(Carregando Índice)... (Carregando Índice)... |
Última revisão: 11/09/2018
Comentários de assinantes: 0
Nathan T. Connell, MD, MPH
Instrutor de Medicina no Departamento de Medicina da Harvard Medical School. Médico Associado na Divisão de Hematologia do Brigham and Women’s Hospital (Boston, MA).
|
Artigo original: Connell, N. MD, MPH. Microangiopathic and Vascular Disorders, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon.
|
Resumo
As microangiopatias trombóticas (MATs) se caracterizam pela presença de anemia hemolítica microangiopática e trombocitopenia e se classificam em autoimunes, induzidas pelo uso de medicamentos, mediadas por complementos, infecciosas e outros tipos diversos. O diagnóstico definitivo desses distúrbios pode se transformar em um grande desafio por causa da semelhança dos sintomas e das descobertas laboratoriais. Os distúrbios específicos apresentados nesta revisão incluem púrpura trombocitopênica trombótica (PTT), síndrome hemolítico-urêmica (SHU), MATs da gravidez (incluindo pré-eclampsia e síndrome HELLP), coagulação intravascular disseminada (CID) e síndrome antifosfolipídica (SAF). Os distúrbios vasculares que produzem anormalidades hematológicas também serão discutidos nesta revisão. As figuras mostram as principais classificações das MATs; a atividade do gene ADAMTS13 no plasma normal e no plasma com PTT; eritrócito fragmentado (seta), também conhecido por esquistócito ou célula em capacete; considerações relevantes no tratamento inicial de PTT e opções para pacientes refratários, assim como considerações sobre o tratamento após a descontinuação da troca de plasma; e um diagrama da via do complemento mostrando as proteínas reguladoras, bem como o sítio de ação do eculizumab, um anticorpo monoclonal. Os quadros apresentam uma lista das medicações associadas à PTT, critérios diagnósticos para HELLP, classificações mais importantes e exemplos das causas de CID, critérios diagnósticos para a SAF, púrpuras vasculares e critérios diagnósticos para telangiectasia hemorrágica hereditária (THH).
As microangiopatias trombóticas (MATs) formam um grupo variado de distúrbios que se caracterizam pela presença de anemia hemolítica microangiopática e trombocitopenia. Os pacientes se apresentam com a doença em vários graus de gravidade, embora, com frequência, os sintomas e as anormalidades laboratoriais tornem o diagnóstico definitivo um grande desafio. Na realidade, o diagnóstico geralmente não é muito claro até que sejam feitas tentativas de múltiplas intervenções terapêuticas.
Os distúrbios discutidos nesta revisão incluem condições como púrpura trombocitopênica trombótica (PTT), síndrome hemolítico-urêmica (SHU), síndrome de pré-eclampsia/hemólise, níveis elevados de enzimas hepáticas e contagem baixa de plaquetas (HELLP), coagulação intravascular disseminada (CID) e distúrbios autoimunes, incluindo a síndrome antifosfolipídica (SAF). Esta revisão conclui com a apresentação de vários distúrbios vasculares que produzem anormalidades hematológicas.
Púrpura Trombocitopênica Trombótica
Sob a perspectiva clínica, a PTT e a SHU são distúrbios semelhantes que se caracterizam pela deposição generalizada de trombos de plaqueta-fibrina em artérias pequenas, arteríolas e vasos capilares. A patogênese subjacente é diferente, embora a MAT seja uma característica comum da PTT e da SHU. A PTT familiar (também conhecida por síndrome de Upshaw-Schulman) é uma condição rara, geralmente diagnosticada na infância ou na adolescência, embora haja relatos de ocorrência de alguns casos na vida adulta. Na maior parte dos casos, a PTT é idiopática ou secundária a uma grande variedade de condições.
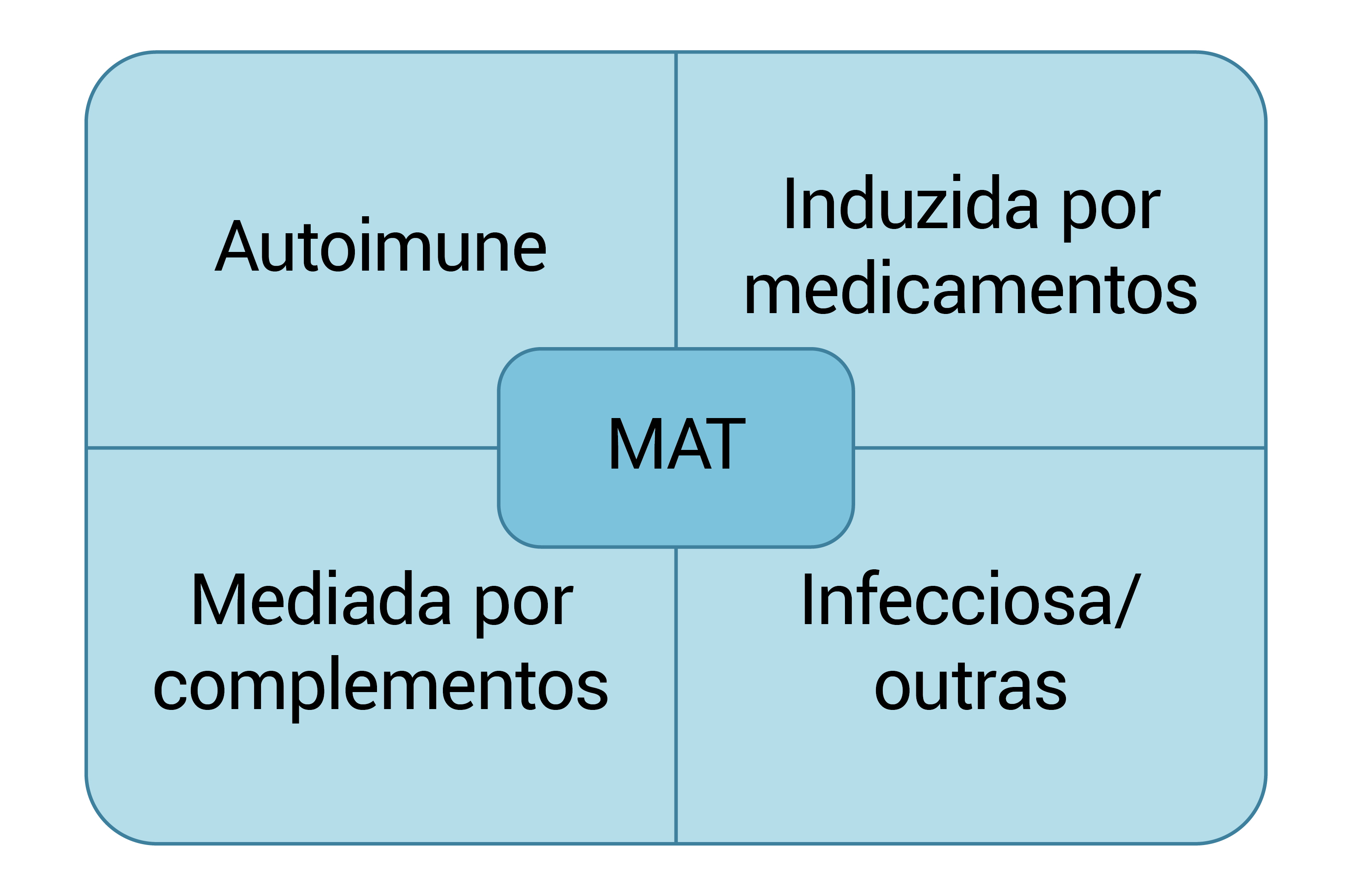
A Figura 1 mostra a classificação das MATs.

MAT: microangiopatia trombótica.
Figura 1 Classificações principais de MAT.
Etiologia
De um modo geral, a PTT ocorre de forma espontânea, embora possa estar associada a alguns fatores tais como gravidez, malignidades, transplantes de células-tronco hematopoiéticas, doenças autoimunes e medicamentos. Nas situações em que estiver associada à gravidez, a PTT geralmente ocorre no terceiro trimestre.
No período logo após a concepção, as manifestações no sistema nervoso central (SNC) podem ser inicialmente confundidas com depressão pós-parto, cujos resultados poderão ser trágicos. Existem relatos de casos depois de partos normais e com deslocamento da placenta e pré-eclampsia. Nos casos de transplante de células-tronco hematopoiéticas, as manifestações se limitam basicamente à MAT renal que, atualmente, é conhecida por MAT pós-transplante.
Aparentemente, diversos medicamentos causam PTT. Esses fármacos incluem medicamentos quimioterápicos (por exemplo, mitomicina C, bleomicina e cisplatina), agentes imunossupressivos (por exemplo, ciclosporina e tacrolimo), contraceptivos orais e quinino. Há casos anedóticos de PTT associada ao agente antiplaquetário clopidogrel em associação com a ticlopidina.1
Existem duas formas distintas de PTT induzida por medicamentos: uma delas tem início agudo, provavelmente mediado por algum mecanismo imune, conforme tipificado na PTT induzida por quinino, e a outra tem início mais gradual, talvez relacionado ao acúmulo da toxicidade farmacológica que afeta sobretudo o fígado. Os medicamentos quimioterápicos são exemplos típicos nesta última forma de PTT induzida por fármacos.
O Quadro 1 apresenta as medicações associadas à púrpura trombocitopênica trombótica.
Quadro 1
|
Medicações Associadas à Púrpura Trombocitopênica Trombótica |
|
Antibióticos Ampicilina Claritromicina Metronidazol Penicilina Rifampicina |
|
Agentes antiplaquetários Clopidogrel Defibrotida Dipiridamol Ticodipina |
|
Agentes quimioterápicos 5-Fluorouracil Cisplatina Citarabina Daunorubicina Gencitabina Hidroxiureia Mitomicina C |
|
Agentes imunossupressivos Ciclosporina Tacrolimo Sirolimo |
|
Hormônios Estrogênios conjugados Danazol Etinil estradiol Levonorgestrel Noretisterona |
O lúpus eritematoso sistêmico (LES) é um dos fatores de risco para o desenvolvimento de anticorpos para o gene ADAMTS13 e poderá ocorrer na presença ou na ausência de anticorpos antifosfolipídicos.2,3 A PTT que ocorrer de forma espontânea, na ausência dessas categorias clínicas, é considerada idiopática e representa, aproximadamente, 50% dos casos no registro do Oklahoma-TTP-HUS.4
PATOGÊNESE
O processamento inapropriado dos multímeros do fator de von Willebrand (vWF) é extremamente importante na patogênese da PTT. O vWF é uma proteína abundante no plasma que faz a mediação da adesão de plaquetas no subendotélio e age como molécula transportadora para o fator VIII. O fator de von Willebrand é sintetizado por megacariócitos e células endoteliais.
Os monômeros do vWF (280.000 Da) se ligam por meio de ligações de dissulfeto para formar os multímeros do fator de von Willebrand, que são liberados na circulação pelas células endoteliais e armazenados no interior dos grânulos-a das plaquetas e nos corpos de Weibel-Palade nas células endoteliais. Os multímeros do vWF armazenados são liberados por meio de estimulação plaquetária ou endotelial.
Os multímeros do vWF liberados são maiores que os multímeros do vWF no plasma, e são conhecidos como multímeros ultra grandes do vWF (ULvWF), cujo tamanho molecular atinge até 20 milhões de daltons. Sob o ponto de vista funcional, esses são os multímeros mais reativos do vWF.
Em 1982, foram encontrados multímeros do vWF no plasma de pacientes com recidiva crônica de PTT, dando origem à hipótese de que a PTT pode ser o resultado da deficiência de protease de clivagem do vWF (depolimerase) que, por sua vez, estimula a circulação dos multímeros ULvWF, contribuindo para o desenvolvimento de trombose.5 Essa hipótese foi comprovada com a identificação da protease de clivagem do vWF e com a demonstração da associação da deficiência da atividade da protease de clivagem do vWF com PTT.
A protease de clivagem do vWF foi identificada como o gene ADAMTS13 (semelhante à desintegrina e à metaloproteinase com trombospondina tipo motif 13).6 Trata-se de uma nova metaloproteinase que faz a clivagem do monômero do vWF em sítios específicos (842Tyr-843Met) no domínio A2. A secreção dos multímeros do ULvWF é feita a partir de células endoteliais estimuladas como uma “cadeia” longa que se prende na superfície das células endoteliais.
Em condições de fluxo no sangue, o gene ADAMTS13 plasmático poderá aderir aos multímeros do ULvWF ligados na superfície celular (através do domínio A3 no monômero do vWF) e fazer a clivagem nos multímeros do ULvWF que são encontrados normalmente no plasma.7 O desdobramento parcial dos multímeros do ULvWF através do estresse por cisalhamento melhora a clivagem enzimática.
Os pacientes com PTT familiar têm deficiência hereditária de ADAMTS13,8 enquanto que muitos pacientes com PTT idiopática possuem anticorpos que inibem a atividade de ADAMTS13.9,10 Em qualquer um desses casos, a presença de multímeros do ULvWF na superfície estimulada das células endoteliais leva à adesão e à subsequente agregação de plaquetas que, por sua vez, resulta na formação de trombos plaquetários (presumivelmente, as plaquetas não se ligam aos multímeros menores do vWF no plasma, tendo em vista que esses sítios de ligação de plaquetas não são expostos a esses múltimeros).
Além de produzirem lesões isquêmicas no sítio de formação de trombos, provavelmente os trombos plaquetários resultantes da agregação de múltimeros do vWF irão se decompor e embolizar no sentido do fluxo, resultando em danos teciduais isquêmicos adicionais.
O gene que codifica ADAMTS13 se localiza no cromossomo 9q34. Mais de 50 mutações nesse gene foram identificadas em pacientes com PTT familiar, sendo que muitas delas resultam na secreção reduzida in vitro de ADAMTS13.6 Na maioria dos casos de PTT adquirida, a produção de autoanticorpos IgG contra o ADAMTS13 resulta na deficiência grave da atividade de ADAMTS13.
Nos casos de PTT familiar, é possível detectar os multímeros do ULvWF no plasma na linha de base que, com frequência, desaparecem durante os episódios agudos, enquanto que na maior parte dos pacientes com PTT adquirida é comum a presença de multímeros do ULvWF nos episódios agudos, embora isso não ocorra após a recuperação.5
Diagnóstico
Manifestações Clínicas
A descrição clássica de PTT envolve a ocorrência de cinco fatores: anemia hemolítica microangiopática, trombocitopenia, febre, alterações neurológicas e insuficiência renal. A anemia hemolítica microangiopática está associada à elevação nos níveis séricos da lactato desidrogenase (LDH) e a esfregaços de sangue que mostram a presença típica de esquizócitos e de células em forma de capacete.
No início, as manifestações no SNC podem ser leves, evidenciadas por agitação transitória, cefaleia e desorientação, embora os sinais possam evoluir para hemiparesia, afasia, convulsões, déficits focais e coma. Normalmente, a disfunção renal é branda e produz elevações moderadas nos níveis de creatinina sérica e de proteínas urinárias.
A maior parte dos pacientes não apresenta todos esses cinco fatores e, nos dias atuais, o consenso geral sobre a definição mais apropriada de PTT é a presença de anemia hemolítica microangiopática e trombocitopenia sem uma causa alternativa.11,12 As características remanescentes dos cinco fatores não são necessárias para o diagnóstico e representam eventos secundários no curso da doença. De um modo geral, os pacientes com PTT familiar (atribuível à deficiência congênita de ADAMTS13) se apresentam na infância e têm um curso recorrente crônico.
Exame Físico
O exame físico provavelmente mostre sequelas da anemia e trombocitopenia. Os pacientes talvez tenham taquicardia, e a pele poderá apresentar evidências de petéquias.
Testes laboratoriais
O hemograma completo mostra uma combinação de anemia com trombocitopenia e, com frequência, a contagem de plaquetas tem apenas um dígito. Levando-se em consideração que a anemia é ocasionada por um processo hemolítico microangiopático, a quantidade total de bilirrubina é elevada, sendo que maior parte é de bilirrubina indireta.
O nível da LDH é excessivamente elevado por causa da anemia hemolítica e da isquemia tecidual decorrentes da trombose microvascular. A análise dos esfregaços de sangue periférico mostra a presença de esquizócitos que representa mais de 1% dos eritrócitos. Provavelmente haja eritrócitos nucleados de acordo com a gravidade da doença.
O número de plaquetas é bastante reduzido nos esfregaços de sangue periférico. O teste direto da antiglobulina (TDA ou teste direto de Coombs) é negativo, e o tempo de protrombina (TP) e o tempo de tromboplastina parcial ativado (TPPa) são normais, excetuando-se os casos em estágio final em que há falência múltipla de órgãos. A medição da atividade de ADAMTS13 deve ser feita em amostras de troca de pré-plasma, embora, tradicionalmente, tempos prolongados de resposta impedem que essas fontes de informações tenham alguma utilidade nas decisões clínicas.
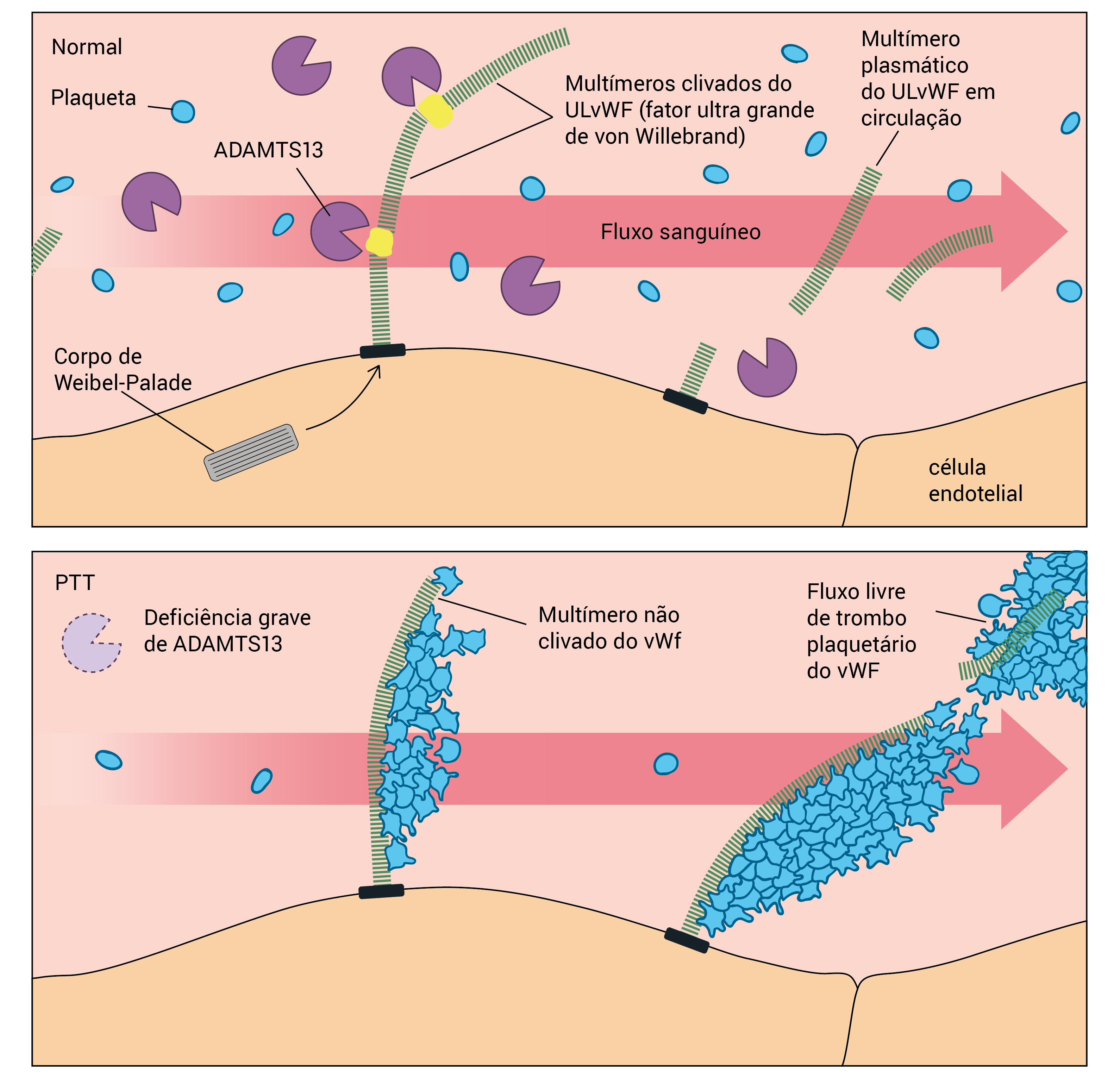
A Figura 2 mostra a atividade de ADAMTS13 no plasma normal e na PTT.
PTT: púrpura trombocitopênica trombótica.
Figura 2 - Atividade de ADAMTS13 no plasma normal e na PTT. (A) Em pessoas normais, as moléculas da enzima ADAMTS13 do plasma aderem e fazem a clivagem dos multímeros dos fatores ultra grandes do fator de von Willebrand (ULvWF), cuja secreção é feita através de cadeias longas a partir de células endoteliais estimuladas. (B) Em pacientes com PTT, a deficiência de ADAMTS13 impede a clivagem dos multímeros do ULvWF produzidos pelas células endoteliais. As plaquetas transportadas pelo fluxo de sangue aderem aos multímeros do ULvWF que não sofreram clivagem, resultando no desenvolvimento de trombos plaquetários.
Ainda que a deficiência grave de ADAMTS13 (<5%) seja específica de PTT, os pacientes com esse nível de deficiência, provavelmente, tenham períodos assintomáticos prolongados.13 Está se tornando cada vez mais evidente que a perda de atividade de ADAMTS13, com aumento associado nos multímeros do ULvWF em circulação, é necessária, porém insuficiente, para produzir episódios clínicos agudos de PTT.
Os dados mais recentes dão suporte à hipótese de deficiência grave de ADAMTS13, familiar ou adquirida, que predispõe o paciente para trombose, sendo que um segundo estímulo inflamatório vascular, como infecção, cirurgia ou gravidez, poderá aumentar a liberação do estoque de multímeros do ULvWF pelo endotélio.
O quadro de processamento excessivamente danificado aumenta os trombos plaquetários do ULvWF na microcirculação e eleva o risco de trombose clínica. As síndromes de anemia hemolítica microangiopática secundária, como aquelas que ocorrem em associação com o transplante de células-tronco hematopoiéticas ou com uso de ciclosporina, não estão associadas à deficiência grave de ADAMTS13.
Diagnóstico Diferencial
O diagnóstico diferencial para trombocitopenia com um único dígito inclui outros distúrbios microangiopáticos tais como pré-eclampsia/HELLP, CID e SAF. Existem também síndromes relacionadas à gravidez, como a síndrome de pré-eclampsia grave e HELLP, que podem ser distinguidas de PTT. Tanto a PTT como a SHU podem ser diferenciadas da síndrome de Evans através da análise de esfregaços de sangue periférico.
Fatores como hemólise microangiopática, leucocitose neutrofílica e teste direto de Coombs negativo (TDA) sugerem fortemente a presença de PTT ou SHU. Normalmente, os testes de coagulação não revelam a presença de anormalidades significativas (isto é, nenhuma evidência de CID); em geral, o nível sérico da LDH é elevado. Não é necessário fazer biópsia da medula; porém, caso seja feita para avaliação de citopenias, ela pode revelar a presença de trombos típicos de plaqueta-fibrina hialina, porém não patognomônicos, em artérias pequenas e arteríolas.
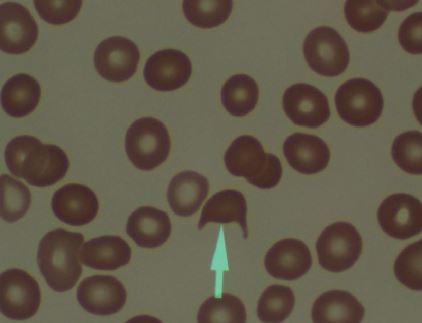
A Figura 3 mostra o eritrócito fragmentado.
Figura 3 - Eritrócito fragmentado (seta) também conhecido por esquistócito ou célula em forma de capacete.
Gerenciamento
A PTT é uma emergência hematológica e seu tratamento exige início imediato de troca terapêutica de plasma.14 Esse tipo de procedimento é executado diariamente até a recuperação na contagem de plaquetas e a normalização, ou quase, dos parâmetros da hemólise. A mera suspeita de que algum autoanticorpo seja a causa subjacente da apresentação da PTT é suficiente para iniciar a administração de corticosteroides para diminuir a produção de anticorpos.
O início imediato da terapia à base de rituximabe é uma grande promessa para evitar a recorrência de PTT e uma opção a ser considerada para aplicação em todos os pacientes que não conseguirem remissão da doença com troca de plasma e corticosteroides.15
Em um teste randomizado de grande porte realizado pelo Canadian Apheresis Group, a troca intensiva de plasma foi mais eficaz que a infusão de plasma em termos de sobrevida dos pacientes (78 versus 63%).16 Em geral, os pacientes clinicamente estáveis com trombocitopenia moderada e nenhum dano neurológico significativo iniciam com troca diária de um volume único.
Entretanto, nos casos em que a situação clínica se agravar, a recomendação é fazer trocas de plasma mais intensivas com volume duplo (5.000 a 6.000mL/dia ou, aproximadamente, 80mL/kg/dia). Levando-se em consideração que os multímeros do vWF se precipitam por congelamento, o crio-sobrenadante (isto é, plasma fresco congelado do qual se removeu o crioprecipitado) poderá ser substituído como líquido de reposição se o paciente não estiver respondendo à troca de plasma rotineira.
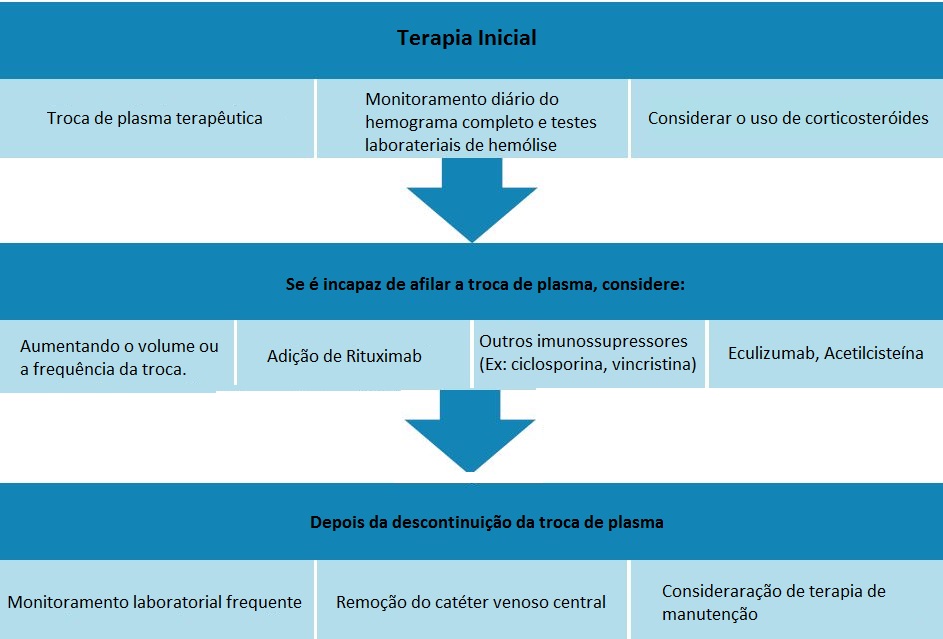
A Figura 4 apresenta considerações importantes sobre o tratamento inicial de PTT e opções para pacientes refratários.
CBC: hemograma completo; PTT: púrpura trombocitopênica trombótica.
Figura 4 - Considerações importantes sobre o tratamento inicial de PTT e opções para pacientes refratários, assim como considerações sobre o tratamento após a descontinuação da troca de plasma.
Um estudo não controlado mostrou que houve aumento nos benefícios com essa preparação em comparação com plasma fresco congelado.17 Após o benefício terapêutico (medido pela recuperação da função normal do SNC, elevando-se as contagens de plaquetas e abaixando-se os níveis de LDH), a intensidade e a frequência da troca de plasma podem ser reduzidas para trocas de volume único.
Em geral, define-se resposta como a recuperação na contagem normal de plaquetas (que é o parâmetro mais comum) e a remissão como contagem normal de plaquetas por 30 dias após a interrupção na troca de plasma. Cabe observar que o número de tratamentos com trocas de plasma necessário para atingir a remissão é extremamente variável, variando de 3 a 89.18
Embora se reconheça que a troca de plasma imediata seja importante, o uso de corticosteroides ou de outras terapias, como ácido acetilsalicílico ou dipiridamol, ainda não chegou a ser avaliado em testes prospectivos.19 Com fundamento na observação de que os autoanticorpos contra o ADAMTS13 sejam causas significativas de PTT idiopática adquirida, fez-se um teste com rituximabe (375mg/m2, IV, 1x/semana, durante 4 semanas) cujo resultado mostrou que se trata de um medicamento eficaz.20?22
Até o presente momento, existem relatos de, aproximadamente, 75 pacientes com PTT idiopática recorrente e com deficiência grave de ADAMTS13; a grande maioria desses pacientes atingiu remissão completa em algumas semanas com o tratamento com rituximabe; o índice de recidiva foi baixo, embora o tempo de acompanhamento tenha sido muito curto.
De um modo geral, o rituximabe é bem tolerado, embora seja importante que os pacientes se conscientizem de seu potencial para reativar infecções virais latentes, incluindo hepatite B, citomegalovírus e vírus varicela-zóster.23 A tendência da aférese de abaixar a contagem de plaquetas em pacientes que já sejam trombocitopênicos levanta a questão sobre a transfusão de plaquetas.
Alguns investigadores observaram que a infusão de plaquetas poderá exacerbar a PTT, enquanto que outros especialistas utilizam a transfusão de plaquetas conforme a necessidade.24 Os dados mais recentes sugerem que os pacientes com PTT que recebem transfusão de plaquetas correm um risco maior de incidência de trombose arterial e de morte, ainda que não haja aumento no risco de trombose venosa.25
Complicações
A troca de plasma é intensa em recursos e tem suas próprias morbidades, tais como risco infeccioso e morte. Uma revisão de dados feita pelo Oklahoma TTP Registry mostrou que, em um período de 3 anos, 17% dos pacientes que fizeram troca de plasma para PTT tiveram complicações atribuídas diretamente ao processo de troca, em comparação com a enfermidade subjacente.26 Essas complicações incluíam problemas produzidos pela colocação da linha, infecções e exposição ao plasma sanguíneo do doador.
Prognóstico
O início imediato da troca de plasma reduz o índice de mortalidade por PTT de, aproximadamente, 90% para algo em torno de 15%. O início em da troca de plasma em tempo hábil reduziu a mortalidade em pacientes com PTT idiopática, com ou sem deficiência grave de ADAMTS13, para 15 a 20%, ao passo que a mortalidade em pacientes com MAT foi muito mais elevada, ou seja, 55 a 60%.27,28
Muitos pacientes do último grupo não apresentaram nenhuma doença e comorbidades subjacentes sérias, como, por exemplo, transplante de células-tronco hematopoiéticas, que provavelmente tenham contribuído para a alta taxa de mortalidade. Os níveis de ADAMTS13 no momento do diagnóstico, aparentemente, carregam informações prognósticas importantes.12,28,29
Anticorpos anti-ADAMTS13 com titulação alta talvez estejam associados a doenças mais refratárias e a índices mais elevados de mortalidade.30 A razão pela qual não é possível detectar um inibidor em todos os casos de deficiência grave de ADAMTS13 ainda não está muito clara. Possivelmente, as várias técnicas de ensaio não sejam suficientemente sensíveis; como alternativa, talvez isso possa envolver algum anticorpo não neutralizante que se ligue ao ADAMTS13 e acelere sua eliminação.31,32
Síndromes Urêmico-Hemolíticas
Epidemiologia
A SHU se apresenta de uma forma semelhante à PTT. Existem duas formas de apresentação: SHU clássica e SHU atípica, também conhecida por SHU mediada pelo complemento. As duas formas de SHU compartilham características clínicas e descobertas laboratoriais semelhantes, embora existam diferenças específicas que facilitam o diagnóstico.
Etiologia e Genética
Um estudo recente feito na França avaliou a epidemiologia da SHU atípica e chegou à conclusão que, embora se acreditasse anteriormente que sua apresentação inicial ocorresse na infância, na realidade, a maior parte dos casos (58%) se apresenta na vida adulta. A mortalidade foi mais elevada em crianças.33 A desregulação da via do complemento provavelmente seja resultado de alguma mutação genética em uma entre as várias proteínas reguladoras, como o fator H,CD46 (anteriormente conhecido por proteína cofator de membrana), fator I,C3, trombomodulina ou fator B.
Patogênese
De um modo geral, a SHU clássica é o resultado de alguma infecção causada pelo organismo Escherichia coli e da liberação subsequente de uma toxina que leva à disfunção endotelial e à liberação de citocinas. Os rins são particularmente suscetíveis porque as células endoteliais renais expressam o glicolipídio Gb3, que tem preferência pela ligação à toxina em circulação.
O organismo E. coli 0157:H7, ou outras cepas, elabora verotoxinas (também conhecidas por toxinas de Shiga) que se ligam a receptores específicos na superfície endotelial, provocando danos celulares e mesmo a morte de células.27 A verotoxina-1 (VT-1) induz a suprarregulação de várias moléculas adesivas pró-trombóticas e pró-inflamatórias nas células endoteliais.32
As células endoteliais microvasculares são particularmente suscetíveis porque expressam níveis elevados de receptores VT-1, o que talvez explique a propensão para trombose na microcirculação. O tratamento de crianças com infecção por E. coli 0157:H7 com antibióticos aumenta ao invés de diminuir o risco de SHU, possivelmente porque libera as verotoxinas das bactérias intestinais afetadas, disponibilizando as toxinas para absorção. Logo, não se recomenda o tratamento de rotina com antibióticos.34
Nos casos de SHU atípica, os pacientes apresentam desregulação da via do complemento, levando à formação do complexo de ataque à membrana (MAC). Por conseguinte, ocorrem danos endoteliais subsequentes, ativação do sistema de coagulação com formação de microtrombos e desenvolvimento de anemia microangiopática com trombocitopenia.
Provavelmente, essa desregulação do complemento se deva a alterações ou a deficiências funcionais em qualquer uma das proteínas reguladoras do complemento. Como alternativa, em situações raras, poderá ocorrer formação de autoanticorpos contra essas proteínas na mesma síndrome clínica.
Diagnóstico
Manifestações Clínicas
A SHU em adultos tem características semelhantes às da PTT, embora a fisiopatologia não seja idêntica. As características comuns entre PTT e SHU incluem anemia hemolítica microangiopática, trombocitopenia e presença de trombos de fibrina plaquetária nos vasos menores. Da mesma forma que a PTT familiar, a SHU atípica (atribuível à deficiência congênita de proteínas reguladoras do complemento) geralmente se apresenta na infância e tem recidivas frequentes ao longo da vida.35
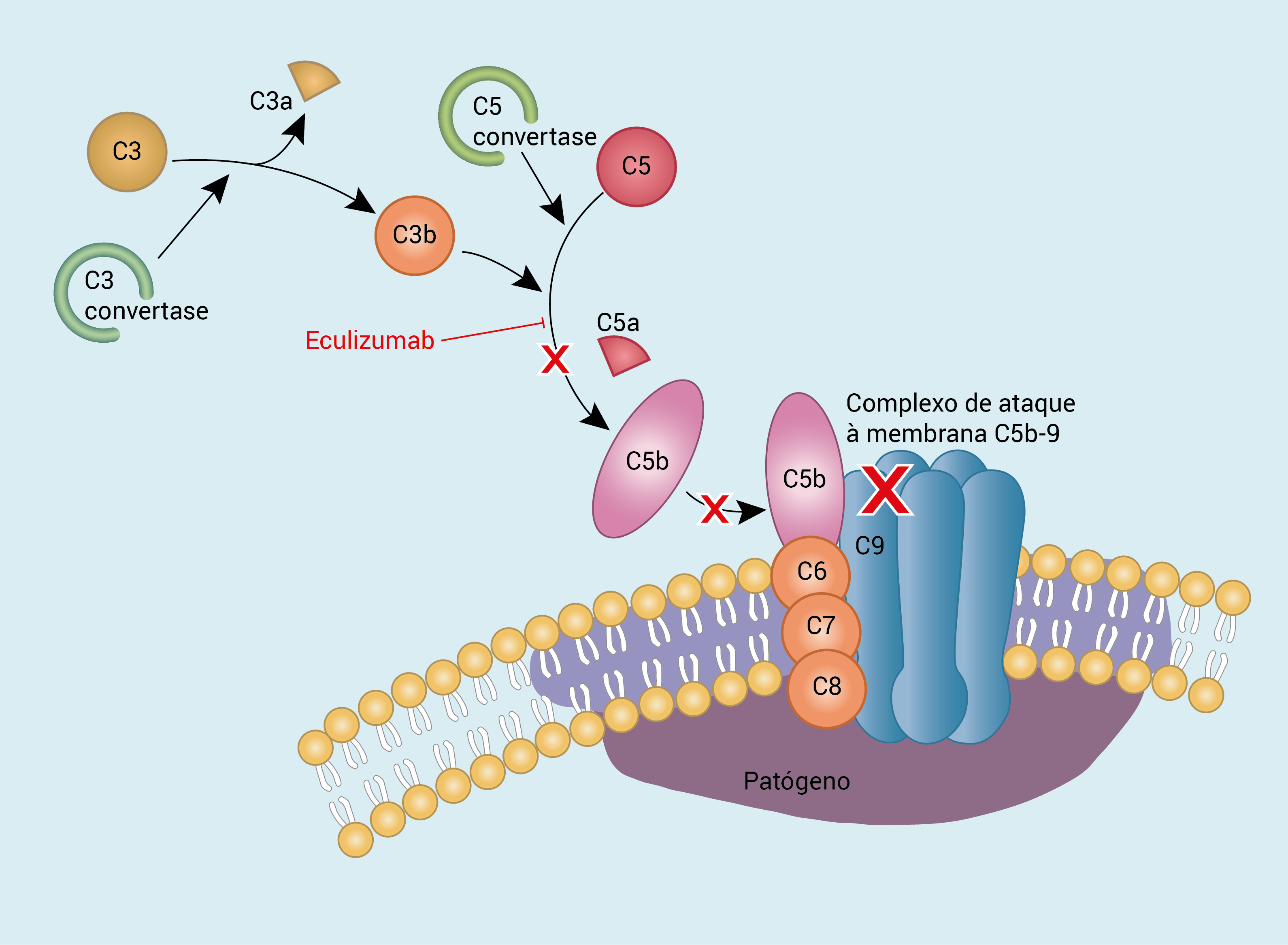
A Figura 5 apresenta o diagrama da vida do complemento mostrando as proteínas reguladoras e o sítio de ação do anticorpo monoclonal eculizumab.
Figura 5 - Diagrama da via do complemento mostrando as proteínas reguladoras e o sítio de ação do anticorpo monoclonal eculizumab.
Exame Físico
As descobertas do exame físico se assemelham às de outras MATs, incluindo equimoses e petéquias. As alterações neurológicas podem ser proeminentes nos casos em que os pacientes tiverem disfunção renal grave e uremia.
Diagnóstico Diferencial
A PTT é o diagnóstico alternativo mais importante a ser considerado no contexto de SHU, embora outras MATs possam ser levadas em consideração dependendo do contexto clínico. Os pacientes com SHU atípica geralmente apresentam um componente maior de disfunção renal e têm menor probabilidade de apresentar manifestações no SNC, ainda que esses critérios não possam ser usados isoladamente para fazer o diagnóstico.
Gerenciamento
A maior parte dos especialistas trata os adultos com SHU da mesma forma que os pacientes adultos com PTT. Entretanto, a resposta à troca de plasma talvez seja menos favorável nos casos de SHU, em comparação com os casos de PTT, o que é consistente com o fato de que o nível de ADAMTS13 geralmente não é baixo na SHU. O eculizumab, um anticorpo monoclonal contra C5 que impede a formação de C5MAC, foi aprovado para tratamento de SHU atípica.
Complicações
É importante observar que o eculizumab foi associado ao aumento no risco de infecções por Neisseria meningitis com risco de vida. Os pacientes que recebem eculizumab devem ser vacinados contra meningococos, preferencialmente 2 semanas antes do início da terapia. Caso isso não seja possível, os pacientes devem receber antibióticos profiláticos com atividade antimeningocócica.
Prognóstico
Alguns dados retrospectivos sugerem que a mortalidade durante os eventos iniciais poderá chegar a 8 a 10%, sendo que, aproximadamente, 25% dos pacientes se tornam dependentes de hemodiálise.36 A recuperação renal pode ser prolongada, e talvez os pacientes não saiam da hemodiálise por algumas semanas ou alguns meses.
Aproximadamente, 30% dos pacientes com mutações no fator H do complemento morreram durante o primeiro episódio antes de iniciar a terapia à base de eculizumab, ao passo que 22% apresentaram disfunção renal e tiveram que fazer hemodiálise.37 Dois testes menores que avaliaram o uso de eculizumab mostraram que houve melhoras na doença renal crônica e chegaram à conclusão que até 80% dos pacientes tenham condições de sair da diálise em 26 semanas.38
Microangiopatias Trombóticas da Gravidez
Epidemiologia
A trombocitopenia é uma anormalidade hematológica comum que ocorre durante a gravidez. Embora a trombocitopenia gestacional leve, normalmente na faixa de 100 a 150 x 109/L e raramente abaixo de 70 x 109/L, ocorra em 5 a 8% de mulheres grávidas (trombocitopenia gestacional), a trombocitopenia mais grave deve ser diferenciada de PTT, PTT associada à gravidez e pré-eclampsia/HELLP.
A síndrome HELLP se refere a um distúrbio que ocorre durante a gravidez e se caracteriza pela presença de hemólise, nível elevado de enzimas hepáticas e contagem baixa de plaquetas. É provável que essa síndrome represente uma forma bastante grave de eclampsia, ainda que existam alguns casos em que não ocorre hipertensão ou proteinúria prévia. Metade dos dados da síndrome HELLP ocorre em mulheres multíparas.
Patogênese
Ainda não está suficientemente clara a patogênese que associa insuficiência placentária às anormalidades hematológicas resultantes. Alguns dados sugerem a existência de uma associação com desregulação do complemento, mesmo que a confirmação desse fato dependa da realização de mais pesquisas.39
Diagnóstico
Manifestações Clínicas
As manifestações clínicas da síndrome HELLP, tais como fadiga e hipertensão, são inespecíficas, embora possam resultar em anormalidades graves, em alterações no estado mental e em convulsões.
Testes Laboratoriais
Em algum ponto entre a 23ª e a 30ª semanas de gravidez, as pacientes afetadas se apresentam com trombocitopenia marcada por contagens de plaquetas inferiores a 100.000/µL, hemólise microangiopática e testes anormais da função hepática. Os resultados dos testes padrões para CID são normais, embora possam ocorrer elevações no nível de dímero-D. O nível de ADAMTS13 é normal.
Embora não exista nenhum critério padrão para o diagnóstico definitivo da síndrome HELLP, há duas estratégias de classificação. A classificação de Tennessee exige evidências de anemia hemolítica microangiopática, contagem de plaquetas inferior a 100 x 109/L, bilirrubina total igual ou superior a 12mg/dL e nível sérico de aspartato-aminotransferase (AST) igual ou acima de 70 IU/L.40
A classificação de Mississipi é uma abordagem alternativa e exige a presença de anemia com evidências de hemólise; LDH acima de 600IU/L; presença de AST superior a 40IU/L e alanina aminostranferase acima de 40IU/L, ou ambos; e trombocitopenia definida como nadir plaquetário inferior a 150 x 109/L.41
Diagnóstico Diferencial
O diagnóstico diferencial inclui gordura hepática aguda da gravidez e PTT/SHU. De um modo geral, nos casos de gordura hepática aguda da gravidez, as pacientes apresentam TP prolongado e TTPa, juntamente com níveis baixos de fibrinogênio; hipertensão e proteinúria geralmente estão ausentes. Nos casos de PTT/SHU, as enzimas hepáticas são normais ou apenas ligeiramente elevadas. As outras causas de trombocitopenia na gravidez não estão associadas a alterações microangiopáticas.
Gerenciamento
A síndrome HELLP é tratada após o término da gravidez, em geral depois do parto, através de cuidados meticulosos de suporte. Em uma grande série de pacientes com a síndrome HELLP, o nadir de trombocitopenia ocorreu em 1 a 2 dias após o parto.42 O nadir poderá também desenvolver pela primeira vez em 24 a 48 horas pós-parto.40 Embora ainda haja muitas controvérsias sobre o tratamento da síndrome HELLP, o uso de corticosteroides aparentemente produz alguns benefícios.43
O Quadro 2 mostra a comparação entre esses dois critérios diagnósticos.
Quadro 2
|
Critérios Diagnósticos para Hemólise, Enzimas Hepáticas Elevadas e Plaquetas Baixas | ||
|
|
Tennessee |
Mississipi |
|
Anemia |
Anemia hemolítica microangiopática com esquistócitos.
|
Anemia progressiva com LDH elevado. |
|
Enzimas hepáticas |
Bilirrubina total =1,2mg/dL; nível sérico de AST =70 IU/L. |
LDH >600IU/L; AST >40IU/L; ALT >40IU/L, ou ambos.
|
|
Plaquetas |
= 100 x 109/L |
<150 x 109/L; HELLP classe I: <50 x 109/L; HELLP classe II: <100 x 109/L; HELLP classe III: <150 x 109/L.
|
ALT: alanina aminotransferase; AST: aspartato-aminotransferase; HELLP: hemólise, enzimas hepáticas elevadas e plaquetas baixas; LDH: lactato desidrogenase.
Complicações
Com frequência, os pacientes com a síndrome HELLP são pessoas gravemente enfermas, com insuficiência circulatória, respiratória e renal; hemorragia pós-parto; hemorragia intra-hepática; e convulsões. A trombocitopenia persistente com microangiopatia ou a presença de falência em múltiplos órgãos sugerem PTT/SHU pós-parto; nesses casos, a terapia com troca de plasma é uma opção a ser considerada.
Prognóstico
Com cuidados pré-natais regulares e intervenção imediata, raramente, a síndrome HELLP resulta em morbidade e mortalidade materna ou fetal significativa, embora a recorrência em gestações subsequentes geralmente seja muito rara.44 Aparentemente, a função renal retorna ao normal, ainda que as mulheres tenham a tendência de ter pressão arterial elevada mesmo vários anos após o parto.45
O Quadro 3 contém as classificações mais importantes e exemplos de causas de CID.
Quadro 3
|
Classificações Mais Importantes e Exemplos de Causas de Coagulação Intravascular Disseminada |
|
Anatômicas Aneurisma na aorta abdominal Hemangioma gigante (síndrome de Kasabach-Merritt)
Hematológicas Reações agudas a transfusões hemolíticas (i.e., incompatibilidade ABO) Hemoglobinúria noturna paroxísmica Púrpura fulminante
Infecciosas Bacterianas (gram-positiva e gram-negativa) Virais
Traumatismos Lesão na cabeça Lesão por esmagamento Queimaduras
Malignidades LMA, em particular a leucemia pró-mieloide aguda. Tumores sólidos (em particular câncer na próstata e CID crônica) Medicamentos Anfetaminas Anti-D IVIg
Relacionadas à gravidez Embolismo de líquido amniótico Placentas abruptas Eclampsia/Pré-eclampsia Síndrome HELLP
Classificações diversas Doença hepática Veneno de cobras |
CID: coagulação intravascular disseminada; HELLP: hemólise, enzimas hepáticas elevadas e plaquetas baixas; IVIg: imunoglobulina intravenosa; LMA: leucemia mieloide aguda.
Coagulação Intravascular Disseminada
Etiologia e Genética
A CID é o resultado de um desequilíbrio nos sistemas de coagulação e de fibrinogênio que leva à geração excessiva de trombina. Trata-se de um processo secundário e, de um modo geral, CID aguda é o resultado de sepse ou trauma, ao passo que a CID crônica provavelmente resulte de alguma malignidade e, em situações raras, do uso de medicações.
A remoção de plaquetas também poderá ser feita por meio de algum leito vascular anormal. Nos casos de hemangiomas gigantes, o fluxo sanguíneo é lento através dos canais endotelizados impropriamente e, por conseguinte, essas superfícies poderão produzir CID de baixo grau.
Patogênese
A CID, independentemente da causa, resulta da ativação do sistema de coagulação, em geral pela exposição do fator tecidual às proteínas inativas da coagulação em circulação. O sistema regulador da coagulação é sobrepujado ativando a fibrinólise por causa da geração de trombina. Esse processo continua em um círculo vicioso, que resulta em trombose microvascular e isquemia de órgãos em estado terminal.
Diagnóstico
Manifestações Clínicas
Qualquer sistema de órgãos poderá se envolver nesse processo e precipitar sinais clínicos de acordo com o nível de gravidade da CID. De um modo geral, é extremamente difícil separar quais sinais refletem CID versus a causa subjacente. Além das manifestações clínicas relacionadas ao processo primário, os pacientes podem se apresentar com hemorragia grave, trombose, ou ambas as condições.
Exame Físico
De maneira geral, os pacientes com CID se apresentam com equimoses e petéquias, assim como com sangramentos nos sítios mucosos e nas linhas intravenosas. A presença de trombose é uma possibilidade.
Testes Laboratoriais
A contagem de plaquetas nos casos de CID aguda geralmente é inferior a 100 x 109/L, e os esfregaços de sangue periférico mostram a presença de alterações microangiográficas típicas observadas em outras MATs, incluindo evidências de esquistócitos. Considerando a depleção dos fatores de coagulação, o TP e TTPa são prolongados, sendo muito comum a presença de hipofibrinogenemia com redução no fibrinogênio. O processo de degradação dos produtos da fibrina e dímero-D é elevado.
Diagnóstico Diferencial
O diagnóstico diferencial para CID inclui muitos das MATs discutidas nesta revisão. Além disso, a disfunção hepática poderá diminuir a contagem de plaquetas e prolongar os tempos de coagulação. A deficiência de vitamina K também pode produzir anormalidades na coagulação, sendo que muitos pacientes se apresentam com uma combinação dessas condições.
Gerenciamento
O tratamento mais importante para CID é identificar e corrigir a causa subjacente. Caso contrário, o tratamento será de suporte, com tentativas de controlar as anormalidades de coagulação até que a abordagem do processo subjacente consiga recuperar o equilíbrio. No caso de sepse, o paciente deve receber tratamento imediato com antibióticos, fazendo-se tentativas de controle da fonte.
Com frequência, faz-se a transfusão de diversos produtos derivados do sangue para manter níveis adequados de fibrinogênio e diminuir os tempos de coagulação, embora diversos caminhos ainda não tenham sido estudados prospectivamente; portanto, os caminhos são arbitrários. As recomendações gerais são de tentar manter a contagem de plaquetas igual ou superior a 50 x 109/L e os níveis de fibrinogênio iguais ou superiores a 100mg/dL, em combinação com TP e TTPa inferior a 1,5 vezes o limite superior da normalidade para o laboratório que estiver fazendo o teste.
Complicações
Sem o reconhecimento e a correção imediata, os pacientes poderão ter hemorragia grave com risco de vida. Trombose arterial e venosa é uma ocorrência provável, e seu gerenciamento pode se tornar muito difícil, tendo em vista a necessidade de anticoagulação nos quadros de coagulação descontrolada e de fibrinólise.
Prognóstico
A presença de CID nos casos de sepse está associada a um índice significativo de mortalidade, variando de 30 a 80% dependendo da experiência publicada.46,47 CID com diagnóstico de câncer é um fator de mau prognóstico independentemente do estágio da doença.48
Síndrome Antifosfolipídica
Epidemiologia
A SAF se caracteriza pela combinação de eventos clínicos e a presença de evidências laboratoriais de anticorpos contra os fosfolipídeos. Os anticorpos antifosfolipídicos mais importantes são o anticoagulante lúpico, os anticorpos anticardiolipinas e os anticorpos anti-ß2-glicoproteína-I.
Etiologia e Genética
Algumas pesquisas em curso procuram delinear a causa de SAF, embora o consenso geral seja de que os pacientes são expostos a agentes infecciosos que levam ao desenvolvimento de anticorpos com reatividade contra os fosfolipídeos. Esse tipo de distúrbio é comum entre pacientes com outros fenômenos autoimunes. Os pacientes que já têm risco trombótico elevado devido à presença de mutações no fator V de Leiden ou da mutação G202010A na protrombina, aparentemente, têm risco mais elevado de desenvolver anticorpos antifosfolipídicos.49
Patogênese
Os anticorpos antifosfolipídicos têm efeitos em diversos níveis do sistema de coagulação por meio da potencialização dos efeitos pró-coagulantes das plaquetas e da inibição da proteína C.50,51 Ocorre um aumento no tônus vascular.52 A presença de outros anticorpos também é muito comum, incluindo anticorpos contra os fatores VII e VIIa, que, por outro lado, interagem com os anticorpos antifosfolipídicos para aumentar ainda mais o risco trombótico.53
Diagnóstico
O Quadro 4 mostra os critérios diagnósticos para a síndrome antifosfolipídica com base no Eleventh International Congress on Antiphospholipid Antibodies.54
Quadro 4
|
Critérios Diagnósticos Para a Síndrome Antifosfolipídica |
|
O diagnóstico de SAF exige, ao menos, a presença de um entre os critérios clínicos e um entre os critérios laboratoriais: Critérios clínicos Trombose vascular confirmada por estudos de imagens ou histopatologia Morbidade na gravidez a. Um ou mais abortos espontâneos sem explicação na 10ª semana de gestação ou depois; ou b. Um ou mais nascimentos prematuros antes da 34ª semana causados por eclampsia ou pré-eclampsia grave; ou c. Três ou mais abortos espontâneos consecutivos sem explicação antes da 10ª de gestação.
Critérios laboratoriais (nota: presença com intervalo de, ao menos, 12 semanas) Anticoagulante lúpico Anticardiolipina IgG ou IgM (>40 GLP ou MLP ou >99º percentil para laboratório) Anti-ß-glicoproteína-I IgG ou IgM (>99º percentil) |
GPL: fosfolipídeo IgG; MPL: fosfolipídeo IgM.
Manifestações Clínicas
As manifestações clínicas da SAF inclui tromboembolismo arterial e venoso. É possível que as pacientes apresentem histórico de aborto espontâneo sem explicação. No exame físico, as pacientes poderão se apresentar com livedo reticular, um padrão cutâneo reticular com manchas observado em uma grande variedade de condições autoimunes.
Testes Laboratoriais
Um subgrupo de antifosfolipídeos que atua como anticoagulante lúpico, detectado como um tempo de tromboplastina parcial ativada que não corrige ao ser misturado com plasma normal, sugerindo a presença de algum inibidor. Recomenda-se também fazer o rastreamento das isoformas IgG e IgM dos anticorpos da anticardiolipina e da anti-ß2-glicoproteína-I. Os esfregaços de sangue periférico talvez mostrem alterações microangiográficas e trombocitopenia, da mesma forma como se observa em qualquer MAT.
Diagnóstico Diferencial
As taxas de testes falsos positivos para anticorpos antifosfolipídicos são elevadas, de modo que os testes deverão ser repetidos em intervalos de, pelo menos, 12 semanas para confirmar a persistência da anormalidade laboratorial. Possivelmente, o tromboembolismo não seja causado por anticorpos antifosfolipídicos em circulação, mas por outros fatores de risco para trombose. Em alguns casos, as alterações microangiopáticas nos esfregaços de sangue periférico, em combinação com disfunção renal, poderão ensejar considerações sobre a presença de PTT.
Gerenciamento
A abordagem padrão aos pacientes com SAF e trombose é iniciar a anticoagulação com heparina (a heparina de baixo peso molecular é preferível à heparina não fracionada). Após a estabilização, deve-se passar para varfarina com dose ajustada, mantendo-se uma meta de razão normalizada internacional de 2 a 3.55 Embora o uso de dabigatrana, um inibidor oral direto da trombina, rivaroxabana e apixabana, inibidores anti-Xa, tenha sido aprovado para anticoagulação em outros distúrbios, ainda não há dados suficientes para dar suporte ao uso rotineiro nos casos de SAF.56
Complicações
A despeito do reconhecimento e do tratamento imediato, as pacientes provavelmente continuem a apresentar eventos trombóticos recorrentes e abortos espontâneos. A anticoagulação tem risco de hemorragia e exige monitoramento laboratorial constante, assim como ajustes na dosagem. A ligação em ponte se faz necessária em pacientes que tenham se submetido a algum procedimento cirúrgico e possam ter risco elevado de trombose no momento do procedimento, considerando os múltiplos riscos pós-operatórios versus tromboembolismo.
Prognóstico
A mortalidade corresponde a, aproximadamente, 5 a 6%, e não há mortalidade fetal significativa associada à SAF não tratada.57 A perda precoce de gravidez talvez ocorra em 15 a 20% de pacientes, sendo que 35% de mulheres têm parto prematuro.
O Quadro 5 apresenta as púrpuras vasculares.
Quadro 5
|
Púrpuras Vasculares |
|
Distúrbios congênitos THH. Distúrbios hereditários nos tecidos conjuntivos: síndrome de Ehlers-Danlos; síndrome de Marfan; pseudoxantoma elástico.
Distúrbios adquiridos Escorbuto Amiloidose Excesso de glicocorticoides Distúrbios da imunoglobulina Macroglobulinemia de Wadenström Crioglobulinemia Hepatite B Púrpura de Henoch-Schönlein Embolia (i.e., dano microvascular causado por êmbolos sépticos e fracos nas válvulas cardíacas, embolia gordurosa, embolia colesterólica) Púrpura senil |
THH: telangiectasia hemorrágica hereditária.
Distúrbios Vasculares
As púrpuras vasculares formam um grupo heterogêneo de distúrbios que se caracterizam por hemorragia cutânea e, ocasionalmente, estão associados a hemorragias na mucosa. O vazamento ocorre em arteríolas terminais, vasos capilares e vênulas pós-capilares. Os resultados dos testes de contagem e de função das plaquetas e os testes da função pró-coagulante são normais.
Telangiectasia Hemorrágica Hereditária
A telangiectasia hemorrágica hereditária (THH) é transmitida como uma doença autossômica dominante, e estima-se que sua incidência seja de 5.000 a 8.000 pessoas.58 A análise de ligação identificou dois genes importantes para THH: endoglina, responsável pela THH-1, e o receptor da quinase semelhante à ativina resultando em THH-2. Ambas as proteínas se expressam nas células endoteliais vasculares e podem atuar como receptoras para o fator de crescimento transformador ß (TGF-ß).
O TGF-ß tem um papel complexo na coordenação de respostas entre as células endoteliais e a matriz extracelular. A mutação no gene da endoglina e do receptor da quinase semelhante à ativina resulta em uma redução de 50% na quantidade normal de proteína nas células endoteliais e leva ao desenvolvimento de vasos sanguíneos anormais e de malformações arteriovenosas (MAVs) de uma forma autossômica dominante.59
Normalmente, a THH não se apresenta no nascimento e se manifesta com a idade. Tipicamente, epistaxe recorrente é o primeiro sinal da doença e, em geral, ocorre na infância. As MAVs pulmonares, que ocorrem em, aproximadamente, 30% de pacientes com THH, se tornam aparentes depois da puberdade e se apresentam como dispneia, dor torácica e hemoptise. O exame físico revela a presença de ruídos no tórax e hipocratismo digital.
As telangiectasias microcutâneas, que ocorrem na maioria de pacientes e em sítios específicos (por exemplo, lábios, cavidade oral, dedos e nariz), geralmente se tornam perceptíveis na terceira década de vida e aumentam em número e dimensões ao longo do processo de envelhecimento. O diagnóstico de THH deve se fundamentar nos quatro critérios descritos nesta seção.60
De um modo geral, os resultados dos testes de coagulação são normais. As MAVs pulmonares podem estar associadas a condições como hipoxemia e policitemia secundária. O diagnóstico poderá ser confirmado por meio de angiografia pulmonar. Nos casos em que forem muito grandes e significativas em termos clínicos, as derivações poderão ser tratadas com embolização por balão.61
O êmbolo paradoxal com acidente vascular cerebral é uma ocorrência provável em pacientes com THH com derivações e malformações arteriovenosas nos pulmões. Defende-se a tese de que os pacientes com THH sintomática sejam rastreados para verificar a eventual presença de MAVs e que sejam tratados profilaticamente com embolização nos casos em que forem encontradas malformações arteriovenosas.62
O tratamento de epistaxe recorrente deve ser conservador e, com frequência, envolve o desenvolvimento de métodos de tamponamento nasal. Recomenda-se evitar cauterização para impedir a ocorrência de lesões na mucosa nasal, que poderão resultar em novo crescimento vascular. Nas situações em for possível, faz-se o gerenciamento das hemorragias gastrintestinais com preparações de ferro.
Alguns testes clínicos de pequeno porte randomizados e controlados por placebo demonstraram que tanto a terapia com estrogênio (50µg de etinil estradiol mais 1mg de noretisterona) como a terapia à base de tamoxifeno (20mg/dia durante 6 meses) diminuíram bastante a necessidade de transfusão nessa população de pacientes.63,64 Existem alguns relatos de casos sobre a eficácia do medicamento bevacizumab, um antagonista do fator de crescimento do endotélio vascular (VEGF).65
O Quadro 6 contém os critérios diagnósticos para telangiectasia hemorrágica hereditária.*
Quadro 6
|
CRITÉRIOS DIAGNÓSTICOS PARA TELANGIECTASIA HEMORRÁGICA HEREDITÁRIA |
|
Epistaxe espontânea e recorrente Telangiectasias mucocutâneas múltiplas Evidências de telangiectasias viscerais e MAVs (p.ex., MAVs no trato gastrintestinal, pulmonares, hepáticas ou cerebrais) Histórico familiar positivo de THH |
MAV: malformação arteriovenosa.
*3 a 4 critérios = diagnóstico definitivo; 2 critérios = diagnóstico possível; 0 a 1 critério = diagnóstico improvável.
Distúrbios Adquiridos em Vasos Sanguíneos que Poderão Provocar Hemorragias.
Escorbuto
A administração de vitamina C normaliza o metabolismo de colágeno, folato e, talvez, do ferro. Os pacientes com escorbuto sofrem sobretudo da síntese anormal de colágeno. A falta da quantidade adequada de colágeno para dar suporte à microvasculatura produz hemorragias perifoliculares, sangramento nas gengivas e hematomas teciduais profundos.
Presumivelmente, os defeitos de colágeno resultam em pelos do tipo saca-rolhas e hiperqueratose, sendo que ambas as condições estão associadas a esse tipo de distúrbio.66 O quadro clínico típico em indivíduos malnutridos sugere o diagnóstico. Os níveis de ácido ascórbico no plasma ou na camada leucoplaquetária são baixos, geralmente com a presença de deficiência de outras vitaminas. A terapia mais eficaz consiste de 1g de ácido ascórbico todos os dias, em doses divididas.
Excesso de Glicocorticoides
O excesso de glicocorticoides, seja de causa endógena, seja de causa exógena, produz hemorragias cutâneas, provavelmente por causa do catabolismo induzido pelos glicocorticoides das proteínas que se localizam nos tecidos de suporte vascular.
Amiloidose
A amiloidose se apresenta como equimoses subcutâneas com predileção pelo pescoço e pela parte superior do tórax. As biópsias feitas no sítio da doença mostram infiltração com deposição de amiloides, que poderá enfraquecer as paredes dos vasos ou interferir na ativação superficial de plaquetas, pró-coagulantes, ou ambos.
A angiopatia amiloide cerebral, embora seja sintomática, é uma das causas mais importantes de hemorragias intracerebrais lobares primárias em pessoas idosas. Estima-se que a angiopatia amiloide cerebral, variando de moderada a grave, esteja presente em 8% de pessoas com idade entre 75 e 84 anos. A deposição de amiloides nas paredes dos vasos sanguíneos cerebrais de tamanho médio, primários ou secundários, poderá provocar hemorragia no cérebro.67
Em um pequeno subgrupo de pacientes (2 a 3%) com amiloidose sistêmica primária de cadeia leve, em especial se for acompanhada por esplenomegalia maciça, o amiloide poderá adsorver uma quantidade de fator X suficiente para ocasionar deficiência grave deste fator (<25%) e hemorragia clínica.68 Comumente, as infusões de plasma fresco congelado são ineficazes. Embora seja bastante eficaz, o fator recombinante VIIa tem custo excessivamente elevado, e seu benefício é apenas temporário.
Algum sucesso foi obtido com o uso de concentrados do complexo da protrombina, ainda que esses concentrados não tenham passado por avaliações sistemáticas. O tratamento agressivo da amiloidose subjacente com quimioterapia, com doses elevadas de melfalano, seguido por transplante de células-tronco autólogas, poderá produzir melhoras significativas; entretanto, o risco de hemorragias durante o período peritransplante é bastante elevado.68
Experiências anedóticas sugerem que a esplenectomia poderá gerar benefícios no longo prazo, sendo, portanto, uma opção a ser considerada para uso em pacientes com hemorragia clínica recorrente séria.69
Distúrbios com Imunoglobulinas
Recomenda-se levantar a hipótese de lesões purpúricas (púrpura palpável) em pacientes portadores de distúrbios com imunoglobulinas (por exemplo, crioglobulinemia, macroglobulinemia de Waldenström, púrpura de Henoch-Schönlein e melomas múltiplos). Nas biópsias, essas lesões podem apresentar desgranulação de mastócitos e, com aplicação de coloração apropriada, deposição do complexo imune.
Presumivelmente, os complexos imunes produzem estimulação quimiotática com acúmulo de granulócitos. Os danos na microvasculatura são causados pelo complexo de ataque do complemento e pela liberação de conteúdos granulares por leucócitos infiltrantes. Esse complexo inflamatório produz púrpuras palpáveis.
|
Informações Financeiras: Nathan T. Connell, MD, MPH, não possui informações financeiras relevantes a declarar. Esta revisão foi anteriormente elaborada por Lawrence L. K. Leung, MD, cuja divulgação foi feita à época da publicação inicial. Este texto foi revisado, atualizado e republicado pelos autores mencionados. |
Referências
1. Bennett CL, Connors JM, Carwile JM, et al. Thrombotic thrombocytopenic purpura associated with clopidogrel. N Engl J Med 2000;342:1773–7.
2. Amoura Z, Costedoat-Chalumeau N, Veyradier A, et al. Thrombotic thrombocytopenic purpura with severe ADAMTS-13 deficiency in two pa-tients with primary antiphospholipid syndrome. Arthritis Rheum 2004;50:3260–4.
3. Devinsky O, Petito CK, Alonso DR. Clinical and neuropathological findings in systemic lupus erythematosus: the role of vasculitis, heart emboli, and thrombotic thrombocytopenic purpura. Ann Neurol 1988;23:380–4.
4. George JN. The thrombotic thrombocytopenic purpura and hemolytic uremic syndromes: overview of pathogenesis (Experience of The Oklahoma TTP-HUS Registry, 1989–2007). Kidney Int Suppl 2009;(112):S8–10.
5. Moake JL. Thrombotic microangiopathies. N Engl J Med 2002;347:589–600.
6. Levy GG, Motto DG, Ginsburg D. ADAMTS13 turns 3. Blood 2005;106:11–7.
7. Dong JF, Moake JL, Bernardo A, et al. ADAMTS-13 metalloprotease interacts with the endothelial cell-derived ultra-large von Willebrand factor. J Biol Chem 2003;278:29633–9.
8. Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001;413:488–94.
9. Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med 1998;339:1578–84.
10. Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998;339:1585–94.
11. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010;116:4060–9.
12. Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008;112:11–8.
13. Bianchi V, Robles R, Alberio L, et al. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood 2002;100:710–3.
14. von Baeyer H. Plasmapheresis in thrombotic microangiopathy-associated syndromes: review of outcome data derived from clinical trials and open studies. Ther Apher 2002;6:320–8.
15. Lim W, Vesely SK, George JN. The role of rituximab in the management of patients with acquired thrombotic thrombocytopenic purpura: evi-dence-based focused review. Blood 2015;125:1526–31. DOI: 10.1182/blood-2014-10-559211.
16. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991;325:393–7.
17. Rock G, Shumak KH, Sutton DM, et al. Cryosupernatant as replacement fluid for plasma exchange in thrombotic thrombocytopenic purpura. Members of the Canadian Apheresis Group. Br J Haematol 1996;94:383–6.
18. Vesely SK, George JN, Lammle B, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood 2003;102:60–8.
19. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical ex-perience in 108 patients. N Engl J Med 1991;325:398–403.
20. Zheng X, Pallera AM, Goodnough LT, et al. Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Ann Intern Med 2003;138:105–8.
21. Yomtovian R, Niklinski W, Silver B, et al. Rituximab for chronic recurring thrombotic thrombocytopenic purpura: a case report and review of the literature. Br J Haematol 2004;124:787–95.
22. Fakhouri F, Vernant JP, Veyradier A, et al. Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13-deficient thrombotic thrombocytopenic purpura: a study of 11 cases. Blood 2005;106:1932–7.
23. Elliott MA, Heit JA, Pruthi RK, et al. Rituximab for refractory and or relapsing thrombotic thrombocytopenic purpura related to immune-mediated severe ADAMTS13-deficiency: a report of four cases and a systematic review of the literature. Eur J Haematol 2009;83:365–72.
24. Harkness DR, Byrnes JJ, Lian EC, et al. Hazard of platelet transfusion in thrombotic thrombocytopenic purpura. JAMA 1981;246:1931–3.
25. Goel R, Ness PM, Takemoto CM, et al. Platelet transfusions in platelet consumptive disorders are associated with arterial thrombosis and in-hospital mortality. Blood 2015;125:1470–6. DOI: 10.1182/blood-2014-10-605493.
26. Som S, Deford CC, Kaiser ML, et al. Decreasing frequency of plasma exchange complications in patients treated for thrombotic thrombocytopenic purpura-hemolytic uremic syndrome, 1996 to 2011. Transfusion 2012;52:2525–32; quiz 2524.
27. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med 1995;333:364–8.
28. Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood 2004;103:4043–9.
29. Ferrari S, Scheiflinger F, Rieger M, et al. Prognostic value of anti-ADAMTS 13 antibody features (Ig isotype, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS 13 activity. Blood 2007;109:2815–22.
30. Coppo P, Wolf M, Veyradier A, et al. Prognostic value of inhibitory anti-ADAMTS13 antibodies in adult-acquired thrombotic thrombocytopenic purpura. Br J Haematol 2006;132:66–74.
31. Kokame K, Matsumoto M, Fujimura Y, Miyata T. VWF73, a region from D1596 to R1668 of von Willebrand factor, provides a minimal substrate for ADAMTS-13. Blood 2004;103:607–12.
32. Whitelock JL, Nolasco L, Bernardo A, et al. ADAMTS-13 activity in plasma is rapidly measured by a new ELISA method that uses recombinant VWF-A2 domain as substrate. J Thromb Haemost 2004;2:485–91.
33. Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series com-paring children and adults. Clin J Am Soc Nephrol 2013;8:554–62.
34. Wong CS, Jelacic S, Habeeb RL, et al. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med 2000;342:1930–6.
35. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009;361:1676–87.
36. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypi-cal hemolytic uremic syndrome. J Am Soc Nephrol 2007;18:2392–400.
37. Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006;108:1267–79.
38. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013;368:2169–81.
39. Salmon JE, Heuser C, Triebwasser M, et al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med 2011;8:e1001013. DOI: 10.1371/journal.pmed.1001013.
40. Sibai BM, Ramadan MK, Usta I, et al. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low plate-lets (HELLP syndrome). Am J Obstet Gynecol 1993;169:1000–6.
41. Martin JN Jr, Rinehart BK, May WL, et al. The spectrum of severe preeclampsia: comparative analysis by HELLP (hemolysis, elevated liver en-zyme levels, and low platelet count) syndrome classification. Am J Obstet Gynecol 1999;180:1373–84.
42. Martin JN Jr, Blake PG, Perry KG Jr, et al. The natural history of HELLP syndrome: patterns of disease progression and regression. Am J Obstet Gynecol 1991;164:1500–9; discussion 1509–13.
43. Sibai BM. Diagnosis, controversies, and management of the syndrome of hemolysis, elevated liver enzymes, and low platelet count. Obstet Gyne-col 2004;103:981–91.
44. van Oostwaard MF, Langenveld J, Schuit E, et al. Recurrence of hypertensive disorders of pregnancy: an individual patient data metaanalysis. Am J Obstet Gynecol 2015;212:624.e1–624.e17. DOI: 10.1016/j.ajog.2015.01.009.
45. Jacquemyn Y, Jochems L, Duiker E, et al. Long-term renal function after HELLP syndrome. Gynecol Obstet Invest 2004;57:117–20.
46. Stephan F, Hollande J, Richard O, et al. Thrombocytopenia in a surgical ICU. Chest 1999;115:1363–70.
47. Garcia-Avello A, Lorente JA, Cesar-Perez J, et al. Degree of hypercoagulability and hyperfibrinolysis is related to organ failure and prognosis after burn trauma. Thromb Res 1998;89:59–64.
48. Sallah S, Wan JY, Nguyen NP, et al. Disseminated intravascular coagulation in solid tumors: clinical and pathologic study. Thromb Haemost 2001;86:828–33.
49. Brouwer JL, Bijl M, Veeger NJ, et al. The contribution of inherited and acquired thrombophilic defects, alone or combined with antiphospholipid antibodies, to venous and arterial thromboembolism in patients with systemic lupus erythematosus. Blood 2004;104:143–8.
50. Urbanus RT, Derksen RH, de Groot PG. Platelets and the antiphospholipid syndrome. Lupus 2008;17:888–94.
51. Forastiero R, Martinuzzo M. Prothrombotic mechanisms based on the impairment of fibrinolysis in the antiphospholipid syndrome. Lupus 2008;17:872–7.
52. Mackworth-Young CG. Antiphospholipid syndrome: multiple mechanisms. Clin Exp Immunol 2004;136:393–401.
53. Bidot CJ, Jy W, Horstman LL, et al. Factor VII/VIIa: a new antigen in the anti-phospholipid antibody syndrome. Br J Haematol 2003;120:618–26.
54. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
55. Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003;349:1133–8.
56. Chighizola CB, Moia M, Meroni PL. New oral anticoagulants in thrombotic antiphospholipid syndrome. Lupus 2014;23:1279–82.
57. Cervera R, Khamashta MA, Shoenfeld Y, et al. Morbidity and mortality in the antiphospholipid syndrome during a 5-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis 2009;68:1428–32.
58. Begbie ME, Wallace GM, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Post-grad Med J 2003;79:18–24.
59. Abdalla SA, Pece-Barbara N, Vera S, et al. Analysis of ALK-1 and endoglin in newborns from families with hereditary hemorrhagic telangiectasia type 2. Hum Mol Genet 2000;9:1227–37.
60. Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66–7.
61. White RI Jr, Lynch-Nyhan A, Terry P, et al. Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radi-ology 1988;169:663–9.
62. Shovlin CL. Molecular defects in rare bleeding disorders: hereditary haemorrhagic telangiectasia. Thromb Haemost 1997;78:145–50.
63. van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. Lancet 1990;335:953–5.
64. Yaniv E, Preis M, Hadar T, et al. Antiestrogen therapy for hereditary hemorrhagic telangiectasia: a double-blind placebo-controlled clinical trial. Laryngoscope 2009;119:284–8.
65. Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 2009;360:2143–4.
66. Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol 1999;41:895–906; quiz 907–10.
67. Greenberg SM, Vonsattel JP. Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke 1997;28:1418–22.
68. Choufani EB, Sanchorawala V, Ernst T, et al. Acquired factor X deficiency in patients with amyloid light-chain amyloidosis: incidence, bleeding manifestations, and response to high-dose chemotherapy. Blood 2001;97:1885–7.
69. Rosenstein ED, Itzkowitz SH, Penziner AS, et al. Resolution of factor X deficiency in primary amyloidosis following splenectomy. Arch Intern Med 1983;143:597–9.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.