(Carregando Índice)... (Carregando Índice)... |
Última revisão: 14/07/2016
Comentários de assinantes: 0
Allan C. Halpern, MD
Chefe do Departamento de Dermatologia, Memorial Sloan-Kettering Cancer Center, New York, NY.
Patricia L. Myskowski, MD
Médica Atendente no Serviço de Dermatologia, Memorial Sloan-Kettering Cancer Center, New York, NY.
Artigo original: Halpern AC, Myskowski PL. Malignant Cutaneous Tumors.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Paulo Henrique Machado.
Revisão técnica: Dr. Lucas Santos Zambon.
Os tumores malignos podem surgir a partir de células de qualquer camada da pele – queratinócitos, melanócitos, fibroblastos, células endoteliais ou adipócitos – assim como a partir de células como os linfócitos, que normalmente transitam através da pele. Além disso, as metástases cutâneas podem se originar em outros sítios primários. Neste capítulo faremos uma revisão dos tumores cutâneos malignos mais comuns, de acordo com a ordem de frequência.
Os cânceres de pele epidérmicos são os tipos mais comuns de câncer em seres humanos. Esse tipo de câncer surge nos queratinócitos e melanócitos da epiderme. Os cânceres de pele epidérmicos apresentam uma única oportunidade para intervenções efetivas, com detecção imediata e prevenção primária. Eles são suscetíveis a diagnósticos clínicos por meio da simples inspeção visual, assim como a diagnósticos patológicos por biópsias minimamente invasivas.
Os carcinomas basocelulares (CBCs) e os carcinomas de células escamosas (CCEs) têm origem nos queratinócitos da epiderme. Levando-se em consideração que esses dois tipos de câncer compartilham muitas características, com frequência eles se juntam no mesmo termo câncer de pele não melanoma (CPNM).
Melanoma maligno é um tipo de malignidade que surge a partir de um melanócito do corpo, incluindo os olhos, sendo que a esmagadora maioria ocorre na pele. Os melanomas cutâneos malignos se classificam em quatro tipos histogenéticos principais: lentigo maligno, melanoma com disseminação superficial, melanoma nodular e melanoma lentiginoso acral.
Várias linhas de evidências implicam a radiação ultravioleta (UV) na patogênese de todos os três principais tipos de câncer de pele epidérmico.1 Os dados epidemiológicos relacionam a exposição cumulativa aos raios solares no longo prazo ao desenvolvimento de carcinoma de células escamosas e a exposição intermitente intensa aos raios solares ao desenvolvimento de carcinomas basocelulares e melanomas. Alguns estudos laboratoriais indicam que tanto a radiação UVA (320 a 400 nm) quanto a radiação UVB (290 a 320 nm) dos raios solares pode danificar o DNA diretamente e através de danos oxidativos. Além disso, a radiação UV poderá suprimir o sistema imune cutâneo.2 A associação de alguns CCEs com carcinógenos químicos e com a ocorrência de melanomas lentiginosos acrais e mucosos em áreas não expostas do corpo ressalta a necessidade de novos estudos para identificar a ação de outros agentes etiológicos.
O reconhecimento do importante papel desempenhado pelos raios solares na etiologia de câncer de pele permite fazer prevenção primária através do uso de protetores solares. Infelizmente, o momento e a dose exata de exposição UV envolvida no desenvolvimento de câncer de pele em seres humanos não são conhecidos e provavelmente variem entre esses tipos de câncer. Da mesma forma, os pacientes devem ser orientados sobre os efeitos danosos da exposição aos raios solares e do bronzeamento de pele. Os esforços de proteção contra os raios solares devem ser direcionados para a redução total da exposição ao sol procurando-se evitar comportamentos de busca do sol e estimulando o uso de roupas que protegem contra a ação dos raios solares. Os filtros solares de espectro amplo devem oferecer proteção contra as radiações UVB e UVA, porém não devem se usados para estender o tempo de exposição direta aos raios solares.4
|
As informações financeiras estão no final deste capítulo, antes das referências. |
O uso consistente de filtros solares com fator de proteção solar (FPS) UVB diminui a incidência de carcinoma de células escamosas4, sendo que um estudo preliminar demonstrou que possivelmente diminua a incidência de melanoma.5 Recomenda-se evitar o uso de camas de bronzeamento. Embora o uso de agentes de bronzeamento artificial seja seguro, o escurecimento da pele não oferece proteção significativa contra os raios UV. Nos casos de indivíduos que são assíduos nos esforços de proteção contra os raios solares, recomenda-se tomar muito cuidado com a ingestão adequada de vitamina D através de dietas ou de suplementos.6
Tipicamente, o câncer de pele não melanoma (CPNM) ocorre como lesões róseas na superfície da pele exposta ao sol. Qualquer lesão rósea que persistir ou ocorrer no mesmo local, principalmente se forem facilmente irritáveis com traumas menores, devem levantar suspeitas de CPNM. Algumas formas de CPNM desaparecem com a mudança de estação (i.e., com redução na exposição aos raios solares) ou com aplicação de esteroides tópicos, sendo que os médicos deverão alertar os pacientes de que a recorrência de qualquer lesão exige atenção especial. A incidência de CPNM varia em todo o mundo, sendo mais elevada nas populações de pele clara que vivem em latitudes mais baixas.7 As taxas mais elevadas de CPNM ocorrem na Austrália (> 1.000 em 100.000 pessoas por ano para carcinomas basocelulares) e as taxas mais baixas ocorrem na África (< 1 em 100.000 pessoas por ano para carcinomas basocelulares).7 As taxas médias de incidência na Inglaterra eram de 76,2 em 100.000 pessoas por ano para carcinomas basocelulares e de 22,65 em 100.000 pessoas por ano para carcinoma de células escamosas.7
Carcinoma basocelular (CBC) é um tumor cutâneo maligno que surge a partir de queratinócitos basais da epiderme.
Epidemiologia. CBC é o câncer de pele mais comum e representa a maior parte da estimativa de 2.100.000 pessoas diagnosticadas com novos casos de câncer de pele não melanoma que surgiram nos Estados Unidos em 2006.8 A incidência registrada varia de 3,4 por 100.000 pessoas por ano em afro-americanos a aproximadamente 1.100 por 100.000 pessoas por ano em Townsville, Queensland, Austrália.9 Embora sejam raras, poderão ocorrer metástases a partir de carcinomas basocelulares.
Etiologia e fatores de risco. Aparentemente, a radiação UV – especificamente com exposição intermitente intensa aos raios solares – desempenha papel importante no desenvolvimento de carcinomas de células basais. Estudos envolvendo a síndrome do nevo de células basais (síndrome de Gorlin) produziram insights dramáticos na genética dos carcinomas basocelulares. O gene patched, que foi reconhecido pela primeira vez como gene evolutivo na mosca de frutas Drosophila, tem um papel importante no desenvolvimento de carcinomas basocelulares (CBCs). Aparentemente, quase todos os pacientes com a síndrome do nevo de células basais herdam uma cópia mutante do gene patched, sendo que estudos de CBCs esporádicos sugerem que as mutações no caminho do gene patched (i.e., o caminho do hedgehog sônico) são inevitáveis e, com frequência, são a etapa suficiente no desenvolvimento da maior parte dos CBCs.10
Diagnóstico. A maior parte dos casos de CBCs ocorre na cabeça e no pescoço. Esses carcinomas basocelulares se apresentam nas formas nodular e superficial, assim como em uma grande variedade de formas menos comuns.
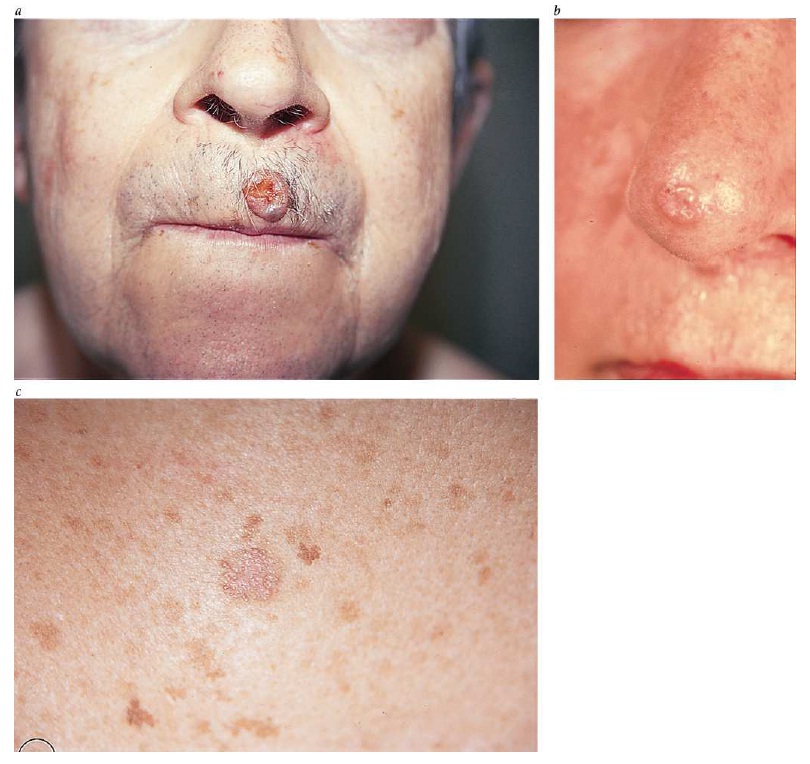
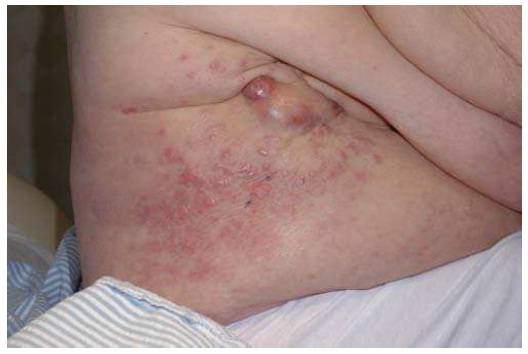
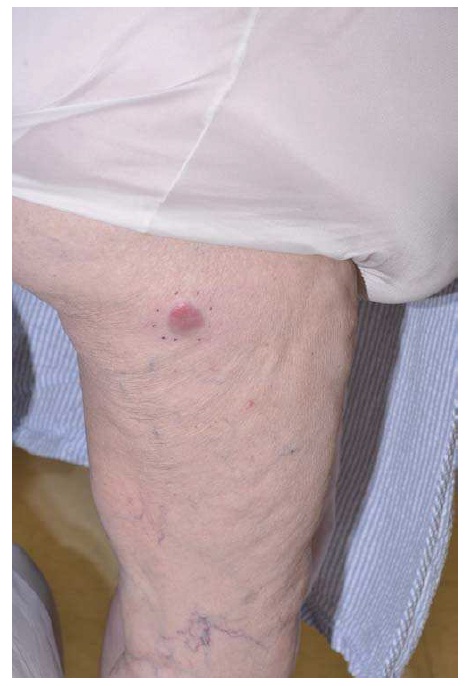
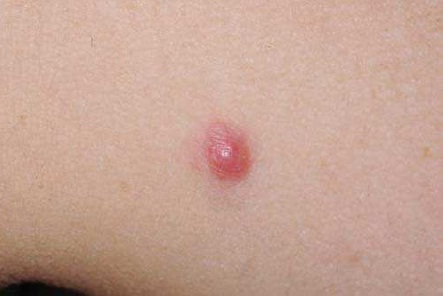
O CBC nodular surge na superfície da pele como uma protuberância rósea, elevada, perolada e translúcida. De maneira geral, essa protuberância se irrita facilmente, é frágil e está associada a episódios de ulceração superficial ou hemorragia. Nos casos em que for proeminente esse tipo de ulceração, apresenta uma aparência conhecida por úlcera de roedores, em que a borda translúcida perolada quase não é perceptível. Aparentemente, algumas lesões causadas por CBCs são mais brancas do que róseas e, quando observadas de perto, geralmente apresentam pequenas telangiectasias. Elas tendem a apresentar superfícies mais lisas e mais brilhantes, além de uma textura mais firme em comparação com os nevos dérmicos comuns [ver a Figura 1].
Os carcinomas basocelulares se apresentam como uma mancha rósea na pele. Em inspeções mais cuidadosas, a maior parte dos CBCs superficiais apresenta bordas filamentosas e translúcidas, com áreas de pele aparentemente normal ou ligeiramente fibrosas no interior da lesão. De maneira geral, encontram-se CBCs superficiais na parte superior do tronco, nos braços e nas pernas.
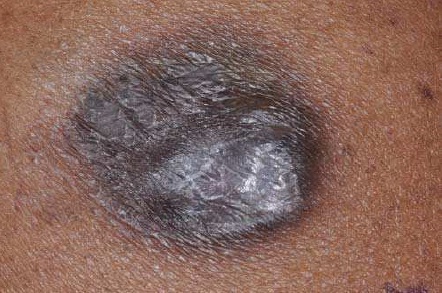
As variantes clínicas menos comuns de CBC incluem lesões morfeaformes, pigmentadas e císticas. Os carcinomas basocelulares morfeaformes apresentam um padrão infiltrativo que se assemelha a uma cicatriz, sob os pontos de vista clínico e histológico. Tipicamente, os carcinomas basocelulares pigmentados possuem manchas com pigmentos azul e preto, embora possam ser pigmentados em toda sua extensão. De maneira geral, as lesões pigmentadas são variantes dos CBCs. Os carcinomas basocelulares císticos tendem a ser mais macios do que os CBCs nodulares típicos e sua aparência varia de uma cor clara para um padrão cinza azulado.
O histórico dos pacientes desempenha papel importante no diagnóstico de carcinoma basocelular. Com frequência os pacientes alertam os médicos sobre a presença de lesões precoces que, em outras circunstâncias, poderiam dificultar a detecção, no momento em que fossem questionados sobre lesões facilmente irritáveis ou que sangram com pequenos traumas. Nos casos de suspeitas de lesões, os médicos devem obter biópsias mantendo os pacientes em anestesia local.
Diagnóstico diferencial. Os carcinomas basocelulares nodulares podem ser confundidos com angiofibromas, nevos dérmicos, melanomas amelanóticos, metástases cutâneas, dermatofibromas e uma grande variedade de tumores anexiais benignos (p.ex., tricoepitelioma). Os carcinomas basocelulares superficiais podem imitar diversos tipos de dermatose inflamatória (p.ex., eczema e tínea) e compartilham várias características clínicas com as queratoses actínicas. Os carcinomas basocelulares pigmentados podem ser facilmente confundidos com neoplasmas melanocíticos primários. Os carcinomas basocelulares císticos podem ser confundidos com tumores anexiais císticos e com lesões inflamatórias.
Tratamento. O objetivo terapêutico é erradicar adequadamente a lesão e assegurar os melhores resultados funcionais e cosméticos. Diversos fatores – como tamanho, localização e subtipo histológico das lesões e atributos do paciente, incluindo idade, estado geral de saúde, cor e flacidez da pele – devem ser levados em consideração na escolha da terapia ideal.
A grande maioria dos carcinomas basocelulares pode ser tratada por meios cirúrgicos. As opções principais incluem eletrodissecação e curetagem, excisão e cirurgia micrográfica de Mohs. Um grupo pequeno, porém significativo, de CBCs pode ser tratado efetivamente com a cirurgia micrográfica de Mohs, que inclui exame microscópico de secções de toda a parte de baixo da superfície do espécime que foi excisado no momento da cirurgia. Essa técnica é indicada para casos de lesões recorrentes e de lesões com grande probabilidade de recorrência. Essas lesões incluem lesões mal definidas, lesões de grande porte (> 2 cm), lesões com histologia de alto risco (p.ex., padrão de crescimento agressivo, padrão esclerosante ou envolvimento perineural) e lesões sobrejacentes a planos de fusão embrionária (p.ex., cantos dos olhos ou sulco nasofacial). A taxa de cura da técnica micrográfica de Mohs é significativamente mais elevada do que as taxas de cura de outros tratamentos para essas lesões de alto risco.11
A radioterapia possivelmente seja uma alternativa eficaz, indolor e bem tolerada que, tipicamente, é reservada para pacientes idosos que não sejam candidatos ideais para tratamento cirúrgico. No entanto, recomenda-se evitar o uso de radioterapia em pacientes com a síndrome do nevo de células basais. A crioterapia é outra opção terapêutica para tratamento de carcinomas basocelulares em pacientes que sejam bons candidatos cirúrgicos.
A terapia tópica em combinação com farmacoterapia usando o imiquimode, um modificador de respostas autoimunes, cinco vezes por semana durante seis semanas, foi aprovada pela Food and Drugs Administration (FDA) para tratamento de CBCs superficiais no tronco e nas extremidades. Uma embalagem (250 mg) de creme de imiquimode a 5% é suficiente para aplicar em 25 cm2 da pele afetada.
As terapias experimentais que se encontram em fase de investigação incluem quimioterapia intralesional, imunomodulação tópica de uma próxima geração e terapia fotodinâmica.
Todos os pacientes tratados para CBC correm o risco de recorrência local e um risco significativo de desenvolvimento de cânceres cutâneos adicionais. Todos os pacientes deverão ser orientados para fazer autoexame da própria pele e para a proteção da pele contra os efeitos dos raios solares. Além disso, esses pacientes devem receber acompanhamento profissional de rotina.
Prognóstico. O risco de recorrência local se relaciona ao tamanho, à localização e à histologia das lesões locais. As metástases são muito raras: uma prevalência de 0,0028% foi documentada em uma série de 50.000 australianos,12 embora outras séries tenham sugerido que a incidência poderá ser de até 0,55%.13 As metástases ocorrem através das vias linfáticas e hematogênicas; os fatores de risco incluem síndrome do nevo de células basais, imunossupressão e exposição prévia à radiação ionizante. As metástases que não estiverem sujeitas ao tratamento cirúrgico estão associadas a resultados pouco satisfatórios, embora o desenvolvimento recente de agentes que têm como alvo a via de sinalização hedgehog tenha resultado em avanços significativos no tratamento de CBCs localmente avançados, não ressectáveis e metastáticos.13-15 O medicamento vismodegib, um inibidor oral de moléculas pequenas da via hedgehog, foi aprovado pela FDA em 2012 para tratamento de CBC metastático e avançado que não sejam tratáveis por meios cirúrgicos ou por radioterapia. Existem relatos de taxas de resposta de 30% em casos de CBC metastático e de 43% em casos de CBC local em estado avançado, com um tempo mediano de duração de resposta de 7,6 meses.15 Eventos adversos como disgeusia, alopecia, fadiga e perda de peso foram comuns e ocorreram em aproximadamente 30% de pacientes.15

Figura 1: O carcinoma basocelular nodular – nesta figura aparece acima do lábio do paciente, com uma úlcera conhecida por úlcera de roedores (a) – geralmente se apresenta como uma protuberância rósea elevada, perolada e translúcida na superfície da pele (b). A forma superficial aparece como uma mancha rósea na pele (c).
Da mesma forma que os carcinomas basocelulares, os carcinomas de células escamosas (CCEs) surgem a partir de queratinócitos da epiderme. Sob a perspectiva histológica, as células de CCEs bem diferenciadas se assemelham às células da porção superior da epiderme.
Epidemiologia. Estima-se que entre 150.000 a 250.000 casos novos de carcinoma de células escamosas cutâneo tenham sido diagnosticados nos Estados Unidos em 1994.16 Entretanto, levando-se em consideração que a incidência de câncer de pele não melanoma (CPNM) tenha sido estimada em mais de 3.300.000 casos em 2006, e tendo em vista que os CCEs representam aproximadamente um quarto desse tipo de câncer, na realidade a incidência atual pode ser quatro vezes maior.8 A mortalidade estimada por CCE nos Estados Unidos em 1998 era de aproximadamente 0,5 em 100.000 pessoas. Várias linhas de dados sugerem que houve um aumento significativo na incidência de CCE. Na Austrália, por exemplo, a incidência de CCE aumentou 51% entre 1985 e 1990.17 Nos Estados Unidos, algumas das taxas mais elevadas de CPNM foram detectadas na região sudoeste. Uma pesquisa com base na população realizada no Novo México chegou à conclusão que a incidência de CCE duplicou em homens e mulheres entre 1978 e 1999.18
Etiologia e fatores de risco. Além dos raios solares, outros agentes etiológicos conhecidos que contribuem para o desenvolvimento de CCE cutâneo são radiação ionizante, carcinógenos químicos, queimaduras térmicas e feridas crônicas que não cicatrizam. Os carcinomas de células escamosas relacionados à exposição aos raios solares têm risco mais baixo de metástases e de morte em comparação com os casos de CCEs relacionados a outros tipos de exposição. Os fatores envolvidos na predisposição de CCEs causados pela exposição aos raios solares incluem pele clara, tendência para queimaduras e incapacidade para bronzear a pele.
Fisiopatologia e patogênese. Com frequência, os casos de CCE relacionados à exposição solar estão associados a lesões precursoras conhecidas por queratoses actínicas. Essas lesões ocorrem no couro cabeludo, na face, nas superfícies extensoras dos antebraços e na parte de trás das mãos. Elas têm a tendência de se apresentar como máculas ou pápulas róseas, com superfície grosseira e de forma irregular, em geral com menos de 5 mm de diâmetro. Com frequência, a sensibilidade é mais rápida do que a observação e geralmente são assintomáticas. A maioria dos pacientes com queratose actínica apresenta lesões múltiplas. Estima-se que o risco de CCE nesses pacientes seja de até 20%.19 Os carcinomas de células escamosas também podem ocorrer em peles aparentemente normais.
Os carcinomas de células escamosas (CCEs) na mucosa oral ou genital surgem em lesões precursoras que se denominam leucoplaquia ou eritroplaquia. Os CCEs mucosos estão associados a um risco significativo de metástases. A vigilância imunológica afeta a progressão dos carcinomas de células escamosas. A imunossupressão, tal como ocorre em receptores de transplantes e em pacientes com linfoma, está associada a uma alta incidência de CCE.20 Nesses pacientes, aparentemente, as infecções causadas pelo papilomavírus humano desempenham um papel etiológico juntamente com a exposição aos raios solares. Os CCEs tendem a ser mais agressivos em indivíduos imunocomprometidos.
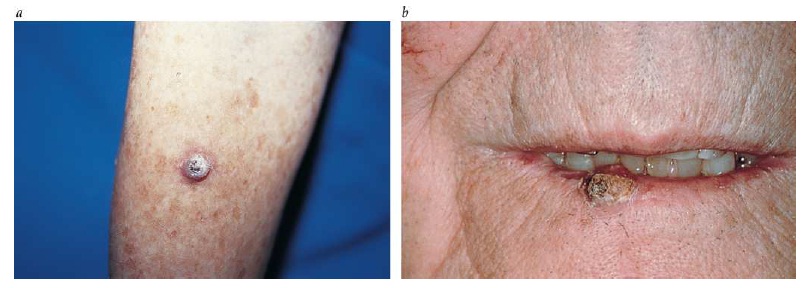
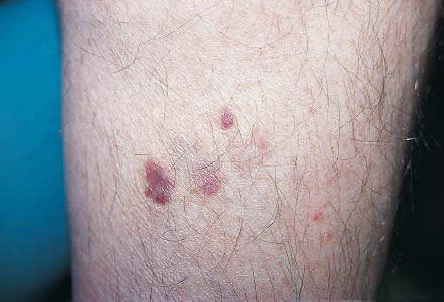
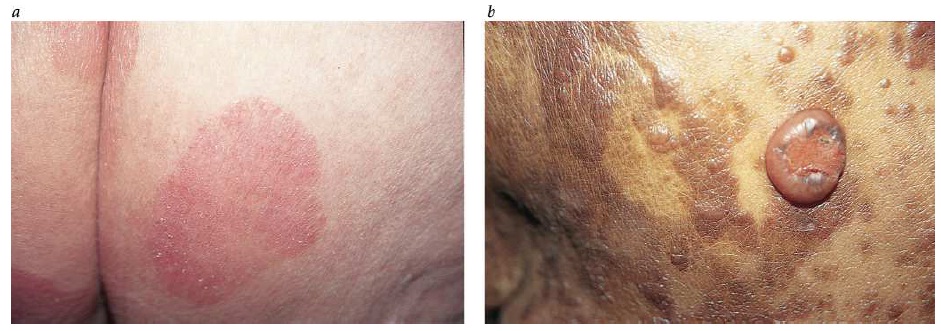
Diagnóstico. A maior parte das lesões ocorre em áreas do corpo que geralmente são expostas ao sol. As lesões se caracterizam pela presença de placas róseas firmes que, com frequência, têm superfícies grossas e escamosas [ver a Figura 2]. O diagnóstico definitivo depende de biópsias.
Figura 2: A figura mostra um carcinoma de células escamosas em um dos braços (a) e no lábio inferior (b).
Diagnóstico diferencial. O diagnóstico diferencial de CCE inclui queratoacantoma, doença de Bowen, carcinoma verrucoso, queratose actínica hipertrófica e verrugas comuns.
Sob os pontos de vista clínico e histológico, os queroacantomas compartilham muitas características com os carcinomas de células escamosas. Eles ressurgem em peles de aparência normal e crescem muito rapidamente. Tipicamente, os queroacantomas são protuberâncias róseas, brilhantes em forma de cúpula, com um plugue ceratótico central em forma de cratera que ocorre na superfície da pele. Eles podem atingir grandes proporções. Embora não estejam associados ao risco de metástase, os queroacantomas podem ser destrutivos em termos locais. Existem relatos indicando que houve regressão espontânea de queroacantomas durante o período de alguns meses.
A doença de Bowen é um carcinoma de células escamosas que se confina na epiderme. Esse tipo de carcinoma se apresenta como placas minimamente elevadas, vermelhas, escamosas e com bordas irregulares bem definidas. A associação documentada entre doença de Bowen e alguma malignidade interna não foi muito explicada em exames mais minuciosos.21
Carcinomas de células escamosas (CCEs) que não tiverem uma superfície ceratótica escamosa podem ser confundidos com hospedeiros de outros tumores cutâneos anexiais e dérmicos. O diagnóstico de CCE poderá ser sugerido, em vez de queratose actínica, em lesões semelhantes sob o ponto de vista clínico, pelas dimensões maiores e por sintomas como sensibilidade ou dor.
Tratamento. Carcinomas de células escamosas que evoluem a partir de queratose actínica podem ser tratados adequadamente com curetagem simples e eletrodessecação. A excisão cirúrgica definitiva, com confirmação de margens negativas, é o melhor tratamento para lesões actínicas maiores, assim como para lesões em áreas da pele não expostas aos raios solares. Em geral, as lesões mal definidas de alto risco, em especial aquelas que ocorrem em áreas da face sensíveis a cirurgias, órgãos genitais, mãos e pés são mais bem tratadas pela cirurgia micrográfica de Mohs.
Radioterapia fracionada é um tratamento alternativo para CCEs em pacientes idosos que sejam candidatos cirúrgicos fracos. Os benefícios da radioterapia adjuvante são menos evidentes, assim como os benefícios da biópsia de nodo linfático sentinela e dissecção eletiva de nodo linfático, para pacientes com CCE de alto risco na cabeça e no pescoço.
A quimioterapia citotóxica e os modificadores de respostas biológicas foram utilizados em pacientes com CCE em estado avançado; os relatos indicam que essa abordagem terapêutica apresentou taxas de respostas completas em até 68% dos casos, embora tenha havido poucos sobreviventes no longo prazo.22 As queratoses actínicas são tratadas com crioterapia, curetagem, terapias tópicas (p.ex., fluorouracil, imiquimode, diclofenaco ou ingenol), terapia fotodinâmica e resurfacing com laser para evitar a progressão para carcinomas de células escamosas.23 A National Comprehensive Cancer Network (NCCN) disponibiliza orientações atualizadas regularmente para tratamento de carcinomas de células escamosas e de carcinomas basocelulares.24
Prognóstico. Seja qual for a terapia utilizada, as lesões de alto risco apresentam uma taxa significativa de recorrência local dentro de cinco anos. Os carcinomas de células escamosas de alto risco incluem aqueles que se localizam em sítios anatômicos específicos (p.ex., orelhas, lábios, órgãos genitais e outras áreas não expostas aos raios solares), aqueles com mais de 2 cm de diâmetro, aqueles com características histológicas agressivas (profundidade > 4 mm, nível IV de Clarke ou acima, envolvimento perineural e histologia mal diferenciada), em pacientes imunossuprimidos.18,25 A via primária de metástases de CCEs é através da disseminação linfática para linfonodos regionais. As taxas de metástases documentadas variam de 0,3% em lesões pequenas causadas pela exposição ao sol, a 33% em lesões maiores não muito bem diferenciadas.25 As taxas globais documentadas de sobrevida de cinco anos para pacientes com CCE e metástases regionais variam de 25 a 47%.25
Nos Estados Unidos, o risco de uma pessoa desenvolver melanoma ao longo da vida é de aproximadamente de 1 em 58 indivíduos.4 Estima-se que nos Estados Unidos, no ano de 2002 ocorreram 76.250 casos novos de melanoma in situ e 9.180 mortes causadas pela doença.26 Entre os anos de 1973 e 1994 a incidência de melanoma aumentou 121% e a taxa de mortalidade aumentou 39%.27 As tendências encorajadoras recentes incluem mudança para a detecção da doença em sua fase inicial, assim como estabilização das taxas de incidência em alguns segmentos da população. Entretanto, em termos de morbidade e mortalidade, a carga de doenças relacionadas ao melanoma continua a aumentar. Embora o melanoma possa ocorrer em qualquer pessoa, trata-se principalmente de uma doença que acomete indivíduos de cor branca. Melanomas da variedade lentiginosa acral ocorrem mais frequentemente em indivíduos de cor negra.
Exposição aos raios solares. Embora fatores epidemiológicos e fortes evidências científicas básicas deem suporte a uma associação entre melanoma e exposição aos raios solares, aparentemente esse tipo de relação é muito complexo.28 Melanoma maligno do tipo lentigo está associado à exposição cumulativa aos raios solares no longo prazo. Aparentemente os melanomas com disseminação superficial e os melanomas nodulares estão associados à exposição solar intermitente intensa, em especial na juventude. A exposição em camas de bronzeamento aumenta a taxa de desenvolvimento de melanomas em mulheres jovens.29 Até o momento, o único teste controlado randomizado envolvendo filtros solares mostrou que há um impacto dos filtros solares 16 SPF sobre a incidência de melanomas.5 Os melanomas lentiginosos acrais não têm nenhuma associação aparente com exposição aos raios solares. Estudos científicos básicos e modelos animais implicaram comprimentos de onda UV diferentes na incidência de carcinogênese causada por melanomas; o comprimento de onda UV varia entre os vários tipos de melanoma.
Cor da pele. Os melanomas podem ocorrer em todos os grupos raciais ou étnicos, embora sejam muito mais comuns em indivíduos de pele mais clara. Foram identificados vários fatores de risco adicionais entre pessoas de pele branca, incluindo compleição clara, tendência para queimaduras, incapacidade para bronzear, formação de sardas e histórico familiar de melanoma.28 O rastreamento de membros da família de pacientes com melanoma (em particular melanomas múltiplos) possivelmente seja uma medida importante sob a ótica preventiva e diagnóstica.30
Moles e nevos displásicos. Os moles (nevos) são os marcadores fenotípicos mais fortes de risco de melanoma – mais especificamente, o aumento na quantidade de moles e a presença de moles atípicos (nevos displásicos). Os melanomas podem surgir em moles pré-existentes ou podem surgir novamente em peles aparentemente normais.
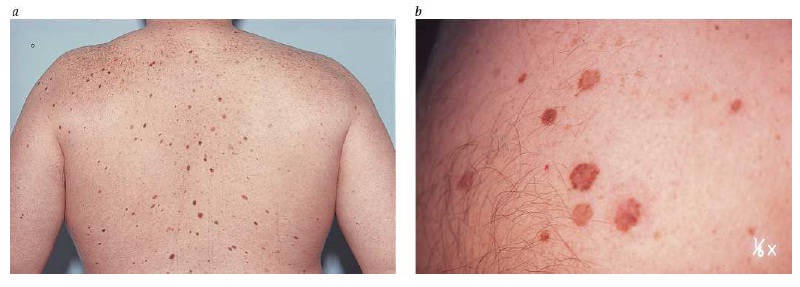
Diversos estudos epidemiológicos correlacionaram os nevos displásicos com o risco de ocorrência de melanomas. Sob a perspectiva clínica, os nevos displásicos são moles de grandes proporções (> 5 mm) com pigmentação variada e bordas mal definidas [ver a Figura 3]. Sob o ponto de vista histológico, os nevos displásicos se caracterizam pela presença de arquitetura atípica e citologia atípica randômica. O grau de risco de melanomas associados a nevos displásicos depende do contexto genético. Aparentemente o fenótipo de moles anormais é herdado de uma forma autossômica dominante em famílias com a síndrome familiar de nevo displásico e melanoma. O risco de melanoma se aproxima de 100% ao longo da vida nos membros dessas famílias com nevos displásicos.31 Fora do contexto de melanomas familiares, os nevos displásicos ocorrem em aproximadamente 5 a 15% da população branca. Nessa população geral, os nevos displásicos são marcadores de elevação no risco de melanoma [ver a Tabela 1].32
Aproximadamente 5% de pacientes com melanomas têm histórico familiar da doença. As mutações no gene p16 regulador do ciclo celular (inibidor da quinase 2a dependente da ciclina) estão associadas a melanomas em aproximadamente 40% das famílias com melanoma familiar.33 Mutações somáticas ativadoras altamente específicas no proto-oncogene BRAF (um dos membros da família RAF de quinases) são encontradas na maior parte dos melanomas e nevos benignos, sugerindo um papel extremamente importante para essa via genética na evolução de tumores melanocíticos.34 As análises genômicas estão começando a distinguir subgrupos biologicamente distintos de melanomas.35
Nos estágios facilmente curáveis, como as lesões pigmentadas que se localizam na superfície da pele, os melanomas são detectáveis precocemente pela simples inspeção visual. Caso não sejam tratados, os melanomas são as formas de câncer mais mortíferas e mais insensíveis sob a ótica terapêutica.
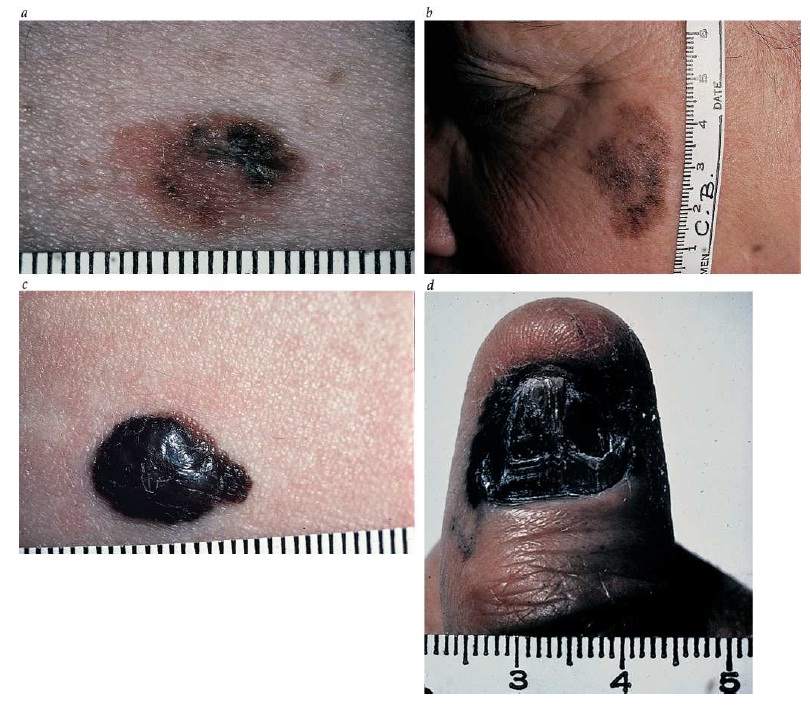
Exame físico. O reconhecimento precoce de um melanoma exige atenção especial às lesões pigmentadas em todas as superfícies do corpo. A despeito da forte associação de melanoma com exposição aos raios solares, esse tipo de câncer poderá ocorrer em qualquer parte da pele ou das mucosas. Portanto, o autoexame pelos próprios pacientes, assim os exames realizados por médicos, deve incluir todas as superfícies cutâneas, incluindo partes do corpo como couro cabeludo, órgãos genitais e sola dos pés. Qualquer lesão cutânea pigmentada com alterações recentes ou com as características descritas no método mnemônico ABCDE (do inglês, asymmetry, border irregularity, color variation, diameter > 6 mm, evolution [assimetria, bordas irregulares, variação na cor, diâmetro > 6 mm, evolução]) talvez sejam indicações sobre a possibilidade de melanoma. Embora quaisquer moles possam se alterar gradualmente ao longo do tempo, o mole que alterar a cor, a forma ou as dimensões em relação aos outros moles de um paciente precisam de uma atenção especial [ver a Figura 4].36 Os nevos displásicos são ao mesmo tempo uma oportunidade e um desafio no processo de detecção de melanomas. Por um lado, sua identificação permite focar com eficiência os grupos de alto risco. Por outro lado, podem complicar as tentativas de detecção de melanomas ao simular clinicamente melanomas precoces. Embora alguns nevos displásicos possam evoluir para melanomas, a esmagadora maioria permanece benigna. Além disso, nem todos os melanomas que surgem em pacientes com nevos displásicos desenvolvem de acordo com um modo pré-existente. A remoção de grandes quantidades de nevos displásicos não é uma abordagem muito prática para prevenção de melanomas. A detecção de melanomas em pacientes com nevos displásicos poderá ser prevista através de exames visuais específicos, com auxílio de autoexames e de acompanhamento profissional para identificar a presença de alterações em lesões.37
Figura 3: Tipicamente, os nevos displásicos são maiores que os moles comuns (a) e têm pigmentação variada e bordas mal definidas (b).
Assistência ao diagnóstico. Diversos tipos de assistência ao diagnóstico de melanoma em pacientes com nevos displásicos estão atualmente em fase de desenvolvimento. A dermoscopia envolve o uso de um dispositivo manual semelhante a um otoscópio que amplia as lesões pigmentadas com aplicação de pressão e de óleo sobre as superfícies. Esse tipo de técnica possibilita a visualização de padrões de pigmentos e de características que não são perceptíveis com a simples inspeção visual. Com experiência e treinamento a dermoscopia pode ser bastante útil para fazer a distinção entre melanoma e lesões pigmentadas benignas; no entanto, se for usada por pessoas inexperientes, a dermoscopia poderá diminuir a precisão diagnóstica.37-39 Acompanhamento assistido por fotografias é outra técnica que facilita a detecção de melanomas em indivíduos de alto risco.40 O uso de uma série de fotografias da pele de todo o corpo na linha de base durante os autoexames e os exames de acompanhamento profissional ajudam a avaliar as alterações que ocorrem nas lesões. Esse procedimento ajuda a evitar excisões desnecessárias de lesões estáveis e melhora a sensibilidade dos exames para detectar alterações. Novas tecnologias de imagens como a microscopia de rastreamento confocal a laser in vivo são as grandes promessas para aprimoramentos futuros no diagnóstico não invasivo de melanoma.41
|
Tabela 1: Estimativa de Riscos Relativos Ajustados de Melanoma por Tipo e Número de Nevos.32 | ||
|
Tipo |
Número |
Risco Relativo Ajustado*
|
|
Nevos > 2 mm e < 5 mm |
0-24 |
1,0 |
|
|
25-49 |
1,8 (1,3-2,5) |
|
|
50-99 |
3,0 (2,1-4,4) |
|
|
>= 100 |
3,4 (2,0-5,7)
|
|
Nevos não displásicos > 5 mm |
0 |
1,0 |
|
|
1 |
0,9 (0,7-1,3) |
|
|
2,4 |
1,3 (1,0-1,8) |
|
|
5-9 |
1,7 (1,0-2,7)
|
|
Nevos displásicos |
Nenhum |
1,0 |
|
|
Indeterminado |
1,0 (0,7-1,6) |
|
|
1 |
2,3 (1,4-3,6) |
|
|
2-4 |
7,3 (4,6-12,0) |
|
|
5-9 |
4,9 (2,5-9,8) |
|
|
>= 10 |
12,0 (4,4-31,0) |
*Mutuamente ajustados e ajustados pela idade, padrão de encaminhamento, nevos morfológicos displásicos com menos de 5 mm, queimaduras solares, sardas, danos solares, cicatrizes, excisões de nevos e histórico familiar de melanona (intervalo de confiança = 95%).
Excisão de espessura total e biópsia. Qualquer lesão que levantar suspeita clínica de melanoma exige diagnóstico definitivo. Excisão de espessura total é a técnica preferida para biópsias nos casos de suspeita de lesões pigmentadas. Biópsias parciais poderão levar a diagnósticos errôneos através de erros de amostragem ou impedindo que o patologista tenha uma visão de toda a arquitetura e citologia da lesão. Entretanto, as biópsias incisionais com boa correlação clínico-patológica possivelmente sejam apropriadas na avaliação de lesões maiores e de lesões que ocorrem em áreas cirurgicamente sensíveis. Não há evidências sugerindo que as biópsias incisionais aumentam o risco de metástase.
Os nevos displásicos compartilham muitas características com a fase inicial de disseminação de melanomas superficiais. Outras lesões comuns que poderão imitar melanoma incluem lentigo, sardas produzidas por queimaduras solares, nevos traumatizados, angiomas com trombose, carcinomas basocelulares, doença pigmentada de Bowen, dermatofibromas e queratoses seborreicas atípicas. Dois outros desafios no diagnóstico diferencial de melanoma merecem atenção especial. Os melanomas amelanóticos (melanomas sem pigmento) se apresentam como lesões róseas que poderão ser diagnosticados incorretamente como carcinomas basocelulares ou como nevos de Spitz. É muito difícil fazer a distinção entre nevos de Spitz e melanoma sob os pontos de vista clínico e histológico. Os nevos de Spitz ocorrem com mais frequência em crianças, embora também possa ocorrer em adultos. Os nevos de Spitz tendem a ocorrer repentinamente na cor vermelha variando a marrom com tom avermelhado.
Sítio primário. Os melanomas cutâneos primários são tratados cirurgicamente com novas excisões definitivas. As excisões amplas do passado deram lugar a novas excisões com margens mais modestas. Testes prospectivos múltiplos e randomizados investigaram ressecções cirúrgicas de melanomas cutâneos primários usando margens diferentes de ressecção; esses estudos tinham como foco populações variadas e sobrepostas de pacientes. Com embasamento nesses dados, a NCCN recomenda margens de ressecção de 1 cm para melanomas com menos de 1 mm de espessura, margens de 1 a 2 cm para melanomas entre 1 e 2 mm de espessura e margens de 2 cm para melanomas com mais de 2 mm de espessura. O fechamento primário e os retalhos reconstrutivos são preferíveis para enxertos cutâneos, sob os pontos de vista cosmético e funcional, e deverão ser usados ao invés de enxertos sempre que for possível.42,43
Linfonodos linfáticos. Os pacientes com doenças linfonodais regionais clinicamente evidentes devem ser tratados com dissecção terapêutica dos Linfonodos.42 A dissecção nodular eletiva em pacientes com melanomas primários e sem evidências clínicas de envolvimento dos linfonodos foi abandonada com base na incapacidade de muitos testes randomizados múltiplos para demonstrar o benefício de sobrevida total com esse procedimento.
A biópsia de linfonodos sentinelas vem sendo utilizada cada vez mais em pacientes com melanomas cutâneos primários. Essa técnica se fundamenta no uso de linfocintilografia para identificar bacias de linfonodos regionais que estiverem drenando para a pele de sítios de melanomas primários. No momento da nova excisão definitiva do melanoma, injetam-se um corante azul e um radioisótopo na derme ao redor do sítio do melanoma. A seguir, faz-se uma pequena incisão no ponto que foi identificado na linfocintilografia como área proximal de drenagem da bacia do linfonodo regional. Em seguida faz-se a excisão do primeiro linfonodo no momento em que se retirar o corante azul e o radioisótopo.
Figura 4: Os melanomas malignos com disseminação superficial iniciam como uma pequena lesão marrom de forma irregular (a). Variações na cor e no contorno são típicas de melanomas malignos tipo lentigo (b). Com frequência, os melanomas nodulares crescem mais em termos de espessura do que no diâmetro (c). Os melanomas lentiginosos acrais se assemelham a um hematoma sob a unha (d).
A seguir, avalia-se o linfonodo sentinela, em geral com auxílio de técnicas imunohistoquímicas e, ocasionalmente, com reação em cadeia da polimerase, que é um método mais sensível. A ausência de melanoma no linfonodo sentinela é altamente sensível para excluir a presença de metástases no remanescente da bacia do linfonodo, nas situações em que o procedimento for executado por equipes experientes. De maneira geral, no momento em que se perceber que o linfonodo é positivo para melanoma, “conclui-se” a dissecção da bacia afetada. Alguns estudos prospectivos demonstraram que o estado de um linfonodo sentinela está fortemente correlacionado com a sobrevida de cinco anos.44 Pacientes com linfonodos sentinelas são considerados candidatos apropriados para terapias adjuvantes [ver Terapias Adjuvantes mais adiante]. Vários testes com múltiplos centros encontram-se atualmente em andamento com a finalidade de avaliar a utilidade clínica desse tipo de procedimento.45 Os relatórios iniciais do primeiro desses testes não conseguiram identificar vantagens globais de sobrevida associadas à aplicação desse procedimento.46
Metástases em trânsito. Metástases em trânsito são metástases que produzem tumores no interior de linfonodos dérmicos e subcutâneos antes de atingirem linfonodos regionais. As metástases em trânsito podem se confinar em um único membro por períodos prolongados de tempo. Nesse contexto, as amputações aparentemente não produzem benefícios de sobrevida no longo prazo.42 As metástases em trânsito com crescimento individual lento podem receber tratamento cirúrgico. Doenças mais extensivas poderão ser tratadas com terapia de sensibilização com dinitroclorobenzeno, interferon intralesional ou agentes tópicos, para modificar a resposta imune. Tratamentos como perfusão isolada de membro ou terapia com infusão, nos casos de metástases em trânsito confinadas em uma extremidade, poderão resultar em uma mitigação dramática e no salvamento do membro. O procedimento implica no isolamento dos vasos da extremidade envolvida em relação aos vasos sistêmicos e na perfusão do membro isolado com agentes quimioterápicos, agentes biológicos, ou ambos, em doses toleráveis se forem administradas de forma sistêmica.47 A radioterapia também desempenha um papel paliativo importante.
Metástases distantes. No passado, a terapia sistêmica para tratamento de melanomas metastáticos oferecia poucas esperanças de respostas sustentadas significativas. A monoterapia com dacarbazina (2 a 4,5 m/kg por 10 dias, repetida a cada 4 semanas) ou com interleucina-2 recombinante (IL-2) (600.000 IU/kg em intervalos de 8 horas em até 14 doses) era o único regime de tratamento aprovado pela FDA para casos de melanomas metastáticos. Observam-se respostas objetivas à dacarbazina em aproximadamente 5 a 20% de pacientes; as respostas completas de longa duração são raras.48 As respostas objetivas à IL-2, um agente muito mais tóxico, são observadas em aproximadamente 15% de pacientes; as respostas de longa duração são observadas em torno de 5% de casos. A radioterapia possivelmente tenha um papel paliativo importante. Entretanto, dois avanços importantes foram aprovados pela FDA em 2011: ipilimumab e vemurafenib. Observou-se que o uso de ipilimumab, que bloqueia o antígeno 4 associado ao linfócito T citotóxico (CTLA4, do inglês cytotoxic T lymphocyte, antigen 4) melhora a sobrevida em pacientes com melanoma metastático.49,50 O vemurafenib, que tem como alvo a mutação BRAF V600E encontrada em cerca de 40% de melanomas metastáticos, foi associado a taxas de melhora na sobrevida total e sem progressão em pacientes com melanoma metastático e com esse tipo de mutação.51 Agentes mais recentes e combinações dessas “imunoterapias” e “terapias com alvo nas vias” entraram na etapa de testes clínicos e são bastante promissores para futuras melhoras nas taxas de resposta de longa duração.52
Terapias adjuvantes. Pacientes com doenças cutâneas ou regionais ou que não apresentarem doenças causadas por meios cirúrgicos, mas que tenham alto risco de recorrência de metástases são candidatos potenciais para terapias adjuvantes. Diversos tipos de terapias adjuvantes foram aplicados em casos de melanoma, incluindo estimulantes imunes como o bacilo Calmette-Guérrin, Corynebacterium parvum e levamisol. Vários agentes quimioterápicos também foram testados. Mais recentemente, foram realizados alguns estudos envolvendo imunoterapias com citocinas, como os interferons e imunização ativa com vacinas. O regime com doses elevadas de interferon-alfa (administração intravenosa diária de 20 milhões de unidades/m2 durante um mês, seguida pela administração subcutânea de 10 milhões de unidades/m2 três vezes por semana durante 48 semanas) foi aprovado pela FDA como terapia adjuvante nos casos de melanoma, assim como o interferon peguilado.53 Alguns estudos demonstraram que houve uma melhora pequena, porém significativa sob o ponto de vista estatístico, na sobrevida total com aplicação desse regime.50 Diversos estudos não conseguiram demonstrar melhoras na sobrevida total no longo prazo com a terapia adjuvante à base de interferon em regimes com doses variando de intermediária a baixa.49,54
Uma grande quantidade de novas estratégias, tais como a imunização ativa, incluindo imunização passiva e uma miríade de terapias biológicas, encontra-se atualmente em fase de estudo e poderá criar oportunidades para pacientes que sejam candidatos apropriados para testes.55 As estratégias especialmente promissoras são as tentativas atualmente em curso para utilizar medicamentos recentes comprovadamente eficazes nos casos de doença metastática avançada no contexto de terapias adjuvantes.
Estágio. O único fator prognóstico mais forte para melanoma é o estágio da doença. Várias classificações de fases têm sido utilizadas ao longo do tempo. Todos os sistemas de classificação de fases aplicáveis aos melanomas levam em consideração a classificação clássica TNM que inclui o tamanho de tumores (T), o envolvimento de linfonodos (N) e as metástases distantes (M). Em grande parte, as diferenças entre os sistemas de classificação de fases se relacionam à determinação das fases do sítio primário. Os sistemas de classificação de fases mais recentes tentam usar os atributos do tumor primário que se correlaciona fortemente com o resultado. Esses atributos incluem espessura, ulceração, taxa mitótica e nível de invasão de Clark no caso de melanomas finos com menos de 1 mm de espessura.56
O advento da biópsia de nodo sentinela levou à inclusão do microestagiamento de linfonodos nos sistemas de classificação de fases [ver as Tabelas 2 e 3].56-58
|
Tabela 2: Classificação da AJCC TNM57 | |||
|
Classificação T |
Espessura (mm) |
Estado de Ulceração/Mitoses
| |
|
T1 |
>=1 |
a: Sem ulceração e mitose < 1/mm2. b: Com ulceração ou mitose >= 1/mm2.
| |
|
T2 |
1,01 a 2,00 |
a: Com ulceração. b: Sem ulceração ou mitose >= 1/mm2.
| |
|
T3 |
2,01 a 4,00 |
a: Sem ulceração. b: Com ulceração.
| |
|
T4 |
> 4,00 |
a: Sem ulceração. b: Com ulceração. | |
|
Classificação N |
Número de Linfonodos Metastáticos |
Carga Metastática Nodal
| |
|
N0 |
0 |
Não aplicável.
| |
|
N1 |
1 |
a: Micrometástase* b: Macrometástase l
| |
|
N2 |
2 a 3 |
a: Micrometástase* b: Macrometástase l c: Metástases em trânsito/satélites sem Linfonodos metastáticos.
| |
|
N3 |
4 + Linfonodos metastáticos ou Linfonodos emaranhados, metástases em trânsito/ satélites sem nodos metastáticos. |
| |
|
Classificação M |
Sítio |
LDH sérico
| |
|
MO |
Metástases não distantes. |
Não aplicável.
| |
|
M1a |
Metástases nodais cutâneas ou subcutâneas distantes. |
Normal
| |
|
M1b |
Metástases pulmonares. |
Normal
| |
|
M1c |
Todas as outras metástases viscerais. Qualquer metástase distante. |
Normal
Elevado | |
|
AJCC = American Joint Committee on Cancer; LDH (lactate dehydrogenase) = lactato desidrogenase. |
| ||
*As micrometástases são diagnosticadas depois de biópsias no linfonodo sentinela.
l As macrometástases são definidas como metástases nodais detectáveis clinicamente e confirmadas patologicamente.
Atributos de tumor primário. Diversos atributos de tumores primários foram identificados como preditores de resultados a partir de melanomas cutâneos primários. A espessura tumoral de Breslow é um forte preditor de resultados, que é medida em milímetros desde a camada granular da epiderme até a célula tumoral mais profunda. Outros parâmetros histológicos importantes são o nível de invasão tumoral de Clark, ausência ou presença de ulceração, taxa de mitose, presença de linfócitos com infiltração tumoral e invasão vascular. A fase de crescimento é um dos preditores mais fortes de resultado nos casos de melanomas primários.59 Aparentemente, os melanomas na fase de crescimento radial não metastizam, enquanto que os melanomas na fase de crescimento vertical (que se caracterizam pela formação de um nódulo tumoral na derme) estão associados a um risco significativo de metástase, mesmo em lesões com menos de 1 mm de espessura.60 As características dos pacientes associadas à melhora na sobrevida nos casos de melanoma incluem idade mais jovem (< 60 anos), sexo feminino e localização do melanoma em uma das extremidades, ao invés das palmas das mãos ou solas dos pés. Foram desenvolvidos modelos multivariáveis para previsão de resultados de melanomas [ver a Tabela 4].60 O desenvolvimento de cálculos eletrônicos ajudou a prever prognósticos para pacientes individuais com base nos dados de grandes grupos de dados colaborativos.61
As metástases cutâneas ocorrem em aproximadamente 5% de pacientes com tumores sólidos e geralmente estão associadas a doenças generalizadas. A frequência relativa de metástases cutâneas é específica de gênero e reflete as taxas de cânceres primários.62 Em mulheres, dois terços das metástases são de câncer de mama, embora também sejam frequentes a partir de câncer de pulmão, câncer colorretal, melanoma e câncer de ovário. Em homens, o câncer de pulmão é mais comum, seguido pelo câncer do intestino grosso, melanoma, carcinoma de células escamosas na cabeça e no pescoço, e câncer renal.62 A distribuição anatômica das metástases cutâneas não é aleatória. De maneira geral, as metástases cutâneas de câncer de mama envolvem a parede torácica e poderão se apresentar como nódulos, linfedema ou celulite.
|
Tabela 3: Sistema de Determinação de Fases da AJCC TNM e Taxas de Sobrevida57 | |||
|
Estágio Patológico
|
TNM |
Sobrevida de 5 Anos |
Sobrevida de 10 Anos |
|
IA |
T1a |
95,3 ± 0,4 |
87,9 ± 1,0
|
|
IB |
T1b |
90,9 ± 1,08 |
83,1 ± 1,5
|
|
|
T2a |
9,0 ± 0,7 |
79,2 ± 1,1
|
|
IIA |
T2b |
77,4 ± 1,7 |
64,4 ± 2,2
|
|
|
T3a |
78,7 ± 1,2 |
63,8 ± 1,7
|
|
IIB |
T3b |
63,0 ± 1,5 |
50,8 ± 1,7
|
|
|
T4a |
67,4 ± 2,4 |
53,9 ± 3,3
|
|
IIC |
T4b |
45,1 ± 1,9 |
32,3 ± 2,1
|
|
IIIA |
N1a |
69,5 ± 3,7 |
63,0 ± 4,4
|
|
|
N2a |
63,3 ± 5,6 |
56,9 ± 6,8
|
|
IIIB |
N1a |
52,8 ± 4,1 |
37,8 ± 4,8
|
|
|
N2a |
49,6 ± 5,7 |
35,9 ± 7,2
|
|
|
N1b |
59,0 ± 4,8 |
47,7 ± 5,8
|
|
|
N2b |
46,3 ± 5,5 |
39,2 ± 5,8
|
|
IIIC |
N1b |
29,0 ± 5,1 |
24,4 ± 5,3
|
|
|
N2b |
24,0 ± 4,4 |
15,0 ± 3,9
|
|
|
N3 |
26,7 ± 2,5 |
18,4 ± 2,5
|
|
IV |
M1a |
18,8 ± 3,0 |
15,7 ± 2,9
|
|
|
M1b |
6,7 ± 2,0 |
2,5 ± 1,5
|
|
|
M1c |
9,5 ± 1,1 |
6,0 ± 0,9 |
AJCC = American Joint Committee on Cancer.
O couro cabeludo é um sítio comum de metástases, principalmente de câncer pulmonar e renal (em homens) e câncer de mama (em mulheres). Os cânceres de cabeça e pescoço podem invadir a pele por extensões locais, dando origem a edemas cutâneos firmes e de cor vermelha escura que se assemelham a celulite. As metástases na parede abdominal, conhecidas por nódulos da Irmã Mary Joseph, podem ocorrer juntamente com malignidades gastrointestinais ou ovarianas.62 Sob a perspectiva clínica, em geral as metástases cutâneas são nódulos ou pápulas dérmicas minimamente sintomáticas de cor pastel ou rosa; sua disseminação ocorre através dos vasos linfáticos ou de vias vasculares [ver a Figura 5]. As metástases cutâneas podem refletir clinicamente a histologia de tumores primários (p.ex., nódulos de cor negra, marrom ou cinza com melanomas metastáticos e nódulos vasculares com células renais ou carcinomas tireoideos).
As malignidades primárias da derme podem se desenvolver a partir de uma miríade de estruturas cutâneas, incluindo glândulas sebáceas (carcinoma sebáceo), tecidos conjuntivos (dermatofibrossarcoma protuberante), tecidos lisos (leiomiossarcoma) e outros tecidos anexiais (carcinomas écrinos). A ocorrência da maior parte dos neoplasmas dérmicos primários é muito rara; esses neoplasmas podem apresentar comportamento biológico agressivo. Embora apresentem diversas histologias, muitos desses neoplasmas compartilham a apresentação clínica comum dos nódulos subcutâneos de crescimento rápido, variando da cor pastel a rosa que, ocasionalmente, se assemelham a cistos sebáceos.
Este tipo de neoplasma é uma malignidade dérmica com origem neuroendócrina que se apresenta como uma pápula ou nódulo dérmico cuja coloração varia da cor vermelha à cor violácea na cabeça ou pescoço de pessoas idosas e/ou de pacientes imunocomprometidos, embora outros sítios [ver a Figura 6] e outros grupos etários também sejam afetados.63,64 O tratamento de escolha é a excisão local ampla, com ou sem linfodenectomia. Alguns especialistas propuseram biópsia de Linfonodos sentinelas para avaliação de linfonodos regionais.
|
Tabela 4: Probabilidade Estimada de Sobrevida de 10 anos em Pacientes com Melanoma Cutâneo Primário60 | ||||
|
Espessura Tumoral (mm)/Idade (anos) dos Pacientes |
Probabilidade de Sobrevida de 10 Anos*
| |||
|
Tumor Localizado nas Extremidades |
Tumor Com Localização Axiall
| |||
|
Mulheres |
Homens |
Mulheres |
Homens
| |
|
< 0,76 |
|
|
|
|
|
<= 60 |
0,99 (0,98-1,0) |
0,98 (0,95-0,99) |
0,97 (0,93-0,99) |
0,94 (0,88-0,97) |
|
>60 |
0,98 (0,95-0,99) |
0,96 (0,89-0,98) |
0,92 (0,82-0,96) |
0,84 (0,70-0,93)
|
|
0,76-1,69 |
|
|
|
|
|
<= 60 |
0,96 (0,92-0,98) |
0,93 (0,85-0,97) |
0,86 (0,76-0,92) |
0,75 (0,62-0,84) |
|
>60 |
0,90 (0,80-0,95) |
0,81 (0,64-0,91) |
0,67 (0,50-0,81) |
0,50 (0,33-0,67) |
|
1,70-3,60 |
|
|
|
|
|
<= 60 |
0,89 (0,64-0,91) |
0,80 (0,65-0,89) |
0,65 (0,50-0,77) |
0,48 (0,35-0,61) |
|
>60 |
0,73 (0,57-0,85) |
0,57 (0,38-0,75) |
0,38 (0,24-0,55) |
0,24 (0,14-0,37)
|
|
>3,60 |
|
|
|
|
|
<= 60 |
0,74 (0,53-0,87) |
0,58 (0,36-0,77) |
0,39 (0,21-0,60) |
0,24 (0,13-0,40) |
|
>60 |
0,48 (0,28-0,69) |
0,32 (0,16-0,53) |
0,18 (0,08-0,35) |
0,10 (0,04-0,20) |
*Intervalo de confiança = 95%.
? A localização axial inclui tronco, cabeça, pescoço e sítios volares e sublinguais.
A radioterapia adjuvante é uma opção a ser considerada. As recorrências locais são frequentes, sendo que as metástases distantes ocorrem em mais de um terço de pacientes. De um modo geral os resultados da quimioterapia são decepcionantes.63,64 Recentemente descobriu-se que um novo poliomavírus está integrado no genoma do carcinoma da célula de Merkel humana, sugerindo que esse tipo de tumor poderá ser incluído nas listas de cânceres com associação viral.65
Doença de Paget é uma malignidade cutânea rara associada a um adenocarcinoma subjacente que, usualmente, se apresenta como uma dermatite eritematosa unilateral secretora nos seios envolvendo os mamilos e as aréolas. O diagnóstico diferencial inclui eczema, psoríase, dermatite de contato e impetigo. Por essa razão, as biópsias de dermatite inflamatória sem solução dos mamilos e aréolas são imprescindíveis. Na doença de Paget as biópsias revelam a presença de células de Paget com coloração clara típica na epiderme. O tratamento de escolha é a ressecção cirúrgica de neoplasmas cutâneos e subjacentes; com frequência ocorrem metástases de linfonodos.63
Figura 5: Metástases cutâneas de carcinoma ovariano.
Figura 6: Carcinoma da célula de Merkel.
A doença extramamária de Paget é ainda menos comum do que a doença de Paget. Em geral ela se apresenta como placas ulceradas, geralmente de cor vermelha, nas áreas perineais de pessoas idosas.62,63 As lesões podem ser pruríticas ou assintomáticas, com frequência têm longa duração, e poderão ser diagnosticadas incorretamente como psoríase, dermatite de contato ou infecção fúngica crônica. Os tumores subjacentes associados incluem carcinomas retais e genitourinários. Mesmo sem uma malignidade interna associada, a doença extramamária de Paget é difícil de tratar e está associada a uma taxa elevada de recorrência local.63
Com frequência, angiossarcoma é uma malignidade vascular rara e altamente agressiva63 que se apresenta como manchas ou nódulos multicêntricos vermelho-púrpura em membros linfadematosos, como, por exemplo, em um braço linfadematoso depois de uma mastectomia (síndrome de Stewart-Treves). Outras apresentações são manchas ou placas violáceas na cabeça ou pescoço (principalmente no couro cabeludo) de pessoas idosas. As abordagens cirúrgicas raramente são eficazes em lesões precoces.64 O prognóstico de pacientes com angiossarcoma não é bom, sendo comum o desenvolvimento de metástases pulmonares, mesmo com cirurgia ou aplicação de radiação.63-65 Os pacientes com angiossarcoma local avançado ou metastático apresentam alguma resposta ao bevacizumab, um anticorpo humano recombinante contra o fator de crescimento do endotélio vascular (VEGF, do inglês vascular endothelial growth factor), administrado isoladamente65 ou em combinação com radioterapia.66,67
Dermatofibrossarcoma protuberante é uma malignidade local agressiva com crescimento lento que raramente metastiza, porém, em geral, é recorrente. Tipicamente, as lesões se apresentam como nódulos firmes de cor púrpura ou avermelhada com tonalidades de marrom e, usualmente, ocorrem no tronco ou nas extremidades não expostas aos raios solares, embora possam ser pigmentadas em indivíduos de pele mais escura [ver a Figura 7].
Figura 7: Dermatofibrossarcoma protuberante em um paciente de pele mais escura.
O diagnóstico diferencial inclui queloides e dermatofibroma benigno. Os adultos jovens são afetados com mais frequência, embora esse tipo de tumor possa ocorrer em qualquer idade. As excisões locais amplas, com ou sem cirurgia micrográfica de Mohs, oferecem melhores chances de cura.51 O imatinib, um inibidor da quinase tirosina de moléculas pequenas, foi aprovado pela FDA em 2009 para uso local em dermatofibrossarcomas protuberantes metastáticos abrigando t(17;22). Esse agente, que inibe o fator de crescimento-ß derivado das plaquetas (PDGFß, do inglês platelet-derived growth factor-ß), produziu taxas de respostas totais de 46% em pacientes com doença avançada.68
O sarcoma de Kaposi (SK) é um neoplasma cutâneo multicêntrico com quatro variantes clínicas distintas.69,70 A despeito do nome, o SK não é um sarcoma autêntico. Embora a célula de origem não tenha sido bem definida,69,70 as células do sarcoma de Kaposi compartilham marcadores fenotípicos com o endotélio linfático, assim como células de músculos lisos vasculares, sugerindo uma origem de células mesenquimatosas vasculares ou pluripotentes.70 Na forma clássica, o sarcoma de Kaposi é uma doença indolente que acomete homens idosos de origem mediterrânea ou provenientes da Europa Oriental, nos quais nódulos e placas violáceas se desenvolvem nas extremidades inferiores.69,70 Uma segunda variante, o sarcoma de Kaposi linfadenopático, é endêmica em algumas áreas da África. O sarcoma de Kaposi africano, que tipicamente afeta adultos jovens e crianças, segue um curso mais agressivo do que o SK clássico, com envolvimento frequente dos ossos, linfonodos e vísceras.69,70 Uma terceira variante de sarcoma de Kaposi ocorre em pacientes com imunossupressão iatrogênica, principalmente receptores de transplantes de órgãos.57 Nessa variante, os homens são afetados com uma frequência ligeiramente maior que as mulheres.69,70 A quarta variante corresponde a um tipo agressivo de sarcoma de Kaposi que ocorre em pacientes aidéticos.
Antes do advento da AIDS, o sarcoma de Kaposi era uma doença rara nos Estados Unidos, com incidência anual ajustada pela idade de 0,29 por uma população de 100.000 em homens e de 0,07 por uma população de 100.000 em mulheres.66 Nos primeiros anos da epidemia de HIV, o sarcoma de Kaposi era uma enfermidade que definia a presença de AIDS em 30 a 40% de pacientes.66 Durante aquele período, a incidência de SK em homens homossexuais infectados pelo HIV era 73.000 vezes mais elevada que na população norte-americana em geral; em mulheres infectadas pelo HIV e em homens heterossexuais infectados pelo HIV a incidência era 10.000 mais elevada.66,69 A incidência de SK declinou significativamente desde a introdução da terapia antirretroviral altamente ativa (HAART, do inglês highly active antiretroviral therapy). Por exemplo, em um grande estudo europeu envolvendo pacientes infectados pelo HIV houve uma redução anual estimada de 39% na incidência de sarcoma de Kaposi entre 1994 e 2003, de modo que a incidência de SK em 2003 foi 10% menos do que os casos registrados em 1994.71
Herpesvírus humano tipo 8. Há bastante tempo a epidemiologia de SK sugere a presença de um cofator ou de um agente infeccioso transmissível.66,69 O herpevírus associado ao sarcoma de Kaposi, também conhecido por herpesvírus humano tipo 8 (HHV-8, do inglês human herpes vírus type 8), foi detectado em todas as variantes do sarcoma de Kaposi.71 O HHV-8 foi encontrado também em pacientes com linfoma nas cavidades corporais, doença de Castleman e linfadenopatia angioblástica, assim como em determinados tipos de lesão na pele de receptores de transplantes de órgãos.63 Embora o mecanismo através do qual as infecções causadas pelo HHV-8 levam à tumorigênese por SK não seja muito claro, provavelmente envolva uma combinação complexa de inflamação, angiogênese e proliferação neoplásica.69,70 Em grande parte, a prevalência de sarcoma de Kaposi se compara à taxa de infecção por HHV-8 em várias populações.66 Embora a incidência de infecções causadas pelo HHV-8 atinjam 2 a 10% na população em geral, a incidência de sarcoma de Kaposi é muito baixa, sugerindo que a maior parte das infecções é subclínica.66,69
Fatores de hospedeiro. Os fatores de hospedeiro, particularmente a imunossupressão, são cruciais em algumas populações com sarcoma de Kaposi.66,69 O HIV pode ter um papel indireto no desenvolvimento de SK através da depleção das células T CD4+ e da estimulação da produção de fatores de crescimento e de citocinas como a IL-1 e a IL-6.71-73 Os medicamentos imunossupressivos, em especial ciclosporina, azatioprina e prednisona, aumentam o risco de SK, principalmente em receptores de transplantes de rim e fígado.69
A despeito da prevalência do sarcoma de Kaposi em alguns grupos étnicos, o papel de quaisquer fatores genéticos possíveis não é muito claro. Há alguns debates sobre aumentos na incidência de HLA-DR5 em pacientes com SK clássico.66 Sarcoma de Kaposi familiar é extremamente raro, sugerindo que os fatores genéticos isoladamente não são responsáveis pela doença.
Para finalizar, o gênero aparentemente é um fator de risco significativo, em especial nos casos de sarcoma de Kaposi clássico, em que a razão entre homem e mulher varia de 3:1 a 10:1.66 As razões dessa predominância masculina permanece inexplicável.69,70
Manifestações clínicas. As manifestações clínicas de sarcoma de Kaposi diferem entre as variantes do distúrbio.66 Nos casos de máculas ou manchas vermelho-púrpura clara ou nódulos de cor púrpura aparecem primeiramente nos pés, principalmente nas solas dos pés. A presença de linfadenopatia (principalmente inguinal) ocorre em ocasiões raras. Eventualmente, as lesões poderão ocorrer também nos braços e nas áreas genitais. Na medida em que a doença evolui, as lesões coalescem em placas violáceas.
Usualmente, os casos de sarcoma de Kaposi se apresentam como lesões cutâneas, embora as primeiras lesões possivelmente surjam na mucosa oral ou em linfonodos. Ao contrário das lesões clássicas causadas pelo sarcoma de Kaposi, as lesões causadas por SK associado ao HIV geralmente iniciam na parte superior do corpo (face, tronco ou braços). De uma forma mais geral, as lesões por SK associadas ao HIV se apresentam na cor púrpura avermelhada, geralmente com pápulas ovais que seguem uma distribuição semelhante à da pitiríase rósea.53-55 As lesões variam de manchas róseas a placas púrpuras [ver a Figura 8] ou podem se assemelhar a equimoses, principalmente em pacientes com contagem baixa de células T CD4+. Tipicamente, as lesões orais se caracterizam pela presença de placas vermelho-púrpura ou nódulos no palato, nas gengivas ou na mucosa bucal. Os pacientes com pele mais escura podem apresentar lesões variando da cor vermelho-púrpura à cor negra ou placas hiperpigmentadas.66
Figura 8: As lesões associadas ao HIV causadas pelo sarcoma de Kaposi variam de manchas róseas (mostradas na figura) as placas de cor roxa escura.
Na medida em que o sarcoma de Kaposi associado ao HIV evolui, possivelmente ocorram linfedemas nos pés, escroto, órgãos genitais e regiões periorbitais, com provável ocorrência de linfadenopatia (principalmente inguinal). Usualmente, as lesões gastrointestinais são submucosas e assintomáticas, embora possam provocar hemorragia gastrointestinal.66
Estudos laboratoriais. O exame laboratorial completo de pacientes com sarcoma de Kaposi inclui testes de anticorpos de HIV, hemograma completo e radiografia torácica. As contagens de células T CD4+são indicadas nos casos de pacientes com HIV positivo. É extremamente importante a obtenção de históricos e exames físicos completos, com atenção especial na presença de infecções oportunistas em pacientes infectados pelo HIV ou em pacientes imunossuprimidos. As biópsias da pele são importantes em pacientes com suspeita de sarcoma de Kaposi. A histopatologia de SK se caracteriza pela presença de células em forma de fuso na derme e pela presença de eritrócitos extravasados nas fendas entre espaços vasculares irregulares.69
O diagnóstico diferencial de sarcoma de Kaposi inclui dermatofibroma, púrpura, granuloma piogênico, angiomatose bacilar, melanoma metastático e carcinomas basocelulares. Outras entidades histopatológicas que podem se assemelhar ao sarcoma de Kaposi incluem angiossarcoma e dermatite de estase.69
Sarcoma de Kaposi clássico. As terapias para tratamento de sarcoma de Kaposi são paliativas. Nos casos de sarcoma de Kaposi clássico, em que a doença é indolente e os pacientes são idosos, raramente se aplica alguma terapia sistêmica agressiva.66,69 Em vez disso, a radioterapia é o tratamento de escolha. O sarcoma de Kaposi é uma doença radiossensível: doses únicas de cGy têm sido usadas em tratamentos paliativos rápidos em pacientes com maus prognósticos. Doses totais de 800 a 3.500 cGy produziram 50% de respostas completas e 46% de respostas parciais, sendo que mais da metade dos pacientes precisam fazer tratamento de acompanhamento por um período de tempo de 13 anos.74 Os especialistas defendem um regime de tratamento equivalente a 3.000 cGy em 10 frações durante duas semanas.74
Nos casos de pacientes com sarcoma de Kaposi clássico com apenas uma ou duas pápulas, a biópsia excisional possivelmente seja suficiente tanto para o diagnóstico como para o tratamento. Nos casos de pápulas isoladas talvez a crioterapia com nitrogênio líquido seja uma opção bastante útil. Terapias sistêmicas para tratamento de sarcoma de Kaposi clássico são indicadas nos casos de doença cutânea extensiva ou de envolvimento visceral. A administração de terapias com um único agente à base de alcaloides vinca (i.e., vincristina ou vimblastina) é muito comum. Embora baixas doses de interferon-alfa recombinante também sejam eficazes nos casos de sarcoma de Kaposi clássico, possivelmente os efeitos colaterais (p.ex., febre, calafrios, mialgias e fadiga) não sejam bem tolerados por pacientes idosos.66,69
Sarcoma de Kaposi associado a transplantes. A regressão espontânea do sarcoma de Kaposi foi observada em receptores de transplantes após a retirada de ciclosporina e corticosteroides.66 O sirolimo (rapamicina), um medicamento imunossupressivo com propriedades antineoplásicas antiangionênicas, foi usado com muito sucesso em 15 receptores de transplante renal que haviam desenvolvido sarcoma de Kaposi. Logo após o diagnóstico da doença, interrompeu-se o uso de ciclosporina e micofenolato mofetil, iniciando-se, em seguida, a administração de sirolimo. O sarcoma de Kaposi cutâneo foi resolvido em todos os pacientes, sem episódios de rejeição aguda ou alterações na função do enxerto renal.71
Sarcoma de Kaposi associado ao HIV. Embora o sarcoma de Kaposi seja mais agressivo em pacientes infectados pelo HIV, a extensão da imunossupressão e a presença de infecções oportunistas ou de outras enfermidades sistêmicas pode ser igualmente importante na definição de estágios, na determinação de prognósticos e na escolha da terapia mais apropriada.66,69 As características clínicas que foram tradicionalmente associadas a resultados mais favoráveis incluíam contagens de células T CD4+ acima de 200 células/µL, ausência de enfermidade sistêmica, sarcoma de Kaposi limitado à pele ou linfonodos, sarcoma de Kaposi oral mínimo (i.e., não nodular), fatores de risco inexpressivos incluindo contagens de células T CD4+ abaixo de 200 células/µL, linfedema associado ao sarcoma de Kaposi, sarcoma de Kaposi visceral, sarcoma de Kaposi ulcerado, sarcoma de Kaposi nodular oral e infecções oportunistas.75 Entretanto, com o advento da terapia HAART, os médicos que tratam pacientes com sarcoma de Kaposi associado ao HIV passaram a ter a oportunidade de influenciar ou mesmo reverter a imunossupressão afetando a carga viral de HIV e a contagem de células T CD4+. Observou-se a regressão do sarcoma de Kaposi após o início da HAART, geralmente durante os primeiros meses da terapia70,76; consequentemente, trata-se da terapia de primeira linha para aplicação em pacientes com sarcoma de Kaposi cutâneo limitado associado ao HIV.69
A terapia local é uma abordagem razoável em pacientes com sarcoma de Kaposi com doença limitada, em pacientes com complicações infecciosas e em pacientes que não conseguem tolerar terapias sistêmicas.76 A radioterapia é eficaz no tratamento de sarcoma de Kaposi associado ao HIV em doses semelhantes àquelas usadas nos casos de sarcoma de Kaposi clássico (ver acima). Entretanto, de maneira geral, as respostas nos casos de sarcoma de Kaposi associado ao HIV são curtas.66 Possivelmente a aplicação tópica de gel de alitretinoina (ácido 9-cis-retinoico) seja eficaz nos casos de sarcoma de Kaposi associado ao HIV, e foi aprovada pela FDA para esse tipo de aplicação.76 Injeções intralesionais de vimblastina ou de interferon também foram úteis em casos selecionados.66 A crioterapia com nitrogênio líquido é uma boa opção para lesões pequenas76; no entanto, a crioterapia é contraindicada para pacientes de pele escura, nos quais a hiperpigmentação pós-tratamento talvez tenha uma aparência cosmética pior que a lesão original.
A terapia sistêmica inclui a quimioterapia convencional e modificadores de respostas biológicas. Nos casos de pacientes com sarcoma de Kaposi cutâneo com progressão lenta (< 25 lesões) provavelmente não seja necessário aplicar terapia antitumoral sistêmica; nesses casos talvez a HAART, com ou sem terapia local, seja suficiente.51 Entretanto, a HAART isoladamente não foi considerada o tratamento de escolha nos casos avançados de sarcoma de Kaposi associado ao HIV.77 As antraciclinas lipossômicas (p.ex., doxorubicina, daunomicina) foram aprovadas pela FDA como terapia de primeira linha para tratamento de sarcoma de Kaposi associado ao HIV.78,79 O uso de uma combinação de HAART e antraciclinas lipossômicas, seguido de uma combinação de HAART mais paclitaxel, se a resposta ao primeiro regime não for adequada, é uma abordagem razoável para tratamento dos casos avançados de sarcoma de Kaposi associado ao HIV.77,78 Algumas abordagens investigacionais promissoras para tratamento de sarcoma de Kaposi associado ao HIV incluem compostos antiangiogênicos, talidomida, inibidores da matriz de metaloproteinase e retinoides. Uma das formas de prevenção de sarcoma de Kaposi associado ao HIV é a terapia antiviral do HHV-8.76
Infecções bacterianas e sepse são ocorrências comuns em pacientes com sarcoma de Kaposi e podem estar associadas a tumores ulcerados nas pernas e nos pés. Possivelmente ocorram infecções oportunistas, principalmente em pacientes com contagens muito baixas de células T CD4+.
A contagem total de células T CD4+ é o preditor mais importante de sobrevida nos casos de sarcoma de Kaposi associado ao HIV.61 Grandes cargas tumorais, linfedema e sarcoma de Kaposi pulmonar também são indicações de resultados mais fracos.75,79
Os linfomas cutâneos podem ser das linhagens de células B ou de células T e provavelmente envolvam a pele primária ou secundariamente. Os linfomas de células B, em particular os linfomas não Hodgkin, podem envolver a pele secundariamente nos casos de doença em estado avançado. Tipicamente, eles se apresentam como nódulos ou placas subcutâneas vermelho-violetas. Os linfomas cutâneos de células B primários também são ocorrências prováveis e representam aproximadamente 20% de todos os linfomas cutâneos primários. Esses linfomas se apresentam como nódulos ou placas de cor avermelhada ou violácea na pele [ver a Figura 9]. De maneira geral, os linfomas cutâneos primários de zonas marginais e os linfomas foliculares cutâneos primários centrais apresentam comportamento indolente e permanecem localizados na pele; com frequência, esses linfomas são tratados com radioterapia, excisão cirúrgica ou observação rigorosa. Os linfomas de células B grandes difusos, do tipo que predomina nas pernas, são os linfomas cutâneos de células B primários mais agressivos, geralmente com episódios de recorrência depois de radioterapia e, ao final, é necessário aplicar quimioterapia sistêmica.80 Entretanto, a grande maioria de linfomas cutâneos primários se enquadra no espectro de linfomas de células T cutâneos (LCTC).81,82
Figura 9: Linfoma de célula B cutâneo primário do tipo zona marginal. Pápula vermelha no braço.
Os LCTCs incluem micose fungoide (MF) e síndrome de Sézary, que é uma variante leucêmica da MF.81,82 A MF é o maior subgrupo de LCTC; entretanto, às vezes os dois termos são intercambiáveis. Outra variante de LCTCs está associada ao vírus linfotrópico de células T humanas tipo 1 (HTLV-1, do inglês human T-cell lymphotropic vírus type I) e faz parte dos espectros do linfoma/leucemia de células T de adultos e do linfoma de células T periféricas.81
O linfoma de células T cutâneo (LCTC) é um distúrbio raro. Nos Estados Unidos, aproximadamente 1.200 casos novos de LCTC são diagnosticados anualmente.83 No período entre 1973 e 2002, incidência de LCTC aumentou 2,9% por década, atingindo uma incidência anual ajustada pela idade de 6,4 por 1.000.000 de pessoas. O LCTC afeta principalmente adultos na meia-idade; a idade mediana de apresentação é de 50 anos.83 A razão de incidência entre homens e mulheres é de aproximadamente 2:1; a população de cor negra tem uma probabilidade duas vezes maior de desenvolver LCTC em relação à população de cor branca.83
Acredita-se que a suscetibilidade de hospedeiro e um antígeno ambiental, possivelmente viral, tenham papel importante na patogênese de LCTC.81 Fatores genéticos podem estar relacionados a antígenos de histocompatibilidade importantes, como um aumento no HLA-DRB1*11 (anteriormente conhecido por HLA-DR5) e no HLA-DQB1*03.84 A estimulação antigênica crônica (p.ex., infecção) possivelmente tenha algum papel etiológico.81 Por exemplo, a infecção causada pelo HTLV-1 possivelmente seja um fator etiológico no desenvolvimento da variante do linfoma de células T periféricas.81
Tipicamente, as manifestações clínicas de micose fungoide (MF) evoluem durante períodos que podem variar de alguns meses a alguns anos. Um estudo clássico mostrou que a duração média dos sintomas, antes do diagnóstico, foi 7,5 anos.85 Manchas eritematosas planas, geralmente com formação de escamas e ocasionalmente atróficas, iniciam no tronco e nas coxas, especialmente na distribuição no tronco [ver a Figura 10]. As lesões são assintomáticas ou levemente pruríticas e poderão desaparecer espontaneamente ou responder à terapia com aplicação tópica de corticosteroides. Eventualmente os pacientes podem apresentar alguma melhora depois da exposição aos raios solares. Na medida em que ficam maiores, as manchas ficam mais espessas e apresentam formação de placas. A cor poderá se tornar vermelha escura; em pessoas de pele escura, as lesões podem ser hiperpigmentadas ou hipopigmentadas na fase inicial da doença e adquirir uma tonalidade eritematosa ou violácea. Os casos de MF em estado avançado poderão apresentar desenvolvimento de tumores ou transformação em linfoma de células grandes.81,82,86
Figura 10: Linfoma de células T cutâneo no estágio de uma grande mancha (a) e como micose fungoide em estágio tumoral (b).
Em aproximadamente 10% de casos os tumores são a apresentação inicial de LCTC (tumeurs d’emblé). Eritroderma generalizado com células T atípicas em circulação (na síndrome de Sézary) é a apresentação em 5% de pacientes com LCTC.81,82
O exame de pacientes com suspeita de LCTC inclui exame cutâneo completo com a classificação das lesões (mancha, placa ou tumor) e extensão da área da superfície do corpo envolvida. É muito importante fazer a palpação de linfonodos, fígado e baço.
É necessário obter biópsias da pele para o diagnóstico definitivo de LCTC. A presença de células linfoides atípicas com núcleos cerebriformes convolutos em agrupamentos na epiderme (microabscessos de Pautrier) e de infiltrado linfocítico semelhante a bandas na derme superior, é diagnóstica de LCTC.81,82 A célula T é a célula maligna, sendo que a maior parte das células expressa marcadores CD2, CD3 e CD5 de células pan-T, assim como deleção frequente de CD7, CD26, ou ambos.81,82,87 A utilização de estudos de reorganização genética de receptores de células T para confirmar a clonicidade na fase inicial da doença possivelmente facilite a determinação do diagnóstico.81 Nem os estudos imunofenotípicos ou a microscopia eletrônica pode ser considerada definitivamente diagnóstica de LCTC. É imprescindível fazer a correlação clínico-patológica.
A avaliação laboratorial de LCTC inclui hemograma completo, contagem de eosinófilos, contagem de células de Sézary, nível de lactato desidrogenase e testes da função hepática. A biópsia da medula óssea é desnecessária na ausência de células leucêmicas em circulação. Os testes de HTLV-1 devem ser considerados em pacientes com fatores de risco ou apresentações atípicas. A biópsia de linfonodos é uma hipótese a ser levada em consideração nos casos de Linfonodos palpáveis, principalmente linfonodos com mais de 2 cm. A tomografia computadorizada abdominal ou as radiografias torácicas pode ser importante em pacientes com tumores ou com suspeita de envolvimento visceral.
Nos estágios iniciais, o linfoma de células T cutâneo (LCTC) pode se assemelhar a qualquer um dos vários distúrbios inflamatórios benignos (p.ex., reação a medicamentos, eczema, psoríase ou dermatite de contato). Esses distúrbios deverão ser excluídos antes de contemplar a terapia.
A determinação de fases de LCTC se fundamenta na avaliação do tipo, na extensão das lesões na pele, na extensão do envolvimento de linfonodos, do sangue periférico e das vísceras.85,86 No estágio inicial a doença se caracteriza pela presença de manchas ou placas limitadas (estágio IA) ou de manchas ou placas generalizadas, sem evidências de envolvimento extracutâneo (estágio IB e IIA); a doença em estado mais avançado se caracteriza pela presença de tumores cutâneos (estágio IIB), de doença extracutânea (estágio III) e de doença extracutânea envolvendo linfonodos (estágio IVA) ou vísceras (estágio IVB). Alterações recentes na classificação de determinação de fases feitas pela International Society for Cutaneous Lymphoma (ISCL) e pela European Organisation for Research and Treatment of Cancer (EORTC) incluíram também uma classificação para envolvimento sanguíneo, assim como da pele, linfonodos e vísceras.
A terapia tópica é a base do tratamento de doenças nos estágios iniciais (estágio IA, IB e IIA) [ver a Tabela 5]. As terapias iniciais agressivas com radiação e quimioterapia comprovadamente não são superiores às abordagens locais para controlar a doença ou melhorar a sobrevida em pacientes com doença limitada.81,82 A aplicação tópica de corticosteroides é uma abordagem racional para tratamento de doença inicialmente limitada (ou equívoca sob o ponto de vista histológico).88 A mostarda de nitrogênio tópica, na forma aquosa ou de pomada, é a quimioterapia tópica usada com mais frequência. Em uma das séries, a taxa global de resposta à mostarda de nitrogênio foi de 83%, a taxa completa de resposta foi de 50%, depois de um tempo mediano de tratamento de 12 meses.89 O tempo mediano de recidiva também foi de 12 meses.89
|
Tabela 5: Visão Geral de Terapias para Linfoma de Célula T Cutâneo |
|
Terapia direcionada para a pele Corticosteroides tópicos Mostarda de nitrogênio para uso tópico: mecloretamina Gel de bexaroteno para uso tópico* Carmustina tópica Fototerapia NBUVB PUVA Radioterapia Feixe de elétrons para a pele total Feixe de elétrons localizado
Terapia sistêmica Bexaroteno* Vorinostat* Romidepsina* Metotrexato Interferon Fotoforese extracorpórea Denileucina diftitox* Alentuzumabe Quimioterapia de combinação |
NBUVB (narrowband ultravioleta B) = raios ultravioleta B de banda estreita; PUVA (psoralen with ultraviolet A light) = psoraleno com raios ultravioleta A.
*Aprovados pela Food and Drug Administration.
O gel de bexaroteno a 1%, um retinoide tópico, comprovou que é eficaz no tratamento de LCTC; este medicamento foi aprovado pela FDA para uso em casos de LCTC.90 A solução de carmustina (BCNU), aplicada diariamente nas lesões, é outro regime potencialmente útil. De maneira geral, o tempo de duração do tratamento varia de 8 a 16 semanas, embora em alguns casos tenha continuado por até seis meses. Recomenda-se monitorar os hemogramas completos levando-se em consideração que a absorção sistêmica poderá resultar na supressão da medula óssea com a solução de carmustina.82
A radioterapia para LCTC tem várias formas, de raios UV à radiação ionizante. Os raios UVB são muito úteis no primeiro estágio da doença. Em um estudo retrospectivo envolvendo 21 pacientes, os raios UVB de banda estreita resultaram na remissão completa em 81% dos pacientes e na remissão parcial em 19%; o intervalo médio sem recidivas foi de 24,5 meses.76 Nos casos de micose fungoide, os raios UVB de banda estreita são preferíveis em relação aos raios UVB de banda larga.91
Outra abordagem eficaz para tratamento de LCTC é a combinação de psoraleno e raios ultravioleta A (PUVA, do inglês psoralen with ultraviolet light in the A range). Em um estudo, 69% de pacientes no primeiro estágio de LCTC apresentaram eliminação completa, com um intervalo médio sem recidivas de 43 meses; as taxas de sobrevida sem doença após 5 e 10 anos para o estágio IA foram, respectivamente, de 56% e 30%.92 Em outro estudo, a remissão completa foi observada em 71% de pacientes nos estágios iniciais; neste estudo, o intervalo médio sem recidivas foi de 22,8 meses.76 Os raios UVB de banda estreita substituíram plenamente a combinação de psoraleno e raios ultravioleta A por causa da semelhança na eficácia93 e por causa da preocupação com o aumento na incidência de câncer de pele com a terapia à base de PUVA.
A radiação com feixes de elétrons para a pele total (TSEB, do inglês total skin electron beam) libera radioterapia para a superfície da pele sem doses internas significativas. Esse tipo de radiação é especialmente útil nos casos de doença com formação de placas. As doses típicas variam de 2.400 a 3.600 cGy fracionadas em várias semanas com radiação de feixes de elétrons de 4 a 9 MeV.94 As respostas ao tratamento estão relacionadas ao estágio do linfoma de células T cutâneo (LCTC);79 os pacientes no estágio inicial (estágio IA) têm taxa de resposta de 95%, embora 50% poderão ter recidiva dentro de dez anos. A radiação com TSEB também é uma opção útil de tratamento no estágio IB da doença (taxa de remissão de 90%), porém dois terços de pacientes com essa modalidade poderão ter recidiva dentro de cinco anos.88 Os pacientes com LCTC no estado tumoral (estágio IIB) poderão ter algum resultado paliativo com TSEB, principalmente em combinação com outras terapias.95
A terapia sistêmica foi considerada como terapia primária em casos avançados de LCTC (estágios III a IVB); nos estágios iniciais da doença, a terapia sistêmica é usada como parte integrante de uma terapia sequencial para promover respostas mais duráveis.82,83
A administração de bexaroteno por via oral produziu taxas de respostas de até 45% em casos avançados de LCTC e foi aprovada pela FDA para uso nesse tipo de doença.96 Outra terapia sistêmica usada no tratamento de casos avançados de LCTC é denileucina diftitox [DAB(389) IL-2].97 Essa proteína de fusão citotóxica com alvo em receptores liga o receptor de IL-2 nas células T; essa terapia atingiu uma taxa de resposta de 30% em pacientes que haviam recebido tratamento pesado.97 O vorinostat, um inibidor oral de histona deacetilase, foi aprovado pela FDA para aplicação em pacientes com LCTC que haviam apresentado evolução ou recorrência depois ou durante duas terapias sistêmicas.98 Da mesma forma, a romidepsina, uma inibidora da histona deacetilase que é administrada por via intravenosa semanalmente, foi aprovada pela FDA para uso em casos de LCTC refratário.99
A fotoforese, que é uma terapia aceita para casos avançados de LCTC, aparentemente é mais útil em LCTC eritrodérmico e na síndrome de Sézary.66,67 Nesse tipo de tratamento administra-se um medicamento fotoativador (8-metoxipsoraleno) no paciente, os leucócitos são coletados através de leucoferese e irradiados com raios UVA, sendo que as células irradiadas retornam para o paciente por via intravenosa. Os casos avançados de LCTC que se caracterizarem pela presença de tumores cutâneos (estágio III) ou por envolvimento visceral (estágio IV) também foram tratados com um único agente e quimioterapia de combinação usando metotrexato, análogos de adenosina, interferon alfa e retinoides.98, 100
Tratamentos iniciais agressivos com TSEB seguidos de quimioterapia de combinação não oferecem nenhuma vantagem sobre a sobrevida em comparação com as terapias tópicas sequenciais.101 Em um teste controlado randomizado, 103 pacientes com micose fungoide (MF) receberam TSEB e, em seguida, quimioterapia parenteral com ciclofosfamida, doxorubicina, etoposida ou tratamento tópico sequencial. Os pacientes que receberam terapia combinada apresentaram taxas significativamente mais altas de respostas completas, em comparação com os pacientes que receberam terapia tópica sequencial; no entanto, não houve nenhuma diferença no índice de sobrevida sem doença e no índice total entre os dois grupos, após um tempo médio de acompanhamento de 75 meses.100 Em um estudo não controlado, a terapia multimodal foi analisada em pacientes com doença precoce e avançada.96 Neste estudo, 95 pacientes com LCTC receberam TSEB em fases consecutivas da terapia com interferon alfa e isotretinoina oral e na terapia de manutenção consistindo da aplicação tópica de mostarda de nitrogênio e interferon alfa. Os pacientes com doença em estado avançado receberam também seis ciclos de quimioterapia de combinação antes de receberem TSEB. Embora a terapia multimodal tenha resultado em altas taxas de respostas (85% de respostas, 60% de respostas completas), o estudo não produziu nenhuma evidência de que essa forma de combinação terapêutica poderia melhorar as taxas totais de sobrevida atualmente conseguidas com a terapia tópica sequencial. De maneira geral, a heterogeneidade dos regimes documentados de combinação de terapias em casos de LCTC torna praticamente impossível fazer comparações entre os resultados.
Inúmeras abordagens experimentais ao LCTC estão atualmente em fase de investigação, incluindo transplante alogênico de medula óssea, anticorpos monoclonais e toxinas de fusão.81 Outras modalidades investigativas incluem citocinas como a IL-12 e IL-2 recombinantes.81
Infecções são as complicações mais sérias de LCTC. Sepse causada por tumores cutâneos ulcerados é uma causa comum de morte. Linfoma de células T cutâneo visceral também é uma ocorrência comum, assim como a possível transformação em linfomas de células grandes em alguns pacientes com LCTC (probabilidade de 39% depois de 12 anos).86 Em sobreviventes no longo prazo com doença nos estágios iniciais, as terapias locais (p.ex., TSEB ou PUVA) podem contribuir para o desenvolvimento de outros tipos de câncer de pele (p.ex., carcinoma basocelular ou carcinoma de células escamosas) e catarata.102
Muitas tentativas diferentes foram feitas com o objetivo de classificar o LCTC em grupos prognósticos úteis. Um dos estudos iniciais, e que ainda é válido, que utilizou o sistema TNM identificou três grupos principais: pacientes com risco bom (estágio IA, IB e IIA, com doença cutânea apenas com placas e nenhum envolvimento de linfonodo, sangue ou de vísceras [sobrevida mediana > 12 anos]); pacientes com risco intermediário (estágio IIB, III e IVA, com tumores cutâneos, eritroderma ou envolvimento de doença plaquetária e nodular ou sanguínea, porém nenhuma doença visceral ou eliminação de linfonodo [sobrevida mediana > 5 anos]); e pacientes com risco baixo (estágio IVB, com envolvimento visceral ou eliminação de linfonodo [sobrevida mediana > 2,5 anos]).85
No caso de doença nos estágios IA e IB, nos pacientes com doença que apresentam apenas manchas a sobrevida é melhor em comparação com os pacientes com placas; além disso, os pacientes com micose fungoide foliculotrópica apresentam os piores prognósticos, sendo que a eosinofilia também está associada a uma sobrevida curta.70 Outros estudos de longo prazo revelaram que os pacientes no estágio IA da doença não têm expectativa reduzida de vida e que menos de 10% desses pacientes apresentam progressão de doença para estágios mais avançados.103-105 A sobrevida mediana de 11,7 anos de pacientes com micose fungoide com mancha/placa generalizada (estágio IB ou IIA) é significativamente pior do que a de uma população de controle formada com base na raça, idade e sexo.82 Aparentemente, características como gênero e raça não têm nenhum efeito sobre a sobrevida, porém os pacientes mais velhos (> 58 anos) têm sobrevida específica da doença mais curta.83
|
Allan C. Halpern, MD recebeu apoio da Quintiles como membro do grupo Data Safety Monitoring Board para o medicamento vermurafinibe.
Patrícia L. Myskowski, MD recebeu subvenção ou apoio de Merck & Co., Inc., Genmab, Allos Therapeutics, Schering-Plough, Orthobiotech, Bristol Myers Squibb e Ligand Pharmaceuticals Inc. (atualmente Eisai).
Corticosteroides tópicos, mostarda de nitrogênio tópica e BCNU foram aprovados pela FDA para tratamento de linfomas de células T cutâneo; interferon alfa, sirolimo e HAART não foram aprovados pela FDA para tratamento de sarcoma de Kaposi. |
1. Green A, Whiteman D, Frost C, et al. Sun exposure, skin cancers and related skin conditions. J Epidemiol 1999;9(6 Suppl):S7.
2. Ullrich S. Mechanisms underlying UV-induced immune suppression. Mutat Res 2005;571:185.
3. Burnett ME, Wang SQ. Current sunscreen controversies: a critical review. Photodermatol Photoimmunol Photomed 2011;58:67.
4. van der Pols, JC, Williams GM, Pandeva N et al. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol Biomarkers Prev 2006;15:2546–8.
5. Green AC, Williams GM, Logan V, Strutton GM. Reduced melanoma risk after regular sunscreen use: randomized trial follow-up. J Clin Oncol 2011;29:257–63.
6. Halpern AC. Vitamin D: a clinical perspective. Pigment Cell Melanoma Res 2012 Sep 7. DOI: 10.111/pcmr.12016. [Epub ahead of print]
7. Lomas A, Leonardi-Bee J, Bath-Hextall F. A systemic review of worldwide incidence of nonmelanoma skin cancer. Br J Dermatol 2012;165:1069–80.
8. Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 2010;146:283–7.
9. Buettner PG, Raasch BA. Incidence rates of skin cancer in Townsville, Australia. Int J Cancer 1998;78:587.
10. High A, Zedan W. Basal cell nevus syndrome. Curr Opin Oncol 2005;17:160.
11. Shriner DL, McCoy DK, Goldberg DJ, et al. Mohs micrographic surgery. J Am Acad Dermatol 1998;39:79.
12. Paver K, Poyzer K, Burry N, et al. The incidence of basal cell carcinoma and their metastases in Australia and New Zealand [letter]. Australas J Dermatol 1973;14:53.
13. Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72.
14. Weiss GJ, Korn RL. Metastatic basal cell carcinoma in the era of hedgehog signaling pathway inhibitors. Cancer 2012. DOI: 10.1002/cncr.27532. [Epub ahead of print]
15. Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012;366:2171–9.
16. Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol 1994;30:774.
17. Staples M, Marks R, Giles G. Trends in incidence of non-melanocytic skin cancer (NMSC) treated in Australia 1985-1995: are primary prevention programs starting to have an effect? Int J Cancer 1998;78:144.
18. Athas WF, Hunt WC, Key CR. Changes in nonmelanoma skin cancer incidence between 1977–1978 and 1998–1999 in northcentral New Mexico. Cancer Epidemiol Biomarkers Prev 2003;12:1105.
19. Kirkham N. Tumors and cysts of the epidermis. In: Elder DE, Murphy GF, Johnson BL, et al, editors. Lever’s histopathology of the skin. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 805.
20. Lindelof B, Sigurgeirsson B, Gabel H, et al. Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol 2000;143:513.
21. Sarmiento JM, Wolff BG, Burgart LJ, et al. Perianal Bowen’s disease: associated tumors, human papillomavirus, surgery, and other controversies. Dis Colon Rectum 1997;40:912.
22. Guthrie TH Jr, Porubsky ES, Luxenberg MN, et al. Cisplatin-based chemotherapy in advanced basal and squamous cell carcinomas of the skin: results in 28 patients including 13 patients receiving multimodality therapy. J Clin Oncol 1990;8:342.
23. 23 Jorizzo JL. Current and novel treatment options for actinic keratosis. J Cutan Med Surg 2004;8 Suppl 3:13.
24. National Comprehensive Cancer Network. Basal cell and squamous cell skin cancers. Clinical practice guidelines in oncology, version 1. Available at: http://www.nccn.org/professionals/physician_gls/PDF/nmsc.pdf (accessed February 12, 2009).
25. Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol 1992;26:976.
26. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012;62:10–29.
27. Hall HI, Miller DR, Rogers JD. Update on the incidence and mortality from melanoma in the United States. J Am Acad Dermatol 1999;40:35.
28. Jemal A, Devesa SS, Hartge P, et al. Recent trends in cutaneous melanoma incidence among whites in the United States. J Natl Cancer Inst 2001;93:678.
29. Ting W, Schultz K, Cac NN, et al. Tanning bed exposure increases the risk of malignant melanoma. Int J Dermatol 2007;46:1253–7.
30. Blackwood MA, Holmes R, Synnestvedt M, et al. Multiple primary melanoma revisited. Cancer 2002;94:2248.
31. Carey WP Jr, Thompson CJ, Synnestvedt M, et al. Dysplastic nevi as a melanoma risk factor in patients with familial melanoma. Cancer 1994;74:3118.
32. Elder DE, Clark WH Jr, Elenitsas R, et al. The early and intermediate precursor lesions of tumor progression in the melanocytic system: common acquired nevi and atypical (dysplastic) nevi. Semin Diagn Pathol 1993;10:18.
33. Pho L, Grossman D, Leachman SA. Melanoma genetics: a review of genetic factors and clinical phenotypes in familial melanoma. Curr Opin Oncol 2006;18:173–9.
34. Tuveson DA, Weber BL, Herlyn M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell 2003;4:95.
35. Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
36. Abbasi NR, Shaw HM, Rigel DS, et al. Early diagnosis of cutaneous melanoma: revisiting the ABCD criteria. JAMA 2004;292:2771.
37. Naeyaert JM, Brochez L. Clinical practice. Dysplastic nevi. N Engl J Med 2003;349:2233.
38. Braun RP, Rabinovitz HS, Oliviero M, et al. Dermoscopy of pigmented skin lesions. J Am Acad Dermatol 2005;52:109.
39. Vestergaard ME, Macaskill P, Holt PE, Menzies SW. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol 2008;159:669–76.
40. Halpern AC. The use of whole body photography in a pigmented lesion clinic. Dermatol Surg 2000;26:1175.
41. Psaty EL, Halpern AC. Current and emerging technologies in melanoma diagnosis: the state of the art. Clin Dermatol 2009;27:35–45.
42. National Comprehensive Cancer Network. Melanoma. Clinical practice guidelines in oncology, version 2. Available at: http://www.nccn.org/professionals/physician_gls/PDF/melanoma.pdf (accessed April 8, 2009).
43. Cook J. Surgical margins for resection of primary cutaneous melanoma. Clin Dermatol 2004;22:228.
44. Roberts AA, Cochran AJ. Pathologic analysis of sentinel lymph nodes in melanoma patients: current and future trends. J Surg Oncol 2004;85:152.
45. Reintgen D, Pendas S, Jakub J, et al. National trials involving lymphatic mapping for melanoma: the Multicenter Selective Lymphadenectomy Trial, the Sunbelt Melanoma Trial, and the Florida Melanoma Trial. Semin Oncol 2004;31:363.
46. Morton DL, Thompson JF, Cochran AJ, et al. Interim results of the Multicenter Selective Lymphadenectomy Trial (MSLT-I) in clinical stage I melanoma [abstract]. In: Proceedings of the American Society of Clinical Oncology Annual Meeting; 2005 Jan 20–22; New Orleans.
47. Fraker DL. Management of in-transit melanoma of the extremity with isolated limb perfusion. Curr Treat Options Oncol 2004;5:173.
48. Buzaid AC. Management of metastatic cutaneous melanoma. Oncology (Williston Park) 2004;18:1443.
49. Ribas A. Update on immunotherapy for melanoma. Natl Compr Canc Netw 2006;4:687–94.
50. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
51. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
52. Coit DG, Andtbacka R, Anker CJ, et al. Melanoma. J Natl Compr Canc Netw 2012;10:366–400.
53. Eggermont AM, Suciu S, MacKie R, et al. Post-surgery adjuvant therapy with intermediate doses of interferon alfa 2b versus observation in patients with stage Iib/III melanoma (EORTC 18952): randomized controlled trial. Lancet 2005;366:1189–96.
54. Kirkwood JM, Strawderman MH, Ernstoff MS, et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 1996;14:7.
55. 55 Hersey P. Adjuvant therapy for high-risk primary and resected metastatic melanoma. Int Med J 2003;33:33.
56. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of the 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27:6199–206.
57. Balch CM, Soong SJ, Atkins, MB, et al. An evidence-based staging system for cutaneous melanoma. CA Cancer J Clin 2004;54:131.
58. Balch CM, Soong SJ, Gershenwald JE, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 2001;19:3622.
59. Guerry D 4th, Synnestvedt M, Elder DE, et al. Lessons from tumor progression: the invasive radial growth phase of melanoma is common, incapable of metastasis, and indolent. J Invest Dermatol 1993;100:342S.
60. Schuchter L, Schultz DJ, Synnestvedt M, et al. A prognostic model for predicting 10-year survival in patients with primary melanoma. The Pigmented Lesion Group. Ann Intern Med 1996;125:369.
61. Soong SJ, Ding S, Coit D, et al, AJCC Melanoma Task Force. Predicting survival outcome of localized melanoma: an electronic prediction tool based on the AJCC Melanoma Database. Ann Surg Oncol 2010;17:2006–14.
62. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol 1995;33:161.
63. Demetrius RW, Randle HW. High-risk nonmelanoma skin cancers. Dermatol Surg 1998;24:1272.
64. Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 2005;23:2300.
65. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319:1096–100.
66. Myskowski P, Krown S. Kaposi’s sarcoma. In: Sober AJ, Haluska FG, editors. Skin cancer. American Cancer Society Atlas of Clinical Oncology Series. Philadelphia: BC Decker; 2001. p. 142–62.
67. Koontz BF, Miles EF, Rubio MA, et al. Preoperative radiotherapy and bevacizumab for angiosarcoma of the head and neck: two case studies. Head Neck 2008;30:262–6.
68. Rutkowski P, van Glabbeke M, Rankin CJ. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 2010;28:1772–9.
69. Schwartz RA, Micali G, Nasca MR, Scuderi L. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol 2008;59:179–206.
70. Cheung TW. AIDS-related cancer in the era of highly active antiretroviral therapy (HAART): a model of the interplay of the immune system, virus, and cancer. “On the offensive—the Trojan Horse is being destroyed.” Part A: Kaposi’s sarcoma. Cancer Invest 2004;22:774.
71. Mocroft A, Kirk O, Clumeck N, et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer 2004;100:2644.
72. Moore PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and without HIV infection. N Engl J Med 1995;332:1181.
73. Stallone G, Schena A, Infante B, et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med 2005;352:1317.
74. Cooper JS, Sacco J, Newall J. The duration of local control of classic (non–AIDS-associated) Kaposi’s sarcoma by radiotherapy. J Am Acad Dermatol 1988;19:59.
75. Krown SE, Testa MA, Huang J. AIDS-related Kaposi’s sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 1997;15:3085.
76. Cattelan AM, Trevenzoli M, Aversa SM. Novel pharmacological therapies for the treatment of AIDS-related Kaposi’s sarcoma. Expert Opin Investig Drugs 2004;13:501.
77. Krown SE. Highly active antiretroviral therapy in AIDS-associated Kaposi’s sarcoma: implications for the design of therapeutic trials in patients with advanced, symptomatic Kaposi’s sarcoma. J Clin Oncol 2004;22:399.
78. Northfelt DW, Dezube BJ, Thommes JA, et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi’s sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 1998;16:2445.
79. Welles L, Saville MW, Lietzau J, et al. Phase II trial with dose titration of paclitaxel for the therapy of human immunodeficiency virus–associated Kaposi’s sarcoma. J Clin Oncol 1998;16:1112.
80. Sokol L, Naghashpour M, Glass LF. Primary cutaneous B-cell lymphomas: recent advances in diagnosis and management. Cancer Control 2012;19:236–44.
81. Li JY, Horwitz S, Moskowitz A, et al. Management of cutaneous T cell lymphoma: new and emerging targets and treatment options. Cancer Manag Res 2012;4:75–89.
82. Hwang ST, Janik JE, Jaffe ES, Wilson WH. Mycosis fungoides and Sézary syndrome. Lancet 2008;371:945–57.
83. Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol 2007;143:854–9.
84. Hodak E, Klein T, Gabay B, et al. Familial mycosis fungoides: report of 6 kindreds and a study of the HLA system. J Am Acad Dermatol 2005;52:393.
85. Sausville EA, Eddy JL, Makuch RW, et al. Histopathologic staging at initial diagnosis of mycosis fungoides and the Sézary syndrome: definition of three distinctive prognostic groups. Ann Intern Med 1988;109:372.
86. Diamandidou E, Colome-Grimmer M, Fayad L, et al. Transformation of mycosis fungoides/Sézary syndrome: clinical characteristics and prognosis. Blood 1998;92:1150.
87. Kim EJ, Hess S, Richardson SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest 2005;115:798.
88. Zackheim HS, Kashani-Sabet M, Amin S. Topical corticosteroids for mycosis fungoides. Experience in 79 patients. Arch Dermatol 1998;134:949.
89. Kim YH, Martinez G, Varghese A, et al. Topical nitrogen mustard in the management of mycosis fungoides: update of the Stanford experience. Arch Dermatol 2003;139:165.
90. Breneman D, Duvic M, Kuzel T, et al. Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphoma. Arch Dermatol 2002;138:325.
91. Diederen PV, van Weelden H, Sanders CJ, et al. Narrowband UVB and psoralen-UVA in the treatment of early-stag¬e mycosis fungoides: a retrospective study. J Am Acad Dermatol 2003;48:215.
92. Querfeld C, Rosen ST, Kuzel TM, et al. Long-term follow-up of patients with early-stage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Arch Dermatol 2005;141:305.
93. Ponte P, Serrao V, Apetato M. Efficacy of narrowband UVB vs. PUVA in patients with early-stage mycosis fungoides. J Eur Acad Dermatol Venereol 2010;24:716–21.
94. Jones GW, Kacinski BM, Wilson LD, et al. Total skin electron radiation in the management of mycosis fungoides: consensus of the European Organization for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Project Group. J Am Acad Dermatol 2002;47:364.
95. Duvic M, Apisarnthanarax N, Cohen DS, et al. Analysis of long-term outcomes of combined modality therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol 2003;49:35.
96. Duvic M, Hymes K, Heald P, et al. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol 2001;19:2456.
97. Olsen E, Duvic M, Frankel A, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol 2001;19:376.
98. Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T cell lymphoma. Oncologist 2007;12:1247–52.
99. Coiffer B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 2012;30:631–6.
100. Kaye FJ, Bunn PA Jr, Steinberg SM, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med 1989;321:1784.
101. Apisarnthanarax N, Talpur R, Duvic M. Treatment of cutaneous T cell lymphoma: current status and future directions. Am J Clin Dermatol 2002;3:193.
102. van Praag MC, Tseng LN, Mommaas AM, et al. Minimising the risks of PUVA treatment. Drug Saf 1993;8:340.
103. Kim YH, Jensen RA, Watanabe GL, et al. Clinical stage IA (limited patch and plaque) mycosis fungoides: a long-term outcome analysis. Arch Dermatol 1996;132:1309.
104. Kim YH, Chow S, Varghese A, et al. Clinical characteristics and long-term outcome of patients with generalized patch and/or plaque (T2) mycosis fungoides. Arch Dermatol 1999;135:26.
105. Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sezary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol 2010;28:4730–9.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.