(Carregando Índice)... (Carregando Índice)... |
Última revisão: 24/04/2019
Comentários de assinantes: 0
|
Artigo original: Amato A A, MD. Salajegheh, M K, MD. Neuromuscular Junction Disorders, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Anthony A. Amato, MD
Chefe da Divisão de Doenças Neuromusculares do Departamento de Neurologia do Brigham and Women’s Hospital. Professor de Neurologia na Harvard Medical School (Boston, MA).
Mohammad Kian Salajegheh, MD
Diretor da Clínica de Nervos Periféricos, Neurologista Associado no Departamento de Neurologia do Brigham and Women’s Hospital. Professor Assistente de Neurologia na Harvard Medical School (Boston, MA).
Resumo
Os três componentes principais da junção neuromuscular (JNM) incluem a região pré-sináptica, a fenda sináptica e a região pós-sináptica. A JNM age como interface entre os nervos motores e os músculos por meio da conversão das correntes elétricas dos nervos motores em sinais químicos e, a seguir, retornando para correntes elétricas nos músculos. Este artigo apresenta uma revisão dos testes eletrodiagnósticos em distúrbios na JNM, incluindo repetição da estimulação nervosa e eletromiografia (EMG) de uma única fibra. Temas como miastenia grave (MG), síndromes miastênicas congênitas (SMCs), síndrome miastênica de Lambert-Eaton (SMLE), botulismo e envenenamento por organofosforados e outras toxinas também serão discutidos, incluindo informações sobre epidemiologia, etiologia/genética, patogênese, diagnóstico, diagnóstico diferencial, gerenciamento, complicações e prognóstico. Os quadros incluem uma visão geral dos distúrbios neuromusculares, medicamentos com efeitos adversos sobre a JNM, agentes imunomoduladores mais comuns usados no tratamento de MG, SMCs e toxinas e venenos. As figuras ilustram a estrutura e a função da JNM, a estrutura das regiões pré-sináptica e pós-sináptica, estudos eletrodiagnósticos em distúrbios na JNM e disfunção da JNM nos casos de MG causada pelo receptor da acetilcolina.
Os três componentes principais da junção neuromuscular (JNM) incluem:
região pré-sináptica;
fenda sináptica;
região pós-sináptica.
A JNM age como interface entre os nervos motores e os músculos por meio da conversão das correntes elétricas dos nervos motores em sinais químicos (acetilcolina [ACh]) e, a seguir, retornando para correntes elétricas nos músculos. A despolarização dos terminais nervosos permite a entrada de íons de cálcio (Ca2+) no interior da região pré-sináptica através dos canais de cálcio controlados por voltagem (VGCCs).
Esse processo resulta na ativação de diversas proteínas, incluindo a sinaptotagmina, a sinaptofisina e várias proteínas Snare, levando à fusão de vesículas de ACh na membrana pré-sináptica e à liberação na fenda sináptica.1,2 A seguir, as moléculas de ACh se espalham sobre as dobras juncionais pós-sinápticas e se ligam aos receptores da acetilcolina (AChRs), resultando na abertura dos canais iônicos seletivos de cátions e na entrada de Na+ no interior da região pós-sináptica.
A liberação de cada vesícula resulta em um potencial de despolarização local que se denomina potencial da placa motora em miniatura (PPMM), com o efeito adicional da criação, por todas as vesículas liberadas, de um potencial da placa motora (PPM) maior e final. Nas situações em que o PPM excede um determinado limite (-65mV), cria-se um potencial de ação da fibra muscular (PAFM) que contribui para o potencial de ação muscular combinada (PAMC) final medido e para a força de contração muscular.
Em pacientes normais, o PPM criado excede o limite pelo que se considera uma margem segura, que garante a geração de um PAFM em cada ciclo de liberação. Nos casos de distúrbios na JNM, o PPM gerado fica abaixo do limite ou tem uma margem de segurança estreita, caindo abaixo do limite com contrações sucessivas, resultando em uma amplitude e força reduzida do PAMC.
Diversas proteínas, como a rapsina, uma quinase específica do músculo (MuSK), entre outras, são importantes para corrigir o agrupamento e a função dos AChRs na membrana pós-sináptica.1,2 Na sequência, interrompe-se a ação da ACh por sua rápida hidrolização em acetila e colina por meio da ação da colinesterase.
Testes Eletrodiagnósticos em Distúrbios na JNM
Estimulação Nervosa Repetitiva
Estes testes se baseiam em alterações na amplitude do PAMC do músculo depois da ativação do respectivo nervo supridor em várias taxas de estimulação, no estado de repouso e logo após a execução de exercícios físicos. As duas formas principais incluem:
1. Estimulação nervosa repetitiva (ENR) lenta. A estimulação de baixa frequência nos nervos motores com menos de 5Hz (normalmente, 2 a 3Hz) leva à depleção de vesículas de ACh no terminal nervoso e à redução no PPM durante os primeiros 1 a 2 segundos. Ao contrário do que ocorre em pacientes normais, que têm margens de segurança adequadas, a margem de segurança mais estreita em pacientes com distúrbios na JNM resulta na queda do PPM abaixo do nível normal, com a consequente redução na amplitude no PAMC (>10% para ser chamada de decremento anormal), acompanhada por um platô e melhora depois de quatro a cinco respostas.1
|
*Os autores e editores agradecem a contribuição do autor anterior, Marinos C. Dalakas, MD, pelo desenvolvimento e pela redação deste artigo. |
2. ENR rápida. Em pacientes com distúrbios pré-sinápticos na JNM, ocorre uma liberação reduzida de vesículas de ACh do terminal pré-sináptico, levando alguns PPMs a permanecerem abaixo do limite. Isso resulta em um número menor de PAFMs e em amplitudes menores de PAMC. A estimulação de alta frequência dos nervos motores (>10 Hz e, normalmente, em 30Hz), ou a contração isométrica do músculo (equivalente a ˜30 a 50Hz), resulta no aumento da liberação de vesículas de ACh. A razão para a ocorrência desse fenômeno é que, levando-se em consideração que o cálcio leva, aproximadamente, 100ms para ser bombeado ativamente para fora do terminal pré-sináptico, as estimulações mais rápidas que 10Hz levarão a esse acúmulo e a um aumento na liberação de vesículas de ACh. Esse processo resulta em um PPM mais elevado e na elevação da amplitude do PAMC nos distúrbios pré-sinápticos na JNM (>100% para ser chamada de facilitação anormal).1
Eletromiografia de Fibra Única
Embora as fibras musculares que pertencem à mesma unidade motora se contraiam ao mesmo tempo, há uma pequena variação no início dos respectivos PAFMs individuais, conhecida por tremedeira, que poderá ser medida com auxílio da eletromiografia de fibra única (SFEMG). O tempo de transição anormal da JNM observado em pacientes com distúrbios na JNM resulta no aumento da tremedeira e, em algumas situações, no bloqueio de alguns PAFMs, que poderá ser determinado por meio da SFEMG.1
Apesar de os resultados normais na ENR e no teste de SFEMG em músculos clinicamente fracos reduzirem consideravelmente a possibilidade de distúrbio na JNM, outros distúrbios neuromusculares (incluindo esclerose lateral amiotrófica [ELA], reinervação depois de lesão nervosa e determinados tipos de doenças musculares) que possam levar a resultados anormais nos testes, deverão ser excluídos com base nesses estudos antes da confirmação do diagnóstico de distúrbio na JNM.1
Distúrbios na Junção Neuromuscular
Esses distúrbios formam um grupo heterogêneo de condições adquiridas ou hereditárias, que produzem fraqueza muscular como resultado de alterações na estrutura ou função da JNM. O defeito poderá ocorrer:
na região pré-sináptica (síntese defeituosa ou enchimento das vesículas com ACh ou liberação reduzida de ACh);
na fenda sináptica (colinesterase reduzida ou disfuncional);
na região pós-sináptica (número reduzido ou reatividade do receptor de ACh ou agrupamento anormal).
As formas adquiridas possivelmente sejam resultado de algum ataque autoimune contra os componentes da JNM causado por um processo paraneoplásico ou pelo efeito de determinados medicamentos ou toxinas.
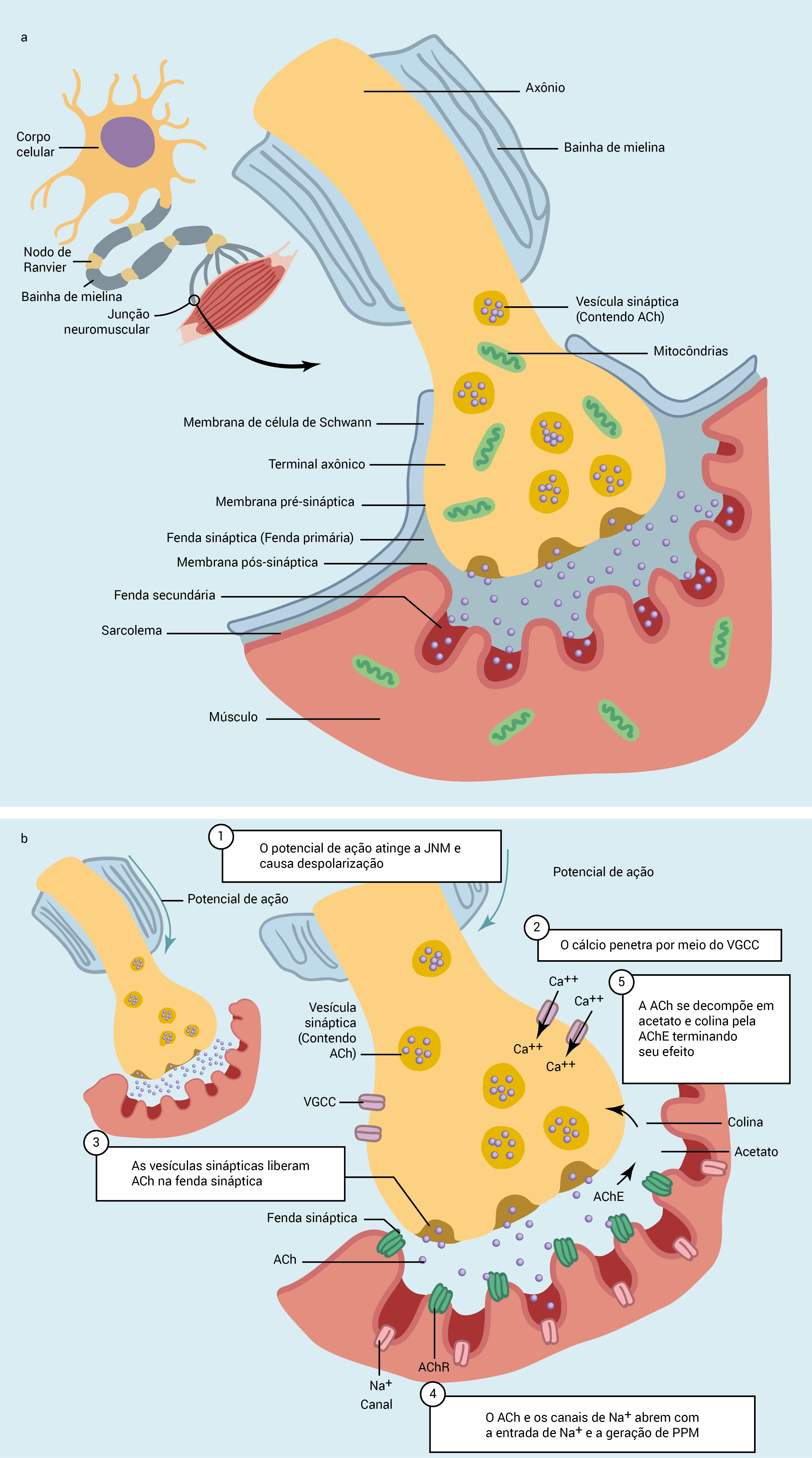
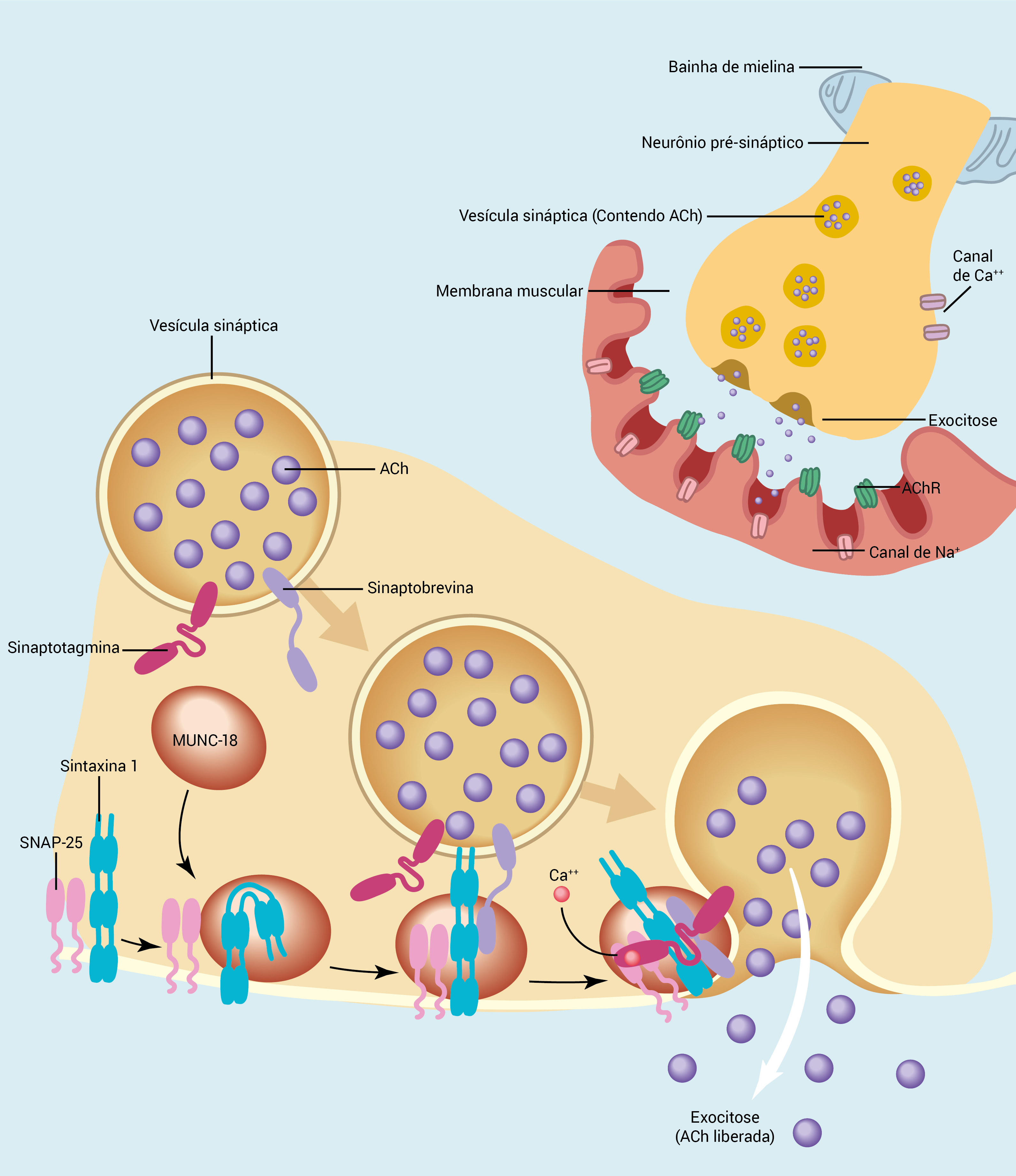
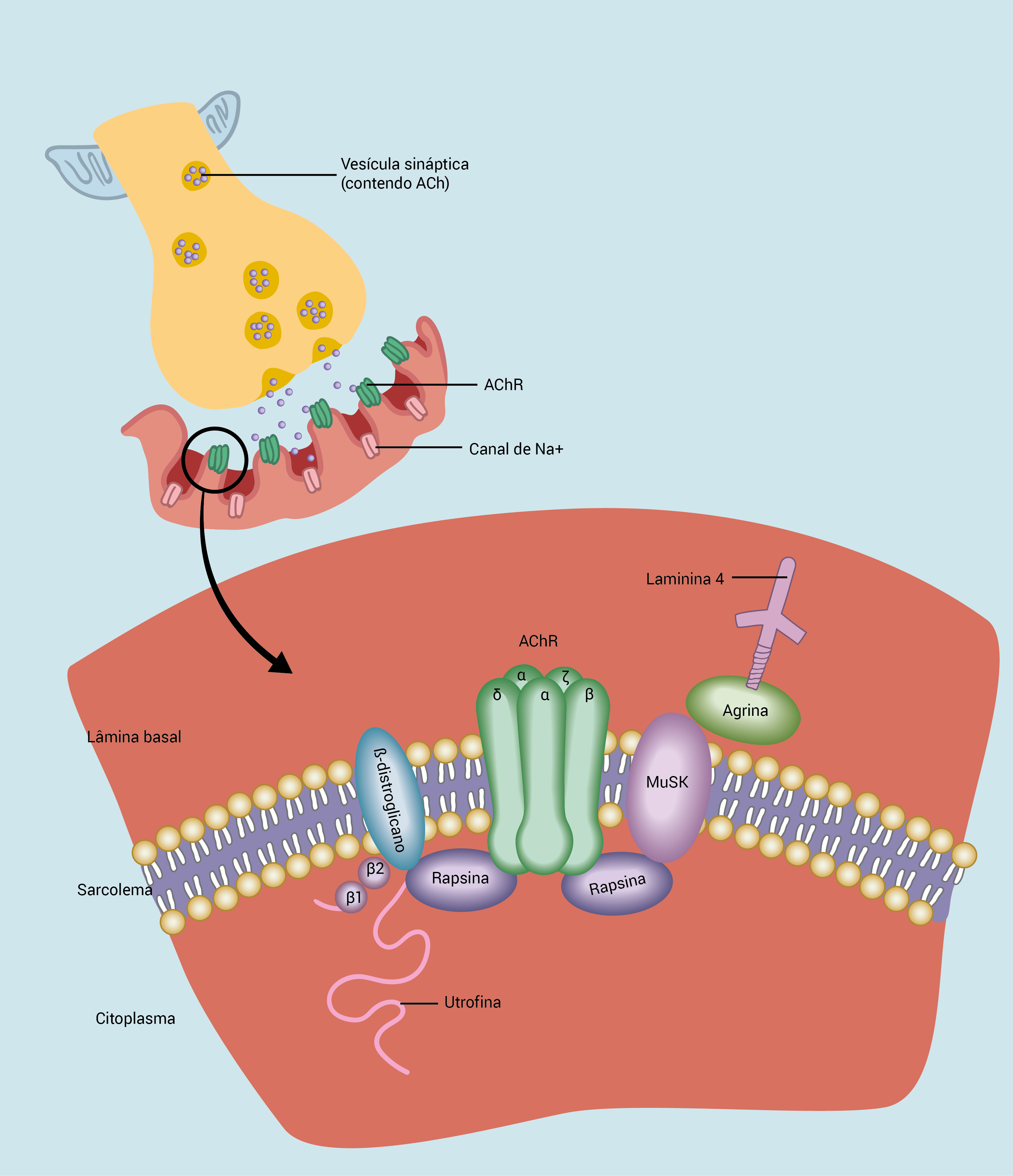
A Figura 1 ilustra a estrutura e a função da JNM; a Figura 2, a estrutura da região pré-sináptica; a Figura 3, a estrutura da região pós-sináptica.
JNM: junção neuromuscular; PPM: potencial da placa motora; VGCCs: canais de cálcio controlados por voltagem.

Figura 1 - Estrutura e função da JNM. A transferência de estimulação das terminações dos nervos motores para as fibras musculares ocorre na JNM. (A) Ela se compõe de: (1) uma região pré-sináptica onde as vesículas de acetilcolina (ACh) são produzidas e armazenadas; (2) uma fenda sináptica no interior da qual as vesículas são liberadas; e (3) uma região pós-sináptica onde a ACh se liga aos receptores da acetilcolina (AChR). (B) Depois que o potencial de ação atingir o terminal nervoso (1), ele permite a abertura dos VGCCs e a entrada de Ca2+ na região pré-sináptica (2). Esse procedimento leva à fusão das vesículas de ACh na membrana pré-sináptica resultando na exocitose na fenda sináptica (3) e na ligação com o AChR na crista das dobras pós-sinápticas. A ativação desses receptores e a abertura dos canais de Na+ na profundidade das dobras resultam na entrada de Na+ na região pós-sináptica e na geração de PPM (4). O PPM se espalha pelos arredores da membrana muscular, provocando a contração das fibras musculares. A ação da ACh termina com sua hidrólise para acetila e colina por meio da ação da acetilcolina esterase - AChE (5).
ACh: acetilcolina; AChR: receptor da acetilcolina.
Figura 2 - Estrutura da região pré-sináptica. A migração, o desvio e a exocitose de vesículas de ACh formam um processo complexo, no qual inúmeras proteínas com várias funções precisam interagir. Essas proteínas incluem as sinaptotagminas (papel importante como sensores de cálcio), as proteínas de mamíferos não coordenados 18 (Munc-18; componentes essenciais do complexo de proteínas de fusão das vesículas sinápticas) e as proteínas do receptor de fixação do NSF solúvel (SNARE). As três principais proteínas Snare incluem a sinaptobrevina (localizada na superfície das vesículas sinápticas), a sintaxina 1 (liga-se à proteína Munc-18) e a proteína 25 com associação sinaptossômica (SNAP-25), sendo que as duas últimas se localizam na membrana plasmática pré-sináptica. Essas proteínas agem em combinação para facilitar a ligação, fusão e liberação de vesículas de ACh na fenda sináptica.
ACh: acetilcolina; AChRs: receptores de acetilcolina; JNM: junção neuromuscular; MuSK: cinase específica para os músculos.
Figura 3 - Estrutura da região pós-sináptica. Na membrana pós-sináptica, a agregação correta dos AChRs na crista das dobras da membrana é imprescindível para otimizar a função da JNM. Diversas proteínas com estrutura na forma de andaimes são importantes nesse processo, incluindo a proteína de apoio rapsina, que se liga à proteína transmembrânica ß-distroglicano, que, por sua vez, liga-se ao citoesqueleto por meio da utrofina. A agrina faz a conexão entre esse complexo e a matriz extracelular e se localiza concomitantemente com a MuSK e os AChRs na membrana pós-sináptica. A MuSK induz a fosforilação de subunidades do AChR e contribui para sua estabilização e agrupamento. Os AChRs propriamente ditos são canais que regulam a entrada de cátions na região pós-sináptica. Eles formam complexos de proteínas transmembrânicas com cinco subunidades (duas subunidades a e uma subunidade ß, d e e nos músculos esqueléticos de adultos), sendo que os sítios de ligação da ACh se localizam em suas superfícies extracelulares.
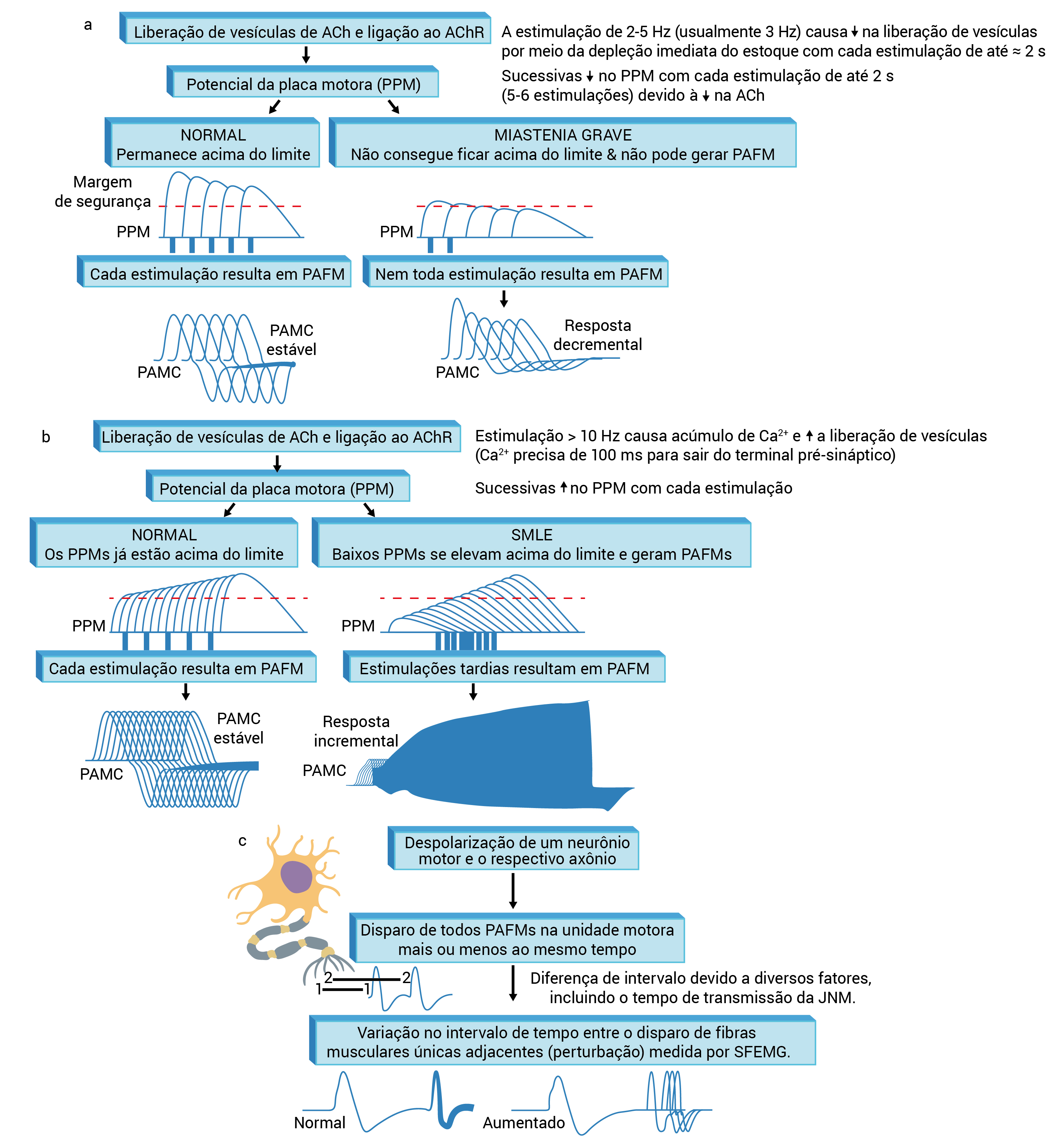
ACh: acetilcolina; ENR: estimulação nervosa repetitiva; JNM: junção neuromuscular; PAFM: potencial de ação da fibra muscular; PAMC: potencial de ação muscular combinada; PPM: potencial da placa motora; SFEMG: eletromiografia de fibra única; SMLE: síndrome miastênica de Lambert-Eaton.
Figura 4 - Estudos eletrodiagnósticos em distúrbios na JNM. (A) A ENR lenta do músculo de 2 a 5Hz (normalmente, 3Hz) libera um número menor de vesículas de ACh em cada estimulação devido à depleção imediata de estoques de ACh. Esse processo resulta no declínio progressivo do PPM por 1 até 2 segundos nas situações em que a reposição de ACh ocorre por meio de estoques secundários. Em indivíduos normais, como decorrência de uma ampla margem de segurança (diferença entre o PPM e o limite exigido para a estimulação de miofibras), todos os PPMs resultam na geração de PAFM e em um PAMC estável. Em pacientes com distúrbios na JNM, a margem estreita de segurança resulta na redução de alguns PPMs abaixo do limite, na incapacidade de gerar PAFM e no declínio sucessivo na amplitude do PAMC para até 2 segundos (resposta decremental). (B) Na ENR rápida do músculo de mais de 10Hz (comumente, 30Hz), cada estimulação libera uma quantidade maior de vesículas de ACh no PPM. Em pacientes com distúrbios pré-sinápticos na JNM, esse processo permite que, inicialmente, pequenos PPMs se elevem acima do limite e gerem PAFM com aumentos sucessivos na amplitude do PAMC (>100% para ser considerada facilitação anormal). Levando-se em consideração que, no início da estimulação, os PPMs permanecem acima do limite em indivíduos normais, deveria ocorrer uma alteração mínima, ou nenhuma alteração, na amplitude do PAMC (aumento máximo de até 40% em relação à linha de base). (C) A SFEMG mede a variação (perturbação) entre os inícios do PAFM da mesma unidade motora. As anormalidades de transmissão na JNM aumentam o tamanho da perturbação ou mesmo o bloqueio de alguns PAFMs.
Sempre há suspeita de algum distúrbio na JNM nas situações em que os pacientes se apresentam com fraqueza oscilante nos músculos dos membros, oculares, bulbares ou respiratórios. Na maior parte dos casos, a fraqueza nos membros é simétrica, proximal e causa fadiga (agrava com os exercícios e melhora após o repouso). Os pacientes, em geral, se queixam de dificuldades para subir escadas ou levantar-se de cadeiras com assentos fundos, pisar em calçadas, erguer e segurar objetos pesados ou pentear os cabelos.
Entretanto, em algumas situações, o estado de fraqueza poderá ser assimétrico, multifocal, ou mesmo focal e fixo.1 Os sintomas oculares incluem ptose e visão dupla. O envolvimento dos músculos faciais e bulbares dificulta a mastigação e a deglutição, assim como pode causar alterações na fala. A fraqueza nos músculos respiratórios se manifesta por meio de dispneia durante o esforço físico, ortopneia e, nos casos mais graves, falta de ar no estado de repouso.
Algumas doenças podem causar disfunção autônoma, sugerida por visão turva, olhos secos e boca seca, alterações na sudorese, saciedade precoce, diarreia e constipação, urgência ou incontinência urinária e disfunção erétil. De modo geral, é comum a presença de reflexos tendinosos profundos, excetos em músculos gravemente enfraquecidos, e de sensibilidade, sendo que são preservadas a sensação, a função do esfíncter e a cognição.1
Embora a doença neuronal motora e a miopatia também sejam presenças prováveis concomitantemente com a fraqueza, essas condições são basicamente estáticas (não apresentam oscilações) e, em geral, produzem atrofia muscular. Nos distúrbios na JNM, em particular a miastenia grave (MG), em decorrência da natureza de sua fisiopatologia exclusiva, os sintomas são dinâmicos (oscilam durante o dia e com as atividades físicas).
Miastenia Grave
A MG é o protótipo de doença autoimune. O antígeno (AChR) é conhecido, a presença de anticorpos direcionados contra ele é comum nos pacientes afetados e pode transmitir a doença para os animais; sua remoção resulta na melhora clínica.1
Epidemiologia
Uma grande metanálise que reuniu estudos epidemiológicos de MG estimou que a taxa de prevalência para a doença varia entre 15 a 179 por um milhão e a incidência anual, entre 1,7 a 21,3 por milhão, sendo que há uma variação acentuada entre as populações.3 Aparentemente, a prevalência de MG aumentou desde a década de 1950; esse aumento foi atribuído à melhora no processo diagnóstico, à longevidade dos pacientes como decorrência de terapias mais eficazes e ao aumento na população de risco como decorrência do envelhecimento.4
A idade de início de MG parece ter uma distribuição bimodal. O início precoce de MG (<40 anos) é maior nas mulheres (pico de incidência na terceira década de vida) em comparação com os homens (pico de incidência na quarta década de vida),1 sendo que o início tardio da doença tem uma predominância igual ou ligeiramente mais elevada nos homens (pico de incidência depois da sexta década de vida).1,5,6
A MG familiar foi documentada em casos raros e nenhum gene foi identificado com confiabilidade como causa da doença.5 A MG foi associada a outras doenças autoimunes, com associação mais frequente à artrite reumatoide e aos distúrbios na tireoide, sugerindo uma predisposição genética comum e a influência de fatores ambientais.5,7
Etiologia e Genética
Devido à baixa prevalência e à heterogeneidade da doença, a base genética de MG é quase totalmente desconhecida e tem inúmeros poliformismos; acredita-se que cada um deles tenha um pequeno efeito na contribuição para a predisposição à doença.5 Existem relatos de uma associação de vários alelos do antígeno leucocitário humano (HLA) à MG, incluindo associação com o haplótipo do HLA-A1-B8-DR3 estendido (também conhecido por haplótipo ancestral 8.1; AH8.1).5,8
Esse haplótipo foi também associado a outras doenças autoimunes, incluindo lúpus eritematoso celíaco e sistêmico.5 Múltiplos genes não HLA também estão associados à MG e a outros distúrbios autoimunes, incluindo a tirosina fosfatase 22 (PTPN22) e o antígeno 4 tardio de células T citotóxicas (CTLA4), o que dá suporte à hipótese da existência de fatores de risco genéticos compartilhados nas doenças autoimunes.5
Alguns estudos realizados em gêmeos mostraram que, além da predisposição genética para a MG, há uma forte influência de fatores ambientais nesse tipo de doença.5 Suspeita-se que as infecções virais tenham desempenhado algum papel nesse processo, em especial o vírus de Epstein-Barr (VEB), embora ainda não tenha sido estabelecida uma ligação definitiva.9,10
Estudos mais recentes sugeriram que há uma influência de microRNAs na regulação imune de MG, embora sejam necessárias investigações adicionais para determinar seu lugar efetivo na fisiopatologia da doença.9,11 Os relatos indicam que a penicilamina-D produz MG em 1 a 7% de pacientes.12 Comumente, os sintomas começam dentro de alguns meses após o início do uso da medicação e desaparecem após a descontinuação.12
Nesses pacientes, a MG está associada à elevação no nível do anticorpo anti-AChR e, raramente, anti-MuSK.13 Ainda que alguns especialistas tenham sugerido a presença de suscetibilidade genética na região do HLA de classe II, o mecanismo exato ainda não está suficientemente claro.12 Diversos relatos indicam que os medicamentos interferon a14 e interferon ß12 também são desencadeadores de MG.
Além disso, muitos medicamentos podem revelar ou agravar a MG, em especial alguns antibióticos, sendo que os aminoglicosídeos estão no topo da lista. Embora nem todos sejam contraindicados para uso nesses pacientes, os prós e os contras devem ser discutidos com os próprios pacientes, sendo imprescindível o monitoramento para verificar a exacerbação dos sintomas.1 Uma revisão ampla pode ser encontrada no site http://myasthenia.org/LinkClick.aspx?fileticket=JuFvZPPq2vg= da Myasthenia Gravis Foundation.
Patogênese
Acredita-se que a MG seja um distúrbio dependente de células T e ativado por células B, cujos anticorpos são direcionados contra diversos componentes da JNM, incluindo o AChR (80 a 85% de casos), MuSK (5 a 8%) e proteína 4 relacionada às lipoproteínas (LRP4; 2 a 3%).9,12 Os alvos antigênicos do ataque imune são desconhecidos no grupo remanescente de pacientes.
Nos pacientes com AChR-MG (soropositivos), a ligação dos autoanticorpos aos respectivos alvos leva à destruição mediada por complemento da membrana pós-sináptica, interferindo na geração de PPM adequado e na transmissão neuromuscular.9
Nos casos de MG, os anticorpos contra o AChR são gerados especificamente contra os receptores nicotínicos, mas não contra os receptores muscarínicos, fazendo com que doença se manifeste com fraqueza nos músculos esqueléticos ao invés de sintomas muscarínicos. O agrupamento de AChRs na membrana pós-sináptica é importante para sua função, sendo que os anticorpos com alvo nas proteínas envolvidas nesse processo (como a MuSK) poderão levar à disfunção da JNM; entretanto, sua fisiopatologia exata não é muito clara.9
Há registros de anormalidades tímicas em, aproximadamente, 80% dos casos de AChR-MG.12 Ainda que as células autorreativas T anti-AChR e as células B produtoras de autoanticorpos estejam presentes no timo, a causa de sua autorreatividade ainda não está clara.15 Alguns estudos sugeriram a presença de um “ambiente inflamatório” no timo desses pacientes, levando ao desencadeamento de respostas imunes contra o AChR pela alteração de sua expressão e pela perda de autotolerância.12
A observação de centros germinais ectópicos no interior do timo de pacientes com AChR-MG, mesmo com algumas características de órgãos linfoides secundários, dá suporte adicional ao papel do timo no processo autoimune.9 Nos casos de pacientes com MuSK-MG e LRP4-MG, o papel do timo na patogênese da doença ainda permanece obscuro.12
O Quadro 1 apresenta os distúrbios neuromusculares e o Quadro 2, os medicamentos com efeitos adversos na JNM.
Quadro 1
|
Distúrbios Neuromusculares | |
|
Distúrbio |
Mecanismo e exemplo |
|
Miastenia grave |
Pós-sináptico: anti-AChR Ab, anti-MuSK-Ab, medicamentos. |
|
Miastenia grave neonatal (transitória) |
Pós-sináptico: transferência de anticorpos da mãe. |
|
Síndromes de miastenia congênita |
Pré-sináptico: defeito na colina acetiltransferase; sináptico: deficiência de AChE na placa terminal; pós-sináptico: deficiência de AChR primário. |
|
Síndrome miastênica de Lambert-Eaton |
Pré-sináptico: anti-VGCC Ab. |
|
Botulismo e toxina botulínica |
Pré-sináptico: exotoxina Clostridium botulinum. |
|
Toxinas e venenos |
Pré-sináptico: organofosforados; sináptico: a-latroxina; pós-sináptico: curare. |
Ab: anticorpo; AChE: acetilcolina esterase; AChR: receptor da acetilcolina; MuSK: quinase específica dos músculos; VGCC: canal de cálcio controlado por voltagem.
Quadro 2
|
Medicamentos com Efeitos Adversos na Junção Neuromuscular |
|
Medicamentos que revelam ou exacerbam MG Antimicrobianos Aminoglicosídeos, clindamicina polimixina, fluoroquinolonas, azitromicina, eritromicina, quinino, tetraciclinas, sulfonamidas, penicilinas, nitrofurantoína. Agentes antiarritmicos Lidocaína, quinidina, procainamida Corticosteroides Magnésio (parenteral) Agentes de bloqueio neuromuscular Agentes despolarizantes e não despolarizantes
Medicamentos que possivelmente revelam ou exacerbam MG Sedativos e anestésicos Diazepam, cetamina Anticonvulsivantes Fenitoína, etosuximida, barbitúricos, carbamazepina, gabapentina ß-bloqueadores Propranolol, timolol, atelonol, labetalol, metoprolol, nadolol BCC Verapamil Drogas de abuso Cocaína Gastrintestinais Cimetidina Oftálmicos Ecotiofato, tropicamida Medicamentos psiquiátricos Fenotiazidas, lítio, amitriptilina, imipramina, anfetaminas, haloperidol Agentes de contraste iodados Outros L-carnitina, trihexifenidil |
BCC: bloqueadores do canal de cálcio; MG: miastenia grave.
Diagnóstico
Manifestações Clínicas
A marca registrada da MG é a presença de fraqueza muscular indolor que oscila ao longo do dia, agravando-se com a atividade e melhorando com repouso.6 O padrão e a intensidade da fraqueza nos casos de MG variam de paciente para paciente. Embora qualquer músculo esquelético possa ser afetado, os músculos controlados pelos nervos cranianos possivelmente sejam os mais suscetíveis, levando a sintomas oculares (comumente assimétricos), disartria, disfagia e dificuldade para deglutir.1
Aproximadamente, 50% dos pacientes se apresentam apenas com sintomas oculares, sendo que 50 a 90% têm ptose e 15%, visão turva ou diplopia. De modo geral, 90 a 95% dos pacientes com MG podem apresentar sintomas oculares em algum momento durante o curso da doença.1 As alterações na fala incluem hipofonia (fraqueza nas cordas vocais ou nos músculos expiratórios), disartria (fraqueza nos lábios, na língua ou na face) ou fala anasalada (fraqueza nos músculos palatais).1
As dificuldades para mastigar e engolir incluem regurgitação nasal de sólidos e líquidos, incapacidade para manipular o bolo de alimentos na boca ou através da faringe e aspiração de alimentos. Os pacientes talvez apresentem força reduzida para tossir, assoar o nariz e limpar a garganta, assim como defeitos inalatórios.1
Na ausência de sintomas oculares, alguns pacientes se apresentam com disfagia e disartria. A fraqueza facial resulta na incapacidade de fechar completamente as pálpebras, assoviar ou encher as bochechas. O sorriso pode se assemelhar a um “rosnado”, tendo em vista a incapacidade de erguer os cantos da boca juntamente com a retração labial.1
Normalmente, a fraqueza nos músculos dos membros é simétrica, com predominância proximal (“padrão cíngulo-límbico”) e pode ser observada em 20 a 30% de pacientes.1 Em situações raras, a fraqueza é predominantemente distal, focal ou multifocal.1 Alguns pacientes se apresentam com cabeça caída como sintoma inicial. Embora a insuficiência ventilatória seja rara como sintoma inicial ou isolado, ela geralmente ocorre nos casos de MG não tratada ou refratária generalizada, e poderá ser suficientemente grave para colocar a vida dos pacientes em risco em 15 a 20% de casos.6
Os pacientes, em geral, se queixam de diversos fatores que podem exacerbar a fraqueza, incluindo temperatura elevada, infecções sistêmicas, menstruações, gravidez, estresse e ansiedade.1 Alguns pacientes se sentem mais à vontade quando ingerem e bebem alimentos e líquidos frios. As variações na gravidade da doença ao longo do tempo também ocorrem na ausência de fatores identificáveis.1
A MG não resulta em queixas sensoriais, nem chega a produzir sintomas na bexiga ou nos intestinos, disfunção autonômica ou alterações cognitivas. A presença desses sintomas poderá levantar suspeitas sobre os efeitos colaterais da terapia, sobre a presença de doença concomitante ou condições paraneoplásicas, incluindo encefalite límbica, síndrome de Isaacs (neuromiotonia), síndrome da pessoa rígida, miosite e pseudo-obstrução intestinal, que se observa em 4 a 7% de pacientes com MG e timoma.16
Outros distúrbios associados à MG incluem anormalidades tímicas e doenças autoimunes. O envolvimento tímico (incluindo 50 a 60% de hiperplasia e 15% de timoma) foi documentado em, aproximadamente, 80% de pacientes com MG e AChR positivo.12 Na maior parte dos casos, os timomas são capsulados, de crescimento lento e ocorrem em pessoas idosas; entretanto, existem relatos de formas invasivas locais em grupos mais jovens.12
Embora o risco de ocorrência de timoma seja mais baixo em pacientes com MuSK-MG, há relatos de incidência nesse grupo.17 Em um estudo de grande porte que reuniu pacientes com LRP4-MG, os relatos indicam que houve incidência de hiperplasia tímica em 32% dos casos avaliados; no entanto, em nenhum dos casos houve ocorrência de timoma.18 As doenças autoimunes associadas à MG incluem doenças nos tecidos conjuntivos, aplasia eritrocitária, colite ulcerativa, sarcoidose, doença de Addison e doença tireoideia autoimune.1
A ocorrência de distúrbios neuromusculares autoimunes também é muito comum e inclui condições como polineuropatia desmielinizante inflamatória crônica (CIDP), neuropatia autonômica, miopatias inflamatórias e canelopatias autoimunes.1
MuSK-MG. Aparentemente, os pacientes com MuSK-MG (39 a 49% de pacientes com MG generalizada e sem anticorpos contra o AChR) se apresentam com características distintas.17 Esses pacientes são predominantemente mulheres, e o pico de incidência ocorre na quarta década de vida. Embora os registros indiquem que 36% dos casos iniciam com sintomas oculares, quase todos se tornam generalizados, com fraqueza proeminente no pescoço e nos ombros, e com fraqueza oculobulbar e crises miastênicas frequentes.
Um número pequeno de pacientes possivelmente desenvolva atrofia e fraqueza fixa na face e na língua. Muitos desses pacientes deterioram rapidamente logo na fase inicial do curso da doença, com respostas fracas à terapia, em comparação com os pacientes com AChR-MG.17 A apresentação algo atípica e os resultados negativos dos testes para o anticorpo do AChR poderão levar a diagnósticos errôneos ou tardios.
LRP4-MG. Embora haja menos informações disponíveis sobre a LRP4-MG, esse tipo de MG foi documentado em 7 a 32% de pacientes com MG e que haviam apresentado resultados negativos nos testes para anticorpos de AChR e MuSK.18 Nesse grupo, a prevalência da doença é mais alta em mulheres, com início do pico da doença na quarta década de vida. A maioria apresenta simplesmente MG ocular ou MG leve, sendo que a resposta global ao tratamento se assemelha à resposta da AChR-MG.18
MG juvenil. Aproximadamente, 10% de MG autoimune ocorrem antes dos 18 anos de idade. Os dados sobre essa subpopulação de pacientes são muito limitados, levando-se em consideração que a ocorrência desse tipo de doença é rara; entretanto, acredita-se que a apresentação seja mais ou menos semelhante à da MG em adultos.1
Exame Físico
Além dos sintomas que confirmam de forma objetiva a situação dos pacientes, o exame físico poderá mostrar também o agravamento ou desenvolvimento de fraqueza com a prática de exercícios. Isso inclui ptose depois do olhar vertical (olhar para cima) prolongado, oftalmoparesia com fixação ocular excêntrica sustentada,1 e fraqueza na abdução do braço ou flexão do quadril depois de mantê-los nas respectivas posições.
Observa-se em alguns pacientes a contração espasmódica de Cogan (repuxo nas pálpebras superiores, acompanhado pelo retorno da ptose ao trazer os olhos para a posição primária depois de 10 a 15 segundos de fixação do olhar para baixo). A combinação do envolvimento motor de múltiplos nervos cranianos, na ausência de dor ou de alterações sensoriais, como a combinação de fraqueza no fechamento dos olhos e ptose, levanta a suspeita de MG.1
A resistência dos músculos respiratórios pode ser avaliada ao lado do leito pedindo ao paciente para contar em voz alta pelo máximo de tempo possível depois de respirar fundo. A multiplicação do número obtido por 100 é uma estimativa grosseira da capacidade vital funcional expressa em mililitros. O exame sensorial e os reflexos nos tendões profundos são normais, exceto nos músculos com fraqueza muito grave.1
Testes Laboratoriais
Exames de sangue. A identificação de autoanticorpos contra as proteínas da JNM e dos músculos é o grande pilar do diagnóstico de MG; entretanto, como se aplica a todos os outros testes, o valor desse procedimento se baseia na sensibilidade e na especificidade dos métodos utilizados, que foram aprimorados ao longo dos anos.19,20
Anticorpos contra o AChR. Os anticorpos contra o AChR são positivos em 80 a 85% de pacientes com MG generalizada e 50 a 75% de pacientes com MG ocular. Esses anticorpos se classificam em ligantes, bloqueadores e moduladores.21 Anticorpos de ligação com o AChR são aqueles medidos com mais frequência e são diagnósticos para MG em pacientes com a apresentação típica da doença. Os níveis são mais elevados na MG com início precoce (< 40 anos), nos casos de MG generalizada e em pacientes com timoma, e menos frequentes em pacientes com doença leve ou com fraqueza restrita.21
Esses anticorpos são altamente específicos para MG, porém raramente são encontrados em pacientes com doença hepática autoimune, lúpus sistêmico, artrite reumatoide, timoma, neuromielite óptica e doença do enxerto versus hospedeiro (DEVH).21 Os anticorpos de bloqueio são uma minoria entre os anticorpos contra o AChR, e seu valor diagnóstico é mínimo, levando-se em conta que raramente estão presentes na ausência de anticorpos de ligação com o AChR.21
Os anticorpos moduladores do AChR são positivos em 3 a 4% de pacientes com resultados negativos no ensaio de anticorpos de ligação.21 O valor das medições seriais dos níveis do anticorpo anti-AChR para monitorar respostas aos tratamentos não é muito claro, tendo em vista que elas não conseguem prever com confiabilidade o curso da doença ou as respostas terapêuticas.21
Uma grande revisão de pacientes com MG documentou um valor preditivo negativo de 99,7% para timoma em pacientes sem anticorpos contra o AChR. Entretanto, é sempre bom lembrar alguns estudos que foram usados para excluir timoma nos casos em que o teste de anticorpos contra o AChR for negativo, considerando as suas limitações.27
Anticorpos contra MuSK. Os anticorpos anti-MuSK são encontrados em, aproximadamente, 50% de casos de MG generalizada e com anticorpos anti-AChR negativos. Sua presença significa que o diagnóstico de MuSK-MG com resultados positivos falsos não foi documentado e que os anticorpos anti-MuSK raramente são encontrados nos casos de AChR-MG21 ou nos casos com anticorpos anti-LRP4.18
Anticorpos contra LRP4. Em um estudo de grande porte que reuniu pacientes com MG negativos para os anticorpos contra AChR e MuSK, a reatividade contra LRP4 foi identificada em 7 a 32% de pacientes. Embora nenhum anticorpo contra LRP4 tenha sido documentado em controles saudáveis, 4% de pacientes com outras doenças neurológicas autoimunes eram positivos.18
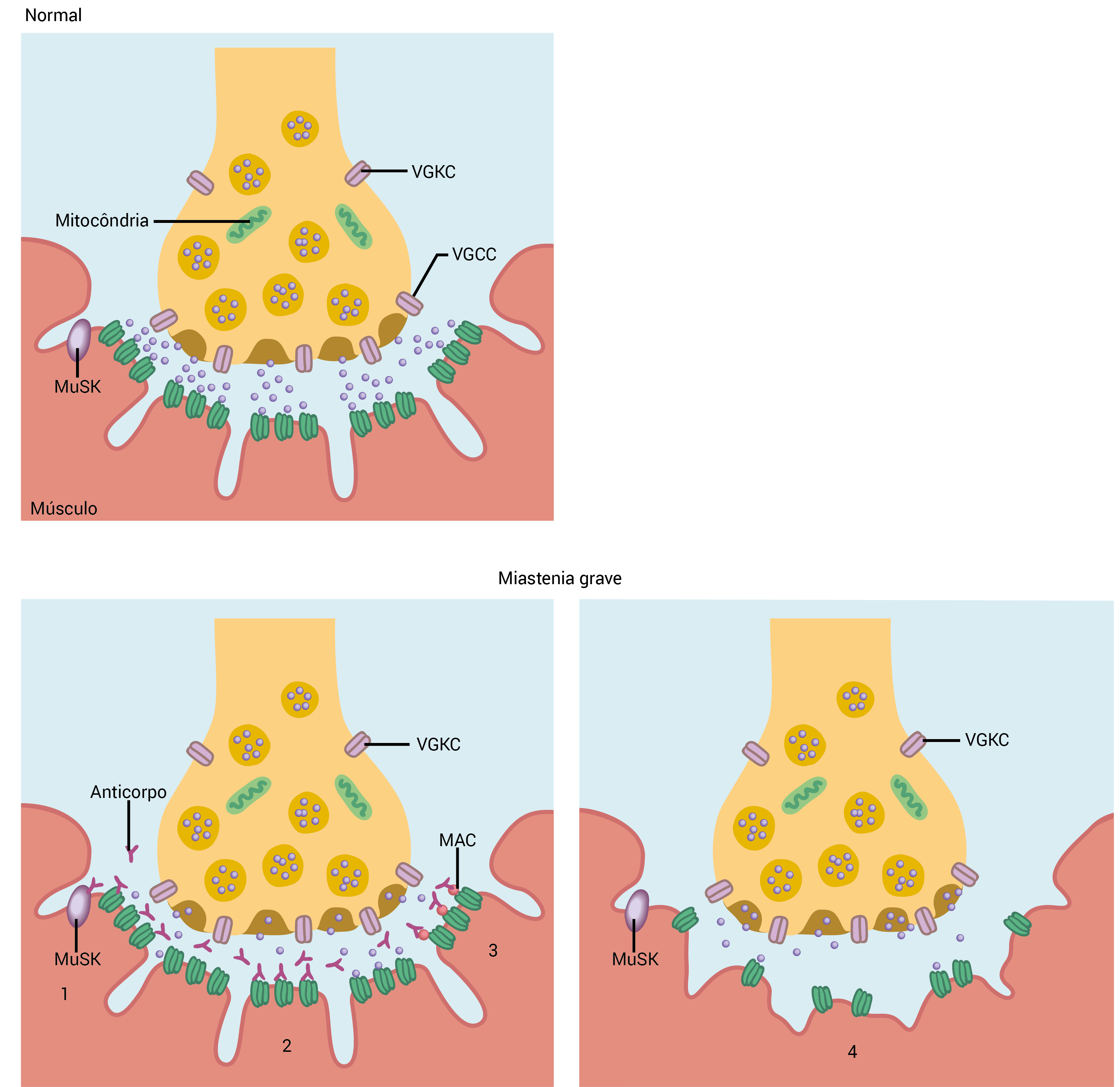
A Figura 5 mostra a disfunção da JNM no AChR- MG.
ACh: acetilcolina; AChR: receptor da acetilcolina; JNM: junção neuromuscular; MG: miastenia grave MuSK: quinase específica do músculo; VGCC: canal de cálcio dependente de voltagem; VGKC: canal de potássio dependente de voltagem.
Figura 5 - Disfunção da JNM no AChR-MG. Os autoanticorpos do AChR podem alterar a transmissão da JNM das seguintes formas: (1) bloqueando os AChRs e impedindo sua ligação com a ACh; (2) fazendo a ligação cruzada entre os AChRs e acelerando sua internalização e destruição; (3) fixando o complemento e a deposição final do complexo de ataque à membrana (MAC); e (4) destruindo a membrana pós-sináptica com uma redução no número e na profundidade das dobras (simplificação) e com a perda de AChRs.
Anticorpos contra AChRs agrupados. Alguns pacientes com MG generalizada e negativos para anticorpos anti-MuSK e anti-AChR revelam a presença de anticorpos de baixa afinidade em circulação que poderão se ligar a AChRs agrupados, em ensaios com base em células e com sensibilidade diagnóstica mais alta.23
Anticorpos (estriacionais) contra músculos estriados. Até 95% de pacientes com MG e timoma e 50% de pacientes com MG de início tardio sem timoma poderão apresentar anticorpos contra músculos estriados (incluindo receptores da titina, miosina, actina e rianodina). Observa-se também a presença desses anticorpos nos casos de doenças autoimunes com timoma e sem MG.21 Raramente, ocorrem na ausência de anticorpos anti-AChR, e seu valor diagnóstico é bastante limitado, exceto para levantar suspeitas futuras de timoma em pacientes com AChR-MG de início precoce (<40 anos).21
Estudos de imagens. As imagens torácicas, por tomografia computadorizada (TC) ou imagens por ressonância magnética (IRMs), são imprescindíveis para verificar a eventual presença de timoma.4
Testes fisiológicos. Esses testes se baseiam na função alterada da JNM.
Testes eletrofisiológicos. Os resultados dos estudos rotineiros de condução nervosa e da eletromiografia (EMG) com agulha são normais na maior parte dos pacientes com MG. Os testes especiais, incluindo estimulação nervosa repetitiva (ENR) e SFEMG, que foram descritos anteriormente, mostram as anormalidades. Esses testes têm altíssima produtividade quando feitos em músculos mais proximais e em músculos sintomáticos específicos.1 É extremamente importante que os pacientes evitem tomar inibidores da colinesterase antes dos testes, tendo em vista que esses medicamentos podem aumentar a probabilidade de resultados falsos negativos.
Teste com compressa de gelo. Uma das avaliações rápidas para o diagnóstico de MG ao lado do leito, em pacientes com ptose ou diplopia, é a demonstração de redução dos sintomas após a aplicação de uma compressa de gelo sobre as pálpebras fechadas; todavia, a precisão diagnóstica desse teste não é muito clara.19,24
Teste com edrofônio (Tensilon). O edrofônio, um agente anti-colinesterase, pode melhorar temporariamente a função da JNM. A administração desse medicamento deve ser feita com monitoramento cardíaco e em condições que permitam fazer ressuscitação cardíaca imediata por causa do risco de bradicardia. Esse tipo de teste deve ser considerado positivo somente nos casos em que há melhora objetiva na resistência, como, por exemplo, a demonstração de redução na ptose ou na oftalmoparesia. Há relatos de respostas modestas ao edrofônio em outros distúrbios neuromusculares.1
Diagnóstico Diferencial
A predileção da MG para envolver músculos inervados por nervos cranianos poderá imitar neuropatias cranianas; no entanto, o envolvimento nos casos de MG é puramente motor e não se restringe a nervos individuais. Por exemplo, a oftalmoparesia que se observa nos casos de MG não é facilmente localizável em relação à disfunção no terceiro, quarto ou sexto nervo craniano.1
O envolvimento do músculo bulbar pode ser confundido com ELA com início bulbar, doença de Kennedy ou miopatias inflamatórias; entretanto, condições como ptose ou oftalmoparesia não estão presentes nesses distúrbios. Distrofia muscular oculofaríngea, miopatias mitocondriais, distrofia miotônica e formas raras de miopatia congênita poderão se apresentar com os sintomas bulbares e oculares.1
Um padrão “cíngulo-límbico” de fraqueza em pacientes com MG poderá ser confundido com alguma doença muscular, incluindo miosite inflamatória, distrofia muscular cíngulo-límbica e miopatias endócrinas e tóxicas. A fraqueza distal, focal ou multifocal poderá ser confundida com neuropatia motora, radiculopatia ou mesmo doença nos neurônios motores.1
É imprescindível considerar as síndromes miastênicas congênitas (SMCs) em pacientes com MG e com resultado negativo nos testes de anticorpos, em especial os pacientes mais jovens que não respondem aos agentes imunomoduladores.
Gerenciamento
O tratamento de MG melhorou drasticamente ao longo das últimas décadas devido ao número crescente de agentes imunomoduladores, assim como pelo aprimoramento no gerenciamento dos tratamentos intensivos. O objetivo do tratamento é possibilitar que os pacientes retornem à função normal o mais rapidamente possível.
Medicações
Os dois grupos principais de medicações utilizadas no tratamento de MG incluem os inibidores da colina esterase, para terapias sintomáticas, e os agentes imunomoduladores, para suprimir os ataques contra os AChRs.
Embora a exacerbação dos sintomas durante as terapias imunossupressoras exija o uso de doses mais elevadas da mesma medicação ou a adição ou substituição por outros agentes, é fundamental excluir e tratar os fatores provocadores como, por exemplo, alteração nos níveis eletrolíticos, infecção sistêmica, enfermidade coincidente e causas iatrogênicas, assim como considerar a não complacência dos pacientes.
Inibidores da colesterase. Essas medicações são usadas nas terapias sintomáticas para tratamento de MG. Esses inibidores são as únicas medicações usadas nos casos de MG ocular e em combinação com agentes imunomediadores enquanto se aguarda a plenitude dos efeitos terapêuticos.
A piridostigmina é o inibidor da colesterase prescrito com mais frequência; comumente, ele é administrado na dose de 30 a 60mg a cada 4 a 6 horas (no máximo, 600mg/dia) e a melhor forma de usá-lo é 30 minutos antes das refeições para melhorar a ingestão. Os efeitos colaterais incluem náusea, vômito, cólicas abdominais, diarreia, aumento nas secreções orais e brônquicas, bradicardia e, raramente, confusão ou psicose.6
Agentes imunomoduladores. Embora a escolha desses agentes varie entre os médicos, a maioria deles tende a iniciar a terapia com corticosteroides e usar outros agentes com base na resposta aos esteroides e na capacidade para reduzir gradativamente a dosagem até a dose mais baixa.
Corticosteroides. A despeito da ausência de testes randomizados controlados de alta qualidade,25 os corticosteroides são considerados os agentes imunossupressores orais mais eficazes.6 Geralmente, se inicia o uso de prednisona com uma dose baixa (20mg/dia), no ambiente ambulatorial, para impedir a exacerbação da doença; a dose poderá ser aumentada lentamente até o desaparecimento dos sintomas ou até os pacientes atingirem a dose máxima de 1 a 1,5mg/kg/dia por 3 a 4 semanas.
A seguir, a dose deverá ser reduzida gradualmente para a quantidade mais baixa tolerável. Alguns médicos defendem o início do tratamento com uma dose alta de prednisona (1mg/kg/dia), embora alguns médicos reservem esse regime apenas para o ambiente hospitalar, tendo em vista que, nesse ambiente, há a possiblidade de monitorar os pacientes.
Ainda que um estudo retrospectivo tenha demonstrado uma generalização reduzida de MG em pacientes com MG ocular que haviam sido tratados com dose baixa de prednisona,26 não existem estudos de alta qualidade que deem suporte a essa hipótese.27 O uso prolongado de corticosteroides é limitado pelos efeitos colaterais.
Outros agentes imunossupressores. De modo geral, por causa do “efeito preservador” de esteroides, os agentes secundários são adicionados à prednisona em pacientes resistentes aos esteroides. A maior parte desses fármacos compartilha os efeitos colaterais comuns de supressão da medula óssea, infecções oportunísticas, teratogenicidade e oncogenicidade, que precisam de monitoramento e devem ser discutidos com os pacientes.
A azatioprina é o agente secundário usado com mais frequência, sendo que sua eficácia foi demonstrada em alguns testes clínicos.28 Uma das limitações desse fármaco é o início tardio da ação, que poderá chegar a muitos meses, e é utilizado nos casos de deficiência de tiopurina metiltransferase (TPMT), que deverá ser excluída antes do início da terapia.6
A ciclosporina, embora seja menos tolerada, também foi eficaz em testes clínicos de MG; o início da resposta foi mais precoce em comparação com a azatioprina (comumente, 2 a 3 meses), porém com os efeitos colaterais comuns de hipertensão e nefrotoxicidade.6
Em estudos retrospectivos, o micofenolato de mofetila apresentou algum sucesso inicial no tratamento de MG e na redução da necessidade de prednisona, com início de 2 a 3 meses;29,30 entretanto, dois testes randomizados controlados não conseguiram demonstrar se houve algum benefício adicional sobre a prednisona nos casos de MG generalizada ou nos efeitos preservadores de esteroides em um período de 9 meses.31,32
As razões sugeridas para isso incluem curta duração dos testes, desfechos primários insensíveis e limitados, bem como resposta à prednisona acima do previsto.30 O metotrexato é usado com menos frequência no tratamento de MG, embora alguns estudos retrospectivos tenham apresentado respostas positivas,33 e um estudo simples-cego demonstrou um efeito preservador de esteroides semelhante em comparação com a azatioprina.34
Encontra-se, atualmente, em curso um teste de grande porte, multicêntrico, duplo-cego e controlado por placebo.33 O rituximabe foi comprovadamente eficaz no tratamento de MG refratária, em especial a MuSK-MG, em algumas séries de casos e em estudos retrospectivos.35 As respostas favoráveis de pacientes com MG ao tacrolimo, como monoterapia e como agente preservador de esteroides, foram demonstradas em algumas séries de casos, em estudos retrospectivos e em um estudo não cego randomizado controlado.6,36
A ciclofosfamida também foi usada em pacientes com MG refratários a outras formas de agentes imunomoduladores, embora os riscos e benefícios devam ser ponderados cuidadosamente, levando-se em conta a toxicidade.6 O eculizumabe é um novo agente imunomodulador que apresentou benefícios em pacientes com MG refratária generalizada em um pequeno estudo duplo-cego controlado por placebo,37 sendo que um estudo de grande porte de fase III está atualmente em andamento.
Imunoglobulina intravenosa (IVIg) e troca de plasma (PLEX). As duas opções foram igualmente eficazes em pacientes com MG moderada a grave38 em testes randomizados controlados. Embora, em geral, elas sejam utilizadas em pacientes com crise miastênica ou para reforçar a resistência dos pacientes em antecipação à timectomia,39 são também usadas como terapias de manutenção em pacientes refratários ou intolerantes a outros tipos de fármacos.6
A IVIg é administrada na dose de 2g/kg/dia, dividida em 2 a 5 dias, com infusões mensais acompanhadas de redução gradual até a menor dose e ao intervalo de tempo mais longo tolerável. Geralmente, a FLEX é utilizada à base de cinco trocas em 10 dias (conforme o peso corporal), sendo que os principais efeitos colaterais são sepse, pneumotórax, tromboflebite e instabilidade cardiovascular com transferência de grandes volumes.1
Timectomia
A presença de timoma é uma indicação clara para timectomia.6 Ainda que na ausência de timoma, um efeito benéfico em pacientes com MG generalizada com idade inferior a 50 a 60 anos tenha sido sugerido, as indicações para cirurgia não são definitivas,6 e estão sendo avaliadas em um teste multicêntrico internacional atualmente em curso.40,41 As complicações incluem exacerbação da fraqueza acompanhada de insuficiência respiratória, infecção e lesões recorrentes nos nervos faríngeo e frênico ou no plexo braquial.1
Circunstâncias Especiais
MuSK-MG. Embora mais da metade dos pacientes com MuSK-MG respondam aos inibidores da colesterase, os efeitos colaterais são comuns e incluem fasciculações acentuadas, cólicas e agravamento dos sintomas em um pequeno percentual de casos.17 A grande maioria desses pacientes responde bem e rapidamente à PLEX, sendo que mais da metade se beneficiam com a IVIg.17 Alguns estudos menores mostraram que houve uma resposta muito boa ao rituximabe, com efeitos de longa duração.42
MG juvenil. Embora os inibidores da colesterase e os agentes imunomodulares sejam utilizados no tratamento de MG juvenil, existem algumas diferenças em relação à MG adulta.43 Em crianças, os efeitos colaterais do uso crônico de esteroides são muito preocupantes, tendo em vista que se assemelham muito àqueles observados em adultos, além de ocorrer uma redução no crescimento linear, que não é totalmente reversível.44
Outras medicações imunossupressoras também não são usadas com muita frequência devido aos efeitos colaterais. Alguns estudos retrospectivos mostraram que a timectomia, incluindo cirurgias toracoscópicas menos invasivas,45 é bastante eficaz.44,46 Todavia, levando-se em consideração essas observações, em geral reserva-se a timectomia somente para os casos de doença generalizada refratária aos tratamentos médicos. Há controvérsias sobre o momento exato de aplicar a timectomia, tendo em vista seu papel no desenvolvimento do sistema imune das crianças mais jovens.44
Gravidez. As pacientes grávidas com miastenia são consideradas de alto risco e, por conseguinte, essas mulheres devem ser acompanhadas por um médico neuromuscular, um obstetra e um neonatologista. O efeito da gravidez sobre a MG é variável e seu curso poderá variar de uma gravidez para outra. As exacerbações são mais comuns no primeiro trimestre, nas quatro últimas semanas de gestação e continuam no puerpério.47
As mulheres com MG devem procurar orientação antes de engravidar para que seja possível determinar a necessidade de timectomia, para otimizar o controle da doença e revisar o plano terapêutico. A piridostigmina e a prednisona foram classificadas como medicamentos para gravidez de categoria C, embora sejam razoavelmente seguros para uso de rotina.
A IVIg e a PLEX podem ser usadas no gerenciamento de sintomas graves ou de crises de MG.47 A azatioprina e o micofenolato de mofetila (categoria D) e o metotrexato (categoria X) apresentam riscos significativos para os fetos e, por isso, não se recomenda o uso desses medicamentos.47 O sulfato de magnésio, usado no gerenciamento de eclampsia, deve ser usado com extrema cautela, tendo em vista que poderá agravar os sintomas de MG. A mãe e o feto devem ser monitorados de perto durante o trabalho de parto e, caso seja necessário, a anestesia regional é preferível à anestesia geral.47
MG neonatal e artrogripose múltipla congênita. A miastenia neonatal transitória ocorre em, aproximadamente, 10% de bebês nascidos de mulheres com MG, mesmo nos casos em que a mãe for assintomática, devido à transferência transplacentária de anticorpos maternos contra o AChR.44 Em geral, os sintomas iniciam alguns dias após o nascimento e incluem ptose, hipotonia, fraqueza generalizada, dificuldade para alimentar e até mesmo problemas respiratórios.48
Tipicamente, os sintomas desaparecem no período médio de 3 semanas, embora possivelmente tenham que ser tratados com inibidores da colinesterase e com suporte ventilatório em vários casos.47 Em casos raros, a MG materna está associada à AMC e se caracteriza por múltiplas contraturas nas articulações, assim como por características dismórficas e outros tipos de anomalias.47
Crise miastênica. A crise miastênica se refere à exacerbação de MG que pode colocar a vida em risco devido à fraqueza nos músculos respiratórios ou à disfunção bulbar, e ocorre em 15 a 20% de casos, predominantemente nos primeiros 2 anos após o início da doença.6 Os pacientes precisam ser monitorados de perto em uma unidade de terapia intensiva, tendo sempre em mente a possibilidade de intubação eletiva precoce. O tratamento requer o uso de PLEX ou IVIg acompanhado de altas doses de corticosteroides. De modo geral, suspende-se o uso de inibidores da colinesterase enquanto os pacientes permanecem intubados.6
O Quadro 3 contém os agentes imunomoduladores mais comuns usados no tratamento de MG.
Quadro 3
|
Agentes Imunomoduladores Mais Comuns Usados no Tratamento de Miastenia Grave | |||||
|
Medicação |
Dose inicial |
Ajuste da dose |
Efeitos colaterais graves |
Monitoramento |
Comentários |
|
Prednisona |
15?20mg/dia |
Aumentar em 5mg a cada 2?4 dias até os sintomas se estabilizarem ou 1?1,5mg/kg/dia. Mudar para dias alternados (se possível) e reduzir gradual e lentamente até a dose mínima. |
Hipertensão, diabetes melito, glaucoma, osteoporose, necrose asséptica nas articulações, catarata, glaucoma, úlceras GI, transtornos psicológicos, miopatia causada por esteroides, infecção. |
K+, glicose, densidade óssea, pressão arterial, exame nos olhos. |
A administração de prednisona poderá iniciar em doses de 1?1,5mg/kg, embora seja necessário um monitoramento rigoroso por 7?10 dias (preferencialmente, em ambiente hospitalar) devido ao risco de exacerbação. |
|
Azatioprina |
50mg/dia |
Aumentar em incrementos de 50mg, de acordo com a necessidade, a cada 2?4 semanas, até 2?3mg/kg/dia. |
Reação sistêmica (febre, náusea, vômito, dor abdominal), mielossupressão, pancreatite, hepatotoxicidade, infecção. |
CBC, TFHs. |
Considerar o ensaio da tiopurina metiltransferase (TPMT) antes do tratamento; não usar com alopurinol. |
|
Ciclosporina |
3?4mg/kg/dia em 2 doses divididas |
Aumentar lentamente até o total de 6mg/kg/dia, de acordo com a necessidade. |
Hipertensão, nefrotoxicidade, tremor, SEPR, convulsão, hepatotoxicidade, infecção. |
CBC, TFHs, BUN, Cr, eletrólitos, níveis mínimos de medicamentos, pressão arterial. |
Há diferença de bioequivalência entre as preparações; evitar o uso de outros medicamentos contra nefrotoxicidade. |
|
Micofenolato de mofetila |
500mg, 2x/dia |
Aumentar para 1.500mg, 2x/dia, de acordo com a necessidade. |
Diarreia, vômito, mielossupressão, infecção. |
CBC, Cr. |
Não houve benefícios adicionais em relação à prednisona em dois testes. |
|
Metotrexato |
7,5 mg/semana |
Aumentar 2,5 mg a cada semana até 25 mg/semana, de acordo com a necessidade. |
Mielossupressão, alopecia, estomatite, doença pulmonar intersticial, infecção, mielossupressão. |
CBC, BUN, Cr |
Administrar diariamente com folato; encontra-se em curso um teste controlado por placebo. |
|
Rituximabe |
375mg/m2 SC/semana, por 4 semanas (alternativa: 1.000IV, com 2 semanas de intervalo) |
Repetir a infusão de acordo com a resposta do paciente a cada 6?24 meses. |
Reação à infusão, anafilaxia, mielossupressão, infecção, disritmia cardíaca. |
CBC, ECG durante e depois da infusão. |
Pré-medicar com acetaminofeno e anti-histaminas. |
|
Globulina imune intravenosa (IVIg) |
2g/kg por 2?5 dias |
Administrar infusão mensal e diminuir gradualmente até a dose mais baixa (0,4?1g/kg). |
Reação à infusão, meningite asséptica, nefrotoxicidade, AVC, sobrecarga de líquidos. |
BUN, Cr. |
Excluir deficiência de IgA (risco de anafilaxia) antes do tratamento, pré-medicar com acetaminofeno e anti-histaminas. |
BUN: nitrogênio ureico sanguíneo; CBC: hemograma completo; Cr: creatinina; ECG: eletrocardiograma; GI: gastrintestinal; SC: superfície corporal; SEPR: síndrome de encefalopatia posterior reversível; TFH: teste da função hepática.
Complicações
As complicações principais estão relacionadas a doenças graves que levam ao comprometimento respiratório e à disfunção bulbar, assim como aos efeitos colaterais da terapia.
Prognóstico
Nos casos de pacientes que se apresentam com sintomas oculares, a fraqueza poderá se restringir aos músculos oculares em 15 a 20% de casos (MG ocular pura); entretanto, a maioria dos pacientes desenvolve fraquezas em outras partes do corpo (MG generalizada).16
No último grupo, 90% dos pacientes chegam a desenvolver doença generalizada nos primeiros 12 meses.16 A remissão espontânea poderá ocorrer em 10 a 15% de casos, normalmente no primeiro ou segundo ano da doença.16 Isso enfatiza a importância da retirada lenta e gradual da medicação imunossupressora após o controle da doença. A taxa anual de mortalidade causada por MG foi estimada em 0,06 a 0,89 por um milhão de acordo com os dados de uma grande metanálise que reuniu vários estudos epidemiológicos de MG.3
Nos casos de AChR-MG, concluiu-se que a correlação entre a gravidade da doença e as titulações do anticorpo anti-AChR é inexpressiva; portanto, elas não podem ser usadas para determinar o prognóstico ou a resposta ao tratamento.21 Os resultados da MuSK-MG no longo prazo se assemelham aos da AChR-MG, embora o início seja agudo com progressão rápida para a gravidade máxima em um curto espaço de tempo.17
Além disso, os níveis de anticorpos anti-MuSK parecem se correlacionar com a gravidade da doença e com a resposta ao tratamento.21 Os anticorpos contra Kv1.4 (canal de potássio muscular controlado por voltagem [VKGC]), documentados em 12 a 28% de pacientes japoneses com MG,49 foram associados à MG (sintomas bulbares e crise miastênica), timoma, miocardite e tempo prolongado de QT nos eletrocardiogramas.50 Entretanto, em um estudo posterior, os pacientes brancos com esse anticorpo se apresentaram apenas com MG ocular ou MG leve.5
Síndromes Miastênicas Congênitas
As SMCs formam um grupo heterogêneo de distúrbios que causam falhas na transmissão neuromuscular como resultado de defeitos genéticos em proteínas pré-sinápticas, sinápticas ou pós-sinápticas, que são importantes para a estrutura ou função da JNM.
Epidemiologia
Essas síndromes são consideradas distúrbios raros. Em uma grande série documentada por Engel, as SMCs foram as anormalidades mais comuns, sendo que as mutações em subunidades do AChR foram responsáveis por mais da metade de todos os casos.52
Etiologia e Genética
Nesse processo, é imprescindível questionar os parentes que tenham sido afetados pela doença, em especial a determinação do grau de consanguinidade dos pais, levando-se em conta que a maioria tem padrão autossômico recessivo de herança (excetuando-se as síndromes de canal lento que são basicamente autossômicas dominantes).1
Patogênese
O defeito unificador nos casos de SMC é a margem de segurança reduzida na transmissão neuromuscular. O mecanismo exato depende da anormalidade genética específica que afeta a síntese ou da concentração de ACh no interior das vesículas, a rota de entrada de cálcio e de liberação de ACh, a função da ACh esterase e a densidade e afinidade do AChR em relação à ACh.52
Diagnóstico
Manifestações Clínicas
As suspeitas de SMC ocorrem nas situações em que os lactentes, as crianças ou mesmo os adultos apresentam fraqueza oscilante nos músculos oculares, nos músculos bulbares ou nos músculos dos membros. Os neonatos podem apresentar hipotonia, sucção fraca e choro fraco, em combinação com estridor, sensação de asfixia e dificuldades ventilatórias, e talvez se apresentem até com artrogripose múltipla congênita (AMC).
Os lactentes, em geral, apresentam eventos motores importantes tardiamente e alguns adultos poderão se apresentar com fraqueza proximal fixa.1 Características específicas ajudam a identificar distúrbios específicos.52
Exame Físico
Em geral, os pacientes apresentam fraqueza nos músculos inervados do crânio ou dos membros, que se agrava com a prática de exercícios ou com a ativação sustentada. Alguns indivíduos se apresentam com um padrão cíngulo-límbico de fraqueza muscular. Os exames cognitivos e sensoriais são normais.
Testes Laboratoriais
Exames de sangue. Os testes de anticorpos para AChR, MuSK e VGCC são negativos. A análise de mutações no DNA isolado do corpo possivelmente identifique os genes responsáveis e deverá se basear na suspeita de características clínicas específicas ou na frequência das mutações genéticas.52
Testes psicológicos. Provavelmente, os pacientes apresentem descobertas anormais na ENR, na SFEMG e no teste com edrofônio, semelhantes às descobertas descritas para os casos de MG.
Diagnóstico Diferencial
A SMC deve ser diferenciada de outras causas de lactentes flácidos, incluindo distúrbios no sistema nervoso central (SNC), atrofia nos músculos espinais, neuropatia hipomielinizante congênita, miastenia neonatal, distrofia miotônica congênita, distrofia muscular e miopatias metabólicas. É imprescindível fazer a distinção entre fraqueza com padrão cíngulo-límbico, com início na vida adulta, de várias outras formas de miopatia como as distrofias musculares.1
Gerenciamento
Os inibidores da colinesterase precisam ser testados no gerenciamento inicial de todos os pacientes, sendo que os testes deverão continuados profilaticamente mesmo nos casos assintomáticos; todavia, alguns pacientes podem não responder ou a doença poderá até agravar com o uso crônico. Alguns pacientes respondem favoravelmente em vários graus a medicamentos como a 3,4-diaminopiridina (3,4-DAP), albuterol ou efedrina.52
Síndrome Miastênica de Lambert-Eaton
A síndrome miastênica de Lambert-Eaton (SMLE) é o segundo distúrbio mais comum na JNM e resulta do envolvimento de VGCCs pré-sinápticos.1
Epidemiologia
A SMLE é um distúrbio raro com incidência documentada de mais ou menos 0,4 por milhão nos Países Baixos; no entanto, a incidência e a prevalência reais talvez não sejam precisas, tendo em vista que esse tipo de doença pode facilmente passar despercebida.53 Aproximadamente, 50 a 60% de pacientes com SMLE têm um tumor e, nesse grupo, 65% são homens com idade média de início da doença de 60 anos.54 Os casos de SMLE não paraneoplásica têm um pico de início da doença aos 35 anos de idade e outro pico aos 60 anos, sendo que mais de 50% de pacientes são mulheres.54
Etiologia e Genética
Acredita-se que os pacientes com SMLE e tumor tenham alguma condição paraneoplásica, sobretudo associada ao câncer de pulmão de pequenas células (CPPC) e, menos frequentemente, a outros tipos de câncer. A SMLE não paraneoplásica é um processo imunomediado e está associado a outras doenças autoimunes, assim como ao HLA-B8-DR3.54
Patogênese
A fraqueza nos casos de SMLE pode ser causada pela entrada reduzida de cálcio no terminal pré-sináptico, secundária aos efeitos bloqueadores dos anticorpos contra o VGCC (sobretudo o tipo P/Q e, em menor proporção, o tipo N).54 Esse processo diminui a liberação de vesículas de ACh na fenda sináptica, reduz o PPM e diminui a geração de energia muscular.
O Quadro 4 contém as síndromes miastênicas congênitas.
Quadro 4
|
Síndromes Miastênicas Congênitas | ||
|
Síndrome miastênica congênita |
Gene alterado |
Características sugestivas |
|
Defeitos pré-sinápticos Defeito na colina acetiltransferase (ChAT) |
CHAT |
Episódios apneicos (espontâneos ou disparados por febre, vômito ou excitação); agravam-se em temperaturas frias. |
|
Escassez de vesículas sinápticas |
Desconhecido |
? |
|
Síndrome congênita semelhante à de Lambert-Eaton |
Desconhecido |
Facilitação na amplitude de PAMC com ENR. |
|
Defeitos sinápticos Deficiência de AChE na placa terminal |
COLQ |
PAMCs repetidos e teste de edrofônio negativo; refratária ao CEI, mas pode responder à efedrina e albuterol. |
|
Deficiência de laminina-ß2 |
LAMB2 |
Pode ter síndrome nefrótica e anormalidades oculares (síndrome de Piearson); refratária ao CEI. |
|
Defeitos pós-sinápticos Anormalidade cinética primária |
- |
- |
|
A. Síndromes do canal lento |
Subunidades de AChR |
Herança AD; fraqueza proeminente nos músculos cervical, do punho e do extensor dos dedos, porém fraqueza leve nos músculos cranianos inervados; PAMCs repetidos e teste de edrofônio negativo; agravam-se com CEI, mas respondem à quinidina ou à fluoxetina. |
|
B. Síndromes do canal rápido |
Subunidades de AChR |
A maioria responde à combinação de 3,4-DAP e CEI. |
|
Deficiência primária de AChR |
Subunidades de AChR (principalmente, a subunidade e) |
Oftalmoparesia proeminente; responde parcialmente ao CEI com benefício adicional do 3,4-DAP ou albuterol. |
|
SMC causada pela rapsina |
RAPSN |
Contraturas articulares múltiplas ou características dismórficas em alguns casos; estrabismo e fraqueza facial e bulbar são comuns; responde ao CEI com benefício adicional do 3,4-DAP. |
|
SMC causada pela plectina |
PLEC |
Possivelmente se apresente com epidemiólise bolhosa simples ou distrofia muscular progressiva. |
|
Miastenia do canal de Na |
SCN4A |
Pode apresentar ataques abruptos de paralisia respiratória e bulbar. |
|
Defeitos no desenvolvimento e na manutenção da placa terminal |
|
|
|
SMC causada por Dok-7 |
DOK7 |
Possível estridor e paralisa nas cordas vocais em neonatos e lactentes; fraqueza basicamente cíngulo-límbica e axial com fraqueza facial leve e ptose e movimento ocular normal; alguns casos com envolvimento bulbar podem se agravar com piridostigmina, mas respondem à efedrina e ao albuterol. |
|
SMC causada por MuSK |
MUSK |
Existem alguns relatos de respostas a doses baixas de piridostigmina e 3,4-DAP. |
|
SMC causada pela agrina |
AGRN |
Pode ter ptose com fraqueza nos músculos faciais e nos músculos da cintura pélvica. |
|
SMC causada por GFPT1 |
GFPT1 |
Fraqueza predominantemente cíngulo-límbica e axial; as biópsias musculares agregam algo na maior parte dos casos. |
AChR: receptor da acetilcolina; AChE: acetilcolina esterase; AD: autossômico dominante; CEI: inibidor da colina esterase; 3,4-DAP: 3,4-diaminopiridina; Dok-7: saída da tirosina quinase; ENR: estimulação nervosa repetitiva; GFPT1: deficiência de glutamina-frutose-6-fosfato transaminase; MuSK: quinase específica dos músculos; PAMC: potencial de ação muscular combinada; SMC: síndrome miastênica congênita.
Diagnóstico
A tríade formada por fraqueza nos músculos proximais, envolvimento autonômico e arreflexia deve levantar suspeitas de SMLE e levar ao teste confirmatório com estudos eletrofisiológicos e testes de anticorpos.
Manifestações Clínicas
Os pacientes se apresentam predominantemente com fraqueza proximal nas pernas, acompanhada de perto por fraqueza proximal nos braços, cuja disseminação envolve os músculos oculobulbares e os músculos dos membros mais distais, agravando-se com a atividade.54 Observa-se que a velocidade de progressão para SMLE paraneoplásica é mais rápida.55
A disfunção autonômica é comum, mas não é muito debilitante, sendo que boca seca é a queixa mais comum, seguida por disfunção erétil e constipação.55 Os músculos cranianos e ventilatórios talvez sejam envolvidos, porém não tanto como nos casos de MG. Os sintomas são exacerbados por temperaturas elevadas e banhos quentes.1
Exame Físico
Os pacientes com resposta pupilar lenta à luz durante o exame e com fraqueza inicial e reflexos reduzidos poderão melhorar temporariamente após uma contração muscular, como resultado da facilitação pós-exercício.54 Em estágios mais avançados da doença, os pacientes poderão desenvolver vários graus de atrofia muscular. Sintomas sensoriais e cerebelares também se desenvolvem como parte da síndrome paraneoplásica (anti-Hu) ou do envolvimento autoimune (anemia perniciosa).1
Testes Laboratoriais
Exames de sangue. Os anticorpos contra VGCC tipo P/Q são observados em 85 a 90% de pacientes com SMLE e, conforme alguns relatos, em quase 100% de casos de SMLE paraneoplásica.54,56 A presença de anticorpos contra o gene da região determinante do sexo de Y-box 1 (SOX1) é altamente sugestiva de câncer de pulmão de pequenas células (especificidade de 95%).54,57
Estudos de imagens. Os pacientes precisam ser rastreados para que o médico possa verificar a eventual presença de alguma malignidade, em especial câncer no pulmão.54
Testes fisiológicos. Os pacientes possivelmente demonstrem respostas decrementais na ENR lenta e, ainda mais importante, facilitação anormal durando menos de 1 minuto, e ENR rápida particularmente em músculos com amplitude inicial baixa do potencial de ação muscular combinada (PAMC). Levando-se em consideração que esses estudos podem ser normais na fase inicial do processo da doença, recomenda-se repeti-los em caso de progressão dos sintomas.58
Diagnóstico Diferencial
Com frequência, se confunde MG com SMLE, especialmente nos casos de envolvimento dos músculos oculobulbares. A fraqueza muscular proximal simétrica pode ser confundida com miopatia, sobretudo com miosite de corpúsculo de inclusão em pacientes mais velhos ou, menos frequentemente, com formas motoras puras de CIDP por causa dos reflexos reduzidos.1
Gerenciamento
O tratamento de SMLE paraneoplásica se baseia na identificação do tumor e na escolha da terapia. A administração de 3,4-DAP é a primeira escolha para terapia sintomática, tendo em vista que é um medicamento bem tolerado na dose de 10 a 20mg, 3 a 4x/dia.54 Os efeitos colaterais mais comuns incluem parestesias perioral e acral; no entanto, doses muito elevadas (>100mg) poderão resultar em convulsões.59
Na ausência de controle dos sintomas, pode ser necessário aplicar terapias imunossupressoras, como, por exemplo, a combinação de azatioprina e prednisona. Um teste cruzado comprovou que a IVIg produz alguns benefícios e que, assim como a troca de plasma, poderá ser usada em casos agudos ou graves.54
Prognóstico
A presença de câncer de pulmão de pequenas células (CPPC) é preditiva de um curso mais rápido da doença,55 sendo que a sobrevida depende do tratamento do tumor. É interessante observar que os pacientes com CPPC e SMLE aparentemente apresentam prognósticos melhores em comparação com os casos sem SMLE.54
Botulismo
O botulismo é o resultado dos efeitos causados pela neurotoxina do organismo Clostridium botulinum sobre os terminais pré-sinápticos da JNM.1
Epidemiologia
Embora a incidência de várias formas de botulismo dependa da idade e de outros fatores de risco, a forma transmitida por alimentos e a forma infantil são as mais comuns.1
Etiologia e Genética
O botulismo se divide em:
clássico ou transmitido por alimentos: contaminados consumidos em restaurantes ou enlatados;
infantil: devido à inoculação de esporos entéricos de Clostridium botulinum (em especial, no mel) em lactentes com 1 a 6 meses de idade;60
oculto: semelhante à forma infantil, porém em crianças com mais de 1 ano de idade e com anormalidades gastrintestinais pré-existentes ou com histórico de uso prolongado de antibióticos;
causado por feridas: observa-se essa forma de botulismo em usuários de drogas intravenosas, em lesões no seio maxilar (em pessoas que cheiram cocaína) ou em vítimas de trauma;
inadvertido: injeções de toxina botulínica.1
Patogênese
As neurotoxinas produzidas por Clostridium botulinum, uma bactéria gram-positiva anaeróbica formadora de esporos, levam à clivagem enzimática das proteínas necessárias para a exocitose da ACh do terminal pré-sináptico, resultando no potencial reduzido da placa motora e na geração de força muscular.1 Até o presente momento, foram identificadas oito toxinas imunologicamente distintas (A, B, C1, C2, D, E, F e G). A maior parte dos casos em seres humanos é causada pelos tipos A, B e E, sendo que o tipo A produz os sintomas mais graves e de mais longa duração.61
Diagnóstico
Manifestações Clínicas
Tipicamente, o botulismo apresenta início agudo com progressão em 12 a 36 horas e disseminação no sentido craniano para caudal. No início da doença, os pacientes apresentam envolvimento dos nervos cranianos e visão turva, diplopia, ptose, disfagia e disartria. Essas condições são seguidas de fraqueza nas extremidades superiores e, a seguir, nas extremidades inferiores, assim como falta de ar. Os pacientes poderão se apresentar com sintomas autonômicos, tais como boca seca, redução na sudorese, sintomas ortostáticos, constipação e retenção urinária. Não há sintomas sensoriais.1
O botulismo transmitido por alimentos pode afetar um determinado grupo de pessoas, sendo que algumas delas possivelmente se lembrem da ingestão de alimentos contaminados. Nesse grupo, os sintomas geralmente iniciam em 2 a 36 horas, sendo que os sintomas gastrintestinais costumam preceder os neurológicos, e incluem náusea, vômito e diarreia seguida de constipação.
Nos casos de botulismo causado por feridas, possivelmente os pacientes não se recordem de algum trauma, e o período de incubação poderá ser de 4 a 14 dias. Sempre há suspeitas de botulismo em usuários de drogas intravenosas e com fraqueza progressiva de etiologia desconhecida. A detecção de botulismo infantil talvez seja muito difícil e pode variar de doença leve a morte súbita. A incidência de constipação é comum e pode ser o único sintoma que se apresenta no início da doença.1,61
Exame Físico
Os pacientes em geral parecem ansiosos e se apresentam com condições como ptose, oftalmoparesia e paralisa focal, além de reflexo de vômito e fraqueza na língua. Os reflexos nos tendões profundos são normais ou reduzidos de acordo com a gravidade da fraqueza. O envolvimento autonômico resulta em resposta pupilar fraca à luz, ausência de resposta vasomotora às mudanças posturais e hipotermia. O exame sensorial geralmente é normal.1
O botulismo infantil se caracteriza por apatia e lactentes “flácidos”, com ptose, expressão facial reduzida, sucção anormal e reflexo de vômito. Excesso de baba e choro fraco são sinais preocupantes e exigem observação constante por causa do risco de agravamento bulbar e da função respiratória.1
Testes Laboratoriais
Testes dos líquidos corporais e de materiais contaminados. A toxina ou o organismo pode ser identificado pela análise ou culturas de alimentos, fezes ou conteúdos gástricos, e pelo soro ou pelo aspirado de feridas, dependendo da forma de botulismo.60
Testes fisiológicos. O monitoramento da capacidade vital funcional permite avaliar se o paciente irá ou não precisar de suporte ventilatório. Os estudos eletrodiagnósticos demonstram facilitação anormal nas estimulações nervosas repetitivas rápidas e devem abranger os músculos distais, proximais e bulbares; todavia, os resultados devem ser normais na fase inicial da doença e precisam ser repetidos.
De um modo geral, o grau de facilitação é menor do que nos casos de SMLE (podendo ser inferior a 100%), embora persista nos casos de duração mais longa (alguns minutos). A EMG com agulha, em geral, revela a presença de unidades motoras miopáticas, potenciais raros de fibrilação e ondas agudas positivas.1
Diagnóstico Diferencial
É imprescindível diferenciar botulismo de outros distúrbios na JNM e de outras causas de fraqueza progressiva, tais como a síndrome de Guillain-Barré (SGB) e suas variantes, paralisia causada por carrapatos, miopatias tóxicas causadas por neuropatia diftérica e canelopatias musculares. ainda que o padrão descendente seja facilmente distinguível das formas clássicas de SGB (em geral, ascendentes), a variante de Miller-Fisher, com anormalidades oculares e bulbares, poderá se tornar um grande desafio diagnóstico.61
Gerenciamento
É muito importante monitorar os pacientes em UTIs em relação à intubação seletiva e outros cuidados de suporte. As antitoxinas (sobretudo as de origem equina) podem ser usadas logo na fase inicial, sendo que a situação ideal seria dentro de 24 horas após o início da doença,62 enquanto a toxina ainda estiver em circulação, tendo sempre em mente o risco de respostas alérgicas.
A globulina botulínica imune humanizada (BIg) está disponível para uso nos casos de botulismo infantil e, aparentemente, reduz o tempo de hospitalização.60 Nos casos de botulismo causado por feridas, a excisão do tecido desvitalizado e o uso de antibióticos são opções de tratamento a serem consideradas; no entanto, é imprescindível aplicar primeiramente a antitoxina para minimizar os efeitos da toxina liberada pela manipulação tecidual e pela lise do organismo causador da doença.1
Um fator muito importante a ser levado em conta é a prevenção da contaminação por meio do preparo correto dos alimentados enlatados. Ao contrário das toxinas, que são desativadas a 85ºC, para matar os esporos são necessárias temperaturas mais elevadas de até 120ºC. O ato de simplesmente ferver o alimento em altas temperaturas antes de ser enlatado talvez não produza temperatura suficientemente elevada para destruir os esporos.61
Complicações
As principais complicações do botulismo se relacionam a condições como paralisia generalizada prolongada grave e disfunção bulbar e respiratória, assim como as complicações produzidas por tratamentos de suporte avançados de longo prazo.
Prognóstico
A recuperação do botulismo é comum depois de um gerenciamento apropriado do tratamento intensivo; todavia, poderá levar várias semanas ou até alguns meses dependendo do tipo de neurotoxina e da gravidade da doença. De um modo geral, os pacientes precisam de reabilitação e de cuidados de suporte de longo prazo.61
Envenenamento por Organofosforados e Outras Toxinas
Existe uma grande variedade de toxinas e venenos na natureza que afetam adversamente a JNM, agredindo suas funções nos níveis pré-sináptico, sináptico e pós-sináptico.1
Epidemiologia
Embora, em muitos países em desenvolvimento, os pesticidas, incluindo os organofosforados, sejam a causa principal de envenenamento,63 a incidência parece ter declinado nos EUA devido à eliminação gradual dos agentes mais comuns de uso doméstico e de aplicação na agricultura.64 A intoxicação humana é basicamente acidental, podendo ocorrer por exposição pulmonar ou dérmica ou intencional (homicídio ou suicídio) nos casos de consumo oral.63
Etiologia e Genética
Os organofosforados foram utilizados como armas (tabun, serina e soman) ou inseticidas/pesticidas (paration).63
Patogênese
Os organofosforados se ligam irreversivelmente à AChE, chegando até a bloquear sua ação, causando despolarização contínua na membrana pós-sináptica, assim como danos e disfunção na JNM.
Diagnóstico
Manifestações Clínicas e Exame Físico
Normalmente, a exposição aguda começa com efeitos muscarínicos, que são lembrados mais facilmente pelo método mnemônico DUMBBELS (defecation/defecação, urination/micção, miosis/miose, bronchorrhea/broncorreia, bradycardia/bradicardia, emesis/êmese, lacrimation/lacrimejamento, salivation/salivação). A broncorreia poderá resultar em edema pulmonar com risco de vida.63
Os sintomas nicotínicos ocorrem com fraqueza, fadiga, fasciculações e até mesmo paralisia nos músculos respiratórios. Os pacientes apresentam alterações sensoriais causadas pelos efeitos colinérgicos no SNC. Os sintomas sensoriais e a fraqueza distal desenvolvem mais tarde como parte de uma polineuropatia axonal sensorimotora dependente do tempo de duração.1
Testes Laboratoriais
Exames de sangue. Ainda que os ensaios que medem a atividade da butirilcolinesterase no plasma ou a acetilcolinesterase nos eritrócitos confirmem o diagnóstico e orientem a terapia, raramente os resultados são disponibilizados em tempo hábil para que os médicos possam tomar as decisões clínicas.65
Testes Fisiológicos
Testes eletrodiagnósticos. Os estudos de condução nervosa talvez sejam normais nas primeiras 24 a 48 horas. Os potenciais pós-descarga, observados nos estudos de condução motora, são os indicadores mais sensíveis de toxicidade precoce. Observam-se amplitudes reduzidas no potencial de ação muscular combinada (PAMC) nos casos de intoxicação grave, assim como degeneração nos axônios sensoriais e motores mais tarde no curso da doença.
A ENR lenta mostra respostas decrementais com intoxicação grave que poderá se agravar com a administração de edrofônio. A ENR rápida pode mostrar respostas decrementais iniciais e respostas incrementais subsequentes, retornando para o PAMC na linha de base.1
Diagnóstico Diferencial
O Quadro 5 contém as toxinas e os venenos e seus mecanismos.
Quadro 5
|
Toxinas e Venenos | |
|
Toxinas e venenos |
Mecanismo |
|
03-conotoxina produzida pelos caracóis marinhos do tipo Conus |
Bloqueiam o VGCC pré-sináptico |
|
ß-bungarotoxina produzida pela serpente marinha tipo Krait com múltiplas listras |
Altera a liberação de ACh |
|
Toxina botulínica |
Altera a liberação de ACh |
|
Crotoxina produzida pela cascavel brasileira |
Aumenta e, em seguida, altera a liberação de ACh |
|
a-latroxina produzida pela aranha viúva negra |
Liberação maciça de neurotransmissores |
|
Compostos organofosforados |
Ligam-se irreversivelmente à AChE |
|
Curare |
Liga-se ao AChR e bloqueia sua ativação |
|
a-conotoxina de pequenos caracóis marinhos |
Liga-se ao AChR e bloqueia sua ativação |
|
a-bungarotoxina produzida pela serpente marinha tipo Krait com listras |
Liga-se ao AChR e bloqueia sua ativação |
|
a-cobratoxina produzida pela cobra siamesa |
Liga-se ao AChR e bloqueia sua ativação |
ACh: acetilcolina; AChE: acetilcolina esterase; AChR: receptor da acetilcolina; VGCC: canal de cálcio controlado por voltagem.
Nos casos em que o histórico de exposição à toxina não for muito claro, o diagnóstico diferencial deve incluir as causas de fraqueza muscular aguda descritos para os casos de botulismo.
Gerenciamento
A descontaminação é imprescindível antes do início do tratamento. Esse procedimento inclui tirar as roupas do paciente e lavar com sabão e água nos casos de exposição dérmica ou nas situações em que a roupa tiver sido contaminada com vômito causado pela ingestão da toxina. As secreções gástricas também devem ser contidas apropriadamente nos casos de ingestão.
Todo o pessoal de atendimento deverá vestir jalecos, luvas e protetores oculares para evitar a contaminação dérmica ou por vapor.63 As vítimas de intoxicação devem ser internadas em uma UTI. Nesses casos, é extremamente importante administrar uma combinação de pralidoxima, atropina e suporte ventilatório.1
Complicações
As complicações se relacionam à paralisia generalizada prolongada grave, à disfunção bulbar e respiratória e advêm dos cuidados avançados de suporte de longo prazo. A intoxicações graves poderão resultar em lesões cerebrais hipóxicas.65
Prognóstico
O gerenciamento médico pode ser tornar muito difícil, considerando que muitos casos ocorrem em áreas rurais. O resultado do envenenamento por organofosforados depende da potência do composto, da quantidade da exposição e do tempo para aplicação da terapia, resultando em uma ampla faixa de taxas de caso-fatalidade.65 Alguns pacientes podem necessitar de tratamento de suporte prolongado.
|
Informações Financeiras: Mohammad Kian Salajegheh, MD, e Anthony A. Amato, MD, não têm informações financeiras relevantes a declarar. A autoria anterior deste artigo foi de Marinos C. Dalakas, MD, cujas informações financeiras foram apresentadas à época da publicação inicial. Este artigo foi revisado, atualizado e reeditado pelos autores mencionados. |
Referências
1. Amato AA, Russell JA. Neuromuscular disorders. 1st ed. New York: McGraw-Hill; 2008.
2. Takamori M. Structure of the neuromuscular junction: function and cooperative mechanisms in the synapse. Ann N Y Acad Sci 2012;1274:14–23.
3. Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol 2010;10:46.
4. Phillips LH. The epidemiology of myasthenia gravis. Semin Neurol 2004;24:17–20.
5. Avidan N, Le Panse R, Berrih-Aknin S, Miller A. Genetic basis of myasthenia gravis - a comprehensive review. J Autoimmun 2013 Dec 19. DOI: 10.1016/j.jaut.2013.12.001. [Epub ahead of print]
6. Silvestri NJ, Wolfe GI. Myasthenia gravis. Semin Neurol 2012;32:215–26.
7. Ramanujam R, Piehl F, Pirskanen R, et al. Concomitant autoimmunity in myasthenia gravis—lack of association with IgA deficiency. J Neuroimmunol 2011;236:118–22.
8. Giraud M, Beaurain G, Eymard B, et al. Genetic control of autoantibody expression in autoimmune myasthenia gravis: role of the self-antigen and of HLA-linked loci. Genes Immun 2004;5:398–404.
9. Le Panse R, Berrih-Aknin S. Autoimmune myasthenia gravis: autoantibody mechanisms and new developments on immune regulation. Curr Opin Neurol 2013;26:569–76.
10. Cavalcante P, Serafini B, Rosicarelli B, et al. Epstein-Barr virus persistence and reactivation in myasthenia gravis thymus. Ann Neurol 2010;67:726–38.
11. Cheng Z, Qiu S, Jiang L, et al. MiR-320a is downregulated in patients with myasthenia gravis and modulates inflammatory cytokines production by targeting mitogen-activated protein kinase 1. J Clin Immunol 2013;33:567–76.
12. Cavalcante P, Cufi P, Mantegazza R, et al. Etiology of myasthenia gravis: innate immunity signature in pathological thymus. Autoimmun Rev 2013;12:863–74.
13. Poulas K, Koutsouraki E, Kordas G, et al. Anti-MuSK- and anti-AChR-positive myasthenia gravis induced by D-penicillamine. J Neuroimmunol 2012;250:94–8.
14. Stubgen JP. Interferon alpha and neuromuscular disorders. J Neuroimmunol 2009;207:3–17.
15. Cavalcante P, Le Panse R, Berrih-Aknin S, et al. The thymus in myasthenia gravis: site of “innate autoimmunity”? Muscle Nerve 2011;44:467–84.
16. Tormoehlen LM, Pascuzzi RM. Thymoma, myasthenia gravis, and other paraneoplastic syndromes. Hematol Oncol Clin North Am 2008;22:509–26.
17. Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011;44:36–40.
18. Zisimopoulou P, Evangelakou P, Tzartos J, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun 2013 Dec 24. DOI: 10.1016/j.jaut.2013.12.004. [Epub ahead of print]
19. Benatar M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord 2006;16:459–67.
20. Leite MI, Waters P, Vincent A. Diagnostic use of autoantibodies in myasthenia gravis. Autoimmunity 2010;43:371–9.
21. Meriggioli MN, Sanders DB. Muscle autoantibodies in myasthenia gravis: beyond diagnosis? Expert Rev Clin Immunol 2012;8:427–38.
22. Decroos EC, Hobson-Webb LD, Juel VC, et al. Do acetylcholine receptor and striated muscle antibodies predict the presence of thymoma in patients with myasthenia gravis? Muscle Nerve 2014;49:30–4.
23. Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008;131:1940–52.
24. Chatzistefanou KI, Kouris T, Iliakis E, et al. The ice pack test in the differential diagnosis of myasthenic diplopia. Ophthalmology 2009;116:2236–43.
25. Schneider-Gold C, Gajdos P, Toyka KV, Hohlfeld RR. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev 2005;(2):CD002828.
26. Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003;60:243–8.
27. Benatar M, Kaminski HJ, Quality Standards Subcommittee of the American Academy of Neurology. Evidence report: the medical treatment of ocular myasthenia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2007;68:2144–9.
28. Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998;50:1778–83.
29. Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003;61:1438–40.
30. Hehir MK, Burns TM, Alpers J, et al. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010;41:593–8.
31. Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008;71:400–6.
32. Muscle Study G. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008;71:394–9.
33. Pasnoor M, He J, Herbelin L, et al, Muscle Study G. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012;1275:23–8.
34. Heckmann JM, Rawoot A, Bateman K, et al. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011;11:97.
35. Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012;46:687–91.
36. Ponseti JM, Gamez J, Azem J, et al. Tacrolimus for myasthenia gravis: a clinical study of 212 patients. Ann N Y Acad Sci 2008;1132:254–63.
37. Howard JF Jr, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 2013;48:76–84.
38. Barth D, Nabavi Nouri M, Ng E, et al. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011;76:2017–23.
39. Jensen P, Bril V. A comparison of the effectiveness of intravenous immunoglobulin and plasma exchange as preoperative therapy of myasthenia gravis. J Clin Neuromuscul Dis 2008;9:352–5.
40. Wolfe GI, Kaminski HJ, Jaretzki A 3rd, et al. Development of a thymectomy trial in nonthymomatous myasthenia gravis patients receiving immunosuppressive therapy. Ann N Y Acad Sci 2003;998:473–80.
41. Newsom-Davis J, Cutter G, Wolfe GI, et al. Status of the thymectomy trial for nonthymomatous myasthenia gravis patients receiving prednisone. Ann N Y Acad Sci 2008;1132:344–7.
42. Diaz-Manera J, Martinez-Hernandez E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012;78:189–93.
43. Ionita CM, Acsadi G. Management of juvenile myasthenia gravis. Pediatr Neurol 2013;48:95–104.
44. Liew WK, Kang PB. Update on juvenile myasthenia gravis. Curr Opin Pediatr 2013;25:694–700.
45. Christison-Lagay E, Dharia B, Vajsar J, Kim PC. Efficacy and safety of thoracoscopic thymectomy in the treatment of juvenile myasthenia gravis. Pediatr Surg Int 2013;29:583–6.
46. Tracy MM, McRae W, Millichap JG. Graded response to thymectomy in children with myasthenia gravis. J Child Neurol 2009;24:454–9.
47. Guidon AC, Massey EW. Neuromuscular disorders in pregnancy. Neurol Clin 2012;30:889–911.
48. Chaudhry SA, Vignarajah B, Koren G. Myasthenia gravis during pregnancy. Can Fam Physician 2012;58:1346–9.
49. Suzuki S, Utsugisawa K, Nagane Y, Suzuki N. Three types of striational antibodies in myasthenia gravis. Autoimmune Dis 2011;2011:740583.
50. Suzuki S, Satoh T, Yasuoka H, et al. Novel autoantibodies to a voltage-gated potassium channel Kv1.4 in a severe form of myasthenia gravis. J Neuroimmunol 2005;170:141–9.
51. Romi F, Suzuki S, Suzuki N, et al. Anti-voltage-gated potassium channel Kv1.4 antibodies in myasthenia gravis. J Neurol 2012;259:1312–6.
52. Engel AG. Current status of the congenital myasthenic syndromes. Neuromuscul Disord 2012;22:99–111.
53. Wirtz PW, van Dijk JG, van Doorn PA, et al. The epidemiology of the Lambert-Eaton myasthenic syndrome in the Netherlands. Neurology 2004;63:397–8.
54. Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol 2011;10:1098–107.
55. Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol 2008;201-202:153–8.
56. Farrugia ME, Vincent A. Autoimmune mediated neuromuscular junction defects. Curr Opin Neurol 2010;23:489–95.
57. Sabater L, Titulaer M, Saiz A, et al. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology 2008;70:924–8.
58. Oh SJ, Kurokawa K, Claussen GC, Ryan HF Jr. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve 2005;32:515–20.
59. Quartel A, Turbeville S, Lounsbury D. Current therapy for Lambert-Eaton myasthenic syndrome: development of 3,4-diaminopyridine phosphate salt as first-line symptomatic treatment. Curr Med Res Opin 2010;26:1363–75.
60. Pifko E, Price A, Sterner S. Infant botulism and indications for administration of botulism immune globulin. Pediatr Emerg Care 2014;30:120–4.
61. Cherington M. Botulism: update and review. Semin Neurol 2004;24:155–63.
62. Sobel J. Botulism. Clin Infect Dis 2005;41:1167–73.
63. Rusyniak DE, Nanagas KA. Organophosphate poisoning. Semin Neurol 2004;24:197–204.
64. Sudakin DL, Power LE. Organophosphate exposures in the United States: a longitudinal analysis of incidents reported to poison centers. J Toxicol Environ Health Part A 2007;70:141–7.
65. Eddleston M, Buckley NA, Eyer P, Dawson AH. Management of acute organophosphorus pesticide poisoning. Lancet 2008;371:597–607.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.