(Carregando Índice)... (Carregando Índice)... |
Última revisão: 06/02/2019
Comentários de assinantes: 0
|
Artigo original: H W. Farber, MD, FAHA, FCCP. Moll M, MD. Sardana M, MBBS. Pulmonary Hypertension, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Harrison W. Farber, MD, FAHA, FCCP
Professor de Medicina no Centro Pulmonar da Boston University School of Medicine (Boston, MA).
Matthew Moll, MD
Residente no Departamento de Medicina Interna da Boston Medical Center (Boston, MA).
Mayank Sardana, MBBS
Bolsista na Divisão de Cardiologia da University of Massachusetts Medical School (Worcester, MA).
Resumo
Esta revisão faz uma abordagem às doenças que afetam diretamente a rede de vasos dos pulmões. Essas condições incluem hipertensão pulmonar (HP); hipertensão arterial pulmonar (HAP); hipertensão pulmonar tromboembólica crônica (HPTC); HP atribuída a alguma doença cardíaca no lado esquerdo, doença pulmonar e/ou hipoxemia e outros distúrbios; cor pulmonale; malformações arteriovenosas pulmonares; e aneurismas pulmonares. As figuras mostram as alterações nos vasos pulmonares em casos de HP, rotas envolvidas no desenvolvimento de HP, orientações gerais para avaliação de suspeitas de HP, artérias pulmonares proximais aumentadas com desbaste nos vasos pulmonares distais (situação típica em casos de HAP em estado avançado), remodelação do coração e resultados de um estudo Doppler de ondas contínuas observados na HP crônica, varreduras por ventilação e perfusão nos pulmões com resultados típicos de HPTC, e uma abordagem geral ao tratamento de pacientes com HAP. Os quadros apresentam uma lista da nomenclatura e classificação revisadas de HP, classificação da capacidade funcional da Organização Mundial da Saúde em pacientes com HP, medicações vasculares avançadas para HAP, e gerenciamento perioperatório de HAP.
hipertensão pulmonar, Cor Pulmonale e Outras Condições Vasculares nos Pulmões
Esta revisão faz uma abordagem dos processos que afetam diretamente a rede vascular dos pulmões de forma crônica, incluindo hipertensão pulmonar (HP) e malformações vasculares e aneurismas nos pulmões, assim como cor pulmonale com características agudas e crônicas independente da causa.
hipertensão pulmonar
A HP se refere a uma pressão arterial pulmonar média (mPAP) acima de 25mmHg no estado de repouso. A Organização Mundial da Saúde (OMS) classifica clinicamente a HP em cinco grupos:1
Grupo 1: consiste de hipertensão arterial pulmonar (HAP).
Grupo 2: consiste de HP relacionada à doença cardíaca no lado esquerdo (congênita e adquirida) e a anormalidades venosas pulmonares.
Grupo 3: consiste de HP relacionada a várias doenças pulmonares (por exemplo, doença pulmonar obstrutiva crônica [DPOC], doenças pulmonares intersticiais [DPIs].
Grupo 4: consiste de HP relacionada à doença pulmonar tromboembólica.
Grupo 5: consiste de HP relacionada a outros tipos de distúrbios, incluindo problemas sistêmicos, estados hemolíticos e doenças metabólicas, entre outras.
Os vários grupos de HP compartilham muitas características idênticas; portanto, o diagnóstico geral, assim como a abordagem ampla do gerenciamento a pacientes com HP são apresentados primeiramente no contexto de HAP. As seções subsequentes apresentam informações clínicas complementares exclusivas sobre as várias formas de HP. Web sites úteis sobre as doenças vasculares pulmonares poderão ser encontrados na sidebar.
O Quadro 1 contém web sites úteis sobre doença vascular pulmonar.
Quadro 1
|
Web Sites Úteis Sobre Doença Vascular Pulmonar |
|
Pulmonary Hypertension Association (Associação de) HP
Pulmonary Vascular Research Institute (Instituto de Pesquisa Vascular Pulmonar)
Hereditary Hemorrhagic Telangiectasia Foundation International (Fundação Internacional de Telangiectasia Hemorrágica Hereditária)
|
HP: hipertensão pulmonar.
Hipertensão Arterial Pulmonar
A HAP inclui um grupo de distúrbios que se caracterizam por alterações pré-capilares na rede vascular dos pulmões. Esses distúrbios são definidos pela presença de mPAP acima de 25mmHg em repouso; pressão capilar pulmonar em cunha (PCPC), pressão atrial esquerda ou pressão diastólica final no ventrículo esquerdo (PDFVE) igual ou inferior a 15mmHg; e resistência vascular pulmonar (RVP) acima de 3 unidades de Wound (240dyn-s/cm5).1,2
Epidemiologia
A HAP pode ser idiopática (HAPI), hereditária (HAPH), induzida por medicamentos ou toxinas, ou associadas a outros distúrbios sistêmicos (HAPA). A HAPI se refere a alguma doença esporádica em que não há histórico familiar de HAP e nem de algum fator de risco identificado ou de alguma condição clínica associada.
Estimativas recentes sugerem uma prevalência de HAP de 15 por 1 milhão, sendo que, aproximadamente, um terço desses pacientes tem HAPI. A HAPH é responsável por 6 a 10% de pacientes com HAP. Algo em torno de 50 a 90% de pacientes com HAPH têm mutações no gene BMPR2.3,4 Inúmeros medicamentos foram associados ao desenvolvimento de HAP.
Embora as associações clássicas tenham sido descritas com aminorex (década de 1960) e com fenfluramina (década de 1990), mais recentemente a HAP foi associada ao benfluorex (um agente anorético)5 e ao dasatinibe (um inibidor da tirosina quinase usado no tratamento de leucemia mieloide crônica).6 Embora sejam bem menos documentadas, foram propostas algumas associações entre o uso de metanfetamina e cocaína e HAP.
Entre todos os casos descritos, a HAP associada às doenças nos tecidos conjuntivos (DTCs) é responsável por 15 a 25% dos casos. A esclerose sistêmica é a etiologia mais comum (em especial, esclerose sistêmica limitada, síndrome CREST [calcinose, fenômeno de Raynaud, dismotilidade esofágica, esclerodactilia e telangiectasia]) e lúpus eritematoso sistêmico (LES).
Cabe observar que HAP associada a DTCs tem prognóstico muito pior em comparação com HAPI (mortalidade em 1 ano de 30% em comparação com 15%),7 sendo que a implementação de um programa de rastreamento sistemático de HAP provavelmente melhore os resultados no longo prazo nessa população de pacientes.
Ao contrário da esclerose sistêmica limitada, o escleroderma difuso está associado à doença pulmonar intersticial (DPI) e ao grupo 3 de HP resultante. Além disso, na ausência de DPI, há relatos pouco frequentes de HAP com a síndrome de Sjögren, polimiosite e artrite reumatoide.
A HAP é uma complicação rara, mas bem documentada, de infecção por HIV e ocorre em 0,5% de pacientes infectados.8 O desenvolvimento de HAP em associação com pressões elevadas na circulação portal é conhecido por hipertensão portopulmonar (HPOP). Aproximadamente, 5% de pacientes com hipertensão portal encaminhados para transplante de fígado são diagnosticados com HAP.
A HPOP está associada não apenas a piores prognósticos em comparação com a HAPI (sobrevida de 3 anos de 40% versus 83%),9 mas também ao aumento na mortalidade perioperatória em pacientes que tenham feito transplante de fígado. Uma proporção significativa de pacientes com doença cardíaca congênita não corrigida, em especial aqueles com desvios sistêmicos para as áreas pulmonares, poderá desenvolver HAP e a síndrome de Eisenmenger.
Nos países em desenvolvimento, a esquistossomíase é uma causa frequente de HAP, com uma estimativa de 200 milhões de pessoas infectadas em todo o mundo. Aproximadamente, 5% de pacientes com esquistossomíase hepatoesplênica desenvolvem HAP e, provavelmente, correspondam a até 20% de todos os diagnósticos recentes dos casos de HAP em países endêmicos.10
O Quadro 2 apresenta a nomenclatura revisada e a classificação de HP (2013).1
Quadro 2
|
Nomenclatura Revisada e Classificação de hipertensão pulmonar (2013) | ||
|
Grupo |
Categoria de doença |
Exemplos |
|
HAP |
HAP idiopática
|
? |
|
|
HAP hereditária |
BMPR2, ALK1, SMAD9, CAV1, KCNK3, outros |
|
|
Induzida por medicamentos e toxinas |
- |
|
|
Condições associadas à HAP |
Doença vascular colagenosa, derivações congênitas sistêmicas para áreas pulmonares, hipertensão portal, infecção por HIV, esquitossomíase. |
|
|
Associada a um envolvimento venoso ou capilar significativo |
Distúrbio pulmonar veno-oclusivo, hemangiomatose nos capilares pulmonares. |
|
|
HPPRN |
- |
|
HP causada por doença cardíaca no lado esquerdo |
Disfunção sistólica |
- |
|
|
Disfunção diastólica |
- |
|
|
Doença cardíaca valvular |
- |
|
|
Fluxo cardíaco interno congênito/adquirido/obstrução no trato de fluxo externo e cardiomiopatias congênitas |
- |
|
HP causada por doenças pulmonares e/ou hipoxemia |
DPOC |
- |
|
|
DPI |
- |
|
|
Outras doenças pulmonares com restrição mista e padrão obstrutivo |
- |
|
|
Distúrbio na respiração durante o sono |
- |
|
|
Distúrbios alveolares de hipoventilação |
Síndrome de hipoventilação da obesidade, distúrbios neuromusculares e na caixa torácica. |
|
|
Exposição crônica a grandes altitudes |
- |
|
|
Anormalidades evolucionárias |
- |
|
HP tromboembólica crônica |
HP tromboembólica crônica HPTC |
- |
|
|
EP não trombótica (tumores, parasitas) |
- |
|
HP com mecanismos multifatoriais obscuros |
Distúrbios hematológicos |
Anemias hemolíticas crônicas, distúrbios mieloproliferativos, esplenectomia. |
|
|
Distúrbios sistêmicos |
Sarcoidose, histiocitose pulmonar da célula de Langerhans, neurofibromatose. |
|
|
Distúrbios metabólicos |
Doenças de armazenamento de glicogênio, doença de Gaucher, distúrbios tireóideos. |
|
|
Outros |
Obstrução tumoral, mediastinite fibrosante, HP segmentar. |
ALK1: quinase 1 semelhante ao receptor da ativina; BMPR2: proteínas morfogênicas do osso tipo 2; CAV1: caveolina 1; DPI: doença pulmonar intersticial; DPOC: doença pulmonar obstrutiva crônica; EP: embolia pulmonar; HAP: hipertensão arterial pulmonar; HP: hipertensão pulmonar; HPPRN: hipertensão pulmonar persistente em recém-nascidos; KCNK3: cadeia de potássio, membro 3 da subfamília K com domínio de dois poros; SMAD9: mães contra o homólogo 9 decapentaplégico.
Outras duas categorias também são classificadas no grupo HP 1, tendo em vista que compartilham muitas semelhanças clínicas com HAP. Essas categorias incluem doenças pulmonares veno-oclusivas (DPVOs) e hipertensão pulmonar persistente do recém-nascido (PPHN).1
Fisiopatologia
A circulação pulmonar normal tem capacidade para acomodar todo o débito cardíaco em pressões de perfusão que correspondem a, aproximadamente, 20% em comparação com a circulação sistêmica, mesmo nos casos em que o débito cardíaco aumentar durante os exercícios físicos.
A circulação pulmonar consegue fazer isso por meio da dilatação da rede de vasos que já estiver recebendo o débito cardíaco e fazendo o recrutamento da rede de vasos que não estiver sendo utilizada (isto é, arteríolas e capilares); por meio desses mecanismos, a circulação pulmonar minimiza as elevações na pressão de perfusão e maximiza a área superficial de troca de gases. A produção local de diversos mediadores humorais, incluindo o óxido nítrico (ON) e a prostaciclina, contribui para a manutenção de um tônus vasomotor baixo.11
Os fatores mobilizadores da HP se classificam da seguinte forma:
1. Aumento no fluxo sanguíneo (hiperdinâmica).12 Observa-se esse fato nos desvios cardíacos (do lado esquerdo para o lado direito) ou em condições associadas ao aumento no fluxo sanguíneo através da circulação sistêmica (por exemplo, hipertireoidismo, anemia, cirrose). A diferenciação poderá ser feita calculando-se a proporção do fluxo sanguíneo pulmonar em relação ao fluxo sistêmico (Qp/Qs). Observa-se uma razão Qp/Qs maior que 1 nos desvios cardíacos do lado esquerdo para o lado direito, enquanto que Qp=Qs (isto é, Qp/Qs=1) se relaciona às condições associadas à circulação sistêmica hiperdinâmica.
2. Aumento na resistência ao fluxo venoso pulmonar externo para o lado esquerdo do coração (passiva). Observa-se esse fato na disfunção ventricular esquerda (sistólica, diastólica, ou uma combinação das duas), na elevação das pressões no átrio esquerdo (por exemplo, regurgitação mitral, estenose mitral), na obstrução ao fluxo externo no ventrículo esquerdo (por exemplo, estenose aórtica, cardiomiopatia hipertrófica), estenose nas veias pulmonares (ocasionalmente, trata-se de uma complicação produzida por algum procedimento para isolar as veias pulmonares) ou compressão extrínseca (por exemplo, fibrose mediastinal ou neoplasma).
3. Aumento na resistência ao fluxo sanguíneo na rede vascular pulmonar externa para o lado esquerdo do coração (resistente). As alterações primárias na circulação pulmonar estão associadas à HAP, hipertensão pulmonar tromboembólica crônica (HPTC) e à HP do grupo 5 da OMS. Sob o ponto de vista histológico, a presença de alguma lesão plexiforme é a descoberta patológica clássica nos casos de HAP. Embora não seja sensível nem específica para HAP, a lesão plexiforme, uma proliferação localizada de células endoteliais, de células de músculos lisos, de fibroblastos e da matriz extracelular, é encontrada nos vasos pulmonares pré-capilares e intra-acinares. Em combinação com a hiperatrofia medial e com o espessamento da íntima por células de músculos lisos e fibroblastos, a resistência ao fluxo sanguíneo no interior das lesões plexiformes é secundária aos canais revestidos com endotélio que estreitam o lúmen vascular.13 A ativação e a expressão de moléculas de adesão por células endoteliais criam um estado pró-coagulante, com deposição de trombina e adesão de plaquetas. Essas anormalidades na vasculatura pulmonar foram resumidas sucintamente por McGoon e Kane como uma predisposição para vasoconstrição em relação à vasodilatação, hiperplasia nas células endoteliais e de músculos lisos fora de proporção em relação à apoptose, e uma diátese trombótica.14
Hipoteticamente, sob a perspectiva genética, a HAP, em geral, e a HAPI, em particular, resultam de algum fator desencadeador em indivíduos suscetíveis. Alguns estudos sobre hereditariedade sugeriram a presença de um padrão autossômico dominante com penetrância incompleta da hipertensão arterial pulmonar hereditária (HAPH). Diversos genótipos foram associados à HAP.
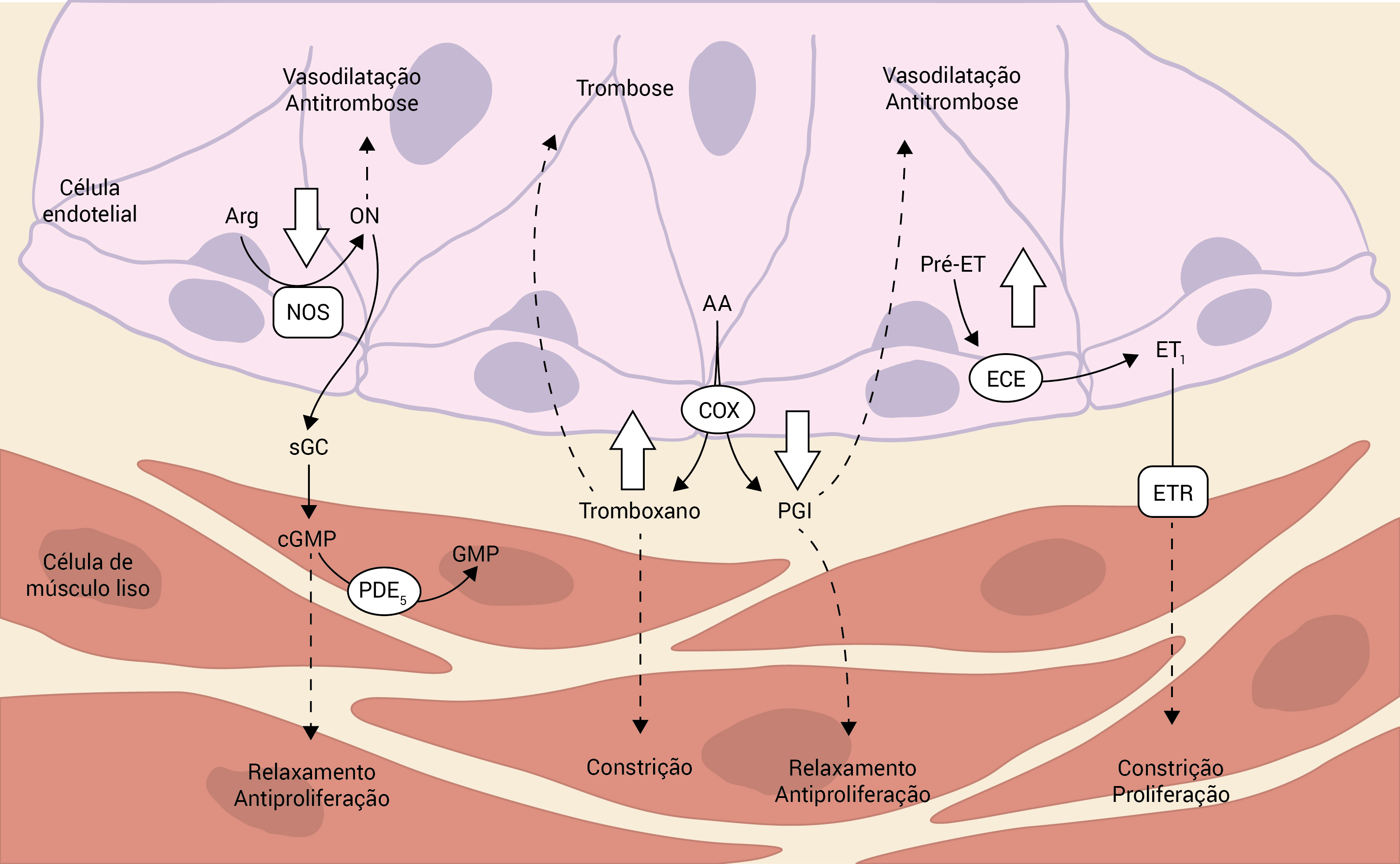
Até 80% de pacientes com HAPH e 20% com HAP esporádica apresentam variantes alélicas da proteína morfogênica do osso tipo 2 (BMPR2), um gene que codifica um receptor do fator de crescimento transformador ß (TGF-ß). Variantes de outro receptor do TGF-ß, a quinase 1 semelhante ao receptor da ativina (ALK1), também foram encontradas em algumas famílias com telangiectasia hemorrágica hereditária (THH) e HAP.3 A Figura 1 mostra a progressão da doença.
AA: ácido aracdônico; Arg: arginina; cGMP: monofosfato de guanosina cíclico; COX: ciclo-oxigenase; ECE: enzima conversora da endotelina; ET: endotelina; ETR: receptor da endotelina; GMP: monofosfato de guanosina; HAP: hipertensão arterial pulmonar; NOS: óxido nítrico-sintetase; ON: óxido nítrico; PDE5: fosfodiesterase tipo 5; pre-ET: pré-endotelina; sGC: gualinato ciclase solúvel.

Figura 1 - Independentemente do(s) evento(s) desencadeador(es), a progressão da doença resulta em vasoconstrição, hiperplasia nos músculos lisos e hipertrofia fora de proporção em relação à apoptose e em diátese trombótica. Os níveis de ON e de prostaciclina (PGI) são reduzidos, enquanto que a atividade da ET e do tromboxano é elevada na HAP.
Mais recentemente, foram descritas associações com a caveolina (CAV1) e o gene KCNK3.15 Alguns estudos sobre a rede vascular pulmonar sugerem que a lesão e a disfunção endotelial ocorrem na fase inicial do processo. Nos casos de hipertensão arterial pulmonar idiopática (HAPI), há uma redução na expressão das sintetases do ON e da prostaciclina, ao passo que ocorre uma elevação nos níveis de endotelina-1 e de tromboxano.
A prostaciclina e o ON são vasodilatadores potentes e inibidores da ativação de plaquetas e da proliferação vascular de músculos lisos. A endotelina-1 e o tromboxano são mitógenos e vasoconstritores pulmonares potentes. Outros mediadores vasoativos como a serotonina, que é outro vasoconstritor pulmonar, também têm alguma importância, em especial nos casos de HP associada aos inibidores do apetite, que inibem a reabsorção da serotonina e aumentam a expressão de seu transportador.
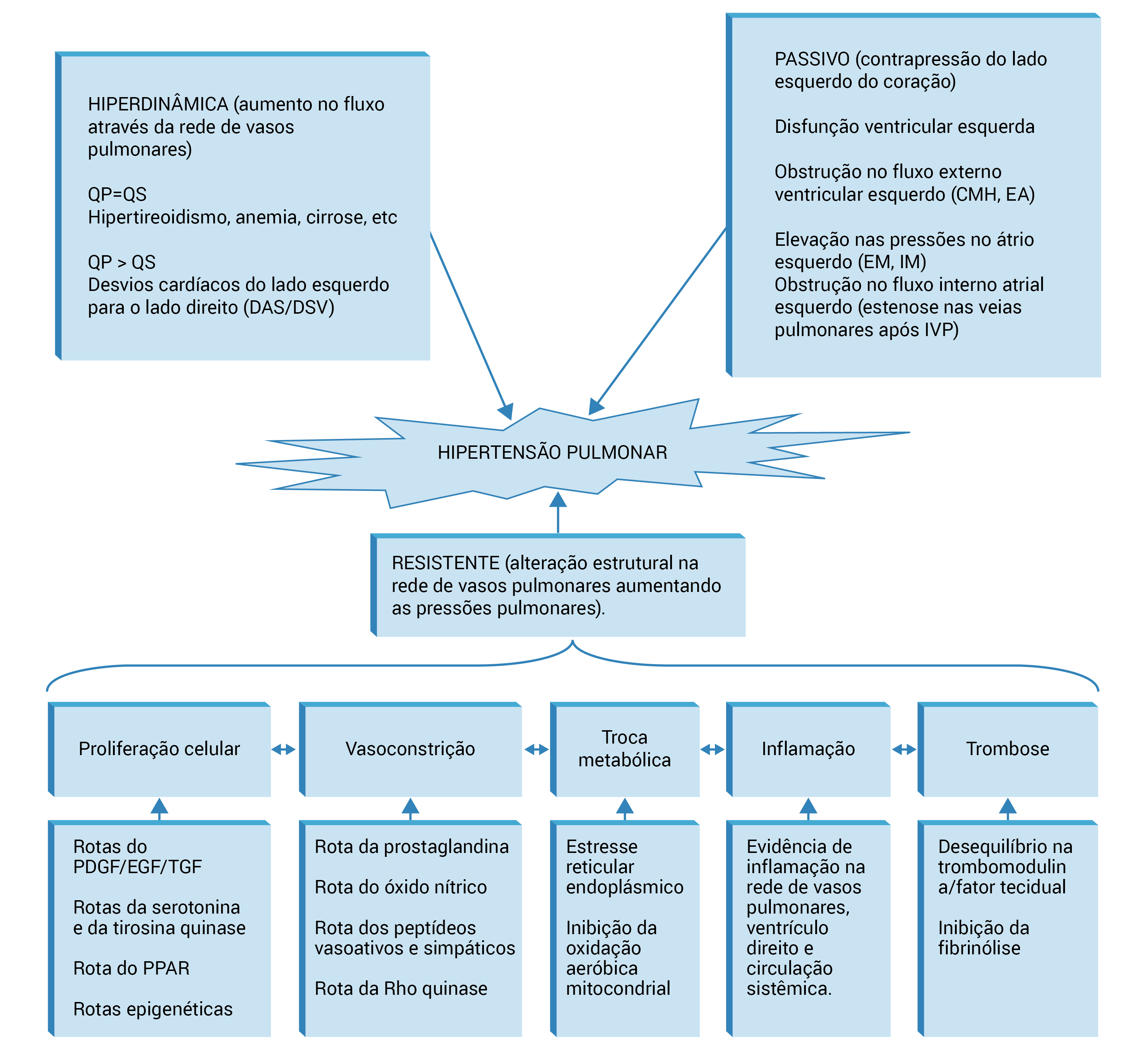
A Figura 2 apresenta as rotas envolvidas no desenvolvimento de HP, contendo a descrição de outros mecanismos que provavelmente expliquem o desenvolvimento de HAP.
CMH: cardiomiopatia hipertrófica; das: defeito no septo atrial; DSV: defeito no septo ventricular; EA: estenose aórtica; EGF: fator de crescimento epidérmico; EM: estenose mitral; FGF: fator de crescimento de fibroblastos; IM: insuficiência mitral; IVP: isolamento das veias pulmonares; PDGF: fator de crescimento derivado das plaquetas; PPAR: receptor do proliferador de peroxissomos; Qp: fluxo através da circulação pulmonar; Qs: fluxo através da circulação sistêmica; TGF: fator de crescimento transformador; DSV: defeito no septo ventricular.
Figura 2 - Rotas envolvidas no desenvolvimento de HP.
Essas rotas não são mutuamente exclusivas e interagem em diversos níveis de desenvolvimento da HP. A descrição dessas rotas está fora do escopo dessa revisão; entretanto, conforme sugere a presença de um número limitado de agentes terapêuticos com foco nessas rotas, entende-se que muitas delas ainda estejam nos estágios iniciais.
Diagnóstico
A avaliação diagnóstica de HP inicia com um histórico detalhado e um exame físico cuidadoso. As indicações históricas que poderão sugerir a presença de HP incluem histórico de insuficiência cardíaca congestiva (ICC) ou de doença cardíaca valvular, doença pulmonar crônica ou hipóxia, tromboembolismo venoso prévio, histórico médico de DTCs, anemia hemolítica crônica, uso de inibidores do apetite, ou exposição conhecida ou suspeita à infecção pelo vírus da imunodeficiência adquirida (HIV).
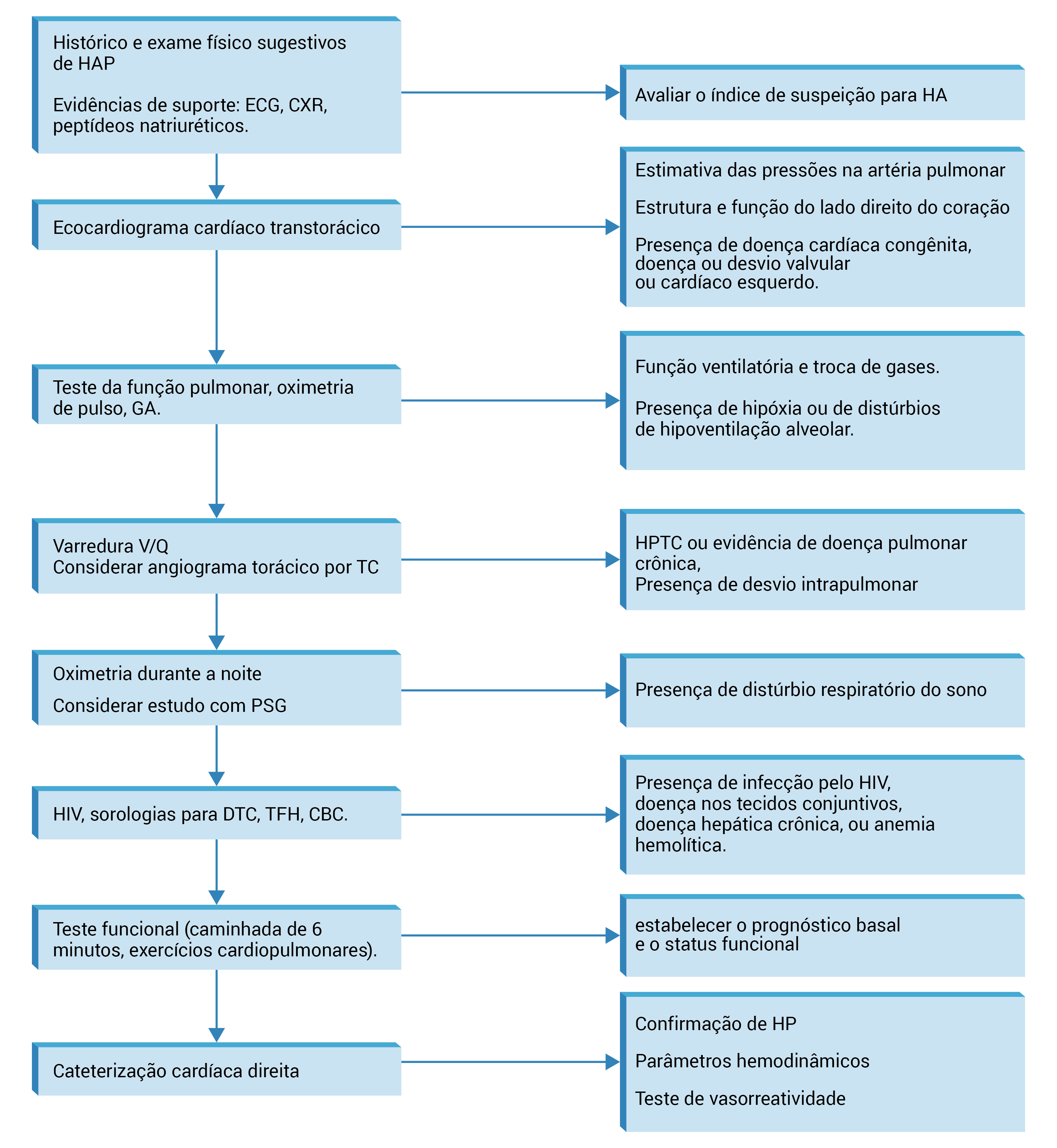
Recomenda-se rastrear históricos familiares de HP. Nos casos de suspeita de HP com base na apresentação clínica e nas descobertas do exame, as novas avaliações deverão iniciar com estudos menos invasivos, comumente um ecocardiograma transtorácico (ETT), seguidos de testes mais específicos que permitam confirmar o diagnóstico, determinar o nível de gravidade e avaliar a etiologia subjacente da HP, conforme a Figura 3. A cateterização cardíaca direita (CCD) ainda é o padrão de ouro para o diagnóstico de HP.
CBC: hemograma completo; CXR: radiografia torácica; DTC: doença nos tecidos conjuntivos; ECG: eletrocardiograma; GA: gases arteriais; HAP: hipertensão arterial pulmonar; HIV: vírus da imunodeficiência adquirida; HP: hipertensão pulmonar; HPTC: hipertensão pulmonar tromboembólica crônica; PSG: polissonograma; TC: tomografia computadorizada; TFH: teste da função hepática; V/Q: ventilação/perfusão.
Figura 3 - Orientações gerais para a avaliação de suspeita de HP. A sequência de testes varia de acordo com a situação clínica, embora a ecocardiografia cardíaca seja o teste de rastreamento inicial na maioria dos cenários; a CCD ainda é o padrão diagnóstico. A HAP idiopática é diagnosticada na ausência de outros distúrbios associados.
Manifestações Clínicas
Muitas características clínicas de HP são semelhantes entre si, sejam quais forem as causas subjacentes da doença. Na fase inicial do processo, os sintomas de HP são mínimos e inespecíficos. A presença de fadiga, fraqueza e dispneia de esforço é comum, assim como dor torácica que poderá imitar angina. Síncope, que, em geral, é consequência de exercícios físicos ou surge depois de exercícios físicos, ocorre na fase final da HP; a síncope é um sinal de mau prognóstico porque implica na incapacidade de aumentar o débito cardíaco durante os exercícios.
A rouquidão possivelmente seja resultado da compressão do nervo laríngeo recorrente (síndrome de Ortner), e a hemoptise poderá ocorrer como resultado do rompimento de pequenos vasos pulmonares ateroscleróticos hipertensivos. Os sintomas de insuficiência no ventrículo direito (VD), como edema e ascite, são relativamente tardios nesse tipo de doença; assim como na síncope, esses sintomas indicam a presença de algum tipo de problema na função ventricular direita e significam maus prognósticos.
A avaliação quantitativa de tolerância às atividades, com embasamento na classificação de capacidade funcional da OMS, é bastante útil para prever prognósticos, para decidir sobre estratégias mais apropriadas de gerenciamento e para monitorar as respostas ao tratamento.16
Exame Físico
O exame físico completo ajuda não apenas a avaliar a gravidade clínica da HP, mas também a identificar quaisquer dicas sobre as condições associadas. As descobertas do exame físico que foram incluídas em vários modelos prognósticos para HAP são os sinais vitais (frequência cardíaca >92BPM e pressão arterial sistólica [PAS] <110mmHg) e as caminhadas por 6 minutos (caminhada normal >440m).16
O Quadro 3 contém a classificação de capacidade funcional da OMS em pacientes com HP.16
Quadro 3
|
Classificação de Capacidade Funcional da Organização Mundial da Saúde em Pacientes com hipertensão pulmonar | |
|
Classe |
Descrição
|
|
I |
Pacientes com HP sem nenhuma restrição às atividades físicas usuais; normalmente, as atividades físicas não produzem dispneia, dor torácica ou pré-síncope.
|
|
II |
Pacientes com HP e restrições leves às atividades físicas; não sentem desconforto no estado de repouso, embora as atividades físicas produzam dispneia, fadiga, dor torácica ou pré-síncope.
|
|
III |
Pacientes com HP e grandes restrições às atividades físicas; não sentem desconforto no estado de repouso, embora ele produza dispneia, fadiga, dor torácica ou pré-síncope com menos intensidade que as atividades físicas comuns.
|
|
IV |
Pacientes com HP e incapazes de executar qualquer atividade física; presença de dispneia e/ou fadiga no estado de repouso, que são agravadas com qualquer atividade física; síncope poderá ocorrer com o esforço físico.
|
HP: hipertensão pulmonar.
As descobertas adicionais do exame físico incluem veias jugulares distendidas com ondas A proeminentes (indicando regurgitação na válvula tricúspide). A palpação no tórax possivelmente detecte alguma agitação na HP na área paraesternal ou, em pacientes com DPOC, na área subxifoidea. A auscultação do coração geralmente revela um aumento no componente P2, um clique de ejeção pulmônica, o ruído suave de regurgitação tricúspide na borda esternal direita inferior que aumenta na inspiração (sinal de Carvallo) e, ocasionalmente, regurgitação pulmônica (sopro de Graham Steell).
Condições como hepatomegalia, ascite e edema nas extremidades inferiores são indicadoras de insuficiência cardíaca (IC) direita. Sinais de IC, incluindo sopros, ponto deslocado de impulso máximo e hipoperfusão, também poderão ser identificados. A auscultação dos pulmões possivelmente indique a presença de algum distúrbio pulmonar subjacente. Locais como pele, unhas e articulações devem ser examinados em busca de evidências de DTCs.
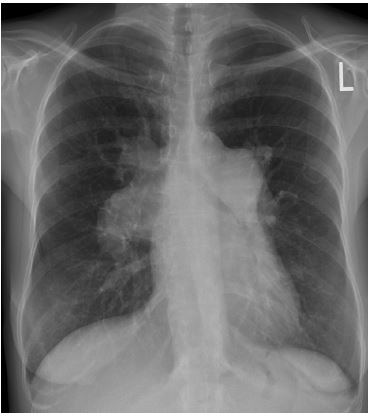
Estudos de imagens. Radiografia torácica. Embora não se conheça a precisão das radiografias torácicas para detecção de HP, essa técnica de imagens poderá sugerir o diagnóstico e a etiologia de HP. Aumentos simétricos nas artérias pulmonares, com afinamento rápido dos vasos distais (desbaste), assim como aumentos no ventrículo direito evidenciados pela supressão do espaço claro retroesternal na projeção lateral, possivelmente sejam localizados pelas radiografias, embora, normalmente, não sejam visualizados até os estágios finais do distúrbio, conforme a Figura 4. As descobertas radiográficas que sugerem a causa subjacente de HP incluem congestão venosa pulmonar (por exemplo, insuficiência ventricular esquerda ou DPVO), hiperinflação (DPOC) e DPI.
Figura 4 - Aumento nas artérias pulmonares proximais, com desbaste na rede vascular distal dos pulmões, o que é típico de HAP avançada.
Eletrocardiografia. A eletrocardiografia (ECG) geralmente é normal nos estágios iniciais, ainda que, com a progressão da doença, o exame mostre alterações hipertróficas no ventrículo direito, desvio no eixo direito e aumento no átrio direito.
Ecocardiografia transtorácica (ETT). A ETT com estimativas Doppler de pressões arteriais pulmonares é o estudo inicial de escolha para avaliar pacientes com sintomas sugestivos de HP.17 A ETT bidimensional avalia alterações consistentes com sobrecargas crônicas no ventrículo direito, como, por exemplo, aumento no átrio direito, hipertrofia no ventrículo direito e redução na atividade do ventrículo direito.
O achatamento sistólico do septo interventricular sugere a presença de HP (sobrecarga pressórica no ventrículo direito). A ETT produz também evidências diretas da função ventricular esquerda e da morfologia valvular, além de fornecer dicas sobre a etiologia de HP. Em pacientes nos quais a hipóxia não responde à suplementação de oxigênio, recomenda-se considerar a presença de desvios arteriovenosos intracardíacos ou pulmonares (por exemplo, em casos de telangiectasia hemorrágica hereditária, síndrome hepatopulmonar, etc.).
O uso da ultrassonografia com Doppler colorido e injeção de solução salina agitada durante a ETT permite detectar e localizar quaisquer desvios. A ecocardiografia com Doppler também possibilita estimar as pressões arteriais pulmonares. Enquanto a estimativa da pressão arterial pulmonar sistólica (sPAP) se baseia na velocidade máxima da regurgitação tricúspide (Vt), a pressão arterial pulmonar diastólica (dPAP) poderá ser estimada utilizando-se a velocidade da regurgitação pulmonar (Vp).
Essas estimativas se baseiam na equação de Bernoulli (sPAP˜4Vp2+RAP e dPAP˜4Vt2+RAP, em que RAP é a pressão atrial direita estimada por meios ecocardiográficos). A HP é considerada possível nas situações em que a sPAP estimada for maior que 40mmHg ou a mPAP for superior a 25mmHg. A presença de tempos curtos de aceleração arterial pulmonar (<90ms), estimados por ecocardiografia com Doppler, também se correlaciona com a presença de HP.18
A ETT é um estudo apropriado para rastrear populações de pacientes com alta incidência de HP; esses pacientes incluem indivíduos com o espectro de escleroderma da doença, indivíduos com histórico familiar de HAP, pacientes com cirrose que estiverem sendo avaliados para transplante e pacientes com anemia crônica.
A ecocardiografia cardíaca é o teste inicial de escolha para avaliação de HP, porém a ETT tem algumas limitações. Em comparação com a CCD, que é o padrão de ouro para medir a pressão arterial pulmonar, de 40 a 60% das sPAPs estimadas por ETT discordam em mais de 10% em relação aos valores medidos.19
Tomografia computadorizada. As tomografias computadorizadas (TCs) do tórax são bastante úteis para visualizar o parênquima pulmonar e detectar enfisema ou DPI não tenha sido percebido nas radiografias torácicas de rotina e, além disso, fornecem dicas importantes sobre a DPVO, que se apresenta com evidências de linhas septais produzidas por edema intersticial.
A angiografia por TC provavelmente sugira a presença de HPTC, evidenciada pela descoberta de material trombótico crônico nas artérias pulmonares centrais e pela colateralização dos vasos brônquicos. Embora não sejam diagnósticas, as evidências de HP identificadas nas TCs torácicas incluem hipertrofia no ventrículo direito e aumento nas artérias pulmonares, uma apresentação frequente de HP com a proliferação das varreduras por TC.
A Figura 5 mostra uma visão apical bidimensional das quatro câmaras mostrando a remodelagem do coração observada com HP crônica.
Figura 5 - (A) A visão apical bidimensional das quatro câmaras mostra a remodelagem do coração observada com HP crônica, incluindo hipertrofia ventricular direita, achatamento do septo interventricular e aumento no septo atrial. (B) O estudo de ondas contínuas com Doppler mostra a velocidade do jato tricúspide regurgitante, que é o meio não invasivo mais comum para estimar a pressão sistólica na artéria pulmonar.
Imagens cardiopulmonares por ressonância magnética. As imagens cardiopulmonares por ressonância nuclear magnética (RNM) permitem fazer a avaliação direta do ventrículo direito e da morfologia e dinâmica das artérias pulmonares. O papel desempenhado pela RNM cardíaca foi investigado para fins diagnósticos e prognósticos em casos de hipertensão arterial pulmonar (HAP).
Comprovou-se que a queda nos índices de pulsatilidade arterial pulmonar (<40%) tem sensibilidade de 93% e especificidade de 60% para o diagnóstico de HAP.20 Volume reduzido no acidente vascular cerebral (AVC), aumento de volume no final da diástole ventricular direita e redução de volume no final da diástole ventricular esquerda, medidos na linha de base, estão associados a maus prognósticos.21 É interessante observar que a cateterização guiada por RNM também poderá ser utilizada para construir com precisão os loops de fluxo-volume para o ventrículo direito.22
Estudos fisiológicos. Teste da função pulmonar. As medições feitas pelo teste da função pulmonar podem ser muito úteis para avaliar os pacientes com HP. A apresentação clássica é a de pacientes com dispneia de esforço significativa, espirometria e volumes pulmonares normais, porém com capacidade reduzida de difusão pulmonar de monóxido de carbono (DLCO). A detecção de defeitos obstrutivos ou restritivos graves é uma indicação de que toda a HP, ou uma porção substancial dela, seja causada por doença pulmonar intrínseca.
As medições dos gases arteriais em estado de repouso e a oximetria de pulso com exercícios devem ser avaliadas em todos os pacientes com HP, com a finalidade de detectar complicações em repouso ou hipoxemia produzida por exercícios, que poderiam ser solucionadas terapeuticamente com suplementação de oxigênio.
A descoberta de hipercapnia é mais compatível com obstrução grave no fluxo de ar, doença pulmonar restritiva ou com alguma síndrome de hipoventilação alveolar. A presença de DLCO inferior a 32% é considerada um péssimo marcador prognóstico nos casos de HAP.16
Polissonografia. Os pacientes com suspeita de transtorno do sono relacionado à respiração, com base no exame clínico e/ou na oximetria noturna, que possa estar causando ou contribuindo para a HP, deverão fazer polissonografia noturna formal. De um modo geral, a HP no contexto de apneia do sono é leve e causada por doença cardíaca concomitante no lado esquerdo.23
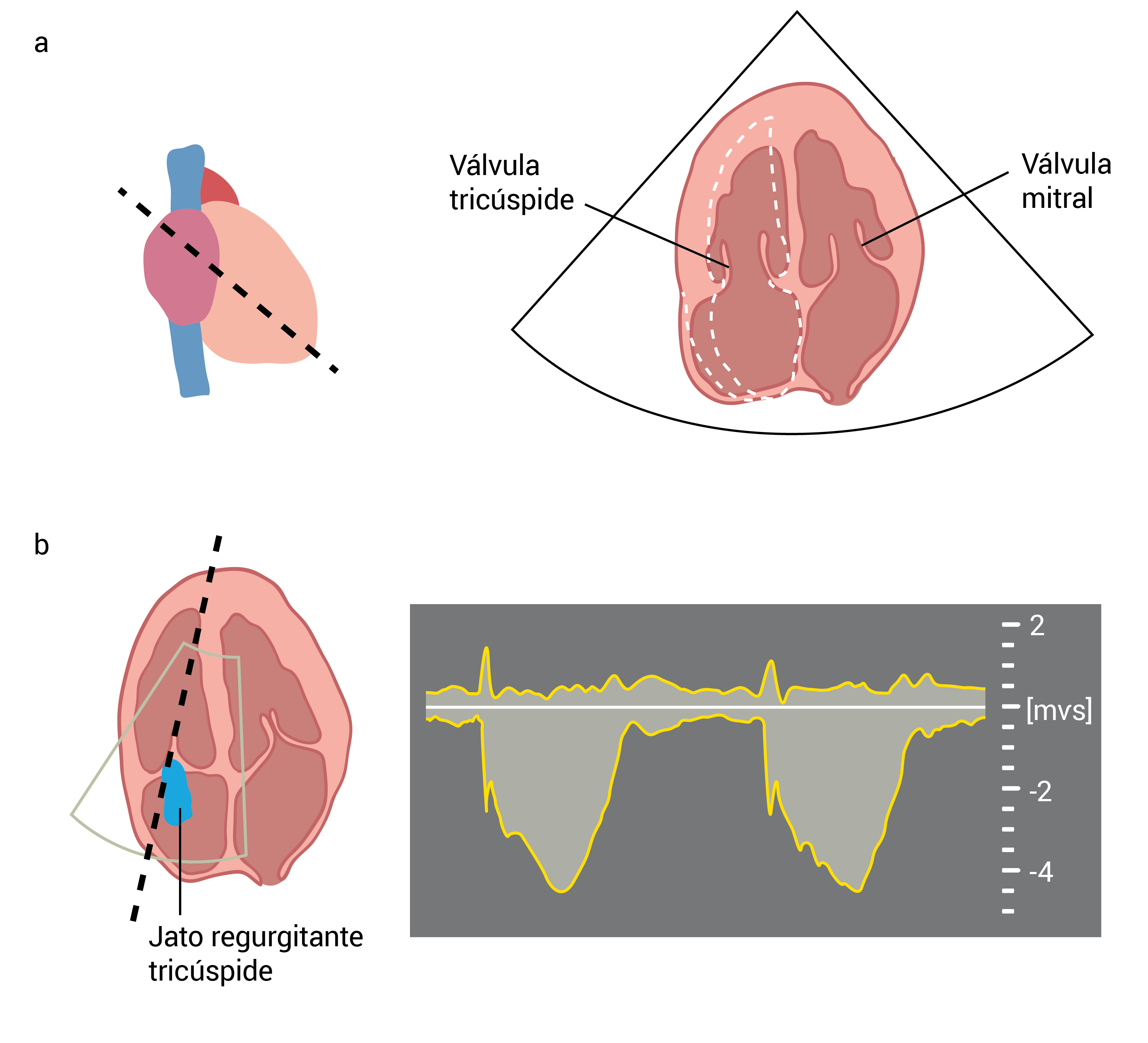
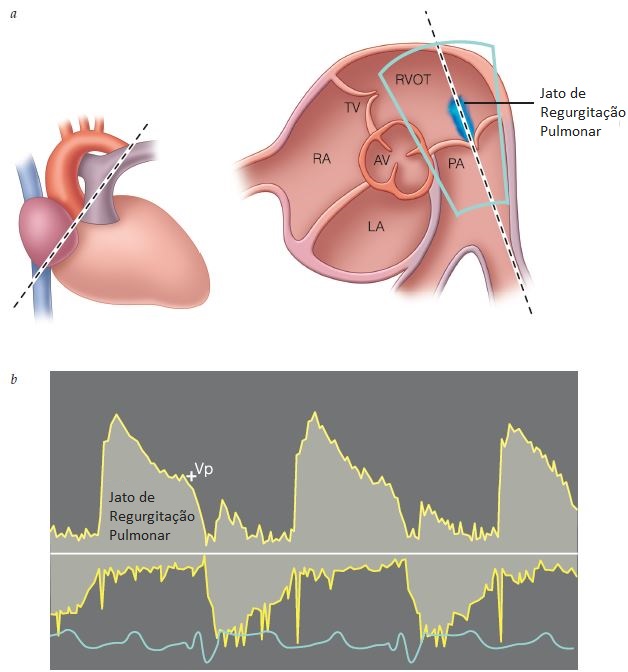
AD: átrio direito; AE: átrio esquerdo; AP: artéria pulmonar; VT: válvula tricúspide.
Figura 6 - (A) Visão bidimensional do eixo curto paraesternal ao nível da válvula aórtica (VA) e ao nível do trato do fluxo ventricular externo direito (RVOT). (B) O estudo Doppler com ondas contínuas mostra a velocidade do jato de regurgitação pulmonar (Vp), que pode ser usado para estimar a pressão arterial pulmonar diastólica.
Varredura pulmonar por ventilação-perfusão. A varredura pulmonar por ventilação-perfusão (V/Q) é um teste importante para avaliar pacientes com HP. Aproximadamente, a metade dos pacientes com HPTC não apresenta histórico de embolia pulmonar (EP) prévia.24 Portanto, a varredura V/Q deve ser considerada em todos os pacientes com HP de causa desconhecida.
Os pacientes com HPTC apresentam um ou mais defeitos de perfusão incompatíveis com o tamanho do segmento. Embora as varreduras V/Q normais mostrem a improbabilidade de HPTC, as varreduras que sugerem a presença da doença requerem confirmação adicional dos resultados falsos positivos no contexto de outros distúrbios pulmonares.25
As varreduras pulmonares por V/Q ainda são o padrão de ouro para confirmação de HPTC, embora a angiografia por TC possivelmente assuma esse papel no futuro. As varreduras por V/Q mostram também evidências de desvio intrapulmonar secundário a malformações arteriovenosas (MAVs) pulmonares, que poderão estar associadas à HAP em distúrbios hereditários como telangiectasia hemorrágica hereditária. A probabilidade de varreduras V/Q normais ou baixas exclui efetivamente a hipótese de HPTC, cuja sensibilidade varia de 90 a 100% e a especificidade, de 94 a 100%.24
Cateterização Cardíaca
A cateterização cardíaca direita (CCD) com arteriografia pulmonar concomitante e cateterização cardíaca esquerda, ou ambas as técnicas, devem ser aplicadas conforme a indicação clínica em todos os pacientes com HP depois de avaliações não invasivas. As medições obtidas por CCD incluem pressão atrial direita, pressão arterial pulmonar, pressão por oclusão (por cunha) na artéria pulmonar e débito cardíaco.
As medições nas saturações de oxigênio detectam a presença de desvios. A cateterização cardíaca não apenas gera informações em relação à presença e à gravidade da HP, mas também ajuda a excluir doença cardíaca esquerda, a doença valvular significativa e desvios. Em pacientes que provavelmente tenham HAP, o teste vasodilatador com agentes como ON inalatório ou epoprostenol intravenoso dá suporte às decisões prognósticas e de tratamento.26
Exames de sangue. Sorologias para DTCs. Levando-se em consideração que a HP possivelmente seja a manifestação inicial, qualquer paciente com suspeita da doença deverá ser avaliado para DTC. Embora, na ausência de DPI, a HAP tenha sido observada em todos os casos de DTC, ela ocorre com mais frequência no espectro de esclerose sistêmica dos distúrbios, em especial nos casos de escleroderma restrito (síndrome CREST).
Se o exame físico não fornecer nenhuma dica, recomenda-se fazer avaliação para DTC, em particular a esclerose sistêmica, com anticorpos antinucleares, anticentroméricos e anti-topoisomerase I (Scl-70). Testes complementares poderão ser apropriados se o teste inicial sugerir a presença de algum distúrbio subjacente.27
A Figura 7 mostra a HPTC.
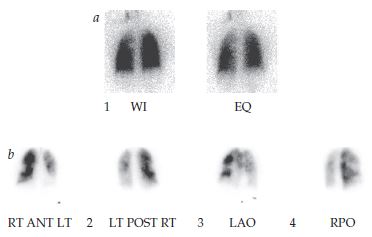
EQ: fase de equilíbrio; HPTC: hipertensão pulmonar tromboembólica crônica; LAO: oblíquo anterior esquerdo; LT POST RT: visões posterior esquerda e direita; por: oblíquo posterior direito; RT ANT LT: visão anterior direita e esquerda; WI: fase de lavagem interna.
Figura 7 - A varredura por ventilação (A) é normal, ainda que a varredura por perfusão (B) apresente múltiplos defeitos segmentares de perfusão, típicos da HPTC.
Rastreamento de HIV. As infecções pelo HIV estão associadas a um modesto aumento na incidência de HAP. Os pacientes com suspeita de HP devem ser avaliados para fatores de risco de HIV e rastreados caso seja necessário.8
Peptídeos natriuréticos. Os peptídeos natriuréticos cerebrais (BNPs) e um dos produtos da clivagem proteolítica, o peptídeo natriurético N-terminal-pró-cérebro (NT-proBNP), são liberados dos miócitos cardíacos em resposta à sobrecarga de pressão e volume. Tanto os BNPs como os NT-proBNP são elevados na doença cardíaca direita e produzem evidências indiretas de HP.28
Biópsia. Biópsia do pulmão. A biópsia do pulmão raramente é exigida para determinar a causa de HP. As únicas exceções ocorrem nos pacientes em que há suspeita de que uma entre as doenças pulmonares intersticiais ou arterite pulmonar seja a causa da HP. As biópsias pulmonares com auxílio de broncoscópio são contraindicadas em pacientes com HP grave atribuída ao risco elevado de hemorragia; nesses casos, as biópsias abertas ou assistidas por vídeo são a técnica de escolha. Esse tipo de biópsia coloca em risco os pacientes com HP em comparação com pacientes sem HP e deverá ser feito com muita cautela.
Gerenciamento
O gerenciamento de HP depende da causa subjacente e se fundamenta no tratamento ideal da doença subjacente. Os distúrbios que afetam a circulação pulmonar de forma aguda (por exemplo, EP, edema pulmonar e síndrome do desconforto respiratório agudo) serão discutidos em algum outro trabalho e são ICC, defeitos nas válvulas cardíacas, defeitos cardíacos congênitos que possam produzir HP, e doenças que afetam as vias respiratórias pulmonares e o parênquima, tais como DPOC, doenças pulmonares intersticiais e transtornos respiratórios do sono.
De um modo geral, as terapias vasculares avançadas para HP, incluindo medicações intravenosas e orais com alvo na prostaciclina, endotelina e nas rotas do ácido nítrico, foram investigadas e aprovadas para uso apenas em pacientes com HAP.
Independentemente da causa subjacente, o tratamento de suporte deverá ter como foco as consequências da HP. Os pacientes devem evitar fazer exercícios físicos pesados que possam agravar as pressões pulmonares e provocar síncope; os exercícios aeróbicos de nível baixo são uma opção para oferecer aos pacientes. Na realidade, um estudo randomizado recente sugeriu que a reabilitação cardiopulmonar cuidadosamente monitorada melhorou a capacidade física para exercícios e a qualidade de vida em pacientes com HAP.29
A hipóxia é um vasoconstritor potente observado em pacientes com HP por várias razões que incluem capacidade reduzida de difusão, desvio da direita para a esquerda e débito cardíaco baixo, resultando na saturação mista baixa do oxigênio venoso. Como consequência, todos os pacientes com HP devem ser avaliados para hipóxia, em repouso e com esforço, e suplementação de oxigênio, de acordo com a necessidade, para manter a normoxia.
No grupo 3, nos pacientes com HP, em particular aqueles com DPOC, a administração de oxigênio é a única terapia que diminui o índice de mortalidade.1,30 Os pacientes deverão ser alertados sobre a exposição a grandes altitudes ou sobre viagens em voos comerciais, sendo que ambos poderão agravar a hipóxia e aumentar a vasoconstrição pulmonar.
O gerenciamento de IC no lado direito com restrição no consumo de sódio é imprescindível. Possivelmente, o uso de diuréticos seja necessário para controlar o volume intravascular e edema, embora tenha de ser de forma equilibrada para assegurar o débito cardíaco, tendo em vista que o ventrículo direito requer pré-carga adequada. A digoxina poderá ser usada como suporte inotrópico no ventrículo direito, embora sua eficácia nos casos de HP ainda não tenha sido comprovada.
Embora a anticoagulação terapêutica com varfarina venha sendo recomendada há muito tempo para todos os pacientes com HPA, uma análise recente feita pelo Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) mostrou que a anticoagulação melhora a sobrevida nos casos de HPAI, embora os dados tenham sido inconclusivos para outros tipos de HAP.31
Dados mais recentes do Registry to Evaluate Early And Long-term PHA Disease Management (REVEAL Registry) demonstrou que não há benefícios na sobrevida com a anticoagulação em pacientes com HPAI e que há um efeito adverso em pacientes com esclerose sistêmica associada à HP. Em pacientes com HPAI sem contraindicações, uma das opções é considerar a anticoagulação com varfarina para atingir a meta de razão normalizada internacional de 1,5 a 2,5.
O benefício da anticoagulação em pacientes com formas associadas de HP não chegou a ser claramente definido. As oscilações hemodinâmicas na gravidez, no trabalho de parto e no parto são catastróficas em pacientes com HAP, sendo que há registros de índices de mortalidade de até 50%.32
Recomenda-se evitar a gravidez, assim como é essencial evitar a contracepção em mulheres com HAP e com potencial para concepção. Os contraceptivos contendo estrogênio podem aumentar o risco de tromboembolismo venoso em mulheres com HAP; os métodos cirúrgicos ou os de barreira são outros métodos alternativos.
Os avanços terapêuticos mais significativos no tratamento de HAP ocorreram na última década. Atualmente, existem diversas medicações para tratar pacientes com de HAP; entretanto, nenhuma dessas terapias é curativa para essa doença progressiva. As medicações disponíveis no mercado têm como alvo a rota da prostaciclina, a rota do ácido nítrico e a rota da endotelina.
Antes de iniciar alguma terapia específica para HP, é importante medir a gravidade inicial da doença e acompanhar as respostas à terapia. O nível de gravidade da doença poderá ser medido pela classe funcional e pelo grau de danos hemodinâmicos na CCD.
As medicações tradicionalmente disponíveis foram estudadas apenas como monoterapias em pacientes com predominância de HAPI ou HAP associada a doenças no tecido conjuntivo; porém, recentemente, alguns testes clínicos começaram a determinar a segurança e a eficácia da combinação de agentes farmacológicos com mecanismos de ação diferentes.
A extrapolação para outras populações de pacientes com HAP ou o uso dos agentes disponíveis em combinação deve ser considerada com cautela pelos médicos mais experientes no tratamento de HP ou no contexto de testes clínicos. Nos casos de pacientes que não respondem ou que progridem a despeito da terapia médica, recomenda-se considerar as opções cirúrgicas, incluindo transplante.
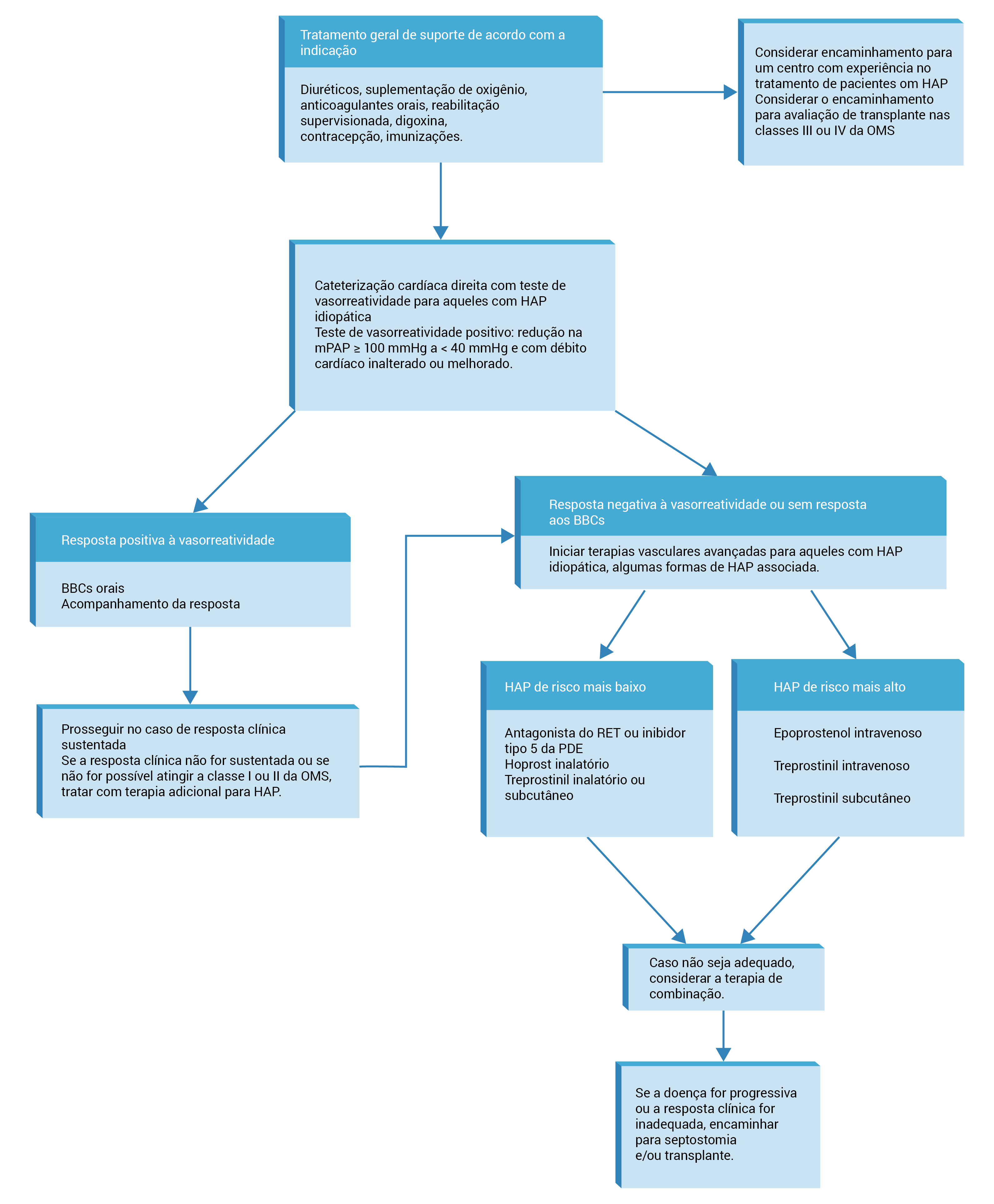
A Figura 8 apresenta uma abordagem ao tratamento de pacientes com HAP.
BCC: bloqueador do canal de cálcio; IC: insuficiência cardíaca; mPAP: pressão arterial pulmonar média; OMS: Organização Mundial da Saúde; PDE: fosfodiesterase; RET: receptor endotelial.
Figura 8 - Abordagem geral aos pacientes com hipertensão arterial pulmonar (HAP). HAP de risco mais baixo se refere aos pacientes sem evidências de IC direita, estados funcionais II ou III da OMS e níveis minimamente elevados de peptídeos natriuréticos. Os pacientes com risco mais elevado de HAP apresentam evidências ecocardiográficas ou clínicas de IC direita, estado funcional IV da OMS e níveis significativamente elevados de peptídeos natriuréticos.
A avaliação inicial de pacientes com HAP deverá incluir avaliação invasiva da hemodinâmica por meio de CCD. Os pacientes com HAPI, e talvez outras populações de pacientes, devem fazer o teste de vasorreatividade aguda em um laboratório de cateterização.
Aproximadamente, 10% de pacientes com HAPI, e uma quantidade menor de pacientes com outras formas de HAP associada, apresentam respostas vasodilatadoras favoráveis, definidas como uma queda na mPAP de 10mmHg a menos de 40mmHg, nas situações com débito cardíaco inalterado ou melhorado, em resposta a algum agente como o ON inalatório ou o epoprostenol intravenoso.
Os pacientes com respostas positivas de vasorreatividade poderão ser tratados inicialmente com um antagonista do canal de cálcio por via oral, como, por exemplo, a nifedipino de ação prolongada, anlodipino ou diltiazem.33,34 Os pacientes tratados dessa forma devem ser acompanhados de perto para verificar se houve alguma melhora clínica, cuja meta é atingir a classe funcional I ou a II.
Nas situações em que os pacientes não responderem à terapia oral com bloqueador do canal de cálcio (BCC), recomenda-se a administração de uma terapia adicional para HAP. Após o início da terapia com um BCC, os pacientes devem ser acompanhados de perto, tendo em vista que alguns deles deterioram a despeito do tratamento. O uso empírico da terapia com BCC deverá ser evitado na ausência de vasorreatividade.34
Os pacientes que não responderem à terapia vasodilatadora, ou que deteriorarem enquanto estiverem na terapia com BCC, devem iniciar uma entre as várias terapias vasculares disponíveis; essas terapias incluem os antagonistas orais e medicamentos como os receptores de endotelina (bosentan, ambrisentana ou macientan); inibidores orais da fosfodiesterase tipo 5 (PDE5) ? sildenafila ou tadalafila; agonistas orais solúveis da guanilina ciclase (riociguat); treprostinil subcutâneo, intravenoso ou oral; iloprost inalatório ou treprostinil inalatório; e epoprostenol intravenoso.
A escolha da terapia de primeira linha é feita com fundamento em vários fatores, incluindo gravidade da HAP, facilidade de administração, toxicidades associadas e custo. De um modo geral, não existem comparações diretas entre os vários agentes para orientar as decisões clínicas.
O epoprostenol é um vasodilatador potente comprovadamente eficaz que melhora a hemodinâmica, a tolerância aos exercícios e a sobrevida.35,36 Por causa da meia-vida de 3 a 5 minutos, o epoprostenol deve ser administrado por infusão IV contínua.
Levando em conta a probabilidade de HP de rebote, caso a terapia seja interrompida, mesmo por períodos curtos de tempo, os planos contingenciais devem ser mantidos à disposição para cada paciente tratado com epoprostenol. Os efeitos colaterais menores incluem cefaleia, dor mandibular, diarreia, dor articular e um tipo específico de erupção cutânea. Recomenda-se aumentar a dose de forma gradual ao longo do tempo para maximizar os benefícios.
Aparentemente, o epoprostenol tem efeitos que vão além da vasodilatação e incluem redução na produção de substâncias vasoconstritoras endógenas e nas propriedades antiplaquetárias e antiproliferativas, que parecem melhorar o que anteriormente pareciam alterações vasculares irreversíveis. O epoprostenol ainda é o único agente aprovado que tem o benefício da sobrevida documentado nos casos de HAP, além de ser o medicamento de escolha para uso em pacientes com HAP de classe IV.37
O treprostinil, um análogo do epoprostenol, poderá ser administrado pelas vias subcutânea, intravenosa ou oral.38?40 Mais recentemente, o treprostinil foi aprovado na forma inalatória. As vantagens do treprostinil em comparação com o epoprostenol incluem a opção da administração oral ou subcutânea contínua, meia-vida mais longa que torna a interrupção da infusão menos arriscada, e sem necessidade de refrigeração, sendo que todas elas dão maior flexibilidade e facilitam a administração do medicamento.
O perfil de efeitos colaterais do treprostinil se assemelha ao do epoprostenol, embora a alta incidência de dor e eritema na infusão subcutânea imponha algumas restrições a este método de administração. Embora a administração intravenosa de epoprostenol e de treprostinil exija acesso venoso central, um estudo retrospectivo sugeriu que o uso intravenoso de treprostinil possivelmente esteja associado a taxas mais elevadas de infecções na corrente sanguínea em comparação com o epoprostenol.41
O iloprost inalatório é um prostanoide que evita a necessidade de administração IV do medicamento; entretanto, o iloprost deve ser administrado por nebulização em torno de seis a nove vezes por dia. Os efeitos colaterais mais comuns incluem tosse, cefaleia, ruborização e dor mandibular.42,43 O treprostinil inalatório é administrado a cada 4 horas. Os efeitos colaterais são semelhantes aos do iloprost e incluem tosse, cefaleia e diarreia.
Os medicamentos bosentan, ambrisentana ou macicentan são antagonistas orais da endotelina. O bosentan é um antagonista duplo do receptor da endotelina, enquanto que o ambrisentana é um antagonista seletivo A do receptor da endotelina. Nenhum teste chegou a fazer uma comparação direta entre eles, embora ambos tenham sido aprovados para o tratamento de HAP moderada a grave.44,45
A via oral de administração torna esses agentes uma alternativa atrativa aos medicamentos subcutâneos ou intravenosos. Dados observacionais disponíveis dos resultados de longo prazo com o bosentan sugerem que houve melhora na sobrevida em pacientes com HAP que usaram esse medicamento em comparação com controles históricos.46
O antagonista duplo do receptor da endotelina macicentan foi desenvolvido através de modificações no bosentan com o objetivo de aumentar a segurança e eficácia; o macicentan melhorou a ligação ao receptor e a penetração tecidual.47 Em um teste randomizado recente controlado por placebo que reuniu 742 pacientes com HAP (Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve cliNical Outcome [SERAPHIN]), o macicentan melhorou o desfecho final da combinação de morbidade e mortalidade de uma forma dose dependente.48
O bosentan e o ambrisentana são contraindicados para aplicação em pacientes com disfunção hepática moderada ou grave. É imprescindível fazer testes mensais da função hepática enquanto os pacientes estiverem usando essas medicações. Não foram observadas interações medicamentosas entre o bosentan e a ciclosporina, contraceptivos orais e gliburida. Os efeitos colaterais, além da toxicidade hepática, incluem edema e anemia. Essas duas medicações são potencialmente teratogênicas, sendo que a contracepção efetiva é essencial.
O perfil de efeitos colaterais do macicentan é mais leve e não produz hepatotoxicidade ou edema periférico com mais frequência que placebo; os efeitos colaterais mais comuns são nasofaringite, cefaleia e anemia. Presume-se que as interações medicamentosas e a teratogenicidade se assemelham às do bosentan e ambrisentana, pelo menos até que seja apresentada uma quantidade maior de dados.48
Os medicamentos sildenafila e tadalafila são inibidores cíclicos da fosfodiesterase tipo 5 (PDE5) administrados por via oral e que foram aprovados para uso em pacientes com HAP moderada.49,50 A atividade do ON é mediada pelo monofosfato de guanosina cíclico (cGMP), um segundo mensageiro que é degradado pela PDE5. O antagonismo à PDE5 por esses agentes prolonga os efeitos vasodilatadores do ON.
A sildenafila deve ser administrada por via oral, 3x/dia, ao passo que a tadalafila deve ser administrada 1x/dia; os efeitos colaterais de ambos os medicamentos incluem cefaleia, refluxo gastroesofágico, ruborização e epistaxe. Recomenda-se evitar o uso de nitratos em pacientes que estiverem tomando inibidores da PDE5, tendo em vista que o efeito aditivo dos medicamentos poderá resultar em hipotensão sistêmica.
O Quadro 4 contém as medicações vasculares avançadas para hipertensão arterial pulmonar.
Quadro 4
|
Medicações Vasculares Avançadas para Hipertensão Arterial Pulmonar | |||
|
Medicamento |
Via de administração |
Dose* |
Comentários |
|
Bloqueadores do canal de cálcio? | |||
|
Nifedipino |
Oral |
30?90mg/dia |
Os BCCs são usados apenas em pacientes com vasorreatividade aguda documentada por cateterização cardíaca. |
|
Anlodipino |
Oral |
2,5?10mg/dia | |
|
Diltiazem |
Oral |
90?360mg/dia | |
|
Análogos da prostaciclina | |||
|
Epoprostenol |
IV |
2?200ng/kg/min |
O epoprostenol e o treprostinil IV são infusões contínuas administradas através de cateteres venosos centrais. |
|
Treprostinil |
IV, SC, oral |
0,625?40ng/kg/min | |
|
Treprostinil |
Inalatório |
18?54µg por inalação q. 4h | |
|
Hoprost |
Inalatório |
2,5?5µg por inalação q. 2?4h | |
|
Antagonistas do RET | |||
|
Bosentan |
Oral |
62,5?125mg, b.i.d. |
O bosentan e o ambrisentana exigem monitoramento mensal com teste hepático e são teratogênicos; contracepção efetiva é essencial.
|
|
Ambrisentana |
Oral |
5?10mg/dia | |
|
Macitentan |
Oral |
5?10mg/dia | |
|
Inibidores da PDE tipo 5 | |||
|
Sildenafila |
Oral |
20mg, t.i.d. |
Os nitratos e os inibidores da PDE tipo 5 em combinação podem causar hipotensão sistêmica.
|
|
Tadalafila |
Oral |
40mg/dia | |
|
Estimuladores da guanilina ciclase | |||
|
Riociguat |
Oral |
0,5?2,5mg, t.i.d. |
A combinação de riociguat e sildenafila causa hipotensão. |
BCC: bloqueador do canal de cálcio; IV: intravenoso; RET: receptor endotelial; SC: subcutâneo.
*As doses devem ser tituladas com base na tolerância e na resposta clínica.
? Não foi aprovado para tratamento de HAP.
O riociguat, um estimulador da guanilina ciclase solúvel administrado por via oral, foi aprovado recentemente para uso em pacientes com HAP e HPTC.24,51 Esse agente evita o aumento dos níveis de ON e estimula a formação do cGMP do segundo mensageiro do ON, provocando vasodilatação pulmonar.
No teste randomizado, duplo-cego e controlado por placebo Pulmonary Arterial Hypertension Soluble Guanylate Cyclase-Stimulator Trial (PATENT), o riociguat melhorou a distância da caminhada de 6 minutos e a hemodinâmica depois de 12 semanas em pacientes que haviam feito terapia com o antagonista do receptor de endotelina ou terapia sem prostanoide IV na linha de base.51
O riociguat deve ser administrado por via oral, iniciando-se com uma dose de 0,5 a 1,0mg, 3x/dia, titulando-a em seguida até o máximo de 2,5mg, 3x/dia. Os efeitos adversos mais comuns relacionados a esse medicamento incluem síncope, gastrite, insuficiência renal aguda e hipotensão. A terapia de combinação é um conceito bastante atrativo, levando-se em consideração a disponibilidade de medicações com alvo em diferentes rotas da doença e a natureza progressiva da HAP.
Recentemente, um estudo randomizado, duplo-cego, orientado por eventos que reuniu 500 pacientes com classe funcional II e III da OMS (ambrisentana e tadalafila em pacientes com HAP [AMBITION]) documentou que a terapia inicial com ambrisentana e tadalafila resultou em uma redução significativa de 50% no risco de falhas clínicas (definidas como uma mistura de morte, hospitalização pelo agravamento da HAP, progressão da doença ou resposta clínica insatisfatória no longo prazo), em comparação com a monoterapia com qualquer um desses agentes.52
A adição de sildenafila nos casos de pacientes com regimes estáveis à base de epoprostenol (Pulmonary Arterial Hypertension Combination Study of Epoprostenol and Sildenafil [PACES]) resultou na melhora da capacidade física para exercícios e dos parâmetros hemodinâmicos; uma análise post hoc demonstrou que houve melhora na sobrevida em comparação com o regime que usou apenas epoprostenol.53
O iloprost inalatório foi estudado como agente complementar em pacientes que estavam usando bosentan, e o resultado foi a melhora na hemodinâmica e no estado clínico.43 A despeito desses avanços, ainda não se conhece a combinação terapêutica ideal para uso em pacientes com HAP que permanecem sintomáticos com a monoterapia.
Além disso, a segurança da combinação de determinados medicamentos não é muito clara, tendo em vista que um teste recente que adicionou riociguat à sildenafila apresentou índices elevados de descontinuação do riociguat por causa da hipotensão.54 A recomendação é não tentar essa combinação até que o perfil de segurança seja definido mais claramente.
A grande maioria dos testes clínicos que investiga o uso de terapias médicas em pacientes com HAP utilizou resultados alternativos tais como estado funcional, parâmetros hemodinâmicos ou hospitalizações ao invés da mortalidade.
Embora a sobrevida não seja um resultado primário na maior parte das investigações, uma metanálise recente de testes importantes sobre HAP apresentou uma redução de 43% na mortalidade por todas as causas utilizando as terapias médicas atualmente disponíveis no mercado, em um número necessário para tratar, aproximadamente, 50 casos e evitar uma morte.55
Da mesma forma, as hospitalizações entre os pacientes tratados com terapia para HAP foram reduzidas em cerca de 40%, mantendo-se um número necessário para tratar algo em torno de 20 casos. A capacidade para fazer exercícios, o estado funcional e os parâmetros hemodinâmicos melhoraram de forma significativa nos pacientes que foram tratados com terapias cujo alvo era a HAP.
Apesar dos avanços no tratamento, muitos pacientes apresentam declínio clínico progressivo, em geral atribuído ao agravamento da IC direita. Nesses pacientes, recomenda-se levar em consideração as opções terapêuticas cirúrgicas e intervencionistas, incluindo septostomia atrial e combinação de transplante de coração e de pulmão. A septostomia atrial com balão percutâneo promove a descompressão do ventrículo direito por meio da criação de um desvio interatrial da direita para a esquerda, reduzindo as pressões de enchimento cardíaco direito e melhorando a função cardíaca nos lados direito e esquerdo.
Embora o desvio diminua a saturação arterial sistêmica de oxigênio, o débito cardíaco melhorado resultante aumenta a liberação sistêmica total do gás. As indicações para septostomia atrial incluem IC direita refratária e/ou síncope recorrente, apesar da maximização da terapia médica, incluindo terapia com alvo na HAP, ou como procedimento-ponte enquanto o paciente estiver na fila para transplante.
Levando-se em consideração que a oxigenação arterial se agrava por causa do desvio da direita para a esquerda, as saturações da oximetria de pulso no ar ambiente antes do procedimento devem ser superiores a 90%. Nos casos de septostomia atrial, a mortalidade total relacionada ao procedimento foi estimada em 16%; pressões no átrio direito antes do procedimento acima de 20mmHg estão fortemente associadas ao aumento no risco de mortalidade pós-procedimento.56
O transplante de pulmão é indicado para pacientes nos quais a HAP tenha progredido apesar da aplicação de terapias médicas e cirúrgicas ideais.1 Nos dias atuais, aproximadamente 5% dos transplantes anuais de coração e de pulmão feitos nos EUA se relacionam à HAP. Os resultados de longo prazo para pacientes com HAP se comparam aos de outros pacientes que fazem transplante de coração e de pulmão, cuja sobrevida após 5 anos é de 50%.57
A maioria dos centros de transplante faz transplantes pulmonares duplos para limitar o grau da lesão por reperfusão pós-transplante que ocorre em pacientes que fazem transplantes simples de pulmão. A decisão de fazer transplante de coração em combinação com transplante de pulmão se baseia na gravidade da descompensação cardíaca ou na presença de doença cardíaca congênita subjacente.
O transplante como opção terapêutica potencial deve ser discutido com todos os pacientes com HAP no momento do diagnóstico. As indicações para encaminhamento para transplante incluem classes III ou IV da OMS na apresentação ou no início do regime com um análogo intravenoso para a prostaciclina.37
Prognóstico
O registro de pacientes com HAP do National Institutes of Health publicado na década de 1980 definiu o histórico natural da doença.56 A expectativa média de vida a partir do diagnóstico era de 2,8 anos e a sobrevida de 5 anos variava entre 22 e 38%. O prognóstico também é influenciado pela etiologia subjacente.
Os pacientes com HAP associada à esclerose sistêmica apresentam piores prognósticos em comparação com pacientes com HAP,59 ao passo que os pacientes com HAP associada à doença cardíaca congênita tendem a apresentar melhores prognósticos.60 Os preditores independentes de mortalidade em pacientes com HAP incluem classe funcional III ou IV da OMS, elevação na pressão no átrio direito, redução no débito cardíaco e elevação na mPAP ou presença de efusão pericardíaca ecocardiográfica.16
Os pacientes com respostas vasodilatadoras positivas são os que apresentam melhores prognósticos.1,61 Os níveis de BNP e de NT-proBNP se correlacionam com a sobrevida nos casos de HAPI.62,63 O advento das terapias vasculares avançadas mudou o histórico natural da doença. Os testes observacionais de longo prazo com terapia médica para tratamento de HAP sugerem que há melhorias na sobrevida, sendo que a sobrevida de 2 anos varia de 70 a 84%.7,64
O Quadro 5 apresenta o gerenciamento perioperatório de HAP.
Quadro 5
|
Gerenciamento Perioperatório de Hipertensão Arterial Pulmonar | |
|
Fatores Operatórios que Afetam a RVP |
Prevenção e Gerenciamento |
|
Descontinuação rápida de terapias para HP com prostenoide inalatório ou parenteral |
A. Evitar a descontinuação rápida de terapias com ação curta e considerar as terapias de ponte de ação prolongada como a sildenafila. B. Gerenciamento agudo com ON inalatório ou com prostanoides inalatórios/parenterais.
|
|
Débito simpático (dor, ansiedade e intubação) |
A. Analgesia adequada com opioides para evitar resposta simpática reflexa. B. Administrar baixas doses de benzodiazepinas para ansiedade na fase pré-operatória.
|
|
Hipercapnia e hipóxia |
A. Pré-oxigenação com 100% de oxigênio para atingir concentrações de oxigênio expirado acima de 90% antes da indução. B. Alcalose respiratória permissível (PCO2 =30?35mmHg).
|
|
Acidose, hipotermia |
Monitorar e corrigir rapidamente; manter pH >7,4.
|
|
PEEP (>15mmHg), pressão inspiratória (>30mmHg)
|
Otimizar os ambientes ventilatórios. |
|
IC esquerda, SDRA, EP |
Monitorar por linha arterial, ecocardiografia transesofágica ou uso de cateter na artéria pulmonar.
|
|
Isquemia-reperfusão, lesão relacionada ao uso de protamina |
A. Minimizar o tempo de desvio e considerar a terapia sem bomba. B. ON inalatório, prostaciclina ou milrinona enquanto o paciente estiver na bomba ou durante o período de saída gradual da bomba.
|
|
Óxido nítrico e propofol |
O etomidato é preferível para indução de anestesia.
|
|
Pneumectomia, oclusão transitória da artéria pulmonar |
Óxido nítrico inalatório profilático e terapia com prostaciclina. |
EP: embolia pulmonar; HP: hipertensão pulmonar; IC: insuficiência cardíaca; ON: óxido nítrico; PEEP: pressão positiva no final da expiração; ON: óxido nítrico; PCO2: tensão do dióxido de carbono; RVP: resistência vascular pulmonar; SDRA: síndrome do desconforto respiratório agudo.
Gerenciamento Perioperatório de Hipertensão Arterial Pulmonar
A adequação entre o uso de analgesia e de ansiólise é extremamente importante em pacientes com HP. Analgesia e/ou ansiólise inadequada poderá resultar em um aumento no débito simpático, aumentando a RVP e a demanda de oxigênio no ventrículo direito. Essas alterações na hemodinâmica ventricular direita em pacientes com ventrículo direito já comprometido poderá criar um círculo vicioso de fisiologia ventricular direita descompensada.
Por outro lado, a sedação excessiva poderá produzir hipoventilação com hipercapnia e hipóxia subsequentes. Ambos os fatores poderão resultar em um aumento potencialmente grave na RVP, agravando, consequentemente, a hemodinâmica ventricular direita.65
A seguir, são descritos alguns fatores perioperatórios comuns que possivelmente afetem a RVP e as estratégias de gerenciamento e que poderão ser usados para sua solução.
hipertensão pulmonar Tromboembólica Crônica
Epidemiologia
A HPTC é a única causa grave de HAP cuja cura potencial poderá ser obtida sem a necessidade de transplante pulmonar; portanto, o reconhecimento e o diagnóstico dessa entidade são extremamente importantes. A HPTC é uma complicação de longo prazo da EP. Aproximadamente, 4% dos pacientes que sofrem de EP apresentam desenvolvimento subsequente de HPTC.24,66
Entretanto, a maioria dos pacientes com HPTC não tem histórico de tromboembolismo venoso. Nos EUA, ocorrem cerca de 2.500 casos novos de HPTC por ano, embora o número estimado de casos não registrados ou não reconhecidos provavelmente seja muito maior. Essa doença foi observada em adultos de todas as idades, porém mais de 50% dos pacientes têm idade inferior a 45 anos.
Embora a grande maioria dos pacientes com HPTC possivelmente tenha algum distúrbio trombótico, como, por exemplo, lúpus eritematoso, a maior parte desses indivíduos não tem fatores de risco tromboembólicos venosos clássicos ou trombofilia hereditária identificável.24 Assim como nos casos de HAP, várias condições médicas sistêmicas foram associadas ao desenvolvimento de HPTC, incluindo esplenectomia, doença intestinal inflamatória e osteomielite crônica.
Fisiopatologia
A fisiopatologia de HPTC é muito complexa e pouco compreendida. Provavelmente, a obstrução das artérias pulmonares proximais por material tromboembólico organizado e o aumento na resistência ao fluxo sejam os insultos iniciais. Subsequentemente, ocorre uma remodelagem vascular nos vasos grandes e pequenos dos pulmões, com espessamento da íntima, deposição de colágeno e calcificação.
A arteriopatia nos vasos pequenos que se observa nos casos de HPTC se assemelha àquela que produz HAPI e reflete a remodelagem vascular em resposta à intensificação do fluxo nas partes do leito arterial pulmonar distal que não foram obstruídas por algum material tromboembólico proximal.
Diagnóstico
O diagnóstico de HAP foi anteriormente. A possível presença de HPTC deve ser levada em consideração em todos os pacientes com HAP. A varredura dos pulmões por V/Q é o teste diagnóstico de escolha. Nos casos de HPTC, as varreduras por V/Q quase sempre são anormais e demonstram pelo menos um defeito de perfusão segmentar ou maior.
Observa-se também esse padrão nos tumores, em DPVO ou nos casos de mediastinite fibrosante. As varreduras por V/Q normais excluem efetivamente a hipótese de HPTC. Embora as angiografias torácicas por TC sejam precisas no diagnóstico de EP aguda, seu papel é muito limitado no diagnóstico de HPTC por causa da baixa sensibilidade.
Os defeitos de enchimento da artéria pulmonar geralmente observados nos casos de EP aguda sem sempre estão presentes na HPTC; perda de contraste nos vasos pulmonares cheios é a marca registrada de HPTC nas angiografias. Uma revisão retrospectiva recente comparou a capacidade da angiografia multidetectora por TC para diagnosticar HPTC com varredura dos pulmões por V/Q, em comparação com a angiografia pulmonar que é o padrão de ouro.26
A varredura dos pulmões por V/Q apresentou uma sensibilidade de 96% e uma especificidade de 90%, ao passo que a angiografia por TC apresentou uma sensibilidade de 51% e uma especificidade de 99%. Portanto, embora a resolução da angiografia por TC esteja melhorando e possa sugerir HPTC, o teste de rastreamento de escolha ainda continua sendo a varredura dos pulmões por V/Q.
A angiografia pulmonar é o teste definitivo nas situações em que houver suspeita de HPTC com base na varredura dos pulmões por V/Q. Essa modalidade serve não apenas para confirmar o diagnóstico, mas também para avaliar doenças corrigíveis por meios cirúrgicos.
Gerenciamento
A endarterectomia pulmonar (EAP) é o tratamento definitivo de HPTC, embora nem todos os pacientes diagnosticados com a doença sejam candidatos cirúrgicos.67 A EAP é a endarterectomia autêntica e envolve o descolamento da camada íntima enferma das artérias pulmonares proximais; esse procedimento é diferente da embolectomia feita em alguns casos de EP aguda. A decisão sobre a viabilidade da EAP para aplicação em pacientes específicos deverá ser tomada em um centro especializado nesse tipo de procedimento.
As considerações gerais incluem evidências de trombos acessíveis por meios cirúrgicos, o grau de RVP de acordo com os resultados das medições feitas durante a CCD, e as comorbidades dos pacientes. A EAP é feita através de uma esternotomia mediana no desvio cardiopulmonar com interrupção hipotérmica profunda para minimizar a perda sanguínea.
Na maior parte dos pacientes, a redução pós-carga no ventrículo direito, por meio da remoção do material obstrutivo da rede de vasos pulmonares, resulta na redução imediata e significativa nas pressões das artérias pulmonares.24,67 Aproximadamente, 10% de pacientes continuam a ter HP persistente depois da endarterectomia pulmonar, provavelmente como consequência da arteriopatia nos vasos menores.24
No caso de pacientes com HP persistente depois de EAP, ou de pacientes com HPTC que não sejam candidatos cirúrgicos, a terapia médica é a melhor opção; o riociguat é um estimulador solúvel da guanilina ciclase que aumenta os níveis de cGMP que foi aprovado recentemente para aplicação nos casos de HPTC inoperável.
Em um estudo randomizado, duplo cego e controlado por placebo que reuniu 261 pacientes com HPTC inoperável, o riociguat melhorou a capacidade para fazer exercícios e a RVP depois em 16 semanas.58 Todos os pacientes com HPTC devem ser tratados com anticoagulação por toda a vida, sendo que a maioria dos pacientes que faz endarterectomia pulmonar recebe filtros na veia cava inferior.
Recentemente, no Japão, uma série de casos sugeriu que os pacientes selecionados com doença periférica, e que estiverem fazendo angiografia pulmonar percutânea, poderão ter benefícios com a angioplastia pulmonar percutânea,69 embora sejam necessários mais dados antes que essa técnica passe a ser utilizada clinicamente. Assim como nos pacientes com HAP, o transplante de pulmão é uma opção para pacientes com HPTC grave refratária às terapias médicas ou cirúrgicas.
Prognóstico
Sem cirurgia, a sobrevida de pacientes com HPTC é baixa. A sobrevida de 5 anos em pacientes com mPAP de 40 a 50mmHg é de 30%; a sobrevida de 5 anos é de 10% em pacientes cujas pressões na artéria pulmonar forem superiores a 50mmHg.70 A mortalidade associada à tromboendarterectomia pulmonar varia de 5 a 24% e é mais baixa nas instituições com maior experiência. Os pacientes que conseguem sobreviver apresentam melhoras significativas no estado funcional e, além disso, a pressão arterial pulmonar poderá continuar a cair em até 1 ano após a cirurgia.24,70
Outras Causas de Hipertensão Arterial Pulmonar
Anemias hemolíticas crônicas, incluindo a doença da célula falciforme, talassemias e esferocitoses, foram associadas a casos de HAP.1 Um teste prospectivo de rastreamento descobriu que 32% dos pacientes com doença da célula falciforme apresentavam sPAPs ligeiramente elevadas que foram medidas por ultrassonografia Doppler não invasiva, e 9% apresentavam elevações moderadas a graves nas sPAPs.71
Alguns estudos de CCD sugerem que entre 6 a 8% de pacientes com células falciformes têm mPAP acima de 25mmHg, cujas etiologias se dividem igualmente entre HAP e doença cardíaca esquerda.72 A patogênese de HAP nos casos de hemólise crônica provavelmente seja multifatorial. A hemoglobina livre desativa o ON, um vasodilatador endógeno, enquanto que a hemólise também libera arginase, uma enzima que degrada a arginina, que é o substrato da síntese do ON.73,74
A disfunção esplênica que ocorre com a hemólise crônica também é um fator; a perda da função esplênica estimula a ativação plaquetária e aumenta a taxa de hemólise intravascular.75 Os pacientes com anemias hemolíticas crônicas mantêm a liberação sistêmica de oxigênio por meio de um aumento no débito cardíaco; portanto, esses pacientes são menos tolerantes aos aumentos na RVP. Embora, de um modo geral, a dispneia em pacientes com hemólise crônica seja atribuída à anemia, e talvez a alguma doença pulmonar, a HP deve ser incluída no diagnóstico diferencial.
O tratamento de HAP em pacientes com anemias hemolíticas crônicas é de suporte e deverá otimizar o controle do processo hemolítico subjacente; as recomendações mais recentes orientam que os pacientes com células falciformes e RVP normal e com pressão elevada (em cunha) de oclusão capilar pulmonar não podem receber terapias vasculares avançadas, embora alguns pacientes com RVP elevada e pressão em cunha normal possam se beneficiar com as terapias específicas para HAP.76
Em pacientes com doença da célula falciforme, a pressão da HP está forte e independentemente associada ao aumento no risco de morte.72 A HPPO é uma complicação incomum de doença hepática em estado terminal. Aproximadamente, 6% de pacientes com cirrose desenvolvem HAP e manifestam HAP elevada, pressões normais de oclusão arterial pulmonar e RVP elevada.
A RVP elevada é uma característica típica importante, levando-se em consideração que muitos pacientes com doença hepática crônica apresentam pressões arteriais pulmonares elevadas secundárias a débitos cardíacos elevados e, mesmo assim, conseguem manter uma RVP baixa. Por conseguinte, em pacientes com doença hepática crônica e sPAP elevada estimada por ecocardiografia transtorácica, a CCD é fundamental para avaliar o débito cardíaco e determinar a RVP.
Não é possível fazer a diferenciação histológica entre HPPO e outras formas de HAP; a presença de hiperplasia íntima e de lesões vasculares plexiformes é uma ocorrência típica. É imprescindível considerar a hipótese de HPPO em pacientes com doença hepática crônica, tendo em vista que está associada aos piores resultados depois de transplantes de fígado.77
A DPVO é uma forma rara, porém distinta, de HP que se caracteriza pela obstrução de pequenas veias intrapulmonares.79 Estima-se que a taxa de incidência de DPVO seja de 0,1 caso por 1 milhão, com uma idade mediana de 40 anos no diagnóstico.80 Embora a maior parte dos casos seja idiopática, alguns pacientes desenvolvem DPVO no ambiente de infecção pelo HIV, uso de medicamentos quimioterápicos ou transplante de medula óssea.
Há também relatos de uma forma hereditária de DPVO.81 Os pacientes com esse tipo de distúrbio apresentam dispneia crescente, às vezes com hemoptise. As descobertas feitas pelas radiografias torácicas sugerem a presença de insuficiência ventricular esquerda; essas descobertas incluem artérias pulmonares aumentadas, linhas B de Kerley, edema pulmonar e efusões pleurais.
Deve-se considerar o diagnóstico de DPVO nas situações em que essas descobertas radiográficas não estiverem associadas a nenhuma evidência ecocardiográfica ou hemodinâmica de disfunção ventricular esquerda, doença na válvula mitral ou obstrução de fluxo no átrio esquerdo. As varreduras por TC no tórax dão suporte para o diagnóstico;82 essas varreduras ajudam também a excluir a hipótese de obstrução de veias pulmonares no mediastino (mediastinite fibrosante ou tumor).
A CCD revela pressões arteriais pulmonares elevadas; paradoxalmente, a pressão de oclusão nas artérias pulmonares (pressão em cunha) costuma ser normal.83 Nos casos em que forem aplicáveis, as biópsias do pulmão mostram uma combinação de congestão crônica, alterações hipertensivas nas artérias pulmonares e estreitamento ou oclusão nas veias pulmonares produzido por fibrose íntima excêntrica ou concêntrica.
Entretanto, não se recomenda fazer biópsias nos casos em que houver fortes suspeitas clínicas de que o procedimento não esteja sendo bem tolerado e tenha um risco substancial de morte. As terapias usuais administradas nos casos de HAP idiopática raramente são bem-sucedidas no tratamento de doença veno-oclusiva.
Mais especificamente, os relatos indicam o desenvolvimento de edema pulmonar em pacientes com DPVO nas situações em que forem tratados com agentes utilizados nos casos de HAP, incluindo o epoprostenol e o bosentan.83 Da mesma forma, o transplante de pulmão também deverá ser considerado no momento do diagnóstico. O prognóstico de DPVO não tratada não é bom, com uma mortalidade estimada 70% depois de 1 ano.79
A hemangiomatose nos vasos capilares dos pulmões, um distúrbio que imita clinicamente ou pode ser um componente de DPVO, é uma causa rara de HP.84 Nesses pacientes, os espécimes de biópsias no pulmão têm regiões irregulares de congestão grave contendo vasos sanguíneos do tamanho de capilares que parecem invadir as paredes das veias pulmonares e, em menor escala, as artérias dos pulmões. As varreduras por TC ajudam a fazer a distinção entre este distúrbio e HAP idiopática.85 Transplante de pulmão é o único tratamento eficaz.
hipertensão pulmonar Atribuída à Doença Cardíaca Esquerda
A doença cardíaca esquerda é uma das causas mais comuns de HP. A IC crônica, atribuída à disfunção sistólica ou diastólica, e a doença valvular são as etiologias mais frequentes de doença cardíaca esquerda que resulta em HP. Até 60% dos pacientes com insuficiência sistólica ventricular esquerda grave e 80% dos com insuficiência diastólica ventricular esquerda grave desenvolvem HP.86
A presença de HP está associada aos piores prognósticos em pacientes com doença cardíaca esquerda. A patogênese de HP nos casos de doença cardíaca esquerda é predominantemente passiva, hipertensão venosa pulmonar secundária a pressões atriais esquerdas elevadas, embora alguns pacientes apresentem vasoconstrição arterial pulmonar concomitante.87
O diagnóstico de HP em casos de doença cardíaca esquerda segue o algoritmo descrito na Figura 3 e, normalmente, se torna óbvio com base em fatores de risco clínicos e em características ecocardiográficas. A CCD documenta a presença e a gravidade da HP; gradientes transpulmonares elevados, a diferença entre mPAP e pressão de oclusão arterial pulmonar (pressão em cunha), superiores a 12mmHg sugerem um componente pré-capilar da HP.
O tratamento de HP em pacientes com doença cardíaca esquerda é o controle ideal da IC subjacente ou de doença valvular. São muito limitados e misturados os dados dos testes randomizados sobre a eficácia das terapias vasculares avançadas para tratar HP no contexto de doença cardíaca esquerda. Os resultados dos estudos iniciais sobre os prostanoides e os antagonistas do receptor de endotelina foram desalentadores e levaram a um aumento na mortalidade e nas exacerbações de IC.
Mais recentemente, a sildenafila ou a inibição da fosfodiesterase tipo 5 surgiram como opções mais bem toleradas e mais eficazes para o tratamento desses pacientes. O riociguat, um inibidor da guanilina ciclase, embora seja bem tolerado, não conseguiu melhorar a mPAP nos testes iniciais feitos em pacientes com IC sistólica e diastólica.88 Os médicos deveriam usar as terapias vasculares avançadas não apenas em pacientes com doença cardíaca esquerda e HP não responsiva aos tratamentos padrões, mas também em testes clínicos.
hipertensão pulmonar Atribuída à Doença Pulmonar e/ou Hipoxemia
A HP é uma complicação conhecida de DPOC. Em uma grande coorte de pacientes com DPOC com um espectro da gravidade da doença, a incidência de HP diagnosticada por CCD foi de, aproximadamente, 50%.89 Alguns estudos indicam que a incidência de HP aumenta em pacientes com doenças mais graves.
Scharf e colaboradores revisaram alguns dados de CCD em pacientes que haviam sido encaminhados para cirurgia de redução no volume pulmonar; nesta coorte, o percentual médio previsto para o volume expiratório forçado em um segundo (FEV1) foi de 27%.90 Nesse grupo, 86% dos pacientes tiveram HP moderada, definida como uma mPAP acima de 20mmHg, enquanto que 5% tiveram HP grave, definida como uma mPAP acima de 35mmHg.
Em outra coorte de pacientes com DPOC grave, 50% apresentaram medição invasiva de mPAP superior a 25mmHg, enquanto que 14% apresentaram uma mPAP acima de 35mmHg.91 Cabe observar que, embora a gravidade da HP tenha a tendência de aumentar com o aumento na gravidade da obstrução no fluxo de ar, há também um subgrupo de pacientes com DPOC com HP “fora de proporção” cuja obstrução de fluxo é apenas moderada, porém com capacidade difusora acentuadamente reduzida, hipoxemia em estado de repouso e pressões pulmonares elevadas.
Em uma coorte, esse último grupo correspondia a 6% dos pacientes com DPOC encaminhados para transplante ou cirurgia de redução no volume pulmonar.91 Embora a HP observada em pacientes com DPOC seja predominantemente leve a moderada em termos de gravidade, a carga de DPOC na população é alta; em relação às outras etiologias de HP, os números absolutos de pacientes com DPOC e HP são bastante significativos.
Além disso, a HP é uma descoberta prognóstica negativa em pacientes com DPOC. Pacientes com mPAP acima de 25mmHg tiveram sobrevida total de 5 anos de 36% em comparação com uma sobrevida de 5 anos de 62% em pacientes com grau semelhante de gravidade de DPOC, porém sem HP.92
A fisiopatologia de HP em pacientes com DPOC se assemelha àquela observada em outras etiologias de HP. Hipóxia alveolar é um fator muito importante, assim como a perda de área transversal à vasculatura pulmonar secundária à destruição parenquimatosa. Da mesma forma como se observa nos casos de HAP, demonstrou-se a ocorrência de remodelagem vascular, inflamação e níveis elevados de vasoconstritores como a endotelina.89
A incapacidade de diminuir a RVP durante os exercícios é um aspecto exclusivo da HP em pacientes com DPOC. Durante os exercícios, o débito cardíaco aumenta e a RVP diminui por meio do recrutamento de vasos pulmonares para acomodar o aumento no fluxo e manter baixas as pressões arteriais pulmonares.
Esse aumento na RVP não ocorre em pacientes com DPOC; além disso, há uma elevação anormal nas pressões pulmonares, mesmo em pacientes com pressões pulmonares normais no estado de repouso. A razão dessa resposta anormal à RVP em pacientes com DPOC provavelmente envolva a perda de leito vascular recrutável e hiperinflação dinâmica relacionada aos exercícios, com elevação nas pressões alveolares transmitidas para os vasos pulmonares.93
A elevação nas pressões pulmonares associada aos exercícios significa que mesmo atividades rotineiras diárias como subir escadas, por exemplo, podem agravar acentuadamente a dispneia na linha de base em pacientes com DPOC.
O diagnóstico de HP em pacientes com DPOC segue o algoritmo descrito na Figura 3. A ecocardiografia se tornou o meio mais útil para detectar alterações cardíacas causadas por HP no lado direito, embora tenha pouca utilidade em pacientes com doença pulmonar crônica, em comparação com pacientes com HAPI, levando-se em conta que as imagens subótimas são mais frequentes.
Em um estudo de grande porte, apenas 44% dos pacientes apresentaram visualização adequada do jato tricúspide regurgitante que permitiram estimar a sPAP.94 Os valores preditivos, positivos e negativos, da ecocardiografia Doppler para diagnóstico de HP em pacientes com doença pulmonar obstrutiva correspondem a 32 e 93%, respectivamente.94
O tratamento de pacientes com HP associada à DPOC é basicamente de suporte. A terapia com oxigênio em pacientes com hipoxemia documentada é primordial. A terapia contínua com oxigênio estabiliza a progressão da HP, chegando mesmo a revertê-la em alguns casos, embora as pressões pulmonares raramente retornem aos níveis normais.95,96
Embora existam alguns agentes aprovados para uso em casos de HAPI, esses medicamentos devem ser usados com cautela em pacientes com doença pulmonar crônica, tendo em vista que poderão agravar a incompatibilidade V/Q. Os medicamentos bosentan e sildenafila foram usados em pacientes com HP associada à DPOC em testes não controlados; não existem testes com desenhos adequados.
Um pequeno teste randomizado de bosentan em pacientes com DPOC grave encontrou uma séria deterioração no conteúdo do oxigênio arterial, sem qualquer melhora nos sintomas clínicos ou na capacidade para fazer exercícios.97 Observa-se também a HP em pacientes com DPI, embora a prevalência geral não tenha sido bem definida e varie significativamente com o tipo específico de DPI. Em um estudo prospectivo, 32% dos pacientes com vários tipos de DPI que haviam feito CCD tinham HP significativa, definida como mPAP superior a 35mmHg.98
A presença de HP é comum, ou talvez seja uniforme, em casos de histiocitose pulmonar de células de Langerhans em estado terminal (granuloma eosinofílico), um distúrbio nas células de Langerhans em proliferação que poderá afetar diversos sistemas de órgãos e que costuma estar associado ao tabagismo. Existem também relatos de HP com outras DPIs, incluindo diversos tipos de pneumoconioses e de sarcoidoses.
Atribui-se a patogênese de HP no contexto de DPI a diversos mecanismos, tais como vasoconstrição hipóxica e obstrução vascular associada à fibrose parenquimatosa progressiva. O tratamento de HP no contexto de DPI depende do distúrbio subjacente. Assim como nos casos de DPOC, o tratamento de suporte e a terapia com oxigênio são indicados de acordo com a necessidade.98
Não existem testes randomizados de grande porte sobre terapia vascular avançada aplicável a casos de HP secundária a DPI; entretanto, alguns estudos-piloto mostraram que a sildenafila e o bosentan são bem tolerados pelos pacientes com DPI, sem efeitos adversos na troca de gases e sugestão de RVP reduzida.99,100.
Da mesma forma, não foi realizado nenhum teste prospectivo com terapias vasculares avançadas em casos de sarcoidose, embora alguns estudos retrospectivos com epoprostenol, sildenafila e bosentan tenham registrado alguma melhora no estado funcional e nos parâmetros hemodinâmicos.101,102
A residência em grandes altitudes possivelmente esteja associada ao desenvolvimento de HP.103 Esse tipo de resposta é inadequado; ocorre apenas em uma pequena parte da população que vive em regiões de grandes altitudes e provavelmente seja secundário à ocorrência de hipóxia crônica. Nesses pacientes, a mudança para altitudes menores diminui a pressão arterial pulmonar.
hipertensão pulmonar Atribuída a Outros Distúrbios
Fatores como compressão ou estenose nas veias ou artérias pulmonares no mediastino por algum tumor ou mediastinite fibrosante ou estenose com múltiplos segmentos periféricos da artéria pulmonar poderão produzir HP pré-capilar ou pós-capilar. Comumente, esses distúrbios são diagnosticados por TC helicoidal ou por angiografia pulmonar.
Cor Pulmonale
Cor pulmonale é o termo usado para IC direita e HP resultante de distúrbios no parênquima pulmonar, na caixa torácica ou no sistema neuromuscular, excluindo doença cardíaca congênita e distúrbios no lado direito do coração. Cor pulmonale poderá ocorrer na forma aguda em ambientes com sobrecarga pressórica no ventrículo direito com início rápido, ou de forma crônica com o início lento da HP.
Nos EUA, DPOC é a causa mais comum de cor pulmonale crônica; as outras causas conhecidas de cor pulmonale incluem DPI, doenças neuromusculares como distrofia muscular, defeitos na caixa torácica como cifoescoliose grave e a síndrome de hipoventilação da obesidade (SHO).104
Cor Pulmonale Aguda
Patogênese
O ventrículo direito normalmente bombeia contra pós-cargas baixas, mesmo quando o débito cardíaco aumentar de forma dramática pelos exercícios. Em resposta a um aumento agudo na pós-carga vascular pulmonar, o ventrículo direito se distende e aumenta o volume ventricular direito sistólico e diastólico, embora não consiga produzir pressões elevadas (a pressão máxima gerada de forma aguda pelo ventrículo direito é de, aproximadamente, 40mmHg).
Nas situações em que o ventrículo direito não conseguir compensar adequadamente, ocorrem elevações na pressão ventricular direita no fim da diástole (RVEDP) produzindo IC direita aguda. Além disso, o débito cardíaco reduzido do ventrículo direito diminui o enchimento ventricular esquerdo, resultando em hipotensão sistêmica.
A redução comum na perfusão causada por pressão diastólica sistêmica baixa diminui ainda mais a capacidade do ventrículo direito para superar a resistência adicional, produzindo um declínio que poderá levar à morte do paciente.105
Os distúrbios que causam cor pulmonale são doenças que produzem obstrução súbita na rede de vasos pulmonares (por exemplo, tromboembolismo pulmonar maciço),106 embolia aguda causada por outros materiais (por exemplo, ar, medula óssea, gordura, líquido amniótico ou tumor), ou obstrução na rede microvascular causada por pressão elevada nas vias respiratórias ou sua destruição, a exemplo do que ocorre na síndrome do desconforto respiratório agudo.107
Diagnóstico
No contexto de insuficiência ou choque respiratório agudo, com evidências de IC direita aguda, a ecocardiografia imediata ao lado do leito geralmente demonstra a presença de cor pulmonale aguda. As características observadas incluem HP (normalmente leve), dilatação ventricular direita sem hipertrofia, regurgitação tricúspide, achatamento septal ou movimento septal paradoxal e disfunção diastólica no ventrículo esquerdo.108
Gerenciamento
Nas situações em que for possível, o reconhecimento rápido da cor pulmonale aguda e o alívio da obstrução nos vasos pulmonares são essenciais para a sobrevida de pacientes com esse tipo de doença. Por exemplo, a terapia trombolítica em pacientes com EP maciça aguda poderá resolver completamente a cor pulmonale aguda.109
Cor Pulmonale Crônica
Patogênese
O ventrículo direito se distende em resposta ao aumento crônico na RVP e se atrofia. Em geral, ocorrem elevações na RVEDP e na pressão atrial direita nas situações em que o ventrículo perder a capacidade de compensação resultando, consequentemente, em IC direita.
A HP que tipifica cor pulmonale crônica é produzida por aumentos na RVP causados por variações nas combinações de destruição e obstrução nos vasos pulmonares, por vasoconstrição causada por hipóxia e pela remodelagem na rede de vasos pulmonares. Algumas dessas alterações (por exemplo, vasoconstrição hipóxica) se revertem com a terapia, enquanto que outras (por exemplo, destruição do leito vascular) não são reversíveis.110
Diagnóstico
A detecção imediata de cor pulmonale crônica talvez seja difícil, tendo em vista que as manifestações de doença pulmonar subjacente dominam o quadro clínico. As características e a avaliação clínica são as mesmas descritas para os casos de HP.
Gerenciamento
A natureza e a gravidade da doença pulmonar subjacente determinam os resultados de pacientes com cor pulmonale crônica. Por exemplo, cor pulmonale secundária à apneia do sono talvez seja totalmente reversível com o tratamento direcionado para o distúrbio ventilatório subjacente, enquanto que a cor pulmonale causada por fibrose pulmonar idiopática geralmente é irreversível. Em pacientes com DOPC, a presença de HP se correlaciona com o aumento na gravidade da disfunção pulmonar.
A abordagem mais importante ao tratamento de cor pulmonale crônica é o tratamento da doença pulmonar subjacente. Para manter a saturação de O2 acima de 90% em pacientes com hipoxemia, a suplementação de oxigênio deve ser feita em estado de repouso, durante os exercícios e durante o sono.
Os diuréticos são úteis para diminuir condições associadas à cor pulmonale como edema, ascite e congestão hepática; entretanto, esses agentes devem ser usados cuidadosamente para evitar quedas nas pressões de enchimento do ventrículo direito, que poderão levar a uma redução no débito cardíaco.
Na era atual da terapia de suplementação de oxigênio, raramente é necessário usar flebotomia em pacientes com policitemia secundária. Possivelmente, em pacientes com insuficiência respiratória crônica a ventilação mecânica não invasiva também melhore a hemodinâmica.111
A síndrome de hipoventilação da obesidade (SHO) é um distúrbio de hipoventilação alveolar que se caracteriza pela presença de hipersonolência, dispneia e hipoxemia em repouso. Os testes dos gases arteriais que mostram hipercapnia durante o dia confirmam o diagnóstico. Enquanto a maioria dos pacientes com apneia obstrutiva do sono (AOS) não tem SHO, a maior parte dos pacientes com SHO tem AOS.
Embora a apneia do sono seja uma causa conhecida de HP, tipicamente ela produz apenas pequenas elevações nas pressões pulmonares e geralmente não está associada à cor pulmonale. Por outro lado, a cor pulmonale poderá se desenvolver em pacientes com SHO, provavelmente como decorrência da hipoxemia persistente necessária para produzir HP significativa e a insuficiência ventricular direita resultante. A ventilação noturna não invasiva é o grande pilar da terapia para AOS e SHO. A terapia adicional para SHO inclui medidas para perda de peso.110
As doenças pulmonares restritivas poderão resultar de alguma doença neuromuscular, de lesão na medula espinal ou de fatores que limitam a expansão da parede torácica, como, por exemplo, cifoescoliose grave. Fatores como fraqueza nos músculos respiratórios e complacência anormal da parede torácica diminuem a capacidade vital; nos casos mais avançados, é comum a ocorrência de hipoxemia e hipercarbia.
A cor pulmonale poderá se desenvolver em todos esses distúrbios como resultado de hipoventilação, em especial a hipoventilação noturna. Assim como em pacientes com SHO, o grande pilar do tratamento nesses pacientes é o suporte respiratório, inicialmente com ventilação noturna não invasiva.
A ventilação noturna é uma opção a ser considerada nos casos em que a pressão parcial arterial do dióxido de carbono (PACO2) exceder 45mmHg ou na hipótese de ocorrer hipoxemia noturna sustentada, com sinais e sintomas de transtorno do sono documentado por meio de polissonografia. Alguns pacientes provavelmente precisem de suporte ventilatório contínuo na medida em que avançar o processo da doença subjacente.108
Malformações Arteriovenosas Pulmonares
Epidemiologia e Patogênese
As comunicações anormais entre ramos das artérias e veias pulmonares, produzindo um desvio da direita para a esquerda, denominam-se malformações arteriovenosas (MAVs) pulmonares. Essas lesões podem ser adquiridas (por exemplo, como resultado de algum trauma ou de alguma complicação da cirrose) ou congênitas.
As MAVs congênitas podem ser simples ou múltiplas. Elas se restringem aos pulmões ou, mais comumente, ocorrem como parte de telangiectasia hemorrágica hereditária (THH) - também conhecida como doença de Weber-Rendu, uma síndrome autossômica dominante de MAVs generalizadas na pele, membranas mucosas, pulmões e outros órgãos internos.
A THH possivelmente esteja associada às mutações da endoglina ou aos genes ALK1.112 Aproximadamente, 80% de todas as MAVs pulmonares ocorrem no contexto de THH. As MAVs pulmonares podem apresentar complicações graves como resultado dos desvios da direita para a esquerda que contornam o leito dos vasos capilares e facilitam a passagem de êmbolos trombóticos e de bactérias para a circulação sistêmica.
Existem registros de abscessos cerebrais em 5 a 9% dos pacientes com THH, assim como de outros abscessos extracerebrais. Os pacientes com THH correm grande risco de infartos cerebrais. Acidentes vasculares cefálicos isquêmicos ocorrem em 10 a 20% de pacientes com THH, sendo que um número maior de pacientes apresenta sintomas isquêmicos transitórios.
A idade média no momento do diagnóstico de MAVs pulmonares é de 42 anos, com uma leve predominância feminina de 2 por 1. Estima-se que a prevalência de THH seja de 1 em cada 5 mil casos, o que a torna uma das doenças monogênicas mais comuns. Embora os sintomas diários sejam dominados por epistaxe, a gravidade da doença está mais relacionada às malformações arteriovenosas pulmonares.113
Diagnóstico
A maior parte dos pacientes é assintomática, embora as MAVs pulmonares produzam sintomas ou complicações graves. Dispneia é um dos sintomas mais comuns que ocorre primeiramente com os exercícios e, mais tarde, no estado de repouso.
Dispneia que se agrava na posição ereta e melhora na posição reclinada (platipneia) é um sintoma clássico e incomum descrito nessa população de pacientes. Esse tipo de dispneia possivelmente seja causado pelo agravamento do desvio da direita para a esquerda nas bases dos pulmões com o paciente na posição ereta, produzindo hipoxemia (ortodeoxia).
A hemoptise, comumente leve e relacionada à telangiectasia nas mucosas brônquicas, é outro sintoma muito comum. Os pacientes com MAVs pulmonares têm hipoxemia em repouso, sendo que o grau da hipoxemia é proporcional à magnitude do desvio. Como consequência, alcalose respiratória crônica e policitemia são ocorrências comuns. Os testes da função pulmonar são normais, excetuando-se a redução na capacidade difusora.114
Em pacientes com THH concomitante, as descobertas físicas adicionais incluem telangiectasia nas membranas mucosas e hipocratismo digital. Os pacientes com THH podem sofrer hemorragias causadas por MAVs fora dos pulmões, em especial no trato gastrintestinal, que geralmente é suficiente para produzir anemia.
Com frequência, o diagnóstico de MAVs pulmonares se baseia na descoberta nas radiografias torácicas de um nódulo pulmonar solitário bem definido ou de vários nódulos com uma artéria alimentadora, veia de drenagem, ou ambas. A ecocardiografia com contraste de solução salina agitada é extremamente sensível ao desvio da direita para a esquerda, além de sugerir o diagnóstico.
A TC helicoidal com contraste confirma a natureza vascular das lesões e poderá detectar outras lesões que não tenham sido óbvias nas radiografias padronizadas. As radiografias torácicas, seguidas de ecocardiografia com contraste, são a abordagem sugerida para rastrear MAVs pulmonares em pacientes com THH suspeita ou confirmada, nos casos em que nenhuma MAV for aparente nas radiografias simples iniciais. A confirmação poderá ser feita com angiografia por TC com contraste nos casos em que qualquer um dos estudos sugerir a presença de MAVs pulmonares.114
Gerenciamento
O foco principal do tratamento de MAVs pulmonares é evitar a ocorrência de complicações graves, em especial abscesso cerebral e AVC, mesmo em indivíduos assintomáticos. Embolização feita com cateter nas MAVs pulmonares, com auxílio de espirais de aço destacáveis, é a terapia de primeira linha.
A taxa global de sucesso documentada é de 75%, embora talvez sejam necessários vários procedimentos para embolizar todas as MAVs visíveis.115 Os pacientes que tenham feito embolização das MAVs pulmonares precisam ser monitorados constantemente para evitar o desenvolvimento de novas malformações. O tratamento cirúrgico via ressecção é uma das opções nos casos em que as MAVs não forem acessíveis à terapia com uso de cateter.107
Aneurismas Pulmonares
Os aneurismas nas artérias pulmonares são ocorrências muito raras.116 Esses aneurismas são considerados anomalias congênitas, geralmente em combinação com anormalidades em outros grandes vasos e no coração; como parte de distúrbios nos tecidos conjuntivos (por exemplo, síndrome de Marfan); como consequência de trauma (os pseudoaneurismas produzidos por cateteres Swan-Ganz poderão ser confundidos com aneurismas pulmonares), de infecção (sífilis, tuberculose, bactérias piogênicas ou fungos), ou de distúrbios imunológicos (doença de Behçet117 ou síndrome de Hughes-Stovin); e em associação com doenças pulmonares como HP ou bronquiectasia.
Em muitos casos, os aneurismas nas artérias pulmonares são assintomáticos. Entretanto, observa-se a presença de condições como tosse, dispneia e, principalmente, hemoptise. De um modo geral, o rompimento de algum aneurisma no interior de uma via respiratória está associado a uma hemoptise súbita, maciça e normalmente fatal. Dissecção na artéria pulmonar também é uma ocorrência provável. O diagnóstico poderá ser feito por TC realçada por contraste ou por angiografia pulmonar.
Os aneurismas na artéria principal e nas artérias proximais direita e esquerda dos pulmões podem ser reparados por meios cirúrgicos. Os aneurismas em artérias pulmonares menores podem ser removidos por ressecção cirúrgica da área envolvida do pulmão.118
Fundamentos Básicos
HAP é definida pela presença de mPAP em repouso acima de 25mmHg; de pressão em cunha nos vasos capilares dos pulmões, pressão atrial esquerda, ou pressão ventricular no fim da diástole igual ou inferior a 15mmHg; e RVP maior que 3 unidades de Wood (240dyn-s/cm5).
HAPH é responsável por aproximadamente 6 a 10% dos pacientes com HAP, e 50 a 90% de pacientes com HAPH têm mutações no gene BMPR2.
Os fatores estimuladores de HP incluem aumento no fluxo sanguíneo pulmonar (hiperdinâmica), aumento na resistência ao fluxo venoso pulmonar externo para o lado esquerdo do coração (passiva) e aumento na resistência ao fluxo sanguíneo na rede vascular dos pulmões (resistiva).
As descobertas no exame físico que foram incluídas em vários modelos prognósticos para HAP são os sinais vitais (frequência cardíaca >92BPM e PAS <110mmHg) e caminhada de 6min (distância normal >440m).
No grupo 3, os pacientes com HP, em particular aqueles com DPOC, o oxigênio é a única terapia que diminui a mortalidade.
Em comparação com CCD, o padrão de ouro para medição das pressões arteriais pulmonares, aproximadamente 40 a 60% das pressões arteriais pulmonares sistólicas estimadas por ETT discordam em mais de 10mmHg em relação aos valores efetivamente medidos.
Os pacientes que apresentarem respostas positivas de vasorreatividade (queda na mPAP =10mmHg a 40mmHg) poderão ser tratados inicialmente com um antagonista do canal de cálcio por via oral como, por exemplo, nifedipino de ação prolongada, anlodipino ou diltiazem.
Varreduras por V/Q normais ou de baixa probabilidade excluem efetivamente a hipótese de HPTC, com uma sensibilidade de 90 a 100% e uma especificidade de 94 a 100%.
Embora a tromboendarterectomia pulmonar seja o único tratamento definitivo para HPTC, o riociguat, um estimulador solúvel da guanilina ciclase que eleva os níveis de cGMP, melhorou a capacidade para fazer exercícios e a RVP em pacientes com HPTC inoperável.
A terapia inicial para HAP com ambrisentana e tadalafila resultou na diminuição significativa de 50% no risco de falha clínica (definida como uma mistura de morte, hospitalização para HAP em fase de agravamento, progressão da doença ou resposta clínica insatisfatória no longo prazo), em comparação com a monoterapia com qualquer um desses agentes.
|
Informações Financeiras: Matthew Moll, MD, and Mayank Sardana, MBBS, não têm informações financeiras relevantes a declarar. Harrison W. Farber, MD, FAHA, FCCP, atuou como palestrante nas empresas Actelion, Bayer e Gilead; trabalhou como consultor nas empresas Actelion, Bayer, Gilead, Bellerophon e United Therapeutics; e firmou contrato de pesquisas com a Gilead e United Therapeutics. Jason Stamm, MD, FACP, e Mark Gladwin, MD, FACP, foram os autores da revisão anterior, cujas informações financeiras foram apresentadas por ocasião da publicação inicial. Este trabalho foi revisado, atualizado e reeditado pelos autores mencionados anteriormente. |
Referências
1. Galiè N, Humbert M, Vachiery JL, et al; Authors/Task Force Members. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Socie-ty for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015;46:1855–6.
2. Hoeper MM, Barbera JA, Channick RN, et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hyperten-sion. J Am Coll Cardiol 2009;54(1 Suppl):S85–96.
3. Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62:D13–21.
4. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173:1023–30.
5. Savale L, Chaumais M-C, Cottin V, et al. Pulmonary hypertension with benfluorex exposure. Eur Respir J 2012;40:1164–72.
6. Montani D, Bergot E, Gunther S, et al. Pulmonary hypertension in patients treated by dasatinib. Circulation 2012;125:2128–37.
7. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122:156–63.
8. Sitbon O, Lascoux-Combe C, Delfraissy JF, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med 2008;177:108–13.
9. Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the U.S.-based REVEAL registry. Chest 2012;141:906–15.
10. Lapa M, Dias B, Jardim C, et al. Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation 2009;119:1518–23.
11. Guignabert C, Tu L, Le Hiress M, et al. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev 2013;22:543–51.
12. Wood P. Pulmonary hypertension with special reference to the vasoconstrictive factor. Br Heart J 1958;20:557–70.
13. Tuder RM, Abman SH, Braun T, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 Suppl):S3–9.
14. McGoon MD, Kane GC. Pulmonary hypertension: diagnosis and management. Mayo Clin Proc 2009;84:191–207.
15. Ma L, Roman-Campos D, Austin E, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013;369:351–61.
16. Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012;141:354–62.
17. Bossone E, Bodini BD, Mazza A, Allegra L. Pulmonary arterial hypertension: the key role of echocardiography. Chest 2005;127:1836–43.
18. Tossavainen E, Soderberg S, Gronlund C, et al. Pulmonary artery acceleration time in identifying pulmonary hypertension patients with raised pulmonary vascular resistance. Eur Heart J Cardiovasc Imaging 2013;14:890–7.
19. Farber HW, Foreman AJ, Miller DP, McGoon MD. REVEAL Registry: correlation of right heart catheterization and echocardiography in patients with pulmonary arterial hypertension. Congest Heart Fail 2011;17:56–63.
20. Sanz J, Kariisa M, Dellegrottaglie S, et al. Evaluation of pulmonary artery stiffness in pulmonary hypertension with cardiac magnetic resonance. JACC Cardiovasc Imaging 2009;2:286–95.
21. van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28:1250–7.
22. Kuehne T, Yilmaz S, Schulze-Neick I, et al. Magnetic resonance imaging guided catheterisation for assessment of pulmonary vascular resistance: in vivo validation and clinical application in patients with pulmonary hypertension. Heart 2005;91:1064–9.
23. Atwood CW Jr, McCrory D, Garcia JG, et al. Pulmonary artery hypertension and sleep-disordered breathing: ACCP evidence-based clinical practice guidelines. Chest 2004;126:72S–7S.
24. Kim N, Delcroix M, Jenkins D, et al. Chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol 2013;62:D92–9.
25. Hoeper MM, Madani MM, Nakanishi N, et al. Chronic thromboembolic pulmonary hypertension. Lancet Respir Med 2014;2:573–82.
26. Tunariu N, Gibbs SJR, Win Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboem-bolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med 2007;48:680–4.
27. McLaughlin V, Shah S, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol 2015;65:1976–97.
28. Warwick G, Thomas PS, Yates DH. Biomarkers in pulmonary hypertension. Eur Respir J 2008;32:503–12.
29. Mereles D, Ehlken N, Kreuscher S, et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 2006;114:1482–9.
30. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Nocturnal Oxygen Therapy Trial Group. Ann Intern Med 1980;93:391–8.
31. Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Pro-spective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014;129:57–65.
32. Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol 1998;31:1650–7.
33. Badesch DB, Abman SH, Simonneau G, et al. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest 2007;131:1917–28.
34. Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54(1 Suppl):S78–84.
35. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med 1996;334:296–302.
36. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106:1477–82.
37. Farber H, Miller D, Meltzer L, et al. Treatment of patients with pulmonary arterial hypertension at the time of death or deterioration to functional class IV: insights from the REVEAL Registry. J Heart Lung Transplant 2013;32:1114–22.
38. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arteri-al hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165:800–4.
39. Tapson VF, Gomberg-Maitland M, McLaughlin VV, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest 2006;129:683–8.
40. Tapson VF, Jing Z-CC, Xu K-FF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endo-thelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest 2013;144:952–8.
41. Bloodstream infections among patients treated with intravenous epoprostenol or intravenous treprostinil for pulmonary arterial hypertension—seven sites, United States, 2003–2006. MMWR Morb Mortal Wkly Rep 2007;56:170–2.
42. Hoeper MM, Schwarze M, Ehlerding S, et al. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med 2000;342:1866–70.
43. McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:1257–63.
44. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896–903.
45. Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010–9.
46. McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005;25:244–9.
47. Gatfield J, Mueller Grandjean C, Sasse T, et al. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antago-nists in pulmonary arterial smooth muscle cells. PLoS One 2012;7:e47662.
48. Pulido T, Adzerikho I, Channick R, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809–18.
49. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148–57.
50. Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119:2894–903.
51. Ghofrani H-AA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330–40.
52. Galiè N, Barberà JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 2015;373:834–44.
53. Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521–30.
54. Galiè N, Müller K, Scalise A-VV, et al. PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arteri-al hypertension. Eur Respir J 2015;45:1314–22.
55. Galie N, Manes A, Negro L, et al. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009;30:394–403.
56. Keogh AM, Mayer E, Benza RL, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol 2009;54(1 Suppl):S67–77.
57. Trulock EP, Edwards LB, Taylor DO, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-third Official Adult Lung and Heart-Lung Transplantation Report-2006. J Heart Lung Transplant 2006;25:800–92.
58. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343–9.
59. Stupi AM, Steen VD, Owens GR, et al. Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum 1986;29:515–24.
60. Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996;15:100–5.
61. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327:76–81.
62. Fijalkowska A, Kurzyna M, Torbicki A, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006;129:1313–21.
63. Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hyperten-sion. Circulation 2000;102:865–70.
64. Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the RE-VEAL Registry. Chest 2012;142:448–56.
65. Moll M, Sardana M, Farber HW. Anesthesia for patients with severe pulmonary hypertension and right heart failure: effects of anesthetic and re-suscitation drugs. ISHLT Monograph Series 2015; Volume 9: chapter 17. London: Elsevier; 2015.
66. Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004;350:2257–64.
67. Jamieson SW, Kapelanski DP, Sakakibara N, et al. Pulmonary endarterectomy: experience and lessons learned in 1,500 cases. Ann Thorac Surg 2003;76:1457–62; discussion 62–4.
68. Ghofrani H-A, D’Armini A, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369:319–29.
69. Mizoguchi H, Ogawa A, Munemasa M, et al. Refined balloon pulmonary angioplasty for inoperable patients with chronic thromboembolic pulmo-nary hypertension. Circ Cardiovasc Interv 2012;5:748–55.
70. Fedullo PF, Auger WR, Kerr KM, Rubin LJ. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2001;345:1465–72.
71. Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886–95.
72. Anthi A, Machado RF, Jison ML, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med 2007;175:1272–9.
73. Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002;8:1383–9.
74. Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005;294:81–90.
75. Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med 2008;359:2254–65.
76. Klings ES, Machado RF, Barst RJ, et al. An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification, and man-agement of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med 2014;189:727–40.
77. Souza R, Fernandes CJ, Jardim CV. Other causes of PAH (schistosomiasis, porto-pulmonary hypertension and hemolysis-associated pulmonary hypertension). Semin Respir Crit Care Med 2009;30:448–57.
78. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment sub-groups. Am J Transplant 2008;8:2445–53.
79. Mandel J, Mark EJ, Hales CA. Pulmonary veno-occlusive disease. Am J Respir Crit Care Med 2000;162:1964–73.
80. Montani D, Price LC, Dorfmuller P, et al. Pulmonary veno-occlusive disease. Eur Respir J 2009;33:189–200.
81. Runo JR, Vnencak-Jones CL, Prince M, et al. Pulmonary veno-occlusive disease caused by an inherited mutation in bone morphogenetic protein receptor II. Am J Respir Crit Care Med 2003;167:889–94.
82. Resten A, Maitre S, Humbert M, et al. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol 2004;183:65–70.
83. Montani D, Achouh L, Dorfmuller P, et al. Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore) 2008;87:220–33.
84. Eltorky MA, Headley AS, Winer-Muram H, et al. Pulmonary capillary hemangiomatosis: a clinicopathologic review. Ann Thorac Surg 1994;57:772–6.
85. Dufour B, Maitre S, Humbert M, et al. High-resolution CT of the chest in four patients with pulmonary capillary hemangiomatosis or pulmonary venoocclusive disease. AJR Am J Roentgenol 1998;171:1321–4.
86. Guazzi M, Borlaug BA. Pulmonary hypertension due to left heart disease. Circulation 2012;126.8:975–90.
87. Guazzi M. Pulmonary hypertension in heart failure preserved ejection fraction prevalence, pathophysiology, and clinical perspectives. Circulation Heart Fail 2014;7.2:367–77.
88. Bonderman D, Pretsch I, Steringer-Mascherbauer R, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension asso-ciated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest 2014;146:1274–85.
89. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J 2008;32:1371–85.
90. Scharf SM, Iqbal M, Keller C, et al. Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med 2002;166:314–22.
91. Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung trans-plantation. Chest 2005;127:1531–6.
92. Oswald-Mammosser M, Weitzenblum E, Quoix E, et al. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest 1995;107:1193–8.
93. Holverda S, Rietema H, Westerhof N, et al. Stroke volume increase to exercise in chronic obstructive pulmonary disease is limited by increased pulmonary artery pressure. Heart 2009;95:137–41.
94. Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med 2003;167:735–40.
95. Timms RM, Khaja FU, Williams GW. Hemodynamic response to oxygen therapy in chronic obstructive pulmonary disease. Ann Intern Med 1985;102:29–36.
96. Weitzenblum E, Sautegeau A, Ehrhart M, et al. Long-term oxygen therapy can reverse the progression of pulmonary hypertension in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 1985;131:493–8.
97. Stolz D, Rasch H, Linka A, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J 2008;32:619–28.
98. Ryu JH, Krowka MJ, Pellikka PA, et al. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc 2007;82:342–50.
99. Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002;360:895–900.
100. Gunther A, Enke B, Markart P, et al. Safety and tolerability of bosentan in idiopathic pulmonary fibrosis: an open label study. Eur Respir J 2007;29:713–9.
101. Fisher KA, Serlin DM, Wilson KC, et al. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest 2006;130:1481–8.
102. Barnett CF, Bonura EJ, Nathan SD, et al. Treatment of sarcoidosis-associated pulmonary hypertension. A two-center experience. Chest 2009;135:1455–61.
103. Naeije R. Pulmonary circulation at high altitude. Respiration 1997;64:429–34.
104. Budev MM, Arroliga AC, Wiedemann HP, Matthay RA. Cor pulmonale: an overview. Semin Respir Crit Care Med 2003;24:233–44.
105. Tsapenko MV, Tsapenko AV, Comfere TB, et al. Arterial pulmonary hypertension in noncardiac intensive care unit. Vasc Health Risk Manag 2008;4:1043–60.
106. Vieillard-Baron A, Page B, Augarde R, et al. Acute cor pulmonale in massive pulmonary embolism: incidence, echocardiographic pattern, clinical implications and recovery rate. Intensive Care Med 2001;27:1481–6.
107. Vieillard-Baron A, Schmitt JM, Augarde R, et al. Acute cor pulmonale in acute respiratory distress syndrome submitted to protective ventilation: incidence, clinical implications, and prognosis. Crit Care Med 2001;29:1551–5.
108. Goldhaber SZ. Echocardiography in the management of pulmonary embolism. Ann Intern Med 2002;136:691–700.
109. Todd JL, Tapson VF. Thrombolytic therapy for acute pulmonary embolism: a critical appraisal. Chest 2009;135:1321–9.
110. Han MK, McLaughlin VV, Criner GJ, Martinez FJ. Pulmonary diseases and the heart. Circulation 2007;116:2992–3005.
111. Schonhofer B, Barchfeld T, Wenzel M, Kohler D. Long term effects of non-invasive mechanical ventilation on pulmonary haemodynamics in patients with chronic respiratory failure. Thorax 2001;56:524–8.
112. Bayrak-Toydemir P, McDonald J, Markewitz B, et al. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A 2006;140:463–70.
113. Cottin V, Dupuis-Girod S, Lesca G, Cordier JF. Pulmonary vascular manifestations of hereditary hemorrhagic telangiectasia (Rendu-Osler dis-ease). Respiration 2007;74:361–78.
114. Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66–7.
115. Remy-Jardin M, Dumont P, Brillet PY, et al. Pulmonary arteriovenous malformations treated with embolotherapy: helical CT evaluation of long-term effectiveness after 2-21-year follow-up. Radiology 2006;239:576–85.
116. Bartter T, Irwin RS, Nash G. Aneurysms of the pulmonary arteries. Chest 1988;94:1065–75.
117. Erkan F, Gul A, Tasali E. Pulmonary manifestations of Behcet’s disease. Thorax 2001;56:572–8.
118. Kuwaki K, Morishita K, Sato H, et al. Surgical repair of the pulmonary trunk aneurysm. Eur J Cardiothorac Surg 2000;18:535–9.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.