(Carregando Índice)... (Carregando Índice)... |
Última revisão: 05/07/2018
Comentários de assinantes: 0
|
Artigo original: Bartlett H, Haynes S, Lamers L, Skorton D. Congenital Heart Disease in Adults, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Resumo
Com os avanços nos tratamentos médicos e cirúrgicos, um número cada vez maior de crianças com doenças congênitas no coração e na vasculatura passou a sobreviver até a vida adulta. Acredita-se que a proporção de adultos afetados por doença cardíaca congênita continue a aumentar ao longo do tempo. Por conseguinte, é extremamente importante que os médicos se familiarizem com o tratamento desses pacientes. Esta revisão apresenta uma análise dos distúrbios acianóticos (derivações e lesões vasculares), anomalias vasculares, distúrbios cianóticos e temas relacionados à saúde das mulheres. As figuras mostram a anatomia de defeitos nos septos atriais, um corte anatômico transversal mostrando o septo atrioventricular, as posições anatômicas dos defeitos no septo atrioventricular (DSAVs), as posições anatômicas dos defeitos no septo ventricular (DSVs), uma varredura por computadorizada (TC) de uma coarctação aórtica, angiograma de uma drenagem persistente na veia cava superior no interior do átrio direito, derivações sistêmicas de uma artéria para a artéria pulmonar, imagem por ressonância magnética (IRM) de um paciente com reparo tetralógico de Fallot e insuficiência permanente na válvula pulmonar, tratamento de regurgitação na válvula pulmonar com uma válvula pulmonar transcateter, imagens por TC de um paciente com cuidados paliativos de desvio atrial para transposição das grandes artérias e múltiplas obstruções defletoras, estágios no reparo de ventrículos funcionais simples e ecocardiogramas (ECOs) de um paciente com anomalia de Ebstein. Os quadros apresentam uma lista de recomendações para reposição da válvula pulmonar em casos de tetralogia reparada de Fallot, condições em que a gravidez representa um alto risco, e indicações cardíacas para ECO fetal.
Doenças congênitas no coração e na vasculatura são os defeitos de nascimento mais comuns. Com os avanços nos tratamentos médicos e cirúrgicos, a maioria dos pacientes com doenças congênitas no coração sobrevive até a vida adulta.1,2
Nos dias atuais, dois terços de pacientes com doença cardíaca congênita são adultos.3 Considerando a evolução dos cuidados cardíacos e as orientações práticas de suporte fisiopatológico, recomenda-se que o tratamento seja coordenado pelos centros regionais de Doença Cardíaca Congênita em Adultos.4 Essa população crescente de pacientes se defronta com uma carga cada vez maior de doenças crônicas.5,6
Com base no índice atual de mortalidade inferior a 5%, estima-se que um em 150 adultos poderá ter doença cardíaca na próxima década.7 Um número cada vez maior de médicos deve se familiarizar com os cuidados desses pacientes. Neste texto,é apresentada uma revisão dos defeitos cardíacos congênitos (DCGs) encontrados com maior frequência em pacientes adultos.
Distúrbios Acianóticos: Desvios
Defeitos no Septo Atrial
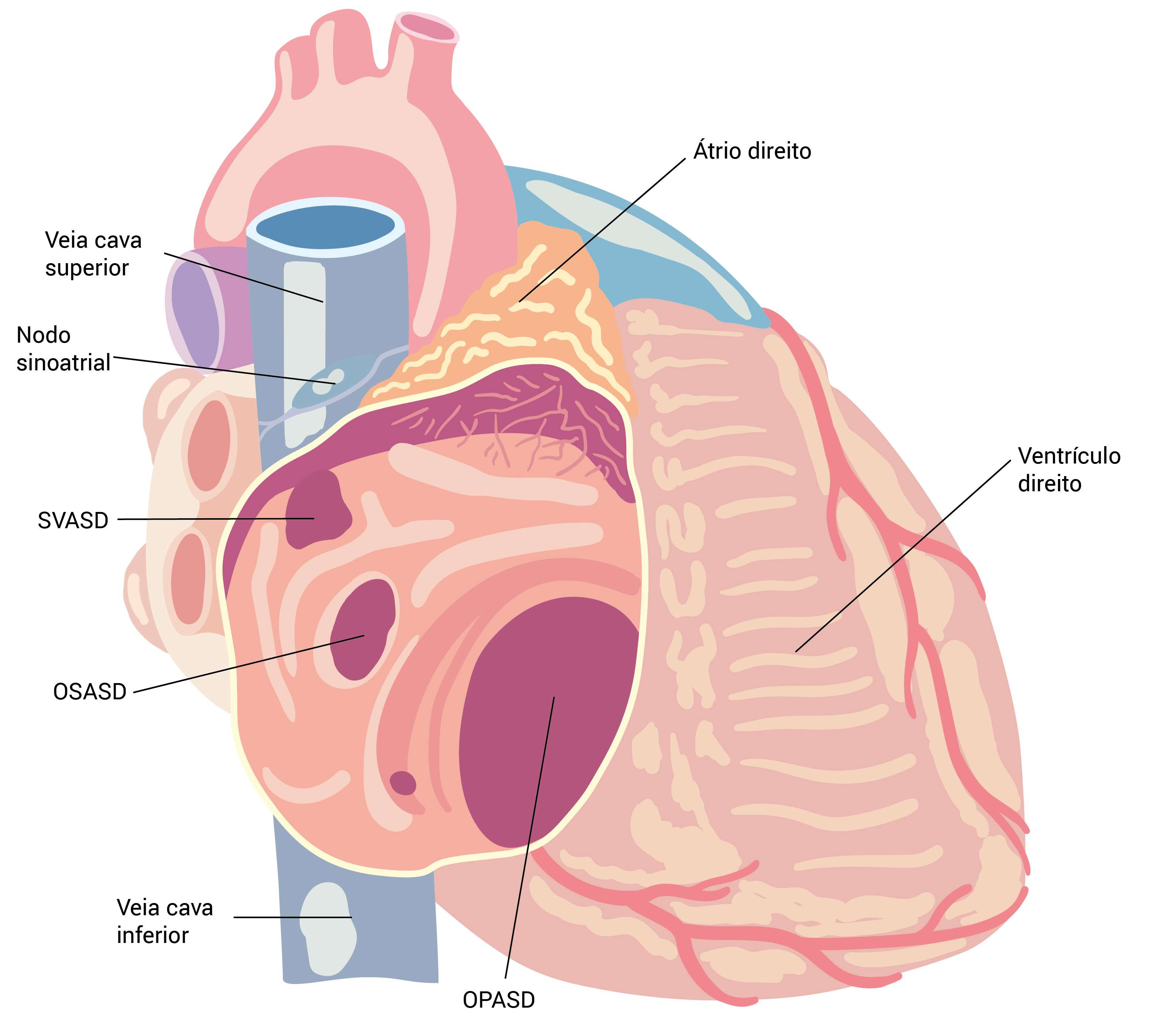
Os defeitos no septo atrial (DSAs) ocorrem em três locais principais, conforme a Figura 1: região da fossa oval (DSAs no forame interatrial do tipo secundum); porção superior do septo atrial nas proximidades da junção com a veia cava superior ?VCS (DSAs no seio venoso); e porção inferior do septo atrial nas proximidades do anel da válvula tricúspide (DSAs no forame interatrial do tipo primum).8 Os DSAs no forame interatrial do tipo primum fazem parte do espectro de DSAVs.
DSA: defeito no septo atrial; OPASD: defeito no septo atrial do forame interatrial do tipo primum; OSASD: defeito no septo atrial do forame interatrial do tipo secundum; SVASD: defeito no septo atrial do seio venoso.

Figura 1 - Anatomia dos DSAs.
Os DSAs no forame interatrial do tipo secundum são a variedade mais comum, sendo responsável por cerca da metade dos casos de DSAs. Os defeitos no seio venoso são relativamente menos prevalentes. Retorno anômalo nas veias pulmonares é uma anormalidade associada muito comum. A proximidade do nodo sinoatrial a esse tipo de DAS pode ocasionar disfunção nodal e arritmias atriais.
Fisiopatologia
Os DSAs estão associados aos desvios da esquerda para a direita em vários graus. Os principais determinantes da direção e da magnitude dos desvios de fluxo são o tamanho do defeito e a complacência relativa dos ventrículos esquerdo e direito.9
Apresentação Clínica
A maior parte dos pacientes com DSA no forame interatrial do tipo secundum ou com DSA no seio venoso permanece assintomática ao longo da vida adulta, embora o uso imediato do ecocardiograma (ECO) possibilite obter diagnóstico de imediato. Os pacientes com DSAs se apresentam com edema periférico, em particular durante os períodos de débito cardíaco mais elevado, como, por exemplo, durante a gravidez.
Na medida em que os pacientes se aproximam da meia idade, a complacência do ventrículo esquerdo possivelmente diminua, aumentando a magnitude do desvio da esquerda para a direita. A dilatação de longa duração no átrio direito geralmente cria arritmias atriais, incluindo palpitação e fibrilação atrial, e poderá ser o sintoma que se apresenta. Um número substancial de pacientes na meia idade se queixa de dispneia, em especial com esforço físico, mesmo que não tenham hipertensão pulmonar.
Aproximadamente, 10% de pacientes com DSAs no forame interatrial do tipo secundum progridem para hipertensão pulmonar associada à doença vascular pulmonar obstrutiva (síndrome de Eisenmenger). Na medida em que a pressão pulmonar se eleva, o desvio da esquerda para a direita diminui e é substituído por um desvio da direita para a esquerda, resultando em cianose.
DAS: Defeito no Septo Atrial.
Figura 2 - Fechamento transcateter de um DSA. (A) A figura mostra o DSA do forame interatrial do tipo secundum com um grande desvio da esquerda para a direita. (B) O fechamento do DSA está bem posicionado com o disco atrial esquerdo maior e o disco atrial direito menor abraçando o septo atrial com um desvio residual trivial da esquerda para a direita.
O ponto mais importante do exame físico de DSA é a divisão ampla e fixa do som do segundo batimento cardíaco. A presença de um sopro sistólico resultante do aumento no fluxo pulmonar é comum e, caso ocorra um grande desvio da esquerda para a direita, o fluxo adicional através da válvula tricúspide poderá ocasionar um ruído remanescente da estenose tricúspide.
Gerenciamento
Grandes DSAs são definidos como aqueles com uma relação pulmonar/fluxo sistêmico [Qp:Qs] de, aproximadamente, 1,5:12 e devem ser fechados para evitar insuficiência cardíaca (IC) direita e o desenvolvimento de hipertensão pulmonar, assim como diminuir o risco êmbolos paradoxais. A ECO ajuda a determinar o tipo e tamanho do defeito, a calcular o fluxo de desvios e a identificar quaisquer anomalias associadas.
Fechamento cirúrgico direto é o método tradicional, embora os dispositivos atualmente existentes no mercado permitam fazer fechamentos com base na cateterização em muitos defeitos no forame interatrial do tipo secundum.10,11 Alguns estudos demonstraram que houve melhora na capacidade para fazer exercícios físicos, redução na incidência de arritmias e melhora na qualidade de vida em pacientes com idade em torno de 50 anos que tenham feito fechamento de DAS com algum tipo de dispositivo.12
Essas descobertas dão suporte ao fechamento de defeitos significativos sob o ponto de vista hemodinâmico, mesmo em adultos mais velhos. O gerenciamento pós-fechamento inclui avaliação periódica para verificar o eventual desenvolvimento de arritmias atriais.13
Os casos raros de ocorrência de erosão cardíaca tardia ou de fratura na estrutura dos dispositivos levou a Food and Drug Administration a recomendar o acompanhamento anual ao longo da vida de alguns oclusores do septo atrial.14 Dor torácica repentina ou síncope sem explicação justificam fazer nova avaliação ecocardiográfica para verificar a possibilidade de perfuração cardíaca.15
Defeitos no Septo Atrioventricular
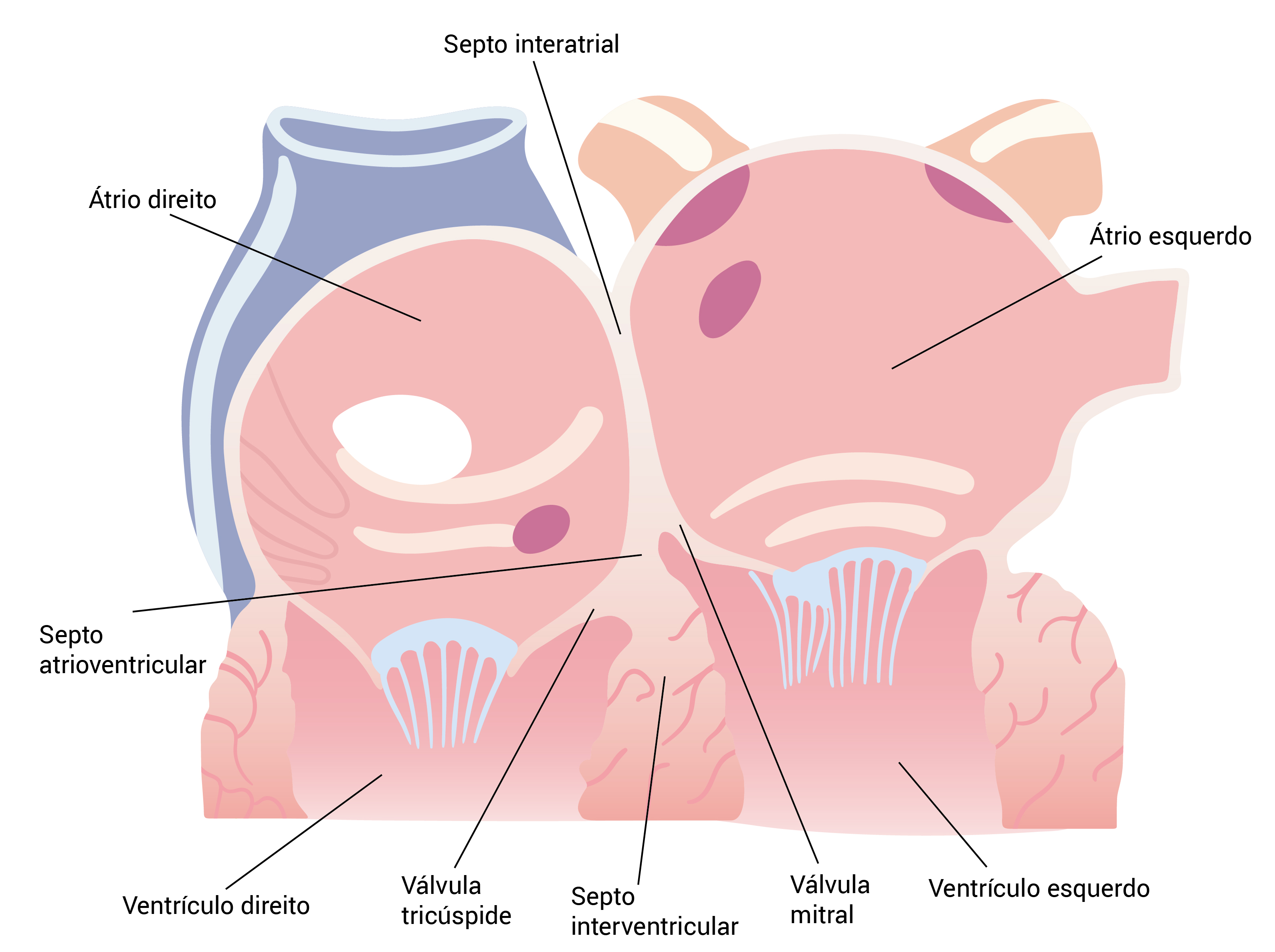
De um modo geral, o folheto septal da válvula tricúspide se insere no septo mais perto do ápice em comparação com o folheto septal da válvula mitral, conforme a Figura 3. Consequentemente, a pequena porção do tecido septal que se localiza em uma posição superior ao ponto de inserção do folheto septal tricúspide separa o átrio direito do ventrículo esquerdo e, por essa razão, denomina-se átrio atrioventricular. O termo defeito no septo atrioventricular (DSAV) se refere a um espectro complexo de distúrbios envolvendo anormalidades no septo atrioventricular e, com frequência, as válvulas atrioventriculares.
SAV: septo atrioventricular.
Figura 3 - Corte transversal anatômico mostrando o SAV. Observa-se que o folheto septal da válvula tricúspide se insere mais próximo do ápice, em comparação com o folheto septal da válvula mitral; consequentemente, a SAV separa o átrio direito do ventrículo direito.
A nomenclatura aplicada a esse espectro de distúrbios é bastante variável. Os termos sinônimos incluem defeito no canal atrioventricular e defeito da almofada endocárdica.
Fisiopatologia
O espectro dos DSAVs varia de DAS simples no forame interatrial do tipo primum a DSAV completo, que permite a livre comunicação entre as quatro câmaras cardíacas. As variações na anatomia do folheto anterior da válvula mitral e do folheto septal da válvula tricúspide incluem a presença de fissura, de qualquer outra anormalidade, ou de ambas nesses folhetos; cordas que se inserem em locais anômalos e podem alterar a função dos folhetos valvulares ou produzir obstrução no trato do fluxo externo ventricular direito; assim como folhetos valvulares atrioventriculares que fazem ponte com o defeito septal.
As consequências fisiológicas variam de acordo com a extensão da anomalia; por exemplo, a adição de um folheto anterior na válvula mitral ajuda a variar os graus de regurgitação mitral. Os defeitos maiores que também envolvem o septo ventricular, assim como DSAVs completos, podem estar associados aos desvios torrenciais da esquerda para a direita ou à mistura de sangue venoso e arterial.
Os pacientes com DSAVs completos que não tenham sido reparados correm grande risco de desenvolver hipertensão pulmonar. A síndrome de Eisenmenger é particularmente comum em pacientes com DSAV que têm também síndrome de Down (trissomiado 21).16
Apresentação Clínica
No exame físico, a maioria dos pacientes com DSAV apresenta sopros causados por desvios, regurgitação valvular ou obstrução no trato do fluxo externo. As descobertas clínicas em pacientes com DAS isolado no forame interatrial do tipo primum se assemelham às de pacientes com DAS no forame interatrial do tipo secundum, e possivelmente permaneçam assintomáticos até a vida adulta e, a seguir, poderão apresentar sintomas como fadiga e dispneia ou sintomas relacionados a arritmias atriais.
Regurgitação grave em qualquer válvula atrioventricular poderá produzir IC ou arritmias. Os desvios maiores da esquerda para a direita com DSAV completo produzem IC congestiva na infância. Os sintomas relacionados à hipertensão pulmonar ocorrem em pacientes que desenvolvem a síndrome de Eisenmenger.
Gerenciamento
Os casos raros de pacientes que se apresentam com DSAV completo na vida adulta devem ser avaliados para verificar a eventual presença de hipertensão pulmonar. Nas situações em que o grau de hipertensão pulmonar não seja proibitivo (isto é, se a resistência dos vasos pulmonares for menos de 50%, em comparação com a resistência vascular sistêmica), é imprescindível fazer o fechamento cirúrgico do defeito e o reparo em anomalias nas válvulas atrioventriculares.
A maior parte dos adultos já fez reparo cirúrgico. No período pós-operatório, os pacientes devem ser avaliados para verificar a adequação do reparo na válvula atrioventricular e monitorados para verificar a eventual presença de algum desvio residual. Em pacientes com regurgitação mitral residual, o gerenciamento deve focar a avaliação e o momento de fazer nova operação, que poderá envolver reparo ou reposição da válvula mitral. Além disso, os pacientes devem ser acompanhados para verificar o eventual desenvolvimento de arritmias atriais.
Defeitos no Septo Ventricular
Os defeitos no septo ventricular (DSVs) estão entre os distúrbios cardíacos congênitos mais comuns no nascimento, embora sejam encontrados com menos frequência como lesões isoladas na vida adulta. Isso ocorre porque a maior parte dos DSVs em lactentes é: grande ou não restritiva (isto é, permite o equilíbrio de pressões entre os ventrículos) e, portanto, ocasiona IC que precisa de fechamento cirúrgico imediato; ou pequena, que fecha espontaneamente.
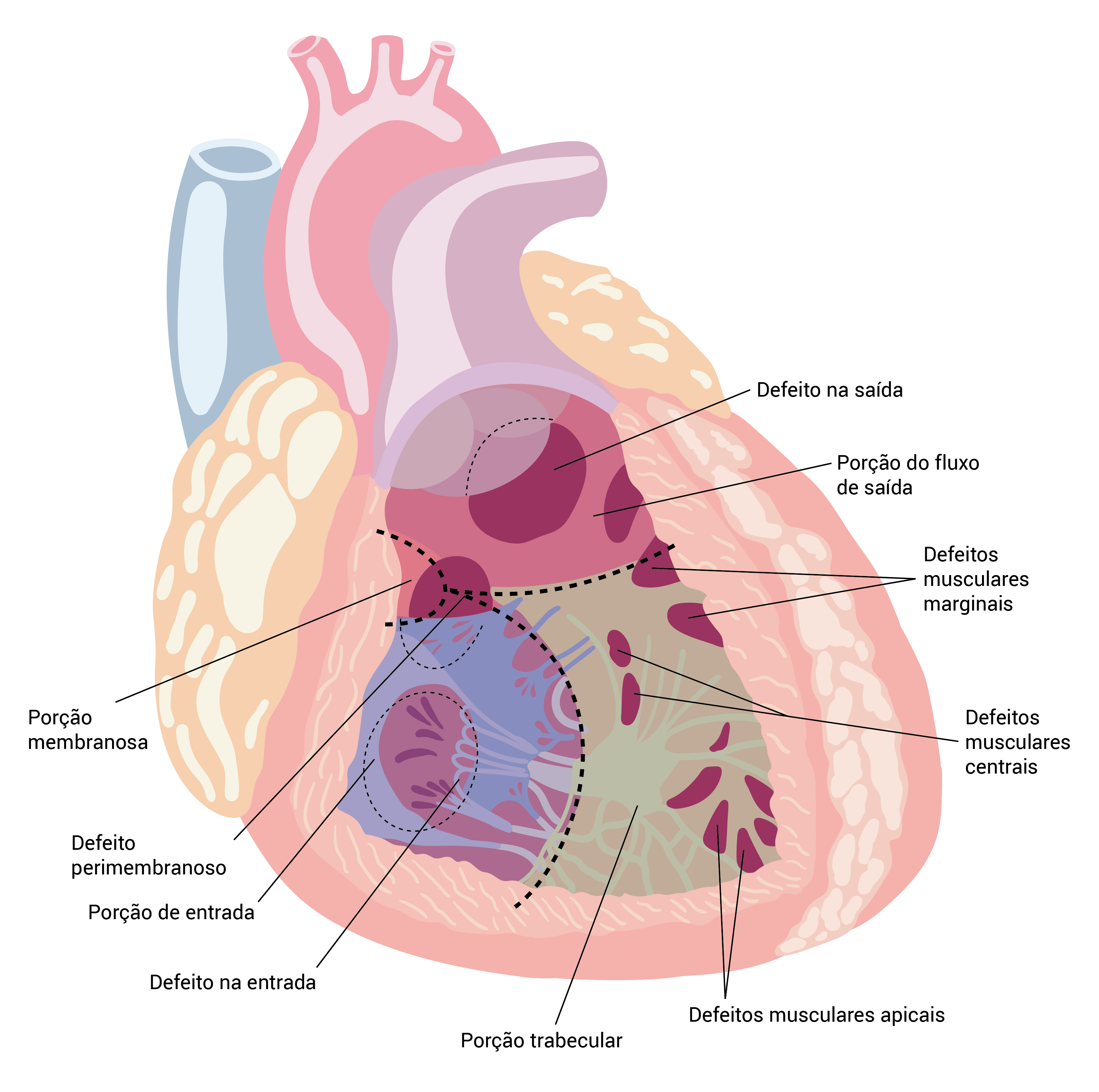
DSV: defeito do septo ventricular.
Figura 4 - Posições anatômicas dos DSVs. As subdivisões anatômicas principais do septo ventricular são as porções membranosas, o fluxo externo, o fluxo de entrada e trabecular. Os DSVs típicos incluem defeito na saída, defeito perimembranoso, defeito muscular marginal, defeito muscular central, defeito na entrada e defeito no músculo apical.
Os sistemas de classificação de DSVs variam bastante, embora, normalmente, se refiram às divisões embriológicas do septo ventricular em porções de entrada, saída, musculares e membranosas. Os defeitos perimembranosos são os mais comuns. Os DSVs na entrada se localizam em uma posição mais posterior e, possivelmente, façam parte do espectro dos DSAVs.
Defeitos simples ou múltiplos podem ocorrer no septo muscular (DSV muscular). Para finalizar, os DSVs na saída incluem os defeitos subpulmonares, que permitem o prolapso de cúspides na aorta, resultando em regurgitação aórtica.
Fisiopatologia
Os defeitos não restritivos no septo ventricular permitem o equilíbrio das pressões entre os ventrículos direito e esquerdo, ao passo que os pequenos defeitos produzem amplo gradiente de pressão ao longo do defeito, de modo que as pressões no lado direito do coração permanecem normais. A magnitude do fluxo do desvio através de DSVs moderados ou grandes depende das resistências relativas do leito de vasos sistêmicos versus do leito de vasos pulmonares.
Em raras situações, os médicos encontram pacientes adultos com defeitos maiores e não restritivos, na ausência de outros tipos de lesão. A estenose pulmonar moderada, ao nível valvular ou subvalvular, possivelmente aumente uma resistência ao fluxo externo ventricular direito suficiente para diminuir o desvio da esquerda para a direita.
Consequentemente, os pacientes com estenose pulmonar variando de leve a moderada provavelmente cheguem à vida adulta sem apresentar sintomas. Os adultos com DSV de longa duração e grandes desvios podem desenvolver a síndrome de Eisenmenger.
Apresentação Clínica
Os pacientes com DSV pequena são assintomáticos, excetuando-se os que contraem endocardite infecciosa ou aqueles com a síndrome de Eisenmenger. A descoberta física clássica de DSV restritiva é a presença de um sopro pansistólico grave, e frequentemente palpável, que pode ser ouvido com mais nitidez na borda esternal inferior esquerda.
Os pacientes com grandes defeitos que permitam equilibrar as pressões ventriculares podem se apresentar com sopros menos expressivos que os pacientes com pequenos defeitos. O sopro da regurgitação aórtica pode ser audível nos casos em que ocorrer prolapso da cúspide da aorta.
Gerenciamento
Os pacientes com DSVs em que a proporção Qp:Qs for maior que 1,5:1 devem ser considerados candidatos para fechamento cirúrgico. Os pacientes com hipertensão pulmonar poderão fazer o fechamento se a resistência pulmonar não ultrapassar, aproximadamente, 50% da resistência sistêmica.
O prolapso da cúspide aórtica possivelmente diminua a magnitude do desvio, embora a presença de prolapso seja uma indicação adicional potencial para fechamento. A ECO é o estudo de escolha para identificar a localização, o tamanho e a significância hemodinâmica de um DSV ou de quaisquer defeitos associados.
Os fechamentos cirúrgicos precoces de DSVs foram feitos por meio de a ventriculotomias direitas, embora, atualmente, a maior parte dos defeitos - em particular aqueles com septo membranoso - é fechada com uma abordagem transatrial; esse tipo de abordagem cria menos problemas com disfunção ventricular direita e arritmias.
O progresso no desenvolvimento de dispositivos transcateter é constante. Entretanto, nos dias atuais, o fechamento cirúrgico de DSVs ainda é a abordagem mais comum. O gerenciamento pós-fechamento envolve avaliação para verificar a hipótese de DSV residual ou recorrente e de arritmias atriais ou ventriculares, assim como para avaliar a função ventricular direita.
Ducto Arterioso Patente
Durante a vida fetal, o ducto arterioso faz a conexão entre a artéria pulmonar e a aorta. Logo após o nascimento, como resultado de alterações nos níveis de prostaglandina em circulação e na saturação do oxigênio arterial, o ducto se contrai e, em seguida, ocorre o fechamento permanente. A impossibilidade de fechar o ducto arterioso ocasiona uma condição que se denomina ducto arterioso patente (DAP).
Fisiopatologia
O desvio da aorta para a artéria pulmonar aumenta o fluxo de sangue pulmonar e retorna para o lado esquerdo do coração. O tamanho do defeito e as resistências relativas dos leitos vasculares pulmonares e sistêmicos determinam o grau do desvio. Os adultos com DAPs se apresentam com um pequeno desvio da esquerda para a direita ou com fluxo maior e, finalmente, com a síndrome de Eisenmenger.
Apresentação Clínica
À exceção de pacientes com a síndrome de Eisenmenger, a maior parte dos adultos com DAPs variando de pequenas a moderadas será assintomática, a não ser nos casos de endarterite.
A descoberta física patognomônica de DAP é um sopro contínuo. Sopro contínuo é aquele audível em toda a sístole e a diástole. O sopro clássico de DAP se assemelha ao som de uma máquina e se estende por toda a sístole e, em vários graus, na diástole, atingindo o pico de intensidade no momento de S2. O escoamento de sangue para a artéria pulmonar na diástole produz uma ampla pressão de pulso por causa da baixa pressão diastólica na aorta. A ECO transtorácica ajuda a identificar o DAP e a quantificar o tamanho do desvio.
Gerenciamento
Com o advento de meios confiáveis para o fechamento transcateter de DAPs, a prática usual é recomendar o fechamento da maioria dos ductos arteriosos patentes.17 Em raras ocasiões, os ductos precisam ser fechados por meios cirúrgicos caso o fechamento transcateter não tenha sido bem-sucedido. Embora não seja muito comum, o gerenciamento pós-procedimental inclui avaliação de algum desvio residual.
Os pacientes com DAP que desenvolvem hipertensão pulmonar são gerenciados da mesma forma que os indivíduos com síndrome de Eisenmenger, ainda que possam se beneficiar clinicamente com os dispositivos de fechamento de DAP com terapias agressivas para hipertensão pulmonar.
Distúrbios Acianóticos: Lesões valvulares
Válvula Aórtica Bicúspide e Outras Causas de Estenose Aórtica
As anormalidades no trato do fluxo externo ventricular esquerdo são os distúrbios cardíacos congênitos mais comuns. Em particular, até 2% da população têm válvula aórtica bicúspide congênita. A presença de válvula bicúspide pode ser uma descoberta incidental no exame físico ou em ECOs feitas por outros motivos, embora possa se tornar estenótica ou regurgitante ao longo do tempo. As válvulas aórticas bicúspides poderão também estar associadas à coarctação da aorta.
Fisiopatologia
Algumas válvulas aórticas bicúspides não se tornam estenóticas ou regurgitantes. No entanto, as válvulas aórticas bicúspides poderão produzir cargas pressóricas no ventrículo esquerdo, resultando em hipertrofia ventricular esquerda e, nos casos graves, em IC, angina ou morte súbita por taquiarritmias.
Os pacientes com válvulas aórticas bicúspides incompetentes apresentam dilatação no ventrículo esquerdo, inicialmente com função sistólica normal. Essa condição poderá evoluir para IC. Mesmo sem obstrução significativa, a dilatação da aorta ascendente geralmente ocorre em associação com válvula aórtica bicúspide.18
Apresentação Clínica
No exame físico, o sinal fundamental de válvula aórtica bicúspide é um clique precoce na ejeção sistólica. A qualidade e o tempo de duração dos sopros geralmente se correlacionam com a gravidade da estenose ou da regurgitação aórtica. Não se ouve nenhum sopro ou sopro de ejeção suave na ausência de anormalidades hemodinâmicas significativas. A presença de sopro diastólico audível sugere, ao menos, a presença de regurgitação aórtica variando de leve a moderada.
Gerenciamento
Os pacientes com regurgitação aórtica causada por válvula bicúspide que são assintomáticos e têm função sistólica normal devem ser acompanhados clinicamente em intervalos regulares. A ECO serial é muito útil para acompanhar a progressão da lesão, assim como o tamanho e a função do ventrículo esquerdo. As dimensões da aorta ascendente podem ser medidas normalmente.19
Nas situações em que as imagens acústicas comprometerem a confiabilidade das medições ecocardiográficas, uma das opções é usar imagens por ressonância magnética (IRMs). Nos casos em que houver sintomas de IC, evidências de redução na função sistólica ou dilatação progressiva do ventrículo esquerdo, recomenda-se fazer a reposição cirúrgica da válvula aórtica.
Intervenção cirúrgica ou valvuloplastia com balão (em pacientes mais jovens) é uma opção de tratamento a ser considerada em pacientes com estenose aórtica grave, em especial quando for acompanhada de condições como IC, síncope ou desconforto torácico.
Uma grande variedade de procedimentos cirúrgicos encontra-se à disposição, incluindo reparo valvular direto, reposição com bioprótese ou prótese mecânica, reposição valvular e da raiz aórtica proximal, e o procedimento de Ross, em que se remove a válvula bicúspide anormal por meios cirúrgicos, substituindo-a pela válvula pulmônica (também conhecida por válvula pulmonar) nativa do paciente que, por sua vez, é substituída por uma válvula tecidual.
O procedimento de Ross elimina a necessidade de colocar uma válvula protética na posição aórtica. O foco principal do gerenciamento pós-operatório é a avaliação da estenose recorrente ou da regurgitação aórtica progressiva.
Estenose Pulmônica
Os pacientes com estenose pulmônica geralmente têm malformação valvular, com fusão de uma ou mais comissuras, resultando na aparência em forma de redoma. A maior parte dessas válvulas é fina e flexível, embora as válvulas de alguns pacientes sejam displásicas e espessas.
Fisiopatologia
As válvulas pulmonares estenóticas fazem pressão sobre o ventrículo direito, ocasionando hipertrofia ventricular direita (HVD) e, em um subgrupo de pacientes, insuficiência ventricular direita. Em pacientes com ventrículos direitos gravemente hipertróficos, o desequilíbrio entre o suprimento e a demanda de oxigênio miocárdico possivelmente ocasione isquemia com desconforto concomitante no tórax anginal e arritmias.
Apresentação Clínica
Os pacientes com estenose pulmônica de gravidade moderada podem permanecer assintomáticos por várias décadas. Os sintomas finais incluem desconforto torácico remanescente de angina, respiração ofegante, fadiga e sintomas de insuficiência ventricular direita. Em pacientes com estenose pulmonar leve, não é comum a progressão da doença e dos sintomas além da adolescência em pacientes com estenose pulmonar leve; a grande descoberta física da estenose pulmônica é a presença de um sopro sistólico crescente-decrescente na válvula estreitada, precedido por um clique de ejeção pulmônica.
O comportamento do clique de ejeção pulmônica durante a respiração provavelmente ajude a diferenciá-lo do clique da válvula aórtica bicúspide. O clique de ejeção pulmônica apresenta uma redução seletiva de intensidade com a inspiração normal e pode até desaparecer completamente com a inspiração. Por outro lado, o clique da válvula aórtica bicúspide não apresenta essa redução seletiva.
Gerenciamento
As abordagens iniciais à estenose pulmônica consistiam de valvotomia cirúrgica fechada ou aberta. Nos últimos 20 anos, o advento de métodos confiáveis de valvuloplastia pulmonar com balão criou melhores oportunidades para abordagem nesses casos.10,20 A primeira linha de tratamento de estenose na válvula pulmonar é a valvuloplastia com balão nos casos de estenose significativa (definida por um gradiente de fluxo externo ventricular direito >50mmHg).
É extremamente difícil tratar adequadamente algumas válvulas displásicas por valvuloplastia com balão que, ao final, acabam por precisar de reparo ou substituição. O gerenciamento pós-intervenção inclui monitoramento para recorrência de estenose ou regurgitação progressiva, assim como avaliação do tamanho e função do ventrículo direito.21 Finalmente, os pacientes possivelmente tenham que fazer reposição valvular no quadro de regurgitação grave.
Anomalias Vasculares
Coarctação da Aorta
Coarctação da aorta é um DCG relativamente comum que pode ocorrer isoladamente ou em associação com outros defeitos (em especial, válvula aórtica bicúspide ou DSV), além de ser uma descoberta usual em pacientes com síndrome de Turner. Coarctação é uma das causas de hipertensão secundária.
Fisiopatologia
A patologia essencial na coarctação da aorta é o estreitamento do lúmen aórtico, normalmente nas proximidades do ligamento arterioso, em uma localização distal em relação à origem da artéria subclávia esquerda. O estreitamento da aorta no sítio da coarctação divide a circulação sistêmica em uma zona de alta pressão em um ponto proximal a ela.
A hipertensão pode acelerar o desenvolvimento de doença aterosclerótica na artéria coronária e ocasionar acidente vascular cerebral (AVC); o AVC é um risco particular na presença de aneurismas no círculo de Willis, como ocorre com o aumento na incidência em pacientes com coarctação.22
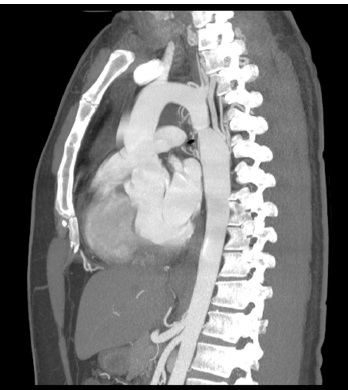
TC: tomografia computadorizada.
Figura 5 - TC de coarctação aórtica. Observa-se o estreitamento agudo do istmo da aorta com leve dilatação pós-estenótica da aorta descendente e vasos torácicos dilatados, acomodando o fluxo lateral que contorna a obstrução.
Apresentação Clínica
Embora a claudicação nas extremidades inferiores seja uma ocorrência provável, mesmo os pacientes com coarctação significativa na aorta podem ser completamente assintomáticos. A característica principal no exame físico é a diferença nos pulsos e nas pressões sanguíneas acima versus a coarctação abaixo.
A palpação nas artérias radial e femoral em pacientes normais revela a chegada simultânea ou, talvez, a chegada ligeiramente precoce do pulso até a artéria femoral. Nos casos de coarctação da aorta, o pulso femoral ocorre mais tardiamente que o pulso radial e, com frequência, tem amplitude mais baixa.
A pressão arterial deve ser avaliada em ambos os braços e nas duas pernas em busca de coarctação da aorta causada por variações anatômicas no arco e nas artérias subclávias. Além das pressões arteriais diferenciais, o exame poderá revelar também a presença de um sopro sistólico através da coarctação, que poderá ser ouvido com mais clareza na área infraescapular esquerda.
Gerenciamento
Qualquer coarctação suficiente para produzir hipertensão sempre deve ser tratada por meios cirúrgicos ou por intervenção transcateter. A ECO e/ou IMR cardíaca é eficaz para identificar coarctações e a circulação colateral. A experiência mais longa é com excisão cirúrgica da coarctação e anastomose de extremidade a extremidade ou interposição de enxertos.
Nos anos mais recentes, a angioplastia com stent comprovou ser uma alternativa viável tanto para o tratamento inicial de coarctação como para o tratamento de restenose no sítio da coarctação, que poderá surgir após o reparo ou angioplastia.10 A angioplastia com stent de coarctação nativa ou recorrente poderá resultar em uma quantidade menor de complicações em comparação com a cirurgia ou com a angioplastia com balão.23
O foco do gerenciamento da coarctação no longo prazo é o tratamento da hipertensão, que é comum mesmo após intervenções cirúrgicas ou transcateter bem-sucedidas. As imagens seriais são indicadas para avaliar a recoarctação e para a vigilância de aneurimas no sítio do reparo.24
Veia Cava Superior Esquerda Persistente
A veia cava superior esquerda (VCSE) é uma estrutura venosa embrionária persistente que não regride normalmente durante o desenvolvimento fetal. Esse fato ocorre em até 10% indivíduos com doença cardíaca congênita e pode ocorrer como anomalia isolada em 0,3% da população.25
Fisiopatologia
A veia cardinal anterior esquerda se oblitera e forma o ligamento de Marshall ao longo do curso do desenvolvimento embriológico. Caso isso não ocorra, a VCSE persistente drena para o interior do seio coronariano. Normalmente, há também uma veia cava superior (VCS) direita.26
Sob a perspectiva do paciente, não há nenhum risco de sintomas. Entretanto, poderão surgir algumas dificuldades durante o procedimento de inserção de cateteres centrais periféricos a partir da extremidade superior esquerda ou durante a implantação de um sistema de marca-passo transvenoso. Na presença de seio coronariano sem teto, a VCSE persistente drena efetivamente para o interior do átrio esquerdo, resultando em um grande potencial de embolização sistêmica de trombos venosos.
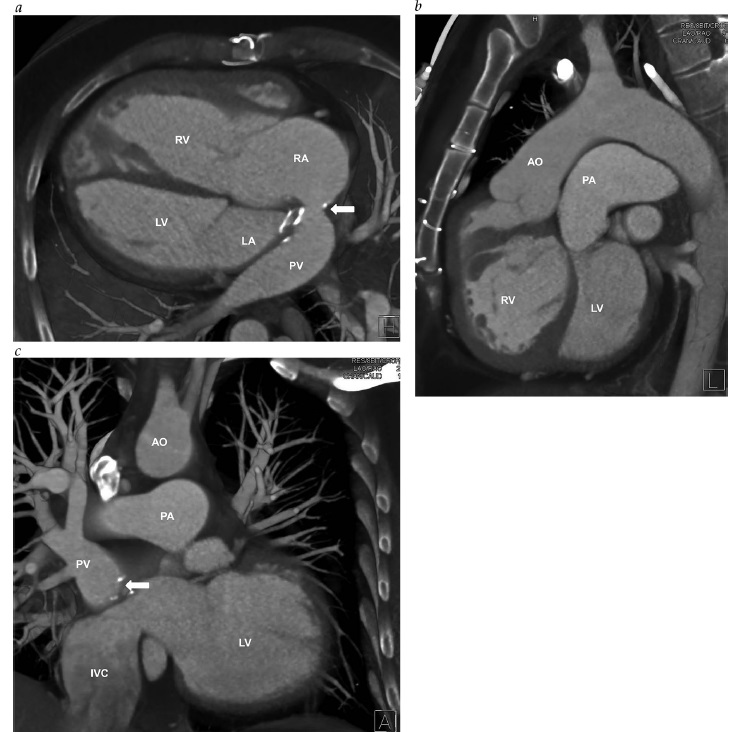
A Figura 6 mostra um angiograma de uma veia cava superior esquerda persistente.
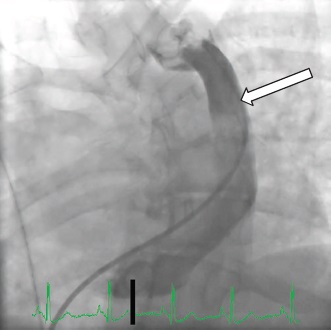
VCSE: veia cava superior esquerda.
Figura 6 - Angiograma de uma VCSE persistente (seta branca) drenando para o interior do átrio direito.
Apresentação Clínica
A menos que sejam instrumentados, os pacientes provavelmente serão totalmente assintomáticos e não apresentarão nenhuma descoberta física sugerindo a presença de veia cava superior esquerda (VCSE). Os pacientes com seio coronariano sem teto (desvio da direita para a esquerda) poderão apresentar vários graus de dessaturação que, em geral, são detectados de forma incidental. A ECO ajuda a detectar a VCSE e, além disso, poderá mostrar a dilatação no seio coronariano, que recebe o aumento de fluxo proveniente da veia cava superior esquerda.
Gerenciamento
De um modo geral, não há nenhum tipo de tratamento para VCSEs persistentes que drenam para seios coronarianos intactos. A melhor opção de tratamento é o reparo cirúrgico na presença de seio coronariano sem teto. Na presença de uma veia ponte, a VCSE poderá ser ligada sem nenhuma abordagem ao seio coronariano. Na ausência de uma veia ponte, o seio coronariano poderá defletir para o interior do átrio direito.25
Distúrbios Cianóticos
Definição e Mecanismos
A cianose central é ocasionada por um desvio intracardíaco ou intrapulmonar da direita para a esquerda. A cianose se torna evidente a partir do momento que a hemoglobina capilar reduzida (não oxigenada) ultrapassar, aproximadamente, 5mg/dL, embora isso dependa do nível de concentração da hemoglobina geral: a cianose se torna aparente mais rapidamente em pacientes com policitemia e menos aparente em pacientes com anemia.
A cianose não se torna visualmente aparente até a saturação cair abaixo de 85% (presumindo que o nível de hemoglobina seja normal). Os pacientes com dessaturação arterial de longa duração possivelmente desenvolverão hipocratismo nos dedos das mãos e dos pés. O hipocratismo digital se caracteriza pelo espessamento e pelo alargamento dos leitos das unhas por causa da perda angular entre a unha e o leito ungueal, produzindo unhas convexas. Observa-se também a escoliose com alguma frequência em associação com doença cardíaca cianótica.
A classificação dos defeitos cardíacos cianóticos (DCCs) de acordo com seu efeito sobre o fluxo sanguíneo pulmonar é bastante útil. Os defeitos que diminuem o fluxo sanguíneo pulmonar incluem tetralogia de Fallot, atresiana válvula tricúspide, anomalia de Ebstein e atresia pulmonar.
Os defeitos associados ao aumento no fluxo sanguíneo pulmonar incluem tronco arterioso persistente, dextrotransposição (D-TGA) das grandes artérias com ou sem DSV, retorno venoso pulmonar anômalo total e ventrículo simples ou ventrículo comum (incluindo a síndrome hipoplásica no lado esquerdo do coração). Os pacientes acianóticos com grandes desvios da esquerda para a direita podem desenvolver doença oclusiva nos vasos do pulmão (síndrome de Eisenmenger), resultando em cianose.
Pacientes adultos com DCC cianótico correm grande risco de hiperviscosidade secundária à eritrocitose. A eritrocitose desenvolve-se como um mecanismo compensatório para a dessaturação do oxigênio dos eritrócitos: é necessário um aumento significativo na massa eritrocitária para liberar um volume adequado de oxigênio para os tecidos periféricos.
A ocorrência de trombose venosa e arterial é uma probabilidade nos casos de DCC e foi atribuída ao aumento na massa eritrocitária e associada à anemia por deficiência de ferro, que também aumenta a viscosidade do sangue. Esse risco aumenta na presença de hipertensão ou de fibrilação atrial, assim como em pacientes com histórico de flebotomia e microcitose, sugerindo a necessidade de abordagens mais conservadoras para flebotomia e de tratamentos agressivos para deficiência de ferro.
Tetralogia de Fallot
Fisiopatologia
A tetralogia de Fallot é a forma mais comum de doença cardíaca cianótica congênita. Classicamente, esse tipo de síndrome inclui estenose pulmonar (subvalvular, valvular, supravalvular ou uma combinação dos três tipos), hipertrofia ventricular direita, DSV subaórtico e dextroposição da aorta de modo que substitua o septo interventricular.
As anomalias associadas incluem arco aórtico (25%), DSA (10%) e anomalias na artéria coronária (10%).27 Aproximadamente, 15% de pacientes com tetralogia de Fallot têm a síndrome de deleção do 22q11 (também conhecida por outras denominações, tais como síndrome de DiGeorge, síndrome de Takao e síndrome velocardiofacial).28
Reparo Cirúrgico na Infância
A prática atual utiliza reparo cirúrgico precoce, normalmente no primeiro ano de vida. Sem cirurgia, não é comum a sobrevida além dos 20 anos de idade. O reparo cirúrgico consiste de fechamento do DSV com adesivo e alívio na obstrução no trato do fluxo externo ventricular direito utilizando-se de um ou mais entre os seguintes métodos: ressecção no músculo infundibular, valvotomia pulmonar, aumento no trato do fluxo externo ou no adesivo transanular e aumento no adesivo da artéria pulmonar principal ou proximal.
Em alguns casos, é necessário colocar um conduto desde o ventrículo direito até a artéria pulmonar. O conduto pode ter válvula ou não e pode ser bioprotético ou um homoenxerto.
Embora, nos dias atuais, o reparo imediato seja a rotina, muitos pacientes se submetem a procedimentos paliativos. A paliação cirúrgica inclui desvio da circulação sistêmica para a artéria pulmonar para aumentar o fluxo pulmonar. Esse dispositivo poderá ser uma derivação de Blalock-Taussig, uma derivação de Potts ou uma derivação de Waterston.
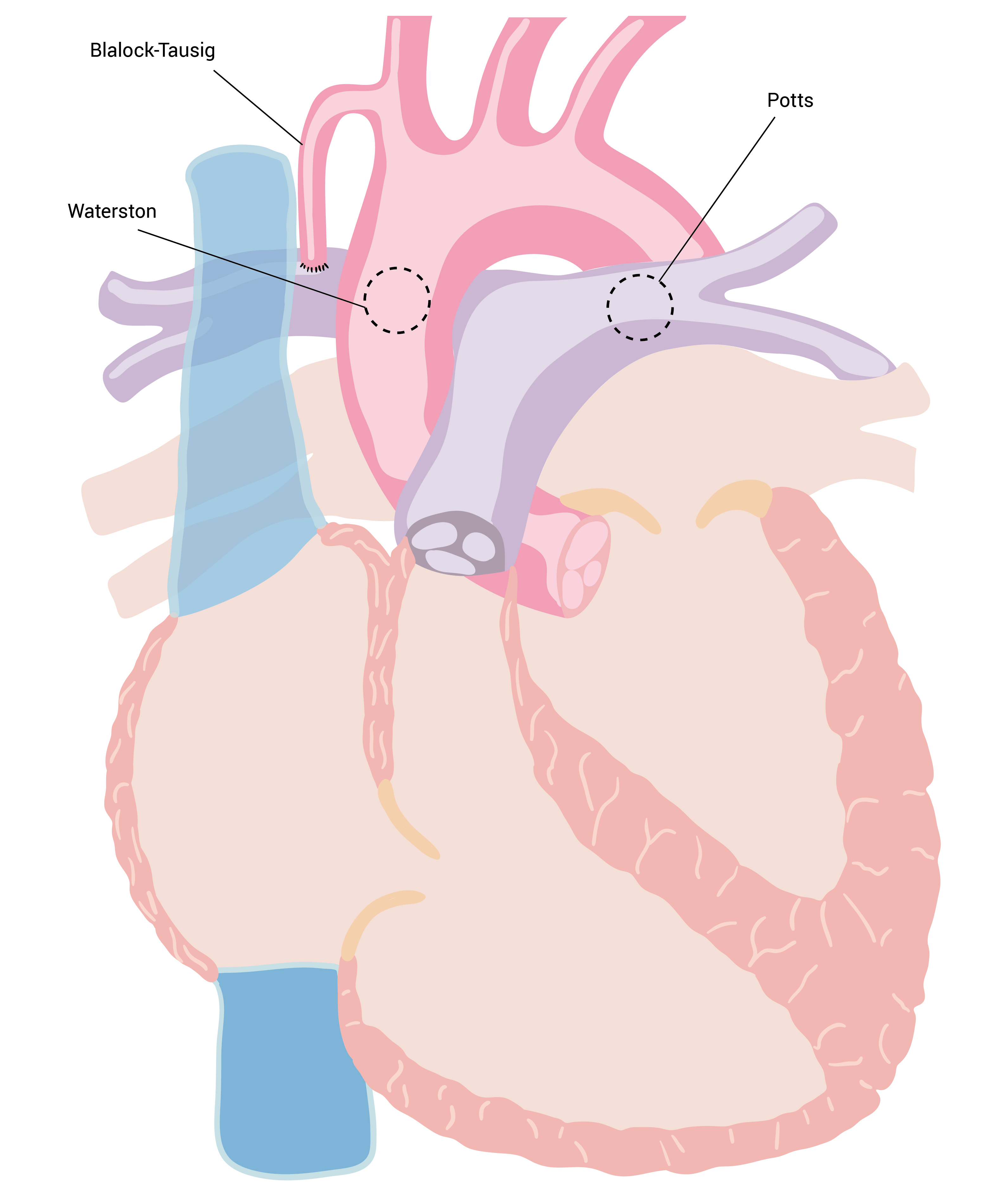
A derivação clássica de Blalock-Taussig conecta a artéria subclávia à artéria pulmonar; a forma modificada engloba a interposição de um tubo, normalmente de politetrafluoetileno (PTFE). A derivação de Potts conecta a aorta descendente à artéria pulmonar esquerda. A derivação de Waterston conecta a aorta ascendente à artéria pulmonar direita, conforme a Figura 7.
Figura 7 - Derivações sistêmicas de uma artéria para a artéria pulmonar. A derivação de Blalock-Taussig conecta a artéria subclávia a uma artéria pulmonar; a derivação de Waterston conecta a aorta ascendente à artéria pulmonar direita e a derivação de Potts conecta a aorta descendente à artéria pulmonar esquerda.
Apresentação Clínica Após o Reparo
Os defeitos residuais são o foco principal do exame físico em pacientes que tenham feito reparo cirúrgico para tetralogia de Fallot. Comumente, esses pacientes apresentam sopro relacionado à obstrução no trato do fluxo externo e regurgitação pulmonar, variando de leve a grave, produzindo um sopro de um lado para o outro. Os sopros por ejeção sistólica podem ser proeminentes e se relacionam mais à técnica utilizada para fazer o reparo do que a quantidade da obstrução.
Os desvios patentes produzem sopro contínuo. O grau de cianose depende da adequação do fluxo de sangue pulmonar criado pela derivação. Defeito residual no septo atrial ou no septo ventricular ou estenose na ramificação da artéria pulmonar são condições que poderão ser detectadas. Com o aumento na sobrecarga volumétrica no ventrículo direito causado por obstrução ou insuficiência no fluxo externo, o paciente poderá apresentar intolerância aos exercícios físicos, IC direita e arritmia atrial ou ventricular.
Gerenciamento
Os pacientes que tenham feito reparo cirúrgico para tetralogia de Fallot devem ser monitorados regularmente para verificar a progressão dos defeitos residuais, particularmente regurgitação pulmonar e obstrução de conduto. As IRMs cardíacas se tornaram o padrão de ouro para avaliação quantitativa do tamanho e da função biventricular, da quantificação da regurgitação pulmonar e da obtenção de imagens da anatomia do trato do fluxo externo ventricular direito, das artérias do pulmão e da aorta, como mostra a Figura 8.
IRM: imagem por ressonância magnética; VD: ventrículo direito; VE: ventrículo esquerdo.
Figura 8 - IRM de um paciente com reparo na tetralogia de Fallot e insuficiência de longa duração na válvula pulmonar. Observa-se que o VD está dilatado em comparação com o VE.
A estenose na ramificação da artéria pulmonar pode ser abordada com angioplastia com balão ou com stent. A repetição da cirurgia ou a intervenção com cateter são opções a serem consideradas em pacientes com defeito residual no septo ventricular, em pacientes cuja pressão ventricular direita está acima de dois terços da pressão sistêmica por causa da obstrução residual, em pacientes com aumento no ventrículo direito secundário a regurgitação pulmonar grave, e em pacientes com baixa tolerância aos exercícios físicos.
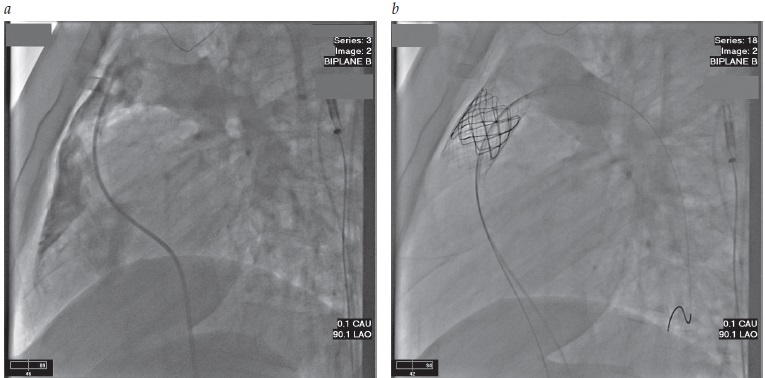
A reposição valvular no pulmão melhora a função ventricular direita, arritmias e a capacidade para fazer exercícios.29 A reoperação em adultos tem baixo risco. Os pacientes que tiveram um conduto ligando o ventrículo direito à artéria pulmonar ou reposição prévia da válvula pulmonar são candidatos à reposição transcateter da válvula pulmonar, que cria uma válvula competente ao mesmo tempo em que alivia a obstrução, conforme ilustra a Figura 9.
Figura 9 - Tratamento de regurgitação na válvula pulmonar com uma válvula pulmonar transcateter. (A) Este angiograma da artéria pulmonar delineia a estenose e a regurgitação no trato do fluxo externo ventricular direito após o reparo na tetralogia de Fallot na infância. (B) Repetição do angiograma após a implantação de uma válvula pulmonar transcateter no trato do fluxo externo ventricular direito, mostrando o calibre normal do trato do fluxo externo ventricular direito e uma válvula pulmonar competente.
A abordagem transcateter tem alta taxa de sucesso procedimental e durabilidade favorável no tempo médio.30,31 Ocasionalmente, é necessário fazer o reparo ou a reposição da válvula aórtica por causa da regurgitação aórtica progressiva.32 A dilatação da aorta é uma descoberta associada comum, mas a dissecção aórtica é uma ocorrência rara.33
As arritmias atriais ocorrem em 20% de pacientes e foram associadas à regurgitação tricúspide e pulmônica e a uma idade mais avançada no momento do reparo.34,35 As arritmias ventriculares, que são detectadas em 40 a 50% de pacientes, foram associadas à idade avançada no reparo primário, à sobrecarga volumétrica no ventrículo direito e à prolongação no intervalo QRS.
Alargamentos acentuados no intervalo QRS para mais de 180ms e disfunção no ventrículo esquerdo foram identificados como fatores de risco para morte cardíaca súbita. Nesses casos, recomenda-se levar em consideração a colocação profilática de um desfibrilador cardíaco implantável.
Caso não tenha sido previamente avaliada, a análise cromossômica talvez seja extremamente útil. A tetralogia de Fallot pode estar associada à deleção do 22Q11, que é uma mutação autossômica dominante e, consequentemente, tem implicações nas crianças de pacientes afetados.28
Os critérios sugeridos para calcular o momento exato de fazer a reposição da válvula pulmonar se baseiam em diversas medições por IRM, como mostra o Quadro 1.36
Quadro 1
|
Recomendações para Reposição da Válvula Pulmonar em Tetralogia De Fallot Reparada |
|
A. Regurgitação pulmonar moderada ou grave (fração regurgitante >25%) por IRM e dois ou mais entre os seguintes fatores: 1. Índice volumétrico VD no fim da diástole =160mL/m2 (pontuação z>5). 2. Índice volumétrico VD no fim da sístole =70mL/m2. 3. Índice volumétrico VE no fim da sístole =65mL/m2. 4. Fração de ejeção VD =45mL/m2. 5. Aneurisma no RVOT. 6. Critérios clínicos: intolerância aos exercícios, sintomas e sinais de IC, medicações cardíacas, síncope, taquicardia ventricular sustentada. B. Outras lesões significativas sob o ponto de vista hemodinâmico que justifiquem o encaminhamento para cirurgia em pacientes com regurgitação pulmonar variando de moderada a grave. C. Pacientes com mais de 3 anos de idade que tenham se submetido a reparo para TOF devem ser considerados candidatos para reposição da válvula pulmonar mais cedo, assim como na presença de dilatação e disfunção no VD. |
IRMs: imagens por ressonância magnética; RVOT: trato do fluxo externo ventricular direito; TOF: tetralogia de Fallot; VD: ventricular direito; VE: ventricular esquerdo.
Dextrotransposição das Grandes Artérias
Fisiopatologia
Na forma mais comum de transposição das grandes artérias (TGA), a D-TGA, a aorta se eleva em uma posição anterior a partir do ventrículo direito e a artéria pulmonar se eleva em uma posição posterior a partir do ventrículo esquerdo. Há uma separação total entre a circulação pulmonar e a circulação sistêmica: o fluxo sanguíneo sistêmico atravessa o lado direito do coração e penetra na aorta, ao passo que o fluxo de sangue pulmonar atravessa o lado esquerdo do coração e penetra na artéria pulmonar.
Aproximadamente, um terço de pacientes apresenta anomalias associadas, incluindo DSA e DSV. A obstrução no trato do fluxo externo ventricular esquerdo é comum. A sobrevida além do primeiro ano não é comum, a menos que ocorra uma melhora no mix intracardíaco.
Reparo Cirúrgico na Infância
A septostomia atrial com balão (procedimento de Rashkind) pode ser necessária no período logo após o nascimento para melhorar a mistura ao nível atrial.37 A cirurgia inicialmente consistia em redirecionar o retorno venoso sistêmico para o ventrículo esquerdo e o retorno venoso pulmonar para o ventrículo direito. As operações conhecidas por desvio atrial (procedimento de Mustard ou de Senning), que utilizavam um defletor no interior dos átrios, embora recuperassem a circulação fisiológica, exigiam que o ventrículo direito funcionasse como o ventrículo sistêmico.
A operação de desvio arterial foi substituída pela operação de desvio atrial, ao menos em pacientes com função normal nas duas válvulas semilunares. Na operação de desvio arterial, a artéria pulmonar e a aorta são as primeiras a serem transectadas acima das válvulas semilunares e das artérias coronárias, e são desviadas logo em seguida.
A aorta se desloca no sentido posterior e é conectada acima da válvula pulmônica, que surge a partir do ventrículo esquerdo, e a artéria pulmonar se desloca no sentido anterior e é conectada acima da válvula aórtica, que surge a partir do ventrículo direito.
Os pacientes com D-TGA e com grande DSV são candidatos ao procedimento de Rastelli. O fluxo proveniente do ventrículo esquerdo deflete através do DSV na direção da válvula aórtica. A artéria pulmonar é dividida e costurada e, a seguir, coloca-se um conduto ligando o ventrículo direito à artéria pulmonar para permitir o fluxo de sangue para o pulmão.
Apresentação Clínica e Reparo
As descobertas clínicas se referem à presença de anomalias associadas (por exemplo, sopro de DSV ou estenose pulmônica). Os pacientes que fazem tratamento paliativo por meio de um procedimento de desvio atrial correm o risco de arritmias, regurgitação da válvula tricúspide sistêmica e disfunção no ventrículo direito sistêmico.
A ocorrência de disfunção no nodo sinusal é comum, assim como taquiarritmias, resultando nos sintomas de fadiga.38 As taquiarritmias atriais foram também associadas ao aumento no risco de morte súbita. De um modo geral, a disfunção no ventrículo direito se manifesta com sintomas de fadiga e sopro holossistólico de alta frequência ocasionado por insuficiência tricúspide.
Problemas adicionais poderão envolver obstrução no retorno venoso sistêmico ou pulmonar através dos defletores atriais, resultando em asceíte/edema periférico ou congestão pulmonar, respectivamente.39 Os pacientes que têm marcapassos transvenosos correm grande risco de obstrução por deflexão e têm o benefício da avaliação antes da colocação de uma derivação.40
Aqueles que receberam tratamento paliativo com desvio arterial poderão ter sopros relacionados à obstrução no trato do fluxo externo residual, que produzirão um sopro causado pela ejeção sistólica. Provavelmente, seja detectado algum DSA ou no septo ventricular.
Gerenciamento
A sobrevida após o desvio atrial é excelente, mas, mesmo assim, o envelhecimento populacional justifica a avaliação para verificar a eventual presença de arritmias e de IC. Os pacientes devem ser monitorados para a presença de um aumento progressivo no ventrículo direito e regurgitação tricúspide, levando à disfunção ventricular.
É possível que esses pacientes precisem se submeter a procedimentos paliativos agressivos ou a transplante cardíaco nas situações em que a terapia médica não for eficaz. As arritmias atriais, incluindo a síndrome do seio enfermo e palpitação atrial, são bastante comuns.38 Morte súbita pode ocorrer em 6 a 17% de pacientes.41A deflexão atrial pode causar obstrução venosa sistêmica ou pulmonar, que poderá ser tratada por meio de nova operação ou por angioplastia com balão e colocação de stent.
O prognóstico de longo prazo de pacientes com desvio arterial é menos conhecido, e essa abordagem foi usada somente por três décadas, embora os estudos iniciais mostrem que há um impacto positivo sobre a função cardíaca, na capacidade para fazer exercícios e na qualidade de vida.42,43
Estenose na artéria pulmonar principal ou em suas ramificações (a complicação mais comum) ou estenose nos sítios de anastomose aórtica também são ocorrências prováveis. Embora seja uma ocorrência comum, a dilatação radicular na aorta raramente é significativa. As lesões coronarianas relacionadas a reparos cirúrgicos são raras na vida adulta.44
As complicações ocasionadas pelo procedimento de Rastelli incluem obstrução subaórtica (deflexão ou obstrução causada por DSV), estenose causada por conduto (com ou sem regurgitação), vazamento em defletores e estenose em alguma ramificação da artéria pulmonar. Os defeitos residuais significativos podem precisar de algum tipo de intervenção.39 Os pacientes que receberam um conduto também poderão ser candidatos à reposição transcateter da válvula pulmonar para tratar a disfunção ocasionada pelo conduto.
Coração Univentricular
O coração univentricular é a forma mais complicada de doença cardíaca congênita. A grande expectativa é que população de adultos com fisiologia de ventrículo único duplique na próxima década. Esses pacientes apresentam sequelas em diversos órgãos e precisam de cuidados multidisciplinares durante toda a vida.
Fisiopatologia
Ventrículos únicos funcionais podem ser o resultado de defeitos como atresia tricúspide, que é um DSAV em desequilíbrio, ocasionando o desenvolvimento preferencial de apenas um ventrículo, atresia pulmonar com septo ventricular intacto, ou síndrome cardíaca esquerda hipoplásica.
Reparo Cirúrgico na Infância
A apresentação inicial de coração univentricular na infância inclui cianose grave associada a uma queda acentuada no fluxo do sangue pulmonar, cianose leve e IC associada a um fluxo excessivo de sangue pulmonar e mistura intracardíaca de circulações, ou fluxos sanguíneos sistêmicos e pulmonares quase em equilíbrio e cianose leve.45
De um modo geral, os pacientes que sobrevivem até a vida adulta se submeteram a um ou mais procedimentos cirúrgicos paliativos. Os procedimentos mais comuns incluem operação de Norwood, anastomose cavopulmonar superior bidirecional (procedimento de Glenn) e anastomose cardiopulmonar completa (procedimento de Fontan). Nos dias atuais, geralmente as operações são feitas por estágios durante a infância.
AO: aorta; IVC: veia cava inferior; LA: átrio esquerdo; LV: ventrículo esquerdo subpulmonar; PA: artéria pulmonar; PV: veia pulmonar; RA: átrio direito; RV: ventrículo direito sistêmico; TC: tomografia computadorizada.
Figura 10 - Imagens por TC de um paciente com tratamento paliativo com desvio atrial de transposição das grandes artérias e obstruções ocasionadas por múltiplas deflexões. (A) Estrutura atrial estática e apresentação tridimensional da posição craniana à posição caudal. Observa-se a calcificação do enxerto de tereftalato de polietileno circundando a obstrução por deflexão. A drenagem das veias pulmonares deflete para o átrio direito nativo, que drena para o interior do ventrículo direito morfológico, que, em seguida, se conecta com a aorta. Os seios coronarianos e as veias coronarianas são dilatados, assim como a veia cava inferior. (B) Estrutura sagital estática e apresentação tridimensional da esquerda para a direita. Observa-se que a localização da aorta é anterior à artéria pulmonar. A descompressão de uma grande veia ázigos permite que o sangue contorne a obstrução causada pela deflexão venosa sistêmica superior. (C) Estrutura coronariana estática e apresentação tridimensional da posição posterior para a posição anterior. A seta branca identifica a estenose da deflexão venosa pulmonar com calcificação no enxerto de tereftalato de polietileno. Há também uma estenose grave associada na deflexão venosa sistêmica superior.
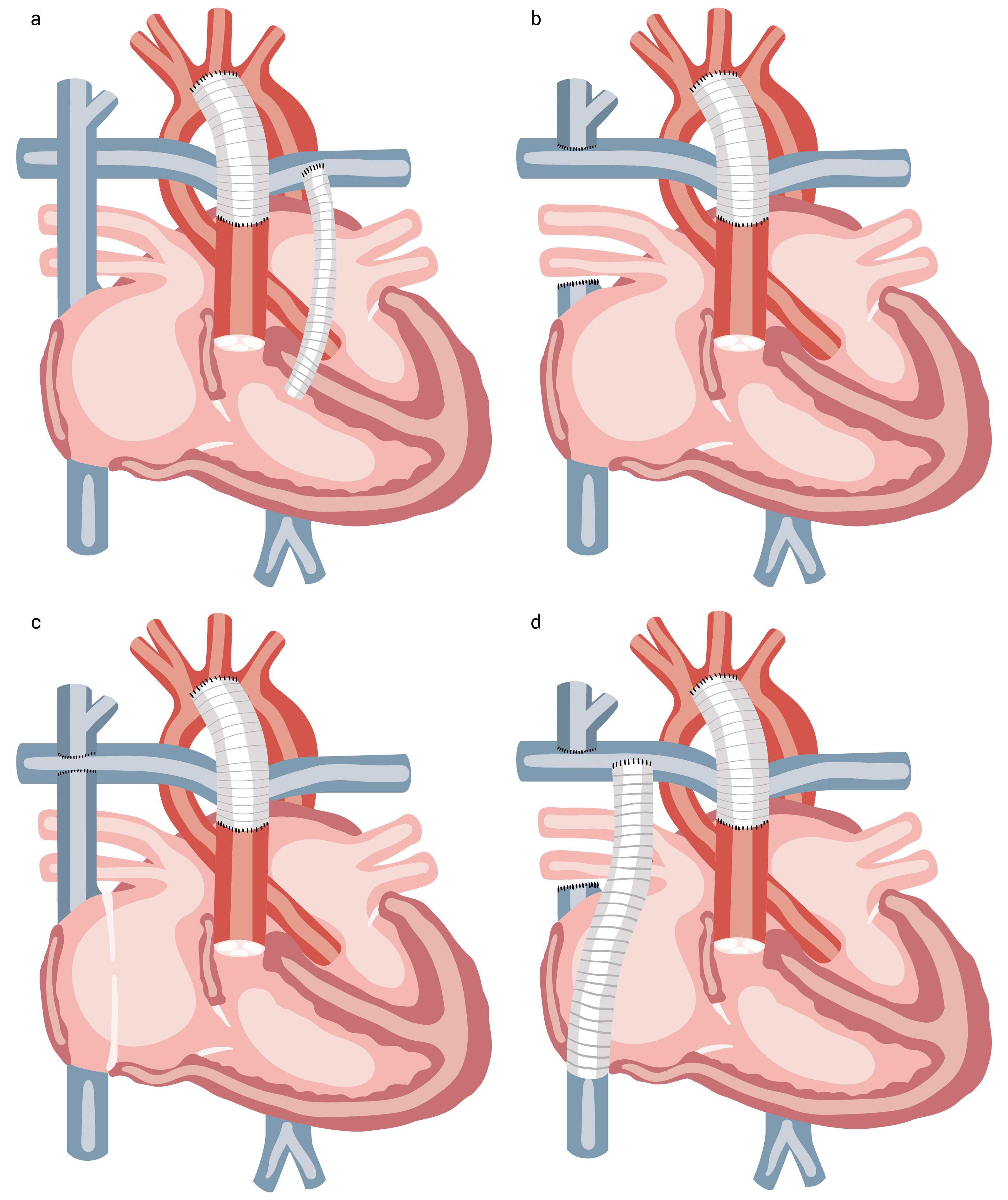
Paliação neonatal. Os procedimentos neonatais paliativos variam substancialmente; todos eles oferecem uma fonte estável de fluxo sanguíneo pulmonar, aliviam a obstrução para o fluxo externo sistêmico e garantem a mistura ao nível atrial.
A operação de Norwood para tratamento da síndrome do coração esquerdo hipoplásico é um exemplo usual. Logo após o nascimento, a aorta é reconstruída com a anastomose da aorta ascendente hipoplásica para a artéria pulmonar principal, aumentando-se o arco da aorta hipoplásica com adesivo.
A septectomia atrial permite colocar uma derivação na artéria pulmonar distal, entre a artéria sistêmica e a artéria pulmonar, para criar um fluxo sanguíneo pulmonar, normalmente uma derivação modificada de Blalock-Taussig ou a derivação de Sano, um conduto ligando o ventrículo direito às artérias pulmonares, como mostra a Figura 11.
Figura 11 - Estágios no reparo de ventrículos únicos funcionais (ver os detalhes no texto). (A) Procedimento de Norwood. (B) Anastomose cavopulmonar superior bidirecional (procedimento de Glenn). (C) Anastomose cavopulmonar total no túnel lateral. (D) Anastomose cavopulmonar total com conduto extracardíaco.
Procedimento de Glenn (anastomose cavopulmonar superior). O procedimento bilateral de Glenn é o segundo procedimento por estágios atualmente executado aos 4 a 6 meses de idade. Embora estabilize o fluxo sanguíneo pulmonar, o procedimento de Glenn diminui a carga volumétrica sobre o ventrículo único. Esse procedimento envolve anastomose da veia cava superior para a artéria pulmonar e inclui a remoção de alguma derivação previamente colocada e o reparo da estenose em alguma ramificação da artéria pulmonar.
O termo bidirecional se refere ao fato de que a artéria pulmonar direita permanece em continuidade com a artéria pulmonar esquerda. Isso diverge do procedimento clássico de Glenn, que envolve anastomose da veia cava superior para a artéria pulmonar direita que havia sido desconectada da artéria principal e da artéria pulmonar esquerda.
Procedimento de Fontan (anastomose cavopulmonar total). O procedimento de Fontan é o procedimento paliativo final que faz a conexão direta entre o fluxo proveniente da veia cava superior para o circuito pulmonar. Nas iterações originais do procedimento, ao invés de intervenções por estágios, costumava-se fazer uma operação que envolvia a ligação do átrio direito à artéria pulmonar ou ao trato do fluxo externo ventricular direito em crianças mais velhas.
A prática atual consiste em reconfigurar o fluxo sanguíneo pulmonar por estágios, conforme descrito anteriormente, seguido pelo direcionamento do fluxo da veia cava inferior diretamente para as artérias pulmonares no período entre 2 a 3 anos de idade (conclusão de Fontan). Direciona-se o fluxo da veia cava inferior para a artéria pulmonar através de um túnel lateral colocado ao longo do átrio direito, ou colocando-se um conduto extracardíaco conectando diretamente a veia cava inferior à artéria pulmonar.
Com qualquer uma dessas vias de fluxo, é possível fazer uma pequena comunicação (fenestração) entre o conduto cavopulmonar e o átrio esquerdo funcional para descomprimir o circuito de Fontan na eventualidade de uma elevação na pressão arterial venosa central/pulmonar.
O fluxo de sangue dos pulmões pode ser obtido através de retorno venoso passivo sem auxílio de câmara de bombeamento ventricular. Qualquer alteração leve ou resistência na pressão pulmonar poderá prejudicar a adequação do fluxo sanguíneo pulmonar. Portanto, a situação ideal é a eliminação da estenose em qualquer ramificação da artéria pulmonar e o gerenciamento consciente de qualquer doença pulmonar concomitante.
Apresentação Clínica Após o Reparo
As características clínicas são variáveis. O tratamento paliativo pode ser bom para alguns pacientes, com saturação quase normal de oxigênio e com níveis aceitáveis de atividade e descobertas inexpressivas no exame cardíaco. Outros pacientes apresentam IC progressiva, como ventrículo único (em especial, no contexto da síndrome do coração esquerdo hipoplásico), e não suportam pressão elevada e sobrecarga volumétrica secundária à regurgitação progressiva na válvula atrioventricular e disfunção miocárdica.46
Arritmias atrial e ventricular são comuns. Fluxos sanguíneos pulmonares lentos poderão predispor os pacientes a trombose e embolia pulmonar in situ, que, por sua vez, impede o fluxo sanguíneo pulmonar por meio da elevação da pressão na artéria pulmonar.
Gerenciamento Cirúrgico
Após a correção cirúrgica, os pacientes apresentam limitações significativas à tolerância aos exercícios, tendo em vista que dependem do fluxo passivo de sangue pulmonar que não chega a atingir o nível máximo com o esforço físico. As arritmias atriais são comuns; elas exigem gerenciamento cuidadoso com técnicas de ablação e de marcapasso para complementar a terapia médica, considerando-se que a sincronia atrioventricular é favorável.
As intervenções transcateter facilitam o tratamento de estenoses em ramificações da artéria pulmonar, fenestrações, colaterais venosos ou colaterais arteriovenosos. Recomenda-se evitar rigorosamente a introdução de ar nas linhas intravenosas, considerando que as fenestrações e os vasos colaterais poderão resultar em desvios da direita para a esquerda.
As complicações trombóticas são comuns e, portanto, recomenda-se aplicar o procedimento de anticoagulação, em especial no contexto de desvios residuais da direita para a esquerda, arritmias atriais e IC. A disfunção hepática é progressiva com o avanço da idade e é um dos marcadores mais confiáveis de IC nessa população.
O monitoramento laboratorial serial do soro e estudos de imagens são as técnicas recomendadas.47 Alguns pacientes mais velhos se beneficiam com a conversão do procedimento clássico de Fontan para anastomose total na artéria cavopulmonar. O transplante cardíaco poderá ser necessário nos casos de IC, embora o risco seja muito elevado.48
A contracepção é um componente importante do tratamento. Recomenda-se evitar o uso de contraceptivos orais, que aumentam o risco de trombose. De um modo geral, os dispositivos intrauterinos com progesterona são a modalidade preferida.
Anomalia de Ebstein na Válvula Tricúspide
Fisiopatologia
Essa anomalia consiste da aderência do folheto posterior e do folheto septal ao miocárdio, ocasionando um deslocamento descendente do anel funcional na direção do ápice do ventrículo direito, assim como um aumento no folheto anterior. O resultado é a atrialização de uma porção do ventrículo direito e da insuficiência tricúspide.
Em pacientes com nascimento precoce, a anomalia de Ebstein costuma ocorrer juntamente com outros defeitos, incluindo DSA e estenose pulmônica. As vias acessórias e as evidências clínicas de pré-excitação são frequentes. Arritmias são as características mais comuns em adultos.
A Figura 12 mostra o ECO de um paciente com anomalia de Ebstein.
aRV: ventrículo direito atrializado; ECO: ecocardiograma; LA: átrio esquerdo; LV: ventrículo esquerdo; RA: átrio direito; RV: ventrículo direito.
Figura 12 - ECO de um paciente com anomalia de Ebstein. (A) Visão apical das quatro câmaras. A seta indica o deslocamento apical do folheto tricúspide. (B) Imagem do fluxo com Doppler colorido mostrando regurgitação tricúspide (TR) grave, com origem nas profundezas do ventrículo direito a partir do folheto da válvula tricúspide deslocada.
Apresentação Clínica
A anomalia de Ebstein pode se tornar clinicamente evidente em qualquer idade. O histórico natural dessa lesão varia de morte no início da vida à sobrevida adulta sem cirurgia, dependendo do grau de regurgitação e da gravidade das lesões associadas. A cianose em neonatos ou adultos é uma ocorrência secundária ao desvio da direita para a esquerda ao nível atrial.
Geralmente, os pacientes adultos se queixam de fadiga, dificuldade para respirar, palpitações ou síncope. As arritmias são detectadas em quase 50% de pacientes.49,50 Nas auscultações, o sopro da regurgitação tricúspide é aparente e, com frequência, está associado a um ritmo galopante, sons de ejeções sistólicas múltiplas e um segundo som amplamente dividido.
Gerenciamento
A cirurgia é recomendada para pacientes com IC sintomática e cardiomegalia, cianose ou arritmias. A valvuloplastia tricúspide é preferida à reposição valvular. Não se recomenda fazer cirurgia em pacientes assintomáticos; no entanto, deve-se considerar a hipótese de intervenção nos casos de deterioração funcional antes do desenvolvimento de sintomas significativos.51 Pode ser necessário fazer avaliação eletrofisiológica e ablação das vias acessórias ou de arritmias atriais.52
Síndrome de Eisenmenger
O desenvolvimento de hipertensão arterial pulmonar grave e irreversível, conhecida por síndrome de Eisenmenger, é uma complicação séria de derivações permanentes da esquerda para a direita na aorta, nos ventrículos ou nas grandes artérias.
Fisiopatologia
Normalmente, a resistência vascular pulmonar é bem mais baixa que a resistência vascular sistêmica, o que facilita a ocorrência de desvios significativos da esquerda para a direita a partir de grandes comunicações intracardíacas ou entre grandes artérias. Na medida em que aumenta a resistência vascular pulmonar, a resistência ao fluxo na circulação pulmonar acaba por exceder a resistência ao fluxo na circulação sistêmica, levando a um desvio da direita para a esquerda.
Isso resultará em graus variados de cianose e de outras descobertas da hipertensão pulmonar. Ao contrário dos pacientes com policitemia vera ou policitemia ocasionada por doença pulmonar obstrutiva crônica, os pacientes com síndrome de Eisenmenger geralmente precisam de hematócritos aos 60 anos, ou mesmo abaixo dos 70 anos, para liberar oxigênio suficiente para os tecidos e evitar a ocorrência de sintomas isquêmicos.
Apresentação Clínica
Os pacientes com síndrome de Eisenmenger podem ser assintomáticos, exceto para cianose. Ao final, muitos pacientes percebem que há uma redução na tolerância aos exercícios e no desconforto torácico, geralmente uma reminiscência de angina. Nas situações em que a eritrocitose atingir níveis graves, os pacientes poderão desenvolver sintomas de hiperviscosidade, incluindo distúrbios visuais e cefaleia.
No exame físico, a síndrome de Eisenmenger revela a presença de manifestações de hipertensão pulmonar, incluindo um componente sonoro pulmonar alto no segundo som cardíaco e um sopro diastólico alto da regurgitação pulmonar (sopro de Graham Steell). As descobertas adicionais incluem cianose, hipocratismo digital e elevação ou levantamento ventricular direito.
Gerenciamento
Os pacientes com síndrome de Eisenmenger podem viver durante décadas após o diagnóstico.53 Alternativamente, talvez ocorram mortes súbitas causadas por arritmias ventriculares. Levando-se em consideração que nesses pacientes a resistência pulmonar é alta e fixa, é extremamente importante tomar o cuidado de evitar determinadas situações que possam perturbar o equilíbrio entre a resistência pulmonar e sistêmica, como, por exemplo, nas anestesias ou no uso de medicamentos vasodilatadores.
A gravidez é extremamente perigosa para mães com hipertensão pulmonar, assim como para os fetos; portanto, ela é absolutamente contraindicada na síndrome de Eisenmenger. A deficiência de ferro deve ser tratada em caso de necessidade. Raramente, é necessário usar a flebotomia para aliviar os sintomas de hiperviscosidade.
Medicamentos como prostaciclina, antagonistas da endotelina e inibidores da fosfodiesterase-5 foram utilizados para abaixar a resistência arteriolar pulmonar.54?56 Transplantes de coração-pulmão ou transplantes de pulmão foram feitos com sucesso em alguns pacientes com síndrome de Eisenmenger.57
Problemas de Saúde nas Mulheres
Contracepção
Apesar da alta eficácia contraceptiva, os anticoncepcionais contendo estrogênio aumentam o risco de trombose arterial e venosa. Portanto, os contraceptivos contendo somente progestina poderão ser usados em mulheres com preocupações trombóticas; as fórmulas de ação prolongada melhoram a eficácia.58 A seleção de métodos contraceptivos deve ser feita após consulta ao obstetra e ao cardiologista.
Risco da Gravidez para Mães com Doença Cardíaca Congênita
Levando-se em consideração o aumento na sobrevida das pessoas nascidas com doença cardíaca congênita, cresce cada vez mais o número de mulheres em idade concepcional com DCGs. As malformações congênitas são a forma mais comum de doença cardíaca em mulheres grávidas. As alterações fisiológicas durante a gravidez resultam no aumento gradual do volume de sangue e no débito cardíaco, assim como na redução da resistência vascular periférica, que se revertem relativamente rápido após o parto.
Esses ajustes podem ser pouco tolerados pelas mulheres com redução pré-existente na função cardíaca.59 As complicações cardíacas, incluindo arritmias e IC, ocorrem em 10% dos casos de gravidez e normalmente são transitórias e passíveis de gerenciamento médico.60 Algumas condições cardíacas têm alto risco de mortalidade materna ou de morbidade grave, conforme mostra o Quadro 2.58 Entretanto, a maior parte das mulheres com doença cardíaca congênita tolera bem a gravidez. O Quadro 3 contém indicações cardíacas para ECO fetal,61?63 e o Quadro 4, os recursos selecionados na Internet para doença cardíaca congênita.
Quadro 2
|
Condições de Alto Risco para Gravidez |
|
Hipertensão na artéria pulmonar por qualquer causa. Disfunção ventricular sistêmica definida como NYHA III-IV ou LVEF <30%. Cardiomiopatia periparto prévia com qualquer dano residual na função ventricular esquerda. Obstrução cardíaca esquerda grave. Síndrome de Marfan com dilatação da aorta >40mm. |
LVEF: fração de ejeção ventricular esquerda; NYHA: New York Heart Association.
Quadro 3
|
Indicações Cardíacas para Ecocardiograma Fetal |
|
Indicações fetais Bradicardia ou taquiarritmias Visão ou eixo cardíaco anormais nas quatro câmaras Indicações familiares Parente de primeiro grau com DCG (mãe, irmãos ou pai) Síndromes mendelianas (esclerose tuberosa, síndrome de Noonan, síndrome de DiGeorge |
DCG: defeito cardíaco congênito.
Quadro 4
|
Recursos Selecionados na Internet para Doença Cardíaca Congênita
|
|
Adult Congenital Heart Association Informações para médicos e pacientes Cove Point Foundation Educação de pacientes em inglês e espanhol Canadian Adult Congenital Heart Network (CACH) Informações para médicos e pacientes International Society for Adult Congenital Heart Disease (ISACHD) Recursos profissionais, informações para pacientes e newsletters The Somerville Foundation Site inglês com informações e suporte para pacientes e suas famílias |
Risco Para os Descendentes de Mães com Doença Cardíaca Congênita
As mulheres com doença cardíaca congênita poderão ser tratadas com medicamentos anti-hipertensivos ou anticoagulantes contraindicados na gravidez. Portanto, as orientações pré-concepção devem ser iniciadas durante a adolescência, de modo que as medicações possam ser ajustadas antes da concepção. As complicações neonatais ocorrem em 25% dos casos de gravidez.
As complicações mais comuns são nascimento prematuro e pequeno porte para a idade gestacional.59,60 As mães com doença cardíaca congênita provavelmente terão filhos com a doença. Esse risco aumenta nas mães com obstrução cardíaca esquerda ou com alguma etiologia genética conhecida, como a síndrome de deleção do 22Q11.
|
Recomenda-se o uso de ECO fetal nas situações em que um dos pais tiver doença cardíaca congênita, nos casos de gravidez em que tiverem sido identificadas outras anomalias ou por outras indicações apresentadas neste trabalho.61?63 O ideal é que os ECOs fetais sejam feitos entre a 18ª e a 22ª semanas de gestação. |
Referências
1. Marelli AJ, Mackie AS, Ionescu-Ittu R, et al. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 2007;115:163–72.
2. Khairy P, Ionescu-Ittu R, Mackie AS, et al. Changing mortality in congenital heart disease. J Am Coll Cardiol 2010;56:1149–57.
3. Marelli AJ, Ionescu-Ittu R, Mackie AS, et al. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation 2014;130:749–56.
4. Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults with Congenital Heart Disease). Circulation 2008;118:e714–833.
5. Rodriguez FH 3rd, Marelli AJ. The epidemiology of heart failure in adults with congenital heart disease. Heart Fail Clin 2014;10(1):1–7.
6. Dimopoulos K, Diller GP, Koltsida E, et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation 2008;117:2320–8.
7. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation 2014;129:e28-e292.
8. Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006;114:1645–53.
9. Driscoll DJ. Left-to-right shunt lesions. Pediatr Clin North Am 1999;46:355–68, x.
10. Inglessis I, Landzberg MJ. Interventional catheterization in adult congenital heart disease. Circulation 2007;115:1622–33.
11. Fischer G, Stieh J, Uebing A, et al. Experience with transcatheter closure of secundum atrial septal defects using the Amplatzer septal occluder: a single centre study in 236 consecutive patients. Heart 2003;89:199–204.
12. Komar M, Przewlocki T, Olszowska M, et al. The benefit of atrial septal defect closure in elderly patients. Clin Interv Aging 2014;9:1101–7.
13. Gatzoulis MA, Freeman MA, Siu SC, et al. Atrial arrhythmia after surgical closure of atrial septal defects in adults. N Engl J Med 1999;340:839–46.
14. Food and Drug Administration. Rare serious erosion events associated with St. Jude Amplatzer Atrial Septal Occluder (ASO). FDA Safety Communication 2013. Available at: http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm371145.htm (accessed Septem-ber 30, 2015).
15. Divekar A, Gaamangwe T, Shaikh N, et al. Cardiac perforation after device closure of atrial septal defects with the Amplatzer septal occluder. J Am Coll Cardiol 2005;45:1213–8.
16. Suzuki K, Yamaki S, Mimori S, et al. Pulmonary vascular disease in Down’s syndrome with complete atrioventricular septal defect. Am J Cardiol 2000;86:434–7.
17. Goyal VS, Fulwani MC, Ramakantan R, et al. Follow-up after coil closure of patent ductus arteriosus. Am J Cardiol 1999;83:463–6, A10.
18. Lewin MB, Otto CM. The bicuspid aortic valve: adverse outcomes from infancy to old age. Circulation 2005;111:832–4.
19. Campens L, Demulier L, De Groote K, et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am J Cardiol 2014;114:914–20.
20. Stanger P, Cassidy SC, Girod DA, et al. Balloon pulmonary valvuloplasty: results of the Valvuloplasty and Angioplasty of Congenital Anomalies Registry. Am J Cardiol 1990;65:775–83.
21. Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation 2007;115:1933–47.
22. Donti A, Spinardi L, Brighenti M, et al. Frequency of intracranial aneurysms determined by magnetic resonance angiography in children (mean age 16) having operative or endovascular treatment of coarctation of the aorta (mean age 3). Am J Cardiol 2015;116:630–3.
23. Forbes TJ, Kim DW, Du W, et al. Comparison of surgical, stent, and balloon angioplasty treatment of native coarctation of the aorta: an observational study by the CCISC (Congenital Cardiovascular Interventional Study Consortium). J Am Coll Cardiol 2011;58:2664–74.
24. Oliver JM, Gallego P, Gonzalez A, et al. Risk factors for aortic complications in adults with coarctation of the aorta. J Am Coll Cardiol 2004;44:1641–7.
25. Moss AJ, Allen HD. Moss and Adams’ heart disease in infants, children, and adolescents: including the fetus and young adult. 7th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008.
26. Ratliff HL, Yousufuddin M, Lieving WR, et al. Persistent left superior vena cava: case reports and clinical implications. Int J Cardiol 2006;113:242–6.
27. Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000;342:334–42.
28. Goldmuntz E, Clark BJ, Mitchell LE, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol 1998;32:492–8.
29. Henkens IR, van Straten A, Schalij MJ, et al. Predicting outcome of pulmonary valve replacement in adult tetralogy of Fallot patients. Ann Thorac Surg 2007;83:907–11.
30. McElhinney DB, Hellenbrand WE, Zahn EM, et al. Short- and medium-term outcomes after transcatheter pulmonary valve placement in the expanded multicenter US melody valve trial. Circulation 2010;122:507–16.
31. Cheatham JP, Hellenbrand WE, Zahn EM, et al. Clinical and hemodynamic outcomes up to 7 years after transcatheter pulmonary valve replacement in the US melody valve investigational device exemption trial. Circulation 2015;131:1960–70.
32. Niwa K, Siu SC, Webb GD, Gatzoulis MA. Progressive aortic root dilatation in adults late after repair of tetralogy of Fallot. Circulation 2002;106:1374–8.
33. Mongeon FP, Gurvitz MZ, Broberg CS, et al. Aortic root dilatation in adults with surgically repaired tetralogy of Fallot: a multicenter cross-sectional study. Circulation 2013;127:172–9.
34. Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study. Lancet 2000; 356:975–81.
35. Khairy P, Aboulhosn J, Gurvitz MZ, et al. Arrhythmia burden in adults with surgically repaired tetralogy of Fallot: a multi-institutional study. Circulation 2010;122:868–75.
36. Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:11–22.
37. Rashkind WJ, Miller WW. Creation of an atrial septal defect without thoracotomy. A palliative approach to complete transposition of the great arteries. JAMA 1966;196:991–2.
38. Dos L, Teruel L, Ferreira IJ, et al. Late outcome of Senning and Mustard procedures for correction of transposition of the great arteries. Heart 2005;91:652–6.
39. Warnes CA. Transposition of the great arteries. Circulation 2006;114:2699–709.
40. Aldoss O, Von Bergen N, Law I, Divekar A. Hemodynamic assessment with interventional support should be routine for primary elec-trophysiology procedures after atrial switch procedure. Congenit Heart Dis 2015;10:E83–8.
41. Wilson NJ, Clarkson PM, Barratt-Boyes BG, et al. Long-term outcome after the mustard repair for simple transposition of the great arteries. 28-year follow-up. J Am Coll Cardiol 1998;32:758–65.
42. Ruys TP, van der Bosch AE, Cuypers JA, et al. Long-term outcome and quality of life after arterial switch operation: a prospective study with a historical comparison. Congenit Heart Dis 2013;8:203–10.
43. Hutter PA, Kreb DL, Mantel SF, et al. Twenty-five years’ experience with the arterial switch operation. J Thorac Cardiovasc Surg 2002;124:790–7.
44. Kempny A, Wustmann K, Borgia F, et al. Outcome in adult patients after arterial switch operation for transposition of the great arteries. Int J Cardiol 2013;167:2588–93.
45. Khairy P, Poirier N, Mercier LA. Univentricular heart. Circulation 2007;115:800–12.
46. d’Udekem Y, Iyengar AJ, Galati JC, et al. Redefining expectations of long-term survival after the Fontan procedure: twenty-five years of follow-up from the entire population of Australia and New Zealand. Circulation 2014;130(11 Suppl 1):S32–8.
47. Assenza GE, Graham DA, Landzberg MJ, et al. MELD-XI score and cardiac mortality or transplantation in patients after Fontan surgery. Heart 2013;99:491–6.
48. Therrien J, Warnes C, Daliento L, et al. Canadian Cardiovascular Society Consensus Conference 2001 update: recommendations for the management of adults with congenital heart disease part III. Can J Cardiol 2001;17:1135–58.
49. Celermajer DS, Bull C, Till JA, et al. Ebstein’s anomaly: presentation and outcome from fetus to adult. J Am Coll Cardiol 1994;23:170–6.
50. Brown ML, Dearani JA, Danielson GK, et al. Functional status after operation for Ebstein anomaly: the Mayo Clinic experience. J Am Coll Cardiol 2008;52:460–6.
51. Attenhofer Jost CH, Connolly HM, Dearani JA, et al. Ebstein’s anomaly. Circulation 2007;115:277–85.
52. Sherwin ED, Triedman JK, Walsh EP. Update on interventional electrophysiology in congenital heart disease: evolving solutions for complex hearts. Circ Arrhythm Electrophysiol 2013;6:1032–40.
53. Oya H, Nagaya N, Uematsu M, et al. Poor prognosis and related factors in adults with Eisenmenger syndrome. Am Heart J 2002;143:739–44.
54. Mukhopadhyay S, Sharma M, Ramakrishnan S, et al. Phosphodiesterase-5 inhibitor in Eisenmenger syndrome: a preliminary observational study. Circulation 2006;114:1807–10.
55. Galie N, Beghetti M, Gatzoulis MA, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, ran-domized, placebo-controlled study. Circulation 2006;114:48–54.
56. Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation 2007;115:1039–50.
57. Franke UF, Wahlers T, Wittwer T, et al. Heart-lung transplantation is the method of choice in the treatment of patients with end-stage pulmonary hypertension. Transplant Proc 2002;34:2181–2.
58. Thorne S, MacGregor A, Nelson-Piercy C. Risks of contraception and pregnancy in heart disease. Heart 2006;92:1520–5.
59. Fernandes SM, Arendt KW, Landzberg MJ, et al. Pregnant women with congenital heart disease: cardiac, anesthetic and obstetrical im-plications. Expert Rev Cardiovasc Ther 2010;8:439–48.
60. Drenthen W, Boersma E, Balci A, et al. Predictors of pregnancy complications in women with congenital heart disease. Eur Heart J 2010;31:2124–32.
61. Small M, Copel JA. Indications for fetal echocardiography. Pediatr Cardiol 2004;25:210–22.
62. Rychik J, Ayres N, Cuneo B, et al. American Society of Echocardiography guidelines and standards for performance of the fetal echocar-diogram. J Am Soc Echocardiogr 2004;17:803–10.
63. AIUM practice guideline for the performance of fetal echocardiography. J Ultrasound Med 2011;30:127–36.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.