(Carregando Índice)... (Carregando Índice)... |
Última revisão: 26/05/2015
Comentários de assinantes: 0
Sashank Prasad, MD
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon.
Os distúrbios neuroftálmicos conseguem causar morbidade significativa, exigindo do clínico o desenvolvimento e manutenção de habilidades diversas que facilitem o estabelecimento do diagnóstico no momento certo e a instituição de tratamento efetivo. Praticamente qualquer doença neurológica pode se manifestar com sintomas de disfunção das vias visuais aferentes ou eferentes. Este capítulo aborda especificamente a perda visual monocular aguda decorrente de doenças envolvendo o nervo óptico e a retina, papiledema, neuropatias ópticas crônicas, déficits de campo visual, perturbações do movimento ocular supra- e infranuclear e nistagmo.
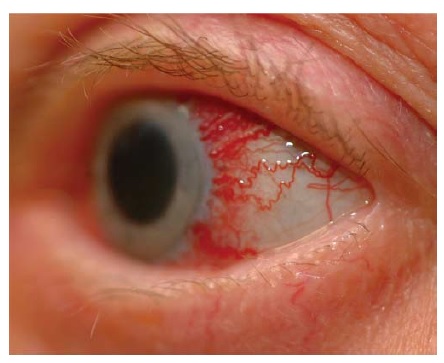
A perda visual aguda de um olho é um sintoma comum que requer avaliação tratamento urgentes. Nestas circunstâncias, é essencial identificar se a perda visual é devida a um distúrbio ocular (especialmente, a doença da retina) ou a uma neuropatia óptica [ver Figura 1]. A presente seção discute os achados característicos dos distúrbios agudos da retina [ver Tabela 1].
A descrição dada pelo paciente dos sintomas pode fornecer uma sugestão preliminar sobre a perda visual ser resultante de patologia ocular ou de nervo óptico. A perda visual monocular associada com metamorfopsia (imagens onduladas, deformadas) e fenômenos positivos (luzes piscando ou coloridas) frequentemente indica lesão de retina. O aumento súbito de “flutuantes” também é sugestivo de patologia de retina.
A acuidade visual central diminuída que não pode ser corrigida com furo de alfinete (pinhole) nem com refração pode ocorrer em condições envolvendo o nervo óptico e a retina. A grade de Amsler, que consiste em uma série de linhas horizontais e verticais semelhante ao papel milimetrado, pode parecer distorcida para um paciente com doença macular. Os campos visuais podem ser avaliados por meio de técnicas de confrontação, mas o teste de perimetria detalhado é mais sensível e pode revelar padrões de perda de campo sugestivos de uma localização específica e do diagnóstico diferencial.1 Um exame de fundo detalhado subsequente à dilatação pupilar farmacológica é essencial para a identificação precisa de um distúrbio de retina, muitas vezes requerendo consulta com oftalmologista. O exame pode revelar evidência de hemorragia vítrea, infarto venoso ou arterial retinal, descolamento da retina ou outra patologia macular.
Em determinadas circunstâncias, os exames adicionais de imagem e eletrofisiologia se mostram úteis. A tomografia de coerência óptica (OCT) fornece imagens de alta resolução de cortes transversais da retina, que ajudam a distinguir ente patologia de retina e neuropatia óptica.2 A angiografia com fluoresceína é usada em alguns casos com suspeita de maculopatia oculta, como uma lesão isquêmica associada ao diabetes. Um eletrorretinograma pode auxiliar no diagnóstico de disfunção retinal externa aguda, em particular na ausência de lesões evidentes e quando a suspeita de disfunção de retina é sugerida pela história.
A obstrução da artéria retinal é caracterizada pelo branqueamento da retina com aparecimento de uma mancha vermelho-cereja macular (onde a circulação coroidal intacta é vista mais prontamente) [ver Figura 2a]. No caso da obstrução de um ramo da artéria retinal, existe uma demarcação evidente entre as áreas afetada e preservada da retina [ver Figura 2b]. A isquemia macular diabética costuma produzir um edema macular que é evidente ao exame de fundo dilatado, tipicamente associada a micro-hemorragias retinais, manchas algodonosas e vasos atenuados. A obstrução de uma veia retinal produz múltiplas hemorragias pequenas na retina e ingurgitamento das veias retinais [ver Figura 2c]. O descolamento de retina, que causa profunda perda visual quando há envolvimento da mácula, é diagnosticado pela observação de elevação ondulante da retina [ver Figura 2d]. A retinopatia serosa central se manifesta com perda visual indolor e aguda causada por líquido subretinal macular [ver Figura 2e]. O edema macular cistoide também causa diminuição da acuidade visual, com leve espessamento ou elevação da mácula que pode ser dificilmente evidente ao exame de fundo dilatado, mas é prontamente evidente à OCT. As retinopatias paraneoplásicas incluem a retinopatia associada ao câncer (mais comumente com anticorpos contra a proteína recoverina) e a retinopatia associada ao melanoma (com anticorpos contra os bastonetes bipolares). Também, ocorrem retinopatias autoimunes na ausência de câncer subjacente.3 As doenças da retina externa incluem a síndrome do alargamento da mancha cega idiopática aguda e a síndrome dos múltiplos pontos brancos evanescentes, em que os paciente podem descrever fotopsias brilhantes e perda visual em curso na região que circunda a mancha cega fisiológica. O exame pode revelar alterações sutis de pigmentação retinal, tipicamente na ausência de inchaço discal.
Pacientes com neuropatia óptica frequentemente descreverão obscurecimento da visão e cores que parecem “lavadas” ou “desbotadas” [ver Figura 3]. O tempo de perda visual fornece um indício decisivo para o diagnóstico. Exemplificando, em pacientes com neurite óptica, a perda visual pode evoluir rapidamente e, depois, melhorar; na neuropatia óptica isquêmica, pode ocorrer de forma súbita e bastante estática; em pacientes com lesões compressivas, sua descoberta pode ser repentina, mas sua ocorrência tende mais a ser insidiosa e de progressão lenta. Adicionalmente, é importante determinar a presença ou ausência de sintomas orbitais, neurológicos ou sistêmicos associados. A qualidade e gravidade da dor devem ser caracterizadas. A dor tipicamente está presente em distúrbios como neurite óptica ou arterite de célula gigante (ACG), mas está ausente na neuropatia óptica isquêmica não arterítica. O conhecimento da idade e da história médica do paciente, em particular da história de fatores de risco vasculares, câncer ou distúrbios autoimunes é essencial para estabelecer um diagnóstico diferencial razoável.

Figura 1 - Algoritmo para avaliação de perda visual monocular aguda. Notas: 1. Com doença de retina acentuada, é possível que um pequeno defeito pupilar aferente esteja presente. 2. Obstrução da artéria retinal central, obstrução de ramo da artéria retinal ou isquemia macular. 3. É possível que não haja anormalidades de retina. 4. Pode haver leve inchaço do disco óptico, tipicamente na ausência de hemorragias papilares. 5. A perda visual pode ser bilateral, recorrente e frequentemente grave. 6. Pode haver pseudoinchaço do disco óptico e telangiectasias peripapilares. 7. Alguns podem apresentar uma estrela macular. Reimpresso com permissão de Prasad S e Galetta SL.164 ACG = arterite de célula gigante; NOHL = neuropatia óptica hereditária de Leber; NOINA = neuropatia óptica isquêmica não arterítica; NMO = neuromielite óptica; DPAR = defeito pupilar aferente relativo.
A visão colorida, que pode ser avaliada com pranchas coloridas especializadas ou por meio de testes para dessaturação de cores percebida, costuma ser afetada de modo desproporcional por uma neuropatia óptica. Testes detalhados de campo visual podem demonstrar uma variedade de padrões que refletem a causa subjacente. A depressão do campo visual central ocorre com frequência em quase todos os tipos de neuropatia óptica. Por outro lado, um “defeito de campo de feixe nervoso” (oriundo da mancha cega fisiológica e dos arcos superior e inferiormente, em relação ao meridiano horizontal) é mais específico para neuropatia óptica isquêmica ou compressiva.
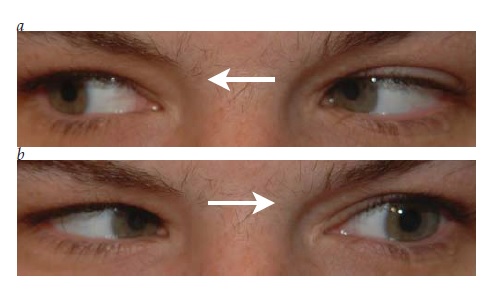
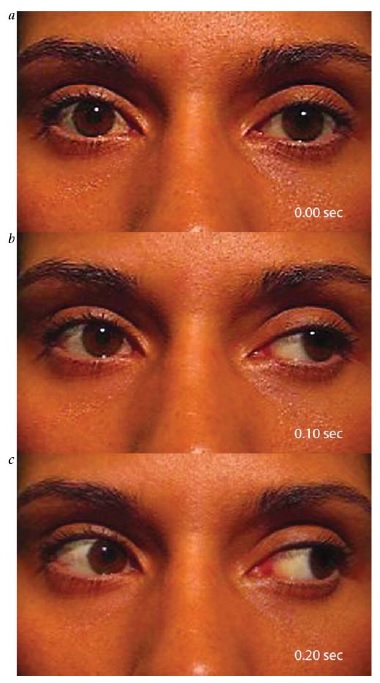
A principal característica da neuropatia óptica unilateral é o defeito pupilar aferente relativo (DPAR) [ver Figura 4].4 O DPAR é determinado pelo teste do flash de luz oscilante, durante o qual a luz é alternadamente dirigida para cada pupila. Quando a luz é dirigida para o olho não afetado, ambas as pupilas devem se contrair normalmente. Quando a luz é direcionada para dentro do olho afetado, a dilatação de ambas as pupilas indica a presença de DPAR. Embora um DPAR sutil possa ser observado no contexto de disfunção retinal moderada ou grave, esse defeito é prontamente visível até mesmo na presença de neuropatia óptica assimétrica ou unilateral branda.
Em um paciente com suspeita de neuropatia óptica, a aparência da cabeça do nervo óptico deve ser julgada como estando inchada, pálida ou normal. 5 O inchaço do disco óptico de um paciente com perda visual monocular aguda pode indicar isquemia ou inflamação da cabeça do nervo óptico. Espera-se um disco óptico normal com processos retrobulbares, como neurite óptica típica. A palidez do disco óptico indica compressão crônica ou lesão prévia do nervo óptico.
|
Tabela 1 - Etiologias da perda visual monocular e seus achados característicos | |
|
Etiologias |
Achados característicos |
|
Doença da retina Obstrução da artéria retinal central |
Branqueamento da retina, mancha vermelho-cereja |
|
Obstrução de ramo da artéria retinal |
Branqueamento segmentar da retina |
|
Obstrução da veia retinal |
Hemorragias de retina e veias ingurgitadas |
|
Descolamento de retina |
Ondeamento e elevação da retina |
|
Retinopatia serosa central |
Líquido subrretinal macular Confirmada por OCT |
|
Edema macular cistoide |
Elevação macular sutil Confirmado por OCT |
|
Síndrome do alargamento da mancha cega idiopática aguda e síndrome dos múltiplos pontos brancos evanescentes |
Fotopsias, alargamento de mancha cega Alterações retinais peripapilares sutis podem ser evidentes |
|
Neuropatia óptica Neurite óptica |
Dor à movimentação ocular Nervo óptico de aparência normal ou com inchaço leve Nadir em 7–10 dias Recuperação espontânea |
|
Neuropatia óptica isquêmica não arterítica |
Indolor Déficit de campo visual de altitude Edema de disco óptico; pode ser por setor Razão cálice: disco pequena (no outro olho) Idade avançada e fatores de risco vasculares Possível hipotensão noturna |
|
Neuropatia óptica isquêmica arterítica |
Sintomas sistêmicos (mialgias, claudicação mandibular, febres, sensibilidade do couro cabeludo, perda de peso) Edema de disco óptico ± manchas algodonosas Idade avançada |
|
Condições inflamatórias |
Possível associação com uveíte, sintomas inflamatórios sistêmicos |
|
Infecções |
“Neurorretinite” com estrela macular, em alguns casos |
|
Condições hereditárias (NOHL) |
Perda visual sequencial indolor Tipicamente, homens jovens Pseudoinchaço do disco óptico Telangiectasias peripapilares |
NOHL = neuropatia óptica hereditária de Leber; OCT = tomografia de coerência óptica.
As sequências (idealmente, incluindo imagens T2-ponderadas coronais e imagens pós-gadolínio gordura-saturadas) de ressonância nuclear magnética (RNM) orbitais tipicamente fornecem as melhores vistas do nervo óptico periquiasmático. Apesar de não totalmente específica, a RNM pode ser útil para separar as neuropatias ópticas inflamatórias, isquêmicas e neoplásicas. 6 Embora o sinal T2 aumentado seja comum a numerosas etiologias de neuropatia óptica, a intensificação patológica do nervo óptico distingue com segurança entre inflamação ou desmielinização do nervo óptico e neuropatia óptica isquêmica não arterítica (NOINA) anterior. Embora as características de imagem do nervo óptico possam não distinguir, diretamente, as causas inflamatórias e desmielinizantes, pode haver evidência do envolvimento adicional das meninges, glândula hipófise ou glândulas lacrimais indicando uma condição inflamatória. Embora os potenciais evocados visuais não sejam usados de forma rotineira no diagnóstico de neurite óptica desmielinizante, o achado de uma resposta P100 com latência prolongada fornece uma boa evidência de desmielinização do nervo óptico.
A gama de condições que podem produzir neuropatia óptica é ampla e inclui a neurite óptica desmielinizante idiopática, neuropatia óptica isquêmica, condições inflamatórias e infecciosas, processos infiltrativos ou neoplásicos, e neuropatias ópticas hereditárias [ver Tabela 1]. As lesões compressivas tipicamente causarão perda visual de progressão lenta, em vez de aguda, embora, algumas vezes, este sintoma venha à tona de forma bastante súbita. Em adição, o declínio agudo da visão ocasionalmente pode ser sobreposto à progressão crônica devido à rápida expansão de uma lesão em forma de massa, como pode ocorrer na apoplexia da hipófise ou no aneurisma.
A neurite óptica desmielinizante idiopática ocorre com mais frequência na faixa etária de 20-50 anos, sendo três vezes mais frequente em mulheres. 7 A perda visual em geral ocorre rapidamente, atinge o nadir entre sete e dez dias, e começa a se recuperar em um mês. A dor retrorbital, particularmente com os movimentos oculares, está presente em quase todos os casos e pode preceder a perda visual em vários dias, persistindo tipicamente entre uma e duas semanas. O teste de perimetria revelará mais frequentemente um escotoma central ou perda de campo difusa [ver Figura 5a]. O inchaço de disco óptico leve está presente em 1/3 dos pacientes [ver Figura 5b], enquanto no restante a cabeça do nervo óptico parece estar normal. A RNM muitas vezes confirma o sinal T2 aumentado e a intensificação de gadolínio patológica do nervo óptico retrorbital. Embora o prognóstico de recuperação da visão geralmente seja favorável (com a maioria dos pacientes retomando 20/20 de acuidade8), a disfunção residual branda é comum e pode ser capturada por medidas como sensibilidade ao contraste e estereoacuidade. 9,10
Figura 2 - Distúrbios da retina. (a) Obstrução da artéria retinal central. Palidez retinal grave e mancha vermelho-cereja macular. (b) Obstrução de ramo da artéria retinal. Palidez retinal superior com retina normal inferiormente. (c) Obstrução da veia retinal central. Inchaço discal leve e hiperemia, hemorragias retinais extensivas, veias retinais dilatadas e manchas algodonosas. (d) Descolamento da retina. Retina temporal dobrada ou enrugada (setas) (foto cortesia de Nicholas Volpe, MD). (e) Retinopatia serosa central. Elevação da retina interna com líquido subjacente (seta) na região da mácula. Figuras a e reimpressas com permissão de Prasad S et al.165 Figuras b, c e d reimpressas com permissão de Prasad S e Galetta SL.164
A probabilidade de neurite óptica progredindo para esclerose múltipla (EM) é mais bem prevista por RNM cerebral realizada no momento do diagnóstico. No Optic Neuritis Treatment Trial, o risco de desenvolvimento de EM em 15 anos foi de 72% entre os pacientes que apresentavam uma ou mais lesões cerebrais características, e de 25% entre os pacientes com RNM normal.11 Os pacientes com neurite óptica tratados com corticosteroides intravenosos parecem ter risco diminuído de desenvolvimento de EM ao longo dos 2 anos subsequentes.12 A longo prazo, todavia, o tratamento agudo com esteroides não afeta a probabilidade de progressão para EM.13 O tratamento com corticosteroides intravenosos, também, pode acelerar a recuperação da função visual, em particular, no caso dos campos visuais e sensibilidade ao contraste, embora não afete significativamente os resultados visuais a longo prazo.8,14,15 O tratamento com corticosteroides orais, por outro lado, pode estar associado ao risco aumentado de recorrência de neurite óptica, de modo que esta terapia deve ser evitada.14 Além dos corticosteroides intravenosos, vários estudos sustentam o uso de tratamentos imunomoduladores (incluindo interferon b-1a, interferon b-1b ou acetato de glatirâmero) para diminuir a probabilidade de progressão para EM entre dois e cinco anos, em pacientes de alto risco.16–19
Os achados que são atípicos de neurite óptica devem conduzir imediatamente a uma investigação rigorosa de outras causas. 7 Os “sinais de alerta” incluem um perfil temporal incomum (progressão além de duas semanas ou ausência de recuperação em um mês), ausência de dor, escotoma atípico (como um defeito altitudinal) ou um exame de fundo atípico (incluindo um nervo acentuadamente inchado ou atrófico, ou anormalidades de retina como hemorragias, inflamação ou exsudatos).
Figura 3 (a) Visão normal. (b) Simulação de visão turva e dessaturação da cor na neuropatia óptica.
A neuromielite óptica (NMO) ou doença de Devic, tipicamente causa lesões desmielinizantes necrotizantes dos nervos óptico e medulares espinais. A perda visual pode ser unilateral, mas é frequentemente bilateral, mais grave (acuidade <20/200) e tende mais à recorrência do que os déficits associados à neurite óptica típica.20 Em adição, a dor periorbital que é bastante característica da neurite óptica pode estar ausente na NMO. A RNM orbital e espinal pode mostrar extensas regiões de anomalia de sinal e intensificação patológica. Os autoanticorpos IgG anti-NMO parecem ser úteis como marcador, com sensibilidade relatada de 76% e especificidade de 91% em uma população norte-americana.21 As imagens de OCT, frequentemente, mostram o desenvolvimento de profunda perda da camada de fibras nervosas da retina, com espessura inferior a 60 mícrons no olho afetado.22 Os tratamentos imunossupressores agressivos (incluindo, por exemplo, o anticorpo monoclonal depletor de célula B rituximabe) pode ser particularmente benéficos para este grupo de pacientes.23,24
A NOINA é causa comum de neuropatia óptica unilateral em adultos com idade acima de 50 anos. 25 A cabeça do nervo óptico normalmente recebe suprimento sanguíneo via múltiplas pequenas artérias ciliares posteriores que surgem da artéria oftálmica. Dada a escassez de anastomoses neste suprimento sanguíneo, a hipoperfusão pode produzir isquemia em zona limítrofe. Os potenciais fatores de risco de NOINA incluem diabetes, hipertensão hipotensão noturna (precipitada possivelmente por terapia anti-hipertensiva),26 apneia do sono, uso de inibidores de fosfodiesterases27 e uma cabeça de nervo óptico amontoada (com uma razão cálice:disco baixa).28 O inchaço de um nervo óptico amontoado junto ao canal escleral pode provocar um ciclo de compressão vascular adicional, isquemia e inchaço.
Figura 4 - Defeito pupilar aferente relativo esquerdo. Uma fonte luminosa é oscilada para frente e para trás. (a) Ambas as pupilas se contraem quando a luz ilumina o olho direito. (b) Ambas as pupilas dilatam quando a fonte de luz ilumina o olhos esquerdo.
A NOINA é um diagnóstico clínico, firmemente baseado nos achados de edema de disco moderado a grave (muitas vezes com distribuição em forma de cunha), tipicamente com hemorragias de fibra nervosa [ver Figura 6a]. A perda visual de campo altitudinal é comum [ver Figura 6b] e a dor quase universalmente está ausente. A inspeção do outro olho mostra uma cabeça de nervo óptico amontoada (com razão cálice: disco baixa). Os perfis clínicos da NOINA e da neurite óptica, ocasionalmente, podem se sobrepor, sendo necessário tomar o cuidado de distinguir ambas as condições. 29 Com frequência, uma forma confiável de distinguir estas duas entidades é por observação, porque a recuperação visual é acentuadamente melhor no caso da neurite óptica do que para a NOINA. Muitos pacientes com NOINA têm déficit visual estável. Pode haver melhora espontânea nos primeiros 6 meses, embora em muitos pacientes isto reflita uma habilidade melhorada com fixação excêntrica.30 A imagem de OCT pode mostrar perda segmentar da camada de fibras nervosas da retina.31
Figura 5 - Neurite óptica. (a) Escotoma central em perimetria automática. (b) Edema leve de disco óptico, maior na porção temporal (seta). Reimpresso com permissão de Prasad S et al.165
Figura 6 - Neuropatia óptica isquêmica não arterítica. (a) Inchaço da cabeça do nervo óptico (setas pretas), hemorragias causadas por imobilizador (setas brancas) e hiperemia da cabeça do nervo óptico (asterisco). (b) Déficit de campo altitudinal arqueado superior no olho direito. Reimpresso com permissão de Prasad S et al.165
A neuropatia óptica isquêmica anterior arterítica (NOIAA) geralmente está relacionada com ACG e pode causar perda visual grave se não for diagnosticada e tratada antecipadamente.32 A prevalência da ACG aumenta com a idade e é rara antes dos 60 anos. A condição está associada à polimialgia reumática, consistindo de artralgia e mialgia proximal, bem como claudicação mandibular, febre, mal estar e sensibilidade no couro cabeludo. Quando há perda visual grave decorrente de ACG, o disco tipicamente aparece edemaciado e com aspecto de giz branco [ver Figura 7]. A coexistência de isquemia retinal (manchas algodonosas) e inchaço discal é altamente sugestiva de NOIAA. O diagnóstico de ACG é sugerido por uma alta velocidade de sedimentação eritrocitária e proteína C reativa, sendo confirmado por evidências de células gigantes e inflamação endovascular em biópsia de artéria temporal. Nos casos suspeitos, o tratamento com corticosteroides deve ser iniciado imediatamente, sem demora na obtenção da biópsia. Os corticosteroides ajudam a mitigar perdas visuais adicionais e diminuem a probabilidade de envolvimento do outro olho. O prognóstico de recuperação do olho afetado, todavia, é desfavorável mesmo com o tratamento do paciente.
A neuropatia óptica isquêmica posterior (NOIP) é uma entidade rara que se manifesta como disfunção do nervo óptico agudo e grave, na ausência de inchaço. 25 No contexto clínico correto, a NOIP pode ser resultado de uma grave perda de sangue ou de procedimentos cirúrgicos demorados (notavelmente, a cirurgia espinal). A ACG é outra consideração importante. A NOIP não artrítica pode ocorrer, ainda que raramente.
Figura 7 - Arterite de célula gigante. Palidez e inchaço da cabeça do nervo óptico (seta), com manchas algodonosas na retina (asteriscos). Reimpresso com permissão de Prasad S et al.165
As condições inflamatórias são causa importante de neuropatia óptica subaguda, mas ocasionalmente exibem manifestação aguda. O envolvimento do nervo óptico ocorre na neurossarcoidose,33 lúpus eritematoso sistêmico e síndrome de Sjögren 34, podendo ser acompanhado de uveíte anterior ou vitrite de segmento posterior [ver Figura 8]. A perda visual decorrente dessas condições costuma ser não só responsiva como, também, dependente de esteroides, com a recidiva da perda visual coincidindo com a retirada dos esteroides.35
As condições infecciosas são outra causa frequente de neuropatia óptica.36 A neurorretinite, em que a neuropatia óptica coexiste com exsudatos maculares ou peripapilares característicos, muitas vezes se deve à doença da arranhadura do gato (Bartonella henselae) [ver Figura 9]. Os exsudatos retinais característicos da neurorretinite podem formar um padrão de estrela parcial ou completo. Os pacientes podem reconhecer uma história de arranhaduras de gato, bem como episódios precedentes de febre e linfadenopatia. Outras neuropatias ópticas infecciosas incluem a sífilis tardia (Treponema pallidum), varicela zoster, doença de Lyme (Borrelia burgdorferi), infecção por HIV e infecções oportunistas, incluindo toxoplasmose, citomegalovírus, criptococos, mucormicose e aspergilose. Pacientes com sífilis secundária podem não se lembrar dos cancros que acompanham a infecção primária e, como se trata de uma condição passível de tratamento, as sorologias devem ser checadas sempre que houver suspeita. A neurite óptica associada à doença de Lyme é relativamente rara, mas pode ser considerada em paciente com história de exposição a carrapatos, eritema migratório, artralgias ou sorologias positivas (tipicamente confirmadas com Western blot). A neuropatia óptica associada ao zóster também é rara, mas ocorre ocasionalmente após o zóster oftálmico (erupção vesicular na distribuição V1). A sinusite paranasal ou mucocele pode levar à neuropatia óptica compressiva ou inflamatória. Recomenda-se manter alto grau de suspeita em casos de pacientes idosos, pacientes com história de doença sinusal grave e indivíduos com oftalmoparesia e febre associada.
Figura 8 - Neurossarcoidose. Imagem de ressonância magnética pós-gadolínio coronal, mostrando intensificação do nervo óptico esquerdo (seta). Reimpresso com permissão de Prasad S et al.165
Figura 9 - Neurorretinite. Inchaço discal acentuado com hiperemia, manchas algodonosas circundantes e exsudação macular em padrão de hemi-estrela. Reimpresso com permissão de Prasad S e Galetta SL.164
Entre as causas neoplásicas de perda visual aguda, o gliobastoma maligno primário em adultos ocasionalmente se origina no nervo óptico e pode mimetizar a neurite óptica nos estágios iniciais do curso clínico. Outras causas neoplásicas de neuropatia óptica incluem a leucemia ou linfoma disseminado, meningite carcinomatosa e metástases diretas para o nervo óptico (dentre as quais as mais comuns são metástases de tumores de mama e pulmão). A neuropatia óptica paraneoplásica tende mais a causar perda visual bilateral do que unilateral, embora ambas tenham sido relatadas. 37 Pode preceder a manifestação de malignidade sistêmica e se manifestar concomitantemente com retinite, vitrite, encefalite límbica, neuropatia periférica ou ataxia. O anticorpo mais comumente identificado é dirigido contra a proteína mediadora da resposta à colapsina-5 (CRMP-5).
A neuropatia óptica pode ocorrer como efeito tardio de radioterapia e, às vezes, pode ser difícil distingui-la de uma recidiva tumoral. 38 A neuropatia óptica da radioterapia é sugerida pela exposição (tipicamente à dose de 50 Gy), um intervalo temporal característico de seis a vinte e quatro meses para o aparecimento dos sintomas, e alterações em tecidos proximais induzidas por radiação. A progressão se dá ao longo de semanas a meses e a recuperação espontânea é rara. Os corticosteroides podem ajudar a diminuir o edema no nervo óptico afetado.
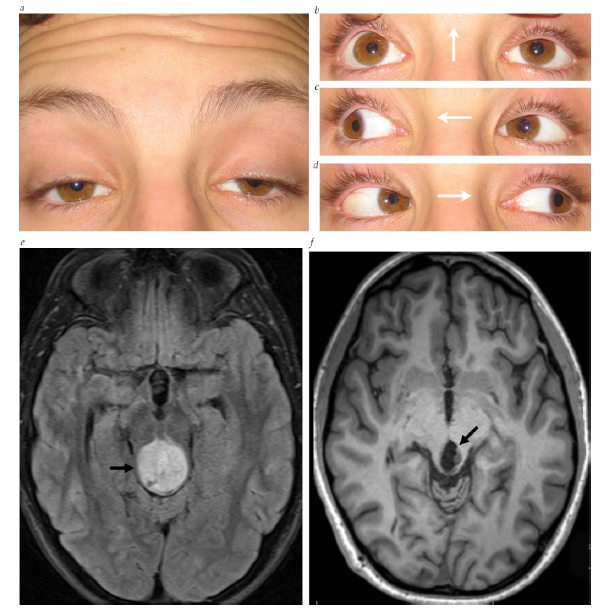
A neuropatia óptica hereditária de Leber (NOHL) se manifesta como perda visual subaguda a aguda, tipicamente na 2ª ou 3ª década da vida. 39 A condição surge a partir de mutações no DNA mitocondrial, que causam disfunção da cadeia de transporte de elétrons. Há herança materna com penetrância incompleta nas famílias. Cerca de 90% dos pacientes com NOHL são homens. A perda da visão inicialmente é bilateral em aproximadamente 50% dos casos, enquanto nos demais pacientes o outro olho se torna afetado dentro de nove meses. Embora não ocorra edema discal verdadeiro na NOHL, o disco óptico pode parecer hiperêmico e levemente inchado na fase aguda. Os vasos telangiectásicos peripapilares são bastante característicos, embora nem sempre estejam presentes [ver Figura 10]. A maioria dos pacientes sofre perda permanente da visão, embora o prognóstico dependa da mutação específica abrigada. Os pacientes com mutação T14484C no DNA mitocondrial tendem a apresentar recuperação espontânea, em comparação aos pacientes com mutações G11778A ou G3460A.39 Falta tratamentos efetivos para NOHL, ainda que o antioxidante idebenona consiga minimizar as perdas visuais subsequentes no outro olho.40 Adicionalmente, pode ser prudente que os pacientes com NOHL evitem potenciais toxinas, incluindo álcool ou tabaco.41
A neuropatia óptica por traumatismo direto pode incluir avulsão ou transecção do nervo, e é facilmente identificada pela história relevante de lesão [ver Figura 11].42 O exame de fundo pode mostrar hemorragias intraoculares extensas. Por outro lado, a neuropatia óptica por traumatismo indireto posterior estará presente com perda visual na ausência de anormalidades de fundo significativas. Esta condição pode resultar de forças de cisalhamento e subsequente edema junto ao canal óptico. Até metade destes pacientes pode apresentar melhora espontânea. 43 Há evidências fracas de que a terapia com corticosteroide possa ser útil durante as primeiras 8 horas. Nenhuma outra intervenção médica ou cirúrgica é comprovadamente efetiva. 44,45
O glaucoma de ângulo fechado é uma consideração importante em um paciente com perda visual dolorosa aguda. 46 É frequentemente distinguido de entidades como a neurite óptica, pela intensidade da dor (que pode ser excruciante) e por um olho avermelhado com pupila aumentada e não reativa.
Alguns casos de perda visual monocular aguda não terão base orgânica identificável. 47 Nestes casos, a acuidade visual e os campos visuais serão acompanhados de retina e disco óptico de aparência normal. De modo significativo, não haverá DPAR. Os índices de confiabilidade dos campos visuais automatizados podem ser baixos, e um padrão em “folha de trevo” de constrição de campo é comum. À confrontação ou exame de tela tangente, o paciente pode apresentar campos constritos “tubulares” não fisiológicos que não se expandem apropriadamente conforme a distância do teste é aumentada.
Figura 10 - Neuropatia óptica hereditária de Leber. Telangiectasias peripapilares (em 1 e 5 horas). Reimpresso com permissão de Prasad S e Galetta SL.164
Figura 11 - Neuropatia óptica traumática. Varredura de tomografia computadorizada axial (janelas ósseas) de um paciente com neuropatia óptica traumática esquerda direta decorrente de avulsão por projetil tipo BB pellet. Reimpresso com permissão de Prasad S et al.165
O papiledema refere-se especificamente ao edema de disco óptico causado por pressão intracraniana aumentada. O inchaço discal observado no papiledema resulta do fluxo axoplásmico comprometido nas fibras nervosas, aumentando o volume de axoplasma no disco óptico. 48 O papiledema pode ser uma manifestação de emergência neurológica e sua presença, portanto, requer avaliação diagnóstica imediata. 49
Os sintomas de pressão intracraniana elevada que acompanham o papiledema incluem cefaleia, zumbido pulsátil, náusea, vômito e diplopia. Pacientes com papiledema podem se queixar de perda visual, ter disfunção visual não percebida ou apresentar função visual normal. Os obscurecimentos visuais transitórios são comuns, consistindo em apagões rápidos e indolores que ocorrem com frequência ao mudar de posição.
O papiledema tem vários estágios, cada um dos quais associado a achados característicos que podem ser observados durante um exame oftalmoscópico [ver Figura 12]. O primeiro sinal de edema de disco é o obscurecimento das margens do disco óptico. Conforme o edema de disco óptico evolui, afeta primeiro as partes inferior e superior do nervo, em seguida a parte nasal e, por fim, a parte temporal.50 Um nervo óptico com inchaço patológico tipicamente apresenta ingurgitamento venoso e hiperêmico. Os vasos retinais que atravessam o disco muitas vezes se tornam parcialmente obscurecidos pela camada de fibras nervosas edemaciada e inchada. É comum haver hemorragias em torno da cabeça do nervo óptico. Pode haver isquemia peripapilar focal da camada de fibras nervosas, produzindo manchas algodonosas brancas características. À medida que o papiledema se torna crônico, a aparência da cabeça do nervo óptico muda. A hiperemia, hemorragias peripapilares e manchas algodonosas tipicamente resolvem, e o disco se torna pálido e gliótico [ver Figura 13].
As pulsações venosas retinais se referem a movimentos rítmicos da parede vascular que correspondem ao ciclo cardíaco, e que pode ser úteis como barômetro da pressão intracraniana. 51 Embora a presença de pulsações venosas seja evidência de pressão intracraniana normal, 52 a ausência de pulsações venosas não necessariamente indica pressão intracraniana aumentada (estas pulsações normalmente estão ausentes em 10% da população). A melhor localização para observar as pulsações é o segmento venoso que se volta na margem do cálice óptico.
A acuidade visual frequentemente está normal quando há papiledema—um achado decisivo que distingue a condição de outras causas de edema de disco óptico. A acuidade se torna comprometida quando o dano ao nervo óptico é grave ou o inchaço peripapilar retinal se estende para a própria mácula em si.
A avaliação de campo visual automatizado é essencial à avaliação e tratamento de pacientes com papiledema. A ampliação das manchas cegas é típica e pode ser causada pela elevação dos elementos retinais em torno do nervo óptico inchado. 53 Perdas de campo visual adicionais ocorrem com frequência primeira no campo visual nasal periférico e, em seguida, invadem o centro do campo visual à medida que a lesão axonal evolui. A progressão das anormalidades de campo visual é um parâmetro decisivo que afeta o tratamento.
Embora o papiledema tipicamente seja bilateral, há casos raros em que pode ser unilateral. 54 O papiledema unilateral é hipoteticamente atribuído a diferenças anatômicas envolvendo a bainha do nervo óptico ou lâmina cribrosa, que levam à transmissão assimétrica de pressão elevada às cabeças do nervo óptico. 54,55
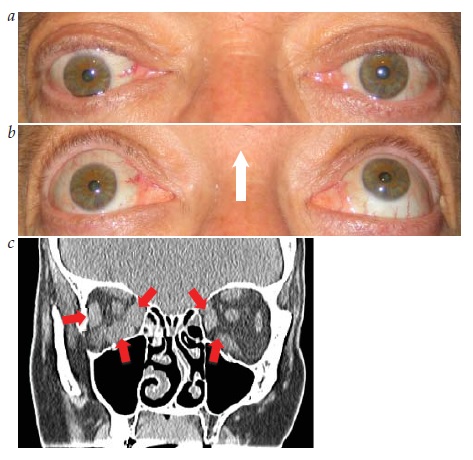
O papiledema verdadeiro deve ser diferenciado de entidades como a elevação anômala congênita dos discos ópticos e a drusa de disco óptico [ver Figura 14].56 Vários achados essenciais ajudam a distinguir o papiledema do conhecido “pseudopapiledema”: (1) aparência de disco hiperêmico; (2) ingurgitamento venoso; (3) obscurecimento da vasculatura ao longo das margens do disco óptico; e (4) hemorragia, manchas algodonosas ou dobras coroidais ao redor do nervo óptico.
Exames de imagem. Quando a incerteza prevalece na diferenciação entre elevação anômala do disco óptico e papiledema, pode ser útil realizar exames diagnósticos adicionais. A angiografia com fluoresceína demonstrará vazamento de corante na cabeça do nervo óptico no papiledema.57 A tomografia computadorizada (TC) ou ultrassonografia podem identificar drusas na ou embaixo da cabeça do nervo óptico [ver Figura 15]. Durante a ultrassonografia ocular, o “teste de 30°” é realizado por meio da obtenção de medidas do diâmetro do nervo óptico durante a fixação do olhar primária e no olha fixo excêntrico. Uma diminuição do diâmetro durante esta manobra indica a presença de líquido na bainha do nervo óptico e é sugestiva de papiledema.58
Figura 12 - Papiledema agudo. O inchaço da camada de fibras nervosas do nervo peripapilar obscurece a vista dos vasos retinais subjacentes (p. ex., setas pretas). As hemorragias por imobilização também sugerem papiledema verdadeiro, em vez de pseudopapiledema (setas brancas). Reimpresso com permissão de Prasad S et al.165
Uma vez levantada a suspeita de papiledema, torna-se necessário realizar uma avaliação eficiente para identificar a causa da pressão intracraniana elevada. Entre os exames de imagem apropriados, estão a TC ou RNM do encéfalo. Em adição, a TC ou a venografia de ressonância magnética fornece informação útil para avaliar a possibilidade de trombose venosa.
Figura 13 - Papiledema crônico. Observe a aparência pálida e gliótica do disco, sem hemorragias retinais. Reimpresso com permissão de Prasad S et al.165
Punção lombar. A punção lombar fornece informação diagnóstica essencial sobre o paciente com papiledema e, em geral, pode ser realizada com segurança (ausência de lesão espaçosa que imponha risco aumentado de herniação). Os exames básicos de líquido cerebrospinal (LCS) são realizados em todos os pacientes, sendo que os exames adicionais para condições infecciosas, inflamatórias e neoplásicas se baseiam nos resultados iniciais e na suspeita clínica. Uma punção lombar também fornece uma medida útil da pressão intracraniana, que é obtida em posição de inclinação lateral, com o paciente relaxado e com as pernas estendidas. Pressão intracraniana normal é tipicamente inferior a 250 mmH2O, embora em alguns paciente um valor inferior também possa ser patológico.59 Embora muitos pacientes sejam submetidos à punção lombar orientada por fluoroscopia, este procedimento tipicamente é realizado em posição pronada, sendo que não há dados normativos confiáveis.60 O posicionamento em pronação pode aumentar a pressão intrabdominal e criar elevação espúria da pressão intracraniana medida.
Figura 14 - Drusa de disco óptico. Excrescências refráteis visíveis sobre o disco óptico (setas).
Figura 15 - Drusa de disco óptico. Drusa enterrada identificada como uma massa hiperecoica à ultrassonografia ocular (seta).
O diagnóstico diferencial de pressão intracraniana elevada inclui (1) uma lesão em forma de massa intracraniana ou intraspinal, ou malformação vascular; (2) trombose cerebrovenosa; (3) meningite; (4) hemorragia subaracnoide; (5) hipertensão intracraniana secundária à medicação ou condições médicas sistêmicas; e (6) hipertensão intracraniana idiopática. As lesões em massa comuns incluem os tumores cerebrais primários ou metastáticos, hemorragia intraparenquimal ou extraxial, abscesso, malformação arteriovenosa, edema cerebral e hidrocefalia obstrutiva. A trombose cerebrovenosa pode ocorrer no contexto de gravidez, hipercoagulabilidade ou desidratação, ou na ausência de fatores de risco identificáveis [ver Figura 16].61 O estreitamento dos seios venosos transversais é observado com frequência em pacientes com hipertensão intracraniana idiopática, mas pode ser consequência (em vez de causa) de pressão intracraniana elevada [ver Figura 17].62,63 A pressão intracraniana elevada decorrente de meningite pode ser devida a infecções fúngicas, virais ou bacterianas agudas ou crônicas; condições inflamatórias autoimunes (tais como sarcoidose, lúpus eritematoso ou doença de Behçet); infiltração neoplásica; ou irritação química. A hemorragia subaracnóidea, diagnosticada por xantocromia ou eritrocitose no líquido espinal, pode produzir acentuada elevação da pressão intracraniana. A hemorragia vítrea simultânea é diagnóstica da síndrome de Terson. Algumas medicações associadas à pressão intracraniana elevada incluem as tetraciclinas (como a doxiciclina e a minociclina), doses altas de derivados de vitamina A (como os retinoides) e corticosteroides (tipicamente, quando da retirada). Embora diversas medicações tenham sido associadas à pressão intracraniana elevada, os dados disponíveis são fracos e existe carência de estudos controlados prospectivos. Por fim, o diagnóstico de hipertensão intracraniana idiopática (pseudotumor cerebral) é estabelecido após uma avaliação diagnóstica completa, com exames de neuroimagem e do líquido espinal, que não forneça outra explicação para a pressão intracraniana elevada.64 A hipertensão intracraniana idiopática exibe predominância feminina e está altamente associada à obesidade ou ganho de peso recente.
Dependendo do diagnóstico específico, o tratamento deve ser dirigido para a causa subjacente da pressão intracraniana elevada. As lesões em forma de massa, como tumores ou hematomas, podem requerer evacuação cirúrgica. A hidrocefalia pode necessitar de desvio ventricular. Os processos infecciosos e inflamatórios provavelmente receberão tratamentos médicos dirigidos. A trombose sinusal venosa é tratada com anticoagulação e muitas vezes hidratação.
Como a hipertensão intracraniana idiopática costuma estar associada à obesidade, a perda de peso é um componente decisivo do tratamento efetivo. Quando as modificações da dieta e exercícios são insuficientes, a cirurgia bariátrica pode ser considerada em casos cuidadosamente selecionados. Entretanto, como a perda de peso tipicamente não ocorre de forma suficientemente rápida, tratamentos médicos ou cirúrgicos adicionais frequentemente são necessários a curto prazo.
A base da terapia é a acetazolamida, um inibidor de anidrase carbônica que diminui a produção de LCS. 65 As parestesias periorais e de membros são um efeito colateral frequente. O topiramato promove inibição moderada da anidrase carbônica e pode ser considerado um auxiliar ou uma alternativa à acetazolamida.66 Os potenciais benefícios adicionais do topiramato incluem a profilaxia conta enxaqueca crônica e a perda de peso, contudo um efeito colateral comum é um leve retardo cognitivo.
Quando a perda visual evolui mesmo com os tratamentos médicos para papiledema em curso, os tratamentos cirúrgicos muitas vezes são justificáveis. Tentativas ocasionais de punções lombares (em particular no contexto da gravidez) podem ser feitas, mas costumam ser inefetivas em consequência da velocidade de reposição do LCS. As opções cirúrgicas incluem os procedimentos de desvio cerebrospinal (lombo-peritoneal e ventrículo-peritoneal) e fenestração da bainha do nervo óptico. A longo prazo, um desvio se torna propenso a complicações, inclusive com falha e infecção do desvio. A curto prazo, porém, com o declínio da visão, o desvio constitui a forma mais imediata de evitar o avanço da progressão clínica.
Assim como uma neuropatia óptica aguda, a neuropatia óptica de progressão crônica frequentemente será identificada por perda visual, dessaturação percebida da cor e DPAR. As considerações diagnósticas são acentuadamente diferentes, tornando relevantes outros achados salientes fornecidos pela história e exame.
Com a perda visual insidiosa, os déficits podem progredir de modo significativo antes de serem notados pelo paciente. A maioria das etiologias da neuropatia óptica de progressão crônica está associada à dor, com exceção da presença de uma massa compressiva que também pode produzir cefaleias. Uma lesão intraorbital pode produzir proptose e diminuição da motilidade ocular, além da perda visual. Em pacientes com perda visual insidiosa, é essencial estabelecer uma história médica de desnutrição, absorção comprometida ou exposição a agentes potencialmente tóxicos. Uma história familiar de déficits visuais semelhantes é bastante pertinente.
Figura 16 - Venografia de ressonância magnética (VRM) em pacientes com hipertensão intracraniana. (a) Trombose sinusal venosa extensiva demonstrada por VRM (setas). (b) No mesmo paciente, um coágulo intraluminal aparece brilhando nas imagens T1 ponderadas sem intensificação (setas). Cortesia de Kathy Chuang, MD.
Figura 17 - Estreitamento sinusal transverso direito (seta), que é um achado comum na hipertensão intracraniana idiopática e não necessariamente patológico.
As principais causas da neuropatia óptica de progressão crônica são classificadas conforme a aparência característica do disco óptico produzida por cada uma. A ampliação do cálice fisiológico com relativa preservação da borda neurorretinal é característica da neuropatia óptica glaucomatosa. Em contraste, a palidez global do nervo, incluindo a borda neurorretinal pode indicar lesão por compressão, agressão tóxica, deficiência nutricional, perturbação metabólica hereditária ou condições neoplásicas.
Um exame de imagem é obrigatório para pacientes com neuropatia óptica unilateral ou bilateral de progressão crônica não atribuída a glaucoma. Um protocolo de aquisição com intensificação por contraste e cortes finos da órbita e sela hipofisária proporcionam maior sensibilidade para detecção de lesões compressivas. Uma varredura de TC sem contraste oferece a vantagem adicional de identificar calcificações ou hiperostose que possam estar associadas com meningioma.
Diversas lesões em forma de massa compressiva podem causar neuropatia óptica progressiva. O disco óptico estará inchado nos casos de compressão intraorbital, mas se tornará progressivamente pálido nos casos de compressão retrorbital. Ocasionalmente, um tumor muito grande, como um meningioma de sulco olfatório, produzirá palidez de um nervo óptico em decorrência de compressão, ao mesmo tempo em que indiretamente causará inchaço do outro disco óptico como resultado da pressão intracraniana elevada. Entre as causas relevantes de neuropatia óptica compressiva, estão a neoplasia (incluindo o meningioma da base do crânio ou da bainha do nervo óptico, adenoma hipofisário e craniofaringioma), lesões sinusais, processos ósseos (como displasia fibrosa), alargamento de músculos extraoculares (como na oftalmopatia de Graves) ou aneurismas [ver Figura 18]. Os meningiomas da bainha do nervo óptico ocorrem primariamente em mulheres e podem causar perda da acuidade associada à atrofia ou ao inchaço discal. Os vasos de desvio optociliar (que abrem rotas adicionais de drenagem a partir da circulação venosa retinal para a coroide) têm forte associação com o meningioma da bainha do nervo óptico, mas podem ser decorrentes de qualquer causa de edema discal crônico por compressão [ver Figura 19].
A neuropatia óptica tóxica ou nutricional é sugerida por uma perda visual simétrica e indolor afetando a visão central. 67 Ocasionalmente, o envolvimento de um olho pode se manifestar antes do envolvimento do outro. Classicamente, as neuropatia ópticas tóxicas/nutricionais estão associadas a escotomas centro-cecais (escotomas centrais que se conectam à mancha cega normal) [ver Figura 20]. Os nervos ópticos podem aparecer normais ou atróficos. A ambliopia por tabaco-álcool refere-se a uma neuropatia óptica putativamente relacionada à desnutrição combinada ao consumo crônico de álcool e tabaco. A deficiência de vitamina B12, como a que pode ocorrer com a anemia perniciosa ou após a cirurgia de desvio gástrico, é causa comum de neuropatia óptica nutricional. A toxicidade por etambutol causa neuropatia óptica com uma relação dose-efeito bastante previsível.68 O envenenamento por metanol é uma neuropatia óptica tóxica incomum, que se manifesta com inchaço discal acentuado.
A neuropatia óptica glaucomatosa está entre as causas mais comuns de perda visual no mundo inteiro. É tipicamente fácil de distinguir pelo sangramento de disco óptico e pressão intraocular elevada [ver Figura 21].69 O glaucoma normotenso é mais difícil de identificar, mas se manifesta com sangramento de disco óptico e constrição progressiva do disco óptico, apesar das pressões intraoculares normais.70
As neoplasias primárias de baixo grau do nervo óptico (i.e., astrocitoma pilocítico juvenil) têm associação com a neurofibromatose de tipo 1 e tendem mais a se manifestarem durante a infância [ver Figura 22]. Estas lesões tipicamente são tratadas de modo conservativo, com exames oftálmicos frequentes ao longo da adolescência. Nos casos em que a progressão clínica ou radiográfica é detectada, a quimioterapia pode ser recomendada, às vezes seguida de radioterapia e, ainda que raramente, de cirurgia. 71
Outra neuropatia óptica que pode ter manifestação insidiosa na infância é a atrofia óptica predominantemente hereditária. 39 A acuidade subnormal pode ser descoberta por acaso, durante um exame de visão de triagem. Estes pacientes tipicamente exibem uma aparência discal impressionante, com palidez acentuada e escavação da porção temporal do disco [ver Figura 23]. O distúrbio é causado por mutações no gene OPA1, com herança autossômica e penetrância variável. O produto do gene OPA1 é dirigido à mitocôndria e sustenta a estabilidade da membrana. Mais de 90 mutações patogênicas diferentes de OPA1 foram descritas, sendo que o sequenciamento genético é disponibilizado para confirmação da suspeita diagnóstica.
Figura 18 - Neuropatia óptica compressiva. (a) Imagem de ressonância magnética pós-contraste coronal, mostrando um meningioma de sela turca comprimindo o nervo óptico direito. (b) Varredura de tomografia computadorizada (janelas ósseas) revelando extensiva displasia fibrosa com estreitamento do canal óptico esquerdo e proptose esquerda.
Figura 19 - Meningioma de bainha do nervo óptico produzindo inchaço discal crônico, com margens discais obscuras (setas pretas) e um vaso de desvio optociliar (seta branca). Reimpresso com permissão de Prasad S et al.165
Outras causas raras de neuropatia óptica hereditária incluem a síndrome de Wolfram (DIDMAOS), caracterizada por diabetes insípido, diabetes melito juvenil, atrofia óptica e surdez, encefalopatia mitocondrial com acidose lática e episódios similares a acidentes vasculares encefálicos (EMALAVE), epilepsia mioclônica com fibras vermelhas esfarrapadas (EMFVE) e encefalomielopatia necrotizante subaguda de Leigh.39 A neuropatia óptica não ocorre de forma isolada sob estas condições e o diagnóstico depende do reconhecimento dos desarranjos multifocais.
Os axônios de cada nervo óptico decussam parcialmente no quiasma óptico para reunir informação oriunda das metades de cada retina, que veem a mesma porção do campo visual. Desta forma, os axônios das células ganglionares nasais atravessam e se unem aos axônios das células ganglionares temporais do olho contralateral. Cada trato óptico contém axônios da retina temporal ipsilateral e da retina nasal contralateral. A maioria das fibras dos tratos ópticos faz sinapse no núcleo geniculado lateral ipsilateral (NGL). Os neurônios de segunda ordem da via visual se estendem do NGL para o córtex estriado (calcarino) no lobo occipital. Estes neurônios estão agrupados em dois feixes principais: as radiações temporais (que seguem um curso anterior pelo polo temporal, denominadas alça de Meyer, antes de se voltarem para a direção oposta) e as radiações parietais. O arranjo retinotópico é preservado; as radiações temporais representam o campo superior contralateral e as radiações parietais representam o campo inferior contralateral. As radiações ópticas surgem na superfície mesial do lobo occipital, no córtex calcarino. Os fascículos oriundos das radiações parietais fazem sinapse no banco superior do córtex calcarino, enquanto aquelas oriundas das radiações temporais surgem do banco inferior. O suprimento sanguíneo principal para o córtex visual é fornecido pelas artérias cerebrais posteriores e seus ramos. No polo occipital, todavia, pode haver um suprimento sanguíneo duplo para a área que serve a função central, com anastomoses entre os ramos das artérias cerebrais posteriores e ramo temporoccipital superior da artéria cerebral média.
A probabilidade de um paciente perceber um déficit de campo visual tipicamente depende da acuidade da alteração. Embora a perda visual súbita tende a ser observada, a lenta perda progressiva pode não ser notada. Os sintomas de déficit de campo visual podem incluir a lesão incidental decorrente de golpes com objetos sobre o lado afetado. Quando um déficit de campo visual inclui a visão central, a fluência da leitura pode ser comprometida.
A forma ideal de testar os campos visuais é avaliar cada olho separadamente, em particular porque uma comparação dos resultados de cada olho, muitas vezes, é útil em termos de valor de localização. Várias técnicas de confrontação podem ser usadas de forma combinada, incluindo a contagem de dedos e a detecção de estímulo cinético, como um dedo oscilando ou um estímulo vermelho. Existem três métodos valiosos como testes de triagem, contudo, técnicas mais sensíveis, frequentemente, são requeridas quando há suspeita de déficit de campo visual sutil.1,72 A perimetria automatizada fornece medidas confiáveis e reproduzíveis da função visual em numerosos pontos dentro de 30° de fixação.
Um déficit de campo visual bitemporal é a principal característica de uma lesão de quiasma. A compressão inferior do quiasma, como ocorre tipicamente no macroadenoma hipofisário, em geral produz uma perda bitemporal que surge nos quadrantes superiores [ver Figura 24]. Em contraste, a compressão superior, muitas vezes, ocorre com lesões como o craniofaringioma, frequentemente produzindo déficits bitemporais inferiores [ver Figura 25]. Outras lesões produzindo perda visual quiasmática incluem os meningiomas oriundos da sela turca, aneurismas amplos do seio cavernoso e lesões quiasmáticas intrínsecas, como neoplasia ou desmielinização inflamatória.
Os déficits de campo homônimo se referem ao meridiano vertical e afetam o mesmo hemicampo a partir de cada olho. Um déficit de campo homônimo indica uma lesão da via visual retroquiasmal. Padrões específicos podem ajudar a localizar a lesão no trato óptico, radiações ópticas ou córtex visual.
Trato óptico. Uma lesão parcial do trato óptico tipicamente produz um déficit de campo homônimo contralateral, de modo que um olho mostra maior perda de hemicampo do que outro [ver Figura 26]. A razão pela qual uma lesão parcial de trato óptico pode produzir déficits de campo incongruentes é que, no trato óptico, as fibras que cruzam a partir da retina nasal contralateral se tornam justapostas à fibra oposta correspondente oriunda da retina temporal ipsilateral. Uma lesão incompleta do trato óptico pode afetar desproporcionalmente as fibras que chegam de cada olho, resultando em déficits assimétricos incongruentes. Em contraste, as lesões das vias visuais posteriores são caracterizadas por déficits de campo homônimo altamente congruentes. Uma advertência a esta regra é a impossibilidade de julgar a congruência de um déficit de campo no contexto de hemianopia homônima completa. Deste modo, a localização clínica correta do déficit pode ser impraticável. Indícios adicionais de que um déficit de campo homônimo se deve a uma lesão de trato óptico são a presença de DPAR ou a palidez do nervo óptico. Estes achados estão ausentes nas lesões das radiações ópticas ou do córtex visual.
Figura 20 - Neuropatia óptica pode ser deficiência de vitamina B12. Escotoma centro-cecal do olho direito (envolvendo a visão central estendendo-se na direção de uma mancha cega aumentada), demonstrado por campos visuais de Goldmann.
Radiações ópticas. Os déficits visuais causados por lesões das radiações ópticas são bastante diretos. Uma lesão da radiação óptica inferior, incluindo uma lesão do lobo temporal anterior, produz quadrantanopia superior contralateral [ver Figura 27]. Em contraste, uma lesão afetando as radiações parietais produz quadrantanopia inferior contralateral [ver Figura 28].
Córtex visual. Uma hemianopia homônima que divide a mácula pode ocorrer com qualquer lesão retroquiasmática. Entretanto, a hemianopia que preserva a mácula tem alto valor de localização, sugerindo fortemente um infarto de lobo occipital contralateral na distribuição da artéria cerebral posterior (ACP) [ver Figura 29]. A representação da mácula no córtex occipital está no polo occipital, que, muitas vezes, conta com um suprimento sanguíneo colateral duplo a partir da ACP e dos ramos da artéria cerebral média. Uma hemianopia, com visão macular intacta, portanto, é consequência da preservação do polo occipital no contexto de um infarto da ACP. Outas causas de lesão occipital, como traumatismo ou infiltração neoplásica, tendem menos a poupar seletivamente o polo occipital e, de forma típica, causarão uma hemianopia que divide a mácula.
Figura 21 - Neuropatia óptica glaucomatosa. Cálice óptico aumentado (razão aproximada de cálice:diâmetro discal = 0,6).
Figura 22 - Glioma óptico. Imagens de ressonância magnética T2-ponderadas, revelando ampliação e sinuosidade aumentada do nervo óptico direito (seta) em uma criança com neurofibromatose tipo 1.
A RNM é essencial para estabelecer a causa da maioria dos déficits de campo visual. Embora campos visuais detalhados possam sugerir uma localização específica, em muitos casos, a imagem serve para estabelecer se a lesão responsável é vascular, neoplásica ou inflamatória.
O refluxo à luz pupilar normal é consensual, implicando que a luz direcionada para dentro do olho produz contração pupilar bilateral. As fibras de nervo óptico destinadas a mediar o reflexo de luz pupilar saem do trato óptico para fazer sinapse nos núcleos pré-tetais do mesencéfalo dorsal, que, então, se conecta bilateralmente aos núcleos de Edinger-Westphal junto ao complexo nuclear oculomotor. Os núcleos de Edinger-Westphal emitem fibras parassimpáticas que seguem com o fascículo do III nervo para sair do mesencéfalo e, então, repousar superficialmente sobre o nervo (tornando-os vulneráveis à compressão extrínseca). As fibras pupilares fazem sinapse no gânglio ciliar, que dá origem aos nervos ciliares curtos que inervam o corpo ciliar (para acomodação) e o músculo do esfíncter da íris (para constrição pupilar). As fibras que seguem para o corpo ciliar são mais numerosas do que aquelas que inervam o esfíncter da íris, a uma razão de 30:1.
A dilatação pupilar é mediada estimulação simpática do olho por uma via de três neurônios que começa no hipotálamo posterolateral. A partir do hipotálamo, o neurônio de primeira ordem se projeta pelo tronco encefálico até a medula espinal cervical inferior. Após fazer sinapse, o neurônio de segunda ordem sai da medula espinal e entra na cadeia simpática, para então seguir por sobre o ápice do pulmão, onde repousa próximo ao plexo braquial inferior. Estas sinapses neuronais no gânglio cervical superior, e depois no neurônio de terceira ordem, ascendem ao longo da artéria carótida interna. Estas fibras entram no seio cavernoso, onde ficam em proximidade com nervo abducente e, em seguida, seguem o ramo oftálmico do nervo trigêmeo (V1). As estruturas importantes inervadas por estas fibras simpáticas incluem os músculos dilatadores da pupila e os músculos de Müller das pálpebras superior e inferior.
Um novo episódio de anisocoria, ocasionalmente, é observado pelo paciente ou por seus familiares. Entretanto, por ser assintomática, a anisocoria, frequentemente, é detectada primeiro durante um exame físico minucioso realizado pelo médico.
Na distinção entre anisocoria fisiológica e outras formas de anisocoria patológica, é essencial avaliar o tamanho da pupila em condições de iluminação e escuridão. A assimetria pupilar maior quando há iluminação (com uma pupila maior que responde de forma lenta à estimulação com luz direta) indica disfunção parassimpática da pupila. Por outro lado, uma anisocoria maior na escuridão (com reatividade pupilar normal à luz, mas com dilatação lenta no escuro) sugere paresia oculossimpática no lado da pupila menor. Na anisocoria fisiológica, a extensão da anisocoria permanece bastante constante, independentemente do nível de iluminação, e as pupilas respondem normalmente à luz e à estimulação de perto. É preciso lembrar que a anisocoria patológica representa uma anormalidade pupilar eferente e não resulta de lesão em via aferente com envolvimento do nervo óptico, quiasma ou trato óptico.
Figura 23 - Atrofia óptica dominante. Palidez temporal extrema, com aparência escavada (asteriscos). Reimpresso com permissão de Prasad S et al.165
Figura 24 - Hemianopia bitemporal. (a) Imagem de ressonância magnética pós-contraste sagital, mostrando um macroadenoma hipofisário. (b) Hemianopia bitemporal superior decorrente de compressão quiasmática inferior.
Figura 25 - Hemianopia bitemporal. (a) Imagem de ressonância magnética T2-ponderada, mostrando craniofaringioma cístico. (b) Hemianopia bitemporal predominantemente inferior decorrente de compressão quiasmática superior.
Figura 26 - Hemianopia homônima incongruente a partir do trato óptico, desmielinizando a lesão. (a) Imagem de ressonância magnética de recuperação por inversão líquido-atenuada, mostrando sinal aumentado no trato óptico esquerdo (seta). Numerosas lesões de substância branca adicionais foram observadas. (b) Hemianopia homônima direita incongruente (maior envolvimento do olho esquerdo).
Figura 27 - Quadrantanopia superior. (a) Imagem de ressonância magnética subsequente à ressecção de cavernoma temporal direito. (b) Quadrantanopia homônima superior resultante de lesão nas radiações ópticas inferiores junto à alça de Meyer.
Figura 28 - Quadrantanopia inferior. (a) Hemorragia parietal esquerda. (b) Quadrantanopia predominantemente inferior direita.
Figura 29 - Hemianopia homônima poupadora de mácula. (a) Imagem difusão-ponderada, mostrando infarto do lobo occipital esquerdo na distribuição da artéria cerebral posterior, poupando o polo occipital. (b) Hemianopia homônima direita com preservação macular.
A anisocoria fisiológica de 0,4 mm ou mais pode ser vista em cerca de 20% dos indivíduos e, quando indivíduos normais são observados frequentemente por um período de 5 dias, a incidência sobe para 40%.73 O exame de fotos antigas costuma ser útil para documentar a anisocoria fisiológica de longa duração.
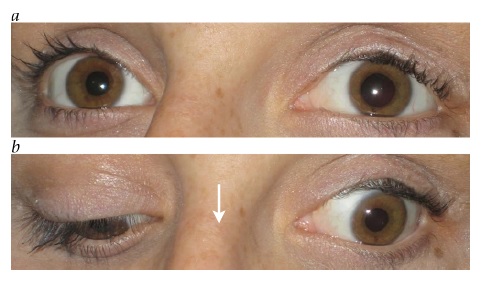
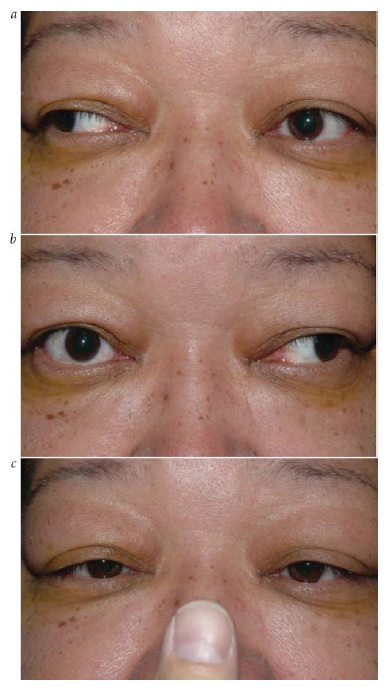
Além da miose (pupila pequena), os outros achados essenciais resultantes da perda da inervação simpática na síndrome de Horner incluem ptose ipsilateral leve e anidrose facial.74 Injeção conjuntival unilateral e pressão intraocular baixa são outros potenciais sinais de interrupção simpática. A ptose da pálpebra superior associada à síndrome de Horner tipicamente mede apenas alguns milímetros, porque a inervação dos levantadores da pálpebra permanece intacta, embora a inervação dos músculos de Müller seja perturbada. Com frequência, a síndrome de Horner também inclui “ptose para cima e para baixo”, referente à elevação discreta da pálpebra inferior. Isto pode dar a falsa impressão de que o olho está afundado dentro da órbita (pseudoenoftalmos).
O padrão de anidrose que acompanha a síndrome de Horner depende de esta ser devida a uma lesão de neurônio de 3ª ordem ou a uma lesão mais proximal.75 Após a sinapse no gânglio cervical superior, a maioria das fibras sudomotoras da face segue ao longo da artéria carótida externa, e não com as fibras destinadas ao olho junto à artéria carótida interna. Sendo assim, com lesões de neurônio de 3ª ordem, pode haver anidrose em um pequeno trecho do lado do nariz, mas, tipicamente, a maior parte da face é poupada. Em contraste, as lesões de neurônios de 1ª ou 2ª ordem podem produzir anidrose em toda a metade ipsilateral da face.
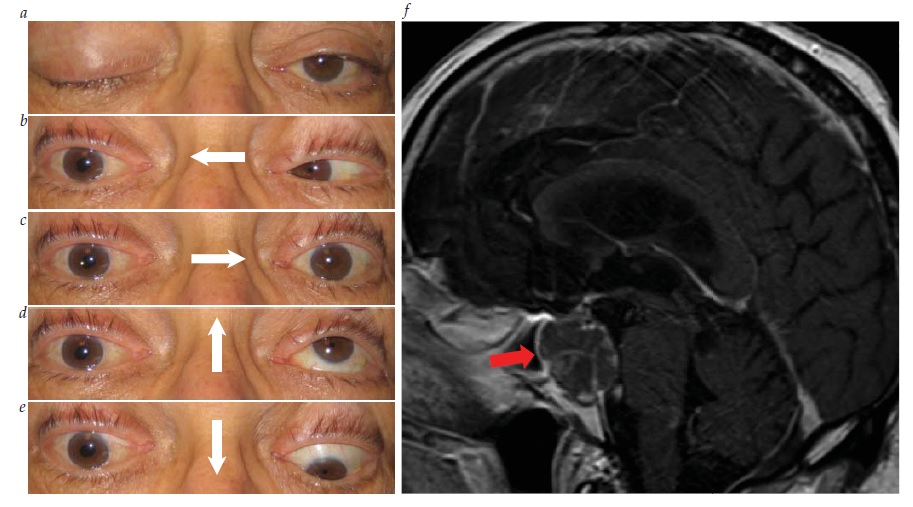
Os sinais clínicos observados ao exame físico, muitas vezes, constituem a melhor forma de localizar a síndrome de Horner. Os sinais tronco encefálicos e medulares espinais sugerem envolvimento de neurônio de 1ª ordem. Uma causa clássica da síndrome de Horner é o infarto medular dorsolateral (síndrome de Wallenberg), que comumente também inclui entorpecimento facial ipsilateral cruzado com entorpecimento corporal contralateral, ataxia, disartria e visão dupla decorrente de desvio enviesado [ver Figura 30].76 Uma síndrome de Horner de 2ª ordem é sugerida por dor no braço, enfraquecimento da mão ou história de cirurgia ou traumatismo cervical. O tumor de Pancoast, envolvendo o ápice do pulmão, é causa importante, ainda que infrequente, de síndrome de Horner de 2ª ordem.77 Os achados de dor cervical e síndrome de Horner ipsilateral, particularmente após o traumatismo, devem ser considerados dissecção de carótida até que se prove o contrário [ver Figura 31].78 Outras causas de síndrome de Horner de 3ª ordem pode incluir obstrução da carótida, lesão em seio cavernoso ou cefaleias autônomas (como a cefaleia em salvas). Entre as causas pediátricas da síndrome de Horner, pode estar o traumatismo ao nascimento, mas é extremamente importante considerar a possibilidade de neuroblastoma oculto. A heterocromia da íris é um sinal sugestivo de síndrome de Horner congênita.
Teste farmacológico para síndrome de Horner. O teste farmacológico se faz necessário quando o diagnóstico da síndrome de Horner é incerto. Embora a cocaína seja usada menos comumente por causa da disponibilidade limitada, é importante rever seu mecanismo de ação [ver Figura 32]. A cocaína bloqueia a recaptação de noradrenalina pelo neurônio de 3ª ordem e, na pupila normal, causa dilatação pupilar. Quando há perturbação simpática envolvendo a pupila (em qualquer um dos três neurônios da via), a noradrenalina não é liberada pelo neurônio de 3ª ordem e, com isso, seu efeito não é potencializado pela presença da cocaína. Uma pupila com disfunção simpática decorrente de síndrome de Horner, portanto, falha em dilatar em resposta à cocaína. Os tamanhos pupilares devem ser avaliados no momento basal, em 40 minutos e em 60 minutos após a instilação de uma gota de solução de cocaína a 10% em cada olho. Em geral, a presença de pelo menos 1,0 mm de anisocoria em resposta ao teste é um resultado positivo confiável.79 Embora a cocaína confirme a presença da síndrome de Horner (p. ex., distinguindo-a da anisocoria fisiológica), não define adicionalmente qual neurônio da via simpática está afetado.
Figura 30 - Síndrome de Horner de 1ª ordem. (a) Miose e ptose de lado direito. Havia entorpecimento na lateral direita da face e no lado esquerdo do corpo. Havia disartria e ataxia de lado direito. (b) Imagem difusão-ponderada mostrando infarto medular lateral direito (síndrome de Wallenberg).
Figura 31 - Síndrome de Horner de 3ª ordem. (a) Miose e ptose de lado esquerdo associadas com dor cervical subsequentes a um acidente de veículos motorizados. (b) Imagem de ressonância magnética do pescoço T1-ponderada com saturação gordurosa, mostrando dissecção carótica direita com trombo mural em crescente (seta).
Figura 32 - Teste de cocaína para síndrome de Horner. (a) Miose e ptose de lado direito. (b) Após a instilação de cocaína (inibidor de recaptação de noradrenalina) em ambos os olhos, a pupila direita falha em dilatar, enquanto a pupila esquerda dilata normalmente.
O teste com gotas de apraclonidina (0,5 ou 1%) emergiu recentemente como um método extremamente útil para detectar a síndrome de Horner [ver Figura 33].80 A apraclonidina é um agonista de receptor a-1 que, normalmente, exerce efeito mínimo ou nulo sobre o tamanho da pupila. Pacientes com síndrome de Horner, entretanto, apresentam midríase da pupila afetada, devido à supersensibilidade da desnervação, em resposta à apraclonidina. Assim, após a instilação de uma gota de apraclonidina em cada olho, a anisocoria, frequentemente, é revertida em síndrome de Horner, com a pupila menor se tornando maior do que a pupila controle não afetada. Um alerta importante com relação ao teste com apraclonidina é a ocorrência frequente de resultados falso-negativos quando o teste é usado no início do curso da anisocoria aguda, porque pode demorar 3-7 dias para que haja supersensibilidade.81
A distinção entre síndrome de Horner de 2ª ordem (pré-ganglionar) e de 3ª ordem (pós-ganglionar) é importante, porque a primeira pode ser sinal de neoplasia subjacente. O teste de hidroxianfetamina pode permitir ao clínico distinguir essas possibilidades.82 A hidroxianfetamina intensifica a liberação de noradrenalina a partir do terminal de 3ª ordem, desde que este esteja intacto. Desta forma, se a síndrome de Horner for pós-ganglionar (3ª ordem), a dilatação pupilar será precária em resposta à hidroxianfetamina. Em contraste, no contexto de síndrome de Horner de 2ª ou 3ª ordem, a pupila miótica irá se dilatar normalmente em resposta à hidroxianfetamina.
Tratamento da síndrome de Horner. As decisões referentes ao tratamento para pacientes adultos síndrome de Horner dependem amplamente da localização. Na maioria dos casos, a imagem deve ser direcionada para a região apropriada.83 Quando a localização é imperfeita após o exame clínico e teste farmacológico, uma síndrome de Horner isolada e inexplicável pode requerer exames de imagem do encéfalo, pescoço e tórax. Quando há dor cervical, é particularmente importante obter RNM ou angiografia convencional do pescoço, a fim de excluir as hipóteses de trombose ou dissecção carótica.84 As RNMs T1-ponderadas axiais são bastante valiosas e podem mostrar a anormalidade de dinal intenso em crescente (trombo mural) na parede do vaso afetado. Crianças com síndrome de Horner, sem história de traumatismo, devem ser testadas quanto à possibilidade de neuroblastoma ou outra lesão em forma de massa. Para estes casos, é sugerida a RNM do encéfalo, pescoço, tórax e abdome, aliada à triagem de catecolamina na urina.85
Figura 33 - Teste de apraclonidina para síndrome de Horner. (a) Miose e ptose de lado direito. (b) Decorridos 60 minutos da instilação de apraclonidina (agonista simpático fraco) em ambos os olhos, a pupila direita dilata e a ptose se resolve, exibindo hipersensibilidade de desnervação. A pupila esquerda não está afetada.
A dilatação pupilar patológica pode resultar de uma lesão no mesencéfalo, III nervo craniano, gânglio ciliar ou músculo da íris. Uma pupila dilatada de origem mesencefálica usualmente está associada a outros sinais de disfunção referíveis a esta região (hemiparesia, ataxia, perda sensorial, retração palpebral ou nistagmo de convergência-retração). A midríase relacionada à disfunção do III nervo ocorre com frequência no contexto de ptose ou déficits de motilidade ocular. Por fim, uma pupila dilatada isolada pode ocorrer com a lesão do gânglio ciliar, bloqueio neuromuscular ou lesão direta à íris (por traumatismo, inflamação ou glaucoma de ângulo fechado).
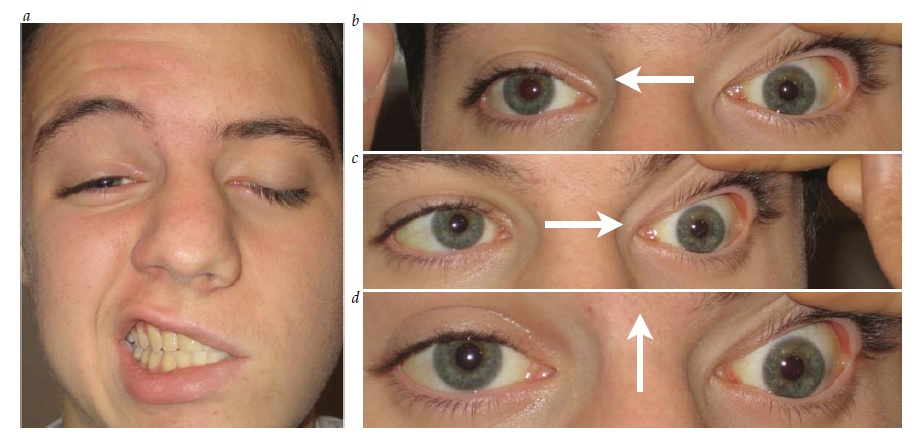
Lesões de III nervo. O envolvimento pupilar, frequentemente, acompanha certas etiologias de paralisia de III nervo. Como as fibras pupilares estão localizadas superficialmente (dorsomedialmente) no nervo, são vulneráveis a processos compressivos como aneurisma ou herniação uncal.
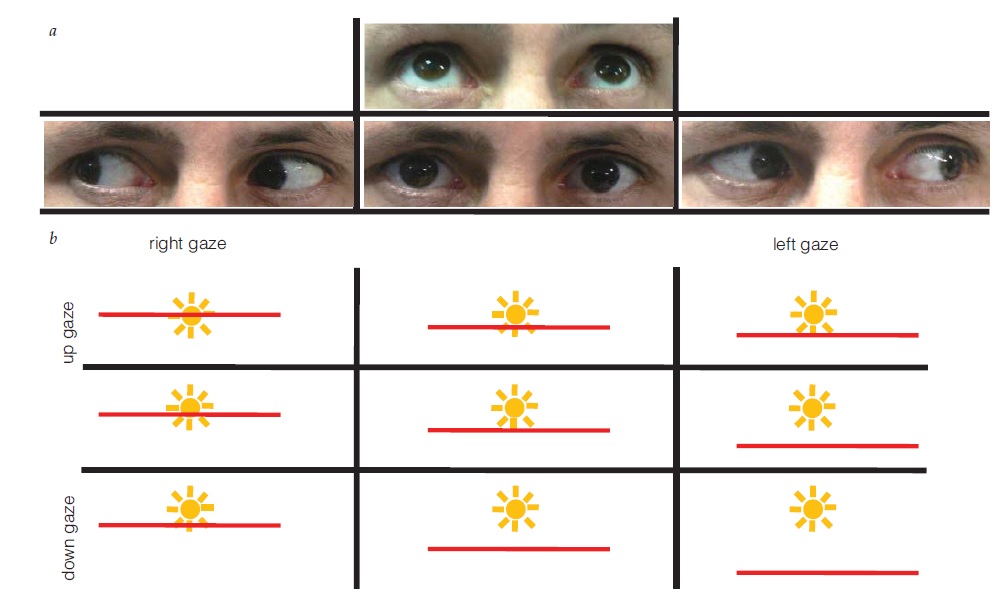
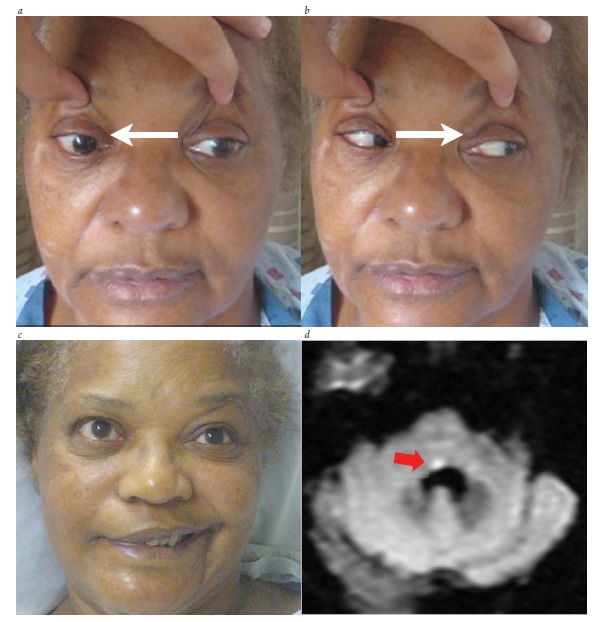
A avaliação de um paciente com paralisia de III nervo com envolvimento de pupila isolada deve incluir exames de imagem para avaliação da possibilidade de compressão a partir de uma lesão como um aneurisma de artéria comunicante posterior (ACoP) [ver Figura 34; discutido adicionalmente também na seção subsequente, sobre paralisia do III nervo].
A regeneração aberrante das estruturas inervadas pelo III nervo, frequentemente, ocorre vários após a aquisição de lesões por traumatismo ou compressão. Um sinal de sincinesia anormal é a contrição pupilar que acompanha a adução do olho. Isto ocorre porque as fibras destinadas ao músculo reto medial inervam inadvertidamente os músculos constritores pupilares.86
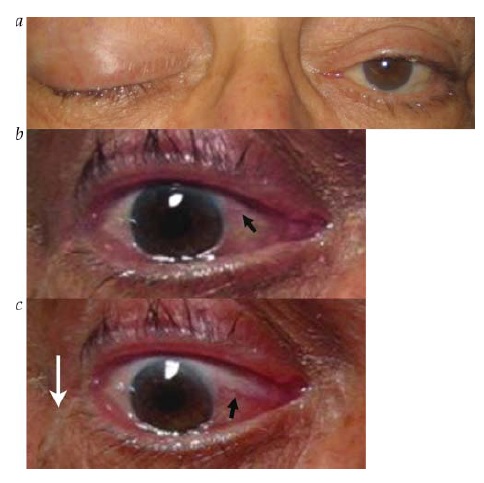
Pupila tônica. Uma pupila tônica resulta da interrupção do suprimento parassimpático que chega do gânglio ciliar [ver Figura 35].87 Neste distúrbio, a pupila tipicamente apresenta contrição precária à luz com constrição relativamente preservada a estímulos próximos, seguida de redilatação “tônica” lenta. A dissociação luz-proximidade de uma pupila tônica se deve à existência de uma proporção da ordem de 30:1 entre as fibras mediadoras da acomodação e as fibras mediadoras da constrição pupilar no gânglio ciliar.87 Desta forma, o dano ao gânglio ciliar tende mais a comprometer a constrição pupilar à luz do que a miose associada à acomodação. A paralisia segmentar da íris é outro achado típico de uma pupila tônica, que ocorre devido à interrupção parcial de seu suprimento parassimpático.
A maioria dos casos de pupila tônica é idiopática e ocorre em mulheres jovens na faixa etária de 20-40 anos. A síndrome de Adie refere-se à combinação de pupila tônica com reflexos tendinosos profundos diminuídos (em particular, de percussão do joelho), provavelmente devido à lesão de ambos os gânglios, da raiz dorsal e ciliar. As pupilas tônicas também podem ocorrer a partir de vários processos locais afetando o gânglio ciliar, incluindo traumatismo, tumor, isquemia ou infecção. Em alguns casos, as pupilas tônicas uni- ou bilaterais são manifestação de um processo autonômico mais amplamente disseminado.
Figura 34 - Paralisia do III nervo com envolvimento pupilar. (a) Ampliação pupilar esquerda, ptose e hipotropia. (b) Olhar fixo para cima comprometido no lado esquerdo. (c) Olhar fixo para baixo comprometido no lado esquerdo. As setas indicam a direção da tentativa de olhar fixo. (d) A angiografia revelou um aneurisma comunicante posterior comprimindo o III nervo.
Uma pupila tônica mostra hipersensibilidade de desnervação que a faz montar uma resposta exagerada à pilocarpina diluída (0,125%).88 Esta solução pode ser preparada misturando até 0,1 cc de pilocarpina a 1% com 0,7 cc de salina estéril em uma seringa de tuberculina de 1 cc. O teste é considerado positivo quando a pupila afetada apresenta contrição maior que a da pupila normal decorrida 1 hora da instilação de uma gota em cada olho. Na maioria dos casos de pupila tônica unilateral, a anisocoria é revertida com o teste de pilocarpina diluída, porque a pupila tônica maior se contrai e se torna menor do que a outra pupila.
Figura 35 - Pupila tônica. (a) Pupila ampliada à esquerda, mostrando fraca reatividade à luz. (b) Constrição pupilar notavelmente preservada à acomodação (visão de alvo próximo).
Midríase farmacológica. Uma pupila farmacologicamente dilatada não responde à estimulação luminosa nem à estimulação de perto. Uma pupila farmacológica é distinguida de uma pupila tônica pela falta de resposta à estimulação de perto; e é distinguida da paralisia de III nervo pela ausência dos demais déficits de motilidade desse nervo. O bloqueio farmacológico ocorre a partir da exposição a fármacos oftálmicos, como atropina, tropicamida ou ciclopentolato. Os adesivos de escopolamina e ipratrópio em aerossol, comumente, causam midríase farmacológica. Os contaminantes ambientais com efeito análogo ao da atropina incluem o estramônio e blue nightshade.89
Na fase aguda, uma pupila farmacologicamente dilatada pode ser confirmada avaliando a resposta a gotas de pilocarpina a 1%. Este agente é um forte constritor pupilar que reverterá quase todas as causas de midríase, contudo, as pupilas com midríase farmacológica falharão em se contrair.90 Estes testes não podem ser interpretados com segurança se tiverem sido realizados após atraso, quando os efeitos do bloqueio farmacológico forem incompletos.
Anormalidades pupilares em outros distúrbios. A midríase isolada transitório, às vezes, pode ser acompanhada de enxaqueca.91 A pupila dilatada pode ser produzida por hiperatividade simpática do dilatador da íris ou disfunção parassimpática.92 Em alguns casos, a pupila pode exibir características de pupila tônica93 ou assumir um formato ovoide.94 As pupilas em forma de girino referem-se aos achados de midríase e distorção pupilar segmentar. Este fenômeno dura alguns minutos e pode ser acompanhado de dor ou sensação anormal no olho afetado. Este achado pupilar pode ser devido ao espasmo segmentar do músculo dilatador da íris. Muitos pacientes com convulsões tônico-clônicas generalizadas apresentarão midríase bilateral. Todavia, também pode haver midríase ou miose ictal unilateral, seja ipsilateral ou contralateral ao foco cortical.95,96
Os movimentos oculares, enfim, são atendidos pelos nervos oculomotores (nervos cranianos III, IV e VI), que inervam os seis músculos extraoculares de cada olho. O nervo oculomotor (III) inerva os músculos reto medial, reto inferior, reto superior e oblíquo inferior, além do levantador da pálpebra. Seu complexo nuclear reside no mesencéfalo dorsal, anterior ao aqueduto cerebral. Em geral, os axônios que chegam dos subnúcleos oculomotores seguem no nervo ipsilateral, com exceção dos axônios oriundos do subnúcleo do reto superior, que seguem através do complexo do III nervo contralateral e se unem ao III nervo desse lado.97,98 Adicionalmente, um único núcleo caudado central emite fibras que unem ambos os III nervos para inervar bilateralmente os músculos levantadores da pálpebra.99
O nervo troclear (IV) inerva o músculo oblíquo superior. Seu núcleo está assentado na junção pontomesencefálica, ventralmente ao aqueduto cerebral. Diferente de todos os outros nervos cranianos, estes axônios saem dorsalmente do tronco encefálico e, então, decussam posteriormente. Por fim, inervam o músculo oblíquo superior contralateral.
O nervo abducente (VI) inerva o músculo reto lateral. Seu núcleo repousa na ponte dorsal, em estreita proximidade com o fascículo do nervo facial (VII). O fascículo do VI nervo segue ventralmente, ao longo dos tratos corticospinais, antes de sair anterolateralmente na junção pontomedular. Em seguida, ascende seguindo pelo canal de Dorello no clivo, até alcançar o seio cavernoso.
No seio cavernoso, o III e o IV nervos estão situados ao longo da parede lateral, enquanto o nervo abducente está em posição mais medial e adjacente à artéria carótida interna. Estes três nervos oculomotores saem do seio cavernoso via fissura orbital superior e, então, atravessam o ápice orbital para alcançar seus músculos-alvo.
A diplopia binocular é o sintoma clínico cardinal do desalinhamento dos olhos. A diplopia que persiste mesmo com um dos olhos fechados (diplopia monocular) mais frequentemente representa patologia junto ao próprio olho em si (p. ex., afetando a córnea, lentes ou retina). Uma vez identificada a diplopia como sendo binocular, indícios localizadores podem ser determinados perguntando se a diplopia é horizontal ou vertical e se piora em determinada direção do olhar fixo ou com a fixação do olhar à distância. Alguns pacientes com desalinhamento ocular podem não descrever a diplopia. Uma razão em potencial para isto pode ser a visão precária em um dos olhos (incluindo as possibilidades de neuropatia óptica concomitante, ptose ou ambiopia prévia de um dos olhos). Adicionalmente, com o desalinhamento ocular grave, um paciente pode não descrever a experiência de visão dupla porque a segunda imagem falsa é percebida tão longe que acaba sendo facilmente ignorada.
O primeiro passo para determinar a causa da diplopia binocular é examinar ducções oculares (movimentos de cada olho individualmente) e versões (movimentos dos olhos juntos). Os movimentos dos olhos devem ser cuidadosamente examinados em todas as direções, para identificar as anormalidades de enfraquecimento ou hiperatividade muscular. O enfraquecimento em determinada direção do olhar fixo pode ser parcial ou completo e resultar de disfunção ao nível do nervo craniano, músculo ocular ou junção neuromuscular. A possibilidade de restrição mecânica (p. ex., a partir de uma massa orbital ou fibrose muscular extraocular) pode ser testada avaliando as ducções forçadas, usando um aplicador com algodão na ponta para girar o globo após a aplicação de anestesia tópica. Em pacientes com paresia não restritiva, o olho pode ser movido em toda a extensão de uma ducção normal. Por fim, a fadigabilidade dos movimentos oculares deve ser examinada (p. ex., sustentar o olhar fixo para cima) para avaliar a possibilidade de distúrbio da junção neuromuscular.
Uma perturbação leve da motilidade pode ser sintomática e, mesmo assim, difícil de detectar com o simples exame direto de ducções e versões. Nesta situação, o teste de cobertura alternada é uma técnica extremamente útil para identificar e caracterizar o desalinhamento ocular. Enquanto o indivíduo fixa um alvo com ambos os olhos, o examinador cobre alternadamente cada olho. No contexto de desalinhamento ocular, cada vez que um olho for descoberto exibirá uma sacada corretiva para refixação no alvo. Esta técnica interrompe a visão binocular do paciente, permitindo que um pequeno desvio dos olhos se torne manifesto.
O desalinhamento vertical dos olhos também pode ser avaliado com o bastão de Maddox, colocado por convenção sobre o olho direito. Este dispositivo também previne a fusão binocular, porque o observador vê, ao mesmo tempo, imagens desiguais (um ponto de luz com o olho esquerdo e uma linha vermelha, com o direito). Se os olhos estiverem desalinhados, a linha vermelha não interceptará o ponto de luz; em vez disso, será deslocada na direção oposta ao desalinhamento. Exemplificando, a linha vermelha é percebida embaixo do ponto de luz no contexto de hiperforia direita. O bastão de Maddox proporciona um método sensível para avaliar pequenos desalinhamentos que podem não ser evidentes no teste de cobertura alternada.
O teste de alinhamento por cobertura alternada ou com bastão de Maddox tipicamente é repetido nas posições cardinais do olhar fixo. Deste modo, um desalinhamento dos olhos pode ser identificado como hiperforia (elevação relativa de um olho), exoforia (posição relativa para fora de um olho) ou esoforia (posição relativa para dentro de um olho). O desalinhamento é considerado concomitante se for aproximadamente igual nas diferentes direções do olhar fixo, sendo considerado não concomitante se for maior em determinada direção do olhar. Um desalinhamento ocular não concomitante se for maior na direção da ação do(s) músculo(s) enfraquecido(s).
A paralisia completa e isolada do III nervo causa enfraquecimento de elevação, depressão e adução do olho, combinado com ptose e midríase. Dependendo da causa específica, a paralisia completa do III nervo pode envolver a pupila (causando midríase) ou poupá-la [ver Figuras 34 e 36]. Na paralisia parcial do III nervo, diferentes padrões de motilidade comprometida podem ocorrer com ou sem envolvimento pupilar. Um achado característico é o olho afetado se mostrar hipotrópico no olhar fixo para cima, mas hipertrópico no olhar fixo para baixo, devido ao enfraquecimento combinado dos músculos reto superior e inferior.
Em oposição às lesões de nervo ou fascículo do III nervo, uma lesão do núcleo do III nervo causará anormalidades bilaterais. Especificamente, há ptose bilateral (porque o núcleo caudal central supre ambos os músculos levantadores da pálpebra) e déficits de elevação bilaterais (porque o subnúcleo do reto superior envia fibras através do núcleo do III nervo contralateral para se unirem ao nervo oposto) [ver Figura 37].100 O quadro clínico clássico de paralisia unilateral nuclear do III nervo, portanto, é a midríase ipsilateral; enfraquecimento ipsilateral dos músculos reto medial, reto inferior e oblíquo inferior; ptose bilateral; e enfraquecimento bilateral do reto superior.
Ao seguir, ventralmente, pelo mesencéfalo, o fascículo do III nervo está vulnerável a lesões intraparenquimais. Existe a possibilidade de déficits parciais, de acordo com o arranjo topográfico das fibras junto ao fascículo do nervo.101 Nestes casos, déficits neurológicos, muitas vezes, acompanham a paralisia do III nervo. Exemplificando, uma lesão que também afete os tratos corticospinais no pedúnculo cerebral causará hemiparesia contralateral (síndrome de Weber); uma lesão envolvendo o núcleo rubro causará tremor no membro contralateral (síndrome de Benedikt); e uma lesão envolvendo o brachium conjunctivum (pedúnculo cerebelar superior) causará ataxia contralateral (síndrome de Claude).102
,
Figura 36 - Paralisia microvasculopática do III nervo. (a) Ptose direita completa. (b) Os olhos estão exotrópicos. (c) Olhar fixo para cima de lado direito limitado. (d) Olhar fixo para baixo de lado direito limitado. A pálpebra está sendo mantida aberta. Note que a pupila direita é discretamente maior do que a esquerda, porém a constrição à luz estava normal e simétrica. As imagens estavam normais, sem lesão abrangente. As setas indicam a direção da tentativa de olhar fixo.
A regeneração aberrante diz respeito à ligação errada de estruturas inervadas pelo III nervo, que leva a padrões de cocontrações.103 São manifestações comuns dessa condição a contração do levantador da pálpebra durante a adução ou depressão do olho, ou a miose de uma pupila dilatada durante a adução (conforme discutido antes) [ver Figura 38]. Este fenômeno ocorre nas formas primária e secundária. A regeneração aberrante primária sugere compressão insidiosa, tipicamente em consequência de uma lesão em expansão no seio cavernoso, como um meningioma, aneurisma, tumor ou outra massa.104 A regeneração aberrante secundária, por outro lado, ocorre durante a fase de recuperação subsequente à paralisia aguda do III nervo, geralmente após traumatismo, mas também após enxaqueca oftalmoplégica, apoplexia hipofisária ou inflamação. Não ocorre regeneração aberrante após a paralisia vasculopática do III nervo.
Etiologias. A paralisia do III nervo nuclear ou fascicular é tipicamente devida ao infarto do mesencéfalo decorrente da obstrução de uma pequena artéria penetrante oriunda da ACP proximal. Outras possíveis causas de doença mesencefálica incluem os tumores, malformações vasculares, abscessos, desmielinização e distúrbios inflamatórios. No espaço subaracnóideo, um aneurisma em expansão da artéria ACoP é causa importante de paralisia do III nervo. Cerca de 90% dos pacientes com aneurisma sintomático sem ruptura apresentam paralisia do III nervo.105
A paralisia de III nervo microvascular comumente está associada a fatores de risco vasculares, tais como hipertensão, diabetes, hiperlipidemia, idade avançada e tabagismo. A microcirculação comprometida leva à desmielinização isquêmica focal de axônios no núcleo do nervo.106–108 A maioria destes pacientes exibe preservação pupilar, porque as fibras pupilares têm localização periférica e mais próxima do suprimento sanguíneo fornecido pelos vasos dos nervos circundantes. Há certo grau de envolvimento pupilar, todavia, com cerca de 40% dos casos demonstrando menos de 1 mm (e até o máximo de 2,5 mm) de anisocoria.109 A paralisia microvascular do III nervo, frequentemente, está associada à dor orbital, que pode ser bastante intensa e possivelmente resulta da isquemia das fibras sensoriais do trigêmeo que se unem ao III nervo junto ao seio cavernoso.110 Os déficits de motilidade a partir da paralisia microvascular do III nervo têm prognóstico excelente de recuperação, tipicamente em 8-12 semanas.111
O traumatismo grave é outra causa comum de paralisias de III nervo, envolvendo tração na base do crânio ou fratura dos ossos da órbita ou da base do crânio.112 Por outro lado, uma paralisia de III nervo que segue a um traumatismo craniano mínimo pode indicar uma lesão estrutural subjacente.113,114 Apesar do prognóstico favorável para recuperação após a paralisia traumática do III nervo, existe uma alta incidência de regeneração aberrante secundária.
Ocasionalmente, as paralisias de III nervo progressivas são causadas pelo crescimento de algum tumor primário do nervo ou de sua bainha. Estas lesões incluem neurinomas, neurofibromas e schwannomas.115 Nestes casos, os exames de neuroimagem identificarão um nervo aumentado e intensificado. Menos comumente, um meningioma maligno, glioblastoma multiforme ou linfoma podem afetar diretamente o III nervo.
A herniação uncal pode causar compressão direta do III nervo contra a borda livre do tentório. Além dos déficits do III nervo, estes pacientes apresentarão depressão do estado mental, entre outros déficits neurológicos proeminentes.
Figura 37 - Paralisia nuclear do III nervo. (a) Ptose bilateral grave (com contração compensatória do músculo frontal). (b) Limitação grave da elevação bilateral. A limitação do olhar fixo vertical não foi superada pela manobra oculocefálica. As setas indicam a direção da tentativa de olhar fixo. (c) Adução esquerda diminuída. (d) Adução direita discretamente diminuída. (e) A imagem de ressonância magnética (RNM) pré-operatória axial cerebral de recuperação de inversão atenuada por líquido mostrou a presença de uma ampla massa heterogênea na linha média comprimindo o mesencéfalo dorsal (seta). (f) RNM obtida 2 anos após a ressecção cirúrgica, mostrando atrofia focal no mesencéfalo dorsal (seta). Reimpresso com permissão de Prasad S e Volpe NJ.166
Na população pediátrica, a enxaqueca oftalmoplégica pode causar paralisia de III nervo isolada transitório. Esta forma rara de enxaqueca complicada tipicamente se manifesta antes dos 10 anos de idade.116 Cefaleia ipsilateral e náusea frequentemente acompanham os movimentos oculares anormais. Por razões desconhecidas, o envolvimento do III nervo é mais comum, ocorrendo em 95% dos casos. A etiologia pode estar relacionada a uma isquemia transitório ou à compressão do nervo junto ao seio cavernoso por uma artéria carótida edemaciada e dilatada.117 Esta condição é um diagnóstico de exclusão, estabelecido depois que uma investigação, com exames de imagem, sangue e, muitas vezes, punção lombar, não mostra nenhum achado revelador. A enxaqueca oftalmoplégica deve ser considerada extremamente improvável em um adulto sem história de episódios similares na infância.
Figura 38 - Regeneração aberrante do III nervo. (a) Dilatação pupilar discreta de lado esquerdo. (b) Olhar fixo para baixo de lado esquerdo deficiente, com elevação espontânea da pálpebra esquerda (decorrente da ligação errada das fibras destinadas ao reto inferior inervando o levantador da pálpebra). As setas indicam a direção da tentativa de olhar fixo. Reimpresso com permissão de Prasad S e Volpe NJ.166
Tratamento. Imagem. A investigação apropriado para um paciente com paralisia de III nervo isolada depende da idade desse paciente e da função pupilar. Em adultos com paralisia de III nervo parcial ou completa envolvendo a pupila, não há controvérsias quanto à investigação: estes pacientes precisam com urgência de exames de imagem para excluir a hipótese de aneurisma de ACoP ou outra massa.118 Tanto a angiografia por TC como a angiografia por ressonância magnética são úteis, mas a exata sensibilidade e disponibilidade destes exames varia entre as instituições.119 Mesmo assim, se estes exames resultarem negativos, é possível que continue havendo necessidade de realizar um angiograma de cateterismo nestes pacientes, porque os aneurismas pequenos são potencialmente perdidos nos exames de imagem não invasivos.
Em pacientes com paralisia de III nervo parcial com preservação pupilar, o limiar para obtenção de imagem também é baixo. Historicamente, uma abordagem razoável tem sido observar estes pacientes por vários dias. Nos casos de paralisia aguda do III nervo em evolução decorrente de aneurisma, a midríase ocorreria em quase todos os casos dentro desse período de observação.120 Havendo desenvolvimento de midríase, torna-se necessária a obtenção urgente de imagens. Na era moderna, entretanto, devido aos altos riscos de perda do diagnóstico de aneurisma e à disponibilização aumentada de RNM ou TC, tornou-se adequada a estratégia da obtenção antecipada de imagens de todos os pacientes com paralisia de III nervo parcial isolada.
Para pacientes com paralisia de III nervo completa com preservação pupilar, que tenham idade acima de 50 anos e apresentem fatores de risco vasculares, uma etiologia microvasculopática é incrivelmente provável, e a observação clínica pode ser uma medida razoável. Se estes pacientes falharem em se recuperar espontaneamente dentro de 12 semanas, então torna-se necessária a obtenção de neuroimagens detalhadas. Todavia, o limiar apropriado para obtenção de neuroimagem nesta população de paciente continua sendo uma fonte de controvérsias. Existe um número crescente de relatos de lesões diagnosticadas por RNM em pacientes que mimetizam a paralisia microvasculopática do III nervo.121 Desta forma, na prática clínica, pode ser prudente obter exames de imagem também destes pacientes.
A obtenção de imagens deve ser considerada no caso de pacientes com paralisia de III nervo causada por traumatismo, em especial se a extensão do traumatismo era relativamente menor, devido à incidência das lesões em forma de massa subjacentes (incluindo aneurismas). As imagens obtidas destes pacientes também avaliarão o aprisionamento muscular decorrente da fratura da parede orbital.
Exames laboratoriais. Nos casos com suspeita de causa infecciosa, inflamatória ou neoplásica e com RNM negativa ou inespecífica, a investigação adicional pode incluir sorologias para doença de Lyme, sífilis e velocidade de sedimentação eritrocitária para exclusão da hipótese de arterite temporal. Por fim, a análise do LCS incluindo contagens celulares, proteína, glicose, citologia e títulos de Lyme e VDRL pode ser necessária.
A paralisia do IV nervo se manifesta com diplopia vertical e comumente é acompanhada de inclinação contralateral compensatória da cabeça.122 A identificação de uma paralisia do IV nervo em paciente com diplopia vertical envolve aplicação do teste de Parks-Bielchowsky de três etapas. Primeiro, a hipertropia sugere enfraquecimento dos músculos oblíquo superior, reto inferior ipsilateral, oblíquo inferior contralateral ou reto superior contralateral. Em seguida, a hipertropia aumentada no olhar fixo contralateral estreita as possibilidades de enfraquecimento dos músculos oblíquo superior ipsilateral ou reto superior contralateral. Por fim, a hipertropia aumentada na inclinação ipsilateral da cabeça diminui ainda mais as possibilidades, identificando, enfim, o enfraquecimento do oblíquo superior ipsilateral.
Embora ducções anormais possam ser detectadas por observação direta, é comum que os pacientes com desalinhamento vertical apresentem pequenos desvios produtores de comprometimento não visível da motilidade ocular. Desta forma, a avaliação do alinhamento usando cobertura alternada ou bastão de Maddox pode ser particularmente útil para demonstrar o padrão característico de motilidade comprometida [ver Figura 39].
A razão pela qual a hipertropia é exacerbada no olhar fixo contralateral está no fato de a paralisia do oblíquo superior causar enfraquecimento da depressão em adução. A razão pela qual a hipertropia piora com a inclinação ipsilateral da cabeça é que o reflexo ocular de contrarrolagem estimula os torcedores internos ipsilaterais (oblíquo superior e reto superior) e torcedores externos contralaterais (oblíquo inferior e reto inferior). Quando o oblíquo superior está fraco, este reflexo produz aumento compensatório da ação do reto superior, resultando em hipertropia adicional (porque o reto superior é levantador).
A diplopia torsional, resultante de torsão ocular cíclica, muitas vezes acompanha a diplopia vertical na paralisia do IV nervo adquirida. Esta condição pode ser rapidamente avaliada fazendo o paciente olhar uma linha reta horizontal, como a borda inferior de uma porta. Um paciente com torsão cíclica a partir da paralisia do IV nervo unilateral verá tanto uma linha horizontal como uma segunda linha inclinada acima ou abaixo da outra, interceptando-a no lado do olho afetado. A torsão cíclica também pode ser avaliada com o bastão de Maddox duplo, que refrata a fonte de luz em uma linha vermelha (vista pelo olho direito) e em uma linha branca (vista pelo olho esquerdo). O grau de torsão cíclica relativo é medido com a rotação dos filtros até o indivíduo relatar que as linhas estão paralelas. Por fim, a torsão cíclica também pode ser avaliada durante o exame de fundo dilatado, por meio da avaliação da posição da mácula em relação ao disco óptico. A torsão cíclica para fora do olho hipertrópico sugere paralisia do IV nervo, devido à torsão para dentro enfraquecida. Em contraste, a torsão para dentro do olho hipertrópico ocorre no desvio enviesado decorrente da estimulação diminuída do subnúcleo oblíquo inferior (discutida adiante).
Figura 39 - Paralisia de IV nervo de lado direito. (a) Ao exame das ducções oculares, é bastante difícil observar uma anormalidade definida (embora uma hipertropia direita sutil pior no olhar fixo esquerdo possa estar presente). (b) Estimulação da visão do paciente com auxílio de um bastão Maddox, em cada direção do olhar fixo. Quando a linha horizontal é vista atravessando a luz branca, os olhos exibem alinhamento vertical normal. Quando a linha vermelha é vista embaixo da luz branca, significa que há hiperforia direita. Este padrão de achados mostra uma hiperforia direita com a maior separação vertical no olhar fixo para baixo e de lado esquerdo, consistente com uma paralisia do IV nervo de lado direito. Reimpresso com permissão de Prasad S et al. 122
Avaliar a torsão cíclica e o desalinhamento vertical em posição vertical e também na posição de supinação pode ser útil para distinguir entre paralisia do IV nervo e desvio enviesado.123 O desalinhamento se mantém bastante constante entre estas posições com a paralisia do IV nervo, mas é frequentemente reduzido na posição supinada com desvio enviesado, possivelmente devido ao desequilíbrio utricular que faz o desvio enviesado diminuir.
Um indício final da etiologia do desalinhamento vertical advém da amplitude fusional (habilidade de fundir imagens diferentes), que sugere a cronicidade do estrabismo. A amplitude fusional é medida solicitando ao paciente que relate a visão dupla enquanto prismas progressivamente maiores são colocados uns sobre os outros. Uma capacidade fusional maior que 8-10 dioptrias sugere a presença de mecanismos compensatórios encontrados em lesões de longa duração ou congênitas.
A paralisia de IV nervo bilateral, mais comumente resultante de traumatismo, é caracterizada por uma constelação de achados exclusiva.124 O alinhamento vertical de posição primária pode ser muito bom, por causa do efeito de cancelamento das paralisias bilaterais. Pode haver esotropia, dificultando o estabelecimento do diagnóstico inicial por potencialmente sugerir a existência de paralisias do VI nervo. Com um exame minucioso, todavia, as paralisias de IV nervo bilaterais são prontamente identificadas. Primeiro, há hiperdesvios de direita e esquerda, cada um ocorrendo com o olhar fixo contralateral e com a inclinação ipsilateral da cabeça. Em segundo lugar, há uma esotropia que é maior com o olhar fixo para baixo (conhecida como “esotropia de padrão V”, com uma diferença > 15 dioptrias prismáticas entre os olhares fixos para cima e para baixo), devido à abdução enfraquecida na depressão (onde o oblíquo superior atua como abdutor). Por último, muitas vezes há um amplo ângulo de torsão cíclica para fora (> 10°), acompanhado de uma proeminente diplopia torsional. Em casos raros, pode haver paralisia bilateral congênita do IV nervo.
Identificar a paralisia do IV nervo no contexto de paralisia do III nervo concomitante pode ser difícil, porque a falha da adução impede a conclusão da prova de função do oblíquo superior. Neste contexto, o oblíquo superior pode ser avaliado por meio da avaliação de sua função secundária: torsão para dentro do olho abduzido na tentativa de olhar fixo para baixo. O movimento de torsão que indica a função do oblíquo superior é melhor apreciado por meio da observação da rotação de um vaso conjuntival ao longo das diferentes posições do relógio [ver Figura 40].
Etiologias. A causa mais comum de paralisia do IV nervo adquirida é o traumatismo.112,124 O nervo troclear é o mais longo e mais fino dentre todos os nervos cranianos, seguindo ao longo da borda livre do tentório pela cisterna pré-pontina, onde é vulnerável a lesões por esmagamento ou cisalhamento. Nos casos de paralisia de IV nervo bilateral traumática, ambos os nervos frequentemente são lesados no véu medular anterior, onde decussam.124 As paralisias traumáticas do IV nervo podem ocorrer após lesões relativamente pequenas na cabeça e sem perda de consciência nem fratura de crânio.
Figura 40 - Teste de função do IV nervo no contexto de paralisia do III nervo. (a) Ptose completa decorrente de paralisia do III nervo. (b) Um vaso conjuntival é observado (seta preta) com o olhar fixo primário. (c) Na tentativa de olhar fixo para baixo, observa-se que o vaso conjuntival se move da posição de 2 horas para a posição de 3 horas (seta preta), demonstrando a torsão do olho para dentro a partir de um músculo oblíquo superior intacto. A seta branca indica a direção da tentativa de olhar fixo. Reimpresso com permissão de Prasad S e Volpe NJ.166
A paralisia do IV nervo congênita descompensada também é comum e pode se manifestar na fase adulta. Frequentemente, há o aparecimento insidioso de diplopia vertical intermitente e inclinação prolongada da cabeça, que podem ser observados durante a inspeção de fotografias anteriores. Os achados característicos da paralisia do IV nervo congênita incluem a ação exagerada do oblíquo inferior, amplitude fusional vertical ampla e diplopia torsional mínima. A etiologia precisa da paralisia do IV nervo congênita é indeterminada, mas pode incluir hipoplasia de núcleo, traumatismo ao nascimento, anomalia de inserção muscular, fibrose muscular, anormalidades estruturais do tendão ou anormalidades do músculo oblíquo inferior.125 A posterior descompensação ao longo da vida provavelmente está relacionada à quebra da fusão vertical levando à diplopia sintomática, em vez da disfunção progressiva do oblíquo superior.
A isquemia microvascular pode causar paralisia do IV nervo, tipicamente em pacientes com mais de 50 anos que tenham fatores de risco vasculares. A dor periorbital contínua, frequentemente, está presente no momento da apresentação e pode ser intensa. A probabilidade de recuperação espontânea em pouco meses é excelente. Outras causas de paralisia do IV nervo incluem schwannoma, compressão aneurismática, meningite, hidrocefalia e herpes-zoster oftálmico.
A mioquimia oblíqua superior consiste em um microtremor que causa episódios característicos de oscilopsia torsional monocular ou diplopia transitório.126 Pode ocorrer na posição primária ou com movimentos opostos à direção da ação do oblíquo superior. Alguns casos podem ser devidos à compressão do IV nervo por um vaso sanguíneo sobrejacente.
Entre as causas intraparenquimatosas de paralisia do IV nervo, estão a hemorragia mesenfálica, infarto ou desmielinização.127 Dada a proximidade em relação a outras estruturas no mesencéfalo, uma lesão no núcleo ou fascículo proximal do IV nervo pode causar enfraquecimento contralateral do oblíquo superior associado com síndrome de Horner ipsilateral,128 oftalmoplegia internuclear ipsilateral (OII)129 ou DPAR contralateral sem perda visual (por afetar o braço do colículo superior).130
O enfraquecimento do reto lateral devido à paralisia do VI nervo leva a uma diplopia horizontal que é pior no lado afetado e à distância [ver Figura 41]. Muitas vezes, a ducção anormal é facilmente observada, mas, nos casos mais sutis, é necessário demonstrar uma esotropia não concomitante por meio do teste de alinhamento binocular.
Uma lesão do fascículo do VI nervo junto à ponte produz, similarmente, um déficit de abdução ipsilateral. Uma lesão na ponte ventral pode afetar o fascículo do VI nervo e o trato corticospinal descendente, produzindo déficit de abdução ipsilateral com hemiparesia contralateral. Em contraste, uma lesão do núcleo do VI nervo afeta tanto o VI nervo ipsilateral como os interneurônios destinados ao subnúcleo do reto medial contralateral. Esta lesão, portanto, causa déficit de abdução no olho ipsilateral e déficit de adução no olho contralateral. Tomados em conjunto, trata-se de uma paralisia do olhar fixo conjugado. Dada a proximidade entre o fascículo do VII nervo e o núcleo do VI nervo (junto ao colículo facial), uma lesão isolada nesta localização tipicamente produzirá paralisia de olhar fixo ipsilateral e enfraquecimento facial superior e inferior [ver Figura 42].
As lesões pontinas costumam ser isquêmicas, devido à obstrução de um ramo penetrante paramediano oriundo da artéria basilar, contudo, o diagnóstico diferencial também inclui malformação vascular, desmielinização ou neoplasia.131
Figura 41 - Paralisia do VI nervo de lado esquerdo. (a) Olhar fixo direito normal. (b) Abdução limitada do olho esquerdo. As setas indicam a direção da tentativa de olhar fixo. Reimpresso com permissão de Prasad S e Volpe NJ.166
Figura 42 - Síndrome do colículo facial direito. (a) Limitação grave do olhar fixo direito (ambas, abdução do olho direito e adução do olho esquerdo). (b) Olhar fixo esquerdo normal. As setas brancas indicam a direção da tentativa de olhar fixo. (c) Enfraquecimento facial inferior e superior de lado direito. (d) Imagem de ressonância magnética difusão-ponderada mostrando infarto na ponte dorsal direita (seta vermelha). Reimpresso com permissão de Prasad S e Galetta SL.167
Conforme o VI nervo surge ao longo do clivo sob o ligamento petroso no interior do canal de Dorello, situado na base do crânio, é vulnerável à lesão do efeito de massa descendente decorrente da pressão intracraniana elevada. Sendo assim, as paralisias unil- ou bilaterais do VI nervo podem ocorrer no contexto de uma massa supratentorial, trombose sinusal venosa,132 hidrocefalia ou pseudotumor cerebral. Por outro lado, uma paralisia de VI nervo também pode surgir em casos de baixa pressão intracraniana, como pode ocorrer em decorrência de vazamento de LCS.133 As paralisias de VI nervo, nestas circunstâncias, tendem a melhorar logo após a normalização da pressão intracraniana. Massas que crescem na base do crânio, como os meningiomas ou cordomas, também podem lesionar o VI nervo.134
A isquemia microvascular é outra causa comum de paralisia do VI nervo, especialmente em pacientes idosos com fatores de risco vasculares. Similarmente a outras paralisias microvasculopáticas, estas podem ser acompanhadas de dor periorbital contínua substancial. A progressão do déficit de abdução ao longo da 1ª semana não é incomum.135 O prognóstico de recuperação dentro de 3 meses é excelente.
Existem várias causas adicionais de paralisia do VI nervo decorrente de forças de cisalhamento ou fratura da base do crânio ou dos ossos orbitais.112 A síndrome de Gradenigo se refere à paralisia do VI nervo combinada à perda auditiva ipsilateral e dor facial, que ocorre quando a mastoidite infecciosa envolve as estruturas do ápice petroso.136 Em crianças, pode ocorrer uma forma benigna e recorrente de paralisia do VI nervo idiopática, na qual a esotropia dura dias a meses.137
As anormalidades congênitas do VI nervo incluem a síndrome de retração de Duane. As diversas variedades de síndrome de Duane são caracterizadas por uma cocontração paradoxal dos músculos reto lateral e reto medial, causando retração visível do globo e estreitamento da fissura palpebral durante as tentativas de ducção horizontal.138 Um paciente com síndrome de Duane pode se mostrar esotrópico ao olhar fixo lateral ao lado afetado, contudo, é possível distinguir estes pacientes daqueles que apresentam déficits de abdução adquiridos, por apresentarem alinhamento normal (em vez de esoforia) no olhar fixo primário. Estudos de patologia sobre pacientes com síndrome de Duane demonstram a hipoplasia do núcleo do VI nervo e anormalidades do fascículo, com ramos do III nervo suprindo o músculo reto lateral.139
As doenças do espaço subaracnoide, incluindo os processos infecciosos, inflamatórios e neoplásicos, podem afetar múltiplos nervos cranianos. Os processos infecciosos incluem infecções virais, fúngicas ou bacterianas (incluindo tuberculose, sífilis e doença de Lyme). As doenças inflamatórias incluem a sarcoidose e a paquimeningite idiopática. Os processos neoplásicos incluem as formas de meningite carcinomatosa e linfomatosa. Os distúrbios desmielinizantes periféricos, inclusive a síndrome de Guillain-Barré, a variante de Miller Fisher [ver Figura 43], a polineuropatia desmielinizante inflamatória crônica e as neuropatias cranianas idiopáticas, são outras possibilidades a considerar quando há envolvimento de múltiplos nervos oculomotores.
Um processo envolvendo o seio cavernoso ou a base do crânio pode afetar qualquer combinação entre o III, IV ou VI nervo e causar disfunção na 1ª e 2ª divisões do nervo trigêmeo. Entre as considerações neoplásicas, estão incluídos o meningioma, o adenoma hipofisário, o craniofaringioma, o linfoma, as metástases, o cordoma , o condrossarcoma e o carcinoma nasofaríngeo. O aparecimento rápido de cefaleia e oftalmoplegia é fortemente sugestivo de apoplexia de macroadenoma hipofisário [ver Figura 44].140 O herpes-zoster oftálmico pode levar à oftalmoplegia à base de vasculite secundária ou inflamação direta dos nervos oculomotores.141 A mucormicose ou aspergilose pode se disseminar a partir dos seios para dentro do seio cavernoso ou da órbita, particularmente em pacientes imunocomprometidos. A síndrome de Tolosa-Hunt consiste na inflamação idiopática do seio cavernoso ou fissura orbital superior, causando oftalmoplegia dolorosa.142
As lesões vasculares incluem os aneurismas expansíveis e as fístulas cavernosas caróticas (FCC).143 Os aneurismas caróticos podem se manifestar com dor e diplopia decorrente do envolvimento de qualquer nervo oculomotor, mais frequentemente do VI nervo. As FCCs são caracterizadas como sendo de alto fluxo (diretas) ou de baixo fluxo (indiretas), com base na fonte do vaso alimentador e na velocidade do fluxo. A maioria das FCCs de alto fluxo resulta de traumatismo grave na cabeça. Pacientes com FCC de alto fluxo apresentam cefaleia, diplopia, proptose, quemose grave e arteriolização das veias epiesclerais. Um ruído pode ser detectado por auscultação sobre a pálpebra fechada ou sobre o mastoide [ver Figura 45]. As FCCs de baixo fluxo (ou malformações arteriovenosas durais) são conexões anormais entre o seio cavernoso e as artérias que suprem a dura-máter. São mais frequentes em mulheres idosas ou associadas à gravidez, hipertensão, doença do tecido conectivo ou traumatismo craniano. Os sintomas produzidos dependem da via de drenagem venosa e podem incluir diplopia, proptose e quemose com progressão insidiosa.
A OII refere-se à adução retardada ou limitada durante tentativas de sacada horizontal.144–146 A adução conjugada durante as sacadas horizontais normais é facilitada por um subgrupo de interneurônios junto ao núcleo abducente. Estas fibras cruzam a linha média e seguem pelo fascículo longitudinal medial (FLM) contralateral, desde a ponte até o subnúcleo do reto medial do complexo oculomotor, no mesencéfalo. O FLM é altamente mielinizado para sustentar a rápida transmissão neural necessária à abdução de um olho e adução do outro, de maneira quase sincronizada. Até mesmo um leve comprometimento das velocidades de transmissão pelo FLM produz sintomas por comprometer essa sincronicidade, resultando em desalinhamento ocular durante as sacadas horizontais. Diferente de outros tratos mielinizados, em que um comprometimento discreto pode não produzir déficits clínicos evidentes, o sistema de sacadas horizontais coordenadas é extremamente sensível às velocidades de transmissão, tornando a OII uma manifestação frequente de condições desmielinizantes como a EM.
O déficit de adução na OII pode se manifestar como retardo durante a ducção horizontal com excursão total do olho [ver Figura 46] ou como adução incompleta produzindo exotropia. Apesar da adução deficiente durante as sacadas horizontais, a função normal do reto medial na OII tipicamente pode ser demonstrada testando a convergência dos olhos [ver Figura 47]. A convergência é mediada por estímulos separados ao subnúcleo do reto medial, que diferem dos estímulos que chegam via FLM. A dissociação entre adução limitada nas sacadas horizontais e adução preservada durante a convergência destaca a natureza supranuclear do déficit de adução na OII, com os componentes nuclear e infranuclear da adução intactos. A OII frequentemente está associada ao nistagmo horizontal associado mais proeminente no olho em abdução. A fase lenta do nistagmo é oposta à direção da tentativa de olhar fixo, com as sacadas rápidas na direção da tentativa de olhar fixo.
O desequilíbrio dos estímulos utriculares, muitas vezes devido a uma lesão tronco encefálica ou cerebelar, produz desalinhamento ciclovertical dos olhos, tipicamente com um desvio vertical concomitante que não segue um padrão característico de paralisia do III ou IV nervo.147 Como regra geral, em presença de uma lesão pontina, o olho ipsilateral se torna mais baixo, enquanto na presença de uma lesão mesencefálica, o olho ipsilateral fica mais alto. Tipicamente, há uma relativa torsão para dentro do olho mais alto (em contraste com a paralisia do IV nervo, em que o olho mais alto mostra uma relativa torsão para fora). Na reação de inclinação ocular, a hipertropia ocular e a torsão para dentro são acompanhadas de inclinação da cabeça afastando-se do olho mais alto.
Figura 43 - Síndrome de Miller Fisher. (a) Ptose bilateral e enfraquecimento facial maior à esquerda. (b) Olhar fixo direito gravemente limitado. (c) Olhar fixo esquerdo seriamente limitado. (d) Olhar fixo para baixo gravemente limitado. Os reflexos tendinosos profundos estavam deprimidos e o conteúdo de proteína do líquido cerebrospinal estava levemente aumentado. A resolução completa ocorreu em 2 meses. As setas indicam a direção da tentativa de olhar fixo. Reimpresso com permissão de Prasad S e Volpe NJ.166
Figura 44 - Oftalmoplegia complexa decorrente de apoplexia hipofisária (paralisia do II nervo direita, do IV nervo direita e do VI nervo esquerda). (a) Ptose completa direita. (b) Déficit de abdução direito. (c) Déficit de abdução esquerda e déficit de adução direita. (d) Déficit de elevação direita. (e) Déficit de depressão direita. As setas indicam a direção da tentativa de olhar fixo. (f) Imagem de ressonância magnética sagital intensificada por contraste, mostrando um amplo macroadenoma hipofisário com hemorragia. Reimpresso com permissão de Prasad S e Volpe NJ.166
Figura 45 - Arteriolização de vasos epiesclerais associada à fístula carótica-cavernosa direta. Reimpresso com permissão de Prasad S e Volpe NJ.166
Os distúrbios da junção neuromuscular ou dos músculos oculares representam causas adicionais de desalinhamento ocular e diplopia. A miastenia grave é o protótipo da doença da transmissão da junção neuromuscular, produzida, na maioria dos casos, por um anticorpo dirigido contra o receptor nicotínico da acetilcolina. Cerca de metade dos pacientes com miastenia grave inicialmente apresenta sintomas visuais. O exame pode demonstrar enfraquecimento de vários músculos do olho, com falta de conformação a qualquer distribuição nervosa isolada. A associação com ptose variável é comum. Os déficits, muitas vezes, pioram com a fadiga e melhoram após o repouso. Embora a miastenia grave possa exibir padrões complexos de movimentos oculares limitados que mimetizam fielmente uma disfunção de nervo craniano, estes pacientes costumam ser distinguidos pela presença de respostas pupilares normais.
A doença ocular tireoidiana é o distúrbio mais comum dos músculos oculares. Nesta condição, a resposta autoimune anormal faz o músculo ocular se tornar aumentado e fibrótico. Os pacientes costumam apresentar retração palpebral e proptose, além de desalinhamento ocular resultante da restrição dos movimentos oculares [ver Figura 48]. Embora muitos pacientes tenham hipertireoidismo, não é incomum os pacientes serem eutireoidianos ou até terem hipotireoidismo.
Tratamento. Imagem. Muitos pacientes com paralisia de III, IV ou VI nervo requerem RNM para excluir causas estruturais, inflamatórias ou neoplásicas.121 Entretanto, um paciente de idade mais avançada, no qual uma causa microvasculopática seja fortemente considerada, deve permanecer sob observação por várias semanas para verificar a ocorrência de melhora espontânea e, assim, evitar a necessidade de obter imagens. A TC ou RNM das órbitas de pacientes com suspeita de doença ocular tireoidiana, muitas vezes, demonstra músculos oculares anormalmente espessados.
Figura 46 - Oftalmoplegia internuclear direita. (a) Olhar fixo primário. (b) Retardo de adução do olho direito no início de uma sacada para a esquerda. (c) Conclusão da sacada para esquerda. Reimpresso com permissão de Prasad S e Galetta SL.168
Exames laboratoriais. A investigação adicional para pacientes pode incluir sorologias e análise do LCS. Quando há suspeita clínica de miastenia grave, é útil determinar os títulos de anticorpo antirreceptor de acetilcolina. No contexto de miastenia grave ocular pura, todavia, a sensibilidade deste teste é bastante limitada (em torno de 50%). Os exames de condução nervosa com estimulação repetitiva e eletromiografia de fibra única apresentam sensibilidade significativamente maior para distúrbios de junção neuromuscular, mas nem sempre são específicos apenas para miastenia grave.
Figura 47 - Preservação da convergência na oftalmoplegia internuclear (bilateral). (a) Oftalmoplegia internuclear direita. (b) Oftalmoplegia internuclear esquerda. (c) Preservação da função do reto medial em ambos os olhos, demonstrada quando se pede ao paciente para cobrir os olhos e fixar o olhar em um alvo próximo. Reimpresso com permissão de Prasad S e Galetta SL.168
Tratamento da diplopia. O tapa-olho monocular é o tratamento mais básico para diplopia binocular. Sem a visão através de um olho, a visão binocular dupla deixa de existir. Alternativamente ao tapa-olho, um adesivo semi-transparente colado na lente do óculos também elimina efetivamente a diplopia.
Se o desalinhamento ocular permanecer estável, é possível usar prismas para diminuir a diplopia no olhar fixo primário. A quantidade correta de prisma necessária para eliminar a visão dupla é estabelecida no consultório e, em seguida, uma massa de prisma de uso temporário pode ser aplicada na lente do óculos. Por fim, se a correção do prisma for bem-sucedida, poderá ser consolidada diretamente em um óculos novo. Entretanto, é preciso alertar os pacientes para o fato de que os prismas não tendem a aliviar a diplopia em casos de olhar fixo excêntrico, quando o desalinhamento ocular não é concomitante. Além disso, a diplopia torsional (como a que frequentemente acompanha a paralisia do IV nervo) não pode ser corrigida com prismas.
Figura 48 - Doença ocular tireoidiana. (a) Retração palpebral e hipertropia esquerda. (b) Olhar fixo para cima comprometido bilateralmente, pior no lado direito. A seta indica a direção da tentativa de olhar fixo. (c) Varredura de tomografia computadorizada coronal das órbitas, mostrando espessamento acentuado dos músculos reto medial, inferior e lateral direito, bem como dos músculos reto inferior e medial esquerdo.
Uma vez que o desalinhamento ocular por paralisia oculomotora tenha permanecido estável por 6-12 meses, é possível considerar a correção cirúrgica. A cirurgia para estrabismo pode usar uma combinação de técnicas, incluindo a retração de alguns músculos (procedimento de enfraquecimento) e procedimentos de ressecção de outros (procedimento firmador para intensificar a ação do músculo). Alguns procedimentos de retração podem ser executados com suturas ajustáveis, de modo a permitir que o realinhamento ocular seja finamente ajustado com base na experiência subjetiva do paciente consciente. A meta final da cirurgia é estabelecer um campo amplo de visão binocular única na posição primária.
O nistagmo se refere às oscilações involuntárias e rítmicas dos olhos para frente e para trás, que podem ser caracterizadas como reflexo ou ondulação pendular. O reflexo do nistagmo é mais comum, no qual há desvios lentos dos olhos em uma direção e movimentos rápidos compensatórios dos olhos na direção oposta. O nistagmo pendular é caracterizado por movimentos oscilatórios que têm a mesma velocidade e duração.
O nistagmo pode ser fisiológico ou patológico. Um exemplo de nistagmo fisiológico é o nistagmo de posição final, caracterizado por alguns batimentos de nistagmo horizontal ocorrendo depois de os olhos terem se movido para os extremos do olhar fixo horizontal. O nistagmo optocinético é outra forma de nistagmo fisiológico que ocorre quando os olhos perseguem uma série de alvos em movimento numa direção e são geradas sacadas corretivas na direção oposta para detectar o próximo alvo.
Por fim, o nistagmo patológico pode ser subclassificado como central ou periférico, com base na localização da anormalidade no sistema nervoso central ou periférico [ver Tabela 2].149 Os achados sugestivos de etiologia central incluem o nistagmo torsional puro ou vertical puro; nistagmo não suprimido com fixação visual; e nistagmo que dificilmente entra em fadiga. Em adição, o nistagmo central pode mudar de direção, dependendo da posição do olhar fixo.
Pacientes com nistagmo adquirido frequentemente relatam oscilopsia, que se refere à percepção ilusória de movimento visual. Dependendo da causa do nistagmo, outros sintomas associados podem incluir vertigem, ataxia, perda auditiva ou outros déficits neurológicos motores ou sensoriais.
Muitos tipos de nistagmo podem ser caracterizados por meio da observação cuidadosa dos movimentos de fase lenta e de fase rápida, em posição primária e em diferentes posições de olhar fixo. Em alguns casos, são usadas manobras provocativas para tentar deflagrar o nistagmo. Estas manobras podem incluir posicionamento específico (i.e., manobra de Dix-Hallpike), agitação da cabeça, hiperventilação ou vibração dos processos mastoide. O teste do reflexo vestibulocular (executando arremetidas horizontais da cabeça enquanto o paciente mantém a fixação visual) muitas vezes fornece informação essencial.150,151 As sacadas de alcance, vistas quando o reflexo vestibulocular está deficiente, implicam em disfunção vestibular periférica. Em contraste, as arremetidas horizontais da cabeça são normais (sem sacadas de alcance) em pacientes com formas centrais de nistagmo.
Um eletronistagmograma e testes vestibulares detalhados podem auxiliar o diagnóstico de distúrbios vestibulares específicos. Um eletronistagmograma consiste em registros do movimento ocular realizados em repouso, com movimentação da cabeça, durante a rotação da cadeira, durante a observação de um objeto em movimento e com a estimulação calórica fria das orelhas internas. Outros exames adicionais podem incluir a posturografia e as avaliações de audiometria.
A vertigem posicional benigna é uma condição comum que se deve à otocônia deslocada mais comumente alojada no canal semicircular posterior.152 Uma forma exclusiva de nistagmo acompanha a vertigem posicional benigna e confirma seu diagnóstico. A manobra de Dix-Hallpike, em que o paciente é deslocado da posição sentada para a posição de decúbito dorsal com a cabeça lateralmente virada em 45° (na posição preferida do canal semicircular posterior ipsilateral), resulta em um nistagmo vertical-torsional misto com fases rápidas dirigidas para cima e na direção da parte inferior da orelha. Este nistagmo característico tipicamente começa após uma latência de cerca de 15 segundos e desaparece após cerca de 90 segundos.
O nistagmo induzido por olhar fixo é uma forma de nistagmo patológico que somente ocorre no olhar fixo excêntrico e não em posição primária.149 Resulta da falha dos “integradores neurais” (núcleo intersticial de Cajal, núcleo propositus hipoglossus, núcleo vestibular medial e suas conexões com o cerebelo) que normalmente mantêm a fixação visual em posição excêntrica. Por definição, este tipo de nistagmo tem uma direção de fase rápida que muda de acordo com a direção do olhar fixo (i.e., o nistagmo será de batimento direito no olhar fixo direito, de batimento esquerdo no olhar fixo esquerdo, de batimento para cima no olhar fixo para cima etc). As lesões estruturais das vias cerebelares, desarranjos metabólicos e algumas medicações (p. ex., antiepiléticos, lítio) podem causar nistagmo induzido por olhar fixo. A deficiência de tiamina (encefalopatia de Wernicke) é uma causa importante potencialmente reversível.
O desequilíbrio no sistema vestibular, surgindo no sistema nervoso central ou periférico, é outra causa comum de nistagmo patológico.149 Uma lesão vestibular unilateral ou assimétrica (como uma neuronite vestibular) produz sinais que causam movimentos oculares lentos na direção do lado comprometido, os quais são seguidos de movimentos de fase rápida na direção oposta. Diferente do nistagmo induzido por olhar fixo, o nistagmo vestibular periférico será unidirecional independentemente da direção do olhar fixo, embora a amplitude do nistagmo venha a ser maior ao olhar na direção oposta a da lesão. Em adição, o nistagmo vestibular pode estar presente no olhar fixo primário.
O nistagmo vertical puro (batimento para cima ou batimento para baixo) pode resultar de estímulo desequilibrado a partir dos canais semicirculares anteriores ou posteriores, respectivamente. Exemplificando, os canais semicirculares posteriores normalmente estão ativos na inclinação da cabeça para trás, causando movimentação reflexa dos olhos para baixo. O comprometimento do canal posterior (i.e., uma lesão na junção cervicomedular) produz o desvio tônico dos olhos para cima e um nistagmo corretivo de batimento para baixo. Uma situação similar ocorre com as lesões do floculo cerebelar, comprometendo as projeções inibitórias normais para as vias do canal anterior. A desinibição das vias do canal anterior também produz desvio dos olhos para cima e nistagmo corretivo de batimento para baixo.
|
Tabela 2 - Achados de nistagmo central e periférico | ||
|
|
Periférico |
Central |
|
Manifestação |
Sintomas >> sinais |
Sinais > sintomas |
|
Direcionalidade |
Unidirecional (fase rápida na direção de uma lesão destrutiva) |
Mudança de direção |
|
Efeito da fixação visual |
Atenuada |
Não afetada |
|
Ondulação |
Alta frequência, baixa amplitude |
Alta amplitude, baixa frequência |
|
Reflexo vestibulocular |
Comprometido (sacadas de alcance) |
Intacto |
Diversas formas adicionais de nistagmo podem ocorrer no contexto de disfunção cerebelar.153 O nistagmo de rebote se refere ao nistagmo reflexo transitório que ocorre no retorno a partir do olhar fixo excêntrico para a posição primária, com a fase rápida distante da direção prévia do olhar fixo lateral. No nistagmo alternante periódico adquirido, a direção muda a cada 90-120 segundos.154 Esta condição pode resultar do dano aos centros cerebelares que, normalmente, modulam e estabilizam as características temporais do reflexo vestibulocular. O nistagmo reflexo de ir-e-vir em forma de onda, no qual há torsão para dentro e elevação de um olho com torsão para fora sincronizada e depressão do outro olho, pode se seguir às lesões de mesencéfalo que interrompem os estímulos para os centros de manutenção do olhar fixo vertical.155
O nistagmo pendular pode resultar do dano às interconexões entre os integradores neurais tronco encefálicos e os centros mantedores do olhar do fixo das tonsilas cerebelares.156 A desmielinização ao longo destas vias, que é comum na EM, muitas vezes contribui para padrões anormais de disparo espontâneo. O nistagmo pode surgir vários meses após a aquisição de uma lesão, sugerindo que a desaferenciação neural pode contribuir para a fisiopatologia. Exemplificando, uma lesão no triângulo de Guillain-Mollaret (núcleo dentado, pedúnculo cerebelar superior, núcleo rubro, trato tegmentar central, oliva inferior e, por fim, o pedúnculo cerebelar inferior) pode originar nistagmo pendular e tremor palatal.157 O tremor oculopalatal costuma estar associado à hipertrofia olivar inferior que, por sua vez, pode ser evidente à RNM.
A manobra de Epley é uma técnica de reposicionamento de partícula que pode ser extremamente efetiva para acelerar a resolução dos sintomas de vertigem posicional benigna.158 A manobra de Epley é realizada fazendo o paciente assumir uma série de posições diferentes, conforme descrito a seguir:
1. O paciente começa na posição sentada e com as duas pernas em extensão total.
2. O paciente, então, deita rapidamente, com a cabeça levemente estendida e virada, de modo que a orelha afetada fique para voltada para baixo.
3. Decorridos cerca de 30 segundos, a cabeça é virada em 90°, de modo que a orelha não afetada fique voltada para o chão.
4. Decorridos cerca de 30 segundos, o paciente rola sobre o ombro do lado da orelha não afetada, virando a cabeça.
5. Passados aproximadamente 30 segundos, o paciente é lentamente trazido para a posição vertical sentada e com o pescoço discretamente flexionado.
Se os sintomas recorrerem, os pacientes podem precisar repetir a manobra de Epley periodicamente.
De modo geral, os esforços para tratar o nistagmo farmacologicamente são frustrantes, mas certos fármacos podem proporcionar benefício sintomático para alguns pacientes.159 Clonazepam, baclofeno, gabapentina e memantina são fármacos razoavelmente bem tolerados e podem ser efetivos.160 As aminopiridinas podem atenuar algumas formas de nistagmo patológico, intensificando a inibição dos núcleos vestibulares pelas fibras de Purkinje cerebelares. Ambas, 4-amino-piridina161 e 3,4-diaminopiridina162 têm mostrado eficácia em estudos controlados, porém seu uso é limitado pelos efeitos colaterais, incluindo náusea, vômito e convulsões.
A terapia de reabilitação vestibular emprega exercícios destinados à melhora do olhar fixo e à estabilização da marcha.163 Estes métodos podem incluir exercícios de estimulação do reflexo vestibulocular para melhorar a fixação visual; exercícios oculomotores destinados a melhorar a perseguição visual e a precisão da sacada; e exercícios de equilíbrio. Tem sido demonstrado que estas técnicas diminuem as sensações subjetivas de tontura, minimizam a ansiedade associada à perda vestibular e melhoram objetivamente o equilíbrio.
O autor não mantém relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
Agradecimentos
O autor deseja agradecer aos residentes participantes do Partners Neurology Program (Brigham and Women’s Hospital e Massachussetts General Hospital) por seu entusiasmo pela neuroftalmologia. Sua natureza inquisitiva estimulou muitas discussões que melhoraram diretamente o conteúdo deste capítulo. Partes do texto deste capítulo foram reimpressas com permissão de Prasad,49 Prasad e Galetta,164 Prasad et al,165 Prasad e Volpe,166 Prasad e Galetta,167 e Prasad e Galetta.168
Figuras 4 e 20 – Christine Kenney.
1. Kerr NM, Chew SS, Eady EK, et al. Diagnostic accuracy of confrontation visual field tests. Neurology 2010; 74:1184–90.
2. Mirza RG, Johnson MW, Jampol LM. Optical coherence tomography use in evaluation of the vitreoretinal inter- face: a review. Surv Ophthalmol 2007; 52:397–421.
3. Damek DM. Paraneoplastic retinopathy/optic neuropathy. Curr Treat Options Neurol 2005; 7:57–67.
4. Levitan P. Pupillary escape in disease of the retina or optic nerve. Arch Ophthalmol 1959;62:768–79.
5. O’Neill EC, Danesh-Meyer HV, Connell PP, et al. The optic nerve head in acquired optic neuropathies. Nat Rev Neurol
2010; 6:221–36.
6. Becker M, Masterson K, Delavelle J, et al. Imaging of the optic nerve. Eur J Radiol 2010; 74:299–313.
7. Balcer LJ. Clinical practice. Optic neuritis. N Engl J Med 2006;354:1273–80.
8. Group ONS. Visual function 15 years after optic neuritis: a final follow-up report from the Optic Neuritis Treatment Trial. Ophthalmology 2008; 115:1079–82 e5.
9. Fleishman JA, Beck RW, Linares OA, Klein JW. Deficits in visual function after resolution of optic neuritis. Ophthalmology 1987; 94:1029–35.
10. Cleary PA, Beck RW, Bourque LB, et al. Visual symptoms after optic neuritis. Results from the Optic Neuritis Treatment Trial. J Neuroophthalmol 1997; 17:18–23; quiz 4–8.
11. Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol 2008;65:727–32.
12. Beck RW, Cleary PA, Trobe JD, et al. The effect of cortico- steroids for acute optic neuritis on the subsequent devel- opment of multiple sclerosis. The Optic Neuritis Study Group. N Engl J Med 1993; 329:1764–9.
13. Optic Neuritis Study Group. The 5-year risk of MS after optic neuritis. Experience of the Optic Neuritis Treatment Trial. Optic Neuritis Study Group. Neurology 1997;49:1404–13.
14. Beck RW, Cleary PA, Anderson MM Jr, et al. A random- ized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med 1992; 326:581–8.
15. Beck RW, Cleary PA. Optic Neuritis Treatment Trial. One- year follow-up results. Arch Ophthalmol 1993;111:773–5.
16. Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo- controlled trial. Lancet 2009; 374:1503–11.
17. Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med 2000; 343:898–904.
18. Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 2006; 67:1242–9.
19. Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclero- sis: a randomised study. Lancet 2001;357:1576–82.
20. Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology 2007;114:810–5.
21. Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology
2006;66:1485–9.
22. Naismith RT, Tutlam NT, Xu J, et al. Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis. Neurology 2009;72:1077–82.
23. Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 2008; 65:1443–8.
24. Sato D, Callegaro D, Lana-Peixoto MA, Fujihara K. Treatment of neuromyelitis optica: an evidence based review. Arq Neuropsiquiatr 2012;70:59–66.
25. Fontal MR, Kerrison JB, Garcia R, Oria V. Ischemic optic neuropathy. Semin Neurol 2007; 27:221–32.
26. Hayreh SS, Podhajsky P, Zimmerman MB. Role of noctur- nal arterial hypotension in optic nerve head ischemic disorders. Ophthalmologica 1999; 213:76–96.
27. Thurtell MJ, Tomsak RL. Nonarteritic anterior ischemic optic neuropathy with PDE-5 inhibitors for erectile dys- function. Int J Impot Res 2008;20:537–43.
28. Arnold AC. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol 2003; 23:157–63.
29. Rizzo JF 3rd, Lessell S. Optic neuritis and ischemic optic neuropathy. Overlapping clinical profiles. Arch Ophthalmol 1991;109:1668–72.
30. Hayreh SS, Zimmerman MB. Nonarteritic anterior ischemic optic neuropathy: natural history of visual outcome. Ophthalmology 2008; 115:298–305 e2.
31. Bellusci C, Savini G, Carbonelli M, et al. Retinal nerve fiber layer thickness in nonarteritic anterior ischemic optic neuropathy: OCT characterization of the acute and resolving phases. Graefes Arch Clin Exp Ophthalmol 2008; 246:641–7.
32. Kale N, Eggenberger E. Diagnosis and management of giant cell arteritis: a review. Curr Opin Ophthalmol 2010; 21:417–22.
33. Prasad S, Moss HE, Lee EB, et al. Clinical reasoning: a 42-year-old man with sequential monocular visual loss. Neurology 2008; 71:e43–9.
34. Delalande S, de Seze J, Fauchais AL, et al. Neurologic man- ifestations in primary Sjogren syndrome: a study of 82 patients. Medicine (Baltimore) 2004; 83:280–91.
35. Kidd D, Burton B, Plant GT, Graham EM. Chronic relapsing inflammatory optic neuropathy (CRION). Brain 2003; 126:276–84.
36. Golnik KC. Infectious optic neuropathy. Semin Ophthalmol 2002;17:11–7.
37. Ko MW, Dalmau J, Galetta SL. Neuro-ophthalmologic manifestations of paraneoplastic syndromes. J Neurooph- thalmol 2008;28:58–68.
38. Danesh-Meyer HV. Radiation-induced optic neuropathy. J Clin Neurosci 2008;15:95–100.
39. Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol 2005; 140:517–23.
40. Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011;134: 2677–86.
41. Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene- environment interactions in Leber hereditary optic neuropathy. Brain 2009; 132:2317–26.
42. Sarkies N. Traumatic optic neuropathy. Eye 2004; 18: 1122–5.
43. Levin LA, Beck RW, Joseph MP, et al. The treatment of traumatic optic neuropathy: the International Optic Nerve Trauma Study. Ophthalmology 1999;106:1268–77.
44. Yu Wai Man P, Griffiths PG. Surgery for traumatic optic neuropathy. Cochrane Database Syst Rev 2005;(4): CD005024.
45. Yu-Wai-Man P, Griffiths PG. Steroids for traumatic optic neuropathy. Cochrane Database Syst Rev 2007; (4): CD006032.
46. Ng WT, Morgan W. Mechanisms and treatment of primary angle closure: a review. Clin Exp Ophthalmol 2012; 40: e218–28.
47. Bruce BB, Newman NJ. Functional visual loss. Neurol Clin 2010; 28:789–802.
48. Hayreh SS. Optic disc edema in raised intracranial pres- sure. V. Pathogenesis. Arch Ophthalmol 1977;95:1553–65.
49. Prasad S. Papilledema. In: Gilman S, editor. Medlink Neurology. San Diego: Medlink Corporation; 2011. Available at: www.medlink.com (accessed December 8, 2012).
50. Hayreh MS, Hayreh SS. Optic disc edema in raised intracranial pressure. I. Evolution and resolution. Arch Ophthalmol 1977; 95: 1237–44.
51. Jacks AS, Miller NR. Spontaneous retinal venous pulsation: aetiology and significance. J Neurol Neurosurg Psychiatry 2003; 74:7–9.
52. Levin BE. The clinical significance of spontaneous pulsations of the retinal vein. Arch Neurol 1978; 35:37–40.
53. Corbett JJ, Jacobson DM, Mauer RC, Thompson HS. Enlargement of the blind spot caused by papilledema. Am
J Ophthalmol 1988;105:261–5.
54. Huna-Baron R, Landau K, Rosenberg M, et al. Unilateral swollen disc due to increased intracranial pressure. Neurology 2001; 56:1588–90.
55. Wall M, White WN 2nd. Asymmetric papilledema in idiopathic intracranial hypertension: prospective interocular comparison of sensory visual function. Invest Ophthalmol Vis Sci 1998; 39:134–42.
56. Auw-Haedrich C, Staubach F, Witschel H. Optic disk drusen. Surv Ophthalmol 2002; 47:515–32.
57. Cartlidge NE, Ng RC, Tilley PJ. Dilemma of the swollen optic disc: a fluorescein retinal angiography study. Br J Ophthalmol 1977; 61: 385–9.
58. Wirostko WJ, Ruttum MS, Lewandowski MF. Evaluating suspected papilledema with ocular echography. J Pediatr Ophthalmol Strabismus 1998; 35: 290–1.
59. Corbett JJ, Mehta MP. Cerebrospinal fluid pressure in normal obese subjects and patients with pseudotumor cerebri. Neurology 1983; 33: 1386–8.
60. Abel AS, Brace JR, McKinney AM, et al. Practice patterns and opening pressure measurements using fluoroscopically guided lumbar puncture. AJNR Am J Neuroradiol 2012; 33: 823–5.
61. de Freitas GR, Bogousslavsky J. Risk factors of cerebral vein and sinus thrombosis. Front Neurol Neurosci 2008; 23:23–54.
62. Baryshnik DB, Farb RI. Changes in the appearance of venous sinuses after treatment of disordered intracranial pressure. Neurology 2004; 62:1445–6.
63. Higgins JN, Pickard JD. Lateral sinus stenoses in idiopathic intracranial hypertension resolving after CSF diversion. Neurology 2004; 62:1907–8.
64. Fraser C, Plant GT. The syndrome of pseudotumour cerebri and idiopathic intracranial hypertension. Curr Opin Neurol 2011;24:12–7.
65. Vogh BP, Godman DR, Maren TH. Effect of AlCl3 and other acids on cerebrospinal fluid production: a correction. J Pharmacol Exp Ther 1987; 243:35–9.
66. Celebisoy N, Gokcay F, Sirin H, Akyurekli O. Treatment of idiopathic intracranial hypertension: topiramate vs acet- azolamide, an open-label study. Acta Neurol Scand 2007; 116:322–7.
67. Orssaud C, Roche O, Dufier JL. Nutritional optic neuropathies. J Neurol Sci 2007; 262: 158–64.
68. Fraunfelder FW, Sadun AA, Wood T. Update on ethambutol optic neuropathy. Expert Opin Drug Saf 2006;5:615–8.
69. Jonas JB, Budde WM. Diagnosis and pathogenesis of glaucomatous optic neuropathy: morphological aspects. Prog Retin Eye Res 2000; 19:1–40.
70. Anderson DR, Drance SM, Schulzer M. Natural history of normal-tension glaucoma. Ophthalmology 2001; 108:247–53.
71. Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic path- way gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 2007;61:189–98.
72. Prasad S, Cohen AB. Diagnostic accuracy of confrontation visual field tests. Neurology 2011; 76: 1192–3; author reply 3.
73. Lam BL, Thompson HS, Corbett JJ. The prevalence of simple anisocoria. Am J Ophthalmol 1987; 104 :69–73.
74. Wilkins RH, Brody IA. Horner’s syndrome. Arch Neurol 1968;19:540–2.
75. Morris JG, Lee J, Lim CL. Facial sweating in Horner’s syndrome. Brain 1984;107(Pt 3):751–8.
76. Wallenberg A. Acute Bulbäraffection (Embolie der Art. cerebellar. post. inf. sinistr.). Arch Psych Nervenheilkd 1895; 27:504–40.
77. Balcer LJ, Galetta SL. Images in clinical medicine. Pancoast’s syndrome. N Engl J Med 1997; 337:1359.
78. Baumgartner RW, Bogousslavsky J. Clinical manifestations of carotid dissection. Front Neurol Neurosci 2005; 20:70–6.
79. Kardon RH, Denison CE, Brown CK, Thompson HS.Critical evaluation of the cocaine test in the diagnosis of Horner’s syndrome. Arch Ophthalmol 1990;108:384–7.
80. Morales J, Brown SM, Abdul-Rahim AS, Crosson CE.Ocular effects of apraclonidine in Horner syndrome. Arch Ophthalmol 2000; 118:951–4.
81. Dewan MA, Harrison AR, Lee MS. False-negative apraclonidine testing in acute Horner syndrome. Can J Ophthalmol 2009;44:109–10.
82. Cremer SA, Thompson HS, Digre KB, Kardon RH.
Hydroxyamphetamine mydriasis in Horner’s syndrome. Am J Ophthalmol 1990; 110:71–6.
83. Digre KB, Smoker WR, Johnston P, et al. Selective MR imaging approach for evaluation of patients with Horner’s syndrome. AJNR Am J Neuroradiol 1992;13:223–7.
84. Patel RR, Adam R, Maldjian C, et al. Cervical carotid artery dissection: current review of diagnosis and treatment. Cardiol Rev 2012; 20:145–52.
85. Mahoney NR, Liu GT, Menacker SJ, et al. Pediatric horner syndrome: etiologies and roles of imaging and urine stud- ies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol 2006; 142:651–9.
86. Czarnecki JS, Thompson HS. The iris sphincter in aberrant regeneration of the third nerve. Arch Ophthalmol 1978; 96:1606–10.
87. Loewenfeld I. Lesions in the ciliary ganglion and short ciliary nerves: the tonic pupil (“Adie’s” syndrome). In: The pupil anatomy, physiology, and clinical applications. Detroit (MI): Wayne State University Press; 1993. p. 1080–
130.
88. Bourgon P, Pilley FJ, Thompson HS. Cholinergic supersen- sitivity of the iris sphincter in Adie’s tonic pupil. Am J Ophthalmol 1978;85:373–7.
89. McCrary JA 3rd, Webb NR. Anisocoria from scopolamine patches. JAMA 1982; 248:353–4.
90. Thompson HS, Newsome DA, Loewenfeld IE. The fixed dilated pupil. Sudden iridoplegia or mydriatic drops? A simple diagnostic test. Arch Ophthalmol 1971; 86:21–7.
91. Woods D, O’Connor PS, Fleming R. Episodic unilateral mydriasis and migraine. Am J Ophthalmol 1984;98:229–34.
92. Jacobson DM. Benign episodic unilateral mydriasis. Clinical characteristics. Ophthalmology 1995; 102:1623–7.
93. Purvin VA. Adie’s tonic pupil secondary to migraine. J Neuroophthalmol 1995; 15:43–4.
94. Thompson HS, Zackon DH, Czarnecki JS. Tadpole-shaped pupils caused by segmental spasm of the iris dilator muscle. Am J Ophthalmol 1983;96:467–77.
95. Zee DS, Griffin J, Price DL. Unilateral pupillary dilatation during adversive seizures. Arch Neurol 1974; 30:403–5.
96. Rosenberg ML, Jabbari B. Miosis and internal ophthalmoplegia as a manifestation of partial seizures. Neurology 1991; 41: 737–9.
97. Bienfang DC. Crossing axons in the third nerve nucleus. Invest Ophthalmol 1975;14:927–31.
98. Kwon JH, Kwon SU, Ahn HS, et al. Isolated superior rectus palsy due to contralateral midbrain infarction. Arch Neurol 2003; 60: 1633–5.
99. Saeki N, Yamaura A, Sunami K. Bilateral ptosis with pupil sparing because of a discrete midbrain lesion: magnetic resonance imaging evidence of topographic arrangement within the oculomotor nerve. J Neuroophthalmol 2000;20: 130–4.
100. Bogousslavsky J, Maeder P, Regli F, Meuli R. Pure mid- brain infarction: clinical syndromes, MRI, and etiologic patterns. Neurology 1994; 44:2032–40.
101. Ksiazek SM, Slamovits TL, Rosen CE, et al. Fascicular arrangement in partial oculomotor paresis. Am J Ophthalmol 1994; 118:97–103.
102. Liu GT, Crenner CW, Logigian EL, et al. Midbrain syndromes of Benedikt, Claude, and Nothnagel: setting the record straight. Neurology 1992; 42:1820–2.
103. Boghen D, Chartrand JP, Laflamme P, et al. Primary aberrant third nerve regeneration. Ann Neurol 1979;6:415–8.
104. Lepore FE, Glaser JS. Misdirection revisited. A critical appraisal of acquired oculomotor nerve synkinesis. Arch Ophthalmol 1980; 98:2206–9.
105. Locksley HB. Natural history of subarachnoid hemor- rhage, intracranial aneurysms and arteriovenous malfor- mations. J Neurosurg 1966; 25:321–68.
106. Krisht A, Barnett DW, Barrow DL, Bonner G. The blood supply of the intracavernous cranial nerves: an anatomic study. Neurosurgery 1994; 34:275–9; discussion 9.
107. Weber RB, Daroff RB, Mackey EA. Pathology of oculomotor nerve palsy in diabetics. Neurology 1970; 20:835–8.
108. Asbury AK, Aldredge H, Hershberg R, Fisher CM. Oculomotor palsy in diabetes mellitus: a clinico-pathological study. Brain 1970; 93: 555–66.
109. Jacobson DM. Pupil involvement in patients with diabetes- associated oculomotor nerve palsy. Arch Ophthalmol 1998;116: 723–7.
110. Bortolami R, D’Alessandro R, Manni E. The origin of pain in ‘ischemic-diabetic’ third-nerve palsy. Arch Neurol 1993; 50:795.
111. Capo H, Warren F, Kupersmith MJ. Evolution of oculomotor nerve palsies. J Clin Neuroophthalmol 1992; 12:21–5.
112. Baker RS, Epstein AD. Ocular motor abnormalities from head trauma. Surv Ophthalmol 1991; 35:245–67.
113. Eyster EF, Hoyt WF, Wilson CB. Oculomotor palsy from minor head trauma. An initial sign of basal intracranial tumor. JAMA 1972; 220: 1083–6.
114. Walter KA, Newman NJ, Lessell S. Oculomotor palsy from minor head trauma: initial sign of intracranial aneurysm. Neurology 1994; 44:148–50.
115. Tanriover N, Kemerdere R, Kafadar AM, et al. Oculomotor nerve schwannoma located in the oculomotor cistern. Surg Neurol 2007; 67:83–8; discussion 8.
116. Friedman AP, Harter DH, Merritt HH. Ophthalmoplegic migraine. Arch Neurol 1962;7:320–7.
117. Walsh JP, O’Doherty DS. A possible explanation of the mechanism of ophthalmoplegic migraine. Neurology 1960;
10:1079–84.
118. Schultz KL, Lee AG. Diagnostic yield of the evaluation of isolated third nerve palsy in adults. Can J Ophthalmol 2007; 42:110–5.
119. Vaphiades MS, Cure J, Kline L. Management of intracranial aneurysm causing a third cranial nerve palsy: MRA, CTA or DSA? Semin Ophthalmol 2008; 23:143–50.
120. Okawara SH. Warning signs prior to rupture of an intracranial aneurysm. J Neurosurg 1973; 38: 575–80.
121. Chou KL, Galetta SL, Liu GT, et al. Acute ocular motor mononeuropathies: prospective study of the roles of neuroimaging and clinical assessment. J Neurol Sci 2004; 219:35–9.
122. Prasad S, Volpe NJ, Tamhankar MA. Clinical reasoning: a 36-year-old man with vertical diplopia. Neurology 2009; 72: e93–9.
123. Parulekar MV, Dai S, Buncic JR, Wong AM. Head positiondependent changes in ocular torsion and vertical misalignment in skew deviation. Arch Ophthalmol 2008; 126:899–905.
124. von Noorden GK, Murray E, Wong SY. Superior oblique paralysis. A review of 270 cases. Arch Ophthalmol 1986; 104:1771–6.
125. Helveston EM, Krach D, Plager DA, Ellis FD. A new classification of superior oblique palsy based on congenital variations in the tendon. Ophthalmology 1992; 99: 1609–15.
126. Brazis PW, Miller NR, Henderer JD, Lee AG. The natural history and results of treatment of superior oblique myokymia. Arch Ophthalmol 1994; 112:1063–7.
127. Keane JR. Trochlear nerve pareses with brainstem lesions. J Clin Neuroophthalmol 1986; 6:242–6.
128. Muri RM, Baumgartner RW. Horner’s syndrome and contralateral trochlear nerve palsy. Neuroophthalmology 1995; 15:161.
129. Vanooteghem P, Dehaene I, Van Zandycke M, Casselman J. Combined trochlear nerve palsy and internuclear ophthalmoplegia. Arch Neurol 1992;49:108–9.
130. Elliot D, Cunningham ET, Miller NR. Fourth nerve paresis and ipsilateral relative afferent pupillary defect without visual sensory disturbance: a sign of contralateral dorsal midbrain disease. J Clin Neuroophthalmol 1991; 11: 169–72.
131. Thomke F. Isolated abducens palsies due to pontine lesions. Neuroophthalmology 1998; 20: 91–100.
132. Prasad S, Liu GT, Abend NS, Ichord RN. Images in paediatrics: sinovenous thrombosis due to mastoiditis. Arch Dis Child 2007; 92:749.
133. Thomke F, Mika-Gruttner A, Visbeck A, Bruhl K. The risk of abducens palsy after diagnostic lumbar puncture. Neurology 2000; 54:768–9.
134. Volpe NJ, Lessell S. Remitting sixth nerve palsy in skull base tumors. Arch Ophthalmol 1993;111:1391–5.
135. Jacobson DM. Progressive ophthalmoplegia with acute ischemic abducens nerve palsies. Am J Ophthalmol 1996; 122:278–9.
136. Dave AV, Diaz-Marchan PJ, Lee AG. Clinical and magnetic resonance imaging features of Gradenigo syndrome. Am J Ophthalmol 1997; 124:568–70.
137. Mahoney NR, Liu GT. Benign recurrent sixth (abducens) nerve palsies in children. Arch Dis Child 2009; 94:394–6.
138. Duane A. Congenital deficiency of abduction, associated with impairment of adduction, retraction movements, con- traction of the palpebral fissure and oblique movements of the eye. 1905. Arch Ophthalmol 1996; 114: 1255–6; discus- sion 7.
139. Miller NR, Kiel SM, Green WR, Clark AW. Unilateral Duane’s retraction syndrome (type 1). Arch Ophthalmol 1982; 100:1468–72.
140. Reid RL, Quigley ME, Yen SS. Pituitary apoplexy. A review. Arch Neurol 1985;42:712–9.
141. Marsh RJ, Dulley B, Kelly V. External ocular motor palsies in ophthalmic zoster: a review. Br J Ophthalmol 1977;61: 677–82.
142. Kline LB. The Tolosa-Hunt syndrome. Surv Ophthalmol 1982;27:79–95.
143. Ringer AJ, Salud L, Tomsick TA. Carotid cavernous fistulas: anatomy, classification, and treatment. Neurosurg Clin N Am 2005; 16:279–95, viii.
144. Baloh RW, Yee RD, Honrubia V. Internuclear ophthalmoplegia. II. Pursuit, optokinetic nystagmus, and vestibulo- ocular reflex. Arch Neurol 1978;35:490–3.
145. Sauvineau C. Un nouveau type de paralyse associee des mouvements horizments horizontaux des yeux. Bull Soc Ophthalmol Fr 1895; 13:524–34.
146. Zee DS. Internuclear ophthalmoplegia: pathophysiology and diagnosis. Baillieres Clin Neurol 1992; 1:455–70.
147. Sharpe JA, Kumar S, Sundaram AN. Ocular torsion and vertical misalignment. Curr Opin Neurol 2011;24:18–24.
148. Nihalani BR, Hunter DG. Adjustable suture strabismus surgery. Eye 2011;25: 1262–76.
149. Thurtell MJ, Leigh RJ. Nystagmus and saccadic intrusions.
Handb Clin Neurol 2011; 102: 333–78.
150. Newman-Toker DE, Kattah JC, Alvernia JE, Wang DZ. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology 2008; 70:2378–85.
151. Weber KP, Aw ST, Todd MJ, et al. Head impulse test in unilateral vestibular loss: vestibulo-ocular reflex and catch-up saccades. Neurology 2008; 70: 454–63.
152. Fife TD. Benign paroxysmal positional vertigo. Semin
Neurol 2009; 29:500–8.
153. Kheradmand A, Zee DS. Cerebellum and ocular motor control. Front Neurol 2011;2:53.
154. Keane JR. Periodic alternating nystagmus with downward beating nystagmus. A clinicoanatomical case study of multiple sclerosis. Arch Neurol 1974;30:399–402.
155. Halmagyi GM, Aw ST, Dehaene I, et al. Jerk-waveform see-saw nystagmus due to unilateral meso-diencephalic lesion. Brain 1994;117(Pt 4):789–803.
156. Averbuch-Heller L, Zivotofsky AZ, Das VE, et al. Investi- gations of the pathogenesis of acquired pendular nystag- mus. Brain 1995; 118(Pt 2):369–78.
157. Guillain G. The syndrome of synchronous and rhythmic palato-pharyngo-laryngo-oculo-diaphragmatic myoclonus. Proc Roy Soc Med 1938; 31: 1031–8.
158. Fife TD, Iverson DJ, Lempert T, et al. Practice parameter: therapies for benign paroxysmal positional vertigo (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2008;70:2067–74.
159. Rucker JC. Current treatment of nystagmus. Curr Treat Options Neurol 2005; 7:69–77.
160. Shery T, Proudlock FA, Sarvananthan N, et al. The effects of gabapentin and memantine in acquired and congenital nystagmus: a retrospective study. Br J Ophthalmol 2006; 90: 839–43.
161. Kalla R, Glasauer S, Schautzer F, et al. 4-Aminopyridine improves downbeat nystagmus, smooth pursuit, and VOR gain. Neurology 2004; 62:1228–9.
162. Strupp M, Schuler O, Krafczyk S, et al. Treatment of down- beat nystagmus with 3,4-diaminopyridine: a placebo- controlled study. Neurology 2003; 61:165–70.
163. Boyer FC, Percebois-Macadre L, Regrain E, et al. Vestibu- lar rehabilitation therapy. Neurophysiol Clin 2008; 38: 479–87.
164. Prasad S, Galetta SL. Approach to the patient with acute monocular visual loss. Neurol Clin Pract 2012; 2:14–23.
165. Prasad S, Volpe NJ, Balcer LJ. Approach to optic neuropathies: clinical update. Neurologist 2010; 16:23–34.
166. Prasad S, Volpe NJ. Paralytic strabismus: third, fourth, and sixth nerve palsy. Neurol Clin 2010; 28:803–33.
167. Prasad S, Galetta SL. The facial nerve. In: Tasman W, Jaeger EA, editors. Duane’s ophthalmology. Philadelphia: Lippincott Williams & Wilkins; 2012. p. 1–44.
168. Prasad S, Galetta SL. Eye movement abnormalities in multiple sclerosis. Neurol Clin 2010; 28:641–55.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.

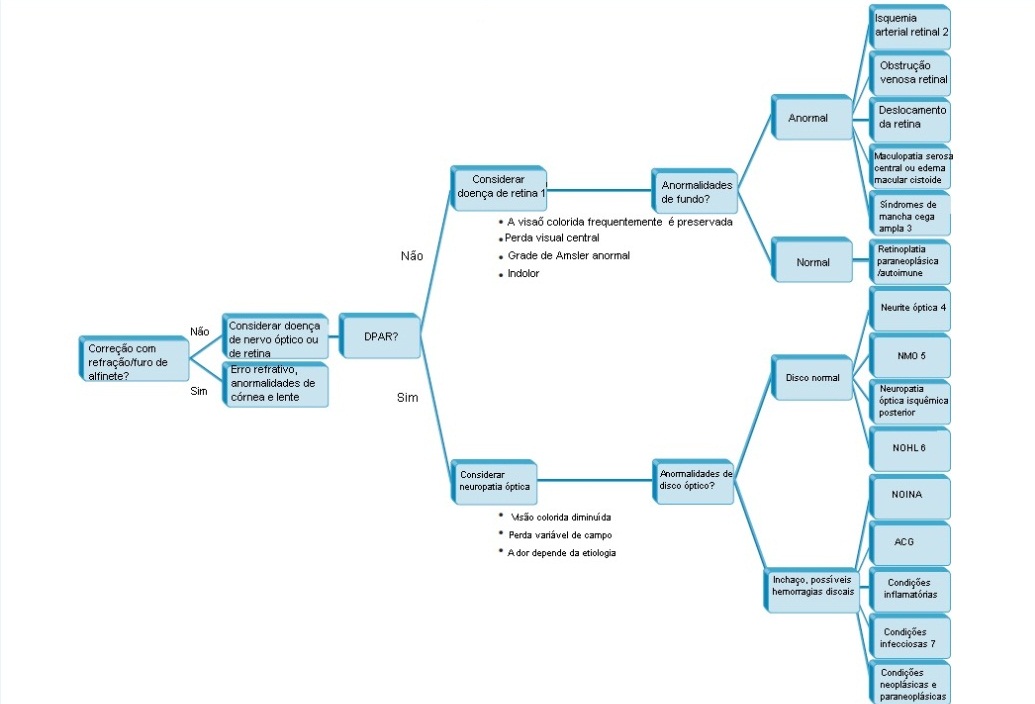
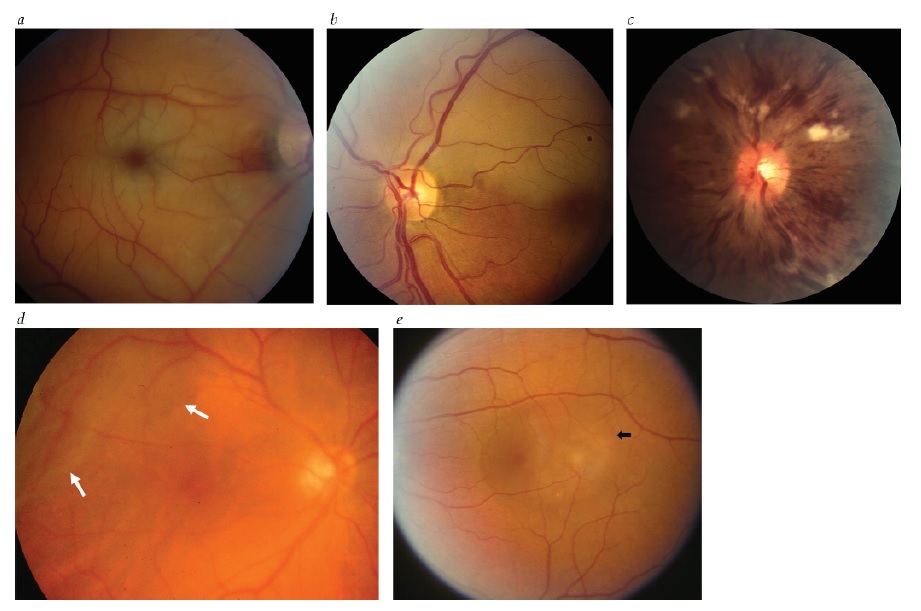

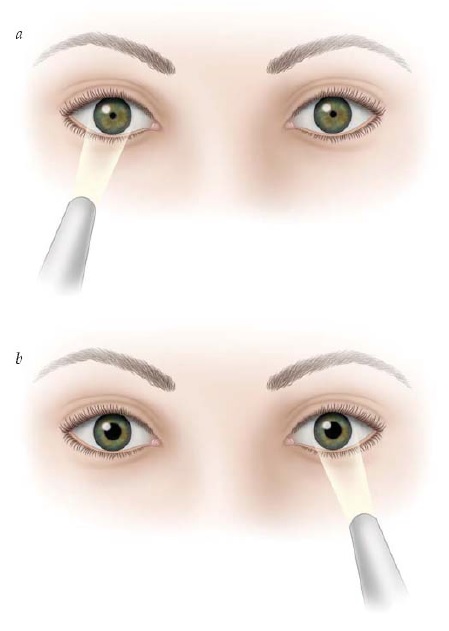
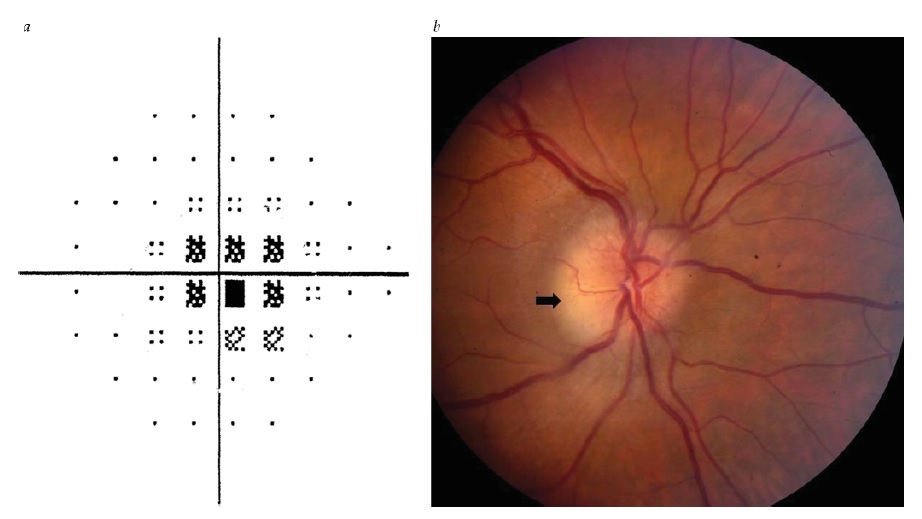
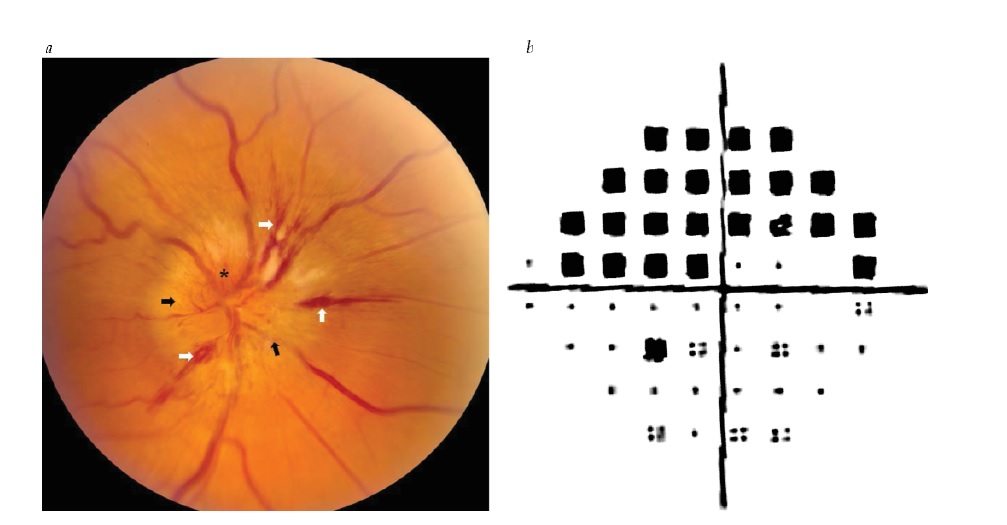
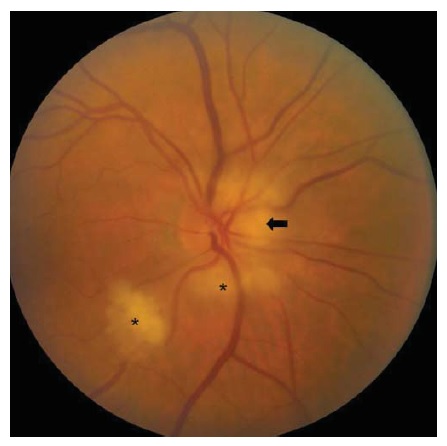
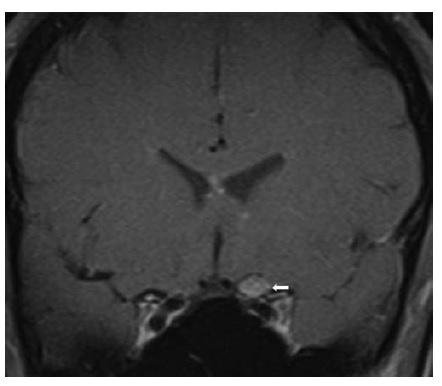
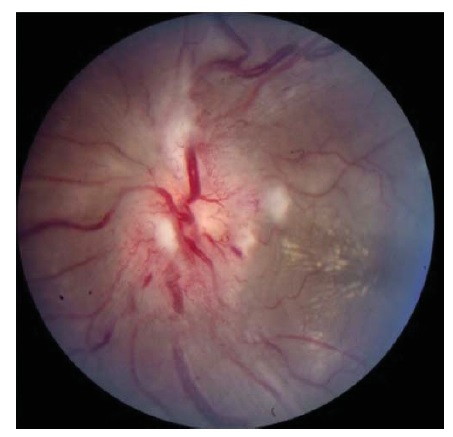
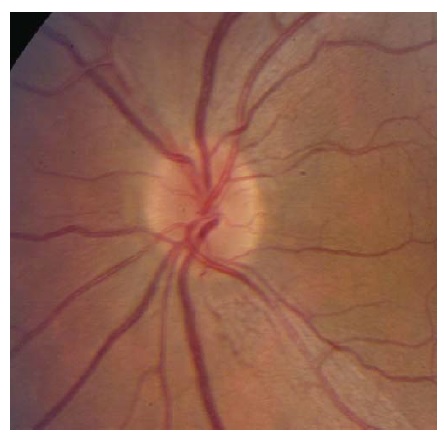
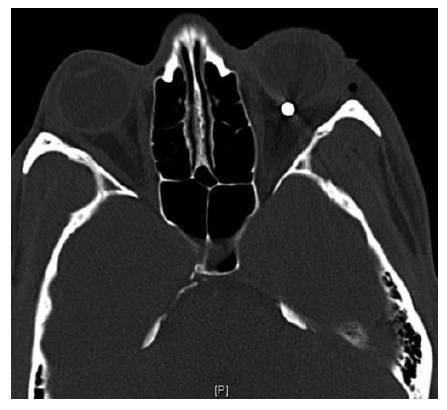
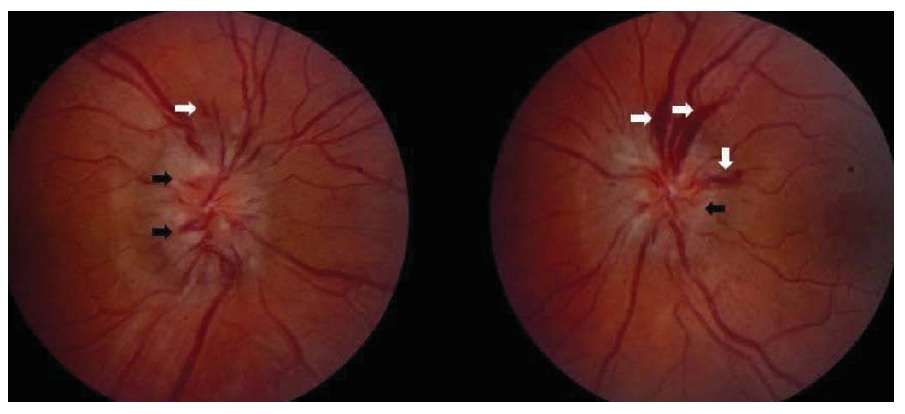
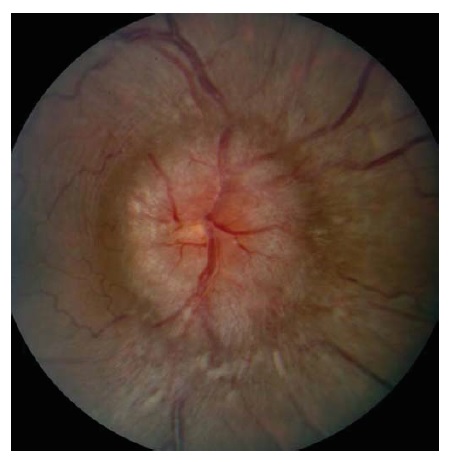
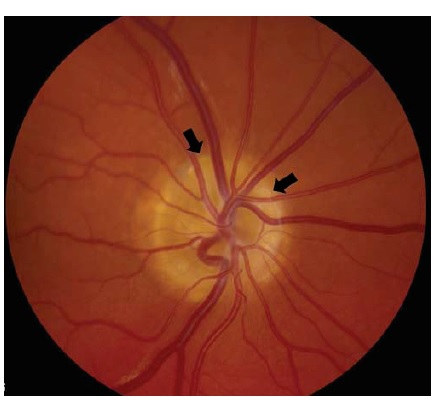
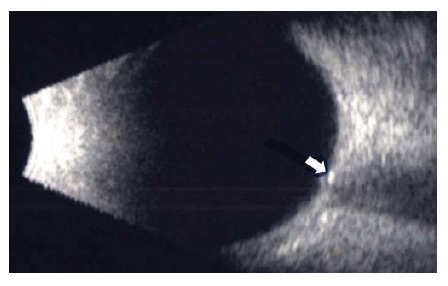
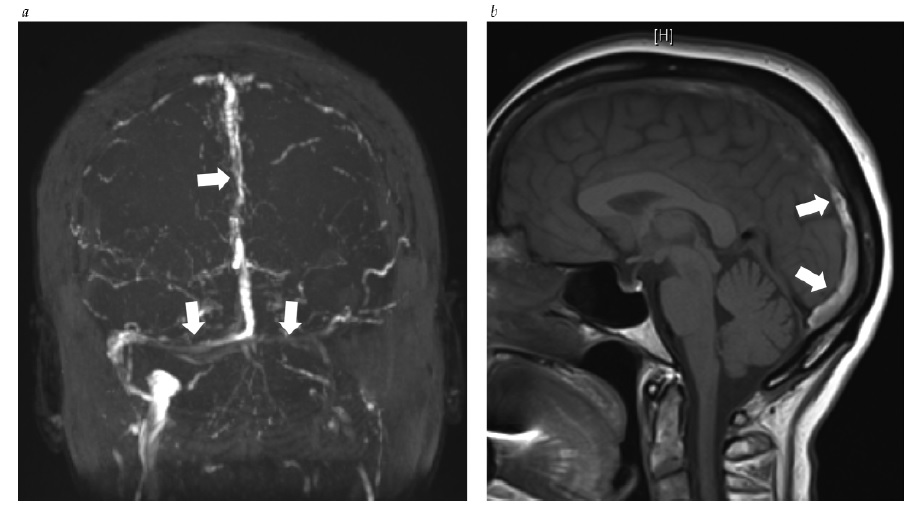
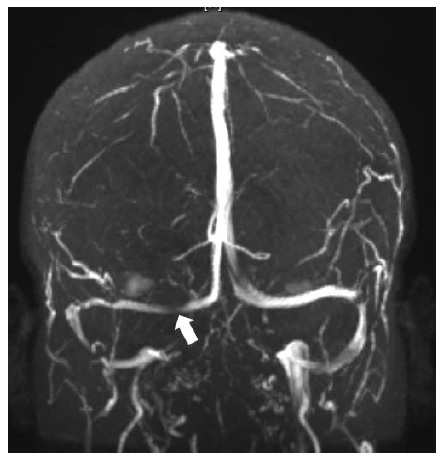
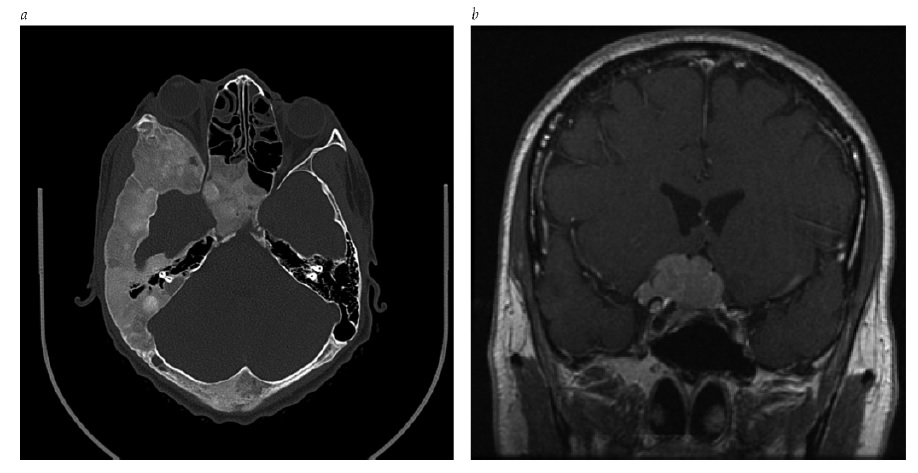
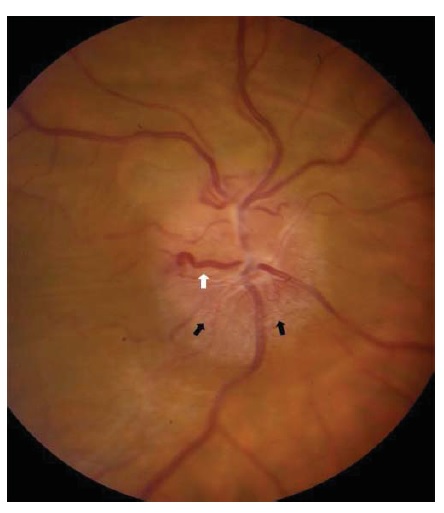
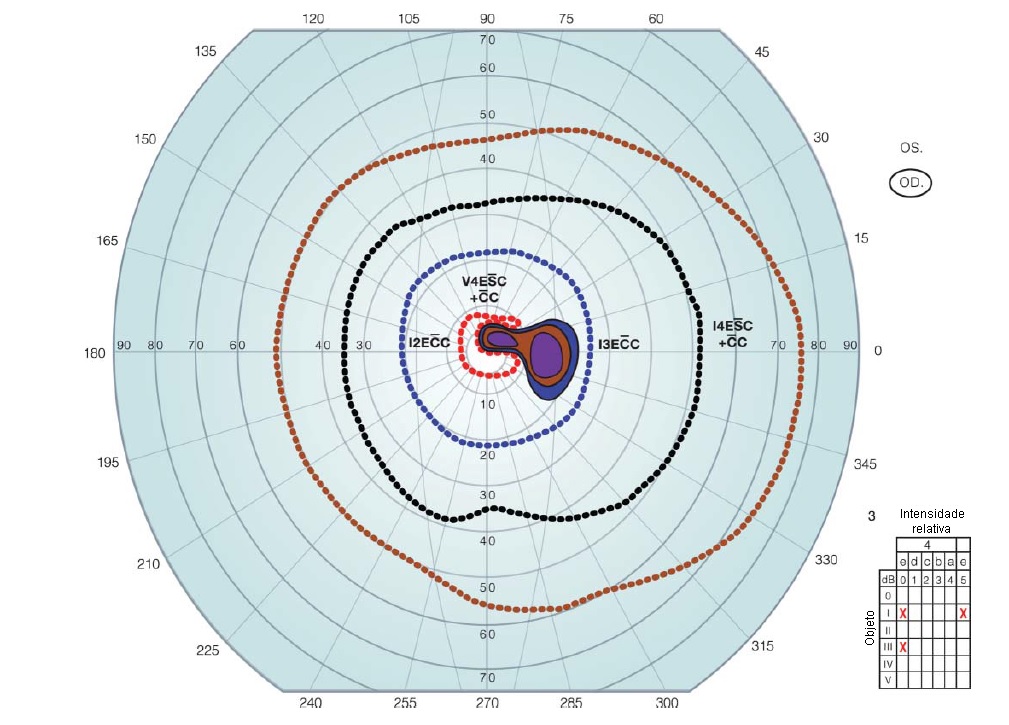
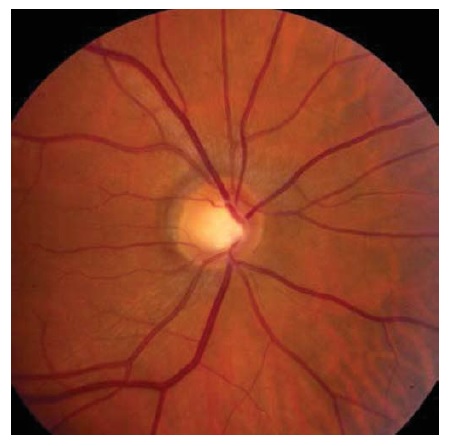
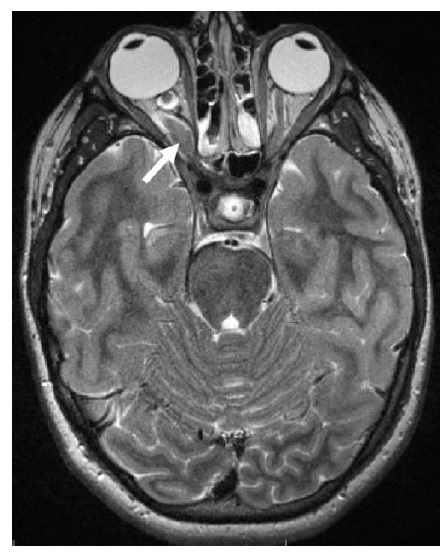
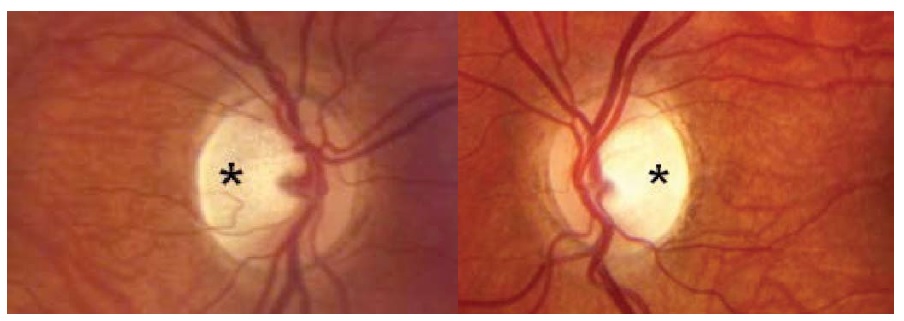

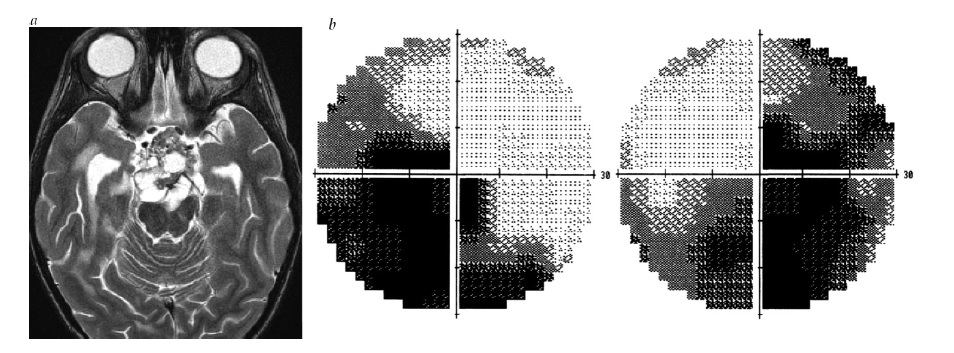
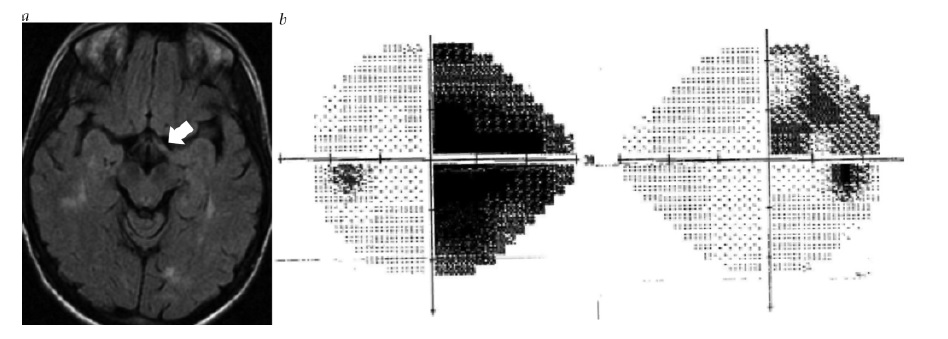
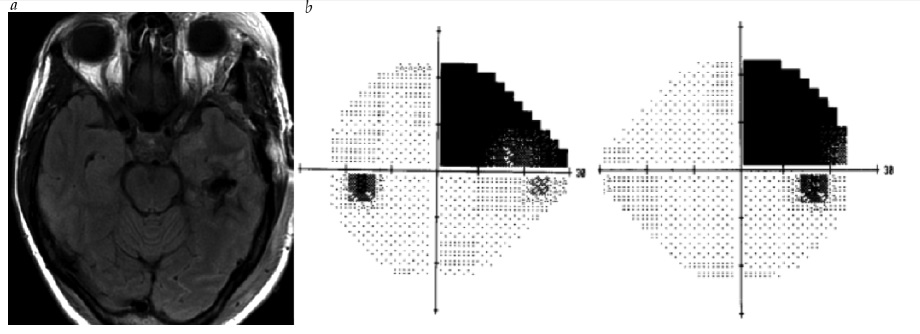
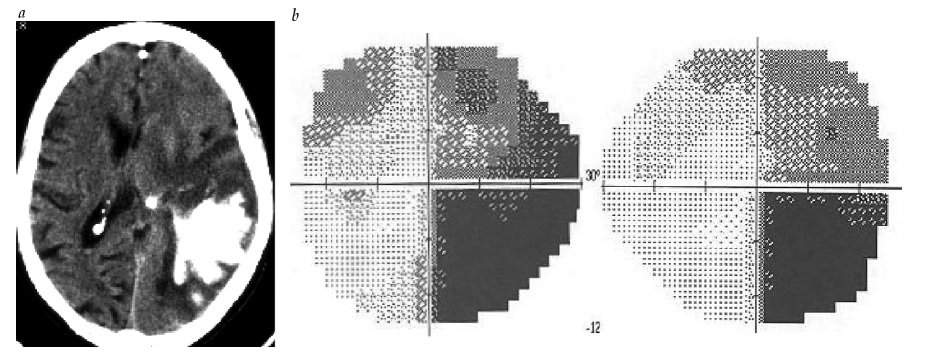
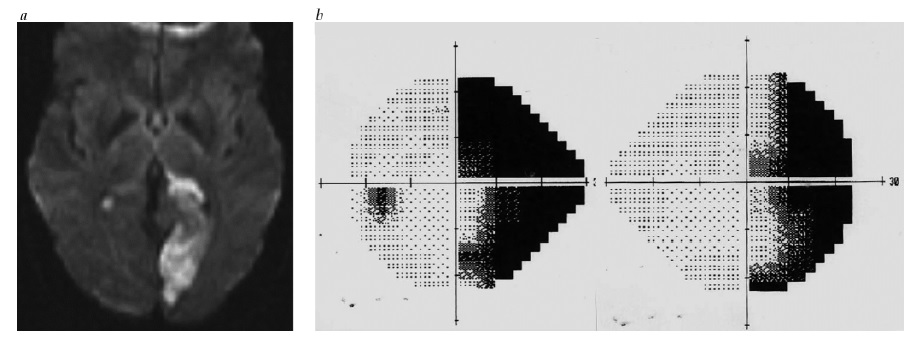
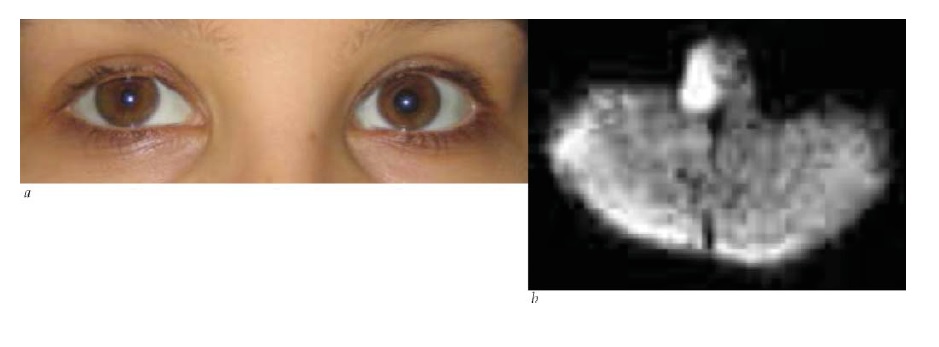
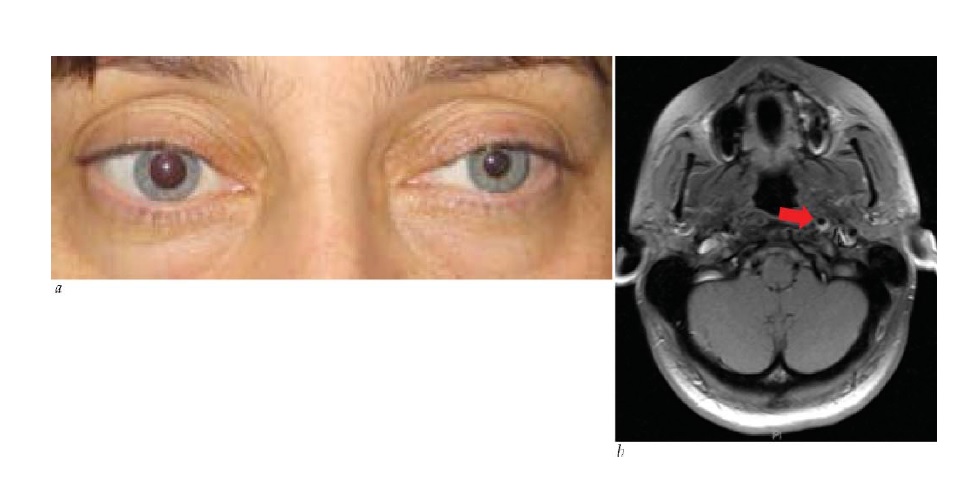
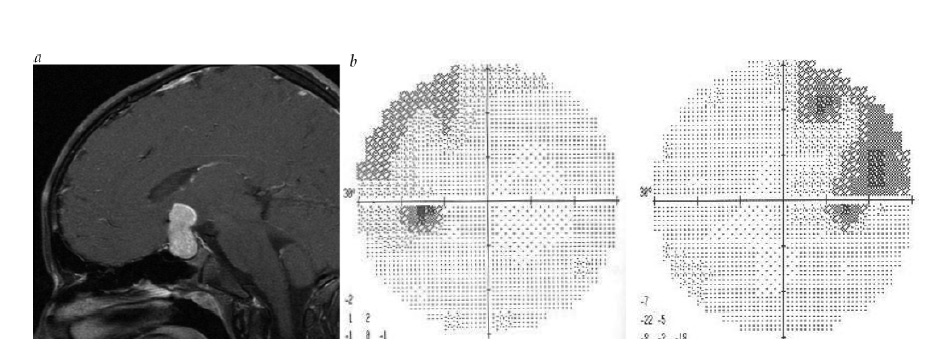
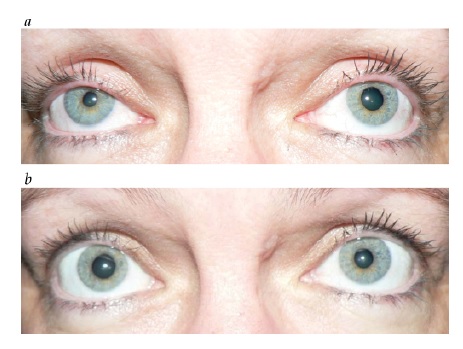
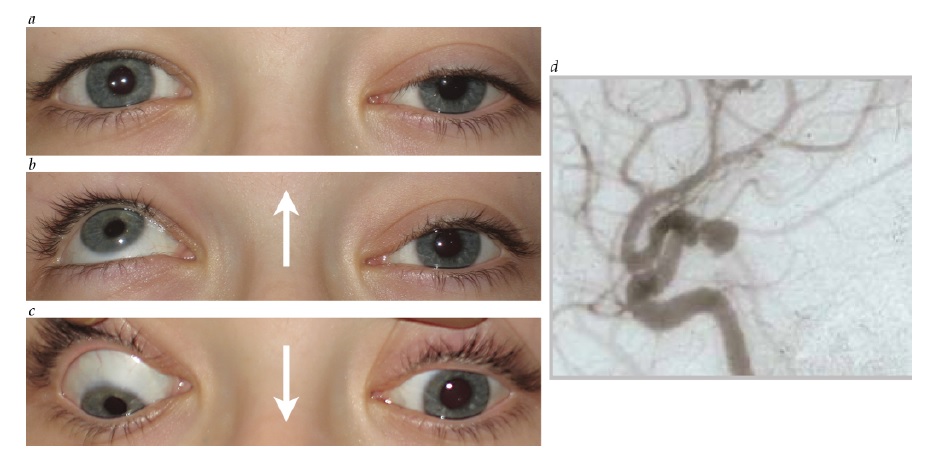
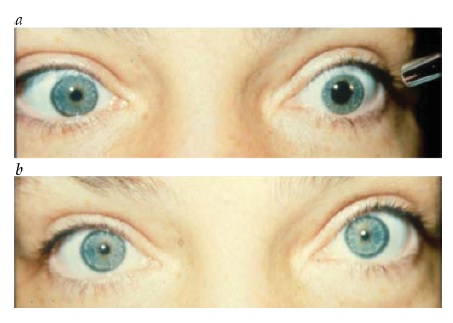
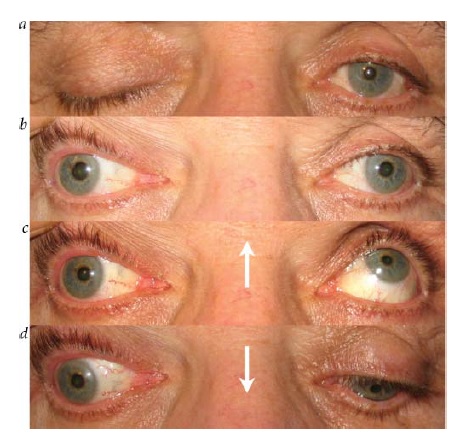 ,
,