(Carregando Índice)... (Carregando Índice)... |
Última revisão: 29/01/2016
Comentários de assinantes: 0
Leif W. Ellisen, MD, PhD*
Associate Professor of Medicine, Harvard Medical School, Boston, MA
Artigo original: Ellisen LW, MD, PhD. Molecular genetics of cancer. ACP Medicine.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Paulo Henrique Machado.
Revisão técnica: Dr. Lucas Santos Zambon.
A expansão clonal descontrolada de uma célula, que geralmente provoca a invasão dos tecidos adjacentes e disseminação metastática, produz câncer. As alterações genéticas iniciais nessa célula que desencadeiam proliferação aberrante são acompanhadas pelo acúmulo de mutações adicionais entre os descendentes genéticos. No final ocorre um processo seletivo em que subclones com propriedades intensificadas de crescimento se tornam dominantes no interior do tumor – fenômeno conhecido por progressão tumoral. Definiu-se para alguns tumores uma clara evolução genética histológica e molecular de lesões pré-cancerígenas para câncer maligno e invasivo (p.ex., câncer no cólon e câncer na bexiga); entretanto, em muitos tipos de câncer esse processo talvez não seja clinicamente evidente. Os genes que se alteram no desenvolvimento e na progressão do câncer ainda estão sendo objeto de uma profunda investigação. Em muitos casos, a identificação desses genes levou a uma visão mais detalhada dos mecanismos fisiológicos normais que controlam a proliferação celular. Os produtos desses genes estão envolvidos em atividades como regulação do ciclo celular básico, transmissão de sinais de crescimento, regulação da diferenciação e da morte das células, e estabelecimento da imortalidade celular. De maneira geral, o rompimento desses genes ocorre exclusivamente nas células somáticas cujo destino é se tornarem cancerígenas. Entretanto, em casos raros, possivelmente ocorram mutações que sejam transmitidas pela linha germinal, resultando em uma predisposição genética para o câncer (i.e., síndromes de câncer familiar).
Acredita-se que fatores ambientais também contribuam para o desenvolvimento de câncer. Em alguns casos, interações entre fatores ambientais e variações genéticas sutis na linha germinal que distinguem os indivíduos podem constituir um determinante importante de risco de câncer na população em geral.1 Em outros casos há uma ligação direta entre o efeito do carcinógeno sobre o DNA somático e mutações específicas que promovem tumores, como aquelas que ocorrem no gene supressor tumoral p53 (p.ex., exposição ao tabaco ou à aflatoxina, uma toxina fúngica). A exposição à radiação ionizante é outra fonte de câncer com ligação direta aos danos genéticos. A incidência de leucemia e de vários tumores sólidos nos sobreviventes da bomba atômica produziu uma grande quantidade de dados para os estudos clássicos sobre a dosagem de radiação e suas consequências. Os dados mais recentes surgiram de casos de câncer como complicações tardias de radioterapias e de quimioterapias radiomiméticas usadas no tratamento de malignidades em fase inicial.
Para finalizar, as infecções virais foram vinculadas ao desenvolvimento de tipos específicos de câncer. As ligações câncer-vírus incluem carcinoma cervical e orofaríngeo de células escamosas com subtipos específicos de papilomavírus humano; carcinoma hepatocelular com infecção crônica causada pelo vírus da hepatite B; carcinoma nasofaríngeo e linfomas com vírus de Epstein-Barr em hospedeiros imunossuprimidos; o caso raro de leucemia aguda de células T transformando o vírus linfotrópico de células T humanas tipo I; e sarcoma de Kaposi com herpesvírus humano tipo 8. Cabe observar que esses casos são exceções à regra; a maior parte dos casos de câncer em seres humanos não é consequência de infecções virais. Entretanto, grande parte de nossos conhecimentos sobre os genes humanos envolvidos nos casos de câncer surgiu originalmente do estudo de vírus que produzem tumores em galinhas e roedores. A apropriação indevida dos vírus dos genes de hospedeiro envolvidos na proliferação celular por esses vírus resultou na identificação de oncogenes e forneceu a primeira pista para os eventos genéticos que causam câncer em seres humanos.
|
*O autor e os editores expressam seus sinceros agradecimentos às contribuições do autor da versão anterior, Daniel A. Haber, MD, PhD no desenvolvimento e na redação deste capítulo. |
As informações financeiras estão no final deste capítulo, antes das referências.
Na maior parte dos casos, as lesões genéticas que promovem tumorigênese são adquiridas por meios somáticos e não envolvem alterações na linha germinal. Essas alterações genéticas podem resultar na ativação ou no ganho funcional (de oncogenes) e na perda funcional (de supressores tumorais). Em situações mais raras, as anormalidades hereditárias na linha germinal contribuem para a patogênese do câncer. Tipicamente, nessas circunstâncias, herda-se apenas uma única cópia defeituosa de um gene, sendo que a transformação exige a perda do segundo alelo (não mutante) em uma célula somática. Levando-se em consideração que a velocidade de uma perda alélica somática simples é exponencialmente mais elevada que a mutação independente de dois alelos no interior da mesma célula, a incidência de tipos específicos de câncer nos portadores da mutação é dramaticamente elevada, em comparação com a incidência na população em geral. De maneira geral (mas nem sempre), nos casos de predisposição para câncer hereditário a perda funcional, e não o ganho funcional, desses genes promove a carcinogênese. Consequentemente, por definição, esses genes são supressores tumorais.
Tipos histológicos diferentes de câncer estão associados a padrões sobrepostos e distintos de alterações genéticas. Alguns eventos genéticos, em particular translocações cromossômicas somáticas específicas, estão associados exclusivamente a um tipo de câncer. Por exemplo, a translocação cromossômica EWS-FLI 1 é específica para o sarcoma de Ewing; consequentemente, essa translocação é um marco diagnóstico bastante útil para esse tipo de tumor.2 Outras anormalidades genéticas somáticas, como a ativação mutacional do oncogene KRAS ou a desativação do gene supressor tumoral p53, estão associadas a muitos tipos diferentes de câncer. De maneira geral, no caso de defeitos genéticos na linha germinal a predisposição para um padrão tecidual específico de câncer é o resultado da herança de alguma anormalidade em um gene supressor tumoral específico. Em muitos casos, a base biológica dessa especificidade tecidual ainda permanece obscura. Por exemplo, o gene BRCA1 é importante para a estabilidade genômica e para o reparo do DNA em todos os tipos de células, porém, mesmo assim, as mutações herdadas do BRCA1 estão associadas quase que exclusivamente à predisposição para câncer de mama e ao câncer de ovário. Uma das descobertas relacionadas a esse fato é que os genes associados à predisposição hereditária para o câncer possivelmente também estejam sujeitos à desativação somática, embora esse conceito se aplique somente a um subgrupo distinto de tipos de câncer. Consequentemente, os carcinomas de pulmão de pequenas células (CPPCs) se caracterizam pela desativação somática do gene supressor do tumor retinoblastoma (RB1).3 Todavia, a mutação de RB1 na linha germinal não está associada a uma incidência elevada de CPPC, mas está relacionada à incidência de retinoblastoma e osteossarcoma. Teoricamente, esse paradoxo aparente reflita papeis contextuais específicos desempenhados pelo gene RB1, de modo que sua ativação seja suficiente para estimular a proliferação de alguns tipos de câncer, enquanto que em outros tipos a ativação talvez seja necessária, porém insuficiente para a tumorigênese, e ainda em outros tipos a ativação possivelmente desencadeie respostas compensatórias, incluindo morte celular. Seja qual for o caso, em geral temos uma compreensão relativamente limitada sobre a razão pela qual anormalidades genéticas específicas estão associadas a padrões teciduais específicos de tumorigênese.
A maior parte dos tipos de câncer em seres humanos, principalmente o câncer comum em adultos, abriga uma quantidade equivalente a dezenas ou mesmo centenas de anormalidades genéticas somáticas. Em um dos estudos, os investigadores preparam uma sequência ampla de carcinomas na mama e no cólon com base no DNA.4 Uma mediana de aproximadamente 80 mutações, que deram origem a uma truncagem proteica precoce ou a uma alteração na sequência proteica, foram encontradas em cada tumor. Todavia, acreditava-se que apenas uma pequena minoria dessas mutações (em torno de 15 por tumor) era “significativa”, tendo em vista a recorrência em vários tipos de câncer. Essas mutações, observadas com maior frequência em genes cancerígenos bem definidos como o p53 e o KRAS, foram denominadas mutações “condutoras” porque se acreditava que estimulassem transformações e progressões malignas. Entretanto, de um modo geral, a maior parte das mutações somáticas específicas de tumores não foram observadas de forma repetitiva em vários tipos de tumor. Presume-se que essas mutações ocorram casualmente e não reflitam qualquer instabilidade genética associada ao processo da tumorigênese. Portanto, passaram a ser conhecidas como mutações do tipo “passageiro”. A distinção entre anormalidades genéticas associadas a tumores do tipo condutor e passageiro pode se tornar um grande desafio e envolve análises de larga escala de genomas tumorais e estudos funcionais das vias cancerosas. A identificação das anormalidades do tipo condutor está se tornando cada vez mais relevante em face do desenvolvimento de uma nova geração de terapias para tratamento de câncer com foco nessas vias específicas.
Os genes causadores de câncer, ou oncogenes, foram descobertos quando os pesquisadores observaram que os genes específicos de galinhas e os retrovírus de roedores poderiam transformar as células normais de mamíferos em culturas. Comprovou-se que esses genes transformadores virais eram homólogos ativados de genes de mamíferos (conhecidos por proto-oncogenes) que foram roubados da célula de hospedeiro durante a evolução viral por sua capacidade de estimular a proliferação celular.5 Embora não sejam causados por infecções virais, descobriu-se que os cânceres humanos primários abrigam alelos ativados semelhantes de proto-oncogenes. Entre os primeiros oncogenes que foram descobertos estavam aqueles que codificavam as proteínas diretamente envolvidas na transmissão de sinais de proliferação celular. Isso inclui os receptores de fatores de crescimento (p.ex., receptor do fator de crescimento derivado das plaquetas [PDGR, do inglês platelet-derived growth factor receptor] ou o receptor do fator de crescimento epidérmico [EGFR, do inglês epidermal growth factor receptor]) que se tornam constitutivamente ativados como se estivessem respondendo à presença contínua de um fator de crescimento e de moléculas sinalizadoras que se localizam no sentido do fluxo (p.ex., HRAS, KRAS e NRAS) que normalmente respondem à sinalização do fator de crescimento e alternam rapidamente entre os estados ativado e desativado, mas sofrem mutações para células cancerígenas em uma posição permanentemente ativada. Os mecanismos através dos quais esses proto-oncogenes celulares normais são ativados nos cânceres humanos incluem pontos de mutação, amplificação genética e translocações cromossômicas [ver a Tabela e a Figura 1]. Essas mutações são conhecidas como mutações de ganho funcional, considerando que resultam em propriedades funcionais novas ou alteradas para a proteína codificada e são geneticamente dominantes sobre o segundo alelo normal.
Embora a ativação de oncogenes possa promover uma proliferação anormal e/ou sobrevivência de células cancerígenas nascentes, esses efeitos têm um custo. De maneira geral, a religação da sinalização de células cancerígenas que resultar da ativação de oncogenes torna as células cancerígenas atipicamente dependentes de rotas controladas pelo(s) oncogene(s) ativado(s) no interior daquelas células. Acredita-se que esse fenômeno, às vezes conhecido como “dependência de oncogene”, reflita estresses metabólicos específicos associados à proliferação desenfreada. Consequentemente, embora o resultado da inibição química do receptor do fator de crescimento epidérmico (EGFR) nas células normais seja apenas modesto, o bloqueio da sinalização do EGFR nas células cancerígenas do pulmão, que abriga as mutações ativadoras somáticas do EGFR, poderá induzir morte celular imediata. Da mesma forma, as células cancerígenas que não apresentarem ativação de um oncogene específico podem ser relativamente insensíveis a um agente terapêutico com alvo específico que, em outras circunstâncias, seria altamente eficaz nos tipos de câncer com a mutação relevante. Por exemplo, em um grande teste randomizado que fez a comparação entre quimioterapia e inibidor específico para o EGFR para tratamento de adenocarcinomas pulmonares, os pacientes com tumores com mutação no EGFR obtiveram benefícios substancialmente maiores com o inibidor com alvo específico, enquanto que os pacientes sem essas mutações obtiveram melhores resultados com a quimioterapia.6 Observações como essas resultaram em uma ênfase maior sobre a análise da genética tumoral na prática oncológica clínica.
|
Tabela 1 Mutações Selecionadas de Oncogenes no Câncer Humano
| ||||
|
Gene |
Mecanismo de Ativação |
Função do Produto Proteico |
Tipos de Câncer |
Mutação na Linha Germinal |
|
KRAS |
Ponto de mutação |
p21 GTPase |
Pancreático, colorretal, pulmonar (adenocarcinoma), endometrial, outros carcinomas.
|
Síndrome de Noonan (SN) |
|
NRAS |
Ponto de mutação |
p21 GTPase |
Leucemia mielocítica
|
ND |
|
NRAS |
Ponto de mutação |
p21 GTPase |
Bexiga
|
Síndrome de Costello |
|
BRAF |
Ponto de mutação |
Quinase na rota de RAS/MAPK |
Melanoma, leucemia, mieloma, câncer colorretal
|
NS, síndrome cardiofaciocutânea |
|
EGFR |
Amplificação, ponto de mutação. |
Receptor de EGF |
Gliomas, carcinoma escamoso e outros carcinomas.
|
ND |
|
ERBB2 (HER-2) |
Amplificação |
Receptor do fator de crescimento |
Carcinomas de mama, ovário, gástrico e outros carcinomas.
|
ND |
|
PIK3CA |
Ponto de mutação |
Subunidade da quinase catalítica PI3 |
Câncer de mama, colorretal e pulmonar, HNSCC.
|
ND |
|
MYC (c-myc) |
Amplificação, translocação cromossômica. |
Fator de transcrição |
Linfoma de Burkitt, CPPC, outros carcinomas.
|
ND |
|
MYCN (N-myc) |
Amplificação |
Fator de transcrição |
Neuroblastoma, CPPC.
|
ND |
|
MYCL1 (L-myc) |
Amplificação |
Fator de transcrição
|
CPPC |
ND |
|
BCL2 |
Translocação cromossômica |
Proteína antiapoptose |
Linfoma de células B (tipo folicular)
|
ND |
|
Tabela 1 Mutações Selecionadas de Oncogenes no Câncer Humano
| ||||
|
Gene |
Mecanismo de Ativação |
Função do Produto Proteico |
Tipos de Câncer |
Mutação na Linha Germinal |
|
CCND1 |
Amplificação, translocação cromossômica. |
Ciclina D, controle do ciclo celular. |
Carcinoma de mama e outros carcinomas, linfoma de células B, adenomas paratireoideos.
|
ND |
|
BCR-ABL |
Translocação cromossômica |
Tirosina quinase quimérica não receptora.
|
LMC, LLA (células T) |
ND |
|
RET |
Translocação cromossômica |
Receptores de tirosina quinase para GDNF. |
Câncer tireoideo (tipo papilar), câncer tireoideo (tipo medular).
|
NEM II |
|
CDK4 |
Amplificação, ponto de mutação. |
Quinase dependente da ciclina.
|
Sarcoma |
Melanoma familiar |
|
MET |
Ponto de mutação, amplificação. |
Receptores de tirosina quinase para HGF |
Câncer gástrico, câncer pulmonar. |
Câncer renal hereditário, tipo papilar.
|
|
SMO |
Ponto de mutação |
Molécula de sinalização transmembranar em rotas sônicas hedgehog.
|
Pele de células basais. |
ND |
|
CTNNB1 (catenina-ß) |
Ponto de mutação, mutação na fase de leitura. |
Coativador transcricional; liga a caderina-E ao citoesqueleto.
|
Melanoma, câncer colorretal. |
ND |
|
HST |
Amplificação |
Fator de crescimento (semelhante ao FGF).
|
Câncer gástrico |
ND |
|
PML-RAR-a |
Translocação cromossômica |
Fatores de transcrição quiméricos.
|
LPA |
ND |
|
W2A-PBX1 |
Translocação cromossômica |
Fatores de transcrição quiméricos.
|
Pré-B LLA |
ND |
|
MDM2 |
Amplificação |
Proteína de ligação com p53 |
Sarcoma |
ND |
|
Tabela 1 Mutações Selecionadas de Oncogenes no Câncer Humano
| ||||
|
Gene |
Mecanismo de Ativação |
Função do Produto Proteico |
Tipos de Câncer |
Mutação na Linha Germinal
|
|
GLI1 |
Amplificação |
Fator de transcrição.
|
Sarcoma, glioma |
ND |
|
NOTCH1 |
Translocação cromossômica, ponto de mutação. |
Receptor de superfícies celulares, fator de transcrição.
|
LLA de células T |
ND |
|
ERG/ETV |
Translocação cromossômica |
Fator de transcrição.
|
Carcinoma na próstata |
ND |
LLA = leucemia linfocítica aguda; LPA = leucemia promielocítica aguda; EGF (epidermal growth factor) = fator de crescimento epidérmico; FGF (fibroblast growth factor) = fator de crescimento de fibroblastos; GDNF (glial cell-derived neurotrophic factor) = fator neurotrófico derivado das células gliais; GTPase (guanosine triphosphatase) = guanosina trifosfatase; HGF (hepatocyte growth factor) = fator de crescimento dos hepatócitos; HNSCC (head and neck squamous cell carcinoma) = carcinoma de células escamosas da cabeça e pescoço; MAPK (mitogen-activated protein kinase) = proteinoquinase ativada por mitógeno; NEM =neoplasia endócrina múltipla; ND = não determinada; PI3 kinase =fosfoinositol-3 quinase; CPPC =câncer de pulmão de pequenas células.
Os pontos de mutações capazes de ativar um produto genético são pouco usuais e, tipicamente, são observados em um grupo altamente restrito de sítios codificadores de proteínas no interior do gene. Por exemplo, somente alterações específicas em três códons das proteínas da família RAS levaram a uma ativação constitutiva da sinalização RAS.7 Presume-se que outras mutações possam ocorrer no interior desses genes, embora sejam funcionalmente silenciosas ou resultem em proteínas inativas e, consequentemente, não sejam selecionadas nas transformações malignas. Da mesma forma, alterações em aminoácidos específicos nos receptores do fator de crescimento ou outras moléculas sinalizadoras produzem um efeito ativo indutor do crescimento ou impedem a infrarregulação por sinais fisiológicos apropriados. Há um interesse especial nas mutações no gene RET, que codifica um receptor do fator de crescimento, além de ser o proto-oncogene raro que é modificado na linha germinal de pacientes com predisposição para câncer. As substituições de aminoácidos dentro de domínios funcionais distintos da proteína estão associadas à neoplasia endócrina múltipla tipo IIA das síndromes de predisposição para câncer familiar ou ao câncer tireoideo medular. Por outro lado, a desativação das mutações no gene RET causa a doença de Hirschsprung, um defeito evolutivo que afeta as inervações colônicas.8 Essa ligação impressionante entre mutações diferentes no gene RET e tipos distintos de câncer e as síndromes de desenvolvimento possivelmente seja o resultado de propriedades funcionais diferentes que são mediadas pelos vários domínios da proteína RET e de rotas diferentes associadas à RET que podem ser ativadas em tipos diferentes de tecidos.

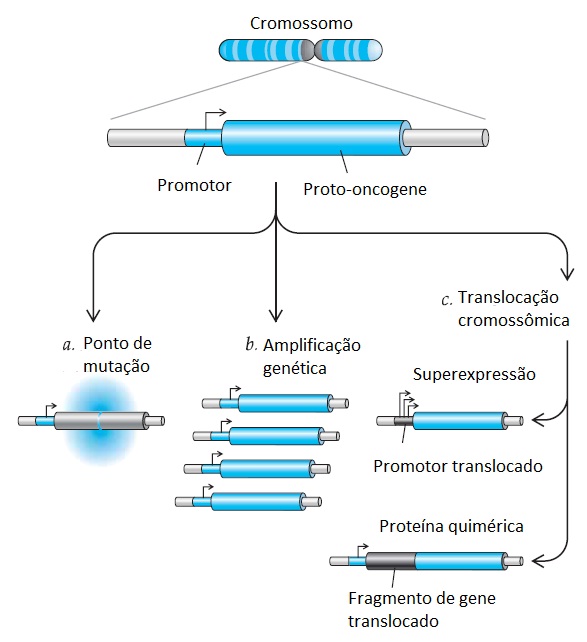
Figura 1 - Mecanismos de ativação de proto-oncogenes. Os genes celulares podem ser ativados no câncer como resultado de (a) pontos de mutações que alteram a sequência de aminoácidos, codificando a proteína possível de ser ativada sob o ponto de vista constitutivo; (b) amplificações do gene celular resultando em níveis mais elevados de expressão proteica; ou (c) translocações cromossômicas que levam à justaposição de um promotor forte, aumentando a expressão proteica, ou que produzam uma proteína quimérica nova, derivada de fragmentos de genes normalmente presentes em cromossomos diferentes.
Além das substituições de aminoácidos resultando em produtos genéticos ativados, os proto-oncogenes podem ser convertidos nas respectivas formas oncogênicas por alterações cromossômicas macroscópicas. A amplificação de genes – replicação excessiva de grandes fragmentos cromossômicos – é um mecanismo comum para superexpressão aberrante de genes específicos em tumores.9 Os genes amplificados , que ocasionalmente podem chegar a algumas centenas de cópias de genes diploides normais, poderão ser integrados em uma única localização cromossômica ou, menos frequentemente, poderão estar presentes em pequenos elementos extracromossômicos (cromossomos de minuto duplo). O MYCN (N-myc) é o mais conhecido entre os oncogenes sujeitos a amplificação nas células cancerígenas. Esse gene codifica o fator de transcrição que desempenha um papel fisiológico na estimulação da proliferação celular, sendo usualmente amplificado no neuroblastoma de câncer pediátrico; o prognóstico clínico para esses pacientes não é bom.10 A superexpressão de um oncogene pode também ser o resultado de alguma translocação ou reorganização cromossômica, que remove o gene celular de seu promotor regulado fisiologicamente e passa a controlá-lo através de um promotor estranho e mais ativo.11 No linfoma de Burkitt, por exemplo, o lócus cromossômico contendo o MYCN (N-myc) é reorganizado de modo que as regiões reguladoras negativas que se localizam no contrafluxo do MYC se perdem e, por outro lado, a expressão do gene é direcionada pelo intensificador forte da cadeia pesada de imunoglobulina, que é altamente ativo nas células B.12,13 Consequentemente, a desregulação da expressão do MYC nessas células é uma grande força estimuladora da proliferação celular. Da mesma forma, em um subgrupo de leucemias linfocíticas de células T, o fragmento de um gene conhecido por NOTCH1, que é extremamente importante para o desenvolvimento normal de muitos tipos de células, apresenta uma superexpressão como resultado da translocação nas proximidades do intensificador genético do receptor ß de células T.14 Entretanto, a situação mais comum é aquela em que o gene NOTCH1 não seja nem translocado nem superexpresso em leucemias causadas por células T, mas, em vez disso, seja o alvo do ponto de ativação de mutações.15 Acredita-se que a sinalização aberrante do gene NOTCH1 promova uma diferenciação e proliferação anormais de células leucêmicas, não importando se o gene seja ativado por translocação ou por ponto de mutação.
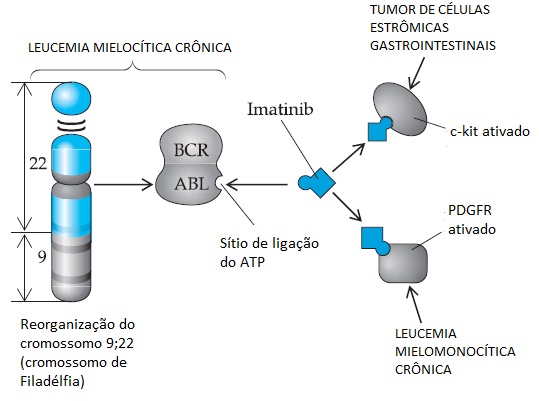
A primeira translocação cromossômica específica identificada no câncer humano foi o cromossomo de Filadélfia, que é subjacente à leucemia mielocítica crônica (LMC). A fusão dos cromossomos 9 e 22 resulta na união de dois genes não relacionados, o gene ABL1 (c-abl), que codifica a tirosina quinase e se localiza no cromossomo 9, e o gene BCR (breakpoint cluster region), que se localiza no cromossomo 22.16 Uma proteína quimérica com novas propriedades transformadoras forma-se a partir dessa reorganização cromossômica específica. A fase acelerada ou imatura da LMC geralmente está associada à duplicação do cromossomo de Filadélfia, sugerindo que o aumento nas cópias desse gene aberrante produz um efeito transformador dependente da dose. A fusão entre os genes BCR e ABL1 também demonstra uma visível especificidade do tipo celular; o ponto clássico de ruptura cromossômica está associado à proliferação mielocítica, enquanto que um ponto de ruptura variante, resultando em uma alteração sutil na proteína quimérica, leva a uma leucemia linfoide pediátrica cujo prognóstico não é bom.17 A importância da translocação BCR-ABL1 como evento genético inicial nos casos de LMC tem o suporte de um modelo de camundongo em que a expressão transgênica da proteína quimérica BCR-ABL1 nas células hematopoiéticas é suficiente para desencadear a proliferação mielocítica e linfoide.18 A descoberta do imatinib mesilato, um inibidor eficiente da quinase BCR-ABL1, levou a respostas dramáticas na leucemia mielocítica crônica e revolucionou o tratamento desse tipo de leucemia19 [ver Genética Tumoral e Resposta à Terapia-alvo para o Câncer, abaixo].
As translocações cromossômicas que geram proteínas quiméricas novas foram associadas a outros tipos de leucemia.20 Nos casos de leucemia promielocítica aguda (LPA), uma reorganização cromossômica se une a um novo gene – LPM – ao gene do receptor-alfa do ácido retinoico (RARA, do inglês retinoic acid receptor-a). Embora as propriedades funcionais exatas da molécula quimérica, LPM-RARA, sejam desconhecidas, essa translocação é subjacente à responsividade dramática desse tipo de leucemia ao tratamento com ácido transretinoico, que revolucionou os cuidados clínicos de pacientes com leucemia promielocítica aguda (LPA).21 Identificou-se também um novo produto da translocação cromossômica, o TEL-AML1, que é subjacente a uma forma comum e responsiva ao tratamento de leucemia linfoblástica aguda pediátrica.22
A capacidade de cultivar células leucêmicas em culturas suficientemente longas para permitir a realização de análises citogenéticas facilitou a caracterização de translocações cromossômicas nos casos de leucemia. Entretanto, translocações cromossômicas específicas também foram observadas em tumores sólidos. Entre os casos mais dignos de nota estão o sarcoma de Ewing e a família de tumores neuroectodérmicos primitivos (PNETs, do inglês primitive neuroectodermal tumors). Esses tumores que anteriormente eram distintos, atualmente são definidos por uma translocação cromossômica, fundindo o gene EWS a um número de fatores de transcrição da família do gene ETS (a proteína quimérica mais comum é a EWS-FLI1).23 Presume-se que esse produto quimérico tenha uma ação direta sobre os promotores-alvo para direcionar a expressão de genes indutores da transformação celular. A identificação das translocações do gene EWS possibilitou o agrupamento celular de uma classe de tumores cuja proliferação é acionada por alterações genéticas semelhantes que respondem a regimes quimioterápicos semelhantes.
Até muito recentemente, acreditava-se que as translocações cromossômicas fossem bastante incomuns nos carcinomas que ocorrem em adultos. Entretanto, uma série de estudos recentes demonstrou que a maioria dos carcinomas da próstata abriga translocações entre genes que codificam um ou dois fatores de transcrição relacionados ao gene ETS, ERG ou ETV1, e o gene TMPRSS2 regulado por androgênios.24 Como resultado dessas translocações, ERG ou ETV1 é expresso de uma forma dependente de androgênios, criando, consequentemente, um mecanismo tecidual específico de transformação celular. Embora as implicações terapêuticas dessa descoberta ainda não tenham sido definidas, a descoberta de que ETV1 é superexpresso por mecanismos independentes de androgênios em um subgrupo de casos25 sugere que esses últimos tumores podem ser resistentes às terapias antiandrogênicas comuns.
Anteriormente conhecidos por antioncogenes, os supressores tumorais formam uma família de genes celulares cuja desativação durante a tumorigênese promove o crescimento maligno [ver a Tabela 2]. Uma das descobertas iniciais que sugeriam a presença de genes supressores de tumores foi que a fusão de uma célula maligna a uma célula não maligna resultou em uma célula híbrida que acabou perdendo as propriedades malignas.26 Essa descoberta inesperada indicou que o estado maligno era recessivo, sugerindo que os genes das células não malignas poderiam recuperar o controle do crescimento normal de uma célula maligna que, presumidamente, havia perdido as informações genéticas.
O estudo dos vírus de tumores do DNA também propiciou uma visão importante dos genes supressores de tumores em seres humanos. Ao contrário dos retrovírus que ativaram versões dos proto-oncogenes celulares incorporados em seus próprios genomas, os vírus de tumores do DNA tais como o adenovírus, o vírus símio 40 (SV40, do inglês simian virus 40) e o papilomavírus codificam proteínas específicas de vírus com capacidade para transformar células de roedores, macacos e seres humanos. Uma descoberta importante sobre a etiologia do câncer foi que os genes transformadores derivados desses três vírus tumorais não relacionados têm a mesma função – ou seja, a desativação dos genes supressores de tumores celulares p53 e RB1.27 Consequentemente, o câncer surge do ganho de sinais proliferativos e da perda de genes que inibem a proliferação celular.
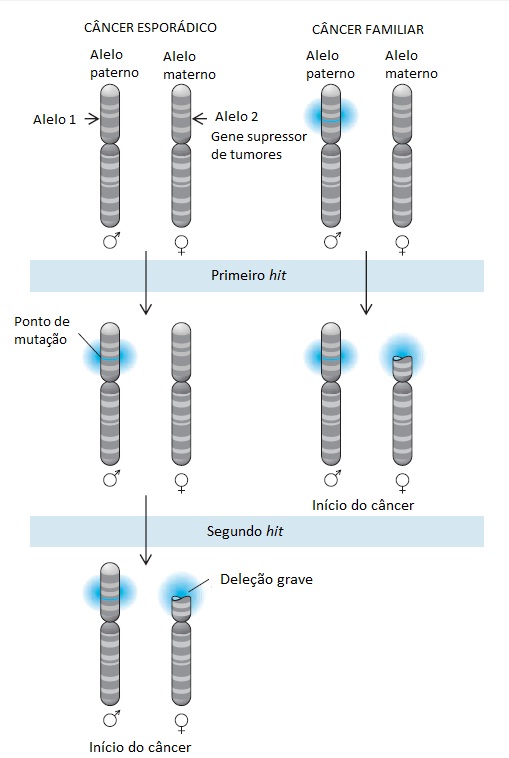
A incidência dos tipos mais comuns de câncer aumenta com o avanço da idade, provavelmente refletindo o acúmulo de danos genéticos somáticos nas células-tronco em processo de envelhecimento. Os cânceres pediátricos, embora sejam raros na população em geral, podem ocorrer com grande frequência em famílias com propensão para o câncer. Nesses casos, os tumores que surgem frequentemente são bilaterais ou multicêntricos e se desenvolvem mais cedo que os casos que surgem esporadicamente no seio da população em geral. Usando a distribuição de Poisson para calcular a probabilidade do desenvolvimento de câncer como uma função da idade em cânceres familiares versus cânceres pediátricos esporádicos, Alfred Knudson propôs o modelo que atualmente é a base da genética do câncer humano [ver a Figura 2].28
O modelo de Knudson prevê que as crianças com tumores familiares herdaram um hit genético inicial (i.e., um supressor de tumor mutante/não funcional) e precisam apenas de um hit genético adicional limitador de velocidade para iniciar a tumorigênese. Por outro lado, crianças com tumores esporádicos precisam adquirir dois hits genéticos independentes dentro da mesma célula, um evento pouco provável que explica a apresentação unilateral menos frequente e o início tardio de tipos esporádicos de câncer. Estudos genéticos subsequentes envolvendo dois dos tumores estudados por Knudson identificaram os genes supressores de tumores críticos dessas famílias: o gene RB1 em retinoblastomas29 e o gene WT1 no tumor de Wilms.30 Além disso, o modelo explica o paradoxo de que as mutações de genes supressores de tumores são mutações com perda funcional ou mutações recessivas [ver a Tabela 2], embora o câncer familiar se apresente como uma característica autossômica dominante. Embora a perda de um único alelo de um gene supressor de tumores seja funcionalmente silenciosa na presença de um segundo alelo normal, a frequência de mutações espontâneas é suficientemente elevada para assegurar que pelo menos uma célula no interior do tecido-alvo provavelmente perca o segundo alelo e inicie uma transformação maligna [ver a Figura 2]. Além disso, modelos sofisticados de animais e estudos genéticos em seres humanos revelaram que, em alguns casos, a perda de ambos os alelos do gene supressor de tumor não seja necessária para a transformação maligna. Em contextos celulares específicos e em combinação com outros eventos genéticos somáticos, a perda de um único alelo pode ser suficiente para desencadear a tumorigênese. O gene supressor de tumores com essa propriedade se denominam “haploinsuficientes” e incluem os genes p53 e PTEN.31
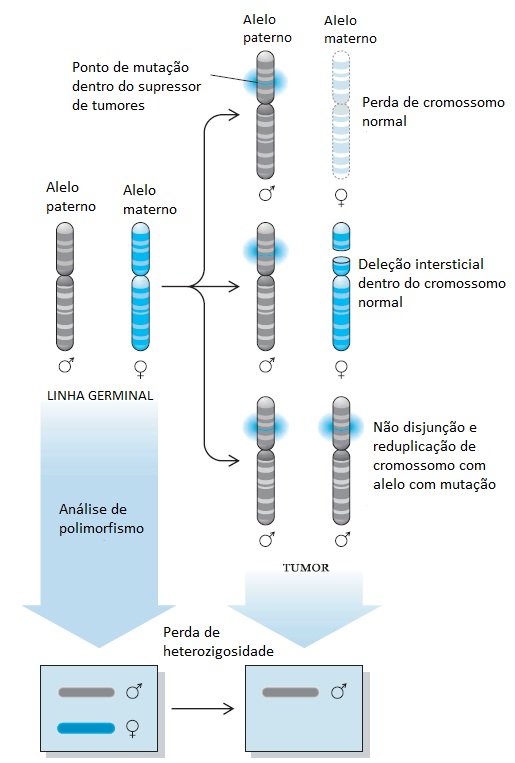
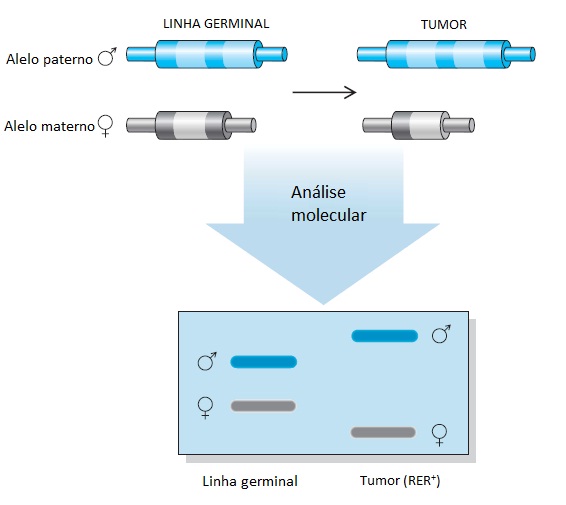
Embora a mutação de linha germinal inicial que desativa um alelo de um gene supressor de tumores seja tipicamente um ponto de mutação no interior do próprio gene, a perda do segundo alelo em uma célula somática geralmente resulta de uma deleção ou reorganização cromossômica básica.32 Como resultado, as análises moleculares dos tecidos tumorais mostram a perda de marcadores genéticos polimórficos intimamente ligados ao gene alvo. Esse fenômeno, conhecido por perda de heterozigosidade (PDH), é uma indicação da desativação do gene supressor de tumores mais próximo em um câncer [ver a Figura 3]. Essas perdas alélicas, que poderão ser mapeadas em todos os cromossomos localizados no interior de um determinado tumor, poderão ser usadas para identificar as localizações de genes supressores de tumores potencialmente novos.
O primeiro supressor tumoral identificado foi o gene RB1, que está envolvido nos retinoblastomas, que são tumores pediátricos nos olhos.29 De várias maneiras o gene RB1 continua sendo o protótipo dessa classe de genes, e sua estreita conexão com o mecanismo básico da progressão do ciclo celular ilustra a íntima conexão entre proliferação celular normal e transformação maligna. Conforme previa o modelo de Knudson,28 um alelo de RB1 é desativado na linha germinal de crianças com retinoblastoma familiar; seus tumores demonstram perda de heterozigosidade, indicando que há uma perda somática do alelo remanescente. As crianças que abrigam uma mutação na linha germinal em um alelo RB1 também têm propensão para osteossarcomas, embora, conforme observamos acima, elas não demonstrem qualquer aumento na suscetibilidade para outros tipos de câncer em que a perda somática tenha sido comprovada, como, por exemplo, o câncer de pulmão de pequenas células (CPPC). A despeito da importância desses casos familiares, as crianças com retinoblastoma representam 90% dos casos; nessas crianças ambos os alelos RB1 são desativados no interior de uma única célula somática.
A proteína codificada por RB1 é uma fosfoproteína nuclear que se liga aos produtos de uma família genética que se denomina E2F que, por sua vez, estão associados às proteínas da família de genes DP.33
|
Tabela 2 Genes Supressores de Tumores Selecionados, Associados a Síndromes Predisponentes na Linha Germinal e a Mutações de Câncer Somático
| |||
|
Gene |
Função do Produto Proteico |
Cânceres com Mutações Somáticas |
Síndrome de Mutação na Linha Germinal |
|
RB1 |
Regulador transcricional, ligação com E2F1 |
Retinoblastoma, osteossarcoma, CPPC, câncer de mama, próstata e bexiga.
|
Retinoblastoma familiar |
|
TP53 (p53) |
Fator de transcrição |
˜50% de todos os cânceres (raro em alguns tipos de câncer, p.ex., carcinoma da próstata, neuroblastoma).
|
Síndrome de Li-Fraumeni |
|
CDKN2A (P16) |
Inibidor da quinase dependente da ciclina. |
˜20 a 25% de muitos tipos diferentes de câncer (p.e., câncer de mama, pulmão, pâncreas, bexiga).
|
Melanoma familiar, carcinoma pancreático familiar. |
|
APC |
Regula a função da beta-catenina, ligação com microtúbulos. |
Mutações colorretais, raro ou ausente na maioria dos outros tipos de câncer. |
Polipose adenomatosa familiar, síndrome de Gardner, síndrome de Turcot, doença desmoide familiar.
|
|
MSH2, MLH1, MSH16, PM52 |
Reparo de DNA incompatível |
Colorretal, endométrico, gástrico. |
Câncer colorretal hereditário sem polipose.
|
|
WT1 |
Fator de transcrição |
Tumor de Wilms. |
WAGR e síndromes de Denys-Drash.
|
|
NF1 |
P21-RAS-GTPase, ligação com microtúbulos. |
Melanoma, neuroblastoma.
|
Neurofibromatose tipo 1 |
|
NF2 |
Ligação da justamembrana com o citoesqueleto. |
Schwanomas, meningiomas, ependimomas.
|
Neurofibromatose tipo 2 |
|
VHL |
Regulador da estabilidade proteica. |
Hemangioblastoma (tipo células claras) renal. |
Doença de von Hippel-Lindau.
|
|
Tabela 2 Genes Supressores de Tumores Selecionados, Associados a Síndromes Predisponentes na Linha Germinal e a Mutações de Câncer Somático
| |||
|
Gene |
Função do Produto Proteico |
Cânceres com Mutações Somáticas |
Síndrome de Mutação na Linha Germinal |
|
BRCA1, BRCA2 |
Reparo de DNA, complexo com Rad51, regulação transcricional (BRCA1,).
|
Raro |
Carcinoma hereditário de mama e ovário, pancreático. |
|
MEN1 |
Desconhecida |
Adenomas paratiroideos, adenomas hipofisários, tumores endócrinos no pâncreas.
|
Neoplasia endócrina múltipla tipo 1. |
|
PTCH |
Receptor transmembranar para a via de sinalização hedgehog sônica; regulador negativo de proteína smoothened.
|
Pele de células basais, meduloblastoma. |
Síndrome de nevo de células basais. |
|
PTEN |
Tirosina fosfatase |
Glioma; câncer de mama, próstata, cabeça e pescoço; câncer de células escamosas; câncer tireóideo folicular.
|
Doença de Cowden, síndrome tumoral com vários hamartomas. |
|
STK11 |
LKB1 quinase que regula o crescimento e a polaridade das células. |
Carcinoma pulmonar, carcinoma de células escamosas.
|
Síndrome de Peutz-Jeghers. |
|
TSC1, TSC2 |
Complexo de proteínas ativadoras da GTPase.
|
Raro |
Esclerose tuberosa. |
|
CDH1 |
Molécula de adesão celular mediada pela transmembrana E-caderina. |
Câncer gástrico difuso e câncer de mama lobular; mutações raras em outros tipos de câncer (p.ex., câncer endometrial e ovariano).
|
Câncer gástrico difuso hereditário. |
|
DPC4 |
Sinalização no sentido do fluxo na rota de TGF-ß. |
Mutações pancreáticas raras em outros tipos de câncer (p.ex., câncer no cólon e câncer gástrico).
|
Desconhecida |
|
IDH1, IDH2 |
Conversão de intermediários do ciclo de Krebs.
|
Glioma, LMA. |
Desconhecida |
|
Tabela 2 Genes Supressores de Tumores Selecionados, Associados a Síndromes Predisponentes na Linha Germinal e a Mutações de Câncer Somático
| |||
|
Gene |
Função do Produto Proteico |
Cânceres com Mutações Somáticas |
Síndrome de Mutação na Linha Germinal |
|
TGF-ß II R |
TGF-ß do receptor transmembranar. |
Câncer colorretal e gástrico, 7 outros tipos de câncer.
|
Desconhecida |
LMA = leucemia mielocítica aguda; GTPase = guanosina trifosfatase; CPPC = câncer de pulmão de pequenas células; TGF-ß (transformating growth factor-beta) = fator de crescimento transformador ß; WAGR (Wilm’s tumor, aniridia, genitourinary anomalies, and mental retardation) = tumor de Wilms, aniridia, malformações geniturinárias e deficiência intelectual.
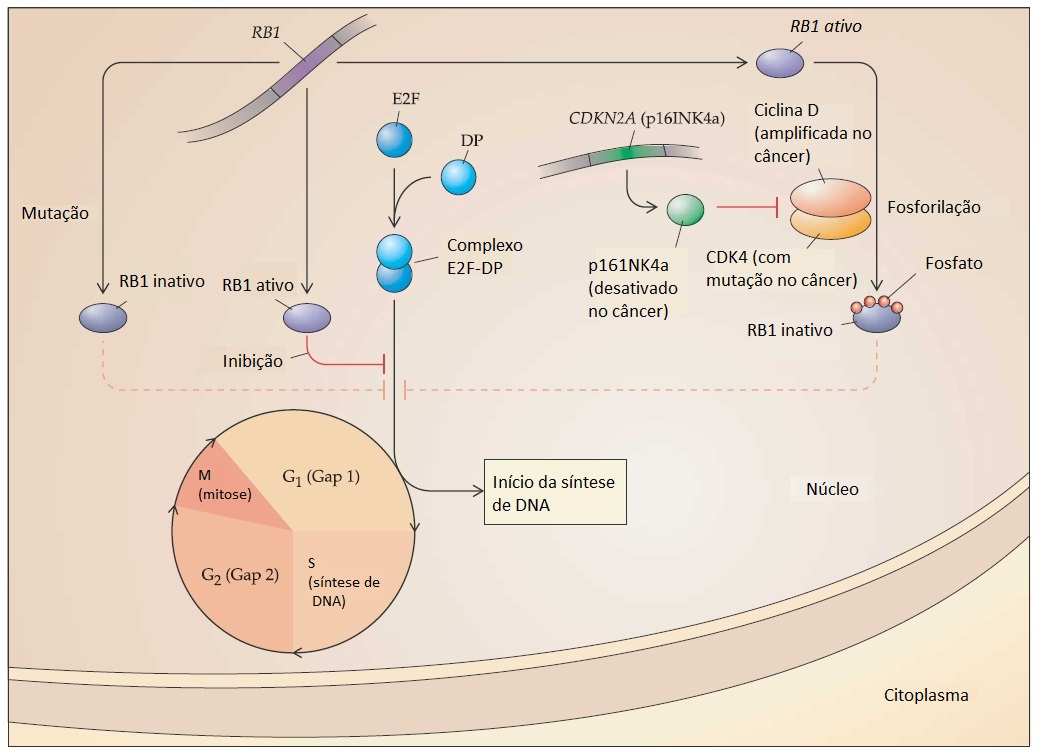
Os complexos E2F-DP desempenham um papel importante na ativação da transcrição dos genes exigidos na síntese do DNA; a ligação a RB1 suprime esse efeito, essencialmente bloqueando células na fase G1 do ciclo celular. Durante a proliferação celular normal, os complexos quinase regulados pelo ciclo celular, incluindo um que contém ciclina D e quinase 4 dependente da ciclina (CDK4, do inglês cyclin dependent kinase 4), fosforilatos RB1, provocam sua liberação de E2F-DP; este fato, por sua vez, permite que as células entrem na fase (S) da síntese do DNA do ciclo celular. Embora a desativação fisiológica de RB1 através de hiperfosforilação seja transitória e reversível, a desativação mutacional de RB1, comum a muitos tipos diferentes de tumor, assegura um estímulo contínuo para a proliferação celular. Da mesma forma, os vírus oncogênicos do DNA codificam proteínas que desativam, especifica e irreversivelmente, o produto do gene RB1, incluindo a proteína E1A de adenovírus, o antígeno T de SV40 e a proteína E7 de papilomavírus. O próprio gene RB1 é um membro de uma família de genes, sendo que dois deles são genes intimamente relacionados denominados RBL1 (p107) e RBL2 (p130). Ainda não se conhece a razão pela qual RB1 é o alvo frequente de mutações nos cânceres humanos, enquanto esses genes semelhantes não sofrem nenhuma mutação. Uma melhor compreensão das diferenças sutis na função fisiológica dos membros dessa família de genes possivelmente explique essa especificidade impressionante.
Figura 2 - Modelo two-hit de Knudson do início tumoral. De acordo com a formulação original, esse modelo prevê a desativação de ambos os alelos de um gene supressor de tumores e limita a velocidade da fase inicial do câncer. No caso de câncer esporádico, a desativação desses dois alelos no interior da mesma célula depende de dois eventos genéticos raros e independentes. Por outro lado, as pessoas que têm um alelo do gene supressor de tumores com mutação em sua linha germinal, herdado de um dos pais ou como resultado de nova mutação na linha germinal, precisa apenas de um evento genético (i.e., perda do segundo alelo) para o início tumoral. Essa única mutação possivelmente ocorra no interior de alguma célula do órgão-alvo, o que explica a alta frequência da incidência de câncer, o início precoce e a frequente apresentação multicêntrica em pessoas com predisposição genética para o câncer.
Figura 3 - Perdas de alelos em tumores e perda de heterozigosidade (PDH). A perda de material genético que acompanha o desenvolvimento tumoral foi utilizada para mapear a localização cromossômica dos genes de supressão de tumores. Embora, tipicamente, o hit genético inicial seja o resultado de um ponto de mutação no interior do gene supressor de tumores, o evento de desativação do segundo gene geralmente resulta de uma perda cromossômica grave. Esse processo poderá incluir o cromossomo normal inteiro, uma pequena deleção que remove um segmento cromossômico que contém o gene, ou uma duplicação do cromossomo que carrega o alelo mutante, com a perda do cromossomo normal. Esses eventos cromossômicos podem ser mapeados com auxílio de polimorfismos do comprimento do fragmento da restrição, polimorfismos com um único nucleotídeo, ou outros métodos moleculares que fazem a distinção entre os alelos herdados da mãe e os alelos herdados do pai. A identificação do lócus cromossômico sujeito à perda de heterozigosidade em tipos específicos de câncer é o ponto de partida para a identificação dos genes com predisposição para câncer.
A compreensão da regulação normal de RB1 propiciou uma visão importante de outros genes cancerígenos que desempenham algum tipo de papel na regulação do ciclo celular [ver a Figura 4]. O CCND1 (ciclina D1), gene cuja expressão leva à hiperfosforilação e desativação de RB1, geralmente é amplificado e superexpresso no câncer de mama. Por outro lado, o CDKN2A, gene que codifica o inibidor p16INK4a de CDK4 que favorece a forma ativa e hipofosforilada de RB1, é deletado em uma grande variedade de tumores. As mutações de CDKN2A na linha germinal são umas das causas de melanoma familiar.34 Para finalizar, a presença do gene CDK4 dependente da quinase, que codifica a proteína insensível à inibição de p16INK4a, favorecendo a desativação de RB1, também foi confirmada em cânceres humanos e em famílias de melanomas. Uma das características marcantes de mutações nesses componentes diferentes da rota do ciclo celular é que, aparentemente, eles são mutuamente exclusivos.
Figura 4 - Rota do ciclo celular do gene do retinoblastoma (RB1). A divisão celular das células de mamíferos é desencadeada por um ponto de controle crítico entre a fase G1 e a síntese de DNA (fase S). O complexo proteico E2F-DP, que ativa os genes necessários para a síntese de DNA, é inibido diretamente pelo produto do gene supressor de tumores RB1. A progressão normal do ciclo celular exige a desativação reversível da proteína RB1 por fosforilação induzida pela ciclina D e CDK4. As mutações de RB1 impedem a inibição normal da progressão do ciclo celular, contribuindo para a desregulação da divisão de células. A amplificação da ciclina D, ativando mutações em CDK4, e a desativação do inibidor p16INK4a do CDK4 (codificado por CDKN2a) são outros mecanismos por meio dos quais a rota de RB1 é interrompida nos cânceres humanos.
De maneira geral, um único tumor apresenta alguma anormalidade em apenas um desses genes – evidência de que essas mutações são funcionalmente equivalentes e que o acúmulo de mutações complementares em componentes da mesma rota não produz nenhuma vantagem adicional no crescimento.35
Embora o gene supressor de tumores RB1 seja um componente central da regulação do ciclo celular, o gene p53 tem um papel importante na manutenção da integridade genômica – que deu origem à designação popular de “guardião de genoma”.36 O gene p53 geralmente é expresso em baixos níveis em todas as células. Entretanto, lesões genéticas, como aquelas que ocorrem através de radiação ionizante, disparam a estabilização e a ativação de p53.
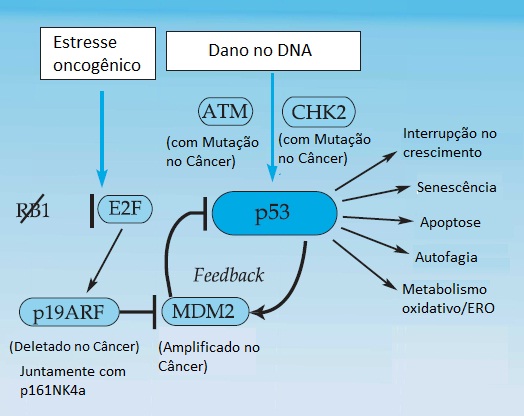
Figura 5 - Rota da resposta dos danos ao DNA e ao estresse celular de p53. A proteína p53 é ativada em resposta aos danos no DNA, ao estresse oncogênico (p.ex., desativação de RB1) e a outros estresses celulares. A proteína p53 induz a transcrição de uma grande variedade de genes que fazem a mediação de suas diversas funções, incluindo a interrupção do ciclo celular da fase G1 e a morte celular programada. Em tumores com mutações de p53, o reparo nos danos no DNA não é adequado, levando a um acúmulo de mutações e de reorganizações genéticas. Dois genes celulares importantes regulam a ativação de p53: MDM2 e p19ARF. O gene MDM2 induz a degradação normal da proteína p53 e, em alguns tumores, a amplificação de MDM2 resulta na perda de função de p53. O gene p19ARF, que é induzido por E2F, inibe os efeitos de MDM2 e contribui para a ativação de p53 após a desativação de RB1. A proteína p19ARF geralmente se perde nos cânceres humanos juntamente com p161NK4a, considerando que essas proteínas são expressas a partir do sítio genético de CDKN2a. ERO = espécies reativas de oxigênio.
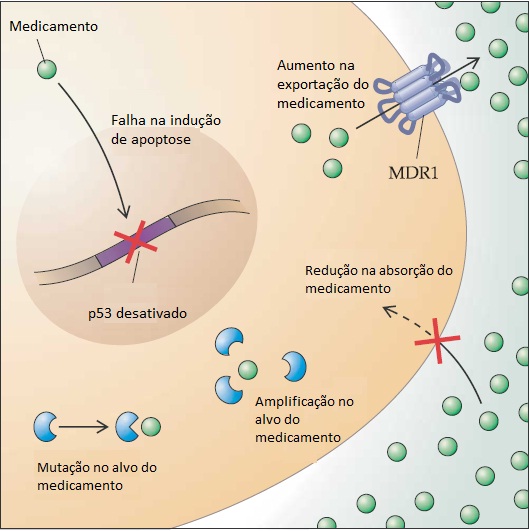
As funções da proteína p53 como fator de transcrição direcionam a expressão de uma grande variedade de genes que controlam a resposta celular aos danos no DNA e a outros estresses celulares [ver a Figura 5]. Entre os genes ativados por p53, o CDKN1A (p21), que codifica um inibidor de quinases que depende da ciclina, é o gene que regula o ciclo celular.37 A ativação de p53 provoca uma interrupção na fase G1 do ciclo celular, permitindo que as células façam o reparo nos danos do DNA antes de passarem para a fase S e da replicação do DNA. Nas outras células, a ativação de p53 resulta na ativação de múltiplos efetores, causando apoptose – um programa de suicídio celular cujo DNA possivelmente tenha sido irreparavelmente danificado. Não é de causar nenhuma surpresa que as mutações de p53 sejam comuns nos cânceres em seres humanos, fato que é demonstrado em torno de 50% de casos.38 Ao contrário das maioria dos supressores tumorais que estão sujeitos principalmente a mutações truncadas (mutações sem sentido), grande parte das mutações de p53 são substituições de aminoácidos dentro do domínio de p53 que faz ligação com o DNA. Essas alterações resultam na estabilização e na consequente expressão de alto nível da proteína mutante p53, explicando o paradoxo de que níveis elevados da proteína p53 em espécimes tumorais geralmente são considerados evidências da ocorrência de uma mutação em p53. Acredita-se que a proteína mutante p53 propriamente dita contribua para a tumorigênese através do rompimento da função de p53 do tipo selvagem (efeito conhecido como negativo-dominante) e através de outros mecanismos que ainda se encontram atualmente em fase de investigação.
Em alguns casos, as constelações de mutações de p53 em tumores possivelmente reflitam o mecanismo presumido de carcinogênese. Por exemplo, mutações específicas de p53 em cânceres hepáticos foram encontradas em populações que vivem em uma região da China onde há exposição a níveis elevados de aflatoxina, um carcinógeno hepático fúngico muito potente, sendo que as mutações que foram observadas no câncer no fígado estão correlacionadas com aquelas induzidas pelos carcinógenos policíclicos do tabaco. Por outro lado, as mutações de p53 deletadas nos carcinomas colorretais frequentemente são observadas em nucleotídeos suscetíveis a metilação que, aparentemente, é um mecanismo comum das mutações espontâneas.39
A mutação de um alelo na linha germinal de p53 é responsável pelo fenótipo de múltiplos cânceres conhecido por síndrome de Li-Fraumeni.40 As famílias afetadas por essa rara característica autossômica dominante demonstram uma predisposição altamente penetrante para tipos distintos de câncer, incluindo sarcomas, câncer de mama, tumores cerebrais e leucemia. Acredita-se que o mecanismo através do qual a desativação da função de p53 leva ao desenvolvimento de câncer seja a perda de um ponto de verificação que monitora a integridade do material genético antes da replicação do DNA. A proteína p53 também desempenha um papel importante no desencadeamento do suicídio celular, em resposta a sinais de crescimento inadequado, como aqueles induzidos pela perda de outros supressores tumorais ou pela ativação de oncogenes. Consequentemente, na ausência de p53, os danos genéticos persistem e, em última análise, induzem a tumorigênese. Em alguns tipos de câncer, como aqueles que surgem como consequência da síndrome de Li-Fraumeni, a perda de p53 inicia a tumorigênese; em outros, como o câncer colorretal, a desativação de p53 é um evento genético tardio importante para a progressão do fenótipo maligno.
Da mesma forma como o R1, o p53 é alvo específico de produtos de oncogenes virais, incluindo a proteína E1B do adenovírus, o antígeno T de SV40 e a proteína E6 do papilomavírus. Consequentemente, os vírus tumorais de DNA agem com uma capacidade dupla: induzem a proliferação celular através da desativação de RB1 enquanto interrompe a capacidade de p53 para desencadear a morte de células em resposta a esse sinal proliferativo aberrante.41 Observou-se que as proteínas celulares também regulam a função de p53. Entre elas, a proteína MDM2, induzida por p53 e ela própria intensifica a degradação de p53, produzindo um ciclo de realimentação negativo crítico [ver a Figura 5]. O fato de p53 e MDM2 fazerem parte de uma rota celular comum tem o suporte de alterações mutuamente exclusivas nos cânceres em seres humanos: os osteossarcomas resultam de uma mutação que desativa o p53 ou de uma amplificação do gene MDM2, levando a uma superprodução do produto genético.42 Os camundongos sem ambos os alelos de MDM2 morrem logo no início do desenvolvimento embrionário, a menos que não tenham também p53, sendo que, nessa hipótese, eles se desenvolvem normalmente para a vida adulta – evidência de que MDM2 é importante para impedir atividade sem oposição de p53 durante o crescimento normal.43
Muitas proteínas que regulam a função de p53 têm envolvimento no câncer humano [ver a Figura 5]. A ativação de p53 em resposta à radiação ionizante ocorre através da ATM (ataxia-telangiectasia mutante) quinase em resposta ao dano no DNA e as respectivas quinases no sentido do fluxo, CHK1 e CHK2.44 Aparentemente, a fosforilação de p53 reduz a ligação pelo MDM2 e aumenta a estabilidade da proteína p53 e intensifica sua atividade. Um segundo regulador importante de p53 é o CDKN2A, um gene atípico que codifica os produtos de duas proteínas distintas, p19ARF e p16INK4a.45 A proteína p19ARF interage com MDM2 para regular a renovação de p53; o seu papel no câncer humano é sugerido por tumores com delações genômicas frequentes que desativam p19ARF e p16INK4a. Acredita-se que a ativação de p53 através da rota de p19ARF seja disparada principalmente por sinais aberrantes de proliferação associados com a indução de E2F, um regulador transcricional de p19ARF.46 Consequentemente, os danos no DNA e os sinais aberrantes de proliferação podem ativar p53 através de rotas distintas, porém complementares. Cabe observar que dois membros da família do gene p53 que passaram despercebidos por muito tempo, TP73 (p73) e TP63 (p63), compartilham algumas das mesmas propriedades funcionais do p53 in vitro, embora as respectivas participações no câncer humano ainda estejam em fase de investigação.47,48
Embora o p53 seja o gene supressor de tumores estudado com mais intensidade no processo de estabilidade genômica, aparentemente os genes associados a doenças adicionais funcionam em rotas semelhantes ou paralelas. O gene da ataxia-telangiectasia mutante (ATM, do inglês ataxia telangiectasia mutated) é responsável por uma síndrome autossômica recessiva que se caracteriza pela presença de condições como degeneração cerebelar, disfunção imunológica e predisposição para o câncer; conforme observamos acima, esse tipo de gene funciona no contrafluxo da rota de p53 e é muito importante na ativação de p53 após a radiação ionizante.49,50 Dois outros genes que têm função importante na estabilidade genômica – BRCA1 e BRCA2 – são responsáveis por uma fração substancial do câncer de mama e de ovário familiar de início precoce.51,52 Os produtos desses genes agem em parte para mediar um mecanismo de reparo de DNA conhecido por recombinação homóloga.53 Os tumores que surgem nos transportadores de mutações de BRCA1 e BRCA2 apresentam defeitos no reparo de DNA, assim como características clínicas e respostas distintas à terapia, em comparação com os cânceres esporádicos de mama e de ovário mais comuns. Recentemente, alguns estudos bioquímicos descobriram a existência de uma ligação funcional entre os genes BRCA1/2 e a anemia de Fanconi (AF). Essa síndrome de predisposição para o câncer resulta de mutações na linha germinal em um dos vários genes da anemia de Fanconi, que codifica proteínas de reparo que atuam em rotas sobrepostas aos genes BRCA1/2.54
Nos casos de síndrome de Lynch ou de câncer colorretal hereditário sem polipose (CCHSP), uma rota distinta para reparos de DNA está sujeita a mutações na linha germinal associadas à incidência de câncer. Essa síndrome, que inclui predisposição para cânceres no cólon, ovário e endométrio, é o resultado de mutações nos genes, tais como MSH2 e MLH1, que são imprescindíveis para os reparos de incompatibilidades de DNA.55
Figura 6 - Instabilidade de microssatélites e reparo de incompatibilidades de DNA. Microssatélites são sequências curtas repetidas de DNA que se distribuem ao longo do genoma e são altamente variáveis, facilitando a distinção entre alelos de herança materna e alelos de herança paterna. Ao contrário dos tumores com perda de heterozigosidade, que têm perda de um alelo em comparação com a linha germinal, os tumores com instabilidade de microssatélites (erro positivo de replicação [RER+, do inglês replication error positive) mostram marcadores de microssatélites com alteração no comprimento. Isso resulta de erros no reparo das incompatibilidades de DNA. Na presença de sequências de microssatélites dentro da região de codificação de um gene, esses erros produzem mutações e perda de função genética.
Uma das marcas registradas dos tumores com defeitos no reparo de incompatibilidades de DNA denomina-se instabilidade de microssatélites (MSI, do inglês microsatellite instability) [ver a Figura 6]. Os microssatélites são prolongamentos de nucleotídeos repetitivos cujos comprimentos variam entre indivíduos. Os tumores com MSI possivelmente tenham microssatélites com tamanhos diferentes em comparação com os microssatélites de células normais de um mesmo paciente.56 Levando-se em consideração que as variações de microssatélites resultam de erros introduzidos durante a replicação de DNA, esse tipo de tumor é conhecido por erro positivo de replicação (RER+ do inglês replication error positive). Além dos tumores associados ao câncer colorretal hereditário sem polipose (CCHSP), aproximadamente 10% dos cânceres colorretais esporádicos apresentam instabilidade de microssatélites, indicando que o rompimento de algum reparo de incompatibilidades também contribui para a incidência de câncer colorretal na ausência de predisposição familiar.57 Embora a MSI seja um marcador de deficiência em reparos de incompatibilidades, as alterações nos microssatélites propriamente ditas não são oncogênicas. Em vez disso, acredita-se que a deficiência em reparos de incompatibilidades produza mutações em outros genes associados às principais rotas de sinalização celular. Nos casos de câncer no cólon, por exemplo, a MSI possivelmente esteja associada a mutações que ocorrem no interior do gene receptor do fator de crescimento transformador ß (TGF-ß, do inglês transformating growth factor-ß), que abriga uma repetição de microssatélites dentro da sequência de codificação.58 A mutação dessa sequência, resultante de problemas nos reparos de incompatibilidades, induz a interrupção na sinalização do TGF-ß, um evento precoce importante na carcinogênese do cólon. Uma caraterística importante que faz a distinção entre defeitos nos reparos de incompatibilidades e outros mecanismos supressores de tumores é que os defeitos nos reparos de incompatibilidades produzem tumores indiretamente através de mutações em outros genes. Embora a expectativa seja que a reintrodução de p53 em tumores com deficiência de p53 induza uma interrupção no crescimento e/ou apoptose, teoricamente a recuperação do gene com reparo de incompatibilidades não tenha nenhum valor terapêutico. Provavelmente esse fenômeno ocorra muito tarde e não permita reverter o fenótipo maligno. Os genes supressores de tumores geralmente são descritos como “cuidadores” de genoma em vez de “guardiães” de rotas específicas que estimulam a proliferação de células cancerígenas.57
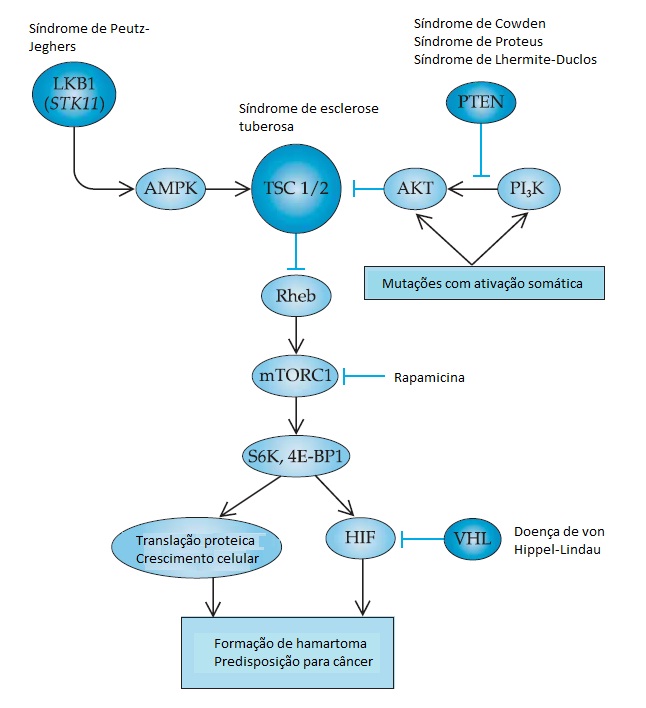
A identificação de genes supressores de tumores envolvidos nas síndromes de predisposição para o câncer criou visões importantes nos mecanismos de sinalização celular que regulam o metabolismo das células. Por outro lado, a compreensão detalhada desses mecanismos revelou que vários genes de supressão tumoral agem dentro de uma rota reguladora comum. Um exemplo típico desse princípio é a rota que controla a atividade de uma quinase conhecida como alvo da rapamicina em mamíferos (mTOR, do inglês mammalian target of rapamycin) [ver a Figura 7]. A quinase mTOR, que está presente em diversos complexos de proteínas distintas, é uma integradora mestre de sinais de fatores de crescimento e uma reguladora do crescimento e da proliferação celular através do controle da translação proteica, autofagia (autocatabolismo) e angiogênese. Diversos genes envolvidos na regulação da atividade do complexo da quinase mTOR 1 (mTORC1) em células normais são mutantes nas síndromes humanas de predisposição tumoral [ver a Figura 7].59
Figura 7 - Oncogenes múltiplos e função de supressores tumorais em uma rota molecular comum que converge na regulação do complexo da quinase 1 alvo da rapamicina em mamíferos (mTORC1). O gene STK11 sofre uma mutação na síndrome de Peutz-Jeghers e codifica a quinase LKB1 que atua através da quinase AMPK e do complexo TSC 1/2 como inibidor de mTORC1. O gene PTEN – que sofre mutações em diversas síndromes de predisposição tumoral, incluindo a síndrome de Cowden – codifica uma fosfatase que bloqueia a ativação de mTORC1 através da inibição da rota da quinase AKT/PI3. Essa rota é também ativada em tumores por meio da mutação somática de genes que codificam as próprias quinases AKT e PI3. Os genes que codificam as proteínas inibidoras de mTORC1 – TSC1 e TSC2 – sofrem mutações na síndrome de esclerose tuberosa. O complexo mTORC1 promove a translação proteica e o crescimento celular por meio da fosforilação de substratos, incluindo as proteínas quinases S6 ribossômicas (S6K) e a proteína de ligação eIF-4E do fator de início da translação (4E-BP1). A ativação de mTORC1 suprarregula o fator induzível por hipoxia (HIF, do inglês hypoxia-inducible fator), que também é suprarregulado pela mutação do supressor tumoral de von Hippel-Lindau (VHL). Todas essas síndromes compartilham uma predisposição para hamartomas benignos e uma incidência variável de tumores malignos. Análogos da rapamicina, um inibidor de pequenas moléculas de mTORC1, encontram-se atualmente em fase de testes clínicos para uma grande variedade de tumores benignos e malignos. Os genes de supressão tumoral estão indicados na cor azul escuro na figura.
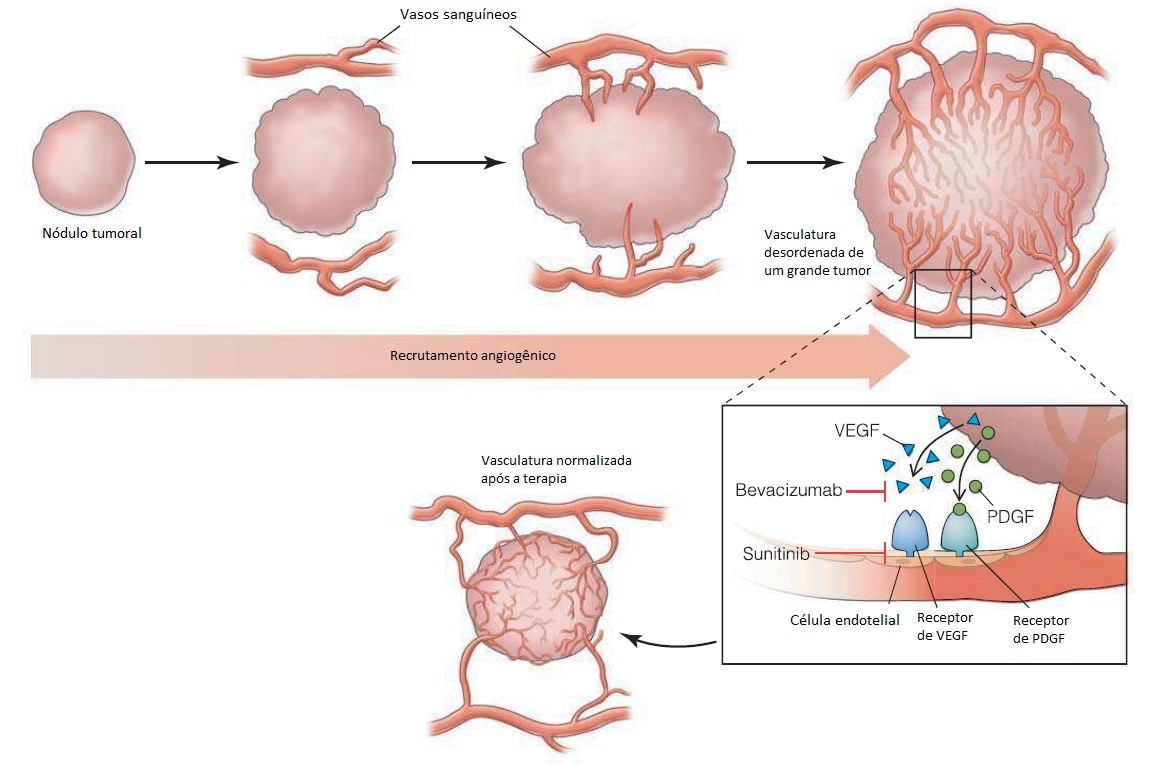
A mutação do gene PTEN na linha germinal está associada a diversas síndromes, como, por exemplo, a doença de Cowden, que se caracteriza pela presença de tumores benignos conhecidos como hamartomas e pela predisposição para câncer de mama.60 Mutações no gene PTEN também são encontradas em muitos tipos diferentes de câncer esporádico. O gene PTEN codifica um tipo de fosfatase que catalisa a remoção de moléculas de fosfato dos lipídeos principais (fosfoinositídeos), regulando, consequentemente, de forma negativa a rota da fosfoinositídeo 3-quinase (PI3K, do inglês phosphoinositide 3- kinase) que é muito importante para a ativação de mTORC1. A mutação do gene STK11 (LKB1) na linha germinal é uma das causas da síndrome de Peutz-Heghers, que está associada aos hamartomas intestinais e aos tumores malignos no intestino, pâncreas e em outros sítios.61 A quinase LKB1 codificada por STK11 ativa a quinase detectora de energia AMPK, que age como reguladora negativa de mTORC1. A síndrome da esclerose tuberosa é o resultado de mutações em TSC1 (hamartina) ou TSC2 (tuberina). Embora não tenha sido associada a um risco elevado de incidência de tumores malignos, a esclerose tuberosa está relacionada a uma taxa elevada de morbidade em decorrência de hamartomas no sistema nervoso central, nos rins e em outros tecidos.62 As proteínas de TSC1 e TSC2 formam um complexo que atua como um guardião essencial para restringir a atividade de mTORC1. Outro supressor tumoral envolvido nessa rota é o gene de von Hippel-Lindau (VHL) que, com frequência, sofre mutações nos cânceres de células renais adultas e na linha germinal de pessoas com alguma síndrome que inclua tumores vasculares benignos e malignos. Embora não seja um regulador no contrafluxo do mTORC1, o produto do gene VHL controla a degradação de determinadas proteínas, incluindo o fator induzível por hipoxia (HIF, do inglês hypoxia-inducible fator), um regulador importante da angiogênese.63 Levando-se também em consideração a regulação positiva do HIF pelo mTORC1, as síndromes mencionadas acima compartilham algumas semelhanças fenotípicas com a doença de von Hippel-Lindau; a atividade do HIF é elevada, levando a tumores altamente vasculares. Talvez o fato mais importante seja a previsão de que os tumores benignos e malignos que abrigam uma quantidade elevada de atividade mTOR/ HIF possam ser responsivos aos inibidores da atividade de mTOR, em especial os derivados de compostos da rapamicina, que atualmente estão em fase de testes clínicos.
Diversas outras síndromes de supressão tumoral estão associadas a tumores malignos, tumores benignos (hamartomas) ou a ambos os tipos de tumor e, consequentemente, apresentam semelhanças fenotípicas com as síndromes descritas acima, embora ainda não tenha sido comprovada a existência de alguma ligação direta com a sinalização de mTOR. Essas outras síndromes incluem polipose adenomatosa familiar (PAF), síndrome que se caracteriza pelo desenvolvimento de inúmeros pólipos colônicos e por um risco muito elevado de transformações malignas. As mutações no gene APC na linha germinal produzem esta síndrome, enquanto que as mutações somáticas no gene APC são a etapa mais precoce do desenvolvimento de câncer colorretal esporádico. O gene APC é um regulador importante na rota de sinalização de WNT através dos efeitos sobre a catenina-ß, um cofator importante de transcrição que atua por meio da mediação da família de LEF/TCF de fatores de transcrição.64 A alteração no gene SMAD4 está associada à polipose juvenil e às malignidades gastrointestinais.65 As proteínas SMAD atuam fazendo a mediação da sinalização de TGF-ß. Outros genes de supressão tumoral ligados às rotas de sinalização celular são o gene NF1, que é uma das causas de neurofibromatose (doença de von Recklinghausen) e cujo produto gênico infrarregula o proto-oncogene RAS66; o gene NF2, que sofre frequentes mutações em mesoteliomas e schwanomas e que codifica uma proteína estrutural que possivelmente esteja envolvida na aderência e na proliferação celular67; e o gene PTCH que codifica um receptor para o fator de crescimento que se denomina hedgehog, cuja mutação representa um alto risco de câncer de pele na síndrome do nevo de células basais.68
Algumas das rotas descritas acima, incluindo WNT, TGF-ß, hedgehog e outras, contribuem para os processos evolutivos normais. O gene WT1 é o paradigma de um gene supressor de tumores envolvido no desenvolvimento normal, que codifica um regulador de transcrição expresso especificamente em podócitos e no glomérulo em desenvolvimento. As mutações em WT1 produzem o tumor de Wilms, um tipo de câncer embrionário nos rins; as mutações na linha germinal causam defeitos no desenvolvimento genitourinário. Aparentemente, os genes que são normalmente regulados pelo produto gênico WT1 estão envolvidos na proliferação e na diferenciação.69 Foi identificado um segundo gene associado ao tumor de Wilms que se localiza no cromossomo X.70 Esse gene, conhecido por WTX, aparentemente controla a sinalização da catenina-ß/WNT, uma descoberta que evidencia a conservação de rotas evolutivas lingadas à tumorigênese.71
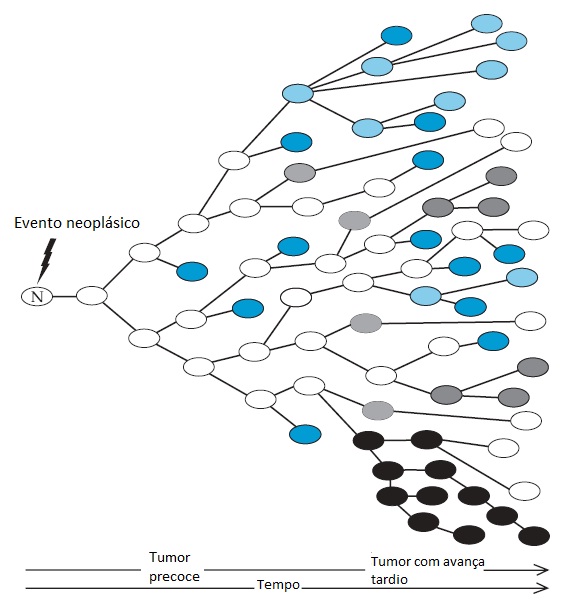
A presença de uma lesão genética no interior de um único gene limitante de velocidade é imprescindível para iniciar uma transformação maligna. Entretanto, a progressão do fenótipo maligno depende da aquisição de mutações adicionais que confiram uma vantagem complementar de crescimento e, consequentemente, sejam selecionadas durante a expansão do clone maligno [ver a Figura 8]. Um determinado tipo de gene poderá ser limitante de velocidade para transformação em um tipo de célula, embora possa ter uma participação secundária em outro tipo. O câncer colorretal é o modelo de progressão tumoral estudado com mais intensidade, para o qual Kinzler e Vogelstein estabeleceram uma correlação entre a progressão histológica de pólipo para carcinoma e o acúmulo de eventos genéticos.72
Figura 8 - Após o início de um evento genético neoplásico e limitante de velocidade, a proliferação de células cancerígenas é estimulada por vantagens seletivas de crescimento que são confirmadas por mutações cumulativas. A divisão celular constante e a perda gênica exigida para manter a estabilidade genômica facilitam a ocorrência de mutações. Propriedades como perda de morte celular, invasão, angiogênese e resistência aos medicamentos tipificam a progressão fenotípica de células tumorais. As cores indicam os descendentes clonais de células individuais.
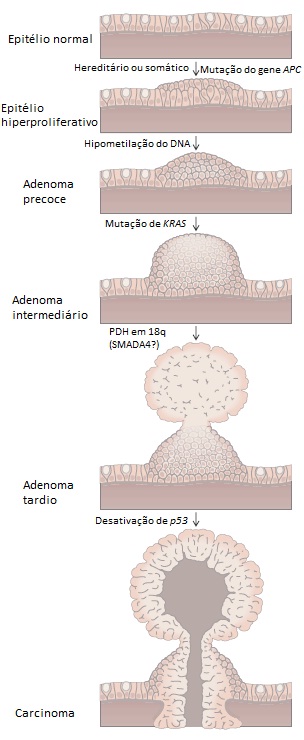
A mutação que desativa o gene APC está associada ao desenvolvimento de hiperplasia epitelial. As mutações que ativam KRAS e alteram a metilação do DNA se correlacionam com a progressão para pólipos adenomatosos; as alterações em um ou mais genes que se localizam no cromossomo 18q denotam a transição para adenomas de alto grau; e, finalmente, a desativação de p53 acompanha a evolução para carcinoma maligno [ver a Figura 9]. As lesões pré-neoplásicas que produzem câncer colorretal são definidas imediatamente porque ocorrem no interior da mucosa colônica e são acessíveis para coleta de biópsias colonoscópicas. Outros modelos de progressão tumoral estão em fase de estudos e incluem cânceres no esôfago, na cabeça e pescoço, e na bexiga.
Além dos eventos genéticos, a progressão tumoral é também acompanhada de alterações dramáticas no perfil epigenético das células cancerígenas em seres humanos. As alterações epigenéticas se referem às alterações cromossômicas não hereditárias que influenciam a expressão gênica. Acredita-se que os dois tipos de modificações que foram identificados tenham uma participação significativa no processo de progressão tumoral. Essas modificações envolvem a metilação do próprio DNA e a metilação ou acetilação das proteínas que compactam o DNA conhecidas por histonas.73 A metilação do DNA ocorre no interior de regiões reguladoras de muitos genes nos sítios denominados ilhas de CpG, que são sequências de DNA enriquecidas pelo dinucleotídeo citosina/guanina, que é o alvo das enzimas DNA metiltransferases. Nas células normais, essas regiões são metiladas com pouca frequência, levando a uma expressão gênica ativa. Entretanto, nas células cancerígenas, as ilhas de CpG de muitos genes são hipermetiladas, resultando no silenciamento de genes específicos, incluindo genes supressores de tumores bem estabelecidos como os genes CDKN2A, MLH1 e muitos outros.73 Paradoxalmente, apesar de terem aumentado a metilação de ilhas de CpG específicas, as células tumorais se apresentam com hipermetilação global, que se torna mais pronunciada durante a progressão, como, por exemplo, de pólipos colônicos para carcinoma [ver a Figura 9].
Figura 9 - Alterações genéticas durante a progressão de câncer colorretal. Este modelo, proposto por Bert Vogelstein, geralmente conhecido por “Vogelgrama”, liga características histológicas da progressão de câncer colorretal a lesões genéticas específicas. A mutação do gene APC da polipose familiar é o evento inicial do câncer colorretal familiar. Lesões genéticas adicionais estão associadas a pólipos de gravidade crescente, enquanto que p53 geralmente marca a transição para câncer colorretal invasivo. A progressão de outros tipos de tumor pode ser desencadeada pela perda de genes supressores de tumores diferentes ou por uma ordem cumulativa diferente, incluindo o gene p53. PDH = perda de heterozigosidade.
Um segundo mecanismo que contribui para o silenciamento gênico epigenético nas células tumorais envolve metilação e acetilação de resíduos de histonas específicas associadas ao DNA. Essas modificações ocorrem em resíduos específicos de aminoácidos de determinadas histonas, sendo que seu padrão está intimamente correlacionado com a expressão gênica, de modo que, às vezes, esses padrões são conhecidos como “código de histonas”, análogo ao código genético (DNA) propriamente dito. As modificações nas histonas e a metilação do DNA são mecanismos reguladores interdependentes dramaticamente alterados nas células cancerígenas.74 O fato de que essas alterações epigenéticas nas células tumorais são potencialmente reversíveis abre as portas para novas oportunidades terapêuticas. Por exemplo, atualmente o vorinostat, um inibidor da histona desacetilase, é usado no tratamento de linfomas cutâneos de células T. Este agente e mais de uma dúzia de compostos relacionados estão atualmente em fase de testes para aplicação em uma grande variedade de cânceres.75
Trata-se de um paradigma completamente novo na regulação gênica, relevante para células normais e células tumorais, que foi criado após a descoberta de microRNAs (miRNAs). Essas moléculas extremamente pequenas de RNA (comprimento de 16 a 29 nucleotídeos, em comparação com o RNA mensageiro com uma média de 1.500 nucleotídeos) não codificam proteínas, porém, em vez disso, agem como reguladores potentes da expressão gênica e da translação proteica através da capacidade de fazer par com as moléculas de RNA que codificam proteínas celulares. Embora se acredite que tenham evoluído inicialmente como um sistema de defesa contra espécies estranhas de RNA associadas a infecções virais, reconhecidamente os miRNAs atuam como reguladores importantes de uma ampla variedade de processos celulares, incluindo proliferação, diferenciação e sobrevivência de células.76 As células tumorais apresentam expressão alterada de muitos miRNAs em comparação com as células normais. A significância dessas alterações não é totalmente compreendida, porém há sugestões bastante claras de que os próprios miRNAs possivelmente atuem como oncogenes ou como genes supressores de tumores. Um subgrupo de casos de leucemia linfocítica crônica de células ß (LLC-ß) mostra uma perda na região cromossômica 13q14, sendo que os únicos genes expressos nessa região são dois miRNAs conhecidos como miR-15a e miR-16-1.77 Atualmente, acredita-se que esses miRNAs funcionem como inibidores do fator de sobrevivência BCL2 e sejam silenciados na maioria dos casos de LLC-ß, assim como em diversos outros tipos de câncer, potencializando a sobrevivência das células tumorais. Da mesma forma, a família de let-7 dos miRNAs pode atuar também como supressor tumoral através da inibição do proto-oncogene RAS; na realidade, a expressão desses miRNAs tem uma correlação inversa com a expressão do RAS em câncer de pulmão de células não pequenas.78 Entre os miRNAs potencialmente oncogênicos estão o miR-372 e o miR-373, que são superexpressos nos carcinomas testiculares e aparentemente interrompem o ciclo celular dependente de p53, explicando possivelmente porque o gene p53 propriamente dito raramente sofre mutações nesses tumores.79 Além de dar uma visão mais profunda da genética tumoral, a caracterização em curso da expressão do miRNA no câncer humano poderá levar a novos meios de determinar prognósticos e de prever respostas terapêuticas.
O estímulo inicial à proliferação celular que ocorre após algum evento transformador geralmente é acompanhado de um aumento na morte celular ou apoptose, um mecanismo compensatório que impede o crescimento rápido de algum tipo de câncer. A desativação do supressor tumoral p53 é a lesão genética mais comum que anula a resposta da morte celular, permitindo o crescimento rápido no número de células cancerígenas. O BCL2 é outro gene envolvido na regulação da apoptose que codifica uma proteína mitocondrial que evita o desencadeamento da cascata de protease exigida para o suicídio celular.80 O papel do gene BCL2 no câncer humano é mais bem ilustrado no linfoma folicular de células B, um tipo de tumor indolente em que a translocação cromossômica coloca o gene BCL2 sob controle do ativador do gene das imunoglobulinas.81 O aumento na expressão de BCL2 nessas células linfoides impede a programação de sua morte celular, resultando no aumento da massa de células linfoides que tipificam esse linfoma de crescimento lento. Conforme observamos acima, a função de BCL2 pode ser mais intensa em alguns tumores, incluindo a leucemia linfocítica crônica (LCC), através do silenciamento de microRNAs cuja função normal é inibir sua expressão. O BCL2 faz parte de uma grande família de genes em que alguns membros apresentam efeitos pró-apoptóticos e outros antagonizam a morte celular programada.80 O gene BAX, membro pró-apoptótico da família do BCL2, contém uma sequência repetitiva de nucleotídeos que é alvo de mutações em tumores com instabilidade nos microssatélites.81,82 Consequentemente, a superexpressão de membros antiapoptóticos da família do gene BCL2 ou a desativação de genes pró-apoptóticos possivelmente contribua para o fenótipo maligno.
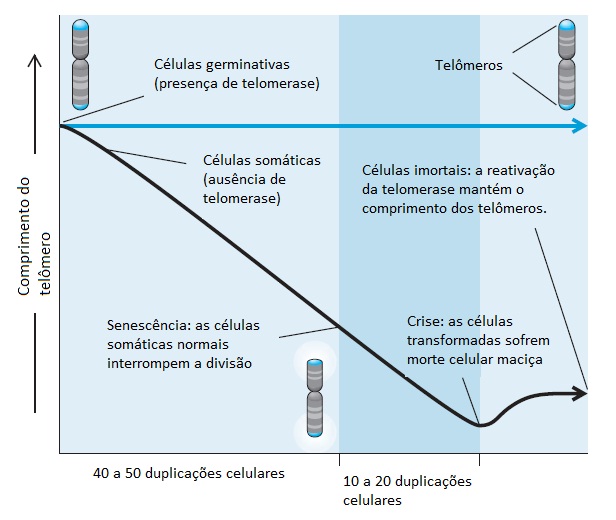
Além de evitar os controles sobre a proliferação e os mecanismos de morte celular, as células tumorais finalmente atingem a imortalidade celular, isto é, a capacidade de sofrer divisões celulares ilimitadas. As células somáticas são programadas para sofrer apenas um número limitado de divisões celulares, após o que elas entram em um estado que se denomina senescência. Embora os mecanismos moleculares subjacentes à senescência celular ainda não sejam bem compreendidos, a participação dos genes supressores de tumores CDKN2A e p53 é teoricamente possível em modelos de camundongos. As células normais que são direcionadas para a proliferação por meio da transformação oncogênica evitam a senescência e entram em crise, ou em morte celular maciça, na medida em que se aproximam do final do espectro de vida. Consequentemente, o relógio biológico das células somáticas normais é a salvaguarda mais potente contra transformações malignas e devem ser dominadas por todas as células cancerígenas para crescerem além do tempo de vida originalmente previsto. Esse relógio biológico está ligado aos telômeros, que são as extremidades dos cromossomos que precisam ser protegidas para manter a integridade cromossômica [ver a Figura 10].
Figura 10 - Senescência celular e ativação da telomerase. A divisão celular normal exige a manutenção de telômeros, que são as extremidades dos cromossomos. Nas células germinativas programadas para a vida eterna, a manutenção desses telômeros é o resultado da atividade da enzima telomerase. O crescimento de células somáticas em culturas resulta no encurtamento progressivo dos telômeros, até o momento em que cessar a divisão celular e se atingir a senescência. As células que forem estimuladas a proliferar além daquele ponto pela expressão de genes transformadores continuam a encolher os respectivos telômeros até entrarem em crise, ponto em que os cromossomos se tornam instáveis provocando morte celular maciça. Em números pequenos de células, a telomerase poderá ser ativada no momento da crise e produzir linhas de células imortais que poderão crescer indefinidamente in vitro. A maior parte dos cânceres humanos expressam altos níveis de telomerase, sugerindo que uma pressão seletiva semelhante in vivo contribui para seu potencial ilimitado de crescimento.
Os telômeros se compõem de uma prolongação de nucleotídeos repetidos e de um “tampão” de proteína que impede que as extremidades dos cromossomos sejam reconhecidas como quebras de DNA.83 Os telômeros funcionam como um mecanismo de contagem de células, tendo em vista que se encurtam ligeiramente em cada divisão celular, resultante da perda de nucleotídeos necessária para a força motora durante o início da síntese do DNA. A crise inicia a partir do momento em que os telômeros ficarem tão curtos que o tampão de proteína se rompa, induzindo uma resposta aos danos no DNA na medida em que tenta fazer o reparo ou recombinar o DNA telomérico. O pequeno número de células que sobreviver à crise estabiliza os respectivos telômeros, geralmente através da suprarregulação da enzima telomerase. No caso da ribonucloproteina com atividade reversa da transcriptase, a telomerase utiliza um modelo de DNA para adicionar repetições apropriadas de nucleotídeos nas extremidades dos telômeros, acompanhando, consequentemente, o encurtamento progressivo que acompanha a divisão celular [ver a Figura 10].84,85 Ao contrário das células somáticas, que têm vida finita e em geral expressam níveis baixos de telomerase, as células germinativas, as células-tronco e as células cancerígenas expressam altos níveis dessa enzima. As propriedades enzimáticas exclusivas da telomerase e seus requisitos na maioria das células tumorais transformaram o desenvolvimento de inibidores específicos dessa enzima em uma abordagem terapêutica potencialmente atraente.
O acúmulo de mutações que estimulam a proliferação celular e a aquisição de períodos de vida infinitos são os componentes essenciais da transformação maligna, embora a invasão tecidual e a metástase sejam as propriedades mais visíveis e ameaçadoras do câncer. As células cancerígenas invadem os tecidos vizinhos normais, penetram na base da membrana epitelial e no revestimento das células endoteliais e percorrem a corrente sanguínea ou as cadeias linfáticas até os órgãos mais distantes, onde saem da vasculatura e estabelecem sítios de crescimento independentes. As células tumorais se adaptam a cada uma dessas etapas no processo metastático; são necessárias alterações genéticas e epigenéticas em diversos genes e, ao final, apenas as células muito raras adquirem potencial metastático total. As alterações na expressão gênica envolvidas na invasão inicial incluem a suprarregulação da família Rho de pequenas guanosina trifosfatases (GTPases, do inglês guanosine triphosphatases), que regula a arquitetura citoesquelética e a mobilidade celular; aumentos nas metaloproteinases matriciais como a metaloproteinase matricial-9 (MPM-9) com capacidade para digerir a membrana basal, facilitando a invasão tecidual e inoculando a corrente sanguínea86; e supressão das moléculas de adesão intercelular como a caderina-E. Paradoxalmente, a sinalização de TGF-ß, que na fase inicial da tumorigênese funciona com um mecanismo supressor de tumores, é ativada e contribui para a progressão metastática através de efeitos parácrinos (por meio da secreção das células estromais) e autócrinos. Os esforços atuais estão focados na inibição da sinalização de TGF-ß como estratégia terapêutica.87 Além dos promotores gerais da metástase, estudos recentes começaram a descobrir alguns fatores que fazem a mediação da metástase para órgãos específicos como os pulmões e os ossos.
Uma das propriedades importantes exigidas para o crescimento de cânceres além de um tamanho mínimo é o recrutamento de vasos sanguíneos [ver a Figura 11].88 O grau de vascularização tumoral é variável, sendo que, mesmo no interior de um determinado tumor, algumas áreas poderão se tornar hipóxicas e francamente necróticas na medida em que superam o crescimento do suprimento sanguíneo.
Figura 11 - Angiogênese tumoral e inibidores da angiogênese terapêutica. Entre as propriedades mais importantes dos tumores está a capacidade de recrutamento de vasos sanguíneos, fornecendo nutrientes que permitem que cresçam além de um tamanho mínimo. O recrutamento angiogênico depende da secreção tumoral de fatores de crescimento endotelial como o fator de crescimento do endotélio vascular (VEGF, do inglês vascular endothelial growth factor) e o fator de crescimento derivado das plaquetas (PDGF, do inglês platelet-derived growth factor). Os vasos tumorais são desorganizados e estão sujeitos a vazamentos, resultando na formação de edemas associados a tumores e em hipóxia que contribuem para a resistência terapêutica. As abordagens clínicas atualmente disponíveis para inibir a angiogênese, como as terapias contra o câncer, incluem o bloqueio do próprio VEGF (bevacizumab) ou a inibição dos receptores de VEGF e de PDGF (sunitinib). As terapias antiangiogênicas normalizam a vasculatura tumoral e melhoram a liberação de medicamentos e a resposta terapêutica.
Esse conceito levou Folkman a propor que os tumores deveriam acionar um programa angiométrico para manter o suprimento de sangue, de modo que a inibição alvo da angiogênese tumoral se tornasse uma estratégia terapêutica eficaz. Estudos subsequentes validaram essa hipótese e abriram caminho para o desenvolvimento de agentes antiangiogênicos clinicamente úteis. Atualmente, sabe-se que as rotas que fazem a mediação de angiogênese específica de tumores envolvem muitos dos mesmos fatores ativados em resposta à hipóxia fisiológica, incluindo o fator induzível por hipoxia (HIF, do inglês hypoxia-inducible factor) e seu alvo ativado no sentido do fluxo, o fator de crescimento do endotélio vascular (VEGF, do inglês vascular endothelial growth factor). Três síndromes distintas de predisposição para tumores resultam de mutações na linha germinal em genes que controlam as rotas metabólicas que regulam a estabilidade e a atividade da proteína do fator induzível por hipóxia; o gene VHL, a subunidade D do gene succinato desidrogenase (SDHD) e o gene da enzima fumarato hidratase (FH).89 Essas descobertas mostram o papel fundamental dos fatores angiogênicos no câncer e enfatizam o potencial da exploração terapêutica da angiogênese tumoral. O bevacizumab, um anticorpo direcionado contra o VEGF, produziu benefícios significativos em alguns ambientes de câncer, assim como o sunitinib, um inibidor de pequenas moléculas do receptor de VEGF.90 Além de induzir a angiogênese, acredita-se que a hipóxia propriamente dita estimule a progressão tumoral através da promoção da instabilidade genética (por meio de espécies alteradas de oxigênio reativo) e da indução das adaptações que promovem a sobrevivência de células tumorais. Estudos recentes mostraram que os tumores angiogênicos possuem vasculatura altamente desordenada e que o efeito dos inibidores terapêuticos não é eliminar a vasculatura, mas “normalizar” sua estrutura, aliviando, consequentemente, a hipóxia tumoral e melhorando a perfusão tumoral através de agentes quimioterápicos.91
Além da ressecção cirúrgica, que é a modalidade de tratamento inicial para a maioria dos casos de tumores sólidos, as abordagens terapêuticas atuais ao câncer tentam explorar vulnerabilidades específicas das células tumorais. A quimioterapia e a radiação agem por meio da indução de várias formas de danos de DNA, inibindo a replicação de DNA ou bloqueando a mitose. As células tumorais são mais sensíveis a esses insultos do que as células normais devido à divisão celular mais rápida, rotas de reparo defeituosas e estresses metabólicos que possam reduzir os respectivos limiares apoptóticos. Embora o foco principal dessas terapias seja as propriedades gerais das células tumorais, uma nova geração de terapias “com foco” inibe rotas específicas que são ativadas em tumores individuais através de mecanismos genéticos específicos. A resistência ao tratamento de câncer humano é o resultado de uma combinação de fatores de hospedeiro (p.ex., variações em enzimas que metabolizam agentes terapêuticos), propriedades intrínsecas de determinados tipos de tumor que refletem sua origem tecidual (resistência geral do epitélio renal aos medicamentos em decorrência de sua capacidade de resistir às toxinas que ocorrem naturalmente) e fatores tumorais específicos.
Em alguns casos, os cânceres altamente sensíveis à quimioterapia na fase inicial podem se tornar resistentes durante o curso do tratamento ou, mais frequentemente, no momento da recorrência da doença. Um dos mecanismos de resistência tumoral específica de uma ampla gama de agentes quimioterápicos é a suprarregulação de genes transportadores com resistência a diversos medicamentos [ver a Figura 12].92 Esses genes codificam o trifosfato de adenosina (ATP, do inglês adenosine triphosphate) dependente de bombas que atravessam a membrana celular e aparentemente estão envolvidos no transporte de grandes compostos orgânicos.
Figura 12 - Mecanismos de resistência aos agentes quimioterápicos. A resistência de cânceres aos medicamentos quimioterápicos pode ser intrínseca, refletindo as propriedades dos respectivos tecidos de origem, ou adquirida, em decorrência de mutações selecionadas durante a progressão tumoral e o tratamento farmacológico anterior. Esses mecanismos incluem mutações no gene p53, que impossibilitam a resposta apoptótica celular fisiológica a lesões no DNA mediadas por esses medicamentos; diminuem a absorção dos medicamentos; aumentam a exportação de medicamentos por proteínas relacionadas ao MDR1; promovem uma amplificação compensatória nas quantidades do alvo celular do medicamento; ou criam mutações no alvo que os tornam resistentes aos efeitos do medicamento.
O gene ABCB1 (MDR1) é o mais intensamente estudados e está envolvido no mecanismo de resistência à quimioterapia, cujo produto gênico tem capacidade para exportar uma ampla gama de compostos, incluindo as antraciclinas e os vinca alcaloides. Na ausência de exposição anterior, a expressão de ABCB1 provavelmente esteja associada à resistência a diversos agentes. Entretanto, aparentemente, em alguns tipos de câncer, como os mielomas múltiplos, a exposição persistente às antraciclinas e aos vinca alcaloides se correlacionam com futuros aumentos na expressão de ABCB1 e com resistência quimioterápica progressiva. Um exemplo bem conhecido de resistência quimioterápica adquirida ocorre após a exposição ao medicamento metotrexato, que inibe especificamente a enzima dihidrofolato redutase (DHFR, do inglês dihydrofolate reductase) que participa na síntese de DNA.93 Os tumores podem se tornar resistentes ao metotrexato ao adquirem defeitos no transporte intracelular do medicamento; por mutações no gene DHFR, resultando em uma enzima que não consegue se ligar ao metotrexato; ou pela amplificação do próprio gene DHFR, aumentando a quantidade de proteína produzida e, consequentemente, superando a concentração intracelular do medicamento inibidor.
O conceito de abordagens terapêuticas com base nos alvos oncogênicos dos medicamentos é mais bem ilustrada pelo sucesso do mesilato de imatinib, um inibidor de pequenas moléculas de quinases proteicas específicas [ver a Figura 13].90 O imatinib foi identificado inicialmente por sua capacidade de inibir a ligação do trifosfato de adenosina (ATP, do inglês adenosine triphosphate) ao domínio da quinase do receptor do fator de crescimento derivado das plaquetas (PDGFR, do inglês platelet-derived growth factor receptor); subsequentemente descobriu-se que também inibe a quinase de Abelson e a quinase KIT (c-kit). Atribui-se a eficácia do imatinib no tratamento de leucemia mielocítica crônica (LMC) à translocação cromossômica subjacente que resulta na proteína quimérica BCR-ABL [ver Amplificação Gênica e Translocações Cromossômicas acima]. A ativação constitutiva da quinase ABL por meio dessa reorganização é essencial para as malignidades, sendo que as células da LMC são extremamente sensíveis a qualquer interrupção nessa rota de sinalização.
Figura 13 - Eficácia do mesilato de imatinib com alvo nas proteínas oncogênicas em diversos tipos de tumor. O imatinib rompe a ligação do trifosfato de adenosina (ATP) com o domínio catalítico de três proteínas quinases: o receptor do fator de crescimento derivado das plaquetas (PDGFR), ABL e KIT (c-kit). Os tipos de tumor em que a ativação dessas proteínas quinases for crítica no crescimento tumoral incluem leucemia mielocítica crônica em que a proteína de fusão quimérica BCR-ABL derivar da reorganização dos cromossomos 9 e 22 (cromossomo de Filadélfia); tumor de células estrômicas gastrointestinais (GIST) contendo mutações ativadoras de c-kit; e leucemia mielomonocítica crônica (LMMC), em que a reorganização do gene PDGFR resultar na sua ativação. Nessas três malignidades diferentes, o tratamento à base de imatinib produz respostas clínicas rápidas e sustentadas.
Consequentemente, a proteína quimérica BCR-ABL é um alvo farmacológico ideal, pois sua função é essencial para a sobrevivência das células leucêmicas, enquanto que a inibição de ABL em células normais tem poucas consequências. Esse medicamento revolucionou o tratamento de leucemia mielocítica crônica (LMC) e, além disso, apresenta uma quantidade relativamente pequena de efeitos colaterais.90 Uma coincidência interessante é a eficácia do imatinib em tumores sólidos sem relação histológica e no tumor de células estrômicas gastrointestinais (GIST), que também depende de uma proteína alvo deste medicamento. Os GISTs são tumores raros que se caracterizam por mutações KIT (c-kit) que provocam a ativação constitutiva da quinase kit. Embora, tipicamente, os GISTs sejam refratários à quimioterapia padrão, observaram-se respostas dramáticas em pacientes que haviam sido tratados com imatinib.94 Da mesma forma, casos de leucemia mielomonocítica crônica (LMMC), em que a lesão genética subjacente envolve a reorganização e a ativação do gene PDGFR, também são responsivos ao imatinib.95 Portanto, o projeto racional de inibidores de pequenas moléculas direcionado para proteínas que sejam componentes importantes de rotas celulares ativadas por oncogenes específicos poderá levar à descoberta de tratamentos novos e eficazes para muitos tipos distintos de câncer.
Outro exemplo impressionante da aplicação de genética molecular na terapia de tipos específicos de câncer envolve o uso de inibidores da tirosina quinase (TKI, do inglês tyrosine kinase) direcionados para moléculas pequenas no tratamento de carcinoma pulmonar de células não pequenas (NSCLC, do inglês non-small cell lung carcinoma). Estudos anteriores demonstraram que o uso de qualquer um dos dois inibidores relacionados do receptor do fator de crescimento epidérmico (EGFR, do inglês epidermal growth factor receptor), gefitinib ou erlotinib, produziu respostas somente em uma pequena minoria de pacientes com NSCLC. Estudos moleculares subsequentes mostraram que esses tumores geralmente abrigam mutações somáticas específicas dentro do domínio intracelular da tirosina quinase do gene EGFR.96,97 Consequentemente, os tumores com mutações em EGFR são “dependentes” da expressão mutante de EGFR, ao contrário dos tumores sem essas mutações. Cabe observar que os tumores com mutações em EGFR ocorrem predominantemente em não fumantes, enquanto que muitos adenocarcinomas pulmonares associados ao tabagismo abrigam mutações no oncogene RAS e são resistentes às terapias atuais. Essas descobertas sugerem uma patogênese molecular distinta para o câncer de pulmão não associado ao tabagismo (menos comum). Os eventos adicionais de estimulação genética observados nos adenocarcinomas pulmonares em não fumantes são translocações cromossômicas distintas envolvendo os genes da quinase ALK e ROS. Cada uma dessas translocações, que são mutuamente exclusivas com as mutações de EGFR em tumores, confere sensibilidade a um inibidor químico da família da quinase ALK, o crizotinib.98 Os adenocarcinomas com mutações em EGFR correspondem a apenas 15% de todos os casos e os tumores associados às translocações de ALK e ROS representam uma fração ainda menor (< 10%) desses tumores. Todavia, com base no sucesso inicial dessa abordagem terapêutica direcionada para oncogenes, a análise genética de tumores para essas e outras alterações moleculares estão se tornando cada vez mais importantes para a tomada de decisões terapêuticas.
O uso de modalidades terapêuticas-alvo eficazes permite selecionar tumores com alterações genéticas que conferem resistência específica a esses agentes. A resistência celular ao imatinib nos casos de leucemia mielocítica crônica (LMC) desenvolve mutações dentro do domínio da quinase de BCR-ABL cujo alvo é esse medicamento. Essas observações levaram à introdução bem sucedida de uma segunda geração de inibidores – dasatinib e nilotinib – que têm maior afinidade com o domínio da quinase e, portanto, são mais potentes, principalmente contra as mutações de BCR-ABL resistentes ao imatinib.99 Outros mecanismos importantes de resistência ocorrem através de alterações genéticas não no alvo terapêutico propriamente dito, mas em genes que atuam em paralelo ou no sentido de fluxo da rota. Por exemplo, um mecanismo adicional de resistência ao imatinib nos casos de leucemia mielocítica crônica (LMC) é através da superexpressão de quinases da família de SRC relacionadas à ABL.100 Essas proteínas podem funcionar em caminhos promotores tumorais comuns como BCR-ABL e, em níveis elevados, podem também remover gradualmente o composto inibidor de BCR-ABL. Um exemplo claro de um evento genético em uma rota no sentido do fluxo que dá resistência a um inibidor-alvo foi observado recentemente em casos de câncer no cólon tratado com cetuximab (Erbitux), um anticorpo inibidor do receptor do fator de crescimento epidérmico (EGFR, do inglês epidermal growth factor receptor). A presença de mutações no oncogene KRAS é altamente preditiva da falta de resposta a esse agente, provavelmente porque a mutação em KRAS induz a ativação funcional da rota da proteinoquinase ativada por mitógeno (MAPK, do inglês mitogen-activated protein kinase), independentemente da sinalização de EGFR no contrafluxo.101 Pela mesma razão, as mutações em KRAS também estão associadas à resistência clínica dos cânceres pulmonares aos inibidores do EGFR gefitinib e erlotinib. Com base nessas descobertas, o teste molecular de câncer no cólon e de câncer no pulmão para mutação em KRAS atualmente faz parte da prática clínica padrão para pacientes que forem selecionados para terapia direcionada para o inibidor do receptor do fator de crescimento epidérmico.
Alvos terapêuticos específicos e mecanismos associados de resistência não são tão bem estabelecidos para as rotas de supressão tumoral, em comparação com as rotas de oncogenes descritas acima. Entretanto, uma exceção são os cânceres de mama e de ovário associados às mutações BRCA1 e BRCA2 na linha germinal. As células tumorais deficientes de BRCA1/2 têm um defeito específico em um mecanismo de reparo de DNA conhecido por recombinação homóloga. Portanto, esses tumores dependem da rota de reparo com base na excisão para reparar os danos espontâneos e induzidos no DNA. Esse tipo de dependência poderá ser explorado tratando-se esses tumores com inibidores da poli (ADP-ribose) polimerase (PARP, do inglês poly-A ribose polymerase), um tipo de enzima imprescindível para a execução de reparos com base em excisões, que poderá ser usada isoladamente ou em combinação com quimioterapia. Entretanto, cabe observar que alguns tumores associados à mutação BRCA2 se tornam resistentes aos inibidores da PARP e à quimioterapia através da aquisição de uma pequena deleção de DNA no interior do alelo de BRCA2 em mutação.102 Essa deleção faz a excisão do codão de terminação mutante, recupera a fase de leitura de BRCA2 para translação de proteínas e resulta na produção de uma proteína BRCA2 funcional. Presume-se que essas células tumorais “recuperadas” da mutação BRCA2 sejam competentes para o reparo do DNA e, consequentemente, deixem de ser sensíveis à terapia. Um mecanismo semelhante foi descrito nos casos de tumores associados à mutação BRCA1. Embora seja um mecanismo incomum de resistência, ele destaca a capacidade das células tumorais para se adaptar a pressões seletivas específicas através de eventos genéticos que superam a suscetibilidade ao tratamento.
Dois avanços tecnológicos importantes – sequenciamento de todo o genoma humano e desenvolvimento de tecnologias de microarranjos de alta densidade – possibilitam analisar anormalidades genéticas em células cancerígenas em uma escala genômica ampla. Os tipos mais comuns de análise se enquadram em duas categorias gerais: perfil da expressão genética e análise genômica ampla do DNA. O perfil da expressão genética pode ser usado para avaliar simultaneamente o nível de expressão genética do RNA de todos os genes humanos (aproximadamente 30.000 genes) e um único espécime [ver a Figura 14]. Quantifica-se a expressão genética por meio da hibridização do RNA marcado de um espécime de teste para sequências de amostras complementares em um chip genético; então a hibridização pode ser detectada por varreduras a laser. É importante ressaltar que as amostras para todo o conjunto de genes humanos poderão ser organizadas em um ou dois chips ou slides com o tamanho aproximado de uma moeda. O principal objetivo da análise estatística desses níveis globais de expressão genética é a identificação de padrões que possam se correlacionar com um fenótipo clínico. Por exemplo, o perfil da expressão permitiu dar uma visão mais ampla da classificação de linfomas e de leucemias – classificações que têm implicações clínicas e que não teriam sido possíveis através da análise de um único gene.103
Figura 14 - Análise de perfis de expressão gênica com auxílio de microarranjos de alta densidade. Essa estratégia permite fazer a quantificação simultânea de todas as transcrições de RNA mensageiro (mRNA) no interior de um tecido. Por exemplo, é possível fazer a comparação entre espécimes de tumores que representam tipos distintos de câncer ou cânceres com comportamentos clínicos diferentes. O mRNA celular total é isolado a partir de espécimes tumorais congelados. O RNA é marcado e hibridizado em microarranjos de alta densidade com amostras para aproximadamente 30.000 genes. O sinal de hibridização de cada gene é detectado e comparado com espécimes clínicos diferentes. Técnicas como mineração de dados complexos e abordagens da bioinformática permitem identificar agrupamentos de padrões de expressão que possivelmente estejam correlacionados com medidas clínicas diferentes, tais como prognósticos ou respostas a terapias específicas.
Este método foi utilizado também para desenvolver a técnica conhecida como assinaturas genéticas no câncer de mama e em outros tipos de câncer. Essas assinaturas são grupos de genes com expressão diferencial que se correlacionam com o resultado clínico ou com respostas a terapias específicas.104 Mammaprint, uma assinatura com 70 genes, e Oncotype Dx, uma assinatura com 21 genes, são duas assinaturas genéticas atualmente em uso clínico que ajudam a orientar o tratamento de câncer de mama na fase inicial.105 As novas assinaturas que se encontram em fase de desenvolvimento procuram fornecer informações prognósticas e responsivas-preditivas para muitos tipos de câncer. A análise dos perfis de expressão pode também levar à descoberta de novos marcadores expressos por tipos distintos de câncer. Alguns desses perfis podem codificar a secreção de proteínas que possivelmente sejam importantes na detecção precoce, ao passo que outros poderão ser alvos de novos medicamentos.
Um segundo tipo geral de análise genômica ampla avalia o DNA celular ao invés do RNA celular. As aplicações utilizadas com maior frequência procuram descobrir regiões cromossômicas de ganhos e perdas de células tumorais em alta resolução. Essas análises utilizam dois tipos de abordagem. Uma dessas abordagens, conhecida por hibridização genômica comparativa (CGH, do inglês comparative genomic hybridization), é conceitualmente semelhante ao perfil de expressão no sentido em que envolve a análise direta do número da cópia do DNA.106 O outro método amplamente utilizado faz a análise da perda de heterozigosidade (PDH) através de microarranjos de alta densidade com capacidade para avaliar polimorfismos de nucleotídeo único (SNPs, do inglês single nucleotide polymorphisms) em todo o genoma.107 A maioria dos cânceres epiteliais tem inúmeras regiões de ganho ou perda cromossômica que poderão ser identificadas com relativa precisão por meio dessas técnicas. Essas regiões de ganho ou perda provavelmente contenham oncogenes ou genes supressores de tumores, respectivamente, sendo que a aplicação dessas técnicas confirmou a amplificação do gene ERBB2 (HER-2) em casos de câncer de mama e a perda de PTEN em casos de glioblastoma e câncer na próstata. Assim como no perfil da expressão gênica com base em microarranjos, a CGH e o SNP foram utilizados para identificar com sucesso padrões globais de ganhos ou perdas cromossômicas que aparentam ter alguma correlação com os resultados clínicos.108 A combinação dessas abordagens com base no DNA com o perfil de expressão gênica é uma técnica muito promissora como meio de classificar as inúmeras alterações na expressão cromossômica e genética que ocorrem nas células tumorais e de identificar os eventos genéticos mais relevantes específicos de tumores.
Embora tenham sido necessárias mais de uma dezena de anos e centenas de milhões de dólares para determinar a sequência do primeiro genoma humano completo, avanços tecnológicos importantíssimos estão revolucionando nossa capacidade para fazer análises de sequências detalhadas de DNA. Nos anos recentes, essas novas abordagens, conhecidas coletivamente por “próxima geração” de tecnologias de sequenciamento, permitiram que equipes de pesquisa fizessem sequenciamentos completos do “exoma” (região de codificação de proteínas) do DNA de dezenas de casos individuais de câncer de mama, pulmão, cólon e outros tipos de câncer.109. Inúmeras mutações potencialmente relevantes são encontradas em cada tipo de tumor, ressaltando a complexidade genética dos cânceres humanos [ver Anormalidades Genéticas de Condutor e Passageiro em Patogênese do Câncer, acima]. Todavia, em diversos casos, essas análises já descobriram oncogenes e mutações clinicamente relevantes no gene supressor de tumores. Um dos exemplos é a leucemia de células pilosas (HCL, do inglês hairy cell leukemia), um tipo de leucemia relativamente sensível ao tratamento para o qual ainda não foram identificados oncogenes relevantes. O sequenciamento completo do exoma identificou a ativação de mutações no gene da quinase BRAF, que foi subsequentemente confirmada em 100% de pacientes que eram portadores dessa doença.110 Já se sabia que as mutações no gene BRAF ocorriam em aproximadamente 60% de casos de melanoma cutâneo maligno, sendo que as terapias com foco no BRAF, que são muito promissoras, encontram-se em fase avançada de testes clínicos para aplicação nesse tipo de doença.111 Embora essa descoberta provavelmente não altere de imediato o tratamento de leucemia de células pilosas, que já pode ser curada com quimioterapia, esse exemplo ressalta a importância da aplicação do sequenciamento amplo de DNA nos genes de câncer e na descoberta de alvos terapêuticos. No futuro, a próxima geração de sequenciamento de DNA poderá ser utilizada para descobrir padrões complexos de tumores específicos de mutações como parte das avaliações diagnósticas de rotina. Essas informações poderão então facilitar a tomada de decisões clínicas, em paralelo com a aplicação clínica das assinaturas de expressão gênica que se está em curso.
Apenas uma pequena parte da predisposição genética para o câncer na população em geral é atribuída aos genes de alto risco (alta penetração) como aqueles apresentados na Tabela 2. Entretanto, nesses casos, o uso apropriado da análise de mutações na linha germinal permite identificar pessoas com risco elevado de tipos específicos de câncer que poderão se beneficiar com o monitoramento rigoroso e/ou com a profilaxia cirúrgica.112 Por exemplo, os benefícios cirúrgicos são bem definidos em portadores de mutações APC com polipose de cólon familiar, em que a colectomia profilática impede o desenvolvimento de câncer colorretal.113 Da mesma forma, geralmente se recomenda a tireoidectomia profilática em pacientes com neoplasia endócrina múltipla.114 Após o término do período de gravidez, a ooforectomia profilática é a prática padrão para portadores de mutações BRCA1 e BRCA2 porque diminui o risco de câncer de mama e de ovário nessa população de pacientes.115 O impacto clínico potencial da identificação de indivíduos que abrigam essas mutações de alto risco na linha germinal ressalta a necessidade de orientação genética adequada e do encaminhamento para testes nos casos de pacientes com históricos familiares sugestivos.
A maior parte dos riscos genéticos de câncer na população não está associada a mutações raras de alto risco, porém, ao invés disso, está associada a variações genéticas comuns que aumentam moderadamente o risco de incidência de câncer (conhecidas por variantes ou alelos de penetração lenta) [ver a Figura 15]. Com frequência, essas variantes são identificadas inicialmente através de estudos de associação genômica ampla (GWASs, do inglês genome-wide association studys), em que as análises de DNA de alto rendimento na linha germinal são aplicadas em grandes populações para identificar loci genéticos comuns associados à predisposição para a doença. O resultado de GWASs para tipos comuns de câncer revelaram a existência e um grande número de loci genéticos que não haviam sido suspeitados previamente e que estão potencialmente envolvidos na suscetibilidade para o câncer dentro da população em geral.116 Existem várias considerações importantes relevantes em relação a esses estudos. Em primeiro lugar, na maior parte dos casos, as variantes polimórficas que definem esses loci associados a algum risco provavelmente não tenham relevância direta em relação à suscetibilidade ao câncer propriamente dito, porém identificam uma região genética (conhecida como haplótipo) que abriga uma variante não identificada que causa câncer. Portanto, é necessário fazer análises experimentais consideráveis que permitam compreender as consequências funcionais de novos loci candidatos com predisposição para o câncer. Em segundo lugar, os estudos de associação genômica ampla devem ser interpretados com muita cautela e no contexto de uma grande coorte estudada. Fatores genéticos e ambientais poderão confundir as análises de GWASs, de modo que os loci de risco identificados em uma população possivelmente não sejam relevantes em outras populações.117 Para finalizar, a maioria das variantes com suscetibilidade para o câncer identificadas até o presente momento em GWASs estão associadas a riscos relativos de 1,1 a 1,5 na população em geral (em comparação com 5 a 20 vezes para os alelos tradicionais que predispõem para o câncer, como as mutações de p53 ou BRCA1) [ver a Figura 15]. Por exemplo, aparentemente a mutação I1307K no gene APC, que está presente em 6% da população de judeus asquenazes, está associada a um aumento de duas vezes no risco de câncer colorretal.118 A mutação 11000delC do gene CHK2 no ponto de verificação do ciclo celular, que está presente em 1% da população em geral, foi associada a um aumento semelhante no risco de câncer de mama.119 Esse risco relativamente baixo desperta o interesse epidemiológico, embora, na maioria dos casos, não seja suficiente para orientar indivíduos portadores de uma variante genética específica. Não obstante, a aplicação clínica dessas informações poderá se tornar mais relevante na medida em que “perfis genômicos pessoais” se tornarem disponíveis, incluindo, sem dúvida alguma, muitas dessas variantes de risco relativamente baixo que, coletivamente, poderão se tornar significativas sob o ponto de vista clínico.
Figura 15 - Espectro de alelos de risco para predisposição para câncer de mama. O risco relativo de câncer associado a variantes genéticas identificadas é quase inversamente proporcional à sua frequência dentro da população. Consequentemente, as mutações genéticas de alto risco, como as que ocorrem nos genes BRCA1 e p53, são relativamente raras, enquanto que as variantes comuns em genes como o FGFR2 estão associadas a um aumento de apenas 20% (1,2 vezes) no risco. As metodologias usadas para identificar essas classes diferentes de variantes de risco estão entre parênteses. Adaptação de Foulkes W. Suscetibilidade hereditária aos cânceres comuns. N. Engl J Med 2008; 349:2143-53.
|
O autor não mantém nenhuma relação comercial com os fabricantes de produtos ou com os fornecedores de serviços mencionados neste capítulo. |
1. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer: analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000;343:78.
2. Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294–9.
3. Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol 2001;28(2 Suppl 4):3–13.
4. Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007;318:1108–13.
5. Stehelin D, Varmus HE, Bishop JM, et al. DNA related to transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976;260:170.
6. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
7. Lowy DR, Willumsen BM. Function and regulation of ras. Annu Rev Biochem 1993;62:851.
8. Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific net protooncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET Mutation Consortium analysis. JAMA 1996;276:1575.
9. Schimke RT. Gene amplification, drug resistance, and cancer. Cancer Res 1984;44:1735.
10. Brodeur GM, Seeger RC, Schwab M, et al. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984;224:1121.
11. Rabbitts TH. Chromosomal translocations in human cancer. Nature 1994;372:143.
12. Dalla-Favera R, Bregni M, Erikson J, et al. Human c-myc oncogene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A 1982;79:7824.
13. Taub R, Kirsch I, Morton C, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cell. Proc Natl Acad Sci U S A 1982;79:7837.
14. Ellisen LW, Bird J, West DC, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991;66:649.
15. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269.
16. Shtivelman E, Lifshitz B, Gale RP, et al. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985;315:550.
17. Rowley JD. Biological implications of consistent chromosome rearrangements in leukemia and lymphoma. Cancer Res 1984;44:3159.
18. Daley GQ, Van-Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990;247:824.
19. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344:1038.
20. Look AT. Oncogenic transcription factors in the human acute leukemias. Science 1997;278:1059.
21. Warrell RP Jr, Frankel SR, Miller WH Jr, et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med 1991;324:1385.
22. Golub TR, Barker GF, Bohlander SK, et al. Fusion of the TEL gene on 12p13 to the AMLI gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 1995;92:4917.
23. Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors: a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294.
24. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644.
25. Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007;448:595.
26. Ephrussi B, Davidson RL, Weiss MC, et al. Malignancy of somatic cell hybrids. Nature 1969;224:1314.
27. Whyte P, Buchkovich KJ, Horowitz JM, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988;334:124.
28. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820.
29. Friend SH, Bernards R, Rogelij S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986;323:643.
30. Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990;60:509.
31. Berger AH, Pandolfi PP. Haplo-insufficiency: a driving force in cancer. J Pathol 2011;223:137–46.
32. Cavenee WK, Dryja TP, Phillips RA, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983;305:779.
33. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995;81:323.
34. Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet 1994;8:15.
35. Sherr CJ. Cancer cell cycles. Science 1996;274:1672.
36. Lane DP. Cancer: p53, guardian of the genome. Nature 1992;358:15.
37. El-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75:817.
38. Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science 1991;253:49.
39. Harris CC, Hollstein M. Clinical implications of the p53 tumor suppressor gene. N Engl J Med 1993;329:1318.
40. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990;250:1233.
41. Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 1993;7:546.
42. Oliner JD, Kinzler KW, Meltzer PS, et al. Amplifications of a gene encoding a p53-associated protein in human sarcoma. Nature 1992;358:80.
43. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995;378:203.
44. Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol 2000;1:79.
45. Quelle DE, Zindy F, Ashmun RA, et al. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995;83:993.
46. Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev 1998;12:2984.
47. Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997;90:809.
48. Osada M, Ohba M, Kawahara C, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med 1998;4:839.
49. Kastan MB, Zhan Q, el-Deiry WS, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992;71:587.
50. Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995;268:1749.
51. Miki Y, Swensew J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66.
52. Wooster R, Bignell G, Lancaster J, et al. Identification of breast cancer susceptibility gene BRCA2. Nature 1995;378:789.
53. Scully R, Chen J, Plug A, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997;88:265.
54. Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001;2:446–57.
55. Lynch HT, Smyrk T, Lynch JF. Molecular genetics and clinical-pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome): historical journey from pedigree anecdote to molecular genetic confirmation. Oncology 1998;55:103–8.
56. Kolodner RD. Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends Biochem Sci 1995;20:397–401.
57. Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338:1481–7.
58. Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995;268:1336–8.
59. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005;37:19.
60. Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol 2004;22:2954.
61. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;391:184.
62. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345.
63. Iliopoulos O, Kaelin WG Jr. The molecular basis of von Hippel-Lindau disease. Mol Med 1997;3:289.
64. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet 2001;10:721.
65. Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996;271:350.
66. McCormick F. Ras signaling and NF1. Curr Opin Genet Dev 1995;5:51.
67. Gusella JF, Ramesh V, MacCollin M, et al. Neurofibromatosis 2: loss of merlin’s protective spell. Curr Opin Genet Dev 1996;6:87.
68. Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996;85:841.
69. Haber DA. Wilms tumor. In: Vogelstein B, Kinzler K, editors. The genetic basis of human cancer. New York: McGraw Hill Book Co; 2002. p. 403.
70. Rivera MN, Kim WJ, Wells J, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 2007;315:642.
71. Major MB, Camp ND, Berndt JD, et al. Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 2007;316:1043.
72. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159.
73. Ting AH, McGarvey KM, Baylin SB. The cancer epigenome: components and functional correlates. Genes Dev 2006;20:3215.
74. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007;8:286.
75. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009;27:5459–68.
76. Wiemer EA. The role of microRNAs in cancer: no small matter. Eur J Cancer 2007;43:1529.
77. Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Nat Acad Sci U S A 2002;99:15524.
78. Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell 2005;120:635.
79. Voorhoeve PM, Le Sage C, Schrier M, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006;124:1169.
80. Korsmeyer SJ. Regulators of cell death. Trends Genet 1995;11:101.
81. Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 1986;47:19.
82. Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997;275:967.
83. Greider CW. Telomeres and senescence: the history, the experiment, the future. Curr Biol 1998;8:R178.
84. Meyerson M, Counter CM, Eaton EN, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997;90:785.
85. Nakamura TM, Morin GB, Chapman KB, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997;277:955.
86. Liotta LA, Tryggvason K, Garbisa S, et al. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 1980;284:67–8.
87. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006;6:506–20.
88. Folkman J. Fighting cancer by attacking its blood supply. Sci Am 1996;275:150.
89. Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 2005;5:857–66.
90. Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007;25:884–96.
91. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307:58–62.
92. Ling V. Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol 1997;40 Suppl:S3–8.
93. Kinsella AR, Smith D. Tumor resistance to antimetabolites. Gen Pharmacol 1998;30:623–6.
94. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472.
95. Apperley JF, Gardembas M, Melo JV, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med 2002;347:481.
96. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129.
97. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497.
98. Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693–703.
99. Quintas-Cardama A, Cortes J. Therapeutic options against BCR-ABL1 T315I-positive chronic myelogenous leukemia. Clin Cancer Res 2008;14:4392–9.
100. Jabbour E, Cortes J, O’Brien S, et al. New targeted therapies for chronic myelogenous leukemia: opportunities to overcome imatinib resistance. Semin Hematol 2007;44(1 Suppl 1):S25–31.
101. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. KRAS mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–65.
102. Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008;451:1111–5.
103. Rosenwald AG, Wright WC, Chan JM, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937.
104. Fan C, Oh DS, Wessels L, et al. Concordance among gene-expression–based predictors for breast cancer. N Engl J Med 2006;355:560.
105. Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med 2009;360:790–800.
106. Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet 1999;21:33.
107. Fan JB, Chee MS, Gunderson KL. Highly parallel genomic assays. Nat Rev 2006;7:632.
108. Kim SY, Hahn WC. Cancer genomics: integrating form and function. Carcinogenesis 2007;28:138.
109. Mardis ER, Wilson RK. Cancer genome sequencing: a review. Hum Mol Genet 2009;18(R2):R163–8.
110. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
111. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
112. Ponder B. Genetic testing for cancer risk. Science 1997;278:1050.
113. Kinzler KW, Vogelstein B. Colorectal tumors. In: Vogelstein B, Kinzler K, editors. The genetic basis of human cancer. New York: McGraw Hill Book Co; 2002. p. 583.
114. Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658.
115. Domchek SM, Armstrong K, Weber BL. Clinical management of BRCA1 and BRCA2 mutation carriers. Nat Clin Pract Oncol 2006;3:2.
116. Stadler ZK, Thom P, Robson ME, et al. Genome-wide association studies of cancer. J Clin Oncol 2010;28:4255–67.
117. Galvan A, Ioannidis JP, Dragani TA. Beyond genome-wide association studies: genetic heterogeneity and individual predisposition to cancer. Trends Genet 2010;26:132–41.
118. Laken SJ, Petersen GM, Gruber SB, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet 1997;17:79.
119. Meijers-Heijboer H, van den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002;31:55.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.