(Carregando Índice)... (Carregando Índice)... |
Última revisão: 07/11/2018
Comentários de assinantes: 0
|
Artigo original: George S, MD. Serrano C, MD, PhD. Management of Gastrointestinal Stromal Tumors, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Suzanne George, MD
Diretora Clínica do Centro de Sarcoma e Oncologia Óssea na Dana-Farber Cancer Institute/Harvard Medical School (Boston, MA).
César Serrano, MD, PhD
Chefe do Sarcoma Translational Research Laboratory and Sarcoma Unit do Departamento de Oncologia no Vall d’Hebron Institute of Oncology da Vall d’Hebrón University Hospital (Barcelona, Spain).
Resumo
Tumores de células estromais gastrintestinais (GISTs) são os tumores mais comuns de origem mesenquimal no trato gastrintestinal. Acredita-se que esses tumores tenham origem nas células intersticiais de Cajal. A ativação oncogênica da mutação KIT ou do receptor-a do fator de crescimento derivado de plaquetas é comum nesses tumores, e os GISTs são considerados modelos bem-sucedidos para o desenvolvimento racional de tratamentos personalizados contra mutações oncogênicas estimuladoras nos casos de câncer. Esta revisão aborda a epidemiologia, a etiologia e a genética, a fisiopatologia e a patogênese, o diagnóstico, o diagnóstico diferencial, o tratamento, as complicações e o prognóstico de GISTs. As figuras mostram as mutações KIT primárias e secundárias, a progressão clínica e molecular de GISTs, a varredura por tomografia computadorizada (TC) intensificada por contraste mostrando um GIST gástrico com uma grande massa abdominal, imagem por ressonância nuclear magnética (RNM) mostrando um GIST retal na linha de base e respondendo ao imatinibe neoadjuvante depois de 8 meses de tratamento, e coloração com hematoxilina-eosina de um GIST fusocelular e epitelioide. Os quadros apresentam uma lista de características demográficas e clínicas, síndromes genéticas associadas, diagnóstico diferencial relevante, sistemas de estratificação de risco usados nos casos de GIST e atividade comparativa dos regimes aprovados para tratamento de GISTs.
|
O Estudo |
Somente a partir de 1998 foi possível distinguir os tumores estromais gastrintestinais (GISTs) de outros tumores de origem mesenquimal no trato gastrintestinal (GI) produzidos por imuno-histoquímica pela expressão do gene KIT. No decurso das últimas duas décadas, houve um aumento exponencial na compreensão da biologia dos GISTs, que transformou esses tumores em modelos bem-sucedidos para o desenvolvimento racional de tratamentos personalizados contra mutações oncogênicas nos casos de câncer.
Definição da Doença
GIST é o tumor mais comum de origem mesenquimal no trato GI e acredita-se que tenha origem nas células intersticiais de Cajal (CIC). Esse tipo de tumor abrange um espectro clínico amplo que varia desde lesões sem nenhum potencial metastático e tumores metastáticos que colocam a vida em risco. O estimulador comum e crucial em toda esse processo evolutivo é a ativação oncogênica do gene KIT ou do receptor da tirosina quinase do receptor a do fator de crescimento derivado das plaquetas (PDGFRA).
Epidemiologia
Incidência
Embora os GISTs sejam os tumores não epiteliais mais comuns envolvendo o trato GI, estima-se que correspondam a apenas 1% de todos os neoplasmas gastrintestinais1 e a 2,2% de todos os tumores gástricos malignos.2
Entretanto, a incidência real de GISTs ainda é desconhecida por causa do grande volume de dados gerados pelos estudos epidemiológicos, sendo que alguns dos estudos realizados antes dos GISTs foram caracterizados sob o ponto de vista molecular. Com base nesse fato, estimava-se, tradicionalmente, que a incidência anual de GISTs nos EUA fosse de 4 mil a 6 mil novos casos, significando, aproximadamente, 7 a 20 casos por 1 milhão a cada ano.3
Alguns estudos populacionais europeus recentes determinaram uma taxa de incidência de GISTs entre 1,2 e 1,5 casos/100.000/ano.4,5 Cabe observar que um estudo francês também definiu, pela primeira vez, que o GIST é o subtipo histológico mais frequente de sarcoma.4
Em termos gerais, a despeito da baixa frequência de GIST clínico no contexto oncológico, um estudo realizado no Japão, entre vários outros, concluiu que, nas ressecções estomacais, até um terço da população apresenta GISTs microscópicos (micro-GISTs) - principalmente, GISTs medindo menos de 1cm e abrigando as mesmas mutações oncogênicas no gene KIT descritas em tumores sintomáticos maiores.6
Levando-se em consideração a incidência anual de GISTs detectados clinicamente, apenas alguns micro-GISTs atingirão o tamanho clínico com potencial metastático. Na realidade, geralmente os micro-GISTs se calcificam e não têm atividade mitótica. São necessários mais estudos para determinar os mecanismos moleculares envolvidos na progressão dos micro-GISTs para doenças malignas.
Aspectos Demográficos dos Gists
Os GISTs ocorrem predominantemente em indivíduos na meia-idade e em pessoas idosas, sendo que a incidência máxima ocorre na sétima década de vida.3?5,7 Além disso, os GISTs são considerados os sarcomas mais comuns em adultos com idade acima de 50 anos.4 Os GISTs são distribuídos igualmente em todos os grupos geográficos, raciais e étnicos, sem nenhuma predileção por gênero, excetuando-se os GISTs pediátricos, que serão discutidos separadamente3?5,7
Acredita-se que os GISTs tenham origem nas células intersticiais de Cajal do trato GI8,9 e, em geral, se localizam no estômago e no intestino delgado proximal, embora também possam ser encontrados em qualquer parte do trato alimentar, incluindo o omento e o mesentério.3,5,7 Alguns subtipos moleculares específicos de GISTs podem se manifestar com apresentações clínicas exclusivas, como, por exemplo, apresentações multifocais de tumores primários no estômago (GISTs pediátricos) ou no intestino delgado (neurofibromatose tipo 1 [NF1] e GISTs estimulados pelo BRAF).
O Quadro 1 apresenta os aspectos demográficos e as características clínicas dos GISTs.3,5,7
Quadro 1
|
Aspectos Demográficos e Características Clínicas dos Tumores Estromais Gastrintestinais | |
|
Idade mediana (anos) |
65 |
|
Proporção entre homens e mulheres |
0,97 |
|
Sítio do tumor (%) Estômago Intestino delgado Reto Outros sítios |
57 32 8 3 |
|
Tamanho do tumor (mm) |
63 |
|
Contagem de mitose (50 HPF) (%) =5 >5 e =10 >10 |
78 23 16 |
|
Estado da doença (%) Doença localizada Doença metastática |
83 17 |
|
Apresentação clínica (%) Incidental Sintomática |
23 77 |
HPF: campo de alta potência.
Etiologia e Genética
A grande maioria dos GISTs é esporádica e não possui nenhum fator etiológico conhecido. Todavia, poucas mutações germinais associadas a condições hereditárias raras predispõem para o aumento na probabilidade de GIST em famílias que carregam esses determinantes genéticos.10 O reconhecimento e o diagnóstico imediato das síndromes de GIST são fatores importantes para melhorar o tratamento médico amplo de pacientes com esse tipo de doença e suas famílias.
Gist Familiar
Nos anos recentes, foram descritas diversas famílias com GISTs afetando vários parentes10,11 e que seguem um padrão autossômico dominante de herança com alta penetrância. A maioria das famílias afetadas carregava mutações germinais do gene KIT ou PDGFRA, que obviamente são os eventos iniciadores e responsáveis pelo espectro de diversas manifestações associadas a essa síndrome.
O Quadro 2 contém as síndromes genéticas de GIST.
Quadro 2
|
Síndromes Genéticas de Tumores Estromais Gastrintestinais | |||
|
Características |
Síndrome da GIST familiar |
Neurofibromatose tipo 1 |
Síndrome de Carney-Stratakis |
|
Gene |
KIT/PDGFRA |
NF1 |
SDHB, C ou D |
|
Herança |
Autossômica dominante |
Autossômica dominante |
Autossômica dominante |
|
Outros neoplasmas |
Não |
Sarcomas, gliomas |
Paraganglioma |
|
Descobertas cutâneas |
Ocasionalmente |
Ocasionalmente |
Não |
|
Idade |
<50 anos |
<50 anos |
3ª década de vida |
|
Predominância de sexo |
Não |
Não |
Não |
|
Sítio do tumor |
Estômago |
Intestino delgado |
Estômago |
|
Imatinibe |
Desconhecido |
Resistente |
Desconhecido |
Ao contrário dos pacientes com GISTs esporádicos, os indivíduos com a síndrome de GIST familiar tendem a ter vários parentes afetados com neoplasmas primários múltiplos, lesões multifocais e tumores que se apresentam em idades mais precoces.
Mecanismos distintos de ativação do gene KIT são encontrados em parentescos diferentes de GIST familiar e talvez determinem o fenótipo em cada caso. Portanto, isso provavelmente explique que algumas famílias, mas nem todas, também podem se apresentar com hiperplasia causada por células intersticiais de Cajal, hiperpigmentação cutânea e/ou mastocitose cutânea.
Neurofibromatose Tipo 1
Os indivíduos com neurofibromatose tipo 1 (NF1) correm grande risco de ocorrência de malignidades específicas, sendo que 7% de pacientes com NF1 desenvolvem um ou mais GISTs ao longo da vida.10,12 NF1 é um distúrbio autossômico dominante. A neurofibromina, uma proteína associada à NF1, é um gene supressor tumoral que regula negativamente a ativação da via da proteinoquinase ativada por RAS/mitógeno (MAPK) ao converter o trifosfato de RAS-guanosina (GTP) na sua forma inativa.
As características clínicas e patológicas dos GISTs em pacientes com NF1 são diferentes das tipificidades dos GISTs esporádicos, e é interessante observar que há algumas semelhanças com a síndrome de GIST familiar, incluindo os neoplasmas primários múltiplos (predominantemente, no intestino delgado em casos de NF1) e as apresentações multifocais. Outras características reconhecidas incluem também a ausência de mutações nos genes KIT/PDGFRA, que é um ótimo prognóstico, e resistência ou baixa sensibilidade ao imatinibe.12,13
Síndrome de Carney-Stratakis
Observa-se a presença de GISTs predominantemente em adultos; esse tipo de diagnóstico não é muito comum na população pediátrica. Cabe observar que alguns GISTs pediátricos originalmente são componentes de duas síndromes distintas de câncer: a tríade de Carney e a síndrome de Carney-Stratakis.
A tríade de Carney pode ser diagnosticada nas situações em que, pelo menos, dois entre os seguintes tumores definidores ocorrem de forma simultânea: GIST, condroma pulmonar e paraganglioma. Entretanto, apenas na presença de GIST e paraganglioma, deve-se considerar o diagnóstico de síndrome de Carney-Stratakis.
Se a tríade de Carney é considerada uma síndrome esporádica de câncer com causa desconhecida, a síndrome de Carney-Stratakis é uma forma de herança autossômica dominante, embora com penetrância incompleta.14 Os fundamentos genéticos subjacentes da síndrome de Carney-Stratakis são mutações germinais desativadoras localizadas nas subunidades B, C ou D da succinato desidrogenase (SDH).
As mutações germinais SDHA não foram associadas a nenhuma síndrome de predisposição para o câncer.14 É interessante observar que os GISTs pediátricos esporádicos KIT/PDGFRA do tipo wild-type (WT) não possuem a expressão de SDHB e, portanto, foram definidos como “GISTs deficientes em SDH”.
Os defeitos na respiração celular são um mecanismo oncogênico central em pacientes pediátricos e adultos com GISTs deficientes em SDH.14,15 O espectro do GIST na síndrome de Carney-Stratakis inclui apresentação multifocal, idade jovem (segunda década de vida), predominância gástrica, e crescimento lento e indolente.14
Fisiopatologia e Patogênese
As mutações ativadoras de KIT ou PDG-FRA mutuamente exclusivas são eventos oncogênicos precoces, e possivelmente em fase inicial, na maior parte dos GISTs16 e continuam sendo os estimuladores cruciais do desenvolvimento e da manutenção tumoral considerando que os GISTs sofreram transformações malignas.9
Essa adição extraordinária à sinalização oncológica de KIT/PDGFRA explica o efeito profundo da inibição focada desses RTKs com pequenas moléculas com o imatinibe de primeira linha na viabilidade e no crescimento das células tumorais. Da mesma forma, a maior parte dos pacientes com GIST apresenta respostas clínicas excepcionais nos tratamentos com esses inibidores.17
A Ativação de Kit/Pdgfra é Central na Oncogêse dos Gists
Mutações no Gene KIT
KIT é um receptor transmembranoso que pertence à família do RTK tipo III, que também inclui PDGFRA, PDGFRB, CSF1R e FLT3. A ligação do ligante de KIT, que é o fator da célula-tronco (FCT), à porção extracelular de KIT resulta na homodimerização do receptor e na ativação do domínio da quinase de KIT.
A seguir, as quinases ativadas fazem a fosforilação cruzada dos resíduos da tirosina intracelular levando à ativação adicional do receptor e desencadeando cascatas posteriores de sinalização celular que controlam as funções celulares mais críticas, tais como sobrevida, proliferação e adesão.9 Em alguns tecidos, o gene KIT é expresso fisiologicamente em níveis elevados, incluindo as células intersticiais de Cajal (ICCs).
As ICCs formam uma rede complexa de células inervadas que se localizam na parede do trato GI e agem como marca-passos das contrações peristálticas do trato gastrintestinal. Com base nesses fatos, acredita-se que as ICCs sejam a origem celular dos GISTs.18 Isso levou à descoberta de que os GISTs geralmente expressam CD117 (KIT).19 Portanto, agora está bem definido que 95% dos GISTs são positivos para CD117 sob o ponto de vista imuno-histoquímico.
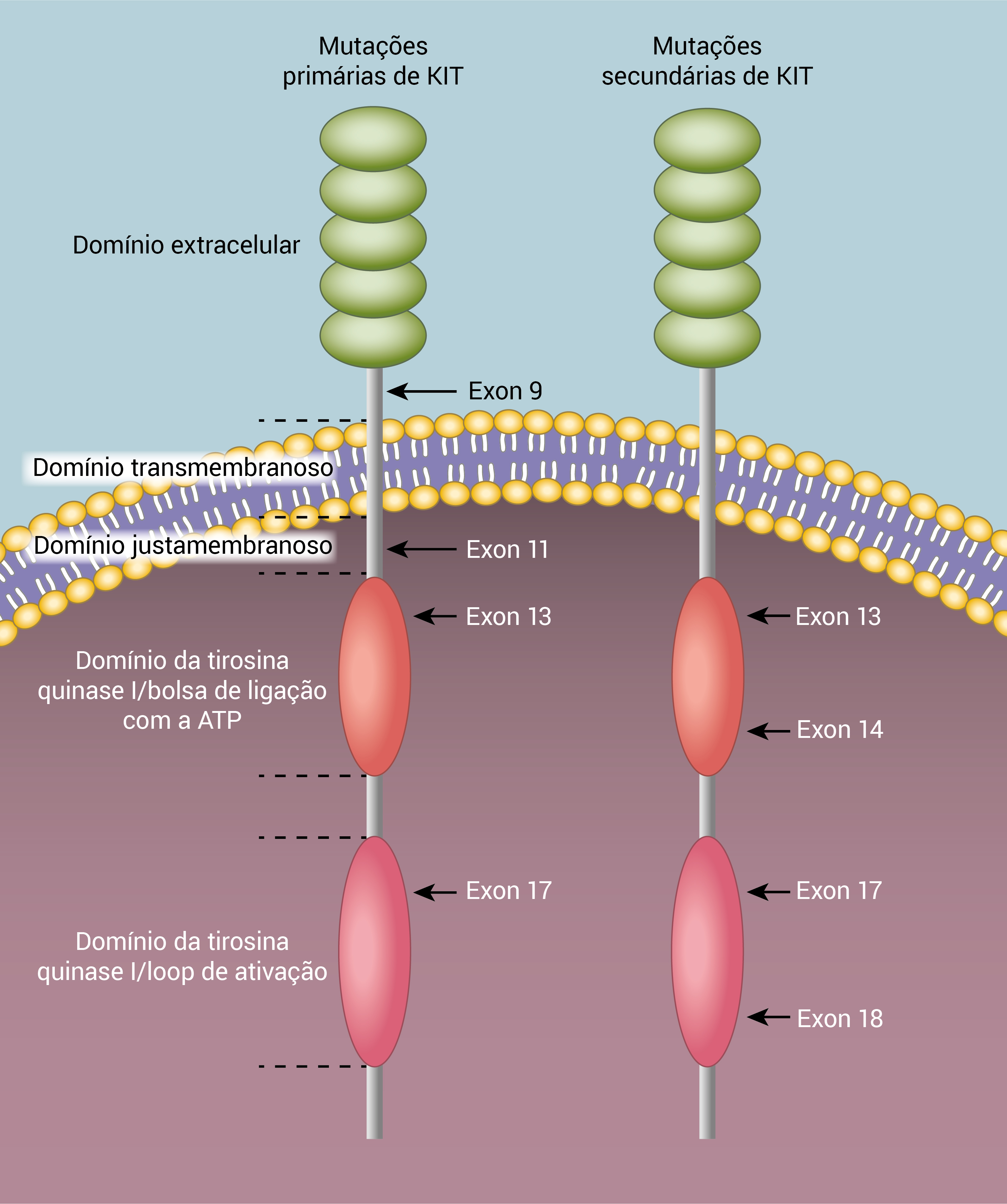
As mutações oncogênicas no gene KIT são encontradas em, aproximadamente, 80% dos GISTs e resultam na ativação da quinase independente de ligantes, que surge como um evento essencial na tumorigênese dos GISTs.9 As mutações mais comuns no gene KIT observadas em pacientes com GISTs afetam o domínio justamembranoso intracelular (67%), codificado por mutações no exon 11. Essa região é responsável pelo estado inativo normal do gene KIT sem ligação com o fator da célula-tronco (FCT).
As mutações de ganhos funcionais no exon 11 do gene KIT rompem o estado autoinibidor normal do gene KIT e resultam na ativação constitutiva. Os GISTs remanescentes possuem mutações ativadoras em outras regiões, como, por exemplo, o domínio extracelular do gene KIT sem ligação com ligantes, codificado por mutações no exon 9 (10%) e, em uma escala menor (<2%), nos domínios da quinase (exons 13 e 17).
Cabe observar que o genótipo do GIST prevê respostas aos medicamentos com atividade contra o gene KIT, sendo que o imatinibe - tratamento padrão de primeira linha em casos de GIST - consegue obter respostas drásticas contra todas as mutações primárias do gene KIT. Entretanto, há algum nível de variação entre as diferentes regiões de KIT, sendo que aquelas codificadas por mutações no exon 9 tendem a ser menos sensíveis à inibição do gene KIT.20,21
A Figura 1 mostra as mutações primárias e secundárias do gene KIT.

Figura 1 - Mutações primárias e secundárias do gene KIT.
O genótipo tumoral primário não apenas determina a sensibilidade ao imatinibe, mas indica também algumas outras características. Os GISTs com mutações primárias no exon 11 do gene KIT, assim como os GISTs com mutações primárias nos exons 13 e 17, não têm predileção por qualquer localização e, geralmente, são encontrados no estômago.
Por outro lado, mutações no exon 9 do gene KIT definem um subgrupo distinto de GISTs, que se localiza predominantemente no intestino delgado e está associado a cursos clínicos desfavoráveis.22 Essa heterogeneidade no comportamento clínico pode estar relacionada ao fato de que os GISTs com mutações no exon 9 do gene KIT apresentam um perfil diferente de expressão genética em comparação com outros GISTs com mutações em KIT, embora ainda não tenham sido descobertos os mecanismos específicos.22,23
Mutações no Gene PDGFRA
As mutações oncogênicas no gene PDGFRA, que levam à ativação do receptor constitutivo, também estão associadas ao início e à patogênese de, aproximadamente, 5 a 8% de GISTs.9 O gene PDGFRA é uma RTK que compartilha uma grande semelhança com o gene KIT e ativa os mesmos sinalizadores intermediários que vêm posteriormente.24,25
As mutações no gene PDGFRA são mutuamente exclusivas em relação às mutações no gene KIT e ocorrem predominantemente no domínio justamembranoso (exon 12) e nos domínios da tirosina quinase 1 (exon 14) e da tirosina quinase 2 (exon 18).
Independentemente do exon que sofreu mutação, os GISTs estimulados por PDGFRA têm características distintas, como, por exemplo, localização gástrica primária, aparência epitelioide ou aparência mista epitelioide e fuselar, expressão variável do gene KIT e potencial maligno reduzido, incluindo dimensões menores e índice mitótico mais baixo, em comparação com os GISTs com mutações em KIT.9
Consequentemente, esse potencial maligno reduzido explica a proporção maior de mutações em PDG-FRA encontradas nos GISTs localizados (10 a 15%) em comparação com a doença em estado avançado (2%).26 A base biológica para o comportamento clínico típico não é muito clara. Entretanto, existem diferenças sutis, porém significativas, nos perfis da expressão genética dos GISTs com mutações em KIT e PDGFRA que poderiam determinar algumas diferenças observadas entre essas duas entidades.23
A mutação D842V é a mutação primária mais comum no exon 18 do gene PDGFRA, sendo responsável por, ao menos, 60% de todas as mutações naquele gene.24,26 Essa substituição simples tem relevância especial porque confere resistência bioquímica aos inibidores de KIT como o imatinibe.
Além de D842V, outras mutações no exon 18 de PDGFRA apresentam uma faixa diversa de sensibilidade ao imatinibe. As mutações nos exons 12 e 14 do gene PDGFRA são homólogas aos exons 11 e 13 do gene KIT, respectivamente; embora sejam incomuns, os dados clínicos in vitro disponíveis sugerem que esses genótipos conseguem respostas duradouras com o imatinibe.24,26
Sinalização Posterior a KIT/PDGFRA
Os caminhos posteriores à sequência PI3K/AKT/mTOR e RAS/RAF/ERK são ativos sob o ponto de vista constitutivo nos casos de GIST, de uma forma dependente de KIT/PDGFRA, por meio da interação direta com os intermediários de sinal PLK e GRB2.27,28
A inibição farmacológica e o nocaute pequeno e sinuoso (hairpin) mediado por RNA do gene KIT resulta na anulação substancial das cascatas de PI3K/AKT/mTOR e RAS/RAF/ERK, dando suporte à dependência oncogênica desses dois caminhos na sinalização de KIT, ao contrário de outros intermediários de sinal que também são encontrados no estado ativado nos casos de GIST (por exemplo, STAT1 e STAT3).
É importante ressaltar que se observa esse efeito nos caminhos posteriores de KIT, independentemente do fato se o estimulador de KIT/PDGFRA no contrafluxo ser sensível ou resistente ao imatinibe, evidenciando o papel crucial dos caminhos PI3K/AKT/mTOR e RAS/RAF/ERK na biologia, na proliferação e na sobrevida dos GISTs.27?29
Em particular, a ativação do KIT no caminho MAPK é extremamente importante para estabilizar a variante 1 de translocação do ETS (ETV1), um fator de transcrição de sobrevida da linhagem indispensável para a transformação oncogênica mediada por KIT.8
Progressão Dos Gist’s
Progressão Citogenética
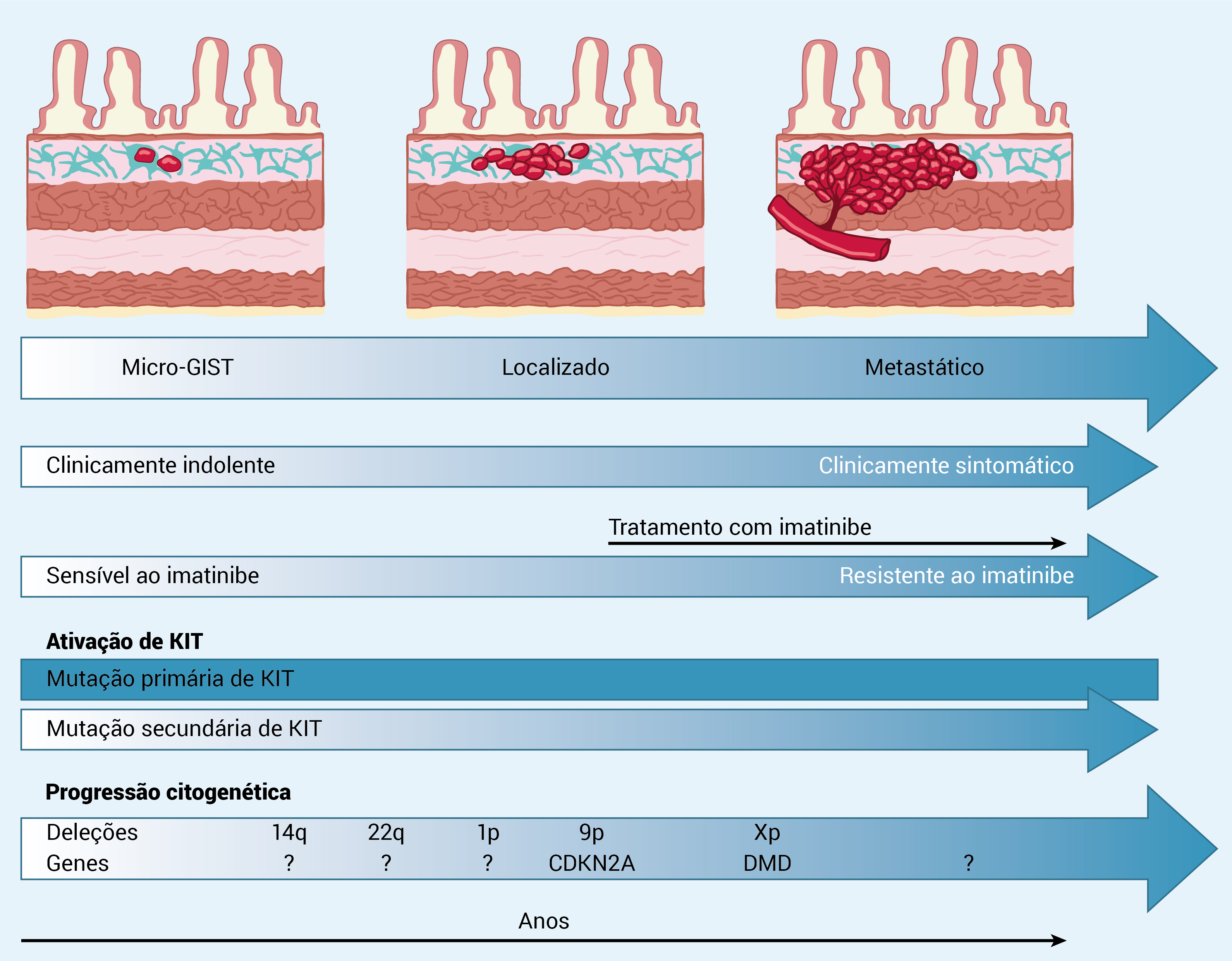
A progressão clínica e biológica dos GISTs representa uma transição gradual que se estende de micro-GISTs a GISTs clinicamente agressivos e metastatizantes. A ativação oncogênica de KIT/PDGFRA é um evento precoce e possivelmente iniciador, sendo que a maior parte dos GISTs permanece na dependência de KIT em toda a progressão tumoral.
Entretanto, as mutações de KIT, consideradas isoladamente, são insuficientes para induzir comportamentos malignos. Consequentemente, são necessários eventos genéticos adicionais para transformar micro-GISTs em tumores com potencial maligno crescente. El-Rifai e colaboradores registraram a progressão citogenética em 95 GISTs: o número médio de aberrações cromossômicas demonstráveis foi de 2,6 em GISTs benignos, 7,5 em GISTs primários malignos e 9 em GISTs metastáticos.30
Deleções dos braços cromossômicos 1p, 14q e 22q são descobertas frequentes em todos os GISTs, seja qual for o grau histológico. Entretanto, outras aberrações, geralmente com alvo na regulação do ciclo celular (isto é, CDKN2A) ? incluindo deleção de 9p, amplificação de 8q e amplificação de 17q ?, são encontradas quase que exclusivamente em GISTs de alto grau (malignos).30
Além disso, há relatos recentes sugerindo que a distrofina é um gene de supressão tumoral, e a presença da deleção do gene (Xp) é quase universal em estágios mais avançados dos GISTs letais de alto grau.31 Provavelmente, a mutação de ativação de KIT ? deleção de 14q ? deleção de 22q ? deleção de 1p ? ganho de 8q ? deleção de 11p ? deleção de 9p ? ganho de 17q ? deleção de Xp seja uma versão simplificada da progressão genética Entretanto, a maior parte dos alvos dessas alterações cromossômicas ainda não foi descoberta pelos investigadores.
Resumindo, dados laboratoriais demonstram que os GISTs representam uma citogenética e uma transição molecular gradual que colaboram de forma progressiva e potencializam a atividade transformadora oncogênica de KIT e PDGFRA.
Progressão dos GISTs para Inibição de KIT: Mutações Secundárias de KIT
A dependência diferenciada dos GISTs em relação à sinalização de KIT/PDGFRA explica as respostas clínicas excepcionais nos tratamentos com imatinibe de primeira linha.17 Todavia, a vasta maioria de pacientes com GISTs em estado avançado, e que se beneficia com o uso de imatinibe, é portadora de doença mensurável persistente que finalmente progride, tipicamente em um período de 2 a 3 anos.
Cabe ressaltar que o gene KIT ainda é o estimulador crucial nos casos de GIST após o insucesso do tratamento com imatinibe. O mecanismo mais comum de resistência aos inibidores da tirosina quinase (TKIs) em pacientes com GISTs implica na expansão de clones tumorais que abrigam uma ampla gama de mutações secundárias no gene KIT que são resistentes ao imatinibe e ocorrem em até 90% de casos.
As mutações resistentes ao imatinibe em agrupamentos de GISTs em duas regiões do domínio da quinase do KIT são as seguintes: a bolsa de ligação com o trifosfato de adenosina (ATP) (codificada pelos exons 13 e 14) e o loop de ativação (codificada pelos exons 17 e 18).9,32
A Figura 2 ilustra a progressão clínica e molecular de um GIST.
GIST: tumor estromal gastrintestinal.
Figura 2 - Progressão clínica e molecular de um GIST.
Portanto, todas as estratégias com foco na ativação de KIT após o insucesso do tratamento com imatinibe são muito úteis nos casos de GIST.33,34 Todavia, há uma heterogeneidade substancial nas mutações secundárias resistentes entre metástases e nas metástases de pacientes individuais; esse fato ressalta a complexidade da população resistente aos TKIs e se apresenta como o principal desafio ao tratamento em pacientes com GISTs após o tratamento com imatinibe de primeira linha.35
Gists Wild Type (WT)
Os GISTs sem mutações no gene KIT ou PDGFRA representam entre 10 e 15% de todos os GISTs e, atualmente, estão agrupados como GISTs Wild Type. Esse tipo de diagnóstico inclui GISTs com uma ampla variedade de eventos genéticos heterogêneos, embora não seja possível distinguir clinicamente entre GISTs WT e GISTs com mutações em KIT/PDGFRA.
Além disso, embora os GISTs possam também apresentar expressão e ativação de KIT, mesmo na ausência de mutações, os mecanismos moleculares subjacentes ainda não são muito claros. Sob a perspectiva biológica, os GISTs WT se subdividem em dois grupos, conforme a presença ou ausência da expressão de SDH.
GIST’S com Deficiência em SDH
O complexo II SDH-ubiquinona é um dos componentes do ciclo de Krebs e da cadeia respiratória mitocondrial formada por quatro subunidades: SDH A, B, C e D. A desativação de qualquer um desses componentes do complexo SDH resulta na desestabilização e na perda funcional de todo o complexo. Dois estudos recentes demonstraram que as mutações no SDH desencadeiam um fenótipo de hipermetilação no DNA recorrente do GIST WT, que difere, de forma surpreendente, sob o ponto de vista mecânico, do GIST estimulado por KIT/PDGFRA.15,36
Da mesma forma, a maior parte dos GISTs WT, ao contrário dos GISTs com mutações em KIT, progride para malignidade sem adquirir aberrações cromossômicas em larga escala,37 dando suporte à hipótese de que a contenção nas alterações epigenômicas, mas não nas alterações genômicas, são o mecanismo crucial nesse subgrupo de tumores.
Os efeitos da hipermetilação sobre a reprogramação e os genes alvos desse fenômeno ainda não são muito claros, embora, tipicamente, os caminhos do fator induzível por hipóxia (HIF) e do receptor do fator de crescimento semelhante à insulina (IGFR) sejam hiperativados em GISTs com deficiência em SDH.9
Os GISTs com deficiência em SDH formam um subgrupo incomum que compreende GISTs pediátricos, a tríade de Carney, a síndrome de Carney-Stratakis e uma pequena proporção de GISTs WT adultos. As mutações de perda de função não são o único mecanismo de desativação da succinato desidrogenase (SDH) e ainda precisam ser compreendidas com maior profundidade.
Independente da subunidade afetada, utiliza-se amplamente a coloração imuno-histoquímica para identificar GISTs com deficiência em SDH.38 Por outro lado, a expressão negativa de SDHA é um preditor exclusivo de mutações neste subgrupo de SDH.39
A grande maioria dos tumores deficientes em SDHB se apresenta em pacientes com idade inferior a 40 anos ? em especial na segunda década de vida ? como tumor gástrico com morfologia epitelioide, padrão de crescimento nodular multifocal, envolvimento frequente de linfonodos locais e predominância em mulheres.40 Além disso, os GISTs WT com deficiência em SDHB correspondem a uma doença com curso crônico, mesmo na presença de metástases peritoneais e hepáticas.40
Os GISTs WT negativos para SDHA são responsáveis por mais de um quarto de GISTs com deficiência em SDH e se caracterizam pela predominância na quarta década de vida, proporção mais baixa entre mulheres e homens e curso mais lento da doença. Ao contrário dos GISTs com deficiência em SDHB, a imunocoloração negativa para SDHA é uma preditora uniforme de mutação de perda de função para a succinato desidrogenase tipo A.39
GIST’S Intactos com SDH
Os GISTs WT que não têm deficiência de SDH abrigam eventos genéticos que ativam a rota RAS/MEK/ERK. O gene NF1 supressor de tumores regula de forma negativa este caminho e a perda de função de NF1 aumenta o risco de incidência de GISTs. Portanto, aproximadamente 7% de pacientes com NF1 desenvolvem GISTs ao longo da vida.12 As mutações em RAS produzem um efeito semelhante e, raramente, foram encontradas em GISTs, provavelmente como evento estimulador principal.41
As mutações oncogênicas BRAF V600E são encontradas em 7 a 15% de GISTs WT positivos para SDH e em 2% de todos os GISTs.42 A preferência pela localização no intestino delgado é única característica distintiva sob o ponto de vista clínico e patológico.
Diagnóstico
Manifestações Clínicas
Não existem sinais ou sintomas específicos ou patognomônicos que confirmem o diagnóstico de GIST e, em grande parte, as manifestações clínicas dependem do tamanho do tumor, da localização do tumor primário e da doença metastática.
Doença Precoce
Muitos pacientes são assintomáticos ou paucissintomáticos nos estágios iniciais da doença, sendo que alguns GISTs são diagnosticados incidentalmente durante as cirurgias ou endoscopias para tratamento de outras condições.43 Isso se deve basicamente ao fato de que o estômago e a cavidade abdominal possuem grandes dimensões e são compatíveis com distensão. Na medida em que evolui, o tumor poderá produzir uma miríade de sintomas associados à porção GI de origem e ao envolvimento das estruturas adjacentes.
Os GISTs ocorrem em todo o trato GI, do esôfago ao ânus, sendo que hemorragias gastrintestinais evidentes ou ocultas, em qualquer ponto, em combinação com a síndrome anêmica subsequente, são as manifestações mais comuns em até dois terços de pacientes.44 A presença de massa e dor abdominal é possível em 40 e 20% de pacientes, respectivamente.
Além disso, os pacientes poderão se apresentar com sintomas de obstrução intestinal em, aproximadamente, 20% de casos, embora a perfuração intestinal não seja uma ocorrência comum. Os outros sintomas são vagos e inespecíficos e incluem o seguinte: alteração no apetite ou nos hábitos alimentares, saciedade pós-prandial, saciedade precoce, sensação de inchaço, náusea, vômito, disfagia, constipação, perda de peso e fadiga. Assim como na maior parte dos sarcomas, a síndrome constitucional (febre, fadiga e perda de peso involuntária) é incomum nos estágios iniciais da doença, mesmo na presença de massas grandes e localizadas.43
Complicações Tardias
Aproximadamente, 15% dos pacientes com GISTs são diagnosticados com doença metastática.5 Entretanto, a disseminação metastática geralmente ocorre durante o curso clínico e é responsável por complicações tardias e, ao final, pela morte dos pacientes. Com frequência, os GISTs metastizam para o fígado e peritônio e raramente para linfonodos regionais, ossos ou pulmões, que são os sítios mais comuns de metástases para a maioria dos sarcomas de tecidos moles.17
O envolvimento peritoneal possivelmente produza ascite, efusão na pleura, dor abdominal ou obstrução intestinal, ao passo que as metástases hepáticas produzem dor abdominal, icterícia ou insuficiência hepática. Nesse estágio, os pacientes se apresentam com a síndrome constitucional.
Descobertas no Exame Físico
De um modo geral, os sinais físicos são eventos tardios e podem refletir a presença de alguma massa primária considerável e/ou talvez sejam consequências de alguma doença metastática, como, por exemplo, envolvimento peritoneal ou hepatomegalia em decorrência de metástases hepáticas.
Essas descobertas não são frequentes no momento da apresentação, e os pacientes talvez tenham aparência saudável no exame físico. Na realidade, a presença de massa abdominal é o sinal mais comum na apresentação clínica, embora ocorra em menos de 10% de pacientes com GISTs. Os sinais remanescentes fazem parte das síndromes descritas nas manifestações clínicas, tais como a síndrome anêmica e a síndrome constitucional.
Testes Laboratoriais
Exames de Sangue
Assim como em outros sarcomas, os testes laboratoriais têm papel limitado nos GISTs porque não há descobertas específicas que levem inequivocamente ao diagnóstico da doença. Além disso, na grande maioria de pacientes, os exames de sangue se enquadram na faixa da normalidade.
Não obstante, a coleta de amostras de sangue ainda é uma parte essencial do exame completo e inclui hematologia, função renal e função hepática. Anemia microcítica causada por deficiência de ferro é a descoberta mais frequente em 9% dos pacientes, embora algumas séries afirmem que esse percentual seja de 85%.43
Estudos Imagiológicos
Os testes radiográficos dos GISTs inicialmente têm como foco as queixas apresentadas pelos pacientes e, em geral, consistem de radiografias simples e/ou tomografia computadorizada (TC) para determinar a origem da dor abdominal ou para excluir a presença de massa abdominal e de obstrução ou perfuração intestinal.
TC abdominal e TC pélvica são os procedimentos de escolha nos casos em que houver suspeita de GIST e devem ser feitos com contrastes orais e intravenosos; isso é extremamente importante para definir a extensão e a localização do tumor primário e das metástases, assim como outras características relacionadas ao comportamento clínico, tais como componentes císticos, ulcerações ou componentes invasivos.
De um modo geral, os GISTs aparecem nas varreduras por TC como massas esféricas bem demarcadas que surgem na camada muscular da parede gastrintestinal. Com frequência, esses tumores se projetam de forma exofítica e/ou intraluminar e possivelmente apresentem ulcerações mucosas sobrejacentes.
Os GISTs maiores quase sempre superam o crescimento do suprimento vascular, criando áreas centrais e extensivas de necrose e hemorragia.45,46 Infelizmente, nos casos de GISTs, embora algumas imagens radiológicas altamente sugestivas sejam inespecíficas e talvez representem diversas entidades, o diagnóstico definitivo é feito por meio de biópsias dos tumores.
Após o diagnóstico de GIST, as imagens são utilizadas na classificação de fases inicial, reclassificação de fases, cirurgia, avaliação, resposta do monitoramento à terapia e acompanhamento de possíveis recorrências.45,46 As metástases pulmonares são distintivamente raras e, portanto, as radiografias torácicas são suficientes para o diagnóstico e acompanhamento; as varreduras por TC também são utilizadas para esse propósito.
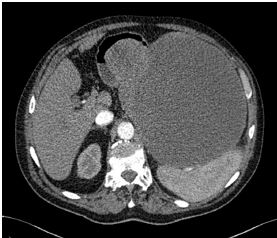
A Figura 3 mostra a varredura por TC realçada por contraste mostrando um GIST.
GIST: tumor estromal gastrintestinal; TC: tomografia computadorizada.
Figura 3 - Varredura por TC realçada por contraste mostrando um GIST gástrico que se apresenta como uma massa abdominal gigantesca. Cabe observar a protrusão no duodeno.
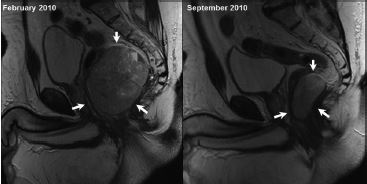
Embora as imagens por TC e as imagens por ressonância nuclear magnética (IRMs) abdominais produzam diagnósticos equivalentes nos casos de GIST,43 a TC é a modalidade imagiológica de escolha,45,46 enquanto que, em geral, reservam-se as IRMs para obtenção de melhores definições anatômicas nas avaliações cirúrgicas em localizações específicas como o reto, conforme a Figura 4.
IRM: imagem por ressonância nuclear magnética; GIST: tumor estromal gastrintestinal.
Figura 4 - IRM mostrando um GIST retal (setas) na linha de base (à esquerda) que responde ao imatinibe adjuvante depois de 8 meses de tratamento (à direita).
Para finalizar, a avaliação da reabsorção da fluorodesoxiglicose (FDG) com auxílio de TC por emissão de pósitrons (PET) é altamente sensível para detectar focos hipermetabólicos e facilita a diferenciação entre tumores ativos e tecidos necróticos ou inativos. Entretanto, não há evidências claras de que a PET forneça informações significativas que não possam ser obtidas por meio das varreduras tradicionais por TC com realce de contraste de boa qualidade.45,46
As respostas à FDG-PET poderão ser observadas em um período de 24 horas após o inicio do tratamento com inibidores de KIT. Consequentemente, a PET talvez seja um instrumento útil para monitorar tratamentos neoadjuvantes nos casos limítrofes de GISTs ressectáveis, em que houver uma janela muito estreita para mudar para alguma terapia alternativa. É imprescindível obter uma PET na linha de base, antes do início da terapia, nas situações em que o médico escolher a PET para monitoramento terapeutico.45,46
Endoscopia e Ultrassonografia Endoscópica
A endoscopia é muito útil para identificar massas gástricas com suspeita de GISTs. Nas endoscopias, os GISTs aparecem como massas submucosas que se projetam no interior do lúmen gástrico, com margens lisas e mucosas sobrejacentes normais. Ulcerações na mucosa também poderão ser encontradas.
Além disso, nas ultrassonografias endoscópicas (USEs), os GISTs se localizam tipicamente na quarta camada do estômago, que corresponde ao próprio músculo, e mais raramente na segunda ou terceira camada. As possíveis características de alto risco das USEs incluem bordas irregulares, espaços císticos, ulcerações, focos ecogênicos e hetererogeneidades.46
Não se recomenda o uso rotineiro de biópsias endoscópicas usando técnicas padrões porque geralmente não coletam tecidos suficientes para fazer diagnósticos definitivos.47 Entretanto, o uso combinado de análise citológica e imuno-histoquímica para CD117 por meio de aspiração por agulha fina (AGF) guiada por USE produz sensibilidade de 82% e especificidade de 100%.47
Biópsia, Patologia e Diagnóstico
Técnica para diagnósticos histológicos. A confirmação histológica de GIST deve ocorrer antes de qualquer manobra terapêutica. As biópsias nos sítios primários, guiadas por USEs bem-sucedidas ou aspiração com agulha fina, são o procedimento preferido se houver acesso aos locais de coleta de material. As biópsias guiadas por imagens percutâneas são apropriadas para a confirmação de doença metastática.46
Nos casos em que as biópsias não fornecerem material suficiente, uma das opções para confirmar o diagnóstico é a incisão laparoscópica ou laparotomia e evitar cirurgia para o tratamento de doenças que não precisam de intervenção cirúrgica (por exemplo, linfoma, tumor de células germinativas, fibrose mesentérica). A amostra dos tumores dever ser fixada em uma proteção de formalina a 4% (a fixação de Bouin não deve ser usada porque dificulta a análise molecular).45
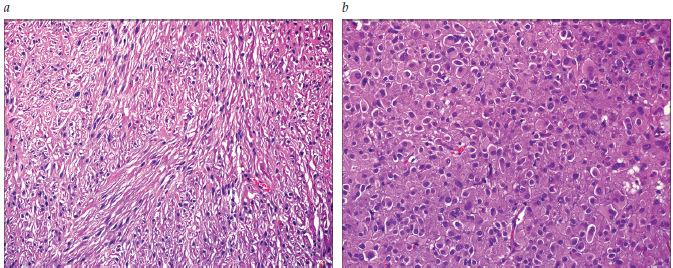
Exame morfológico. Os GISTs se estendem em um amplo espectro morfológico que varia desde a aparência de célula fusiforme (77%) à morfologia epitelioide (8%) ou a uma histologia mista (15%).1 Observa-se a morfologia epitelioide com mais frequência no estômago. A atipia nuclear é relativamente incomum.
Atualmente, obtém-se a taxa mitótica com base no número de mitoses em uma área de 5mm2, que equivale a 40 campos de alta potência em ampliações de 40 vezes nos microscópios com campo de tamanho tradicional. No diagnóstico, a maioria dos GISTs apresenta índices mitóticos baixos ou muito baixos (=5 mitoses/5mm2).1,48 Tipicamente, os tumores com deficiência em SDH têm morfologia epitelioide, multinodularidade e envolvimento mural plexiforme.48
A Figura 5 mostra a coloração com hematoxilina e eosina de um GIST.
GIST: tumor estromal gastrintestinal.
Figura 5 - Coloração com hematoxilina e eosina de um GIST fusocelular (A) e epitelioide (B).
Imuno-histoquímica. O diagnóstico morfológico fundamentado em exames microscópicos de secções histológicas é o padrão utilizado no diagnóstico de GIST, com suporte de imuno-histoquímica para CD117. De um modo geral, aproximadamente 95% das colorações de GISTs para CD117 (KIT) apresentam um padrão citoplasmático difuso e, em menor escala, uma expressão membranosa ou perinuclear.48
O anticorpo DOG1 eventualmente poderá ser incluído no painel inicial, embora seja fortemente recomendado para os casos negativos para CD117, nos quais esse anticorpo se expressa em um terço dos tumores.49 Além disso, 80% também expressam CD34; 30%, actina; 10%, S-100; e 4%, desmina.1,48
Diagnóstico molecular. A análise mutacional das mutações em KIT e PDGFRA é fortemente recomendada, levando-se em consideração que o tipo de mutação em pacientes com GISTs localizados ou metastáticos é um preditor de resposta à terapia com TKI.20,50,51
Além disso, a análise mutacional de mutações conhecidas envolvendo KIT e PDGFRA poderá confirmar o diagnóstico de GIST, principalmente nos casos de tumores no mesênquima gastrintestinal com expressões negativas para CD117 e DOG1.
Todavia, os status das mutações de KIT e PDGFRA não determinam o potencial biológico ou o curso clínico dos GISTs e, portanto, não é recomendado como fator prognóstico. Os casos com suspeita de GIST e que apresentarem características histopatológicas complexas ou atípicas devem ser avaliados em centros especiais com experiência no diagnóstico de sarcomas.
Diagnóstico Diferencial
Os GISTs são os sarcomas mais comuns no trato GI; portanto, é a primeira suspeita diagnóstica de qualquer neoplasma mesenquimatoso maligno nessa localização.1 Entretanto, a ampla variedade morfológica dos GISTs exige diagnóstico diferencial rigoroso de quaisquer tumores subepiteliais que surgem no trato gastrintestinal.
Os GISTs de células fusiformes possivelmente se assemelhem a leiomiossarcomas, tumores malignos na bainha de nervos periféricos (MPNST) ou carcinomas “sarcomatoides”, ao passo que os GISTs epitelioides devem ser distinguidos de tumores perivasculares de células epitelioides (PEComa) ou tumores carcinoides, conforme o Quadro 3.1,48 A interpretação das secções coloridas com hematoxilina e eosina apenas com luz microscópica orientam o diagnóstico, embora o uso de imuno-histoquímica seja imprescindível para fazer o diagnóstico definitivo.
Quadro 3
|
Diagnóstico Diferencial Relevante de Tumor Estromal Gastrintestinal | ||
|
Aparência morfológica |
Benigno |
Maligno |
|
Fusiforme |
Schwanoma, leiomioma |
Fibromatose desmoide, miofibroblástico inflamatório, leiomiossarcoma, DDLPS, angiossarcoma, carcinoma sarcomatoide, MPNST. |
|
Epitelioide |
- |
Tumor carcinoide, paraganglioma, PEComa, melanoma, MPNST. |
|
Outras |
- |
Melanoma. |
DDLPS: lipossarcoma desdiferenciado; MPNST: tumor maligno na bainha de nervos periféricos; PEComa: tumor perivascular de células epitelioides.
Gerenciamento
O gerenciamento de GISTs deve ser feito em centros de referência e/ou em redes de referência que compartilhem especialidades multidisciplinares e que tratem anualmente um grande número de pacientes.
Tumores Estromais Gastrintestinais Localizados
Cirurgia
Ressecção cirúrgica completa é o tratamento padrão e a única terapia potencialmente curativa para GISTs localizados. A meta principal da operação é atingir uma ressecção R0: remoção completa com margens microscopicamente negativas e pseudocápsulas intactas. Para atingir essa meta, a extensão da cirurgia geralmente envolve ressecção por cunha no estômago ou no intestino sem deixar margens largas para adenocarcinomas.
A margem ideal de ressecção de GISTs ainda é desconhecida. Na realidade, sob a ótica microscópica, não há nenhuma diferença em termos de sobrevida sem recorrências (SSRs) depois de ressecções completas, com margens negativas ou microscopicamente positivas (ressecção R0 ou R1, respectivamente), enquanto que as ressecções macroscópicas incompletas certamente estão associadas aos piores resultados.52
As novas excisões com R1 depois de cirurgias não estão bem definidas e deverão ser oferecidas aos pacientes somente se não implicarem em grandes riscos ou em consequências funcionais.46 Além disso, o rompimento de GISTs na cavidade abdominal durante as cirurgias está associado ao risco de recorrência de quase 100% e, portanto, recomenda-se evitar esse tipo de procedimento.53
A linfadenectomia de rotina é desnecessária porque as metástases nodais são raras, exceto nos casos incomuns de GISTs pediátricos. Para finalizar, na aplicação de laparotomia ou laparoscopia, o abdome e o fígado deverão ser totalmente explorados para que seja possível identificar e remover quaisquer deposições metastáticas no peritônio que não haviam sido previamente detectadas.
Todos os GISTs com 2cm ou mais devem ser removidos, levando-se em consideração que nenhum deles é benigno. Entretanto, não se chegou a um consenso sobre o gerenciamento de GISTs menores. A presença de micro-GISTs gástricos incidentais (<1cm) é comum em pacientes adultos6 e deve ser acompanhada periodicamente com endoscopia.
Por outro lado, sem avaliação com biópsias e determinação da taxa mitótica, o comportamento de GISTs entre 1 e 2cm é imprevisível. Os riscos e benefícios da cirurgia versus observação devem ser discutidos com os pacientes nos casos em que o estômago for a localização primária. Todavia, os GISTs no intestino delgado, colo e reto deverão ser removidos seja qual for o tamanho, tendo em vista o risco de recorrência e o grande potencial maligno.
Imatinibe adjuvante/neoadjuvante
A ressecção completa é possível na maior parte dos casos de GISTs localizados; esse procedimento é, atualmente, o padrão de tratamento. Entretanto, de acordo com algumas séries, a despeito da ressecção macroscópica completa, aproximadamente 50% dos indivíduos poderão desenvolver doença recorrente após a cirurgia.
No entanto, esse tipo de risco varia bastante conforme as características relacionadas a tamanho, localização e contagem mitótica. As cirurgias com R0 e R1 estão associadas à taxas de sobrevida geral em 5 anos variando de 34 a 64%, enquanto que a ressecção R2 está associada a uma taxa de sobrevida geral em 5 anos de 8%.54
Consequentemente, o sucesso do imatinibe nos casos de doença em estado avançado, em combinação com a impossibilidade de atender às necessidades médicas para impedir recorrência após a cirurgia, despertou o interesse no uso perioperatório do medicamento, que inclui tratamento pós-operatório de pacientes com risco elevado de recorrência após a ressecção completa de GISTs primários e terapia pré-operatória em pacientes com doença não ressectável ou com tumores no limiar da ressecção.
O Quadro 4 apresenta os sistemas de estratificação de risco usados em GISTs.
Quadro 4
|
Sistemas de Estratificação de Risco Usados em Tumor Estromal Gastrintestinal | |||
|
Grupo de risco |
Características dos GISTs | ||
|
Tamanho do tumor (cm) |
Contagem mitótica (50 HPF) |
Sítio do tumor | |
|
Critérios consensuais da NIH55 |
|
|
|
|
Risco muito baixo |
<2 |
<5 |
- |
|
Risco baixo |
2?5 |
<5 |
- |
|
Risco intermediário |
<5 |
6?10 |
- |
|
|
5?10 |
<5 |
|
|
Risco elevado |
>10 |
Qualquer contagem |
- |
|
|
Qualquer tamanho |
>10 |
- |
|
|
>5 |
>5 |
|
|
Critérios do AFIP57 |
|
|
|
|
Grupo 1 |
<2,0 |
=5 |
- |
|
Grupo 2 |
2,1?5,0 |
=5 |
- |
|
Grupo 3a |
5,1?10,0 |
=5 |
- |
|
Grupo 3b |
>10,0 |
=5 |
- |
|
Grupo 4 |
<2,0 |
>5 |
- |
|
Grupo 5 |
2,1?5,0 |
>5 |
- |
|
Grupo 6a |
5,1?10,0 |
>5 |
- |
|
Grupo 6b |
>10,0 |
>5 |
- |
|
Critérios Consensuais Modificados da NIH56 |
|
|
|
|
Risco muito baixo |
<2 |
=5 |
Qualquer sítio |
|
Risco baixo |
2,1?5,0 |
=5 |
Qualquer sítio |
|
Risco intermediário |
=5 |
6?10 |
Gástrico |
|
|
5,1?10,0 |
=5 |
Gástrico |
|
Risco elevado* |
>10,0 |
Qualquer contagem |
Qualquer sítio |
|
|
Qualquer tamanho |
>10 |
Qualquer sítio |
|
|
>5,0 |
>5,0 |
Qualquer sítio |
|
|
=5 |
>5 |
Não gástrico |
|
|
5?10 |
=5 |
Não gástrico |
AFIP: Armed Forces Institute of Pathology; GIST: tumor estromal gastrintestinal; NIH: National Institutes of Health.
*Qualquer tamanho, sítio ou contagem mitótica na presença de rompimento tumoral.
Existem vários critérios que foram criados especificamente para definir melhor o subgrupo de pacientes de GISTs com maior probabilidade de recorrência e que poderiam ter mais benefícios com o tratamento perioperatório à base de imatinibe.55?57 De um modo geral, e conforme a estratificação de risco e os critérios de inclusão utilizados em testes clínicos recentes, os pacientes com GISTs são classificados como de risco baixo, risco intermediário e risco elevado de recorrência após a cirurgia.
Imatinibe adjuvante. Três testes clínicos randomizados de fase III avaliaram o papel de 400mg/dia de imatinibe no contexto adjuvante; o teste Z9001 do American College of Surgeons Oncology Group (ACOSOG),58 o teste XVIII/AIO (Arbeitsgemeinschaft Internistische Onkologie) do Scadinavian Sarcoma Group (SSG),59 e o teste 62024 da European Organization for Research and Treatment of Cancer (EORTC).60
No teste Z9001, 713 pacientes que haviam feito cirurgia para remoção de GISTs de 3cm ou mais foram distribuídos aleatoriamente para imatinibe ou placebo durante 1 ano após a intervenção cirúrgica.58 O imatinibe adjuvante melhorou a sobrevida sem recorrências (SSR): a SSR depois de 1 ano foi de 98% no grupo de imatinibe e de 83% no grupo de placebo, e a taxa de risco (HR) entre os braços do teste foi de 0,35 (intervalo de confiança [IC] de 95%, 0,22 a 0,53; p <0,001).
Não houve nenhuma diferença na sobrevida total (ST) entre os dois grupos, provavelmente devido ao cruzamento de informações entre o grupo de placebo e o grupo de imatinibe após a detecção de recorrência de GIST, e também porque talvez a concepção original do teste não permita determinar essas diferenças. Uma das maiores críticas que se fez a este estudo foi a inclusão de uma ampla gama de pacientes de GISTs com riscos diferentes.
O teste clínico XVIII/AIO de fase III do SSG investigou a seguir se a duração do imatinibe adjuvante teve algum impacto sobre as recidivas após a cirurgia.59 Nesse estudo, 400 pacientes com GISTs, definidos como de alto risco conforme a classificação consensual do National Institutes of Health ou com rompimento tumoral, foram alocados para o grupo de imatinibe, 400mg/dia, por períodos de 12 ou 36 meses.
Nos pacientes do grupo de 3 anos, a sobrevida de 5 anos sem recorrência foi de 65,6%, em comparação com 47,9% no grupo de 1 ano. É interessante observar que, embora o teste não tenha sido concebido para avaliar diferenças na sobrevida total, a análise mostrou que o tratamento de 3 anos atingiu uma ST de 5 anos em 92% dos casos em comparação com 81,7% no grupo de 1 ano de tratamento.
Em face desses resultados, a National Comprehensive Cancer Network (NCCN) dos EUA e a European Society of Medical Oncology (ESMO) recomendam 3 anos de tratamento adjuvante com imatinibe em pacientes de alto risco.45,46 A NCCN também indica esse tipo de tratamento considerando o uso de imatinibe adjuvante em pacientes de risco intermediário.
Os resultados da primeira análise provisória do estudo EORTC 62024 foram divulgados recentemente.60 Esse estudo de fase III incluiu pacientes com GISTs de risco intermediário e de risco elevado que foram randomizados para tratamento de 2 anos com imatinibe ou para observação após a ressecção cirúrgica.
Cabe observar que esse teste incorporou a sobrevida sem imatinibe (SSI) como desfecho primário, definido como o tempo a partir do qual se iniciou o tratamento com um inibidor da tirosina quinase diferente. Esse novo desfecho para o contexto adjuvante teve como objetivo a incorporação de resistência secundária. O estudo confirmou que, embora o imatinibe adjuvante tenha causado impacto na ausência de recidiva no curto prazo, não conseguiu demonstrar diferenças na sobrevida sem imatinibe e na sobrevida total depois de 5 anos de acompanhamento.
De um modo geral, o benefício da sobrevida sem recorrências (SSR) parece ser mais evidente durante o tratamento com imatinibe e logo após a terapia, ao passo que as curvas tendem a se sobrepor com acompanhamento mais longo, na medida em que aumenta a recorrência da doença em um período de 6 a 12 meses após a descontinuação no uso de imatinibe adjuvante, independentemente do tempo de tratamento.
Portanto, essa descoberta desperta alguma preocupação sobre o mecanismo de ação do imatinibe adjuvante nos casos de GIST, tendo em vista que ainda não se sabe se as recorrências estão sendo realmente evitadas ou apenas adiadas. Os próximos estudos deverão ter como foco principal a definição clara da duração ideal do imatinibe adjuvante.
Outras áreas de incerteza ainda precisam ser avaliadas por meio de estudos prospectivos que deverão questionar a conveniência de usar doses acima de 400mg no contexto adjuvante ou se apenas os pacientes com mutações KIT/PDGFRA têm algum benefício com a terapia à base de imatinibe adjuvante.
Imatinibe neoadjuvante. Baseando-se nas altas taxas de respostas que foram observadas com o imatinibe em pacientes com GISTs metastáticos, o uso pré-operatório de imatinibe tem o objetivo de reduzir o volume tumoral para facilitar a ressecção cirúrgica completa ou de aumentar a probabilidade de preservação do órgão que era inicialmente não ressectável ou de doença no limiar da ressecção, em especial nos casos em que o plano de tratamento prever a realização de cirurgia mutiladora.
Consequentemente, o uso de imatinibe neoadjuvante é uma opção a ser considerada em pacientes com GISTs que tenham tumor retal ou duodenal para evitar ressecções que afetam adversamente a qualidade de vida, assim como em pacientes com GISTs gástricos de grande porte, que provavelmente precisam fazer gastrectomia total ou subtotal.
Entretanto, é extremamente relevante considerar a inexistência de testes randomizados que avaliem os benefícios do imatinibe neoadjuvante e que todas as evidências vêm de séries retrospectivas e de alguns testes prospectivos de fase II com braço único. Esses estudos mostram que a maior parte dos GISTs locais em estado avançado encolhe ou estabiliza as dimensões depois de 6 a 12 meses após o uso pré-operatório de imatinibe, o que possibilita fazer a cirurgia subsequente.61,62
Portanto, a decisão de usar imatinibe na fase pré-operatória terá que ser tomada em bases individuais, e os diversos aspectos precisam ser abordados com muito cuidado: em primeiro lugar, a avaliação de respostas é imprescindível para minimizar os riscos, considerando que qualquer atraso poderá dificultar a cirurgia nas situações em que o imatinibe não conseguir encolher ou estabilizar as dimensões tumorais.
Aparentemente, as técnicas PET/TC na linha de base e nas primeiras 4 semanas de terapia com imatinibe são as opções mais sensíveis.63 Nesse aspecto, a genotipagem na linha de base e a identificação de mutações não responsivas (por exemplo, PDGFRA D842V) são altamente recomendadas.
Em segundo lugar, a estratificação de risco e a indicação da terapia à base de imatinibe adjuvante provavelmente sejam imprecisas, tendo em vista que as biópsias feitas antes do tratamento e os espécimes coletados após o tratamento não permitem fazer avaliações completas das taxas mitóticas tumorais. Além disso, o benefício do imatinibe adjuvante sobre a sobrevida total ainda não está muito claro, considerando que, em alguns estudos, os pacientes receberam esse tipo de medicamento por 2 anos.
Tumores de Células Estromais Gastrintestinais Avançados, Metastáticos ou Recorrentes
Os GISTs são paradigmas de tumores quimicamente resistentes que apresentam menos de 5% de respostas e sobrevida mediana de 14 meses.64 Nos últimos 15 anos, pôde-se verificar como a compreensão biológica dos GISTs e o desenho racional dos medicamentos com foco nos mecanismos oncogênicos mais críticos melhoraram os resultados de forma dramática.
Terapia com Foco
O tratamento de GISTs foi revolucionado pela descoberta de que a ativação mutacional no receptor da tirosina quinase de KIT ou PDGFRA é o evento oncogênico mais importante que ocorre nessas células cancerígenas.19 Portanto, a inibição de KIT/PDGFRA com terapias focadas, na forma de inibidores de pequenas moléculas, produz benefícios rápidos, dramáticos e sustentados em pacientes com GISTs.
Os medicamentos imatinibe, sunitinibe (ambos aprovados em todo o mundo) e regorafenibe (com aprovação da Food and Drug Administration [FDA] apenas para uso nos EUA) são os tratamentos atuais de primeira, segunda e terceira linhas de GISTs avançados/metastáticos.
Imatinibe. O imatinibe (Gleevec nos EUA, Glivec em outros países; Novartis Oncology, Basileia, Suíça), um inibidor de pequenas moléculas, comercializado para uso oral, com atividade contra a mutação KIT, PDGFRA e as quinases ABL, foi o primeiro inibidor da tirosina quinase aprovado pela FDA para tratamento de GISTs metastáticos positivos para a mutação KIT e/ou GISTs não ressectáveis, após a demonstração em um teste de fase III (estudo B2222) da resposta sustentada ao imatinibe (400mg/dia).17
Nesse estudo, 147 pacientes com GISTs foram distribuídos aleatoriamente para receber tratamento com 400 ou 600mg/dia de imatinibe. Depois de 9 meses de um acompanhamento mediano, 54% dos pacientes responderam ao tratamento, 28% apresentaram doença estável e somente 14% tiveram progressão da doença nos primeiros 6 meses de tratamento.17
O Quadro 5 apresenta um resumo da atividade desses medicamentos aprovados.
Quadro 5
|
Atividade Comparativa de Regimes Aprovados para Tumores de Células Estromais Gastrintestinais | ||||
|
Resultado |
Imatinibe65 (n = 147) |
Sunitinibe34 (n = 207) |
Regorafenibe33 (n = 133) |
Nova administração de imatinibe73 (n = 41) |
|
ORR (%) |
68,1 |
7 |
4,5 |
0 |
|
SD12 semanas (%) |
15,6 |
53 |
48,1 |
32 |
|
TTP/PES (meses) |
24 |
6,1 |
4,8 |
1,8 |
ORR: taxa de resposta total (resposta total e resposta parcial); PFS: sobrevida sem progressão; SD: doença estável; TTP: tempo para progressão do tratamento.
Não se observou nenhuma diferença entre as duas doses; portanto, a dose de 400mg/dia de imatinibe foi selecionada como regime padrão para o tratamento de GISTs.
Um estudo posterior de acompanhamento confirmou os benefícios do imatinibe no longo prazo em pacientes com GISTs avançados e GISTs metastáticos, mostrando um tempo mediano de progressão do tratamento (TTP) de 24 meses e uma sobrevida total mediana estimada de 57 meses65 ? claramente superior à sobrevida de 10 a 20 meses que prevalecia na era pré-imatinibe.64 Volumes tumorais mais baixos na linha de base eram preditores de tempo mais longo de progressão do tratamento e de sobrevida total.
Dois testes separados de fase III deram suporte às observações prévias, assim como estudaram a eficácia de doses mais elevadas de imatinibe (800 versus 400mg/dia).66,67 Em ambos os testes, as doses diárias de 400mg versus 800mg foram comparáveis em termos de benefícios clínicos e sobrevida, embora uma pequena vantagem na sobrevida sem progressão (PFS), mas significativa sob a ótica estatística, tenha sido observada com doses mais elevadas no teste europeu.67
É interessante observar que, aproximadamente, 30% de pacientes com GISTs que progrediram com a dose padrão conseguiram respostas parciais ou estabilização da doença, embora para uma PFS mediana de 5 meses.67 Uma metanálise posterior que analisou 1.640 pacientes confirmou a vantagem pequena, porém significativa, na PFS em pacientes que haviam sido tratados com doses mais elevadas, com taxas de risco de 0,89 (IC de 95%; 0,79 a 1,00; p = 0,04)68; entretanto, não houve nenhuma diferença na sobrevida total, e o perfil de toxicidade foi favorável às doses mais baixas.
À luz desses resultados, a administração diária de 400mg de imatinibe é a dose padrão inicial para tratamento de pacientes com GISTs metastáticos ou em estado avançado. A escalada das doses para 800mg/dia é uma opção razoável para aplicação em pacientes que estiverem progredindo com 400mg/dia. Além disso, a menos que ocorram toxicidades significativas, recomenda-se a terapia contínua até a progressão da doença, tendo em vista que a descontinuação no uso de imatinibe poderá resultar na progressão da doença em 12 meses na maior parte dos pacientes.69
Cabe ressaltar que um subgrupo de pacientes com GISTs consegue controlar a doença no longo prazo, conforme demonstraram relatórios preliminares atualizados de um teste inicial de fase III e do teste americano de fase III, em que, aproximadamente, um quinto dos pacientes permaneceram sem progressão da doença depois de 10 anos.70,71
O genótipo do GIST é um preditor da resposta ao imatinibe em pacientes com GISTs metastáticos e, além disso, o imatinibe tem impacto positivo em pacientes com a mutação exon 11 no gene KIT. A presença do genótipo mutante exon 11, em comparação com pacientes que abrigam uma mutação exon 9, ou nenhuma mutação determinável em KIT ou PDGFRA, se correlaciona com melhora nas taxas de respostas (72 versus 44 e 45%, respectivamente).
O TTP é 25 versus 17 e 13 meses, respectivamente, e a sobrevida total, mediana de 60 versus 30 e 49 meses, respectivamente.21 Somente os pacientes com GISTs com a mutação exon 9 no gene KIT se beneficiaram com doses iniciais mais elevadas de imatinibe na taxa de resposta total (OR) e na PFS, novamente sem nenhuma diferença na sobrevida total.68
Outros fatores associados de forma independente com uma melhor sobrevida incluem idade mais jovem, gênero feminino, contagem baixa de neutrófilos, albumina normal, estado de bom desempenho e volume tumoral menor no início do tratamento com imatinibe.65,66,68
Resistência primária e secundária ao imatinibe. Aproximadamente, 10 a 15% de pacientes com GISTs desenvolvem resistência primária ao imatinibe e progridem em um período de 6 meses após o início da terapia.17
A resistência primária foi observada em todos os subtipos de genótipos, embora, mais provavelmente, seja encontrada nos casos de GISTs Wild Type (KIT/PDGFRA não são mais estimulados por tumorigênese), na substituição de FRA D842V (envolve mudanças conformacionais que impedem a ligação do imatinibe) e na mutação exon 9 de KIT, embora, nessa última situação, a dosagem inadequada talvez seja responsável por alguns casos.9
Todavia, a imensa maioria de pacientes com GISTs metastáticos ou em estado avançado apresenta benefícios iniciais e sustentados com o imatinibe. Mesmo assim, 80 a 85% desses pacientes acabam progredindo, tipicamente em 2 a 8 anos. O mecanismo mais comum de resistência ao imatinibe em pacientes com GISTs implica na expansão de clones tumorais que abrigam uma gama de mutações secundárias em KIT ou PDGFRA, e que são resistentes ao imatinibe.
Nos casos de GISTs, as mutações de KIT resistentes ao imatinibe se agrupam em duas regiões do domínio da quinase de KIT: a bolsa de ligação com ATP (codificada pelos exons 13 e 14) e o loop de ativação (codificado pelos exons 17 e 18), sendo que essas mutações ocorrem quase que exclusivamente no mesmo gene e alelo, como a mutação primária com estimulação oncogênica.9
Além disso, há uma heterogeneidade substancial nas mutações secundárias resistentes, seja entre metástases e ou no interior de metástases de pacientes individuais, ressaltando a complexidade da população resistente ao inibidor da tirosina quinase (TKI), e surge como o principal desafio ao tratamento em pacientes com GISTs após o tratamento com imatinibe.35
Sunitinibe. Após o insucesso do tratamento com imatinibe, o sunitinibe (Stent, marca da Pfizer Inc.), um TKI de moléculas pequenas com alvos múltiplos e com atividade potente contra KIT e PDGFRA, entre várias outras quinases, foi aprovado para uso em todo o mundo para tratamento de GISTs metastáticos em pacientes com resistência ou intolerância ao imatinibe.
No teste crítico de fase III, 312 pacientes com GISTs refratários ou intolerantes ao imatinibe foram randomizados para receber sunitinibe ou placebo. O sunitinibe foi administrado na dose diária de 50mg, 4 semanas sim e 4 semanas não. A despeito da baixa taxa de resposta objetiva no grupo de sunitinibe (ORR de 7%), o tempo para progressão do tratamento (TTP), que era o desfecho primário, foi quatro vezes mais elevado com sunitinibe em comparação com placebo, isto é, 27 versus 6 semanas, respectivamente.34
A dosagem diária contínua de 37,5mg de sunitinibe também é ativa no mesmo contexto, conforme mostrou o teste clínico randomizado de fase III que produziu uma taxa de benefícios clínicos de 53% e uma sobrevida sem progressão (PFS) de 34 semanas.72
É interessante observar que, após o insucesso com imatinibe, o genótipo da quinase dos GISTs também se correlaciona com a atividade do sunitinibe. Por outro lado, os benefícios clínicos, a PFS mediana e a sobrevida total foram significativamente mais elevados nos pacientes com exon 9 primária de KIT ou mutação WT em KIT/PDG-FRA.
Além disso, houve também alguma correlação entre mutações secundárias de KIT e a resposta ao sunitinibe, e apenas as mutações secundárias na bolsa de ligação de ATP com KIT aparentavam ser especialmente sensíveis ao sunitinibe.50
Regorafenibe. O regorafenibe (Stirvarga, marca da Bayer HealthCare Pharmaceuticals Inc) é um inibidor oral ativo multiquinase com atividade contra uma grande variedade de quinases, incluindo KIT e PDGFRA. Um teste multicentro internacional de fase III (teste GRI) randomizado e controlado por placebo estudou a atividade do regorafenibe em GISTs.33
Sob o ponto de vista estatístico, a PFS mediana foi significativamente mais longa nos pacientes que haviam sido tratados com regorafenibe por 4,8 meses, em comparação com 0,9 meses no grupo de placebo (HR de 0,27; IC de 95% 0,19 a 0,39; p <0,0001). Não se observou nenhuma diferença na sobrevida total mediana entre os grupos devido ao desenho transversal.
Assim como se observou com o sunitinibe, a taxa de resposta total foi baixa (4,5%), considerando que a maior parte dos benefícios foi atribuída à estabilização da doença. Com base nesses resultados, o regorafenibe obteve aprovação da FDA para tratamento de GISTs metastáticos ou em estado avançado após a falha ou intolerância ao imatinibe e ao sunitinibe, nas doses recomendadas de 160mg, 1x/dia, durante os primeiros 21 dias de cada ciclo de 28 dias.46
Recarga com imatinibe. Apesar dos insucessos anteriores, a retomada no uso de imatinibe com finalidade paliativa tem sido uma prática comum com base na evidência de progressão rápida e agravamento dos sintomas após a descontinuação no uso de Inibidores da tirosina quinase, provavelmente por causa do crescimento externo rápido de clones sensíveis ao imatinibe.63
O teste RIGHT (Rechallenge of Imatinib in GIST Having no Effective Treatment) de fase III, randomizado e controlado por placebo, abordou formalmente essa questão em 81 pacientes com GISTs refratários ao imatinibe.73 A sobrevida sem progressão foi significativamente mais longa no braço do imatinibe em comparação com placebo, 1,8 meses versus 0,9 mês, em combinação com uma taxa de controle da doença de 32% depois de 12 semanas.
Não houve melhora significativa na resposta objetiva de sobrevida total. Portanto, a recarga com imatinibe poderá ser usada como abordagem terapêutica útil para paliação e para minimizar o agravamento dos sintomas nas situações em que não houver outras opções disponíveis; todavia, não existem dados sobre a qualidade de vida.
Novas terapias para tratamento de GISTs refratários ao TKI (imatinibe, sunitinibe, regorafenibe) são necessidades clínicas que ainda não se materializaram, embora haja incentivos para o tratamento desses pacientes em testes clínicos. Nesses casos, recomenda-se o encaminhamento dos pacientes para uma equipe multidisciplinar com experiência no tratamento de GISTs.
Cirurgia
Possivelmente, a ressecção de doenças metastáticas com o objetivo de obter respostas completas produza benefícios em pacientes com GISTs, seja por meio de respostas aos TKIs ou pela progressão focal, porém não é uma opção que poderá ser considerada para uso em pacientes com progressão generalizada da doença enquanto estiverem recebendo um TKI.
Para esses casos, e na ausência de testes controlados, a escolha do tratamento terá que ser feita em bases individuais, preferencialmente em centros de encaminhamento. Além disso, a ressecção, mesmo que seja completa, não elimina a necessidade de tratamento contínuo com terapia à base de TKIs. Não obstante, ao aplicar essas medidas, 33 a 80% de pacientes ficarão livres de progressão por cerca de 1 ano.74
Complicações
Imatinibe
De um modo geral, o imatinibe é bem tolerado; a maior parte dos efeitos colaterais é leve, e a maioria dos pacientes poderá continuar o tratamento por vários anos sem alterações na dosagem ou interrupção no uso do medicamento. Além disso, o perfil de efeitos colaterais tende a melhorar nas terapias prolongadas.17,65
Diarreia leve, retenção de líquidos, náusea, fadiga, cãibras musculares, erupção cutânea, dor abdominal e anemia são as toxicidades mais comuns relacionadas ao tratamento, embora os pacientes que fazem terapias com doses mais elevadas apresentam mais queixas de efeitos colaterais.
Sunitinibe e Regorafenibe
Como decorrência do espectro mais amplo da inibição alvo, a gama de efeitos colaterais do sunitinibe e regorafenibe é maior em comparação com o imatinibe, embora a maior parte desses efeitos varie de leve a moderada em termos de gravidade e de gerenciamento por meio de alterações na dosagem ou da interrupção o uso desses medicamentos.
Os eventos adversos mais comuns relacionados ao tratamento com sunitinibe são fadiga, diarreia, descoloração da pele e náusea.34 Da mesma forma, o perfil de toxicidade do regorafenibe é consistente com o de outros inibidores da quinase com espectro alvo semelhante.
Os eventos adversos mais comuns são reação na pele das mãos e dos pés, hipertensão, diarreia, fadiga e mucosite oral.34 A redução na dosagem foi necessária em 11% dos pacientes que recebem sunitinibe e em 20% que recebem regorafenibe. Entretanto, o índice de descontinuação no tratamento é baixo.
Prognóstico
Todos os GISTs com mais de 2cm têm algum risco de recorrência, embora o comportamento clínico seja altamente variável. Vários critérios prognósticos são utilizados pelos médicos para identificar tumores com maior probabilidade de apresentar comportamentos malignos, tanto no contexto localizado como no metastático.
Doença Localizada
Fatores Prognósticos Clínicos e Patológicos para Recorrência de Tumores de Células Estromais Gastrintestinais
Os fatores prognósticos independentes mais importantes para recorrência pós-cirúrgica de GISTs são taxa de proliferação tumoral, tamanho do tumor e sítio do tumor.55?57 Avalia-se a taxa de proliferação por meio da contagem mitótica usando-se a área total de 5mm2 ou 25 campos de alta potência nos microscópios modernos com amplo campo visual.45,46
O tamanho e o sítio do tumor também são fatores prognósticos importantes. De um modo geral, os GISTs gástricos apresentam resultados mais favoráveis em comparação com os GISTs que surgem em outras localizações tais como intestino delgado, colo ou reto. Além disso, os rompimentos tumorais causados por perfurações ou cirurgias estão associados ao risco de recidiva de virtualmente 100%.53
Existem três esquemas de classificação que estimam o risco de recorrência de GISTs depois de cirurgias. Os critérios do Armed Forces Institute of Pathology (AFIP)57 e os critérios consensuais modificados56 do National Institutes of Health (NIH) incluem proliferação, tamanho e sítio dos tumores, enquanto que os critérios consensuais originais do NIH não incluem o sítio tumorais. Todavia, esses três métodos são comparáveis e preditores razoáveis.54
Análise das Mutações Tumorais
Embora as mutações em KIT e PDGFRA sejam preditoras de resultados clínicos, ainda não está suficientemente claro se sua significância prognóstica é independente de outros fatores prognósticos bem definidos.
Os pacientes com mutações em PDGFRA, e particularmente GISTs com a mutação D824V no exon 18, geralmente apresentam resultados favoráveis, ao passo que os GISTs com a mutação exon 9 de KIT e deleção da mutação exon 11, envolvendo os códons 557 a 558, estão associados a taxas elevadas de recidiva depois da cirurgia.54
Doença Metastática
Além do prognóstico, a análise das mutações tumorais é bastante relevante para identificar as mutações em KIT/PDGFRA que conferem sensibilidade ao imatinibe, que determina o prognóstico em pacientes com GISTs não removíveis.
|
Informações Financeiras: César Serrano, MD, PhD: palestrante da Novartis. Suzanne George: pesquisadora das empresas Ariad Pharmaceuticals, Inc; Bayer AG; Blueprint; e Pfizer, Inc. |
Referências
1. Miettinen M, Lasota J. Gastrointestinal stromal tumors—definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438:1–12.
2. Thomas RM, Sobin LH. Gastrointestinal cancer. Cancer 1995;75(1 Suppl):154–70.
3. Tran T, Davila JA, El-Serag HB. The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1,458 cases from 1992 to 2000. Am J Gastroenterol 2005;100:162–8.
4. Ducimetiere F, Lurkin A, Ranchere-Vince D, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiolog-ical study with central pathology review and molecular testing. PLoS One 2011;6:e20294.
5. Nilsson B, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prog-nostication in the preimatinib mesylate era—a population-based study in western Sweden. Cancer 2005;103:821–9.
6. Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol 2006;37:1527–35.
7. Cassier PA, Ducimetiere F, Lurkin A, et al. A prospective epidemiological study of new incident GISTs during two consecutive years in Rhone Alpes region: incidence and molecular distribution of GIST in a European region. Br J Cancer 2010;103:165–70.
8. Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 2010;467:849–53.
9. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer 2011;11:865–78.
10. Burgoyne AM, Somaiah N, Sicklick JK. Gastrointestinal stromal tumors in the setting of multiple tumor syndromes. Curr Opin Oncol 2014;26:408–14.
11. Li FP, Fletcher JA, Heinrich MC, et al. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J Clin Oncol 2005;23:2735–43.
12. Brems H, Beert E, de Ravel T, et al. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol 2009;10:508–15.
13. Mussi C, Schildhaus HU, Gronchi A, et al. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res 2008;14:4550–5.
14. Janeway KA, Weldon CB. Pediatric gastrointestinal stromal tumor. Semin Pediatr Surg 2012;21:31–43.
15. Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stro-mal tumor. Cancer Discov 2013;3:648–57.
16. Corless CL, McGreevey L, Haley A, et al. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol 2002;160:1567–72.
17. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–80.
18. Huizinga JD, Thuneberg L, Kluppel M, et al. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995;373:347–9.
19. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–80.
20. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–9.
21. Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360–7.
22. Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003;9:3329–37.
23. Subramanian S, West RB, Corless CL, et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene 2004;23:7780–90.
24. Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensi-tivity to imatinib. J Clin Oncol 2005;23:5357–64.
25. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–10.
26. Cassier PA, Fumagalli E, Rutkowski P, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastroin-testinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res 2012;18:4458–64.
27. Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 2004;23:3999–4006.
28. Zhu MJ, Ou WB, Fletcher CD, et al. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene 2007;26:6386–95.
29. Bauer S, Duensing A, Demetri GD, et al. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007;26:7560–8.
30. El-Rifai W, Sarlomo-Rikala M, Andersson LC, et al. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor pro-gression and prognostic significance. Cancer Res 2000;60:3899–903.
31. Wang Y, Marino-Enriquez A, Bennett RR, et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet 2014;46:601–6.
32. Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006;24:4764–74.
33. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:295–302.
34. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tu-mour after failure of imatinib: a randomised controlled trial. Lancet 2006;368:1329–38.
35. Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64–74.
36. Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013;23:739–52.
37. Janeway KA, Liegl B, Harlow A, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res 2007;67:9084–8.
38. Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 2011;108:314–8.
39. Wagner AJ, Remillard SP, Zhang YX, et al. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol 2013;26:289–94.
40. Miettinen M, Wang ZF, Sarlomo-Rikala M, et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol 2011;35:1712–21.
41. Serrano C, Wang Y, Marino-Enriquez A, et al. KRAS and KIT gatekeeper mutations confer polyclonal primary imatinib resistance in GI stromal tumors: relevance of concomitant phosphatidylinositol 3-kinase/AKT dysregulation. J Clin Oncol 2014 Mar 31. [Epub ahead of print]
42. Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008;47:853–9.
43. Scarpa M, Bertin M, Ruffolo C, et al. A systematic review on the clinical diagnosis of gastrointestinal stromal tumors. J Surg Oncol 2008;98:384–92.
44. Chou FF, Eng HL, Sheen-Chen SM. Smooth muscle tumors of the gastrointestinal tract: analysis of prognostic factors. Surgery 1996;119:171–7.
45. ESMO/European Sarcoma Network Working Group. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 Suppl 3:iii21–6.
46. von Mehren M, Randall RL, Benjamin RS, et al. Gastrointestinal stromal tumors, version 2.2014. J Natl Compr Canc Netw 2014;12:853–62.
47. Watson RR, Binmoeller KF, Hamerski CM, et al. Yield and performance characteristics of endoscopic ultrasound-guided fine needle as-piration for diagnosing upper GI tract stromal tumors. Dig Dis Sci 2011;56:1757–62.
48. Fletcher CDM, Bridge JA, Hogendoorn PCW, editors. WHO classification of tumours of soft tissue and bone. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC; 2013.
49. Liegl B, Hornick JL, Corless CL, et al. Monoclonal antibody DOG1.1 shows higher sensitivity than KIT in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes. Am J Surg Pathol 2009;33:437–46.
50. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–9.
51. Serrano-Garcia C, Heinrich M, Zhu M, et al. In vitro and in vivo activity of regorafenib (REGO) in drug-resistant gastrointestinal stromal tumors (GIST) [abstract 10510]. J Clin Oncol 2013;31 Suppl.
52. McCarter MD, Antonescu CR, Ballman KV, et al. Microscopically positive margins for primary gastrointestinal stromal tumors: analysis of risk factors and tumor recurrence. J Am Coll Surg 2012;215:53–9; discussion 59–60.
53. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumour. Br J Surg 2010;97:1854–9.
54. Joensuu H, Vehtari A, Riihimaki J, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled popu-lation-based cohorts. Lancet Oncol 2012;13:265–74.
55. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002;33:459–65.
56. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 2008;39:1411–9.
57. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006;130:1466–78.
58. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:1097–104.
59. Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA 2012;307:1265–72.
60. Casali P, Le Cesne A, Poveda A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial [abstract 10500]. J Clin Oncol 2013;31 Suppl.
61. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and meta-static/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol 2009;99:42–7.
62. McAuliffe JC, Hunt KK, Lazar AJ, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol 2009;16:910–9.
63. Van den Abbeele AD, Gatsonis C, de Vries DJ, et al. ACRIN 6665/RTOG 0132 phase II trial of neoadjuvant imatinib mesylate for operable malignant gastrointestinal stromal tumor: monitoring with 18F-FDG PET and correlation with genotype and GLUT4 expression. J Nucl Med 2012;53:567–74.
64. Joensuu H, Fletcher C, Dimitrijevic S, et al. Management of malignant gastrointestinal stromal tumours. Lancet Oncol 2002;3:655–64.
65. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620–5.
66. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626–32.
67. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004;364:1127–34.
68. MetaGIST. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol 2010;28:1247–53.
69. Patrikidou A, Chabaud S, Ray-Coquard I, et al. Influence of imatinib interruption and rechallenge on the residual disease in patients with advanced GIST: results of the BFR14 prospective French Sarcoma Group randomised, phase III trial. Ann Oncol 2013;24:1087–93.
70. Demetri G, Rankin C, Benjamin RS, et al. Long-term disease control of advanced gastrointestinal stromal tumors (GIST) with imatinib (IM): 10-year outcomes from SWOG phase III intergroup trial S0033 [abstract 10508]. J Clin Oncol 2014;32 Suppl:5s.
71. Von Mehren M, Heinrich M, Joensuu H, et al. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST) [abstract 10016]. J Clin Oncol 2011;29 Suppl.
72. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastroin-testinal stromal tumour after imatinib failure. Eur J Cancer 2009;45:1959–68.
73. Kang YK, Ryu MH, Yoo C, et al. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2013;14:1175–82.
74. Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted sys-temic therapy using kinase inhibitors. J Clin Oncol 2006;24:2325–31.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.