(Carregando Índice)... (Carregando Índice)... |
Última revisão: 06/10/2015
Comentários de assinantes: 0
Esperance A.K. Schaefer, MD, MPH
Research Fellow, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA
Jules L. Dienstag, MD
Carl W. Walter Professor of Medicine and Dean for Medical Education, Harvard Medical School, and Professor of Medicine, Massachusetts General Hospital, Boston, MA
Artigo original: Schaefer EAK, MD, MPH, Dienstag JL, MD. Viral Hepatitis. ACP Medicine. 2012.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon De Oliveira
Revisão técnica: Dr. Lucas Santos Zambon
*Os autores e editores agradecem as contribuições do autor da edição anterior, Marlyn Mayo, MD, para o desenvolvimento e redação deste capítulo atualizado.
Apesar de um tropismo comum para o fígado, os vírus da hepatite englobam um grupo heterogêneo de doenças com diferentes modos de aquisição, sintomas, cronicidade e complicações. Os cinco vírus de hepatite principais—hepatite A, B, C, D e E—podem ser classificados por seus meios de transmissão (parenteral versus entérico) e por seu risco de cronicidade [ver Tabela 1]. As infecções pelo vírus da hepatite A (HAV) e pelo vírus da hepatite E (HEV) são adquiridas pela via fecal-oral e não estabelecem cronicidade. Em comparação, as infecções pelos vírus da hepatite B (HBV), vírus da hepatite C (HCV) e vírus da hepatite D (HDV) são adquiridas por contato direto com sangue ou líquidos corporais, e todas têm o potencial e causar doença crônica. A hepatite viral crônica impõe um importante problema de saúde pública global. Estima-se que mais de 500 milhões de pessoas em todo o mundo tenham infecção crônica com hepatite B ou C e, ao nível mundial, as complicações da hepatite viral crônica são responsáveis por 2,5% das mortes, além de serem causas primárias de cirrose e carcinoma hepatocelular (CHC).1
A apresentação clínica da hepatite aguda varia consideravelmente, de acordo com a gravidade e em função de suas causas. Todavia, em termos mais gerais, é caracterizada por fadiga, mal-estar, desconforto abdominal, hepatomegalia e testes de bioquímica hepática alterados, que podem ser classificados por elevações da atividade de aminotransferase (um padrão hepatocelular), elevações de fosfatase alcalina e bilirrubina (um padrão colestático) ou por uma mistura de ambas. O diagnóstico diferencial para o paciente com hepatite aguda é amplo e inclui lesão hepática fármaco-induzida, lesão hepática isquêmica, hepatite alcoólica, hepatite autoimune e hepatite viral [ver Tabela 2]. A hepatite viral aguda resulta mais comumente da infecção com os vírus das hepatites A, B ou C e, de modo geral, a incidência anual de hepatite viral aguda tem estado em declínio nos Estados Unidos. Exemplificando, o último pico de hepatite A aguda ocorreu em 1995, quando 12 casos em cada 100.000 eram relatados. Em 2007, porém, o número de casos relatados tinha declinado para 1 caso por cada 100.000.2 Esta demografia variável da hepatite A aguda foi atribuída à implementação bem-sucedida de programas de vacinação contra HAV. Uma redução similar na incidência de hepatite B aguda está relacionada primariamente à vacinação, enquanto uma redução concomitante na incidência de hepatite C aguda está relacionada a mudanças de comportamento, sobretudo entre usuários de drogas injetáveis, que minimizam a disseminação de infecções transmitidas pelo sangue. Os números de vigilância relatados ao Centers for Disease Control and Prevention (CDC), entretanto, subestimam os verdadeiros números de casos de hepatite aguda, que em sua maioria não são reconhecidos, podem ser brandos demais para chamar atenção médica ou não são relatados.
A hepatite fulminante é rara, mas pode ocorrer no contexto de hepatite viral aguda e está mais comumente associada à hepatite A e à hepatite B. Em uma série de pacientes com insuficiência hepática aguda, a hepatite viral aguda contribuiu para 12% de todos os casos. Embora HAV e HBV tenham sido a causa viral da maioria dos casos de insuficiência hepática aguda, o vírus do herpes simples (HSV) também foi identificado entre os casos associados com vírus.3
As hepatites B, C e D têm, todas, o potencial de causar hepatite crônica. A hepatite viral crônica é definida pela presença de RNA ou DNA virais detectáveis no soro por mais de 6 meses. De forma bastante semelhante ao observado na hepatite viral aguda, a apresentação clínica da hepatite viral crônica pode variar da elevação assintomática dos níveis de aminotransferase até a doença hepática em estagio terminal. Os sintomas de hepatite crônica podem incluir fadiga, mal-estar e desconforto abdominal. No caso das hepatites B e C, o envolvimento extra-hepático podem resultar em manifestações vasculíticas, como artrite, doença renal e erupções cutâneas. A vasculite generalizada (poliarterite nodosa) tem sido associada à hepatite B crônica, enquanto a crioglobulinemia essencial mista (púrpura palpável, vasculite leucocitoclástica) e os distúrbios linfoproliferativos podem agravar a hepatite C crônica. A hepatite viral crônica pode progredir para cirrose e doença hepática em estágio terminal, em que os sintomas podem ser mais evidentes e graves, bem como incluir fadiga profunda, anorexia, confusão, aumento da medida da cintura abdominal (decorrente de ascite), icterícia, edema e presença de hipertensão porta grave, hemorragia gastrintestinal. Ao exame físico, é possível detectar os estigmas de doença hepática terminal, incluindo teleangiectasias em forma de “aranha”, eritema palmar, ginecomastia, ascite e asterix relacionada à encefalopatia. O diagnóstico diferencial de hepatite crônica também é muito amplo e inclui doença hepática alcoólica, esteatose hepática não alcoólica, doença hepática autoimune, doença de Wilson (deposição de cobre), hematocromatose hereditária e deficiência de a1-antitripsina [ver Tabela 3].
Tabela 1 Características dos tipos de hepatite viral humana
|
|
Hepatite A (HAV) |
Hepatite B (HBV) |
Hepatite C (HCV) |
Hepatite D (HDV) |
Hepatite E (HEV) |
|
Genoma viral |
RNA de 7,5 kb, Hepatovirus |
DNA de 3,2 kb, Hepadnavirus |
RNA de 9,4 kb, Hepacivirus |
RNA de 1,7 kb, semelhante a um virioide vegetal |
RNA de 7,6 kb, Hepevirus |
|
Transmissão |
Fecal-oral |
Perinatal, percutânea, sexual |
Percutânea, sexual, extremamente rara |
Percutânea, sexual, perinatal |
Fecal-oral |
|
Período de incubação (dias) |
15-45 |
30-180 |
15-160 |
30-180 |
14-60 |
|
Progressão para cronicidade |
Não há |
Ocasional: comum antes de 5 anos de idade; rara em adultos sadios |
Comum (85%) |
Comum com superinfecção por HBV; rara com coinfecção aguda por HDV/HBV |
Rara (ocorre somente em imunossuprimidos) |
|
Disponibilidade de vacina |
Sim |
Sim |
Não |
Vacinação somente contra HBV |
Atualmente indisponível nos EUA |
|
Tratamento |
De suporte |
Interferon, interferon peguilado, inibidores nucleosídicos/ nucleotídicos |
Interferon, peguilado interferon, Inibidores de protease NS3/4A |
Interferon, peguilado interferon |
De suporte |
|
Profilaxia |
Ig, vacina inativada |
Vacina recombinante HBIg |
Não há |
Vacina contra hepatite B |
Vacinação (se disponível) |
HBIg = imunoglobulina anti-hepatite B; Ig = imunoglobulina.
Tabela 2 Diagnóstico diferencial de hepatite aguda
|
Vírus hepatotrópicos Vírus da hepatite A (HAV) Vírus da hepatite B (HBV) Vírus da hepatite C (HCV) Vírus da hepatite D (HDV) Vírus da hepatite E (HEV) (raro) Vírus não hepatotrópico Vírus do herpes simples (HSV) Citomegalovírus (CMV) HIV Vírus Epstein-Barr (EBV) Vírus varicela zoster (VZV) Parvovírus Vírus Coxsackie Adenovírus Não infecciosos Lesão hepática fármaco-induzida (LHFI) Doença vascular/hepatite isquêmica Hepatite autoimune Hepatite alcoólica Doença de Wilson Obstrução biliar aguda Outras infecções Legionella Erliquiose Leptospirose Febre maculosa das Montanhas Rochosas Febre Q Toxocariose Sífilis secundária |
Tabela 3 Diagnóstico diferencial de hepatite crônica e cirrose
|
Vírus hepatotrópicos Vírus da hepatite B (HBV) Vírus da hepatite C (HCV) Vírus da hepatite D (HDV) Vírus da hepatite E (HEV) (extremamente raro) Doenças colestáticas Colangite esclerosante primária Cirrose biliar primária Síndrome do ducto biliar evanescente Colangiopatia da Aids Não infecciosos Esteato-hepatite alcoólica Esteato-hepatite não alcoólica Hepatite autoimune Hemocromatose hereditária Doença de Wilson Hepatite crônica fármaco-induzida Deficiência de a1-antitripsina Doença celíaca hipertireoidismo Sarcoidose Hepatite granulomatosa Criptogênico |
Níveis elevados de alanina aminotransferases (ALTs) e aspartato aminotransferases (ASTs) geralmente fornecem os primeiros indícios da presença de hepatite viral. Entretanto, níveis elevados de aminotransferases não são um achado universal na hepatite viral crônica, particularmente em certos estágios da hepatite B crônica e da cirrose. Em adição, as faixas “normais” para AST e ALT foram reexaminadas e redefinidas. Muitos laboratórios relatam níveis abaixo de 40-50 UI/mL como estando dentro da faixa normal. Entretanto, estudos recentes envolvendo voluntários sadios e doadores de sangue demonstraram que, em média, os níveis de ALT eram de 17-20 UI/mL em mulheres e 23-29 UI/mL em homens, e que mais de 95% da população estudada tinha níveis de ALT abaixo do limite normal máximo.4 No contexto de cirrose e doença hepática terminal, outras anormalidades laboratoriais importantes incluem hipoalbuminemia e aumento do tempo de protrombina/razão normalizada internacional (INR) (refletindo comprometimento da função sintética hepática), além de trombocitopenia (secundária ao hiperesplenismo).
Embora o diagnóstico de hepatite viral seja baseado em exames sorológicos, é difícil avaliar de modo não invasivo o grau de inflamação ou fibrose hepática. Os exames de imagem, como ultrassonografia, tomografia computadorizada (TC) e imagem de ressonância magnética (RM), podem fornecer indícios da ocorrência de infiltração gordurosa (esteatose), lesões hepáticas discretas, nodularidade (cirrose) e sequelas da hipertensão porta (p. ex., esplenomegalia e varizes). Entretanto, a sensibilidade e especificidade destas abordagens de imagem para detecção da presença ou ausência de cirrose com precisão são limitadas. Algumas autoridades defendem a obtenção de um painel de exames de sangue ou a medida por imagem da elasticidade hepática para avaliação do grau de fibrose. Entretanto, estas abordagens funcionam melhor na identificação dos extremos dos estágios de fibrose (a fibrose muito branda em um extremo do espectro e, no outro, a cirrose) do que na distinção entre os níveis intermediários de fibrose, além de terem sensibilidade e especificidade limitadas. Nestes testes não invasivos, faltam características. O valor preditivo negativo para ausência de fibrose com o menor escore não invasivo é 85%, enquanto o valor preditivo positivo do escore mais alto é 76%.5 Assim, o padrão ouro de avaliação do grau de fibrose e para exclusão de outras causas de doença no contexto da hepatite viral crônica continua sendo a biópsia de fígado.
O exame histológico do tecido hepático permite examinar duas características importantes da hepatite viral crônica: o grau de atividade necroinflamatória (ou grau) e o grau de fibrose (ou estágio). Vários achados patológicos são considerados ao avaliar o grau de atividade histológica, incluindo a inflamação porta, degeneração de hepatócito, necrose de hepatócito e nível de contenção ou extensão da infiltração inflamatória porta. O sistema de escore histológico mais comumente usado nos EUA é a escala Ishak (grau histológico variando de 0 [sem atividade] a 18 [atividade intensa] e estágio histológico variando de 0 [sem fibrose] a 6 [cirrose estabelecida]), enquanto na Europa é a escala Metavir (escore necroinflamatório que varia de 0 [sem atividade] a 3 [atividade intensa]; e estágio de fibrose variando de 0 [sem cicatriz] a 4 [cirrose, cicatrização avançada]). Os principais riscos associados á biópsia hepática são o desconforto abdominal e o sangramento. Em uma revisão recente de biópsias hepáticas realizadas em pacientes com hepatite C crônica e fibrose, os autores constataram que o risco de sangramento associado ao procedimento era 0,5% e a maioria dos fatores de risco significativos eram uma contagem de plaquetas abaixo de 60.000/mm3 e uma INR >1,3.6 Embora a biópsia hepática possa fornecer informação importante e útil para orientar o tratamento, no caso dos pacientes com trombocitopenia ou coagulopatia grave, o risco aumentado de complicações hemorrágicas pode superar o potencial benefício do procedimento. Em adição, quase todos os pacientes com estes fatores de risco hematológico avançado têm cirrose bem estabelecida e a contribuição dos dados histológicos para o tratamento, neste contexto, é bastante limitada.
A abordagem para tratamento de um paciente com hepatite viral crônica (HVC) inclui a exclusão de outras causas de doença hepática crônica, minimização do risco de lesão hepática futura e monitoramento das complicações da cirrose. Todos os pacientes com HVC devem passar por triagem de imunidade às hepatites A e B e, devido aos modos de transmissão comuns destes dois tipos de hepatite viral e do HIV, também devem ser submetidos a testes de infecção por HIV. Na ausência de evidências de exposição prévia ou de imunidade contra HAV ou HBV, estes pacientes devem ser vacinados. Não existe uma quantidade segura de álcool estabelecida na HVC. Como o álcool acelera a lesão e fibrose hepáticas, os pacientes devem receber aconselhamento sobre a ingesta de carvão. Similarmente, os pacientes devem ser aconselhados acerca dos suplementos e medicações de balcão potencialmente hepatotóxicos. O uso de analgésicos de balcão, especificamente de fármacos anti-inflamatórios não esteroidais (AINEs) e acetaminofeno, deve ser abordado com cautela. Na hepatite crônica e na cirrose, é seguro usar acetaminofeno por períodos curtos e em doses de até 4 g/dia. Entretanto, havendo necessidade de um uso mais prolongado, devem ser consideradas doses menores (<2-3 g/dia).7 Em pacientes com cirrose, o acetaminofeno é preferido aos AINEs, porque estes podem ser nefrotóxicos em indivíduos com cirrose.8 As estatinas são uma classe de medicamentos comumente prescritos e, devido ao potencial de causar hepatotoxicidade, seu uso costuma ser evitado em pacientes com hepatite crônica. Entretanto, numerosas evidências sustentam a segurança do uso destes medicamentos na HVC.9 Na hepatite aguda, estes agentes devem ser categoricamente evitados.
No contexto de cirrose, os pacientes devem ser monitorados quanto ao desenvolvimento de complicações. O monitoramento da presença ou desenvolvimento de varizes por endoscopia superior é recomendado a cada 2 anos.10 Pacientes com doença hepática descompensada devem ser encaminhados para consideração de transplante de fígado.
Pode haver desenvolvimento de CHC em pacientes com cirrose e, conforme discutido adiante, também em pacientes com hepatite B crônica acompanhada ou não de cirrose. A triagem de CHC é recomendada a cada 6-12 meses, por imagem de ultrassom, para todos os pacientes com HVC que tenham cirrose e/ou fibrose avançada.11,12 A TC e a RM podem melhorar significativamente a sensibilidade e a especificidade da detecção do CHC no fígado cirrótico.13 Por outro lado, o alto custo destes procedimentos de imagem limita a sua ampla adoção. Mesmo assim, e sobretudo em pacientes com lesões hepáticas reveladas por ultrassonografia, estas abordagens de imagem sofisticadas têm sido incorporadas aos programas de vigilância de CHC. Se for feita a detecção de CHC, as opções de tratamento incluem ablação percutânea, ressecção cirúrgica, quimioembolização e transplante hepático. Para ser elegível para transplante de fígado, o paciente deve atender aos critérios de Milan: não ter mais do que uma única lesão medindo menos de 5 cm, ou no máximo três lesões medindo menos de 3 cm, na ausência de qualquer evidência de doença extra-hepática.
A HBV continua sendo a forma mais comum de hepatite viral crônica no mundo inteiro e é a causa mais comum de cirrose. Estimativas sugerem que cerca de 1/3 da população global foi exposta ao HBV e 350-400 milhões de pessoas estão cronicamente infectadas pelo vírus.14 Há uma substancial variação geográfica na prevalência da infecção crônica por HBV, a qual varia de menos de 2% nos EUA e Oeste da Europa a 2-8% no Mediterrâneo e América do Sul, aproximando-se de 20% na Ásia, Alaska e África subsaariana.15 Se não houvesse vacinação, modelos sugerem que o HBV teria causado 10 milhões de infecções crônicas e 1 milhão de mortes na coorte de crianças nascidas no ano 2000, destacando a importância da implementação dos programas de vacinação.16
A vacina contra hepatite B foi introduzida nos EUA em 1981 e, hoje, é recomendada para todas as crianças. A vacinação infantil universal foi implementada em 1991. Para os adultos não vacinados, algumas populações merecem ser vacinadas na fase adulta, incluindo os funcionários da área da assistência médica saudáveis, algumas pessoas que praticam sexo de alto risco e os penitenciários [ver Tabela 4]. Os programas de imunização diminuíram drasticamente as taxas de infecção aguda por hepatite B. exemplificando, nos EUA, apenas 4.519 casos de hepatite B foram relatados em 2007, representando uma incidência anual de 1,5 casos em cada 100.000 indivíduos—o menor registro já feito nos EUA, indicando uma queda aguda em relação aos 8,5 casos por 100.000 indivíduos relatados em 1990.2
A infecção por HBV é transmitida por meio da exposição percutânea ou mucosa ao sangue ou aos líquidos corporais. Nas áreas de alta prevalência, a exposição frequentemente ocorre no período perinatal ou início da infância, através do contato próximo. A exposição do adulto é mais comum em áreas de baixa prevalência, sendo que os modos de exposição durante a fase adulta incluem o uso de drogas injetáveis e o contato sexual.
Foram identificados oito genótipos principais de HBV, com distribuição geograficamente variável. O genótipo A é mais comum nos EUA e Europa Ocidental; os genótipos B e C são mais comuns na Ásia; o genótipo D é mais frequente no Mediterrâneo e Oriente Médio; e os genótipos E e F estão localizados na África Ocidental e América do Sul, respectivamente. Embora os testes de genotipagem de HBV de rotina não sejam recomendados, vários resultados clinicamente relevantes foram associados ao genótipo. O genótipo A foi associado a uma resposta sorológica (probabilidade de hepatite B e de soroconversão de antígeno [AgHBe] e de antígeno de superfície da hepatite B) ao tratamento antiviral,17 enquanto os genótipos C e D estão associados a taxas mais altas de resultados adversos, como CHC e cirrose.18
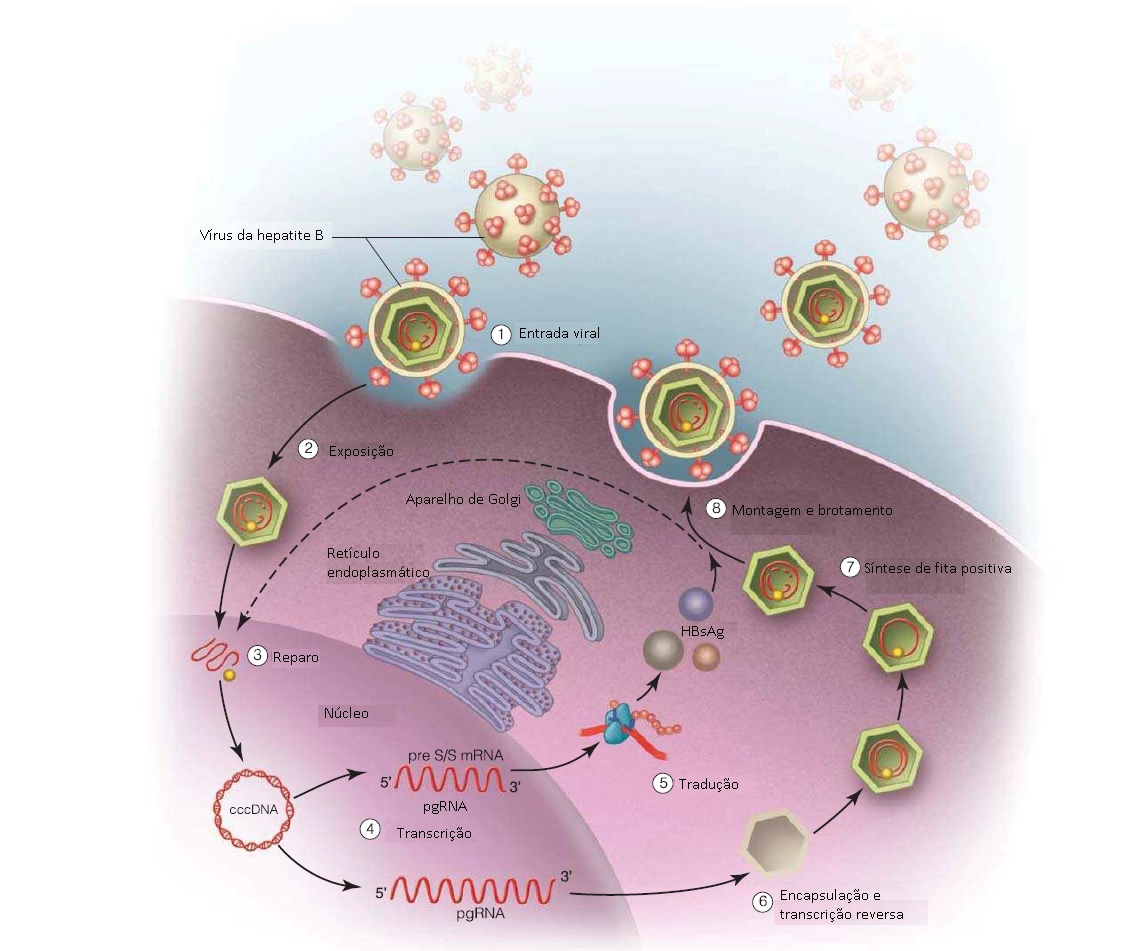
O HBV é um pequeno vírus envelopado contendo DNA, que pertence à família Hepadnaviridae. O vírus circula no sangue na forma de uma pequena partícula infecciosa de 42 nm, que consiste em um nucleocapsídeo viral circundado por um envelope lipídico, constituído por antígenos proteicos de superfície de três tamanhos distintos: pequeno, médio e grande.19 O DNA viral codifica sete proteínas virais: as proteínas do core expressas no antígeno do core da hepatite B (AgHBc); um antígeno pré-core não estrutural expressando AgHBe; DNA polimerase com atividade de polimerase e de transcriptase reversa; as três proteínas de superfície— pequena, média e grande—AgHBs; além das proteínas X, antígeno x da hepatite B e HBxAg [ver Fig. 1].20
O modo como a partícula de Dane entra nos hepatócitos ainda é indeterminado. Contudo, uma vez dentro da célula, o nucleocapsídeo viral é transportado para dentro do núcleo. Uma vez dentro do núcleo, o DNA viral, que é parcialmente uma fita dupla e uma fita única, é “reparado” pelas proteínas virais e do hospedeiro formando então uma fita de DNA totalmente dupla e convalentemente fechada com formato circular (cccDNA).21 Este cccDNA estabelece residência no núcleo (até 50 partículas de cccDNA por hepatócito) assumindo a forma de um microcromossomo altamente estável,22 um importante reservatório nuclear de DNA de HBV e fonte de persistência viral até mesmo nos período de supressão viral resultantes da terapia antiviral ou após a imunocontenção. O cccDNA é responsável pela dificuldade de erradicar definitivamente a infecção por HBV e também pelo potencial de reativação viral, sendo que a estabilidade deste DNA viral no núcleo do hepatócito pode contribuir para o desenvolvimento de CHC.
O cccDNA serve de molde para transcrição de RNA. Um tipo de RNA é traduzido para gerar as principais proteínas virais que são montadas em vírions, no citoplasma, enquanto outro tipo de RNA passa por transcrição reversa em fita de DNA negativa que, por sua vez, servirá de molde para síntese de fita de DNA positiva. O novo DNA de HBV é incorporado ao nucleocapsídeo e encapsidado pelas proteínas do envelope no citoplasma. O HBV e a replicação viral isolados não causam dano aos hepatócitos infectados. É a resposta imune do hospedeiro aos hepatócitos infectados pelos vírus que causa inflamação e lesão hepáticas. As diferenças nesta dependência da responsividade do hospedeiro para haver lesão hepática contribuem para o amplo espectro e para as diferenças individuais das manifestações clínicas associadas à infecção aguda e crônica por HBV.
Tabela 4 Esquemas de imunização contra hepatite B
|
População |
Dose |
Número de doses |
Formulação |
Esquema |
Esquema alternativo |
|
Recombivax HB (Merck) | |||||
|
Bebês <1 anoa |
5 µg (0,5 mL) |
3 |
Pediátrica (10 µg /mL) |
0, 1 e 6 meses |
— |
|
Bebês >1 ano, crianças, adolescentes (0-19 anos) |
5 µg (0,5 mL) |
3 |
Pediátrica (10 µg /mL) |
0, 1 e 6 meses |
0, 1 e 4 meses 0, 2 e 4 meses 1, 12 e 24 meses |
|
Adolescentes (11-15 anos), alternativo |
10 µg (1,0 mL) |
2 |
Adulto (10 µg /mL) |
0 e 4-6 meses |
— |
|
Adultos |
10 µg (1,0 mL) |
3 |
Adulto (10 µg /mL) |
0, 1 e 6 meses |
0, 1 e 4 meses 0, 2 e 4 meses 1, 12 e 24 meses |
|
Hemodiálise |
40 µg (1,0 mL) |
3 |
Hemodiálise (40 µg /mL) |
0, 1 e 6 meses |
— |
|
ENGERIX-B (GlaxoSmithKline) | |||||
|
Bebês <1 anoa |
10 µg (0,5 mL) |
3 |
Duas seringas pré-cheias disponíveis: 10 µg (0,5 mL) |
0, 1 e 6 meses |
0, 1, 2 e 12 meses (se a mãe for AgHBs-positiva) |
|
Bebês >1 ano, crianças, adolescentes |
10 µg (0,5 mL) |
3 |
20 µg (1,0 mL) |
0, 1 e 6 meses |
0, 12 e 24 meses 0, 1, 2 e 12 meses |
|
Adultos |
20 µg (1,0 mL) |
3 |
|
0, 1 e 6 meses |
0, 1, 2 e 12 meses |
|
Hemodiálise |
40 µg (2,0 mL) |
4 |
|
0, 1, 2 e 6 meses |
— |
AgHBs = antígeno de superfície da hepatite B.
aPara bebês prematuros pesando <2 kg com mães AgHBs-negativas, a dose ao nascimento pode ser adiada por até 30 dias. Bebês nascidos de mães AgHBs-positivas também devem receber imunoglobulina anti-hepatite B ao nascimento.

Figura 1: Ciclo de vida do vírus da hepatite B (HBV). O HBV circular como uma pequena partícula viral contendo o nucleocapsídeo viral e um envelope lipídico no qual a proteína de superfície está inserida. A partícula viral entra no hepatócito por mecanismos incertos (etapa 1, entrada). Dentro da célula, o vírus perde o revestimento e há exposição do nucleocapsídeo que contém uma fita de DNA dupla parcial circular relaxada, ou rcDNA (etapa 2, exposição). O nucleocapsídeo então é translocado para o núcleo. Dentro do núcleo, o DNA genômico é “reparado” (etapa 3, reparo) para formar a fita de DNA integralmente dupla e convalentemente fechada circular (cccDNA). O cccDNA é transcrito (etapa 4, transcrição) em dois moldes distintos de RNA. O mais curto, o transcrito de RNA mensageiro (mRNA) pré-S/S menor, é traduzido (estada 5, tradução) em proteínas de superfície. O mais comprido, RNA pré-genômico (pgRNA), é encapsulado com as proteínas do core e a HBV polimerase, e sofre transcrição reversa para gerar novas fitas de DNA genômico (etapa 6, encapsulação e transcrição reversa). A partir da fita negativa, são criados uma fita positiva e um novo rcDNA genômico (etapa 7, síntese de fita positiva). Um novo genoma encapsidado pode ser montado com as proteínas de superfície e liberado a partir da célula como um novo vírion (etapa 8, montagem e brotamento). Alternativamente, pode ser reciclado de volta o núcleo e contribuir para o reservatório celular de cccDNA. AgHBs = antígeno de superfície da hepatite B.
A apresentação e as consequências da exposição ao HBV variam consideravelmente, dependendo da idade em que ocorreu a exposição. A infecção associada à exposição perinatal é clinicamente silenciosa. A reposta imune contra os hepatócitos infectados por vírus é limitada após a exposição ao HBV no início da infância e, do mesmo modo, na vasta maioria (mais de 90%) dos indivíduos assim expostos, a infecção crônica se tornará estabelecida. Similarmente, quando a exposição ao HBV ocorre durante o início da infância, a infecção aguda pelo vírus da hepatite B costuma ser totalmente assintomática e cerca de 30% dos pacientes evoluem para desenvolver a forma crônica da infecção. Entretanto, para adolescentes e adultos sadios, a resposta imune contra os hepatócitos infectados pelo HBV é altamente efetiva, a hepatite aguda sintomática é muito mais comum e a infecção crônica se estabelece em apenas 1-2%.23-25
Os estágios clínicos da infecção crônica pelo vírus da hepatite B refletem a complexa interface entre o vírus e o sistema imune do hospedeiro [ver Tabela 5]. Na maioria dos pacientes com hepatite B crônica, especialmente aqueles infectados no início da vida, durante os primeiros anos e décadas de infecção, o vírus replica rápido sofrendo pouca interferência do sistema imune do hospedeiro. Este período inicial de lesão hepática relativamente limitada foi chamado, por algumas autoridades, fase de imunotolerância. Durante este período, os níveis de replicação viral podem ser bastante altos, porém a atividade necroinflamatória é negligenciável e os níveis de aminotransferases tendem a ser normais. Este período de relativa imunotolerância dura 2-3 décadas em indivíduos que sofreram exposição no início da vida, sendo que o fator deflagrador da imunoativação contra o vírus e a transição para uma fase mais imunologicamente ativa é incerto.
Este período mais imunológica e clinicamente ativo, que alguns chamam fase imunoativa ou fase de imunodepuração, representa um período de vigorosa resposta imune do hospedeiro aos hepatócitos infectados por HBV e é caracterizada do ponto de vista bioquímico por níveis altos de ALT (que podem ser flutuantes), do ponto de vista virológico por uma concentração de DNA de HBV persistentemente aumentada, e do ponto de vista sorológico pela presença de AgHBe. A elevação da ALT reflete a atividade necroinflamatória contínua mediada pela imunodepuração do vírus pelo hospedeiro, e a taxa de soroconversão espontânea de AgHBe está entre 2 e 15% ao ano.15 Como a resposta imune tende a ser apenas parcialmente efetiva em termos de depuração do vírus, a replicação viral persiste e contribui para os ciclos repetidos de lesão hepática. Desta forma, a resposta imune à replicação viral persistente é o fator de contribuição primária para a patogênese da doença e o desenvolvimento de fibrose. Estas distinções entre uma chamada fase imunotolerante e uma fase imunoativa não ocorrem em pacientes com hepatite B iniciada na fase adulta. E, mesmo após infecções adquiridas no período neonatal/início da vida, um nível baixo e intermitente de atividade necroinflamatória ocorre periodicamente durante o período de relativa imunotolerância. Em adição, até mesmo durante o período de relativa imunoatividade, há persistência de um alto nível de imunotolerância.
A perda de AgHBe com a aquisição de anticorpos anti-HBe muitas vezes anuncia a transição de hepatite crônica para um estado de portador de HBV inativo. No portador de HBV inativo, os níveis de ALT tendem a estar normais e os níveis circulantes de DNA de HBV estão baixos, inferiores a 2.000 UI/mL. A replicação viral se mantém abaixo deste limiar, a infectividade é baixa e a lesão hepática permanece quiescente por tempo prolongado na maioria dos portadores de vírus inativo. Entretanto, em uma proporção dos casos, pode haver reativação. Cerca de 20% dos portadores infectivos revertem para hepatite clinicamente ativa AgHBe-positiva. Uma proporção bem menor segue para alcançar uma depuração viram bem-sucedida, enquanto cerca de 1% ao ano sofre perda espontânea de AgHBs e transição para recuperação da infecção por HBV.25 portanto, é essencial fazer o monitoramento sorológico contínuo dos pacientes com infecção por HBV inativo.
Nem todos os indivíduos que perdem AgHBe, todavia, se tornam portadores de HBV inativo. Cerca de 10% entre direto em um estagio clínico de hepatite B crônica AgHBe-negativa, que é caracterizada por inflamação persistente, ALT elevada e alta concentração de DNA de HBV (apesar dos títulos baixos). A maioria tem mutações na região promotora do pré-core ou core do genoma viral, que previne a síntese e liberação de AgHBe. As taxas de hepatite B crônica AgHBe-negativa variam por genótipo e por região geográfica. Exemplificando, os genótipos B e C, que têm uma barreira muito menor às mutações pré-core, são comuns no Leste e no Sul da Europa e na Ásia. Cerca de 80-90% dos casos de hepatite B crônica que ocorrem nestas regiões são negativos para AgHBe.15
As principais complicações da infecção crônica por HBV são o CHC e a cirrose, sendo que o risco aumenta com o alto nível de DNA de HBV;26 coinfecção por HIV, HDV ou HCV; e consumo de álcool. Em pacientes com hepatite B crônica, a incidência de CHC ao longo da vida foi estimada em 21,7% e a de cirrose, em 41,5%.27 Diferente de outros tipos de hepatite crônica, em pacientes com infecção crônica por HBV, o risco de CHC é substancial até mesmo na ausência de cirrose. Os principais fatores preditivos de CHC são o avanço da idade, uma história familiar de CHC e níveis altos de DNA de HBV.28 Em adição, as diferenças genotípicas de HBV parecem influenciar o risco de CHC (p. ex., o genótipo C está associado a um risco maior de CHC, em comparação ao genótipo B).29 Apesar de substancialmente menor, o risco de CHC existe (incidência anual de 0,06%) até mesmo em portadores de hepatite B inativa, nos quais os níveis de DNA de HBV tendem a ser baixos.30
Em pacientes com hepatite B crônica, mesmo na ausência de cirrose, a triagem de CHC é recomendada para homens e mulheres asiáticos com idade acima de 40 e 50 anos, respectivamente, e para africanos com idade acima de 20 anos. Similarmente, a triagem de CHC é recomendada, independentemente da idade, para pacientes com cirrose, qualquer indivíduo com história familiar de CHC e qualquer pessoa infectada por HBV com elevação (persistente ou intermitente) de ALT, e/ou níveis aumentados de DNA de HBV superiores a 2.000 UI/mL.
Tabela 5 Estágios clínicos da infecção por HBV
|
Fase clínica |
Período de relativa imunotolerância |
Período de relativa atividade imunológica |
Portador de HBV inativo |
Recuperação |
Hepatite crônica AgHBe-negativa |
|
AgHBs |
+ |
+ |
+ |
– |
+ |
|
AgHBe |
+ |
+ |
– |
– |
– |
|
Anti-HBs |
– |
– |
– |
+ |
– |
|
Anti-HBe |
– |
– |
+ |
+ |
+ |
|
DNA de HBV |
Alto |
Alto |
<103 UI/mL |
Negativo |
Moderado |
|
ALT |
Normal/ quase normal |
Elevada |
Normal |
Normal |
Normal/alta |
|
Atividade necroinflamatória |
Pouco/ nenhuma |
Alta |
Mínima |
Nenhuma |
± |
|
Risco de CHC |
— |
Moderado/alto |
Baixo |
+, se cirrose |
Moderado/alto |
|
Cirrose |
— |
2-5% do risco anual |
Rara |
Rara |
2-5% do risco anual |
ALT = alanina aminotransferase; anti-HBe = anticorpos anti-hepatite B e; anti-HBs = anticorpo anti-hepatite B de superfície; AgHBe = antígeno de hepatite B e; AgHBs = antígeno da hepatite B de superfície; HBV = vírus da hepatite B; CHC = carcinoma hepatocelular.
Os testes sorológicos são a base do diagnóstico e do tratamento da hepatite B. Atualmente, os testes disponíveis incluem a avaliação de antígenos virais, anticorpos virais específicos e testes baseados no ensaio altamente sensível de reação em cadeia da polimerase (PCR) para DNA de HBV.31 O uso combinado destes testes se faz necessário para estabelecer corretamente o diagnóstico e o estágio clínico da hepatite B [ver Tabela 6].
O AgHBs, que constitui o componente envelope do vírion, também é expresso em partículas circulantes tubulares e esféricas medindo 22 nm. A presença de AgHBs é indicativa de infecção por HBV em curso e constitui a principal característica do diagnóstico de hepatite B (aguda ou crônica). Os ensaios de rotina para AgHBs são qualitativos e, apesar da disponibilidade, a utilidade dos ensaios quantitativos na prática clínica ainda é indeterminada.32 Em geral, os anticorpos contra AgHBs (anti-HBs) refletem a depuração bem-sucedida de AgHBs e a resolução da infecção ou a imunização prévia com vacina contra hepatite B, que consiste em AgHBs puro. Por outro lado, a resolução sorológica não indica que a infecção por HBV foi completamente erradicada do hospedeiro. Mesmo após a resolução sorológica, pequenas quantidades de cccDNA podem persistir nos núcleos dos hepatócitos e o risco de reativação permanece, particularmente no contexto de imunossupressão.
Outro marcador sorológico importante da infecção por HBV é o anticorpo contra o antígeno do core do vírus da hepatite B (anti-HBc), que surge no início do curso da hepatite B aguda e persiste subsequentemente, tanto com a resolução como com a persistência da infecção por HBV. Como a resposta imune inicial ao antígeno do core da hepatite B envolve a classe de anticorpos IgM, enquanto a resposta imune tardia (além de ˜6 meses) envolve a classe IgG, os testes para classe de imunoglobulina anti-HBc distinguem entre infecção recente (IgM) e infecção remota ou crônica (IgG) por HBV. Raramente (sobretudo com os ensaios de última geração altamente sensíveis para AgHBs e anti-HBs), na “janela” entre a perda de AgHBs detectável e a presença de anti-HBs detectável, os anticorpos anti-HBc podem ser o único marcador sorológico da infecção por HBV (IgM durante a evolução da hepatite B aguda; IgG durante a soroconversão de AgHBs associada à hepatite B crônica).
Uma combinação de AgHBs, anti-HBs e anti-HBc constitui a ferramenta de triagem primária para detecção da infecção aguda ou crônica por HBV. Indivíduos com alto risco de exposição prévia devem passar por triagem, incluindo mulheres grávidas, mulheres oriundas de áreas de alta prevalência geográfica e mulheres com fatores de alto risco de exposição na fase adulta (p. ex., atividade sexual de alto risco ou história de doença sexualmente transmissível, uso de droga injetável ou detenção em regime de encarceramento). Cerca de 10-15% dos imigrantes oriundos de áreas de alta prevalência abrigam a infecção crônica por HBV.33 Devido ao perigo de reativação do HBV durante e após a quimioterapia imunossupressora e citotóxica, os pacientes que estão prestes a receber terapia imunossupressora também devem passar por triagem para infecção por HBV.
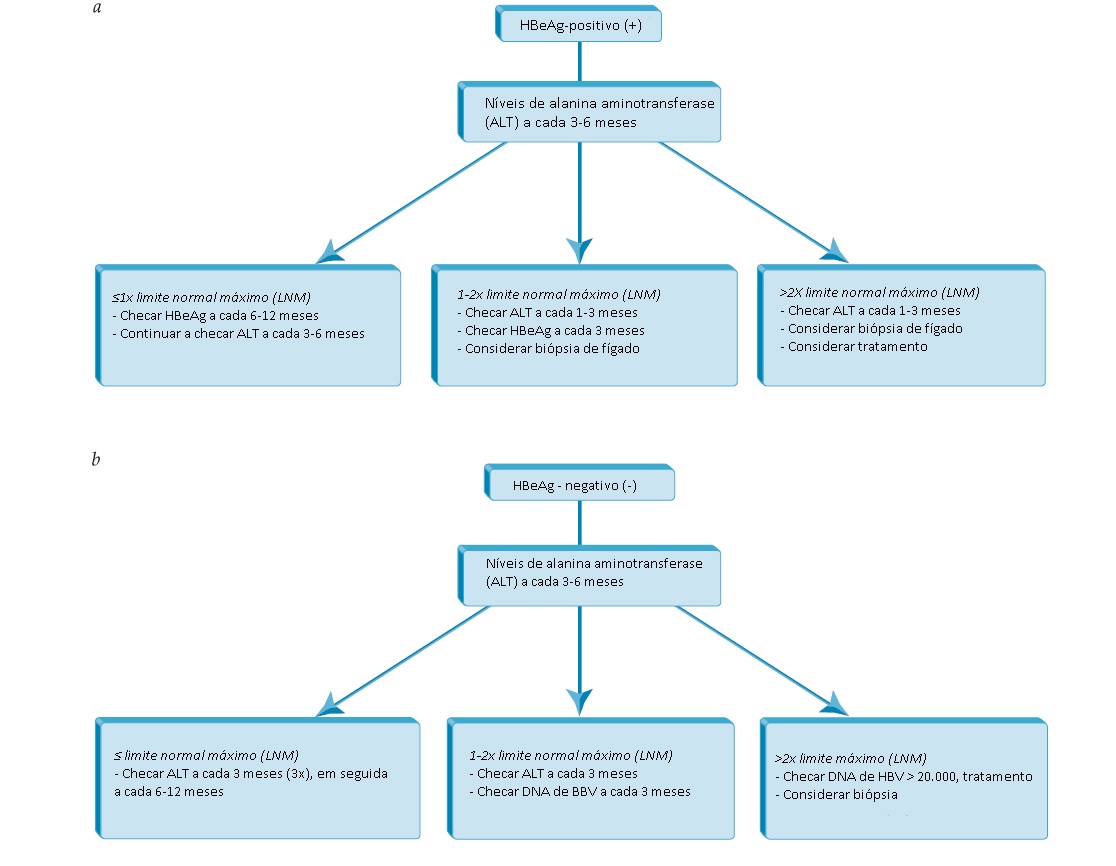
Se o AgHBs for detectado, a realização de testes adicionais é justificada para determinar mais precisamente o estado sorológico, virológico e bioquímico da doença. Este tipo de avaliação inclui exames de bioquímica hepática (AST, ALT), testes de excreção hepática (bilirrubina sérica) e prova de função de síntese (albumina sérica, tempo de protrombina), AgHBe, anti-HBe e DNA de HBV. Ensaios à base de PCR quantitativos e altamente sensíveis para detecção de DNA de HBV fornecem informação sobre a carga viral, que pode flutuar ao longo do curso da doença. A frequência com que o AgHBe e o DNA de HBV são monitorados após o diagnóstico é resumida na Fig. 2.
A necessidade de obter biópsia de fígado de um paciente para avaliar informação histopatológica é determinada pela contribuição destes dados para o tratamento. Uma biópsia de fígado fornece informação sobre o grau de inflamação (i.e., grau) e de fibrose (i.e., estágio), sendo que estes dados, por sua vez, podem ser úteis para determinar se um paciente é candidato à terapia antiviral e a necessidade de realizar triagem de complicações de cirrose e/ou CHC. Entretanto, uma combinação de informações demográficas (p. ex., idade), sorológicas, virológicas e bioquímicas muitas vezes é suficiente, na ausência de informação histológica, para determinar a necessidade de tratamento para infecção crônica por HBV.23,34,35
Tabela 6 Interpretação dos exames sorológicos para infecção por HBV
|
Teste sorológico |
Nunca exposto |
Imunizado |
Recuperação/exposição prévia |
|
AgHBs |
– |
– |
– |
|
Anti-HBs |
– |
+ |
+ |
|
IgM anti HBc |
– |
– |
– |
|
IgG anti-HBc |
– |
– |
+ |
|
AgHBe |
– |
– |
– |
|
Anti-HBe |
– |
– |
+ |
|
DNA de HBV |
– |
– |
– |
Contin..
|
Hepatite B aguda/inicial |
“Período de janela” da exposição aguda |
Hepatite B crônica AgHBe-positiva |
Portador de HBV inativo |
Hepatite B crônica AgHBe-negativa |
|
+ |
– |
+ |
+ |
+ |
|
– |
– |
– |
– |
– |
|
+ |
+ |
– |
– |
– |
|
– |
– |
+ |
+ |
+ |
|
+ |
– |
+ |
– |
– |
|
– |
– |
– |
+ |
+ |
|
++ |
+ |
+++ |
± |
++ |
Anti-HBc = anticorpo anti-core de hepatite B; anti-HBe = anticorpo anti-hepatite B e; anti-HBs = anticorpos anti-hepatite B de superfície; HBV = vírus da hepatite B; AgHBe = antígeno da hepatite B e; AgHBs = antígeno de superfície da hepatite B.
Embora o objetivo definitivo do tratamento da infecção crônica por HBV seja a prevenção de complicações (i.e., cirrose, CHC e morte), as metas a curto prazo incluem a supressão da replicação do HBV e da lesão hepática, bem como o alcance dos marcos de referência sorológicos associados à doença menos progressiva. Na prática, a decisão de iniciar o tratamento depende em grande parte da existência de evidências de inflamação e lesão hepáticas ativas. Embora as recomendações estabelecidas pelas sociedades “versadas” seja discretamente diferentes (p. ex., baseadas em limiares de ALT e de DNA de HBV algo diferentes), há um consenso emergente acerca das indicações para terapia antiviral em geral [ver Tabela 7} e, especificamente, quanto às diferenças de tratamento entre as formas AgHBe-positiva e AgHBe-negativa da hepatite B crônica. Em ambas as formas da doença, positiva e negativa para AgHBe, o tratamento deve ser iniciado para pacientes com altos níveis de DNA de HBV (=2 x 104 UI/mL) e ALT elevada (=2 x o limite normal máximo). Os limiares para tratamento de pacientes AgHBe-positivos e AgHBe-negativos diferem discretamente para os pacientes com atividade de ALT quase normal. Em pacientes AgHBe-positivos com altos níveis de DNA de HBV (HBV DNA =2 x 104 UI/mL) e atividade de ALT quase normal (=2 x o limite normal máximo), ou seja, em pacientes mais jovens com alto nível de imunotolerância ao HBV, o benefício da iniciação da terapia ainda não foi demonstrado. Em vez disto, estes pacientes devem ser monitorados a intervalos regulares de 3-6 meses e a terapia deve ser iniciado quando a doença se torna mais bioquimicamente ativa (ou havendo suspeita sugestiva e/ou se a idade do paciente for maior que 40 anos, uma biópsia de fígado mostre fibrose mais avançada).34-37 Em casos de pacientes com hepatite B crônica AgHBe-negativa com níveis de DNA de HBV =2 x 104 UI/mL e atividade de ALT quase normal (1 a <2 x o limite normal máximo), o tratamento é reservado para aqueles com inflamação mais ativa e/ou fibrose avançada na biópsia de fígado. O tratamento não seria iniciado em nenhuma das formas (positiva e negativa para AgHBe) de hepatite B crônica com níveis de DNA de HBV abaixo destes limiares e ALT normal. a duração do tratamento nem sempre é claramente definida, podendo ser indefinida. Em geral, as duas metas primárias imediatas da terapia são alcançar (1) níveis de DNA de HBV indetectáveis e (2) a perda ou soroconversão de AgHBe. No caso de um paciente que sofre soroconversão de AgHBe durante o tratamento, é possível considerar a suspensão da terapia após 6 meses, contudo o este paciente ainda deve continuar sendo monitorado quanto à reativação. Em contraste, em quase todos os pacientes com hepatite AgHBe-negativa, a duração do tratamento com agente oral tende a ser indefinida.
Atualmente, sete fármacos foram aprovados para uso no tratamento da hepatite B crônica; 5 análogos nucleosídicos dirigidos diretamente contra a polimerase viral, e duas preparações de interferon [ver Tabela 8]. A lamivudina (LMV), o primeiro fármaco oral aprovado para uso no tratamento da hepatite B, e outros dois fármacos orais menos efetivos e mais propensos à resistência, adefovir (ADV) e telbivudina (LdT), não mais são considerados terapia de primeira linha. Agora, o entecavir (ETV) e o fumarato de tenovofir disoproxila (TDF) são os tratamentos orais de primeira linha recomendados para pacientes tratamento-naive com hepatite B crônica. O interferon peguilado, que substituiu o interferon padrão, também é considerado um agente de primeira linha, exceto para pacientes com cirrose avançada.35 No contexto de falha de tratamento anterior ou recidiva após a terapia, a escolha do agente é guiada pelo regime de tratamento anterior e pela existência de mutações determinantes de resistência.
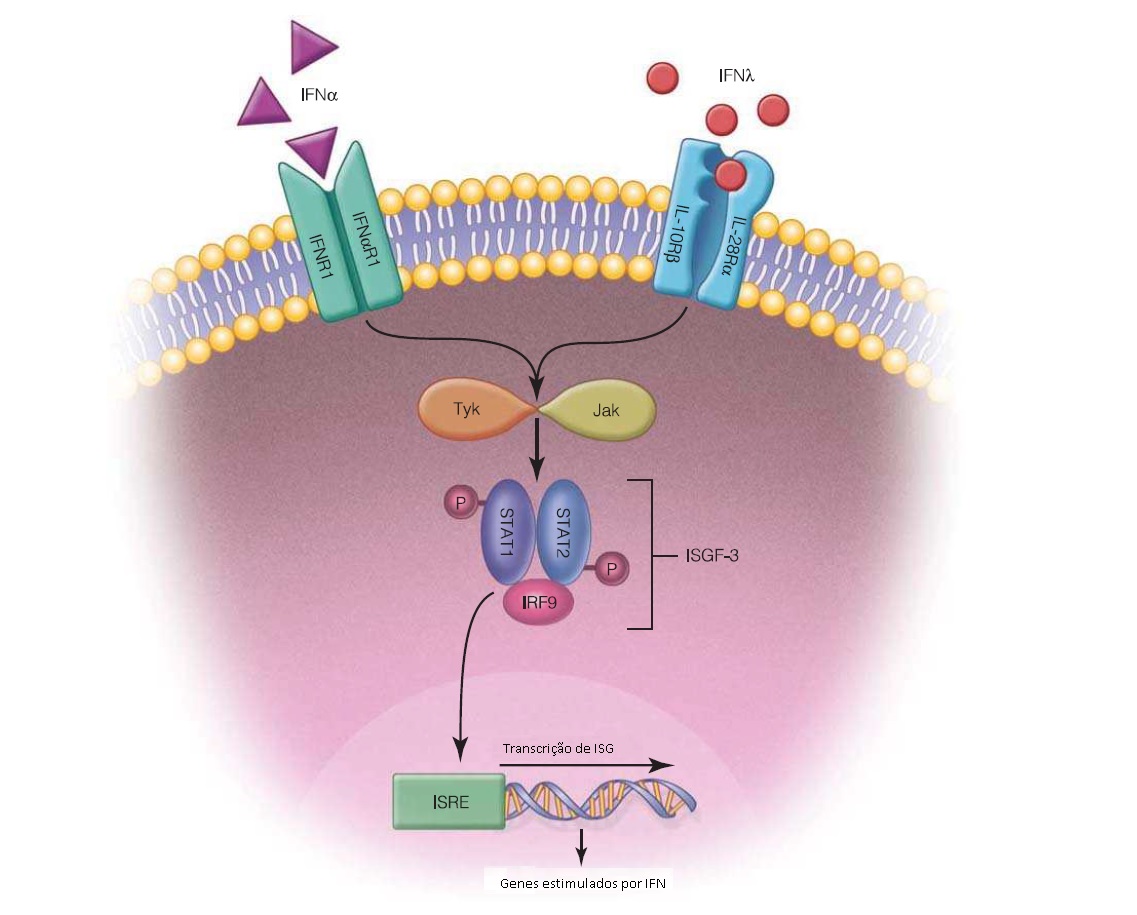
O interferon-a peguilado exerce atividades imunomodulatórias e antiproliferativas, embora não tenha sido determinado se estas propriedades contribuem para a sua atividade antiviral. Ligando-se aos receptores de superfície celular que ativam a cascata de transdução de sinal JAK-STAT, o interferon tem potencial de interferir em múltiplas etapas do ciclo de vida do HBV, entre outros vírus. Embora muitas autoridades favoreçam o uso dos inibidores de polimerase orais e não da terapia com interferon peguilado, dois estudos de referência—um sobre hepatite B AgHBe-reativa e outro sobre hepatite B AgHBe crônica—demonstraram a superioridade de um curso finito de 48 semanas de interferon peguilado, em comparação a um curso de 48 semanas de LMV, o primeiro agente oral, suplantado por seus sucessores mais efetivos e de menor resistência. Em pacientes com hepatite B AgHBe-reativa, decorridos 6 meses do término da terapia, 32% dos indivíduos tratados com interferon peguilado (versus apenas 19% dos indivíduos tratados com LMV) sofreram soroconversão de AgHBe. Apenas 14% dos indivíduos tratados com interferon peguilado continuaram apresentando supressão contínua do DNA.17 Em pacientes AgHBe-negativos, decorridos 6 meses do término do curso de 48 semanas de interferon peguilado ou de LMV, os níveis de DNA de HBV foram suprimidos a menos de 400 UI/mL em 19% e 7% dos indivíduos, respectivamente.38 Em ambos os estudos, a terapia combinada não foi melhor do que a monoterapia com interferon peguilado. Alguns destes resultados originais, todavia, não foram confirmados em estudos subsequentes e os resultados obtidos após o segundo ano de tratamento com agentes orais potentes foram similares ou superiores aos resultados alcançados durante o curso de um ano de interferon peguilado. Nos primeiros estudos realizados com interferon peguilado testado contra os primeiros agentes orais de baixa potência, o interferon peguilado pareceu proporcionar uma vantagem em termos de alcançar a perda de AgHBs. Entretanto, os níveis de análogos nucleosídicos de alta potência de última geração promovem níveis comparáveis de perda de AgHBs. Assim, a utilidade clínica do interferon peguilado é limitada por seus numerosos efeitos colaterais, bem como pela via de administração subcutânea e pela baixa tolerabilidade. As taxas de resposta ao interferon peguilado são mais altas em pacientes com HBV de genótipo A, ALT elevada e níveis baixos de DNA de HBV. Algumas autoridades sugerem o uso de interferon peguilado para pacientes jovens com níveis de DNA de HBV <109 e níveis de ALT no mínimo 2-3 vezes acima do limite normal máximo.36 Entretanto, estas variáveis de pacientes estão altamente associadas à responsividade aos antivirais orais. O interferon peguilado não é recomendado no contexto de cirrose avançada, devido ao alto risco de eventos adversos graves e, inclusive, potencialmente fatais.
Os outros fármacos aprovados para uso no tratamento da hepatite B têm como alvo a polimerase do HBV. Todos os fármacos atualmente disponíveis são inibidores nucleosídicos ou nucleotídicos que são incorporados à fita de DNA em alongamento, levando ao término da replicação. Embora estes fármacos sejam inibidores potentes do novo DNA viral, suprimem o cccDNA sem eliminá-lo (o mesmo se aplica ao interferon peguilado). Ou seja, nenhum dos agentes antivirais contra HBV erradica o vírus. Mesmo assim, a supressão da replicação do HBV está comprovadamente associada à melhora da histologia, inclusive com reversão da fibrose ou cirrose, e à diminuição das complicações da hepatite B crônica.
Os inibidores nucleosídicos são classificados em três grupos estruturais, que têm importância clínica pelo fato de a resistência e a resistência cruzada serem estrutura-específicas. Os L-nucleosídeos são LMV e LdT (outro agente estruturalmente bastante similar à LMV e com atividade indistinguível, a emtricitabina [FTC], é usado no tratamento da infecção por HIV/Aids, mas sem aprovação para uso no tratamento da hepatite B). Os dois alquil-fosfonatos, que são análogos nucleotídicos (e requerem uma única etapa [e não três etapas] de fosforilação para se tornarem ativos), são ADV e TDF. Por fim, ETV, um D-ciclopentano, tem perfil de resistência similar (porém com uma barreira muito maior à resistência) ao dos L-nucleosídeos.
A LMV, primeiro inibidor de análogo nucleosídico a ter aprovação do uso no tratamento da hepatite B, suprime o DNA do HBV em 60-73% dos pacientes e promove perda ou soroconversão de AgHBe em cerca de 20% dos pacientes após 1 ano de tratamento, e em cerca de 50% após 5 anos de terapia.39 Como primeiro agente oral para hepatite B, a LMV mostrou que—e comprovou o princípio de que os inibidores de polimerase—diminui a atividade necroinflamatória hepática, retarda a progressão da fibrose, reverte a fibrose e até a cirrose, recupera pacientes com descompensação hepática, e previne a descompensação em pacientes com fibrose ou cirrose avançada. Entretanto, a utilidade da LMV é limitada por sua barreira muito baixa à resistência. No contexto de monoterapia com LMV, há desenvolvimento de resistência em 23% dos pacientes após 1 anos e, após 5 anos de terapia, em até 80% dos pacientes.40 Há desenvolvimento de uma série característica de variantes de resistência à LMV que conferem resistência cruzada a ETV e LdT.
O ETV, um análogo nucleosídico mais potente, tem uma alta barreira à resistência porque a concentração de fármaco requerida para suprimir o HBV é tão baixa que as concentrações farmacológicas padrão alcançadas são suficientes para suprimir até mesmo as variantes resistentes, além disso é preciso que ocorram múltiplas (e não uma única) mutações para haver progressão.41 Em estudos de fase III envolvendo pacientes com doença AgHBe-positiva e AgHBe-negativa, o uso de ETV alcançou índices mais de supressão de DNA e melhora histológica, em comparação ao uso de LMV, ainda que nenhuma vantagem tenha sido observada entre os pacientes AgHBe-positivos em termos de soroconversão de AgHBe em 1 ano.37 Entretanto, após 1 ano, as taxas de soroconversão de AgHBe aumentaram progressivamente para 42% ao redor do 6º ano. A perda de AgHBs, incomum em pacientes tratados com inibidores nucleosídicos de gerações mais antigas, ocorreu em 2%, 5% e 6% dos indivíduos tratados com ETV em 1, 2 e 5 anos, respectivamente. Nos pacientes tratamento-naive, a emergência de resistência ao ETV é rara. Nos indivíduos estudados após as triagens de registro, houve desenvolvimento de resistência em apenas 1,2% dos casos após 6 anos de monoterapia. Em pacientes previamente tratados e resistentes à LMV, porém, a resistência ao ETV emerge rapidamente e atinge 51% por volta do 5º ano.40 Desta forma, embora seu uso seja aprovado para pacientes resistentes à LMV, o ETV não deve ser usado nesta população de pacientes.
O TDF é o análogo nucleosídico mais recentemente aprovado para uso no tratamento da hepatite B. Embora a soroconversão de AgHBe após 1 ano seja aproximadamente de apenas 20%, após 4 anos de terapia, a perda de AgHBe aumenta para 41%.39 Nos pacientes AgHBe-reativos, a perda de AgHBs aumenta de 3% no 1º ano para 8% no 3º ano. Também foi demonstrado que o TDF tem uma barreira muito alta à resistência e não apresenta resistência cruzada com L-nucleosídeos nem com o ETV. Após cerca de 5 anos de avaliação de segmento, nenhuma resistência foi relatada em pacientes tratados com monoterapia de TDF.42 O uso de TDF, sendo bastante seguro, está associado a um pequeno risco de nefrotoxicidade que requer monitoramento regular periódico (p. ex., a cada 6 meses) de creatinina.
O ADV, introduzido antes e estruturalmente similar ao TDF, é o menos potente dos agentes orais e tem menor barreira de resistência do que o TDF. Assim como este, o ADV não apresenta resistência cruzada com a LMV. Em estudos de registro, apenas 12% dos pacientes AgHBe-reativos alcançaram soroconversão de AgHBe após 1 ano. Mesmo na ausência de resistência, a supressão viral pode ser incompleta. O principal efeito adverso do ADV, assim como o do TDF, é a nefrotoxicidade dose-dependente que ocorre em até 0,5% de todos os pacientes bem compensados e em 6% dos pacientes com doença hepática descompensada. Por sua inferioridade ao TDF, o ADV não é mais considerado um tratamento de primeira linha para hepatite B, mas seu uso requer monitoramento rotineiro da creatinina.
O LdT é estruturalmente similar à LMV, mas sua potência é maior. Entretanto, similar ao LMV, o LdT apresenta alta taxa de resistência—21% após 2 anos de monoterapia40– e não é recomendado como agente de primeira linha. Entretanto, o LdT é classificado na categoria B de gravidez e foi estudado na prevenção da transmissão materno-fetal da infecção por HBV em mães com altos níveis de DNA de HBV. A terapia com LdT iniciada na 20ª a 32ª semana de gestação diminuiu a positividade para AgHBs em recém-nascidos de 8% para 0 aos 8 meses de seguimento.43 Embora, teoricamente, o LdT possa ter utilidade no tratamento da hepatite B durante a gestação, relatos incluídos nos registros de gravidez demonstraram que, até o presente, os outros agentes orais são seguros para uso durante a gestação e, em termos práticos, o LdT exerce papel muito limitado ou nulo no tratamento da hepatite B.
A FTC é estruturalmente similar à LMV, tendo potência limitada e baixa barreira à resistência à LMV. Embora não seja aprovada como terapia para hepatite B, a FTC é disponibilizada na forma combinada com TDF (Truvada), que é comercializada para tratamento da infecção por HIV. Alguns autores prescrevem FTC/TDF para pacientes previamente tratados que apresentem resistência à LMV.
Incomum entre as infecções virais, a infecção por HBV geralmente responde à monoterapia, em especial na época atual, em que os fármacos de primeira linha (ETV e TDF) apresentam uma barreira tão alta à resistência. Devido à preocupação com a emergência de resistência, os pacientes tratados para hepatite B devem ter os níveis de DNA de HBV monitorados regularmente. Entretanto, como o risco de resistência é baixo com o uso dos agentes de última geração, é possível espaçar o monitoramento com intervalos de 6 meses. O monitoramento intensivo é necessário após a descontinuação da terapia antiviral, após as respostas de AgHBe ou AgHBs. É necessário haver aderência estrita à terapia diária para que o desenvolvimento de resistência seja limitado. O primeiro sinal de resistência é o rebote sustentado da viremia após uma supressão adequada, ao qual se segue a elevação dos níveis de aminotransferase. Se a não aderência for excluída, um padrão de viremia reemergente ± elevação de ALT geralmente é suficiente para sugerir resistência e resultar na adição de um segundo fármaco isento de resistência cruzada. Alternativamente, a genotipagem da resistência é uma opção. Um princípio importante do manejo da resistência é o de que a monoterapia sequencial deve ser evitada para minimizar o risco de resistência a múltiplos fármacos, ou seja, quando há desenvolvimento de resistência, o novo fármaco sem resistência cruzada deve ser adicionado e não substituir o fármaco original [ver Tabela 9].
Figura 2: Monitoramento de paciente com positividade para antígeno de superfície da hepatite B (AgHBs) que não recebe terapia antiviral. (a) recomendações de monitoramento para pacientes com hepatite B e positivos para antígeno (AgHBe). (b) Recomendações de monitoramento para pacientes AgHBe-negativos.
Tabela 7 Diretrizes do tratamento para hepatite B crônica
|
HBe/Ag |
DNA de HBV |
ALT |
Biópsia de fígado |
Manejo |
Escolha do agente de primeira linha |
|
Positivo |
>20.000 UI/mL |
<2 x LNM |
Nenhuma |
Considerar a obtenção de biópsia para idade >40 anos, ALT normal alta ou história familiar de CHC |
N/A |
|
Sem inflamação |
“Imunotolerante”, continuar o monitoramento |
N/A | |||
|
Inflamação moderada ou intensa, fibrose |
Tratar |
Entecavir, tenofovir, interferon alfa PEG (x48 semanas) | |||
|
>20.000 UI/mL |
>2 x LNM |
Inflamação ou fibrose |
Tratar |
Entecavir, tenofovir, interferon alfa PEG (x48 semanas) | |
|
Negativo |
>20.000 UI/mL |
>2 x LNM |
Sem inflamação, fibrose |
Tratar |
Entecavir, tenofovir indefinidamente; interferon alfa PEG (x48 semanas) |
|
>2.000 UI/mL |
>2 x LNM |
Nenhuma |
Considerar biópsia |
N/A | |
|
|
|
Inflamação moderada ou intensa, fibrose |
Tratar |
Entecavir, tenofovir indefinidamente; interferon alfa PEG (x48 semanas) | |
|
<2.000 UI/mL |
Normal |
Nenhuma |
Continuar a seguir ALT, DNA |
N/A | |
|
± |
Detectável |
Qualquer |
Cirrose |
Tratar; considerar encaminhamento para centro de transplante em caso de descompensação |
Entecavir, tenofovir indefinidamente |
ALT = alanina aminotransferase; CHC = carcinoma hepatocelular; AgHBe = antígeno da hepatite B e; HBV = vírus da hepatite B; N/A = não aplicável; PEG = peguilado; LNM = limite normal máximo.
Tabela 8 Fármacos antivirais para hepatite B143,144
|
|
Interferon peguilado |
Lamivudina |
Telbivudina |
|
Classe |
Agente biológico |
L-nucleosídeo |
L-nucleosídeo |
|
Dose* |
180 pg, 1x/semana, injeção subcutânea |
100 mg /dia |
600 mg /dia |
|
Duração |
48 semanas |
Variável a indefinida |
Variável a indefinida |
|
Log10 do declínio de DNA de HBV (48 semanas)+ |
4,5 |
5,5 |
6,4 |
|
Perda de AgHBe (48-52 semanas)+ |
27% |
21% |
22% |
|
Perda de AgHBs (48-52 semanas)+ |
3% |
<1% |
<1% |
|
Frequência da resistência |
|
30% em 1 ano 80% em 5 anos |
4,4% em 1 ano |
|
Frequência de resistência (resistente à LMV) |
|
|
|
|
Efeitos adversos/monitoramento |
Pouco tolerado: sintomas semelhantes aos da gripe, mialgias, leucopenia, trombocitopenia; contraindicado na cirrose descompensada |
Altas taxas de resistência |
Elevações assintomáticas de creatinina quinase |
Contin...
|
Adefovir |
Tenofovir |
Entecavir |
|
Nucleotídeo acíclico |
Nucleotídeo acíclico |
Análogo de guanosina |
|
10 mg /dia |
300 mg /dia |
0,5 mg /dia |
|
Variável a indefinida |
Variável a indefinida |
Variável a indefinida |
|
3,5 |
6,2 |
6,9 |
|
12% |
21% |
21% |
|
0% |
3% |
2% |
|
0% em 1 ano 29% em 5 anos |
0% em 1 ano 0% em 5 anos |
0,2% em 1 ano 1,2% em 5 anos |
|
18% em 1 ano |
|
6% em 1 ano (51% em 5 anos) |
|
Função renal comprometida (rara); monitoramento periódico da creatinina recomendado |
Associado com comprometimento renal raro; monitoramento periódico da creatinina recomendado |
Bem tolerado; perfil de segurança similar ao da lamivudina |
AgHBe = antígeno da hepatite B e; HBV = vírus da hepatite B; LMV = lamivudina.
*Ajustes de dose requeridos para o comprometimento renal.
+Hepatite B crônica HBe-Ag-positiva.
Pacientes submetidos à quimioterapia citotóxica e aqueles que necessitam de imunossupressão potente por tempo prolongado (como a necessidade de doses altas de corticosteroides ou inibidores de fator de necrose tumoral-a na artrite reumatoide ou na enteropatia inflamatória) apresentam risco de reativação da hepatite B durante esta quimioterapia ou conforme a quimioterapia é retirada. Historicamente, 4-72% dos pacientes submetidos à quimioterapia apresentam reativação de HBV.44 Em adição, pode ser difícil detectar a reativação durante a imunossupressão, porque os níveis de ALT podem estar normais apesar da rápida replicação de HBV. Entretanto, depois que os agentes quimioterápicos são suspensos e há imunorreconstituição no hospedeiro, uma robusta resposta necroinflamatória aos hepatócitos vírus-infectados se torna evidente e pode haver desenvolvimento de uma hepatite grave e até fulminante.
Este risco de reativação se aplica não só a pessoas com AgHBs detectável (incluindo pacientes com hepatite crônica e estado de portador de AgHBs inativo) como também àquelas com infecção “resolvida”, que têm anticorpos anti-HBs e anti-HBc. Portanto, os pacientes para os quais há planos de imunossupressão devem ser submetidos à triagem de rotina para detecção de evidências de infecção por HBV vigente ou anterior à iniciação do tratamento. Todo paciente positivo para AgHBs deve receber profilaxia antiviral durante a terapia citotóxica ou imunossupressora, enquanto os pacientes AgHBs-negativos com anticorpos anti-HBs e/ou anticorpos anti-HBc devem ser monitorados de perto. Embora a LMV atualmente tenha utilidade clínica limitada como terapia de primeira linha, é o fármaco mais bem estudado na profilaxia da reativação. A resistência à LMV tende a não ocorrer durante o breve período de tratamento profilático, sendo que alguns clínicos ainda a usam com esta indicação. Entretanto, a maioria seleciona agentes mais potentes, de baixa resistência e modernos. Com base nas recomendações atuais, os pacientes AgHBs-positivos devem receber um análogo nucleosídico oral a partir da 1ª semana, mas de preferência por várias semanas antes da imunossupressão. A duração da terapia antiviral após a imunossupressão é indeterminada, mas deve ser estendida por no mínimo 6 meses após a restauração do funcionamento normal do sistema imune.36,44 Em muitos casos, é difícil descontinuar a terapia antiviral devido à hepatite persistente após a retirada do agente antiviral. Se a LMV for usada com esta indicação, as taxas de resistência a este agente durante o tratamento profilático tendem a ser muito baixas, contudo se houver necessidade de um tratamento com duração mais longa, a adição de ADV ou TDF deve ser considerada.
Para a maioria dos pacientes com hepatite B crônica, a atual geração de fármacos antivirais é suficiente para suprimir a replicação viral e limitar profundamente a lesão, bem como para minimizar substancialmente as complicações da hepatite B crônica. Entretanto, a vasta maioria dos pacientes necessita de terapia antiviral indefinida. Embora as terapias atualmente disponíveis sejam altamente efetivas para supressão da viremia, a meta final da erradicação viral sustentada por meio da terapia antiviral continua sendo elusivo. Outras classes de agentes antivirais com outros alvos além da HBV DNA polimerase estão sendo procurados. Um exemplo são os inibidores de encapsidação (que interferem na encapsidação da proteína do core, da polimerase e do RNA pré-genômico viral na montagem do capsídeo viral). Entretanto, embora estes novos agentes aumentem as opções para tratamento da infecção por HBV nucleosídeo-resistente, estes fármacos novos também terão capacidade limitada de promover uma erradicação viral completa e sustentada.41,45 Para atingir esta meta, seria necessário que as novas classes de fármacos fizessem mais do que suprimir a viremia da hepatite B, ou seja, teriam que replicar a resposta imune do hospedeiro associada à recuperação espontânea a partir da infecção por HBV. Ainda é discutido se estas aspirações poderão ser concretizadas.
Tabela 9 Mutações determinantes de resistência ao HBV e manejo da resistência aos fármacos antivirais37,144,145
|
Classe farmacológica |
Mutação |
Lamivudina |
Telbivudina |
Adefovir |
Tenofovir |
Entecavir |
Recomendação |
|
Tipo selvagem |
Não há
|
S |
S |
S |
S |
S |
|
|
L-nucleosídeo |
M204V/I |
R |
R |
S |
S |
I |
Adicionar adefovir ou tenofovir Suspender à lamivudina, trocar por tenofovir |
|
L-nucleosídeo |
A181T/V |
I/R |
R |
R |
I* |
S |
Adicionar tenofovir à lamivudina Trocar por tenofovir Trocar por entecavir |
|
Nucleotídeo acíclico |
A181T/V |
I/R |
R |
R |
I* |
S |
Adicionar tenofovir à lamivudina Trocar por tenofovir Trocar por entecavir |
|
Nucleotídeo acíclico |
N236T |
S |
S |
R |
I* |
S |
Trocar por tenofovir Trocar por entecavir Adicionar lamivudina ao adefovir |
|
D-ciclopentano |
L180M e M204V/I
|
R |
R |
S |
S |
R |
Trocar por ou adicionar tenofovir ou adefovir |
HBV = vírus da hepatite B; I = intermediário; R = resistência; S = sensibilidade.
*Resistência in vitro e não in vivo ao tenofovir observada em seres humanos por até 144 semanas de tratamento.
O vírus da hepatite d ou D (HDV) é um pequeno vírus contendo RNA defeituoso que conta com a coinfecção pelo HBV para poder se replicar, e também com a proteína estrutural do envelope, a AgHBs. A epidemiologia da infecção por HDV, portanto, é paralela (mas não replica exatamente) a da infecção pelo HBV. A infecção pelo HDV está amplamente distribuída ao redor do mundo. Estima-se que cerca de 5% dos indivíduos com hepatite B crônica (i.e., cerca de 15 milhões de pessoas) estão coinfectadas pelo HDV.46 As taxas de prevalência da infecção por HDV entre indivíduos com hepatite B crônica variam ao longo das regiões geográficas, indo de 90% nas ilhas do Pacífico a 5% no Japão.47 A existência da infecção por HDV foi descoberta inicialmente na Itália, durante a década de 1970, coincidindo com um período de migração a partir da Itália sulista, agrária e de alta prevalência para a Itália nortista, industrializada e de baixa prevalência. Entre 1987 e 1997, na Itália, a soropositividade para HDV entre indivíduos com AgHBs, todavia, diminuiu de 23% para 9%.48 Esta redução pode ter resultado dos padrões migratórios alterados, da saturação da base de indivíduos AgHBs-positivos, da implementação de programas de vacinação contra hepatite B e das práticas adotadas pelos usuários de drogas injetáveis para evitar a aquisição de infecção por HIV. O declínio observado na Itália, porém, não foi totalmente sustentado e as taxas de prevalência da infecção por HDV entre indivíduos AgHBs-positivos em algumas áreas da Europa continuam chegando a 8-14%,49 impulsionadas com toda certeza pela imigração a partir de áreas onde a infecção por HDV permanece endêmica. Apesar de as evidências sugerirem a ocorrência de transmissão intrafamiliar e sexual da infecção por HDV, a transmissão percutânea é o modo de exposição mais comum. Conforme sugerido anteriormente, a prevalência da infecção por HDV em muitas regiões é mais alta entre os usuários de drogas injetáveis. A prevalência e a incidência da infecção por HDV são paralelas à ocorrência da infecção por HDV em usuários de drogas injetáveis. Em um estudo conduzido nos EUA, uma alarmante prevalência de infecção por HDV de 50% foi encontrada entre os usuários de drogas injetáveis cronicamente infectados pelo HBV.50 Assim, a hepatite continua sendo um problema de saúde pública substancial em certas populações, particularmente entre os usuários de drogas injetáveis.
Os oito genótipos de HDV identificados estão amplamente distribuídos, mas tendem a seguir padrões geográficos diferentes.51 O genótipo 1, que é o mais comum, é encontrado no mundo inteiro. O genótipo 3 está concentrado na bacia amazônica (onde uma forma particularmente virulenta de infecção por HDV [coinfecção ou superinfecção; ver adiante] resulta em “febre Lábrea”, uma doença caracterizada por hepatite progressiva fulminante), enquanto os genótipos 5 a 8 são comuns na África. Do ponto de vista clínico, o genótipo 3 foi associado a um risco maior de hepatite fulminante, enquanto o genótipo 1 é considerado associado a uma doença de curso mais progressivo e menos responsiva ao tratamento com interferon.46,52
O HDV é um vírus a RNA com genoma contendo apenas 1.700 nucleotídeos—o menor genoma dentre todos os vírus humanos conhecidos. De fato, o HDV se parece mais com um viroide vegetal do que com outros vírus a RNA humanos. Seu genoma codifica uma única proteína, o antígeno d (HDVAg).53,54 O vírus circula como um pequeno vírion de 35-37 nm, que contém o genoma viral encapsulado em um envelope de AgHBs. Conforme já observado, a infecção por HDV conta com a coinfecção pelo HBV. Em indivíduos imunocompetentes, a replicação do HDV requer a proteína do envelope do HBV, o AgHBs, que teoricamente permitiria ao HDV se ligar e entrar no hepatócito. No entanto, o mecanismo preciso da entrada do HDV é desconhecido. Uma vez internalizado na célula, o HDV se transloca para o núcleo e se torna independente da assistência do HBV, passando a roubar as RNA polimerases do hospedeiro para replicar seu genoma. O ciclo de replicação do HDV está bem caracterizado e depende de um mecanismo circular rolante duplo para replicação,55 que permite ao vírus desviar um DNA intermediário. Três formas de RNA viral são os produtos de replicação, um dos quais é o produto do RNA mensageiro de quadro de leitura aberto, que é traduzido no citoplasma nas duas formas de antígeno d (grande e pequeno). O antígeno pequeno volta para o núcleo e promove replicação viral,56 enquanto o antígeno maior atua na montagem do vírion. Modificações pós-tradução (especificamente a prenilação) do antígeno grande são requeridas por sua função na montagem viral.57 Com a dependência de fatores do hospedeiro e do HBV, o HDV tem alvos limitados em seu ciclo de vida que são passíveis de intervenção terapêutica, contudo a prenilação tem sido uma área de interesse. Uma observação interessante é a ocorrência, tipicamente, de níveis muito baixos de HBV em pacientes coinfectados por HDV/HBV, sugerindo que a replicação do HDV inibe a replicação do HBV.58,59
A apresentação clínica e o resultado da infecção por HDV dependem em parte de o vírus ter sido adquirido ao mesmo tempo em que uma infecção aguda por HBV (coinfecção simultânea) ou ter se sobreposto a uma infecção por HBV já crônica (superinfecção). A coinfecção aguda se manifesta mais frequentemente como um episódio agudo e autolimitado de hepatite, embora a coinfecção tenda mais do que a infecção por HBV isolada a causar hepatite fulminante.52 Por depender totalmente da replicação do HBV, a infecção por HDV não pode durar mais do que a infecção por HBV e, como a hepatite B aguda em indivíduos saudáveis é quase sempre autolimitada, a coinfecção simultânea por HDV/HBV aguda raramente progride para cronicidade.
Entretanto, se a exposição do HDV ocorrer depois que infecção persistente por HBV já estiver estabelecida, tanto a apresentação clínica como o resultado serão bastante diferentes. Os achados clínicos de superinfecção em pacientes com hepatite B crônica (incluindo aqueles que são portadores de AgHBs inativo e aqueles com hepatite crônica) variam de uma elevação assintomática ou não detectada dos níveis de aminotransferase a uma hepatite aguda, hepatite fulminante até a descompensação da hepatite B crônica. Em contraste com a coinfecção aguda, a superinfecção resulta em persistência viral em mais de 90% dos casos,46 sendo que o fenótipo viral mais comum na infecção crônica é o de hepatite D ativa com hepatite B inativa. Com o passar do tempo, entretanto, a taxa de replicação de HDV pode enfraquecer, permitindo que ocorra em alguns pacientes a restauração do HBV como vírus dominante.
A história natural de coinfecção por HDV/HBV parece seguir um curso mais progressivo, com taxas maiores de cirrose e CHC. Há desenvolvimento de cirrose em até 70% dos casos, podendo ocorrer dentro de 2 anos de coinfecção em 15%. O risco de progressão para CHC e cirrose está associado à prevalência da replicação do vírus ativo, com o sexo feminino e com o suo de álcool.60 Entretanto, diferente da hepatite B crônica, a progressão da doença na hepatite D crônica não está claramente associada a níveis detectáveis de replicação viral,52 mas pode estar relacionada mais com o genótipo HDV ou, conforme sugerido em vários estudos, aos genótipos de HDV e HBV. Exemplificando, o genótipo I de HDV e o genótipo C de HBV estão associados a taxas mais altas de cirrose e mortalidade associada ao fígado.59 Dados retrospectivos sugerem que os pacientes submetidos ao tratamento antiviral para hepatite D se comportam melhor, com taxas diminuídas de CHC e de descompensação hepática.61
O diagnóstico sorológico de infecção por HDV é baseado na detecção de anticorpos anti-HDV (IgM e IgG circulam, mas não permitem distinguir entre infecção aguda/recente e crônica/remota) e/ou de RNA de HDV. O HDVAg sérico, que não é prontamente detectável, não tem utilidade como marcador da infecção ativa por HDV. Quando presente, o RNA de HDV, medido por ensaios de amplificação altamente sensíveis (p. ex., PCR), é indicativo de infecção ativa e constitui um marcador diagnóstico mais confiável. Conforme observado, os anticorpos IgM anti-HDV podem estar presentes durante a infecção aguda por HDV, mas podem demorar 30-40 dias após a exposição para aparecerem—momento em que anticorpos IgG anti-HDV podem estar presentes também—podendo apresentar títulos baixos e durabilidade limitada. A IgG anti-HDV pode representar uma exposição prévia ou atual, sendo que o teste de RNA de HDV se faz necessário para determinar se há infecção ativa [ver Tabela 10].
Em pacientes com hepatite B aguda ou crônica, o teste para infecção por HDV deve ser considerado para pacientes que apresentam fatores de risco de infecção por HDV (p. ex., uso de drogas injetáveis, hemodiálise), pacientes de áreas de ampla endemicidade da infecção por HDV e pacientes com hepatite incomumente grave ou fulminante, sobretudo em pacientes previamente estáveis com hepatite B crônica que sofreram descompensação precipitada.
Nenhum dos análogos nucleosídicos orais efetivos no tratamento da hepatite B é efetivo no tratamento da hepatite D. Atualmente, a única opção terapêutica para o manejo da hepatite D crônica é o interferon, que parece melhorar o resultado a longo prazo.
O interferon-a e o interferon peguilado são as únicas medicações que comprovadamente tratam a hepatite D de forma efetiva, contudo as taxas de resposta são baixas e um curso terapêutico prolongado se faz necessário. Quando o interferon peguilado é administrado por 48-72 semanas, as taxas de resposta ficam na faixa de 20-30%. Conforme notado antes, não há benefícios quando um análogo nucleosídico oral (p. ex., adefovir, que foi alvo de um estudo) é adicionado ao interferon peguilado.62 A busca de novos alvos farmacológicos é justificada. Duas classes de fármacos ainda em fase inicial de exploração clínica são os inibidores de prenilação, que bloqueiam a modificação pós-tradução do HDVAg grande (necessário à produção dos vírions infecciosos), e os inibidores de entrada de HBV que, teoricamente, deveriam bloquear a entrada celular de ambos os vírus contendo envelope de AgHBs (HBV e HDV).63 Como a terapia antiviral para hepatite D ainda é muito limitada, o tratamento ideal enfoca a prevenção da infecção por HDV por meio de uma combinação de vacinação contra hepatite B (que pode prevenir a coinfecção simultânea por HBV e HDV, mas que não previne a superinfecção por HDV depois de estabelecida a infecção por HBV), triagem e minimização do risco.
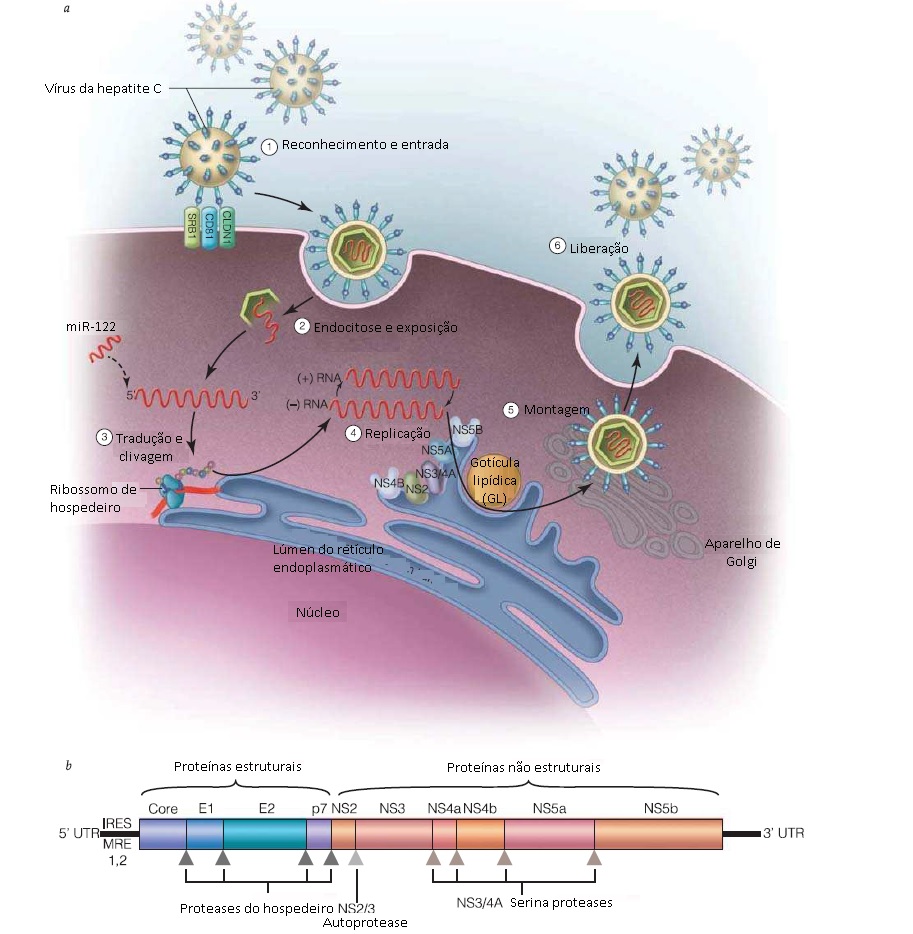
O HCV é a segunda causa vírus-associada mais frequente de CHC e de morte associada ao fígado no mundo inteiro, perdendo apenas para a infecção por HBV. Cerca de 3% da população global (130-170 milhões de pessoas) brigam a infecção crônica por HCV.64 A prevalência do HCV varia geograficamente, com as menores taxas encontradas na Escandinávia (<1%) e as maiores taxas no Egito (15-20%).65 A alta prevalência no Egito é uma consequência triste e não intencional de uma campanha nacional de terapia com injeção de antimônio antiesquistossomal, que usou seringas reutilizáveis, conduzida nas décadas de 1970 e 1980. Nos EUA, cerca de 1-2% da população abriga a infecção crônica por HCV, sendo que a maior carga de infecção atualmente reside na coorte de indivíduos de 40-50 anos de idade, sugerindo a ocorrência de um pico de transmissão há 30-40 anos. De fato, a incidência anual de novos casos relatados ao US Public Health Service caiu drasticamente no período de 1989 a 2002, estabilizando-se subsequentemente em uma taxa bem mais baixa. O período de pico de incidência ocorrido há 30-40 anos coincide com uma época em que houve aumento epidêmico do uso/experimentação de drogas injetáveis, enquanto o declínio ocorrido durante a após a década de 1990 se deu em paralelo a uma era de consciência aumentada—e adoção de comportamentos preventivos—sobre a infecção por HIV/Aids e, em extensão bem menor, à iniciação da triagem de HCV em doadores de sangue a partir de 1992. Embora a hepatite C inicialmente tenha sido identificada em receptores de produtos do sangue, a hepatite C associada à transfusão nunca representou mais do que uma pequena fração de todas as infecções por HCV que, em sua vasta maioria, eram adquiridas pelo uso de drogas injetáveis. Com os ensaios e testes de rotina atualmente disponíveis para análise do sangue doado, o risco de infecção por HCV associada à transfusão caiu para 1 em 2,3 milhões.
O HCV é transmitido efetivamente por contato percutâneo direto com sangue infectado. Os modos de transmissão incluem o uso de drogas injetáveis, uso antigo (mas atualmente suspenso) de produtos do sangue contaminados, além de exposição ocupacional e outras formas de exposição percutânea (p. ex., via picada de agulha no cenário da assistência médica). A transmissão materno-fetal é possível, todavia a taxas bem menores do que para HBV ou HIV. Cerca de 4,3% das mães com viremia de hepatite C transmitem a infecção por HCV aos bebês, porém a coinfecção por HIV aumenta a taxa de transmissão perinatal de HCV para 19%.66 Embora pequenas quantidades de RNA de HCV possam ser detectadas no leite materno, a amamentação não está associada à transmissão da infecção por HCV.
A transmissão sexual da infecção por HCV é ineficiente e as limitadas evidências que sustentam a transmissão sexual são, na melhor hipótese, controversas. No contexto de comportamentos sexuais de alto risco, entretanto, como ocorre com indivíduos que têm múltiplos parceiros sexuais e/ou doenças sexualmente transmissíveis (i.e., em situações associadas à lesão de mucosa, com promoção de contato direto sangue-sangue), pode haver transmissão sexual do HCV de forma mais eficiente. Uma alta frequência de transmissão sexual da infecção por HCV foi relatada em homens HIV-positivos que têm relações sexuais com outros homens.67
Nos Estados Unidos, o maior grupo de risco de infecção por HCV continua sendo o de usuários de drogas injetáveis. Estima-se que quase 50% dos americanos que alguma vez fizeram uso de drogas injetáveis foram expostos ao HCV.68 A coinfecção por HIV/HCV é particularmente preocupante, sendo que alguns estudos demonstraram que, entre os usuários de drogas injetáveis, até 90% também têm infecção por HCV crônica.69
Foram identificados seis genótipos de HCV, numerados de 1 a 6. Distribuição genotípica varia geograficamente e o genótipo do HCV tem implicações clínicas relevantes. Nos EUA, o genótipo 1 é o mais comum, responsável por 70% das infecções crônicas, seguido pelo genótipos 2 e 3, que juntos respondem por 30% dos casos. O genótipo 4 é responsável por uma proporção muito pequena das infecções por HCV nos Estados Unidos, enquanto os outros genótipos são ainda mais raros. Na Europa, assim como nos EUA, os genótipos 1, 2 e 3 prevalecem. Em contraste, os genótipos 4 a 6 são encontrados em áreas de alta endemicidade. No Egito, a vasta maioria das pessoas com hepatite C crônica abriga vírus com genótipo 4. Os genótipos 5 e 6 foram encontrados na África do Sul e Sudeste Asiático, respectivamente. Os genótipos diferem drasticamente quanto às taxas de resposta à terapia à base de interferon, com os genótipos 1 e 4 sendo os mais refratários e os genótipos 2 e 3, os mais responsivos ao tratamento.
Tabela 10 Características clínicas da hepatite D
|
|
Coinfecção aguda por HBV/HDV |
Coinfecção aguda resolvida por HBV/HDV |
Superinfecção por HDV de infecção por HBV crônica |
|
Resultado clínico |
90-95% dos adultos depuram ambos os vírus |
Há desenvolvimento de hepatite D crônica em >80% | |
|
Marcadores sorológicos |
|
|
|
|
AgHBs |
+ |
– |
+ |
|
IgM anti-HBc |
+ |
–/raramente residualmente reativo |
– |
|
Anti-HBs |
– |
+ |
– |
|
DNA de HBV |
++ |
– |
++ (títulos frequentemente baixos) |
|
Ag de HDV |
+ |
– |
+ (pode ser transiente) |
|
RNA de HDV |
+ |
– |
+++ |
|
IgM anti-HDV |
+ |
– |
± |
|
IgG anti-HDV |
± |
+ (título baixo) |
+ (título alto) |
|
Tratamento |
De suporte |
Nenhum |
Interferon peguilado: 180 µg/semana (x48 semanas) |
Anti-HBc = anticorpo anti-core da hepatite B; anti-HBs = anticorpos anti-hepatite B de superfície; AgHBs = antígeno de superfície da hepatite B; HBV = vírus da hepatite B; HDV = vírus da hepatite D.
O HCV, um pequeno vírus envelopado contendo RNA, recebeu o rótulo de Hepacivirus junto à família Flaviviridae. O conhecimento acerca do ciclo de vida do HCV aumentou substancialmente desde os primeiros modelos efetivos de replicação do HCV, baseados em culturas de células, descobertos na última década.70,71 O vírus circula na forma de uma partícula rica em lipídios, denominada partícula lipoviral, que compartilha muitas similaridades com a lipoproteína de densidade muito baixa humana e contém apolipoproteínas humanas e apolipoproteínas de superfície virais.72 Vários receptores de superfície essenciais presentes nos hepatócitos estão envolvidos na entrada do HCV, incluindo a ocludina, claudina, receptor scavenger de classe B do tipo I (SR-BI) e CD81.73 O receptor de lipoproteína de baixa densidade também pode atuar na entrada viral e reconhecer tanto as apolipoproteínas do hospedeiro como as glicoproteínas de superfície virais, levando à internalização do vírus.74
Uma vez que dentro da célula, o HCV é desnudado revelando o genoma viral descoberto. O genoma viral consiste em uma fita única de RNA medindo 9,6 kB, contendo as regiões não traduzidas (RNTs) 5’ e 3’ e um único quadro de leitura aberto que codifica 10 proteínas virais. As RNTs 5’ e 3’ exercem papeis importantes na promoção da replicação e da tradução. A RNT 5’ tem dois elementos essenciais: um sítio de reconhecimento para o microRNA-122 fígado-específico e um sítio de entrada ribossomal interno (IRES), que permite ao vírus proceder diretamente à síntese proteica de modo “capa-independente” (i.e., o sítio de reconhecimento ribossômico usual na extremidade 5’ de um RNA não é requerido para iniciar a tradução). A relação entre o microRNA-122 e a replicação viral é complexa, sendo que o microRNA-122 parece promover a tradução (em contraste com o papel clássico dos microRNAs na inibição da expressão proteica) e potencialmente ajudar o vírus a escapar dos imunossensores inatos intracelulares.75-77
O vírus rouba a maquinaria de tradução do hospedeiro para criar um polipeptídeo único. Em seguida, uma combinação de proteases do hospedeiro e do vírus cliva o polipeptídeo e forma 10 proteínas virais [ver a Fig. 3].78,79 Depois que a autoprotease viral NS2/NS3 cliva a junção entre NS2 e NS3, a protease viral NS3/NS4 se torna necessária para ligar as proteínas não estruturais remanescentes, incluindo RNA-polimerase RNA-dependente NS5B e a proteína NS5A.
A replicação viral ocorre em uma rede vesicular associada à membrana, a chamada rede membranosa, e as proteínas não estruturais se combinam para formar um complexo de replicação e promover a replicação do RNA genômico. Subsequentemente, considera-se que a partícula viral é transportada para o aparelho de Golgi, onde o vírion infeccioso é montado e onde a criação da partícula lipoviral infecciosa ocorre em paralelo à montagem da VLDL.80-82
Após a exposição ao HCV, segue-se um período de incubação de aproximadamente 7 semanas antes do inicio da hepatite aguda. Mais frequentemente, a infecção aguda por HCV é assintomática. Apenas 25% dos casos chega ao conhecimento médico. Quando sintomática, a hepatite C aguda pode se manifestar com desconforto abdominal, mal-estar, fadiga, anorexia, erupção cutânea e, ainda que raramente, icterícia. A hepatite C fulminante é extremamente rara. Após a exposição aguda ao HCV na fase adulta, em contraste com o observado na hepatite B, a depuração espontânea o vírus ocorre em menos de 15% dos pacientes.83,84 Na maioria dos pacientes, a hepatite C crônica se desenvolve após a exposição. Os fatores associados ao êxito da depuração do vírus incluem a pouca idade, o sexo feminino e a presença de icterícia. Também foi demonstrado que a etnia influencia: brancos são mais propensos a apresentar depuração espontânea do vírus, em comparação aos afro-americanos.85 Recentemente, foi descrito um conjunto de polimorfismos de nucleotídeo único (SNPs) localizado em torno do gene IL28B (interferon-l3), que são potentes como fatores preditivos de depuração viral espontânea na hepatite C aguda e na resposta ao tratamento baseado em interferon da hepatite C crônica.86-91 A frequência destes alelos (C/C benéfica; T/T desfavorável) varia entre os grupos étnicos e parece contribuir para uma proporção substancial das diferenças raciais de depuração espontânea e resposta ao tratamento historicamente observadas.
A história natural da hepatite C é de progressão lenta e indolente ao longo de décadas. Estima-se que haja desenvolvimento de cirrose em 25% dos pacientes, após 20 anos de infecção.92 A hepatite C crônica em geral tende a se clinicamente silenciosa, e costuma ser detectada somente depois que os fatores de risco são deflagrados, testes de bioquímica hepática anormais são identificados ou testes virais específicos são realizados associados com doação de sangue ou com a aplicação para seguro de vida. Os fatores associados a uma progressão mais rápida da doença incluem a idade avançada no momento da infecção, consumo concomitante de álcool, obesidade e diabetes tipo 2, além de coinfecção por HIV ou HBV. Depois que há desenvolvimento de cirrose, em geral após 2-3 décadas, 2-4% progridem anualmente para doença hepática terminal descompensada e, anualmente, há desenvolvimento de CHC em 1-4% dos casos.93
A hepatite C crônica continua sendo a principal indicação para transplante de fígado nos EUA. O tratamento bem-sucedido da hepatite C pode mudar o curso da doença e tem sido associado a resultados melhores e diminuição da mortalidade associada ao fígado.94 Para pacientes com fibrose avançada que falham em responder a um ou mais cursos de terapia antiviral baseada em interferon, todavia, a terapia de manutenção comprovadamente não confere nenhum benefício em termos de prevenção do CHC ou progressão da doença.95,96
A infecção crônica por HCV, assim como as infecções aguda e crônica por HBV, podem ser complicadas por manifestações extra-hepáticas que tendem a ser diferentes daquelas associadas à infecção por HBV. A resistência à insulina e a síndrome metabólica têm sido cada vez mais avaliadas como complicações extra-hepáticas extremamente comuns da infecção por HCV crônica, com ou sem cirrose. Em pacientes com hepatite C crônica, a síndrome metabólica foi identificada em 12,4% e a prevalência de resistência à insulina é de 35%.97 A prevalência do diabetes na hepatite C crônica foi estimada em 21-50%, em comparação com apenas 10% associados a outras formas de doença hepática crônica.98 O risco de resistência à insulina foi associado aos genótipos 1 e 4, bem como a níveis mais altos de RNA de HCV. Em adição, foi demonstrado que o diabetes potencializa a fibrose na hepatite C crônica, levando a uma progressão mais rápida da doença.99
A resposta imune à infecção por HCV crônica pode ser caracterizada pela ativação desorganizada e inespecífica da célula B, levando a um amplo espectro de manifestações extra-hepáticas autoimunes e hematológicas. A crioglobulinemia mista de tipo II está fortemente associada à infecção por HCV crônica. As imunoglobulinas precipitadas pelo frio, crioglobulinas, têm sido encontradas em até 50% dos pacientes com hepatite C crônica e, por outro lado, 91% dos pacientes com “crioglobulinemia mista essencial” têm hepatite C crônica.100,101 Vários tipos de linfoma não Hodgkin (LNH) estão associados à infecção crônica por HCV, dos quais o mais comum é o LNH de célula B. dados epidemiológicos sugerem que o risco de linfoma é mais que duas vezes maior em pacientes com hepatite C crônica, apesar da variação geográfica observada neste risco. A resposta imune perturbada ao HCV pode levar à produção de autoanticorpos, incluindo anticorpo antinuclear (ANA), anticorpo antimúsculo liso (ASMA) e anticorpo anticardiolipina, porém estes achados representam uma produção de anticorpos inespecífica e não uma doença autoimune verdadeira concomitante. Como a hepatite C pode estar associada à fadiga e a artralgias, o diagnóstico de comorbidade reumatológica verdadeira a princípio pode ser duvidoso, porém uma avaliação clínica detalhada geralmente é suficiente para distinguir definitivamente os dois distúrbios.
Figura 3: Virologia da hepatite C. (a) Ciclo de vida do vírus da hepatite C (HVC). O vírus circula no hospedeiro na forma de uma partícula lipoviral de lipoproteína de densidade muito baixa. A entrada no hepatócito requer receptores de superfície celulares (etapa 1). Após a internalização, o vírus perde o revestimento (etapa 2, exposição) expondo o nucleocapsídeo e a fita única de RNA genômico. O vírus rouba a maquinaria de tradução do hospedeiro para permitir a tradução do genoma em um polipeptídeo único (etapa 3), que é clivado por uma combinação de proteases do hospedeiro e do vírus nas 10 proteínas virais distintas. A replicação então ocorre em uma estrutura derivada do retículo endoplasmático especializada conhecida como rede membranosa (etapa 4). Uma combinação de proteínas virais (NS4A-NS5B) e fatores do hospedeiro, incluindo ciclofilina A e microRNA-122, se faz necessária à replicação viral. A replicação ocorre intimamente adjacente a uma gotícula lipídica especializada. A fita de RNA recém-sintetizada é então empacotada pelo aparelho de Golgi em uma nova partícula lipoviral infecciosa, que então é liberada da célula (etapas 5 e 6). (b) Genoma do HCV. O HCV tem um RNA genômico de fita única e sentido positivo. O quadro de leitura aberto codifica as 10 proteínas virais que inicialmente são traduzidas pelo hospedeiro em um polipeptídeo único. A região não traduzida (UTR) 5’ tem dois componentes funcionais importantes. O sítio de entrada ribossomal interno (SERI) permite que a translação do polipeptídeo aconteça de modo capa-independente. Os elementos de reconhecimento do microRNA (MRE 1,2) permitem a interação com o microRNA-122 do hospedeiro, facilitando a replicação viral.
Os dois testes sorológicos usados na detecção da infecção por HCV são o imunoensaio enzimático para anticorpos anti-HCV e os ensaios de amplificação altamente sensíveis (p. ex., PCR ou amplificação mediada por transcrição do RNA de HCV). Depois que uma pessoa é exposta ao HCV, o RNA do HCV pode ser detectado no soro em 7-10 dias, várias semanas antes da manifestação de hepatite sintomática. A emergência de anticorpos anti-HCV detectáveis costumava durar até 1-3 meses, entretanto os ensaios altamente sensíveis de última geração mostraram que o anticorpo anti-HCV quase sempre é detectável no início da hepatite C aguda.
O diagnóstico de infecção crônica é estabelecido com base na presença de RNA de HCV e de anticorpos anti-HCV persistentemente detectáveis. A atividade normal de aminotransferase ocorre em até 20% dos pacientes com hepatite C crônica e não exclui a infecção por HCV. Vale a pena destacar que os níveis de RNA de HCV e de aminotransferase tendem a flutuar durante o curso da hepatite C crônica. Um teste reativo para anticorpo anti-HCV na ausência de RNA de HCV reflete um resultado falso-positivo de anticorpos anti-HCV, uma infecção previa por HCV que já depurada, ou a presença intermitentemente indetectável de RNA de HCV durante a infecção crônica. A biópsia hepática é útil para avaliar o nível (grau) de atividade necroinflamatória, bem como o grau (estágio) de fibrose. Entretanto, outras informações clínicas tornam a opção de biópsia percutânea menos atraente. Potencialmente, a instituição de terapia antiviral pode ser suspensa temporariamente para pacientes com estágio e grau histológico muito brandos, sendo que o reconhecimento da presença de fibrose ou cirrose avançada tem implicações para o sucesso antecipado da terapia antiviral e suas complicações.
Na avaliação diagnóstica de um paciente com RNA de HCV detectável, a determinação do genótipo viral, que é disponibilizada como exame laboratorial de rotina, é essencial para a tomada de decisões sobre a potencial eficácia da terapia antiviral e seleção de um regime terapêutico. Apesar da disponibilização de testes para genótipo IL28B de paciente, os testes de rotina para IL28B não são recomendados e, conforme discutido adiante, estes testes podem ter utilidade limitada em um futuro próximo, na era dos antivirais de ação direta e novos agentes terapêuticos.
Diferente da terapia antiviral para hepatite B que, na maioria dos casos é continuada indefinidamente, a terapia antiviral para hepatite C é tempo-limitada, em geral durando 24-48 semanas. Vários marcos referenciais terapêuticos importantes foram definidos após a iniciação da terapia: resposta virológica rápida (RVR); RNA de HCV indetectável na 4ª semana; RVR estendida (eRVR); RNA de HCV indetectável na 4ª e 12ª semanas; resposta virológica inicial (RVI); declínio do RNA de HCV =2 log10 na 12ª semana; RVI completa (cRVI); RNA de HCV indetectável na 12ª semana; resposta de final de tratamento (RFT); RNA de HCV indetectável ao final do tratamento (i.e., em 24-48 semanas); e, o mais importante, resposta virológica sustentada (RVS); RNA de HCV indetectável ao final do tratamento e assim mantido por 24 semanas após a conclusão da terapia. Breakthrough é o retorno do RNA de HCV detectável durante a terapia antiviral (depois que supressão viral é alcançada) e recidiva é o retorno da viremia após a conclusão da terapia em um paciente que tenha alcançado supressão completa do RNA de HCV durante a terapia. Alcançar a RVS é considerada “cura”, uma vez que a erradicação viral é mantida depois que a RVS é alcançada em cerca de 99% dos pacientes. De fato, a viremia recorrente 1 ano após o tratamento tende mais a representar reinfecção do que recidiva.94
Por mais de uma década, a base da terapia para hepatite C de todos os genótipos era uma combinação de interferon peguilado e ribavirina (RBV). Entretanto, a duração da terapia, a dose de RBV e a eficácia da terapia diferiam conforme o genótipo. Duração padrão da terapia é 48 semanas para os genótipos 1 e 4, e 24 semanas para os genótipos 2 e 3. A dose diária de RBV é baseada no peso, sendo igual a 1.000-1.200 mg (modificada subsequentemente para até 1.400 mg) para os genótipos 1 e 4, contudo uma dose baixa fixa de 800 mg para os genótipos 2 e 3. Apenas 40-45% dos pacientes com genótipo 1 alcançam a RVS, porém os genótipos 2 e 3 se saem muito melhor, com respostas mais próximas de 80%.102,103 Os fatores preditivos da resposta à terapia com interferon peguilado e RBV incluem o genótipo, níveis baixos de RNA de HCV (<400.000 UI/mL), etnia não afrodescendente, estágio histológico menos avançado, ausência de esteatose e genótipo IL28B favorável. Em pacientes tratados com interferon peguilado/RBV, a duração da terapia, especialmente para o genótipo 1, pode ser ajustada—descontinuada na 24ª semana, se a RVR for alcançada (com a RVS se aproximando de 90%) , e estendida até a 72ª semana na ausência de RVR (aumento da frequência de RVS em relação àquela alcançada com apenas 48 semanas de terapia).
Em 2011, dois agentes novos dirigidos contra a protease viral NS3/4ª de HCV foram aprovados para uso combinado com interferon peguilado/RBV no tratamento da hepatite C crônica de genótipo 1. O telaprevir e o boceprevir são inibidores de protease orais que melhoram significativamente as taxas de RVS em pacientes com infecção por HCV de genótipo 1. Nas triagens clínicas entre pacientes tratamento-naive, quando o telaprevir era combinado com interferon a-2a peguilado e RBV (com base no peso, 1.000-1.200 mg/dia) por um período de 12 semanas, seguido de mais 12-36 semanas de interferon peguilado e RBV (curso total de 24 semanas para pacientes com eRVR [eRVR, i.e., RNA de HCV indetectável na 4ª e 12ª semanas]; curso total de 48 semanas na ausência de eRVR, na chamada “terapia orientada por resposta”), as taxas de RVS atingiram 73-79%.104 O telaprevir também se mostrou efetivo em pacientes que passaram por falha prévia do tratamento com interferon peguilado/RBV, com a RVS chegando a 88% dos pacientes recidivantes, 59% dos pacientes parcialmente responsivos (supressão do RNA de HCV durante a terapia prévia em =2 log10 e não a níveis indetectáveis), e 31% dos pacientes previamente responsivos nulos (redução <2 log10 do RNA de HCV durante a terapia prévia). Nos controles tratados com interferon peguilado/RBV, as RVSs foram alcançadas apenas em 24%, 15% e 5% destes três grupos, respectivamente.105 Os principais eventos adversos limitantes do tratamento observados com a adição de telaprevir foram erupção cutânea e anemia.
Nos estudos clínicos do boceprevir, tanto em pacientes tratamento-naive como em pacientes submetidos ao tratamento para hepatite C crônica, genótipo 1, o regime de tratamento começou com 4 semanas de administração “introdutória” de interferon a-2b peguilado e RBV (baseado no peso, 600-1.400 mg/dia) antes da adição de boceprevir. Em pacientes tratamento-naive, a duração da terapia tripla com boceprevir/interferon peguilado/RBV continuou por 48 semanas em um ramo ou, no outro ramo, foi orientada pela resposta às últimas 24 semanas, ou seja, uma duração total de 28 semanas (se a RVR era alcançada e o HCV permanecia indetectável subsequentemente) ou 48 semanas (na ausência de RVR, enquanto o RNA de HCV se tornou indetectável em torno da 24 semana). Esta terapia farmacológica tripla à base de boceprevir foi efetiva em até 66% dos participantes do estudo. Nos pacientes previamente submetidos a tratamento, a RVS ocorreu em até 66% daqueles que receberam a terapia de três fármacos à base de boceprevir—até 52% dos respondedores parciais e até 75% dos previamente recidivantes (os respondedores nulos não foram incluídos nas triagens de registro).106 A anemia foi o principal evento adverso, observada em metade dos pacientes tratados com boceprevir. A disgeusia foi o segundo efeito colateral mais comumente relatado.
A terapia com telaprevir e boceprevir é mais efetiva nos respondedores virológicos rápidos, indivíduos não afrodescendentes, não cirróticos, com HCV de genótipo 1b (versus 1a) e pacientes com genótipos IL28B favoráveis.
Embora estes agentes tenham levado a aprimoramentos marcantes no tratamento bem-sucedido de pacientes com hepatite C crônica de genótipo 1, e embora a atividade de telaprevir contra o genótipo 2 (e não o 3) tenha sido demonstrada em um estudo pequeno,107 nenhum destes inibidores de protease tem aprovação para uso no tratamento dos genótipos 2 ou 3. O tratamento padrão para a doença dos genótipos 2 e 3, atualmente, continua sendo interferon peguilado e RBV [ver Tabelas 11 e 12].
A cura bem-sucedida da hepatite C leva a resultados melhores a longo prazo, incluindo uma mortalidade associada ao fígado diminuída, melhora dos escores de fibrose e neuroinflamatório, e risco diminuído de CHC, especialmente em pacientes sem cirrose.94 Dada a progressão lenta da doença em muitos pacientes com hepatite C crônica, os efeitos colaterais substanciais associados à terapia à base de interferon, bem como as baixas taxas de resposta ao tratamento antes da disponibilização da terapia com inibidor de protease NS3/4A, há muito se discute o momento ideal para o tratamento. O consenso geral, entretanto, especialmente no contexto dos inibidores de protease altamente efetivos, é o de que o tratamento deve ser considerado em todos os casos de pacientes com hepatite C crônica e que tenham evidência bioquímica ou histológica de atividade da doença. Para cada paciente, os riscos associados às terapias atualmente disponíveis (incluindo doença psiquiátrica, anemia e outras citopenias) deve ser ponderado contra o potencial de progressão substância e acelerada da doença. Em pacientes bem compensados apresentando fibrose, o tratamento deve ser iniciado se for tolerado. A atual terapia antiviral baseada em interferon não é recomendada para pacientes com cirrose descompensada e está associada a níveis intoleráveis de efeitos colaterais prejudiciais à vida. Estes pacientes devem ser encaminhados para os centros de transplante de fígado.
O tratamento com interferon peguilado por um período mínimo de 12-24 semanas também deve ser considerado para pacientes com hepatite C aguda. Entretanto, a duração ideal da terapia e se a RBV deve ou não ser adicionada são aspectos indeterminados.108 Embora o tratamento da hepatite aguda, em comparação com a hepatite C crônica, leve a uma eficácia significativamente maior ao longos dos genótipos—95% de RVS quando o tratamento é iniciado 8 semanas após o estabelecimento do diagnóstico; 93% em 12 semanas; e 76% em 20 semanas109—a decisão de iniciar o tratamento deve ser temperada pelo reconhecimento de 15-20% dos pacientes com hepatite C aguda eliminam o vírus e se recuperam espontaneamente, em geral em 3 meses. Portanto, de acordo com as diretrizes de tratamento estabelecidas pela American Association for the Study Of Liver Diseases, retardar a terapia por 8-12 semanas é recomendado para permitir a identificação de pacientes que apresentam depuração espontânea do HCV e confinar o tratamento àqueles que permanecem infectados pelo HCV, subsequentemente.110 O papel da checagem de IL28B, como fator preditivo de contenção viral espontânea na hepatite C aguda, ainda foi investigado.
Tabela 11 Regimes de tratamento para pacientes com hepatite C crônica com função renal normal
|
Medicação |
Duração do tratamento (tratamento-naive) (semanas) |
Doses de medicação |
Taxas de TVS (%) |
Efeitos adversos | ||||
|
eRVR |
Sem eRVR |
Inibidor de protease |
Interferon peguilado |
Ribavarina baseada no peso+ |
Tratamento-naive |
Tratamento prévio* | ||
|
Genótipo 1 | ||||||||
|
TPR/PEG interferon/ RBV TPR semanas 0-12 |
24 |
48 |
750 mg, 3x/dia (com refeição gordurosa) |
alfa-2a, 180 µg, 1x/semana |
1.000-1.200 mg/dia |
73-79 |
64-66 |
Erupção, anemia, prurido |
|
BOC/PEG interferon/ RBV OC semanas 4-28 (naive), 4-36 (prévio), ou 4-48 cirrótico |
28 |
48 |
800 mg, 3x/dia, com refeição normal |
Alafa-2b, 1,5 µg /kg/semana |
600-1.400 mg/dia |
63-66 |
59-66 |
Anemia, disgeusia |
|
PEG interferon/ RBV |
48 |
48 |
N/A |
Alfa-2a, 180 µg, 1x/semana Alfa-2b, 1,5 µg /kg/semana |
Para interferon alfa PEG, <75 kg: 1.000 mg/dia; =75 kg: 1.200 mg/dia Para interferon alfa-2b PEG, 600-1.400 mg/dia |
40-50 |
5-20 |
Sintomas similares aos de gripe, mialgias, febre, anemia, leucopenia, trombocitopenia |
|
Genótipo 2/3 | ||||||||
|
PEG interferon/ RBV |
24 |
24 |
N/A |
Alfa-2a, 180 µg, 1x/semana Alfa-2b, 1,5 µg /kg/semana |
800 mg/dia |
70-80 |
55-65 |
Sintomas similares aos de gripe, mialgias, febre, anemia, leucopenia, trombocitopenia |
BOC = boceprevir; eRVR = resposta virológica rápida estendida (RNA de vírus da hepatite C indetectável em 4 e 12 semanas de regimes à base de telaprevir ou semanas 8-24 sob regimes à base de boceprevir); N/A = não se aplica; PEG = peguilado; RBV = ribavarina; SVR = resposta virológica sustentada; TRP = telaprevir.
*As taxas de RVS variam dependendo do regime de tratamento anterior e da resposta prévia (recidiva, resposta parcial, resposta nula).
+Em duas doses divididas.
Tabela 12 Regimes de tratamento para hepatite C crônica: pacientes com doença renal crônica108
|
Estágio da doença renal |
TFG estimada (cc/min) |
Regimes com interferon |
Ribavirina |
|
1 |
>90 |
IFN Alfa-2a/2b PEG nas doses padrão |
RBV nas doses padrão |
|
2 |
60-90 |
IFN Alfa -2a/2b PEG nas doses padrão |
RBV nas doses padrão |
|
3 |
30-50 |
IFN Alfa -2a PEG, 135 µg SC, 1x/semana IFN Alfa -2b PEG, 1 µg/kg, 1x/semana |
RBV, 200-800 mg/dia em doses divididas |
|
4 |
15-29 |
IFN Alfa -2a PEG, 135 µg SC, 1x/semana IFN Alfa -2b PEG, 1 µg/kg, 1x/semana |
RBV, 200-800 mg/dia em doses divididas |
|
5: insuficiência renal e hemodiálise |
<15 |
IFN, 3 mU, 3x/semana IFN alfa-2a PEG, 135 µg SC, 1x/semana IFN alfa-2b PEG, 1 µg/kg, 1x/semana |
Considerar doses acentuadamente reduzidas, se usado |
|
Após o transplante renal |
Não indicado: alto risco de rejeição do enxerto |
N/A | |
TFG = taxa de filtração glomerular; N/A = não se aplica; IFN-PEG = interferon peguilado; RBV = ribavarina; SC = subcutâneo.
O SNP IL28B está localizado na região de codificação genética para interferon l3, um interferon de tipo III que se parece mais com uma citocina (interleucina-10) do que os interferons de outras classes. Apesar de sua diferença estrutural, o interferon l3 compartilha uma via de sinalização com o interferon a [ver Fig. 4] e conduz as mesmas alterações genéticas sucessivas,111-113 como nos caso dos genes interferon-estimulados (ISGs), cuja magnitude da indução está comprovadamente correlacionada com a resposta à terapia.114,115
Os SNPs IL28B foram identificados, a princípio, em um estudo de associação genômica (EAG) de uma coorte bem caracterizada de pacientes (incluídos no estudo “IDEAL”, em que dois tipos de interferon a peguilado foram comparados e considerados similares) que receberam interferon peguilado e RBV para tratamento de hepatite C crônica de genótipo 1.86 Neste estudo, a associação mais forte foi observada entre rs12979860, localizado 3 kb no sentido contrário ao do IL28B. para este SNP, o genótipo favorável (CC) estava associado a um aumento de 2-3 vezes na RVS, em comparação com o genótipo desfavorável (TT). Em adição, diferenças significativas de frequência alélica foram identificadas entre os grupos étnicos, com os afro-americanos apresentando a menor taxa de CC. Entretanto, o genótipo IL28B é responsável somente por cerca da metade das disparidades encontradas na resposta ao tratamento vistas entre os grupos étnicos de afrodescendentes e não afrodescendentes. O haplótipo CC do mesmo SNP também foi associado a taxas mais altas de depuração espontânea após a hepatite C aguda.90
Subsequentemente, vários polimorfismos adicionais no sentido contrário ao do gene IL28B foram identificados, com base nos relatos de EAG, que eram fatores preditivos potentes da resposta ao interferon peguilado e à RBV.88,89 Os estudos apresentaram uma variação discreta quanto à plataforma usada para EAG, tendo sido identificados dois genótipos diferentes como fatores preditivos de resposta mais fortes. O primeiro é rs12979860, para o qual o CC é o genótipo favorável. O segundo é rs8099917, cujo genótipo favorável é TT, associado a uma resposta melhor ao tratamento e a taxas mais altas de depuração viral espontânea após a infecção aguda por HCV.
A aprovação dos inibidores de protease pelo Food and Drug Administration ocorreu logo após a descoberta dos loci IL28B. Embora os genótipos IL28B sejam preditivos de resposta à terapia antiviral com interferon peguilado à base de inibidor de protease, a rapidez e a profundidade da resposta antiviral são significativamente mais preditivas do que o IL28B ou qualquer outra variável do paciente.116 Apesar dos relatos de dados conflitantes,117 o genótipo IL28B também parece ser preditivo da resposta para os genótipos 2 e 3.118 Desta forma, para a terapia combinada com interferon peguilado/RBV/inibidor de protease, o genótipo IL28B parece reter valor preditivo. Entretanto, relatos iniciais de pacientes tratados com novos regimes orais sem interferon (ver adiante) sugerem que, conforme poderia ser previsto com base na localização deste SNP, o genótipo IL28B deixou de ser preditivo da resposta a este tipo de terapia. Apesar da descoberta dos polimorfismos de IL28B terem levado a um maior escrutínio da resposta imune inata ao HCV e à terapia à base de interferon, a utilidade clínica dos testes de rotina para genótipo IL28B ainda não foi estabelecida nem recomendada.
Com a expansão do conhecimento acerca do ciclo de vida do HCV e a disponibilização dos modelos de replicação de HCV in vitro, houve a emergência de um número crescente de agentes terapêuticos dirigidos primariamente contra alvos virais, mas também contra alvos do hospedeiro. Em fase de desenvolvimento clínico avançado, estão os inibidores de protease NS3/4A, que são mais potentes e modernos, podem ser tomados uma vez ao dia e são mais tolerados do que os inibidores de protease de primeira geração, além de serem efetivos com todos os genótipos e terem uma barreira maior à resistência. Adicionalmente, fármacos dirigidos contra outras duas proteínas virais se mostraram promissores em estudos clínicos iniciais: inibidores nucleosídicos/nucleotídicos e não nucleosídicos NS5B RNA-dependentes da RNA polimerase (vários inibidores nucleosídicos com barreiras muito altas à resistência); e inibidores NS5A (estão entre os novos agentes de maior potencia e ação mais rápida). Estudos iniciais, em que os inibidores de protease e inibidores NS5A foram administrados de forma combinada, sem interferon peguilado, comprovaram o princípio de que todos os regimes antivirais orais e de ação direta podem ser bem-sucedidos. Também foi demonstrado que um dos inibidores de polimerase orais cura a hepatite C sem necessidade de interferon peguilado. Em geral, quando administrados de forma isolada, os antivirais de ação direta tendem a promover a emergência antecipada de variantes associadas à resistência. Desta forma, é totalmente provável que, nos próximos anos, os regimes antivirais para hepatite C venham a ser combinações de agentes de alta potência e baixa resistência com ação mais efetiva em termos de supressão duradoura da infecção por HCV, na ausência de resistência farmacológica.
A dependência do HCV dos processos celulares do hospedeiro tem permitido investigar os alvos no hospedeiro que poderiam ser úteis como agentes terapêuticos contra a hepatite C e que, por afetarem as proteínas do hospedeiro e não as virais, teriam uma barreira muito alta à resistência viral e atividade pangenotípica viral. Os inibidores de HMG-CoA redutase (estatinas) são um exemplo. O vírus, que depende das lipoproteínas do hospedeiro para sua replicação, requer um produto da via do mevalonato de metabolização do colesterol para replicação. A depleção deste metabólito com estatinas impede a replicação viral in vitro. Entretanto, a inibição potente da replicação do HCV requer níveis de dosagem muito acima daqueles alcançados clinicamente, e as estatinas não têm ação comprovadamente efetiva para alcançar a RVS no tratamento da hepatite C, apesar de várias observações sugerirem o contrário.119,120 A ciclofilina A é uma proteína do hospedeiro que forma complexo estável com o NS5A e serve de fator essencial do hospedeiro para replicação deste. Foi demonstrado que um inibidor não imunossupressor da ciclofilina A (alisporivir) causa inibição moderada da replicação viral em seres humanos.121 Entretanto, devido ao papel íntimo das ciclofilinas na replicação do HCV, variantes de NS5A podem estar associadas à resistência aos inibidores de ciclofilina A. Por fim, está comprovado que o microRNA-122 (miR-122) é um novo agente terapêutico e que pequenos ácidos nucleicos antagonistas de miR-122 causam inibição potente e durável da replicação viral em chimpanzés e em seres humanos.122 Ainda não foi determinado se estas terapias hospedeiro-dirigidas serão competitivas contra (ou combinadas com) os antivirais de ação direta.
Ao longo dos próximos anos, o tratamento da hepatite C passará por uma evolução drástica e, com toda certeza, a hepatite C será tratada com regimes orais (poupadores de interferon) combinando fármacos com diferentes mecanismos de ação, perfis de resistência e efeitos colaterais, a fim de promover uma cura quase universal da doença. Veja mais informação sobre hepatite C no ACP Medicine.
Figura 4: Sinalização de interferon tipos I e III. Tanto os interferons (IFNs) de tipo I (p. ex., interferon a [IFN-a]) como os de tipo III (p. ex., interferon l [IFN-l]) usam a via de sinalização JAK-STAT na produção de seus efeitos antivirais. Os interferons de tipo I se ligam a um receptor homodimérico, o receptor de interferon a (IFNaR1), enquanto os interferons de tipo III se ligam a um receptor heterodimérico, incluindo o receptor a da interleucina 28 (IL-28Ra) e o receptor b da IL-10 (IL-10Rb). Após a ligação, a via de sinalização JAK-STAT é ativada, levando à fosforilação de STAT-1 e STAT-2, que se combinam ao fator de regulação 9 (IRF9) para formar um complexo conhecido como fator do gene estimulado por interferon 3 (ISGF3), que então é translocado para o núcleo e se liga ao elemento de resposta interferon-sensível (ISRE), levando à transcrição de um subgrupo de genes conhecidos como genes interferon-estimulados (ISGs), que atuam na resposta antiviral.
O HAV é adquirido pela via fecal-oral, ou seja, pela disseminação pessoa-pessoa ou pela ingesta de alimentos e água contaminados. Os mariscos podem abrigar altas concentrações de HAV e servem de reservatório para o vírus. O HAV se mantém estável no meio ambiente por meses, mas pode ser inativado com uma solução de alvejante doméstico diluído a 1:1.000 ou por fervura durante 1 minuto. Casos muito raros de infecção via transfusão de produtos do sangue foram relatados, atribuídos à doação de sangue por indivíduos com viremia transiente de hepatite A durante fase inicial pré-assintomática da hepatite aguda. Em indivíduos saudáveis, o HAV causa infecção aguda e autolimitada, em vez de hepatite crônica ou do estado de portador assintomático.
Cerca de 1,4 milhões de casos novos de hepatite A aguda são identificados a cada ano, no mundo inteiro.123 Em áreas de alta prevalência, a exposição tende a ocorrer de modo universal na infância. Durante esta fase, a doença costuma ser muito branda, quando não assintomática. Por outro lado, em áreas de baixa prevalência, a exposição comumente ocorre durante a fase adulta, quando a hepatite A aguda tende a causar hepatite sintomática, frequentemente grave. A estratégia de vacinação, nos EUA, a princípio tinha como alvo os indivíduos pertencentes a populações de alto risco, como os usuários de fármacos, homens que têm relações sexuais com homens, viajantes internacionais e crianças em áreas de alta prevalência. Nos EUA, a incidência anual de hepatite A aguda tem declinado desde a introdução da vacinação para adultos de alto risco, iniciada em 1996, e da iniciação da vacinação de crianças de alto risco, em 1999. A partir de 2006, como estratégia para reduzir o pequeno número de casos relatados residuais (mais de 50% dos quais ocorridos em comunidades como a dos nativos do Alaska, índios americanos e hispânicos) e os surtos periódicos transmitidos por alimentos, o US Public Health Service recomendou a vacinação universal de crianças de 1-2 anos de idade. Em 2009, apenas 1.987 casos de hepatite A aguda foram relatados ao CDC, representando uma queda de 56% em relação aos 4.488 relatos registrados em 2005.2
As vacinas contra hepatite A inativadas são seguras e conferem imunidade de longa duração. Embora uma única dose de vacina produza excelente resposta de anticorpos a curto prazo, duas doses separadas por um intervalo de 6-18 meses são recomendadas para garantir a proteção a longo prazo. Dentre as várias vacinas atualmente disponíveis, nenhum tem licença para ser usada em crianças com menos de 12 meses. Embora os surtos de transmissão por alimentos ainda causem uma proporção significativa de todos os casos de hepatite A nos EUA, o modo mais comum de aquisição da infecção por HAV é de longe as viagens internacionais, sendo que a vacinação deve ser considerada para adultos em viagem a áreas endêmicas.124
O período de incubação da hepatite A dura 15-45 dias e a infecção resulta em uma hepatite aguda autolimitada que varia amplamente quanto à gravidade clínica, desde uma forma branda ou assintomática até a forma aguda fulminante de hepatite com insuficiência hepática. O pico da liberação fecal do vírus tende a ocorrer ao redor do final do período de incubação e pouco antes do início da lesão hepatocelular, raramente persistindo além da manifestação inicial da doença clínica conhecida, e em geral tendo já desaparecido no momento em que surge a icterícia nos casos ictéricos.125 Portanto, o vírus se dissemina de forma mais efetiva antes de o paciente se tornar sintomático ou procurar atenção médica, do que após a emergência de doença clinicamente evidente (quando, então, as precauções com o contato são desnecessárias).
Crianças pequenas (com menos de 6 anos) tendem a ser totalmente assintomáticas e a gravidade dos sintomas aumenta com o avanço da idade. Adultos na faixa etária a partir de 50 anos têm risco aumentado de doença grave e até de resultado fatal, enquanto indivíduos com hepatite B crônica subjacente, hepatite C ou hepatite alcoólica também são mais propensos a desenvolverem uma hepatite mais grave e, de acordo com alguns relatos e não outros, a progredirem para insuficiência hepática fulminante. A icterícia, que é mais comum em adultos de idade avançada do que em adultos jovens e crianças, segue um pródromo viral inespecífico que inclui fadiga, enfraquecimento, anorexia, náusea, vômito, diarreia, febres, cefaleia, dor abdominal, mialgias e artralgias. A internação por hepatite A aguda raramente se faz necessária, exceto em casos de pacientes idosos ou pacientes com comorbidades graves, dos quais até 1/3 podem requerer internação. Até em casos graves, quase todos os pacientes eventualmente se recuperam. A taxa de mortalidade geral é de apenas 0,6%.
Conforme notado antes, a infecção aguda por HAV não evolui para hepatite crônica. Duas variantes clínicas relativamente raras estão associadas a uma recuperação mais lenta (e não à cronicidade): a hepatite A aguda recidivante (em que uma ou mais crises de replicação viral recorrente, excreção fecal de vírus e lesão hepática seguem a crise aguda ao longo de 6-10 semanas) e a hepatite A colestática (caracterizada por alta atividade de fosfatase alcalina, com ou sem icterícia, e um curso agudo mais prolongado de resolução lenta). A recuperação da infecção por HAV aguda leva à imunidade por toda a vida.
O vírus a RNA HAV é classificado como Hepatovírus pertencente à família Picornaviridae. Após a aquisição através do intestino delgado, o vírus é disseminado via circulação porta para o fígado, onde uma proteína do envelope viral (provavelmente) é reconhecida por um receptor de superfície do hepatócito ainda não identificado. Após a ligação da proteína com este receptor, o vírus é internalizado no hepatócito.
O genoma do RNA tem comprimento aproximado de 7.500 nucleotídeos, com um único quadro de leitura aberto, e é traduzido pelo hospedeiro em um único polipeptídeo que é clivado no pós-tradução pela protease viral e pelas proteases do hospedeiro. Similar ao que ocorre com o HCV, a replicação do HAV ocorre no citoplasma, em vesículas ligadas à membrana, e o RNA recém-sintetizado pode servir de molde para replicação adicional ou ser empacotado dentro de um capsídeo para montagem e exportação viral. Os mecanismos pelos quais o vírus é montado e exportado da célula ainda são indeterminados. É mais provável que o vírus saia do hepatócito por transporte biliar nos canalículos biliares do hepatócito, ganhando acesso ao sistema de ductos biliares en route para o lúmen intestinal e excreção fecal.125
Assim como os outros vírus causadores de hepatite humana, o próprio HAV em si é não citopático e não causa inflamação hepática sem recrutamento da resposta imune do hospedeiro. Durante o período de pródromo, a replicação viral atinge o pico e começa a declinar quando os sintomas de hepatite aguda se manifestam.
Anticorpos anti-HAV, da classe da imunoglobulina M (IgM anti-HAV), estão presentes no momento do aparecimento dos sintomas em quase todos os pacientes. Em casos raros, pode ser necessário repetir os testes para detectar IgM anti-HAV. Este anticorpo continua sendo detectável por 3-6 meses após a infecção aguda e é substituído por anticorpos IgG anti-HAV, que permanecem detectáveis subsequentemente, em geral por toda a vida. A presença de IgG anti-HAV é um excelente marcador de imunidade à reinfecção. A IgG anti-HAV se torna detectável em 2-4 semanas após a administração da dose inicial da vacina contra hepatite A, sendo que os anticorpos anti-HAV permanecem detectáveis por pelo menos 5 anos em 99% dos indivíduos que completam a série de vacinação. Mesmo quando os níveis de anticorpos anti-HAV caem abaixo do nível de detecção após a vacinação, a imunidade de longa duração parece ser preservada.
No Third National Health and Nutrition Examination Survey (NHANES III), que foi conduzido no período de 1988 a 1994, cerca de 1/3 da população dos EUA apresentava evidência sorológica de imunidade ao HAV.126 A prevalência da infecção aumentava com o avanço da idade, sendo que mais de 75% dos indivíduos com idade a partir de 70 anos tinham anticorpos anti-HAV detectáveis. Os dados do NHANES referentes ao período de 1999-2006 mostraram ausência de alteração geral na soroprevalência de anti-HAV, com exceção de um aumento detectado nas crianças que residiam em estados onde a vacinação era recomendada naquela época. Este aumento foi provavelmente um reflexo de índices de vacinação mais altos, em vez de uma incidência aumentada durante os anos em que ocorreram novas infecções por HAV (quando foi registrada a queda do número de casos relatados).127
Nos EUA, os ensaios comerciais para RNA de HAV e para antígeno de HAV não são comercializados e quase nunca são necessários para o diagnóstico e tratamento de rotina da hepatite A. O ensaio de transcrição reversa por reação em cadeia da polimerase (RT-PCR) do RNA de HAV e a pesquisa de antígeno viral nas fezes são usados como ferramentas de pesquisa em poucos laboratórios especializados.
A prevenção por vacinação apropriada é a estratégia mais efetiva para reduzir a morbidade e a mortalidade resultantes da hepatite A. Nos surtos que ocorrem nas comunidades, a vacinação tem sido efetiva para limitar a disseminação da doença. Para indivíduos com exposição recente comprovada ao HAV, a imunoglobulina é administrada por via intramuscular na dose de 0,02 mg/kg. Quando administrada em 2 semanas após a exposição, a imunoglobulina apresenta efetividade de 85% na prevenção da infecção aguda por HAV. A imunidade dura 1-3 meses. Quando a administração é feita após 2 semanas da exposição, a imunoglobulina pode atenuar o curso clínico da doença. Diferente da vacina contra hepatite A, a imunoglobulina pode ser administrada a bebês. Para indivíduos que sustentam uma exposição aguda à hepatite A e recebem imunoglobulina, mas continuam apresentando risco de exposição recorrente, a imunoglobulina deve ser complementada com um curso de vacina contra hepatite A. a imunoglobulina e a primeira dose da vacina contra hepatite A podem ser administradas ao mesmo tempo.
Pacientes que apresentam, infecção por HAV sintomática requerem tratamento de suporte. Não há nenhuma medicação antiviral específica disponível ou que seja necessária. Internação e encaminhamento para transplante de fígado podem ser necessários em casos graves.
O HEV é transmitido pela via fecal-oral. As fontes de água contaminadas continuam sendo causa importante de surtos epidêmicos de infecção por HEV, particularmente nas áreas endêmicas durante as épocas de enchentes, que podem resultar na contaminação da água potável com dejetos humanos. Quase todos os casos clinicamente evidentes de hepatite E são confinados a áreas endêmicas, como o subcontinente da Índia, onde a doença inicialmente foi reconhecida. Entretanto, o conhecimento acerca da epidemiologia do HEV tem evoluído recentemente. Já considerada uma infecção rara causadora de uma grave epidemia de hepatite em regiões geográficas seletas, sabe-se hoje que a infecção por HEV causa doença de gravidade clínica variável e é mais prevalente do que a princípio se admitia nas áreas não endêmicas. Em geral, nas áreas de alta endemicidade, os surtos epidêmicos são responsáveis pela principal carga imposta pela doença, enquanto nas áreas de baixa endemicidade a maioria das infecções são esporádicas e poucas são sintomáticas. Estas diferenças de padrões epidemiológicos estão relacionadas em parte com os genótipos virais.
Quatro genótipos de HEV foram identificados, com distribuições geográficas distintas. Os genótipos 1 e 2, ambos comuns em áreas de alta endemicidade (Índia, Ásia, África, América do Sul), estão associados a surtos epidêmicos de hepatite E aguda. Os genótipos 3 e 4 são encontrados em áreas de baixa endemicidade e doença esporádica (não epidêmica). Os porcos parecem ser o reservatório zoonótico destes genótipos de HEV em áreas não endêmicas, sendo que estes genótipos tendem menos a causar doença grave em seres humanos. O genótipo 3 é encontrado nos EUA e na Europa, enquanto o genótipo 4 é encontrado no Japão, Taiwan e China.
Com base no NHANES III, 21% da população geral dos EUA têm anticorpos anti-HEV. Índices maiores foram encontrados na população masculina do Meio-Oeste, donos de animais de estimação e indivíduos com história de consumo de fígado e órgãos de animais.178 Uma relação entre carne de órgãos de animais e infecção por HEV esporádica foi observada previamente, sendo que um levantamento dos fígados suínos comercializados descobriu RNA de HEV de genótipo 3 quantificável em alguns.129 Conforme notado antes, a transmissão zoonótica é reconhecida como um importante modo de aquisição de doença esporádica.
O HEV, um vírus a RNA pertencente à família Hepeviridae, é hoje o único membro conhecido desta família. O ciclo de vida viral do HEV é o menos bem caracterizado dentre todos os vírus de hepatite humana. O vírion mede 27-32 nm de diâmetro e contém apenas uma única proteína e um genoma viral de fita única.130
O RNA genômico tem três quadros de leitura sobrepostos, que são traduzidos em várias proteínas virais, incluindo uma RNA polimerase dependente de RNA, uma cisteína protease e uma proteína estrutural. A replicação viral ocorre no citoplasma. O novo RNA viral é transcrito a partir de uma fita de RNA negativo que serve de molde. As etapas da replicação do vírion infeccioso e montagem nos vírions não estão totalmente caracterizadas.
O período de incubação da infecção por HEV é 28-40 dias. Os níveis séricos de aminotransferase atingem o pico em 42-46 dias após a exposição. Como em outras formas de hepatite viral, a apresentação clínica da hepatite E varia da elevação assintomática de aminotransferase à hepatite fulminante, enquanto a gravidade clínica parece diferir entre os genótipos 1 e 2 (clinicamente evidentes, graves, epidêmicos) e os genótipos 3 e 4 (clinicamente mais brandos ou assintomáticos, esporádicos). Quando a hepatite E é sintomática, os sintomas incluem um pródromo viral, icterícia, dor abdominal e, em muitos casos, prurido.
Em pacientes expostos durante os surtos epidêmicos de hepatite E, a hepatite ictérica é encontrada em cerca de 15% dos casos, mas as taxas de incidência de hepatite fulminante permanecem muito baixas. Por outro lado, as taxas de incidência de hepatite fulminante são significativamente maiores entre gestantes, sendo que a mortalidade por hepatite E aumenta progressivamente com a evolução da gestação. A mortalidade materna é de aproximadamente 1-2% no primeiro trimestre, de 8-10% no segundo trimestre, e de 20% no terceiro trimestre. Pacientes com doença hepática subjacente também apresentam risco de mortalidade muito alta.131,132
O curso clínico da infecção por HEV esporádica geralmente é mais brando e ainda precisa ser totalmente caracterizado, contudo os dados de soroprevalência sugerem que a vasta maioria dos (se não todos) casos de infecção por HEV associada aos genótipos 3 e 4 pode ser clinicamente silenciosa.133 Recentemente, observou-se que uma proporção muito pequena de pacientes com lesão hepática presumidamente induzida por fármacos apresentava evidência de infecção aguda por HEV de genótipo 3.134
Embora a hepatite E em indivíduos imunocomprometidos seja resolvida espontaneamente após a doença autolimitada aguda, a infecção crônica por HEV tem sido descrita somente em indivíduos imunossuprimidos, incluindo pacientes com infecção por HIV/Aids, receptores de transplante de órgão e pacientes submetidos à quimioterapia. Em adição, a infecção crônica por HEV em indivíduos imunocomprometidos pode causar hepatite ativa, fibrose hepática e, potencialmente, cirrose.135
Os ensaios diagnósticos, sorológicos e virológicos para infecção por HEV não são disponibilizados de forma rotineira nos EUA. Anticorpos da classe imunoglobulina M contra HEV (IgM anti-HEV) geralmente acompanham a elevação da ALT durante a hepatite E aguda e persistem por cerca de 6 meses após a resolução da doença. Como ocasionalmente o aparecimento de IgM anti-HEV é retardado, pode ser necessário repetir o ensaio se um teste inicial resultar negativo e a suspeita clínica for forte. O RNA de HEV é detectável no soro em cerca de 2 semanas após a exposição. Os testes de RT-PCR para RNA de HEV não são disponibilizados de forma rotineira, mas podem ser realizados em laboratórios de referência especializados, podendo ser úteis quando houver suspeita de infecção crônica por HEV em indivíduos imunocomprometidos.
A imunoglobulina G contra HEV (IgG anti-HEV) passa a ser detectável pouco após a IgM anti-HEV e persiste por mais tempo, embora a duração média seja desconhecida. Do mesmo modo, o período de imunidade que se segue à recuperação a partir da infecção aguda é indeterminado.
O tratamento da infecção aguda por HEV continua sendo de suporte. O encaminhamento para transplante de fígado deve ser considerado nos raros casos de pacientes com hepatite fulminante. Cautela especial se faz necessária em casos de gestantes com infecção por HEV aguda.
Na infecção por HEV crônica, diminuir a imunossupressão pode levar à depuração viral.136 Em adição, têm aparecido relatos pouco confiáveis de tratamento bem-sucedido com RBV, interferon peguilado ou ambos de pacientes com infecção por HEV crônica. Entretanto, estes relatos de casos individuais e pequenas séries de casos estão longe de fornecer dados robustos e confiáveis para embasar as decisões terapêuticas.137-139
Além dos vírus de hepatite A a E, outros vírus diferentes podem causar inflamação hepática, ainda que o fígado não seja o sítio primário de replicação viral e lesão [ver a Tabela 1]. Com exceção do HSV e do vírus varicela-zoster (VZV), estes vírus tipicamente causam uma forma de hepatite aguda mais branda, com elevação de 2-10 vezes dos níveis séricos de aminotransferase e, mais raramente, aumento dos níveis séricos de bilirrubina em até 10 mg/dL.
Ambos HSV-1 e HSV-2 podem causar síndrome de hepatite aguda se a infecção for disseminada. A infecção disseminada geralmente ocorre em indivíduos imunocomprometidos, mas também pode ocorrer em indivíduos imunocompetentes, em particular durante a gravidez, bem como em neonatos. Lesões concomitantes na pele e nas membranas mucosas são encontradas em quase 40% dos casos. A hepatite associada ao HSV muitas vezes segue um curso grave e rapidamente fatal, quando o aciclovir não é iniciado logo. Assim, havendo suspeita de hepatite decorrente de infecção por HSV, deve ser iniciado um curso de aciclovir (10-30 mg/kg/dia, a cada 8 h) enquanto a confirmação do diagnóstico é aguardada. Em pacientes com hepatite associada ao HSV, um ensaio de PCR para RNA de HSV no soro geralmente resulta positivo. A biópsia de fígado revela áreas focais de necrose, por vezes com inclusões intranucleares dentro dos hepatócitos. Em adultos imunocompetentes com hepatite esmagadora decorrente de infecção por HSV, o transplante de fígado pode ser considerado.
A infecção primária por VZV (catapora) pode causar hepatite aguda. Em crianças, isto se manifesta primariamente como uma elevação discreta dos níveis séricos de aminotransferase. Entretanto, em adultos, os níveis de aminotransferase podem aumentar para mais de 1.000 unidades/L e a doença pode progredir para insuficiência hepática fulminante. O envolvimento hepático na reativação do zoster é raro, mas pode ocorrer em indivíduos imunocomprometidos. A melhor forma de confirmar o diagnóstico é por meio da detecção de DNA e VZV no soro, através de PCR. A biópsia de fígado pode ser semelhante a de hepatite associada ao HSV, com necrose coagulativa e inclusões intranucleares. Tipicamente, porém, o VZV induz mais de uma resposta inflamatória no fígado. O tratamento é feito com aciclovir, na dose de 30 mg/kg/dia, a cada 8 horas, por 7-10 dias.
O vírus Epstein-Barr (EBV) é um vírus linfotrópico contendo DNA, que causa envolvimento do fígado, baço ou de ambos órgãos em 10-15% das infecções sintomáticas. O EBV é transmitido através de secreções orais (daí o termo popular “beijo da morte”) e geralmente permanece para sempre no corpo. mais de 90% da população mundial está infectada pelo EBV. Muitas infecções primárias ocorrem na infância. As infecções primárias por EBV em adultos com mais de 30 anos de idade tendem mais a resultar em hepatite e icterícia.140 A apresentação típica do envolvimento hepática é a icterícia leve com hepatomegalia. Os achados mais comuns em geral são faringite, linfadenopatia cervical, esplenomegalia e linfocitose. A trombocitopenia frequentemente está presente. O teste de anticorpo heterófilo (i.e., Monospot) é sensível, mas não totalmente específico. A confirmação com um ensaio de anticorpo anti-EBV é sugerida. Quando uma biópsia é obtida, linfócitos atípicos podem ser vistos infiltrando os sinusoides.
Não há tratamento específico disponível nem necessário para a hepatite associada ao EBV, que em geral é uma doença branda e autolimitada. Têm aparecido relatos muito raros de insuficiência hepática primariamente em indivíduos imunocomprometidos.
Assim como o EBV, o citomegalovírus (CMV) é um vírus (encontrado em até 70% da população) linfotrópico contendo DNA, que persiste em estado dormente após a infecção aguda. A hepatite por CMV pode ocorrer com a infecção primária ou, mais comumente, com reativação. Pacientes imunocompetentes podem ter icterícia branda, níveis séricos discreta a moderadamente elevados de aminotransferase e de fosfatase alcalina, e hepatoesplenomegalia.
Por outro lado, em pacientes imunocomprometidos, a doença pode seguir um curso grave e potencialmente fatal. Pacientes imunocomprometidos com hepatite por CMV devem ser tratados com ganciclovir ou foscarnet. A melhor forma de estabelecer o diagnóstico é por detecção do antígeno inicial de CMV ou da DNA polimerase viral. Um ensaio para IgM anti-CMV também pode resultar positivo. A biópsia de fígado ocasionalmente pode mostrar inclusões nucleares conhecidas como “olho de coruja”, mas geralmente mostra células gigantes multinucleadas e necrose focal.
O parvovírus humano B19 é um pequeno vírus contendo DNA, que causa ampla variedade de doenças clínicas, mais comumente o eritema infeccioso da infância (a 5ª doença). Pode haver elevação discreta dos níveis séricos de aminotransferase durante o curso da 5ª doença. O parvovírus humano B19 foi associado à anemia aplásica e, especificamente, tem sido documentado em 50-67% dos pacientes com anemia aplásica e insuficiência hepática associadas à hepatite.141 A terapia é de suporte e o transplante de fígado pode ser considerado para os casos de infecção por parvovírus humano B19 associada à insuficiência hepática. Entretanto, o transplante de fígado pode ser contraindicado, por causa da anemia aplásica acompanhante.
As infecções por adenovírus são extremamente comuns, mas o envolvimento hepático é incomum. Os adenovírus são vírus contendo DNA, geralmente causadores de doença branda no trato respiratório superior ou de síndrome gastrintestinal. Em casos raros, porém, relatos de caso documentam a ocorrência de insuficiência hepática fulminante associada com infecção por adenovírus em pacientes imunocomprometidos e pacientes submetidos à terapia genética tendo como vetor um adenovírus.142 A terapia é de suporte.
O vírus da febre amarela, um dos vírus a RNA causadores de febre hemorrágica, é membro da família Flaviviridae, ao lado do HCV. A febre amarela é transmitida por mosquitos em áreas tropicais (exclusivamente as áreas tropicais da América do Norte). No início do curso da doença, pode haver desenvolvimento de icterícia e os níveis séricos de aminotransferase se tornam anormais. A forma branda da febre amarela pode sofrer resolução espontânea, mas 20% dos pacientes apresentam progressão da condição pata falência de múltiplos órgãos e morte. Não há terapia específica disponível, mas uma vacina com vírus vivo atenuado confere proteção e deve ser ofertada aos viajantes que planejam visitar áreas endêmicas.
Elevações brandas dos níveis séricos de aminotransferase podem acompanhar a rubéola ou o sarampo. Entretanto, desde a iniciação das vacinações da infância bastante efetivas nos EUA, pouquíssimos casos têm sido relatados.
O vírus coxsackie B é um enterovírus que afeta predominantemente crianças com menos de 2 anos, mas que pode ser transmitido para crianças maiores e adultos. Na vasta maioria das novas infecções (>95%), os pacientes são assintomáticos ou sofrem uma breve doença envolvendo o trato respiratório superior. Entretanto, ainda que raramente, a infecção pode levar à encefalite, meningite, miopericardite, erupção (mãos, pés e doença bucal) e às vezes hepatite. Em neonatos, a doença pode progredir para uma síndrome de sepse acompanhada de insuficiência hepática. O diagnóstico de infecção por vírus coxsackie B é melhor estabelecido pela demonstração (com ensaio de PCR) da presença de DNA de enterovírus. Nenhuma medicação antiviral específica é útil.
O dr. Dienstag recebeu suporte ou atuou em conselhos junto às empresas Vertex, Bristol-Myers Squibb, Gilead Sciences, Achillion Pharmaceuticals, Genzyme, Medtronic, Merck, CVS e Ikaria.
O dr. Schaefer não mantém relações comerciais com os fabricantes de produtos ou prestadores de serviços discutidos nesta seção.
1. Perz JF, Armstrong GL, Farrington LA, et al. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol 2006;45:529–38.
2. Centers for Disease Control and Prevention. Surveillance for acute viral hepatitis - United States 2007. Morb Mortal Wkly Rep 2009;58:1–27.
3. Schiodt FV, Davern TJ, Shakil AO, et al. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003;98:448–53.
4. Kariv R, Leshno M, Beth-Or A, et al. Re-evaluation of serum alanine aminotransferase upper normal limit and its modulating factors in a large-scale population study. Liver Int 2006;26:445–50.
5. Rossi E, Adams L, Prins A, et al. Validation of the FibroTest biochemical markers score in assessing liver fibrosis in hepatitis C patients. Clin Chem 2003;49:450–4.
6. Seeff LB, Everson GT, Morgan TR, et al. Complication rate of percutaneous liver biopsies among persons with advanced chronic liver disease in the HALT-C trial. Clin Gastroenterol Hepatol 2010;8:877–83.
7. Chandok N, Watt KD. Pain management in the cirrhotic patient: the clinical challenge. Mayo Clin Proc 2010;85:451–8.
8. Khalid SK, Lane J, Navarro V, Garcia-Tsao G. Use of over-the-counter analgesics is not associated with acute decompensation in patients with cirrhosis. Clin Gastroenterol Hepatol 2009;7:994–9; quiz 13–4.
9. Tandra S, Vuppalanchi R. Use of statins in patients with liver disease. Curr Treat Options Cardiovasc Med 2009;11:272–8.
10. Jensen DM. Endoscopic screening for varices in cirrhosis: findings, implications, and outcomes. Gastroenterology 2002;122:1620–30.
11. Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology 2005;42:1208–36.
12. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology 2011;53:1020–2.
13. Yu NC, Chaudhari V, Raman SS, et al. CT and MRI improve detection of hepatocellular carcinoma, compared with ultrasound alone, in patients with cirrhosis. Clin Gastroenterol Hepatol 2011;9:161–7.
14. Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004;11:97–107.
15. Liaw YF, Brunetto MR, Hadziyannis S. The natural history of chronic HBV infection and geographical differences. Antiviral Ther 2010;15 Suppl 3:25–33.
16. Goldstein ST, Zhou F, Hadler SC, et al. A mathematical model to estimate global hepatitis B disease burden and vaccination impact. Int J Epidemiol 2005;34:1329–39.
17. Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med 2005;352:2682–95.
18. Hatzakis A, Wait S, Bruix J, et al. The state of hepatitis B and C in Europe: report from the Hepatitis B and C Summit Conference. J Viral Hepat 2011;18 Suppl 1:1–16.
19. Ganem D, Varmus HE. The molecular biology of the hepatitis B viruses. Annu Rev Biochem 1987;56:651–93.
20. Ganem D, Prince AM. Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med 2004;350:1118–29.
21. Nassal M. Hepatitis B viruses: reverse transcription a different way. Virus Res 2008;134:235–49.
22. Grimm D, Thimme R, Blum HE. HBV life cycle and novel drug targets. Hepatol Int 2011;5:644–53.
23. Dienstag JL. Hepatitis B virus infection. N Engl J Med 2008;359:1486–500.
24. Lai CL, Ratziu V, Yuen M-F, Poynard T. Viral hepatitis B. Lancet 2003;362:2089–94.
25. McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology 2009;49(5 Suppl):S45–55.
26. Iloeje UH, Yang HI, Su J, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006;130:678–86.
27. Chen CJ, Yang HI. Natural history of chronic hepatitis B REVEALed. J Gastroenterol Hepatol 2011;26:628–38.
28. Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65–73.
29. Yang HI, Yeh SH, Chen PJ, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst 2008;100:1134–43.
30. Chen JD, Yang HI, Iloeje UH, et al. Carriers of inactive hepatitis B virus are still at risk for hepatocellular carcinoma and liver-related death. Gastroenterology 2010;138:1747–54.
31. Bonino F, Piratvisuth T, Brunetto MR, Liaw YF. Diagnostic markers of chronic hepatitis B infection and disease. Antiviral Ther 2010;15 Suppl 3:35–44.
32. Chan HL, Thompson A, Martinot-Peignoux M, et al. Hepatitis B surface antigen quantification: why and how to use it in 2011—a core group report. J Hepatol 2011;55:1121–31.
33. Lin SY, Chang ET, So SK. Why we should routinely screen Asian American adults for hepatitis B: a cross-sectional study of Asians in California. Hepatology 2007;46:1034–40.
34. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009;50:227–42.
35. Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009;50:661–2.
36. Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 update. Clin Gastroenterol Hepatol 2008;6:1315–41; quiz 286.
37. Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007;45:507–39.
38. Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2004;351:1206–17.
39. Ayoub WS, Keeffe EB. Review article: current antiviral therapy of chronic hepatitis B. Aliment Pharmacol Ther 2011;34:1145–58.
40. Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009;137:1593–608 e1-2.
41. Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology 2007;132:1574–85.
42. Marcellin P, Heathcote E, Corsa A, et al. No detectable resistance to tenofovir disoproxil fumarate (TDF) following up to 240 weeks of treatment in patients with HBeAg+ and HbeAg- chronic hepatitis B virus infection. Presentation at the American Association for the Study of Liver Diseases; 2011 Nov 8; San Francisco, CA.
43. Han GR, Cao MK, Zhao W, et al. A prospective and open-label study for the efficacy and safety of telbivudine in pregnancy for the prevention of perinatal transmission of hepatitis B virus infection. J Hepatol 2011;55:1215–21.
44. Mindikoglu AL, Regev A, Schiff ER. Hepatitis B virus reactivation after cytotoxic chemotherapy: the disease and its prevention. Clin Gastroenterol Hepatol 2006;4:1076–81.
45. Billioud G, Pichoud C, Puerstinger G, et al. The main hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antiviral Res 2011;92:271–6.
46. Farci P. Delta hepatitis: an update. J Hepatol 2003;39 Suppl 1:S212–9.
47. Pascarella S, Negro F. Hepatitis D virus: an update. Liver Int 2011;31:7–21.
48. Rizzetto M. Hepatitis D: the comeback? Liver Int 2009;29 Suppl 1:140–2.
49. Wedemeyer H, Heidrich B, Manns MP. Hepatitis D virus infection—not a vanishing disease in Europe! Hepatology 2007;45:1331–2; author reply 2–3.
50. Kucirka LM, Farzadegan H, Feld JJ, et al. Prevalence, correlates, and viral dynamics of hepatitis delta among injection drug users. J Infect Dis 2010;202:845–52.
51. Le Gal F, Gault E, Ripault MP, et al. Eighth major clade for hepatitis delta virus. Emerg Infect Dis 2006;12:1447–50.
52. Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet 2011;378:73–85.
53. Wang KS, Choo QL, Weiner AJ, et al. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986;323:508–14.
54. Kos A, Dijkema R, Arnberg AC, et al. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986;323:558–60.
55. Branch AD, Robertson HD. A replication cycle for viroids and other small infectious RNA’s. Science 1984;223:450–5.
56. Han Z, Alves C, Gudima S, Taylor J. Intracellular localization of hepatitis delta virus proteins in the presence and absence of viral RNA accumulation. J Virol 2009;83:6457–63.
57. Otto JC, Casey PJ. The hepatitis delta virus large antigen is farnesylated both in vitro and in animal cells. J Biol Chem 1996;271:4569–72.
58. Kiesslich D, Crispim MA, Santos C, et al. Influence of hepatitis B virus (HBV) genotype on the clinical course of disease in patients coinfected with HBV and hepatitis delta virus. J Infect Dis 2009;199:1608–11.
59. Su CW, Huang YH, Huo TI, et al. Genotypes and viremia of hepatitis B and D viruses are associated with outcomes of chronic hepatitis D patients. Gastroenterology 2006;130:1625–35.
60. Romeo R, Del Ninno E, Rumi M, et al. A 28-year study of the course of hepatitis delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology 2009;136:1629–38.
61. Niro GA, Smedile A, Ippolito AM, et al. Outcome of chronic delta hepatitis in Italy: a long-term cohort study. J Hepatol 2010;53:834–40.
62. Wedemeyer H, Yurdaydin C, Dalekos GN, et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med 2011;364:322–31.
63. Wedemeyer H. Hepatitis D revival. Liver Int 2011;31 Suppl 1:140–4.
64. Lavanchy D. The global burden of hepatitis C. Liver Int 2009;29 Suppl 1:74–81.
65. Alter M. Epidemiology of hepatitis C virus infection. World J Gastroenterol 2007;13:2436–41.
66. Indolfi G, Resti M. Perinatal transmission of hepatitis C virus infection. J Med Virol 2009;81:836–43.
67. van de Laar T, Pybus O, Bruisten S, et al. Evidence of a large, international network of HCV transmission in HIV-positive men who have sex with men. Gastroenterology 2009;136:1609–17.
68. Armstrong GL, Wasley A, Simard EP, et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006;144:705–14.
69. Strader DB. Coinfection with HIV and hepatitis C virus in injection drug users and minority populations. Clin Infect Dis 2005;41 Suppl 1:S7–13.
70. Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 2005;11:791–6.
71. Lindenbach BD, Meuleman P, Ploss A, et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci U S A 2006;103:3805–9.
72. Herker E, Ott M. Unique ties between hepatitis C virus replication and intracellular lipids. Trends Endocrinol Metab 2011;22:241–8.
73. Ploss A, Evans MJ, Gaysinskaya VA, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009;457:882–6.
74. Mazumdar B, Banerjee A, Meyer K, Ray R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 2011;54:1149–56.
75. Jangra RK, Yi M, Lemon SM. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 2010;84:6615–25.
76. Jopling CL, Schutz S, Sarnow P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008;4:77–85.
77. Roberts AP, Lewis AP, Jopling CL. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 2011;39:7716–29.
78. Gottwein JM, Bukh J. Cutting the gordian knot-development and biological relevance of hepatitis C virus cell culture systems. Adv Virus Res 2008;71:51–133.
79. Poenisch M, Bartenschlager R. New insights into structure and replication of the hepatitis C virus and clinical implications. Semin Liver Dis 2010;30:333–47.
80. Suzuki T. Assembly of hepatitis C virus particles. Microbiol Immunol 2011;55:12–8.
81. Jiang J, Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 2009;83:12680–91.
82. Nahmias Y, Goldwasser J, Casali M, et al. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 2008;47:1437–45.
83. Page K, Hahn JA, Evans J, et al. Acute hepatitis C virus infection in young adult injection drug users: a prospective study of incident infection, resolution, and reinfection. J Infect Dis 2009;200:1216–26.
84. Wang CC, Krantz E, Klarquist J, et al. Acute hepatitis C in a contemporary US cohort: modes of acquisition and factors influencing viral clearance. J Infect Dis 2007;196:1474–82.
85. Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000;284:450–6.
86. Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009;461:399–401.
87. Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009;461:798–801.
88. Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009;41:1100–4.
89. Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009;41:1105–9.
90. Tillmann HL, Thompson AJ, Patel K, et al. A polymorphism near IL28B is associated with spontaneous clearance of acute hepatitis C virus and jaundice. Gastroenterology 2010;139:1586–92, 92 e1.
91. McCarthy JJ, Li JH, Thompson A, et al. Replicated association between an IL28B gene variant and a sustained response to pegylated interferon and ribavirin. Gastroenterology 2010;138:2307–14.
92. Thein HH, Yi Q, Dore GJ, Krahn MD. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infection: a meta-analysis and meta-regression. Hepatology 2008;48:418–31.
93. Thomas DL, Seeff LB. Natural history of hepatitis C. Clin Liver Dis 2005;9:383–98, vi.
94. Kagawa T, Keeffe EB. Long-term effects of antiviral therapy in patients with chronic hepatitis C. Hepat Res Treat 2010;2010:562–78.
95. Di Bisceglie AM, Shiffman ML, Everson GT, et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med 2008;359:2429–41.
96. Di Bisceglie AM, Stoddard AM, Dienstag JL, et al. Excess mortality in patients with advanced chronic hepatitis C treated with long-term peginterferon. Hepatology 2011;53:1100–8.
97. Serste T, Nkuize M, Moucari R, et al. Metabolic disorders associated with chronic hepatitis C: impact of genotype and ethnicity. Liver Int 2010;30:1131–6.
98. Serfaty L, Capeau J. Hepatitis C, insulin resistance and diabetes: clinical and pathogenic data. Liver Int 2009;29 Suppl 2:13–25.
99. Negro F, Clement S. Impact of obesity, steatosis and insulin resistance on progression and response to therapy of hepatitis C. J Viral Hepat 2009;16:681–8.
100. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
101. Jacobson IM, Cacoub P, Dal Maso L, et al. Manifestations of chronic hepatitis C virus infection beyond the liver. Clin Gastroenterol Hepatol 2010;8:1017–29.
102. Hayashi N, Takehara T. Antiviral therapy for chronic hepatitis C: past, present, and future. J Gastroenterol 2006;41:17–27.
103. Shiffman ML, Suter F, Bacon BR, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med 2007;357:124–34.
104. Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011;364:2405–16.
105. Zeuzem S, Andreone P, Pol S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med 2011;364:2417–28.
106. Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011;364:1207–17.
107. Foster GR, Hezode C, Bronowicki JP, et al. Telaprevir alone or with peginterferon and ribavirin reduces HCV RNA in patients with chronic genotype 2 but not genotype 3 infections. Gastroenterology 2011;141:881–9 e1.
108. Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009;49:1335–74.
109. Kamal SM, Fouly AE, Kamel RR, et al. Peginterferon alfa-2b therapy in acute hepatitis C: impact of onset of therapy on sustained virologic response. Gastroenterology 2006;130:632–8.
110. Maheshwari A, Thuluvath PJ. Management of acute hepatitis C. Clin Liver Dis 2010;14:169–76; x.
111. Kotenko SV, Gallagher G, Baurin VV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 2003;4:69–77.
112. Sheppard P, Kindsvogel W, Xu W, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 2003;4:63–8.
113. Gad HH, Dellgren C, Hamming OJ, et al. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem 2009;284:20869–75.
114. Sixtos-Alonso MS, Sanchez-Munoz F, Sanchez-Avila JF, et al. IFN-stimulated gene expression is a useful potential molecular marker of response to antiviral treatment with Peg-IFNalpha 2b and ribavirin in patients with hepatitis C virus genotype 1. Arch Med Res 2011;42:28–33.
115. Honda M, Sakai A, Yamashita T, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology 2010;139:499–509.
116. Chayama K, Hayes CN, Abe H, et al. IL28B but not ITPA polymorphism is predictive of response to pegylated interferon, ribavirin, and telaprevir triple therapy in patients with genotype 1 hepatitis C. J Infect Dis 2011;204:84–93.
117. Moghaddam A, Melum E, Reinton N, et al. IL28B genetic variation and treatment response in patients with hepatitis C virus genotype 3 infection. Hepatology 2011;53:746–54.
118. Mangia A, Thompson AJ, Santoro R, et al. An IL28B polymorphism determines treatment response of hepatitis C virus genotype 2 or 3 patients who do not achieve a rapid virologic response. Gastroenterology 2010;139:821–7, 7 e1.
119. Georgescu EF, Streba L, Teodorescu R, et al. Potential enhancement of both early (EVR) and sustained (SVR) virological response by fluvastatin in chronic hepatitis C treated with standard pegIFN-ribavirin therapy. a pilot study. Presentation at European Association for the Study of the Liver; 2011 Apr 2011; Berlin, Germany.
120. Rao GA, Pandya PK. Statin therapy improves sustained virologic response among diabetic patients with chronic hepatitis C. Gastroenterology 2011;140:144–52.
121. Flisiak R, Feinman SV, Jablkowski M, et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 2009;49:1460–8.
122. Janssen HL, Reesink HW, Zeuzem S, et al. A randomized, double-blind, placebo (PLB) controlled safety and anti-viral proof of concept study of Miravirsen (MIR), an oligonucleotide targeting miR-122, in treatment naïve patients with genotype 1 (GT1) chronic HCV infection. Presentation at American Association for the Study of Liver Diseases; 2011 Nov 7, 2011; San Francisco, CA.
123. World Health Organization. Viral hepatitis: report by the Secretariat. In: Sixty-third World Health Assembly; 2010 March 25, 2010; Geneva, Switzerland. Available at: http://apps.who.int/gb/ebwha/pdf_files/WHA63/A63_15-en.pdf (accessed February 17, 2012).
124. Klevens RM, Miller JT, Iqbal K, et al. The evolving epidemiology of hepatitis a in the United States: incidence and molecular epidemiology from population-based surveillance, 2005-2007. Arch Intern Med 2010;170:1811–8.
125. Martin A, Lemon SM. Hepatitis A virus: from discovery to vaccines. Hepatology 2006;43(2 Suppl 1):S164–72.
126. Centers for Disease Control and Prevention. Viral hepati¬tis, 1994. Available at: http://www.cdc.gov/nchs/data/nhanes/databriefs/viralhep.pdf (access February 17, 2012).
127. Klevens RM, Kruszon-Moran D, Wasley A, et al. Seroprevalence of hepatitis A virus antibodies in the U.S.: results from the National Health and Nutrition Examination Survey. Public Health Rep 2011;126:522–32.
128. Kuniholm MH, Purcell RH, McQuillan GM, et al. Epidemiology of hepatitis E virus in the United States: results from the Third National Health and Nutrition Examination Survey, 1988-1994. J Infect Dis 2009;200:48–56.
129. Feagins AR, Opriessnig T, Guenette DK, et al. Detection and characterization of infectious hepatitis E virus from commercial pig livers sold in local grocery stores in the USA. J Gen Virol 2007;88:912–7.
130. Aggarwal R, Jameel S. Hepatitis E. Hepatology 2011;54:2218–26.
131. Cao X, Ma X, Liu Y, et al. Epidemiological and etiological studies on enterically transmitted non-A, non-B hepatitis in the south part of Xinang. In: Shikata T, Purcell RH, Uchida T, editors. Viral hepatitis C, D, and E. Amsterdam: Excerpta Medica; 1991. p. 297.
132. Ramachandran J, Eapen CE, Kang G, et al. Hepatitis E superinfection produces severe decompensation in patients with chronic liver disease. J Gastroenterol Hepatol 2004;19:134–8.
133. Renou C, Pariente A, Cadranel JF, et al. Clinically silent forms may partly explain the rarity of acute cases of autochthonous genotype 3c hepatitis E infection in France. J Clin Virol 2011;51:139–41.
134. Davern TJ, Chalasani N, Fontana RJ, et al. Acute hepatitis e infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology 2011;141:1665–72 e9.
135. Aggarwal R. Hepatitis E: historical, contemporary and future perspectives. J Gastroenterol Hepatol 2011;26 Suppl 1:72–82.
136. Kamar N, Garrouste C, Haagsma EB, et al. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology 2011;140:1481–9.
137. Dalton HR, Keane FE, Bendall R, et al. Treatment of chronic hepatitis E in a patient with HIV infection. Ann Intern Med 2011;155:479–80.
138. Mallet V, Nicand E, Sultanik P, et al. Brief communication: case reports of ribavirin treatment for chronic hepatitis E. Ann Intern Med 2010;153:85–9.
139. Haagsma EB, Riezebos-Brilman A, van den Berg AP, et al. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon alpha-2b. Liver Transplantation 2010;16:474–7.
140. Hoagland RJ. The clinical manifestations of infectious mononucleosis: a report of two hundred cases. Am J Med Sci 1960;240:55–63.
141. Karetnyi YV, Beck PR, Markin RS, et al. Human parvovirus B19 infection in acute fulminant liver failure. Arch Virol 1999;144:1713–24.
142. Wang WH, Wang HL. Fulminant adenovirus hepatitis following bone marrow transplantation. A case report and brief review of the literature. Arch Pathol Lab Med 2003;127:e246–8.
143. Dienstag JL. Benefits and risk of nucleoside analog therapy for hepatitis B. Hepatology 2009;49(5 Suppl):S112–21.
144. Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009;137:1593–608.
145. Snow Lampart A, Chappell B, Curtis M, et al. No resistance to tenofovir disoproxil fumarate detected after up to 144 weeks of therapy in patients monoinfected with chronic hepatitis B virus. Hepatology 2011;53:763–73.
Figuras 1,3 e 4 — Christine Kenney.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.