(Carregando Índice)... (Carregando Índice)... |
Última revisão: 21/06/2018
Comentários de assinantes: 0
|
Artigo original: Stone, J, H. MD, MPH. LGG4 - Related Disease and Retroperitoneal Fibrosis, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon.
|
John H. Stone, MD, MPH
Diretor da Reumatologia Clínica no Massachusetts General Hospital. Professor de Medicina na Harvard Medical School (Boston, MA).
Resumo
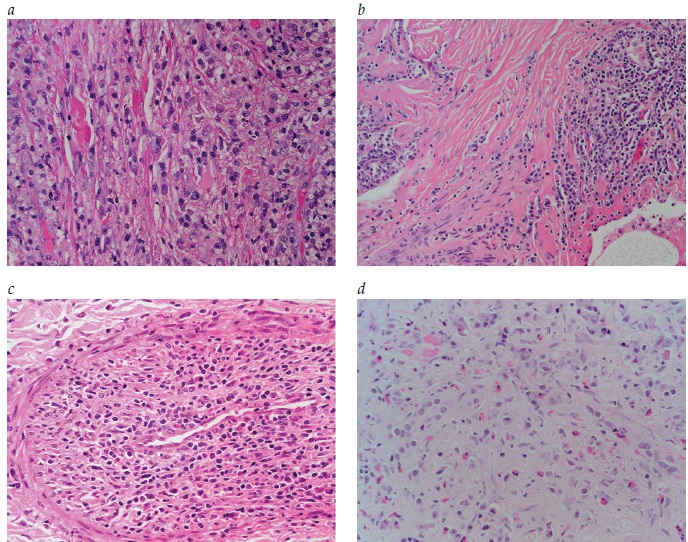
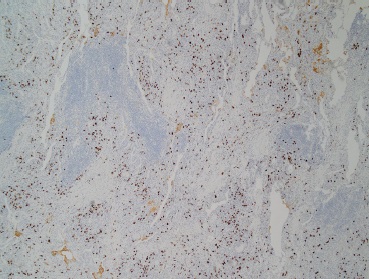
As doenças relacionadas à imunoglobulina G4 (IgG4-RD) afetam quase todos os sistemas de órgãos, sendo que as descobertas histopatológicas são consistentes nos sistemas. A IgG4-RD imita distúrbios malignos, infecciosos e inflamatórios e, portanto, a consideração de características histopatológicas nas biópsias teciduais e as correlações clínico-patológicas rigorosas são essenciais para evitar a obtenção de diagnósticos incorretos. A partir do início dos anos 2000, a IgG4-RD passou a ser reconhecida como causa do que anteriormente se conhecia por fibrose retroperitoneal (FRP) “idiopática”, sendo que, nos dias atuais, considera-se que essa FRP relacionada à IgG4 abrange um subgrupo importante da IgG4-RD. Esta revisão inclui uma visão geral da IgG4-RD e apresenta discussões sobre temas como patologia, fisiopatologia e manifestações clínicas da IgG4. A FRP relacionada à IgG4 também faz parte desta revisão, incluindo tópicos como FRP relacionada à IgG4 versus FRP por outras causas, diferenças entre FRP e outros subgrupos de IgG4-RD, e tratamento de IgG4-RD e de FRP relacionada à IgG4. As figuras mostram as características histopatológicas da IgG4-RD, coloração de tecidos para IgG4, FRP relacionada à IgG4 e periaortite crônica, “doença de “Mikulicz”, IgG4-RD no pulmão, doença renal relacionada à IgG4 e pancreatite tipo 1 (relacionada à IgG4). O quadro apresenta uma lista de condições anteriormente conhecidas por outras denominações, que geralmente se enquadram no espectro da IgG4-RD.
|
Distúrbios Relacionados à LGG4 e Fibrose Retroperitoneal |
Condições Conhecidas Anteriormente por Outras Denominações que Geralmente se Enquadram no Espectro de Doença Relacionada À Imunoglobulina G4 |
FRP
Fibrose mediastinal
Periaortite/periarterite
Pseudotumor inflamatório
|

Tipicamente, o número de células plasmáticas IgG4+ no interior dos tecidos afetados é maior que 30/campo de alta potência (em inglês, high-power field [HPF]), embora esse número possa se alterar de acordo com órgãos ou sítios teciduais específicos.15 As características mais comuns da IgG4-RD são pancreatite autoimune, sialadenite nas glândulas salivares maiores (principalmente, as glândulas submandibulares e parótidas), doença orbital (em particular, a dacrioadenite), linfadenopatia e FRP.
O parênquima cerebral e os músculos esqueléticos estão entre os poucos órgãos nos quais ainda não se identificou a IgG4-RD. A FRP relacionada à IgG4 em geral ocorre simultaneamente com periaortite crônica, aortite abdominal relacionada à IgG4 e fibrose perianeurismática relacionada à IgG4.14 Alguns casos de FRP idiopática que não parecem fazer parte do espectro de IgG4-RD têm apresentações semelhantes e devem ser distinguidos de IgG4-RD por meio de rigorosos estudos histopatológicos e de imunocoloração.
As causas de FRP que não fazem parte do espectro da IgG4-RD incluem medicações, doenças malignas, infecções, radiação e cirurgia prévia. As medicações implicadas com mais frequência na etiologia de FRP são as seguintes: metisergida, ergotamina, ß-bloqueadores, metildopa e hidralazina.
Entretanto, cabe observar que não há provas definitivas da associação de algumas dessas medicações.6 As seguintes malignidades que poderão resultar em FRP são: sarcomas, linfomas de Hodgkin e não Hodgkin, carcinoides e carcinomas no colo, próstata, mama, estômago e ovário. Infecções como histoplasmose, tuberculose e actinomicose; cirurgia abdominal anterior;22 e exposição ao amianto23 também foram associadas à FRP. Camadas hialinizadas de fibrose inespecíficas na aparência são típicas de FRP relacionada à IgG4-RD.
Os relatos implicando as células T auxiliares tipo 1 (Th1) e as células Th2 na fisiopatologia da IgG4-RD são conflitantes. As células T CD4+, que são as células mais abundantes nos tecidos afetados, tipicamente se dispersam em todas as lesões localizadas causadas pela IgG4-RD.32,33 A expansão clonal de uma população expandida de linfócitos T citotóxicos CD4+, tanto no sangue periférico como em lesões fibróticas em pacientes com IgG4-RD, sugere que essas células estão centralizadas em relação à doença.31
Essas células T citotóxicas geram produtos como granzima B e perforina que, em geral, estão associados às células T CD8+. Esses produtos produzem também o fator de crescimento transformador ß, interferon y e interleucina-1 (IL-1), sendo que todos podem ser mediadores importantes de fibrose. Por outro lado, as células Th2 se acumulam apenas no sangue de indivíduos com atopia concomitante e não apresentam a mesma oligoclonalidade que os linfócitos T citotóxicos CD4+.34,35
É provável que as respostas das células T auxiliares foliculares, separadamente dos linfócitos T citotóxicos CD4+, sejam responsáveis pelo desenvolvimento de centros germinais nos linfonodos (assim como nos órgãos envolvidos). Teoricamente, essas células T auxiliares foliculares produzem as citocinas (por exemplo, IL-4), que estimulam a troca de classe da IgG4, culminando na criação de plasmablastos secretores de IgG4 e de células plasmáticas de longa duração.
Manifestações Clínicas de Igg4-Rd
As manifestações da IgG4-RD nos sítios de múltiplos órgãos serão discutidas a seguir.
Sintomas Constitucionais e Musculoesqueléticos
Tipicamente, a IgG4-RD se apresenta na forma subaguda, com a presença de sinais e sintomas de disfunção orgânica por meses ou anos, mesmo antes do diagnóstico. Perdas de peso de 4,5 a 9kg são comuns, mesmo que, paradoxalmente, os pacientes aparentem estar bem.
De maneira geral, a fadiga acompanha a IgG4-RD, em especial nos pacientes com a doença em múltiplos órgãos. No entanto, a presença de febre não é comum. A artrite de Frank é atípica, embora muitos pacientes se queixem de artralgias e de outros sintomas musculoesqueléticos, em especial a entesopatia, cujas características ainda não são muito conhecidas até o momento.
Doença de Mikulicz: Glândulas Lacrimais, Parótidas e Submandibulares
A apresentação clássica de IgG4-RD da tríade formada por dacrioadenite, crescimento da glândula parótida e da glândula submandibular, foi conhecida por mais de 100 anos como “doença de Mikulicz”.36,37 O crescimento isolado da glândula submandibular, quase sempre bilateral, também é uma descoberta nos casos de IgG4-RD. O crescimento da glândula lacrimal é a característica mais comum de doença oftálmica relacionada à IgG4.
Órbitas
A proptose pode ser o resultado de combinações de lesões orbitais, tais como os pseudotumores que não afetam a glândula lacrimal, inflamação e espessamento dos músculos extraoculares.38 Os pseudotumores oculares ocorrem com frequência em regiões anatômicas difíceis de coletar materiais para biópsias, como, por exemplo, o seio cavernoso e o ápice orbital, onde poderão causar danos consideráveis por causa da compressão de estruturas vitais em espaços exíguos.
A doença do ducto nasolacrimal (obstrução) também é uma ocorrência comum nos casos de IgG4-RD; ela imita granulomatose com poliangite. A ocorrência de esclerite é rara, embora haja alguns relatos desse tipo de doença.
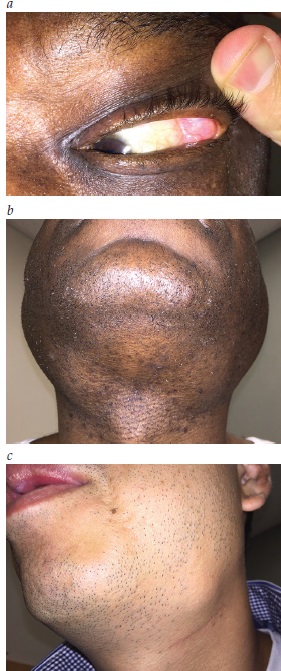
A Figura 4 ilustra a doença de Mikulicz.
Figura 4 - Doença de Mikulicz. (A) Aumento na glândula lacrimal em um paciente com dacrioadenite relacionada à IgG4 e sialadenite esclerosante. (B) Aumento na glândula parótida e submandibular no mesmo paciente que aparece em (A). (C) O aumento isolado na glândula submandibular, quase sempre bilateral, também é uma descoberta comum nos casos de IgG4-RD.
Ouvido/Nariz/Garganta E Glândula Tireoide
Rinite alérgica, pólipos nasais, sinusite crônica, obstrução nasal e rinorreia são comuns em IgG4-RD. Entretanto, as células Th2 de memória em circulação são encontradas apenas em pacientes com históricos pré-existentes de atopia.35 A prevalência de atopia entre pacientes com IgG4-RD aparentemente se assemelha à prevalência na população em geral, embora um subgrupo de pacientes não atópicos tenha eosinofilia no sangue periférico e apresente níveis elevados de IgE.34
Esse fato sugere que, em alguns pacientes, a IgG4-RD propriamente dita, além da atopia, contribui para a eosinofilia e para a elevação nos níveis de IgE. Eosinofilia periférica acentuada e concentrações séricas elevadas de IgE na linha de base são marcadores de pacientes com risco elevado de manifestação súbita da doença após o tratamento.19
Há relatos de lesões por massa que causam destruição substancial dos tecidos nos seios nasais, no ouvido médio e nos ossos faciais.39,40 A IgG4-RD pode também ocasionar inflamação difusa na faringe, na hipofaringe e nas cordas vocais.41 A tireoidite de Riedel foi associada à IgG4-RD de forma convincente.42 Entretanto, ainda permanece incerta a relação entre tireoidite fibrosante de Hashimoto e IgG4-R.
Linfadenopatia
Há inúmeras descrições de variantes histopatológicas de linfadenopatia relacionada à IgG4.43 Uma delas é uma imitação bem próxima da doença de Castleman. Tipicamente, a linfadenopatia associada à IgG4-RD é uma doença generalizada ou localizada adjacente a algum órgão afetado.44 Os linfonodos envolvidos, em geral, têm diâmetro de 1 a 3cm e não são sensíveis.
Aorta Torácica, Ramificações Aórticas E Lesões Coronarianas
Às vezes, a aortite relacionada à IgG4 é uma descoberta cirúrgica ou um diagnóstico inesperado após a identificação de alguma descoberta radiológica incidental. A aortite relacionada à IgG4 poderá resultar em aneurismas ou em dissecções na aorta torácica, embora seja raro o envolvimento das ramificações aórticas primárias, como, por exemplo, as artérias subclávias, ao contrário das células gigantes e da arterite de Takaiasu.45 Existem registros de lesões na artéria coronária em casos de IgG4-RD que, eventualmente, provocam a formação de aneurismas na artéria coronária.46
Pulmão
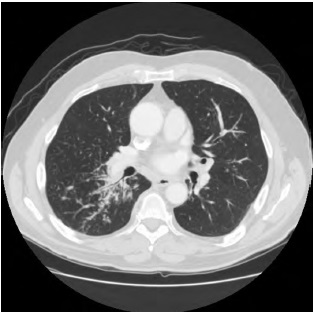
A IgG4-RD atinge o grau máximo de diversidade nas manifestações clínicas e nas características radiológicas nos pulmões. A IgG4-RD tende a acompanhar os feixes broncovasculares desses órgãos, ocasionando o espessamento do feixe broncovascular, que se torna evidente nos estudos por tomografia computadorizada (TC), conforme a Figura 5. Outras características radiológicas da IgG4-RD incluem nódulos pulmonares, opacidades em vidro fosco, espessamento da pleura e doença pulmonar intersticial.
TC: tomografia computadorizada.
Figura 5 - Doença pulmonar relacionada à IgG4. Estudo por TC do pulmão mostrando o espessamento nas vias respiratórias e a presença de infiltrados tipo vidro fosco.
Rins
A nefrite tubular intersticial (NTI) é a forma mais comum de doença renal relacionada à IgG4. O resultado é a presença de disfunção renal em estado avançado ou mesmo de doença renal em estágio terminal. A urinálise em pacientes com as complicações desse tipo de doença geralmente é normal.
De um modo geral, a proteinúria da NTI relacionada à IgG4 é subnefrótica. Na realidade, a presença de proteinúria significativa provavelmente eleve o espectro da glomerulonefropatia membranosa concomitante, que ocorre em uma minoria de pacientes com IgG4-RD e doença renal.
Os estudos por TC em casos de NTI relacionada à IgG4 revelam aumento renal substancial e a presença de lesões hipodensas no interior do parênquima renal. Com frequência, a NTI relacionada à IgG4 está associada à hipocomplementemia profunda, cujos fundamentos ainda não são muito bem compreendidos. A ligação da IgG4 com os complementos não é eficaz e, consequentemente, é pouco provável que seja uma grande contribuinte para a deposição do complexo imune nesse tipo de doença. Todavia, há descrições de complexos imunes contendo IgG4.47
Uma explicação alternativa para a hipocomplementemia em alguns pacientes é que uma proporção substancial de complexos imunes formados contém IgG1 ou IgG3, subclasses que são ligantes eficientes de complementos e, de um modo geral, são também elevados nos casos de IgG4-RD. Rins afetados pela IgG4-RD poderão se atrofiar, mesmo no contexto de boas respostas clínicas à terapia.48
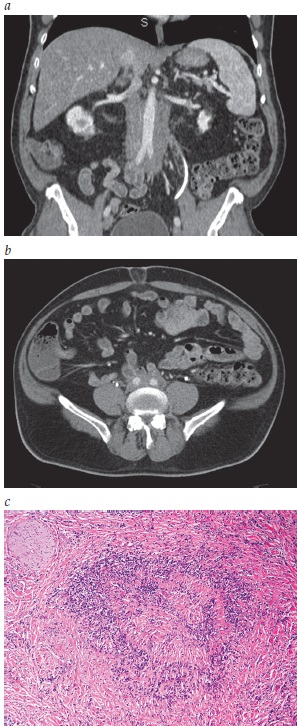
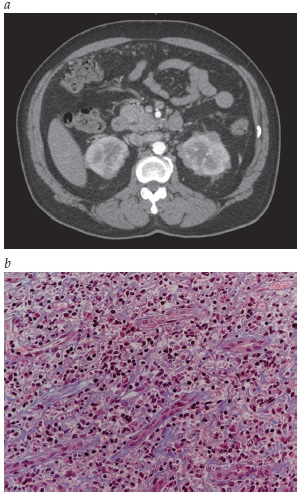
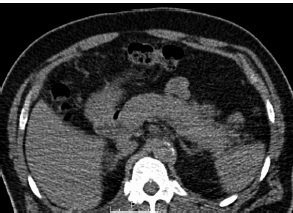
A Figura 6 mostra a doença renal relacionada à IgG4.
TC: tomografia computadorizada.
Figura 6 - Doença renal relacionada à IgG4. (A) Estudo por TC dos rins demonstrando aumento renal e a presença de lesões hipodensas no rim. (B) Biópsia renal mostrando a presença de fibrose estoriforme e de infiltrado linfoplasmacítico no interstício renal (coloração tricromática; ampliação do original em 400 vezes).
Doença no Pâncreas e no Trato Biliar
O pâncreas foi o primeiro órgão a ser reconhecido como tendo associação às concentrações séricas de IgG4.40?51 Nos dias atuais, passou-se a reconhecer a existência de dois subtipos de pancreatite autoimune (PAI), sendo que apenas um deles (tipo 1) está associado à IgG4-RD.
A PAI tipo 1 demonstra as descobertas histopatológicas clássicas de pancreatite linfoplasmacítica esclerosante. A icterícia obstrutiva ? “icterícia indolor” ? é a apresentação clínica mais comum da PAI tipo 1 induzida por colangite esclerosante concomitante relacionada à IgG4-RD. O diabetes melito secundário ocorre em, aproximadamente, 50% de todos os casos.
Tipicamente, as varreduras abdominais por TC em pacientes com PAI revelam a presença de aumento pancreático difuso, geralmente associada à intensificação tardia e a uma borda capsular de baixa densidade.52 De um modo geral, o ducto pancreático apresenta um estreitamento irregular e difuso.
Normalmente, a colangite esclerosante relacionada à IgG4-RD ocorre simultaneamente com a PAI tipo 1 e, com muito menos frequência, isoladamente. A colangite esclerosante relacionada à IgG4-RD deve ser diferenciada de colangite esclerosante primária e de colangiocarcinoma hilar.
A Figura 7 mostra a pancreatite relacionada à IgG4.
TC: tomografia computadorizada.
Figura 7 - Pancreatite relacionada à IgG4. Varredura por TC mostrando aumento em forma de salsicha no pâncreas de um paciente com IgG4-RD.
Doença no Sistema Nervoso
A IgG4-RD está entre as causas mais comuns de paquimeningite hipertrófica “idiopática”, assim como é causa estabelecida de hipofisite,53 embora sejam raros os relatos de envolvimento do parênquima cerebral. A hipofisite relacionada à IgG4-RD pode ocasionar deficiências hormonais na hipófise anterior e na posterior.
As imagens por ressonância nuclear magnética (IRM) mostram aumento selar e espessamento no pedículo hipofisário. Os estudos de IRM geralmente revelam a presença de inflamação perineural nos nervos periféricos na área da órbita, em especial os nervos trigêmeo e infraorbital.54 Essas lesões nos nervos periféricos geralmente são assintomáticas, porém, às vezes, ocasionam disestesias e outros incômodos.
Fibrose Retroperitoneal
Com frequência, a FRP relacionada à IgG4 está associada à periaortite crônica e tende a ter três sítios como alvos:
regiões periaórtica/arterial, envolvendo os tecidos conjuntivos ao redor da aorta abdominal ou suas primeiras ramificações;
áreas periureterais, causando obstrução ureteral e hidronefrose;
massa semelhante a uma placa, que envolve amplamente o retroperitôneo.
As apresentações mais comuns de FRP relacionada à IgG4 e de periaortite crônica são: dor nas costas mal localizada, dor nos flancos, dor na parte inferior do abdome ou coxas, edema nas extremidades inferiores; e hidronefrose resultante do envolvimento ureteral. De um modo geral, essas apresentações são primeiramente sutis, subagudas no início e inespecíficas, o que retarda o diagnóstico.
A dor é o sinal mais comum de apresentação de FRP e ocorre em, aproximadamente, 90% de pacientes.22,23,55 A localização da dor varia entre várias regiões: as costas, os flancos, o(s) testículo(s) ou um ou mais quadrantes abdominais. Outras características comuns incluem sintomas constitucionais como fadiga, perda de peso, febre, calafrios e sudorese noturna, que ocorrem em 20 a 40% dos pacientes.
Sintomas gastrintestinais, incluindo náusea, vômito e constipação, afetam de 25 a 30% dos pacientes e, em alguns casos, refletem outro processo formador de fibras, como, por exemplo, mesenterite esclerosante. Outras condições também foram descritas e incluem hidrocele, claudicação nas extremidades inferiores, anejaculação e frequência urinária.
Diferenças Entre Frp e Outros Subgrupos de LGG4-Rd
Os dados que estão surgindo indicam que os pacientes com FRP relacionada à IgG4 formam um subgrupo distinto de IgG4-RD, com um padrão diferente de envolvimento de órgãos, uma predileção mais baixa por doença sistêmica generalizada e uma quantidade menor de características laboratoriais de inflamação.17
Como exemplos, há os pacientes com doença renal relacionada à IgG4 que não têm probabilidade de desenvolver doença renal intrínseca relacionada à IgG4 ou que têm linfadenopatia difusa. Por outro lado, há descrições da FRP relacionada à IgG4 em associação com tireoidite de Riedel relacionada à IgG4, paquimeningite relacionada à IgG4 e aortite.
Os pacientes com FRP relacionada à IgG4 têm maior probabilidade de apresentar concentrações séricas normais de IgG4, em comparação com os indivíduos com outras manifestações orgânicas dessa doença e, em raras situações, demonstram a presença de hipocomplementemia. As razões para essas diferenças nos subgrupos ainda não são muito claras.
Estudos de Imagens em LGG4-Rd
Não há características imagiológicas típicas relacionadas à IgG4 que ajudem a fazer distinção entre FRP relacionada à IgG4 e FRP idiopática.14 Nos casos de abordagens gerais à IgG4-RD, as varreduras por TC no tórax, abdome e pelve ajudam a detectar o envolvimento de doença assintomática.
Muitos pacientes com FRP fazem TC abdominal e pélvica como parte do exame completo inicial. A inclusão de TC torácica é bastante útil para detectar linfadenopatia intratorácica, envolvimento pericárdico, fibrose mediastinal e envolvimento pulmonar, incluindo nódulos pulmonares, pseudotumores e doença pulmonar intersticial.
Os estudos por TC em pacientes com FRP revelam a presença de espessamento tecidual ao redor da aorta abdominal, desde o nível de vasos renais até a ramificação das artérias ilíacas. A densidade dos tecidos moles pode se estender ao redor da veia cava inferior, ureteres ou da parede pélvica lateral, estendendo-se até as áreas presssacral, retrovesicular ou paracólica.14,22,23
Observa-se a presença de hidronefrose unilateral ou bilateral em, aproximadamente, 50% dos casos. A linfadenopatia pélvica é uma ocorrência comum que acompanha a FRP por TC e não ajuda a fazer distinção entre FRP maligna e idiopática.14,56
A presença de massas mensentéricas também é comum e, em alguns casos, a distinção entre FRP e mesenterite esclerosante é muito difícil. Infelizmente, as características imagiológicas da FRP não distinguem com certeza absoluta as várias etiologias da doença. A biópsia é essencial para excluir malignidades.
Avaliou-se a IRM como modalidade diagnóstica adicional à TC. Uma série retrospectiva de 50 pacientes com FRP maligna ou idiopática comparou as características da IRM nos dois grupos.56 Alguns casos relacionados a malignidades foram associados a uma maior probabilidade de extensão acima das artérias renais e abaixo da bifurcação da aorta. Por outro lado, os pacientes com FRP idiopática tinham mais chances para demonstrar distração ureteral medial em comparação com os pacientes com FRP maligna.
A TC por emissão de pósitrons (PET) é uma modalidade que vem sendo usada com frequência cada vez maior nos casos de FRP. Os métodos para detectar diferenças nas descobertas entre FRP idiopática e FRP secundária usando a técnica PET ainda não foram totalmente esclarecidos.57 Em um estudo, 20 entre 26 pacientes com FRP idiopática demonstraram reabsorção de fluorodesoxiglicose (FDG).58
A utilidade de acompanhar os pacientes após a imunossupressão também chegou a ser avaliada em algumas séries menores. Vaglio e colaboradores acompanharam o curso da FRP por PET-CT em sete pacientes e mostraram que seis deles apresentaram redução na avidez da FDG com terapia imunossupressiva.59
Tratamento de LGG4-Rd e Frp Relacionada à LGG4
Nenhum teste específico chegou a abordar o tratamento de FRP relacionada à IgG4. De um modo geral, a maior parte do que se conhece atualmente sobre o tratamento de IgG4-RD foi extrapolada da literatura sobre pancreatite autoimune e de uma série de casos relativamente pequena envolvendo pacientes com esse tipo de doença.
Recentemente, foi publicada uma declaração de consenso sobre gerenciamento e tratamento de IgG4-RD por uma colaboração internacional de investigadores.60 A literatura sobre pancreatite autoimune estabeleceu o padrão para uso de corticosteroides como terapia de primeira linha.18,61 De um modo geral, a dosagem recomendada de corticosteroides é de 40 ou 30mg de prednisona ao dia, reduzida por 5 a 10mg semanalmente até atingir a dosagem diária de 5mg e, a seguir, continuada durante, pelo menos, 6 meses.18
Aparentemente, os glicocorticoides são altamente eficazes para controlar a doença no contexto de IgG4-RD, embora a crescente experiência longitudinal sugira que apenas uma pequena minoria de pacientes consegue remissões duradouras após a descontinuação no uso de glicocorticoides. A regra geral é continuar o tratamento com prednisona na dosagem de 5 a 10mg/dia.62
Há uma escassez relativa de informações que deem suporte ao uso de medicações convencionais que poupam esteroides como a azatioprina, metotrexato ou micofenolato de mofetila, nos casos de IgG4-RD. Os estudos sobre o micofenolato de mofetila também são muito limitados para aplicação no tratamento de FRP idiopática.63
Em uma série de casos prospectivos envolvendo 28 pacientes, todos eles receberam prednisona na dose inicial de 40mg/dia, que foi reduzida gradualmente durante 6 meses, em combinação com 1.000mg de micofenolato de mofetila, 2x/dia, por uma média de 24 meses.63
Os autores concluíram que, depois de 6 meses, 89% (25 entre 28) de pacientes apresentaram uma redução de 25% no tamanho da massa para-aórtica por TC abdominal, embora tenha sido impossível determinar se a proporção dessa melhora foi atribuída ao micofenolato de mofetila ou somente à prednisona.
O tamoxifeno foi uma grande promessa nas investigações iniciais, sendo que um artigo publicado recentemente aprovou o uso desse medicamento.64 Em um estudo observacional de 55 pacientes com FRP idiopática que haviam sido tratados com 20mg de tamoxifeno, 2x/dia, a maioria deles (85%) apresentou alívio rápido dos sintomas em 3 semanas.
Com base na repetição sucessiva de varreduras por TC, a maioria dos pacientes (39 entre 55; 71%) teve regressão em massa variando de moderada (menos de 50%), significativa (>50%) ou completa depois de 4 meses, seguida de mais regressão depois de 8 meses (47 entre 55; 85%). Entre os 55 pacientes, 19 que haviam usado prednisona em combinação com azatioprina ou micofenolato de mofetila não responderam à terapia à base de tamoxifeno.65
A depleção de células B aparentemente não é muito eficaz nos casos de IgG4-RD.26?28 O rituximabe diminui de forma significativa as pontuações da atividade da doença após o tratamento, assim como um declínio seletivo nos níveis séricos de IgG4, sugerindo que os plasmablastos e as células plasmáticas que produzem IgG4 nessa condição são da variedade de vida curta.
A depleção de células B também tem algum impacto sobre a contribuição das células T para a doença. Existem estudos adicionais sobre depleção de células B e outras estratégias com foco na linhagem de células B (por exemplo, plasmablastos). Vale a pena também considerar as terapias anti-células T.
As intervenções cirúrgicas e urológicas ainda são os grandes pilares do tratamento de FRP.66 Uma das abordagens para otimizar os resultados é a combinação de terapias intervencionistas/cirúrgicas e de terapia médica. Observa-se a presença de hidronefrose em, aproximadamente, 50% de pacientes; portanto, é necessário colocar stents. Com frequência, a colocação de stents é seguida de ureterólise por causa da persistência da hidronefrose ou da dependência da colocação de stent ureteral.
|
Informações Financeiras: John H. Stone, MD, MPH, não tem nenhuma informação financeira relevante a declarar. |
Referências
1. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;366:539–51.
2. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet 2015;385:1460–71. DOI: 10.1016/S0140-6736(14)60720-0.
3. Mahajan VS, Mattoo H, Deshpande V, et al. IgG4-related disease. Annu Rev Pathol 2014;9:315–47.
4. Ormond JK. Bilateral ureteral obstruction due to envelopment and compression by an inflammatory process. J Urol 1948;59:1072–9.
5. Albarran J. Retention renale par periureterite. Liberation externe de le’uretere. Assoc Fr Urol 1905;9:511.
6. Vaglio A, Salvarani C, Buzio C. Retroperitoneal fibrosis. Lancet 2006;367:241–51.
7. Vaglio A, Palmisano A, Corradi D, et al. Retroperitoneal fibrosis: evolving concepts. Rheum Dis Clin North Am 2007;33:803–17, vi–vii.
8. Hamano H, Kawa S, Ochi Y, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet 2002;359:1403–4.
9. Fukuda W, Kimura M, Akaogi T, et al. Multifocal fibrosclerosis: retroperitoneal fibrosis associated with a suprasellar tumor and pach-ymeningitis. Intern Med 2003;42:1006–10.
10. Fukui T, Okazaki K, Yoshizawa H, et al. A case of autoimmune pancreatitis associated with sclerosing cholangitis, retroperitoneal fi-brosis and Sjögren syndrome. Pancreatology 2005;5:86–91.
11. Fukukura Y, Fujiyoshi F, Nakamura F, et al. Autoimmune pancreatitis associated with idiopathic retroperitoneal fibrosis. AJR Am J Roentgenol 2003;181:993–5.
12. Comings DE, Skubi KB, Van Eyes J, Motulsky AG. Familial multifocal fibrosclerosis. Findings suggesting that retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel’s thyroiditis, and pseudotumor of the orbit may be different manifestations of a single disease. Ann Intern Med 1967;66:884–92.
13. Zen Y, Onodera M, Inoue D, et al. Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol 2009;33:1833–9.
14. Khosroshahi A, Carruthers MN, Stone JH, et al. Rethinking Ormond’s disease: “idiopathic” retroperitoneal fibrosis in the era of IgG4-related disease. Medicine (Baltimore) 2013;92:82–91.
15. Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25:1181–92.
16. Khosroshahi A, Cheryk LA, Carruthers MN, et al. Brief Report: Spuriously low serum IgG4 concentrations caused by the phenomenon in patients with IgG4-related disease. Arthritis Rheum 2014;66:213–7.
17. Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: baseline clinical and laboratory features in 125 patients with biopsy-proven disease. Arthritis Rheum 2015 May 18. DOI: 10.1002/art.39205. [Epub ahead of print]
18. Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut 2009;58:1504–7.
19. Wallace ZS, Mattoo H, Mahajan VS, et al. Predictors of disease relapse in IgG4-related disease. [Submitted]
20. Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303–32.
21. Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol 2010;34:1812–9.
22. Scheel PJ Jr, Feeley N. Retroperitoneal fibrosis: the clinical, laboratory, and radiographic presentation. Medicine (Baltimore) 2009;88:202–7.
23. Van Bommel EF, Jansen I, Hendriksz TR, Aarnoudse AL. Idiopathic retroperitoneal fibrosis: prospective evaluation of incidence and clinicoradiologic presentation. Medicine (Baltimore) 2009;88:193–201.
24. Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 2011;23:119–24.
25. Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy 2009;39:469–77.
26. Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 2010;62:1755–62.
27. Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012;91:57–66.
28. Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 2015;74:1171–7. DOI: 10.1136/annrheumdis-2014-206605.
29. Mattoo H, Mahajan VS, Della Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol 2014;134:679–87. DOI: 10.1016/j.jaci.2014.03.034.
30. Wallace ZS, Mattoo H, Carruthers MN, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 con-centrations. Ann Rheum Dis 2015;74:190–5. DOI: 10.1136/annrheumdis-2014-205233.rint.
31. Mattoo H, Mahajan V, Deshpande V, et al. Clonal expansion of IL1-b producing CD4+SLAMF7+ cytotoxic T lymphocytes in a human fibrotic disease. [Submitted]
32. Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000;118:573–81.
33. Zen Y, Fujii T, Harada K, et al. Th2 and regulatory immune reactions are increased in immunoglobulin G4-related sclerosing pancrea-titis and cholangitis. Hepatology 2007;45:1538–46.
34. Della Torre E, Mattoo H, Mahajan VS, et al. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy 2014;69:269–72.
35. Mattoo H, Della-Torre E, Mahajan VS, et al. Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy 2014;69:399–402.
36. Yao Q, Wu G, Hoschar A. IgG4-related Mikulicz’s disease is a multiorgan lymphoproliferative disease distinct from Sjögren’s syn-drome: a Caucasian patient and literature review.?Clin Exp Rheumatol 2013;31:289–94.
37. Himi T, Takano K, Yamamoto M, et al. A novel concept of Mikulicz’s disease as IgG4-related disease. Auris Nasus Larynx 2012;39:9–17.
38. Wallace ZS, Khosroshahi A, Jakobiec FA, et al. IgG4-related systemic disease as a cause of “idiopathic” orbital inflammation, includ-ing orbital myositis, and trigeminal nerve involvement. Surv Ophthalmol 2012;57:26–33.
39. Hu EK, Parrish C, Wrobel B, et al. Immunoglobulin G4-related disease presenting as an ethmoid and maxillary mass. Ann Allergy Asthma Immunol 2013;111:75–7.
40. Schiffenbauer AI, Gahl WA, Pittaluga S, et al. IgG4-related disease presenting as recurrent mastoiditis. Laryngoscope 2012;122:681–4.
41. Reder L, Della Torre E, Stone JH, et al. Clinical manifestations IgG4-related disease in the pharynx: case series and review of the liter-ature. Ann Otol Rhinol Laryngol 2015;124:173–8.
42. Dahlgren M, Khosroshahi A, Nielsen GP, et al. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res 2010;62:1312–8.
43. Sato Y, Kojima M, Takata K, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Cas-tleman’s disease. Mod Pathol 2009;22:589–99.
44. Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol 2012;29:226–34.
45. Stone JR. Aortitis, periaortitis, and retroperitoneal fibrosis, as manifestations of IgG4-related systemic disease. Curr Opin Rheumatol 2011;23:88–94.
46. Inokuchi G, Hayakawa M, Kishimoto T, et al. A suspected case of coronary periarteritis due to IgG4-related disease as a cause of is-chemic heart disease. Forensic Sci Med Pathol 2014;10:103–8.
47. Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011;22:1343–52.
48. Saeki T, Kawano M, Mizushima I, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int 2013;84:826–33.
49. Kamisawa T, Egawa N, Nakajima H. Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol 2003;98:2811–2.
50. Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003;38:982–4.
51. Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–8.
52. Kamisawa T, Takuma K, Egawa N, et al. Autoimmune pancreatitis and IgG4-related sclerosing disease. Nat Rev Gastroenterol Hepatol 2010;7:401–9.
53. Leporati P, Landek-Salgado MA, Lupi I, et al. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endo-crinol Metab 2011;96:1971–80.
54. Inoue D, Zen Y, Sato Y, et al. IgG4-related perineural disease. Int J Rheumatol 2012;29:212–8.
55. Kermani TA, Crowson CS, Achenbach SJ, Luthra HS. Idiopathic retroperitoneal fibrosis: a retrospective review of clinical presentation, treatment, and outcomes. Mayo Clin Proc 2011;86:297–303.
56. Mirault T, Lambert M, Puech P, et al. Malignant retroperitoneal fibrosis: MRI characteristics in 50 patients. Medicine (Baltimore) 2012;91:242–50.
57. Treglia G, Mattoli MV, Bertagna F, et al. Emerging role of fluorine-18-fluorodeoxyglucose positron emission tomography in patients with retroperitoneal fibrosis: a systematic review. Rheumatol Int 2013;33:549–55. DOI: 10.1007/s00296-012-2576-0.
58. Jansen I, Hendriksz TR, Han SH, et al. (18)F-fluorodeoxyglucose position emission tomography (FDG-PET) for monitoring disease activity and treatment response in idiopathic retroperitoneal fibrosis. Eur J Intern Med 2010;21:216–21.
59. Vaglio A, Greco P, Versari A, et al. Post-treatment residual tissue in idiopathic retroperitoneal fibrosis: active residual disease or silent “scar”? A study using 18F-fluorodeoxyglucose positron emission tomography. Clin Exp Rheumatol 2005;23:231–4.
60. Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheum 2015;67:1688–99. DOI: 10.1002/art.39132.
61. Hart PA, Kamisawa T, Brugge WR, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut 2013;62:1771–6.
62. Masaki Y, for the All-Japan IgG4-Related Disease Team. Corticosteroids for the treatment of IgG4-related disease. Presented at: sec-ond international symposium on IgG4-Related Diseases and Associated Conditions. February, 2014. Waikiki, Hawaii.
63. Scheel PJ Jr, Feeley N, Sozio SM. Combined prednisone and mycophenolate mofetil treatment for RPF: a case series. Ann Intern Med 2011;154:31–6.
64. Van Bommel EF, Pelkmans LG, van Damme H, Hendriksz TR. Long-term safety and efficacy of a tamoxifen-based treatment strategy for idiopathic retroperitoneal fibrosis. Eur J Intern Med 2013;24:444–50.
65. Vaglio A, Palmisano A, Alberici F, et al. Prednisone versus tamoxifen in patients with idiopathic retroperitoneal fibrosis: an open-label randomized controlled trial. Lancet 2011;378:338–46.
66. Swartz RD. Idiopathic RPF: a review of the pathogenesis and approaches to treatment. Am J Kidney Dis 2009;54:546–53.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.