Khamashta MA, Font J, et al: Systemic lupus erythematosus: clinical and immunologic patterns of disease expression in a cohort of 1,000 patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore) 72:113, 1993
84. Merkel PA, Chang Y, Pierangeli SS, et al: The prevalence and clinical associations of anticardiolipin antibodies in a large inception cohort of patients with connective tissue diseases. Am J Med 101:576, 1996
85. Love PE, Santoro SA: Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders: prevalence and clinical significance. Ann Intern Med 112:682, 1990
86. Alarcon-Segovia D, Perez-Vazquez ME, Villa AR, et al: Preliminary classification criteria for the antiphospholipid syndrome within systemic lupus erythematosus. Semin Arthritis Rheum 21:275, 1992
87. Lawrence RC, Helmick CG, Arnett FC, et al: Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum 41:778, 1998
88. Silman AJ: Epidemiology of scleroderma. Ann Rheum Dis 50:846, 1991
89. Medsger TA, Masi AT: The epidemiology of systematic sclerosis (scleroderma). Ann Intern Med 74:714, 1971
90. Livi R, Teghini L, Pignone A, et al: Renal functional reserve is impaired in patients with systemic sclerosis without clinical signs of kidney involvement. Ann Rheum Dis 61:682, 2002
91. Kovalchik MT, Guggenheim SJ, Silverman MH, et al: The kidney in progressive systemic sclerosis: a prospective study. Ann Intern Med 89:881, 1978
92. Tuffanelli DL, Winkelmann RK: Systemic scleroderma, a clinical study of 727 cases. Arch Dermatol 84:359, 1961
93. Traub YM, Shapiro AP, Rodnan GP, et al: Hypertension and renal failure (scleroderma renal crisis) in progressive systemic sclerosis: review of a 25-year experience with 68 cases. Medicine (Baltimore) 62:335, 1983
94. Silman AJ, Hochberg MC: Occupational and environmental influences on scleroderma. Rheum Dis Clin N Am 22:737, 1996
95. Haustein UF, Herrmann K: Environmental scleroderma. Clin Dermatol 12:467, 1994
96. Garabrant DH, Dumas C: Epidemiology of organic solvents and connective tissue disease. Arthritis Res 2:5, 2000
97. Ferri C, Bernini L, Cecchetti R, et al: Cutaneous and serologic subsets of systemic sclerosis. J Rheumatol 18:1826, 1991
98. Steen VD, Medsger TA Jr: Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum 41:1613, 1998
99. Steen VD, Medsger TA Jr: Long-term outcomes of scleroderma renal crisis. Ann Intern Med 133:600, 2000
100.Vaughan CJ, Delanty N: Hypertensive emergencies. Lancet 356:411, 2000
101.Platt OS, Brambilla DJ, Rosse WF, et al: Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med 330:1639, 1994
102.Bunn HF: Pathogenesis and treatment of sickle cell disease. N Engl J Med 337:762, 1997
103.Wesson DE: The initiation and progression of sickle cell nephropathy. Kidney Int 61:2277, 2002
104.Thompson J, Reid M, Hambleton I, et al: Albuminuria and renal function in homozygous sickle cell disease: observations from a cohort study. Arch Intern Med 167:701, 2007
105.Addae SK: The kidney in sickle cell disease. V: Clinical manifestations. Ghana Med J 12:352, 1973
106.Flaster S, Lome LG, Presman D: Urologic complications of renal papillary necrosis. Urology 5: 331, 1975
107.McCall JW, Moule N, Desai P, et al: Urologic findings in homozygous sickle cell disease. Radiology 126:99, 1978
108.Tejani A, Phadke K, Adamson O, et al: Renal lesions in sickle cell nephropathy in children. Nephron 39:352, 1985
109.Bhathena DB, Sondheimer JH: The glomerulopathy of homozygous sickle hemoglobin (SS) disease: morphology and pathogenesis. J Am Soc Nephrol 1:1241, 1991
110.Falk RJ, Scheinman J, Phillips G, et al: Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med 326:910, 1992
111.Quinn CT, Rogers ZR, Buchanan GR: Survival of children with sickle cell disease. Blood 103:4023, 2004
112.Modi KS, Rao VK: Atheroembolic renal disease. J Am Soc Nephrol 12:1781, 2001
113.Blankenship JC, Butler M, Garbes A: Prospective assessment of cholesterol embolization in patients with acute myocardial infarction treated with thrombolytic vs conservative therapy. Chest 107:662, 1995
114.Fine MJ, Kapoor W, Falanga V: Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology 38:769, 1987
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.