(Carregando Índice)... (Carregando Índice)... |
Última revisão: 14/03/2014
Comentários de assinantes: 0
Professor of Neurology, Department of Neurology, Mayo Clinic, Rochester, MN
Artigo original: Knopman DS. Alzheimer disease and other major dementing illnesses. ACP Medicine. 2009;1-15.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figuras 3, 4 e 8 – Joseph Parisi, MD, Mayo Clinic. Figura 7 – Keith Josephs, MD, Mayo Clinic.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. José Paulo Ladeira
A doença de Alzheimer (DA) é histopatologicamente definida pela presença de emaranhados neurofibrilares e placas neuríticas no córtex cerebral. A DA também costuma ser usada como diagnóstico clínico para uma síndrome de demência cujo sintoma dominante é uma amnésia anterógrada. A base patológica mais comum para a síndrome clínica com amnésia anterógrada proeminente é de fato a DA patologicamente definida.
Contudo, desde 2009, a perspectiva-padrão considera que um indivíduo tem DA somente quando passa a apresentar demência. Um indivíduo com DA sofre uma perda da função cognitiva que interfere nas atividades sociais e ocupacionais e, em geral, o resultado é a dependência de terceiros para a realização de ao menos uma parte das principais tarefas diárias. O declínio de um nível funcional previamente alto diferencia a demência dos retardos de desenvolvimento ou retardo mental. Na demência, o sensório (isto é, o nível de alerta e vigília do indivíduo) não é afetado. Este aspecto distingue a demência do delírio, em que há comprometimento cognitivo no contexto de um sensório alterado.
A definição de demência implica no comprometimento de mais de uma área de função cognitiva. Na síndrome da DA, isto significa que, além das dificuldades decorrentes da amnésia anterógrada, deve haver também o comprometimento de pelo menos uma das seguintes funções: linguagem, pensamento abstrato, função executiva ou processamento visual-espacial. A definição de DA mais amplamente empregada baseia-se nos critérios estabelecidos pelo grupo National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer Disease and Related Disorders Association (NINCDS-ADRDA), em 1984 [Tabela 1].1
Tabela 1. Critérios para o diagnóstico clínico da DA*
|
A. Evidências fornecidas pela história e exame do estado mental que indiquem a existência de um distúrbio caracterizado por um comprometimento significativo do aprendizado e da retenção de novas informações, aliadas a pelo menos um dos seguintes achados: |
|
1. Comprometimento da execução de tarefas complexas |
|
2. Comprometimento da capacidade de raciocínio |
|
3. Comprometimento da capacidade e orientação espacial |
|
4. Comprometimento da linguagem |
|
B. Os distúrbios referidos em A interferem de maneira significativa no trabalho ou nas atividades ou relacionamentos sociais habituais com outros indivíduos |
|
C. Os distúrbios referidos em A representam um declínio significativo em relação aos níveis prévios de funcionamento |
|
D. Os distúrbios referidos em A são de aparecimento insidioso e evolução progressiva, com base nas evidências fornecidas pela história ou pelos exames seriados do estado mental |
|
E. Os distúrbios não ocorrem exclusivamente durante o curso do delírio |
|
F. Os distúrbios não são mais bem explicados por um diagnóstico psiquiátrico significativo |
|
G. Os distúrbios não são mais bem explicados por uma doença sistêmica nem por outra doença cerebral |
DA = doença de Alzheimer.
*Adaptado de McKhann et al.1 e American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington (DC): American Psychiatric Association; 1994.
O achado cognitivo central na DA é a amnésia anterógrada, que representa a incapacidade de aprender novas informações. Os indivíduos com amnésia anterógrada tipicamente não conseguem se manter atualizados com a data, lembrar-se de conversas recentes ou lembrar onde deixaram algum objeto, e com frequência são autorrepetitivos nas conversações. Embora estes tipos de dificuldade sejam comumente descritos como perda da memória de curta duração, a principal dificuldade associada à amnésia anterógrada da DA está na aquisição ou aprendizado de novas informações. A falha de aprendizado resulta na dificuldade de relembrar a informação recebida, depois de alguns minutos. A DA não é um distúrbio generalizado da recordação, pois sobretudo no início da doença, mas também nos estágios moderados, os indivíduos com DA conseguem recuperar e lembrar as informações aprendidas em um passado remoto.
Bem antes de se manifestar clinicamente como uma síndrome de demência, a DA patológica produz alterações na função de memória recente, além de outras alterações comportamentais e cognitivas sutis. Presume-se que quase todos os indivíduos destinados a desenvolver demência atribuível à DA sofrem alterações cognitivas e comportamentais assintomáticas durante o período de pródromo.2 Em comparação aos indivíduos que não desenvolvem demência, aqueles destinados a sofrer o declínio alcançam escores piores, ainda que dentro da faixa considerada “normal”. Eventualmente, estes indivíduos destinados a desenvolver demência são identificados como tendo comprometimento da memória recente ou outros déficits cognitivos menores, porém continuam atuando quase normalmente no dia a dia. Esta condição é referida como comprometimento cognitivo leve (CCL).3 Assim como na fase sintomática mais inicial da DA, o CCL pode ser detectado por meio de avaliações neuropsicológicas da memória e da agilidade mental. As opiniões em evolução na área levaram alguns entendidos a propor que os pacientes com CCL cujos exames de imagem de ressonância magnética (IRM) ou análise de biomarcadores no líquido cerebrospinal (LCS) sejam consistentes com DA devem, na verdade, receber o diagnóstico de DA mesmo que não apresentem demência.4
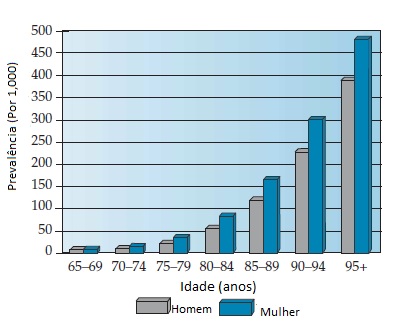
Nos Estados Unidos, a prevalência da DA é de cerca de 8% na população acima de 65 anos de idade.5 Neste país, cerca de 3 milhões de indivíduos têm DA. As estimativas variam um pouco em função das diferentes definições de caso empregadas.5 Se os casos de DA muito leve forem incluídos, o número de indivíduos afetados ultrapassa 5 milhões.6 A prevalência está fortemente associada à idade [Figura 1]: a DA é incomum em indivíduos com menos de 65 anos e, acima desta faixa etária, sua prevalência duplica a cada 5 anos. Até 30 a 40% dos indivíduos com mais de 85 anos de idade têm DA.

Figura 1. Histograma da prevalência da doença de Alzheimer (DA) de acordo com o sexo e a idade.
Adaptado de Hy LX, Keller DM.5
A incidência de DA reflete sua prevalência, sendo muito baixa antes dos 65 anos de idade e, então, aumentando de maneira estável nas faixas etárias de 70 e 80 anos. Ao redor dos 80 anos, a incidência anual de DA se aproxima de 2 em cada 100 indivíduos. Há controvérsias quanto à incidência da DA na 10ª e 11ª décadas da vida. É provável que a incidência continue a aumentar, porém a metodologia e os números de casos nos extremos de expectativa de vida contribuem para a incerteza.
Também há controvérsia quanto ao fato de as mulheres serem mais comumente afetadas do que os homens. A maioria dos estudos sobre prevalência e incidência mostra que as mulheres estão discretamente super-representadas. A diferença é pequena. Esta diferença pode ser importante em termos de propensão biológica ao desenvolvimento de DA entre homens e mulheres, ou talvez seja um artefato associado à menor expectativa de vida geral dos homens.7 As estimativas da prevalência da demência ao nível mundial mostram uma consistência notável entre os grupos raciais e étnicos.
Existem vários fatores associados ao risco aumentado de desenvolvimento de DA. Os fatores de risco de DA mais proeminentes são a idade avançada e a história familiar de demência. Vários fatores de risco vasculares que surgem na meia-idade, como obesidade, diabetes e hipertensão, estão associados ao risco aumentado de desenvolvimento de DA em fases mais tardias da vida.8 Ainda não está claro se os mecanismos de doença vascular generalizada ou os fatores de risco vasculares estão diretamente relacionados aos mecanismos da DA patogênica ou ao efeito aditivo da doença cerebrovascular e DA. A doença cerebrovascular e a DA coexistem em muitos idosos com demência.9 Por este motivo, não surpreende que os fatores de risco vasculares influenciem a expressão clínica da DA.
A terapia anti-hipertensiva, especialmente quando instituída para pacientes de meia-idade ou idosos, foi associada a um risco menor de DA, em numerosos estudos observacionais.10 Além disso, os fármacos anti-hipertensivos apresentaram efeitos comprovadamente protetores em estudos clínicos.11
O uso de outras medicações foi associado a um risco menor de desenvolvimento de DA. Entre estes fármacos, estão o estrogênio, os fármacos redutores de colesterol do tipo estatina e os fármacos anti-inflamatórios não hormonais (AINH). A importância destas observações atualmente é bastante questionada, pois os estudos clínicos sobre cada um destes agentes forneceram resultados desapontadores. No caso do estrogênio e dos AINH, o uso da medicação foi avaliado em pacientes sem demência, e esta estratégia metodológica fortaleceu a crença de que a diminuição do risco observada não foi um artefato do aparecimento de DA precoce. Contudo, os estudos clínicos subsequentes do uso de estrogênio,12,13 AINH14 ou estatinas15 em estudos sobre prevenção falharam em evidenciar os benefícios proporcionados por estes agentes. A conclusão desapontadora parece ser a de que as associações epidemiológicas não se traduzem automaticamente em intervenções terapêuticas bem-sucedidas. A perspectiva alternativa de que os estudos clínicos foram conduzidos de forma incorreta não pode ser excluída. A falha dos estudos clínicos sobre o uso de AINH, terapia de reposição de estrogênios e estatinas na prevenção da demência ou em seu tratamento sintomático significa que estes agentes não podem ser recomendados para uso na prevenção nem no tratamento sintomático da DA.
A capacidade de aprendizado e o desempenho intelectual na infância também parecem estar relacionados ao risco de desenvolvimento de DA em fases posteriores da vida. Exemplificando, indivíduos submetidos a testes de aprendizado acadêmico aos 11 anos de idade foram contatados quando se tornaram adultos e avaliados quanto à demência. Observou-se a existência de uma notável associação entre escores mais altos aos 11 anos e uma menor probabilidade de desenvolvimento de demência em fases tardias da vida. Estudos adicionais realizados com esta coorte sugeriram que poderia haver um risco maior de demência associada à doença cerebrovascular do que de demência associada à DA.16 Numerosos estudos observaram que a capacidade de aprendizado parece estar inversamente relacionada ao risco de DA.
A base genética da DA pode ser dividida em 4 perfis. O 1º perfil consiste em indivíduos com síndrome de Down (trissomia do 21). O 2º perfil inclui as formas raríssimas de DA autossômica dominante. O 3º perfil é o mais comum – a forma de DA de aparecimento tardio – em que a história familiar constitui um risco modestamente forte. O 4º perfil é a DA esporádica, que abrange a maioria dos pacientes com DA, incluindo aqueles com doença de manifestação precoce e, em especial, as formas de aparecimento tardio que parecem ser não genéticas.
O número de indivíduos com DA autossômica dominante é infinitesimal, se comparado ao imenso número de indivíduos com DA esporádica de aparecimento tardio. Estas famílias autossômicas dominantes são centrais, todavia, em termos de fornecimento de hipóteses sobre os mecanismos desta doença. Em geral, os indivíduos afetados das famílias com casos de DA autossômica dominante desenvolvem demência ao redor dos 30 a 60 anos de idade. O distúrbio genético adianta a idade em que a DA se manifesta aproximadamente em 2 a 3 décadas, em relação ao observado na DA esporádica.
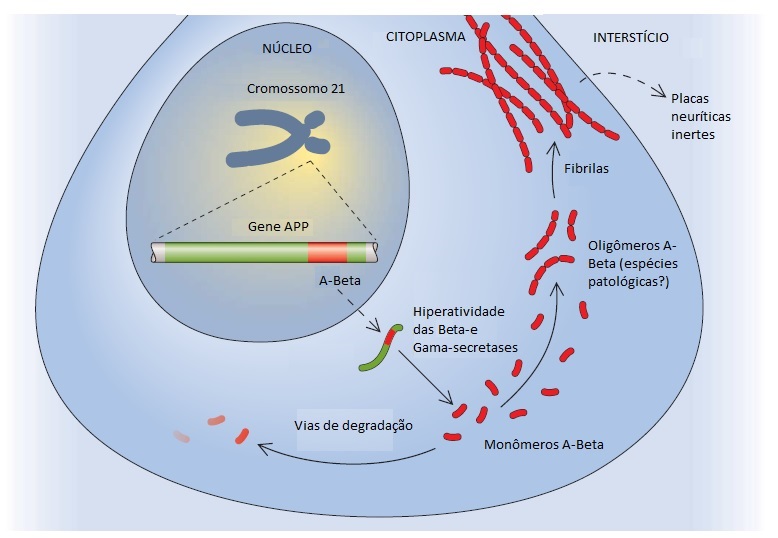
Foram implicados 3 genes na DA autossômica dominante17 (ver uma lista regularmente atualizada no website www.molgen.ua.ac.be/ADMutations/). Um destes genes é o gene da proteína precursora amiloide (APP), localizado no cromossomo 21, que codifica o peptídeo beta-amiloide [Figura 2]. O peptídeo beta-amiloide parece ser uma molécula patogênica essencial. Apenas algumas famílias (85 famílias com um total de 32 mutações discriminadas em junho de 2009) em todo o mundo abrigam as mutações causadoras de DA neste gene. Os outros 2 genes envolvidos na DA autossômica dominante são genes homólogos entre si. Dentre estes genes, o gene da presenilina-1 (PSEN1) é significativamente mais frequente e está localizado no cromossomo 14. Na maioria das famílias com DA autossômica dominante, a doença está ligada à ocorrência de mutações no cromossomo 14.18 Assim como foi feito em junho de 2009, foram enumeradas 176 mutações exclusivas em PSEN1 em 390 famílias. O homólogo menos comum – o gene da presenilina-2 (PSEN-2) – está localizado no cromossomo 1, onde foram identificadas 14 mutações em 23 famílias.
À parte das raras famílias com condição autossômica dominante, uma história familiar de DA é consistente como fator de risco da doença.19 O impacto da história familiar sobre o risco de DA diminui quando o probando tem mais de 75 anos de idade, e é quase nulo após os 80 anos. Subsequentemente, conforme as famílias com DA de aparecimento tardio foram sendo estudadas quanto à genética, constatou-se que o gene da apolipoproteína E (APOE) é responsável por uma fração substancial do risco familiar de DA de aparecimento tardio.20 O gene APOE possui 3 variantes alélicas em seres humanos, que resultam de polimorfismos em 2 nucleotídeos. O alelo APOE eta4 está fortemente associado ao desenvolvimento mais precoce de DA, em comparação aos indivíduos portadores de gene não-eta4.21 Contudo, a presença de um alelo APOE eta4 não garante o desenvolvimento de DA pelos indivíduos portadores de APOE eta4, ao passo que sua ausência não confere proteção contra a DA aos indivíduos portadores de APOE não-eta4. Não há diferenças clínicas entre os pacientes com DA com e sem alelo APOE eta4. Vários estudos de associação genômica foram realizados na DA.22 A análise revelou alguns genes candidatos distintos que pareceram estar associados à DA, embora nenhum tenha se mostrado comprovadamente patogênico. Até o presente, além dos APOE, nenhum outro gene determinante de DA de aparecimento tardio familiar foi identificado como tendo ligação com a doença.
É comum o cérebro de um paciente que morreu com síndrome de demência da DA apresentar atrofia grosseira dos hemisférios cerebrais, em particular na metade posterior do cérebro. Todavia, o exame microscópico do cérebro é necessário para estabelecer o diagnóstico definitivo de DA.
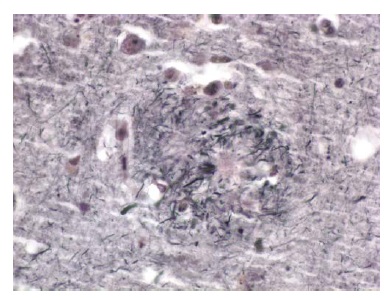
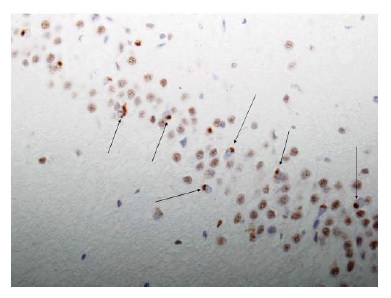
Existem 2 achados histológicos principais encontrados na DA. O primeiro é a placa amiloide. Existem várias formas de placas, sendo as 2 mais importantes a placa difusa e a placa neurítica. As placas difusas são depósitos extracelulares de peptídeo beta-amiloide que não incluem nenhum elemento neural degenerativo. As placas neuríticas são estruturas extracelulares contendo um core constituído por peptídeo beta-amiloide e circundadas por processos dendríticos e axonais degenerados [Figura 3]. Estas placas podem ser visualizadas por meio da coloração de rotina com hematoxilina e eosina, mas são mais nitidamente observadas com colorações de prata. No paciente com DA típico, as placas neuríticas são numerosas23 e estão localizadas em muitas partes do córtex cerebral. Evidências convincentes mostram que as próprias placas contendo beta-amiloide agregado insolúvel são como “lápides” inertes, enquanto algumas formas oligoméricas solúveis de beta-amiloide que surgem mais cedo são neurotóxicas.24
Figura 3. Fotomicrografia de uma placa neurítica no córtex de um paciente com doença de Alzheimer (DA).
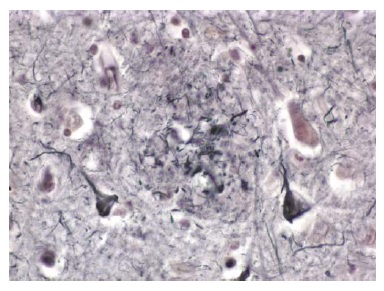
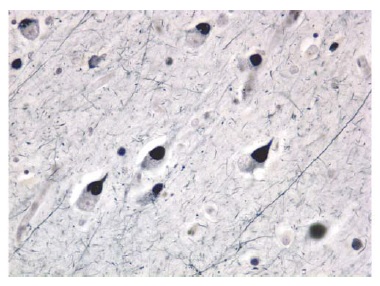
O outro aspecto histológico importante na DA são os emaranhados neurofibrilares intracelulares [Figura 4]. Os emaranhados neurofibrilares são encontrados dentro dos neurônios e são constituídos por uma proteína conhecida como proteína tau microtúbulo-associada (MAPT). A proteína tau é a molécula mais importante presente nos emaranhados neurofibrilares. Esta proteína geralmente está envolvida na estabilização de elementos do citoesqueleto dentro dos neurônios (isto é, microtúbulos). Na DA, a tau está excessivamente fosforilada e agregada.25 A tau hiperfosforilada promove rompimento dos microtúbulos. Os emaranhados neurofibrilares podem ser observados no córtex entorrinal e no hipocampo26 de idosos sem demência. Entretanto, quando os emaranhados neurofibrilares são encontrados em números maiores no neocórtex dos lobos temporal e parietal, o indivíduo quase invariavelmente tem demência de DA.
Figura 4. Fotomicrografia de vários neurônios contendo emaranhados neurofibrilares no córtex de um paciente com doença de Alzheimer (DA).
A ligação existente entre a formação do emaranhado neurofibrilar e a via do peptídeo beta-amiloide patogênico continua indefinida. Além disso, são pouco conhecidos os eventos deflagradores que levam precisamente à formação do emaranhado neurofibrilar ou à invasão da patologia neurofibrilar no neocórtex. Curiosamente, apesar da descoberta das mutações no gene MAPT que causam degeneração lobar frontotemporal (DLFT, ver adiante), nenhuma mutação no gene MAPT foi associada à DA. Sendo assim, é possível que a formação do emaranhado neurofibrilar represente uma resposta fisiológica e, por fim, desastrosa à lesão a partir dos oligômeros de peptídeo beta-amiloide ou algo mais adiante.
Segundo a hipótese do amiloide da DA, o peptídeo beta-amiloide é tóxico para os neurônios de forma dose-dependente27 [Figura 2]. Uma hipótese alternativa ou mais provavelmente complementar é a de que a patologia da tau, seja de forma independente ou facilitada pelo acúmulo excessivo de beta-amiloide no cérebro, é o processo determinante na evolução da neurodegeneração que leva à demência.26,28 A disponibilização de imagens do beta-amiloide tem se concentrado na relação com o excesso de peptídeo amiloide, bem como em sua relação com a demência. O acúmulo de peptídeo beta-amiloide no cérebro é um processo que se desenvolve de modo bastante lento e pode evoluir ao longo de mais de 20 anos antes de chegar ao nível máximo.29 Na ausência de outras patologias, é necessário (ainda que insuficiente) ultrapassar um determinado limiar de beta-amiloidose cerebral para que a demência ocorra. Os processos que possibilitam o acúmulo de peptídeo beta-amiloide mais precocemente na vida incluem mutações nos 3 genes autossômicos dominantes (APP, PSEN1 e PSEN2), bem como a trissomia do 21. As mutações no gene APP causam superprodução do peptídeo beta-amiloide, pois perturbam o equilíbrio das 3 secretases envolvidas no metabolismo de APP. Os genes PSEN1 e PSEN2 parecem exercer papel integral na ação de uma das secretases – a gama-secretase. As mutações em PSEN1 e PSEN2 resultam no aumento da atividade da gama-secretase, que, por sua vez, também acarreta produção excessiva de peptídeo beta-amiloide. O estado de portador do alelo APOE eta4 favorece a ocorrência de amiloidose cerebral em fases mais precoces da vida, embora não tão precocemente como na presença dos genes autossômicos dominantes. A quantidade de amiloidose cerebral não está correlacionada com o grau de comprometimento cognitivo. Em contraste, estudos patológicos demonstraram de modo consistente que a patologia do emaranhado neurofibrilar está correlacionada com a severidade da demência.30 A atrofia cerebral, por sua vez, apresenta correlação com o aparecimento da patologia do emaranhado neurofibrilar nas regiões cerebrais localizadas fora do lobo temporal medial.31 O grau de atrofia cerebral23,29 possui uma relação ainda mais forte com a demência do que os emaranhados neurofibrilares.
Embora seja conveniente referir-se à DA como uma entidade isolada, na verdade, a DA observada em idosos é invariavelmente acompanhada de outras alterações patológicas, entre as quais a doença cerebrovascular e a doença dos corpúsculos de Lewy (DCL; sinucleinopatia).9,32,33 Com o avanço da idade, a patologia cerebrovascular invariavelmente aumenta.33 Talvez seja por isso que a patologia da DA diminui quantitativamente diante de um determinado nível de demência na 9ª e 10ª décadas de vida.23,24 Contudo, é possível que a sinucleinopatia pare de aumentar após a 9ª década da vida.33 A ideia de que a demência clínica em indivíduos com mais de 80 anos de idade deve ser considerada representativa de múltiplas etiologias, em especial da DA e da doença cerebrovascular, está sendo cada vez mais aceita.35
Os pacientes com CCL que apresentam comprometimento da memória recente e continuam realizando as tarefas diárias normalmente representam o estágio sintomático mais inicial da DA.3 É possível demonstrar por meio de avaliações diretas que os pacientes com CCL apresentam comprometimento funcional no dia a dia. Entretanto, é provável que estes indivíduos possuam capacidades compensatórias e reservas cognitivas, e por isso sua independência não é imediatamente comprometida. Nem todos os pacientes que atendem aos critérios para CCL evoluem até desenvolverem demência. Contudo, quase 10 a 15% dos pacientes com CCL a cada ano sofrem um declínio perceptível e passam a se qualificar para o diagnóstico de demência.
A evolução do CCL para a demência leve atribuível à DA quase sempre ocorre de forma bastante gradual e insidiosa. Depois que a condição é diagnosticada, as alterações de memória e as alterações funcionais do cotidiano tipicamente passam a ser evidentes em uma análise retrospectiva de no mínimo 1 ano, se não de 2 a 3 anos. Depois que a demência é diagnosticada, é comum que seu curso clínico siga por 3 a 7 anos até que a demência severa se manifeste. Em média, o tempo decorrido desde o aparecimento dos sintomas até a morte do paciente é de aproximadamente 7 anos. Este intervalo é maior no caso dos pacientes mais jovens e um pouco menor para os pacientes que estão na faixa etária de 80 e 90 anos.
Um dos aspectos mais frustrantes e desafiadores da evolução da síndrome clínica da DA é a intensificação gradativa da perda de autoconsciência e do discernimento. Isto é referido como anosognosia, ou a perda da consciência dos próprios déficits. A demência, cujo exemplo mais comum é a DA, é quase a única dentre as condições médicas em que o próprio paciente geralmente não colabora para levar seu problema ao conhecimento do médico. Às vezes, os indivíduos afetados articulam preocupações acerca da própria memória, mas na maioria dos casos os familiares do paciente forçam a questão.
A amnésia anterógrada da DA tipicamente se manifesta nos afazeres diários do paciente, como uma dificuldade em lembrar de eventos e conversas recentes, uma tendência a repetir o que já disse durante uma conversa, e uma dificuldade em se manter atualizado com a data. A princípio, estes comportamentos podem ser intermitentes ou surgir apenas nos momentos de estresse ou quando o indivíduo é tirado de suas rotinas habituais. Gradualmente, à medida que a condição progride, os episódios de esquecimento vão se tornando mais frequentes e levam a erros da realização dos afazeres diários. Estes erros podem incluir o esquecimento de compromissos, dificuldades para fazer compras e problemas em seguir instruções com múltiplas etapas (p. ex., receitas culinárias) ou para seguir direções durante uma viagem.
Passada a fase de evolução inicial da demência, outros déficits cognitivos emergem e vão sendo introduzidos nos afazeres diários do paciente. Estes déficits podem incluir a perda da facilidade para dizer palavras simples ou uma dificuldade para lembrar os nomes de familiares ou conhecidos. Outros desenvolvimentos comuns incluem a capacidade diminuída de resolver problemas diários ou domésticos comuns; diminuição da capacidade de administrar as finanças; ou maior dificuldade para planejar uma refeição, uma reunião familiar ou uma viagem. As dificuldades de planejamento, agilidade mental, solução de problemas e pensamento abstrato por vezes são referidas como disfunção executiva. A desorientação geográfica também é uma característica da demência emergente da DA e manifesta-se com o paciente ficando perdido em um local que costumava ser familiar. Em uma história típica, o paciente sai de carro para visitar um amigo e começa a ficar confuso em relação às direções. Em alguns casos, o paciente dirige seu carro por várias horas tentando chegar ao destino pretendido. Em outras situações, o paciente simplesmente desiste da viagem e tenta voltar para casa.
Conforme a demência se manifesta, alterações de comportamento e humor começam a ocorrer com frequência, ainda que nem sempre. A perda de interesse por coisas que o paciente gostava e pelos afazeres da família são manifestações da apatia e da perda de iniciativa comumente observadas na DA inicial. De forma menos comum, o indivíduo sofre alterações de humor significativas, como choro, melancolia ou ansiedade. Notavelmente, a depressão é menos comum do que poderia ser previsto, talvez por causa da perda concomitante de discernimento que acompanha a demência da DA inicial.
Costuma-se definir a demência severa como o ponto em que a incapacidade de administrar as atividades básicas do dia a dia (p. ex., tomar banho, vestir-se, ir ao banheiro ou comer) se torna evidente. A progressão dos sintomas de DA varia tremendamente de um indivíduo para outro.
Obter uma história relatada por alguém que esteja diariamente em estreito contato com o paciente constitui uma parte essencial da avaliação dos pacientes com suspeita de CCL e demência atribuível à DA. Uma história típica deve estabelecer a duração dos sintomas e revisar os diversos aspectos do funcionamento diário em que pode haver comprometimento. Uma lista de checagem padronizada é útil como ponto de partida, contudo outros aspectos podem ser levantados por aquele que fornece as informações. Uma avaliação funcional, seja individualizada ou padronizada, é parte fundamental da avaliação longitudinal dos pacientes com DA e outras demências.
As avaliações do funcionamento mental constituem outra base do diagnóstico do CCL e da demência. Podem ser realizadas por um médico da assistência primária no contexto de uma consulta de rotina ou por um neuropsicólogo em um laboratório especializado. Ambos os métodos são concordantes e baseiam-se nos mesmos princípios de avaliação das capacidades cognitivas. A abordagem neuropsicológica é mais demorada e, consequentemente, fornece uma visão mais detalhada da função cognitiva do paciente.
O exame da condição mental é um dos pilares do diagnóstico da DA. O Mini-Mental State Examination (MMSE, mini-exame do estado mental) é amplamente usado e tem normas para idade e nível de instrução. Consiste na aplicação de perguntas que avaliam o nível de orientação e inclui avaliações de linguagem, função visual-motora, agilidade mental e lembrança tardia. Embora o MMSE careça de sensibilidade para a detecção de graus leves de disfunção, é bastante efetivo como ferramenta para o clínico. Na Mayo Clinic, usa-se o Short Test of Mental Status (teste rápido do estado mental), porque este teste foi criado para ser mais sensível à demência em estágio inicial.37 O Montreal Cognitive Assessment representa a tentativa mais recente de arquitetar um exame à beira de leito mais sensível e específico para o CCL.38 Ao usarem um destes testes, os clínicos podem obter uma visão objetiva da função cognitiva do paciente. De forma típica, os pacientes com DA em estágio inicial cometem 1 ou 2 erros de orientação temporal ou espacial e não conseguem se lembrar de nada que lhes tenha sido especificamente apresentado para ser lembrado após alguns minutos. Entretanto, estes indivíduos não cometem erros de leitura, escrita, nomeação de itens simples ou seguir instruções de múltiplas etapas. Os exames de estado mental também integram o seguimento longitudinal dos pacientes com DA e outras demências. Conforme a demência da DA evolui, o desempenho no exame do estado mental declina de maneira importante. No MMSE, os pacientes com DA perdem em média 3 pontos ao ano. Entretanto, a faixa de desempenho de um ano para outro de um determinado paciente em particular pode variar bastante, desde a ausência de perda de pontos até uma perda de 6 ou 7 pontos.
O diagnóstico de DA ainda é clínico, sendo estabelecido com base na história, exame do estado mental, demais exames neurológicos e exame físico geral.39 Nenhum marcador laboratorial de DA é suficientemente acurado para substituir ou suplantar o julgamento de um médico experiente. O médico pondera a evidência fornecida pela história de declínio cognitivo e julga a história com base no exame do estado mental. Para estabelecer o diagnóstico de demência de DA, a história de declínio cognitivo e os achados do exame do estado mental devem ser concordantes. Quando a história é fortemente sugestiva de um distúrbio cognitivo, porém o resultado do exame do estado mental é normal, ainda existe a possibilidade de se tratar de um caso de DA em estágio bem inicial, sobretudo quando o paciente tem alto nível de instrução. Quando o exame do estado mental resulta significativamente anormal, porém os familiares relatam que o paciente não tem dificuldades de funcionamento no dia a dia, a hipótese de DA também deve ser considerada, embora talvez seja mais provável que se trate de um caso de delírio ou de distúrbio de progressão rápida. Os familiares às vezes subestimam o grau de comprometimento apresentado pelo ente querido, seja devido à falta de observação, negação ou falta de conhecimento.
Os exames laboratoriais têm o papel de avaliar os pacientes com demência mesmo quando a hipótese de DA parece ser mais provável do ponto de vista clínico.39 Os exames de sangue, como o hemograma completo, eletrólitos, cálcio sérico, ureia sérica, níveis de tireotropina e níveis de vitamina B12, são necessários para excluir a hipótese de desarranjos hematológicos ou metabólicos prévios negligenciados. Embora ainda seja bastante rara em idosos, a possibilidade de infecção pelo HIV também deve ser considerada. A avaliação para detecção de neurossífilis pode ser considerada nas regiões onde a sífilis primária ou secundária são observadas com alguma frequência. Embora nenhum destes testes esteja relacionado a uma demência comumente encontrada, são exames simples e econômicos que ajudam a excluir hipóteses de condições médicas gerais comuns. O exame do LCS, a urinálise para detecção de toxinas e os eletroencefalogramas somente são necessários quando há indicações específicas.
A avaliação do conteúdo proteico do LCS para estabelecer o diagnóstico específico de DA tem sido incentivada, mas ainda não chegou ao estágio de utilidade clínica definida para fins diagnósticos. Os exames para detecção do peptídeo beta-amiloide e da proteína tau apresentam sensibilidade e especificidade modestas na diferenciação entre DA e outras condições.49 Todavia, na maioria dos casos em que há um nível razoável de suspeita clínica de DA, estes exames acrescentam pouca informação àquelas já fornecidas pela avaliação clínica. Em contraste, em casos de pacientes com sintomas mínimos, incluindo os indivíduos com CCL, a diminuição dos níveis de peptídeo beta-amiloide ou elevação dos níveis de proteína tau no LCS são substancialmente preditivas na identificação de indivíduos destinados a sofrerem declínio associado à demência dentro de alguns anos.41,42
A genotipagem dos pacientes com demência para fins diagnósticos atualmente não é recomendada. O valor aditivo da genotipagem de APOE em relação à história e aos exames é bastante modesto. A avaliação em busca de mutações nos genes APP, PSEN1 e PSEN2 fornece um rendimento mínimo, a não ser nos casos de transmissão autossômica dominante da DA de manifestação precoce.
Os exames de imagem cerebral são necessários para a avaliação diagnóstica inicial dos pacientes com demência. Uma avaliação do cérebro por tomografia computadorizada (TC) pode ser adequada, mas a IRM sem intensificação por contraste fornece informações de maior utilidade clínica. A principal finalidade do exame de imagem cerebral é excluir a possível existência de lesões expansivas, como tumores e hematomas subdurais. A IRM também está se tornando cada vez mais útil para avaliar a ocorrência de infartos cerebrais. Os idosos apresentam risco aumentado de sofrerem pequenos infartos em estruturas cerebrais profundas, que podem ocorrer de forma oculta.43 As imagens do cérebro são desnecessárias no tratamento prolongado de pacientes com DA, a menos que ocorra alguma alteração na condição do paciente que não tenha sido previamente antecipada como parte da progressão da DA.
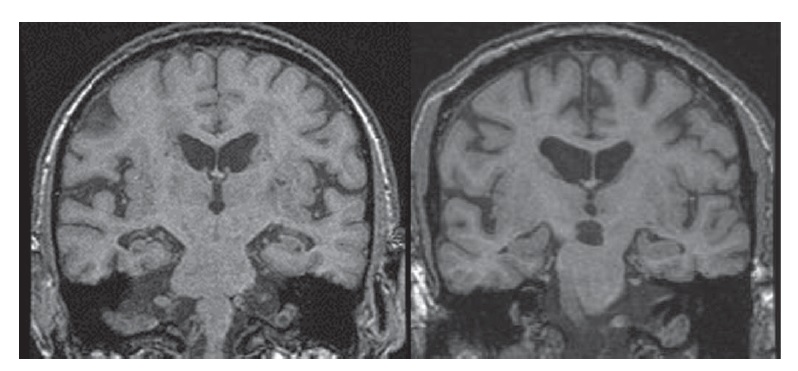
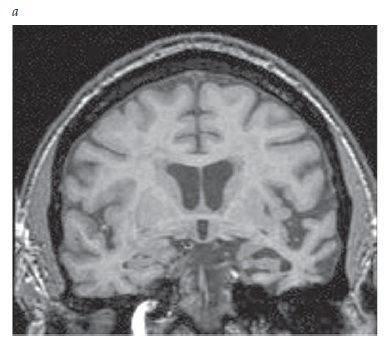
A IRM também pode detectar a atrofia hipocampal com a obtenção de imagens coronais [Figura 5]. A atrofia hipocampal é observada com frequência na DA, porém este achado é mais sugestivo do que diagnóstico de DA, por também ser encontrado em pacientes idosos normais e em indivíduos com outras demências.
Figura 5. Imagem de ressonância magnética (IRM) coronal mostrando a atrofia hipocampal de um paciente clinicamente diagnosticado com doença de Alzheimer (DA) (esquerda), comparada à varredura de um indivíduo normal de idade equivalente (direita).
A quantificação do uso de glicose cerebral com 18fluorodesoxiglicose e tomografia por emissão de pósitrons (FDG-PET) permite distinguir um padrão de hipometabolismo cerebral que é específico para DA.44 A FDG-PET é mais útil para distinguir entre DA e DLFT,45 mas não serve para determinar se um indivíduo tem comprometimento cognitivo nem para distinguir entre DA e demência vascular (DV) ou DCL. Devido ao valor limitado da FDG-PET em relação ao diagnóstico clínico, esta não deve ser solicitada como teste de rotina na avaliação de pacientes com suspeita de DA.39 O desenvolvimento de compostos que permitem a visualização do amiloide cerebral tem sido estimulante, contudo estes agentes, dentre os quais o composto B de Pittsburg de 11carbono (PIB)46 é o mais intensivamente estudado, ainda estão indisponíveis para uso na prática clínica. Os trabalhos iniciais indicam que a PIB-PET é uma técnica bastante sensível para a detecção de DA,47 embora sua especificidade ainda esteja sendo avaliada. A varredura por PIB-PET revela a ocorrência de amiloidose em mais de 20% dos pacientes idosos cognitivamente normais,48 e isto significa que seu uso deve ser cuidadosamente ajustado de acordo com a pergunta clínica.
Ao considerar o diagnóstico de demência da DA, o clínico deve primeiro determinar se o perfil clínico do paciente se ajusta ao de demência – ou seja, se o paciente apresentava um nível alto de funcionamento diário que declinou e agora está comprometido. Para os propósitos desta seção, também se estipula que o paciente apresentava uma função cognitiva intacta em algum momento anterior. Isto elimina a possibilidade de distúrbios do desenvolvimento, como um retardo mental. A existência de níveis amplamente normais de alerta e atenção exclui a hipótese de delírio. Na demência da DA, o aparecimento e a progressão da condição são obrigatoriamente graduais. Em geral, o intervalo temporal de manifestação dos sintomas é de pelo menos 6 meses a 1 ano ou mais. O aparecimento mais rápido da demência (ou seja, ao longo de semanas ou meses) implica a possibilidade de doença de Creutzfeldt-Jakob [ver 11:XVII Doenças do sistema nervoso central causadas por vírus lentos e príons].
As convulsões e cefaleias não fazem parte da manifestação inicial da demência da DA. Um paciente que apresenta demência, convulsões e cefaleia deve ser avaliado quanto à existência de lesões expansivas no cérebro, como um tumor cerebral ou hematoma subdural, ou uma infecção no sistema nervoso central. Os tumores cerebrais, hematomas subdurais e infecções cerebrais tipicamente também progridem ao longo de vários dias a semanas.
Quando os acidentes vasculares cerebrais (AVC) clínicos precedem a demência, e em particular se um AVC ocorreu dentro de 3 meses antes do aparecimento da demência, a hipótese de DV deve ser seriamente considerada. A possibilidade de DV também surge quando os exames de imagem revelam a ocorrência de infartos na substância cinzenta bilateralmente, mesmo que estes infartos não tenham sido associados a um AVC clínico evidente. Nos pacientes que não apresentam nenhum destes aspectos, é improvável que exista um componente cerebrovascular [ver Demência vascular (DV), adiante].
A presença de parkinsonismo ou outros distúrbios proeminentes da função motora aponta no sentido de outros diagnósticos, diferentes da DA. A hipótese de DCL deve ser considerada diante de um paciente com doença de Parkinson que desenvolve demência ou no caso de um indivíduo que já tenha demência e desenvolva uma postura curvada para a frente, comprometimento da marcha e do equilíbrio, além de outros sinais de parkinsonismo. Os aspectos adicionais sugestivos de DCL e não de DA são as alucinações visuais gráficas proeminentes, flutuações da vigília no dia a dia, e um peculiar distúrbio do sono [ver Demência com corpúsculos de Lewy (DCL), adiante].
Os déficits cognitivos necessários ao diagnóstico de DA incluem o comprometimento da memória recente e a perda de pelo menos um dos outros domínios cognitivos. No entanto, quando os distúrbios da fala ou da linguagem predominam em relação aos déficits de memória recente, deve ser considerado o diagnóstico de afasia progressiva ou de um distúrbio conhecido como demência semântica. O diagnóstico de demência frontotemporal é sugerido pela ocorrência de alterações do comportamento ou conduta ou personalidade que obscurecem as dificuldades de memória.
Em indivíduos com mais de 60 anos de idade, a DA representa 50 a 80% de todas as demências.9,32 Desta forma, quando os pacientes pertencem a esta faixa etária, é razoável suspeitar de DA, a menos que algum aspecto da história ou do exame apontem fortemente no sentido de outro diagnóstico específico. A coexistência de características cerebrovasculares e características da DCL implica, em alguns casos, a necessidade de considerar outras etiologias adicionais.
Em contraste com a dominância da DA entre os idosos, o diagnóstico diferencial de demência para pacientes com menos de 60 anos de idade é bastante diferente. Os pacientes mais jovens são mais propensos a apresentar distúrbios em que as vias metabólicas estão alteradas. Algumas destas doenças são as doenças mitocondriais, doença de Wilson e leucodistrofia metacromática. Doenças como a esclerose múltipla devem ser consideradas. Além disso, a sobreposição entre as doenças psiquiátricas e a demência pode ser um desafio diagnóstico proeminente em paciente mais jovens, assim como a sobreposição entre abuso de substâncias e comprometimento cognitivo progressivo.
A terapia da DA pode ser primária ou secundária. A meta das terapias primárias é retardar a manifestação dos sintomas centrais. As terapias atualmente disponíveis melhoram os sintomas, mas não modificam a doença, ou seja, não combatem os mecanismos básicos da DA. As terapias secundárias são destinadas a melhorar problemas como depressão, ansiedade, distúrbios do sono e agitação.
Os inibidores de colinesterase donepezil, galantamina e rivastigmina foram aprovados pelo Food and Drug Administration (FDA) para uso no tratamento da DA. Para cada um destes agentes, os estudos clínicos demonstraram que o uso prolongado resulta em uma estabilização modesta da condição cognitiva e funcional por cerca de 6 a 12 meses, em comparação à ausência de tratamento.49 Em um estudo particularmente iluminador, os pacientes com DA leve a moderada foram randomizados para receber donepezil ou placebo por até 1 ano. O ponto de término da terapia foi a perda de um grau predefinido de funcionamento diário. Uma perda significativa da capacidade funcional diária foi observada após 12 meses em 51% dos pacientes tratados com placebo e em apenas 38% dos pacientes tratados com donepezil.50
A experiência de um estudo clínico com inibidores de colinesterase é limitada à DA de grau leve a moderadamente severo. Estudos envolvendo pacientes com CCL ou DA em estágio bem inicial estão em andamento e poderão esclarecer se os fármacos desta classe são úteis nos estágios mais iniciais da síndrome clínica.
Os efeitos colaterais produzidos pelos inibidores de colinesterase são primariamente gastrintestinais e consistem em náusea, perda do apetite, diarreia e, menos comumente, vômitos. Estes efeitos colaterais possuem uma relação forte e estreita com a dose e muitas vezes se manifestam quando o curso de fármacos é iniciado ou a dose é aumentada.
Os pacientes com DA leve a moderada são candidatos adequados à terapia com inibidor de colinesterase. Estes pacientes precisam de um cuidador para supervisionar o uso da medicação. O tratamento com inibidores de colinesterase deve ser mantido até o paciente atingir o estágio de demência severa, em que um declínio adicional é esperado.
A vitamina E antigamente era recomendada para pacientes com DA. Entretanto, após um estudo sobre os efeitos da vitamina E em pacientes com CCL terem falhado em mostrar benefícios,51 o entusiasmo por seu uso como tratamento para DA enfraqueceu. De modo similar, o interesse pela ginkgo biloba diminuiu, à medida que estudos clínicos de pacientes com DA sintomática e um estudo sobre prevenção envolvendo um amplo grupo de idosos normais falharam em mostrar benefícios.52 Além disso, surgiram preocupações com relação à segurança do uso de vitamina E em doses altas.53
A memantina também foi aprovada pelo FDA para uso no tratamento da DA moderada a severa. Este agente modulador do glutamato, um antagonista não competitivo do receptor de N-metil-D-aspartato, foi submetido a diversos estudos clínicos que relataram resultados positivos na demência moderada a severa.54 Hipotetizou-se que a superestimulação glutamatérgica faz parte de um ciclo patogênico na DA. Em estudos clínicos, a memantina retardou a progressão dos sintomas. Entretanto, não há evidências de que a memantina afete o curso biológico da DA. Os estudos sobre o uso da monoterapia no tratamento de pacientes com DA leve a moderada demonstraram os benefícios promovidos pela memantina.55 Contudo, como a terapia combinada com inibidor de colinesterase e memantina não apresentou benefícios adicionais comprovados em relação ao uso isolado dos inibidores de colinesterase em casos de DA leve a moderada,56 a memantina atualmente não tem aprovação para ser usada no tratamento da DA leve.
O campo da terapêutica na DA está evoluindo rápido. Várias abordagens para diminuir a carga de peptídeo beta-amiloide estão sendo avaliadas, incluindo os inibidores de gama- secretases e beta-secretases e as estratégias imunológicas. Além disso, também estão sendo conduzidos vários esforços antecipados no sentido de atacar o sistema tubulina-tau.
O tratamento da depressão ou da ansiedade para pacientes com DA deve ser tão agressivo quanto para pacientes sem DA, aderindo às melhores práticas da farmacologia geriátrica. A depressão contribui para a morbidade e perda da função em pacientes com DA. Os agentes mais modernos, como citaloprama, paroxetina, sertralina e mirtazapina, geralmente são bem tolerados por pacientes com demência. A dosagem pode começar em um nível mais baixo do que aquela possivelmente usada no tratamento de um adulto jovem; os aumentos das doses também devem ser mais espaçados.
O tratamento da ansiedade em pacientes com DA é mais desafiador, pois os agentes comumente usados no tratamento de pacientes mais jovens – os benzodiazepínicos – produzem efeitos colaterais distintos indesejáveis em indivíduos com DA. Fármacos como lorazepam ou alprazolam podem aumentar a confusão em pacientes com DA. O clonazepam, um agente de ação prolongada, pode ser uma escolha mais adequada. A buspirona é outra alternativa para o tratamento da ansiedade em pacientes com DA.
O tratamento da agitação – que pode se manifestar como agressividade física, agitação verbal perturbadora ou alucinações provocadoras de medo e ansiedade – geralmente requer antipsicóticos, mas há bastante controvérsia quanto ao uso destes agentes.57 A quetiapina proporciona a vantagem significativa de ser bem menos propensa a induzir sinais extrapiramidais, em comparação tanto aos agentes modernos como aos agentes antigos. Um dos desafios impostos pelo uso de qualquer antipsicótico no tratamento de idosos com DA reside no fato de ser necessário usar uma dose baixa no início do tratamento. Vários incrementos de dose podem ser necessários para que a dose terapêutica efetiva seja alcançada. Considerando a natureza por vezes catastrófica da agressividade física ou das graves alucinações apresentadas pelos pacientes com DA, os cuidadores podem se sentir fácil e justificadamente frustrados se o controle dos sintomas for retardado. Contudo, uma abordagem exageradamente agressiva para o uso de antipsicóticos pode acarretar sedação excessiva e consequências adversas sérias. As evidências de mortalidade excessiva entre usuários idosos de fármacos antipsicóticos também forçou os clínicos a se tornarem mais seletivos quanto ao fármacos empregados no tratamento de pacientes idosos, frágeis e agitados com demência.58
O suporte e autorização para cuidadores de pacientes com DA deve ser parte integral do tratamento. A saúde emocional e física dos cuidadores é fundamental para os resultados alcançados a longo prazo. O suporte pode assumir a forma de instrução individual e incentivo à participação em grupos de apoio, envolvimento na Alzheimer’s Association (www.alz.org/), bem como ao uso de cuidados diários e tratamento com pausas. Para os médicos da assistência primária, saber onde encontrar conhecimentos especializados sobre demência e como contatar a Alzheimer’s Association pode ser tão importante quanto estabelecer o diagnóstico correto e iniciar a terapia. A autorização fornecida aos cuidadores lhes permite planejar suas próprias vidas com um maior senso de controle e confiança.
O prognóstico para DA e para todas as doenças cerebrovasculares e degenerativas discutidas neste capítulo é terrível. Contudo, viver por tempo prolongado com demência em estágio que requer cuidado total e a morte precoce são as perspectivas que o paciente com DA tem de encarar. Embora o ritmo da progressão seja bastante variável, a maioria dos pacientes eventualmente sofre um declínio que os torna consideravelmente necessitados de cuidados, os quais, muitas vezes, somente podem ser conseguidos em uma instituição prestadora de terapia prolongada. Mesmo quando são admitidos em unidades de tratamento especializado, os pacientes com DA podem sobreviver por muitos anos. Enfim, a sobrevida dos pacientes diagnosticados com DA é consideravelmente menor, em comparação à dos pacientes da mesma faixa etária que não têm demência, exceto na 10ª década da vida, quando esta diferença é atenuada.
A DV, também conhecida como comprometimento cognitivo vascular, representa o comprometimento cognitivo resultante da doença cerebrovascular. Enfatizando um ponto destacado anteriormente, na seção sobre DA deste capítulo, a doença cerebrovascular no idoso raramente existe de forma isolada em relação à DA coexistente. As definições de DV atualmente vigentes são distorcidas no sentido de considerar a condição como sendo uma entidade circunscrita. Este talvez seja um dos motivos pelos quais jamais se chegou a um consenso sobre a definição de DV. Atualmente, vários conjuntos de critérios diagnósticos estão em uso. Os critérios estabelecidos pelo 1993 National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseigne ment en Neurosciences (NINDS-AIREN) para DV provável são mais amplamente usados e têm sido adotados para estudos clínicos. A essência dos critérios NINDS-AIREN são as seguintes: (1) aparecimento ou piora da demência ocorridos dentro de 3 meses após um episódio de AVC clínico; (2) exames de imagem mostrando evidências de infartos bilaterais nas regiões corticais, gânglios basais, tálamo ou substância branca; e (3) exame neurológico mostrando déficits neurológicos focais. Embora todos os 3 critérios sejam requeridos, o 3º é provavelmente redundante, se os outros critérios forem atendidos. A relação temporal entre o episódio de um AVC e a demência, bem como a presença de infartos demonstrados pelos exames de imagem, são aspectos clínicos essenciais para a DV. Os estudos de correlação clinicopatológica demonstraram que esta definição é bastante específica, e isto implica que os pacientes que atendem a estes critérios são altamente propensos a ter DV, do ponto de vista patológico. Entretanto, os critérios NINDS-AIREN são bastante insensíveis e falham em diagnosticar a maioria dos pacientes com DV comprovada por autópsia.59 Em muitos pacientes com DV, uma característica essencial está presente e falta a outra.
Os déficits cognitivos presentes na DV não seguem um padrão em particular.60 Sendo assim, a DV não possui um perfil de disfunção cognitiva que seja útil para fins diagnósticos. Os pacientes com DV tendem a apresentar mais déficits de função executiva, pois os infartos nos núcleos caudado, tálamos ou substância branca dos lobos frontoparietais tendem a romper os circuitos envolvidos nas funções executivas. Entretanto, alguns pacientes com DV apresentam amnésia anterógrada proeminente, semelhante ao observado na DA, sem um componente executivo proeminente. De modo significativo, o comprometimento cognitivo que ocorre com a doença cerebrovascular pode variar de mínimo, evidenciado somente por exames neuropsicológicos e com alterações mínimas no funcionamento diário, a severo e incapacitante.
A história natural de DV é variável. Alguns pacientes apresentam platôs prolongados quando param de sofrer AVC, enquanto outros inexoravelmente sofrem declínio. Os pacientes com DV cujo distúrbio atende aos critérios NINDS-AIREN apresentam uma sobrevida consideravelmente pior, em comparação aos pacientes com DA.61
A prevalência e incidência da DV correspondem a cerca de 1/5 a 1/10 da prevalência62 e incidência63 da DA. Assim como a DA e a doença cerebrovascular em geral, a DV torna-se mais comum com o avanço da idade. Os fatores de risco de DV são idênticos àqueles da doença cerebrovascular e incluem hipertensão, diabetes e doença cardiovascular.
Não há predileção genética, no sentido tradicional, para as formas típicas de DV. Entretanto, existem formas hereditárias bastante raras de demência por vasculopatia, como a síndrome CADASIL (arteriopatia cerebral autossômica dominante com infartos subcorticais e leucoencefalopatia).64
O diagnóstico de comprometimento cognitivo de origem cerebrovascular baseia-se na história de AVC e em sua relação com o aparecimento e progressão do comprometimento cognitivo, exame neurológico e neuroimagem. Os aspectos clínicos decisivos incluem uma história de AVC, sinais neurológicos focais ao exame, carga de hiperintensidades de substância branca, número de lacunas, número de infartos corticais e presença de infartos em regiões críticas.
A demência da DV frequentemente se torna evidente durante o período de recuperação subsequente a um AVC. Como os déficits motor e sensorial produzidos pelo AVC costumam ser esmagadores, o comprometimento cognitivo a princípio é frequentemente obscurecido. Este tipo de DV é capturado pelos critérios clínicos atualmente disponíveis. A DV também pode começar de maneira insidiosa, porque uma fração substancial desta condição parece resultar do acúmulo de uma série de infartos conhecidos como silenciosos ou ocultos.65 Esta última forma de doença cerebrovascular microvascular não pode ser diagnosticada com as metodologias clínicas disponíveis e, em vez disso, é deduzida a partir dos exames de neuroimagem.
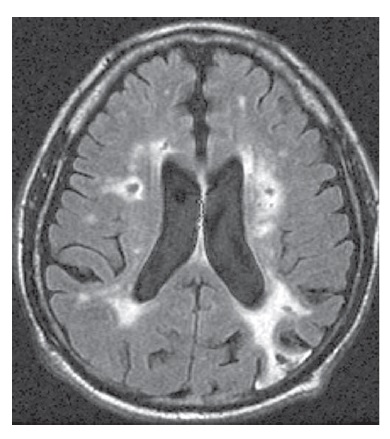
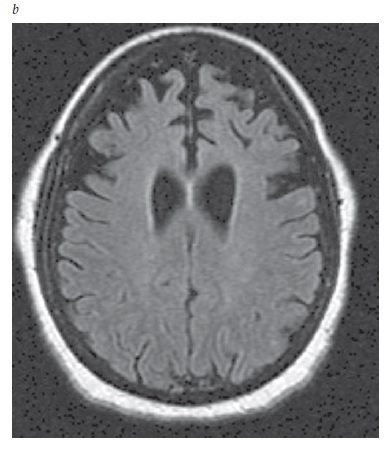
A neuroimagem é essencial ao reconhecimento da doença cerebrovascular que poderia sustentar um diagnóstico de DV. A IRM é preferível à TC, por sua capacidade superior de detectar infartos lacunares. De forma típica, um paciente com DV apresenta infartos difusos [Figura 6]. Quando se trata de DV, o volume do infarto pode não ser tão decisivo quanto sua localização. A bilateralidade da lesão isquêmica também é relevante, embora os infartos posicionados unilateralmente em locais como o núcleo caudado, tálamo, formação hipocampal ou lobo parietal possam produzir sintomas cognitivos estreitamente semelhantes àqueles observados na demência. As hiperintensidades da substância branca, por si só, não são suficientes como evidência para atribuir o comprometimento cognitivo à doença cerebrovascular. As hiperintensidades de substância branca são um provável marcador de doença microvascular e estão associadas (ainda que de forma não tão invariável) ao comprometimento cognitivo66 e ao declínio cognitivo.67
Figura 6. IRM de um paciente com demência vascular (DV), mostrando múltiplos infartos: 1 infarto cortical parieto-occipital e 2 infartos na substância branca.
Existem 2 aspectos da fisiopatologia da DV que contribuem para sua complexidade. O 1º aspecto é o fato de a DV raramente se manifestar como uma forma pura, ou seja, em geral há certo grau de patologia de DA concomitante. Pode ser bastante difícil entender as contribuições de ambas as etiologias.68 O 2º aspecto reside na suspeita (ainda não comprovada de maneira conclusiva) de que a doença microvascular em forma de microinfartos e desmielinização da substância branca de origem provavelmente isquêmica contribua para o comprometimento cognitivo.69,70
A terapia da DV deve ser voltada principalmente para a prevenção, por meio do tratamento da hipertensão, do diabetes melito, abandono do tabagismo e tratamento de doenças comprovadamente associadas a riscos de embolia cerebral. Vários estudos demonstraram que o tratamento da hipertensão diminui a incidência de demência (provavelmente, da demência com um componente vascular).11
A DCL inclui a síndrome em que o distúrbio cognitivo precede ou ocorre simultaneamente ao distúrbio do movimento e a síndrome em que o parkinsonismo antecede a demência em mais de 1 ano.71 A primeira síndrome – DCL – é diagnosticada em pacientes que apresentam espontaneamente (isto é, não fármaco-induzido) parkinsonismo; demência; alucinações visuais bem formadas, recorrentes e proeminentes; e flutuações cognitivas com variação acentuada dos níveis de alerta e atenção [Tabela 2]. A demência da doença de Parkinson (DDP) é diagnosticada em indivíduos que já sofriam de parkinsonismo há mais de 1 ano quando o comprometimento cognitivo surgiu. Quando a demência está presente, ambos os distúrbios são quase idênticos do ponto de vista clínico.72
Tabela 2. Critérios diagnósticos para DCL71
|
A. Demência |
|
B. O distúrbio cognitivo surge de maneira insidiosa e evolui progressivamente, com base nas evidências fornecidas pela história e pelos exames seriados cognitivos |
|
C. Presença de pelo menos 2 dos seguintes achados: |
|
1. Parkinsonismo (rigidez, tremores em repouso, bradicinesia, instabilidade postural, distúrbio da marcha parkinsoniano) |
|
2. Alucinações visuais integralmente formadas e proeminentes |
|
3. Flutuações significativas da vigília ou da cognição |
|
4. Distúrbio do comportamento do sono com movimentos oculares rápidos (REM) [ver 11:XIII Distúrbios do sono] |
|
D. O distúrbio não é mais bem explicado por uma doença sistêmica nem por outra doença cerebral. |
DCL = demência com corpúsculos de Lewy.
Depois da DA e da doença cerebrovascular, a DCL é a próxima entidade patológica mais importante a causar demência nas fases tardias da vida.9 Os estudos sobre a prevalência e incidência da DCL em geral encontraram taxas menores do que aquelas encontradas nas séries patológicas,73 refletindo a atual insensibilidade do diagnóstico clínico deste distúrbio. A demografia da DCL e da DA são bastante parecidas.
A patologia da DCL inclui a típica DCL – frequentemente, dos corpúsculos de Lewy e das neurites de Lewy – presentes no tronco encefálico observada na doença de Parkinson [ver 11:XV Doença de Parkinson e outros distúrbios do movimento] e DCL presentes no sistema límbico e no neocórtex. O principal constituinte proteico dos corpúsculos de Lewy é a alfa-sinucleína.74 Assim como o peptídeo beta-amiloide na DA, a alfa-sinucleína está agregada dentro das fibrilas que formam os corpúsculos de Lewy e as neurites de Lewy. Entretanto, os mecanismos intermediários pelos quais a alfa-sinucleína exerce efeito neurotóxico não estão totalmente esclarecidos.
A DCL não está associada a uma condição genética mendeliana. A DDP foi associada à ocorrência de mutações no gene da alfa-sinucleína localizado no cromossomo 4, bem como à duplicação deste gene.74
O espectro da patologia cortical na DCL varia de números modestos de corpúsculos de Lewy corticais com patologia de DA proeminente a uma profusão de corpúsculos de Lewy com patologia de DA mínima. Os critérios estabelecidos pelo 2005 Consortium for DLB têm excelentes valor preditivo positivo e especificidade para a DCL. Em uma série, a sensibilidade para DCL foi de 87%.75 Nesta mesma série de pacientes com DCL provável clinicamente diagnosticada, a vasta maioria (95%) apresentou uma carga intermediária ou alta de DCL nas regiões límbica e cortical. Ao mesmo tempo, entre estes pacientes clinicamente diagnosticados com DCL, 29% apresentava uma alta carga de patologia de DA.75 Como a DCL é comum em indivíduos com DA concomitante,33 pode não haver uma resposta simples para a sensibilidade dos critérios clínicos da DCL.
O distúrbio cognitivo da DCL pode ser bastante parecido com a DA, todavia existem algumas diferenças notáveis em muitos pacientes.76 Tais diferenças incluem um déficit de aprendizado e memória discretamente menos proeminente e dificuldades bem mais significativas relacionadas às funções visual-espaciais, desempenho com tarefas cronometradas e funções executivas. Contudo, o perfil neuropsicológico não é diagnóstico.
Na DCL, o parkinsonismo pode variar de uma forma relativamente isolada de instabilidade de marcha, com quedas frequentes, a um padrão típico de doença de Parkinson com tremores em repouso, rigidez, bradicinesia e instabilidade postural.
Os pacientes com DCL muitas vezes apresentam flutuações marcantes da vigília e do nível de alerta, bem como do nível de alerta de um dia para outro.77 Estes pacientes costumam piscar excessivamente. O distúrbio de comportamento do sono com movimentos oculares rápidos (DCR) muitas vezes pode preceder em vários anos a demência e o distúrbio do movimento, além de ser altamente específico para DCL.78 Na DCR, os pacientes engajam-se em interpretar sonhos, debatendo-se no leito ou conversando durante o sono. Outro aspecto da DCL que pode estar relacionado ao distúrbio do alerta são as alucinações diurnas frequentes que podem ser bastante vívidas e detalhadas. Foi postulado que as alucinações bem formadas e detalhadas da DCL representam uma atividade do sono que invade indevidamente a vigília.
O diagnóstico de DCL é clínico, sendo estabelecido com base na história e no exame neurológico. O diagnóstico diferencial de uma demência com parkinsonismo em um idoso também inclui distúrbios do movimento menos comuns, como a degeneração corticobasal, paralisia supranuclear progressiva e atrofia multissistêmica. Uma combinação de DA e patologia cerebrovascular também pode mimetizar a DCL, ainda que em raros casos. Em pacientes mais jovens, devem ser consideradas as hipóteses de doença de Wilson e doença de Huntington.
A DCL não possui marcadores sanguíneos ou plasmáticos comprovados.71 As características de imagem estruturais não permitem distinguir entre DCL e DA. A visualização do amiloide em pacientes com DCL também mostra uma proporção bastante alta de casos com padrão do tipo DA.79 As imagens do transportador de dopamina obtidas por TEP revelam uma função dopaminérgica reduzida no estriado e podem ajudar a distinguir entre DCL e DA.71
O tratamento da DCL pode ser complexo, pois envolve a interface de domínios de sintomas múltiplos – demência, distúrbio do movimento, distúrbio do sono e perturbações comportamentais – sendo que as medicações efetivas para um domínio podem exercer impacto sobre outro domínio. Os inibidores de colinesterase parecem ser benéficos na DCL e aparentemente não pioram o parkinsonismo nem o comportamento.80 A levodopa pode ser essencial para o tratamento dos problemas de marcha e equilíbrio, mas também pode intensificar as alucinações ou a confusão. Os agentes antipsicóticos, como a quetiapina ou a clozapina, não exacerbam o parkinsonismo nem a confusão, mas devem ser usados com cautela porque estão associados a uma sobrevida menor.
As DLFT constituem um subgrupo de demências menos comuns. Nas DLFT, os distúrbios cognitivos manifestam-se como um distúrbio dominado por alterações comportamentais e de relacionamento interpessoal (demência frontotemporal de variante comportamental [DFTvc]) ou distúrbios de linguagem (diversas variantes exclusivas de uma afasia progressiva).81 Existem ao menos 2 subtipos patológicos principais de DLFT, incluindo um subtipo associado a anormalidades na proteína tau; um subtipo com anormalidades na proteína TDP43; e vários subtipos adicionais menos comuns.82 Apesar da sobreposição, existem algumas correlações clinicopatológicas distintas entre as síndromes clínicas específicas e as patologias específicas. O termo DLFT atualmente é preferido à antiga denominação epônima, doença de Pick, devido ao significado restrito que a doença de Pick adquiriu como uma das “taupatias”. A identificação dos diferentes tipos clínicos e patológicos de DLFT é importante, porque os principais aspectos do tratamento e os prognósticos são bastante diferentes entre si e daqueles associados à DA, com os quais, todavia, são confundidos com frequência.
A DLFT é bastante incomum após os 75 anos de idade. Geralmente, a idade dos pacientes no momento do aparecimento da condição é de 45 a 65 anos. Nesta faixa etária, a DLFT apresenta quase a mesma taxa de incidência da DA, que é aproximadamente de 3 a 4 casos em cada 100.000 indivíduos ao ano.83
Embora a maioria dos pacientes tenha doença esporádica e não genética, a DLFT possui um componente genético significativo.84 Quase metade dos pacientes com DLFT tem na família mais alguém com doença neurodegenerativa relacionada. Cada uma das mutações que ocorrem em 2 genes é responsável por cerca de 6 a 10% dos casos.85 Estes genes são o gene MAPT e o gene da granulina (GRN), ambos localizados no cromossomo 17 (ver no website www.molgen.ua.ac.be/ADMutations/). O mecanismo pelo qual as mutações no gene de tau causam neurodegeneração parece ser a alteração da capacidade da proteína tau de se ligar aos microtúbulos, com consequente hiperfosforilação seguida de agregação de moléculas de tau em uma forma fibrilar insolúvel. Todas as mutações no gene GRN atualmente conhecidas podem causar a degradação anormal do RNA mensageiro e, em consequência, uma diminuição marcante da produção da proteína progranulina. O mecanismo é referido como haploinsuficiência. Atualmente, não se sabe quais funções da proteína progranulina são úteis, e, desta forma, o modo como a haploinsuficiência conduz à neurodegeneração é obscuro. Contudo, a coloração por imuno-histoquímica de regiões do córtex cerebral de pacientes com mutações na progranulina invariavelmente demonstra o acúmulo citoplasmático anômalo da proteína TDP43 [Figura 7].
Figura 7. Fotomicrografia de um neurônio TDP43+, como se observa em pacientes com degenerações lobares frontotemporais (DLFT). As setas apontam as inclusões TDP43-imunorreativas presentes nas células dentadas hipocampais (x40, ampliação original).
A patologia das DLFT é localizada nas regiões frontal e temporal anterior, e bem menos frequentemente nos lobos parietais anteriores.81,82 Independentemente do tipo molecular, pode haver uma profunda atrofia nestas regiões. As estruturas subcorticais, incluindo o estriado, tálamo e hipocampo, também podem apresentar uma severa atrofia. Uma taupatia é demonstrada em cerca de metade dos pacientes com DLFT. Na taupatias, a coloração imuno-histoquímica revela a presença de material tau-positivo nas regiões afetadas. A lesão a princípio identificada como principal característica da doença de Pick é o corpúsculo de Pick, uma inclusão intracelular tau-positiva [Figura 8]. A outra metade dos pacientes com DLFT não apresenta aparência histoquímica anormal de material tau-positivo e, em vez disso, mostra um acúmulo citoplasmático anormal de TDP43. Na situação normal, a TDP43 é encontrada no núcleo neuronal. Sua migração para o citoplasma, nos estados neurodegenerativos, é indicativa de patologia, porém os mecanismos envolvidos são desconhecidos. Os pacientes com mutações em GRN sempre apresentam inclusões de TDP43.
Figura 8. Fotomicrografia dos corpúsculos de Pick, que são característicos da degeneração lobar frontotemporal (DLFT) envolvendo anormalidades da proteína tau.
O fenótipo clínico mais comum das DLFT é a DFTvc. Esta condição se manifesta como um distúrbio do controle comportamental e perda das habilidades sociais, bem como um distúrbio da função cognitiva executiva. Os pacientes tornam-se apáticos e sem iniciativa. Demonstram profunda perda de empatia e compreensão pelos sentimentos dos outros. Carecem de discernimento em relação ao próprio comportamento desorientado. Tornam-se impulsivos, facilmente distraídos e irresponsáveis. Seu julgamento é bastante precário e, nas situações pessoais ou no trabalho, estes pacientes frequentemente fazem julgamentos desastrosamente limitados. A memória recente pode estar comprometida, mas é bastante modesta ou definitivamente menos invasiva do que o distúrbio comportamental e de conduta social. Qualquer um dos subtipos patológicos pode produzir este fenótipo clínico.
Existem vários modos diferentes pelos quais os distúrbios de linguagem se manifestam nos distúrbios de DLFT. Um deles é uma fala desordenadamente expressiva, referida como afasia não fluente progressiva. A fala expressiva dos pacientes com afasia não fluente progressiva é caracterizada pela diminuição do número de palavras por expressão vocal; alteração melódica da fala; hesitação para falar; articulação difícil e trabalhosa acompanhada de frustração ao falar; e deleção de artigos e preposições (agramatismo). A compreensão é preservada, com exceção dos assuntos mais complexos. A crise de alterações patológicas ocorre nas regiões pré-frontais inferiores dominantes.86,87 A afasia não fluente progressiva geralmente é causada por uma taupatia. Outro tipo de expressão da linguagem nas DLFT é da demência semântica, que consiste em uma condição na qual a mecânica, a melodia e a entonação da fala são preservadas, mas há perda do acesso ao significado das palavras e do conhecimento sobre os objetos. Quando esta síndrome ocorre, invariavelmente existe uma atrofia profunda do lobo temporal anterior dominante.86,87 A demência semântica geralmente está associada às alterações patológicas de uma proteinopatia TDP43. Em uma 3ª variante, a fala espontânea é dominada pelo comprometimento da recuperação de palavras, contudo a mecânica da fala permanece normal e o acesso ao significado das palavras é preservado. Este último subtipo clínico é referido como afasia logopênica.88 Sua base anatômica é o lobo parietal inferior dominante. Em contraste com as outras variantes, embora a afasia logopênica ocorra com as proteinopatias taupática ou de TDP43, também pode estar associada à patologia da DA.
As DLFT podem ser diagnosticadas no momento de sua manifestação inicial, com base no perfil clínico distintivo, testes neuropsicométricos e exames de neuroimagem. A história de alterações do comportamento e da conduta fornecidas por um informante confiável é a chave para o diagnóstico de DFTvc. As alterações neuropsicológicas quase sempre estão presentes em pacientes com DFTvc, embora possam ser sutis e necessitem de avaliação por um neuropsicólogo que esteja familiarizado com este grupo de distúrbios. Os pacientes com DFTvc apresentam desempenho precário nos testes de função executiva, como o teste de Trailmaking, teste de Porteus Mazes, Wisconsin Card Sorting Test, teste de Stroop de cores e palavras, e testes de fluência verbal. O diagnóstico de afasia não fluente progressiva e demência semântica baseiam-se amplamente no exame neurológico da fala, linguagem e cognição. Em geral, estes pacientes também apresentam algumas anormalidades no teste neuropsicológico. Muitos (embora nem todos) pacientes com DLFT têm atrofia lobar frontal focal, insular ou temporal anterior, seja simétrica ou assimétrica [Figura 9]. Além disso, as técnicas de neuroimagem funcional, como PET ou tomografia por emissão fotônica única, são úteis para demonstrar a diminuição do metabolismo ou da perfusão nas regiões temporal anterior ou frontal.45
Figura 9. Varredura de ressonância magnética de atrofia temporal focal (a) ou de atrofia frontal (b) em pacientes com degenerações lobares frontotemporais (DLFT).
As DLFT apresentam uma considerável sobreposição com a paralisia supranuclear progressiva, síndrome da degeneração corticobasal e doença do neurônio motor. As 2 primeiras são, de fato, taupatias cuja manifestação inicial é dominada por uma variedade de manifestações motoras. O comprometimento cognitivo observado na paralisia supranuclear progressiva e na síndrome da degeneração corticobasal pode mimetizar a DFTvc ou uma das afasias progressivas. A doença do neurônio motor também está associada a um distúrbio cognitivo idêntico à DFTvc.89
Atualmente, não há tratamentos comprovadamente efetivos para as DLFT, além do uso de agentes controladores de depressão, ansiedade ou agitação.90 O avanço do nosso conhecimento acerca da biologia molecular e bioquímica das DLFT deixa-nos otimistas de que os estudos clínicos sobre tratamentos candidatos com forte embasamento lógico para as patologias de DLFT estejam em um futuro próximo.
O autor recebeu apoio financeiro para a realização das pesquisas clínicas das empresas Forest Laboratories, Elan Pharmaceuticals e Baxter Laboratories nos últimos 2 anos; atuou como consultor na GlaxoSmithKline; e atua no conselho de monitoramento de segurança de dados da Lilly Pharmaceuticals. Também é editor associado do Neurology e recebe pagamento da American Academy of Neurology por seu trabalho editorial.
1. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–44.
2. Amieva H, Jacqmin-Gadda H, Orgogozo JM, et al. The 9 year cognitive decline before dementia of the Alzheimer type: a prospective population-based study. Brain 2005;128: 1093–101.
3. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–94.
4. Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007;6:734–46.
5. Hy LX, Keller DM. Prevalence of AD among whites: a summary by levels of severity. Neurology 2000;55:198–204.
6. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60:1119–22.
7. Hebert LE, Scherr PA, McCann JJ, et al. Is the risk of developing Alzheimer’s disease greater for women than for men? Am J Epidemiol 2001;153:132–6.
8. Whitmer RA, Sidney S, Selby J, et al. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology 2005;64:277–81.
9. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69: 2197–204.
10. Haag MDM, Hofman A, Koudstaal PJ, et al. Duration of antihypertensive drug use and risk of dementia, a prospective cohort study. Neurology 2009;72:1727–34.
11. Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med 2003;163: 1069–75.
12. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004;291:2947–58.
13. Shumaker SA, Legault C, Thal L, et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: The Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003;289:2651–62.
14. Adapt Research Group, Lyketsos CG, Breitner JC, Green RC, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 2007;68:1800–8.
15. Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002;360:1623–30.
16. McGurn B, Deary IJ, Starr JM. Childhood cognitive ability and risk of late-onset Alzheimer and vascular dementia. Neurology 2008;71:1051–6.
17. Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 2008;9:768–78.
18. Lleo A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS-1 mutations. Am J Geriatr Psychiatry 2004;12:146–56.
19. Silverman JM, Smith CJ, Marin DB, et al. Familial patterns of risk in very late-onset Alzheimer disease. Arch Gen Psychiatry 2003;60:190–7.
20. Coon KD, Myers AJ, Craig DW, et al. A high-density wholegenome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007;68:613–8.
21. Breitner JC, Wyse BW, Anthony JC, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology 1999;53: 321–31.
22. Carrasquillo MM, Zou F, Pankratz VS, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat Genet 2009;41:192–8.
23. Savva GM, Wharton SB, Ince PG, et al. Age, neuropathology, and dementia. N Engl J Med 2009;360:2302–9.
24. Lesne S, Koh MT, Kotilinek L, et al. A specifi c amyloid-beta protein assembly in the brain impairs memory. Nature 2006;440:352–7.
25. Iqbal K, Liu F, Gong CX, et al. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 2009;118:53–69.
26. Lace G, Savva GM, Forster G, et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain 2009;132:1324–34.
27. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002;297:353–6.
28. Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron 2008;60:534–42.
29. Jack CR Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain 2009;132:1355–65.
30. Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004;62:925–31.
31. Whitwell JL, Josephs KA, Murray ME, et al. MRI correlates of neurofi brillary tangle pathology at autopsy: a voxelbased morphometry study. Neurology 2008;71:743–9.
32. Troncoso JC, Zonderman AB, Resnick SM, et al. Effect of infarcts on dementia in the Baltimore longitudinal study of aging. Ann Neurol 2008;64:168–76.
33. Jicha GA, Parisi JE, Dickson DW, et al. Age and apoE associations with complex pathologic features in Alzheimer’s disease. J Neurol Sci 2008;273:34–9.
34. Haroutunian V, Schnaider-Beeri M, Schmeidler J, et al. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch Neurol 2008;65:1211–7.
35. Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology 2009;72: 368–74.
36. Okonkwo OC, Wadley VG, Griffi th HR, et al. Cognitive correlates of fi nancial abilities in mild cognitive impairment. J Am Geriatr Soc 2006;54:1745–50.
37. Tang-Wai DF, Knopman DS, Geda YE, et al. Comparison of the Short Test of Mental Status and the Mini-Mental State Examination in mild cognitive impairment. Arch Neurol 2003;60:1777–81.
38. Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53: 695–9.
39. Knopman DS, DeKosky ST, Cummings JL, et al. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology 2001;56:1143–53.
40. Hampel H, Burger K, Teipel SJ, et al. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement 2008;4:38–48.
41. Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 2009; 65:403–13.
42. Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 2006;5:228–34.
43. Longstreth WT Jr, Dulberg C, Manolio TA, et al. Incidence, manifestations, and predictors of brain infarcts defi ned by serial cranial magnetic resonance imaging in the elderly: the Cardiovascular Health Study. Stroke 2002;33:2376–82.
44. Jagust W, Reed B, Mungas D, et al. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology 2007;69:871–7.
45. Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain 2007;130:2616–35.
46. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–19.
47. Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68: 1718–25.
48. Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 2007; 130:2837–44.
49. Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 2006;(1):CD005593.
50. Mohs RC, Doody RS, Morris JC, et al. A 1-year, placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology 2001;57:481–8.
51. Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005;352:2379–88.
52. DeKosky ST, Williamson JD, Fitzpatrick AL, et al. Ginkgo biloba for prevention of dementia: a randomized controlled trial. JAMA 2008;300:2253–62.
53. Miller ER 3rd, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2004;10:10.
54. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 2004;291:317–24.
55. Peskind ER, Potkin SG, Pomara N, et al. Memantine treatment in mild to moderate Alzheimer disease: a 24- week randomized, controlled trial. Am J Geriatr Psychiatry 2006;14:704–15.
56. Porsteinsson AP, Grossberg GT, Mintzer J, Olin JT. Memantine treatment in patients with mild to moderate Alzheimer’s disease already receiving a cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr Alzheimer Res 2008;5:83–9.
57. Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med 2006;355:1525–38.
58. Ray WA, Chung CP, Murray KT, et al. Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med 2009;360:225–35.
59. Holmes C, Cairns N, Lantos P, Mann A. Validity of current clinical criteria for Alzheimer’s disease, vascular dementia and dementia with Lewy bodies. Br J Psychiatry 1999;174: 45–50.
60. Reed BR, Mungas DM, Kramer JH, et al. Profi les of neuropsychological impairment in autopsy-defi ned Alzheimer’s disease and cerebrovascular disease. Brain 2007;130:731–9.
61. Knopman D, Rocca WA, Cha RH, et al. Survival study of vascular dementia in Rochester, Minnesota. Arch Neurol 2003;60:85–90.
62. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 Suppl 5): S4–9.
63. Fratiglioni L, Launer LJ, Andersen K, et al. Incidence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 Suppl 5): S10–5.
64. Desmond DW, Moroney JT, Lynch T, et al. The natural history of CADASIL: a pooled analysis of previously published cases. Stroke 1999;30:1230–3.
65. Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 2003;348:1215–22.
66. Au R, Massaro JM, Wolf PA, et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol 2006;63:246–50.
67. De Groot JC, De Leeuw FE, Oudkerk M, et al. Periventricular cerebral white matter lesions predict rate of cognitive decline. Ann Neurol 2002;52:335–41.
68. Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol 2006;60:677–87.
69. Kovari E, Gold G, Herrmann FR, et al. Cortical microinfarcts and demyelination affect cognition in cases at high risk for dementia. Neurology 2007;68:927–31.
70. Gold G, Giannakopoulos P, Herrmann FR, et al. Identifi cation of Alzheimer and vascular lesion thresholds for mixed dementia. Brain 2007;130:2830–6.
71. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–72.
72. Galvin JE, Pollack J, Morris JC. Clinical phenotype of Parkinson disease dementia. Neurology 2006;67:1605–11.
73. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing 2005;34:561–6.
74. Lippa CF, Duda JE, Grossman M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007;68:812–9.
75. Fujishiro H, Ferman TJ, Boeve BF, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol 2008;67:649–56.
76. Ferman TJ, Boeve BF, Smith GE, et al. Dementia with Lewy bodies may present as dementia and REM sleep behavior disorder without parkinsonism or hallucinations. J Int Neuropsychol Soc 2002;8:907–14.
77. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol 2006;20: 623–36.
78. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology 2003;61:40–5.
79. Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–8.
80. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351:2509–18.
81. Josephs KA. Frontotemporal dementia and related disorders: deciphering the enigma. Ann Neurol 2008;64: 4–14.
82. Bigio EH. Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol 2008;67:635–48.
83. Mercy L, Hodges JR, Dawson K, et al. Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 2008;71:1496–9.
84. Rademakers R, Hutton M. The genetics of frontotemporal lobar degeneration. Curr Neurol Neurosci Rep 2007;7: 434–42.
85. Seelaar H, Kamphorst W, Rosso SM, et al. Distinct genetic forms of frontotemporal dementia. Neurology 2008;71: 1220–6.
86. Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004;55:335–46.
87. Rohrer JD, Warren JD, Modat M, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology 2009;72:1562–9.
88. Gorno-Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 2008;71:1227–34.
89. Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002;59:1077–9.
90. Boxer AL, Boeve BF. Frontotemporal dementia treatment: current symptomatic therapies and implications of recent genetic, biochemical, and neuroimaging studies. Alzheimer Dis Assoc Disord 2007;21:S79–87.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.