(Carregando Índice)... (Carregando Índice)... |
Última revisão: 13/04/2017
Comentários de assinantes: 0
Transplante de Células Hematopoiéticas
Frederick R. Appelbaum, MD
Diretor na Divisão de Pesquisa Clínica do Fred Hutchinson Cancer Research Center (Seattle, WA)
|
Artigo original: Applebaum, FR, MD. Hematopoietic Cell Transplantation SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2012 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Resumo:
Considerando que é possível fazer a coleta de células-tronco hematopoiéticas no sangue periférico, na medula óssea e no cordão umbilical, o termo transplante de medula óssea foi substituído por “transplante de células hematopoiéticas”. O transplante possibilita substituir o sistema linfo-hematopoiético por sistemas saudáveis, o que torna o procedimento uma forma terapêutica saudável para uma grande variedade de doenças não malignas. Neste artigo, serão apresentados cinco tipos de Transplante de Células Hematopoiéticas: transplante singênico, transplante alogênico, transplante autólogo, transplante de células do sangue periférico e transplante de sangue do cordão umbilical. As especificidades do procedimento, incluindo regime preparatório, coleta e infusão de células-tronco e enxerto, são discutidas com detalhes. Algumas complicações dos transplantes de células hematopoiéticas incluem toxicidades diretas precoces e tardias do regime preparatório ablativo, toxicidades diretas do condicionamento de intensidade reduzida, falência dos enxertos, doença do enxerto contra hospedeiro (DECH) e uma grande variedade de doenças infecciosas. Os transplantes de células hematopoiéticas podem ser usados no combate a enfermidades específicas como doenças de imunodeficiência, anemia aplásica, talassemia, anemia da célula falciforme e inúmeras doenças não malignas. O tratamento de recidivas no período pós-transplante também será objeto de discussão.
|
Transplante de Células Hematopoiéticas |
Frederick R. Appelbaum, MD
Os transplantes de células-tronco hematopoiéticas (TCTHs) conseguem substituir células-tronco hematopoiéticas anormais, porém não substituem células não malignas com células de doadores saudáveis, o que torna a terapia eficaz em uma grande variedade de doenças não malignas no sistema linfo-hematopoiético (como, por exemplo, a doença de imunodeficiência grave combinada, a síndrome de Wiskott-Aldrich, a anemia aplásica, a talassemia, a anemia da célula falciforme e a doença de Gaucher).
Além disso, o TCTH é utilizado para tratar inúmeras malignidades por duas razões: o procedimento permite a administração de doses mais elevadas e potencialmente mais eficazes de quimioterapia (Qt) e radioterapia que, em outras circunstâncias, poderiam produzir mielossupressão inaceitável; os transplantes alogênicos produzem seus próprios efeitos de enxertos imunologicamente mediados versus efeitos tumorais além dos efeitos da quimiorradioterapia. Em 2010, estima-se que 60 mil pacientes fizeram TCTHs em todo o mundo.1
Células-tronco hematopoiéticas
Algumas características do sistema linfo-hematopoiético viabilizam os transplantes. São elas: capacidade regenerativa; abrigo de células-tronco em sítios que promovem a sobrevivência e a proliferação; capacidade de sobrevivência das células-tronco, com uma pequena quantidade de danos; processo de congelamento e descongelamento na criopreservação. Em modelos de camundongos, uma única célula-tronco hematopoiética consegue reconstituir um receptor com radiação letal2
Em seres humanos, o transplante de menos de 10% da medula óssea de um doador resulta na reposição completa e sustentada de todo o sistema linfo-hematopoiético do receptor, incluindo eritrócitos, plaquetas, granulócitos, células T e células B, assim como macrófagos pulmonares alveolares, células de Kupffer do fígado, osteoblastos, células de Langerhans da pele e células microgliais do cérebro. Embora exista alguma controvérsia, as evidências indicam que as células-tronco hematopoiéticas são incapazes de se transdiferenciar em tecidos de origem não hematopoiética.3,4
Ainda que o mecanismo de estabilização não seja compreendido em sua totalidade, pode ser um processo com múltiplas etapas que inclui sinalização pelo microambiente da medula óssea usando o fator-1 derivado de células do estroma (em inglês, stromal cell-derived fator [SDF-1]) e o fator da célula-tronco; essa sinalização direciona a migração de células-tronco para a medula óssea e aumenta as moléculas de adesão (por exemplo, CXCR4 e VLA-1) nas células-tronco.4
As células-tronco hematopoiéticas humanas expressam antígenos superficiais distintos.5 Um deles, o CD34, se expressa somente em 1 a 5% de células adultas da medula óssea, porém, quando cultivada in vitro, todas as colônias derivam da população do antígeno CD34+. Transplantes bem-sucedidos poderão ser feitos em seres humanos apenas com células CD34+ selecionadas de forma positiva.
Cerca de 90% das células CD34+ também expressam CD38, porém os 10% que são CD34+ selecionadas formam a população com melhor capacidade para suportar hematopoiese de longo prazo in vitro e, por conseguinte, são considerados uma população mais primitiva. As células-tronco hematopoiéticas humanas expressam também c-kit e Thy-kit (CD90), mas não possuem marcadores conhecidos da linhagem de células B ou de células T; portanto, são consideradas como linhagens negativas.
Tipos de transplante de células-tronco hematopoiéticas
O TCTH pode ser classificado de acordo com a relação entre doadores e receptores e com a fonte anatômica da célula-tronco. As células-tronco hematopoiéticas para transplantes podem ter origem na medula óssea, no sangue periférico ou no sangue do cordão umbilical, e poderão ser colhidas de doadores singênicos, alogênicos ou autólogos.
Transplante singênico
Transplante singênico é aquele em que o doador e o receptor são gêmeos idênticos. Sempre que se usar doadores singênicos, os receptores não sofrerão rejeição do enxerto ou DECH. Comprova-se a singenicidade através da identidade com base na tipificação do DNA usando locais de repetições curtas em tandem. Somente 1 em cada 100 pacientes que fazem transplantes terá um gêmeo idêntico.
Transplante alogênico
Os transplantes alogênicos, que envolvem doadores com ou sem relação de parentesco, são mais complicados que os singênicos ou autólogos devido às diferenças imunológicas entre doadores e receptores. Nos casos de TCTH, os grandes desafios clínicos são evitar a rejeição de enxertos pelas células hospedeiras que sobreviverem ao regime preparatório antes do transplante e impedir que os doadores de células produzam lesões imunomediadas aos pacientes (isto é, DECH).
Em grande parte, a reatividade imunológica entre doadores e hospedeiros é mediada por células imunocompetentes que reagem com os antígenos leucocitários humanos (em inglês, human leukocyte antigens [HLAs]) codificados por genes do complexo de histocompatibilidade principal. As moléculas do HLA apresentam peptídeos exógenos (por exemplo, de algum vírus infectante) e endógenos, e os apresenta para as células T, um passo importante no início das respostas imunes.
No caso de duas pessoas não compartilharem os mesmos antígenos HLA, as células retiradas de uma pessoa reagirão de forma vigorosa às moléculas do HLA incompatível na superfície das células do outro. Essas reações ocorrem contra os determinantes do HLA principal. Mesmo quando duas pessoas que não sejam gêmeos idênticos tiverem tipos idênticos de HLA, os peptídeos endógenos apresentados pelos antígenos HLA serão diferentes, disparando respostas contra os determinantes do HLA menos importante.
Os genes que codificam o HLA de classe I (HLA–A, HLA–B e HLA–C) e de classe II (HLA–DP, HLA–DQ e HLA–DR) estão fortemente ligados e podem ser herdados juntamente como haplótipos com baixas frequências de recombinação. Para qualquer paciente em particular, há uma probabilidade de 25% de que qualquer irmão tenha herdado o mesmo haplótipo paterno e materno, fato que torna os gêmeos idênticos em relação ao genótipo HLA.
Levando-se em consideração que o número médio de crianças por família é maior que dois nos EUA, a chance média de que um paciente tenha um irmão com HLA compatível é de, aproximadamente, 30%. A fórmula para calcular a probabilidade de um paciente ter um irmão com HLA idêntico é 1 – (0,75)n, onde n é igual ao número de irmãos.
Os transplantes alogênicos têm sido feitos com irmãos doadores com HLA idêntico, com outros doadores compatíveis e incompatíveis nos membros da família e com doadores compatíveis sem nenhuma relação de parentesco. De maneira geral, os melhores resultados foram obtidos com irmãos doadores com HLA idêntico.
Nos casos de transplantes de doadores que faziam parte da família e que eram idênticos para um haplótipo e incompatíveis para um único lócus (isto é, HLA–A, HLA–B ou HLA–C) no outro haplótipo, a taxa de sobrevivência é igual à de doadores com HLA idêntico, embora haja uma incidência maior de doença do enxerto contra hospedeiro (DECH).6 Os transplantes usando doadores membros da família incompatíveis para dois ou mais loci apresentam piores resultados ? incidência mais elevada de DECH e de rejeição de enxertos e probabilidade mais baixa de sobrevivência.6
Por causa da natureza altamente polimórfica dos antígenos HLA, a probabilidade de que duas pessoas sem relação de parentesco sejam compatíveis é extremamente baixa. O transplante de doadores compatíveis sem relação de parentesco foi feito pela primeira vez no final de década de 1970. A aplicação mais ampla dessa técnica tornou-se viável pela criação de grandes registros de doadores no final dos anos 1980. A partir de então, o número de transplantes de doadores sem relação de parentesco aumentou de forma rápida.
Nos dias atuais, somente nos EUA, mais de 14 milhões de pessoas saudáveis se apresentaram como voluntários como doadores de medula óssea, elevando as chances de encontrar doadores compatíveis sem relação de parentesco para HLA–A, HLA–B, HLA–C, HLA–DR e HLA–DQ para algo em torno de 50% (atualmente, a maior parte dos programas de transplantes ignora o HLA–DP).
A probabilidade de encontrar doadores compatíveis sem relação de parentesco é mais elevada na população branca e consideravelmente menor entre hispânicos e negros, não somente pela baixa representatividade de ambos os grupos nos registros de doadores, mas também por causa da maior diversidade de HLA nessas populações. Em média, são necessários 3 meses, contados desde o início da busca, para identificar um doador e iniciar o transplante. Em 2010, nos EUA foram realizados em torno de 3 mil transplantes de doadores sem relação de parentesco.7
Os resultados dos primeiros transplantes mostraram que a DECH foi mais comum e a rejeição de enxertos mais frequente nos casos de doadores sem relação de parentesco, em comparação com doadores sem relação de parentesco com HLA compatível.8 A diferença na incidência de DECH talvez seja explicada, de forma parcial, pelo aumento na disparidade em determinantes menos importantes de HLA em indivíduos sem relação de parentesco.
Além disso, disparidades nos tipos de HLA que não haviam sido detectadas entre pares de doadores e receptores, supostamente compatíveis e sem relação de parentesco, podem ter contribuído para o aumento na incidência de DECH.
Antes de 1998, a tipificação de HLA–A, HLA–B e HLA–C era feita através de métodos sorológicos. A partir de então, o sequenciamento direto automatizado desses genes passou a assegurar a identidade molecular. Nos dias atuais, os resultados de transplantes que utilizam doadores compatíveis identificados pela tipificação moderna se aproximam dos resultados de transplantes com irmãos compatíveis em muitas categorias de doenças.9
Transplante autólogo
Em comparação com os alogênicos, os transplantes autólogos têm a vantagem de evitar a incidência de DECH e das complicações associadas; a grande desvantagem é que as células autólogas não têm os efeitos antitumorais com a mediação imunológica dos transplantes alogênicos e podem conter células tumorais viáveis. A remoção de células tumorais com antibióticos para antígenos específicos de tumores, juntamente com complementos, toxinas ou grânulos imunomagnéticos, é bastante eficaz e reduz em 1 mil a 10 mil vezes o número de células tumorais.10
Outros métodos de purificação de células-tronco que se encontram, atualmente, sob investigação são adesão de anticorpos e técnicas de fluxo que selecionam células-tronco hematopoiéticas normais, deixando para trás as células tumorais; tratamento in vitro das células autólogas com agentes quimioterápicos seletivos; cultivação in vitro de células hematopoiéticas normais com crescimento seletivo.
Estudos de marcação de genes demonstraram que as células tumorais remanescentes podem contribuir para recidivas;11 entretanto, ainda não se sabe quais métodos ? caso exista algum ? de purificação celular podem impedir a ocorrência de recidivas. Além disso, muitas dessas técnicas poderão retardar a recuperação hematológica e imunológica depois de transplantes.
Várias análises retrospectivas sugerem que o tratamento in vitro de medula óssea pode ser eficaz nos casos de leucemia mieloide aguda (LMA) e de linfoma não Hodgkin (LNH) de células B, embora não exista nenhum estudo prospectivo controlado suficientemente amplo.10
Transplante de eritrócitos periféricos
De maneira geral, as células-tronco hematopoiéticas circulam no sangue periférico, embora em números bastante baixos. Os primeiros experimentos em modelos animais mostraram que são necessárias, no mínimo, 10 vezes mais células mononucleares para salvar animais da irradiação corporal total letal nas situações em que as células forem coletadas no sangue periférico, ao invés da medula de animais não tratados.
As tentativas iniciais para usar células-tronco do sangue periférico, como fontes de enxertos hematopoiéticos, foram complicadas pelo grande número de coletas (plasmaférese) ? em geral, sete ou mais ? e pela lentidão do processo de enxerto.
Em seguida, demonstrou-se que ocorre um aumento acentuado no número de progenitores hematopoiéticos no sangue, medidos como unidades formadoras de colônias hematopoiéticas ou como células CD34+, durante a recuperação de Qt anterior ou logo após a exposição aos fatores de crescimento hematopoiéticos.12,13 Esse fato culminou na realização de diversos estudos sobre o uso de células-tronco periféricas como substitutas para a medula óssea.
No início, esses estudos foram conduzidos no ambiente autólogo porque as coletas de células-tronco do sangue periférico contêm um grande número de células T, que poderiam induzir DECH se as coletas fossem usadas em transplantes alogênicos.
No ambiente autólogo, comumente é possível coletar um número suficiente de células para promover o enxerto com uma a três leucofereses com 4 horas de duração após o tratamento do paciente com fator estimulador das colônias de granulócitos (em inglês, granulocyte colony stimulating fator [G-CSF]) ou com fator estimulador das colônias de granulócitos-macrófagos (em inglês, granulocyte-macrophage colony-stimulating fator [GM-CSF]).
Ainda não é conhecido o mecanismo exato pelo qual os fatores de crescimento mielocítico aumentam de forma acentuada o número de células-tronco no sangue periférico; entretanto, alguns estudos sobre a murina sugerem que os fatores de crescimento mielocítico ativam neutrófilos para liberar serina-proteases que, por sua vez, fazem a clivagem proteolítica de moléculas de adesão vascular na medula, liberando células-tronco hematopoiéticas.14
O plerixafor é um antagonista de CXCCR4 e, pelo bloqueio de sua interação com SCF-1, mobiliza as células CD34+a partir da medula. Estima-se que, em 10 a 20% de pacientes que não conseguem mobilizar quantidades suficientes de células CD34+apenas com um fator de crescimento mielocítico, a adição de plerixafor pode produzir alguns benefícios.15
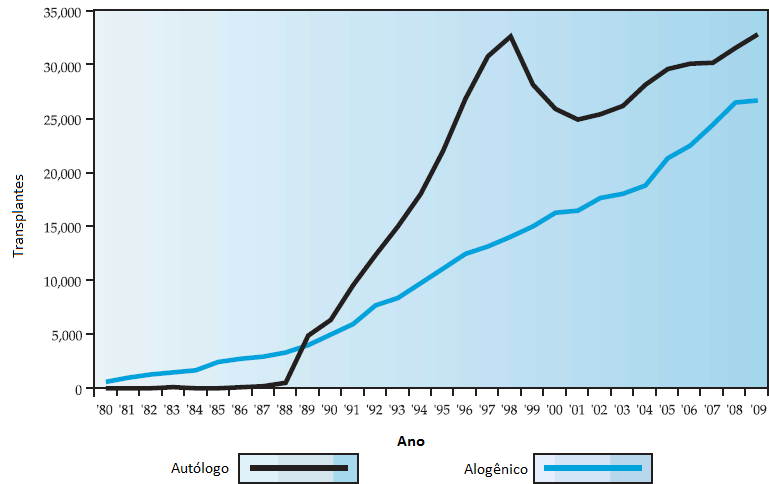
A Figura 1, a seguir, descreve os números totais estimados de TCTHs alogênicas e autólogas feitos em todo o mundo no período de 1980 a 2009. A queda nos transplantes autólogos em 1999 foi atribuída à redução nos transplantes para tratamento de câncer de mama, enquanto que o achatamento na curva de transplantes alogênicos resultou de uma quantidade menor de transplantes para tratamento de anemia mielogênica crônica desde a introdução do mesilato de imatinibe.

TCTH: transplante de células-tronco hematopoiéticas.
Figura 1 - Números totais estimados de TCTHs alogênicas e autólogas feitos em todo o mundo no período de 1980 a 2009, de acordo com estimativas do International Bone Marrow Transplant Registry.
DÍSTICOS DA FIGURA 1
|
Tranplants |
= |
Transplantes
|
|
Year |
= |
Ano
|
|
Autologous |
= |
Autólogos
|
|
Allogeneic |
= |
Alogênicos
|
Nos casos em que são coletadas mais de 2,5 milhões de células CD34+ e, subsequentemente, são usadas para transplantes autólogos, a recuperação de 500 granulócitos/µL, em um período de 12 dias após o transplante, e a recuperação de 20 mil plaquetas/µL, em um período de 14 dias após o transplante, são ocorrências bastante comuns.16 A recuperação é mais rápida do que aquela com células-tronco medulares autólogas.
Mesmo que ainda não se saiba se a probabilidade de contaminação por células tumorais implantáveis é maior ou menor no sangue periférico, em comparação com a medula óssea, a mobilização do sangue periférico substitui quase por completo a medula como fonte de células-tronco para transplantes autólogos.
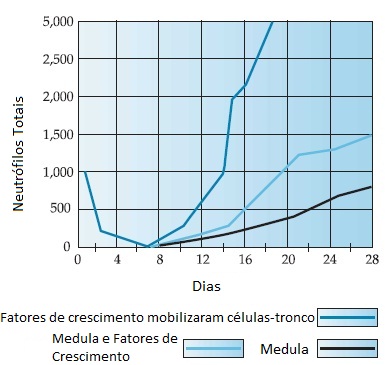
A Figura 2 mostra os padrões típicos de recuperação mielocítica após o TCTH usando apenas a medula óssea, medula óssea mais fatores de crescimento mielocíticos pós-transplante e células-tronco do sangue periférico mobilizadas por fatores de crescimento.
TCTH: transplante de células-tronco hematopoiéticas.
Figura 2 - Padrões típicos de recuperação mielocítica após o TCTH, usando apenas a medula óssea, medula óssea mais fatores de crescimento mielocíticos pós-transplante, e células-tronco do sangue periférico mobilizadas por fatores de crescimento.
DÍSTICOS DA FIGURA 2
|
Total neutophils (cells/µL) |
= |
Quantidade total de neutrófilos (células/µL)
|
|
Days |
= |
Dias
|
|
Growth-factor mobilized peripheral blood stem cells |
= |
Células-tronco do sangue periférico mobilizadas por fatores de crescimento
|
|
Marrow and growth factors |
= |
Medula e fatores de crescimento
|
|
Marrow |
= |
Medula
|
As células-tronco do sangue periférico também foram utilizadas em transplantes alogênicos. Alguns estudos iniciais com células-tronco do sangue periférico mobilizadas pelo G-CSF de doadores compatíveis com HLA idêntico indicaram que enxertam de forma mais rápida e que, aparentemente, a incidência da DECH não é mais elevada do que se poderia esperar com medula óssea, apesar do transplante de uma quantidade, pelo menos, 10 vezes maior de células T maduras.17
Alguns testes randomizados confirmaram que o uso de células-tronco do sangue periférico acelera o processo de enxerto sem aumentar a incidência de DECH.18–20 Ainda que o número de ocorrências de DECH crônica seja um pouco mais elevado com células-tronco do sangue periférico, a incidência de recorrências tumorais é menor e a sobrevida geral sem a doença tende a ser mais longa com o uso de células-tronco do sangue periférico, sobretudo em pacientes com leucemias de risco mais elevado.18–20
Transplante de sangue do cordão umbilical
O sangue do cordão umbilical é rico em células CD34+, as quais poderão servir de fontes alternativas de células-tronco. Os primeiros transplantes de sangue do cordão umbilical em seres humanos foram feitos no final da década de 1980 em indivíduos com anemia de Fanconi. Em uma série de 44 crianças tratadas com sangue do cordão umbilical de irmãos, a velocidade do enxerto mielocítico foi semelhante à observada em transplantes medulares, embora a recuperação das plaquetas tenha sido mais lenta.
A incidência de DECH foi de 6%, sendo considerada lenta. É provável que isso tenha refletido as idades jovens dos receptores e o fato de o sangue do cordão umbilical ser relativamente desprovido de células T maduras. Posteriormente, em vários estudos, o sangue do cordão umbilical de doadores sem relação de parentesco foi colocado em um banco de sangue e usado em transplantes futuros.20
Um sumário dos primeiros 562 transplantes de sangue de cordão umbilical de doadores sem relação de parentesco promovidos pelo New York Blood Center’s Program registrou enxertos em cerca de 80% dos pacientes, embora o tempo para o enxerto tenha sido bastante prolongado ? 24 dias para neutrófilos e 72 dias para plaquetas.21 A incidência geral de DECH aguda grave foi de 23%.
Atualmente, o sangue de cordão umbilical é usado como fonte de células-tronco em, aproximadamente, um terço dos transplantes pediátricos alogênicos. As vantagens do sangue do cordão umbilical como fonte de células-tronco incluem a capacidade aparente de usar doadores com maior disparidade de HLA, risco aparentemente mais baixo de DECH, disponibilidade rápida de unidades e estoques de unidades de doadores sem relação de parentesco. As desvantagens incluem enxerto mais lento, maior incidência de falhas, reconstituição imune mais lenta e tendência para taxas de recorrência mais elevadas das doenças.
Alguns estudos que fizeram a comparação dos resultados de doadores compatíveis sem relação de parentesco com doadores sem relação de parentesco de transplantes de sangue do cordão umbilical em pacientes pediátricos apresentaram, em alguns casos, sobrevida equivalente, ainda que outros estudos tenham mostrado uma tendência para melhorar a sobrevida usando doadores de medula sem relação de parentesco.22 O número total de células nucleadas e do conteúdo de células CD34+ de sangue do cordão umbilical é um preditor da velocidade de recuperação e está altamente associado com o resultado geral dos transplantes.
Nos dias atuais, recomendam-se doses limiares de 2,5x107 células/kg em relação ao peso do receptor ou 1,7x105 células CD34+/kg. As tentativas iniciais de realização de transplantes de sangue do cordão umbilical de doares sem relação de parentesco em crianças e adultos maiores resultaram em uma sobrevida um pouco menor em comparação com o uso de doadores adultos sem relação de parentesco.23,24 A melhora na sobrevida foi associada a doses mais elevadas de células de sangue do cordão umbilical e a um maior grau de compatibilidade.
No esforço de aumentar o número de células de sangue do cordão umbilical, passou-se a explorar transplantes combinando duas unidades de cordão umbilical, sendo que os relatos sugerem que os enxertos em adultos poderão melhorar com essa abordagem.25 Embora, em última análise, apenas um cordão seja enxertado, o uso de dois cordões diminui o risco de falhas e, aparentemente, está associado a uma melhora no efeito enxerto versus tumor.26
Procedimento de transplante
Regimes preparatórios
Os regimes preparatórios são administrados antes dos TCTHs. O objetivo principal deles é eliminar as células anormais ou malignas que causam a doença e, no contexto de transplantes alogênicos, suprimir o sistema imune para impedir a rejeição de enxertos. O regime apropriado para qualquer indivíduo é determinado pela doença que será tratada, pela idade e pelo estado de saúde do paciente, assim como pela fonte de células-tronco a serem enxertadas.
Em um extremo, os pacientes que fazem transplantes para tratamento de doença de imunodeficiência combinada grave com células-tronco de um irmão com HLA compatível não precisam de nenhum regime preparatório, tendo em vista que não há células anormais para eliminar (a doença está sendo causada pela ausência de células normais) e que os sistemas imunes dos pacientes já estão suficientemente deprimidos para evitar a rejeição do enxerto.
O sistema hematopoiético de pacientes com anemia aplásica não é normal, porém são imunocompetentes o bastante para rejeitar medulas ósseas alogênicas caso não seja administrado nenhum tipo de imunossupressão. Nesse quadro, o tratamento com altas doses de ciclofosfamida mais globulina antitimócitos é imunossupressivo o suficiente para assegurar o enxerto, desde que o doador seja um irmão com HLA idêntico. A imunossupressão deverá ser intensificada nas situações em que o transplante for de um doador sem nenhuma relação de parentesco; por conseguinte, costuma-se adicionar ao tratamento baixas doses de irradiação corporal total.
Se o transplante for usado para tratar doenças que se caracterizam pela presença de uma população de células anormais e não malignas, como talassemia e anemia da célula falciforme, deve-se eliminar do regime preparatório a população de células anormais e suprimir o sistema imune do paciente. Para complementar o tratamento, recomenda-se adicionar altas doses de bussulfano (16mg/kg, dividida em 4 dias) à ciclofosfamida durante o regime preparatório.
Ao desenvolver regimes preparatórios para transplantes com a finalidade de tratar malignidades, a maior parte dos investigadores mantém o foco no uso de agentes altamente ativos contra a malignidade que estiver sendo tratada, em que a mielossupressão é a toxicidade limitadora de dose dominante em ambientes sem transplantes. Como resultado, as terapias utilizadas com maior frequência se baseiam na administração de agentes alquilantes (por exemplo, ciclofosfamida, bussulfano, tiotepa, melfalana, carmustina), etoposida, citarabina e irradiação corporal total.
Tipicamente, utilizam-se altas doses de regimes preparatórios nas situações em que a finalidade dos transplantes alogênicos é o tratamento de malignidades; entretanto, o efeito do enxerto alogênico versus o efeito tumoral suprime o crescimento tumoral independentemente do regime preparatório. Esse fato levou alguns investigadores a questionar se os regimes menos intensivos são mais eficazes e menos tóxicos.
A nomenclatura atual define os regimes preparatórios como não mieloablativos se a supressão medular for mínima, mesmo que o transplante não seja realizado ou tenha condicionamento de intensidade reduzida, nos casos em que produzir citopenias significativas com algum tempo de duração.27
Ainda não foram publicadas grandes comparações prospectivas de diferentes regimes não mieloablativos ou com condicionamento de intensidade reduzida. Em comparação com regimes convencionais que utilizam doses elevadas, os com dose reduzida resultam em períodos mais curtos de pancitopenia, em uma quantidade menor de transfusões, menos infecções bacterianas e menor incidência de danos diretos no fígado e nos pulmões.
Embora a aplicação desses regimes implique em taxas de recidiva mais elevadas, essa desvantagem poderá ser contrabalançada pela melhora na segurança e, além do mais, os regimes com doses mais baixas permitem a realização de transplantes alogênicos em pacientes na sexta ou sétima décadas de vida.
As curas no longo prazo de pacientes com uma grande variedade de malignidades hematológicas que, em outras circunstâncias, não seriam curáveis foram documentadas com o uso de condicionamento com dose reduzida, mesmo que ainda não tenha sido definida a escolha de regimes específicos para cada tipo de doença no contexto de cada paciente.
Coleta e infusão de células-tronco
Em geral, faz-se a coleta de medula nas posições anterior e posterior da crista ilíaca, mantendo-se o doador em anestesia espinal ou geral. Retira-se um volume medular total equivalente a 10–15mL/kg; a coleta de medula limita-se ao volume de 3–5mL em cada sítio de aspiração para evitar diluição excessiva com circulação de sangue periférico dentro da crista ilíaca. A medula é filtrada em filtros de 0,3mm e 0,2mm para remover espículas ósseas e gordura.
Eventualmente, a medula pode precisar de tratamento adicional in vitro para remoção de outras células, incluindo eritrócitos de doadores, para evitar reações às transfusões hemolíticas nos casos de incompatibilidade ABO; de células T de doadores para impedir a ocorrência de DECH; de células tumorais de medulas autólogas. Os riscos associados à doação de medula são pequenos; algumas séries apresentaram seis complicações sérias não fatais em um total de 1.220 doações medulares consecutivas.28
De maneira geral, as células-tronco de sangue periférico são coletadas através de aférese em doadores previamente tratados apenas com fator de crescimento hematopoiético ou, nos casos de transplantes autólogos, depois de Qt em combinação com tratamento com fatores de crescimento.
Comumente, as tentativas buscam coletar uma quantidade mínima de 5x106 células CD34+ ? com essa dose, o enxerto é rápido e consistente.16 As infusões de medula e de células-tronco de sangue periférico costumam ser bem toleradas, embora, às vezes, os pacientes desenvolvam febre, tosse ou uma leve falta de ar. Na maior parte dos casos, a lentificação da infusão alivia esses sintomas.
Enxertos
A taxa de enxerto depende da fonte de células-tronco, da escolha da profilaxia contra DECH e da utilização ou não de fatores de crescimento hematopoiéticos. O enxerto mais rápido é com células-tronco de sangue periférico; nesse contexto, ele ocorre, em geral, depois de 12 dias. Nos casos de transplante de medula ou de sangue do cordão umbilical, a contagem de granulócitos costuma atingir 100 células/µL por volta do décimo sexto dia e 500 células/µL no vigésimo segundo dia.
A taxa de recuperação mielocítica poderá ser acelerada em 4 a 6 dias com o uso de G-CSF ou de GM-CSF após os transplantes de medula; entretanto, os fatores de crescimento pós-transplante têm efeitos menores nos casos em que o sangue periférico mobilizado for a fonte das células-tronco transplantadas.29 O uso de metotrexato depois de transplantes alogênicos retarda a recuperação por uma média de 4 dias. De maneira geral, a recuperação de plaquetas ocorre logo após a recuperação dos granulócitos.
Complicações dos transplantes
Toxicidades diretas precoces do regime preparatório ablativo
Os regimes preparatórios ablativos que precedem os transplantes estão associados a uma quantidade substancial de toxicidades, que variam de forma considerável de acordo com o regime específico utilizado. Por exemplo, condições como náusea, vômito e eritema cutâneo leve surgem de imediato em quase todos os pacientes após o regime de irradiação corporal total com ciclofosfamida. Algumas vezes, observa-se a presença de cistite hemorrágica a despeito da irrigação da bexiga ou da terapia com um composto de mesna.
Em circunstâncias muito raras (menos de 2% de casos), ocorre o desenvolvimento de cardite hemorrágica. Com frequência, a mucosite oral se desenvolve dentro de um período de 5 a 7 dias após os transplantes e, de maneira geral, exige a aplicação de analgesia com narcóticos. A analgesia controlada pelos pacientes aumenta a satisfação deles e, de forma surpreendente, resulta em doses cumulativas mais baixas de narcóticos.
O fator de crescimento de queratinócitos (em inglês, keratinocyte growth factor [HGF]), isto é, a palifermina, pode encurtar de forma substancial o tempo de duração da mucosite grave depois de regimes de transplantes ablativos.30 Dentro de 10 dias após o transplante, a maioria dos pacientes desenvolve alopecia e granulocitopenia profunda.
A doença venoclusiva hepática (DVOH), também conhecida por síndrome de obstrução sinusoidal, é uma complicação séria de altas doses de Qt; esse tipo de doença acomete cerca de 10 a 20% dos indivíduos.31 A DVOH, tipificada pelo desenvolvimento de ascite, hepatomegalia sensível, icterícia e retenção de líquidos, pode ocorrer a qualquer momento no primeiro mês após o transplante; o pico de incidência é por volta do décimo sexto dia.
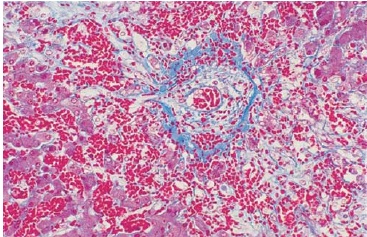
As características histológicas da DVOH incluem estreitamento concêntrico ou obliteração fibrosa das vênulas hepáticas terminais e das veias sublobulares e necrose de hepatócitos da zona 3, conforme se pode ver na Figura 3. Os fatores predisponentes são hepatite por qualquer causa antes do transplante (por exemplo, evidenciada por marcadores elevados de hepatite B e de hepatite C) e uso de regimes condicionadores mais intensivos.32,33
DVOH: doença venoclusiva hepática.
Figura 3 - Fotomicrografia de biópsia de fígado colorida com tricromo (ampliação de 25 vezes do original), com alterações típicas da DVOH. Destaca-se uma veia sublobular pelo tecido conjuntivo denso na cor azul na camada adventícia externa. Há um estreitamento acentuado do lúmen da veia por uma zona subendotelial larga e eritematosa contendo eritrócitos aprisionados e matriz extracelular solta. Os cordões de hepatócitos que circundam a veia estão necrosados, e os sinusoides intervenientes são hemorrágicos. A deposição de coagulantes nos sinusoides e na zona subendotelial da veia obstrui o fluxo externo do fígado, produzindo hipertensão sinusoidal e hepatomegalia.
Fonte: Cortesia do Fred Hutchinson Cancer Research Center.
Apesar de ainda não se conhecer a sequência precisa dos eventos que resultam na apresentação clínica da doença venoclusiva, é muito comum ocorrerem lesões citotóxicas diretas nas vênulas hepáticas e no endotélio sinusoidal, com deposição subsequente de fibrina e desenvolvimento de um estado hipercoagulável local, na fase inicial da condição. As lesões citotóxicas diretas nos hepatócitos da zona 3 são fatores contribuintes.
Aproximadamente, 30% de pacientes que desenvolvem DVOH morrem em decorrência da doença, sendo que a insuficiência hepática progressiva leva a uma síndrome hepatorrenal terminal. A defibrotida, um polidesoxirribonucleotídeo, reduz a trombogenicidade do endotélio vascular e, aparentemente, em testes prospectivos não randomizados, tem apresentado alguns benefícios sem toxicidade significativa.34
A síndrome de pneumonia idiopática acomete até 5% de pacientes, ainda que a maior parte dos casos de pneumonia que ocorre depois de transplantes seja causada por agentes microbianos.35 A maioria dos especialistas considera a síndrome de pneumonia idiopática uma toxicidade direta da Qt intensiva, embora as evidências de um papel importante para as citocinas solúveis estejam crescendo cada vez mais.
As biópsias revelam danos alveolares difusos em alguns casos, ao passo que, em outros, há um componente mais claramente intersticial. O tratamento com altas doses de glicocorticoides, em geral, é uma tentativa válida, embora ainda não tenha sido feito nenhum teste randomizado para avaliar sua eficácia. Os testes atuais têm como foco a função bloqueadora do fator de necrose tumoral.36
Toxicidades diretas tardias do regime preparatório ablativo
As complicações diretas da Qt observadas tardiamente depois de transplantes incluem taxa de crescimento reduzida em crianças e atraso no desenvolvimento das características sexuais secundárias.37 A maior parte das crianças pode apresentar alguma deficiência no fator de crescimento e deve fazer terapia de reposição.
A falência ovariana é uma ocorrência comum na maioria das mulheres na fase pós-puberal, enquanto a azoospermia, nos homens. A osteonecrose asséptica chega a acometer até 10% de pacientes de transplante, em especial indivíduos com DECH crônica que precisam fazer tratamento à base de corticosteroides.
Da mesma forma, o desenvolvimento de catarata é comum em 10 a 20% dos indivíduos, sendo que o risco é mais elevado em pacientes que tomam esteroides para tratamento de DECH crônica.38 A recuperação dos efeitos dos transplantes é um processo gradual, e a recuperação total pode ocorrer dentro de um período de 3 a 5 anos.39
Os pacientes tratados com altas doses de Qt e hematócrito (hct) correm um grande risco de desenvolver malignidades secundárias,40,41 sendo o risco mais elevado nos que recebem transplantes de medula óssea com depleção de células T e naqueles que recebem múltiplos ciclos de medicamentos altamente imunossupressivos depois de transplantes para tratamento de DECH; nesses casos, observa-se uma alta incidência de doença linfoproliferativa associada ao vírus Epstein-Barr.
Observa-se uma incidência menor de tumores sólidos após os transplantes, cuja taxa anual cumulativa é de 2,9% em 10 anos. A incidência atuarial de mielodisplasia depois de transplantes autólogos para tratamento de LNH e doença de Hodgkin (DH) pode chegar ao nível de 10%.42
Toxidades diretas ou regimes de condicionamento com redução de intensidade
O espectro de toxicidade observado depois de transplantes alogênicos usando regime não mieloablativo ou regimes de condicionamento com redução de intensidade difere bastante do observado depois de regimes preparatórios ablativos. Com esses regimes menos intensivos, os pacientes exigem uma quantidade menor de transfusões de eritrócitos e de plaquetas, têm incidência menor de doença venoclusiva e de pneumonia intersticial e apresentam uma quantidade menor de infecções bacterianas.
Entretanto, esses regimes não diminuem a incidência de infecções fúngicas ou virais. A incidência cumulativa de DECH aguda e crônica se assemelha àquela observada depois de regimes ablativos, ainda que o início da DECH aguda possa ser retardado com condicionamentos de intensidade mais baixa. Essas diferenças se traduzem em uma menor incidência total de mortalidade não causada por recidivas, sobretudo em indivíduos com grau mais elevado de comorbidades no início do procedimento de transplante.43–45
Falência de enxertos
Mesmo que os enxertos completos e sustentados sejam a regra geral depois de transplantes, a função medular não retorna em alguns casos; em outros, depois de enxertos temporários, a função da medula óssea acaba se perdendo ao longo do tempo. A falência de enxertos depois de transplantes autólogos pode ser resultado de danos medulares ocorridos antes da colheita, durante o tratamento ex vivo, durante o armazenamento ou após a exposição a agentes mielotóxicos depois de transplantes.46
Infecções por citomegalovírus (CMV) ou por herpes-vírus tipo 6 também poderão resultar no funcionamento inadequado da medula óssea. A falência de enxertos depois de transplantes alogênicos pode ser devido à rejeição de enxertos com mediação imunológica, sendo mais comum depois de regimes de condicionamento menos imunossupressivos em receptores de medula óssea com depleção de células T, assim como em receptores de medula óssea com HLA incompatível.
O tratamento da falência de enxertos inicia com a remoção de todos os agentes potencialmente mielossupressivos. Um segundo passo seria tentar fazer um teste curto com um fator de crescimento mielocítico (GM-CSF ou G-CSF); em torno de 40 a 50% de pacientes respondem ao teste.47
A identificação de linfócitos persistentes de hospedeiro no sangue periférico ou na medula óssea sugere rejeição imunológica. Para esses indivíduos, é indicada imunossupressão adicional antes da realização de um segundo transplante. Vários estudos documentaram segundos transplantes bem-sucedidos depois de um regime à base de ciclofosfamida e de globulina antitimocítica ou fludarabina mais irradiação corporal total em baixas doses.48
Doença do enxerto contra hospedeiro
Doença do enxerto contra hospedeiro aguda
A DECH é o resultado da transferência de células T alogênicas, com reação do enxerto contra alvos de hospedeiros diferentes sob o ponto de vista genético.49 Classifica-se como aguda a DECH que se desenvolve nos primeiros 3 meses após o transplante e que se caracteriza pela presença de erupções cutâneas maculopapulares eritematosas que, ao contrário de muitos tipos de erupção, surgem na palma da mão ou na sola do pé.
Além disso, a DECH se caracteriza pela presença de condições como anorexia ou diarreia persistentes, ou ambas, e de doença hepática, evidenciada pelo aumento nos níveis de bilirrubina, alanina e aspartato aminotransferase, e fosfatase alcalina. Eventualmente, uma síndrome semelhante ocorre além de 3 meses a partir de um transplante (DECH aguda com início tardio) ou somente quando se retira a imunossupressão.
Biópsias da pele, fígado e intestinais endoscópicas são os métodos usuais para determinar o diagnóstico de DECH aguda; essas biópsias podem revelar a presença de danos na epiderme e nos folículos pilosos, rompimento segmentar em pequenos ductos biliares e ulceração na mucosa intestinal causada pela destruição das criptas do intestino.50
O sistema de estágios clínicos de DECH aguda mais amplamente utilizado inclui extensão do envolvimento da pele, do fígado e dos intestinos. Numerosas variações desse sistema foram desenvolvidas na tentativa de melhorar sua utilidade para propósitos específicos.51–53 A incidência de DECH aguda aumenta em indivíduos mais velhos, em receptores de medula óssea de doadores incompatíveis e em pacientes incapazes de receber doses completas dos medicamentos utilizados para impedir a ocorrência da DECH.54
Existem duas abordagens para impedir a ocorrência de DECH aguda:
uso de agentes imunossupressivos durante o período pós-transplante imediato;
remoção de células T da população de células transplantadas.
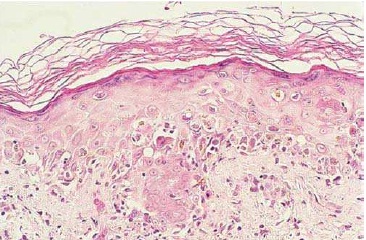
A Figura 4 mostra a fotomicrografia de uma biópsia de pele, a qual mostra as características da DECH na pele.
DECH: doença do enxerto contra hospedeiro.
Figura 4 - Fotomicrografia de uma biópsia de pele, colorida com hematoxilina e eosina (ampliação de 40 vezes do original), mostrando as características da DECH na pele.
Fonte: Cortesia do Fred Hutchinson Cancer Research Center.
Com base na Figura 4, pode-se observar que o edema intercelular (espongiose) e o edema intracelular (degeneração reticular) da epiderme inferior são evidentes. As células mononucleares se espalham em toda a área epidérmica juntamente com muitos corpos que sofreram apoptose, incluindo células epidérmicas mortas com citoplasma hipereosinofílico e núcleos picnóticos basofílicos.
Verifica-se, também, que o processo inflamatório produziu incontinência de pigmento de melanina na derme papilar, assim como bloqueios intraepidérmicos sérios de melanina, levando à hiperpigmentação. Como a apoptose e os danos na camada basal são extensivos, a epiderme se torna muito escamosa e possivelmente necrosada.
O metotrexato e a ciclosporina, considerados isoladamente, são também eficazes em termos de profilaxia, embora o uso em combinação seja menos eficiente.55 Medicamentos como prednisona, FK 506 (tacrolimo), rapamicina e micofenolato também foram usados em várias combinações, assim como altas doses de ciclofosfamida.56 Até o presente momento, nenhum regime simples foi comprovadamente superior à combinação de um inibidor da calcineurina (ciclosporina ou tacrolimo) e metotrexato.
A remoção de células T da medula óssea alogênica é bastante eficaz para evitar a ocorrência de DECH aguda, embora esteja associada a um aumento na incidência de rejeição de enxertos e de recidiva leucêmica, sobretudo no quadro de leucemia mieloide crônica (LMC).
Da mesma forma, diversas terapias potenciais encontram-se, atualmente, em fase de estudos, incluindo depleção parcial de células T e depleção total de células T, acompanhadas pela reintrodução de uma fração de células T. A partir do momento em que começa a se desenvolver, a DECH aguda pode ser tratada com glicocorticoides, globulina antitimocítica e anticorpos monoclonais cujos alvos são as células T ou seus receptores.
O Quadro 1 mostra o estagiamento clínico da DECH aguda.
Quadro 1
|
ESTAGIAMENTO CLÍNICO DA DOENÇA DE ENXERTO CONTRA HOSPEDEIRO AGUDA*
| |||
|
Estágios |
Alterações na pele |
Fígado |
Intestino
|
|
I |
Erupção cutânea maculopapular <25% da superfície corporal |
Bilirrubina: 2–3mg/dL |
Diarreia: 500–1.000mL/dia
|
|
II |
Erupção cutânea maculopapular: 25–30% da superfície corporal |
Bilirrubina: 3–6mg/dL |
Diarreia: 1.000–1.500mL/dia
|
|
III |
Eritrodermia generalizada |
Bilirrubina: 6–15mg/dL |
Diarreia: >1.500mL/dia
|
|
IV |
Descamação e bolhas |
Bilirrubina >15mg/dL |
Dor e íleo
|
*A gravidade geral varia de envolvimento leve da pele (estágio I) a envolvimento grave de múltiplos órgãos, normalmente com resultado fatal (estágio IV).
Doença do enxerto contra hospedeiro crônica
A DECH que persiste por 3 meses ou mais depois de um transplante denomina-se DECH crônica, e possui características em comum com as doenças vasculares colagenosas, incluindo erupção cutânea malar, alterações esclerodermatosas, síndrome de Sicca, artrite, bronquiolite obliterativa e, em alguns casos, degeneração do ducto biliar e colestasia.
A DECH crônica pode também se desenvolver como síndrome sobreposta com características tanto de aguda como de crônica. O desenvolvimento de DECH crônica ocorre em 20 a 40% dos pacientes, sendo mais frequente em pacientes mais velhos e naqueles com DECH aguda precedente.57
O tratamento usual é à base de prednisona, ciclosporina ou de uma combinação entre esses dois medicamentos;58 alguns estudos adicionaram azatioprina ou talidomida, porém estudos randomizados que exploraram a prednisona ou prednisona mais um inibidor da calcineurina apresentaram resultados negativos até o momento.59
Na maior parte dos pacientes, a DECH crônica desaparece de forma espontânea dentro de algum tempo, permitindo a interrupção da terapia imunossupressiva, embora ainda seja necessário fazer o tratamento por um período de 1 a 3 anos. Os pacientes com DECH crônica que estiverem fazendo terapia imunossupressiva são suscetíveis a infecções bacterianas e, portanto, devem receber tratamento com antibióticos profiláticos.
Doenças infecciosas
Nas primeiras 2 a 3 semanas após um transplante, todos os receptores de transplantes mieloablativos se tornam gravemente granulocitopênicos. Para diminuir o risco de infecções bacterianas disseminadas, os pacientes pós-transplante começam a receber terapia com antibióticos de espectro amplo a partir do momento em que se tornam granulocitopênicos.
Além disso, a administração profilática de fluconazol diminui a incidência de infecções causadas pelo organismo cândida albicans.60 Alguns estudos que fizeram testes com outros agentes moderadores como o voriconazol em vez do fluconazol apresentaram resultados inconsistentes e nenhum benefício expressivo. O tratamento de pacientes que se tornam febris, a despeito da terapia antibiótica profilática e da terapia antifúngica, é um grande desafio; nesses casos, a terapia deve ser orientada pelas condições de cada paciente e pela experiência da instituição.
Por exemplo, nos casos em que houver desenvolvimento de febre em pacientes que estiverem fazendo tratamento profilático à base de levofloxacino, a escolha subsequente de antibiótico deverá ser orientada pela suspeita de que a fonte da infecção seja intra-abdominal, sendo que, nessa hipótese, a terapia com meropeném ou imipeném é a mais apropriada. Se a fonte da infecção intra-abdominal não for uma grande preocupação, uma das opções é a seleção da ceftazidima.61
Se a febre persistir por mais de 72 horas, costuma-se adicionar ao regime de tratamento medicamentos como derivados da anfotericina, caspofungina ou voriconazol.62 As transfusões de granulócitos poderão ser eficazes no tratamento de infecções específicas, sobretudo levando-se em conta que, nos dias atuais, os doadores podem ser tratados com G-CSF antes da doação, aumentando de forma substancial o número de granulócitos possíveis de serem coletados e transfundidos.62
Entretanto, não se chegou a atribuir nenhum papel para as transfusões profiláticas de granulócitos. O isolamento do fluxo de ar laminar pode diminuir a incidência de infecções, mesmo que não tenha nenhum impacto na sobrevida de pacientes de transplantes que tenham sido tratados contra malignidades. Com os métodos atuais de tratamento de suporte, o risco de morte por causas infecciosas é inferior a 5% durante o período da granulocitopenia.
Em geral, no passado, as infecções por CMV ocorriam depois dos transplantes, sobretudo em receptores de medula óssea alogênica. Todavia, comprovou-se a possibilidade de evitar infecções primárias por CMV em pacientes soronegativos para CMV com uso de produtos derivados do sangue soronegativos para CMV.
Em pacientes soropositivos para CMV, o tratamento com ganciclovir, logo após a excreção do vírus se tornar evidente, pode reduzir a incidência de doença e de morte associadas ao CMV, ainda que, em alguns indivíduos, a doença se desenvolva antes ou ao mesmo tempo em que se observa a excreção viral.
O início da profilaxia com ganciclovir no momento do enxerto impede o desenvolvimento de infecções por CMV na maior parte dos indivíduos, embora esse medicamento produza supressão medular significativa em, pelo menos, 30% de pacientes.63 A maioria dos centros, após os transplantes, faz o monitoramento do sangue periférico para detecção da antigenemia para citomegalovírus ou do CMV DNA por meio da reação da cadeia de polimerase, iniciando a terapia preventiva com ganciclovir ou foscarnete apenas se, e quando, os pacientes apresentarem resultados positivos nos testes para detectar a presença de CMV.
O foscarnete também é muito eficaz em indivíduos que desenvolvem antigenemia para citomegalovírus ou infecção, apesar da terapia com ganciclovir, ou em pacientes que não toleram o ganciclovir. Caso não sejam evitadas, as infecções causadas pelo herpes-vírus contribuem para a gravidade da esofagite e da mucosite oral precoce. Entretanto, o uso profilático de aciclovir na dosagem de 250mg/m2, por via intravenosa (IV), em intervalos de 8 horas, ou 800mg, por via oral (VO), 2x/dia, impede a reativação do vírus de herpes-vírus em quase todos os pacientes soropositivos.64
O vírus sincicial respiratório (VSR), o vírus da influenza, o metapneumovírus e outros agentes que produzem infecções respiratórias superiores e inferiores adquiridas na comunidade são observados também em receptores de transplantes e carregam um grande risco de morte. É fundamental proteger os pacientes contra visitas e membros da equipe médica infectados e adotar medidas como lavar as mãos de forma adequada, usar máscaras faciais e fazer vacinação anual contra gripe em membros da família e da equipe médica.64
Recomenda-se adiar os transplantes em pacientes com infecções respiratórias virais ativas. A ribavirina inalatória pode ser eficaz nos casos de VSR; a globulina venosa imune (IVIg) ou o palivizumabe também foram usados no tratamento de doenças no trato respiratório inferior. Os inibidores da neuraminidase também são eficazes no tratamento de infecções causadas por influenzavírus. O herpes-vírus humano tipo 6 (HHV–6) foi associado à falência de enxertos, à encefalite, à enterite e à pneumonia.
Níveis elevados de viremia pelo papovavírus BK levam à cistite hemorrágica. As infecções produzidas pelo adenovírus estão associadas a condições como pneumonia, hepatite e insuficiência renal, e são observadas com menos frequência em pacientes que tenham recebido tratamento com ganciclovir para profilaxia de CMV ou como terapia preventiva.
O pneumocystis jirovecii já causou pneumonia em 5 a 10% de pacientes depois de transplantes, embora, atualmente, seja possível prevenir a incidência desse tipo de complicação em todos os pacientes tratando-os, primeiramente, com trimetoprima + sulfametoxazol, VO, durante 1 semana antes dos transplantes, retomando o tratamento com a frequência de 2 dias por semana logo após a colocação do enxerto e prosseguindo durante toda a terapia imunossupressiva.64
As tentativas de dessensibilização são importantes em pacientes com reações alérgicas à combinação trimetoprima + sulfametoxazol. O uso de dapsona (50mg, VO, 2x/dia), embora não seja tão eficaz como a trimetoprima + sulfametoxazol, pode ser uma boa substituição; deve ser evitado, no entanto, em pacientes com deficiência de glicose–6–fosfato desidrogenase. Outros agentes alternativos incluem atovaquona ou pentamidina.
Mesmo depois de 3 meses, ou mais, de transplantes, os pacientes ainda correm o risco de contrair infecções causadas pelo vírus varicela-zóster (VVZ) e, se tiverem DECH crônica, de infecções bacterianas recorrentes. Normalmente, as infecções causadas pelo VVZ ocorrem, de início, como doença localizada (isto é, herpes-zóster ou cobreiro), podendo também ser disseminada. Caso não sejam tratadas, as infecções disseminadas são fatais. Por conseguinte, todos os pacientes com infecções localizadas causadas pelo VVZ devem ser tratados com aciclovir para impedir a disseminação.
Atualmente, muitos centros colocam, de rotina, todos os receptores de transplantes alogênicos em regimes de terapia profilática com aciclovir no primeiro ano após os transplantes. No esforço de reduzir a incidência de infecções bacterianas tardias, muitos centros colocam os pacientes com DECH crônica que recebem tratamento imunossupressivo em terapia diária à base de trimetoprima + sulfametoxazol.
Transplante de células-tronco hematopoiéticas para tratamento de doenças específicas
Tratamento de estados de imunodeficiência
A experiência mais significativa nos cuidados de imunodeficiência com TCTH tem sido no tratamento de doenças combinadas de imunodeficiência.65,66 Nas situações em que se utilizam técnicas modernas de tratamento de suporte, costumam ser excelentes os resultados esperados dos transplantes de doadores que sejam membros da família e tenham HLA idêntico, com uma probabilidade superior a 90% de sobrevida no longo prazo.65,66
Por outro lado, observam-se resultados muito bons (sobrevida de cerca de 80%) usando-se doadores compatíveis sem relação de parentesco. Nos casos de pacientes sem doadores compatíveis, com ou sem relação de parentesco, os transplantes de um dos pais com haplótipo incompatível resultam em enxertos e sobrevidas acima de 2 anos em 50 a 70% de casos.
A experiência no tratamento da síndrome de Wiskott-Aldrich e de outros estados de imunodeficiência é bastante limitada.67 Houve cura em mais de 50% dos indivíduos, sendo que os melhores resultados foram naqueles que haviam feito transplante quando tinham idade inferior a 5 anos.
Tratamento de doenças não malignas de hematopoiese
Anemia aplásica
O transplante de irmãos compatíveis, depois de regimes preparatórios com doses elevadas de ciclofosfamida e globulina antitimocítica, em combinação com uso de metotrexato e ciclosporina, para profilaxia de DECH, é um regime bastante eficaz para aplicação em pacientes com anemia aplásica. Os resultados atuais sugerem que o índice de cura seja superior a 90%.68
Os resultados com doadores incompatíveis ou com doadores compatíveis sem relação de parentesco são um pouco piores; portanto, os indivíduos com anemia aplásica que não têm irmãos doadores, em geral, fazem um teste com terapia imunossupressiva antes do transplante.
O Quadro 2 apresenta a sobrevida sem doença após o TCTH.
Quadro 2
|
SOBREVIDA SEM DOENÇA APÓS TRANSPLANTE DE CÉLULAS-TRONCO HEMATOPOIÉTICAS | ||
|
Doenças |
Sobrevida de 5 anos sem doença (%)
| |
|
LMC |
Fase crônica |
6–70 |
|
Fase acelerada |
30–40 | |
|
Fase blástica |
15–20 | |
|
LMA |
Primeira remissão |
40–70 |
|
Segunda remissão |
30 | |
|
LLC |
50 | |
|
LLA |
30–50 | |
|
Mielomas múltiplos |
35 | |
|
LNH |
Depois da primeira recidiva, tumores quimiossensíveis |
40–50 |
|
DH |
Primeiro tratamento depois do tratamento padrão |
40–70 |
|
Doença em estado avançado |
15–30 | |
|
Síndromes mielodisplásicas |
45 | |
|
Anemia aplásica grave |
>90 | |
|
Talassemia maior |
70–90 | |
|
Anemia de Fanconi |
50–70 | |
|
Doença da célula falciforme |
50–90 | |
|
Doença de imunodeficiência grave combinada |
90 | |
DH: doença de Hodgkin; LLA: leucemia linfoide aguda; LLC: leucemia linfoide crônica; LMA: leucemia mieloide aguda; LMC: leucemia mieloide crônica: LNH: linfoma não Hodgkin.
Talassemia
Os transplantes de medula óssea de irmãos com HLA idêntico, depois de um regime preparatório com bussulfano e ciclofosfamida, podem curar de 70 a 90% de indivíduos portadores de talassemia maior.69 Os melhores resultados foram obtidos em pacientes que haviam feito transplantes antes do desenvolvimento de hepatomegalia ou de fibrose portal e que haviam recebido terapia adequada com quelação de ferro. Em outro estudo envolvendo 121 desses pacientes, as probabilidades de sobrevida e sobrevida de 5 anos sem doença depois dos transplantes foram de 95 e 90%, respectivamente.69
É possível também atingir sobrevida prolongada com terapia agressiva de quelação, embora os transplantes permaneçam apenas como tratamentos curativos. Menos de 30% de pacientes com talassemia têm irmãos com HLA idêntico. A experiência com doadores compatíveis sem relação de parentesco é mais limitada, sendo que a taxa de sobrevida documentada de 5 anos sem doença é de 70%.70 Resultados semelhantes foram registrados com o uso de sangue do cordão umbilical como fonte de células-tronco.71
Anemia da célula falciforme
Embora ainda seja limitada, a experiência com transplantes para tratamento da doença da célula falciforme encontra-se em fase de crescimento. Em um estudo francês de 87 pacientes com doença da célula falciforme que haviam recebido transplantes de irmãos HLA compatível, a taxa de sobrevida de 6 anos foi de 93% e a de sobrevida sem doença, de 86%.72 Um estudo envolvendo 59 pacientes, realizado nos EUA, apresentou taxas semelhantes ? 93 e 84%, respectivamente.73
Outras doenças não malignas
O TCTH vem sendo utilizado com sucesso para tratar uma grande variedade de outras doenças não malignas, porém fatais ? o que inclui distúrbios congênitos de eritrócitos, tais como síndrome de Kostmann, doença granulomatosa crônica, defeitos na actina de neutrófilos, deficiência de adesão leucocitária e síndrome de Chédiak-Higashi.
As anemias congênitas, incluindo anemia de Falconi e anemia de Blackfan-Diamond, também são tratáveis com TCTH.72,74 A osteoporose é um distúrbio hereditário raro causado pela incapacidade de reabsorção óssea dos osteoclastos. Considerando que esses são macrófagos especializados derivados da medula óssea, a osteoporose poderá ser tratada com transplante de medula.75
Uma categoria final de doenças não malignas tratáveis é a de doenças de estocagem causadas por deficiências enzimáticas, incluindo a síndrome de Maroteaux-Lamy, leucodistrofia metacromática, síndrome de Hurler e síndrome de Hunter. Os transplantes para tratamento desses distúrbios ainda não foram universalmente bem-sucedidos, embora o tratamento logo no início do curso da doença, antes da ocorrência de danos irreversíveis nos órgãos-alvo, aumente as chances de resultados favoráveis.76
Foram realizados alguns estudos sobre o uso de transplantes no tratamento de distúrbios autoimunes graves. Esses estudos se fundamentam na demonstração de que os transplantes podem curar doenças autoimunes em alguns modelos animais, assim como na observação de que alguns pacientes com malignidades hematológicas coexistentes e distúrbios autoimunes foram curados de ambas as condições com transplantes.77
Tratamento de doenças malignas
Leucemia mieloide aguda
Os transplantes alogênicos de medula óssea curam de 15 a 20% de pacientes com LMA nos quais a terapia de indução não tenha sido bem-sucedida e, na realidade, é a única terapia que pode curar esses pacientes.78 Logo, recomenda-se determinar o tipo de HLA de todos os pacientes com 60 anos de idade ou menos com diagnóstico recente de LMA, assim como o das respectivas famílias, logo após o diagnóstico, para possibilitar o transplante naqueles em que a terapia de indução não tenha sido bem-sucedida.
No caso de pacientes que conseguem a primeira remissão, a escolha é prosseguir com o transplante ou continuar com a Qt. No que diz respeito aos pacientes com 60 anos de idade ou menos, um grande número de testes prospectivos publicados demonstra que o transplante alogênico usando altas doses de regimes preparatórios em pacientes com irmãos com HLA compatível se compara à Qt ou ao transplante autólogo em pacientes sem esse tipo de parentesco.
Algumas metanálises recentes demonstram que houve melhora na sobrevida geral com TCTH na primeira remissão, em comparação com a Qt e com os transplantes autólogos, cuja taxa de risco é de 1,15.79,80 Essas análises não encontraram nenhum benefício nos transplantes autólogos. O benefício dos transplantes alogênicos foi observado sobretudo em indivíduos com citogenética de baixo risco, o que não ocorreu em pacientes com citogenética de risco favorável.79–82
Os pacientes com cariótipo leucêmico normal (doença de risco intermediário) se dividem em indivíduos cuja leucemia tem a mutação NPM1, sem a mutação FLT3 (que define grupos de melhor risco) e em indivíduos com a mutação FLT3 e/ou sem a mutação NPM1 (um preditor de resultados menos favoráveis).
Alguns estudos prospectivos comprovaram que há vantagens na sobrevida com transplantes alogênicos de células hematopoiéticas na primeira remissão em pacientes com mutações FLT3 e/ou sem mutação NPM1, porém o mesmo não ocorre em indivíduos com cariótipos normais com mutação NPM1 e com mutação FLT3 selvagem.81
Os resultados dos transplantes de medula óssea nos casos de primeira remissão que não havia sido tratada e nos casos de segunda remissão são semelhantes, sobretudo quando os transplantes forem feitos logo no início da primeira recidiva.
Levando-se em conta a possibilidade de se conseguir uma segunda remissão em, aproximadamente, 50% de pacientes com LMA que recebem Qt, e considerando que complicações que impedem a realização de transplantes subsequentes ocorrem em alguns desses pacientes, aparentemente há poucas vantagens em administrar Qt adicional, ou tentar uma segunda remissão, antes do transplante de medula óssea, se a recidiva for detectada logo no início e houver alguma possibilidade de fazer transplante imediato.
Existem poucos estudos envolvendo pacientes com idade em torno de 60 anos que utilizaram condicionamento com doses reduzidas. Nesse contexto, embora os resultados preliminares pareçam animadores, poucos testes prospectivos fizeram a comparação entre os resultados dos transplantes e da Qt.83
Leucemia linfoide aguda
Assim como nos casos de LMA, os transplantes alogênicos conseguem curar entre 15 a 20% de pacientes com leucemia linfoide aguda (LLA) nos quais a terapia de indução não tenha sido bem-sucedida ou naqueles em que ocorre o desenvolvimento de alguma doença resistente à Qt; portanto, esses indivíduos são candidatos ao procedimento.
Os resultados dos transplantes em pacientes com segunda remissão são melhores, cujas taxas de cura variam entre 30 e 50% ? melhor do que se poderia esperar com Qt adicional.84 A sobrevida de longo prazo sem doença ocorre em 45 a 65% de adultos com LLA tratados com transplante alogênico de células hematopoiéticas enquanto o paciente ainda está na primeira remissão.1 Alguns testes prospectivos que fizeram a comparação entre TCTH alogênico e Qt convencional em pacientes com LLA na primeira remissão completa produziram resultados inconsistentes.
De maneira geral, os testes são consistentes com base na descoberta de que é possível melhorar a sobrevida de longo prazo em pacientes de alto risco, incluindo aqueles com t(9;22) e t(4;11), caso sejam tratados com TCTH alogênico, ainda que os resultados sejam menos consistentes em pacientes portadores de doença de risco padrão.
Nessa população, alguns testes sugerem sobrevida equivalente com Qt e transplantes,85 mesmo que os testes maiores e mais recentes indiquem a possibilidade de melhorar a sobrevida de pacientes adultos de risco padrão e de alto risco caso sejam tratados com transplantes alogênicos de irmãos compatíveis enquanto estiverem na primeira remissão.86 Considerando a excelência dos resultados da Qt isoladamente em crianças com LLA, na primeira remissão, o TCTH se restringe, em geral, aos portadores de doenças com risco mais elevado.87
Síndromes mielodisplásicas
De maneira geral, as síndromes mielodisplásicas são consideradas incuráveis, a não ser com transplantes de medula óssea. Em alguns pacientes, elas apresentam curso relativamente indolente, sendo que os transplantes poderão ser mantidos com segurança até a doença começar a progredir.
Entretanto, logo após a doença progredir para o estágio 2 intermediário, de acordo com o Revised International Prognostic Scoring System, ou se o paciente desenvolver granulocitopenia significativa (menos de 1.000 células/µL) ou trombocitopenia (menos de 40.000 células/µL), o transplante deverá ser rigorosamente levado a sério, pois, sem ele, o tempo esperado de sobrevida será bastante curto.88
Nos casos em que houver um irmão com HLA compatível para servir de doador, as chances de sobrevida de longo prazo com o transplante podem chegar a 55%, sendo que os melhores resultados são obtidos em pacientes mais jovens e naqueles que recebem transplante logo no início do curso da doença.89 Há registros de casos semelhantes com transplantes de doadores compatíveis sem relação de parentesco.90 Ainda não foi definido nenhum tipo de papel para os transplantes autólogos nas síndromes mielodisplásicas.
Mielofibrose
Os transplantes alogênicos de células hematopoiéticas têm capacidade para curar pacientes com mielofibrose primária ou com mielofibrose secundária em casos essenciais de trombocitopenia ou policitemia vera. Em um dos estudos sobre o tema, a sobrevida de 5 anos sem progressão da doença foi observada em cerca de dois terços de pacientes com regimes preparatórios combinando bussulfano e ciclofosfamida, seguidos de transplantes alogênicos. A ocorrência de falência dos enxertos foi rara, apesar da presença de esplenomegalia e fibrose medular.91
Leucemia mieloide crônica
Os transplantes alogênicos e singênicos de medula óssea são as únicas formas terapêuticas conhecidas para curar LMC. As taxas de sobrevida de 5 anos sem a doença variam de 15 a 20% em pacientes que fazem transplantes durante a crise de explosão da doença, de 30 a 40% naqueles que fazem transplantes durante a fase acelerada e, aproximadamente, 70% nos que fazem transplantes durante a fase crônica, com resultados semelhantes usando doadores compatíveis com relação de parentesco e doadores compatíveis sem relação de parentesco.92,93
O papel geral dos TCTHs em casos de LMC mudou após a introdução do mesilato de imatinibe e de outros inibidores eficazes da tirosina quinase (em inglês, tyrosine kinase inhibitors [TKI]) relativamente não tóxicos.94 O uso de imatinibe não resulta, necessariamente, em remissões completas no nível molecular na maioria dos pacientes; portanto, alguns especialistas argumentam que os transplantes alogênicos imediatos de doadores compatíveis ainda são o tratamento de escolha em pacientes mais jovens.
Todavia, no caso da esmagadora maioria de pacientes com LMC, de maneira geral, é preferível fazer testes iniciais envolvendo o uso de imatinibe ou de outro TKI. As evidências atuais sugerem que a exposição ao imatinibe antes dos transplantes não impõe nenhum risco adicional aos procedimentos. Foram propostas algumas estratégias de terapia inicial com o mesilato de imatinibe em combinação com monitoramento molecular rigoroso e transplante no momento da progressão da doença.95
Leucemia linfoide crônica
O papel do uso de TCTH em casos de leucemia linfoide crônica (LLC) não foi tão considerado, talvez por causa da natureza indolente da doença e de sua propensão para acometer pacientes mais velhos. Dentre o número limitado de pacientes que receberam transplantes alogênicos, muitos tiveram remissão completa e cerca de 50% permaneceram sem a doença.96 Entretanto, nesse grupo, a mortalidade relacionada aos transplantes foi substancial.
Há relatos de resistência a respostas completas com menos toxicidade associada aos transplantes alogênicos não mieloablativos.97 Ainda que os transplantes autólogos para tratamento de LLC melhorem o tempo de duração da resposta inicial, aparentemente não alongam a sobrevida geral e, por essa razão, não são recomendados.98
Linfoma não Hodgkin
Pacientes com LNH disseminado intermediário ou de grau elevado, nos quais a terapia convencional não tenha sido bem-sucedida, dificilmente serão curados sem transplante. Terapias com altas doses seguidas de transplante de medula óssea, alogênico ou autólogo, poderão curar um número significativo desses indivíduos. Diversos estudos documentaram taxas de cura de 40 a 50% entre aqueles que fazem transplantes depois de recidivas iniciais e cujos tumores permanecem sensíveis à Qt.99
Em um estudo randomizado envolvendo o papel dos transplantes autólogos em casos de LNH intermediário ou de alto grau, a taxa de sobrevida de 5 anos sem a doença em pacientes que haviam feito transplantes autólogos para tratamento de doenças quimiossensíveis foi de 40%, em comparação com 12% em pacientes do grupo de Qt (p = 0,001). Os índices de cura caem de forma substancial na medida em que a doença se torna resistente às doses quimioterápicas normais.99
Situações como mau desempenho e grandes volumes tumorais são fatores de risco adversos adicionais. Ainda não foi definido o papel dos transplantes autólogos em pacientes com primeira remissão. Dentre os estudos randomizados feitos até o presente momento, alguns constataram que houve benefícios significativos; outros não encontraram nenhum benefício; outros, ainda, observaram que houve benefícios apenas no subgrupo de pacientes portadores de doença variando de risco intermediário a alto risco ou de doença com risco elevado.100,101 Duas metanálises desses estudos não conseguiram chegar a conclusões firmes.102,103
Assim como em outras doenças, os pacientes que recebem transplantes alogênicos para tratamento de linfomas intermediários e de alto grau apresentam taxas mais baixas de recidiva, mesmo com risco maior de mortalidade sem recidivas, em comparação com aqueles que recebem transplantes autólogos.104
Para a maior parte das categorias de LNH intermediário e de alto grau, os resultados dos transplantes alogênicos e dos autólogos são semelhantes, embora os fatos indiquem que há uma vantagem no uso de transplantes alogênicos em pacientes com linfoma linfoblástico. Mais recentemente, foram realizados alguns estudos sobre transplantes alogênicos com condicionamento de intensidade reduzida em linfomas de grandes células.
Nos casos de pacientes com LNH recorrente e disseminado de baixo grau, a administração de altas doses terapêuticas com suporte de transplantes autólogos resulta em taxas elevadas de resposta e na melhora da sobrevida sem progressão da doença, em comparação com as doses terapêuticas padrões. Em um estudo europeu, a sobrevida total de 4 anos foi de 46% com Qt, em comparação com 74% com transplantes autólogos.105
O papel dos transplantes autólogos no tratamento inicial de pacientes com linfomas indolentes não é muito claro. Os resultados obtidos até o momento demonstram taxas de respostas completas mais elevadas e melhoria na sobrevida sem eventos, embora as conclusões sobre os efeitos na sobrevida global ainda sejam prematuras e, nos dias atuais, é raro os pacientes serem tratados com transplantes como parte da terapia inicial, a não ser em testes clínicos.106
Os transplantes alogênicos mieloablativos resultam em um maior efeito antitumoral em comparação com os autólogos, ainda que ao custo de uma toxicidade maior. Os regimes preparatórios não mieloablativos ou de intensidade reduzida, seguidos de transplantes alogênicos, resultam em taxas elevadas de resposta com muito menos toxicidade do que com transplantes ablativos.107,108
Doença de Hodgkin
Os resultados de transplantes para tratamento da DH são semelhantes aos obtidos no tratamento de LNH intermediário e de alto grau. No caso de pacientes com DH primária progressiva (definida como progressão durante o tratamento de indução ou dentro de um período de 90 dias após o término da terapia), a aplicação de altas doses de Qt seguidas de transplantes autólogos resultou em uma taxa de sobrevida de 5 anos de 42% sem a doença ? esses resultados parecem ser superiores aos alcançados com a Qt convencional.109
Alguns testes randomizados prospectivos também mostraram algumas vantagens de altas doses de Qt seguidas de transplantes autólogos, em comparação com as doses quimioterápicas convencionais, em pacientes com DH recorrente ou refratária.110,111
Ainda não se definiu nenhum tipo de papel para os transplantes autólogos como parte de estratégias iniciais de tratamento em pacientes com DH. Assim como ocorre nos casos de LNH, os pacientes portadores da DH tratados com regimes ablativos preparatórios, seguidos de transplantes alogênicos, apresentam taxas menores de recidiva e mortalidade mais elevada sem recidiva.112
O uso de regimes preparatórios não mieloablativos ou de intensidade reduzida, seguidos de transplantes alogênicos, resulta, de forma comprovada, em respostas completas e duradouras com níveis aceitáveis de toxicidade em pacientes selecionados com DH recorrente, incluindo indivíduos nos quais os transplantes autólogos precedentes não tenham sido bem-sucedidos.113,114
Mielomas múltiplos
A administração de altas doses de Qt seguida de transplantes autólogos em pacientes com mielomas múltiplos recorrentes pode resultar em uma redução significativa na carga tumoral e, em muitos casos, em remissões completas temporárias. Dois testes randomizados prospectivos demonstraram que a inclusão de altas doses de Qt seguida de transplantes autólogos no tratamento inicial de pacientes com mielomas múltiplos resulta em um prolongamento significativo na sobrevida dos pacientes, sendo considerada o padrão atual de tratamento.115,116
Alguns testes randomizados prospectivos sugeriram que há uma vantagem adicional no tratamento de pacientes com dois ciclos de altas doses terapêuticas, cada uma delas com suporte de transplante autólogo, em comparação com um único transplante, embora, aparentemente, o benefício se restrinja àqueles que não conseguem atingir respostas completas ou parciais satisfatórias com o primeiro transplante.117,118 O transplante alogênico de células hematopoiéticas depois de regimes ablativos preparatórios pode levar à cura no longo prazo, ainda que esteja associado a níveis consideráveis de morbidade e mortalidade.119
A redução na morbidade e na mortalidade associadas a transplantes, sem perda do efeito do enxerto alogênico versus mieloma, é possível com uso de regimes preparatórios não mieloablativos ou de intensidade reduzida. Encontram-se, atualmente, em fase de testes estratégias para tratar pacientes com mielomas múltiplos com um único transplante autólogo, seguido de transplante alogênico não mieloablativo.120,121
Outras malignidades hematológicas
Existem diversos relatos de sobrevida no longo prazo depois de transplantes alogênicos de medula óssea em pacientes com leucemia de células pilosas, várias síndromes mieloproliferativas e outras malignidades hematológicas, apesar de ser pequeno o número de pacientes registrados em qualquer uma das categorias.
Câncer testicular
Embora a Qt com dose padrão seja muito eficaz nos casos de câncer testicular, os regimes convencionais falham em 30 a 40% de casos. Dois ciclos de Qt com doses altas administradas com suporte de células-tronco autólogas podem curar cerca de 50% de pacientes com doença recorrente, além de serem considerados o atual padrão de tratamento.122
Câncer de mama
Com base na hipótese de que o câncer de mama pode responder melhor a altas doses terapêuticas, diversos testes clínicos exploraram os resultados da administração de altas doses quimioterápicas com suporte de células-tronco hematopoiéticas autólogas.
Há registros de resultados encorajadores em testes de fase II em pacientes com doença metastática, câncer de mama inflamatório e doença de alto risco de estágio II.123,124 Entretanto, os testes randomizados subsequentes foram menos encorajadores e, por isso, essa abordagem não costuma ser recomendada para o tratamento desse tipo de câncer.
Outros tumores sólidos
A utilidade de altas doses de Qt com suporte de células-tronco autólogas para tratamento de diversos outros tipos de tumores sólidos, incluindo câncer de ovário, câncer pulmonar de pequenas células, neuroblastoma e sarcomas pediátricos, encontra-se ainda em fase de estudos. Assim como costuma ocorrer em outras situações, os melhores resultados são obtidos em pacientes com volume tumoral limitado, nos quais os tumores permanecem sensíveis à dose convencional de Qt.
Alguns testes randomizados mostraram que crianças com neuroblastomas de alto risco costumam se beneficiar com a terapia mieloablativa seguida de transplante autólogo, embora ainda não haja nenhum registro de testes randomizados sobre a utilidade dessa abordagem nos outros tumores sólidos mencionados anteriormente.125
Tratamento de recidivas pós-transplante
Ocasionalmente, os pacientes com malignidades que têm recidivas depois de transplantes autólogos respondem às doses quimioterápicas convencionais adicionais, em especial nos casos em que o intervalo decorrido entre o transplante e a recidiva é excessivamente longo. Existem mais opções para pacientes que têm recidivas depois de transplantes alogênicos. De maneira geral, os indivíduos com LMC respondem à terapia à base de interferon ou de mesilato de imatinibe, sendo que outros respondem à descontinuação da imunossupressão.
Pode ocorrer de os pacientes que têm recidivas depois de transplantes alogênicos responderem às infusões de linfócitos de doadores não irradiados. Um sumário de 258 pacientes apresentado por um registro europeu observou que houve respostas completas em 75% de indivíduos com LMC, em 38% com mielodisplasia, em 24% com leucemia mielocítica aguda e em 15% com mieloma.126 Resposta em pacientes com LLA foi rara.
As principais complicações das infusões de linfócitos de doadores no período pós-transplante foram DECH e mielossupressão, sendo que ambas podem ser graves e fatais. Iniciar as transfusões com baixas doses de células e aumentar de forma gradual a dose pode diminuir o risco de toxicidade grave. Eventualmente, um segundo TCTH pode ser eficaz, sobretudo em pacientes mais jovens e naqueles com intervalos mais longos entre o primeiro transplante e a recidiva e que não sejam portadores de doença em estado avançado.
|
O autor trabalhou como consultor da Abbott Laboratories, Celator e Igenica, tendo participado também do conselho consultivo da Genzyme Corporation. Este trabalho contou, também, com os suportes financeiros CA-18029, CA-47748 e CA-26386 do National Institutes of Health, US, Department of Health and Human Services. As terapias GCSF, GM-CSF, ciclofosfamida, bussulfano, tiotepa, melfalana, carmustina, etoposida, citarabina, irradiação corporal total, globulina antitimocítica, fludarabina, prednisona, metotrexato, tacrolimo, rapamicina, micofenolato de mofetila, azatioprina e talidomida foram aprovadas pela Food and Drug Administration para as aplicações descritas neste artigo.
|
Referências
1. International Bone Marrow Transplantation Registry/Autologous Blood and Marrow Registry. Available at: http://www.cibmtr.org (accessed June 1, 2011).
2. Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells [published erratum appears in Science 1989;244:1030]. Science 1988;241:58–62.
3. Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 2002;297:2256–9.
4. Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood 2005;106:1901–10.
5. Sprangrude GJ, Perry SS, Slayton WB. Early stages of hematopoietic differentiation. Ann N Y Acad Sci 2003;996:186.
6. Anasetti C, Amos D, Beatty PG, et al. Effect of HLA compatibility on engraftment of bone marrow transplants in patients with leukemia or lymphoma. N Engl J Med 1989;320:197–204.
7. National Marrow Donor Program, Minneapolis, Minnesota. Available at: http://www.nmdp.org (accessed June 1, 2011).
8. Kernan NA, Bartsch G, Ash RC, et al. Analysis of 462 transplantations from unrelated donors facilitated by The National Marrow Donor Program. N Engl J Med 1993;328:593–602.
9. Walter RB, Pagel JM, Gooley TA, et al. Comparison of matched unrelated and matched related donor myeloablative hematopoietic cell transplantation for adults with acute myeloid leukemia in first remission. Leukemia 2010;24:1276–82.
10. Gribben JG, Freedman AS, Neuberg D, et al. Immunologic purging of marrow assessed by PCR before autologous bone marrow transplantation for B-cell lymphoma. N Engl J Med 1991;325:1525–33.
11. Brenner MK, Rill DR, Moen RC, et al. Gene-marking to trace origin of relapse after autologous bone-marrow transplantation. Lancet 1993;341:85–6.
12. Richman CM, Weiner RS, Yankee RA. Increase in circulating stem cells following chemotherapy in man. Blood 1976;47:1031–9.
13. Antman KS, Griffin JD, Elias A, et al. Effect of recombinant human granulocyte-macrophage colony-stimulating factor on chemotherapy-induced myelosuppression. N Engl J Med 1988;319:593–8.
14. Levesque JP, Takamatsu Y, Nilsson SK, et al. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood 2001;98:1289–97.
15. Calandra G, McCarty J, McGuirk J, et al. AMD3100 plus G-CSF can successfully mobilize CD34+ cells from non-Hodgkin’s lymphoma, Hodgkin’s disease and multiple myeloma patients previously failing mobilization with chemotherapy and/or cytokine treatment: compassionate use data. Bone Marrow Transplant 2008;41:331–8.
16. Bensinger WI, Longin K, Appelbaum F, et al. Peripheral blood stem cells (PBSCs) collected after recombinant granulocyte colony stimulating factor (rhG-CSF): an analysis of factors correlating with the tempo of engraftment after transplantation. Br J Haematol 1994;87:825–31.
17. Bensinger WI, Weaver CH, Appelbaum FR, et al. Transplantation of allogeneic peripheral blood stem cells mobilized by recombinant human granulocyte colony-stimulating factor. Blood 1995;85:1655–8.
18. Bensinger WI, Martin PJ, Storer B, et al. Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. N Engl J Med 2001;344:175–81.
19. Couban S, Simpson DR, Barnett MJ, et al. A randomized multicenter comparison of bone marrow and peripheral blood in recipients of matched sibling allogeneic transplants for myeloid malignancies. Blood 2002;100:1525–31.
20. Schmitz N, Beksac M, Hasenclever D, et al. Transplantation of mobilized peripheral blood cells to HLA-identical siblings with standard-risk leukemia. Blood 2002;100:761–7.
21. Rubinstein P, Carrier C, Scaradavou A, et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med 1998;339:1565–77.
22. Rocha V, Cornish J, Sievers EL, et al. Comparison of outcomes of unrelated bone marrow and umbilical cord blood transplants in children with acute leukemia. Blood 2001;97:2962–71.
23. Rocha V, Labopin M, Sanz G, et al. Transplants of umbilical-cord blood or bone marrow from unrelated donors in adults with acute leukemia. N Engl J Med 2004;351:2276–85.
24. Laughlin MJ, Eapen M, Rubinstein P, et al. Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N Engl J Med 2004;351:2265–75.
25. Barker JN, Weisdorf DJ, Defor TE, et al. Transplantation of 2 partially HLA-matched umbilical cord blood units to enhance engraftment in adults with hematologic malignancy. Blood 2005;105:1343–7.
26. Delaney C, Gutman JA, Appelbaum FR. Cord blood transplantation for haematological malignancies: conditioning regimens, double cord transplant and infectious complications. Br J Haematol 2009;147:207–16.
27. Bacigalupo A, Ballen K, Rizzo D, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant 2009;15:1628–33.
28. Buckner CD, Clift RA, Sanders JE, et al. Marrow harvesting from normal donors. Blood 1984;64:630–4.
29. Nemunaitis J, Rabinowe SN, Singer JW, et al. Recombinant granulocyte-macrophage colony-stimulating factor after autologous bone marrow transplantation for lymphoid cancer. N Engl J Med 1991;324:1773–8.
30. Spielberger R, Stiff P, Bensinger W, et al. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med 2004;351:2590–8.
31. Bearman SI. The syndrome of hepatic veno-occlusive disease after marrow transplantation. Blood 1995;85:3005–20.
32. Losasciulli A, Testa M, Valsecchi MG, et al. The role of hepatitis C and B virus infections as risk factors for severe liver complications following allogeneic BMT: a prospective study by the Infectious Disease Working Party of the European Blood and Marrow Transplantation Group. Transplantation 1999;68:1486.
33. Carreras E, Bertz H, Arcese W, et al. Incidence and outcome of hepatic veno-occlusive disease after blood or marrow transplantation: a prospective cohort study of the European Group for Blood and Marrow Transplantation. European Group for Blood and Marrow Transplantation Chronic Leukemia Working Party. Blood 1998;92:3599–604.
34. Richardson PG, Murakami C, Jin Z, et al. Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood 2002;100:4337–43.
35. Crawford SW, Hackman RC. Clinical course of idiopathic pneumonia after bone marrow transplantation. Am Rev Respir Dis 1993;147:1393–400.
36. Panoskaltsis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med 2011;183:1262–79.
37. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med 2006;354:1813.
38. Flowers MED, Deeg HJ. Delayed complications after hematopoietic cell transplantation. In: Blume KG, Forman SJ, Appelbaum FR, editors. Thomas’ hematopoietic cell transplantation. Oxford (UK): Blackwell Publishing; 2004. p. 944–61.
39. Syrjala KL, Langer SL, Abrams JR, et al. Recovery and long-term function after hematopoietic cell transplantation for leukemia or lymphoma. JAMA 2004;291:2335–43.
40. Baker KS, Defor TE, Burns LJ, et al. New malignancies after blood or marrow stem-cell transplantation in children and adults: incidence and risk factors [published erratum appears in J Clin Oncol 2003;21:3181]. J Clin Oncol 2003;21:1352–8.
41. Curtis RE, Rowlings PA, Deeg HJ, et al. Solid cancers after bone marrow transplantation. N Engl J Med 1997;336:897–904.
42. Stone RM, Neuberg D, Soiffer R, et al. Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Oncol 1994;12:2535–42.
43. Banker DE, Mayer SJ, Li HY, et al. Cholesterol synthesis and import contribute to protective cholesterol increments in acute myeloid leukemia cells. Blood 2004;104:1816–24.
44. Sorror ML, Maris MB, Storer B, et al. Comparing morbidity and mortality of HLA-matched unrelated donor hematopoietic cell transplantation after nonmyeloablative and myeloablative conditioning: influence of pretransplant comorbidities. Blood 2004;104:961–8.
45. Diaconescu R, Flowers CR, Storer B, et al. Morbidity and mortality with nonmyeloablative compared to myeloablative conditioning before hematopoietic cell transplantation from HLA matched related donors. Blood 2004;104:1550–8.
46. Rabinowe SN, Neuberg D, Bierman PJ, et al. Long term follow-up of a phase III study of rhGM-CSF after autologous bone marrow transplantation for lymphoid malignancies. Blood 1993;81:1903–8.
47. Nemunaitis J, Singer JW, Buckner CD, et al. Use of recombinant human granulocyte-macrophage colony-stimulating factor in graft failure after bone marrow transplantation. Blood 1990;76:245–53.
48. Gyurkocza B, Cao TM, Storb RF, et al. Salvage allogeneic hematopoietic cell transplantation with fludarabine and low-dose total body irradiation after rejection of first allografts. Biol Blood Marrow Transplant 2009;15:1314–22.
49. Ferrara JLM, Deeg HJ. Graft-versus-host disease. N Engl J Med 1991;324:667–74.
50. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2005;11:945–56.
51. Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HLA-matched sibling donors. Transplantation 1974;18:295–304.
52. Rowlings PA, Przepiorka D, Klein JP, et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol 1997;97:855–64.
53. Leisenring WM, Martin PJ, Petersdorf EW, et al. An acute graft-versus-host disease activity index to predict survival after hematopoietic cell transplantation with myeloablative conditioning regimens. Blood 2006;108:749–55.
54. Nash RA, Pepe MS, Storb R, et al. Acute graft-versus-host disease: analysis of risk factors after allogeneic marrow transplantation and prophylaxis with cyclosporine and methotrexate. Blood 1992;80:1838–45.
55. Sorror ML, Leisenring W, Deeg HJ, et al. Twenty-year follow-up of a controlled trial comparing a combination of methotrexate plus cyclosporine with cyclosporine alone for prophylaxis of graft-versus-host disease in patients administered HLA-identical marrow grafts for leukemia [letter]. Biol Blood Marrow Transplant 2005;11:814–5.
56. Chao NJ. Pharmacology and use of immunosuppressive agents after hematopoietic cell transplantation. In: Thomas ED, Blume KG, Forman SJ, editors. Hematopoietic cell transplantation. 2nd ed. Boston: Blackwell Science; 1999. p. 176–85.
57. Atkinson K, Horowitz MM, Gale RP, et al. Risk factors for chronic graft-versus-host disease after HLA-identical sibling bone marrow transplantation. Blood 1990;75:2459–64.
58. Koc S, Leisenring W, Flowers MED, et al. Therapy for chronic graft-versus-host disease: a randomized trial comparing cyclosporine plus prednisone versus prednisone alone. Blood 2002;100:48–51.
59. Vogelsang GB, Farmer ER, Hess AD, et al. Thalidomide for the treatment of chronic graft versus host disease. N Engl J Med 1992;326:1055–8.
60. Goodman JL, Winston DJ, Greenfield RA, et al. A controlled trial of fluconazole to prevent fungal infections in patients undergoing bone marrow transplantation. N Engl J Med 1992;326:845–51.
61. Pizzo PA. Management of fever in patients with cancer and treatment-induced neutropenia. N Engl J Med 1993;328:1323–32.
62. Bensinger WI, Price TH, Dale DC, et al. The effects of daily recombinant human granulocyte colony stimulating factor administration on normal granulocyte donors undergoing leukapheresis. Blood 1993;81:1883–8.
63. Goodrich JM, Bowden RA, Fisher L, et al. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Ann Intern Med 1993;118:173–8.
64. Centers for Disease Control and Prevention, Infectious Disease Society of America, American Society of Blood and Marrow Transplantation. Guidelines for preventing opportunistic infections among hematopoietic stem cell transplant recipients. MMWR Morb Mortal Wkly Rep 2000;49(RR-10):1–125.
65. Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med 1999;340:508–16.
66. Grunebaum E, Mazzolari E, Porta F, et al. Bone marrow transplantation for severe combined immune deficiency. JAMA 2006;295:508–18.
67. Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol 2006;117:725.
68. Storb R, Leisenring W, Anasetti C, et al. Long-term follow-up of allogeneic marrow transplants in patients with aplastic anemia conditioned by cyclophosphamide combined with antithymocyte globulin [letter]. Blood 1997;89:3890–1.
69. Lucarelli G, Galimberti M, Giardini C, et al. Bone marrow transplantation in thalassemia. The experience of Pesaro. Ann N Y Acad Sci 1998;850:270–5.
70. La Nasa G, Caocci G, Argiolu F, et al. Unrelated donor stem cell transplantation in adult patients with thalassemia. Bone Marrow Transplant 2005;36:971–5.
71. Locatelli F, Rocha V, Reed W, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood 2003;101:2137–43.
72. Segal BH, Veys P, Malech H, Cowan MJ. Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant 2011;17 Suppl 1:S123–31.
73. Walters MC, Storb R, Patience M, et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Blood 2000;95:1918–24.
74. MacMillan ML, Hughes MR, Agarwal S, Daley GQ. Cellular therapy for Fanconi anemia: the past, present, and future. Biol Blood Marrow Transplant 2011;17 Suppl 1:S109–14.
75. Driessen GJ, Gerritsen EJ, Fischer A, et al. Long-term outcome of haematopoietic stem cell transplantation in autosomal recessive osteopetrosis: an EBMT report. Bone Marrow Transplant 2003;32:657–63.
76. Peters C, Steward CG, National MDP, International Bone Marrow Transplant Registry, Working Party on Inborn Errors EBMTG. Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant 2003;31:229–39.
77. Nash RA, Bowen JD, McSweeney PA, et al. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood 2003;102:2364–72.
78. Fung HC, Stein A, Slovak M, et al. A long-term follow-up report on allogeneic stem cell transplantation for patients with primary refractory acute myelogenous leukemia: impact of cytogenetic characteristics on transplantation outcome. Biol Blood Marrow Transplant 2003;9:766–71.
79. Yanada M, Matsuo K, Emi N, Naoe T. Efficacy of allogeneic hematopoietic stem cell transplantation depends on cytogenetic risk for acute myeloid leukemia in first disease remission: a metaanalysis. Cancer 2005;103:1652–8.
80. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: a systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–60.
81. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
82. Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group study. Blood 2000;96:4075–83.
83. Gyurkocza B, Storb R, Storer BE, et al. Nonmyeloablative allogeneic hematopoietic cell transplantation in patients with acute myeloid leukemia. J Clin Oncol 2010;28:2859–67.
84. Barrett AJ, Horowitz MM, Pollock BH, et al. Bone marrow transplants from HLA-identical siblings as compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission. N Engl J Med 1994;331:1253–8.
85. Thiebaut A, Vernant JP, Degos L, et al. Adult acute lymphocytic leukemia study testing chemotherapy and autologous and allogeneic transplantation. A follow-up report of the French protocol LALA 87. Hematol Oncol Clin North Am 2000;14:1353–66.
86. Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood 2008;111:1827–33.
87. Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med 2000;342:998–1006.
88. Oliansky DM, Antin JH, Bennett JM, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of myelodysplastic syndromes: an evidence-based review. Biol Blood Marrow Transplant 2009;15:137–72.
89. Deeg HJ, Storer B, Slattery JT, et al. Conditioning with targeted busulfan and cyclophosphamide for hemopoietic stem cell transplantation from related and unrelated donors in patients with myelodysplastic syndrome. Blood 2002;100:1201–7.
90. Deeg HJ, Gooley TA, Flowers MED, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003;102:3912–8.
91. Kerbauy DMB, Gooley TA, Sale GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant 2007;13:355–65.
92. Oehler VG, Radich JP, Storer B, et al. Randomized trial of allogeneic related bone marrow transplantation versus peripheral blood stem cell transplantation for chronic myeloid leukemia. Biol Blood Marrow Transplant 2005;11:85–92.
93. Hansen JA, Gooley TA, Martin PJ, et al. Bone marrow transplants from unrelated donors for patients with chronic myeloid leukemia. N Engl J Med 1998;338:962–8.
94. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003;348:994–1004.
95. Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006;108:1809–20.
96. Dreger P, Corradini P, Kimby E, et al. Indications for allogeneic stem cell transplantation in chronic lymphocytic leukemia: the EBMT transplant consensus. Leukemia 2007;21:12–7.
97. Sorror ML, Maris MB, Sandmaier BM, et al. Hematopoietic cell transplantation after nonmyeloablative conditioning for advanced chronic lymphocytic leukemia. J Clin Oncol 2005;23:3819–29.
98. Sutton L, Chevret S, Tournilhac O, et al. Autologous stem cell transplantation as a first-line treatment strategy for chronic lymphocytic leukemia: a multicenter, randomized, contolled trial from the SFGM-TC and GFLLC. Blood 2011;117:6109–19.
99. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
100. Haioun C, Lepage E, Gisselbrecht C, et al. Benefit of autologous bone marrow transplantation over sequential chemotherapy in poor-risk aggressive non-Hodgkin’s lymphoma: updated results of the prospective study LNH87-2. Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol 1997;15:1131–7.
101. Gianni AM, Bregni M, Siena S, et al. High-dose chemotherapy and autologous bone marrow transplantation compared with MACOP-B in aggressive B-cell lymphoma. N Engl J Med 1997;336:1290–7.
102. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
103. Greb A, Bohlius J, Trelle S, et al. High-dose chemotherapy with autologous stem cell support in first-line treatment of aggressive non-Hodgkin lymphoma—results of a comprehensive meta-analysis. Cancer Treat Rev 2007;33:338–46.
104. Peniket AJ, Ruiz DEM, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
105. Schouten HC, Qian W, Kvaloy S, et al. High-dose therapy improves progression-free survival and survival in relapsed follicular non-Hodgkin’s lymphoma: results from the randomized European CUP trial. J Clin Oncol 2003;21:3918–27.
106. Lenz G, Dreyling M, Schiegnitz E, et al. Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 2004;104:2667–74.
107. Khouri IF, Saliba RM, Giralt SA, et al. Nonablative allogeneic hematopoietic transplantation as adoptive immunotherapy for indolent lymphoma: low incidence of toxicity, acute graft-versus-host disease, and treatment-related mortality. Blood 2001;98:3595–9.
108. Khouri IF, McLaughlin P, Saliba RM, et al. Eight-year experience with allogeneic stem cell transplantation for relapsed follicular lymphoma after nonmyeloablative conditioning with fludarabine, cyclophosphamide, and rituximab. Blood 2008;111:5530–6.
109. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
110. Linch DC, Hancock B, Winfield D, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
111. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
112. Anderson JE, Litzow MR, Appelbaum FR, et al. Allogeneic, syngeneic, and autologous marrow transplantation for Hodgkin’s disease: the 21 year Seattle experience. J Clin Oncol 1993;11:2342–50.
113. Schmitz N, Sureda A, Robinson S. Allogeneic transplantation of hematopoietic stem cells after nonmyeloablative conditioning for Hodgkin’s disease: indications and results. Semin Oncol 2004;31:27–32.
114. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
115. Attal M, Harousseau J-L, Stoppa A-M, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. N Engl J Med 1996;335:91–7.
116. Child JA, Morgan GJ, Davies FE, et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med 2003;348:1875–83.
117. Attal M, Harousseau JL, Facon T, et al. Single versus double autologous stem-cell transplantation for multiple myeloma. N Engl J Med 2003;349:2495–502.
118. Cavo M, Tosi P, Zamagni E, et al. The “Bologna 96” clinical trial of single vs. double autotransplants for previously untreated multiple myeloma patients [abstract]. Blood 2002;100(11 Pt 1):179a.
119. Gahrton G, Svensson H, Cavo M, et al. Progress in allogeneic bone marrow and peripheral blood stem cell transplantation for multiple myeloma: a comparison between transplants performed 1983 - 93 and 1994 - 98 at European Group for Blood and Marrow Transplantation centres. Br J Haematol 2001;113:209–16.
120. Bruno B, Rotta M, Patriarca F, et al. A comparison of allografting with autografting for newly diagnosed myeloma. N Engl J Med 2007;356:1110–20.
121. Peters WP, Shpall EJ, Jones RB, et al. High-dose combination alkylating agents with bone marrow support as initial treatment for metastatic breast cancer. J Clin Oncol 1988;6:1368–76.
122. Einhorn LH, Williams SD, Chamness A, et al. High-dose chemotherapy and stem-cell rescue for metastatic germ-cell tumors. N Engl J Med 2007;357:340–8.
123. Rosinol L, Perez-Simon JA, Sureda A, et al. A prospective PETHEMA study of tandem autologous transplantation versus autograft followed by reduced-intensity conditioning allogeneic transplantation in newly diagnosed multiple myeloma. Blood 2008;112:3591–3.
124. Dunphy FR, Spitzer G, Buzdar AU, et al. Treatment of estrogen receptor-negative or hormonally refractory breast cancer with double high-dose chemotherapy intensification and bone marrow support. J Clin Oncol 1990;8:1207–16.
125. Matthay KK, Villablanca JG, Seeger RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med 1999;341:1165–73.
126. Kolb HJ, Schattenberg A, Goldman JM, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. European Group for Blood and Marrow Transplantation Working Party Chronic Leukemia. Blood 1995;86:2041–50.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.