(Carregando Índice)... (Carregando Índice)... |
Última revisão: 11/11/2015
Comentários de assinantes: 0
Reproduzido de:
Formulário Terapêutico Nacional 2010: Rename 2010 [Link Livre para o Documento Original]
Série B. Textos Básicos de Saúde
MINISTÉRIO DA SAÚDE
Secretaria de Ciência, Tecnologia e Insumos Estratégicos
Departamento de Assistência Farmacêutica e Insumos Estratégicos
Brasília / DF – 2010
Paulo Sérgio Dourado Arrais
Na Rename 2010: itens 3.2 e 17.2
-Suspensão injetável (3 mg + 3 mg)/mL.
-Prevenção do desenvolvimento da síndrome de angústia respiratória, hemorragia intraventricular e morte do recém-nascido em mulheres grávidas com risco de interrupção prematura da gravidez (idade gestacional de 24 a 34 semanas) e em grávidas com ruptura precoce de membranas (idade gestacional menor que 32 semanas).
-Hipersensibilidade a betametasona ou a outros corticosteroides.
-Infecções fúngicas sistêmicas.
- Exclusivo para via intramuscular.
- Usar com cuidado nos casos de:
– pré-eclampsia e hipertensão arterial sistêmica.
– síndrome de Cushing e supressão adrenocortical.
– diverticulite, colite ulcerativa, hiperglicemia, hipotireoidismo, osteoporose, úlcera péptica, miastenia grave, tendências psicóticas e insuficiência renal.
– herpes simples ocular e infecções sistêmicas não tratadas com antimicrobiano.
-Cursos repetidos de betametasona antes do parto expõem a maior risco de corioamnionite, endometrite, sepse e morte neonatal.
-Pode suprimir o sistema imunitário, predispondo a infecções.
-Categoria de risco na gravidez (FDA): C.
-Dose de 12 mg, por via intramuscular, a cada 24 horas, durante 2 dias (2 doses), entre a 24ª e a 34ª semanas da gravidez. a administração da mesma dose feita 24 horas antes do parto pode ainda trazer algum benefício.
-O efeito ótimo ocorre em 24 horas e permanece por 7 dias.
-Absorção: (intramuscular) é rapidamente distribuída a todos os tecidos corporais.
-Atravessa a barreira placentária.
-Metabolismo: hepático (principalmente)
-Meia-vida de eliminação: cerca de 60 horas.
-Excreção: renal.
-Em doses altas, ou repetidas, podem induzir insuficiência suprarrenal reversiva no recém-nascido.
-Cetoconazol, eritromicina, itraconazol, quinidina, verapamil: pode ocorrer aumento de efeito de betametasona pela redução do metabolismo.
-Contraceptivos orais contendo estrogênios: o estrogênio pode alterar o metabolismo e a ligação proteica dos glicocorticoides, levando a diminuição da depuração renal, aumento do tempo de meia-vida e do efeito terapêutico e tóxico dos corticoides. Ajuste da dose do corticosteroide pode ser necessário.
-Fenitoína, fenobarbital, primidona, rifampicina: pode haver redução de efeito de betametasona.
-Fluoroquinolonas: o uso concomitante com betametasona aumenta o risco de ruptura de tendões.
-Vacinas com vírus vivos ou outros imunizantes: a administração de vacinas com vírus vivos para pacientes recebendo doses imunossupressoras de glicocorticoide pode potencializar a replicação dos vírus e/ou diminuir a resposta dos anticorpos do paciente à vacina.
-Vacina contra rotavírus: aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
-Orientar para evitar vacinas sem consulta prévia.
-Alertar para evitar contato com pessoas acometidas de infecções.
-Não interromper o uso sem contatar o médico.
-Armazenar a suspensão a temperaturas entre 15 e 30 ºC. Proteger da luz.
-Não injetar por via intravenosa.
-Não misturar com solução injetável de anestésico local (lidocaína 1% ou 2%) contendo parabenos, fenol ou outros preservativos.
Paulo Sérgio Dourado Arrais
Na Rename 2010: itens 3.2 e 20.3
-Creme 1%.
t Tratamento sintomático de processos alérgicos e inflamatórios cutâneos, tais como dermatite atópica, eczemas, dermatite de contato, picadas de insetos, eczema de escabiose e pruridos.
t Reações fototóxicas.
t Tratamento de curta duração da psoríase da face e dobras.
t Dermatite esfoliativa, seborreica e facial, pitiriase rósea e líquen plano.
t Hipersensibilidade a qualquer dos componentes da formulação.
t Infecções cutâneas fúngicas, bacterianas ou virais não tratadas com antimicrobiano.
t Rosácea, acne, dermatite perioral, psoríase em placa e urticária.
t Pele com cortes, feridas.
t Infecção no local do tratamento, atrofia pré-existente da pele.
– crianças (maior absorção e maior susceptibilidade a efeitos adversos; limitar o período de tratamento em 5 a 7 dias).
– idosos (maior risco de púrpura e lacerações na pele).
– lactação (ver Apêndice B).
t Evitar tratamentos prolongados, principalmente na face, e também em crianças.
t Manter distante dos olhos.
t Fator de risco na gravidez (FDA): C (ver Apêndice a).
t Aplicar fina camada na área afetada, 1 a 2 vezes ao dia, durante 1 a 2 semanas. Em adultos, pode-se aplicar até 4 vezes ao dia.
t O tratamento deve ser realizado em curtos períodos, mas pode durar 2-4 semanas no caso de psoríase em face e dobras; não deve exceder uma semana nos casos de lesões inflamatórias não infectadas em lábios e região perioral.
t Para minimizar a possibilidade de absorção sistêmica significativa em tratamento prolongado, deve-se interromper o tratamento periodicamente, aplicar pequenas quantidades do creme ou tratar uma área do corpo por vez.
t Em casos mais graves pode ser necessária a oclusão da lesão. Aspectos farmacocinéticos clinicamente relevantes.
t Absorção: rápida.
t a absorção é afetada pelo tipo de veículo utilizado, pela área de aplicação e pelo grau da lesão na pele.
t Os Efeitos adversos são raros, mas podem ser sistêmicos quando aplicado em grande quantidade, em áreas extensas da pele, com curativos oclusivos ou em pele lesionada.
t Hipersensibilidade.
t Quadros acneiformes, afinamento da pele, telangiectasia, estrias, diminuição na cicatrização de feridas, equimose, hirsutismo, ardor, prurido e irritação no local da aplicação, dermatite de contato, rosácea, dermatite perioral, hipertricose, foliculite, furunculose, pústulas, pioderma, hipopigmentação.
t Hiperestesia.
t Catarata, glaucoma.
t Síndrome de Cushing, edema.
t Úlcera gástrica.
t Hipertensão, síndrome hipercalemica.
t Orientar que este medicamento é somente para uso externo e para evitar contato com olhos e mucosas.
t Orientar para não cobrir a pele tratada com curativos oclusivos.
t Orientar para evitar fraldas plásticas ou apertadas caso o creme seja aplicado na área da fralda.
t Alertar para não aplicar com outros medicamentos no mesmo local da pele. Obedecer ao intervalo de pelo menos 30 minutos entre a aplicação de diferentes medicamentos na mesma região.
t Armazenar a temperaturas entre 15 e 30 ºC, em recipientes bem fechados.
Evitar o congelamento. Manter ao abrigo de luz, calor e umidade.
Larissa Niro
Na Rename 2010: itens 6.2.2, 18.1
t Pó para suspensão injetável 3,75 mg.
t Câncer de próstata avançado (tratamento paliativo).
t Endometriose.
t Anemia devido a leiomioma uterino (tratamento pré-operatório associado a ferro).
t Puberdade precoce central.
t Hipersensibilidade a leuprorrelina ou aos análogos do hormônio liberador de gonadotrofina, ou a qualquer componente da formulação.
t Sangramento vaginal não diagnosticado.
t Categoria de risco na gravidez (FDA): X (ver Apêndice a).
t a amamentação é contraindicada com o uso deste medicamento (ver Apêndice B).
t Osteoporose grave.
t Metástase vertebral.
t Obstrução urinária no câncer de próstata.
t Usar com cuidado nos casos de:
– história familiar de osteoporose ou pacientes com doença osteometabólica (pode ocorrer redução da densidade mineral óssea).
– uso prolongado de outros fármacos e produtos que reduzam a densidade dos ossos, incluindo álcool e tabaco.
– idosos (não há estudos apropriados sobre a relação da idade com os efeitos da leuprorrelina, na população geriátrica; entretanto, este medicamento é frequentemente usado neste grupo, especialmente para o tratamento de câncer de próstata).
– diabete melito.
– homens com risco de exacerbação do câncer (devem ser monitorados durante o primeiro mês de tratamento).
t mulheres devem usar métodos contraceptivos não-hormonais durante todo o período do tratamento.
– Dose inicial 0,3 mg/kg, por via intramuscular, a cada 4 semanas, com dose mínima de 7,5 mg. Se necessário, a dose pode ser aumentada em 3,75 mg a cada 4 semanas.
– 3,75 a 7,5 mg, por via intramuscular, a cada 4 semanas, durante no máximo 6 meses;
– 3,75 mg, por via intramuscular, como dose única nos primeiros 5 dias do ciclo menstrual; repetir a cada mês até o máximo de 6 meses;
– Anemia devido a leiomioma uterino (tratamento pré-operatório associado a ferro)
– 3,75 mg por via intramuscular, uma vez por mês até o máximo de 3 meses.
t Absorção: acetato de leuprorrelina não é muito ativo quando dado por via oral, mas é bem absorvido por via subcutânea ou intramuscular.
t Aumento transitório das concentrações de testoterona e estradiol ocorrem nas primeiras semanas de tratamento. Níveis de castração e pós-menopausa ocorrem em 2 a 4 semanas, respectivamente.
t Biodisponibilidade: após injeção intramuscular com a formulação de depósito é estimada em cerca de 90%.
t Amenorreia ocorre frequentemente após 1 a 2 meses de tratamento. No câncer de próstata o início do efeito se dá após 2 a 4 semanas.
t O sistema hipofisário-gonadal restabelece a função normal em 4 a 12 semanas após a suspensão do tratamento e o ciclo menstrual retorna em 60 a 90 dias.
t Excreção: Renal. a eliminação é menor que 5% de uma dose de 3,75 mg.
t Apoplexia hipofisária (estado mental alterado, colapso cardiovascular, diplopia, cefaleia, alterações visuais, vômitos).
t Distúrbio tromboembólico, leucopenia, trombocitopenia, embolia pulmonar.
t Edema periférico, arritmias cardíacas ou palpitações, angina ou enfarte do miocárdio, hipotensão, síncope.
t Eritema (1,7% a 12,5%), dor, enduração, granulomas e abscesso estéril são particularmente associados com injeções de depósito de análogos de gonadorrelina (incidência de 5%); necrólise hemorrágica do macroedema e fraqueza generalizada.
t Alopecia (2,2% a 2,3%).
t Fogachos (5,3%), amenorreia, sangramento vaginal profuso, efeitos androgênicos, aumento de peso, diminuição da libido. Os análogos da gonadorrelina causam Efeitos adversos similares aos da menopausa nas mulheres e orquidectomia nos homens e incluem suores noturnos (2,7% a 3,3%), disfunção sexual, secura vaginal ou sangramento, e ginecomastia ou mudanças no tamanho dos seios.
t Diminuição do tamanho dos testículos, impotência, exacerbação transitória do câncer de próstata
t Dor em ossos, nos músculos e nas articulações, parestesia.
t Exacerbação da depressão, alteração do humor, cefaleia, tontura, anorexia, paralisia, distúrbios de memória.
t Reações de hipersensibilidade, incluindo anafilaxia.
t Recomendar que a injeção intramuscular de depósito seja administrada por profissional habilitado. Os locais de aplicação das injeções devem ser alternados periodicamente.
t Alertar para a possibilidade de, no tratamento de anemia devido à leiomioma uterino ou endometriose, ocorrer amenorreia ou de períodos menstruais irregulares.
t Alertar para notificar caso a menstruação regular ocorre durante o tratamento e também caso não ocorra dentro de 60 a 90 dias após a suspensão do medicamento.
t Orientar para não usar contraceptivos orais durante o tratamento e sim outras formas de contracepção não-hormonais. Suspender o tratamento com acetato de leuprorrelina se houver suspeita de gravidez.
t Alertar que o uso de leuprorrelina pode prejudicar a fertilidade, suprimindo a produção do esperma em homens e causando anovulação na maioria das mulheres, geralmente reversiva depois da suspensão.
t Alertar que o acompanhamento do tratamento pelo médico é muito importante.
t Devido às diferentes características de liberação, uma dose fracionada das formulações de depósito de 3 ou 4 meses não é equivalente à mesma dose da formulação de depósito de 1 mês e não deve ser dada em seu lugar.
t Armazenar sob temperatura ambiente (15 a 30 ºC). Não congelar. Manter ao abrigo da luz.
t Acetato de leuprorrelina para injeção (depósito intramuscular) deve ser reconstituído com volume apropriado do diluente fornecido pelo produtor, a suspensão deve ser agitada até homogeneização completa para dispersar eventuais partículas.
Karen Luise Lang
Na Rename 2010: itens 18.4.2 e 18.4.5
t Comprimido 2,5 e 10 mg.
t Suspensão injetável 150 mg/mL.
t Contracepção (injeção trimestral).
t Distúrbios vasomotores da menopausa.
t Endometriose.
t Hemorragias uterinas.
t Amenorreia secundária.
t Hipersensibilidade ao acetato de medroxiprogesterona ou a qualquer componente da formulação.
t Tumores malignos de mama ou em órgãos genitais.
t Disfunções hepáticas.
t Porfiria.
t Tromboembolismo venoso e doenças arteriais.
t Hemorragia geniturinária não diagnosticada.
t Histórico de abortos espontâneos.
t Histórico de prurido ou icterícia idiopática durante a gravidez.
t Gravidez. Categoria de risco na gravidez (FDA): X (ver Apêndice a).
t Usar com cuidado nos casos de:
– perda de densidade óssea, risco de tromboembolismo, retenção de fluidos orgânicos, asma, depressão, convulsões, epilepsia, enxaqueca, diabetes, hiperlipidemias, disfunções cardíacas, hipertensão, histórico de desenvolvimento de tumor de mama e distúrbios oftálmicos.
– distúrbios de sangramento menstrual.
– insuficiência hepática e renal (ver Apêndice D).
– climatério (sinais e sintomas do climatério podem ser mascarados).
t Evitar terapia prolongada.
– 2,5 a 10 mg, por via oral, durante 5 a 10 dias, iniciando entre o 16º e o 21º dia do ciclo. Repetir por três ciclos em amenorreia secundária.
– 150 mg, por via intramuscular, até o 5º dia do ciclo menstrual, repetidos a cada 3 meses.
– Após o parto, a administração deve ser realizada até o 5º dia posterior ao nascimento ou, em caso de amamentação, até 6 semanas.
– 10 mg, por via oral, a cada 8 horas, durante 90 dias, iniciando no primeiro dia do ciclo.
– 2,5 a 10 mg, por via oral, durante 5 a 10 dias, iniciando entre o 16º e o 21º dia do ciclo. Repetir por dois ciclos em hemorragia uterina disfuncional.
– 5 a 10 mg, por via oral, durante 12 a 14 dias por mês, iniciando no 1º ou 16º dia do ciclo
– Em mulheres com leiomioma, considerar doses menores de 2,5 mg por dia
t Pico de concentração plasmática: 2 a 4 horas, por via oral; aproximadamente 3 semanas, por via intramuscular.
t Metabolismo preponderantemente hepático.
t Eliminação preponderantemente renal.
t Meia-vida: 12 a 17 horas (oral) ou 50 dias (intramuscular).
t Alterações de peso corporal (frequência da reação: acima de 5%)
t Amenorreia, desordens menstruais, maior retardo na volta da fertilidade (forma de depósito), galactorreia.
t Diminuição da massa óssea (frequência da reação: acima de 5%), osteoporose
t Dor abdominal (acima de 5%)
t Astenia, vertigens, cefaleia.
t Trombose venosa profunda, embolia pulmonar.
t Icterícia.
t Anafilaxia.
t Síndrome de Cushing.
t Alprazolam pode ter seu risco de toxicidade aumentado pela inibição do metabolismo hepático. Monitorar o aumento da resposta ao benzodiazepínico.
t Aprepitanto, bexaroteno, bosentana e rifampicina podem induzir o metabolismo da medroxiprogesterona administrada por via oral. Orientar para a utilização de método contraceptivo adicional durante o tratamento.
t Em caso de esquecimento de uma dose oral, esta deve ser ingerida assim que possível, desde que o horário da dose seguinte não esteja próximo. Cuidado para não duplicar a dose.
t Caso o intervalo entre as aplicações da forma injetável ultrapasse 3 meses e 14 dias, a hipótese de gravidez deve ser excluída antes de efetuar a próxima administração, e um método de contracepção alternativo deverá ser utilizado durante os 7 dias posteriores.
t As pacientes devem ser alertadas antes do início do tratamento sobre possíveis irregularidades menstruais e um potencial atraso no retorno da fertilidade após a suspensão do uso do medicamento.
t Conservar à temperatura ambiente (15 a 30 ºC). Manter em recipientes hermeticamente fechados e ao abrigo da luz.
t Agitar bem, antes da utilização por via intramuscular.
Larissa Niro
Na Rename 2010: item 6.2.1
t Comprimido 160 mg
t Carcinoma de mama (tratamento paliativo).
t Carcinoma de endométrio (tratamento paliativo).
t História de hipersensibilidade ao acetato de megestrol ou a qualquer outro componente da formulação.
t Gravidez. Categoria de risco na gravidez (FDA): X (ver Apêndice a).
t Usar com cuidado nos casos de:
– diabetes ou hiperglicemia e história de doença tromboembólica.
– retirada após uso prolongado (possibilidade de insuficiência suprarrenal).
– crianças e idosos (segurança e efetividade do megestrol não foram estabelecidas).
– lactação.
t Concentrações elevadas de megestrol aumentam a taxa de crescimento da microbiota normal, conduzindo a um aumento da inflamação gengival e sangramento. Um programa de limpeza estritamente reforçado por um profissional, combinado com controle de placas pelo paciente, irá minimizar a gravidade.
– 160 mg/dia, por via oral, por dois meses de tratamento contínuo; a dose pode chegar até 800 a 1.600 mg/dia em câncer de mama avançado metastático; a eficácia é observada no mínimo após 2 meses de tratamento contínuo.
– 40 a 320 mg/dia, por via oral, em doses divididas, por dois meses de tratamento contínuo.
t Absorção: bem absorvido oralmente.
t Pico de concentração: 1 a 3 horas.
t Duração de efeito: 3 a 12 meses (em média 7 meses).
t Metabolismo: hepático.
t Excreção: renal (em média 66,4%) e fecal (em média 19,8%).
t Meia-vida de eliminação: 15 a 100 horas (em média 38 horas).
t Efeitos mais graves: insuficiência suprarrenal, anemia (5%), trombose venosa profunda, tromboflebite e embolia pulmonar.
t Hipertensão (8%), edema (1% a 3%), palpitação (1% a 3%), cardiomiopatia (1% a 3%).
t Erupções cutâneas (2% a 12%), prurido, alopecia, sudorese.
t Insônia (6%), cefaleia (10%), confusão (1% a 3%), convulsões (1% a 3%), depressão (1% a 3%).
t Impotência (4% a 14%), mascaramento do início da menopausa, hiperglicemia (16% com doses altas de megestrol).
t Diarreia (6% a 15%), flatulência (10%), indigestão, vômitos (6%), náuseas (5%).
t Aumento de apetite e ganho de peso (mais de 50%).
t Sangramento endometrial na retirada.
t Alterações de humor.
t Retenção hídrica.
t Desenvolvimento de folículos ovarianos.
t Distúrbios visuais, incluindo perda parcial da visão.
t Febre (1% a 6%).
t Ciclosporina pode ter sua hepatotoxicidade aumentada pelo megestrol.
t Alertar para ter cuidado ao dirigir ou desenvolver atividade que requeira porque o medicamento pode causar tontura.
t Orientar para notificar a ocorrência de sangramento uterino continuado por mais de 3 meses.
t Orientar para notificar suspeita de gravidez ou atraso menstrual.
t Orientar para notificar afecções gengivais ao longo do tratamento.
t Alertar para descontinuar o medicamento em caso de perda de visão parcial ou total ou inicio repentino de proptose, diplopia ou enxaqueca.
t Alertar para notificar sobre história de alergia a amendoim ou de hipersensibilidade às progestinas ou a qualquer componente do produto.
t Armazenar os comprimidos a temperatura ambiente, entre 15 a 30 ºC, em recipiente bem fechado.
Letícia Figueira Freitas
Na Rename 2010: item 10.3
t Solução injetável 2 mEq/mL.
t Nutrição parenteral em casos de acidose.
t Alcalose metabólica ou respiratória, hipocalcemia ou hipocloridria.
t Sais de sódio devem ser usados com extrema cautela em pacientes com hipertensão, edema periférico ou pulmonar, insuficiência renal, pré-eclampsia, aldosteronismo ou outras condições associadas a retenção de sódio.
t As doses devem ser estabelecidas com base nas necessidades de cada paciente e na composição do produto utilizado.
t Sofre biotransformação para bicarbonato.
t a concentração plasmática de sódio desejada deve estar entre 135 a 145 mmol/litro.
t Hipopotassemia e alcalose metabólica, com uso excessivo e principalmente em pacientes com prejuízo da função renal.
t Hipernatremia com uso excessivo (sintomas: sonolência, confusão, parada respiratória e coma). Os sintomas no SNC são mais graves quando a hipernatremia se desenvolve rapidamente. Se ocorrer depleção de volume, podem surgir outros sintomas como hipotensão, taquicardia e sintomas de insuficiência respiratória.
t Hipertonicidade muscular, contração muscular e tetania (especialmente em pacientes com hipocalcemia).
t Edema periférico e pulmonar (nos casos de retenção e excesso de sódio).
t Salicilatos, tetraciclinas e barbitúricos: aumento da excreção renal por alcalinização da urina, porque são substâncias ácidas.
t Fármacos alcalinos: têm sua meia-vida prolongada, podendo resultar em toxicidade.
t Composto cristalino, muito solúvel em água e solúvel em álcool.
t Uma solução a 5% em água apresenta pH de 7,5 a 9,0.
t a solução deve ser armazenada em recipiente hermético.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
Atenção: se ocorrer alcalose metabólica por dose excessiva, o tratamento consiste principalmente em correção apropriada de fluido e equilíbrio eletrolítico.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 21.5
t Comprimido 250 mg
t Glaucoma de ângulo aberto crônico (adjuvante).
t Glaucoma secundário.
t Pré-operatório de glaucoma de ângulo fechado agudo.
t Hipersensibilidade a acetazolamida, sulfonamidas ou inibidores da anidrase carbônica, ou a qualquer componente da formulação.
t Glaucoma de ângulo fechado crônico (pode mascarar deterioração).
t Hipopotassemia.
t Hiponatremia.
t Acidose hiperclorêmica.
t Insuficiência renal grave.
t Insuficiência hepática grave.
t Insuficiência adrenocortical (doença de Addison).
t Cirrose hepática.
t Usar com cuidado nos casos de:
– idosos, diabete melito, obstrução pulmonar ou enfizema, insuficiência renal leve ou moderada (ver Apêndice D), insuficiência hepática (leve ou moderada).
– dose elevada (pode aumentar sonolência e/ou parestesia e diminuir diurese).
– uso prolongado (monitorar contagem sanguínea e eletrólitos).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
t Pode diminuir a capacidade de dirigir veículos e operar máquinas.
– 250 mg a 1 g por dia, por via oral, em doses divididas até de 6 em 6 horas. Ajustar de acordo com sintomatologia e pressão intraocular
– 250 mg, por via oral, a cada 4 horas. Alguns pacientes respondem a 250 mg, por via oral, a cada 12 horas.
– Iniciar com 125 mg, por via oral, a cada 6 horas. Se dose bem tolerada sem redução da pressão intraocular, ajustar a dose e aompanhar a pressão.
t Absorção oral.
t Duração da ação: 8 a 12 horas.
t Pico de concentração: 1 a 4 horas.
t Ligação a proteínas plasmáticas: 70% a 90% dependendo da depuração renal.
t Não metabolizada: 90% a 100% da dose é excretada inalterada na urina em até 24 horas após a administração.
t Meia-vida de eliminação: 4-8 horas.
t Dialisável.
t Alimentos não alteram biodisponibilidade.
t Efeito é dose-dependente, mas há pouco efeito adicional com doses maiores.
t perda de apetite e peso, sede, diarreia, melena, anorexia, náusea, vômito, alteração do paladar.
t confusão, parestesia, sonolência, depressão fadiga, irritabilidade.
t alteração na audição, zumbido (frequente no início do tratamento).
t inibição da libido.
t urticária.
t poliúria, glicosúria, hematúria, cálculos renais, cristalúria e cólica renal, aumento da frequência urinária ou do volume da urina.
t função hepática alterada.
t glaucoma de ângulo fechado.
t acidose metabólica e alteração nos eletrólitos se uso prolongado.
t reações adversas semelhantes à das sulfonamidas (anafilaxia, discrasias sanguíneas, eritema multiforme, necrólise hepática fulminante, síndrome de Stevens-Johnson e necrólise epidérmica tóxica.
t Ácido acetilsalicílico e outros salicilatos: aumento do efeito da acetazolamida. Monitorizar toxicidade por salicilato, especialmente em pacientes com disfunção renal ou doses altas de ácido acetilsalicílico.
t Digoxina (por indução de hipopotassemia), ciclosporina e sotalol (por indução de hipopotassemia e hipomagnesemia): aumento da toxicidade. Se for possível, evitar as combinações. Se administrados concomitantemente, monitorar níveis de ciclosporina circulante e ajustar dose de ciclosporina se necessário; também monitorar pacientes para aumento da toxicidade da ciclosporina (disfunção renal e neurotoxicidade).
t Fenitoína e fosfenitoína: aumento do risco de osteomalácia. Monitorar paciente para sinais iniciais de osteomalácia; suspender acetazolamida e administrar vitamina D e fosfato.
t Lítio: pode haver tanto efetividade reduzida como concentração plasmática aumentada, com aumento na toxicidade (fraqueza, tremor, sede, confusão).
t Alertar para não operar máquinas ou dirigir.
t Orientar para ingerir bastante líquido durante o tratamento, para evitar cálculos renais.
t Orientar para usar preferentemente pela manhã e, se possível, evitar doses após as 18 h, devido ao efeito diurético.
t Dose esquecida: orientar para ingerir imediatamente quando lembrar.
t Orientar para não suspender repentinamente, quando o objetivo for o controle de epilepsia.
t Orientar para a ingestão de bastante líquido para evitar cálculos renais.
t Armazenar à temperatura ambiente, entre 15 e 30 °C. Proteger do calor, da umidade e da luz direta.
t Medicamentos produzidos por laboratórios diferentes só podem ser intercambiáveis quando houver informação sobre a bioequivalência.
t Comprimidos de acetazolamida podem ser triturados. São relatadas preparações extemporâneas a partir dos comprimidos, tanto sob forma de suspensões como de soluções (que devem ser tamponadas a pH 4,0). Trituram-se os comprimidos, suspendendo o pó resultante em xarope de gosto forte (cereja, framboesa, chocolate, etc). Pode suspender-se até 500 mg em 5 mL de xarope, mas uma suspensão que contenha 250 mg por 5 mL tem melhor palatabilidade. Esta suspensão é estável por uma semana. a refrigeração pode melhorar o sabor, mas não aumenta a estabilidade. Os elixires ou outros veículos que contenham álcool ou glicerina não proporcionam suspensão de sabor palatável. O armazenamento deve ser feito entre 15 e 30 ºC e ao abrigo da luz direta.
Atenção: usar com cautela na insuficiência renal por risco de acidose.
Vanessa Rocha Machado
Na Rename 2010: item 5.5.1
t Comprimido 200 mg.
t Pó para solução injetável 250 mg.
t Infecção por vírus Herpes simplex (tratamento e profilaxia).
t Infecção por vírus Varicella-zoster em indivíduos imunocomprometidos (tratamento e profilaxia).
t Tratamento de Herpes zoster.
t Hipersensibilidade ao aciclovir ou a qualquer componente da formulação.
t Usar com cuidado nos casos de:
– uso intravenoso (deve ser reservado para tratamento de encefalite, pneumonite e infecções graves e disseminadas, especialmente em neonatos).
– pacientes obesos (calcular a dose com base no peso ideal para altura para evitar dose excessiva).
– idosos
– insuficiência renal (requer ajuste de dose).
– outros agentes nefrotóxicos (evitar uso concomitante).
t Categoria de risco na gravidez (FDA): B.
t 20 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante 14 dias
t 20 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante 21 dias
t 10 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante 7 dias. Dose máxima de 20 mg/kg, por via intravenosa, a cada 8 horas.
t 20 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante pelo menos 10 dias, possivelmente por 14 a 21 dias
t 20 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante 7 dias. Dose máxima de 20 mg/kg, por via intravenosa, a cada 8 horas.
t 200 mg, por via oral, a cada 4 horas, 5 vezes ao dia, durante 5 dias ou período superior se aparecerem novas lesões durante o tratamento ou se não obter cura completa
t 400 mg, por via oral, a cada 4 horas, 5 vezes ao dia, durante 5 dias ou período superior se aparecerem novas lesões durante o tratamento ou se não obter cura completa
t 5 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, por 7 dias. Dose máxima de 20 mg/kg, por via intravenosa, a cada 8 horas.
t 200 mg, por via oral, a cada 4 horas, 5 vezes ao dia, ou 400 mg, por via oral, a cada 8 horas, durante 7 a 10 dias (primeiro episódio) ou 5 dias (recidiva) ou período superior se aparecerem novas lesões durante o tratamento ou se não obter cura completa
t 400 mg, por via oral, a cada 4 horas, 5 vezes ao dia, durante 7 a 10 dias (primeiro episódio) ou 400 mg, por via oral, de 8 em 8 horas, durante 5 a 10 dias (recidiva)
t 5 a 10 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante 2 a 7 dias ou até melhora clínica, seguido de terapia oral para completar pelo menos 10 dias de tratamento em primeiro episódio grave
t 400 mg, por via oral, a cada 12 horas, ou 200 mg, por via oral, a cada 8 horas, aumentado para 400 mg, por via oral, a cada 8 horas se houver recidiva durante o tratamento, interrompendo o tratamento a cada 6 a 12 meses para reavaliar a frequência de recidiva. Considerar retratamento após 2 a 3 recidivas.
t De 200 a 400 mg, por via oral, a cada 6 horas, ou 800 mg, por via oral, a cada 12 horas, interrompendo o tratamento a cada 6 a 12 meses para reavaliar a frequência de recidiva. Considerar retratamento após 2 a 3 recidivas.
t 10 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante pelo menos 10 dias, possivelmente por 14 a 21 dias
t 800 mg, por via oral, a cada 4 horas, 5 vezes ao dia, durante 7 a 10 dias.
t 5 mg/kg, por via intravenosa, em infusão contínua por 1 hora, a cada 8 horas, durante 5 dias.
t 10 mg/kg, por via intravenosa, em infusão contínua durante 1 hora, a cada 8 horas, durante 7 dias. Dose máxima 20 mg/kg, por via intravenosa, a cada 8 horas.
t Início de efeito: 1,5 a 2,5 horas (oral).
t Meia-vida: 2 a 19,5 horas (dependente da função renal).
t Excreção: renal (62% a 91%).
t Náusea e vômito (7%), diarreia (8% a 9%), epigastralgia.
t Erupção cutânea (4% a 5%).
t Cefaleia (13%).
t Tromboflebite (14%), necrólise ao extravasamento.
t Agitação, confusão mental, letargia, tremor, alucinação (1%).
t Neutropenia, trombocitopenia, anemia, leucocitose e neutrofilia (menor que 1%).
t Elevação transitória da creatinina sérica (4% a 5%), insuficiência renal (5% a 10%), hematúria (1%) quando usado em altas doses.
t Ácido valproico, fenitoína, fosfenitoína podem ter seu efeito reduzido por aciclovir. Considerar terapia com outro antiviral.
t Zidovudina: aumento de letargia e fadiga. Alertar paciente quanto ao risco de fadiga excessiva pelo uso concomitante do aciclovir com zidovudina.
t Orientar para a necessidade de aumento da ingestão hídrica durante o tratamento.
t Evitar relações sexuais enquanto estiver com herpes genital.
t Usar preservativo masculino para evitar doença sexualmente transmissível.
t Esclarecer o paciente, que apesar da frequência a cada 4 horas, a dose oral da madrugada não deve ser feita e que não haverá prejuízo no efeito terapêutico.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Armazenar à temperatura ambiente, entre 15 a 25 ºC. Manter ao abrigo do ar, calor, luz direta e umidade.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Após reconstituição com água estéril para injeção, a diluição em glicose a 5% ou solução de cloreto de sódio a 0,9% é estável por 24 horas.
t Incompatibilidades: água bacteriostática para injeção, aztreonam, dobutamina, dopamina, foscarnete, idarrubicina, levofloxacino, meropeném, ondansetrona, piperacilina sódica/tazobactam, sargramostim, vinorelbina.
Atenção: devido ao risco de precipitação do aciclovir nos túbulos renais, recomendar o aumento de ingestão hídrica durante o tratamento. No caso de administração intravenosa, evitar infusão por tempo inferior a uma hora, para prevenir danos renais 3.
Aline Lins Camargo
Na Rename 2010: itens 2.1, 2.3, 3.1, 14.3 e 15.3
t Comprimidos 100 mg e 500 mg.
t Dor leve a moderada.
t Enxaqueca e outros tipos de cefaleia.
t Febre.
t Processos inflamatórios.
t Profilaxia e tratamento de doenças tromboembólicas.
t Prevenção de trombose em cirurgias cardíacas.
t Prevenção secundária de evento vascular encefálico transitório.
t Prevenção secundária de cardiopatia isquêmica.
t Prevenção secundária de enfarte agudo do miocárdio.
t Tratamento adjuvante em angina estável e instável.
t Suspeita de enfarte agudo do miocárdio.
– Tratamento de enfarte agudo do miocárdio em associação com trombolítico.
– Terapia após angioplastia com e sem implantação de stent.
t Hipersensibilidade ao ácido acetilsalicílico ou a anti-inflamatórios não-esteroides.
t Crianças e adolescentes com menos de 16 anos (risco de síndrome de Reye).
t Tratamento de gota.
t Ulceração péptica prévia ou ativa.
t Hemofilia e outras doenças hemorrágicas.
t Usar com cuidado nos casos de:
– asma, pólipos nasais e outras doenças alérgicas, hipertensão não controlada, desidratação, deficiência de glicose-6-fosfato desidrogenase e consumo exagerado de álcool.
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– cirurgias (suspender o uso uma a duas semanas antes do procedimento para reduzir o risco de sangramento excessivo.
– uso de bebida alcoólica (risco de sangramento gastrintestinal).
– ocorrência de zumbidos ou perda de acuidade auditiva (suspender o uso).
– idosos (mais Susceptíveis aos efeitos tóxicos dos salicilatos).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): D (ver Apêndice a).
t Dose de 500 a 1.000 mg, por via oral, a cada 4 a 6 horas. Dose máxima diária: 4 g.
t Dose de 1.000 a 1.500 mg, por via oral, a cada 6 horas. Dose máxima diária: 4 a 6 g.
t Dose de 100 a 200 mg, por via oral, a cada 24 horas. Prevenção de formação de trombo após cirurgia cardíaca t Dose de 100 a 300 mg, por via oral, a cada 24 horas.
t Dose de 100 a 300 mg, por via oral, a cada 24 horas, com início nas primeiras horas após o episódio e mantida durante tempo indeterminado.
t Dose de 100 a 300 mg, por via oral, a cada 24 horas, durante tempo indeterminado.
t Dose de 100 a 300 mg, por via oral, a cada 24 horas, durante tempo indeterminado.
t Dose única de 150 a 300 mg, por via oral, administrada precocemente após o diagnóstico. Dose de manutenção de 100 mg, por via oral, a cada 24 horas.
t Dose de 300 mg, por via oral, pelo menos 2 horas antes da inserção, e após 150 a 300 mg, por via oral, a cada 24 horas.
t Início de efeito: 15 a 30 minutos (analgésico, antipirético e anti-inflamatório), 1 a 7,5 minutos (antiplaquetário).
t Pico de efeito: 1 a 2 horas.
t Duração de efeito: 4 a 6 horas.
t Meia-vida de eliminação: ácido acetilsalicílico: 15 a 20 minutos; salicilatos (dose dependente): 2,5 a 3 horas (500 mg); 5 a 6 horas (acima de 1.000 mg).
t Metabolismo: hepático.
t Excreção: renal.
t Geralmente são leves e infrequentes em doses baixas.
t Indigestão, náuseas, vômitos.
t Úlceras gastrintestinais (6% a 31%)
t Sangramentos.
t Perda auditiva e zumbido no ouvido (uso de doses elevadas e/ou crônico).
t Broncoespasmo, angioedema, reações de hipersensibilidade, síndrome de Reye (crianças).
t Ácido valproico: pode aumentar as concentrações séricas de ácido valproico livre. Com doses repetidas, monitorar concentração plasmática de ácido valproico, sintomas de toxicidade do ácido valproico e enzimas hepáticas.
t Anticoagulantes (anisindiona, femprocumona, heparina, heparinas de baixo peso molecular, varfarina): aumento da atividade anticoagulante e risco de sangramento. a administração combinada deve ser evitada sempre que possível. Quando administrados juntos monitorar paciente cuidadosamente. Ajuste da dose do anticoagulante pode ser necessário.
t Celecoxibe: aumento do risco de sangramento gastrintestinal. Observar pacientes para sinais de sangramento gastrintestinal, quando doses elevadas de ácido acetilsalicílico são utilizadas.
t Cetorolaco: pode resultar em aumento dos Efeitos adversos gastrintestinais (úlcera péptica, sangramento e perfuração gastrintestinal). Uso concomitante é contraindicado.
t Corticoides sistêmicos (por exemplo, prednisona, prednisolona, dexametasona, metilprednisolona): aumento do risco de ulcerações gastrintestinais e concentrações séricas sub-terapêuticas do ácido acetilsalicílico. Monitorar pacientes para Efeitos adversos gastrintestinais excessivos e para diminuição da efetividade do ácido acetilsalicílico.
t Diltiazem, verapamil: pode resultar em prolongamento do tempo de sangramento. Monitorar pacientes para sinais e sintomas de sangramento, especialmente no trato gastrintestinal.
t Estreptoquinase: aumento do risco de complicações hemorrágicas. O uso concomitante com ácido acetilsalicílico deve ser evitado em paciente com acidente vascular isquêmico até trombólise ter sido completada.
t Furosemida: pode diminuir o efeito diurético da furosemida. Em adultos, evitar doses superiores a 650 mg de ácido acetilsalicílico quando administrado com furosemida.
t Ginkgo: aumento do risco de sangramento. Monitorar sinais de sangramento.
t Ibuprofeno: possível redução do efeito antiplaquetário do ácido acetilsalicílico. Uso concomitante deve ser evitado (aumento dos efeitos adversos), mas caso este seja necessário, usar o ácido acetilsalicílico pelo menos 30 minutos antes ou 8 horas após a administração de ibuprofeno.
t Inibidores da enzima conversora de angiotensina (ECA), como captopril e enalapril: o uso concomitante com ácido acetilsalicílico pode diminuir a efetividade do inibidor da ECA. Avaliar a relação risco-benefício do uso combinado.
t Inibidores da recaptação de serotonina (citaprolam, fluoxetina, paroxetina, sertralina): aumento do risco de sangramento. Monitorar pacientes em relação a sinais de sangramento.
t Metotrexato: aumento do risco de toxicidade do metotrexato (leucopenia, trombocitopenia, anemia, ulcerações nas mucosas). Monitorar sinais de toxicidade, especialmente mielossupressão e toxicidade gastrintestinal.
t Nitroglicerina: pode aumentar as concentrações de nitroglicerina e depressão aditiva da função plaquetária. Esta interação pode ser usada terapeuticamente em pacientes com enfarte agudo do miocárdio. Em outros pacientes, monitorar resposta exacerbada à nitroglicerina, evidenciada por cefaleia e síncope.
t Sulfonilureias (exemplo, clorpropamida, tolbutamida): aumento do risco de hipoglicemia. Em pacientes recebendo doses elevadas de ácido acetilsalicílico, monitorar glicose sanguínea e sinais clínicos de hipoglicemia. Ajuste da dose da sulfonilureia pode ser necessário.
t Ticlopidina: aumento do risco de sangramento. Uso concomitante deve ser acompanhado de monitoria cuidadoso da função hematológica.
t Vacina contra varicela: pode aumentar o risco de desenvolvimento de síndrome de Reye. Evitar uso de salicilatos por 6 semanas após administração da vacina.
t Venlafaxina: aumento do risco de sangramento. Monitorar pacientes para sinais de aumento de sangramento quando venlafaxina é iniciada ou descontinuada.
t Orientar para ingerir os comprimidos com 250 mL de água e não deitar dentro de 15 a 30 minutos após a administração.
t Orientar para ingerir o medicamento com alimentos ou grande quantidade de água ou leite para evitar desconforto gastrintestinal.
t Reforçar a importância de evitar o uso de bebidas alcoólicas.
t Alertar que é importante notificar imediatamente ao médico se apresentar os seguintes efeitos adversos: dor de estômago forte, vômito com sangue ou vômito com aparência de grumos de café, sangue nas fezes ou urina, exantema ou bolhas na pele com prurido intenso, inchaço da face ou pálpebras, respiração difícil ou ruidosa, muita tontura ou sonolência, zumbido no ouvido.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC, em embalagens bem fechadas e protegidas de calor excessivo, umidade e luz direta.
t Após exposição à água ou umidade, o fármaco sofre hidrólise, resultando em salicilato e acetato, que possuem odor semelhante a vinagre. Não usar se odor forte estiver presente.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: itens 3.3, 10.1, 12 e 15.1
t Comprimido 5 mg
t Solução oral 0,2 mg/mL
t Anemia megaloblástica por deficiência de ácido fólico.
t Deficiência de folato devido a má-nutrição, gravidez, uso de antiepilépticos ou má-absorção.
t Prevenção de defeito do tubo neural na gravidez.
t Prevenção de Efeitos adversos induzidos pelo metotrexato em doença reumática.
t Anemia megaloblástica não diagnosticada ou outro estado de deficiência de vitamina B12, a não ser que seja associado a vitamina B12, para evitar precipitação de neuropatia (degeneração subaguda da coluna vertebral).
t Hipersensibilidade ao ácido fólico.
t Doença maligna dependente de folato.
t Usar com cuidado nos casos de:
– mulheres que recebem terapia anticonvulsivante (ácido fólico pode reduzir ação do anticonvulsivante).
– anemia perniciosa e deficiências de vitamina B12 (podem ser mascaradas com doses acima de 0,1 mg/dia), especialmente em idosos.
t Categoria de risco na gravidez (FDA): a.
t até 1 mg/dia, via oral, a cada 24 horas, independente da idade, até resolução dos sintomas. Após normalização dos índices sanguíneos, dose de manutenção conforme faixa etária:
– Menores de 1 mês: 0,1 mg, por via oral, a cada 24 horas;
– De 1 mês a 4 anos: até 0,3 mg, por via oral, a cada 24 horas;
– Maiores de 4 anos: 0,4 mg, por via oral, a cada 24 horas.
t 1 mg, por via oral, a cada 24 horas, ou 5 mg, por via oral, uma ou duas vezes por semana.
t até 1 mg, por via oral, a cada 24 horas, até resolução dos sintomas. Após normalização dos índices sanguíneos, dose de manutenção de 0,4 mg (0,8 mg para mulheres grávidas e lactantes), por via oral, a cada 24 horas.
t da primeira ocorrência: 0,4 mg, por via oral, a cada 24 horas, de 4 semanas antes da concepção até as primeiras 12 semanas de gravidez.
t da recorrência: 4 a 5 mg, por via oral, a cada 24 horas, de 4 semanas antes da concepção até as primeiras 12 semanas de gravidez.
t 5 mg, por via oral, uma vez por semana
t Metabolito ativo: 5-metiltetraidrofolato, que é extensamente ligado a proteínas plasmáticas
t Eliminação renal quase completamente sob a forma de metabólitos.
t Removido por hemodiálise, mas não significativamente nos pacientes bem nutridos.
t Reação alérgica, incluindo broncoespasmo, eritema, febre, mal-estar geral, exantema ou prurido (incidência rara)
t Náusea, distensão abdominal, desconforto, flatulência, sabor amargo na boca (doses altas)
t Distúrbios do sono, confusão, irritabilidade, agitação, dificuldade de concentração, depressão (doses altas)
t Fenitoína pode ter suas concentrações reduzidas, com possível redução dos efeitos. Monitorar concentração de fenitoína e ajustar dose, se necessário.
t Alimentos ricos em ácido fólico: vegetais verdes, cereais, frutas e fígado.
t Alertar que o aquecimento destrói o ácido fólico dos alimentos (50% a 90%).
t Orientar para notificar em caso de aparecimento de manifestações neurológicas, gastrintestinais e alérgicas.
t Alertar para usar a dose esquecida o mais breve possível. Se estiver perto da hora regular, ingerir a dose normal e ignorar a dose esquecida. Alertar para não usar duas doses ao mesmo tempo.
t Conservar à temperatura ambiente, entre 15 a 30 ºC. Não congelar.
t Manter ao abrigo de ar e luz.
Cláudia Du Bocage Santos Pinto
Marcela de Andrade Conti
Na Rename 2010: item 20.4
t Pomada 5% (FN).
t Hiperceratoses, como ictioses, ceratose plantar e das mãos.
t Psoríase.
t Dermatite seborreica.
t Verrugas e calosidades (empregam-se formulações contendo 5% a 40% de ácido salicílico).
t Crianças com menos de 2 anos de idade.
t Aplicação sobre feridas ou inflamações cutâneas.
t Hipersensibilidade a salicilatos.
t Usar com cuidado nos casos de:
– crianças e indivíduos com insuficiência hepática ou renal (o risco de salicilismo dever ser monitorado).
– distúrbios circulatórios, diabetes e neuropatia periférica.
t Evitar contato com o rosto, olhos, boca, mucosas e região anogenital.
t Evitar uso prolongado e aplicação em áreas extensas, para minimizar absorção e efeitos sistêmicos.
t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
t aplicar, por via tópica, a cada 24 horas, sobre a área limpa e seca, considerando-se o risco aumentado de toxicidade devido à maior proporção da área tratada em relação à superfície corporal total.
t aplicar, por via tópica, a cada 24 horas, sobre a área limpa e seca, preferentemente à noite, mantendo-a sob oclusão
t aplicar, por via tópica, a cada 12 a 24 horas, durante até 12 semanas.
t Absorção sistêmica: aproximadamente 25%.
t Pode atravessar a barreira placentária.
t Eliminação urinária.
t Pico de concentrarão: 5 horas (com oclusão)
t Irritação local, dermatite, ressecamento da pele, queimadura, ardor, prurido.
t Salicilismo, geralmente após aplicação excessiva, particularmente em crianças.
t Antifúngicos: têm sua absorção facilitada pela redução da camada de queratina (efeito sinérgico).
t Vacina contra varicela: evitar o uso da pomada durante as 6 semanas após a administração da vacina, devido ao risco aumentado de desenvolvimento da síndrome de Reye.
t Orientar para evitar contato com o rosto, olhos, boca, mucosas, região anal ou genital.
t Orientar para evitar o uso de outros produtos tópicos ao mesmo tempo e no mesmo local.
t Lavar as mãos antes e após a aplicação do medicamento.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC, em recipientes bem fechados e protegidos da luz.
Letícia Figueira Freitas
Gabriela Costa Chaves
Na Rename 2010: item 9
t Ampolas de 5 mL e 10 mL.
t Frascos de 100 mL e 500 mL.
t Veículo para preparação de medicamentos para administração parenteral, para ressuspensão, dissolução ou diluição em formulações estéreis.
t Deve ser estéril e livre de pirogênio.
t Quando usada para preparação de soluções parenterais, deve ser esterilizada antes, ou a solução final deve ser esterilizada após o preparo.
t Armazenar sob temperatura ambiente, mantendo o sistema hermeticamente fechado para impedir contaminação.
César Augusto Braum
Na Rename 2010: item 5.6.1
t Comprimido mastigável 400 mg.
t Suspensão oral 40 mg/mL.
t Infestações helmínticas pelos nematódios Ascaris lumbricoides, Ancylostoma duodenale e Necator americanus, Larva migrans visceral, Larva migrans cutânea, Trichuris trichiura, Strongyloides stercoralis, Enterobius vermicularis, Trichinella spiralis, Wuchereria bancrofti.
t Infestações helmínticas pelos cestódios Echinococcus granulosus (cisto hidático), Taenia saginata, Taenia solium (neurocisticercose).
t Giardia intestinalis (Giardia lamblia ou Giardia duodenalis).
t Hipersensibilidade a algum dos componentes da formulação.
t Usar com cuidado nos casos de:
– tratamento para neurocisticercose (tratar previamente com corticosteroides, por vários dias, para minimizar episódios de hipertensão cerebral e minimizar reações alérgicas; pode ser considerado o uso de anticonvulsivantes).
– cisticercose (aumento do risco de lesões na retina).
– tratamento prolongado (monitorar função hepática e toxicidade medular).
t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
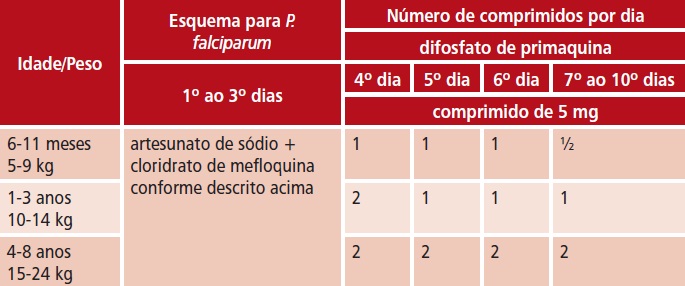
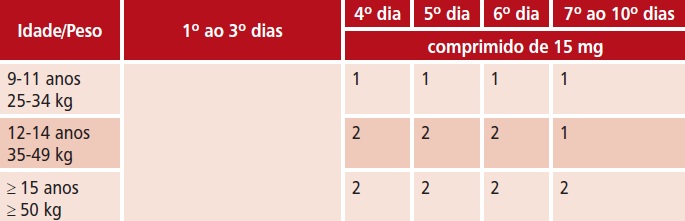
Ascaris lumbricoides, Ancylostoma duodenale, Necator americanus e Enterobius vermicularis
– 200 mg, por via oral, em dose única; o tratamento pode ser repetido após 3 semanas, principalmente em enterobíase.
– 200 mg, por via oral, a cada 24 horas, durante 3 dias.
t 200 mg, por via oral, a cada 24 horas, durante 3 dias.
t 200 mg, por via oral, a cada 24 horas, durante 3 dias.
t Ascaris lumbricoides, Ancylostoma duodenale, Necator americanus e Enterobius vermicularis
t 400 mg, por via oral, em dose única; o tratamento pode ser repetido após 2 a 3 semanas, principalmente em enterobíase.
t 400 mg, por via oral, a cada 24 horas, durante 3 dias.
t 400 mg, por via oral, a cada 12 ou 24 horas, durante 2 a 3 dias.
Echinococcus granulosus (Equinococose cística)
t Menos de 60 kg: 7,5 mg/kg, por via oral, a cada 12 horas, com as refeições, durante 28 dias. Interromper o tratamento por 14 dias e repetir o esquema posológico por até 3 vezes. Dose máxima diária: 800 mg.
t Mais de 60 kg: 400 mg, por via oral, a cada 12 horas, com as refeições, durante 28 dias. Interromper o tratamento por 14 dias e repetir o esquema posológico por até 3 vezes.
Echinococcus multilocularis (Equinococose alveolar)
t Mesmo esquema posológico anterior, mas os ciclos podem continuar por meses ou anos.
t Menos de 60 kg: dose 7,5 mg/kg/dia, por via oral, a cada 12 horas, por 8 a 30 dias; dose máxima diária: 800 mg.
t Mais de 60 kg: dose 400 mg, por via oral, a cada 12 horas, durante 8 a 30 dias.
t O curso de terapia pode ser repetido, se necessário.
t 400 mg, por via oral, a cada 24 horas, durante 3 dias.
t 400 mg, por via oral, a cada 24 horas, durante 5 dias.
t 400 mg, por via oral, a cada 24 horas, juntamente com citrato de dietilcarbamazina, 6 mg/kg, durante 7 dias.
t 400 mg, por via oral, a cada 12 horas, durante 8 a 14 dias.
t 400 mg, por via oral, a cada 12 horas, durante 5 dias.
t 400 mg, por via oral, a cada 12 horas, durante 3 dias.
t Absorção: menos de 5%. Absorção aumenta com alimentação rica em gorduras.
t Pico de concentração sérica: 2 a 4 horas.
t Meia-vida: 8 a 15 horas
t Metabolismo: hepático (metabólito ativo na forma de sulfóxido)
t Excreção: extensivamente pela bile. a excreção renal é baixa.
t Dor epigástrica, náusea, vômitos, anorexia, obstipação, xerostomia, diarreia.
t Cefaleia, tontura (leves e transitórios em terapia de curto prazo), aumento da pressão intracraniana.
t Erupções cutâneas, alopecia, prurido, urticária, edema, Síndrome StevensJohnson.
t Aumento dos níveis séricos das transaminases, icterícia (rara), colestase.
t Fadiga.
t Febre.
t Leucopenia, trombocitopenia, eosinofilia (em tratamentos prolongados), anemia aplástica, neutropenia, agranulocitose, pancitopenia.
t Orientar para ingerir durante as refeições para aumentar a absorção do fármaco e para evitar suco de pomelo, pois o uso concomitante pode aumentar o risco de Efeitos adversos do albendazol
t Alertar para não ingerir as duas doses ao mesmo tempo
t Armazenar os comprimidos em local seco, ao abrigo de luz e calor.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 15.5
t Solução injetável 20%
t Hipoalbuminemia (grandes queimados, sepse, cirrose hepática, quando houver edemas refratários aos diuréticos e que coloquem em risco a vida).
t Hipoproteinemia aguda com edema
t Hipersensibilidade à albumina.
t Pacientes em risco de sobrecarga circulatória aguda (insuficiência cardíaca, edema pulmonar, anemia grave).
t Usar com cuidado nos casos de:
– doença cardíaca ou circulatória (administrar lentamente para evitar rápida elevação na pressão arterial e insuficiência cardíaca; monitorizar função cardiovascular e respiratória).
– administração da solução concentrada (corrigir desidratação).
– insuficiência renal crônica.
– anemia crônica.
– hipovolemia, queimadura e hipoalbuminemia (aumento de 5% no risco de mortalidade).
t 0,5 a 1 g/kg, por via intravenosa, à velocidade de infusão de 0,05 a 0,1 g por minuto, a cada 1 ou 2 dias ou conforme cálculo de reposição da perda de proteína. Dose máxima diária: 6 g/kg.
– 4 a 6 g, por via intravenosa, à velocidade de infusão de até 0,25 mg/minuto com albumina diluída a 5% e 0,2 a 0,4 mg/minuto de albumina a 20%.
t Reações de hipersensibilidade (incluindo anafilaxia).
t Insuficiência cardíaca
t Alumínio tem sido detectado como contaminante em soluções de albumina humana. Seu acúmulo pode produzir efeitos tóxicos (encefalopatia, osteodistrofia com osteomalácia associada, além de fraturas) em pacientes com insuficiência renal e neonatos prematuros.
t Inibidores da enzima conversora de angiotensina (IECA): risco de reações atípicas (rubor, hipotensão). IECA devem ser suspensos pelo menos 24 horas antes da substituição plasmática, quando são administrados grandes volumes de albumina humana.
t Alertar para o possível desenvolvimento de reação alérgica à albumina.
t Armazenar sob refrigeração, entre 2 a 8 ºC. Não usar soluções que foram congeladas, ou se estiverem turvas ou com sedimentos.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Soluções de albumina a 20% apenas devem ser diluídas em soluções de glicose a 5% ou cloreto de sódio a 0,9%.
t Diluição da albumina 20% com água esterilizada para injeção produz solução hipotônica que, se administrada, pode resultar em hemólise potencialmente fatal ou insuficiência renal aguda, ou ainda hiponatremia ou edema cerebral. Pode não ser adequada para administração rápida em grandes volumes (troca de plasma ou plasmaferese)
t Pode ser administrada juntamente ou combinada com sangue total, plasma ou glicose, lactato de sódio ou cloreto de sódio.
t Não administrar soluções se decorridas mais de 4 horas desde a abertura do frasco.
Atenção: devido ao risco associado a administração de soluções hipotônicas, as soluções de albumina humana a 20% devem conter 130-160 mEq de sódio por litro. O rápido aumento na pressão arterial que acompanha a administração de albumina humana, após lesões ou cirurgia, pode revelar pontos de sangramento não aparentes com a pressão mais baixa; observar cuidadosamente o paciente para prevenir hemorragia e choque.
Fernando Genovez de Avelar
Marcela de Andrade Conti
Na Rename 2010: item 20.4
t Pomada 1% (FN).
t Psoríase em placa crônica, em monoterapia ou associada à radiação ultravioleta.
t Fase de liquenificação de eczema atópico crônico.
t Psoríase purulenta, aguda ou inflamada.
t Eczema exsudativo.
t Foliculite ou acne.
t Hipersensibilidade aos componentes da formulação.
t Fotossensibilidade.
t Usar com cuidado nos casos de:
– tratamento que inclua radiação ultravioleta (essa deve ocorrer de 2 a 72 horas após a aplicação da pomada de alcatrão mineral, que deve ser previamente removida da pele).
– lactação.
t Evitar exposição do paciente à luz solar ou à radiação ultravioleta para reduzir as reações de fotossensibilidade.
t Não aplicar em áreas inflamadas ou lesionadas ou nas regiões genital ou retal.
t Evitar contato com os olhos.
t Categoria de risco na gravidez (FDA): C.
t Aplicar sobre a pele limpa, de uma a três vezes ao dia.
t Excreção: urinária. Detecta-se acridina na urina após o uso tópico.
t Mais comuns: foliculite estéril, alteração da cor de pele e pelos, irritação e alergia de contato.
t Pouco frequentes: reações de fotossensibilidade, dermatite de contato, fototoxicidade, lesões herpéticas e verrugas plantares quando associado à radiação ultravioleta.
t Raramente: Hipersensibilidade, psoríase pustular generalizada.
t Orientar para utilizar filtro solar para proteger a área tratada da luz solar.
t Orientar para evitar contato com os olhos e outras mucosas.
t Orientar para evitar contato com áreas da pele que contenham cortes ou arranhões.
t Alertar que a pomada possui odor desagradável e pode manchar a roupa.
t Conservar sob temperaturas entre 15 e 30 ºC, em recipientes bem fechados, longe do calor, umidade e luz direta.
Letícia Figueira Freitas
Gabriela Costa Chaves
Na Rename 2010: item 5.7
t Solução 70% (p/p) (FN).
t Gel 70% (FN).
t Antissepsia da pele antes de injeção, punção venosa ou procedimento cirúrgico.
t Desinfecção e limpeza de mãos, superfícies e equipamentos.
t Usar com cuidado nos casos de:
– lesões e ferimentos abertos.
– aplicação em conjunto com procedimentos diatérmicos, que envolvem geração de calor no tecido por meio de corrente elétrica (podem ocorrer queimaduras graves na pele com este ou outros desinfetantes alcoólicos).
t Líquido inflamável.
t Aplica-se solução não diluída, sob fricção, para assepsia de pele.
t Aplica-se álcool como desinfetante sobre superfícies, equipamentos fixos e artigos que não toleram outros tipos de desinfecção ou esterilização.
t Ressecamento, descamação e irritação cutânea, somente em aplicações frequentes.
t Soluções com iodopovidona e clorexidina proporcionam efeito germicida aumentado.
t Preparo deve ser ajustado com alcoômetro, para garantir a concentração desejada.
Atenção: não é eficaz para a esterilização de instrumentos cirúrgicos ou odontológicos devido à baixa atividade contra os esporos de bactérias e à incapacidade de penetrar em materiais ricos em proteínas.
Não diluir ou misturar a solução ou gel com outros produtos, sob risco de perda da efetividade, exceto quando explicitamente determinado pelo fabricante.
Pode ocorrer necrólise hemorrágica localizada se usado com clorexidina para limpeza de coto umbilical.
Maria Isabel Fischer
Na Rename 2010: item 19
t Comprimido de 70 mg.
t Tratamento da osteoporose.
t Anormalidades esofágicas e outros fatores que retardem o esvaziamento esofágico.
t Hipersensibilidade ao alendronato ou a qualquer componente da fórmula.
t Hipocalcemia.
t Incapacidade de ficar em pé ou sentado de forma ereta por 30 minutos.
t Usar com cuidado nos casos de:
– alterações gastrintestinais.
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C.
t 70 mg, por via oral, a cada 7 dias.
t Absorção: pouco absorvido via oral. a presença de cálcio e outros cátions polivalentes reduz significativamente a absorção de alendronato.
t Biodisponiblididade: 0,7% (mulheres) 0,59% (homens). Alimento reduz a biodisponibilidade em 40%.
t Pico de resposta: 6 a 12 meses, em tratamento de mulheres pós-menopausa
t Tempo para pico plasmático: 1 hora
t Metabolismo: o alendronato não é metabolizado
t Excreção: renal 50%. a depuração renal é reduzida significativamente em pacientes com disfunção renal
t Meia-vida de eliminação: 1,9 horas (ossos: até 10 anos)
t Incidência acima de 10%:
– hipocalcemia (transitória, leve); hipofosfatemia (transitória, leve)
t Incidência entre 1% e 10%:
– cefaleia.
– dor abdominal, refluxo ácido, refluxo gastroesofágico; dispepsia; náusea, flatulência; diarreia, obstipação, úlcera esofágica; úlcera gástrica; distensão abdominal; gastrite; vômito; disfagia.
– dor musculoesquelética; câimbra muscular.
t Incidência abaixo de 1%
– Reações de hipersensibilidade, incluindo angioedema, eritema, prurido, exantema.
– Erosão esofágica com sangramento, úlcera, duodenal, perfuração esofágica, estreitamento esofágico, esofagite,
– Febre,
– Síndrome tipo influenza,
– Hipocalcemia (sintomática),
– Mialgia,
– Osteonecrose de mandíbula,
– Edema periférico,
– Dor óssea, muscular e articular.
t Os comprimidos devem ser engolidos inteiros com um copo cheio de água, não menos do que 60 mL, com o estômago vazio pelo menos 30 minutos antes do café da manhã ou de ingerir outro medicamento; o paciente deve ficar sentado de forma ereta ou ficar em pé por pelo menos 30 minutos após ingerir o medicamento. Evitar o uso de água mineral, pois pode conter grande quantidade de cálcio.
t Se ocorrerem reações esofágicas graves o tratamento deve ser interrompido; procurar o médico se houver sintomas de irritação esofágica nova ou piora na azia, dor ao engolir ou dor retroesternal.
t Consultar o dentista antes de iniciar tratamento com bifosfonatos.
t Conservar sob temperatura ambiente (15 a 30oC) em frasco hermeticamente fechado.
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 17.2
t Líquido 80 mg/Ml
t Tratamento e profilaxia da síndrome do desconforto respiratório em neonatos prematuros.
t Hipersensibilidade ao alfaporactanto.
t Usar com cuidado nos casos de:
– pressão arterial ou transcutânea de CO2 abaixo de 30 torr (reduzir imediatamente a taxa de ventilação).
– criança ficar rosada ou a saturação de O2 ultrapassar 95% (reduzir gradualmente a Fração de Oxigênio Inspirado – FiO2.
– crianças nascidas após três semanas da ruptura das membranas amnióticas (o uso de alfaporactanto pode determinar hipoplasia pulmonar).
– crianças nascidas com má-formação congênitas importantes, ou com hemorragia intraventricular de graus III ou IV.
– diminuição significativa da expansão torácica após o uso de alfaporactanto (reduzir imediatamente o pico de pressão inspiratória do ventilador, mesmo previamente aos resultados da gasometria).
t a ventilação pode ser prejudicada pelo desenvolvimento de muco tamponado intratraqueal após a administração do alfaporactanto; sucção do muco previamente a administração pode minimizar o problema.
t Dose inicial 2,5 mL/kg, via intratraqueal. Até 2 doses adicionais de 1,25 mL/kg a cada 12 horas. Dose máxima total: 5 mL/kg.
t Nota: seguir técnica de administração adotada pelo serviço médico.
t a resposta inicial ocorre entre 3 e 6 horas.
t Observou-se uma melhora de 98% no PaO2/FiO2 com 6 horas depois da administração de doses de 200 mg/kg.
t Melhora significativa (45%) da estabilidade respiratória tem sido alcançada depois de 3 horas de instilação de alfaporactanto (200 mg/kg). a taxa de oxigenação arterial/alveolar teve também uma melhora significativa neste tempo.
t Após administração de dose única 200 mg/kg taxas adequadas de PaO2/FiO2 persistem por mais de 48 horas.
t Risco de hemorragia pulmonar, pneumotórax e hiperoxemia, especialmente em crianças prematuras.
t Bradicardia transitória.
t Obstrução mucosa do tubo endotraqueal.
t Diminuição da atividade elétrica cerebral.
t Conservar à temperatura entre 2 e 8 oC e protegido da luz.
t a solução deve ser administrada à temperatura ambiente, permanece estável por 24 horas. a solução poderá retornar ao refrigerador uma única vez.
t Se necessário o pH da solução de alfaporactanto deve ser ajustado com bicarbonato de sódio até a faixa de 5,5 a 6,5.
t a cor adequada da solução é creme claro. Inspeção visual antes do uso é necessária para detectar alguma descoloração do produto.
t Se a solução mostrar-se ter separado, o frasco deverá ser girado suavemente (SEM AGITAÇÃO) para re-suspender as partículas precipitadas durante o armazenamento.
Atenção: é imprescindível a monitoria do aporte ventilatório e o controle da função respiratória.
Aline Lins Camargo
Na Rename 2010: item 3.4
t Comprimidos de 100 mg e 300 mg
t Profilaxia da gota.
t Profilaxia de hiperuricemia associada à neoplasia.
t Gota aguda.
t Hipersensibilidade ao alopurinol ou a qualquer componente da formulação.
t Usar com cuidado nos casos de:
– hiperuricemia assintomática (uso não indicado).
– ocorrência de exantema (interromper tratamento).
– lactação.
– insuficiência renal (ver Apêndice D) e hepática (ver Apêndice C).
t Assegurar ingestão adequada de líquidos, de 2 a 3 litros/dia.
t Categoria de risco na gravidez (FDA): C.
t 10 a 20 mg/kg/dia, dividido a cada 8 a 12 horas. Dose máxima diária: 400 mg.
t Dose inicial 2 a 3 semanas após controle da crise (ou ataque) aguda de 100 mg, por via oral, a cada 24 horas, preferentemente após alimentos, ajustada de acordo com a concentração sérica ou urinária de ácido úrico, durante tempo indeterminado.
t Dose de manutenção 100 a 200 mg, por via oral, a cada 8 a 12 horas ou 300 mg, por via oral, a cada 24 horas. Gota leve: 100 a 300 mg, por via oral, ao dia. Gota moderada: 300 a 600 mg, por via oral, ao dia. Gota grave: 700 a 900 mg, por via oral, ao dia. Doses superiores a 300 mg, devem ser divididas a cada 8 a 12 horas. Dose máxima: 900 mg/dia.
t 600 a 800 mg, por via oral, a cada 24 horas; iniciar de 12 horas a 3 dias antes do tratamento para o câncer e continuar por 2 a 10 dias após.
t Início de efeito: 2 a 3 dias.
t Pico do efeito: 7 a 10 dias.
t Pico de concentração plasmática: 0,5 a 2 horas.
t Duração de efeito: os efeitos persistem por aproximadamente 6 dias após suspensão da terapia.
t Metabolismo: hepático (metabólito ativo: oxipurinol).
t Meia-vida de eliminação: função renal normal: alopurinol: 1 a 3 horas, oxipurinol: 12 -30 horas. Doença renal em estágio final: prolongada.
t Excreção: renal (76% como oxipurinol, 12% em forma inalterada) e fecal.
t Alopurinol e oxipurinol são dialisáveis.
t Prurido(<1%), exantema (1,5%), síndrome de Stevens-Johnson (menos frequente), alopecia.
t Náusea (1,3%), vômitos (1,2%), alteração ou perda do paladar.
t Insuficiência renal (1,2%).
t Vasculite.
t Cefaleia.
t Sonolência.
t Agranulocitose (menos frequentes), anemia (menos frequentes), anemia aplásica (menos frequentes), trombocitopenia (menos frequentes).
t Mielossupressão (menos frequentes).
t Hepatotoxicidade (menos frequentes).
t Anticoagulantes orais (varfarina, femprocumona): uso concomitante ao alopurinol aumenta o risco de sangramento. Com uso concomitante, monitorar tempo de protrombina ao introduzir ou retirar alopurinol e periodicamente durante uso combinado. Ajuste de dose do anticoagulante oral pode ser necessário.
t Ciclofosfamida: pode ter sua toxicidade aumentada (supressão da medula óssea, náusea, vômito). Evitar a administração concomitante, se possível. Quando administrados juntos, monitorar para aumento de Efeitos adversos da ciclofosfamida, especialmente mielossupressão.
t Ciclosporina: pode ter sua toxicidade aumentada pelo alopurinol (insuficiência renal, colestase, parestesias). Monitorar sinais de toxicidade e níveis plasmáticos de ciclosporina e ajustar sua dose, se necessário.
t Didanosina: alopurinol pode aumentar as concentrações séricas de didanosina. Pacientes devem ser monitorados para Efeitos adversos relacionados à didanosina.
t Hidróxido de alumínio: pode reduzir a efetividade do alopurinol. O hidróxido de alumínio deve ser tomado pelo menos três horas ou após a administração de alopurinol.
t Inibidores da enzima conversora de angiotensina (ECA), como enalapril e captopril: o uso concomitante ao alopurinol pode resultar em reações de hipersensibilidade (síndrome de Stevens-Johnson, erupções na pele, espasmo coronariano anafilático). Monitorar para sinais de hipersensibilidade; se tais manifestações ocorrerem, suspender os dois medicamentos.
t Penicilinas (ampicilina, amoxicilina): uso concomitante ao alopurinol pode aumentar o risco de exantema. Se ocorrer exantema, considerar a redução da dose de alopurinol ou terapia farmacológica alternativa.
t Tiopurinas (azatioprina, mercaptopurina): alopurinol pode aumentar o efeito e a toxicidade das tiopurinas (náuseas, vômitos, leucopenia, anemia). Se possível, evitar o uso combinado. As doses das tiopurinas devem ser reduzidas para 1/3 a 1/4 da dose usual quando usado concomitantemente. Monitorar paciente hematologicamente.
t Vidarabina: uso concomitante com alopurinol pode resultar em neurotoxicidade, tremores e redução da função cognitiva. Se terapia concomitante é requerida, monitorar para sinais de neurotoxicidade. Ajuste da dose de vidarabina pode ser necessário.
t Evitar ingestão de bebidas alcoólicas e de alimentos ricos em purina como anchovas, sardinhas, fígado, rim e lentilha.
t Orientar para tomar o medicamento após as refeições para evitar desconforto estomacal.
t Reforçar a necessidade da ingestão hídrica abundante, cerca de 10 a 12 copos de líquidos por dia.
t Pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Orientar para a importância de comunicar ao perceber qualquer sinal de efeito adverso.
t Orientar para suspender o uso do alopurinol e comunicar imediatamente ao médico se ocorrer exantema na pele, dor ao urinar, sangue na urina, irritação dos olhos ou inchaço dos lábios ou boca.
t Os comprimidos devem ser armazenados em frascos bem fechados, à temperatura entre 15 a 30 °C, em locais secos e protegidos da luz.
t Preparação extemporânea: triturar comprimidos para fazer uma suspensão na concentração de 5 mg/mL em xarope simples; estável por 14 dias sob refrigeração.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item15.7
t Pó para solução injetável 50 mg.
t Acidente vascular cerebral (AVC) isquêmico, restrito para uso hospitalar em centro especializado. Contraindicações 2, 3 t AVC grave.
t Convulsões associadas a AVC.
t História de AVC em pacientes com diabete.
t Acidente vascular cerebral nos últimos três meses.
t Hipoglicemia. t Hiperglicemia. t Coma.
t Diátese hemorrágica, administração de heparina nas 48 horas anteriores ao AVC com tempo de tromboplastina parcial ativada elevada ao encaminhamento médico, contagem de plaquetas menor que 100.000/mm3.
t Doença pulmonar aguda com cavitação.
t Doença hepática grave (ver Apêndice C).
t Hemorragia recente ou hemorragia interna ativa, hemorragia intracraniana, suspeita de hemorragia subaracnoidea, trauma.
t Hipertensão grave descontrolada, dissecção da aorta, endocardite bacteriana.
t História recente de úlcera péptica, varizes esofágicas.
t Neoplasia intracraniana, má-formação arteriovenosa, aneurisma.
t Pancreatite aguda.
t Reações alérgicas prévias a alteplase, estreptoquinase ou anistreplase.
t Uso repetido 4 dias após a primeira administração de estreptoquinase ou anistreplase.
t Usar com cuidado nos casos de:
– compressão torácica externa.
– cirurgia de grande porte recente (últimos três meses): colocação de dispositivo arterial coronariano, parto, biopsia em órgão interno, extração dental.
– defeitos de coagulação.
– insuficiência hepática.
– insuficiência renal.
– doença cerebrovascular, endocardite bacteriana subaguda, pericardite aguda.
– hipertrofia atrial esquerda com fibrilação atrial (risco de dissolução do coágulo e embolização subsequente).
– idosos (não recomendado para maiores de 80 anos).
– retinopatia hemorrágica diabética ou outra condição oftalmológica (risco de hemorragia da retina).
– uso recente ou concomitante de anticoagulantes (ex.: varfarina) ou de outros fármacos que oferecem risco de sangramento.
– acidente vascular cerebral agudo (aumento do risco de sangramento cerebral).
– grave deficiência neurológica (NIHSS maior que 22).
– sangramento geniturinário nos últimos 21 dias.
– tromboflebite séptica ou cânula AV ocluída em sítio gravemente infectado.
– infecção suspeita ou conhecida em cateter.
– sinais de enfarte maior recente à tomografia computadorizada.
t Monitorar para detectar hemorragia intracraniana; monitorar pressão arterial (recomendar anti-hipertensivo se sistólica acima de 180 mm Hg ou diastólica acima de 110 mm Hg).
t Risco de sangramento por punção venosa em sítios não compressíveis ou procedimentos invasivos.
t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
t 900 microgramas/kg, máximo 90 mg por via intravenosa; iniciar com 10% da dose por injeção intravenosa, o restante por infusão intravenosa em 60 minutos. Dose máxima 90 mg.
t a dose deve ser administrada em até 3 horas após o início dos sintomas, mas alguns pacientes podem se beneficiar da administração entre 3 e 4,5 horas.
t Concentração plasmática efetiva mínima recomendada: 0,75 microgramas/mL.
t Tempo para pico de concentração plasmática: 20-40 minutos por via intravenosa.
t Metabolismo hepático, fármaco degradado aos aminoácidos constituintes.
t Depuração: mais de 50% depurados em 5 minutos após término da infusão e aproximadamente 80% em 10 minutos.
t Hemorragia interna gastrintestinal, geniturinária, retroperitoneal ou intra-craniana; superficial, observada principalmente em cortes venosos, punções arteriais, locais de intervenção cirúrgica recente. Interromper se sangramento grave; pode ser necessário o uso de fatores de coagulação ou antifibrinolíticos
t Sangramento na punção de cateterização após administração acelerada de 100 mg de alteplase (15,3%); sangramentos menores, sem necessidade de transfusão (0,5% a 4,3%) e maiores, necessitando transfusão (8,5% a 11%).
t Recorrência clínica de enfarte ou extensão do enfarte (11% a 14%)
t Reação anafilática com angioedema, urticária, hipotensão, exantema, rubor e uveíte por via intravenosa mas não instilada em cateter (1,9%)
t AVC (geral: 1,5%); em pacientes maiores de 75 anos, 3,9%; hemorragia intracraniana.
t Hipotensão (1% a 4%) – pode ser controlada por elevação dos membros inferiores, reduzindo a taxa de infusão ou suspendendo temporariamente
t Arritmias por reperfusão, ritmo idioventricular acelerado, bradicardia, despolarização ventricular prematura.
t Náusea e vômito. (frequentes no enfarte do miocárdio e podem ou não ser atribuidas à terapia com alteplase.)
t Lombalgia
t Febre
t Convulsões
t Pediatria: complicações hemorrágicas mais comuns em pacientes com menor peso.
t Anticoagulantes, trombolíticos e heparinas de baixo peso molecular: risco aumentado de complicações hemorrágicas em caso de uso concomitante. Monitorar resultados laboratoriais e clínicos, observar sinais externos de sangramento.
t Nitroglicerina: menor reperfusão coronariana, maior tempo de perfusão, mais recidiva de oclusão coronariana. Se necessário uso concomitante, empregar a menor dose possível da nitroglicerina.
t Orientar o paciente para a importância de informar sobre náusea, vertigem, ocorrência de sangramento.
t Orientar para evitar se ferir nas atividades diárias.
t Manter à temperatura controlada para não exceder 30 ºC ou sob refrigeração (2 a 8 ºC).
t Proteger o material liofilizado contra excessiva exposição a luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Utilizar a solução no máximo 8 horas após reconstituição.
t Quando diluída em solução salina ou em glicose a 5% a solução é estável por 8 horas sob temperatura ambiente.
t 1 mg de alteplase apresenta potência entre 622.000 e 667.000 unidades USP.
Rogério Hoefler
Na Rename 2010: item 10
t Solução injetável 100 mg/mL (10%) uso adulto.
t Solução injetável 100 mg/mL (10%) uso pediátrico.
t Nutrição parenteral total.
t Anúria.
t Desequilíbrio ácido-base grave.
t Hipovolemia.
t Encefalopatia.
t Coma hepático.
t Hipersensibilidade a aminoácidos presentes na formulação.
t Usar com cuidado nos casos de:
– insuficiência hepática.
– insuficiência renal (ver Apêndice D).
– pacientes com uremia (monitorar a concentração plasmática de nitrogênio).
– hiperamonemia (interromper e reavaliar o uso de aminoácidos).
– neonatos, sobretudo com baixo peso (empregar formulação específica).
t Uso hospitalar e restrito para prescrição apenas por especialista.
t Administração sem carboidratos pode causar cetonemia.
t Categoria de risco na gravidez (FDA): C.
Condição clínica / Dose
Sem estresse / 1,6 a 2,2 g/kg/dia, por via intravenosa
Com baixo estresse / 2,0 a 3,0 g/kg/dia, por via intravenosa
Críticos ou com queimaduras graves / 2,5 a 3,5 g/kg/dia, por via intravenosa
Lactentes de baixo peso
Condição clínica / Dose
Usual / 3,0 g/kg/dia, por via intravenosa
Em insuficiência hepática grave / 2,0 a 3,0 g/kg/dia, por via intravenosa
Em insuficiência renal sem diálise / 1,0 a 1,8 g/kg/dia, por via intravenosa
Em insuficiência renal com diálise / 1,5 a 3,6 g/kg/dia, por via intravenosa
Condição clínica / Dose
Sem estresse / 0,8 a 1,0 g/kg/dia, por via intravenosa
Com estresse / 1,0 a 1,7 g/kg/dia, por via intravenosa
Críticos / 1,5 a 2,0 g/kg/dia, por via intravenosa
Com queimaduras graves / 2,0 a 3,0 g/kg/dia, por via intravenosa
Em insuficiência hepática grave / 0,8-1,1 g/kg/dia, por via intravenosa
Em insuficiência renal sem diálise / 0,6-1,0 g/kg/dia, por via intravenosa
Em insuficiência renal com diálise / 1,2-2,7 g/kg/dia, por via intravenosa
t Os aminoácidos distribuem-se a todos os tecidos.
t O metabolismo se dá em todos os tecidos corporais, sendo aumentado em situação de estresse ou sepse e diminuído em insuficiências hepática ou renal.
t Flebite, tromboflebite, rubor no local da administração.
t Náusea.
t Febre.
t Distúrbios eletrolíticos, síndrome hiperosmolar, hiperamonemia, distúrbios metabólicos, desidratação.
t Aumento das enzimas hepáticas e colestase intra-hepática.
t Distúrbios metabólicos.
t Armazenar a temperatura ambiente (15 a 30 oC) e ao abrigo da luz até a utilização.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade em solução.
t Incompatibilidades associadas aos aminoácidos estão relacionadas principalmente ao armazenamento após mistura com glicose.
t a coloração da solução varia de incolor a palha-límpida. O amarelecimento da solução é indicativo de degradação.
Silvio Barberato Filho e Simone Sena Farina
Na Rename 2010: itens 5.1.1 e 16.3
t Cápsula ou comprimido 500 mg.
t Pó para suspensão oral 50 mg/mL.
t Tratamento de infecções causadas por microrganismos sensíveis no trato urinário e trato respiratório superior, incluindo bronquite, pneumonia, otite média, abscessos dentais e outras infecções orais, osteomielites, doença de Lyme, profilaxia pós-esplenotomia, infecções ginecológicas, antraz, infecções não-graves por Haemophilus influenzae e salmonelose invasiva.
t Profilaxia de endocardite bacteriana.
t Erradicação de Helicobacter pylori (esquema com claritromicina).
t Hipersensibilidade a amoxicilina ou a outras penicilinas.
t Usar com cuidado nos casos de:
– hipersensibilidade às penicilinas (obter história prévia para prevenir novas reações).
– mononucleose infecciosa, leucemia linfocítica aguda ou crônica, infecção por citomegalovírus ou portadores de HIV (há risco elevado de exantema eritematoso).
– uso de altas doses de amoxicilina (manter hidratação adequada para reduzir risco de cristalúria).
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
t Hipersensibilidade cruzada com cefalosporinas (menos de 10%): não utilizar cefalosporinas em pacientes com reações de hipersensibilidade imediata às penicilinas.
t Categoria de risco na gravidez (FDA): B.
t 20 a 90 mg/kg, dividido a cada 8 ou 12 horas. a dose e a duração do tratamento dependem do local e gravidade da infecção.
t 50 mg/kg, por via oral, em dose única, 30 minutos a 1 hora antes de procedimento em que haja sangramento. Dose máxima: 2 g.
t 250 a 500 mg, por via oral, a cada 8 ou 12 horas. a dose e a duração do tratamento dependem do local e gravidade da infecção.
t 2 g, por via oral, em dose única, de 30 minutos a 1 hora antes de procedimento em que haja sangramento.
t 1 g, por via oral, a cada 12 horas, combinada a claritromicina 500 mg e omeprazol 20 mg, ambos por via oral, a cada 12 horas, durante 7 a 14 dias.
t Absorção não é influenciada pela presença de alimentos.
t Pico da concentração plasmática: 1 a 2 horas.
t Meia-vida de eliminação: 1 a 2 horas (3,7 horas em neonatos; prolongada também em idosos e em pacientes com insuficiência renal).
t Excreção: renal (60% em forma inalterada).
t Concentrações após injeção intramuscular são semelhantes àquelas alcançadas com administração oral.
t Dialisável.
t Reações de hipersensibilidade incluindo urticária, febre, dor nas articulações, exantema, angioedema, anafilaxia, doença do soro, anemia hemolítica e nefrite intersticial.
t Diarreia, náusea, vômito.
t Colite pseudomembranosa (raramente) por Clostridium difficile.
t Acenocumarol: pode resultar em risco aumentado de sangramento. Se for necessário o uso concomitante de acenocumarol e amoxicilina, monitorar cuidadosamente o tempo de protrombina ao adicionar ou descontinuar a amoxicilina. Pode ser necessário ajustar a dose do acenocumarol.
t Contraceptivos: estrógenos podem ter reduzida a sua efetividade. Utilizar método adicional para prevenir gravidez.
t Metotrexato: aumento da toxicidade do metotrexato. Evitar o uso simultâneo, mas se o mesmo for necessário, diminuir a dose de metotrexato e monitorar sua concentração sérica. Monitorar o paciente quanto ao aumento dos Efeitos adversos do metotrexato, incluindo leucopenia, trombocitopenia e ulceração cutânea.
t Probenecida: aumenta a concentração plasmática e prolonga o efeito de amoxicilina. Útil quando é necessária elevada concentração plasmática e tecidual do antibiótico.
t Varfarina: pode resultar em risco aumentado de sangramento. Determinar o tempo de protrombina basal antes de iniciar o tratamento com amoxicilina e continuar monitorando durante o tratamento.
t Venlafaxina: risco aumentado de síndrome serotoninérgica. Monitorar cuidadosamente os sintomas da síndrome: anormalidades neuromusculares, hiperatividade autonômica, agitação e delírio. Caso os sintomas apareçam, descontinuar os medicamentos e oferecer tratamento de suporte.
t Orientar para comunicar o aparecimento tardio de exantema com sintomas de febre, fadiga e dor de garganta.
t Orientar que não há restrições quanto ao uso com alimentos nem em jejum.
t Orientar que este medicamento pode causar náusea, vômito, diarreia e exantema.
t Alertar para empregar método alternativo ou adicional para evitar a gravidez se estiver em uso de contraceptivos orais.
t Orientar para agitar o frasco da suspensão oral antes de cada administração.
t Alertar para não interromper o uso antes do final do tratamento, mesmo quando houver melhora dos sintomas após a primeira dose.
t Cada grama de amoxicilina sódica contém 2,6 mmol de sódio.
t Armazenar cápsulas sob temperatura inferior a 20 ºC. Armazenar o comprimido ou pó para suspensão oral (antes da reconstituição) sob temperatura até 25 ºC. Proteger de calor, umidade e luz direta.
t Após reconstituição, a suspensão deve ser preferentemente mantida sob refrigeração (entre 2 e 8 ºC), mas também é estável à temperatura ambiente. Descartar 14 dias após a reconstituição.
Silvio Barberato Filho e Fernando de Sá Del Fiol
Na Rename 2010: item 5.1.1
t Comprimido 500 mg + 125 mg.
t Suspensão oral 50 mg + 12,5 mg/mL.
t Infecções causadas por bactérias produtoras de betalactamase, originalmente sensíveis à amoxicilina.
t Hipersensibilidade à amoxicilina e a outras penicilinas ou ao ácido clavulânico.
t História de icterícia colestática ou disfunção hepática induzidas por penicilina ou pela associação dos fármacos.
t Usar com cuidado nos casos de:
– hipersensibilidade às penicilinas (obter história prévia para prevenir novas reações).
– mononucleose infecciosa, leucemia linfocítica aguda ou crônica, infecção por citomegalovírus ou portadores de HIV (há risco elevado de exantema eritematoso).
– uso de altas doses de amoxicilina (manter hidratação adequada para reduzir risco de cristalúria).
– insuficiência hepática (ver Apêndice C) ou insuficiência renal (ver Apêndice D).
t Hipersensibilidade cruzada com cefalosporinas (menos de 10%): não utilizar cefalosporinas em pacientes com reações de hipersensibilidade imediata às penicilinas.
t Categoria de risco na gravidez (FDA): B.
t 20 a 90 mg/kg de amoxicilina, por via oral, a cada 8 ou 12 horas. a dose e a duração do tratamento dependem do local e gravidade da infecção.
t 250 + 62,5 a 500 + 125 mg, por via oral, a cada 8 ou 12 horas. a dose e a duração do tratamento dependem do local e gravidade da infecção.
t a associação dos fármacos não altera a absorção nem a farmacocinética de nenhum deles.
t Alimentos melhoram a absorção e diminuem a intolerância gastrintestinal.
t Pico de concentração: 40 a 120 minutos.
t Meia-vida de eliminação: 1 hora para ambos os fármacos.
t Metabolismo: hepático.
t Excreção: renal (50% a 70% de amoxicilina e 25% a 40% de clavulanato)
t Dialisável.
t Reações de hipersensibilidade incluindo urticária, febre, dor nas articulações, exantema, angioedema, anafilaxia, doença do soro, anemia hemolítica e nefrite intersticial.
t Hepatite, icterícia colestática. t Síndrome de Stevens-Johnson. Mais comuns:
t Diarreia (3% a 15%), náusea (2% a 3%), vômito (até 2%)
t Micose (3%); vaginite (1%); candidíase (1,4%)
t Exantema (1% a 3%); dermatite das fraldas (3,5%)
t Colite pseudomembranosa (raramente) por Clostridium difficile.
t Acenocumarol: pode resultar em risco aumentado de sangramento. Se for necessário o uso concomitante de acenocumarol e amoxicilina, monitorar cuidadosamente o tempo de protrombina (TP) com a adição ou a retirada da amoxicilina. Pode ser necessário ajustar a dose do acenocumarol.
t Contraceptivos: pode reduzir efetividade do contraceptivo. Durante a administração dos antibióticos, utilizar método adicional para prevenir gravidez.
t Metotrexato: aumento da toxicidade do metotrexato. Evitar o uso simultâneo, porém se isto não for possível, diminuir a dose de metotrexato e monitorar sua concentração sérica. Monitorar o paciente quanto ao aumento dos Efeitos adversos do metotrexato, incluindo leucopenia, trombocitopenia e ulceração de pele.
t Probenecida: aumenta a concentração plasmática e prolonga o efeito de amoxicilina. Útil quando necessária elevada concentração plasmática e tecidual dos antibióticos.
t Vacina febre tifoide: a resposta imunológica à vacina pode ser reduzida. Deixar no mínimo 24 h de intervalo entre a última dose do antibiótico e a administração da vacina.
t Varfarina: pode resultar em risco aumentado de sangramento. Monitorar TP durante o tratamento.
t Venlafaxina: risco aumentado de síndrome serotoninérgica. Monitorar cuidadosamente os sintomas da síndrome: anormalidades neuromusculares, hiperatividade autonômica, agitação e delírio. Caso os sintomas apareçam, descontinuar os medicamentos e oferecer tratamento de suporte.
t Orientar que não há restrições quanto ao uso juntamente com alimentos e para a ingestão no início das refeições de modo a aumentar a absorção do ácido clavulânico.
t Orientar para agitar o frasco da suspensão oral antes de cada administração.
t Alertar para não interromper o uso antes do final do tratamento, mesmo quando houver melhora dos sintomas após as primeiras doses.
t Alertar para empregar método alternativo ou adicional para evitar a gravidez se estiver em uso de contraceptivos orais.
t Armazenar comprimidos abaixo de 25 ºC. Proteger de calor, umidade e luz direta.
t Suspensão oral deve ser mantida sob refrigeração depois de reconstituída. Evitar congelamento. Descartar 10 dias depois de aberto o frasco.
Silvio Barberato Filho e Fernando de Sá Del Fiol
Na Rename 2010: item 5.1.1
t Pó para solução injetável 500 mg e 1 g.
t Infecções por microrganismos sensíveis em pacientes sem disponibilidade de via oral.
t Profilaxia de endocardite bacteriana em pacientes sem disponibilidade de via oral.
t Hipersensibilidade a penicilinas.
t Usar com cuidado nos casos de:
– hipersensibilidade às penicilinas (obter história prévia para prevenir novas reações).
– mononucleose infecciosa, leucemia linfocítica, infecção por citomegalovírus ou portadores de HIV (risco elevado de exantema eritematoso).
– insuficiência renal (ver Apêndice D).
– lactação.
t Hipersensibilidade cruzada com cefalosporinas (menos de 10%): não utilizar cefalosporinas em pacientes com reações de hipersensibilidade imediata à ampicilina.
t Categoria de risco na gravidez (FDA): B.
t 250 mg, por via intramuscular ou intravenosa lenta (10 a 15 minutos) ou infusão intravenosa, a cada 4 a 6 horas.
t 150 a 200 mg/kg/dia, por injeção intravenosa lenta (10 a 15 minutos), dividido a cada 3 a 4 horas.
t abaixo de 7 dias: 50 a 100 mg/kg, por infusão intravenosa, a cada 12 horas.
t de 7 a 21 dias: 50 a 100 mg/kg, por infusão intravenosa, a cada 8 horas.
t de 21 a 28 dias: 50 a 100 mg/kg, por infusão intravenosa, a cada 6 horas.
t de 1 mês a 12 anos: 50 mg/kg, por infusão intravenosa, a cada 4 ou 6 horas. Dose máxima: 2 g a cada 4 horas.
t Dar 50 mg/kg, por via intramuscular ou intravenosa lenta (10 a 15 minutos), 30 a 60 minutos antes do procedimento.
t 500 mg, por via intramuscular ou intravenosa lenta (10 a 15 minutos) ou infusão intravenosa, a cada 4 a 6 horas.
t 1 a 2 g, por injeção intravenosa lenta (10 a 15 minutos), a cada 3 a 6 horas. Dose máxima diária: 14 g.
t 12 g, por infusão intravenosa, dividido a cada 4 a 6 horas, durante 10 a 14 dias.
t 2 g, por via intramuscular ou intravenosa lenta (10 a 15 minutos), 30 a 60 minutos antes do procedimento.
t Pico de concentração: 1 hora (após dose de 500 mg por via intramuscular).
t Meia-vida plasmática: 60 a 90 minutos.
t Metabolismo: 10% do fármaco é metabolizado.
t Excreção: renal.
t Dialisável.
t Reações de hipersensibilidade (10%).
t Diarreia (5%), náusea, vômito.
t Colite pseudomembranosa por Clostridium difficile
t Contraceptivos: uso concomitante com ampicilina diminui a efetividade do contraceptivo. Usar método contraceptivo adicional.
t Alertar para empregar método alternativo ou adicional para evitar a gravidez se estiver em uso de contraceptivos orais.
t Orientar para comunicar o aparecimento tardio de exantema com sintomas de febre, fadiga e dor de garganta.
t Armazenar pó para reconstituição entre 15 e 30 °C.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t O tempo de validade da solução reconstituída pode variar e depende das instruções do produtor. De qualquer modo, soluções reconstituídas devem ser usadas dentro de 24 horas, desde que mantidas sob refrigeração (entre 2 e 8 ºC). Não congelar.
t Soluções para infusões intravenosas são mais estáveis quando se emprega cloreto de sódio a 0,9% como diluente.
t a estabilidade das soluções de ampicilina sódica decai em presença de glicose, frutose, açúcar invertido, dextranos, hetamido, bicarbonato de sódio e lactato.
t Incompatível com aminoglicosídeos em solução.
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 6.2.4
t Comprimido 1 mg
t Adjuvante no tratamento do câncer de mama inicial, localmente avançado ou metastático com receptor de hormônio positivo em mulheres na pós-menopausa.
t Adjuvante no tratamento de câncer de mama avançado em mulheres na pós-menopausa, após terapia com tamoxifeno.
t Hipersensibilidade ao anastrozol ou a algum excipiente da formulação.
t Mulheres que ainda não entraram na menopausa (a segurança e a eficácia ainda não foram estabelecidas).
t Gravidez. Categoria de risco na gravidez (FDA): X (ver Apêndice a).
t Usar com cuidado nos casos de:
– osteoporose ou risco de seu desenvolvimento.
– lactação.
t 1 mg, por via oral, a cada 24 horas, até a progressão do tumor.
t 1 mg, por via oral, a cada 24 horas, por tempo ainda desconhecido (tratamento por 5 anos foi utilizado em ensaio clínico).
t 1 mg, por via oral, a cada 24 horas, até a progressão do tumor
t 1 mg, por via oral, a cada 24 horas, durante 3 meses antes da cirurgia.
t Bem absorvido por via oral com biodisponibilidade de 80%; a absorção pode ser diminuída na presença de alimentos.
t Resposta inicial: 24 horas.
t Pico da resposta: 2 semanas.
t Duração da ação: 7 dias (dose única).
t Metabolismo hepático (85%).
t Eliminação: predominantemente fecal (85%); e renal (11%).
t Meia-vida de eliminação: 40 a 50 horas.
t Distúrbios gastrintestinais, incluindo anorexia, vômito e diarreia.
t Vertigem, sonolência, cefaleia, depressão, astenia, insônia, alteração no humor.
t Rubor associado à menopausa.
t Exantema.
t Edema periférico, vasodilatação.
t Aumento da frequência de tosse e faringite, dispneia.
t Tromboflebite (2% a 5%).
t Hipercolesterolemia (7%).
t Anemia, leucopenia, distúrbios tromboembólicos.
t Fraturas ósseas, artralgia (10% a 15%), redução da densidade mineral óssea, sangramento vaginal (1% a 5%), artrite (17%), lombalgia (10% a 12%).
t Reações de hipersensibilidade (muito raro).
t Tamoxifeno pode reduzir as concentrações plasmáticas de anastrozol em 27%. O uso concomitante não é recomendado.
t Evitar tomar anastrozol próximo ou durante as refeições.
t Não tomar este medicamento em caso de gravidez suspeita ou confirmada.
t O tratamento não deve ser interrompido se surgirem, eventualmente, náuseas, vômitos e diarreia. Em caso de vômito logo após o uso contatar o médico.
t Efeitos indesejados graves ou frequentes devem ser notificados ao médico.
t Armazenar sob temperatura ambiente (15 a 30 ºC), protegido do calor e umidade. Não deve ser refrigerado ou congelado.
Atenção: o uso de anastrozol deve ser reservado para mulheres na pós-menopausa.
Eudiana Vale Francelino
Na Rename 2010: itens 5.3.1 e 5.6.2.5
t Pó para solução injetável 50 mg em desoxicolato de sódio.
t Tratamento agudo de micoses sistêmicas graves, tais como: aspergilose, blastomicose, candidíase sistêmica, coccidioidomicose, criptococose, esporotricose, infecção fúngica grave no sistema nervoso central, nos pulmões ou no trato urinário, mucormicose e paracoccidioidomicose.
t Leishmaniose mucocutânea e visceral (segunda linha de tratamento).
t Histoplasmose.
t Conidiobolomicose (zigomicose).
t Basidiobolomicose.
t Infecção por Rhodotorula spp.
t Hipersensibilidade a qualquer forma de anfotericina B.
t Tratamento de dermatofitoses superficiais.
t Lactação.
t Infecções fúngicas não-invasivas (por exemplo, candidíase vaginal, candidíase esofágica) em pacientes imunodeprimidos com contagem normal de neutrófilos.
t Usar com cuidado nos casos de:
– pacientes com experiência prévia de reações adversas agudas à anfotericina B (caso o tratamento com anfotericina B seja essencial, usar profilaxia com antipirético, anti-histamínico ou hidrocortisona).
– crianças e idosos (usar a menor dose necessária e monitorar cuidadosamente, pois não há estudos adequados nesses grupos etários). Pacientes pediátricos são mais Susceptíveis a Efeitos adversos cardiovasculares (hipotensão, bradicardia e parada cardíaca).
– insuficiência cardíaca, insuficiência renal e ocorrência de reações graves à dose experimental (iniciar o tratamento com dose diária baixa e aumentar gradualmente; ver nota em esquema de administração).
– uso simultâneo de outros medicamentos (atentar para eventual risco de interações que aumentem a toxicidade ou diminuam a atividade de anfotericina B).
t Monitorar função renal (ver Apêndice D), função hepática (ver Apêndice C), eletrólitos séricos e contagens hematológicas durante o tratamento. Diante de alterações importantes dos dados laboratoriais, associadas a manifestações clínicas indesejadas, interromper o tratamento, reduzir as doses ou aumentar o intervalo entre as administrações.
t Monitorar os parâmetros clínicos como resolução da febre e demais sinais e sintomas decorrentes da infecção, bem como cultura negativa para um fungo específico.
t Observar função pulmonar, principalmente nos casos de transfusão leucocitária ou de outros produtos do sangue.
t Realizar teste antes da primeira infusão para detectar possível reação infusional. Não administrar antes de o paciente ser observado clinicamente por 30 minutos. Reações infusionais são mais comuns no início da administração (1 a 3 horas), diminuindo com a continuação da terapia.
t Evitar a administração rápida em função do risco de arritmias e irritação local.
t Dose excessiva aguda pode resultar em desconforto cardiorrespiratório.
t Anfotericina convencional apresenta maior toxicidade do que diversas formas de emulsão lipídica, mas a toxicidade da forma convencional pode ser diminuída com infusões lentas, sem comprometer a eficácia.
t Categoria de risco na gravidez (FDA): B (ver Apêndice a).
t Dose teste inicial 0,1 mg/kg, por infusão intravenosa durante 20 a 30 minutos, 30 a 60 minutos antes da administração da dose plena de 0,25 mg/kg/dia (incluindo a dose inicial), aumentada por 2 a 4 dias, se tolerada, até 1 mg/ kg/dia, por via intravenosa, ou em infecções graves até 1,5 mg/kg/dia ou em dias alternados.
t Tratamento preferencial: 0,7 a 1 mg/kg, por via intravenosa, a cada 24 horas, combinada a flucitosina 25 mg/kg, por via oral, a cada 6 horas, durante no mínimo 2 semanas, seguido de fluconazol por via intravenosa ou oral, durante 8 semanas.
t Terapia alternativa (se flucitosina não for tolerada): 0,7 a 1,5 mg/kg, por via intravenosa, a cada 24 horas, durante no mínimo 2 semanas, seguido de fluconazol por via intravenosa ou oral, durante 8 semanas.
t 1 mg/kg, por via intravenosa, a cada 24 horas, durante 1 a 2 semanas, seguido de itraconazol por via oral até completar um total de 12 semanas de tratamento.
t Monoterapia: 1 mg/kg, por via intravenosa, a cada 24 horas, durante 4 a 6 semanas.
t Tratamento alternativo: 1 mg/kg, por via intravenosa, a cada 24 horas, durante 2 a 4 semanas, seguido de itraconazol por via oral até completar um total de 3 meses de tratamento.
t 1 mg/kg, por via intravenosa, a cada 24 horas, durante 4 semanas.
t Dose recomendada para tratamento inicial: 0,7 mg/kg/dia intravenoso seguida de terapia com itraconazol.
t Dose teste inicial 1 mg, por infusão intravenosa durante 20 a 30 minutos, junto com antipirético, anti-histamínico ou hidrocortisona 50 a 100 mg, por via intravenosa, 30 a 60 minutos antes da administração da dose plena. Dose de 0,25 a 1,5 mg/kg, em infusão intravenosa contínua, a cada 24 horas; ou 1,0 a 1,5 mg/kg, em infusão intravenosa durante 4 a 6 horas, em dias alternados.
t 0,25 a 1 mg/kg, por infusão intravenosa de 2 a 6 horas, a cada 24 horas, durante no máximo 11 meses ou até atingir a dose total de 3,6 g. Dose máxima diária: 1,5 mg/kg quando administrada em dias alternados.
t 0,25 a 1 mg/kg, por infusão intravenosa de 2 a 6 horas, a cada 24 horas. Dose máxima diária: 1,5 mg/kg quando administrada em dias alternados.
t 0,4 a 0,6 mg/kg, por via intravenosa, a cada 24 horas, até 1 a 1,5 mg/kg/dia, durante 7 a 14 dias em pacientes de baixo risco e 6 semanas ou mais em pessoas com alto risco de morbidade e mortalidade.
t 1 mg/kg, por via intravenosa, a cada 24 horas, associada com flucitosina 100 mg/kg/dia, por via oral.
t 0,3 mg/kg, por via inatravenosa, a cada 24 horas, durante 5 a 7 dias.
t 0,5 a 1 mg/kg, por via intravenosa, a cada 24 horas, durante 4 a 12 semanas conforme resposta clínica. Dose máxima diária: 1,5 mg/kg.
t 0,5 a 1 mg/kg, por via intravenosa, a cada 24 horas.
t 0,7 a 1 mg/kg, por via intravenosa, a cada 24 horas, associada com flucitosina 25 mg/kg/dia, por via oral, a cada 6 horas, durante 2 semanas, seguido de fluconazol 400 mg, por via oral, a cada 24 horas, durante no mínimo 10 semanas.
t 0,7 a 1 mg/kg, por via intravenosa, a cada 24 horas, durante 6 a 10 semanas.
t 0,7 a 1 mg/kg, por via intravenosa, a cada 24 horas, durante por 4 a 6 semanas. Dose máxima diária: 1,5 mg/kg quando administrada em dias alternados.
t 0,7 a 1 mg/kg, por via intravenosa, a cada 24 horas, até que o paciente demonstre uma resposta favorável, seguida pela retirada gradual da terapia, associada com itraconazol durante um total de 12 meses. Dose máxima diária: 1,5 mg/kg quando administrada em dias alternados.
t 0,5 a 0,6 mg/kg, por via intravenosa, a cada 24 horas, durante 4 a 8 semanas. Em casos graves, 0,7 a 1 mg/kg, por via intravenosa, a cada 24 horas, até obter resposta clínica, seguido de tratamento com um agente azólico por via oral. Dose máxima diária: 1,5 mg/kg em dias quando administrada em dias alterandos.
t Dose teste inicial 1 mg, por infusão intravenosa em 20 a 30 minutos, seguida por 5 a 10 mg, aumentada gradualmente em 5 a 10 mg, a cada 24 horas, até 0,5 a 1 mg/kg/dia, passando a ser administrada em dias alternados, até uma dose acumulada total de 1 a 3 g.
t 0,4 a 0,5 mg/kg, por via intravenosa, a cada 24 horas. Em casos graves 1 a 1,5 mg/kg, por via intravenosa, a cada 24 horas.
t 1 a 1,5 mg/kg, por via intravenosa, a cada 24 horas, durante 2 a 3 meses. Dose total de 3 a 4 g, por via intravenosa, é recomendada para o tratamento de ficomicose rinocerebral.
t Nota: insuficiência cardíaca, insuficiência renal e ocorrência de reações graves à dose experimental (iniciar o tratamento com dose diária de 5 a 10 mg; dependendo do estado do paciente cardiopata ou nefropata, a dose pode ser gradualmente aumentada em 5 a 10 mg por dia até alcançar uma dose diária final de 0,5 a 0,7 mg/kg).
t Absorção: mal absorvida no TGI.
t Pico de concentração plasmática: 1 hora.
t Excreção: renal (40%).
t Meia-vida de eliminação bifásica: 15 a 48 horas (inicial) e 15 dias (terminal).
t Pode atravessar a placenta e em baixas concentrações atinge o líquido amniótico.
t Detecção no sangue por até 4 semanas e na urina por até 4 a 8 semanas.
t Anfotericina B não é removida por hemodiálise.
t Anemia, hemólise, agranulocitose, coagulopatia, tempo de protrombina diminuído ou aumentado, trombocitopenia, leucopenia, eosinofilia, leucocitose.
t Hipopotassemia, hipomagnesemia, azotemia.
t Cansaço, fraqueza, dor generalizada, parestesias, cãibras.
t Febre, calafrios, cefaleia, mal-estar.
t Náusea, vômitos, indigestão, perda de apetite, diarreia, dor epigástrica, perda de peso, xerostomia, estomatite, dispepsia, gastroenterite hemorrágica, hematêmese.
t Arritmia, tromboflebite, insuficiência cardíaca, cardiomiopatia, hipertensão.
t Choque, edema pulmonar, dispneia, taquipneia, broncoespasmo, hipóxia.
t Anafilaxia, reações anafilactoides.
t Síndrome do homem vermelho, exantema, erupção cutânea (incluindo erupção cutânea maculopapular ou vesicobolhosa), púrpura, prurido, urticária, sudorese, dermatite esfoliativa, eritema multiforme, alopecia, pele seca, descoloração da pele ou úlcera.
t Neuropatia periférica, convulsão, encefalopatia.
t Visão borrada, diplopia.
t Insuficiência renal e nefrotoxicidade.
t Depressão, confusão, tonturas, insônia, sonolência, coma, ansiedade, agitação, nervosismo, pensamentos anormais, alucinações, tremores, apreensões, miastenia, perda auditiva, zumbido, vertigem, acidente vascular cerebral, síndrome extrapiramidal, leucoencefalopatia.
t Distonia muscular, dor nos ossos ou nas articulações.
t Aumento das concentrações séricas de fosfatase alcalina, bilirrubina, gamaglutamiltransferase.
t Insuficiência hepática aguda, hepatite, icterícia, hiperglicemia, hipoglicemia. t Hipostenuria, acidose tubular renal, nefrocalcinose, aumento da concentração sérica de ureia, diminuição da taxa de filtração glomerular, do fluxo plasmático renal, aumento da excreção de ácido, anúria, oligúria, disúria, hematúria.
t Ciclosporina: aumento da atividade/toxicidade de anfotericina B. Se clinicamente possível, evitar essa combinação. Monitorar parâmetros de função renal e concentrações séricas de ciclosporina.
t Deslanosídeo, digitoxina, digoxina e metildigoxina: aumento do risco de toxicidade digitálica (náusea, vômitos, distúrbios visuais e arritmias cardíacas), devido à hipopotassemia induzida. Monitorar a concentração de potássio e, se necessário, administrar cloreto de potássio.
t Tacrolimo: aumento da atividade/toxicidade de anfotericina B. Se a administração concomitante for necessária, monitorar testes de função renal.
t Trióxido de arsênio: aumento do risco de prolongamento do intervalo QT. O trióxido de arsênio deve ser administrado com extrema cautela em pacientes que possam apresentar risco para o desenvolvimento da síndrome do prolongamento do intervalo QT.
t Alertar para suspender o uso e notificar imediatamente caso haja urticária, edema em face, mãos ou boca, dispneia, erupção cutânea, náusea, vômito e diarreia.
t Alertar quando ocorrer arritmias, hipotensão e alterações no sangue.
t Coloração da anfotericina B convencional: pó amarelo a laranja, estéril.
t Armazenamento da anfotericina B convencional em pó: 2 a 8 °C.
t Reconstituir somente com água estéril para injeção, sem conservantes. Não reconstituir com água bacteriostática.
t a solução-mãe é estável à temperatura ambiente (25 ºC) por 24 horas e sob refrigeração (+ 4 °C) por 2 dias. Deve ser protegida da luz.
t O diluente padrão é solução de glicose a 5%. O pH da solução de glicose não deve ser abaixo de 4,2. Diluir a dose em 250 a 500 mL.
t Soluções diluídas não necessitam proteção contra iluminação típica de hospital se a administração ocorrer dentro das 24 horas após a preparação.
t Incompatibilidades (com precipitação): álcool benzílico, cloreto de sódio ou soluções eletrolíticas.
t Misturas da anfotericina B convencional em emulsão comercial lipídica têm sido considerada instável, contudo algumas fontes mencionam que a estabilidade é satisfatória.
t Não se recomenda misturar em solução com quaisquer outros fármacos.
Atenção: anfotericina B em grávidas: o uso por via intravenosa pode ser preferencial para terapia supressiva ou de manutenção contra coccidioidomicose, criptococose, histoplasmose ou infecção fúngica oportunista associada à infecção pelo HIV, especialmente durante o primeiro trimestre, devido à preocupação relativa a antifúngicos azólicos, como fluconazol e itraconazol.
Elaine Silva Miranda
Na Rename 2010: item 5.6.2.5
t Solução injetável 300 mg/mL (81 mg Sb5+/mL).
t Leishmaníase (tratamento das formas visceral, cutânea, cutânea difusa, mucocutânea e de lesões nodulares iniciais).
t Insuficiência renal grave (ver Apêndice D).
t Doença cardíaca.
t Hipersensibilidade a compostos antimoniais.
t Hepatopatias graves (ver Apêndice C).
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– pneumonia, tuberculose e alterações eletrocardiográficas, desnutrição e infecção por HIV avançada.
– crianças com menos de 18 meses (uso não recomendado). t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
t 20 mg/kg, por via intramuscular profunda, a cada 24 horas, durante 20 a 28 dias. Essa dose pode ser repetida ou continuada dependendo da evolução do paciente. Dose máxima diária: 6 g.
t 10 a 20 mg/kg, por via intramuscular profunda, a cada 24 horas, durante no mínimo 4 semanas. Dose máxima diária: 6 g. Em caso de recidiva deve-se continuar o tratamento com pentamidina.
t 20 mg/kg, por via intramuscular profunda, a cada 24 horas, até alguns meses após a melhora clínica. Dose máxima diária: 6 g.
t 20 mg/kg, por via intramuscular profunda, a cada 24 horas, durante no mínimo 4 semanas. Dose máxima diária: 6 g. Em caso de recidiva, repetir o tratamento por 8 semanas. Caso o tratamento não seja efetivo, tratar com pentamidina ou anfotericina B.
t 1 a 3 mL da solução, por injeção intralesional, repetida uma ou duas vezes se necessário, a cada 24 ou 48 horas.
t a injeção intralesional é contraindicada em casos de: lesões próximas aos olhos; maiores que 3 cm de diâmetro; nas articulações; lesões super infectadas; lesões produzidas por L. brazilensis, L. guyanensis e L. tropica, formas esporotricoides e quando o número de lesões for maior ou igual a 3.
Nota:
t Doses superiores a 3 ampolas devem ser fracionadas em 2 aplicações para administração intramuscular.
t Recomenda-se a continuação do tratamento por duas semanas após a cura parasitológica.
t Absorção rápida após injeção intramuscular.
t Pobremente absorvido pelo trato gastrintestinal.
t Meia-vida de eliminação bifásica: 2 horas e 76 horas.
t Metabolismo hepático.
t Excreção: renal (maior que 90%).
t Acumula-se no cabelo, o que pode servir para monitoria durante o tratamento.
t Cardiotoxicidade (alterações ao eletrocardiograma: aumento do espaço QT, distúrbios da repolarização ventricular, inversão da onda T).
t Náusea, vômito, dor abdominal, pancreatite, hepatotoxicidade, aumento de enzimas hepáticas.
t Febre, anorexia, cefaleia, vertigem.
t Dispneia, tosse.
t Nefrotoxicidade.
t Exantema, reação anafilática, edema facial, suor, rubor.
t Leucopenia.
t Letargia, mialgia, artralgia, dor no lugar da injeção intramuscular.
t Sangramento nasal.
t Álcool pode potencializar o risco de hepatotoxicidade.
t Antiarrítmicos, antidepressivos tricíclicos e outros agentes que prolonguem o intervalo QT podem aumentar o risco de arritmia.
t Orientar para adoção preferencial de dieta rica em proteína durante o tratamento.
t Manter ao abrigo de luz e à temperatura de 15 a 30 ºC.
t a solução pode deteriorar-se com o tempo.
t a solução resiste ao calor e pode sofrer esterilização em autoclave.
Atenção: a dose preconizada é sempre expressa em antimônio pentavalente. Outras formas de antimônio (p.ex. trivalente), por serem mais tóxicas, não têm o mesmo esquema de administração.
a injeção intramuscular é dolorosa e deve ser administrada lentamente.
É recomendado monitoria periódica das funções hepáticas, cardíacas e renais durante o tratamento.
Se possível corrigir deficiência de ferro e outras carências nutricionais.
O tratamento exitoso da leishmaniose mucocutânea pode induzir inflamação grave ao redor das lesões (risco de morte se envolver faringe ou traqueia), o que pode requerer o uso de corticosteroide.
Cláudia Du Bocage Santos Pinto
Gabriela Costa Chaves
Na Rename 2010: item 5.6.2.2
t Solução injetável 80 mg/mL.
t Malária grave por Plasmodium falciparum, incluindo forma cerebral, em área resistente a sulfato de quinina.
t Hipersensibilidade a compostos derivados de artemisinina.
t Primeiro trimestre da gravidez. (ver Apêndice a).
t Usar com cuidado nos casos de:
– crianças com menos de 5 anos de idade.
– operação de maquinários e direção de veículos (perigo de acidentes; o arteméter prejudica a capacidade de realizar atividades que exijam atenção).
– doença cardiovascular.
– insuficiência renal.
– insuficiência hepática.
t Não deve ser utilizado em situações diferentes das indicadas, para que não se desenvolva resistência adquirida com Plasmodium falciparum.
t Não deve ser usado em profilaxia devido a meia-vida curta e indefinida segurança em indivíduos sadios, além de evidência de altas taxas de recrudescimento após 14 a 28 dias de tratamento monoterápico.
t 3,2 mg/kg por via intramuscular, em dose única. Após 24 horas aplicar 1,6 mg/kg, a cada 24 horas, durante mais 4 dias (totalizando 5 dias de tratamento). Adicionalmente, administrar clindamicina 20 mg/kg, por via intravenosa, a cada 24 horas, diluída em solução glicosada a 5% (1,5 mL/kg de peso), infundida gota a gota em 1 hora, durante 7 dias. Se o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t Pico de concentração plasmática: 4 a 8 horas (intramuscular).
t Início do efeito clínico: 1 a 3 dias.
t Meia-vida: 4 a 11 horas.)
t Ligação à proteínas plasmáticas: 95,4%.
t Metabolismo: hepático (metabólito ativo).
t Excreção: urinária.
t Dor no lugar da injeção intramuscular
t Reações alérgicas
t Cefaleia, vertigens, zumbidos.
t Vômito, náusea, dor abdominal, diarreia.
t Aumento transitório da temperatura corporal após redução da parasitemia.
t Cardiotoxicidade com uso de altas doses, com bradicardia ou taquicardia supraventricular.
t Redução da contagem de reticulócitos e neutrófilos.
t Aumento de níveis de enzimas hepáticas.
t Aurotioglicose: aumenta o risco de indução de discrasia sanguínea. O uso concomitante é contraindicado.
t Lumefantrina (em doses fixas): a associação é sinérgica e útil em áreas de grande resistência do plasmódio.
t Mefloquina: a associação aumenta a eficácia e reduz a resistência potencial, evitando a recidiva.
t Alertar que a tontura provocada pelo medicamento pode impedir o desenvolvimento de atividades que exijam atenção como, por exemplo, operar máquinas e dirigir veículos.
t Armazenar à temperatura ambiente (15 a 30 ºC), em frasco escuro hermeticamente fechado.
t Mantém atividade por tempo longo nessas condições.
Elaine Silva Miranda
Na Rename 2010: item 5.6.2.2
t Pó para solução injetável 60 mg.
t Malária grave, incluindo forma cerebral, por Plasmodium falciparum.
t Hipersensibilidade a derivados da artemisina.
t Primeiro trimestre da gravidez (ver Apêndice a).
t Usar com cuidado nos casos de:
– uso como monoterapia em pacientes não imunes (risco de recrudescência).
– operação de maquinários e direção de veículos (perigo de acidentes; o artesunato prejudica a capacidade de realizar atividades que exijam atenção).
– doença cardiovascular, neurológica, betatalassemia, ou história de convulsão.
– insuficiência renal.
– insuficiência hepática (ver Apêndice C)
– malária causada por Plasmodium vivax, que é normalmente responsiva a cloroquina.
t Deve ser utilizado em combinação com outros antimaláricos.
t Pode induzir supressão de medula óssea.
t Não deve ser utilizado em situações diferentes das indicadas, para que não se desenvolva resistência adquirida com Plasmodium falciparum.
t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
t 2,4 mg/kg, por via intravenosa, seguido de 1,2 mg/kg administrado após 12 e 24 horas. Depois, 1,2 mg/kg, a cada 24 horas, durante 6 dias. Adicionalmente, administrar clindamicina 20 mg/kg, por via intravenosa, diluída em solução glicosada a 5% (1,5 mL/kg de peso), infundida gota a gota em 1 hora, durante 7 dias. Se o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t Pico de concentração plasmática via oral: 15-30 minutos.
t Pico de concentração plasmática via intravenosa ou intramuscular: 7-12 minutos.
t Início do efeito clínico: redução da parasitemia e da febre após 24 a 48 horas do início do tratamento.
t Meia-vida de eliminação plasmática: 40 a 95 minutos
t Metabolismo: hepático (metabólito ativo: di-hidroartemisinina, responsável pela maior parte dos efeitos antimaláricos in vivo).
t Excreção: renal.
t Exantema, prurido.
t Náusea, vômito, dor abdominal, diarreia.
t Cefaleia, vertigem, zumbido, tinitus, convulsões, disfunção cerebelar.
t Perda de cabelos.
t Bradicardia, bloqueio atrioventricular de primeiro grau (quando administrado via intravenosa), alterações ao eletrocardiograma: prolongamento do intervalo QT.
t Diminuição de reticulócitos e neutrófilos (pode ser limitante de dose).
t Elevação de enzimas hepáticas.
t Reação anafilática, ocorre raramente. Recomenda-se tratamento imediato com anti-histamínicos, esteroides ou agentes pressores.
t Agentes que prolonguem o intervalo QT, tais como determinados antiarrítmicos e antidepressivos tricíclicos, devem ser evitados.
t Alertar que a tontura provocada pelo medicamento pode impedir o desen-
volvimento de atividades que exijam atenção como, por exemplo, operar máquinas e dirigir veículos.
t a solução injetável deve ser reconstituida em bicarbonato de sódio 5%; após, diluír em glicose 5% ou soro fisiológico até obter o volume de administração desejado.
t a solução deve ser preparada no momento da administração.
Atenção: recomenda-se terapia antimalárica alternativa para todos os pacientes que desenvolveram exantema e urticária durante exposição inicial ao artesunato.
Elaine Silva Miranda
Na Rename 2010: item 5.6.2.2
t Comprimido 25 mg + 55 mg.
t Comprimido 100 mg + 220 mg.
t Tratamento de primeira linha para Malária por Plasmodium falciparum não-complicada.
t Tratamento de infecções mistas por Plasmodium falciparum e P. vivax ou P. ovale (com tratamento subsequente de suas formas hipnozoitas).
t Tratamento de infecções mistas por Plasmodium falciparum e P. malariae.
t Hipersensibilidade à mefloquina, quinina, quinidina ou artesunato, e a outros derivados da artemisinina.
t Primeiro trimestre da gravidez.
t Crianças com menos de 6 meses de idade e/ou pesando menos que 5 kg.
t Histórico de doenças neuropsiquiátricas, incluindo depressão, distúrbio afetivo bipolar, neurose, ansiedade grave e convulsão.
t Malária grave.
t Tratamento recente ou concomitante com halofantrina.
t Epilepsia (a mefloquina pode aumentar o risco de convulsões).
t Lactação
t Usar com cuidado nos casos de:
– distúrbios da condução cardíaca, arritmia cardíaca.
– uso concomitante com quinina ou cloroquina (pode aumentar o risco de convulsões).
– operação de maquinários e direção de veículos (perigo de acidentes; o uso de mefloquina prejudica a capacidade de realizar atividades que exijam atenção).
– Categoria de risco na gravidez (FDA): D (ver Apêndice a).
t 1 comprimido 25 mg + 55 mg, por via oral, a cada 24 horas, durante três dias.
t 2 comprimidos 25 mg + 55 mg, por via oral, a cada 24 horas, durante três dias.
t 1 comprimido 100 mg + 220 mg, por via oral, a cada 24 horas, durante três dias.
t 2 comprimidos 100 mg + 220 mg, por via oral, a cada 24 horas, durante três dias.

t Sempre dar preferência ao peso para a escolha da dose.
t Administrar os medicamentos preferentemente com alimentos.
t Se surgir icterícia, suspender a primaquina.
t Metabolizado por esterases plasmáticas e intestinais no metabólito ativo, di-hidroartemisinina (DHA).
t Biodisponibilidade: alta.
t Excreção: a DHA é excretada principalmente pela urina, com menos de 1% de artesunato inalterado na urina e fezes.
t É parcialmente metabolizada no fígado em carboximefloquina e vários outros metabólitos.
t Pico de concentração plasmática: 6 a 24 horas.
t Meia-vida: 1533 dias.
t Excreção: principalmente pelas fezes, mas mefloquina inalterada (9%) é também encontrada na urina.
t O medicamento leva em média cerca de uma hora para início de sua ação farmacológica.
t Náuseas, vômitos, dor abdominal, diarreia, anorexia.
t Sonolência, insônia, sonhos anormais, dispneia, alucinação visual
t Fraqueza muscular, mialgia, artralgia.
t Prurido
t Palpitações
t Diminuição da audição
t Hepatite, hiperbilirrubinemia.
t Manifestações neuropsiquiátricas graves, incluindo cefaleia, vertigem, dificuldade de equilíbrio, distúrbios visuais, visão turva
t Recrudescência de malária.
t Nenhum estudo formal de Efeitos adversos foi conduzido para a associação artesunato de sódio + cloridrato de mefloquina. Consultar as monografias individuais do artesunato de sódio e do cloridrato de mefloquina.
t Pode ocorrer vômito após ingestão. Se o vômito ocorrer dentro de 30 minutos após ingestão do medicamento, deve-se readministrar a mesma dose. Se o vômito ocorrer após 30 a 60 minutos, ingerir meia dose e prosseguir com as doses programadas.
t Alertar para a necessidade de notificar efeitos adversos, para que sejam prescritos antimaláricos alternativos.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Alertar que a tontura provocada pelo medicamento pode impedir o desenvolvimento de atividades que exijam atenção como, por exemplo, operar máquinas, dirigir veículos ou realizar tarefas que exijam um alto grau de destreza psicomotora e/ou manual.
t Alertar que o paciente deve tomar os comprimidos inteiros com um pouco de líquido.
t No caso de crianças ou pacientes que não sejam capazes de engolir comprimidos, estes devem ser dissolvidos em uma colher das de sopa com água para administração.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC, ao abrigo de luz, calor e umidade.
Atenção: paciente em risco de morte: se existirem alternativas limitadas de medicamentos eficazes contra a malária, artesunato + mefloquina deve ser considerado como opção terapêutica mesmo na presença de contraindicações. O tratamento sintomático da febre e uso de antieméticos pode reduzir o risco de vômitos associado ao medicamento. Testes de função hepática e oftamológicos são recomendados principalmente em pacientes que recebem mefloquina há mais de 1 ano. Monitoramento com ECG é necessário em pacientes com distúrbio cardíaco.
Maurício Fábio Gomes
Na Rename 2010: itens 6.1.6
t Solução injetável 10.000 UI.
t Leucemia linfoblástica aguda.
t Pancreatite ativa ou história desta doença.
t Hipersensibilidade a asparaginase ou a outro componente da formulação.
t Usar com cuidado nos casos de:
– insuficiência hepática e coagulopatias.
– infecções preexistentes.
– lactação (ver Apêndice B).
t Reações alérgicas graves podem ocorrer, tendo como fatores de risco administração intravenosa, terapia prolongada, terapia prévia com asparaginase e terapia intermitente. Recomenda-se fazer teste intradérmico com 0,1 mL de solução com concentração de 20 UI/mL, antes da primeira dose do tratamento. Observar o paciente por 60 minutos. Podem ocorrer falsos-negativos.
t Monitorar regularmente os níveis glicêmicos e de amilase sérica.
t Pode alterar testes de função da tireoide.
t Categoria de risco na gravidez (FDA): C (ver Apêndice a).
t Administrar 1000 UI/kg/dia, por infusão intravenosa, durante 10 dias, iniciando no 22º dia do período de tratamento com vincristina 1,5 mg/m2/dia nos dias 1, 8 e 15, prednisona 40 mg/m2/dia, por via oral, três vezes ao dia, por 15 dias, depois reduzir a dose de prednisona em 50% a cada 2 dias até 2,5 mg/m2/dia e, então, descontinuar. Ou então, a asparaginase pode ser administrada por via intramuscular profunda, na dose de 6000 UI/m2/dia, nos dias 4, 7, 10, 13, 16, 19, 22, 25 e 28.
t Como agente de indução isolado, administrar 6000 a 10000 UI/m2/dia, por infusão intravenosa, a cada 7 dias, num total de 2 doses para induzir remissão; 6000 a 10000 UI/m2/dia, por infusão intravenosa, durante 14 dias para consolidação remissão.
t Administrar 500 UI/kg/dia, por infusão intravenosa, dos dias 22 a 32, com daunorrubicina 45 mg/m2/dia, por via intravenosa, nos dias 1, 2, e 3, vincristina 2 mg/dia, por via intravenosa, nos dias 1, 8, e 15 e prednisona 40 mg/m2/ dia, por via oral, do dia 1 ao 22.
t Como agente de indução isolado, administrar 200 UI/kg/dia, por infusão intravenosa, durante 28 dias; ou 5000 a 10000 UI/m2/dia, por infusão intravenosa, durante 7 dias, a cada 3 semanas, ou 10000 a 40000 UI, por infusão intravenosa, a cada 2 a 3 semanas
Notas:
t a injeção intramuscular profunda deve ser feita em músculo de grande volume. O volume em único local deve ser limitado a 2 mL; se o volume for maior, 2 locais devem ser utilizados; a injeção intramuscular oferece menor risco de reacoes anafilaticas.
t Um teste intradérmico deve ser procedido antes da administração da primeira dose do medicamento e por ocasião da segunda dose, se esta for administrada uma semana ou mais após a primeira.
t Regime de dessensibilização pode ser feito em pacientes que reagem à dose-teste, ou que não possua outra terapia alternativa. Iniciar com 1 unidade e dobrar a dose a cada 10 minutos até que a quantidade total acumulada alcance a dose programada para aquele dia de tratamento, conforme exemplo na tabela 1.
Tabela 1. Esquema de dessensibilização à asparaginase
t Pico sérico de concentração: 14 a 24 horas (intramuscular); 50% mais baixas na administração intramuscular em comparação à infusão intravenosa.
t Duração do efeito: 6 a 21 semanas (via intravenosa); baixas concentrações sanguíneas de asparaginase permanecem por 7 a 10 dias após a suspensão do tratamento.
t Meia-vida de eliminação: 39 a 49 horas (intramuscular), 8 a 30 horas (intravenosa).
t Metabolismo: degradação sistêmica.
t Excreção: renal (traços).
t Efeitos imediatos: febre, dores, náuseas, vômitos (50-60%).
t Coma (25%), estupor, confusão mental, desorientação, convulsões (10-60%), sonolência, fadiga, depressão, alucinações.
t Náuseas e vômitos, perda de apetite, estomatite, cólica abdominal (70%), pacreatite aguda (15%), pseudocisto do pâncreas.
t Hepatotoxicidade.
t Uremia (66%), falência renal aguda.
t Diabetes, hiperglicemia, alterações lipídicas.
t Anemia, leucopenia, hipofibrinogenemia, depressão dos fatores V, VII, VIII e IX da coagulação, trombose venosa profunda, hemorragia cerebral, trombocitopenia.
t Anafilaxia, angustia respiratória, faringite, flebites, erupção cutâneas
t Vacina rotavírus humano G1P1[8] (atenuada): aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Se a vacina for necessária, administrar a vacina depois de três meses da descontinuação da quimioterapia.
t Alertar para não utilizar este medicamento concomitantemente ao ácido acetilsalicílico.
t Alertar para não tomar qualquer tipo de vacina sem antes notificar.
t Orientar para interromper lactação durante o uso deste medicamento.
t Reforçar a necessidade de aumentar a ingestão de líquidos.
t Alertar para evitar atividades que requerem coordenação ou atenção.
t Orientar para utilizar um efetivo método contraceptivo enquanto utilizar asparaginase.
t Utilizar os procedimentos adequados para o manuseio e descarte de quimio-terápicos.
t a solução de asparaginase é incompatível com borracha.
t Estocar frascos estéreis de asparaginase (não-reconstituída) a temperaturas entre 2 e 8oC. Não congelar.
t Solução não-reconstituída é estável por 48 horas sob temperatura ambiente (de 15 a 30oC).
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Soluções reconstituídas e não utilizadas devem ser estocadas entre 2 e 8oC e descartadas após 8 horas ou se estiverem turvas. Não congelar.
t Preparo para administração intravenosa: a solução reconstituída deve ser diluída com solução de cloreto de sódio a 0,9% ou solução aquosa de glicose 5%. Infundir dentro de 8 horas e somente se estiver límpida.
t Preparo para administração intramuscular: reconstituir frasco de 10000 UI com 2 mL de solução de cloreto de sódio a 0,9%; utilizar dentro de 8 horas somente se estiver límpida; a agitação normal do frasco durante a reconstituição não inativa a enzima.
t Preparo de solução para teste de hipersensibilidade: Reconstituir frasco de 10000 UI em 5 mL de diluente. Transferir 0,1 mL desta solução e misturar em outro frasco com 9,9 mL de diluente (concentração final de 20 UI/mL).
Atenção: recomenda-se que asparaginase seja administrada em ambiente hospitalar sob estreita supervisão do médico oncologista e com equipamentos e medicamentos para manejo de reação anafilática. Adultos tem demonstrado maior susceptibilidade à reações anafilática que crianças.
Rosa Martins
Na Rename 2010: itens 14.3 e 14.4.2
t Comprimidos de 50 mg e 100 mg.
t Cardiopatia isquêmica: enfarte agudo do miocárdio, angina.
t Hipertensão arterial sistêmica: uso não recomendado para pacientes com mais de 60 anos, gravidas e aqueles com intervalo QT longo.
t Hipersensibilidade ao atenolol.
t Choque cardiogênico.
t Bradicardia sinusal grave.
t Insuficiência cardíaca congestiva descompensada.
t Bloqueio atrioventricular de 2º e 3º graus.
t Asma brônquica ou doença pulmonar obstrutiva crônica.
t Acidose metabólica.
t Usar com cuidado nos casos de:
– uso de anestésicos que diminuam a função do miocárdio.
– interrupção do tratamento (risco de hipertensão rebote; suspender o fármaco gradualmente, no decurso de 1 a 2 semanas).
– uso concomitante com clonidina (interromper o uso do atenolol alguns dias antes da retirada da clonidina).
– história de doença broncoespástica, diabete melito (pode mascarar os sintomas de hipoglicemia), hipertireoidismo, doença vascular periférica e insuficiência cardíaca congestiva.
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): D (ver Apêndice a).
t Angina: dose inicial 50 mg, por via oral, a cada 24 horas. Se não obtiver resposta dentro de 1 semana, aumentar para 100 mg, por via oral, a cada 24 horas. Dose máxima diária: 200 mg.
t Enfarte agudo do miocárdio: 5 mg, por via intravenosa lenta (5 minutos), nas primeiras 12 horas após o enfarte, podendo repetir mais 5 mg após 10 minutos; seguido por 50 mg, por via oral, 15 minutos após o término da primeira dose por via intravenosa; e mais 50 mg, por via oral, após 12 horas; dose de manutenção 100 mg, por via oral, a cada 24 horas, durante pelo menos 10 dias.
t 25 a 100 mg, por via oral, a cada 24 horas. Dose máxima diária: 100 mg.
t Biodisponibilidade: 46 a 60%. a presença de alimento diminui a biodisponibilidade do atenolol.
t Início da ação: 3 horas.
t Pico de concentração: 2 a 4 horas.
t Duração da ação: 24 horas.
t Não é metabolizado no fígado
t Tempo de meia-vida: 6 a 7 horas.
t Excreção renal (40 a 50%) e fezes (50%), na forma inalterada.
t É removido por diálise.
t Bradicardia, extremidades frias (2,6%), hipotensão (4%), insuficiência cardíaca (0,4%).
t Diarreia (3%), náusea (3%).
t Tontura (13%), fadiga (26%), insônia, depressão (12%).
t Hipoglicemia em pacientes com diabetes tipo I, evitar uso; e hiperglicemia em pacientes com diabetes tipo II.
t Fadiga muscular (4%)
t Amiodarona, bloqueadores de canal de cálcio diidropiridínicos, diltiazem, fentanila, quinidina, verapamil: podem aumentar o efeito hipotensor, bradicardiazante do atenolol e o risco de parada cardíaca. Monitorar função cardíaca, particularmente em pacientes predispostos a insuficiência cardíaca, pode ser necessário ajuste de dose.
t Bloqueadores alfa-1-adrenérgicos (na primeira dose) e digoxina: podem ter seu efeito aumentado pelo atenolol. Monitorar paciente para sinais e sintomas específicos.
t Clonidina e moxonidina: em uso concomitante com atenolol, pode ocorrer crise hipertensiva durante a suspensao desses medicamentos. Suspender o betabloqueador antes de retirar a clonidina ou moxonidina, monitorar pressão arterial.
t Erva-de-são-joão (Hypericum perforatum) pode diminuir efeito do betabloqueador. Monitorar para sinais e sintomas de hipertensão e angina.
t Hipoglicemiantes podem ter os sintomas de hipoglicemia mascarados pelo atenolol e causar hiper ou hipoglicenia. Evitar uso concomitante, preferir betabloqueador cardiosseletivo, monitorar para sinais e sintomas específicos.
t Orientar para a importância de comunicar ao perceber qualquer sinal de efeito adverso.
t Alertar para não ingerir juntamente a suplementos de cálcio, antiácidos e suco de laranja.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se o horário da próxima dose for a menos de 8 horas, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Deve ser mantido ao abrigo de luz e umidade e à temperatura de 20 a 25 ºC.
Atenção: atenolol é um betabloqueador cardiosseletivo sem atividade simpaticomimética intrinseca e propriedades estabilizantes de membrana. Substituido pelo metoprolol no tratamento de arritmia. Em hipertensão não é recomendado para pacientes com mais de 60 anos, grávidas e aqueles com intervalo QT longo. a segurança e eficácia não está estabelecido em crianças.
Maurício Fábio Gomes
Na Rename 2010: Item 7.1
t Comprimido 50 mg.
t Prevenção de rejeição de transplantes
t Hipersensibilidade a azatioprina ou mercaptopurina.
t Uso prévio de agentes alquilantes.
t Gravidez: categoria de risco na gravidez (FDA): D (ver Apêndice a).
t Usar com cuidado nos casos de:
– toxicidade hematológica ou outra (pode ser necessário interromper o tratamento).
– supressão de medula óssea (identificar sinais ou sintomas e monitorar neutropenia ou trombocitopenia, semanalmente, nas primeiras 4 semanas, depois reduzir a frequência de monitoria para no mínimo a cada 3 meses).
– idosos (reduzir dose devido a diminuição da função renal).
t Lactação (ver apêndice B).
t Insuficiência renal (ver apêndice D).
t Insuficiência hepática (ver apêndice C).
t Dar 3 a 5 mg/kg, por via oral, no dia da cirurgia; no pós-operatório: 1 a 4 mg/ kg/dia, por via oral, de acordo com a resposta, após, reduzir a dose em 0,5 mg/kg/dia, a cada 4 semanas, até alcançar doses efetivas menores.
t Meia-vida de eliminação: 5 horas.
t Metabolismo: hepático (metabólito ativo: mercaptopurina).
t Excreção: urina e bile.
t Reações de hipersensibilidade: mal-estar, tontura, náuseas (12%), vômitos (12%), diarreia, febre, calafrio, dores musculares, artralgia, exantema, hipotensão, bradicardia, oligúria (raro), ou nefrites intestinais.
t Supressão da medula óssea (dose-dependente), leucopenia (5% a 16%) e trombocitopenia (reversíveis com a retirada), anemia megaloblástica.
t Hepatotoxicidade (3% a 10%), icterícia colestática; doença hepática venoclusiva.
t Aumento a susceptibilidade a infecções e colites em pacientes em uso concomitante de corticosteroides.
t Pancreatites (2% a 12%), pneumonites, alopecia, infecções por herpes zoster.
t a imunossupressão crônica aumenta o risco de neoplasias em humanos (raro).
t Alopurinol: aumento da toxicidade da azatioprina. Monitorar efeitos tóxicos e reduzir a dose da azatioprina (que deve ficar entre 1/3 a 1/4 da dose habitual).
t Captopril e outros inibidores da ECA: aumento do risco de mielossupressão. Evitar o uso concomitante.
t Ciclosporina: pode ter suas concentrações plasmáticas diminuídas. Monitorar níveis plasmáticos quando a ciclosporina for adicionada ao esquema terapêutico ou descontinuada.
t Ribavirina: aumenta o risco de mielotoxicidade. Evitar o uso concomitante. Caso a coadministração seja necessária, monitorar alterações hematológicas.
t Varfarina e outros anticoagulantes cumarínicos: têm efetividade diminuída. Monitorar paciente ao introduzir ou suspender a azatioprina e durante o uso concomitante. Ajustar a dose se necessário.
t Vacinas de vírus vivos: aumentam o risco de infecção pela vacina. a vacinação só deve ser realizada se o potencial benefício superar o risco.
t Orientar a ingestão do medicamento após as refeições para prevenir náuseas.
t Alertar para não tomar vacinas sem aprovação prévia.
t Orientar para notificar imediatamente a ocorrência de febre ou calafrios, tosse ou rouquidão, dores, hemorragias ou lesões inexplicáveis, lombalgia, melena, hematúria, enterorragia, ou petéquias, infecção ou dor de garganta.
t Orientar para adotar extremo cuidado para não se ferir ao proceder higiene pessoal.
t Não tocar os olhos ou mucosa nasal sem prévia lavagem das mãos.
t Armazenar à temperatura entre 15 e 25oC, ao abrigo da umidade, calor e luz.
Simone Sena Farina e Maria Inês de Toledo
Na Rename 2010: itens 5.1.6 e 5.2.1
t Comprimido de 500 mg.
t Pó para suspensão oral de 40 mg/mL.
t Infecção genital por Chlamydia trachomatis não complicada.
t Tracoma.
t Profilaxia para endocardite em pacientes alérgicos a penicilina ou em criança em substituição a clindamicina.
t Hipersensibilidade a azitromicina e outros macrolídeos.
t Insuficiência hepática (ver apêndice C).
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– crianças com menos de 6 meses de idade (não foi estabelecida a segurança do medicamento).
– suspeita de gonorreia concomitante (evitar uso de azitromicina devido ao rápido aparecimento de resistência).
– lactação (ver Apêndice B).
– miastenia grave.
t Categoria de risco na gravidez (FDA): B.
t Acima de 45 kg ou maiores de 8 anos: 1 g, por via oral, em dose única.
t 20 mg/kg, por via oral, em dose única.
t 15 mg/kg, por via oral, 30 a 60 minutos antes do procedimento.
t Acima de 45 kg: 1 g, por via oral, em dose única.
t Abaixo de 45 kg: 20 mg/kg, por via oral, em dose única.
t Acima de 45 kg: 1 g, por via oral, em dose única.
t Abaixo de 45 kg: 20 mg/kg, por via oral, em dose única.
t 500 mg, por via oral, 30 a 60 minutos antes do procedimento.
t Absorção adequada com e sem alimentos.
t Pico de concentração: 2,2 a 3,2 horas.
t Meia-vida de eliminação: 68 horas
t Metabolismo: hepático.
t Excreção: biliar e renal.
t Pode promover prolongamento do intervalo QT no eletrocardiograma.
t Diarreia (5%), dor abdominal (2,7% a 3%), náusea, vômito, alteração no paladar.
t Erosão córnea (menor que 1%).
t Cefaleia, tontura.
t Trombocitopenia.
t Ergotamina e análogos: aumento do risco de ergotismo agudo (náusea, vômito, isquemia vasospástica). Uso concomitante contraindicado.
t Digoxina: pode ter sua toxicidade aumentada. Monitorar clinicamente crianças recebendo digoxina.
t Disopiramida: pode resultar em arritmias cardíacas (prolongamento QTc, taquicardia ventricular). Evitar uso concomitante. Caso necessário, monitorar níveis de disopiramida.
t Fentanila: pode haver aumento ou prolongamento dos efeitos opioides, que devem ser monitorados. Se necessário, ajustar dose da fentanila.
t Nelfinavir: aumento do risco de Efeitos adversos da azitromicina, os quais devem ser monitorados.
t Pimozida: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsade de pointes, parada cardíaca). Uso concomitante contraindicado.
t Rifabutina: pode ter sua toxicidade aumentada. Se uso concomitante for necessário, utilizar com muita cautela.
t Teofilina: pode ter sua concentração plasmática aumentada. Monitorar concentrações plasmáticas.
t Varfarina: aumento do risco de sangramento. Monitorar tempo de protrombina e RNI. Caso necessário, ajustar a dose da varfarina.
t Orientar que pode ser administrada com alimentos.
t Continuar usando o medicamento pelo tampo determinado, mesmo após desaparecimento dos sintomas.
t Alertar para não administrar simultaneamente com antiácidos contendo alumínio ou magnésio.
t Orientar para a necessária agitação do frasco da suspensão oral antes de cada administração.
t Manter ao abrigo de luz e calor.
t Suspensão: manter à temperatura entre 5 e 30 °C após reconstituição. Descartar após 10 dias.
t Comprimidos: manter à temperatura entre 15 e 30 °C.
Consta no documento:
“Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial.”
O objetivo do site MedicinaNet e seus editores é divulgar este importante documento. Esta reprodução permanecerá aberta para não assinantes indefinidamente.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.