(Carregando Índice)... (Carregando Índice)... |
Última revisão: 05/02/2014
Comentários de assinantes: 0
Professor of Medicine, Pritzker School of Medicine, University of Chicago, Chicago, IL
Artigo original: Larson RA. Acute leukemia. ACP Medicine. 2010;1-19.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon
As leucemias agudas são caracterizadas pela diferenciação e proliferação aberrante de células progenitoras hematopoiéticas que sofreram transformação maligna. Estas células acumulam-se na medula óssea e, após a formação de uma carga substancial de células leucêmicas, suprimem a produção de células sanguíneas normais. Os sintomas resultam de vários graus de anemia, neutropenia e trombocitopenia, ou da infiltração nos tecidos. Embora quase todos os sistemas orgânicos possam apresentar envolvimento após a entrada das células leucêmicas no sangue periférico, os sítios mais comumente detectados na clínica são os linfonodos, fígado, baço, sistema nervoso central (SNC) e pele.
O 1º reconhecimento da leucemia como entidade distintiva em geral é concedido, de maneira independente, a Virchow (Berlin) e Bennett (na Escócia), no ano de 1845. Em 1847, Virchow elegeu o termo “leucemia” (do grego leuk, que significa “células brancas”, e emia, que significa “sangue”) para substituir o termo alemão weisshäme. Os distúrbios leucêmicos atualmente são classificados de acordo com a provável célula de origem. A leucemia mieloide aguda (LMA) resulta da transformação maligna de uma célula progenitora de medula óssea (mieloide) ou de uma célula-tronco, que é a precursora normal de granulócitos, eritrócitos e megacariócitos.1 A leucemia linfoblástica aguda (LLA) é uma doença maligna de células precursoras iniciais das linhagens linfocíticas de células B e T. A síndrome mielodisplásica (SMD) é um dos grupos heterogêneos de distúrbios hematológicos malignos que frequentemente envolvem todas as 3 linhagens de células mieloides e cuja origem provável é uma célula bastante primitiva ou uma célula-tronco totipotente.2,3 A SMD combina a insuficiência da medula óssea a numerosos aspectos de uma leucemia crônica ou da conhecida leucemia latente, que pode evoluir para LMA.
Embora a leucemia possa ocorrer em qualquer fase da vida, sua incidência está mais fortemente relacionada ao avanço da idade. A LLA predomina em crianças, enquanto a LMA corresponde à maioria dos casos envolvendo adultos. A média da idade dos pacientes no momento do diagnóstico de LMA está em torno de 72 anos.4 De acordo com um registro populacional sueco, a incidência da condição aumenta de 5 casos em 100.000 indivíduos na faixa etária de 16 a 24 anos para quase 160 casos em 100.000 indivíduos na faixa etária de 80 a 84 anos. A incidência de LLA atinge o pico na faixa etária de 3 a 5 anos, sendo que a LLA da infância é manifestamente diferente da LLA de fase adulta. Embora a média da idade dos indivíduos adultos com LLA incluídos nas avaliações clínicas seja de 35 a 40 anos, é bastante provável que os indivíduos de idade mais avançada estejam sub-representados nestes relatos. Existem diferenças marcantes entre os vários subtipos de leucemia que ocorrem em crianças, adultos jovens e adultos de idade mais avançada. A SMD é observada com pouca frequência em indivíduos com menos de 50 anos e passa a ser cada vez mais comum entre os indivíduos de faixas etárias mais altas. Existem numerosas similaridades biológicas e clínicas entre a SMD e os subtipos mais comuns de LMA observados em indivíduos adultos de idade mais avançada. Além disso, alguns pacientes com LMA podem ter uma doença que evoluiu a partir de uma SMD não identificada.
A leucemia aguda é mais comum em indivíduos brancos do que em afrodescendentes, em todas as idades.5 A leucemia aguda também é mais comum em homens do que mulheres, em parte devido à maior incidência entre meninos e homens de idade mais avançada. A exposição ocupacional e ambiental pode contribuir para a disparidade observada entre os sexos.
As variações geográficas da incidência provavelmente estão relacionadas a alguns fatores, incluindo condição socioeconômica, etnia e contexto urbano ou rural, podendo contribuir para as maiores frequências relatadas em países industrializados e áreas urbanas. Algumas anormalidades citogenéticas são relatadas com maior frequência em determinados países.
Embora a causa da leucemia aguda em seres humanos seja desconhecida, existem diversos fatores hereditários e ambientais que parecem exercer algum papel etiológico. A patogênese da leucemia aguda envolve interações complexas entre suscetibilidade do hospedeiro, dano cromossômico secundário à exposição física ou química, e possivelmente a incorporação de informação genética transmitida por vírus a células progenitoras suscetíveis.6 Grande parte das evidências é indireta e foi inferida a partir de estudos epidemiológicos. Os pesquisadores baseiam-se pesadamente nos modelos de experimentação animal de leucemogênese. Em certas espécies não humanas, por exemplo, a exposição a agentes como radiação ionizante ou um carcinógeno químico aumenta incidência de leucemia.
Os distúrbios caracteristicamente leucêmicos podem ser produzidos em modelos experimentais animais, por meio da transfecção de genes como BCR/ABL em células germinativas. O gene de fusão BCR/ABL resulta da translocação ou justaposição do gene ABL no cromossomo 9 ao gene BCR no cromossomo 22. Este novo gene de fusão produz uma proteína que está ausente nas células normais. A translocação t(9;22) origina um cromossomo 22 anormal, que é denominado cromossomo Philadelphia (Ph). Este rearranjo é encontrado em cerca de 1/3 dos adultos com LLA, bem como em todos os pacientes com leucemia mieloide crônica (LMC).
A LMA é uma doença genética e fenotipicamente heterogênea. O espectro de mutações identificado sugere que os alelos determinantes da leucemia podem ser agrupados em 2 amplos grupos complementares: um grupo de alelos que conferem vantagem proliferativa ou de sobrevida aos progenitores hematopoiéticos; e um grupo de alelos de comprometem a diferenciação hematopoiética e conferem propriedades de autorrenovação às células hematopoiéticas que estejam em um determinado estágio de diferenciação.7
O potencial leucemogênico da radiação ionizante foi bem reconhecido durante vários anos. As leucemias associadas à radiação em geral derivam da linhagem mieloide, mas frequentemente apresentam aspectos de tripla linhagem característicos de células-tronco multipotentes. Uma parte significativa das evidências epidemiológicas deriva de observações dos resultados da exposição humana à radiação oriunda de explosões nucleares, fontes de radiação terapêuticas ou diagnósticas, e de fontes de radiação ocupacionais. Após os ataques de bomba atômica às cidades de Hiroshima e Nagasaki, os sobreviventes expostos sofreram um aumento marcante da incidência das formas aguda e crônica de leucemia.8 A probabilidade de desenvolvimento de leucemia nos sobreviventes destes ataques estava relacionada à intensidade da exposição do indivíduo à radiação, sendo que as exposições a menos de 100 cGy foram associadas a um risco relativamente baixo.9
Os pacientes tratados com irradiação de feixe-externo em decorrência de espondilite anquilosante ou menorragia, bem como aqueles tratados com fósforo radioativo (32P) em decorrência de policitemia vera apresentaram incidência aumentada de leucemia aguda.10,11 O risco de desenvolvimento de leucemia mieloide aguda associada à terapia (LMA-t) após o uso apenas de radioterapia para tratamento de doença de Hodgkin ou câncer de mama parece ser baixo.12 Atualmente, o uso do diagnóstico por imagem de raios X não parece estar associado a nenhum risco aumentado de leucemia entre os pacientes. Entretanto, a exposição fetal intrauterina aos raios X aumenta o risco de desenvolvimento subsequente de leucemia na infância.13
O risco de desenvolvimento de leucemia em indivíduos expostos à radiação no local de trabalho foi comprovado por estudos antigos sobre mulheres que trabalhavam na pintura de mostradores de relógio de pulso contendo rádio, bem como pela incidência aumentada de leucemia entre radiologistas observada no início do século XX.14,15 As práticas de proteção contra radiação vigentes parecem ter diminuído significativamente este risco.
Existem dados epidemiológicos substanciais associando o desenvolvimento de leucemia aguda à exposição de agentes químicos terapêuticos ou industriais. A exposição a agentes alquilantes, como melfalana ou mostarda nitrogenada, está associada ao desenvolvimento de neoplasias mieloides relacionadas à terapia (NM-t) após um período de latência de 5 a 7 anos.12,16 Esta doença é caracterizada por pancitopenia, mielodisplasia de tripla linhagem e anormalidades citogenéticas complexas, mais frequentemente envolvendo perdas parciais ou totais dos cromossomos 5 ou 7.17,18 A NM-t é uma complicação bem conhecida do tratamento da doença de Hodgkin, mieloma múltiplo e câncer de ovário ou mama, bem como do transplante de células-tronco hematopoiéticas (CTH) autólogas. O risco é maior em pacientes submetidos a quimio e radioterapia. Outro subtipo de LMA-t foi observado em pacientes submetidos ao tratamento de certas neoplasias hematológicas e tumores sólidos com fármacos epipodofiloxina (etoposídeo e teniposídeo), e também foi associado ao uso de agentes inibidores da atividade de topoisomerase II, como doxorrubicina e mitoxantrona.12,16,19 Esta leucemia apresenta um período de latência bem mais curto, frequentemente de 1 a 2 anos, e uma manifestação mais aguda, com contagem de leucócitos elevada, morfologia monoblástica e, em geral, anormalidade citogenética envolvendo o braço longo do cromossomo 11 na banda q23. O rearranjo do gene MLL (leucemia de linhagem mista) neste locus ocorre com frequência.20
No início do século XX, a leucemia aguda foi observada em trabalhadores expostos ao benzeno. Exemplificando, a incidência de leucemia aguda em sapateiros turcos submetidos à exposição prolongada ao benzeno foi 2 a 3 vezes maior do que a incidência de leucemia aguda na população em geral.21 Com o advento das leis de segurança do trabalho mais severas, a leucemia associada ao benzeno quase desapareceu nos países ocidentais, embora continue presente nos países em desenvolvimento.22,23
Alguns vírus são comprovadamente causadores de leucemia aguda em espécies não humanas. O papel em potencial dos vírus na leucemogênese humana tem sido explorado há décadas. Não existem dados conclusivos que indiquem a origem viral de qualquer um dos tipos comuns de leucemia aguda humana. Entretanto, o genoma do vírus Epstein-Barr (EBV) está presente na maioria das células do linfoma/leucemia de Burkitt. A leucemia de células T do adulto é uma forma incomum de leucemia humana, amplamente restrita ao sul do Japão e ao Caribe, a qual apresenta uma associação estreita com a infecção por um vírus leucemogênico – o vírus linfotrópico da célula T humana de tipo 1 (HTLV-1). Entretanto, a maioria dos indivíduos que expressam anticorpos contra este vírus jamais exibe evidências de malignidade.
As observações de incidência aumentada da leucemia aguda entre membros de algumas famílias com alta suscetibilidade, a alta frequência de leucemia concordante em gêmeos monozigóticos, e a associação da leucemia aguda em indivíduos com distúrbios genéticos estabeleceram que os fatores hereditários atuam no desenvolvimento da leucemia.5,6 Em gêmeos idênticos, quando um dos irmãos tem leucemia, o outro também apresenta risco de desenvolvimento de leucemia, frequentemente antes de 8 anos de idade.24 É comum a doença se desenvolver no segundo gêmeo dentro de um período de 1 ano após o diagnóstico do primeiro. Irmãos não idênticos de indivíduos afetados pela leucemia apresentam risco menor, contudo este risco continua sendo maior do que aquele observado na população em geral. Evidências moleculares da presença do gene de fusão MLL/AF4 foram detectadas em recém-nascidos que posteriormente desenvolveram LLA com t(4;11) durante a lactância ou infância.25
A incidência tanto da LMA como da LLA está aumentando entre os pacientes com síndrome de Down, uma doença caracterizada pela trissomia do 21 resultante de não disjunção cromossômica.26 Cerca de 5 a 10% dos casos de LMA da infância ocorrem em crianças com síndrome de Down. O prognóstico destes pacientes é mais favorável do que o prognóstico de crianças na mesma faixa etária com LMA e sem síndrome de Down.27 Uma evidência adicional da relação existente entre leucemia e anormalidades cromossômicas é a incidência aumentada entre os pacientes com síndrome de Bloom e anemia de Fanconi. Ambos os distúrbios são condições hereditárias caracterizadas pelo aumento da quebra cromossômica e defeito do reparo do DNA. Outras condições genéticas normalmente não associadas a anomalias cromossômicas, como a ataxia-telangiectasia e a agamaglobulinemia congênita, também estão associadas à leucemia aguda. As deficiências de imunidade celular e humoral que caracterizam estas condições podem aumentar a suscetibilidade ao desenvolvimento de leucemia. A incidência de 10 anos da LMA ou da SMD entre pacientes com neutropenia congênita severa, um distúrbio de transmissão autossômica associado à resposta defeituosa de células precursoras ao fator estimulador de colônia de granulócitos (G-CSF), ultrapassa 20%.28 O distúrbio plaquetário familiar é uma rara condição autossômica dominante caracterizada por anormalidades qualitativas e quantitativas e por mutações monoalélicas em RUNX1 na linhagem germinativa. A LMA é observada em indivíduos afetados que adquirem um 2º evento genético envolvendo RUNX1.29
A classificação tradicional das leucemias agudas é baseada na descrição morfológica, refletindo o tipo celular predominante na população da medula óssea e associando a célula a sua contraparte hematopoiética normal. Em 1976, o grupo de hematopatologistas French-American-British (FAB) estabeleceu um sistema de classificação para leucemias agudas que separou a LMA e a LLA em distúrbios distintos.30 Este sistema baseava-se somente na avaliação microscópica de esfregaços de sangue e de medula tratados com colorações de rotina e suplementados por um número limitado de procedimentos citoquímicos. Dada a facilidade do uso e a aplicabilidade na comparação de resultados de tratamentos entre instituições, este sistema teve ampla aceitação. Em 1985, o sistema foi revisado para fins de esclarecimento e ampliação para abrangência de novas técnicas diagnósticas.31 Em 2008, um comitê da Organização Mundial de Saúde (OMS) atualizou um esquema de classificação mais abrangente, que usa morfologia, imunofenotipagem, etiologia e citogenética, e faz uma distinção mais nítida entre LMA, SMD e distúrbios mieloproliferativos crônicos [Tabela 1].18 Além disso, um painel de especialistas do European LeukemiaNet publicou diretrizes detalhadas para uso da classificação da OMS em estudos clínicos e no tratamento risco-adaptado individualizado.32
Tabela 1. LMA e neoplasias de precursores correlatas, e leucemias agudas de linhagem ambígua18
|
LMA com anormalidades genéticas recorrentes |
|
LMA com t(8;21)(q22;q22); RUNX1-RUNX1T1 |
|
LMA com inv(16)(p13.1q22) ou t(16;16)(p13.1;q22); CBFB-MYH11 |
|
LPMA com t(15;17)(q22;q12); PML-RARA |
|
LMA com t(9;11)(p22;q23); MLLT3-MLL |
|
LMA com t(6;9)(p23;q34); DEK-NUP214 |
|
LMA com inv(3)(q21q26.2) ou t(3;3)(q21;q26.2); RPN1-EVI1 |
|
LMA (megacarioblástica) com t(1;22)(p13;q13); RBM15-MKL1 |
|
Entidade temporária: LMA com NPM1 mutante |
|
Entidade temporária: LMA com CEBPA mutante |
|
LMA com alterações relacionadas à mielodisplasia |
|
Neoplasias mieloides relacionadas à terapia* |
|
LMA, SOE |
|
LMA com diferenciação mínima |
|
LMA sem maturação |
|
LMA com maturação |
|
Leucemia mielomonocítica aguda |
|
Leucemia monocítica/monoblástica aguda |
|
Leucemia eritroide aguda |
|
Leucemia eritroide pura |
|
Eritroleucemia, eritroide/mieloide |
|
Leucemia megacarioblástica aguda |
|
Leucemia basofílica aguda |
|
Panmielose aguda com mielofibrose (mielofibrose aguda, mielosclerose aguda) |
|
Sarcoma mieloide (tumor mieloide extramedular, sarcoma granulocítico, cloroma) |
|
Proliferações mieloides relacionadas à síndrome de Down |
|
Mielopoiese anormal transiente (distúrbio mieloproliferativo transiente) |
|
Leucemia mieloide associada à síndrome de Down |
|
Neoplasia de célula dendrítica plasmacitoide blástica |
|
Leucemias agudas de linhagem ambígua |
|
Leucemia indiferenciada aguda |
|
Leucemia aguda de fenótipo misto com t(9;22)(q34;q11.2); BCR-ABL1 |
|
Leucemia aguda de fenótipo misto com t(v;11q23); rearranjo de MLL |
|
Leucemia aguda de fenótipo misto, B/mieloide, SOE |
|
Leucemia aguda de fenótipo misto, T/mieloide, SOE |
|
Entidade temporária: leucemia linfoblástica de célula natural killer/linfoma |
*A NM-t inclui o espectro de qualquer SMD ou LMA que ocorra subsequentemente à terapia citotóxica prévia para distúrbio não mieloide.
LMA = leucemia mieloide aguda; LPMA = leucemia promielocítica aguda; NM-t = neoplasia mieloide relacionada à terapia; SMD = síndrome mielodisplásica; SOE = sem outras especificações.
Os blastos dos pacientes com LMA costumam ser maiores do que os linfoblastos e exibem maior heterogeneidade em termos de tamanho e forma. As células blásticas da LMA possuem citoplasma abundante e em muitos casos contêm grânulos citoplasmáticos. Os corpúsculos de Auer (acúmulos do tipo cristalino, azurofílicos, de grânulos lisossômicos anormais, que podem ser vistos no citoplasma pela coloração de Wright) são detectados em cerca de 10% dos pacientes com LMA.
Um diagnóstico de LMA é estabelecido quando pelo menos 20% de todas as células nucleadas da medula são blastos.18,32 Uma forma de LMA apresenta diferenciação mínima e não pode ser diagnosticada apenas com base nas características morfológicas ou citoquímicas, mas requer o uso adicional de coloração imuno-histoquímica ou imunofenotipagem por citometria de fluxo.33 Estes blastos expressam antígenos mieloides na superfície, mas não exibem reatividade à mieloperoxidase.
O exame morfológico de esfregaços de sangue ou medula óssea às vezes falha em fornecer um diagnóstico inequívoco. No entanto, a identificação de vários antígenos de diferenciação na superfície das células anômalas por citometria de fluxo pode fornecer rapidamente esta informação decisiva34,35 [Tabela 2]. A expressão aberrante dos antígenos de superfície ao diagnóstico também fornece um marcador do clone maligno, que pode ser usado para detecção de doença residual mínima após o tratamento.36
Tabela 2. Expressão de marcadores de superfície celular e citoplasmáticos para diagnóstico da LMA e LAFM
|
Diagnóstico de LMA |
|
|
Estágio precursor |
CD34, CD38, CD117, CD133, HLA-DR |
|
Marcadores granulocíticos |
CD13, CD15, CD16, CD33, CD65, cMPO |
|
Marcadores monocíticos |
NSE, CD11c, CD14, CD64, lisozima, CD4, CD11b, CD36, homólogo NG2 |
|
Marcadores megacariocíticos |
CD41 (glicoproteína IIb/IIIa), CD61 (glicoproteína IIIa), CD42 (glicoproteína 1b) |
|
Marcador eritroide |
CD235a (glicoforina A) |
|
Diagnóstico de LAFM* |
|
|
Linhagem mieloide |
MPO ou evidência de diferenciação monocítica (pelo menos 2 dos seguintes marcadores: NSE, CD11c, CD14, CD64, lisozima) |
|
Linhagem B |
CD19 (forte) com pelo menos 1 dos seguintes marcadores: CD79a, cCD22, CD10 ou CD19 (fraco) com pelo menos 2 dos seguintes marcadores: CD79a, cCD22, CD10 |
|
Linhagem T |
cCD3 ou CD3 de superfície |
*Note que a LAFM pode ser diagnosticada diante da existência de populações separadas de blastos linfoides e mieloides.
cMPO = mieloperoxidase citoplasmática; LAFM = leucemia aguda de fenótipo misto; LMA = leucemia mieloide aguda; MPO = mieloperoxidase; NSE = esterase inespecífica.
Até agora, nenhum antígeno de superfície LMA-específico foi identificado. Em vez disso, as células da LMA frequentemente coexpressam antígenos de superfície que não são coexpressos em suas contrapartes mieloides normais. De uma forma geral, as tentativas de correlacionar a imunofenotipagem à classificação morfológica da LMA ou ao resultado clínico têm sido imprecisas. Todavia, a expressão de certos antígenos, como o CD34, um marcador de célula-tronco inicial, pode fornecer informação prognóstica significativa.37 Além disso, o CD33, que é expresso na maioria dos mieloblastos, tornou-se um alvo importante da terapia dirigida com anticorpos monoclonais.38
A leucemia promielocítica aguda (LPMA) é uma forma clínica, morfológica, patológica e geneticamente distintiva de leucemia, que hoje é considerada à parte da LMA.39 Os aspectos característicos incluem uma proliferação intensa de promielócitos malignos contendo o gene de fusão PML/RARA e um distúrbio hemorrágico parcialmente relacionado à coagulação intravascular disseminada (CID). Os contornos nucleares das células da LPMA são tipicamente bilobados ou reniformes. Embora a condição seja caracterizada pela presença de grânulos citoplasmáticos azurofílicos amplos, também existe uma variante de LPMA microgranular.
A SMD faz parte de um grupo heterogêneo de distúrbios de CTH caracterizados por displasia citológica na medula óssea e no sangue, e por várias combinações de anemia, neutropenia e trombocitopenia.2,3,40 Estes distúrbios compartilham a evolução progressiva da população monoclonal de células hematopoiéticas, geralmente envolvendo múltiplas linhagens, na maioria das vezes com a acompanhante supressão da hematopoiese normal. A história natural destas síndromes apresenta uma ampla variação, que vai da anemia crônica com baixa propensão à conversão leucêmica até as síndromes com distúrbios hematológicos severos e alto risco de progressão para LMA. O exame de esfregaços devidamente preparados de sangue periférico, esfregaços de aspirado de medula óssea e amostras de biópsia de medula óssea em geral permitem confirmar o diagnóstico de SMD. A medula é tipicamente normocelular ou hipercelular para a idade do paciente. A avaliação citológica de esfregaços de sangue e de aspirado de medula deve mostrar a ocorrência de displasia em pelo menos 1 linhagem celular. É preciso enfatizar que estas anormalidades são inespecíficas para SMD e podem ser observadas em diversos distúrbios hematológicos, tanto hereditários como adquiridos. É preciso ter o cuidado de excluir a hipótese destes e de outros processos, antes de estabelecer o diagnóstico de SMD. Existe uma sobreposição entre a SMD hipocelular e a anemia aplásica autoimune.
Diversos termos, entre os quais “pré-leucemia”, “leucemia latente ou subaguda”, “síndrome dismielopoiética” e “mielodisplasia”, são empregados para descrever a SMD. Evidências consideráveis indicam que estes distúrbios são clonais e neoplásicos desde a 1ª vez em que são detectados. Sendo assim, o termo “pré-leucemia” parece inadequado, pois implica em uma condição pré-maligna. Em vez disso, estes distúrbios são mais apropriadamente considerados uma forma de leucemia de células-tronco crônica ou latente. Contudo, as técnicas diagnósticas e a nomenclatura ainda são inadequadas para discriminar os subgrupos clinicamente distintos junto a estas síndromes, com raras exceções. Existe uma sobreposição considerável com outras doenças morfologicamente determinadas, em particular aquelas classificadas como neoplasias mieloproliferativas (NMP) crônicas. Enquanto a diferenciação hematopoiética é qualitativamente não pareada na NMP, pelo menos durante os estágios iniciais, a SMD caracteriza-se brevemente por uma hematopoiese inefetiva, intensificação da apoptose e comprometimento da diferenciação. Desta forma, é possível especular que a SMD resulta de alterações em genes que controlam a transcrição, diferenciação e sobrevida celulares, em vez dos genes controladores da proliferação celular.
Em 1982, o grupo FAB tentou padronizar a classificação dos pacientes com SMD empregando critérios baseados na citologia e número de blastos na medula e no sangue periférico.41 Em 1996, outro subgrupo importante foi descrito: a citopenia refratária com displasia de linhagens múltiplas (CRDM).42 A classificação da SMD criada pela OMS atualmente tem ampla aceitação [Tabela 3].18 A leucemia mielomonocítica crônica é hoje classificada como uma categoria à parte de SMD/distúrbios NMP. Em alguns pacientes, observa-se uma progressão natural da doença entre as categorias, à medida que o amadurecimento celular se torna mais lento e os blastos se acumulam. Em outros pacientes, contudo, a categoria diagnóstica não muda ao longo da expectativa de vida do paciente. Estes distúrbios causam uma considerável morbidade e mortalidade, devido à insuficiência medular e à progressão para LMA.
Tabela 3. Classificação da OMS da SMD primária2,18
|
Subtipo da OMS |
Sangue periférico |
Medula óssea |
|
ARF |
Hemoglobina < 10 g/dL; < 1% blastos; reticulocitopenia; macrocítica ou normocrômica/ normocítica |
Geralmente, hiperplasia eritroide com diseritropoiese; < 5% blastos; < 15% sideroblastos anelares |
|
Neutropenia refratária |
Neutrófilos < 1.800/mcL; displasia |
< 5% blastos |
|
Trombocitopenia refratária |
Plaquetas < 100.000/mcL; displasia |
< 5% blastos |
|
ARSA |
Anemia; somente diseritropoiese; morfologia de hemácias dimórficas |
Como na ARF, porém com sideroblastos anelares = 15% dos precursores eritroides |
|
ARF com del(5q) isolada |
Anemia; < 5% blastos |
< 5% blastos; del(5q) isolada; sem corpúsculos de Auer |
|
CRDM |
< 1% blastos; citopenias; displasia de múltiplas linhagens; sem corpúsculos de Auer |
< 5% blastos; displasia de múltiplas linhagens (= 10% Das células em = 2 linhagens celulares mieloides); independente da contagem de sideroblastos anelares |
|
ARF com excesso de blastos–1 (ARFEB-1) |
2 a 4% blastos |
5 a 9% blastos; displasia de uma ou múltiplas linhagens |
|
ARF com excesso de blastos–2 (ARFEB-2) |
5 a 19% blastos; pode haver corpúsculos de Auer |
10 a 19% blastos ou corpúsculos de Auer |
|
SMD-NC |
< 1% blastos; citopenias |
< 5% blastos; displasia de linhagem única; sem corpúsculos de Auer |
ARF = anemia refratária; ARSA = anemia refratária com sideroblastos anelares; CRDM = citopenia refratária com displasia de linhagens múltiplas; OMS = Organização Mundial de Saúde; SMD = síndrome mielodisplásica; SMDNC = síndrome mielodisplásica não classificável.
O uso aumentado da análise citogenética e o desenvolvimento de novas técnicas para avaliação da clonalidade estão se mostrando úteis não só para fins diagnósticos como também para discernir a patogênese e os padrões de responsividade aos fatores de crescimento e possíveis agentes indutores de diferenciação.40,43 Dados significativos sugerem que a SMD resulta do aparecimento de defeitos combinados nas CTH e células estromais. Hoje em dia, é possível definir várias síndromes que podem estar associadas a uma história natural mais previsível.2,3 Exemplificando, a deleção de um braço longo do cromossomo 5 pode ser detectada em alguns pacientes de idade mais avançada, sobretudo entre as mulheres, com anemia refratária (ARF) macrocítica. A contagem de plaquetas costuma estar normal ou aumentada. Na ARF com síndrome do 5q-, a condição da medula óssea é caracterizada pela presença de micromegacariócitos mono e bilobulados. Uma deleção hemizigota de uma cópia do gene RPS14 foi implicada como causa deste distúrbio.44 Um total de 2/3 destes pacientes têm ARF ou anemia refratária com sideroblastos anelares (ARSA), enquanto o restante apresenta anemia refratária com excesso de blastos (AREB). Em pacientes cuja única anormalidade citogenética é a del(5q), a SMD tende a seguir um curso mais benigno, embora possa progredir para LMA.45 Apesar de a SMD não ser comumente encontrada em pacientes com menos de 50 anos de idade, existem 2 síndromes pediátricas distintas consideradas importantes: a leucemia mielomonocítica juvenil (LMMJ) e a síndrome da monossomia 7.46
O sistema de classificação do FAB descrevia 3 subtipos de LLA (L1, L2 e L3), definidos por características citológicas individuais, como tamanho, padrão de cromatina nuclear, nucléolos e intensidade da basofilia citoplasmática.47 O subtipo L1 representa mais de 80% dos casos de LLA em crianças e consiste predominantemente de células pequenas, com diâmetro máximo equivalente a 2 vezes o diâmetro de um linfócito pequeno. A maioria dos casos de pacientes adultos com LLA é classificada como sendo de subtipo L2. As células encontradas em L2 são maiores do aquelas encontradas em L1 e costumam ser heterogêneas em termos de tamanho. Entretanto, esta distinção frequentemente é arbitrária e fornece pouca orientação para o manejo de pacientes individuais.48 Em consequência, acabou caindo em desuso. A classificação da OMS identifica apenas os subgrupos de precursores de célula B e precursores de célula T.18
A forma menos comum de LLA, observada em cerca de 3 a 4% dos pacientes (crianças e adultos), foi denominada L3 e agora é chamada linfoma/leucemia de Burkitt. Neste subtipo, as células neoplásicas são morfologicamente idênticas, não importa se a manifestação primária da doença ocorre nos linfonodos (linfonodos) ou no sangue e medula (leucemia). As células são amplas e uniformes, com uma cromatina finamente pontilhada e núcleo com formato regular. Os nucléolos costumam ser proeminentes. O citoplasma é moderadamente abundante e apresenta uma profunda basofilia. É importante reconhecer esta variante, pois o tratamento inicial difere de forma marcante do tratamento destinado aos outros subtipos de LLA.
As avaliações citoquímicas dos blastos, na LLA, revelam padrões característicos. Por definição, as colorações para enzimas lisossomais (p. ex., mieloperoxidase ou sudão negro) devem resultar negativas para sustentar o diagnóstico de LLA. A reação com ácido periódico de Schiff revelará uma positividade aglomerada, produzida pelo glicogênio nos blastos da LLA (exceto no tipo Burkitt, com reação negativa) e que é considerada fraca como discriminador de linhagem celular, pois muitas células de LMA também apresentarão reação positiva. As colorações para cloroacetato esterase e lisozima resultam negativas na LLA, porém a coloração para alfanaftil acetato esterase pode resultar positiva nos linfoblastos T. Na LLA, os blastos contêm a enzima terminal desoxinucleotidil transferase (TdT), que, quando presente na maioria das células, é bastante confiável como marcador de LLA. As células da LLA de Burkitt frequentemente apresentam coloração positiva com oil red O, devido à presença do lipídio neutro central junto aos vacúolos citoplasmáticos.
Cerca de 80% dos casos de LLA têm origem na linhagem de células B. As células de LLA expressam antígenos de diferenciação da célula B (CD19, CD20 ou ambos) e apresentam rearranjo dos genes da cadeia pesada e da cadeia leve da imunoglobulina.49 Os linfoblastos de pacientes com LLA de progenitor de células B em estágio inicial não expressam o antígeno da leucemia linfoblástica aguda comum (CALLA), que é denominado CD10. As células de LLA CD10+ podem ser adicionalmente classificadas com base na presença da imunoglobulina de cadeia pesada mu citoplasmática (cmu). Na maioria dos casos de LLA, a cmu não é expressa nos blastos, e a condição é denominada LLA de progenitor B (CD10-) ou LLA comum (CD10+). Cerca de 20% dos casos CD10+ expressam cmu e são denominados LLA pré-B. Embora os genes de imunoglobulina estejam sempre clonalmente rearranjados na LLA da linhagem de células B, a expressão da imunoglobulina de superfície ocorre em apenas 2 a 5% de todos os casos de LLA, que em geral são casos de LLA do tipo Burkitt com morfologia característica.
Cerca de 15 a 20% de todos os casos de LLA têm origem na linhagem de células T.34,35,49 Estas células expressam antígenos de célula T, como CD2, CD5 e CD7. O CD10 também pode ser expresso, mas nunca há expressão de CD19 nem de CD20. Na maioria dos casos, um ou mais genes de receptor de célula T estão rearranjados. A subclassificação adicional da LLA de células T nos tipos inicial, intermediário e de timócitos maduros é baseada na expressão de vários antígenos de diferenciação da célula T.
Alguns antígenos normalmente expressos apenas nas células mieloides podem estar expressos de modo aberrante em linfoblastos malignos de origem B ou T. Este distúrbio é denominado LLA positiva para antígeno mieloide.50 Um pequeno percentual dos casos de LLA que não apresentam características de célula B nem de célula T recebe a denominação “leucemia indiferenciada aguda”. Os perfis de imunofenotipagem distintivos são clinicamente úteis para a detecção de doença residual mínima durante e após o tratamento.51
A aplicação da imunofenotipagem e das sondas moleculares tem revelado casos em que as células leucêmicas exibem características tanto de células mieloides como de células linfoides. Nestes casos, uma única célula neoplásica pode coexpressar características de linhagens distintas (bifenotípica), ou, ainda, 2 subpopulações de células leucêmicas podem expressar separadamente características mieloides ou linfoides (leucemia bilinhagem). Foi proposto o uso de um sistema de escores para definir a leucemia bifenotípica52 [Tabela 4]. Várias teorias foram propostas para explicar a ocorrência destas leucemias híbridas. Uma hipótese, denominada “infidelidade de linhagem”, estabelece que a célula leucêmica exibe expressão genética aberrante em decorrência de sua transformação neoplásica. Outra teoria, denominada “promiscuidade de linhagem”, propõe que as células em diferenciação normal podem expressar características de mais de uma linhagem distinta e a célula leucêmica é meramente um reflexo desta fase particular do desenvolvimento celular. Por fim, uma teoria denominada “troca de linhagem” propõe que, em certos casos, a leucemia é uma malignidade de célula-tronco pluripotente capaz de se diferenciar em células de linhagem mieloide ou linfoide. Desta forma, qualquer caso individual pode expressar um ou ambos os fenótipos. O significado prognóstico desta “infidelidade” de linhagem é incerto, contudo estes pacientes costumam apresentar resultados desfavoráveis independentemente da terapia (para LMA ou LLA) usada.
Tabela 4. Sistema de escores para leucemia bifenotípica52
|
Escore |
Linhagem | ||
|
Linfoide B |
Linfoide T |
Mieloide | |
|
2 |
cCD79A cIgM cCD22 |
cCD3 anti-TCR |
MPO |
|
1 |
CD19 CD20 CD10 |
CD2 CD5 CD8 CD10 |
CD117 CD13 CD33 CD65 |
|
0,5 |
TdT CD24 |
TdT CD7 CD1a |
CD14 CD15 CD64 |
É necessário marcar um total > 2 pontos para considerar o envolvimento de uma linhagem.
cIgM = imunoglobulina M citoplasmática; MPO = positividade para mieloperoxidase; TCR = receptor da célula T; TdT = desoxinucleotidil transferase terminal.
As anormalidades citogenéticas ocorrem comumente nas leucemias agudas e são usadas com frequência para identificar e definir o clone hematopoiético maligno.18,20,53 Desde a aplicação das técnicas de bandeamento cromossômico, no início da década de 1970, a análise citogenética passou a fornecer a abordagem de maior utilidade clínica para a subclassificação das leucemias agudas. As anormalidades cromossômicas não aleatórias particulares estão especificamente correlacionadas com subtipos morfológicos e perfis clínicos.18,20,53-57
Estas anormalidades citogenéticas são mutações somáticas (e não de linhagem germinativa) que frequentemente resultam de translocações do DNA cromossômico e levam à produção de proteínas novas (anormais) a partir dos genes de fusão. Admite-se que as proteínas codificadas por estes genes de fusão são responsáveis pela desregulação celular que acarreta a condição maligna. Estas anormalidades cromossômicas recorrentes são decisivas para determinar a estratégia terapêutica e têm fornecido informação independente importante sobre a resposta à terapia e o prognóstico geral [Tabelas 5 e 6].
Tabela 5. Subgrupos citogenéticos e moleculares genéticos de importância prognóstica na LMA32
|
Resultado clínico |
Subgrupos genéticos |
|
Favorável |
t(8;21)(q22;q22); RUNX1-RUNX1T1 inv(16)(p13.1q22) ou t(16;16)(p13.1;q22); CBFB-MYH11 NPM1 mutante sem FLT3-ITD (cariótipo normal) CEBPA mutante (cariótipo normal) |
|
Intermediário-I |
NPM1 mutante e FLT3-ITD (cariótipo normal) NPM1 selvagem e FLT3-ITD (cariótipo normal) NPM1 selvagem sem FLT3-ITD (cariótipo normal) |
|
Intermediário-II |
t(9;11)(p22;q23); MLLT3-MLL Anormalidades citogenéticas não classificadas como favoráveis nem adversas |
|
Adverso |
inv(3)(q21q26.2) ou t(3;3)(q21;q26.2); RPN1-EVI1 t(6;9)(p23;q34); DEK-NUP214 t(v;11)(v;q23); rearranjo de MLL -5 ou del(5q); -7; (17p) anormal; cariótipo complexo* |
*O cariótipo complexo é definido pela ocorrência de 3 ou mais anormalidades cromossômicas na ausência de uma das translocações ou inversões recorrentes designadas pela OMS, ou seja, t(15;17), t(8;21), inv(16) ou t(16;16), t(9;11), t(v;11)(v;q23), t(6;9), inv(3)/t(3;3).
LMA = leucemia mieloide aguda; OMS = Organização Mundial de Saúde.
Tabela 6. Subtipos citogenéticos e moleculares da LLA
|
Subtipo |
Cariótipo |
Frequência (%) |
Sobrevida livre de doença* | ||
|
Crianças |
Adultos |
Crianças |
Adultos | ||
|
Célula de Burkitt |
t(8;14); t(2;8); t(8;22) |
2 |
3 a 5 |
75 a 85 |
60 a 70 |
|
Hiperdiploide |
> 50 cromossomos |
25 |
2 a 5 |
80 a 90 |
40 a 50 |
|
TEL/AML1 |
t(12;21) |
20 a 25 |
1 a 3 |
85 a 90 |
? |
|
E2A/PBX1 |
t(1;19) |
5 a 6 |
1 a 3 |
70 a 80 |
10 a 50 |
|
MLL/AF4 |
t(4;11) |
2 a 3 |
3 a 6 |
10 a 35 |
10 a 40 |
|
BCR/ABL |
t(9;22) |
3 a 4 |
25 a 30 |
80 |
50 |
|
Hipodiploide |
< 45 cromossomos |
7 |
4 a 5 |
25 a 40 |
10 |
|
LLA de célula T e outras |
t(14q11) |
15 |
20 |
65 a 75 |
60 |
*Percentual em 3 a 5 anos.
LLA = leucemia linfoblástica aguda.
A análise citogenética tem aumentado nossa compreensão acerca da leucemogênese.18,53 Uma análise detalhada dos pontos de quebra cromossômica associados a anormalidades citogenéticas leucemia-específicas tem permitido identificar alguns genes que parecem exercer papel integral na leucemogênese.58-60 Foi identificada a localização cromossômica de amplo número destes oncogenes conhecidos. As funções destes oncogenes estão sendo investigadas, sendo que muitos deles estão envolvidos nas vias de sinalização intracelulares e no controle da proliferação e diferenciação. As alterações cromossômicas estruturais podem levar à ativação ou perturbação da expressão dos oncogenes, com consequente perturbação da regulação celular e, eventualmente, transformação maligna. As deleções ou perda de DNA podem eliminar genes com funções de supressão tumoral. As alterações epigenéticas podem resultar no silenciamento de certos genes necessários à diferenciação hematopoiética normal ou à apoptose. Os genes que comprovadamente afetam os resultados alcançados pelos pacientes com LMA são FLT3, NPM1, CEBPA, KIT, BAALC, ERG e MN1 [Tabela 5].61-63 Os arranjos de expressão genética identificam outras vias críticas nas células de leucemia aguda.64 O sequenciamento do DNA genômico integral está em andamento, com o objetivo de identificar todo o complemento de mutações responsáveis pela patogênese da leucemia aguda.65,66
A meta da quimioterapia de indução de remissão é promover a rápida restauração da função normal da medula óssea. O termo “remissão completa” (RC) é reservado para os pacientes que apresentam recuperação total das contagens sanguíneas periféricas normais e celularidade da medula óssea com menos de 5% de blastos residuais [Tabela 7]. A terapia de indução objetiva diminuir a população de células leucêmicas total do corpo, que deve cair de uma concentração aproximada de 1012 células para uma concentração inferior aos níveis citologicamente detectáveis (cerca de 109 células). As células leucêmicas de alguns pacientes apresentam altos níveis de resistência farmacológica primária. Nestes indivíduos, a leucemia é refratária aos cursos quimioterápicos de indução de remissão. Entretanto, até mesmo quando há RC, admite-se que uma carga substancial de células leucêmicas permanece não detectada e leva à recorrência em algumas semanas ou meses, caso nenhuma terapia adicional seja instituída. A terapia de consolidação pós-indução ou pós-remissão, que geralmente consiste em vários cursos extras de quimioterapia, destina-se a erradicar a leucemia residual e possibilitar a cura. A administração de múltiplos quimioterápicos em doses altas é tipicamente realizada para prevenir a emergência de subclones resistentes e limitar toxicidades cumulativas e sobrepostas. A terapia de manutenção da remissão prolongada, com duração de 1 a 3 anos e empregando doses menores de fármacos, tem sido usada com algum sucesso na LLA. Entretanto, o valor desta terapia auxiliar na LMA é incerto.
Tabela 7. Terminologia usada no tratamento da leucemia
|
Termo |
Definição |
|
Terapia de indução de remissão |
Tratamento quimioterápico inicial, voltado para a promoção da RC |
|
Terapia de consolidação (intensificação) da remissão |
Terapia pós-remissão, voltada para a eliminação da doença clinicamente oculta |
|
Terapia de manutenção (continuação) |
Doses menores de quimioterapia, administradas com o objetivo de prevenir a reemergência da leucemia |
|
RC |
Desaparecimento da leucemia após o tratamento, com regeneração total da hematopoiese normal |
|
RC citogenética (ou molecular) |
Incapacidade de detectar a leucemia residual utilizando métodos genéticos |
|
Doença residual mínima |
Leucemia persistente, indetectável por microscopia óptica |
|
Doença refratária |
Leucemia que não entra em RC |
|
Doença recidivante |
Recorrência clinicamente evidente de leucemia após a RC |
|
Imunofenótipo |
Padrão de expressão de marcadores de superfície celular ou citoplasmáticos |
|
Citogenética |
Análise de cromossomos em células em metáfase, para detecção de adições, perdas ou rearranjos |
|
Resistência multifarmacológica |
Característica biológica que protege as células contra certas classes de fármacos quimioterápicos, por meio de diferentes mecanismos de membrana |
|
Transplante de célula-tronco |
Transfusão de CTH do paciente (autóloga) ou de um doador normal (alogênica), após a quimioterapia com doses altas |
CTH = células-tronco hematopoiéticas; RC = remissão completa.
As complicações da terapia citotóxica incluem a síndrome da lise tumoral com nefropatia por urato e desequilíbrio eletrolítico (hipercalemia, hipocalcemia e hiperfosfatemia), lesão gastrintestinal (mucosite e diarreia), sangramento trombocitopênico, e infecções neutropênicas.67 A adoção de medidas profiláticas e tratamento de suporte é essencialmente importante durante o período de tratamento. Os pacientes precisam receber hidratação endovenosa adequada. A hiperuricemia responde rápido à rasburicase. A administração de alopurinol, a hidratação parenteral e a alcalinização da urina para pH 7 são medidas que diminuem a probabilidade de precipitação de ácido úrico nos túbulos renais. Os produtos do sangue são transfundidos para manter uma contagem de plaquetas superior a 10.000/mcL em pacientes não hemorrágicos com hematócrito acima de 25%. O curso de antibióticos de amplo espectro é iniciado de modo empírico, sempre que a temperatura corporal exceder 38,5°C em pacientes neutropênicos (< 500 neutrófilos/mcL). A terapia antifúngica é acrescentada em casos de pacientes com febre persistente.
A terapia mieloablativa seguida de transplante de CTH oriundas da medula óssea ou do sangue de um doador de HLA idêntico constitui uma modalidade terapêutica já estabelecida para pacientes com leucemia aguda. Esta terapia é indicada para pacientes de alto risco adequados na 1ª remissão e para pacientes jovens ou de meia-idade na 1ª ou 2ª remissão.68 O transplante de células-tronco hematopoiéticas alogênicas (alo-CTH) possui 2 componentes terapêuticos. Primeiro, a terapia mieloablativa intensiva é usada para erradicar todas as células tumorais, quando possível. Em 2º lugar, as células T presentes entre as células-tronco do doador produzem uma resposta imune de enxerto vs. leucemia que destrói as células leucêmicas remanescentes. Este efeito foi correlacionado à melhora da sobrevida livre de doença. Contudo, esta resposta imune benéfica possui uma relação estreita com o desenvolvimento de doença do enxerto vs. hospedeiro (DEVH) aguda e crônica, uma das principais causas de morbidade e mortalidade após o transplante de alo-CTH. A DEVH pode ser minimizada por meio da depleção das células T presentes entre as células-tronco do doador, todavia somente à custa de taxas maiores de falha de transplante e recidiva da leucemia. Como a mortalidade associada ao tratamento aumenta com o avanço da idade, muitos centros restringem o transplante de CTH a pacientes com menos de 60 anos de idade. Para pacientes com idade superior a 60 anos, são usados regimes preparatórios não mieloablativos.69 Esta abordagem é mais bem tolerada e busca maximizar o efeito imunológico de enxerto vs. leucemia. O uso do transplante de alo-CTH também é limitado, em parte, pela disponibilidade de doadores. A probabilidade de um dado paciente e cada um de seus irmãos herdarem alelos HLA idênticos é de apenas 25 a 30%. Voluntários sem grau de parentesco podem fazer doações bem-sucedidas de células-tronco HLA-compatíveis para transplantes de CTH. As células-tronco periféricas restauram as contagens sanguíneas mais rapidamente do que as células de medula, sem aumentar o risco de DEVH.70
O transplante autólogo de CTH permite instituir a terapia mieloablativa na ausência de um doador alogênico, bem como para pacientes de idade mais avançada.71 O papel apropriado para esta modalidade de tratamento é incerto. A morbidade e mortalidade associadas ao tratamento são relativamente baixas (< 5%), viabilizando assim o uso para pacientes de idade mais avançada. Entretanto, os índices de recaída são altos, e os resultados gerais não são nitidamente melhores do que os resultados alcançados por pacientes submetidos à terapia não ablativa intensiva.
Tipicamente, os sintomas antecedentes (p. ex., fadiga) são breves nos pacientes com LMA. Cerca de 1/3 dos pacientes apresentam contusões ou hemorragia. O equivalente a 1/4 dos pacientes apresentam infecções sérias envolvendo os pulmões, tecidos moles ou a pele. A esplenomegalia e a hepatomegalia não são comuns, ocorrendo em menos de 25% dos pacientes. A linfadenopatia é ainda menos comum. A hipertrofia gengival ou infiltração da pele pela leucemia (denominada cútis leucêmica) são observadas em 50% dos pacientes com leucemia monocítica. Os pacientes com LPMA frequentemente apresentam sangramento mais severo decorrente de CID.
A contagem de leucócitos está aumentada acima da faixa normal em 50% dos pacientes com LMA. O sangue periférico contém alguns blastos leucêmicos em 85 a 90% dos casos. Contagens acima de 100.000/mcL são relatadas em menos de 10% dos pacientes que, todavia, podem apresentar efeitos severos envolvendo o SNC ou sofrimento pulmonar por leucostase. A leucaférese emergencial e a quimioterapia podem salvar vidas. Na LMA, a contagem de neutrófilos absoluta quase sempre está deprimida e, no momento do diagnóstico, está abaixo de 1.500/mcL em 50% dos pacientes. É comum haver um grau moderado de anemia. A contagem de plaquetas é tipicamente inferior a 100.000/mcL e muitas vezes pode chegar a menos de 20.000/mcL.
Elevações discretas a moderadas dos níveis séricos de ácido úrico são comuns e tipicamente refletem a intensificação da renovação celular. Os níveis séricos de lactato desidrogenase (DHL) podem estar aumentados, mas não tão comumente quanto na LLA. A concentração de muramidase (lisozima) está elevada no soro ou urina de pacientes com leucemia mielomonocítica aguda ou leucemia monoblástica, podendo contribuir para o desenvolvimento de disfunção renal.
A LMA não se limita à medula óssea e ao sangue periférico. Anormalidades envolvendo um ou mais sistemas orgânicos podem resultar da infiltração por células leucêmicas ou de complicações metabólicas relacionadas à leucemia. Em raros casos, um paciente com LMA desenvolve uma massa sólida de células leucêmicas denominada sarcoma granulocítico ou mieloide (cloroma). A pele e os ossos (em particular o esterno, as costelas e a órbita) são mais comumente envolvidos, porém os sarcomas mieloides podem ocorrer em qualquer órgão. O sofrimento respiratório em pacientes com LMA é mais frequentemente causado por infecção. No entanto, pacientes com números muito altos (> 100.000/mcL) de blastos circulantes podem apresentar dispneia severa e hipoxemia, em decorrência da leucostase junto à vasculatura pulmonar. A disfunção cardíaca, incluindo murmúrios, insuficiência cardíaca e disritmias, é mais frequentemente secundária à anemia.
As hemorragias de retina comumente são causadas pela trombocitopenia. Contudo, pacientes com hiperleucocitose extrema podem desenvolver as conhecidas manchas algodonosas, como resultado de isquemia retinal. A ocorrência de franco envolvimento do SNC e de neuropatias cranianas é incomum no momento em que o diagnóstico inicial de LMA é estabelecido.
As anormalidades cromossômicas clonais podem ser detectadas na maioria dos casos de LMA.1,18,53,57 Estas anormalidades incluem ganhos ou perdas de cromossomos inteiros ou perdas de braços longos (q) ou curtos (p) de cromossomos (deleções), bem como uma variedade de rearranjos estruturais (isto é, translocações, inversões ou inserções).
É fortemente recomendado que a análise citogenética seja realizada antes do início da terapia, para cada paciente recém-diagnosticado, porque os exames de importância prognóstica para as anormalidades citogenéticas recorrentes na LMA têm fornecido resultados consistentemente similares.1,54 A caracterização citogenética tornou-se o fator preditor mais forte da resposta à terapia e da duração da remissão. Desta forma, em muitos centros, o planejamento da terapia pós-remissão baseia-se fortemente na análise citogenética realizada no momento do diagnóstico [Tabela 5]. Com mais frequência, o cariótipo volta ao normal à medida que os pacientes entram em remissão morfológica. A persistência do clone citogeneticamente anormal é preditiva de uma remissão de menor duração.72
Os dados de citogenética são empregados no mapeamento dos pontos de quebra cromossômica ao nível molecular, viabilizando o uso de sondas para hibridização in situ fluorescente (FISH), bem como de primers para reação em cadeia da polimerase com transcriptase reversa (RT-PCR) como métodos de detecção de células tumorais. A FISH e a RT-PCR podem detectar rearranjos genéticos moleculares que são invisíveis ao exame das bandas cromossômicas pelos métodos convencionais. Entretanto, ambos os métodos avaliam apenas a presença de mutações genéticas definidas específicas e não podem ser usados inicialmente para fins de avaliação geral ou abrangente. A análise de FISH é mais sensível do que a análise cariotípica convencional e pode ser realizada em células em metáfase ou intérfase. A morfologia das células positivas pode ser determinada simultaneamente, sendo possível avaliar o envolvimento leucêmico proporcional de todas as células hematopoiéticas.
A RT-PCR é o método mais sensível para detecção de células leucêmicas ocultas (cerca de 1 em cada 105 células). Os métodos mais modernos são quantitativos, e o significado clínico de um resultado positivo subsequente ao tratamento está sendo investigado. Um ensaio que resulte positivo confirma a presença de células portadoras da anormalidade genética específica, mas não necessariamente aponta o potencial de desenvolvimento neoplásico destas células. Exemplificando, um ensaio de RT-PCR que resulte positivo após o tratamento parece predizer com segurança a recorrência da leucemia em pacientes com LPMA e t(15;17), mas não em pacientes com LMA-M2 e t(8;21).73,74
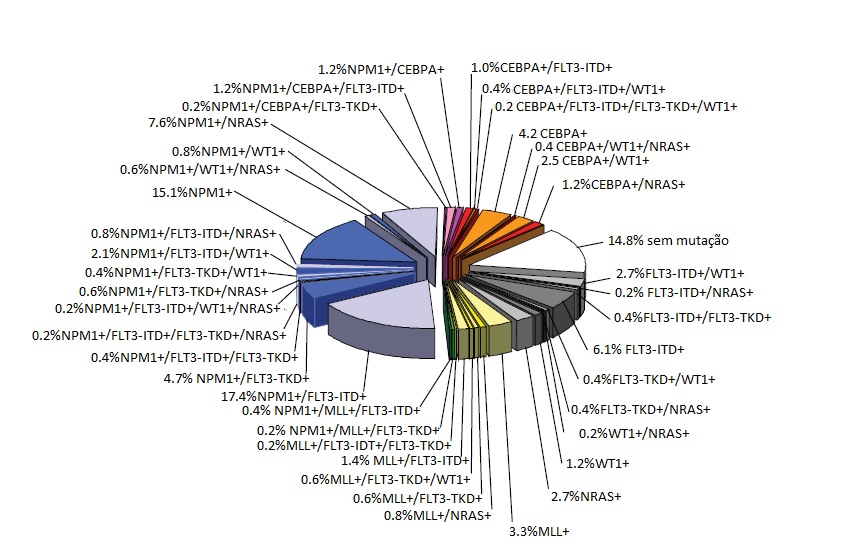
A leucemia mieloide aguda citogeneticamente normal (LMA-CN) constitui o maior grupo, dentre os pacientes com LMA mais jovens e de idade mais avançada, e o mais bem caracterizado ao nível molecular [Figura 1]. Ao longo dos últimos 15 anos, mutações recorrentes de importância prognóstica nos genes FLT3, NPM1, CEBPA, WT1 e MLL foram identificadas na LMA-CN de novo [Tabela 5].75-78 Estes marcadores não são mutuamente exclusivos e por vezes ocorrem em combinações que podem refinar ainda mais o prognóstico. Exemplificando, os pacientes cujos blastos de LMA contêm mutações em NPM1 e não possuem duplicação em tandem interna (ITD) de FLT3 pertencem à categoria de baixo risco molecular e alcançam um resultado mais satisfatório do que aquele obtido pelos pacientes classificados no grupo de alto risco molecular, que possuem alelos de NPM1 do tipo selvagem e/ou FLT3-ITD. Entretanto, se este último grupo também abrigar uma mutação CEBPA, os pacientes apresentarão prognóstico similar àquele dos pacientes com alelos NPM1 do tipo selvagem e ausência de FLT3-ITD. Embora a maioria dos pacientes com LMA-CN abrigue pelo menos 1 destas mutações, nenhuma mutação é detectada em cerca de 15% dos casos.

Figura 1. Gráfico de torta ilustrando a heterogeneidade molecular da leucemia mieloide aguda citogeneticamente normal (LMA-CN), com base em mutações nos genes NPM1, CEBPA, MLL, FLT3 (mutações de duplicação em tandem interna [ITD] e no domínio da tirosina quinase [TKD] nos códons D835 e I836), NRAS e WT1. As cores azuladas denotam os subgrupos de NPM1 mutante; as cores laranja/vermelha indicam os subgrupos de CEBPA mutante; e as cores amarela/verde denotam os subgrupos de MLL mutante. Em cinza estão representados os subgrupos sem mutação hipotética de classe II. Os setores em branco mostram o subgrupo isento de quaisquer uma das mutações genéticas mencionadas. Os dados derivam de uma análise mutacional de 485 pacientes jovens com LMA-CN realizada pelo AMLSG.
Adaptado de Döhner H et al.32
O regime de indução de remissão mais comumente usado para pacientes com LMA consiste na administração de citarabina como infusão endovenosa contínua diária, por um período de 7 dias, aliada à administração diária daunorubicina por 3 dias (regime 7 + 3). Dependendo da idade e seleção do paciente, 50 a 80% dos pacientes alcançam a RC.1,79 Estudos recentes sugerem que doses maiores de daunorubicina (90 mg/m2/dia) produzem resultados melhores do que os resultados obtidos com doses menores.80,81 De modo geral, o resultado não melhora com a substituição de outras antraciclinas (p. ex., idarubicina ou mitoxantrona) pela daunorubicina, nem com o aumento da dose de citarabina ou com a adição de um 3º ou 4º fármaco citotóxico.82 Os estudos atualmente em curso buscam explorar novas terapias alvo-dirigidas, por exemplo, contra a FLT3 tirosina quinase.83
A instituição de quimioterapia adicional após uma indução de remissão bem-sucedida é obrigatória para a cura da LMA. Em média, a sobrevida livre de doença dos pacientes que não recebem terapia adicional é de apenas cerca de 4 meses. Quando vários cursos de quimioterapia de consolidação são fornecidos, a sobrevida de 4 anos é de 40 a 50% para pacientes adultos jovens e de meia-idade. Para os pacientes com menos de 60 anos, a terapia de consolidação resulta em sobrevida significativamente maior do aquela alcançada com o uso apenas da terapia de manutenção.84-88 Para fins de consolidação, a mesma quimioterapia de indução pode ser repetida por 1 ou mais ciclos, com ou sem intensificação da dose, ou, como alternativa, podem ser usados fármacos sem reatividade cruzada. Há evidências de que os cursos de doses altas de citarabina (HiDAC) promovem a melhor sobrevida para pacientes com prognóstico favorável e intermediário.86,88
A terapia de manutenção com doses relativamente não mielossupressoras de fármacos citotóxicos promove benefícios incertos no tratamento da LMA. Atualmente, estudos clínicos estão testando os benefícios das terapias imunomoduladoras, como a interleucina-2 (IL-2) ou os inibidores de DNA metiltransferase (p. ex., azacitidina ou decitabina), para pacientes com LMA em remissão.
Os estudos que descrevem o uso do transplante alogênico ou autólogo de CTH em pacientes com LMA na 1ª RC são na maioria randomizados, e muitos são retrospectivos.68,69 Uma considerável tendenciosidade da seleção resulta da demora entre a indução da remissão e o transplante, bem como dos requisitos de entrada para desempenho satisfatório da maioria dos estudos. Os estudos prospectivos randomizados que compararam a quimioterapia de consolidação intensiva ao transplante de CTH falharam em demonstrar a existência de uma vantagem nítida de sobrevida para os pacientes com LMA de risco favorável. Entretanto, está claro que a sobrevida melhora com o uso do transplante de alo-CTH no tratamento de pacientes que apresentam fatores de risco citogenéticos adversos.90-92
Existe um número limitado de agentes efetivos no tratamento da LMA, e o tratamento da doença resistente ou recidivante é difícil. Recomenda-se que estes pacientes sejam encaminhados para centros especializados para realização de exames clínicos. Pacientes com remissão inicial duradoura (> 1 ano) apresentam um índice de reindução de 50 a 60% com o uso de daunorubicina e citarabina ou HiDAC, porém a 2ª remissão geralmente dura menos que a 1ª. O gemtuzumabe ozogamicina, um imunoconjugado anti-CD33, é ativo como agente único em casos de recidiva de LMA.38 Muitos fármacos novos atualmente estão em fase de estudo clínico, porém a LMA recidivante apresenta múltiplos mecanismos de resistência. O transplante de CTH deve ser considerado para todos os pacientes que apresentam recaída após terem passado por um programa de tratamento intensivo inicial.
A LPMA é uma doença biologicamente distinta, que possui características clínicas, morfológicas e citogenéticas próprias.39,73,93 Esta condição representa aproximadamente 8 a 15% dos casos de LMA. As células malignas apresentam uma t(15;17) que justapõe o gene RARa no cromossomo 17 ao gene PML no cromossomo 15. A CID que ocorre no momento da manifestação da doença ou logo após o início da quimioterapia citotóxica pode causar hemorragia pulmonar ou cerebrovascular em até 40% dos pacientes, e está associada a uma alta taxa de mortalidade. Os grânulos citoplasmáticos encontrados nos blastos leucêmicos contêm fatores que possuem atividade fibrinolítica e pró-coagulante.94
O ácido all-trans-retinoico (ATRA ou tretinoína) foi empregado pela 1ª vez no tratamento da LPMA na China, em 1986, e mostrou-se altamente efetivo como agente indutor de remissão.39,93,95 O ATRA acelera a diferenciação terminal dos promielócitos malignos em neutrófilos maduros, causando apoptose e RC sem hipoplasia da medula óssea. Este efeito é uma consequência exclusiva do rearranjo dos genes PML/RARa resultante da t(15;17), que define a LPMA.39 O uso combinado do ATRA com daunorubicina e citarabina ou idarubicina na terapia de indução promove taxas de RC de 80 a 95% tanto em pacientes sem tratamento prévio como em pacientes com recaída.95 O trióxido de arsênico é um tratamento efetivo para pacientes com recaída de LPMA e é comprovadamente benéfico quando usado na consolidação da 1ª RC.96,97 Para os pacientes com contagens de leucócitos inferiores a 10.000/mcL, a sobrevida livre de eventos é superior a 80%. Os pacientes com contagens de leucócitos maiores no momento do diagnóstico apresentam taxas de indução de morte mais altas. Entretanto, depois que alcançam a RC, estes pacientes apresentam excelente sobrevida ao receberem trióxido de arsênico durante a terapia de consolidação. O trióxido de arsênico também é usado combinado ao ATRA, no tratamento inicial.98
O ATRA não apresenta as toxicidades geralmente associadas à quimioterapia citotóxica. A toxicidade farmacológica intrínseca geralmente é mínima. O ATRA não é imunossupressor nem mielossupressor. Contudo, o tratamento da LPMA com ATRA está associado à possível ocorrência de 2 complicações sérias.93 Em 25 a 40% dos pacientes, a conhecida “síndrome do ácido retinoico” (também denominada síndrome da diferenciação) desenvolve-se dentro de um período de 2 a 21 dias após o início do tratamento. Esta síndrome é caracterizada por febre, edema periférico, infiltração pulmonar e sofrimento respiratório, hipotensão, disfunção renal e hepática, e serosite, com consequente produção de efusões pleurais e pericárdicas. A síndrome possivelmente resulta da infiltração dos tecidos por promielócitos malignos em maturação e dos efeitos sistêmicos da liberação de citocinas. O reconhecimento antecipado e tratamento agressivo com dexametasona são efetivos. A hiperleucocitose ocorre em até 50% dos pacientes tratados com ATRA e é provavelmente secundária à indução da maturação celular.
O tratamento da coagulopatia associada à LPMA pode ser difícil e deve ser conduzido de maneira esperançosa.93,95 Os parâmetros de coagulação, incluindo os níveis de fibrinogênio, D-dímero e plaquetas, devem ser monitorados intensivamente. As transfusões de plaquetas e crioprecipitados ou de plasma fresco congelado são usadas para manter os níveis de fibrinogênio acima de 100 mg/dL e a contagem de plaquetas acima de 20.000/mL. Os inibidores de fibrinólise devem ser considerados apenas em casos de hemorragia prejudicial à vida. A coagulopatia da LPMA tipicamente melhora rápido após o início do tratamento com ATRA.
Não há nenhuma terapia específica e uniformemente efetiva para a SMD.99 Os fatores prognósticos clínicos, incluindo a citogenética e a morfologia da medula óssea, devem ser avaliados no momento do diagnóstico.45 Para os pacientes assintomáticos, recomenda-se um período de observação com seguimento intensivo das contagens sanguíneas seriadas, como forma de manejo inicial, sendo que um 2º exame de medula óssea após vários meses é útil para avaliar a velocidade da progressão da doença.
A ARF e a ARSA costumam seguir um curso clínico indolente. Os pacientes afetados podem dispensar tratamento por períodos de tempo variáveis, mas aqueles com anemia severa (hemoglobina < 7 a 8 g/dL) devem receber transfusão de hemácias. O fornecimento de unidades de hemácias lavadas raramente é necessário. Em geral, a recorrência de fadiga ou angina é mais útil como indicador da necessidade da próxima transfusão de hemácias, se comparada a qualquer medida dos níveis de hemoglobina. Os requerimentos para transfusão aumentam com o avanço da SMD, podendo haver sobrecarga de ferro e hemocromatose. O benefício proporcionado pela terapia à base de ferro quelante oral está sendo estudado.
As transfusões de plaquetas profiláticas não são administradas de forma rotineira aos pacientes trombocitopênicos com SMD que não apresentam hemorragia, devido ao custo, à inconveniência e ao risco de aloimunização. Este risco aumenta com a frequência das transfusões de plaquetas e pode limitar as opções terapêuticas futuras. O suporte de plaquetas deve ser reservado para tratamento da hemorragia aguda ou profilaxia antes da cirurgia. Os pacientes com SMD trombocitopênicos devem ser cuidadosamente orientados a evitar o uso de produtos contendo aspirina e outros fármacos anti-inflamatórios não hormonais, que interferem na função plaquetária.
As infecções são um problema comum em pacientes com SMD e podem ser prejudiciais à vida. A utilidade das vacinas (p. ex., contra penumococos, gripe e hepatite B) não devem ser negligenciadas. Febre, infecções localizadas ou até mesmo um mal-estar geral devem ser detalhadamente avaliados em pacientes com neutropenia quantitativa e funcional. Enquanto se tenta localizar a fonte de infecção, é essencial que uma terapia antibiótica de amplo espectro seja prontamente instituída. Esta terapia deve ser mantida por 7 a 10 dias, mesmo na ausência de uma fonte evidente de infecção ou de hemoculturas que resultem positivas. Se um paciente neutropênico falhar em responder aos agentes antibacterianos ou apresentar episódios repetidos de febre na ausência de uma fonte de infecção evidente, então deve ser levantada a suspeita de infecção fúngica e o tratamento apropriado deve ser iniciado.
O papel dos fármacos citotóxicos no tratamento da SMD é incerto para a maioria dos pacientes, mas deve ser considerado em casos de pacientes mais jovens e saudáveis com formas mais agressivas da doença, como aqueles com CRDM, AREB e SMD associada à terapia (t-SMD). Em geral, a terapia citotóxica com regimes-padrão empregada no tratamento da LMA (p. ex., regimes contendo daunorubicina e doses padrão ou altas de citarabina) apresentam sucesso limitado em termos de extensão da sobrevida para maioria dos pacientes com SMD.100,101 Este resultado precário não é surpreendente, por diversos motivos: (1) a maioria dos pacientes com SMD tem idade avançada e é pouco tolerante ao tratamento agressivo; (2) o número de CTH normais disponíveis para regenerar a medula óssea após a hipoplasia terapia-induzida provavelmente será reduzido; e (3) a maioria dos fármacos antileucemia pode ser relativamente inefetiva diante das condições de baixa proliferação observadas na SMD, pois são fármacos cuja atuação primária ocorre durante a divisão celular. Por estes motivos, a quimioterapia intensiva foi associada a taxas de RC inferiores a 40%, dependendo da idade e das comorbidades, com remissões que em geral duram menos de 1 ano. A toxicidade da terapia e a consequente hipoplasia e citopenia produzidas na medula óssea na verdade podem encurtar as vidas dos pacientes de idade mais avançada. Por isso, é importante direcionar esta opção terapêutica essencialmente aos pacientes de SMD mais jovens, que conseguem tolerá-la melhor.
O transplante de alo-CTH foi testado como terapia curativa em pacientes com SMD mais jovens que dispunham de um doador histocompatível.102 Cerca de 45% dos pacientes submetidos ao transplante alcançaram sobrevida livre de doença. Os pacientes com fatores citogenéticos favoráveis ou intermediários alcançam resultados melhores do que aqueles obtidos por pacientes com cariótipos desfavoráveis ou doença em estágio mais avançado. Estes dados sugerem que os pacientes corretamente selecionados podem ser curados com o transplante de CTH.
Como na maioria dos casos os pacientes com SMD não são candidatos ao transplante de CTH e o resultado da terapia citotóxica convencional geralmente é precário, passou-se a dar maior atenção a uma variedade de agentes capazes de induzir diferenciação de linhagens celulares de leucemia aguda in vitro, retardar a proliferação e restaurar a aparente normalidade da maturação e função. A azacitidina e a decitabina, dois inibidores de DNA metiltransferase, podem ativar genes silenciados e promovem benefícios clínicos comprovados na SMD.103 A lenalidomida, um agente imunomodulador oral, pode produzir um aumento marcante dos níveis de hemoglobina em pacientes com ARF que apresentam del(5q), além de promover remissões citogenéticas completas.104 Vários agentes (p. ex., baixas doses de citarabina, talidomida, amifostina, inibidores de histona desacetilase e fatores de crescimento hematopoiéticos) foram testados em estudos clínicos da SMD. Os estudos clínicos não demonstraram nenhum benefício decorrente do uso prolongado de fatores estimuladores de colônias mieloides, como G-CSF ou fator estimulador de colônias de granulócitos-macrófagos (GM-CSF), embora estes agentes possam ter papel auxiliar a curto prazo com antibióticos no tratamento de infecções. A terapia com eritropoetina é benéfica para pacientes anêmicos com níveis de eritropoetina endógena inadequadamente baixos (< 500 U/L) no sangue. Os agentes mimetizadores de trombopoetina, como o romiplostim, são comprovadamente capazes de elevar as contagens plaquetárias de pacientes com SMD em estágio inicial.
Recentemente, o tratamento de adultos de idade avançada com LMA tem recebido atenção considerável.105 A combinação de 2 fatores explica em grande parte o resultado precário alcançado pelos pacientes de idade avançada leucêmicos.106 O aspecto mais evidente é a incapacidade de muitos destes pacientes de resistir aos rigores da quimioterapia intensiva e das complicações esperadas. Os pacientes com distúrbios renais, pulmonares ou cardíacos crônicos relacionados à idade sofrem uma toxicidade aguda mais intensa decorrente da quimioterapia. Em pacientes idosos, a capacidade regenerativa da medula óssea pode ser menor, mesmo após uma citorredução de leucemia bem-sucedida. A incapacidade de tolerar longos períodos de pancitopenia e desnutrição continuam sendo as principais barreiras ao sucesso do tratamento.
Um fato menos evidente é o de que as mutações genéticas mais frequentemente associadas à falha terapêutica em pacientes jovens (p. ex., anormalidades nos cromossomos 5 ou 7 na LMA ou [t99;22] na LLA) são mais comuns em pacientes de idade avançada. Ao contrário, todas as conhecidas anormalidades citogenéticas favoráveis, como t(8;21), t(15;17) e inv(16) na LMA, são mais comuns em adultos jovens e respondem em parte pela melhor sobrevida livre de doença alcançada pelos pacientes jovens e de meia-idade. O fenótipo de resistência multifarmacológica, que resulta em parte da superexpressão da glicoproteína P responsável pelo efluxo dos quimioterápicos das células de LMA, também é mais comum em pacientes idosos.107
A SMD é mais resistente à terapia e também é mais frequente entre os pacientes idosos. Em muitos casos envolvendo pacientes de idade avançada com LMA associada à displasia, é provável que a LMA tenha progredido mediante uma fase mielodisplásica. A SMD é caracterizada pelo acúmulo gradual de anormalidades genéticas, de modo análogo à evolução de novas anormalidades cromossômicas que ocorre com o avanço da LMC para a fase blástica terminal. O fenótipo de resistência multifarmacológica também pode emergir durante este processo evolucionário. Ao mesmo tempo, a hematopoiese normal vai sendo cada vez mais inibida, e o compartimento de células-tronco normal pode ser perdido. O resultado líquido é uma hematopoiese inefetiva e células sanguíneas disfuncionais. No momento da manifestação da LMA, estes pacientes muitas vezes apresentam infecções produzidas pela flora patogênica. Além disso, estes pacientes frequentemente apresentam episódios recorrentes de sangramento e muitas vezes dependem de transfusões. Após a quimioterapia, estes pacientes alcançam resultados bastante precários. Todos estes fatores se combinam para resultar em uma taxa de indução de morte mais alta após a quimioterapia convencional, bem como em uma menor sobrevida livre de doença.108 Uma variedade de agentes novos e terapias dirigidas atualmente estão sendo avaliadas quanto aos possíveis benefícios clínicos junto a esta população de idade avançada.109
Nem todos os pacientes são beneficiados pela quimioterapia intensiva, e isto é particularmente válido para os pacientes idosos. Tentativas bem intencionadas de induzir remissão podem, na verdade, diminuir a sobrevida. Os pacientes que provavelmente não sobreviverão ao tratamento podem ser identificados pela condição funcional precária ou pelas comorbidades que apresentam. Séries de casos relatados por grandes instituições de referência indicam que a quimioterapia de indução de remissão não é oferecida a 25 a 50% dos pacientes com LMA na faixa etária acima dos 60 anos.
Em alguns poucos pacientes idosos com leucemia aguda (determinada pelos critérios quantitativos usuais de medula óssea contendo > 20% de blastos), a doença segue um curso significativamente mais latente, sobretudo quando estes pacientes já tiveram SMD. Estes indivíduos sofrem mais frequentemente de falha de medula óssea e pancitopenia, do que de hiperleucocitose. Eles podem alcançar sobrevida igualmente longa e melhor qualidade vida, se forem tratados com suporte transfusional e antibióticos, em vez de quimioterapia intensiva. Este achado pode ser particularmente válido para pacientes com LMA hipoplásica. A azacitidina e a decitabina, que são agentes hipometilantes aprovados para uso no tratamento da SMD, produzem benefícios clínicos comprovados para alguns pacientes adultos de idade avançada com LMA latente.
A quimioterapia curativa deve ser oferecida aos pacientes idosos que possuem apenas leucemia aguda, especialmente aqueles com características citogenéticas favoráveis. Com um regime padrão 7 + 3 de citarabina e a administração de antraciclina, cerca de 50% dos pacientes com mais de 60 anos de idade alcançam a RC. Contudo, até mesmo com o uso da quimioterapia de consolidação pós-remissão, a sobrevida geral dos pacientes deste grupo é inferior a 10% após 4 anos.98,108-111 Os resultados alcançados a longo prazo parecem ser melhores quando estes pacientes são submetidos ao transplante de alo-CTH não mieloablativo logo após alcançarem a RC.
Vários estudos controlados amplos sobre o uso de GM-CSF ou G-CSF principalmente em pacientes idosos com LMA demonstraram que a duração da neutropenia foi apenas minimamente diminuída com a administração destes fatores de crescimento após a quimioterapia de indução de remissão.112,113 Ainda que uma recuperação mais rápida dos neutrófilos tenha sido observada em alguns estudos, o nadir não foi afetado. Sendo assim, a incidência de infecção severa continua alta. Ao mesmo tempo, a estimulação da repetição do desenvolvimento da leucemia por ação dos fatores de crescimento mieloides parece ser incomum in vivo. É possível que benefícios maiores sejam alcançados com o uso dos fatores de crescimento após a quimioterapia de consolidação, quando os pacientes já entraram em remissão, do que se estes fatores forem usados antecipadamente durante a indução da remissão.114 Até agora, os fatores de crescimento não produziram impacto significativo sobre a sobrevida ou duração da remissão em pacientes com LMA.
A LLA representa cerca de 20% das leucemias agudas do adulto. Em contraste, a LLA é de longe a malignidade mais comum na infância.
A apresentação clínica da LLA em adultos é mais frequentemente aguda. Os sintomas em geral se manifestam ao longo de poucas semanas, antes do diagnóstico. Mal-estar, letargia, perda de peso, febres e sudorese noturna podem estar presentes, mas tipicamente não são severos. A dor óssea e as artralgias ocorrem ocasionalmente, porém são bem menos frequentes em adultos do que nas crianças. Infecção e hemorragia ocorrem em 1/3 dos pacientes no momento do diagnóstico, mas não costumam ser mais severas do que aquelas observadas na LMA. Linfadenopatia, esplenomegalia e hepatomegalia são mais comuns do que na LMA, afetando metade do universo de pacientes adultos com LLA. As radiografias de tórax podem revelar a presença de uma massa tímica em 10 a 15% dos adultos. A maioria destes pacientes tem LLA de células B.
O envolvimento leucêmico do SNC ocorre em 5 a 10% dos pacientes adultos, contudo é pouco comum no momento do diagnóstico. As paralisias de nervo craniano mais frequentemente envolvem o VI e VII nervos cranianos. Cefaleia e papiledema podem resultar da pressão intracraniana aumentada produzida pela infiltração meníngea e obstrução do orifício de saída de líquido cerebrospinal (LCS). As hemorragias retinais podem ser resultantes da trombocitopenia.
Graus variáveis de neutropenia, anemia e trombocitopenia são detectados ao exame de sangue periférico. Em uma série de mais de 1.200 casos de pacientes adultos, a contagem de granulócitos era inferior a 1.500/mcL em apenas 1/5 dos pacientes. Reduções leves a moderadas dos níveis de hemoglobina foram achados típicos, porém quase 1/3 dos pacientes apresentavam níveis de hemoglobina inferiores a 8 g/dL. A trombocitopenia era frequente, sendo que a contagem de plaquetas estava abaixo de 50.000/mcL em mais de 50% dos pacientes. A contagem de leucócitos total estava diminuída em cerca de 1/3 dos pacientes, mas estava normal ou moderadamente aumentada em quase metade dos pacientes. Os linfoblastos característicos podem ser identificados no sangue periférico em mais de 90% dos casos. Uma leucocitose marcante (> 100.000/mcL) estava presente no momento do diagnóstico em 16% dos pacientes, todavia a leucostase sintomática é incomum na LLA, mesmo nestes níveis.
Atualmente, é possível caracterizar os subgrupos de LLA com base em características genéticas distintivas.48,115,116 Estes subgrupos mais homogêneos de LLA apresentam diferenças clínicas importantes em termos de resposta a regimes terapêuticos específicos.56,117,118 Entre os subgrupos estão a leucemia de precursor de célula B e de célula de Burkitt, e a LLA de precursor de célula T [Tabela 6]. O subgrupo de precursor de célula B pode ser subclassificado de acordo com as características citogenéticas e moleculares. Existem 2 subgrupos importantes associados a um resultado mais favorável. A LLA hiperdiploide, envolvendo mais de 50 cromossomos, representa 25% dos casos de LLA na infância. Estes casos estão associados a um índice de cura de 80 a 90%. De modo similar, em 20 a 25% dos casos de LLA na infância, os pacientes expressam o gene da fusão TEL/AML1 (também chamado ETV6/CBFA2), que resulta da t(12;21) críptica. Estes casos também estão associados a índices de cura de 85 a 90%. Entretanto, estes subtipos genéticos raramente são encontrados em séries de pacientes adultos. O único subgrupo mais amplo de pacientes adultos com LLA é aquele em que há expressão do gene de fusão BCR/ABL, resultante da t(9;22). O subtipo resultante de LLA, também conhecido como leucemia linfoblástica aguda cromossomo Philadelphia-positiva (Ph+), representa até 25 a 30% dos casos de LLA do adulto, e sua frequência aumenta com o avanço da idade. Em contraste, pacientes com translocação envolvendo a banda cromossômica 14q11 ou que superexpressam o gene HOX11 geralmente têm LLA de célula T e apresentam prognóstico favorável quando tratados com os regimes multifarmacológicos convencionais. Os pacientes com LLA de células de Burkitt geralmente apresentam t(8;14) (gene de fusão MYC/IGH) e um prognóstico igualmente favorável, porém necessitam de uma quimioterapia bastante diferente. As outras anormalidades cromossômicas recorrentes e genes de fusão [Tabela 3] são menos comuns. Estas diversas anomalias cromossômicas estão associadas a prognósticos distintos.
É importante notar que as diferenças consideráveis em termos de resultado a longo prazo apresentadas pelos vários subgrupos citogenéticos não são tão evidentes quando apenas a taxa de RC inicial é analisada. Ou seja, com os atuais programas de indução intensiva, 80% dos pacientes com citogenética de risco desfavorável conseguem alcançar a remissão, em comparação aos cerca de 90% dos pacientes que apresentam cariótipos normais ou outras anormalidades. A importância clínica deste dado está no fato de os pacientes de risco desfavorável responderem ao tratamento inicial de modo suficientemente satisfatório para se tornarem candidatos a um transplante antecipado de alo-CTH.
Os objetivos dos regimes modernos de tratamento da LLA são a rápida restauração da função da medula óssea com o uso de múltiplos fármacos quimioterápicos de toxicidades aceitáveis, para prevenir a emergência de subclones resistentes; o tratamento profilático adequado de “sítios-santuário”, como o SNC; e a instituição de terapias de consolidação pós-remissão para eliminar a doença residual mínima (indetectável).116 A terapia de pós-remissão tradicionalmente consiste no tratamento de intensificação ou consolidação seguido de terapia prolongada de manutenção ou continuação. A capacidade dos pacientes e de seus médicos de adesão ao exigente esquema terapêutico pode ser decisiva para alcançar resultados ideais.119
Tipicamente, são utilizados 4 ou 5 fármacos (p. ex., vincristina, prednisona ou dexametasona, daunorubicina, L-asparaginase e ciclofosfamida) na indução da remissão. Após a indução da remissão, regimes similares que também incluem antimetabólitos são empregados no tratamento de consolidação da remissão.50,116,117,119-121 A vasta maioria (80 a 90%) dos adultos jovens entram em RC. Cerca de 30 a 50% dos pacientes permanecem vivos e livres de doença decorridos 3 anos da entrada em RC. A LLA é uma doença heterogênea, e os resultados variam de acordo com a idade, imunofenótipo e características clínicas, citogenéticas e moleculares do paciente. As estratégias de tratamento modernas adotam uma abordagem risco-adaptada. Para adolescentes e adultos jovens, o risco melhora com o uso de regimes de tratamento pediátrico.119,122
Na LLA, os fatores prognósticos adversos significativos exercem influência importante sobre as taxas de RC, duração da remissão e sobrevida.50,115-118 Em análises de múltiplas variáveis, os pacientes com contagens de leucócitos acima de 30.000/mcL apresentam remissão significativamente menos duradoura do que os pacientes com contagem de leucócitos mais baixas. Entretanto, a leucocitose extrema não exerce efeito negativo sobre o resultado alcançado por pacientes com LLA de célula T.50 A idade avançada (> 60 anos) constitui outra característica desfavorável. No momento do diagnóstico, o cariótipo atua como fator preditivo independente do resultado e é essencial ao sucesso do planejamento terapêutico.56,117,118 Os fatores menores, ou fatores que possuem alguma importância relacionada a certos regimes terapêuticos, são o percentual de blastos circulantes; o grau de envolvimento da medula óssea; a presença de hepatomegalia, esplenomegalia ou linfadenopatia; os níveis de DHL; o envolvimento do SNC no momento da apresentação; e o tempo necessário para alcançar a RC (p. ex., > 4 a 6 semanas).
O tratamento de consolidação da remissão destina-se a erradicar as células neoplásicas de proliferação rápida, que são consideradas responsáveis pelas recidivas precoces. Em geral, os fármacos administrados durante este período são antimetabólitos específicos para fases do ciclo celular. Ao menos um curso de reindução intensiva administrado durante a terapia de consolidação parece ser necessário para se alcançar a cura. O benefício relativo de qualquer regime de consolidação em particular é provavelmente uma função da intensidade da terapia de indução inicial e sua eficácia na redução rápida da massa de células leucêmicas. A terapia pós-remissão ideal para adultos com LLA ainda é obscura. A morbidade e mortalidade relacionadas ao tratamento são maiores com o transplante de alo-CTH do que com a quimioterapia, embora as recidivas sejam menos comuns.123-125 Os dados disponíveis indicam que não há um consenso claro quanto ao transplante de alo-CTH ser mais vantajoso do que a quimioterapia no tratamento de adultos com LLA com características de risco padrão durante a 1ª RC.123,124,126,127
Entretanto, o transplante de alo-CTH é recomendado durante uma 1ª RC para pacientes com alto risco de desenvolvimento de LLA e para aqueles em 2ª RC.123,124,128-130 Em um futuro próximo, pode ser possível identificar os pacientes que apresentam alto risco de recidiva por meio da detecção de doença residual mínima ainda durante a remissão.131
Um período de tratamento prolongado com baixas doses de quimioterápicos – denominado terapia de continuação ou manutenção da remissão – é a abordagem-padrão em casos de LLA. Esta abordagem contrasta acentuadamente com o tratamento da maioria dos outros cânceres considerados curáveis, como o linfoma de Hodgkin, linfoma de células grandes ou câncer de testículo, em que a cura é consequência da instituição de uma terapia citorredutora intensiva inicial e a quimioterapia de manutenção com doses baixas não promove nenhum benefício adicional. A necessidade de uma terapia de manutenção prolongada para adultos com LLA também pode ser uma função da intensidade e do sucesso da quimioterapia inicial. Tradicionalmente, é instituído um tratamento de 2 a 3 anos de duração, à base de 6-mercaptopurina e metotrexato, muitas vezes com pulsos mensais de vincristina e prednisona (ou dexametasona). Até agora, a necessidade de terapia de manutenção ainda não foi comprovada em pacientes adultos, mas é provavelmente importante para a maioria dos tipos de LLA.
O SNC é um sítio importante de envolvimento pela LLA.132 Embora não seja comumente detectado no momento do diagnóstico, o envolvimento do SNC ocorre com frequência na recaída. As meninges podem abrigar células leucêmicas e a barreira hematoencefálica pode protegê-las contra a quimioterapia sistêmica. A recidiva junto ao SNC geralmente coincide com a recidiva sistêmica. A probabilidade de uma recidiva envolvendo isoladamente o SNC em pacientes adultos com LLA parece estar em torno de 5%. O tratamento preventivo do SNC durante a terapia de pós-remissão – denominado profilaxia contra o envolvimento do SNC – tornou-se parte integrante de quase todos os protocolos de tratamento de LLA vigentes. Embora o valor real desta profilaxia para pacientes adultos seja controverso, estudos demonstraram que os pacientes adultos que recusaram ou estavam impossibilitados de receber profilaxia contra o envolvimento do SNC apresentaram taxas mais altas de recidiva com envolvimento do SNC do que os pacientes que receberam profilaxia. A leucemia com envolvimento do SNC é mais facilmente prevenida do que tratada. Depois que esta condição se desenvolve, existe alta probabilidade de recidiva com envolvimento do SNC subsequente, mesmo com a instituição do tratamento.
A profilaxia para o SNC tipicamente consiste na irradiação craniana aliada à administração intratecal de metotrexato. A citarabina e a hidrocortisona às vezes são adicionadas ao metotrexato na terapia intratecal tripla. Em vez da irradiação craniana, alguns pesquisadores usam a quimioterapia com doses altas de metotrexato ou citarabina, porque é possível atingir os níveis terapêuticos destes fármacos no SNC com a administração de doses altas por via endovenosa.121 De uma forma geral, a superioridade de qualquer terapia profilática ainda não foi estabelecida.
Dentre os subgrupos de alto risco, o 1º a merecer atenção especial é o de LLA de células de Burkitt, também conhecida como FAB L3 ou LLA de células B maduras.133 Este subgrupo representa até 3 a 5% de todos os casos de LLA em adultos. A condição manifesta-se como uma leucemia ou linfoma. Alguns pacientes adquirem infecção pelo HIV. As características biológicas ubíquas são a presença de uma imunoglobulina de superfície monoclonal, a t(8;14) ou uma de suas variantes – t(2;8) ou t(8;22) –, e a expressão constitutiva do oncogene MYC. O reconhecimento da LLA da célula de Burkitt no momento do diagnóstico é relativamente fácil e baseia-se nos achados clínicos característicos de hepatoesplenomegalia e linfadenopatia. Os níveis de DHL e ácido úrico em geral estão acentuadamente elevados, e com frequência há envolvimento leptomeníngeo. Os linfoblastos geralmente não apresentam reatividade TdT, mas costumam apresentar forte expressão de CD20. Antigamente, poucos ou nenhum destes pacientes sobreviviam aos regimes terapêuticos padrão para LLA. Agora, porém, os programas quimioterápicos intensivos e de curta duração para LLA de células de Burkitt promovem uma alta taxa de RC e um platô de sobrevida na faixa de 50 a 70%.134,135 Estes regimes, que podem requerer 16 a 18 semanas de tratamento, empregam altas doses de metotrexato, citarabina e ciclofosfamida ou ifosfamida combinadas a outros fármacos usados no tratamento da LLA. Mais recentemente, a adição do anticorpo anti-CD20 rituximabe à quimioterapia tem promovido benefícios consideráveis.136
A LLA Ph+ é identificada pela t(9;22)(q34;q22) ou pela expressão do gene de fusão BCR/ABL.137 Em cerca de 2/3 dos casos, há expressão do transcrito de RNA mensageiro de p190, enquanto o transcrito p210 é expresso em 1/3 dos casos, de modo idêntico ao observado na LMC. A LLA Ph+ tem representado um desafio significativo à cura da LLA, pois representa até 25 a 30% de todos os casos envolvendo pacientes adultos e talvez 40 a 50% dos casos de LLA de linhagem B em idosos.56,137,138 Avanços consideráveis foram alcançados com a adição dos inibidores de tirosina quinase (TKI) imatinibe ou dasatinibe à terapia de indução de remissão. Taxas de RC superiores a 90% atualmente são rotineiras.139,140 Até o presente, nenhum regime quimioterápico isolado parece ser capaz de curar este grupo de pacientes, embora remissões prolongadas tenham sido observadas com o uso combinado de um TKI e quimioterapia.141 Quando o imatinibe é incorporado durante a indução da remissão para melhorar a condição da remissão, a realização de um transplante de alo-CTH subsequente cura mais da metade dos pacientes com LLA Ph+.142-144 Entretanto, há uma probabilidade de recidiva pós-transplante aproximada de 20%, e isto atesta a natureza terapia-resistente desta doença. Por enquanto, o tratamento da LLA Ph+ deve incluir um programa quimioterápico de indução de remissão intensivo com imatinibe ou dasatinibe, seguido de transplante de alo-CTH na 1ª RC, caso haja disponibilidade de doador.
Os pacientes com LLA com t(4;11)(q21;q23) constituem outro subgrupo citogenético em que a terapia convencional promove resultados precários e que, se possível, deve ser submetido ao transplante alogênico. O gene envolvido no cromossomo 11 foi chamado MLL.20,53 A incidência é de 3 a 6% nas séries citogenéticas de pacientes adultos.56 O imunofenótipo é de progenitor de célula B (CD19+, CD22+ e HLA-DR+, e CD10-).145 No caso dos adultos, estes pacientes frequentemente são mulheres de idade avançada (mais da metade tem idade > 50 anos) que apresentam contagem de leucócitos elevada.
Apesar dos avanços significativos ocorridos no tratamento de adultos com leucemia aguda ao longo da última década, muitos pacientes continuam morrendo em decorrência desta doença ou das complicações associadas a seu tratamento [Tabela 8]. Contudo, algumas abordagens clínicas e experimentais novas trazem a promessa de índices de cura melhores. A aplicação das modernas tecnologias moleculares projetadas para detectar a leucemia residual mínima podem ajudar os clínicos a monitorar a doença durante e após a quimioterapia.146 Estas tecnologias poderiam, por exemplo, possibilitar a detecção antecipada dos pacientes propensos a recidivas, para os quais uma terapia adicional ou um transplante de alo-CTH inicial poderiam ser benéficos. Atualmente, estão sendo investigados novos métodos que permitam evitar a resistência multifarmacológica, explorar os mecanismos imunes ou alterar o controle do crescimento, diferenciação e apoptose nas células malignas.
Tabela 8. Resultado do tratamento para pacientes adultos com leucemia aguda
Subgrupo da doença |
RC (%) |
Sobrevida livre de doença em 3 anos (%) |
|
LMA, geral |
60 a 80 |
20 a 40 |
|
Cariótipo favorável |
90 |
60 a 70 |
|
< 60 anos |
70 a 80 |
40 |
|
= 60 anos |
45 a 55 |
10 a 20 |
|
LPMA, t(15;17) |
90 |
80 |
|
LLA, geral |
75 a 90 |
30 a 40 |
|
LLA de célula T |
90 a 95 |
60 a 65 |
|
LLA de precursor de célula B |
75 a 85 |
30 a 40 |
|
LLA Ph+ |
85 a 95 |
50 |
|
LLA de célula de Burkitt |
75 |
70 |
|
= 60 anos |
60 a 80 |
10 a 25 |
LLA = leucemia linfoblástica aguda; LMA = leucemia mieloide aguda; LPMA = leucemia promielocítica aguda; Ph+ = cromossomo Philadelphia-positiva; RC = remissão completa.
Richard A. Larson, MD, recebeu apoio financeiro para pesquisa clínica das empresas Amgen, Ambit Bioscience, Chroma Therapeutics, Celgene, Sanofi-Aventis, Millennium, Novartis e Bristol-Myers Squibb; recebeu apoio financeiro para realização de atividades de ensino das empresas Novartis e Bristol-Myers Squibb; e atua como consultor junto à Amgen, Novartis, Bristol-Myers Squibb, Hana Bioscience, Antisoma e Caremark.
1. Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006;368:1894–907.
2. Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med 2009;361:1872–85.
3. Nimer SD. Myelodysplastic syndromes. Blood 2008;111: 4841–51.
4. Juliusson G, Antunovic P, Derolf A, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood 2009;113:4179–87.
5. Linet MS. The leukemias: epidemiologic aspects. New York: Oxford University Press; 1985.
6. Sandler DP, Ross JA. Epidemiology of acute leukemia in children and adults. Semin Oncol 1997;24:3.
7. Gilliland DG, Griffi n JD. The roles of FLT3 in hema topoiesis and leukemia. Blood 2002;100:1532.
8. Brill AB, Tomonoga M, Heyssel RM. Leukemia in man following exposure to ionizing irradiation: summary of fi ndings in Hiroshima and Nagasaki and comparison to other human experience. Ann Intern Med 1962;56:590.
9. Kato H, Schull WJ. Studies of the mortality of A-bomb survivors. 7. Mortality, 1950–1978: part I. Cancer mortality. Radiat Res 1982;90:395.
10. Smith PG, Doll R. Mortality among patients with ankylosing spondylitis after a single treatment course with x-rays. Br Med J 1982;284:449.
11. Modan B, Lilienfeld AM. Polycythemia vera and leukemia: the role of radiation treatment. Medicine (Baltimore) 1965; 44:305.
12. Godley LA, Larson RA. Therapy-related myeloid leukemia. Semin Oncol 2008;35:418–29.
13. Graham S, Levin ML, Lilienfeld AM, et al. Preconception, intrauterine, and postnatal irradiation as related to leukemia. Monogr Natl Cancer Inst 1966;19:347.
14. Polednak AP, Stehney AF, Rowland RE. Mortality among women fi rst employed before 1930 in the U.S. radium dial painting industry. Am J Epidemiol 1978;107:179.
15. March HC. Leukemia in radiologists. Radiology 1944;43: 275.
16. Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 2003;102:43.
17. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classifi cation
a. of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937–51.
18. Swerdlow SH, Campo E, Harris NL, et al, editors. World Health Organization classifi cation of tumours of haematopoietic and lymphoid tissues. Lyon (France): IARC Press; 2008.
19. Pui CH, Ribeiro RC, Hancock ML, et al. Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 1991;325: 1682.
20. Le Beau MM, Larson RA. Cytogenetics and neoplasia. In: Hoffman R, Benz EJ Jr, Shattil SJ, et al, editors. Hematology: basic principles and practice. 3rd ed. New York: Churchill Livingstone; 2000. p. 848.
21. Aksoy M, Erdem S, Din Col G. Leukemia in shoe workers chronically exposed to benzene. Blood 1974;44:837.
22. Hayes RB, Yin SN, Dosemeci M, et al. Benzene and the dose-related incidence of hematologic neoplasms in China. J Natl Cancer Inst 1997;89:1065.
23. Lan Q, Zhang L, Li G, et al. Hematotoxicity in workers exposed to low levels of benzene. Science 2004;306(5702): 1774–6.
24. Miller RW. Deaths from childhood leukemia and solid tumors among twins and other sibs in the United States, 1960–67. J Natl Cancer Inst 1971;46:203.
25. Eguchi M, Eguchi-Ishiae M, Greaves M. The role of the MLL gene in infant leukemia. Int J Hematol 2003;78:390.
26. Malinge S, Shai Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood 2009;113:2619–28.
27. Lange BJ, Kobrinsky N, Barnard DR, et al. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down’s syndrome: Children’s Cancer Group studies 2861 and 2891. Blood 1998;91:608.
28. Rosenberg PS, Alter BP, Bolyard AA, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood 2006;107:4628–35.
29. Preudhomme C, Renneville A, Bourdon V, et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009;113:5583–7.
30. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classifi cation of the acute leukemias. Br J Haematol 1976;33:451.
31. Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classifi cation of acute myeloid leukemia: a report of the French-American-British cooperative group. Ann Intern Med 1985;103:620.
32. Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010;115:453–74.
33. Bennett JM, Catovsky D, Daniel MT, et al. Proposal for the recognition of minimally differentiated acute myeloid leukemia (AML-M0). Br J Haematol 1991;78:325.
34. Craig FE, Foon KA. Flow cytometric immunophenotyping for hematologic neoplasms. Blood 2008;111:3941–67.
35. Terstappen LWMM. Cell differentiation and maturation in normal bone marrow and acute leukemia. In: Macey MG, editor. Flow cytometry clinical applications. Oxford (UK): Blackwell Scientif c Publications; 1995. p. 101.
36. Buccisano F, Maurillo L, Spagnoli A, et al. Monitoring of minimal residual disease in acute myeloid leukemia. Curr Opin Oncol 2009;21:582–8.
37. Geller RB, Zahurak M, Hurwitz CA, et al. Prognostic importance of immunophenotyping in adults with acute myelocytic leukemia: the signif cance of the stem-cell glycoprotein CD34 (My10). Br J Haematol 1990;76:340.
38. Larson RA, Sievers EL, Stadtmauer EA, et al. Final report of the eff cacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in f rst recurrence. Cancer 2005;104:1442.
39. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
40. Steensma DP, editor. Myelodysplastic syndromes: pathobiology and clinical management. 2nd ed. New York: Informa Healthcare USA, Inc.; 2009.
41. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classif cation of the myelodysplastic syndromes. Br J Haematol 1982;51:189.
42. Rosati S, Mick R, Xu F, et al. Refractory anemia with multilineage dysplasia: further characterization of an “unclassif able” myelodysplastic syndrome. Leukemia 1996;10:20.
43. Olney HJ, Le Beau MM. The cytogenetics of myelodysplastic syndromes. Best Pract Res Clin Haematol 2001;14: 479–95.
44. Ebert BL, Pretz J, Bosco J, et al. Identif cation of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008;451(7176):335–9.
45. Greenberg P, Cox C, Le Beau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1996;89:2079.
46. Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classif cation and treatment options. Br J Haematol 2008;140:610–24.
47. Bennett JM, Catovsky D, Daniel MT, et al. The morphologic classif cation of acute lymphoblastic leukemia: concordance among observers and clinical correlations. Br J Haematol 1981;47:553.
48. Kebriaei P, Anastasi J, Larson RA. Acute lymphoblastic leukemia: diagnosis and classif cation. Best Pract Res Clin Hematol 2003;15:597.
49. Czuczman MS, Dodge RK, Stewart CC, et al. Value of immunophenotype in intensively treated adult acute lymphoblastic leukemia: Cancer and Leukemia Group B Study 8364. Blood 1999;93:3931.
50. Larson RA, Dodge RK, Burns CP, et al. A f ve-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: Cancer and Leukemia Group B Study 8811. Blood 1995;85:2025.
51. Bene MC, Kaeda JS. How and why minimal residual disease studies are necessary in leukemia: a review from WP10 and WP12 of the European LeukemiaNet. Haematologica 2009;94:1135–50.
52. Matutes E, Morilla R, Farahat N, et al. Def nition of acute biphenotypic leukemia. Haematologica 1997;82:64.
53. Rowley JD. Chromosomal translocations: revisited yet again. Blood 2008;112:2183–9.
54. Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002;100:4325.
55. Mrozek K, Heerema NA, Bloomf eld CD. Cytogenetics in acute leukemia. Blood Rev 2004;18:115–36.
56. Wetzler M, Dodge RK, Mrozek K, et al. Prospective karyotype analysis in adult acute leukemia: the Cancer and Leukemia Group B experience. Blood 1999;93:3983.
57. Mrózek K, Bloomf eld CD. Clinical signif cance of the most common chromosome translocations in adult acute myeloid leukemia. J Natl Cancer Inst Monogr 2008;(39): 52–7.
58. Caligiuri MA, Strout MP, Gilliland DG. Molecular biology of acute myeloid leukemia. Semin Oncol 1997;24:32.
59. Thandla S, Aplan PD. Molecular biology of acute lymphocytic leukemia. Semin Oncol 1997;24:45.
60. Baldus CD, Mrózek K, Marcucci G, Bloomf eld CD. Clinical outcome of de novo acute myeloid leukaemia patients with normal cytogenetics is affected by molecular genetic alterations: a concise review. Br J Haematol 2007;137: 387–400.
61. Mrózek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classif cation? Blood 2007;109:431–8.
62. Baldus CD, Bullinger L. Gene expression with prognostic implications in cytogenetically normal acute myeloid leukemia. Semin Oncol 2008;35:356–64.
63. Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol 2005;23:9234.
64. Radmacher MD, Marcucci G, Ruppert AS, et al. Independent conf rmation of a prognostic gene expression signature in adult acute myeloid leukemia with a normal karyotype: a Cancer and Leukemia Group B study. Blood 2006;108:1677–83.
65. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1–9.
66. Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukemia. Nature 2007;446:758–64.
67. Tosi P, Barosi G, Lazzaro C, et al. Consensus conference on the management of tumor lysis syndrome. Haematologica 2008;93:1877–85.
68. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in f rst complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
69. Hegenbart U, Niederwieser D, Sandmaier BM, et al. Treatment for acute myelogenous leukemia by low-dose, total-body, irradiation-based conditioning and hematopoietic cell transplantation from related and unrelated donors. J Clin Oncol 2006;24:444.
70. Bensinger WI, Martin PJ, Storer B, et al. Transplantation of bone marrow as compared with peripheral blood cells from HLA-identical relatives in patients with hematologic cancers. N Engl J Med 2001;344:175.
71. Seshadri T, Keating A. Is there a role for autotransplants in AML in fi rst remission? Biol Blood Marrow Transplant 2009;15(1 Suppl):17–20.
72. Marcucci G, Mrozek K, Ruppert AS, et al. Abnormal cytogenetics at date of morphologic complete remission predicts short overall and disease-free survival, and higher relapse rate in adult acute myeloid leukemia: results from Cancer and Leukemia Group B study 8461. J Clin Oncol 2004;22:2410.
73. Lo Coco F, Diverio D, Falini B, et al. Genetic diagnosis and molecular monitoring in the management of acute promyelocytic leukemia. Blood 1999;94:12.
74. Nucifora G, Larson RA, Rowley JD. Persistence of the 8;21 translocation in patients with acute myeloid leukemia type M2 in long-term remission. Blood 1993;82:712.
75. Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005;352:254–66.
76. Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 2005;106:3740–6.
77. Fröhling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 2004;22:624–33.
78. Marcucci G, Maharry K, Radmacher MD, et al. Prognostic signifi cance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B study. J Clin Oncol 2008;26:5078–87.
79. Ravandi F, Burnett AK, Agura ED, Kantarjian HM. Progress in the treatment of acute myeloid leukemia. Cancer 2007;110:1900–10.
80. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensifi cation in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
81. Löwenberg B, Ossenkoppele GJ, van Putten W, et al; Dutch-Belgian Cooperative Trial Group for HematoOncology (HOVON); German AML Study Group (AMLSG); Swiss Group for Clinical Cancer Research (SAKK) Collaborative Group. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
82. Kimby E, Nygren P, Glimelius B, et al. A systematic overview of chemotherapy effects in acute myeloid leukaemia. Acta Oncol 2001;40:231.
83. Sanz M, Burnett A, Lo-Coco F, Löwenberg B. FLT3 inhibition as a targeted therapy for acute myeloid leukemia. Curr Opin Oncol 2009;21:594–600.
84. Stone RM. Postremission therapy in adults with acute myeloid leukemia. Semin Hematol 2001;38:17.
85. Cassileth PA, Lynch E, Hines JD, et al. Varying intensity of postremission therapy in acute myeloid leukemia. Blood 1992;79:1924.
86. Buchner T, Hiddemann W, Wormann B, et al. Double induction strategy for acute myeloid leukemia: the effect
a. of high-dose cytarabine with mitoxantrone instead of standard-dose cytarabine with daunorubicin and 6- thioguanine: a randomized trial by the German AML Cooperative Group. Blood 1999;93:4116.
87. Marcucci G, Mrozek K, Ruppert AS, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol 2005;23:5705.
88. Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N Engl J Med 1994;331:896.
89. Oliansky DM, Appelbaum F, Cassileth PA, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute myelogenous leukemia in adults: an evidence-based review. Biol Blood Marrow Transplant 2008;14:137–80.
90. Cornelissen JJ, van Putten WL, Verdonck LF, et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in fi rst remission acute myeloid leukemia in young and middle-aged adults: benefi ts for whom? Blood 2007;109:3658–66.
91. Cassileth PA, Harrington DP, Appelbaum FR, et al. Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in fi rst remission. N Engl J Med 1998; 339:1649.
92. Burnett AK, Goldstone AH, Stevens RMF, et al. Randomised comparison of addition of autologous bone-marrow transplantation to intensive chemotherapy for acute myeloid leukaemia in first remission: results of the MRC AML10 trial. Lancet 1998;351:700.
93. Sanz MA, Grimwade D, Tallman MS, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
94. Menell JS, Cesarman GM, Jacovina AT, et al. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med 1999;340:994.
95. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
96. Soignet SL, Frankel SR, Douer D, et al. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol 2001;19:3852.
97. Powell BL, Moser B, Stock W, et al. Consolidation with arsenic trioxide (As2O3) signifi cantly improves event-free survival and overall survival among patients with newly diagnosed acute promyelocytic leukemia: North American Intergroup Protocol C9710 [abstract]. J Clin Oncol 2007; 25:185 abstr 2.
98. Hu J, Liu YF, Wu CF, et al. Long-term effi cacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 2009;106:3342–47.
99. Larson RA. Myelodysplasia: when to treat and how. Best Pract Res Clin Haematol 2006;19:293.
100.Stone RM. How I treat patients with myelodysplastic syndromes. Blood 2009;113:6296–303.
101.Estey E. Acute myeloid leukemia and myelodysplastic syndromes in older patients. J Clin Oncol 2007;25: 1908–15.
102.de Witte T, Suciu S, Verhoef G, et al. Intensive chemotherapy followed by allogeneic or autologous stem cell transplantation for patients with myelodysplastic syndromes and acute myeloid leukemia following MDS. Blood 2001; 98:2326.
103.Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol 2006;24: 3895–903.
104.List A, Kurtin S, Roe DJ, et al. Eff cacy of lenalidomide in myelodysplastic syndromes. N Engl J Med 2005;352:549.
105.Sekeres MA. Treatment of older adults with acute myeloid leukemia: state of the art and current perspectives. Haematologica 2008;93:1769–72.
106.Appelbaum FR, Gundacker H, Head DR, et al. Age and acute myeloid leukemia. Blood 2006;107:3481–5.
107.Leith CP, Kopecky KJ, Chen IM, et al. Frequency and clinical signif cance of the expression of the multidrug resistance proteins MDR1/P-glycoprotein, MRP1, and LRP in acute myeloid leukemia: a Southwest Oncology Group Study. Blood 1999;94:1086.
108.Kantarjian H, O’Brien S, Cortes J, et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer 2006;106:1090–8.
109.Kuendgen A, Germing U. Emerging treatment strategies for acute myeloid leukemia in the elderly. Cancer Treat Rev 2009;35:97–120.
110.Stone RM, Berg DT, George SL, et al. Postremission therapy in older patients with de novo acute myeloid leukemia: a randomized trial comparing mitoxantrone and intermediate-dose cytarabine with standard-dose cytarabine. Blood 2001;98:548.
111.Estey EH. General approach to, and perspectives on clinical research in, older patients with newly diagnosed acute myeloid leukemia. Semin Hematol 2006;43:89.
112.Schiffer CA. Hematopoietic growth factors as adjuncts to the treatment of acute myeloid leukemia. Blood 1996;88: 3675.
113.Wadleigh M, Stone RM. The role of myeloid growth factors in acute leukemia. J Natl Compr Canc Network 2009;7:84–91.
114.Heil G, Hoelzer D, Sanz MA, et al. A randomized, double-blind, placebo-controlled, phase III study of f lgrastim in remission induction and consolidation therapy for adults with de novo acute myeloid leukemia. Blood 1997;90: 4710.
115.Campana D. Molecular determinants of treatment response in acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Progr 2008;366–73.
116.Laport GF, Larson RA. Treatment of adult acute lymphoblastic leukemia. Semin Oncol 1997;24:70.
117.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354:166.
118.Moorman AV, Harrison CJ, Buck GA, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC)
a. UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 2007;109:3189–97.
119.Stock W, La M, Sanford B, et al. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood 2008;112: 1646–54.
120.Linker C, Damon L, Ries C, et al. Intensif ed and shortened cyclical chemotherapy for adult acute lymphoblastic leukemia. J Clin Oncol 2002;20:2464.
121.Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (hyperCVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer 2004;101:2788–801.
122.Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol 2009;27:911–8.
123.Hahn T, Wall D, Camitta B, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 2005;11:823.
124.Hahn T, Wall D, Camitta B, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in adults: an evidence-based review. Biol Blood Marrow Transplant 2006;12:1.
125.Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benef t is achieved from a matched sibling allogeneic transplantation in f rst complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: f nal results of the International ALL Trial (MRC UKALL XII/ ECOG E2993). Blood 2008;111:1827–33.
126.Weisdorf D, Forman S. Allogeneic hematopoietic stem cell transplantation for adult acute lymphoblastic leukemia. Cancer Treat Res 2009;144:1–14.
127.Larson RA. Allogeneic hematopoietic cell transplantation is not recommended for all adults with standard-risk acute lymphoblastic leukemia in f rst complete remission. Biol Blood Marrow Transpl 2009;15 Suppl 1:11–6.
128.Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome–positive acute lymphoblastic leukemia. N Engl J Med 2000;342: 998.
129.Yanada M, Matsuo K, Suzuki T, Naoe T. Allogeneic hematopoietic stem cell transplantation as part of postremission therapy improves survival for adult patients with high-risk acute lymphoblastic leukemia. A metaanalysis. Cancer 2007;106:2657–63.
130.Hoelzer D, Gokbuget N. Allogeneic stem cell transplant in acute lymphoblastic leukemia: who and when? ASCO Educ Book 2009;355–9.
131.Patel B, Rai L, Buck G, et al. Minimal residual disease is a signif cant predictor of treatment failure in non T-lineage adult acute lymphoblastic leukaemia: f nal results of the international trial UKALL XII/ECOG2993. Br J Haematol 2010;148:80–9.
132.Cataland SR, Larson RA. Treatment of adult acute lymphoblastic leukemia: perspective 1. In: Pui C-H, editor. Treatment of acute leukemias: new directions for clinical research. Totowa (NJ): Humana Press; 2003. p. 131.
133.Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
134.Hoelzer D, Ludwig WD, Thiel D, et al. Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 1996; 87:495.
135.Rizzieri DA, Johnson JL, Niedzwiecki D, et al. Intensive chemotherapy with and without cranial radiation for Burkitt leukemia and lymphoma: fi nal results of Cancer and Leukemia Group B study 9251. Cancer 2004;100:1438.
136.Thomas DA, Faderl S, O’Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 2006;106:1569.
137.Ravandi F, Kebriaei P. Philadelphia chromosome-positive acute lymphoblastic leukemia. Hematol Oncol Clin North Am 2009;23:1043–63.
138.Gleissner B, Gokbuget N, Bartram CR, et al. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confi rmed polymerase chain reaction analysis. Blood 2002; 99:1536.
139.Thomas DA, Faderl S, Cortes J, et al. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 2004; 103:4396.
140.Wassmann B, Pfeifer H, Goekbuget N, et al. Alternating versus concurrent schedules of imatinib and chemo therapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ALL). Blood 2006;108:1469–77.
141.Ottmann OG, Pfeifer H. Management of Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL). Hematology Am Soc Hematol Educ Program 2009; 371–81.
142.Yanada M, Takeuchi J, Sugiura I, et al. High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol 2006;24:460–6.
143.de Labarthe A, Rousselot P, Huguet-Rigal F, et al; Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL). Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood 2007;109: 1408–13.
144.Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a Children’s Oncology Group study. J Clin Oncol 2009;27:5175–81.
145.Ludwig WD, Rieder H, Bartram CR, et al. Immunophenotypic and genotypic features, clinical characteristics, and treatment outcome of adult pro-B acute lymphoblastic leukemia: results of the German multicenter trials GMALL 03/87 and 04/89. Blood 1998;92:1898.
146.Campana D. Minimal residual disease in acute lymphoblastic leukemia. Semin Hematol 2009;46:100–6.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.