(Carregando Índice)... (Carregando Índice)... |
Última revisão: 11/11/2015
Comentários de assinantes: 0
Reproduzido de:
Formulário Terapêutico Nacional 2010: Rename 2010 [Link Livre para o Documento Original]
Série B. Textos Básicos de Saúde
MINISTÉRIO DA SAÚDE
Secretaria de Ciência, Tecnologia e Insumos Estratégicos
Departamento de Assistência Farmacêutica e Insumos Estratégicos
Brasília / DF – 2010
Karen Luise Lang
Na Rename 2010: item 18.6
t Comprimido 0,5 mg.
t Hiperprolactinemia.
t Inibir a lactação em mulheres HIV positivas.
t Hipersensibilidade ao fármaco ou a ergotamina e análogos.
t Hipertensão arterial sistêmica não controlada.
t Valvulopatias cardíacas.
t História de fibrose pulmonar, pericárdica, valvar cardíaca ou retroperitoneal.
t Usar com cuidado nos casos de:
– insuficiência hepática grave (pode elevar as concentrações plasmáticas de cabergolina) (ver Apêndice C).
– tratamento prolongado (eleva o risco de desenvolvimento de efusão pleural, fibrose pulmonar e valvapatia).
t Doses iniciais superiores a 1 mg podem provocar hipotensão ortostática, especialmente em pacientes em uso de anti-hipertensivos.
t Pode aumentar o risco de hipertensão, derrame e convulsões, especialmente em grávidas.
t Categoria de risco na gravidez (FDA): B (ver Apêndice A).
t Dose inicial: 0,5 mg, por via oral, 1 vez por semana ou dividida 2 vezes por semana em dias separados, com aumento de 0,5 mg a cada 4 semanas, até obtenção de resposta terapêutica, mantida posteriormente durante 6 meses. Doses maiores que 1 mg por semana, devem ser divididas 2 vezes por semana em dias separados. Dose máxima semanal: 2 mg.
t 1 mg, por via oral, 24 a 27 horas após o parto.
t Início da ação na hiperprolactinemia: 3 horas; para inibição da lactação: 24 horas.
t Pico de concentração: 3 horas.
t Metabolismo: preponderantemente hepático.
t Meia-vida: 63 a 69 horas.
t Excreção fecal (60%) e renal (22%).
t Obstipação (10%), náuseas (29%), diarreia, flatulência, dor abdominal (5%), dispepsia, xerostomia.
t Vertigens (17%), cefaleia (30%), crises de calor (3%), parestesia, sonolência (5%), depressão, insônia, confusão
t Fadiga (10%)
t Astenia (9%), dores musculares (2%)
t Hipotensão ortostática (10%), edema periférico, arritmias, distúrbios valvares
t Prurido (1%), rinite, reações alérgicas cutâneas, alopecia
t Dismenorreia
t Efusão pleural, fibrose pulmonar.
t Orientar para o caso de esquecimento de uma dose: ingerir assim que a paciente lembrar. Se o horário já estiver próximo da dose seguinte, não usar a dose do horário anterior e manter a escala regular de dose. Não dobrar a dose. O esquecimento de doses deve sempre ser comunicado ao médico.
t Alertar para notificar rapidamente a suspeita de gravidez durante o tratamento.
t Orientar para a necessidade de cautela ao realizar atividades que exijam atenção, como dirigir veículos e operar máquinas, pois o medicamento pode provocar sonolência.
t Orientar para executar lentamente movimentos como sentar e levantar para evitar vertigens.
t Acompanhar constantemente a pressão arterial.
t Manter ao abrigo de ar e luz e à temperatura ambiente, de 20 a 25 ºC. Proteger do calor, luz direta e umidade.
Fabiana Whal Hennigen
Na Rename 2010: itens 12 e 19
t Cápsula 0,25 microgramas.
t Hipocalcemia em hipoparatireoidismo e pseudohipoparatireoidismo.
t Hipocalcemia em pacientes sob diálise renal crônica.
t Hiperparatireoismo secundário em pacientes com insuficiência renal crônica moderada a grave.
t Tratamento e prevenção da osteoporose (uso restrito para pacientes com insuficiência renal).
t Hipercalcemia.
t Hipersensibilidade a calcitriol.
t Hipervitaminose D.
t Calcificação metastática.
t Usar com cuidado nos casos de:
– hipoparatireoidismo e diálise (instituir rotina de monitoria de cálcio e fosfato séricos).
– idosos com comprometimento coronário, hepático ou renal (ver Apêndice D).
– doença hepática grave (evitar o uso de calcitriol). (ver Apêndice C).
– síndromes de má-absorção (resposta terapêutica pode ser limitada ou imprevisível).
– lactação (ver Apêndice B).
t Avaliar a quantidade de vitamina D ingerida na dieta e em suplementos alimentícios.
t Adequada resposta a calcitriol depende de adequada ingestão de cálcio na dieta.
t Iniciar tratamento com a menor dose possível (monitoria do cálcio sérico é necessário para orientar aumento da dose).
t Doses excessivas podem levar a hipercalcemia crônica, calcificação vascular generalizada, nefrocalcinose e calcificações em outros tecidos.
t Manter adequada ingestão de água.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t 1 a 5 anos: dose inicial 0,25 microgramas, por via oral, a cada 24 horas, pela manhã; a dose pode ser aumentada em intervalos de 2 a 4 semanas; dose usual 0,25 a 0,75 microgramas, por via oral, a cada 24 horas.
t Maiores de 6 anos: dose inicial 0,25 microgramas, por via oral, a cada 24 horas, pela manhã; a dose pode ser aumentada em intervalos de 2 a 4 semanas; dose usual 0,5 a 2 microgramas, por via oral, a cada 24 horas.
t Com hemodiálise: dose inicial 0,25 a 2 microgramas, por via oral, a cada 24 horas; aumento da dose deve ser feito em intervalos de 4 a 8 semanas.
t Sem hemodiálise: dose inicial 0,014 a 0,041 microgramas/kg/dia; aumento da dose deve ser feito em intervalos de 4 a 8 semanas.
t Pré-diálise:
– maiores de 3 anos: dose inicial 0,25 microgramas, por via oral, a cada 24 horas; a dose pode ser aumentada para 0,5 microgramas, por via oral, a cada 24 horas;
– menores de 3 anos: dose inicial 0,01 a 0,015 microgramas/kg por via oral a cada 24 horas.
t Diálise: 0,25 a 2 microgramas, por via oral, a cada 24 horas.
t Dose inicial 0,25 microgramas, por via oral, a cada 24 hora, pela manhã; a dose pode ser aumentada em intervalos de 2 a 4 semanas; dose usual 0,5 a 2 microgramas, por via oral, a cada 24 horas.
t Dose inicial 0,25 microgramas, por via oral, a cada 24 ou 48 horas; aumentos de 0,25 microgramas por dia podem ser feitos em intervalos de 4 a 8 semanas; dose usual 0,5 a 1 microgramas, por via oral, a cada 24 horas.
t Pré-diálise: dose inicial 0,25 microgramas, por via oral, a cada 24 horas; a dose pode ser aumentada para 0,5 microgramas, por via oral, a cada 24 horas.
t Diálise: dose inicial 0,25 microgramas, por via oral, a cada 24 ou 48 horas; aumentos de 0,25 microgramas por dia podem ser feitos em intervalos de 4 a 8 semanas; dose usual 0,5 a 1 microgramas, por via oral, a cada 24 horas.
t 0,25 microgramas, por via oral, a cada 12 horas.
t Absorção: rápida.
t Início de ação: 2 a 6 horas
t Pico de concentração: 3 a 6 horas.
t Duração de ação: 3 a 5 dias.
t Meia-vida de eliminação: 5 a 8 horas
t Hipercalcemia (33%), hipercalciúria, hipermagnesemia, hiperfosfatemia.
t Arritmia cardíaca, hipertensão, hipotensão.
t Cefaleia, irritabilidade, sonolência, psicose. t Prurido, eritema multiforme, dermatite.
t Anorexia, fraqueza, náusea, vômito, diarreia, obstipação, xerostomia, paladar metálico, polidipsia, pancreatite.
t Calcificação tecidual, dor óssea, mialgia, distrofia.
t Conjuntivite, fotofobia.
t Nefrotoxicidade, insuficiência renal, poliúria.
t Aumento das enzimas hepáticas.
t Hipertermia.
t Diminuição da libido.
t Diuréticos tiazídicos: aumento dos níveis séricos de cálcio resultando em hipercalcemia. Monitorar os níveis de cálcio e, se necessário, descontinuar um ou ambos os fármacos.
t Carbonato de magnésio: risco de hipermagnesemia. A administração concomitante de calcitriol e antiácidos contendo magnésio não é recomendada. Observar o paciente quanto a intoxicação por magnésio (letargia, fraqueza, hiporreflexia e hipertensão).
t Orientar para administrar junto das refeições para redução dos efeitos gastrintestinais.
t Orientar ao paciente em diálise para evitar o uso de antiácidos contendo magnésio.
t Orientar para não usar suplementos vitamínicos ou outras formas de vitamina D.
t Estimular a ingestão de alimentos ricos em cálcio, como leite e derivados.
t Estimular para aumentar ou adotar exposição diária ao sol.
t Orientar para a necessidade de fazer hidratação abundante.
t Armazenar a temperatura ambiente entre 20 e 25 °C, em recipiente fechado e protegido da luz.
Atenção: sinonímia: 1,25-dihidroxicolecalciferol, vitamina D3 ativa
Rosa Martins
Na Rename 2010: item 14.4.5
t Comprimido 25 mg.
t Urgência hipertensiva.
t Hipersensibilidade ao captopril ou outros inibidores da ECA.
t Hipersensibilidade a sulfonamidas.
t História de angioedema
t Usar com cuidado nos casos de:
– uso concomitante com diurético (pode causar hipotensão mesmo com a primeira dose; reduzir a dose do diurético e iniciar o captopril em dose baixa e acompanhar pressão arterial).
– elevação das enzimas hepáticas ou ocorrência de icterícia durante o tratamento (monitorar função hepática; retirar imediatamente o captopril nesses casos).
– doença vascular periférica; cardiomiopatia hiperrarófica; estenose de arteria aórtica ou renal; angioedema intestinal, de cabeça e de pescoço; cirurgia/anestesia.
– história de alergias (atenção, pode ocorrer angioedema mesmo com a primeira dose).
– crianças (segurança e eficácia não estabelecidas).
– insuficiência renal (ver Apêndice D)
– lactação (ver Apêncice B).
t Monitorar níveis de potássio, especialmente se houver insuficiência renal.
t Categoria de risco na gravidez (FDA): C, para o primeiro trimestre; e D, para segundo e terceiro trimestres (ver Apêndice A).
Adultos
t Urgência hipertensiva.
t 25 mg, por via oral. Repetir em uma hora se necessário.
t Alimentos diminuem a absorção de captopril.
t Biodisponibilidade: 70 a 75%
t Início da ação: 15 a 30 minutos.
t Pico de concentração: 30 a 90 minutos.
t Duração da ação: 6 horas.
t Metabolismo hepático (50%), metabólitos inativos.
t Meia-vida de eliminação: 1,9 horas.
t Excreção: renal (predominantemente em forma inalterada).
t Dialisável (20-50%).
t Hipotensão (> 1%), taquicardia (1%), palpitação (1%).
t Tosse (0,5 a 2%)
t Cefaleia
t Prurido sem exantema (2%), exantema (4 a 7%), angioedema (0,1%)
t Hiperpotassemia > 5,1 mmol/L (11%)
t Proteinúria (0,7%)
t Alfainterferona 2, alopurinol, azatioprina, diuréticos poupadores de potássio, suplementos de potássio podem ter a efetividade/toxicidade aumentada pelo captopril. Acompanhar sinais e sintomas específicos.
t Bupivacaína, clorpromazina, diuréticos de alça (primeira dose), diuréticos tiazídicos (primeira dose) podem aumentar o efeito do captopril. Acompanhar sinais e sintomas específicos.
t Ácido acetilsalicílico ou anti-inflamatórios não-esteroides: podem diminuir a efetividade do captopril. Acompanhar sinais e sintomas específicos.
t Alertar que alimentos reduzem a absorção.
t Alertar que pode causar tosse.
t Orientar para evitar medicamentos que aumentem o potássio sérico.
t Alertar para recorrer a atendimento médico caso surjam edema de face, transtorno para respirar ou deglutir e rouquidão.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar entre 15 e 30 °C, proteger do calor, umidade e luz direta.
t Comprimidos podem apresentar leve odor sulfuroso.
t Existe descrita formulação extemporânea para uso em criança.
Tatiana Aragão Figueiredo
Rachel Magarinos-Torres
Na Rename 2010: itens 13.1 e 13.2
t Comprimido 200 mg.
t Suspensão oral 20 mg/mL.
t Crises convulsivas parciais simples e complexas (primeira escolha) e secundariamente generalizadas.
t Convulsões tônico-clônicas generalizadas.
t Transtorno bipolar, durante a latência ou em ausência de resposta ou intolerância ao lítio.
t Antecedentes de mielossupressão.
t Alterações hematológicas, como agranulocitose, leucopenia e porfiria.
t Anomalias na condução atrioventricular.
t Hipersensibilidade a carbamazepina ou a antidepressivos tricíclicos.
t Uso de inibidores da monoamina oxidase, concomitante ou nos últimos 14 dias.
t Usar com cuidado nos casos de:
– hepatopatia, alterações hematológicas relacionadas à utilização de medicamentos, reações cutâneas, glaucoma, dependência ao álcool, diabete melito, antecedentes de crises de ausência atípica, antecedentes de distúrbio de condução cardíaca.
– porfiria hepática, pelo risco de crise de porfiria.
– lactação (ver Apêndice B).
– idosos (reduzir a dose inicial definida para adultos).
– suspensão do tratamento (deve ser gradual para reduzir o risco de recidiva e estado de mal epiléptico).
t Verificar concentração plasmática até regularidade do efeito e depois uma a duas vezes ao ano. A medida deve ser realizada em jejum, antes da dose matinal.
t Hipersensibilidade cruzada com anticonvulsivantes como fenitoína e fenobarbital.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t 100 a 200 mg, por via oral, divididos a cada 8 horas.
t 200 a 400 mg, por via oral, divididos a cada 8 horas.
t Dose inicial 10 a 20 mg/kg/dia, por via oral, divididos a cada 6 horas (solução oral) ou 8 horas (comprimido), aumentada semanalmente até obter a resposta clínica desejada. Dose máxima diária: 35 mg/kg.
t Dose inicial 200 mg, por via oral, divididos a cada 6 horas (solução oral) ou 12 horas (comprimido), aumentada semanalmente em 100 mg por dia, administrado por via oral, a cada 6 a 8 horas (solução oral ou comprimido) até obter resposta clínica desejada. Dose de manutenção usual 400 a 800 mg, por via oral, a cada 6 a 8 horas. Dose máxima diária: 1000 mg.
t Dose inicial 400 mg, por via oral, divididos a cada 6 horas (solução oral) ou 12 horas (comprimido), aumentada semanalmente em 200 mg por dia, administrado por via oral, a cada 6 a 8 horas até obter resposta clinica. Dose de manutenção usual 800 a 1.000 mg, por via oral, a cada 6 a 8 horas para crianças até 15 anos, e até 1.200 mg, por via oral, a cada 6 a 8 horas para crianças acima de 15 anos.
t Dose inicial de 100 a 200 mg, por via oral, uma a duas vezes ao dia.
t Aumentar a dose conforme a resposta; dose de 5 a 9 mg/kg/dia determinam níveis efetivos.
t Dose de manutenção: 400 a 1.200 mg/dia (excepcionalmente pode ser necessária dose de 1.600 a 2.000 mg/dia), fracionada em 3 tomadas.
t Dose inicial 400 mg, por via oral, divididos a cada 12 horas, aumentados até controle dos sintomas. Dose de manutenção usual 400 a 600 mg, por via oral, a cada 8 a 12 horas. Dose máxima diária: 2.000 mg.
t Absorção oral aumentada na presença de alimentos.
t Biotransformação hepática, originando metabólito mais ativo. Carbamazepina induz seu próprio metabolismo em 3-5 semanas de um regime de dose fixa.
t Pico sérico: 4 horas. Níveis plasmáticos regulares são atingidos em 2-10 dias.
t Meia-vida de eliminação: 12 a 17 horas.
t Excreção renal do metabólito (72%) e da forma ativa (menos de 3%). Parcialmente excretada nas fezes após administração oral (28%).
t Náuseas e vômitos (acima de 10%), diarreia (1 a 10%).
t Sonolência, vertigens, cefaleia, ataxia, diplopia, nistagmo, confusão, tremor, prejuízo cognitivo (acima de 10%)
t Hipertermia e síndrome neuroléptica maligna (abaixo de 1%).
t Síndrome de Stevens-Johnson e necrólise epidérmica tóxica (1% a 10%)
t Erupção cutânea, acne, eritema multiforme, alopecia (abaixo de 1%).
t Hiponatremia (4% a 22% dos pacientes), diaforese (1% a 10%), síndrome de secreção inapropriada de hormônio antidiurético (1% a 10%).
t Discrasias sanguíneas, anemia aplástica e agranulocitose, hepatotoxicidade, anormalidades cardíacas, insuficiência renal aguda, hipersensibilidade pulmonar aguda, neurite periférica, hipotireoidismo, porfiria, ganho de peso, pancreatite, visão turva, retinopatia, osteomalácia (todos abaixo de 1%)
t Artralgia, febre, linfonodomegalia, discinesias, paraestesia, depressão, impotência, infertilidade masculina, ginecomastia, galactorreia, psicose, fotossensibilidade, angioedema (frequência desconhecida).
t Amoxapina, amitriptilina: têm sua concentração plasmática reduzida.
Verificar adequada resposta clínica aos fármacos, sinais de toxicidade da carbamazepina e níveis séricos de ambos os agentes; ajustar doses quando necessário.
t Aripiprazol: tem sua concentração plasmática reduzida. Aumentar a dose do aripiprazol.
t Clozapina: aumento do risco de supressão da medula óssea, tremores no pulso (asteríxis) ou redução dos níveis séricos de clozapina. Acompanhar resposta clínica ao uso da clozapina e o aparecimento de agranulocitose. Considerar redução de dose tanto da clozapina como da carbamazepina.
t Delavirdina: tem seu nível sérico reduzido. O uso concomitante não é recomendado.
t Doxepina: tem sua efetividade reduzida e ocorre aumento da toxicidade da carbamazepina (diplopia, visão turva, tonturas, tremores). Acompanhar resposta clínica à doxepina e sinais de toxicidade da carbamazepina. Pode ser necessário ajuste de doses.
t Efavirenz: redução das concentrações plasmáticas do efavirenz e/ou da carbamazepina. Considerar opção anticonvulsivante para pacientes que receberam efavirenz, uma vez que não há recomendação de ajuste de dose para este caso.
t Erva-de-são-joão (Hypericum perforatum): alteração nas concentrações plasmáticas da carbamazepina. Usar dose consistente de erva-de-são-joão de produto confiável com concentrações consistentes. Acompanhar concentrações de carbamazepina se o paciente informar perda do controle das convulsões ao utilizar erva-de-são-joão concomitante. Interrupção do uso da erva-de-são-joão exige monitoria de níveis e sintomas de toxicidade da carbamazepina (sonolência, ataxia, fala arrastada, nistagmo, reações distônicas, alucinações e vômito).
t Felbamato: redução da eficácia da carbamazepina ou do felbamato.
Acompanhar as concentrações séricas de carbamazepina
t Fenitoína, fosfenitoína: aumento das concentrações de fenitoína e redução das concentrações de carbamazepina. Medir níveis séricos tanto da fenitoína como da carbamazepina após o início ou a interrupção de um ou de outro agente, com o adequado ajuste posológico. Verificar níveis séricos após ajustes de dose e periodicamente.
t Fluconazol, flunarizina, macrolídeos, propoxifeno, vigabatrina: aumento do risco de toxicidade por carbamazepina (ataxia, nistagmo, diplopia, cefaleia, vômitos, apneia, convulsões, coma). Verificar concentrações séricas de carbamazepina, sinais de toxicidade e ajustar a dose caso seja necessário uso concomitante.
t Haloperidol: redução da eficácia do haloperidol. Recomenda-se observação dos pacientes para verificar adequada resposta clínica ao haloperidol. Pode ser necessário um aumento de dose do haloperidol.
t Irinotecano: tem sua eficácia reduzida, em razão do aumento do metabolismo pela carbamazepina. Considerar a substituição por anticonvulsivante que não seja indutor enzimático. Iniciar substituição por 2 semanas antes da utilização do irinotecano.
t Lapatinibe: redução das concentrações plasmáticas de lapatinibe, pelo aumento do metabolismo pela carbamazepina. Verificar as concentrações séricas de lapatinibe e considerar aumento da dose.
t Lopinavir, ritonavir: redução da ação do lopinavir e aumento nos níveis e toxicidade da carbamazepina. Uso concomitante a lopinavir/ritonavir pode induzir o metabolismo do lopinavir. Coadministração a lopinavir/ritonavir pode resultar em aumento nos níveis e toxicidade da carbamazepina. Se necessário uso concomitante, reduzir dose de carbamazepina em 25-50% e acompanhar o paciente quanto a níveis de carbamazepina 3-5 dias antes de iniciar inibidor de protease.
t Midazolam: tem sua eficácia reduzida. Pode ser necessário aumento da dose de midazolam para obtenção de resposta hipnótica.
t Nefazodona: redução das concentrações plasmáticas e da eficácia da nefazodona e de seu metabólito ativo. Aumento do risco de toxicidade pela carbamazepina (ataxia, nistagmo, diplopia, cefaleia, vômitos, apneia, convulsões, coma). O uso concomitante é contraindicado.
t Oxcarbazepina: tem sua concentração plasmática reduzida por aumento do metabolismo pela carbamazepina. Acompanhar os pacientes para adequada resposta clínica à oxcarbazepina.
t Sertralina: aumento do risco de toxicidade (ataxia, nistagmo, diplopia, cefaleia, vômitos, apneia, convulsões, coma). Por causa do aumento em potência dos níveis de carbamazepina, os pacientes devem ser cuidadosamente observados quanto a qualquer sinal de toxicidade. Medir níveis séricos de carbamazepina em 2-3 semanas após início da associação ou descontinuação da sertralina, podendo ser necessário ajuste de dose. Pode ocorrer perda de eficácia da sertralina.
t Tramadol: redução da eficácia tramadol e aumento do risco de convulsões, pelo aumento do metabolismo pela carbamazepina. O uso concomitante não é recomendado.
t Vecurônio: tem a duração da ação reduzida, em razão do aumento de sua depuração endógena pela carbamazepina. Observar o paciente quanto a adequada resposta clínica ao bloqueador neuromuscular. Pode ser necessária a administração de doses menores de vecurônio ou em intervalos maiores.
t Orientar a procura de serviço de saúde na ocorrência de febre, dor de garganta, erupções cutâneas, úlceras bucais, hematoma ou hemorragia.
t Orientar quanto a possibilidade de afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Alertar para não suspender abruptamente o tratamento.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Conservar sob temperaturas entre 15 e 30 ºC, em recipientes bem fechados e protegidos da luz. Proteger de umidade, já que um terço ou mais da eficácia pode ser perdida se armazenado nessas condições.
t Agitar bem o solução oral antes de utilizar. Não deve ser administrado simultaneamente com outros medicamentos ou diluentes líquidos.
Atenção: antes do início e durante o tratamento, a cada 6 meses, devem ser realizados hemograma (especialmente plaquetas e reticulócitos), ferro plasmático e testes de função hepática. Este medicamento possui um número elevado de Efeitos adversos : avaliar em particular cada uma.
Fabiana Wahl Hennigen
Na Rename 2010: item 11
t Comprimido 1.250 mg (equivalente a 500 mg Ca2+).
t Tratamento e prevenção de deficiência de cálcio.
t Tratamento de hiperfosfatemia em pacientes com insuficiência renal avançada ou associada a hiperparatireoidismo.
t Prevenção de pré-eclampsia com risco elevado de hipertensão e ingestão pobre em cálcio.
t Hipercalcemia.
t Cálculo renal.
t Hipofosfatemia.
t Hipercalciúria.
t Usar com cuidado nos casos de:
– acloridria ou hipocloridria, comum em idosos (a absorção de carbonato de cálcio pode ser reduzida; administrar junto das refeições ou considerar o uso de outro sal de cálcio).
– hipoparatireoidismo tratado por período prolongado, junto de altas doses de vitamina D (podem ocorrer hipercalcemia e hipercalciúria).
– insuficiência renal (ver Apêndice D), sarcoidose e história de nefrolitíase.
t A administração é seguida por aumento da secreção ácida gástrica em até 2 horas da administração.
t Observação: os esquemas de administração estão baseados em dose de cálcio elementar.
t 50 a 150 mg/kg, por via oral, divididas a cada 4 a 6 horas. Dose máxima diária: 1 g.
t 45 a 65 mg/kg, por via oral, divididas a cada 6 horas.
t 1 mês a 1 ano: 120 mg, por via oral, a cada 6 ou 8 horas, antes ou durante as refeições, ajustadas se necessário.
t 1 a 6 anos: 300 mg, por via oral, a cada 6 ou 8 horas, antes ou durante as refeições, ajustadas se necessário.
t 6 a 12 anos: 600 mg, por via oral, a cada 6 ou 8 horas, antes ou durante as refeições, ajustadas se necessário.
t 12 a 18 anos: 1,25 g, por via oral, a cada 6 ou 8 horas, antes ou durante as refeições, ajustadas se necessário.
t 1 a 2 g/dia, por via oral, dividido a cada 6 a 8 horas, junto de refeições. Tratamento de hiperfosfatemia associada a doença renal crônica ou hiperparatiroidismo secundário
t 2,5 g, por via oral, em doses divididas, aumentado até 17 g/dia, em doses divididas, se necessário.
t 1,0 a 2,0g, em doses divididas.
t Absorvido no intestino delgado, dependendo da presença de vitamina D, pH no lúmen, idade, dose e presença ou ausência de alimentos. Alimentos podem aumentar a absorção do cálcio. A absorção é mínima a menos que doses altas e por tempo prolongado sejam administradas. Cálcio é absorvido na forma solúvel, ionizada. A solubilidade é aumentada em ambiente ácido.
t com frequência entre 1% e 10%:
– Hipercalcemia, hipofosfatemia.
– Cefaleia.
– Obstipação, efeito laxativo, hipersecreção gástrica e rebote ácido (doses altas ou uso prolongado), náusea, vômito, dor abdominal, flatulência, anorexia e xerostomia
– Síndrome do leite alcalino com doses altas ou por tempo prolongado (cefaleia, náusea, irritabilidade, fraqueza, alcalose, hipercalcemia, insuficiência renal).
– Calculose urinária.
t Amprenavir: pode haver redução da eficácia do antiviral. Separar a administração em pelo menos uma hora.
t Dasatinibe: redução da exposição ao dasatinibe e da sua concentração plasmática. Evitar o uso concomitante ou administrar o carbonato de cálcio duas horas antes ou após o dasatinibe.
t Digoxina: aumento do risco de toxicidade (arritmia e colapso cardiovascular). A administração concomitante não é recomendada.
t Erlotinibe: redução da absorção do erlotinibe. Separar a administração por algumas horas.
t Fosfatos orais: redução da absorção do fosfato. Separar a administração em pelo menos uma hora.
t Hidroclorotiazida/clorotiazida: aumento do efeito do diurético tiazídico. Risco de síndrome do leite alcalino. Evitar a ingestão excessiva de cálcio (antiácidos, laticínios). Verificar o cálcio sérico e/ou a função da paratireoide.
t Itraconazol: redução da sua eficácia. Administrar o produto contendo cálcio pelo menos 1 hora antes ou 2 horas após o itraconazol.
t Lansoprazol: redução da biodisponibilidade do lansoprazol. Administrar pelo menos uma hora após o carbonato de cálcio.
t Levotiroxina: redução da absorção da levotiroxina. Separar a administração em pelo menos 4 horas. Acompanhar os níveis séricos do hormônio estimulante da tireoide.
t Poliestirenossulfonato de sódio: risco de alcalose metabólica. Separar a administração tanto quanto possível. Considerar a administração do poliestirenosssulfonato por via retal. Reconhecer manifestação de alcalose.
t Propranolol: redução da biodisponibilidade do propranolol. Separar a administração tanto quanto possível. Avaliar a eficácia do propranolol.
t Quinolonas (como ciprofloxacino): redução da sua eficácia. Evitar a administração concomitante. Administrar a quinolona pelo menos 2 horas antes ou após o produto que contenha cálcio. Avaliar a eficácia antibiótica.
t Tetraciclinas: redução da sua eficácia. A administração concomitante não é recomendada. Separar a administração em pelo menos 3 a 4 horas. Verificar a eficácia antibiótica.
t Ticlopidina: redução da sua eficácia. A administração concomitante não é recomendada. Administrar a ticlopidina pelo menos 1 a 2 horas antes do carbonato de cálcio.
t Tipranavir: redução da sua eficácia. Administrar uma hora antes ou duas horas após o carbonato de cálcio.
t Zalcitabina: redução da sua eficácia. Separar a administração tanto quanto possível.
t Orientar para a administração com refeições.
t Estimular a prática de exercícios físicos, pela importância na construção e manutenção da massa óssea e prevenção da osteoporose.
t Explicar que adequadas quantidades de vitamina D ou exposição solar auxiliam na absorção de cálcio.
t Orientar para evitar uso concomitante de alimentos ricos em fibras, álcool, fumo ou cafeína.
t Estocar sob temperatura entre 15 e 30 ºC, em recipiente bem fechado.
Fabiana Wahl Hennigen
Na Rename 2008: item 19
t Comprimido 500 mg de carbonato de cálcio + 400 UI de colecalciferol
t Tratamento e prevenção de osteoporose.
t Prevenção de fraturas não-vertebrais em idosos com baixa ingestão de cálcio.
t Hipersensibilidade ao colecalciferol, ergocalciferol ou a metabólitos da vitamina D, como calcitriol.
t Hipercalcemia.
t Hipervitaminose D.
t Calcificação metastática.
t Ver demais contraindicações na monografia do carbonato de cálcio.
t Usar com cuidado nos casos de:
– arteriosclerose e condições cardíacas (exacerbação em potência relacionada aos efeitos hipercalcêmicos persistentes durante o uso terapêutico).
– uso concomitante de produtos contendo cálcio, outras preparações contendo vitamina D ou seus análogos ou diuréticos tiazídicos (pode aumentar o risco de hipercalcemia).
– hiperlipidemia (aumento em potência dos níveis de LDL).
– hiperfosfatemia (risco de calcificação metastática; tornar normais os níveis de fosfato antes do início da terapia).
– hepatopatia (ver apêndice C).
– insuficiência renal (ver Apêndice D).
– sarcoidose e outras doenças granulomatosas (aumento em potência da sensibilidade ao colecalciferol).
– doses diárias de colecalciferol acima de 400 UI, por períodos prolongados (fazer monitoria de cálcio sérico e urinário; cálcio sérico deve ser mantido entre 9 e 10 mg/dL).
– idosos (maior risco de deficiência de vitamina D, especialmente durante o inverno ou em indivíduos institucionalizados).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): não classificado (ver apêndice A)
t Ver demais precauções na monografia do carbonato de cálcio
t 1 comprimido, por via oral, a cada 12 horas durante as refeições.
t Boa absorção após administração oral. A absorção é reduzida na presença de disfunção hepática ou biliar ou síndromes de má-absorção.
t Requer hidroxilação via renal para ser ativada, formando o metabólito 1,25-dihidroxicolecalciferol (calcitriol).
t Apresenta início de ação lento e duração prolongada.
t Início de resposta: 10 a 14 dias (deficiência de vitamina D).
t Duração de ação: até 6 meses, após múltiplas doses. Vitamina D é armazenada no fígado e tecido adiposo, prolongando os efeitos hipercalcêmicos.
t Meia-vida de eliminação: 19 a 48 horas.
t Ver demais aspectos farmacocinético na monografia do carbonato de cálcio
t Os Efeitos adversos da sobredose de vitamina D incluem anorexia, cansaço, náusea e vômito, diarreia ou obstipação, perda de peso, noctúria, polidipsia, poliúria, transpiração, cefaleia, vertigem, alterações mentais e aumento da concentração de cálcio e fosfato no plasma e na urina.
t Manifestações crônicas incluem proteinúria e disfunção renal; calcificação tecidual (nefrolitíase e nefrocalcinose); hipertensão e possivelmente arritmias; piora dos sintomas gastrintestinais; pancreatite; e psicose.
t Efeitos dislipidêmicos, caracterizados por redução dos níveis de HDL e aumento dos de LDL.
t Ver demais reações adversas na monografia do carbonato de cálcio
t Cimetidina: redução da concentração sistêmica do colecalciferol. Considerar suplemento de vitamina D. Acompanhar o paciente quanto a reações adversas relacionadas a deficiência de vitamina D, incluindo sinais e sintomas de hipocalcemia e hiperparatireoidismo secundário.
t Ver demais interações na monografia do carbonato de cálcio.
t Orientar ao paciente que a dose diária recomendada de vitamina D em adultos pode ser obtida pela exposição à luz solar e por dieta.
t Orientar para administrar junto de refeições ou com leite.
t Ver demais orientações na monografia do carbonato de cálcio
t Armazenar sob temperatura entre 2 e 8 ºC, em recipiente bem fechado.
Proteger da luz.
Atenção: colecalciferol = vitamina D3.
Cláudia Du Bocage Santos Pinto
Gabriela Costa Chaves
Na Rename 2010: item 13.2
t Comprimido 300 mg.
t Tratamento da mania.
t Profilaxia da mania no transtorno bipolar (prevenção de recidivas).
t Hipersensibilidade ao lítio.
t Insuficiência renal (ver Apêndice D), insuficiência cardíaca e pacientes muito enfraquecidos.
t Desidratação e depleção de sódio.
t Usar com cuidado nos casos de:
– ocorrência de diarreia, vômito e infecção intercorrente, especialmente se estiver associada à sudorese intensa (reduzir a dose ou interromper o uso).
– quatro ou mais episódios de doença bipolar por ano (cicladores rápidos) e em obesos (profilaxia com lítio pode falhar).
– idosos (administrar doses mais baixas e manter maior vigilância em virtude da função renal diminuída).
– crianças com menos de 12 anos de idade (eficácia e segurança não foram determinadas).
– psoríase (risco de exarcebação), miastenia grave e submetidos a cirurgias.
– lactação (ver Apêndice B).
t Os efeitos relacionam-se às concentrações séricas, sendo indispensável a monitoria.
t A capacidade de tolerar o lítio é maior durante a fase aguda, decaindo com a diminuição dos sintomas.
t Toxicidade pode ocorrer mesmo com litemia normal, se houver fatores de descompensação da homeostasia orgânica.
t Fazer monitoria de níveis séricos de lítio, para atingir e manter concentração sérica emtre 0,4 a 1 mEq/L, com retirada de sangue entre 8 e 12 horas após a dose precedente:
– 4 dias após início do tratamento.
– semanalmente, até que haja controle.
– pelo menos a cada 3 meses, após controle.
t Evitar retirada abrupta.
t Cerca de 20 a 30% dos pacientes são refratários ao tratamento.
t Acompanhar função renal (depuração de creatinina endógena) (ver Apêndice D) e função tireoidiana a cada 6-12 meses em esquemas que obtiveram controle.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t As doses são inicialmente divididas ao longo do dia, mas prefere-se a administração única diária quando a concentração sérica do lítio está sob controle.
t 600 a 1800 mg, por via oral, divididos a cada 6 a 8 horas.
t 600 a 1200 mg, por via oral, divididos a cada 12 a 24 horas.
t Dose inicial 300 a 600 mg, por via oral, divididos a cada 12 ou 24 horas, aumentado semanalmente em 300 mg, até a dose capaz de fazer cessar os sintomas. Raramente são necessários mais de 900 a 1.200 mg por dia.
t 300 a 900 mg, por via oral, divididos a cada 12 ou 24 horas.
t Rápida absorção; completa em 6 a 8 horas.
t Pico de concentração plasmática: 0,5 a 2 horas.
t Concentração plasmática terapêutica para controle do distúrbio bipolar: 0,4 a 1 mEq/L, excepcionalmente em casos agudos até 1,5 mEq/L. Acima de 1,5 mEq/L podem ocorrer efeitos tóxicos.
t Início da ação terapêutica e melhora clínica: 1 a 3 semanas.
t Meia-vida de eliminação: 14 a 24 horas (adultos), 18 horas (adolescentes) e acima de 36 horas (idosos).
t Eliminação renal (89 a 98%), em forma ativa.
t Cerca de metade da concentração sanguínea de lítio é excretada pelo leite materno.
t Os efeitos e gravidade estão relacionados a sensibilidade do indivíduo e alta concentração sérica do lítio.
t Eletrocardiograma (ECG) anormal, arritmia, hipotensão, edema, bradicardia, síncope, bradiarritmia (grave), arritmia cardíaca, hipotensão, disfunção do nodo sinusal.
t Diarreia, náusea (branda), vômito, xerostomia.
t Irritabilidade muscular, fraqueza muscular, miastenia grave.
t Tremor fino, hiperreflexia, reflexo tendinoso profundo, sonolência, vertigem, confusão, fadiga, letargia, cefaleia, ataxia, disartria, coma, pseudotumor cerebral, aumento da pressão intracraniana, papiledema, convulsões.
t Escotoma transitório, visão turva, nistagmo.
t Insuficiência renal, albuminúria, glicosúria, oligúria, poliúria, polidipsia, incontinência urinária.
t Leucocitose, trombocitose.
t Hipo e hipertireoidismo, bócio atóxico, hiperglicemia, diabetes insípido (sinal de toxicidade grave), aumento da concentração do hormônio antidiurético.
t Disfunção sexual
t Antagonistas da angiotensina II (candesartana cilexetila, losartana, telmisartana, valsartana): aumento das concentrações e da toxicidade do lítio (fraqueza, tremor, sede excessiva, confusão). Verificar concentrações e sinais de toxicidade pelo lítio.
t Antagonistas da dopamina: fraqueza, discinesia, sintomas extrapiramidais, encefalopatia e dano cerebral. Recomenda-se monitoria cuidadosa dos pacientes quanto a sintomas extrapiramidais, especialmente se altas doses de antagonistas da dopamina são administradas. Níveis plasmáticos de lítio devem ser periodicamente avaliados.
t Anti-inflamatórios não-esteroides (como ácido mefenâmico, cetoprofeno, cetorolaco, diclofenaco, piroxicam, valdecoxibe): aumento das concentrações de lítio, levando a toxicidade. Durante o uso concomitante desses medicamentos, o paciente deve ser observado quanto ao aparecimento de efeitos tóxicos do lítio e deve-se intensificar a monitoria da concentração plasmática de lítio.
t Calcitonina: redução das concentrações de litio, com perda da sua eficácia.
t Carbamazepina: risco de neurotoxicidade aditiva.
t Diltiazem: risco aumentado de neurotoxicidade e psicose. Observar o surgimento de efeitos neurotóxicos. Deve-se fazer monitoria da concentração sérica de lítio.
t Dipirona, mazindol e metronidazol: risco de aumento das concentrações de lítio, levando à toxicidade.
t Diuréticos tiazídicos (como bendroflumetiazida, hidroclorotiazida, politiazida) e de alça (exemplo, furosemida): aumento das concentrações e da toxicidade do lítio (fraqueza, tremor, sede excessiva, confusão). Verificar concentrações e sinais de toxicidade pelo lítio. Fazer monitoria das concentrações de lítio nos primeiros 5 a 7 dias após a introdução ou descontinuação do diurético e na sequência do tratamento; pode ser necessário reduzir a dose do lítio durante a terapia concomitante.
t Fenelzina e outros inibidores não-seletivos da MAO: aumento do risco de hiperpirexia maligna. Evitar o uso concomitante. Esperar 2 semanas após a descontinuação do inibidor da MAO para introduzir o lítio.
t Inibidores da ECA (alacepril, benazepril, cilazapril, espirapril, fosinopril, lisinopril, perindopril, quinapril, trandolapril, zofenopril): aumento das concentrações de lítio, levando a toxicidade e/ou nefrotoxicidade. Recomendase avaliar a concentração plasmática de lítio durante o tratamento concomitante e atentar para o surgimento dos efeitos tóxicos do lítio. Pode ser necessária diminuição da dose de lítio.
t Inibidores seletivos de recaptação de serotonina: o uso com lítio pode resultar em aumento das concentrações de lítio e/ou síndrome serotoninérgica. Avaliar concentrações de lítio e observar pacientes quanto a sinais e sintomas, como alterações neuromusculares e de estado mental (inclusive delírio).
t Ioimbina: aumento do risco de mania.
t Linezolida e sibutramina: aumento do risco de síndrome serotoninérgica (hipertensão, hipertermia, alterações no estado mental). Evitar coadministração com lítio.
t Suxametônio: prolongamento do bloqueio neuromuscular.
t Verapamil: perda do controle da mania, neurotoxicidade e bradicardia. Verificar cuidadosamente os níveis séricos de lítio. Os pacientes devem ser observados quanto ao surgimento de sinais de mania ou psicose, assim como sintomas de neurotoxicidade como ataxia, tremores, zumbidos, náusea, vômito ou diarreia.
t Alertar para manter adequada ingestão de líquidos durante o uso do medicamento.
t Orientar que os comprimidos devem ser ingeridos com muito líquido, a fim de garantir trânsito intestinal.
t Orientar que os comprimidos devem ser ingeridos logo após as refeições para propiciar aumento na absorção.
t Alertar para a necessidade de suplemento de sal nos períodos de muito calor, quando há perda de água e sais por sudorese.
t Alertar para evitar mudanças na alimentação que possam reduzir ou aumentar a ingestão de sódio.
t Alertar para evitar bebidas com alto teor de cafeína.
t Alertar sobre a importância de comunicar o aparecimento de sintomas de hipotireoidismo, como sensação de frio e letargia (é maior o risco em mulheres).
t Alertar para evitar a realização de atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Em caso de esquecimento de uma dose, não tomar duas doses juntas.
t Não se deve escolher outra formulação ou preparação do medicamento sem conhecimento do médico.
t Conservar sob temperaturas inferiores entre 15 e 30 oC, em recipientes bem fechados.
Atenção: a biodisponibilidade de lítio pode mudar segundo a formulação farmacêutica.
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 6.1.5
t Solução injetável 10 mg/mL
t Câncer de ovário avançado (tratamento inicial em combinação com outros quimioterápicos).
t Câncer de ovário avançado (tratamento paliativo de doença recorrente, incluindo pacientes previamente tratados com cisplatina).
t Carcinoma pulmonar de não-pequenas células metastático.
t Carcinoma pulmonar de pequenas células avançado.
t Hipersensibilidade a cisplatina/platina ou manitol.
t Mielossupressão ou hemorragia significante.
t Risco de leucemia secundária à cisplatina.
t Idosos e indivíduos previamente tratados com cisplatina (maior risco para neuropatia periférica induzida por carboplatina).
t Tratamento prévio com aminoglicosídeos (maior risco de nefrotoxicidade).
t Evitar extravasamento durante a administração.
t Requer hidratação intravenosa intensiva.
t Agulha ou outro material que contenha alumínio (não utilizar; o alumínio reage com carboplatina e forma um precipitado inativo.
t Insuficiência renal (ver Apêndice D).
t Lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Administrar 300 mg/m2, por via intravenosa, no dia 1, a cada 4 semanas, durante 6 ciclos, em combinação com a ciclofosfamida 600 mg/m2 no dia 1 dia, a cada 4 semanas, durante 6 ciclos; As doses subsequentes de carboplatina pós-tratamento devem ser ajustadas de acordo com a contagem de plaquetas e neutrófilos; A dose (em mg) de carboplatina pode ainda ser calculada, de acordo com a fórmula de Calvert, pelo produto da Área sob a Curva ( mg/ mL/minuto) e o Índice de Filtração Glomerular (mL/minuto) somada a 25; nas siglas em inglês, area under curve e glomerular filtration rote – D = AUC x (IFG + 25).
t Administrar 360 mg/m2, por via intravenosa, no dia 1 a cada 4 semanas ou dose baseada na fórmula de Calvert, com alvo de Área sob a Curva (AUC) de 4-6 mg/mL/min.
t Administrar por via intravenosa, AUC-alvo de 6 mg/mL/min, em combinação com 200 mg/m2 de paclitaxel e 15 mg/kg de bevacizumabe a cada 3 semanas por 6 ciclos; continuar bevacizumabe até progressão da doença.
t Como monoterapia, administrar 250 a 450 mg/m2, por via intravenosa, a cada 4 semanas
t Administar 300 mg/m2, por via intravenosa, em combinação com 50 mg/m2 de epirrubicina, no dia 1, e 100 mg/m2 de etoposídeo nos dias 1 a 3; repetir a cada 4 semanas.
t A carboplatina é degrada em platina no organismo
t A platina não se distribui adequadamente no tecido adiposo e nervoso
t De 60 a 80% da platina é eliminada pelos rins
t Meia-vida de eliminação total da platina do plasma após administração intraperitoneal da carboplatina é de 6,7 a 7,7 dias
t O fármaco é removido eficientemente por hemodiálise
t Náuseas e vômitos
t Colite neutropênica
t Anemia; leucopenia (15%), em pacientes com pré-tratado de câncer de ovário (26%); neutropenia (16%), em pacientes com pré-tratamento de câncer de ovário (21%)
t Desequilíbrio eletrolítico
t Mielossupressão
t Reações de hipersensibilidade (2% a 9,2%).
t Neuropatia periférica
t Distúrbios visuais (raro)
t Vacina rotavírus humano G1P1[8] (atenuada): aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Se a vacina for necessária, administrá-la depois de três meses da descontinuação da quimioterapia.
t Topotecano: pode resultar em mielossupressão. Evitar o uso concomitante ou em sequência.
t Fenitoína: pode resultar na redução da efetividade da fenitoína; determinar os níveis plasmáticos da fenitoína e ajustar a dose se necessário, durante e após o tratamento com carboplatina.
t Varfarina: pode resultar na redução da efetividade da varfarina com risco aumentado de hemorragias. Verificar tempo de protrombina e ajustar a dose se necessário.
t Antes de iniciar o tratamento é importante identificar: história prévia de hipersensibilidade a carboplatina, manitol e produtos de platina, gravidez e lactação, doenças renais, problemas auditivos, infecções.
t Utilizar métodos contraceptivos efetivos enquanto fazer uso de carboplatina. Em casos da ocorrência de gravidez, informar seu médico.
t Evitar vacinas a não ser que aprovadas pelo médico.
t Informar ao médico se ocorrer sangramento incomum.
t Cuidado ao escovar os dentes, passar fio ou palito nos dentes.
t Não tocar os olhos ou o interior do nariz a não ser que esteja com as mãos limpas.
t Cuidado para não se cortar com barbeador ou cortador de unhas.
t Evitar esportes de contato ou outras situações as quais pode ocorrer sangramento ou lesão.
t Evitar pessoas que apresentem infecções ou que tenham recebido vacina oral contra poliomielite.
t Armazenar os frascos de carboplatina sob temperatura ambiente, entre 15 e 30 °C, proteger da luz.
t Observar orientação específica do produto quanto a diluição, compatibilidade e estabilidade da solução.
t Carboplatina em solução, quando preparada de acordo com as instruções, apresenta estabilidade por 8 horas sob temperatura ambiente (25°C) e tem pH de 5 a 7. Soluções devem ser descartadas após 8 horas, pois não existe qualquer conservante antibacteriano na formulação.
t Carboplatina pode ser diluída com água para injeção, ou solução injetável de glicose 5% ou cloreto de sódio 0,9%. A concentração final após diluição deve ser de 10 mg/mL.
t Cerca de 5% da concentração inicial do fármaco é perdida durante 24 horas quando a solução for diluída em cloreto de sódio 0,9% e sob controle a 25 °C, portanto, uma solução para infusão contínua por períodos superiores a 24 horas não deve ser preparada com solução injetável de cloreto de sódio 0,9%.
t A carboplatina reage com alumínio formando precipitado, assim, agulhas, seringas, cateteres ou equipos contendo alumínio não devem ser utilizados para administrar carboplatina.
t Carboplatina é incompatível com soluções de fluoruracila, mesna, bicarbonato de sódio, anfotericina B, cloridrato de clorpromazina, diazepam, tiopental sódico, cloridrato de procainamida, fenitoína sódica, lansoprazol, folinato de cálcio.
Atenção: mielossupressão grave relacionada à dose, resultando em infecção ou sangramento. Anemia secundária ao acúmulo de doses pode requerer transfusão sanguínea. Considerar, em potência, a ocorrência de reações anafiláticas
Isabella Campagnuci Knust
Na Rename 2010: item 8.1
t Pó para uso oral.
t Antídoto inespecífico usado em intoxicações exógenas agudas por: acetilcisteína, fenotiazinas, ácido acetilsalicílico, ácido mefenâmico, indometacina, ácido valproico, iodetos, anfetaminas, ipeca, antidepressivos tricíclicos, antimônio, cloreto de mercúrio, arsênio, morfina, atropina, cloreto de metiltionínio (azul de metileno), ópio, barbitúricos, organofosforados, cânfora, paracetamol, carbamazepina, paraquat, clordiazepóxido, paration, clorfeniramina, penicilina, cloroquina, prata, cocaína, primaquina, colchicina, probenecida, propoxifeno, propantelina, diazepam, mepacrina, digitálicos, quinidina, estricnina, quinina, etclorvinol, salicilato de metila, fenilbutazona, selênio, sulfonamidas, fenitoína, teofilina, fenol e tetraciclinas.
t Intoxicação por hidrocarbonetos.
t Intoxicação por substâncias corrosivas.
t Íleo paralítico.
t Perfuração gastrintestinal.
t Obstrução intestinal.
t Cirurgia recente.
t Risco de hemorragia gastrintestinal.
t Usar com cuidado nos casos de:
– diminuição do grau de consciência (risco de aspiração; recomenda-se intubação nasogástrica).
t Gravidez e lactação (falta de prova de segurança).
t Carvão ativado não é efetivo em intoxicações por álcool, DDT, cianetos, ferro, lítio, potássio, ácido bórico, carbamatos, bases e ácidos fortes.
t Não há prova quanto à efetividade de doses repetidas.
t 1g/kg, ou até 50 g, por sonda nasogástrica, administrado até 1 hora ou o mais breve possível após a intoxicação.
t 1 a 2 g/kg, ou até 50 a 100 g, por sonda nasogástrica, administrado até 1 hora ou o mais breve possível após a intoxicação.
t O carvão vegetal ativado não é absorvido pelo trato gastrintestinal.
t Presença de alimentos prejudica a capacidade de ligação do carvão ativado.
t O tempo médio de trânsito intestinal do carvão é de 25 horas, contra 1,1 hora para carvão em sorbitol.
t Mais frequentes: fezes enegrecidas, vômitos, obstipação.
t Menos frequentes: aspiração de partículas de carvão, causando lesão pulmonar.
t Em razão de possibilidade de redução da absorção gastrintestinal durante o tratamento com carvão vegetal ativado, recomenda-se que medicamentos de uso contínuo sejam administrados por via parenteral.
t Laxantes: associação pode ocasionar hipopotassemia, hipernatremia, hipermagnesemia e acidose metabólica.
t Alertar para a frequência do aparecimento de fezes enegrecidas.
t Comprimidos de carvão não contêm carvão “ativado”, não devendo ser utilizados em casos de intoxicações.
t Carvão ativado na forma de suspensão possui grande superfície de adsorção disponível, adequada para adsorção de tóxicos.
t Suspensões com sorbitol ou edulcorantes são preferidas por mascarar o gosto desagradável.
t A mistura com sorbitol a 70% proporciona suspensão espessa e doce, mais palatável que carvão em água, e tem efeito laxativo que corrige o efeito constipante do carvão. O carvão pode ser misturado a xarope de cereja ou chocolate, sacarina, frutose e sacarose, sem perda de eficácia.
t Suspensões mantidas em frascos lacrados à temperatura ambiente, de 15 a 30ºC, podem ser armazenadas por até 1 ano.
Atenção: restringir o uso repetido a substâncias que diminuem o esvaziamento gástrico: carbamazepina, dapsona, quinina, fenobarbital, amitriptilina, dextropropoxifeno, digoxina, disopiramida, nadolol, fenilbutazona, fenitoína, piroxicam, sotalol e teofilina. 1, 3, 4
Rosa Martins
Na Rename 2010: item 14.1
t Comprimido 3,125 mg, 6,25 mg, 12,5 mg e 25 mg.
t Insuficiência cardíaca congestiva (ICC).
t Hipersensibilidade ao carvedilol ou a outros betabloqueadores.
t Bloqueio atrioventricular de segundo ou terceiro grau.
t Bradicardia grave.
t Asma brônquica ou broncoespasmo.
t Choque cardiogênico.
t Insuficiência cardíaca descompensada e necessitando de terapia inotrópica intravenosa.
t Insuficiência hepática grave (ver Apêndice C).
t Síndrome do nó sinoatrial.
t Usar com cuidado nos casos de:
– retirada do medicamento (deve ser gradual, principalmente em pacientes com doença da artéria coronária; retirada abrupta pode exacerbar angina e pode desencadear enfarte do miocárdio e arritmia ventricular).
– história de reação anafilática a vários alérgenos (pode aumentar a reatividade e diminuir a resposta à epinefrina).
– bradicardia – abaixo de 55 batimentos por minuto (reduzir dose, se necessário).
– diabete melito (o carvedilol pode mascarar sintoma de hipoglicemia, como a taquicardia, e pode piorar a hiperglicemia em pacientes com ICC).
– doença cardíaca isquêmica, doença vascular difusa, insuficiência renal e hipotensão (pressão sistólica abaixo 100 mmHg) pode piorar função renal de paciente com ICC (reduzir dose ou interromper o uso).
– feocromocitoma.
– tirotoxicose (o carvedilol pode mascarar sinais de hipertireoidismo).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C e D (ver Apêndice A).
t Dose inicial: 3,125 mg, por via oral, a cada 12 horas, com alimentos. Dobrar a dose a cada 2 semanas até a maior dose tolerada. Dose máxima diária: 50 mg para pacientes com menos de 85 kg e 100 mg com mais de 85 kg.
t Biodisponibilidade: 25 a 35%
t Início da ação: 1 hora (em hipertensão)
t Pico de concentração: 1 a 1,5 horas.
t Duração da ação: 24 horas (em hipertensão)
t Metabolismo hepático, extenso (reações de fase I e II). Há extenso metabolismo de primeira passagem. Metabolitos ativos.
t Meia-vida de eliminação: 6 a 10 horas.
t Excreção: 16% renal, 60% fezes.
t Dialisável: não.
t Hipotensão (2 a 20%), angina (2 a 6%), bloqueio atrioventricular (1 a 3%), bradiarritmia (2 a 10%), edema (5%), edema periférico (1 a 7%), palpitação (1 a 3%), sincope (0,1 a 4%).
t Prurido sem exantema (até 1%), exantema (até 1%).
t Diabetes (até 3%), hipoglicemia (até 3%), hiperglicemia (5 a 12%), hiperco-
lesterolemia (até 4%), hiperpotassemia (até 1%), ganho de peso (10 a 12%).
t Diarreia (1 a 12%), náusea (4 a 9%), vômito (1 a 6%)
t Trombocitopenia (até 3%)
t Artralgia (1 a 6%)
t Astenia (7 a 11%), tontura (6 a 33%), cefaleia (5 a 8%)
t Visão anormal (5%)
t Fadiga (24%)
t Aumento sérico da ureia nitrogenada (6%)
t Disfunção erétil (13%), impotência (até 3%),
t Tosse (5 a 8%), crepitações respiratórias (4%)
t Amiodarona, bloqueadores de canal de cálcio diidropiridínicos, cimetidina, diltiazem, fentanila, mebefradil, verapamil: podem aumentar o efeito hipotensor, bradicardizante do carvedilol e risco de parada cardíaca. Acompanhar a função cardíaca, particularmente em pacientes predispostos à insuficiência cardíaca. Pode ser necessário ajuste de dose.
t Antagonistas de receptores alfa-adrenérgicos (na primeira dose), digoxina: podem ter seu efeito aumentado pelo carvedilol. Acompanhar o paciente quanto a sinais/sintomas específicos.
t Hipoglicemiantes: sintomas de hipoglicemia podem ser mascarados pelo carvedilol. Pode surgir hiper ou hipoglicemia. Evitar uso concomitante, preferir betabloqueador cardiosseletivo, acompanhar quanto a sinais/sintomas específicos.
t Erva-de-são-joão (Hypericum perforatum), rifampicina, rifapentina: podem reduzir efeito do carvedilol. Estar atento ao surgimento de hipertensão e angina.
t Epinefrina, arbutamina e dobutamina podem ter a efetividade diminuída pelo carvedilol. Evitar uso concomitante; suspender o carvedilol 48 horas antes do uso da arbutamina); verificar pressão arterial (hipertensão), bradicardia reflexa e resistência a epinefrina em anafilaxia.
t Alertar para sinal/sintoma de hipotensão, principalmente tontura.
t Recomendar uso com alimento para diminuir o risco de hipotensão ortostática.
t Não interromper o uso do medicamento abruptamente.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar entre 15 e 30 °C, proteger do calor, umidade e luz direta.
Atenção: início do efeito na ICC após 3 meses; não é tratamento de emergência.
Fernando de Sá Del Fiol e Silvio Barberato Filho
Na Rename 2010: item 5.1.3
t Cefalexina: cápsula ou comprimido 500 mg.
t Cefalexina monoidratada: suspensão oral 50 mg/mL.
t Tratamento de infecções por microrganismos sensíveis (cocos gram-positivos aeróbios, exceto enterococos; Staphylococcus aureus produtor de penicilinase, mas não contra os oxacilina-resistentes; Escherichia coli; Proteus mirabilis e Klebsiella pneumoniae).
t Hipersensibilidade às cefalosporinas.
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– insuficiência hepática.
– hipersensibilidade a penicilinas (pode apresentar hipersensibilidade cruzada).
t Pode induzir colite pseudomembranosa por Clostridium difficile.
t Pode causar diminuição da atividade de protrombina.
t Categoria de risco na gravidez (FDA): B.
t 25 a 50 mg/kg, por via oral, dividido a cada 6 horas. Em infecções graves, 50 a 100 mg/kg, por via oral, dividido a cada 6 horas. Dose máxima diária: 4 g.
t 250 a 500 mg, por via oral, a cada 6 horas. Em infecções graves, 1 g, por via oral, a cada 6 horas. Dose máxima diária: 4 g.
t Boa absorção pelo trato digestório, mesmo em presença de alimentos.
t Pico de concentração plasmática: 1 a 1,5 hora.
t Meia-vida: 1 a 2 horas; 20 a 24 horas (insuficiência renal).
t Excreção: renal (90% em forma inalterada).
t Diarreias, náuseas e vômitos
t Hipersensibilidade cruzada em 10% dos pacientes alérgicos às penicilinas.
t Hepatotoxicidade transitória.
t Possibilidade de desenvolvimento de colite pseudomembranosa
t Urticárias e dermatites (2%)
t Aminoglicosídeos: intensificação da toxicidade renal pelos dois fármacos.
Fazer monitoria da função renal do paciente, especialmente nefropatas.
t Colestiramina: diminuição da efetividade da cefalexina por diminuição de sua absorção. Administrar a cefalexina uma hora antes ou quatro a seis horas depois da colestiramina.
t Metformina: aumento dos níveis plasmáticos de metformina, por diminuição de sua excreção. Acompanhar a associação, com especial atenção aos Efeitos adversos da metformina (diarreia, náuseas, vômitos, cefaleias). Considerar redução na dose de metformina.
t Probenecida: aumento do tempo de meia-vida da cefalexina por competição na excreção (secreção tubular). Combinação indicada para aumentar os níveis plasmáticos de cefalexina.
t Orientar que pode ser tomada com alimento ou leite para evitar desconforto gástrico.
t Orientar agitar a suspensão antes de usar. Alertar para a observação cuidadosa da validade da suspensão após a reconstituição.
t Orientar para notificar imediatamente manifestações alérgicas.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas, sob risco de desenvolvimento de resistência bacteriana.
t Conservar à temperatura ambiente, entre 15 e 30 ºC, em recipientes bem fechados.
t Depois da reconstituição, a suspensão mantém-se estável por 7 a 14 dias, se refrigerada. Não congelar. Observar instruções do produtor.
Fernando de Sá Del Fiol e Silvio Barberato Filho
Na Rename 2010: item 5.1.3
t Cefalotina sódica: pó para solução injetável 1 g
t Uso restrito para tratamento de infecções por microrganismos Susceptíveis a cefalosporinas de 1ª geração e para preservar o uso de cefazolina para quimioprofilaxia cirúrgica.
t Hipersensibilidade às cefalosporinas.
t Meningites (não penetra a barreira hematoencefálica).
t Infecções por anaeróbios (sem atividade significante).
t Usar com cuidado nos casos de:
– história de hipersensibilidade aguda às penicilinas.
– história de colite pseudomembranosa.
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C). t Categoria de risco na gravidez (FDA): B.
t 25 mg/kg, por via intravenosa, a cada 6 horas.
t 80 a 160 mg/kg, por via intravenosa, dividido a cada 4 a 6 horas. Dose máxima diária: 10 a 12 g.
t 500 mg, por via intravenosa, a cada 6 horas. Em infecções graves, com risco de morte, 2 g, por via intravenosa, a cada 4 horas. Dose máxima diária: 12 g.
t Pico sérico: 30 a 45 minutos (intramuscular).
t Meia-vida de eliminação: 30 a 60 minutos (aumentada na insuficiência renal).
t Metabolismo: hepático.
t Excreção: predominantemente renal (70%).
t Hipersensibilidade cruzada com penicilina (10%).
t Tromboflebites, dor no lugar da injeção, especialmente em altas doses (maiores que 6 g/dia).
t Diarreia, náuseas.
t Trombocitopenia, neutropenia, leucopenia, trombocitose.
t Febre.
t Nefrotoxicidade.
t Aminoglicosídeos: intensificação da toxicidade renal pelos dois fármacos.
Fazer monitoria da função renal do paciente, especialmente nefropatas.
t Probenecida: aumento do tempo de meia-vida da cefalexina por competição na excreção (secreção tubular). Combinação indicada para aumentar os níveis plasmáticos de cefalexina.
t Pode aparecer dor no lugar da injeção.
t Caso ocorra edema periorbital ou perioral após o uso do medicamento, procurar serviço de saúde.
t Conservar sob temperatura ambiente e protegido da luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t O diluente padrão é solução de glicose a 5%.
t Após reconstituição, manter refrigerado, com possibilidade de uso até 10 dias. À temperatura ambiente, usar até 48 horas.
t Pode precipitar em contato com aminoglicosídeos, tetraciclinas e barbitúricos.
Fernando de Sá Del Fiol e Maria Inês de Toledo
Na Rename 2010: item 5.1.3
t Cefazolina sódica: pó para solução injetável 1 g.
t Profilaxia de infecções pós-cirúrgicas.
t Hipersensibilidade às cefalosporinas.
t Usar com cuidado nos casos de:
– história de hipersensibilidade imediata às penicilinas.
– história de colite pseudomembranosa.
– insuficiência renal (ver Apêndice D).
– insuficiência hepática.
– teste de Coombs (pode apresentar falso positivo).
– administração em bolo (deve ser lenta, em 3 e 5 minutos).
t Reservar para a profilaxia de infecção pós-cirúrgica (não usar como tratamento de infecções Susceptíveis a fim de manter o emprego em hospital).
t Categoria de risco na gravidez (FDA): B.
t 25 mg/kg, por via intravenosa, em dose única, 30 minutos antes do procedimento. Dose máxima: 1 g.
t 1 g, por via intravenosa, em dose única, 30 minutos antes do procedimento (no momento da indução anestésica). Em transoperatório prolongado, a dose pode ser repetida a cada 3 horas.
t Pico sérico: 12 horas (intramuscular).
t Meia-vida de eliminação: 2 horas (intramuscular) e 1,8 horas (intravenosa).
t Tem penetração biliar, óssea, mas não no sistema nervoso central, mesmo na existência de inflamação.
t Excreção: renal.
t Hipersensibilidade cruzada com penicilina (10%).
t Tromboflebites, dor no lugar da injeção.
t Diarreia, náuseas, vômitos, anorexia, colite, cólica intestinal.
t Hepatite colestática.
t Trombocitopenia, neutropenia, leucopenia, trombocitose.
t Febre, convulsão.
t Insuficiência renal.
t Varfarina: pode aumentar o risco de sangramento. Ajustar a dose de varfarina para manter o efeito anticoagulante desejado ou trocar por outra cefalosporina sem ação na diminuição do tempo de protrombina.
t Não usar se houver antecedentes de reações alérgicas do tipo imediata às penicilinas ou de qualquer tipo às cefalosporinas.
t Caso ocorra edema periorbital ou perioral após o uso do medicamento, procurar serviço de saúde.
t Armazenar à temperatura ambiente (20 a 25 ºC) e ao abrigo de ar e luz.
t Soluções reconstituídas são utilizáveis por 24 horas (temperatura ambiente) ou 10 dias se mantidas sob refrigeração (+ 5 oC).
t Não congelar.
t Não se recomenda emprego de cloreto de sódio 0,9% como diluente pelo perigo de cristalização. Deve-se utilizar água estéril para injeção. Agitar bem após reconstituição.
Fernando de Sá Del Fiol e Silvio Barberato Filho
Na Rename 2010: item 5.1.3
t Pó para solução injetável 500 mg
t Tratamento de infecções causadas por bacilos gram-negativos aeróbios (que não Pseudomonas) e cocos gram-positivos aeróbios (que não enterococos) multirresistentes, em neonatos.
t Hipersensibilidade às cefalosporinas.
t Usar com cuidado nos casos de:
– história de hipersensibilidade às penicilinas.
– imaturidade renal (ajustar as doses).
– insuficiência renal (ver Apêndice D)
t A terapia não deve ser superior a 10 dias, podendo causar granulocitopenia ou agranulocitose.
t De 0 a 4 semanas e menor que 1,2 kg: 50 mg/kg, por via intravenosa durante 3 a 5 minutos, ou intramuscular, a cada 12 horas.
t Menos de uma semana e maior que 2,0 kg: 50 mg/kg, por via intravenosa durante 3 a 5 minutos, ou intramuscular, a cada 8 ou 12 horas.
t Uma semana ou mais e entre 1,2 e 2,0 kg: 50 mg/kg, por via intravenosa durante 3 a 5 minutos, ou intramuscular, a cada 8 horas.
t Uma semana ou mais e maior que 2,0 kg: 50 mg/kg, por via intravenosa durante 3 a 5 minutos, ou intramuscular, a cada 6 ou 8 horas.
t Pico de concentração plasmática: 30 minutos (intramuscular).
t Meia-vida de eliminação em adultos: 0,8 a 1,4 horas
t Meia-vida de eliminação em maiores que 1,5 kg: 4,6 horas
t Metabolismo: hepático.
t Excreção: renal.
t Após administração em bolo podem ocorrer arritmias cardíacas.
t Dor no lugar da injeção (4%).
t Náuseas, vômitos, diarreia (1,4%), colite.
t Reações alérgicas (2,4%).
t Agranulocitose e anemia hemolítica (1%), trombocitopenia e leucopenia (3,8%).
t Não usar se houver antecedentes de reações alérgicas do tipo imediata às penicilinas ou de qualquer tipo às cefalosporinas.
t Caso ocorra edema periorbital ou perioral após o uso do medicamento, procurar serviço de saúde.
t Armazenar à temperatura ambiente (20 a 25 ºC) e ao abrigo de ar e luz.
t A reconstituição pode ser feita com água estéril e solução glicosada (5%), mas o volume utilizado depende da concentração final e da via a ser administrada (intravenosa ou intramuscular). Recorrer a orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Soluções reconstituídas são utilizáveis por 12 a 24 horas (temperatura ambiente) ou de 7 a 10 dias se mantidas sob refrigeração (abaixo de 5oC).
t Não diluir em soluções alcalinas como bicarbonato de sódio ou com aminoglicosídeos.
Fernando de Sá Del Fiol e Simone Sena Farina
Na Rename 2010: item 5.1.3
t Pó para solução injetável 1 g.
t Tratamento de infecções causadas por Pseudomonas ssp
t Hipersensibilidade às cefalosporinas.
t Usar com cuidado nos casos de:
– história de hipersensibilidade as penicilinas (pode apresentar reação cruzada).
– história de colite.
– insuficiência renal – risco de convulsões e encefalopatia (ver Apêndice D).
t Categoria de risco na gravidez (FDA): B.
t 30 mg/kg, por via intravenosa, a cada 12 horas.
t 30 a 50 mg/kg, por via intravenosa, a cada 8 horas. Dose máxima diária: 6 g.
t 30 a 50 mg/kg, por via intravenosa, a cada 8 horas. Dose máxima diária: 6 g.
t Após administração intramuscular, atinge pico de concentração plasmática em uma hora.
t Tem boa penetração biliar e no SNC.
t Há excreção renal (80 a 90% de forma inalterada) em 24 horas.
t Meia-vida de eliminação: 1 a 2 horas.
t Diarreia, náuseas, vômitos, desconforto abdominal.
t Flebite e dor no lugar da injeção.
t Reações alérgicas (2%).
t Cloranfenicol: diminui a efetividade da ceftazidima. Evitar a combinação, mas caso for necessária, acompahar o paciente quanto a evolução da infecção.
t Pode aparecer dor no lugar da injeção.
t Não usar se houver antecedentes de reações alérgicas do tipo imediata às penicilinas ou de qualquer tipo às cefalosporinas.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Após reconstituição, mantém estabilidade por 12 horas à temperatura ambiente (15 a 30oC), ou por 3 dias, sob refrigeração (2 a 8oC).
Fernando de Sá Del Fiol e Silvio Barberato Filho
Na Rename 2010: item 5.1.3
t Pó para solução injetável 500 mg e 1 g.
t Tratamento de infecções causadas por bacilos gram-negativos aeróbios (que não Pseudomonas) e cocos gram-positivos aeróbios multirresistentes.
t Tratamento em dose única de infecções por Neisseria gonorrhoeae.
t Tratamento empírico de meningites.
t Hipersensibilidade às cefalosporinas.
t Neonatos, especialmente com alterações no metabolismo da bilirrubina (ictéricos).
t Usar com cuidado nos casos de:
– história de hipersensibilidade imediata a betalactâmicos.
– história de colite.
– uso prolongado (induz superinfecção).
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– neonatos prematuros (risco de kernicterus). t Categoria de risco na gravidez (FDA): B.
t 20 a 50 mg/kg/dia, por via intravenosa em infusão contínua durante 60 minutos. Dose máxima diária: 50 mg/kg.
t 20 a 50 mg/kg, por via intramuscular profunda ou intravenosa, administrado durante 2 a 4 minutos, dividido a cada 12 ou 24 horas, ou por via intravenosa em infusão contínua. Em infecções graves, até 80 mg/kg/dia. Doses de 50 mg/kg ou mais devem ser admnistradas somente por via intravenosa em infusão contínua. Dose máxima diária: 2 g.
t 100 mg/kg/dia, por via intravenosa, dividido a cada 12 ou 24 horas, durante 7 a 14 dias. Dose máxima: 4 g/dia.
t 1 a 2 g, por via intramuscular profunda ou intravenosa, administrado durante 2 a 4 minutos, a cada 12 ou 24 horas, ou por via intravenosa em infusão contínua. Doses superiores a 1 g por via intramuscular devem ser divididas em mais de um lugar de aplicação. Doses superiores a 1 g por via intravenosa devem ser administradas somente por infusão contínua. Dose máxima diária: 4 g.
t 4 g, por via intravenosa em infusão contínua, dividido a cada 12 ou 24 horas. Dose máxima diária: 4 g.
t 250 mg, por via intramuscular profunda, em dose única. Aspectos farmacocinéticos clinicamente relevantes 3, 4
t Pico sérico: 1 a 3 horas (intramuscular).
t Meia-vida de eliminação: 6 a 8 horas. Em neonatos e crianças, de 4 a 6 horas.
t Excreção: urina (30 a 60%) e bile.
t Náuseas, vômitos, diarreia (3%) e desconforto abdominal, colite.
t Hepatotoxicidade transitória.
t Neonatos: deslocamento da bilirrubina.
t Sedimentos na vesícula biliar (descontinuar o uso do fármaco).
t Reações alérgicas
t Leucopenia, trombocitopenia, eosinofilia, agranulocitose, anemia hemolítica
t Eritema multiforme.
t Soluções contendo cálcio (gliceptato de cálcio, cloreto de cálcio, solução de Ringer, solução de Ringer+lactato, acetato de cálcio e gliconato de cálcio) não devem ser administradas com ceftriaxona sódica (intravenosa) pelo risco de ocorrer precipitação de ceftriaxona cálcica em neonatos. Não misturar nem administrar pela mesma linha de infusão soluções contendo cálcio (nem mesmo nutrição parenteral). Em outros pacientes que não neonatos, pode ser realizada administração em sequencia de ceftriaxona e soluções contendo cálcio se entre as infusões as linhas forem cuidadosamente lavadas com um fluido compatível.
t Alertar para a possibilidade de surgir dor no lugar da injeção.
t O produto deve ser protegido de luz e mantido a temperatura ambiente.
t A cor da solução de ceftriaxona pode ser de amarelo ao âmbar, dependendo do tempo de estoque, concentração e diluente.
t Não administrar com soluções contendo cálcio.
t Quando utilizar lidocaína como diluente na administração intramuscular, consultar Efeitos adversos na monografia da lidocaína.
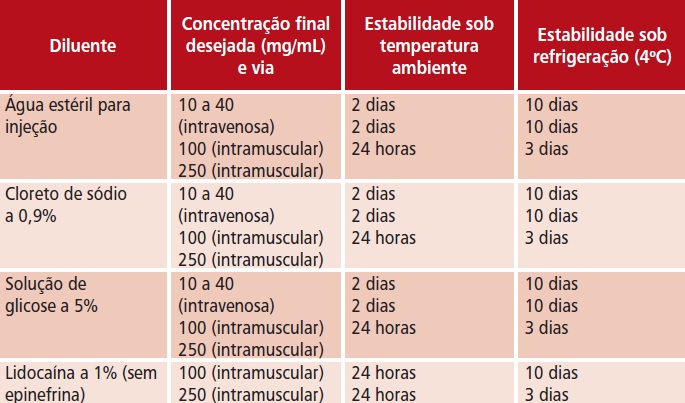
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução. Após reconstituição, seguir a tabela de estabilidade.
t Compatibilidade e estabilidade de ceftriaxona sódica em solução:

Mirian Parente Monteiro
Na Rename 2010: item 5.3.2
t Xampu 2%.
t Dermatite seborreica, inclusive do couro cabeludo (caspa).
t Pitiríase versicolor.
t Hipersensibilidade ao cetoconazol.
t crianças (eficácia e segurança não foram estabelecidas em crianças com menos de 12 anos de idade).
t idosos (alguns pacientes podem ser mais sensíveis aos Efeitos adversos desse medicamento).
t Categoria de risco na gravidez (FDA): C.
t Aplicar a cada 3 a 4 dias durante 2 a 4 semanas (ocasionalmente, o uso pode ser mais prolongado do que 8 semanas), quantidade suficiente sobre os cabelos molhados, massageando o couro cabeludo até formação de espuma abundante. Enxaguar e repetir a operação.
t Aplicar 1 vez ao dia durante 5 dias, quantidade suficiente para umedecer toda a área afetada. Espumar e deixar agir durante 5 minutos, depois enxaguar.
t Não há absorção sistêmica considerável através da pele com o uso do xampu.
t Irritação nos olhos.
t Ressecamento e prurido em pele e couro cabeludo.
t Ressecamento, modificação de textura e aumento da perda normal dos cabelos.
t Ressecamento ou oleosidade do cabelo ou do couro cabeludo; cefaleia, prurido, hiperemia, queimação ou irritação não presentes antes do uso do medicamento.
t Não foram descritas, na literatura consultada, Efeitos adversos com o uso de cetoconazol xampu.
t Informar ao médico a ocorrência de reação alérgica.
t Evitar contato com os olhos, boca, nariz e vagina.
t Evitar o uso em pele com cortes ou arranhões.
t Não usar esse medicamento se houver antecedentes de reação alérgica ao cetoconazol.
t Armazenar à temperatura ambiente que não exceda 25 ºC. Proteger da luz.
t Manter protegido do calor (produto pode ser inflamável).
Rogério Aparecido Minini dos Santos
Na Rename 2010: seções 6.1.1, 7.1
t Pó para solução injetável 200 mg e 1 g
t Comprimido 50 mg
t Câncer de mama.
t Carcinoma de ovário.
t Doença de Hodgkin (estádios III e IV).
t Leucemia linfoide aguda e crônica.
t Leucemia mieloide aguda e crônica.
t Linfoma de Burkitt.
t Linfoma de células de Mantle (estádios III e IV).
t Linfoma maligno (pequenas e grandes células).
t Linfoma maligno nodular ou difuso (pequenas células).
t Linfoma não-Hodgkin.
t Micose fungoide avançada.
t Mieloma múltiplo.
t Neuroblastoma disseminado.
t Retinoblastoma.
t Hipersensibilidade à ciclofosfamida.
t Mielossupressão grave.
t Usar com cuidado nos casos de:
– cistite hemorrágica.
– porfiria aguda (evitar).
– infecções.
– leucopenia ou trombocitopenia.
– quimioterapia ou radioterapia prévia.
– tumor de medula óssea com infiltração celular.
– insuficiência hepática.
– pacientes suprarrenalectonizados.
t Ciclofosfamida está associada a carcinogênese, mutagênese e infertilidade.
t Toxicidade cardíaca.
t Interferência na cicatrização de feridas.
t Reações anafiláticas (possibilidade de sensibilidade cruzada com outros alquilantes).
t Insuficiência renal (ver Apêndice D).
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenoso cada 7 a 10 dias; de 3 a 5 mg/kg intravenoso duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Regime AC: doxorrubicina 60 mg/m2 intravenoso, ciclofosfamida 600 mg/ m2 intravenoso. Repetir em ciclos de 21 dias. Como opção, segue-se com paclitaxel 175 mg a 225 mg/m2 intravenoso OU fluoruracila 500 mg/m2 intravenoso, nos dias 1 e 8 OU docetaxel 75 mg/m2 intravenoso uma hora depois da ciclofosfamida.
t Regime CFC: ciclofosfamida 500 a 600 mg/m2 intravenosa, fluoruracila 500 a 600 mg/m2 intravenoso, mitoxantrona 10 a 12 mg/m2 intravenoso, todos no dia 1.Repetir o ciclo a cada 21 dias.
t Regime CMF: ciclofosfamida 100 mg/m2 V0, nos dias 1 a 14, metotrexato 40 mg/m2 intravenosa e fluoruracila 600 mg/m2 intravenoso nos dias 1 e 8. Repetir o ciclo a cada 28 dias.
t Pacientes idosas com câncer de mama avançado: idarrubicina 35 mg/m2/dia via oral no dia 1 e ciclofosfamida 200 mg/m2/dia via oral nos dias de 3 a 6. Repetir o ciclo a cada 4 semanas, ou até dose máxima acumulada de 400 mg/ m2 de idarrubicina.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Regime CAP: ciclofosfamida na dose de 600 mg/m2, doxorrubicina na dose de 40 mg/m2, cisplatina na dose de 100 mg/m2, a cada 4 semanas.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
Leucemia linfocítica crônica; Leucemia mieloide aguda; Leucemia mieloide crônica; Linfoma de Burkitt; Linfoma maligno (pequenas e grandes células); Linfoma maligno nodular ou difuso (pequenas células); Micose fungoide avançada;
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenoso, divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenoso cada 7 a 10 dias; ou 3 a 5 mg/kg intravenoso duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenoso, divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Regime MACOP-B: doxorrubicina na dose de 50 mg/m2, ciclofosfamida na dose de 350 mg/m2, metotrexato na dose de 400 mg/m2, vincristina na dose de 1.4 mg/m2, bleomicina na dose de 10 U/m2, em associação com sulfatrimexazol-trimetoprima e prednisona.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Regime HiperVAD: ciclofosfamida na dose de 300 mg/m2 intravenosa a cada 12 horas nos dias de 1 a 6, mesna na dose de 600 mg/m2/dia por 3 dias; após 12 horas do término da ciclofosfamida, iniciar vincristina na dose de 2 mg/ m2 e doxorrubicina na dose de 50 mg/m2 como infusão intravenosa por 48 horas; repetir vincristina 2 mg/m2 intravenosa rápida no dia 11; dexametasona na dose de 20 mg/m2/dia via oral nos dias de 1 a 5 e nos dias de 11 a 14.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Regime combinado de ciclofosfamida e doxorrubicina, combinado ou não a vincristina, ou teniposídeo, ou cisplatina.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia por via oral para doses iniciais e manutenção.
t Regime MOPP, mecloretamina, vincristina, procarbazina, prednisona. Leucemia linfocítica crônica; Leucemia linfoide aguda; Leucemia mieloide crônica; Linfoma de Burkitt; Linfoma de células de Mantle (estádios III e IV); Linfoma maligno (pequenas e grandes células); Linfoma maligno nodular ou difuso (pequenas células); Micose fungoide avançada
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia por via oral para doses iniciais e manutenção.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia via oral para doses iniciais e manutenção.
t Regime CAT, ciclofosfamida na dose de 500 mg/m2 a cada 12 horas em 6 doses nos dias de 1 a 3, topotecano na dose de 1.25 mg/m2/dia por infusão intravenosa contínua nos dias de 2 a 6, citarabina na dose de 2 g/m2/dia por infusão intravenosa por 4 horas nos dias de 2 a 6. Repetir o tratamento cada 3 a 4 semanas.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, divida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia por via oral para doses iniciais e manutenção.
t Regime LSA2L2, ciclofosfamida, metotrexato intratecal, vincristina daunorrubicina, prednisona, citarabina, asparaginase, tioguanina, carmustina, e hidroxiureia.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia por via oral para doses iniciais e manutenção.
t Regime HiperVAD, Ciclofosfamida na dose de 300 mg/m2 intravenosa a cada 12 horas nos dias de 1 a 6, mesna na dose de 600 mg/m2/dia por 3 dias; após 12 horas do final da ciclofosfamida iniciar vincristina na dose de 2 mg/m2 e doxorrubicina na dose de 50 mg/m2 como infusão intravenosa por 48 horas; repetir vincristina 2 mg/m2 intravenosa rápida no dia 11; dexametasona na dose de 20 mg/m2/dia por via oral nos dias de 1 a 5 e nos dias de 11 a 14.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia por via oral para doses iniciais e manutenção.
t Regime MADDOC, mecloretamina, doxorrubicina, cisplatina, dacarbazina, vincristina, ciclofosfamida.
t Ciclofosfamida na dose de 40 a 50 mg/kg intravenosa, dividida em 2 a 5 dias; ou 10 a 15 mg/kg intravenosa cada 7 a 10 dias; ou 3 a 5 mg/kg intravenosa duas vezes por semana; ou 1 a 5 mg/kg/dia por via oral para doses iniciais e manutenção.
t Regime combinado de ciclofosfamida e doxorrubicina, associado ou não a vincristina, ou teniposídeo, ou cisplatina.
t Biodisponibilidade > 75%.
t Distribui-se amplamente por todos os tecidos e atravessa as barreiras hematoencefálica e placentária.
t Metabolismo hepático.
t Excreção renal 5 a 25%; é excretada no leite materno.
t Meia-vida de eliminação de 3 a 12 horas
t Alopecia.
t Náuseas e vômitos.
t Mielossupressão, leucopenia.
t Acidose metabólica (31%).
t Cistite hemorrágica, nefrotoxicidade (6%), amenorreia, azospermia, oligozoospermia, outros distúrbios no trato urinário.
t Cardiomiopatias.
t Pneumonia intersticial.
t Doenças infectantes.
t Síndrome de Stevens-Johnson e necrólise epidérmica tóxica (raro).
t Neurotoxicidade (12%).
t Alopurinol: aumento da toxicidade pela ciclofosfamida. Evitar o uso concomitante. Se a combinação for necessária verificar para o aumento dos Efeitos adversos da ciclofosfamida (especialmente mielossupressão).
t Ciclosporina: concentrações plasmáticas diminuídas pela ciclofosfamida. Fazer a monitoria dos os níveis plasmáticos de ciclosporina, a resposta do paciente e ajustar a dose se necessário. Observar sinais de reação imunológica.
t Erva-de-são-joão (Hypericum perforatum): redução da efetividade da ciclofosfamida. Evitar o uso concomitante.
t Etanercepte: aumento da incidência de tumores sólidos não cutâneos. Uso concomitante contraindicado (especialmente na granulomatose de Wegener).
t Hidroclorotiazida: aumento do risco de mielossupressão (granulocitopenia). Observar sinais de mielossupressão.
t Nevirapina: redução das concentrações plasmáticas da ciclofosfamida. Considerar ajuste da dose de ciclofosfamida.
t Ondansetrona: redução da exposição sistêmica a ciclofosfamida. Acompanhar a efetividade da ciclofosfamida.
t Pentostatina: risco aumentado de cardiotoxicidade. Se a combinação for necessária, observar o paciente em relação a sinais preliminares de cardiotoxicidade.
t Tamoxifeno: aumento do risco de tromboembolia. Se a combinação for necessária avaliar a relação risco-benefício.
t Trastuzumabe: aumento do risco de disfunções cardíacas. Avaliar cuidadosamente a função cardíaca. A descontinuação do trastuzumabe é recomendada ao primeiro sinal de disfunção ventricular.
t Vacina rotavírus humano G1P1[8] (atenuada): aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Se a vacina for necessária, administrá-la depois de três meses da descontinuação da quimioterapia.
t Varfarina: aumento do risco de sangramento. Acompanhar o tempo de protrombina e sinais de sangramento. Pode ser necessário reduzir a dose da varfarina.
t Antes de iniciar o tratamento é importante identificar: história prévia de hipersensibilidade a ciclofosfamida, gravidez, lactação, cardiomiopatias.
t Ingerir mais líquido do que o normal, pois ajuda a prevenir problemas renais e na bexiga e a manter os rins funcionando.
t O paciente deve ser alertado quanto a sinais de possível cistite hemorrágica.
t A ciclofosfamida pode causar infertilidade em homens e mulheres.
t Ciclofosfamida pode causar náuseas e vômitos. Entretanto, é muito importante continuar a receber o medicamento mesmo com estes sintomas. Perguntar ao profissional de saúde maneiras para reduzir estes efeitos.
t Enquanto estiver utilizando ciclofosfamida, evitar as vacinas sem a aprovação do médico, pois este medicamento pode reduzir a resistência do organismo havendo maior chance de adquirir a infecção a qual a vacina iria prevenir.
t Ciclofosfamida pode reduzir temporariamente o número de células brancas do seu corpo, aumentando a chance de adquirir infecções. Pode também reduzir o número de plaquetas. Se isto ocorrer, deve-se tomar alguns cuidados:
t Evitar pessoas com infecções.
t Falar com o médico imediatamente se houver sangramento incomum.
t Cuidado ao usar escova, fio ou palito de dentes. O médico, dentista ou enfermeiro pode recomendar outras maneiras de limpar dentes e gengiva.
t Não tocar os olhos ou o interior do nariz ao menos que tenha lavado as mãos.
t Cuidado para não se cortar ao usar objetos cortantes como barbeadores, cortador de unhas.
t Evitar esportes de contato ou outras situações em que possa ocorrer sangramento ou lesão.
t Os comprimidos de ciclofosfamida devem ser armazenados sob temperaturas de até 25ºC. Durante o transporte e administração, os comprimidos são resistentes quando expostos a temperaturas não superiores a 30 ºC por um curto intervalo de tempo.
t Armazenar os frascos com ciclofosfamida liofilizada sob temperaturas controladas (15 e 30 ºC). Não expor à luz ou congelamento.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução. Para infusão intravenosa reconstituir o pó com água estéril para injeção (livre de álcool benzílico); a solução permanece estável por 24 horas sob temperaturas entre 15 e 30 ºC e por 6 dias se mantida sobre refrigeração (2 a 8 ºC).
t Para injeção intravenosa, reconstituir o pó em solução injetável de cloreto de sódio 0,9%.
t A dissolução normalmente ocorre entre 10 e 15 minutos. Se ao fim deste período ainda se observar a presença na solução de cristais, ela deve ser aquecida por 15 minutos sob temperaturas de 50 a 60 ºC.
t Uma solução oral extemporânea de ciclofosfamida pode ser produzida a partir da quantidade desejada do pó liofilizado para injeção diluído em um elixir aromático. A solução permanece estável por 2 semanas se acondicionada em frasco de vidro e mantida sob refrigeração.
t A mistura de ciclofosfamida e mesna é compatível, mas permanece estável por até 24 horas sob temperaturas entre 15 e 30 ºC.
t Ciclofosfamida é compatível quando misturada em solução diversos antineoplásicos; entretanto, parece ser incompatível com álcool benzílico usado como conservante água e outros diluentes para injeções. Ciclofosfamida não deve ser misturada a anfotericina B e lansoprazol.
Atenção: risco de ocorrência de efeitos urotóxicos, especialmente cistite hemorrágica. Pode determinar mielossupressão grave.
Maurício Fábio Gomes
Larissa Niro e Maurício Fábio Gomes
Na Rename 2010: itens 7.1
t Cápsula 25 mg, 50 mg e 100 mg
t Solução oral 100 mg/mL
t Tratamento e profilaxia de rejeição de transplante de órgãos e tecidos.
t Prevenção de doença do enxerto-versus-hospedeiro.
t Anemia aplástica.
t Síndrome nefrótica.
t Artrite reumatoide grave, não-responsiva a metotrexato.
t Colite ulcerativa grave.
t Psoríase grave em placas, recalcitrante, não-responsiva a outros medicamentos sistêmicos há pelo menos um ano.
t Hipersensibilidade a ciclosporina ou a algum constituinte da formulação.
t Hipertensão arterial sistêmica não controlada; infecções não controlas e neoplasias.
t Uso concomitante com os fármacos bosentana e dronedarona.
t Usar com cuidado nos casos de:
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– porfiria (evitar o uso).
– idosos (mais Susceptíveis a hipertensão e problemas renais).
– lactação (ver Apêndice B).
t Monitorar os níveis séricos de potássio, magnésio, ácido úrico e lipídios.
t O uso de ciclosporina aumenta as chances de desenvolver infecções e neoplasias (como linfomas).
t Hipertensão e danos estruturais renais são consequências em potência do uso de ciclosporina.
t Evitar exposição excessiva a luz solar.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Perioperatório: 15 mg/kg, por via oral, de 4 a 12 horas antes do transplante, continuar com a mesma dose por 1 a 2 semanas. Ou então, 5 a 6 mg/kg/dia, por via intravenosa, de 4 a 12 horas antes do transplante, continuar com a mesma dose até que o paciente possa receber o tratamento por via oral.
t Manutenção: reduzir gradualmente a dose para 2 a 6 mg/kg/dia, por via oral, de acordo com a concentração plasmática alvo da ciclosporina e a função renal do paciente.
t Perioperatório: 12,5 a 15 mg/kg/dia, por via oral, desde 1 a 2 dias antes do transplante, continuar com a mesma dose por 2 semanas. Ou então, 3 a 5 mg/kg/dia, por infusão intravenosa, durante 2 a 6 horas no dia anterior ao transplante, e continuar com a mesma dose por 2 semanas.
t Manutenção: 12,5 mg/kg/dia, por via oral, durante 3 a 6 meses, podendo chegar a um ano de tratamento, depois descontinuar o uso.
t 5 mg/kg/dia, por via oral, dividido em 2 doses, durante 6 meses, mantendo a concentração plasmática alvo entre 75 e 200 nanogramas/mL, combinado com 15 mg/kg/dia de globulina antitimocítica, por via intravenosa e 1 mg/kg/ dia de prednisolona, por via oral, durante os primeiros 5 dias. Descontinuar a prednisona gradualmente durante a semana seguinte.
t 5 mg/kg/dia, divididos em 2 doses, por via oral; reduzir à dose mínima efetiva de acordo com a melhora da função renal, baseado em valores de proteinúria e creatinina sérica. Descontinuar após 3 meses de tratamento se não houver melhora na glomerulonefrite ou glomeruloesclerose, e após 6 meses na glomerulonefrite membranosa.
t 2,5 mg/kg/dia, por via oral, dividido em 2 doses; a dose pode ser aumentada em 0,5 a 0,75 mg/kg/dia por 4 a 8 semanas nos pacientes com resposta inadequada, mas com boa tolerabilidade, até o máximo de 5 mg/kg/dia.
t Dose inicial: 2 a 4 mg/kg/dia, por infusão intravenosa, durante 8 dias, com dose ajustada de acordo com a concentração plasmática alvo de ciclosporina; manter com 8 mg/kg/dia, por via oral, durante 3 meses.
t Dose inicial: 2,5 mg/kg/dia, por via oral, dividido em 2 doses; aumentar até o máximo de 5 mg/kg/dia se não houver melhora em 4 semanas; descontinuar se a resposta ainda for insuficiente depois de 6 semanas; doses iniciais de 5 mg/kg/dia justificam-se quando se requer um controle rápido da afecção.
t Perioperatório: dar 15 mg/kg, por via oral, de 4 a 12 horas antes do transplante, continuar a dose por 1 a 2 semanas. Ou então, administrar 5 a 6 mg/ kg/dia, por via intravenosa, de 4 a 12 horas antes do transplante e continuar a dose até o paciente tolerar a formulação oral.
t Manutenção: reduzir em 5% por semana até 5 a 10 mg/kg/dia, por via oral. Ajustar a dose de acordo com a concentração plasmática alvo da ciclosporina
t Perioperatório: 12,5 a 15 mg/kg/dia, por via oral, desde 1 a 2 dias antes do transplante, continuar com a mesma dose por 2 semanas. Ou então, 3 a 5 mg/kg/dia, por infusão intravenosa, durante 2 a 6 horas no dia anterior ao transplante, continuar com a mesma dose por 2 semanas.
t Manutenção: 12,5 mg/kg/dia, por via oral, durante 3 a 6 meses, podendo chegar a um ano de tratamento, depois descontinuar o uso.
t Dose de 6 mg/kg/dia, divididos em 2 doses, por via oral; reduzir à dose mínima efetiva de acordo com a melhora da função renal, baseado em valores de proteinúria e creatinina sérica. Descontinuar após 3 meses de tratamento se não houver melhora na glomerulonefrite ou glomeruloesclerose, e após 6 meses na glomerulonefrite membranosa.
t Pico de concentração plasmática: 1,5-2 horas (oral).
t Metabolismo: fígado.
t Excreção: renal (6%) e fecal.
t Meia-vida de eliminação: 19 horas (oral).
t Hirsutismo (21 a 45%), hipertricose, alopecia, prurido.
t Diarreia (3%), náuseas e vômitos (3%), hiperplasia gengival (5 a 16%), colite.
t Cefaleia (2 a 25%), tremor (3 a 55%), convulsões (1 a 25%), fadiga (3 a 12%), astenia (raro), confusão (2%), tonturas (3 a 8%).
t Hipertensão (frequente), edema (2%).
t Anemia leve (2%), síndrome hemolítico-urêmica (raro).
t Hepatotoxicidade (4 a 19%). t Nefrotoxicidade (frequente).
t Doenças infectantes.
t Pancreatite (raro), distúrbios linfoproliferativas pós-transplante.
t Dismenorreia ou amenorreia.
t Miopatia, mialgia, parestesia (1 a 11%).
t Ganho de peso.
t Ginecomastia (<4%).
t Hipomagnesemia, hiperglicemia, hipercolesterolemia, hiperpotassemia (raro), hiperuricemia.
t Reações alérgicas (2%).
t Actaea racemosa, Cimicifuga racemosa e Medicago sativa podem diminuir a efetividade da ciclosporina e causar rejeição aguda do transplante. Evitar o uso concomitante.
t Alisquireno pode ter aumento acentuado da sua exposição sistêmica e concentrações plasmáticas. Evitar o uso concomitante.
t Bosentana pode ter sua concentração plasmática aumentada e diminuir a de ciclosporina. Contraindicado.
t Caspofungina pode ter sua concentração plasmática e risco de elevação das enzimas hepáticas. Se o uso concomitante for necessário, pesar risco/benefício e avaliar função hepática.
t Ciclofosfamida poder resultar em redução das concentrações plasmáticas de ciclosporina. Se o uso concomitante for necessário, verificar concentrações plasmáticas de ciclosporina, ajustar a dose se necessário e observar sinais de ocorrência de doença enxerto-versus-hospedeiro.
t Colchicina pode resultar no aumento das concentrações plasmáticas de ambos os fármacos. Se o uso concomitante for necessário, a dose diária de colchicina não deve ultrapassar 0,6 mg; identificar sinais de toxicidade para os dois fármacos e as concentrações plasmáticas de ciclosporina.
t Dronedarona pode ter sua concentração plasmática aumentada. Contraindicado.
t Estatinas podem ter risco aumentado de miopatias e rabdomiólise. Se o uso concomitante for necessário, iniciar com baixas doses da estatina e pesquisar sinais de miopatias e rabdomiólise.
t Etoposídeo pode ter aumento da concentração plasmática com risco de leucopenia grave. Se o uso concomitante for necessário, a dose de etoposídeo deve ser reduzida em 50% nos tratamentos com altas doses de ciclosporina.
t Felodipino pode ter sua toxicidade aumentada. Identificar sinais de reações adversas ao felodipino.
t Hypericum perforatum (erva-de-são-joão) pode resultar em redução significante da concentração plasmática de ciclosporina, com rejeição aguda do transplante e perda do enxerto. Se o uso concomitante for necessário, fazê-lo com muita cautela.
t Itraconazol pode aumentar as concentrações plasmáticas de ciclosporina com risco de toxicidade. Monitorar concentrações plasmáticas de ciclosporina e reduzir a dose se necessário.
t Nafcilina e octreotida podem diminuir a efetividade da ciclosporina. Avaliar concentrações plasmáticas de ciclosporina e ajustar a dose se necessário.
t Orlistate poder resultar em redução das concentrações plasmáticas de ciclosporina. Verificar concentrações plasmáticas de ciclosporina e administrá-la 2 horas antes e/ou depois do orlistate.
t Posaconazol pode resultar em aumento da concentração de ciclosporina no sangue total, aumentando o risco de nefrotoxicidade, leucoencefalopatia e morte. Se o uso concomitante for necessário, reduzir a dose de ciclosporina em 75% e aumentar gradualmente após a descontinuação do posaconazol; monitorar concentração de ciclosporina no sangue total.
t Rifampicina e sulfimpirazona podem resultar em redução das concentrações plasmáticas de ciclosporina e aumentar o risco de rejeição do transplante. Se o uso concomitante for necessário, verificar concentrações plasmáticas de ciclosporina e sinais de rejeição.
t Topotecano pode ter aumento acentuado na circulação. Se o uso concomitante for necessário, identificar sinais de reações adversas ao topotecano.
t Voriconazol pode aumentar a concentração plasmática de ciclosporina. Se o uso concomitante for necessário, reduzir a dose de ciclosporina pela metade e aumentar gradualmente após a descontinuação do voriconazol; acompanhar as concentrações plasmáticas de ciclosporina.
t Orientar para ingerir o medicamento diariamente, sempre no mesmo horário.
t Orientar para ingerir preferentemente com alimentos ricos em gorduras para aumentar a absorção da ciclosporina.
t Ensinar a medir a dose da solução oral com dosador graduado.
t Orientar para misturar a solução oral com leite, achocolatado ou suco (laranja ou maçã), sob temperatura ambiente, para melhorar a palatabilidade.
t Evitar aplicação de vacinas de vírus no paciente e em familiares ou outras pessoas que mantenham contato direto com paciente. Caso tenham sido aplicadas em famíliares e outros, o paciente deve utilizar máscara que cubra a boca e nariz.
t Orientar para manter higiene bucal e visitar o dentista frequentemente para limpeza dentária, a fim de prevenir dor, sangramento e crescimento gengival.
t Orientar para armazenar este medicamento em recipientes fechado, à temperatura ambiente, evitar incidência de luz direta, calor e umidade. Não armazenar em refrigerador ou freezer.
t Reforçar para notificar imediatamente se aparecer sangue na urina, assim como instruir a identificar e notificar sinais e sintomas de rejeição do transplante.
t Orientar para não interromper o tratamento abruptamente.
t A formulação modificada de ciclosporina (cápsulas com microemulsão) não é bioequivalente à ciclosporina convencional, portanto não são intercambiáveis.
t Armazenar a solução oral em sua embalagem original, à temperatura ambiente, entre 15 e 30 oC, e ao abrigo de luz direta. Frascos abertos devem ser consumidos de preferência até 2 meses.
t Armazenar aa cápsulas em sua embalagem original, à temperatura ambiente, entre 15 e 30 oC, e ao abrigo de luz direta.
Atenção: a ciclosporina apresenta um número elevado de interações. Deve ser realizada uma pesquisa específica sobre este aspecto quando se considerar a terapia com ciclosporina, bem como ao introduzir ou descontinuar outros medicamentos no esquema terapêutico do paciente.
Karen Luise Lang
Na Rename 2010: item 18.4.3
t Solução injetável 100 mg/mL.
t Hipogonadismo.
t Tumores metastáticos de mama em mulheres.
t Obtenção de efeito anabolizante em infectados por HIV.
t Hipersensibilidade à testosterona.
t Tumores de mama em homens.
t Tumores de próstata e hiperplasia benigna de próstata com obstrução.
t Histórico de tumores hepáticos.
t Hipercalcemia.
t Síndrome nefrótica.
t Lactação (ver Apêndice B).
t Gravidez. Categoria de risco na gravidez (FDA): X (ver Apêndice A).
t Usar com cuidado nos casos de:
– insuficiência hepática (ver Apêndice C), insuficiência renal (ver Apêndice D) ou cardiopatia pré-existente, hipertensão, epilepsia, enxaqueca, apneia do sono, obesidade, porfiria e diabete melito.
– tratamentos prolongados (altas doses de testosterona elevam o risco de icterícia, tumores hepáticos e peliose hepática, em potência fatal; realizar exames regulares de atividade hepática).
– jovens (risco de soldadura das epífises, com permanente cessação de crescimento linear).
– idosos (pode haver aumento da densidade mineral óssea e da massa magra, com decréscimo de massa gorda; também há aumento do risco de desenvolver hiperplasia ou tumores de próstata).
t Exames regulares de próstata e mamas devem ser realizados durante o tratamento.
t A ginecomastia pode ser persistente.
t Dose inicial 200 a 250 mg, por via intramuscular, lentamente, a cada 2 a 3 semanas. Dose de manutenção 250 mg, por via intramuscular, lentamente, a cada 3 a 6 semanas.
t 200 a 400 mg, por via intramuscular, lentamente, a cada 2 a 4 semanas.
t Administração: injeção intramuscular profunda no quadrante superior externo do glúteo maior. Não usar por via intravenosa.
t Duração de efeito: 2-4 semanas.
t Metabolismoo: hepático
t Meia-vida: aproximadamente 8 dias.
t Excreção: preponderantemente renal (90%)
t Acne, prurido (37%), queimação no lugar da aplicação (12%), eritema (7%), hirsutismo, seborreia.
t Irritação na boca e gengivas (9%), gengivite, sabor amargo, distúrbios gastrintestinais, náuseas.
t Cefaleia, depressão, ansiedade, parestesia.
t Anomalias e tumores na próstata, dor e aumento do tamanho das mamas, desenvolvimento sexual precoce em meninos na pré-puberdade, alterações na libido, virilização feminina e supressão da espermatogênese em homens, priapismo.
t Retenção de líquidos, ganho de peso.
t Hipertensão
t Alcaçuz: pode reduzir a efetividade da testosterona. Evitar o uso concomitante.
t Varfarina e demais anticoagulantes orais podem elevar o risco de hemorragias. Se o uso concomitante for necessário, intensificar a monitoria do tempo de protrombina, particularmente ao iniciar ou descontinuar a testosterona.
t Recomendar que sejam feitas consultas frequentes em casos de tratamentos prolongados, para avaliação do progresso do tratamento.
t Alertar pacientes com diabetes melito quanto à possibilidade de hipoglicemia. Alterações nos níveis de glicose sanguínea ou urinária devem ser notificadas.
t Alertar para notificar se ocorrerem ereções frequentes ou persistentes, náuseas, vômitos, mudanças de coloração cutânea, edema de membros inferiores, problemas respiratórios e bucais.
t Armazenar ao abrigo de ar e luz e à temperatura ambiente de 15 a 30 ºC.
t Quando exposto a temperaturas mais baixas, deve-se agitar a ampola e aquecer entre as mãos antes da administração para solubilizar possíveis cristais formados durante o armazenamento.
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 6.1.5
t Solução injetável 1 mg/mL.
t Câncer de testículo metastático.
t Câncer de ovário metastático.
t Câncer de bexiga de células transicionais avançado
t Câncer de endométrio avançado.
t Câncer de pulmão de células não pequenas (não ressecável)
t Câncer de pulmão de pequenas células.
t Câncer de cabeça, pescoço e esôfago.
t Câncer gástrico (adenocarcinoma avançado).
t Hepatoblastoma.
t Hipersensibilidade a cisplatina ou a produtos contendo platina.
t Mielossupressão.
t Deficiênciasr enal ou auditiva pre-existentes.
t Gravidez. Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– idosos (aumenta o risco de mielossupressão, nefrotoxicidade e neurotoxicidade).
– insuficiência renal (ver Apêndice D).
– regime TCF – docetaxel + cisplatina + fluoruracila (requer ajuste da dose).
– plaquetopenia (não utilizar em ciclos repetidos até que a contagem de plaquetas esteja acima de 100.000/mm3.
– extravasamento.
t Eletrólitos devem ser medidos antes de iniciar a terapêutica, e antes de cada curso subsequente. Hidratação adequada e débito urinário devem ser mantidos até 24 horas depois da dose.
t Pré-operatório: administrar 80 mg/m2 de cisplatina, infusão intravenosa contínua durante 24 horas, a cada 2 semanas, por 4 ciclos. Após completa ressecção, estender o tratamento por mais 2 ciclos.
t 20 mg/m2 de cisplatina e 100 mg/m2 de etoposídeo, ambos por via intravenosa, nos dias de 1 a 5, combinado com 30 U de bleomicina, por via intravenosa, nos dias 2, 9 e 16; repetir o ciclo a cada 3 semanas por 4 ciclos.
t 75 a 100 mg/m2, por via intravenosa, combinado na sequência com 600 mg/ m2 de ciclofosfamida, por via intravenosa, uma vez a cada 4 semanas.
t Monoterapia: administrar 50 a 70 mg/m2, por via intravenosa, uma vez a cada 3 a 4 semanas. Iniciar com 50 mg/m2 nos pacientes pré-tratados.
t 45 mg/m2 de doxorrubicina, seguidos imediatamente de 50 mg/m2 de cisplatina, ambos por via intravenosa, no dia 1. No dia 2 administrar 160 mg/m2 de paclitaxel, por infusão intravenosa durante 3 horas; combinado com 5 mg/ kg de filgrastima, por via sub cutânea, nos dias 3 a 12. Repetir o ciclo a cada 3 semanas por 7 ciclos ou até progressão da doença.
t 135 mg/m2 de paclitaxel, por infusão intravenosa durante 3 horas, no dia 1, seguido de 75 mg/m2 de cisplatina, por via intravenosa, no dia 2. Repetir o ciclo a cada 3 semanas por 3 a 4 ciclos.
t De modo alternativo, administrar gencitabina 1 g/m2, por via intravenosa, nos dias 1, 8 e 15, combinado com 100 mg/m2 de cisplatina, na sequência da gencitabina, por via intravenosa, no dia 1. Repetir o ciclo a cada 4 semanas.
t 60 a 120 mg/m2 de cisplatina, por via intravenosa, no dia 1, em combinação com 120 mg/m2 de etoposídeo, por via intravenosa, nos dias de 1 a 3. Repetir o ciclo a cada 3 a 4 semanas. OU 75 mg/m2 de cisplatina, por via intravenosa, no dia 1, em combinação com 100 mg/m2 de etoposídeo, por via intravenosa, no dia 1, e 200 mg/m2 de etoposídeo, por via intravenosa, nos dias de 2 a 4. Repetir o ciclo a cada 3 semanas. OU 80 mg/m2 de cisplatina, por via intravenosa, no dia 1, em combinação com 80 mg/m2 de etoposídeo, por via intravenosa, nos dias de 1 a 3. Repetir o ciclo a cada 3 semanas.
t De modo alternativo, administrar 60 mg/m2 de cisplatina, por via intravenosa, no dia 1, em combinação com 60 mg/m2 de irinotecano, por via intravenosa, nos dias 1, 8 e 15. Repetir o ciclo a cada 4 semanas. OU 30 mg/m2 de cisplatina e 65 mg/m2 de irinotecano, ambos por via intravenosa, nos dias 1 e 8. Repetir o ciclo a cada 3 semanas a partir do dia 1.
t Tratamento pós-cirúrgico seguido de radioterapia: administrar 100 mg/m2 de cisplatina, por via intravenosa, no dia 1, em combinação com 1 g/m2/dia de fluoruracila, infusão intravenosa contínua, nos dias de 1 a 5. Repetir o ciclo a cada 3 a 4 semanas.
t Recorrência: administrar 175 mg/m2, por infusão intravenosa durante 3 horas, no dia 1, em combinação com 1 g/m2 de ifosfamida, por infusão intravenosa durante 2 horas, nos dias de 1 a 3; administrar 400 mg/m2 de mesna, por via intravenosa, antes da ifosfamida e 200 mg/m2, por via intravenosa, 4 horas depois da ifosfamida, 60 mg/m2 de cisplatina, por via intravenosa, no dia 1. Repetir o ciclo a cada 3 a 4 semanas.
t Iniciar no dia zero (0) com 8 mg de dexametasona, por via oral, duas vezes ao dia, continuar por 3 dias, seguido de 75 mg/m2 de docetaxel, infusão intravenosa durante 1 hora, no dia 1, seguido de 75 mg/m2 de cisplatina, infusão intravenosa durante 1 a 3 horas, no dia 1, seguido por 750 mg/m2/ dia, infusão intravenosa contínua por 24 horas, nos dias de 1 a 5. Repetir o ciclo a cada 3 semanas.
t Liga-se em 90% às proteínas plasmáticas.
t Excreção renal de 13 a 45%.
t A cisplatina não é dialisável.
t A meia-vida de eliminação entre 16 e 53 horas.
t Desequilíbrio eletrolítico.
t Náuseas e vômitos.
t Mielossupressão (frequente).
t Reação de hipersensibilidade.
t Hérnia cerebral, encefalopatia, síndrome de leucoencefalopatia posterior reversiva, convulsões.
t Ototoxicidade.
t Nefrotoxicidade (frequente).
t Aldesleucina: pode aumentar o risco de reações de hipersensibilidade.
Verificar sinais e sintomas das reações de hipersensibilidade; intervenção médica pode ser necessária.
t Docetaxel: pode aumentar o risco de neuropatias. Observar sinais de neuropatias periféricas.
t Doxorrubicina: pode resultar em leucemia. O uso concomitante deve ser feito com extrema cautela; avaliar a relação benefício/risco.
t Fenitoína e fosfenitoína: pode resultar na redução da efetividade de fenitoína e fosfenitoína. Considerar a monitoria das concentrações séricas de fenitoína e fosfenitoína durante a quimioterapia. Se necessário a dose deve ser ajustada durante e após o tratamento com cisplatina.
t Paclitaxel e topotecano: pode resultar em mielossupressão grave. Se o uso concomitante ou posterior for necessário, fazer monitoria de mielotoxicidade.
t Rituximabe e tacrolimo: pode exacerbar a nefrotoxicidade. Se o uso concomitante for necessário, acompanhar função renal.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Contraindicado.
t Vinorelbina: pode aumentar o risco de granulocitopenia. Acompanhar quanto a granulocitopenia de graus 3 e 4.
t Antes de iniciar o tratamento é importante identificar: história prévia de hipersensibilidade a cisplatina, manitol e produtos de platina, gravidez e lactação, doenças renais, problemas auditivos, infecções.
t Utilizar métodos contraceptivos efetivos durante o tratamento com cisplatina. Em casos da ocorrência de gravidez, informar seu médico.
t Evitar vacinas ao menos que aprovadas pelo médico.
t Falar com o médico se ocorrer sangramento incomum.
t Cuidado ao escovar os dentes, passar fio ou palito de dentes.
t Não tocar os olhos ou o interior do nariz ao menos que esteja com as mãos limpas.
t Cuidado para não se cortar com barbeador ou cortador de unhas.
t Evitar esportes de contato ou outras situações nas quais pode ocorrer sangramento ou lesão.
t Evitar pessoas que apresentem infecções ou que tenham recebido vacina de poliovírus oral.
t Armazenar os frascos de cisplatina sob temperaturas ambiente, entre 15 a 25 oC, protegidos da luz e não refrigerar.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Soluções de grande volume contendo cisplatina devem ser protegidas da luz quando infundidas por períodos superiores a 6 horas.
t O pH das soluções é o fator que mais afeta a estabilidade da cisplatina, e o seu valor não deve ultrapassar 4,3.
t A cisplatina é compatível com soluções injetáveis de cloreto de sódio 0,9% e em soro glicofisiológico (5% a 0,45%).
t A cisplatina é degradada em soluções contendo bissulfito, metabissulfito, cloreto de potássio.
t A cisplatina é incompatível com fluoruracila, mesna, bicarbonato de sódio, tiotepa, amifostina, anfotericina B, lansoprazol, pantoprazol, cloridrato de cefepima, dantroleno sódico, diazepam, piperacilina/tazobactam, e soluções de NPT.
t A estabilidade da cisplatina pode se alterar em misturas contendo etoposídeo ou paclitaxel, mas isto irá depender das concentrações dos fármacos e da temperatura das soluções
t A cisplatina reage com alumínio formando precipitado, portanto, agulhas, seringas, cateteres ou equipos contendo alumínio não devem ser utilizados para administrar carboplatina.
Atenção: nefrotoxicidade, mielossupressão, náuseas e vômitos ocorrem com frequência e são dose dependentes. Ototoxicidade com perda de audição e surdez é mais intensa em crianças. Existem riscos de reações anafiláticas minutos após a administração de cisplatina.
Maurício Fábio Gomes
Na Rename 2010: Item 6.1.2
t Pó para solução injetável 100 mg, 500 mg e 1g.
t Leucemia linfoblástica aguda.
t Leucemia mieloide aguda (em combinação com outros antineoplásicos).
t Leucemia mieloide crônica (fase blástica).
t Leucemia meníngea.
t Hipersensibilidade a citarabina.
t Usar com cuidado nos casos de:
– mulheres em idade fértil.
– uso concomitante de medicamentos imunossupressores.
– insuficiência hepática (ver Apêndice C) e renal (ver Apêndice D).
– crianças (não utilizar diluentes que contenham álcool benzílico).
– lactação (ver Apêndice B).
t Monitorar cuidadosamente os níveis hematológicos (pode ser necessário interromper o tratamento se a contagem de plaquetas for inferior a 100.000/mm3 ou a de granulócitos for inferior a 1.000/mm3).
t Induz grave toxicidade no trato gastrintestinal, sistema nervoso central e pulmões.
t Doses intravenosas em bolo comportam-se diferentemente da infusão intravenosa; as primeiras são relativamente bem toleradas, embora causem neurotoxicidade; a administração contínua resulta em mielossupressão.
t Evitar extravasamento.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Administrar 300 mg/m2 de citarabina e 165 mg/m2 de teniposídeo, por via intravenosa, duas vezes por semana, por 4 a 5 semanas.
t Tratamento de indução: no primeiro ciclo, administrar citarabina 100 a 200 mg/m2/dia, por infusão intravenosa contínua, nos dias de 1 a 7, em combinação com daunorrubicina 45 mg/m2, por via intravenosa, OU idarrubicina 12 mg/m2, por via intravenosa, OU mitoxantrona 12 mg/m2, por via intravenosa, nos dias de 1 a 3. Nos ciclos seguintes, citarabina deve ser administrada dos dias 1 a 5 e o agente combinado nos dias de 1 a 2.
t Tratamento de reindução/consolidação: administrar citarabina 100 a 200 mg/m2/dia, por infusão intravenosa contínua, nos dias de 1 e 5, em combinação com daunorrubicina 45 mg/m2, por via intravenosa, OU mitoxantrona 12 mg/m2, por via intravenosa, nos dias de 1 a 2. Para iniciar o tratamento de reindução, aguardar por 4 a 6 semanas após o último ciclo de indução.
t Tratamento e profilaxia: administrar citarabina de 5 a 75 mg/m2, por via intratecal, a cada 4 dias, até normalidade do fluido cérebro-espinhal, seguido por um ciclo adicional. Dose de 30 mg/m2 tem sido mais comumente utilizada.
t Tratamento de indução: administrar citarabina 3 g/m2/dia, por infusão intravenosa contínua, por 5 dias; mitoxantrona 80 mg/m2, por via intravenosa, no dia 3; e suporte com fator de estimulação de colônias de granulócitos (G-CSF).
t Tratamento de reindução/consolidação: administrar citarabina 200 mg/m2/ dia, por infusão intravenosa contínua em combinação com metotrexato 25 mg/m2, por via intravenosa, por 5 dias, por 3 ciclos, começando nos dias 1, 36 e 71; alternado com 3 ciclos de citarabina 150 mg/m2, por via intravenosa a cada 12 horas e tioguanina 120 mg/m2, por via oral, por 6 dias, começando nos dias 15, 50 e 85.
t Tratamento de indução: no primeiro ciclo, administrar citarabina 100 a 200 mg/m2/dia, por infusão intravenosa contínua, nos dias de 1 a 7, em combinação com daunorrubicina 45 mg/m2, por via intravenosa, OU idarrubicina 12 mg/m2, por via intravenosa, OU mitoxantrona 12 mg/m2, por via intravenosa, nos dias de 1 a 3. Nos ciclos seguintes, citarabina deve ser administrada dos dias 1 a 5 e o agente combinado nos dias de 1 a 2.
t Tratamento de reindução/consolidação: administrar citarabina 100 a 200 mg/m2/dia, por infusão intravenosa contínua, nos dias de 1 e 5, em combinação com daunorrubicina 45 mg/m2, por via intravenosa, OU mitoxantrona 12 mg/m2, por via intravenosa, nos dias de 1 a 2. Para iniciar o tratamento de reindução, aguardar por 4 a 6 semanas após o último ciclo de indução.
t Como monoterapia, administrar citarabina 1,5 g/m2/dia (altas doses), por infusão intravenosa contínua, por 4 dias.
t 100 mg/m2/dia, por infusão intravenosa contínua, por 7 dias; mitoxantrona 12 mg/m2/dia, em bolo intravenoso, por 3 dias, OU daunorrubicina 45 mg/ m2/dia, em bolo intravenoso por 3 dias.
t Tratamento e profilaxia: administrar citarabina de 5 a 75 mg/m2, por via intratecal, a cada 4 dias, até normalidade do fluido cérebro-espinhal, seguido por um ciclo adicional. Dose de 30 mg/m2 tem sido mais comumente utilizada.
t Pico de concentração plasmática: 50% maior em terapia de altas doses.
t Metabolismo: hepático.
t Excreção: renal (80% como metabólitos, menos de 10% na forma inalterada).
t Meia-vida de eliminação: 1 a 3 horas; 2 horas (intratecal).
t Terapia de altas doses: toxicidade cerebelar (ataxia, disartria), convulsões (via intratecal), conjutivite, ceratite corneana, hiperbilirrubinemia, edema pulmonar, pericardite (raro).
t Alopecia, erupções cutâneas.
t Náusea, vômito, diarreia, hemorragias gastrintestinais, perda de apetite, inflamação e úlcera anal, estomatites, úlcera bucal.
t Pacreatites (raro), hepatotoxicidade, icterícia.
t Vasculites, tromboflebites.
t Hiperuricemia, hiperpotassemia, hipocalcemia, disfunção renal.
t Anemia megaloblástica, hemorragia, leucopenia, trombocitopenia.
t Imunossupressão, anafilaxia, infecções e sepse.
t Vacina rotavírus humano G1P1[8] (atenuada): aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina.Se a vacina for necessária, administrar a vacina depois de três meses da descontinuação da quimioterapia.
t Orientar para evitar vacinas e contato com pessoas que tenham recebido vacina de poliovírus oral.
t Orientar para evitar contato com pessoas com infecções ou, caso imprescindível, utilizar máscara protetora que cubra boca e nariz.
t Evitar situações que determinem cortes e machucaduras, inclusive cautela ao realizar higiene pessoal.
t Orientar para notificar imediatamente o surgimento de sangramento.
t Ingerir grandes volumes de líquidos. Não ingerir álcool.
t Armazenar os frascos à temperatura ambiente, entre 15 e 30 oC.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Citarabina reconstituída com água bacteriostática para injeção com álcool benzílico (0,945%) é estável por 48 horas se estocada sob temperatura ambiente (20 a 25 oC).
t Devem-se descartar soluções que se tornem turvas ou opacas.
t Citarabina permanece estável por 8 dias à temperatura ambiente, quando reconstituída em água para injeção, solução aquosa de glicose 5% ou solução de cloreto de sódio 0,9%.
t Incompatível com soluções de fluoruracila, heparina, insulina regular, nafcilina, oxacilina de sódio, penicilina G potássica e metotrexato; incompatível com alopurinol, amiodarona, anfotericina B, daptomicina, diazepam, nitrato de gálio, ganciclovir, lansoprazol e fenitoína.
t Frascos de 100 mg, 500 mg e 1g podem ser reconstituídos com 5 mL, 10 mL e 10 mL respectivamente, de água para injeção com álcool benzílico 0,945%. Entretanto, diluentes que contêm álcool benzílico não devem ser utilizados para reconstituição de citarabina para administração intratecal.
t Soluções para uso intratecal devem ser utilizadas imediatamente após a preparação.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: a síndrome de citarabina é observada entre a 6 e 12 horas após a administração do fármaco. Caracteriza-se por dor óssea, ocasionalmente dor torácica, exantema maculopapular, conjuntivite, febre, mialgia e mal estar. O uso de corticosteroides é benéfico para a prevenção e tratamento desta síndrome.
Karen Luise Lang
Na Rename 2010: item 18.4.6
t Comprimido 50 mg.
t Tratamento de infertilidade por falha de ovulação quando se deseja engravidar.
t Hipersensibilidade ao clomifeno ou a qualquer componente da formulação.
t Distúrbios hepáticos graves (ver Apêndice C).
t Lesão intracraniana orgânica.
t Disfunções tireoidiana e suprarrenal não controladas na existência de tumor pituitário.
t Cistos ovarianos.
t Tumores hormônio-dependentes.
t Hemorragias uterinas de causa desconhecida.
t Categoria de risco na gravidez (FDA): X (ver Apêndice A).
t Usar com cuidado nos casos de:
– distúrbios visuais (descontinuar ou realizar exame oftálmico).
– síndrome de hiperestimulação ovariana (descontinuar o tratamento imediatamente).
– síndrome do ovário policístico (cistos podem aumentar durante o tratamento).
– fibrose uterina.
– hepatopatias.
– lactação (ver Apêndice B).
t Aumento do risco de gravidez ectópica.
t Primeiro curso: 50 mg, por via oral, a cada 24 horas, durante 5 dias, iniciando nos 5 primeiros dias do ciclo menstrual, preferentemente no segundo dia, ou a qualquer momento se houver amenorreia (normalmente precedido por sangramento induzido por progestagênio). A maioria das pacientes ovula após o primeiro curso de tratamento.
t Segundo curso: 100 mg, por via oral, a cada 24 horas, durante 5 dias, 30 dias após a primeira administração; a ser realizado em caso de ausência de ovulação. Não se deve aumentar a dose ou a duração de uso.
t Terceiro curso: 100 mg, por via oral, a cada 24 horas, durante 5 dias, 30 dias após a segunda administração. Se a ovulação não ocorrer após o terceiro curso, não se recomenda continuar o tratamento e a paciente deve ser reavaliada.
t Início da ação: a ovulação normalmente ocorre em 5 a 12 dias após o término do curso do tratamento, sendo que esse tempo pode mudar de acordo com a paciente ou o ciclo.
t Pico de concentração plasmática: 6 a 7 horas.
t Meia-vida: 5 dias.
t Metabolismo preponderantemente hepático.
t Excreção fecal (37-51%) e renal (8%).
t Exantemas, acne, dermatite, eritema, fogachos (> 10%), hipertricose, alopecia (reversiva), prurido, urticária.
t Desconforto abdominal (> 5%), distensão abdominal, náuseas e vômitos (2,2%), obstipação, diarreia.
t Aumento do ovário, aumento e dor em mamas (2,1%), irregularidades menstruais, síndrome pré-menstrual, ressecamento vaginal.
t Depressão, cefaleia (1,3%), vertigens, insônia, fadiga.
t Tromboembolismo.
t Poliúria.
t Alterações de peso.
t Distúrbios visuais (1,5%)
t Orientar quanto a necessidade de obedecer rigorosamente o horário de administração.
t Orientar para o esquecimento de uma dose: ingerir assim que a paciente lembrar. Não tomar duas doses juntas. O esquecimento de doses deve sempre ser informado ao médico.
t Alertar quanto a possibilidade de nascimentos múltiplos.
t Manter ao abrigo de ar, luz, umidade e à temperatura ambiente, de 15 a 30 ºC.
César Augusto Braum
Na Rename 2010: item 5.6.1
t Comprimido 50 mg.
t Tratamento de filaríase linfática.
t Prevenção de filaríase linfática, em combinação com albendazol.
t Eosinofilia pulmonar tropical por Wuchereria bancrofti.
t Hipersensibilidade ao fármaco.
t Paciente enfraquecidos.
t Gravidez (ver Apêndice A).
t Usar com cuidado nos casos de:
– episódio agudo de linfangite (uso não recomendado).
– doenças cardíacas.
– oncocercose (realizar exames oftalmológicos durante a terapia para determinar o número de microfilárias intraocular e possíveis reações adversas).
– insuficiência renal (ver Apêndice D).
t Terapia precoce, antes da obstrução linfática, atinge melhor resultado terapêutico.
t A prevenção anual só é feita para indivíduos sob alto risco em zonas em que loiíase e oncocercose não sejam coendêmicas.
t Fisioterapia pode auxiliar no manejo do linfedema e da elefantíase.
t A administração concomitante com corticosteroides reduz as manifestações alérgicas secundárias à desintegração das microfilárias.
t Administrar metade da dose dos adultos.
t Nota: Programas de controle da doença em larga escala recomendam o tratamento com 3 mg/kg, em doses divididas, durante 24 horas, uma vez ao ano.
t Dose de 1 mg/kg, por via oral, em dose única no primeiro dia, aumentado gradualmente nos próximos 3 dias até a dose de 6 mg/kg/dia, dividida a cada 12 horas após as refeições por 12 a 21 dias. O uso concomitante com 400 mg/dia de albendazol durante 7 dias pode tornar o tratamento mais efetivo.
t Nota: Programas de controle da doença em larga escala recomendam o tratamento com 6 mg/kg/dia, em doses divididas, uma vez ao ano.
t Dose de 6 a 8 mg/kg/dia, por via oral, divididos em 3 doses, durante 14 a 21 dias. Repetir o tratamento caso os sintomas retornem.
t Apresenta rápida absorção oral.
t Pico de concentração plasmática: 1 a 2 horas.
t Meia-vida plasmática: aproximadamente 8 horas. A alcalinização da urina (pH 7,5 a 8,0) aumenta a meia-vida do fármaco.
t Duração de efeito: 6 meses a 4 anos após dose única.
t Em infecção por Wuchereria bancrofti a resposta inicial ocorre em 5 dias.
t Metabolismo: hepático.
t Excreção: renal (mais de 50% em forma inalterada) e fecal (menos de 5%).
t Prurido, erupções cutâneas.
t Reação de Mazzotti, caracterizada por febre, taquicardia, hipotensão e inflamação ocular.
t Náuseas, vômitos, anorexia.
t Esplenomegalia.
t Artralgia.
t Proteinúria.
t Leucocitose.
t Linfoadenopatia.
t Hemorragias retinianas, uveíte, queratite punctata.
t Asma em pacientes asmáticos.
t Cefaleia, tontura, sonolência, encefalopatia, cansaço, fraqueza.
t Orientar para ingerir o medicamento logo após as refeições.
t Medidas não farmacológicas incluem: higiene, uso de sapatos cômodos para evitar lesão nos pés, exercício e elevação dos pés para evitar estase linfática.
t Armazenar o medicamento em recipiente fechado e à temperatura ambiente.
Maurício Fábio Gomes
Na Rename 2010: itens 1.1.3 e 2.2
t Solução injetável 78,5 microgramas/mL (equivalente a 50 microgramas de fentanila/mL).
t Adjuvante analgésico em anestesia geral.
t Analgesia pós-operatória.
t Depressão/hipoventilação respiratória.
t Intolerância à fentanila ou a outros opioides.
t Hipersensibilidade à fentanila ou aos componentes da formulação.
t Íleo paralítico.
t Uso concomitante com naltrexona.
t Usar com cuidado nos casos de:
– neonatos (a meia-vida do fármaco é longa – aproximadamente 300 minutos – e efeito cumulativo pode ocorrer em uso prolongado).
– altas doses (risco de depressão respiratória; monitorar ventilação no pós-operatório; manter à disposição equipamento para ressucitação e intubação).
– interrupção após uso prolongado (deve ser gradual para diminuir síndrome de abstinência).
– pacientes enfraquecidos, idosos e crianças (risco aumentado de depressão respiratória).
– trauma cefálico, tumor cerebral (aumento da pressão intracraniana, inconsciência ou coma; retenção de dióxido de carbono pode exacerbar os efeitos sedativos dos opioides e aumentar o risco de dose excessiva fatal).
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– hipotireoidismo ou insuficiência supracortical.
– distúrbios convulsivos.
– distúrbios intestinais obstrutivos ou inflamatórios.
– hipertrofia prostática.
– hipotensão ou choque.
– miastenia grave.
– lactação (ver Apêndice B).
t A fentanila tem potência de abuso e causa dependência física e psicológica. t Evitar uso concomitante com álcool ou outros fármacos indutores de abuso. t Pode causar bradicardia (acompanhar função cardíaca).
t Há prejuízo no desempenho de tarefas que exijam coordenação motora, como dirigir e operar máquinas.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t 20 a 50 microgramas/kg em infusão intravenosa contínua, associada com óxido nitroso e oxigênio; doses de até 150 microgramas/kg podem ser requeridas.
t 2 a 50 microgramas/kg em infusão intravenosa contínua; doses de até 150 microgramas/kg podem ser requeridas. De modo alternativo, de 50 a 100 microgramas por via intramuscular, nos 30 a 60 minutos que antecedem a cirurgia; ou 50 microgramas/hora, por infusão epidural contínua (reduzindo para 25 microgramas/kg/hora).
t Dose única de 50 a 100 microgramas por via intramuscular ou por infusão intravenosa lenta, a cada 1 ou 2 horas, quando analgesia adicional for requerida.
t 25 microgramas de fentanila adicionados a 12,5 mg bupivacaína durante anestesia epidural. Se analgesia adicional for necessária administrar de 50 a 100 microgramas, por via intramuscular, ou por via intravenosa lenta (durante 1 a 2 minutos); podendo-se repetir após 1 ou 2 horas.
t 50 a 100 microgramas, por via intramuscular, nos 30 a 60 minutos que antecedem a cirurgia, em associação com droperidol.
t De 2 a 12 anos de idade: 30 microgramas/kg, por via intravenosa em bolo, seguida de infusão contínua de 0,3 microgramas/kg/minuto, combinada a óxido nitroso.
t De 2 a 12 anos de idade: 2 a 3 microgramas/kg/hora, por via intravenosa contínua.
t Neonatos: 0.5 a 2 microgramas/kg/hora, por via intravenosa contínua.
Aspectos farmacocinéticos clinicamente relevantes2-4
t Início de efeito: 7 e 8 minutos (intramuscular), 1 e 2 minutos (intravenosa).
t Pico de efeito: 20 e 30 minutos.
t Duração do efeito: 1 a 2 horas (intramuscular), 30 a 60 minutos (intravenoso).
t Meia-vida de eliminação: 219 minutos.
t Metabolismo: hepático.
t Excreção: renal e fecal.
t Edema periférico (5-32%), taquicardia (1% ou mais), hipotensão, palpitação.
t Desidratação (0-21%), perda de peso (3-11%), hipopotassemia (0-15%).
t Dor abdominal (3-15%), obstipação (8-26%), diarreia (0-16%), perda de apetite (5-11%), náusea (9-24%), vômito (0-37%), disfagia, ulceração da boca, alteração no paladar, xerostomia, espasmos biliares.
t Reação no lugar da aplicação (10%), dores nas costas (0-11%), rigidez muscular.
t Astenia (5-16%), confusão (3-16%), vertigem (6-32%), enxaqueca (2-15%), sedação (0-15%), depressão (3-11%), fadiga (2-20%), insônia (3-11%), mioclonia, ansiedade, alucinações, euforia, disforia, alterações no humor.
t Artralgia (0-8%).
t Retenção urinária, disfunção sexual.
t Visão borrada, ambliopia (<1%), visão anormal (2-3%)
t Tosse explosiva (3-9%), apneia (3-10%), dispneia (0-19%), pneumonia (2-
16%).
t Anemia (9-32%), neutropenia (0-8%).
t Agonistas/antagonistas opioides: pode precipitar sintomas da retirada de opioides. Se o uso concomitante for necessário, considerar, na ocorrência dos sintomas, tratamento de restituição de opioides. Se os sintomas não ocorrerem, a dose de fentanila pode ser aumentada progressivamente até alcançar o efeito desejado.
t Antifúngicos azólicos, antimicrobianos macrolídeos: pode aumentar ou prolongar o efeito opioide. Verificar sinais de toxicidade opioide e considerar a redução da dose de fentanila.
t Antirretrovirais: aumento do risco de toxicidade pela fentanila. Se o uso concomitante for necessário, considerar a redução da dose de fentanila, quando esta for administrada continuamente. Acompanhar sinais de depressão central.
t Barbitúricos, benzodiazepinas e relaxantes musculares de ação central: pode resultar em depressão respiratória aditiva. Se o uso concomitante for necessário, considerar a redução da dose de ambos os fármacos e observar sinais de depressão respiratória.
t Betabloqueadores: pode resultar em hipotensão grave. Descontinuar os betabloqueadores antes e durante a anestesia com fentanila.
t Bloqueadores de canais de cálcio: pode resultar em hipotensão grave. Na ocorrência de hipotensão pode ser necessário incrementar a volemia.
t Naltrexona pode precipitar sintomas da retirada de opioides e diminuir a efetividade de fentanila. Uso concomitante contraindicado.
t Nefazodona: pode aumentar ou prolongar o efeito opioide. Usar com cautela. Monitorar para sinais de toxicidade opioide e considerar a redução da dose de um dos fármacos.
t Sibutramina pode resultar no aumento do risco de síndrome serotoninérgica. Evitar uso concomitante.
t Tranilcipromina pode resultar em instabilidade cardíaca, hiperpirexia, coma e aumento grave e imprevisível dos efeitos opioides. Se o uso concomitante for necessário, descontinuar tranilcipromina duas semanas antes do uso de fentanila e aguardar algumas semanas para sua reintrodução.
t Orientar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Armazenar sob temperaturas ambiente, entre 15 e 25 oC e ao abrigo da luz.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t O citrato de fentanila é compatível com soluções injetáveis de glicose 5% e cloreto de sódio 0,9%.
t Incompatível com: azitromicina, pantoprazol sódico, dantroleno sódico, fenitoína sódica, diazóxido, sulfametoxazol-trimetoprima, fenobarbital sódico, metoexital, cloranfenicol, cefapirina e hidroxocobalamina.
t O citrato de fentanila tem perda por adsorção pelo PVC quando em soluções alcalinas.
t Evitar contato com a pele e a inalação de partículas de citrato de fentanila.
Atenção: o uso de citrato de fentanila está associado ao desenvolvimento de depressão respiratória e hipoventilação no pós-operatório, especialmente em condições clínicas especiais ou uso concomitante de medicamentos que favoreçam a ocorrência desse quadro. Considerar a descontinuação de outros medicamentos no esquema terapêutico do paciente em caso de uso do citrato de fentanila.
Larissa Niro
Na Rename 2010: Item 6.2.3
t Comprimidos 10 mg e 20 mg
t Carcinoma de mama avançado.
t Carcinoma de mama metastático em mulheres e homens.
t Carcinoma de mama intraductal in situ.
t Terapia com varfarina.
t Trombose venosa profunda e embolia pulmonar.
t Hipersensibilidade a tamoxifeno ou outro componente da formulação.
t A eficácia e segurança do citrato de tamoxifeno não foram estabelecidas para menores de 10 anos.
t Usar com cuidado nos casos de:
– carcinoma intraductal in situ (avaliar a relação benefício/risco do tratamento; risco de eventos adversos graves associados ao uso de citrato de tamoxifeno nessa condição).
– hiperlipidemias, leucopenia e trombocitopenia.
– uso concomitante com fármacos antineoplásicos (risco aumentado de tromboembolismo, anormalidades hepáticas e alterações visuais).
– lactação (ver Apêndice B).
– porfiria (evitar o uso).
– mulheres na pré-menopausa (inchaço de cistos ovarianos).
t Acompanhar alterações do endométrio (aumento da incidência de hiperplasia; câncer; sarcoma uterino e pólipos).
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Dose de 20 mg, por via oral, em dose única diária.
t Tratamento adjuvante: 20 a 40 mg, por via oral, diariamente, em dose única ou em 2 vezes, por 5 anos.
t Profilaxia: 20 mg, por via oral, em dose única diária, por 5 anos. Carcinoma de mama intraductal in situ (para evitar risco de doença invasiva) t Dose de 20 mg, por via oral, em dose única diária, por 5 anos.
t Dose de 20 mg, por via oral, em dose única diária, nos dias 2, 3, 4 e 5 do ciclo.
t Pico sérico: 5 horas.
t Início de resposta: entre 4 e 10 semanas.
t Metabolismo: hepático.
t Excreção: fecal (26 a 51%) e renal (9 a 13%).
t Meia-vida de eliminação: 5 a 7 dias; 14 dias (metabólito ativo).
t Fogachos (17-60%), amenorreia, distúrbios menstruais com períodos irregulares, corrimento e/ou sangramento vaginal, dor pélvica, fibrose uterina, prurido vulvar.
t Náuseas (3-21%).
t Acidente vascular cerebral, retenção hídrica (2-25%), trombose (1%).
t Alterações na córnea, catarata (8%), retinopatia, cirurgia de catarata (1,5%).
t Malignidades uterinas (risco aumentado em 2-5 vezes após o tratamento de 5 anos).
t Embolia pulmonar, raramente pneumonite intersticial.
t Alopecia, erupção cutânea, penfigoide.
t Reações de hipersensibilidade, incluindo angioedema e síndrome de Stevens-Johnson.
t Leucopenia, anemia, trombocitopenia, raramente neutropenia.
t Aldesleucina pode resultar em um risco aumentado de reação de hipersensibilidade (eritema, prurido, hipotensão). Observar sinais e sintomas de reações de hipersensibilidade. Intervenção médica pode ser necessária.
t Aminoglutetimida e rifampicina podem resultar em diminuição da concentração plasmática do tamoxifeno. Acompanhar a eficácia do tamoxifeno.
t Anastrozol e letrozol podem ter os níveis plasmáticos reduzidos. O uso concomitante não é recomendado.
t Bexaroteno pode resultar na redução da concentração plasmática do tamoxifeno. O uso concomitante deve ser feito com cautela.
t Ciclofosfamida, fluoruracila e metotrexato podem resultar em risco maior de sangramento tromboembolismo. O uso concomitante deve ser feito com cautela.
t Erva-de-são-joão, fluoxetina, paroxetina e sertralina podem resultar na diminuição da efetividade do tamoxifeno. Evitar o uso concomitante.
t Mitomicina pode resultar em aumento do risco de síndrome urêmica hemolítica. Acompanhar sinais desta síndrome.
t Varfarina tem efeito anticoagulante intensificado, prolongação do tempo de protrombina, podendo causar sangramento. O uso concomitante é contraindicado.
t Recomendar o uso de métodos contraceptivos, com exceção dos anticoncepcionais hormonais.
t Orientar para notificar possível gravidez.
t Orientar para não fumar nem ingerir bebida alcoólica.
t Orientar para ingerir em horários diferentes das refeições, com muita água.
t Orientar para não descontinuar o uso em caso de náuseas e vômitos.
t Seguir a dose e intervalos de tomada definidos pelo médico. Se esquecer uma dose do medicamento, desconsiderar a dose esquecida e voltar para o programa regular de doses. Não dobrar as doses. Chamar o médico ou farmacêutico para instruções.
t O tratamento com citrato de tamoxifeno geralmente é bem tolerado.
t Informar o uso de citrato de tamoxifeno sempre que procurar um serviço de saúde.
t Situações em que a paciente deve contatar imediatamente o profissional de saúde: alterações no corrimento vaginal; alterações visuais; sangramento vaginal; irregularidades na menstruação; dor nas pernas; dor pélvica; dor repentina no peito; tosse com sangue; novo caroço no seio.
t Armazenar sob temperaturas entre 20 e 25 ºC, em recipiente bem fechado e resistente à luz e umidade.
Atenção: a relação benefício/risco do tratamento do carcinoma intraductal in situ deve ser avaliada, pela ocorrência de eventos sérios em potência relacionados ao citrato de tamoxifeno nessa condição. Embora quando comparado com outras terapias, o tamoxifeno é semelhante ou melhor na eficácia e tem um perfil melhor de segurança.
Simone Sena Farina e Maria Inês de Toledo
Na Rename 2010: itens 5.1.6 e 16.3
t Cápsula ou comprimido de 250 mg.
t Erradicação de Helicobacter pylori.
t Infeccções por micobacterioses atípicas (Mycobacterium avium)
t Hipersensibilidade à claritromicina, eritromicina ou qualquer antibiótico macrolídeo.Ver fármacos contraindicados no item interações de medicamentos desta monografia.
t Usar com cuidado nos casos de:
– insuficiência renal grave (ver apêndice D).
– desenvolvimento de diarreia durante o tratamento (considerar diagnóstico de colite pseudomembranosa).
– lactação.
– história de porfiria aguda (uso concomitante com ranitidina não é recomendado).
– ausência de provas ou forte suspeita de infecção (aumento do risco de desenvolver resistência bacteriana).
– neonatos com menos de duas semanas.
– porfiria aguda (evitar uso).
– insuficiência hepática (ver apêndice C).
t Predisposição a prolongamento de intervalo QT no eletrocardiograma.
t Categoria de risco na gravidez (FDA): C.
t 7,5 a 15 mg/kg, por via oral, a cada 12 horas, combinado a etambutol 15 a 25 mg/kg, por via oral, a cada 24 horas. Doses máximas diárias de 1 g para claritromicina e 2,5 g para etambutol.
t 500 mg, por via oral, a cada 12 horas, combinado a omeprazol 20 mg e amoxicilina 1 g ou metronidazol 500 mg, ambos por via oral, a cada 12 horas, durante 7 a 14 dias.
t 500 mg, por via oral, a cada 12 horas, em combinação com etambutol 15 mg/ kg, por via oral, a cada 24 horas.
t Presença de alimento reduz a velocidade de absorção, mas não sua extensão
t Pico de concentração sérica: 2 a 3 horas.
t Meia-vida de eliminação: 5 a 7 horas, para dose de 500 mg a cada 12 horas.
t Metabolismo: hepático.
t Excreção: renal.
t Alteração no paladar (3% a 18,9%) e olfato, diarreia (adulto 3% a 6%, crianças 6%), náusea (3%), indigestão (2%), vômito (6%), dor abdominal (adulto 2%, crianças 3%), estomatite, glossite, descoloração dos dentes e língua.
t Cefaleia (2% a 9%).
t Insuficiência hepática e hepatite.
t Reação de hipersensibilidade grave, anafilaxia, síndrome de StevensJonhson, necrólise epidérmica tóxica.
t Artralgia, mialgia.
t Hipoglicemia.
t Leucopenia, trombocitopenia.
t Pancreatite.
t Insuficiência renal.
t Efeitos transitórios sobre o SNC (insônia, tontura, ansiedade, confusão, psicose, parestesia, convulsões, pesadelos).
t Alfentanila: tem níveis plasmáticos aumentados. Fazer monitoria no paciente.
t Alprazolam, diazepam, midazolam: podem ter a toxicidade aumentada (depressão do sistema nervoso central, ataxia e letargia). Pode ser necessário reduzir dose do benzodiazepínico (entre 50 e 75%) após dois a quatro dias de uso concomitante.
t Atorvastatina, lovastatina, sinvastatina: pode ocorrer aumento do risco de miopatia e rabdomiólise. Muita precaução no uso com atorvastatina. Suspender terapia com sinvastatina. Acompanhar níveis de creatinaquinase e sinais e sintomas de miopatia e rabdomiólise.
t Bepridil, disopiramida, fluconazol, gemifloxacino, quinidina, pimozida, ranolazina, tioridazina, ziprasidona: pode resultar em cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca). Verificar prolongamento do segmento QT no eletrocardiograma e níveis plasmáticos dos fármacos citados. É contraindicado o uso concomitante.
t Bromocriptina: pode ter as concentrações plasmáticas aumentadas. Verificar concentrações plasmáticas de bromocriptina.
t Carbamazepina: pode ter risco de toxicidade aumentado (ataxia, nistagmo, diplopia, cefaleia, vômito, apneia, convulsão e coma). Reduzir dose da carbamazepina em 25% no início do tratamento com claritromicina e ajustar conforme sintomas clínicos e concentrações séricas de carbamazepina.
t Ciclosporina: pode ter risco de toxicidade aumentado (disfunção renal, colestase e parestesia). Realizar concentrações plasmáticas de ciclosporina, ajustar a dose se necessário e acompanhar toxicidade da ciclosporina.
t Cilostazol: pode aumentar a exposição sistêmica do cilostazol. Monitorar toxicidade do cilostazol.
t Colchicina: pode ter concentrações plasmáticas e risco de toxicidade aumentados. Evitar o uso concomitante, mas se este for necessário, observar sinais e sintomas de toxicidade da colchicina (dor abdominal, vômito, diarreia, febre e pancitopenia).
t Darunavir, delavirdina, lopinavir, ritonavir: pode ocorrer aumento das concentrações plasmáticas de claritromicina. Observar pacientes quanto aos Efeitos adversos da claritromicina. Verificar função renal. Em pacientes com depuração de creatinina endógena (DCE) entre 30 e 60 mL/minuto, reduzir a dose de claritromicina em 50%; em pacientes com DCE menor que 30 mL/ minuto, reduzir a dose em 75%.
t Ergotamina e análogos: pode ocorrer aumento do risco de ergotismo agudo (náusea, vômito, isquemia vasoespástica). Uso contraindicado.
t Digoxina: pode resultar em toxicidade da digoxina (náusea, vômito e arritmias). Uso concomitante deve ser evitado, mas se este for necessário, verificar níveis séricos de digoxina e considerar diminuição da dose.
t Efavirenz: pode aumentar o risco de exantema. Considerar o uso de outro antibiótico macrolídeo, como azitromicina.
t Estrogênios: podem ter aumentadas suas concentrações plasmáticas. Observar sinais e sintomas desse aumento (hipertensão, cefaleia, tromboembolismo, depressão, ganho de peso, retenção de líquidos, dor abdominal aguda, flatulência ou distensão abdominal por gases).
t Fenitoína: pode haver aumento do risco de toxicidade (ataxia, hiperreflexia, nistagmo e tremor). Acompanhar as concentrações plasmáticas da fenitoína.
t Fentanila: pode ter os efeitos opioides intensificados ou prolongados (depressão do sistema nervoso central, depressão respiratória). Pode ser necessário ajustar dose da fentanila.
t Fluoxetina: pode resultar em delírios e psicose. Evitar uso.
t Glipizida: pode aumentar o risco de hipoglicemia. Uso com precaução, especialmente em diabéticos com insuficiência renal moderada. Considerar monitoria adicional de glicemia.
t Indinavir: pode ocorrer aumento nas concentrações de claritromicina e/ou indinavir. Precaução no uso.
t Itraconazol: pode aumentar as concentrações de ambos. Verificar toxicidade da claritromicina e do itraconazol.
t Metilprednisolona, prednisona: pode aumentar o risco de sintomas psicóticos. Acompanhar o comportamento do paciente. Considerar uso de outro antibiótico.
t Nevirapina: pode diminuir as concentrações de claritromicina. Utilizar outro antimicrobiano, como azitromicina.
t Paroxetina: pode aumentar o risco de síndrome serotoninérgica (hipertensão, hipertermia, mioclônus, mudanças do estado mental). Acompanhar o paciente e considerar a redução da dose da paroxetina.
t Repaglinida: pode aumentar a exposição e a concentração plasmática de repaglinida. Ajuste de dose da repaglinida pode ser necessário. Avaliar concentração de glicose.
t Rifabutina: pode resultar em concentrações plasmáticas subterapêuticas de claritromicina e aumentar o risco de toxicidade da rifabutina (exantema, distúrbios gastrintestinais, anormalidades hematológicas). Precaução no uso. Avaliar eficácia terapêutica e toxicidade.
t Saquinavir: pode ter níveis séricos aumentados. Observar toxicidade do saquinavir.
t Tacrolimo: pode ter toxicidade aumentada (nefrotoxicidade e hiperglicemia). Verificar concentrações plasmáticas de tacrolimo e sinais de toxicidade dele. Reduzir dose do tracolimo se necessário.
t Tolterodina: pode ter a biodisponibilidade aumentada em indivíduos com atividade deficiente do citocromo P450 2D6.
t Trazodona: pode ter níveis plasmáticos aumentados. Considerar utilização de doses menores de trazodona. Observar Efeitos adversos da claritromicina
t Varfarina: pode aumentar o risco de sangramento. Verificar tempo de protrombina. Pode ser necessário ajustar dose da varfarina.
t Verapamil: pode aumentar o risco de hipotensão e/ou bradicardia. Acompanhar paciente e ajustar a dose do verapamil.
t Orientar que não há restrições quanto ao uso com alimentos.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses para evitar resistência bacteriana.
t Armazenar entre 15 e 30 ºC, em recipientes fechados e protegido da luz.
Atenção: este fármaco apresenta número elevado de Efeitos adversos e deve ser realizada pesquisa específica sobre este aspecto ao introduzir ou descontinuar este ou outros medicamentos no esquema terapêutico do paciente.
Silvio Barberato Filho e Simone Sena Farina
Na Rename 2010: item 5.2.3
t Cápsula de 50 mg e 100 mg.
t Hanseníase, em terapia múltipla (multibacilar).
t Hanseníase com bacilos resistentes a dapsona (adultos).
t Eritema nodoso hansênico.
t Hipersensibilidade à clofazimina.
t Usar com cuidado nos casos de:
– insuficiência renal e hepática (ver apêndice C) e sintomas gastrintestinais prévios.
– lactação (ver Apêndice B).
t O fármaco induz alteração de coloração cutânea (de rosa a castanho-escuro), de mucosas, de líquidos orgânicos e de lentes de contato gelatinosas. É reversiva, mas pode demorar meses ou anos para retornar à cor normal.
t Categoria de risco na gravidez (FDA): C.
t Até 30 kg: 1 mg/kg, por via oral, a cada 24 horas, combinada a dapsona 1,5 mg/kg/dia, por via oral, a cada 24 horas, e uma dose mensal supervisionada de rifampicina 10 a 20 mg/kg, clofazimina 5 mg/kg e dapsona 1,5 mg/kg, todos por via oral, durante 12 meses.
t De 30 kg ou mais: 50 mg, por via oral, a cada 48 horas, com dapsona 50 mg, por via oral, a cada 24 horas, e uma dose mensal supervisionada de rifampicina 450 mg, clofazimina 150 mg e dapsona 50 mg, todos por via oral, durante 12 meses.
t Dose de 100 mg, por via oral, a cada 8 ou 12 horas, por no máximo 3 meses. De 4 a 6 semanas de tratamento podem ser necessárias até o início dos efeitos.
t 50 mg, por via oral, a cada 24 horas, combinada a dapsona 100 mg, por via oral, a cada 24 horas, e uma dose mensal supervisionada de rifampicina 600 mg e clofazimina 300 mg, ambos por via oral, durante 12 meses.
t 100 mg, por via oral, a cada 24 horas, durante 3 anos, com um ou mais hansenostáticos, seguido por monoterapia com clofazimina 100 mg, por via oral, uma vez ao dia.
t Dose de 100 mg, por via oral, a cada 8 ou 12 horas, por no máximo 3 meses. De 4 a 6 semanas de tratamento podem ser necessárias até o início dos efeitos.
t Biodisponibilidade: 70%. Alimentos aumentam a absorção.
t Início de efeito: 3 a 14 dias; em eritema nodoso: 4 a 6 semanas.
t É amplamente armazenado em tecido adiposo.
t Metabolismo: hepático.
t Meia-vida: 70 dias.
t Excreção: fecal (11% a 59%).
t Mais comuns: alteração da coloração da pele e do cabelo, pele seca, prurido e exantema; distúrbios gastrintestinais, náusea e vômito; alteração da coloração da conjuntiva, da córnea e dos fluidos corporais.
t Mais graves: obstrução intestinal, hemorragia gastrintestinal; sintomas gastrintestinais graves; depósito de clofazimina no cristalino; depressão reativa em consequência da alteração da coloração da pele; enfarte esplênico.
t Outros: erupções acneicas, xeroftalmia, fotossensibilidade, hiperglicemia e perda de peso.
t Antiácidos (contendo sais de alumínio e magnésio): podem resultar em diminuição da concentração plasmática de clofazimina. Evitar o uso concomitante.
t Fenitoína: pode resultar em diminuição da concentração sérica de fenitoína e perda de eficácia. Avaliar eficácia e níveis séricos da fenitoína. Pode ser necessário ajustar dose.
t Suco de laranja: pode resultar em diminuição da concentração plasmática de clofazimina. Evitar o uso concomitante.
t Orientar para ingerir com alimentos (exceto suco de laranja) para evitar desconforto estomacal e melhorar a absorção.
t Orientar para procurar uma Unidade de Saúde em caso de sintomas de depressão ou gastrintestinais.
t Alertar para a possibilidade de ocorrer alteração, com reversão, na coloração cutânea e dos olhos. Pode demorar meses ou anos para retornar à cor normal.
t Alertar para a possibilidade de também ocorrer alteração na coloração de fezes, urina, saliva, suor e lágrimas.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Armazenar à temperatura ambiente, protegido da luz, calor e umidade.
Rachel Magarinos-Torres
Na Rename 2010: item 13.1
t Solução oral 2,5 mg/mL
t epilepsia em crianças (epilepsia mioclônica grave na infância, epilepsia mioclônica juvenil e síndrome de Gasteaut-Lennox; tratamento de segunda escolha).
t Hipersensibilidade a clonazepam ou outros benzodiazepínicos.
t Glaucoma de ângulo fechado.
t Insuficiência hepática grave (ver Apêndice C).
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– doença respiratória, porfiria, histórico de dependência de álcool e/ou psicofármacos, depressão, e pacientes em uso de álcool e outros depressores do SNC.
– pacientes com múltiplos tipos de convulsões (pode ocorrer piora das convulsões).
– interrupção do tratamento (deve ser gradual, 0,125 mg, 2 vezes ao dia, a cada 3 dias).
t Encontra-se no leite materno, não sendo recomendado o uso em lactantes (ver apêndice B).
t Até 1 ano: iniciar com dose de 0,25 mg, por via oral, durante 4 dias; aumentar ao longo de 2 a 4 semanas até dose ótima de acordo com a resposta da criança. Dose máxima de manutenção 0,5 a 1,0 mg/dia.
t Entre 1 e 5 anos: iniciar com dose de 0,25 mg, por via oral, durante 4 dias; aumentar ao longo de 2 a 4 semanas até dose ótima de acordo com a resposta da criança. Dose máxima de manutenção de 1 a 3 mg/dia.
t Entre 5 e 12 anos: iniciar com dose de 0,5 mg, por via oral, durante 4 dias; aumentar ao longo de 2 a 4 semanas até dose ótima de acordo com a resposta da criança. Dose máxima de manutenção de 3 a 6 mg/dia.
t Bem absorvido por via oral.
t Resposta inicial ao uso oral: 20 a 40 minutos.
t Pico plasmático: 1 a 2 horas.
t Meia-vida de eliminação: 18 a 33 horas.
t Aumento da secreção salivar e/ou bronquial com risco de problemas respiratórios.
t Amnésia, ataxia, disartria, sonolência, concentração difícil, fadiga, fraqueza muscular, distúrbios de coordenação, labilidade emocional, reação paradoxal (agressividade, ansiedade), vertigem, depressão respiratória, cefaleia.
t Desenvolvimento prematuro de características sexuais secundárias, disfunção sexual.
t Síndrome da boca ardente.
t Incontinência urinária.
t Urticária, prurido, perda de cabelo reversiva, mudanças na pigmentação da pele.
t Distúrbios visuais.
t Trombocitopenia.
t Amiodarona: risco aumentado de toxicidade (confusão, fala indistinta, enurese). Observar sinais de intoxicação. Se presentes, reduzir dose de clonazepam.
t Analgésicos opioides (ex: meperidina, codeína, morfina e fentanila), barbitúricos (ex.: fenobarbital, tiopental): risco de depressão respiratória. Acompanhar os níveis séricos de clonazepam e atentar para sinais e sintomas de toxicidade como sedação, tontura e confusão. Pode ser necessário reduzir a dose de um ou de ambos os medicamentos.
t Carbamazepina, nevirapina, teofilina: podem diminuir concentração plasmática e efeito do clonazepam. Pode ser necessário aumento na dose do clonazepam.
t Desipramina: tem sua efetividade reduzida. Pode ser necessário aumentar doses de desipramina se houver adição do clonazepam e, inversamente, reduzir a dose de desipramina após interrupção do uso do clonazepam.
t Erva-de-são-joão (Hypericum perforatum), teofilina: podem reduzir efetividade do clonazepam pela redução dos níveis plasmáticos. Avaliar alterações nos efeitos terapêuticos e adversos dos benzodiazepínicos. Se houve o uso da erva-de-são-joão antes de cirurgia na qual foi utilizado midazolam, avaliar o paciente quanto a sinais de efetividade reduzida e ajustar dose do benzodiazepínico, se necessário. Rever esquema posológico quando for interrompida a administração simultânea.
t Ritonavir: aumento da concentração plasmática de clonazepam e toxicidade associada (sedação excessiva, confusão). Verificar níveis de clonazepam, avaliar sinais e sintomas de toxicidade. Pode ser necessário reduzir doses de clonazepam.
t Alertar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Alertar para risco de quedas em idosos.
t Alertar para não ingerir bebidas alcoólicas.
t Orientar para não suspender tratamento abruptamente.
t Informar mulheres em idade fértil quanto aos riscos e aconselhar a comunicação quanto a possibilidade de gravidez.
t Conservar sob temperaturas entre 15 e 30 ºC, em recipientes bem fechados e ao abrigo da luz.
Atenção: clonazepam é medicamento de segunda linha para a indicação apontada. Como os demais benzodiazepínicos, causa dependência física.
Maurício Fábio Gomes
Na Rename 2010: itens 6.1.1
t Comprimido 2 mg
t Leucemia linfocítica crônica.
t Linfoma Hodgkin e não-Hodgkin.
t Macroglobulinemia de Waldenström
t Hipersensibilidade ou prévia resistência a clorambucila.
t Usar com cuidado nos casos de:
– crianças (faltam provas conclusivas de eficácia e segurança).
– porfiria aguda (se o uso for justificável, a excreção urinária de porfibilinogênio deve ser verificada regularmente; se houver aumento, o tratamento deve ser interrompido).
– reações de hipersensibilidade com erupções cutâneas (substituir clorambucila por ciclofosfamida).
– após pelo menos 4 semanas de radioterapia ou quimioterapia com outros agentes (evitar o uso ou administrar em doses reduzidas).
– história de convulsões ou traumas cranianos.
– história de epilepsia em crianças com síndrome nefrótica.
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– lactação (ver Apêndice B).
t A elevação de ácido úrico pode ser prevenida com hidratação oral adequada ou, se necessário, com o uso de alopurinol.
t Verificar sintomas de mielossupressão e realizar contagens sanguíneas regularmente.
t Toxicidade pulmonar.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Inicialmente: dose de 100 a 200 microgramas/kg/dia, por via oral, em dose única diária, por 3 a 6 semanas.
t Manutenção: dose de 30 a 100 microgramas/kg/dia, por via oral, em dose única diária, iniciada 4 semanas após o primeiro curso.
t Esquemas alternativos: dose de 400 microgramas/kg/dia, por via oral, em dose única, bi-semanal ou mensalmente. Aumenta-se em 100 microgramas/ kg, por via oral, até controle da linfocitose ou observação de toxicidade.
t Inicialmente: dose de 100 a 200 microgramas/kg/dia, por via oral, em dose única diária, por 3-6 semanas.
t Manutenção: dose de 30 a 100 microgramas/kg/dia, por via oral, em dose única diária.
Observação:
t Na presença de infiltração linfocítica da medula óssea ou quando a mesma se encontrar hipoplásica, a dose de clorambucila não deve exceder 0,1 mg/ kg/dia.
t Inicialmente: dose de 100 a 200 microgramas/kg/dia, por via oral, em dose única diária, por 3 a 6 semanas.
t Manutenção: dose de 30 a 100 microgramas/kg/dia, por via oral, em dose única diária.
t dose de 6 a 12 mg/dia até ocorrência de leucopenia, então reduzir para 2 a 8 mg/dia.
t Absorção rápida e completa pelo trato grastrintestinal, prejudicada pela ingestão com alimentos.
t Pico de concentração: 1 hora.
t Metabolismo: hepático.
t Meia-vida: 1,5 horas.
t Excreção: Renal.
t Anemia, leucopenia, neutropenia (25-33%), pancitopenia, trombocitopenia, leucemia, mielossupressão.
t Eritema multiforme (raro), síndrome de Stevens-Johnson (raro), necrólise epidérmica tóxica (raro), febre.
t Hepatotoxicidade.
t Infertilidade.
t Neuropatia periférica, convulsões; alucinações.
t Pneumonite aguda, toxicidade pulmonar.
t Neoplasia maligna secundária.
t Estomatites.
t Vacina oral contra rotavírus humano: aumento do risco de infecção pela vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pela vacina. Em pacientes com leucemia em remissão, a vacinação é permitida após três meses da descontinuação da quimioterapia.
t Orientar para armazenar este medicamento no refrigerador.
t Orientar para aumentar a ingestão de líquidos.
t Orientar para não interromper o tratamento se houver náuseas e vômitos.
t Evitar proximidade com pessoas resfriadas ou com outras infecções durante o tratamento.
t Orientar para evitar vacinas, especialmente contra poliovírus, ou contato com pessoas próximas que receberam a vacina. Se for imprescindível, usar máscara de proteção.
t Alertar para a necessidade de lavar as mãos com frequência, tomar cuidado para não se envolver em situações que determinem cortes e machucados, e ter inclusive cautela ao realizar higiene pessoal.
t Armazenar sob refrigeração, sob temperaturas entre 2 e 8 oC, livre de umidade.
t Os comprimidos de clorambucila mantêm-se estáveis por 1 semana a temperaturas até 30 oC.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: clorambucila pode causar mielossupressão grave e infertilidade.
Fernando de Sá Del Fiol e Silvio Barberato Filho
Na Rename 2010: item 5.1.11
t Cloranfenicol: cápsula ou comprimido de 250 mg.
t Palmitato de cloranfenicol: xarope 54,4 mg/mL.
t Succinato sódico de cloranfenicol: pó para solução injetável 1 g.
t Tratamento alternativo de infecções graves, por bactérias sensíveis, no sistema nervoso central e epiglotite aguda em crianças.
t Peste (Yersinia pestis).
t Febre tifoide (Salmonella typhi).
t Hipersensibilidade ao cloranfenicol.
t Porfiria (cloranfenicol está relacionado a ataques agudos de porfiria).
t Usar com cuidado nos casos de:
– prematuridade e baixo peso ao nascimento.
– imaturidade hepática, como em recém-nascidos e prematuros (ocorre acúmulo do fármaco que desencadeia a síndrome cinzenta).
– neonatos (risco de aplasia de medula óssea; realizar contagem de células sanguíneas e das concentrações plasmáticas do fármaco).
– insuficiência hepática (ver Apêndice C).
– lactação (ver Apêndice B).
t Evitar tratamentos repetidos e prolongados.
t Categoria de risco da gravidez (FDA): C (Ver Apêndice A).
Neonatos, com menos de duas semanas
t 25 mg/kg/dia, por via oral ou intravenosa fracionada a cada 6 horas.
Com mais de duas semanas a um ano de idade
t 50 mg/kg/dia, por via oral ou intravenosa fracionada a cada 6 horas.
Acima de 1 ano
t 50 a 100 mg/kg/dia, por via oral ou intravenosa, fracionada a cada 6 horas. Reservar a dose maior para infecções graves (meningite, septicemia e epiglotite por Haemophilus).
t 50 a 100 mg/kg/dia, por via oral ou intravenosa, fracionada a cada 6 horas. Reservar a dose maior para infecções graves (meningite, septicemia e epiglotite por Haemophilus).
t Nota: em infecções por Rickettsia, recomenda-se que o tratamento seja mantido por 4 dias adicionais após normalidade da temperatura do paciente, para diminuir o risco de recidiva; no caso de febre tifoide, deve ser mantido por 8 a 10 dias adicionais.
t Absorção por via oral (90-100%).
t Distribuição plena no líquor.
t Pico de concentração sérica: 2 a 4 horas (oral) e 30 minutos (intravenosa).
t Metabolismo: hepático (90%), originando metabólitos inativos.
t Excreção: renal (5% a 15%; metabólitos inativos; não necessita ajuste em insuficiência renal), biliar (2% a 4%) e fecal (2% a 4%).
t Meia-vida de eliminação: 1,6 a 3,3 horas. A meia-vida se prolonga até 28 horas em neonatos e é imprevisível em pacientes com insuficiência hepática.
t Removido por diálise.
t Intolerância digestiva, gosto desagradável.
t Supressão medular reversiva e dependente da dose (anemia, trombocitopenia e leucopenia progressivas).
t Neurite óptica (3,5%).
t Anemia aplástica (reação idiossincrática, em potência fatal, que ocorre com a administração por qualquer via).
t Síndrome cinzenta do recém-nascido (cianose, taquipneia, distensão abdominal, vômitos, diarreia, hipotonia, hipotermia e colapso circulatório agudo). Os sintomas da intoxicação iniciam-se 3 a 4 dias do uso do medicamento.
t Ceftazidima: pode ter sua efetividade reduzida. Evitar a associação. Caso necessário, acompanhar a evolução da infecção.
t Ciclosporina: pode ter sua toxicidade aumentada por inibição de seu metabolismo. Verificar níveis de ciclosporina e ajustar dose se necessário.
t Clorpropamida: pode levar a hipoglicemia (com depressão do SNC e convulsões), por aumentar o tempo de meia-vida da clorpropamida. Avaliar a glicose sanguínea e espaçar as doses de cloranfenicol, se necessário.
t Dicumarol: pode ter sua concentração plasmática aumentada, por inibição de seu metabolismo. Verificar a concentração plasmática e os Efeitos adversos do dicumarol.
t Fenitoína, fosfenitoína: podem ter sua toxicidade aumentada (ataxia, hiperreflexia, nistagmo, tremores) por diminuição de seu metabolismo. Avaliar os níveis plasmáticos, reduzindo dose, se necessário.
t Paracetamol: pode aumentar a toxicidade do cloranfenicol. Acompanhar as concentrações plasmáticas de cloranfenicol e ajustar a dose, se necessário.
t Rifampicina, rifapentina: pode diminuir a efetividade do cloranfenicol, por ter seu metabolismo aumentado. Verificar a concentração de cloranfenicol e ajustar a dose se necessário.
t Tacrolimo: pode ter sua concentração aumentada, com aumento da toxicidade, por redução do metabolismo. Acompanhar os níveis de tacrolimo e seus Efeitos adversos (nefrotoxicidade, hiperglicemia, hiperpotassemia).
t Tolbutamida: pode levar a hipoglicemia (depressão do SNC e convulsões), por aumentar o tempo de meia-vida da tolbutamida. Associação contraindicada. Se necessário, avaliar a glicose sanguínea.
t Voriconazol: pode ter sua concentração plasmática aumentada, por inibição do metabolismo. Acompanhar concentração plasmática e Efeitos adversos do voriconazol.
t Usar com o estômago vazio: uma hora antes ou duas horas após às refeições.
t Orientar para consultar o médico regularmente, por causa da necessidade de acompanhamento de problemas sanguíneos.
t Orientar para procurar assistência médica caso surjam sangramentos.
t Orientar para usar durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Armazenar em recipiente fechado à temperatura ambiente, longe do calor umidade e luz direta.
t Na forma liofilizada, o succinato sódico de cloranfenicol é estável sob temperatura ambiente.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Após reconstituição em solução de glicose 5% (pH de 6 a 7,5) é estável por 24 horas.
t Pequena mudança na coloração não indica redução na atividade. Soluções turvas não devem ser utilizadas.
t Soluções congeladas de succinato sódico de cloranfenicol são estáveis por 6 meses.
Atenção: o risco de aplasia medular pelo cloranfenicol é muito baixo, mas sua gravidade levou ao abandono de seu emprego como primeira escolha.
Isabella Campagnuci Knust
Na Rename 2010: item 8.2
t Solução injetável 10 mg/mL.
t Metemoglobinemia por intoxicações exógenas.
t Metemoglobunemia induzida por clorato ou por nitrito (envenenamento por cianeto).
t Hipersensibilidade ao fármaco.
t Anemia hemolítica.
t Usar com cuidado nos casos de:
– altas doses (acentuam a metemoglobinemia; avaliar o paciente).
– insuficiência renal grave.
– indivíduos com deficiência de glicose-6-fosfato desidrogenase (não há melhora de sintomas e pode resultar em hemólise).
– lactação.
t Evitar injeção subcutânea pelo risco de abscesso necrótico.
t Verificar gasometria, hemólise e nível de metemoglobina.
t Categoria de risco na gravidez (FDA): C
t Dose de 0,1 a 0,2 mL/kg, por via intravenosa, sem diluição (como solução a 1%) ou diluída em soro fisiológico a 0,9%, durante 5 minutos. Pode ser repetido após 1 hora, se necessário.
t Excreção: urina (75%).
t Cefaleia, tontura, confusão mental.
t Sudorese.
t Hipertensão, hipotensão.
t Arritmias.
t Náuseas, vômitos.
t Anemia hemolítica
t Metemoglobinemia com altas doses.
t Coloração azulada dos fluidos corporais.
t Hipertermia maligna.
t Bupropiona, buspirona, clomipramina, mirtazapina ou venlafaxina: se utilizados recentemente, podem provocar efeitos tóxicos no sistema nervoso central, caso o paciente receba administração intravenosa do cloreto de metiltionínio. O uso concomitante é contraindicado.
t Pode haver interação com exame de oximetria, levando a falso resultado.
t Os fluidos orgânicos podem ficar tingidos de azul. Alertar que pode haver tingimento de pele, mucosas e roupas.
t Ampolas devem ser mantidas à temperatura ambiente (15-30 ºC), em frascos bem tampados, de preferência de cor âmbar, ao abrigo da luz.
Elaine Silva Miranda
Na Rename 2010: itens 9 e 10.3
t Solução injetável 19,1% (2,56 mEq/mL).
t Hipopotassemia, com ou sem alcalose metabólica.
t Correção do equilíbrio eletrolítico.
t Hipopotassemia.
t Hipercloremia.
t Adinamia episódica hereditária.
t Cãibras por calor.
t Concentração plasmática de potássio acima de 5 mmol/L.
t Insuficiência renal grave (ver Apêndice D).
t Desidratação aguda.
t Trauma tecidual grave.
t Doença de Addison não tratada.
t Usar com cuidado nos casos de:
– doença cardíaca, insuficiência renal, insuficiência adrenocortical, úlcera péptica, diarreia grave ou prolongada, compressão esofágica, obstrução intestinal, acidose metabólica aguda, diabete melito descontrolada.
– uso de medicamentos que aumentam a concentração plasmática de potássio, tais como diuréticos poupadores de potássio, inibidores da ECA e ciclosporinas.
– destruição tecidual extensa como ocorre em queimaduras graves.
t A concentração da solução para infusão intravenosa não deve exceder a 3,2 g (43 mmol/L).
t A infusão rápida é cardiotóxica.
t Fazer monitoria eletrocardiográfica.
t Categoria de risco na gravidez (FDA): C
t Inicial: dose de 1 mEq/kg por infusão intravenosa, por duas horas, repetidos na medida do necessário; a infusão intermitente não excede 1 mEq/kg/hora ou 40 mEq/hora.
t Administração intravenosa intermitente de 0.5 a 1 mEq/kg/dose; infundida a 0.3 a 0.5 mEq/kg/h; máximo de 1 mEq/kg/h e 30 mEq por dose. Dose máxima: 3 mEq/kg/dia.
t Infusão intravenosa intermitente na velocidade de 5-10 mEq/hora, não excedendo 40 mEq/hora; dose máxima: 400 mEq/dia.
t Potássio sérico menor que 2 mEq/L: 20 a 40 mEq/hora, em infusão intravenosa, com monitoria cardíaca contínua; dose máxima: 400 mEq/dia.
t Potássio sérico maior que 2,5 mEq/L: 10 a 15 mEq/hora, por infusão intravenosa; dose máxima: 200 mEq/dia.
t Excreção: renal (90%).
t Dor no lugar da injeção, necrólise ao extravasamento.
t Hiperpotassemia, alcalose.
t Fraqueza.
t Confusão, dormência nas mãos, pés ou lábios.
t Transtornos respiratórios, dispneia.
t Ansiedade, cansaço.
t Bradicardia, arritmia.
t Amilorida, antagonistas de receptores de angiotensina II, diuréticos poupadores de potássio, indometacina, inibidores da enzima conversora da angiotensina: risco aumentado de hiperpotassemia. Avaliar potássio sérico.
t Sais de cálcio usados concomitantemente induzem risco aumentado de arritmia cardíaca.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução
t Deve ser diluído antes da infusão.
t A solução deve ser armazenada entre 15 e 30 ºC. Evitar o congelamento.
t Incompatível com: anfotericina B, diazepam, ergotamina, estreptomicina, fenitoína, metilprednisolona, nitroprusseto de sódio. Somente utilizar se a solução estiver límpida.
t Usar as preparações dentro de 24 horas.
Rogério Hoefler
Na Rename 2010: itens 9, 10.3
t Solução injetável 0,9% (0,154 mEq/mL)
t Solução injetável 20% (3,4 mEq/mL)
t Solução injetável 0,9%:
– Reposição hídrica e eletrolítica.
– Hipernatremia com depleção de volume.
– Veículo ou diluente para a administração parenteral de fármacos e para manter desobstrução de cateteres e cânulas.
– Fluido para irrigações estéreis, por exemplo, de olho ou bexiga e limpeza geral de pele ou ferimento.
– Veículo para nebulização.
t Solução injetável 20%:
– Reposição eletrolítica.
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D), insuficiência cardíaca, hipertensão, edema pulmonar e periférico, toxemia gravídica, insuficiência circulatória, hipoproteinemia, doença cirrótica (ver Apêndice C), hipervolemia, obstrução do trato urinário e uso de fármacos que causam retenção de sódio (requerem ajuste de dose).
– infusão intravenosa de cloreto de sódio 0,9% durante ou imediatamente após cirurgia (pode resultar em retenção de sódio).
– hiponatremia dilucional aguda grave (embora soluções hipertônicas possam ser usadas em certos pacientes, a correção deve ser lenta para evitar síndrome de desmielinização osmótica).
t Evitar administração excessiva. O aumento da concentração plasmática de sódio não deve exceder 10 mmol/L em 24 horas.
t Observar pressão venosa jugular, crepitações em bases pulmonares e, em idosos, pressão venosa central.
t Categoria de risco na gravidez (FDA): C
t A solução de cloreto de sódio 20% é hipertônica e deve ser diluída antes do uso. Deve ser administrada preferentemente por meio de cateter instalado em veia de maior calibre.
t A concentração e a dose das soluções de cloreto de sódio para uso intravenoso são determinadas por diversos fatores, incluindo idade, peso, condição clínica e, em particular, o estado de hidratação do paciente. As concentrações séricas de eletrólitos devem ser cuidadosamente verificadas.
t Em depleção grave de sódio, 2 a 3 litros de cloreto de sódio 0,9% podem ser administrados durante 2 a 3 horas, seguidos de infusão intravenosa mais lenta. Se houver depleção combinada de água e sódio, uma mistura 1:1 de cloreto de sódio 0,9% e glicose 5% pode ser apropriada.
t As necessidades médias diárias de sódio e cloreto para adultos são alcançadas pela infusão de 1 L de cloreto de sódio 0,9%. As necessidades de fluido devem ser calculadas com manutenção ou reposição de necessidades de fluido.
t Excesso de sódio é excretado principalmente pelos rins, e pequenas quantidades são perdidas em fezes e suor.
t Acúmulo de sódio, edema e acidose hiperclorêmica (doses elevadas).
t Trombose venosa ou flebite.
t Dor local, necrólise tecidual, infecção no lugar da injeção ou em extravasamento.
t Febre.
t Desidratação cerebral que causa sonolência e confusão e resulta em convulsões, coma, transtorno respiratório e morte.
t Sede, redução da salivação e lacrimação.
t Transpiração.
t Taquicardia, hipertensão ou hipotensão.
t Cefaleia, tontura, cansaço, irritabilidade.
t Fraqueza, contração e rigidez muscular.
t Não utilizar soluções turvas e descartar porções não utilizadas.
t Durante infusão contínua, substituir o frasco da solução ao menos a cada 24 horas.
t Proteger a embalagem de extremos de temperatura.
t Armazenar a 25 ºC. Breve exposição até 40 ºC não afeta adversamente a solução.
Ana Cláudia de Brito Passos
Na Rename 2010: item 17.3
t Solução nasal 0,9%
t Alívio de congestão nasal.
t Umidificação da mucosa nasal.
t Fluidificação da secreção nasal. Esquemas de administração 3,11 Neonato
t 1 a 2 gotas em cada narina antes da amamentação
t 2 a 6 gotas em cada narina, a cada duas horas.
t Nesta via de administração, o cloreto de sódio não apresenta risco conhecido à saúde.
t Orientar para evitar contato do conta-gotas com a pele para não contaminá-lo.
t A solução nasal de cloreto de sódio 0,9% contém conservante, geralmente cloreto de benzalcônio. Apesar da proteção antimicrobiana conferida pelo conservante, recomenda-se manuseio higiênico do frasco conta-gotas para evitar contaminação.
t O medicamento deve ser mantido à temperatura ambiente, de 15 a 30 ºC.
Orozimbo Henriques Campos Neto
Na Rename 2010: item 1.3
t Pó para solução injetável 500 mg.
t Relaxamento muscular em procedimentos de curta duração (cirurgia ou ventilação mecânica, endoscopia e eletroconvulsoterapia).
t Intubação endotraqueal, convencional e de sequência rápida.
t Fase aguda em grandes traumas, grandes queimados.
t História pessoal ou familiar de hipertermia maligna.
t Miopatias, miastenia grave.
t Lesão neuronal motora superior.
t Baixa atividade de colinesterase plasmática, incluindo insuficiência hepática grave.
t Imobilização prolongada, pelo risco de hiperpotassemia.
t Distrofia muscular de Duchenne.
t Glaucoma, cirurgia ocular.
t Usar com cuidado nos casos de:
– pacientes com miastenia grave (são resistentes ao suxametônio, mas podem desenvolver bloqueio dual – causado pelo desenvolvimento de bloqueio não despolarizante após o bloqueio despolarizante inicial –, resultando em atraso na recuperação; pode ocorrer paralisia prolongada).
– intoxicação digitálica ou digitalização recente.
– doença cardíaca, respiratória ou neuromuscular, miopatias, arritmias cardíacas, hiperpotassemia, sepse (risco de hiperpotassemia), paraplegia, lesão da medula ou trauma grave.
– elevação da pressão intraocular (prevenir lesão ocular).
– insuficiência hepática grave ou cirrose.
– insuficiência renal (ver Apêndice D).
t Suxametônio deve ser administrado após indução anestésica porque a paralisia é geralmente precedida de fasciculações musculares dolorosas.
t Taquicardia ocorre com dose única; bradicardia com doses repetidas (em adultos) e na primeira dose (em crianças). Pré-tratamento com atropina reduz bradicardia e salivação excessiva associada ao suxametônio.
t Categoria de risco na gravidez (FDA): C.
t Dose de 1 a 2 mg/kg, por via intravenosa, inicialmente; na manutenção, 0,3 a 0,6 mg/kg, a cada 5 a 10 minutos, se necessário.
t Dose de 4 a 5 mg/kg, por via intramuscular, para menores de 1 ano. Acima de 4 mg/kg para maiores de 1 ano.
t Dose máxima: 150 mg.
t Neonatos, lactentes e crianças pequenas: dose de 2 mg/kg, por via intravenosa ou 4 a 5 mg/kg, por via intramuscular.
t Crianças mais velhas e adolescentes: dose de 1 mg/kg, por via intravenosa, ou 2 a 4 mg/kg, por via intramuscular, se veia adequada estiver inacessável.
t Dose máxima total: 150 mg.
t Por infusão intravenosa contínua: dose de 10 a 100 microgramas/kg/minuto, da solução injetável diluída em soro fisiológico ou solução glicosada 5% (concentração de 1 a 2 mg/mL).
t 2,5 a 4 mg/kg, por via intramuscular profunda, preferentemente no músculo deltoide, 1,5 mg/kg, por via intravenosa. Dose máxima total: 150 mg. Usar solução injetável sem diluição.
t 1 mg/kg, por via intravenosa, ou 2 a 4 mg/kg, por via intramuscular, se veia adequada estiver inacessável. Dose máxima total: 150 mg.
t Por infusão intravenosa contínua: dose de 10 a 100 microgramas/kg/minuto, da solução injetável diluída em soro fisiológico ou solução glicosada 5% (concentração de 1 a 2 mg/mL).
t Início de efeito – Injeção intramuscular: 1 minuto e 15 segundos a 3 minutos, em adultos, 3 minutos e 30 segundos em maiores de 1 ano e 4 minutos em menores de 1 ano. Injeção intravenosa: 30 a 60 segundos (intravenosa).
t Duração de efeito: 15 a 20 minutos (intramuscular), 4 a 6 minutos (dose única intravenosa).
t Meia-vida de eliminação: menos de 1 minuto. Metabolismo: rápido, por pseudocolinesterase plasmática.
t Excreção: renal (10%).
t Taquiarritmia, parada cardíaca, bradiarritmia e arritmias cardíacas, especialmente em crianças; hipertensão, hipotensão.
t Hiperpotassemia.
t Hipertermia maligna.
t Bloqueio neuromuscular prolongado, depressão respiratória.
t Rabdomiólise com mioglobinemia em crianças; mialgia pós-operatória (particularmente em pacientes deambulando após cirurgia, mais comum em mulheres),
t Apneia, broncoespasmo,
t Aumento da pressão intraocular,
t Exantema, urticária, choque.
t Mioglobinúria, mioglobinemia,
t Aumento na pressão gástrica, particularmente em pacientes deambulando após cirurgia e mais comum em mulheres.
t Amicacina, gentamicina, tobramicina, netilmicina, canamicina, estreptomicina favorecem a toxicidade do suxametônio (depressão respiratória). Ajustar a dose do suxametônio. Realizar monitoria de pacientes não ventilados quanto à paralisia respiratória.
t Donepezila induz prolongamento do bloqueio neuromuscular. Administrar suxametônio com extremo cuidado antes ou depois da anestesia em pacientes que recebem inibidores de colinesterase. Avaliar sinais de aumento no bloqueio neuromuscular. Reduzir ou interromper ambos os medicamentos se houver sintomas.
t Erva-de-são-joão (Hypericum perforatum) provoca hipotensão e/ou demora na recuperação da anestesia. Manejo: suspender o uso de erva-de-são-joão cinco dias antes de cirurgia.
t Inibidores da colinesterase (distigmina, fisostigmina, neostigmina, piridostigmina): aumento no bloqueio neuromuscular. Evitar suxametônio durante anestesia em pacientes recebendo inibidores colinesterásicos. Pode ocorrer prolongamento da paralisia, requerendo ventilação e suporte até o retorno das funções musculares.
t Lidocaína, pancurônio, quinidina: favorecem a toxicidade do suxametônio, depressão respiratória, apneia. Se for necessário o uso concomitante, fazer monitoria para depressão respiratória e bloqueio neuromuscular prolongado.
t Lítio e metoclopramida levam ao prolongamento do bloqueio neuromuscular. Observar paciente até recuperação.
t Procainamida promove bloqueio neuromuscular excessivo. Reduzir a dose de suxametônio.
t Tiopental: a associação pode causar coagulação intravenosa disseminada. Para evitar esse tipo de coagulação fatal administrar os medicamentos em grandes veias. Se for necessário empregar veias mais finas, insuflar o tubo com salina e esperar dois ou três minutos entre a administração dos fármacos.
t Vancomicina e sais de magnésio promovem a potência do bloqueio neuromuscular. Ajustar a dose de suxametônio. Ajustar naqueles pacientes que recebem doses altas de sais de magnésio.
t Informar sobre os riscos associados aos pacientes também de doenças cardíacas, asma, tumores, que sofrem de processos alérgicos graves, miastenia grave ou síndrome de Eaton-Lambert. Além destes casos é importante certificar-se dos casos de gravidez e lactação.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Solução injetável requer armazenamento sob refrigeração (2-8 oC).
t Sob refrigeração, a solução diluída é estável por 24 horas.
t Incompatibilidade com: tiopental, pentobarbital, amobarbital, meticilina, metoexital e bicarbonato de sódio.
Rosa Martins
Na Rename 2010: item 14.2
t Comprimido de 200 mg.
t Solução injetável 50 mg/mL
t Arritmias supraventriculares
t Fibrilação atrial
t Taquicardia e fibrilação ventricular em parada cardíaca refratária a desfibrilação
t Hipersensibilidade a amiodarona.
t Hipersensibilidade ao iodo.
t Bloqueio atrioventricular de 2º e 3º graus.
t Bradicardia sinusal grave.
t Disfunção grave do nó sinusal e atrioventricular.
t Distúrbio de condução infranodal.
t Choque cardiogênico.
t Episódios de bradicardia com síncope.
t Hipotensão arterial grave.
t Doença pulmonar.
t Gravidez. Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Usar com cuidado nos casos de:
– disfunção da tireoide.
– insuficiência cardíaca.
– porfiria aguda.
– infusão intravenosa (evitar veia pariférica; preferir infusão lenta em cateter central; realizar eletrocardiograma durante infusão).
– crianças (uso não recomendado).
– idosos (aumento de incidência de ataxia e outros efeitos neurotóxicos).
– insuficiência hepática (ver apêndice C) e insuficiência renal grave.
– lactação (ver apêndice B).
t Podem ocorrer microdeposições de cristais de amiodarona na córnea, causando halo visual e visão borrada, reversiva após a suspensão do fármaco.
t 200 mg, por via oral, a cada 8 horas, durante 1 semana, depois reduzida para 200 mg, por via oral, a cada 12 horas por mais 1 semana. Dose de manutenção 200 mg, por via oral, a cada 24 horas ou a menor dose requerida para controle da arritmia.
t Dose inicial 5 mg/kg, por infusão intravenosa, diluída em 250 mL de soro glicosado 5%, durante 20 a 120 minutos sob monitoria eletrocardiográfica. Infusões subsequentes administradas de acordo com a resposta até o máximo de 1,2 g em 24 horas, diluída em até 500 mL de soro glicosado 5%.
t 300 mg ou 5 mg/kg, por via intravenosa, diluído em soro glicosado a 5%, após adrenalina. Dose adicional de 150 mg ou 2,5 mg/kg, por via intravenosa, pode ser administrada se necessário, seguida de 900 mg, por infusão intravenosa, durante 24 horas.
t Absorção incompleta e lenta (latência de 30 minutos a 3 horas).
t Biodisponibilidade oral: 50%. Alimento aumenta tanto a extensão quanto a quantidade absorvida.
t Pico plasmático: 3 a 7 horas.
t Efeito após administração intravenosa: 1 a 30 minutos e persiste por 1 até 3 horas.
t Duração da ação: 40 a 55 dias após a suspensão de tratamento.
t Metabolismo hepático (metabólitos com propriedade antiarrítmica).
t Meia-vida de eliminação com terapia crônica oral é bifásica, com redução inicial de 50% em 2,5 a 10 dias; seguido por uma lenta meia-vida de eliminação terminal de 26 a 107 dias.
t Meia-vida de eliminação com terapia intravenosa: 4,2 a 34,5 dias.
t Excreção renal (menos de 1% em forma inalterada) e biliar.
t Não é removido por diálise.
t A maioria dos Efeitos adversos são dose dependentes e revertidos com redução da dose, entretanto, em razão de meia-vida longa os efeitos podem persistir ou aparecer após interrupção do tratamento.
t Infiltrado pulmonar e/ou fibrose, pneumonite (1 a 23%).
t Neuropatia periférica (20 a 40%), tremor, cefaleia, vertigem, fadiga (20 a 40%), insônia e ataxia (20 a 40%).
t Fotossensibilização (10%).
t Hipotireoidismo ou hipertireoidismo (1 a 14%).
t Depósitos na córnea, com repercussões visuais (3 a 13%).
t Insuficiência cardíaca, bradicardia (59%), hipotensão (16%).
t Intolerância digestiva, anorexia (25%), náusea/vômito (10 a 33%), obstipação (4 a 9%), alteração do paladar (1 a 3%), aumento enzimas hepáticas (40%), hepatite pelo fármaco (1 a 3%).
t Reação no local de infusão: dor e inflamação.
t Alprazolam, anticoagulantes cumarínicos, aripiprazol, betabloqueadores adrenérgicos, bloqueadores de canais de cálcio, budesonida, buspirona, ciclosporina, clonazepam, clopidogrel, corticosteroides, diazepam, ergotamina e análogos, digoxina, docetaxel, estatinas, estrógenos conjugados, fentanila, fenitoína, lidocaína, midazolam, metotrexato, procainamida, quinidina, teofilina, vincristina: podem ter suas concentrações plasmáticas aumentadas pela amiodarona. Ajustar a dose destes fármacos e observar para sinais e sintomas específicos.
t Antiarrítmicos da classe 1A, cisaprida, fluoroquinolonas, inibidores de protease, loratadina, tioridazina, pimozida: contraindicado o uso concomitante, pelo aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca).
t Fenitoína, nevirapina, rifampicina, rifapentina: pode ocorrer diminuição da concentração plasmática da amiodarona, reduzindo o efeito. Acompanhar sinais e sintomas específicos da efetividade da amiodarona.
t Metronidazol, trazodona: pode ocorrer aumento da concentração plasmática e do efeito da amiodarona. Observar sinais e sintomas específicos e toxicidade da amiodarona.
t Orientar que pode ser tomado com ou sem alimento
t Orientar para a importância de informar sobre o aparecimento de qualquer sinal de efeito adverso.
t Recomendar o uso de protetor solar continuamente, pelo risco de fotossensibilidade.
t Reforçar a importância de informar a ocorrência de halo visual e visão borrada.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Comprimidos devem ser mantidos sob temperatura ambiente (de 15 a 30 ºC) e ao abrigo de luz e umidade.
t A solução injetável pode ser diluída em solução de glicose a 5%. Verificar orientação do produtor quanto a informação específica.
t A solução injetável armazenada em recipiente de poliolefina é estável por 24 horas à temperatura ambiente (de 15 a 30 ºC). A solução injetável é estável por 5 dias quando armazenada em frasco escuro, protegido da luz.
t A solução injetável é incompatível com aminofilina, flucloxacilina, heparina, bicarbonato de sódio, ampicilina/sulbactam, ceftazidina, digoxina, furosemida, imipeném, sulfato de magnésio, piperacilina, fosfato de potássio e fosfato de sódio.
Atenção: amiodarona apresenta número elevado de Efeitos adversos , por isso é necessário uma pesquisa específica ao avaliar a terapia com este fármaco. Sinais/sintomas de toxicidade: alteração de ECG e PA, letargia, edema de mãos e pés, perda de peso, alterações na função pulmonar, hepática e da tireoide.
Tatiana Aragão Figueiredo
Isabella Campagnuci Knust
Na Rename 2010: itens 2.3 e 13.2
t Comprimido 25 mg.
t Transtornos e episódios de depressão maior, particularmente quando sedação é necessária.
t Profilaxia de enxaqueca (tratamento intercrises).
t Enfarte do miocárdio recente, arritmias cardíacas.
t Insuficiência hepática grave (ver apêndice C).
t Fase maníaca do transtorno bipolar.
t Porfiria.
t Hipersensibilidade ao fármaco ou a outros antidepressivos tricíclicos.
t Uso de inibidores da monoamina oxidase (IMAO) nos últimos 15 dias (a troca de um IMAO por tricíclico ou vice-versa deve guardar o intervalo mínimo de 15 dias).
t Usar com cuidado nos casos de:
– menores de 12 anos.
– lactação (ver Apêndice B).
– cardiopatia, retenção urinária, insuficiência hepática (ver Apêndice C), insuficiência renal crônica (ver Apêndice D), epilepsia, hipertrofia prostática, hipertireoidismo, glaucoma de ângulo fechado, diabete melito, história de hipertensão intraocular, ideação suicida, sintomas de paranoia, transtorno bipolar, esquizofrenia ou distúrbios cognitivos.
– idosos (reduzir doses).
– suspensão do tratamento (deve ser gradual).
– eletrochoque concomitante (pode aumentar os riscos da terapia com eletrochoque).
– feocromocitoma.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Perigo ao dirigir ou realizar outras tarefas que exijam atenção e coordenação motora.
t Dose de 25 a 50 mg/dia, por via oral, ao deitar ou fracionados em duas doses; aumento gradual até 100 mg/dia
t Dose de 25 mg, por via oral, uma vez à noite; a dose pode ser aumentada gradualmente até 75 mg.
t Incrementos semanais subsequentes de 50 mg até doses terapêuticas médias entre 150 a 300 mg.
t Em geral após 4 a 6 semanas de tratamento, os pacientes se tornam assintomáticos. As doses de resposta devem ser mantidas por 3 a 4 meses, com redução gradual à metade.
t O tratamento deve ser feito durante 6 a 12 meses, para evitar recidivas.
t Na retirada gradual, diminui-se a dose em 25 mg a cada 2 ou 3 dias. Se os sintomas reaparecem, retomam-se os níveis iniciais.
t Dose de 10 a 25 mg, por via oral, ao deitar; dose usual: 75 mg por dia, durante 6 a 12 meses.
t Dose de 10 a 25 mg, por via oral, ao deitar; se bem tolerada, a dose pode ser aumentada em 25 mg a cada semana; dose média: 25 a 150 mg/dia.
t Dose de 10 mg três vezes por dia e 20 mg ao deitar, como início do esquema de administração.
t Período de latência: usualmente pode levar 2 a 3 semanas para o início da resposta terapêutica.
t Pico sérico em torno de 4 horas. Deve ser dado ao deitar (efeito sedativo máximo durante o sono).
t Meia-vida: 9 a 26 horas.
t Distribuição: atravessa a placenta e se excreta no leite materno.
t Metabolismo exclusivamente hepático, gerando o metabólito ativo nortriptilina.
t Eliminação renal (18% em forma ativa) e fecal (pequena proporção).
t Hipotensão ortostática (que pode levar a quedas em idosos), lipotímia, distúrbios na repolarização ventricular, transtornos de condução cardíaca, taquicardia, alterações eletrocardiográficas, hipertensão, enfarte do miocárdio.
t Sedação, tontura, insônia, hipomnésia, fadiga, ansiedade, tremores finos de extremidades, disartria, incoordenação motora, desorientação, visão turva, diminuição do limiar convulsivo, sintomas extrapiramidais, sudorese.
t Secura na boca, estomatite, gosto amargo, aumento do apetite, anorexia, dispepsia, diminuição da função hepática, diarreia, obstipação, náusea, vômito.
t Retenção urinária, especialmente em idosos com hipertrofia prostática.
t Efeitos anticolinérgicos: xerostomia, midríase, cicloplegia, retenção urinária, diminuição da motilidade gastrintestinal, taquicardia, aumento da pressão intraocular.
t Distúrbios comportamentais (especialmente em crianças), confusão, alucinações ou delírio (sobretudo em idosos), cefaleia. Os transtornos confusionais podem ser acompanhados de ansiedade, alteração no sono, tendências suicidas.
t Leucopenia, agranulocitose, eosinofilia, trombocitopenia, púrpura.
t Urticária, angiedema, fotossensibilidade.
t Ganho ou perda de peso, ginecomastia, galactorreia, aumento testicular, alterações dos níveis glicêmicos, diminuição da libido.
t Em dose excessiva aguda ocorrem hipotermia, agitação, confusão, delírio, alucinações, convulsões, hipotensão, taquicardia, acidose metabólica, depressão respiratória e cardíaca, coma e eventualmente morte.
t Álcool e outros depressores do sistema nervoso central, anticoagulantes cumarínicos, fármacos com efeitos anticolinérgicos (anti-histamínicos H1, antiparkinsonianos e neurolépticos): podem ter seus efeitos intensificados. Em pacientes recebendo anticoagulante oral, o tempo de protrombina deve ser cuidadosamente avaliado e ajustes da dose do anticoagulante podem ser necessárias.
t Amiodarona, aprindina, azimilida, bepridil, cinacalcete, cisaprida, disopiramida, dofetilida, dolasetrona, droperidol, espiramicina, fenitoína, fenotiazinas, fluconazol, haloperidol, hidrato de cloral, ibutilida, lidoflazina, mesoridazina, octreotida, pentamidina, pimozida, proclorperazina, sulfametoxazol, tioridazina, trimetoprima, vasopressina, venlafaxina, zolmitriptana: pode levar a aumento da toxicidade da amitriptilina. Acompanhar a concentração plasmática, sinais e sintomas de toxicidade do antidepressivo; ajustes da dose da amitriptilina podem ser necessários.
t Amprenavir, antidepressivos, bloqueadores seletivos da recaptação de serotonina, antipsicóticos, cimetidina, contraceptivos orais, dissulfiram, fenfluramina, fosamprenavir, topiramato: aumento de efeito do antidepressivo. Acompanhar a concentração plasmática, sinais e sintomas de toxicidade do antidepressivo; ajustes da dose da amitriptilina podem ser necessários.
t Anestésicos gerais, anfetaminas, antiarrítmicos, antibióticos macrolídeos e quinolonas, anti-histamínicos, antimaláricos, bloqueadores adrenérgicos: aumento da toxicidade da amitriptilina. Avaliar a concentração plasmática, sinais e sintomas de toxicidade do antidepressivo; ajustes da dose da amitriptilina podem ser necessários.
t Barbitúricos, carbamazepina, erva-de-são-joão (Hypericum perforatum), fenitoína, hidrato de cloral, nicotina (tabaco), rifapentina: diminuição de efeito do antidepressivo.
t Carbamazepina e rifapentina: diminuição de efeito da amitriptilina. Verificar as concentrações plasmáticas da amitriptilina; ajustes de dose podem ser necessários.
t Clonidina, betanidina, guanadrel, guanfacina: podem ter seus efeitos diminuídos. Acompanhar a pressão arterial para possível ajuste de dose dos anti-hipertensivos.
t Diazepam: uso concomitante pode levar ao desenvolvimento de deficiências psicomotoras.
t Inibidores da MAO, linezolida: a associação pode levar a neurotoxicidade, convulsões, ou síndrome serotoninérgica (hipertensão, hipertermia, mioclônus, alterações do estado mental).
t Procarbazina: associação com procarbazina pode levar a neurotoxicidade e convulsões.
t Simpaticomiméticos: o uso concomitante com amitriptilina pode levar a hipertensão, arritmia cardíaca e taquicardia. A vasoconstrição proveniente de fármacos alfa-adrenérgicos e de outros simpaticomiméticos é substancialmente reforçada com a presença de antidepressivos tricíclicos. Se estes fármacos são utilizados em associação, um acompanhamento atento e uma redução da dose dos simpaticomiméticos são necessários.
t Não fazer uso de bebidas alcoólicas.
t Não suspender o uso de maneira repentina.
t Alertar para tempo de latência para início da resposta terapêutica.
t Orientar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Orientar para mudança na frequência cardíaca e a levantar-se mais lentamente para evitar hipotensão ortostática.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas. Caso tome o medicamento antes de deitar, se esquecer não use o medicamento pela manhã e espere até a próxima noite.
t Conservar entre 15 e 30 oC, em recipientes bem fechados e ao abrigo da luz.
Atenção: os efeitos terapêuticos podem demorar até 15 dias para se manifestar. Acompanhamento contínuo de pressão arterial e frequência cardíaca nas semanas iniciais.
Rachel Magarinos-Torres
Na Rename 2010: item 13.4
t Comprimido 2 mg (cloridrato de biperideno)
t Solução injetável 5 mg/mL (lactato de biperideno)
t Distúrbios motores decorrentes do uso de neurolépticos.
t Glaucoma de ângulo fechado.
t Retenção urinária.
t Hipertrofia prostática.
t Miastenia grave.
t Obstrução gastrintestinal, megacólon.
t Usar com cuidado nos casos de:
– discinesia tardia (há piora com o uso de anticolinérgicos).
– idosos (ajustar a dose).
– antes de iniciar tratamento (fazer avaliação cognitiva, urológica e cardiovascular).
– ocorrência de efeitos anticolinérgicos centrais e periféricos (interromper tratamento).
– problemas cardiovasculares, epilepsia e insuficiência renal e hepática.
– retirada (deve ser gradual para reduzir risco de efeito rebote e piora do parkinsonismo).
– lactação (ver Apêndice B).
t Pode induzir problemas psiquiátricos, principalmente na esfera cognitiva.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Dose inicial 1 mg, por via oral, a cada 12 horas, aumentado gradualmente para 2 mg, a cada 8 horas. Dose de manutenção: 2 a 12 mg/dia em doses divididas.
t 2 a 5 mg, por via intramuscular ou injeção intravenosa lenta, a cada 30 minutos. Dose máxima: 20 mg/dia.
t Bem absorvido pelo trato gastrintestinal, mas com biodisponibilidade oral de cerca de 30% (extenso metabolismo de primeira passagem)
t Pico sérico: 1-2 horas
t Meia-vida de eliminação:18-24 horas
t Excreção urinária, em forma ativa e como metabólitos
t Obstipação, náusea, xerostomia
t Visão borrada
t Retenção urinária
t Confusão mental, excitação, delírio, tontura, déficit de memória, alucinações, agitação, sonolência
t Taquicardia, arritmias, hipotensão postural
t Cloreto de potássio (comprimido): risco de lesões gastrintestinais. O uso concomitante é contraindicado.
t Alertar para não ingerir bebidas alcoólicas.
t Alertar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora, como operar máquinas e dirigir.
t Alertar para evitar a realização de atividades que aumentam a temperatura corporal, como exercício físico intenso e exposição a calor extremo, pelo risco de desidratação.
t Orientar para ingerir com alimentos a fim de diminuir irritação gástrica.
t Orientar para adotar dieta rica em fibras e boa hidratação para evitar obstipação.
t Orientar para instituir boa higiene oral e intensificação do controle mecânico de placas dentárias em função da xerostomia.
t Alertar para não suspender abruptamente o tratamento.
t Conservar sob temperatura ambiente, entre 15 e 30 ºC, em recipientes bem fechados e ao abrigo da luz. Evitar o congelamento.
Thais Furtado de Souza
Na Rename 2010: item 1.2
t Solução injetável 0,25% e 0,5%
t Anestesia local (epidural) para procedimentos prolongados.
t Anestesia regional.
t Bloqueio simpático e bloqueio neural periférico.
t Analgesia epidural contínua no parto.
t Controle de dor no pós-operatório.
t Hipersensibilidade a bupivacaína, sulfitos ou outros anestésicos do tipo amida.
t Anestesia de bloqueio paracervical em obstetrícia.
t Anestesia espinhal ou epidural em paciente desidratado ou hipovolêmico.
t Anestesia intravenosa regional (bloqueio de Bier).
t Hemorragia ou anemia grave, uso concomitante de anticoagulante.
t Hipotensão grave ou choque, arritmias, bloqueio cardíaco.
t Infecção cutânea adjacente, septicemia.
t Usar com cuidado nos casos de:
– formulação contendo conservante (não usar em anestesia espinhal, epidural ou regional intravenosa.
– injeção intravenosa acidental (está associada a convulsões e falência cardíaca).
– epilepsia, porfiria, miastenia grave, hipertireoidismo, insuficiência respiratória, renal (ver Apêndice D) ou hepática (ver Apêndice C).
– idosos e enfraquecidos (reduzir dose).
– menores de 12 anos (uso não recomendado).
– crianças e idosos (risco aumentado da ocorrência de efeito tóxico sistêmico).
– lactação (ver Apêndice B).
t Bupivacaína tem maior potência cardiodepressora e cardiotóxica do que lidocaína em doses equiefetivas, podendo causar arritmias ventriculares graves e depressão miocárdica após injeção acidental intravenosa de altas doses.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Crianças acima de 10 anos: 0,3 a 0,4 mL/kg (caudal) ou 0,4 a 0,8 mL/kg (torácica) da solução a 0,25%.
t Crianças acima de 12 anos: epidural, dose de 1,0 a 2,5 mg/kg de solução a 0,25% em dose única.
t Crianças acima de 12 anos: 20 a 50 mL (50 a 125 mg) de solução a 0,25%, repetida a cada 3 horas, se necessário.
t Peso igual ou inferior a 10 kg: caudal ou epidural, dose de 0,1 a 0,2 mg/kg/h em infusão contínua de solução 0,1%, 0,125% ou 0,25%.
t Peso maior que 10 kg: caudal, dose de 0,2 a 0,4 mg/kg/h em infusão contínua de solução a 0,1%, 0,125%, ou 0,25%.
t 15 a 30 mL de solução a 0,5% (75 a 150 mg) ou a 0,25% (37,5 a 75 mg). Repetir a cada 3 horas, se necessário.
t 10 a 20 mL de solução a 0,25% (25 a 50 mg) ou a 0,5% (50 a 100 mg). Repetir a cada 3 horas, se necessário.
t 20 a 50 mL de solução a 0,25% (50 a 125 mg), repetida a cada 3 horas, se necessário.
t Infundir dose de 6,25 a 18,75 mg/h de solução a 0,0625 a 0,125%.
t Nota: não exceder 175 mg em dose única ou 400 mg em 24 horas. O intervalo mínimo entre as doses deve ser de 3 horas.
t Início da ação: 2 a 10 minutos.
t Duração da ação: até 7 horas. A adição de adrenalina diminui o fluxo sanguíneo local, diminui a absorção de bupivacaína e prolonga o efeito anestésico.
t Meia-vida: 1,5 a 5,5 horas (adultos) e 8,1 horas (neonatos).
t Metabolismo: hepático.
t Excreção: renal (5% em forma inalterada).
t Hipotensão, bloqueio cardíaco, bradicardia.
t Depressão ou excitação do SNC, convulsão (dose dependente).
t Falência respiratória (dose excessiva ou administração intravenosa).
t Hipersensibilidade e reações alérgicas (raras).
t Erva-de-são-joão (Hypericum perforatum): aumento do risco de colapso cardiovascular e retardo da anestesia. Suspender o uso da erva-de-são-joão pelo menos 5 dias antes do procedimento.
t Hialuronidase: aumento da incidência de reações sistêmicas ao anestésico. Acompanhar o paciente.
t Inibidores da ECA: bradicardia e hipotensão com perda da consciência. Observar intensivamente e tratar desequilíbrio hemodinâmicos durante o procedimento.
t Propofol: aumento do efeito hipnótico. Reduzir doses do propofol se paciente recebeu anestesia em tecidos moles.
t Propranolol: aumenta toxicidade da bupivacaína. Verificar sinais de toxicidade da bupivacaína.
t Verapamil: pode ocasionar risco aumentado de bloqueio cardíaco. Avaliar função cardíaca.
t Orientar para que o paciente informe se perceber os seguintes sintomas: prurido ou urticária, inchaço no rosto ou nas mãos, inchaço ou formigamento na boca ou garganta, dor no peito, batimento cardíaco irregular, dificuldade para respirar, dormência em lugar diferente da injeção.
t Orientar que o medicamento pode causar dormência no lugar da injeção.
t Armazenar sob temperatura entre 15 e 30°C. Evitar congelamento.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Não utilizar se a solução apresentar turbidez ou precipitação.
Thais Furtado de Souza
Na Rename 2010: item 1.2
t Solução injetável de cloridrato de bupivacaína 0,5% + glicose 8%.
t Anestesia espinhal.
t A anestesia espinhal é contraindicada em paciente desidratado ou hipovolêmico.
t A anestesia espinhal não é recomendada para menores de 18 anos.
t Usar com cuidado em idosos (reduzir as doses).
t A solução de cloridrato de bupivacaína + glicose é hiperbárica e se difunde na direção da cabeça mais extensivamente do que as soluções isobáricas. A densidade da solução no espaço subaracnoideo e a posição do paciente são os principais fatores determinantes da propagação de bupivacaína; a distribuição de bupivacaína hiperbárica demonstra uma relação dose-dependente.
t Considerar demais precauções listadas na monografia do cloridrato de bupivacaína.
t Dose de 7,5 a 10,5 mg.
t Dose de 6 mg em parto normal.
t Dose de 12 (cirurgia abdominal baixa) a 15 mg (cirurgia abdominal alta).
t Dose de 7,5 mg.
t Início da ação: 4 a 15 minutos.
t Duração da ação: 2 horas. A adição de adrenalina diminui o fluxo sanguíneo local, diminui a absorção de bupivacaína e prolonga o efeito anestésico.
t Meia-vida: 1,5 a 5,5 horas (adultos) e 8,1 horas (neonatos).
t Metabolismo: hepático.
t Excreção: renal (5% em forma inalterada).
t Hipotensão, bloqueio cardíaco, bradicardia.
t Depressão ou excitação do SNC, convulsão (dose dependente).
t Falência respiratória (dose excessiva ou administração intravenosa).
t Hipersensibilidade e reações alérgicas (raras).
t Erva-de-são-joão (Hypericum perforatum): aumento do risco de colapso cardiovascular e lenta recuperação da anestesia. Suspender o uso da erva-desão-joão pelo menos cinco dias antes do procedimento.
t Hialuronidase: aumento da incidência de reações sistêmicas ao anestésico. Observar o paciente para sinais de toxicidade.
t Inibidores da ECA: aumentam o risco de bradicardia e hipotensão. A terapia com inibidores da ECA pode ser mantida até a cirurgia, mas os pacientes devem ser intensivamente acompanhados e eventuais desequilíbrios hemodinâmicos durante o procedimento devem ser tratados.
t Propofol: aumento do efeito hipnótico. Reduzir doses do propofol se paciente recebeu bupivacaína em tecidos moles.
t Propranolol: aumento da toxicidade da bupivacaína. Monitorar sinais e sintomas de toxicidade.
t Verapamil: pode ocasionar risco aumentado de bloqueio cardíaco. Avaliar a função cardíaca.
t Orientar que o medicamento pode causar dormência no lugar da injeção.
t A solução de cloridrato de bupivacaína + glicose não contém conservante, portanto, deve ser usada imediatamente após a abertura da ampola, e o restante da solução deve ser descartado.
t Manter a temperatura ambiente, até 30 °C.
Isabella Campagnuci Knust
Na Rename 2010: item 25
t Comprimido de 150 mg
t Tratamento adjuvante na cessação do tabagismo.
t Crises convulsivas.
t História de descontinuação abrupta de álcool ou sedativos, incluindo benzodiazepínicos.
t Bulimia ou anorexia.
t Transtorno bipolar.
t Hipersensibilidade à bupropiona.
t Uso de inibidores da monoamina oxidase (IMAO) nos últimos 14 dias.
t Uso de outros produtos contendo bupropiona.
t Usar com cuidado nos casos de:
– distúrbios mentais e em uso de medicamentos que atuam sobre o sistema nervoso central.
– idosos, crianças e adolescentes (o risco não está bem definido).
– insônia.
– hipertensão, enfarte do miocárdio, tumores do SNC, angina instável, nefropatia (ver Apêndice D), insuficiência hepática (ver Apêndice C) e diabete tratado com hipoglicemiantes orais ou insulina.
– lactação (ver Apêndice B)
t Categoria de risco na gravidez (FDA): C
t dose de 1,4 a 6 mg/kg/dia
t dose de 150 mg por dia, por via oral, nos primeiros 3 dias; aumentar para 300 mg por dia, em duas tomadas. Continuar o tratamento por, no máximo, 7 a 9 semanas.
t Dose diária máxima de 150 mg.
t Absorção é feita em 80% pelo trato gastrintestinal, atingindo pico sérico em 3 horas.
t Metabolismo: hepático.
t Excreção: renal (87%) e fecal (10%), com meia-vida de eliminação é de 21 horas, o que propicia intervalos entre doses de 24 horas.
t A dose deve ser ajustada em caso de insuficiência renal e hepática.
t Tontura, cefaleia, insônia, agitação, ansiedade, confusão, tremores, exacerbação de depressão, comportamento hostil, mania, sonolência, visão borrada, zumbidos.
t Dor abdominal, obstipação, náuseas, faringite, xerostomia.
t Alterações de condução cardíaca, enfarte do miocárdio, taquiarritmia, hipertensão ou hipotensão.
t Prurido, exantema, urticária, sudorese, síndrome de Stevens-Johnson.
t Fogachos, disfunção sexual, alteração menstrual.
t Artralgias, mialgias, parestesias.
t Álcool, antipsicóticos, corticosteroides, testosterona: diminuição do limiar para desencadeamento de crise convulsiva.
t Amantadina, levodopa: risco aumentado de Efeitos adversos (náusea, vômito, excitação).
t Carbimazol: aumento da hepatotoxicidade. Monitorar o paciente para sinais e sintomas de hepatotoxicidade aguda e testes de função hepática.
t Citalopram, desipramina, flecainida, fluoxetina, haloperidol, metoprolol, nortriptilina, paroxetina, propafenona, risperidona, tioridazina: estes fármacos podem ter seus efeitos potencializados.
t Droperidol: aumento da cardiotoxicidade.
t Efavirenz, lopinavir, nevirapina, rifampicina, ritonavir, tipranavir: a eficácia da bupropiona pode ser diminuída.
t Erva-de-são-joão (Hypericum perforatum): aumento do risco de Efeitos adversos dopaminérgicos. O uso concomitante não é recomendado, mas caso ocorra, monitorar o paciente para sinais precoces de Efeitos adversos dopaminérgicos.
t IMAO (ex. selegilina): os níveis séricos da bupropiona podem ser aumentados. A administração concomitante de bupropiona e IMAO é contraindicada; descontinuar os IMAO pelo menos 14 dias antes de introduzir a bupropiona.
t Linezolida: aumento do risco da síndrome serotoninérgica (hipertermia, hiperreflexia, mioclônus, alteração do estado mental). Monitorar o paciente para sinais e sintomas; caso a síndrome ocorrer, descontinuar os medicamentos e providenciar o suporte e terapia necessários.
t Metoclopramida: aumento do risco de Efeitos adversos extrapiramidais; o uso concomitante é contraindicado, mas se o mesmo for necessário, monitorar o paciente em relação aos efeitos extrapiramidais.
t Nicotina (adesivo): aumento do risco de urgência hipertensiva.
t Teofilina: pode ter suas concentrações séricas aumentadas. Reduzir a dose inicial e aumentar gradualmente a dose de teofilina durante o uso concomitante com bupropiona.
t Zolpidem: aumento do risco de alucinações.
t Recomendar para não associar com bebida alcoólica.
t Orientar para ingerir com alimentos para diminuir a possibilidade de irritação gástrica.
t Orientar para engolir o comprimido inteiro, não devendo ser dividido ou triturado.
t Orientar sobre a possibilidade de prejudicar o desempenho de atividades que exijam atenção (como dirigir e operar máquinas perigosas).
t Comunicar ao médico se houver piora dos sintomas de depressão e/ou pensamento suicida e/ou comportamento agressivo.
t Manter ao abrigo de luz e à temperatura ambiente, entre 15 e 30 ºC.
Thais Furtado de Souza
Na Rename 2010: item 1.1.2
t Solução injetável 57,67 mg/mL – equivalente a 50 mg/mL de cetamina.
t Indução e manutenção da anestesia geral.
t Analgesia em procedimentos dolorosos de curta duração.
t Hipersensibilidade ao fármaco.
t Hipertensão arterial sistêmica, insuficiência cardíaca congestiva, angina e doença cardíaca grave.
t Elevação da pressão intracraniana.
t Elevação da pressão intraocular.
t Histórico de acidente cerebrovascular, trauma cerebral e aneurisma.
t Tireotoxicose.
t Porfiria.
t Alucinações e distúrbios psiquiátricos.
t Usar com cuidado nos casos de:
– uso prolongado (pode determinar dependência).
– histórico de uso abusivo de álcool.
– administração muito rápida ou dose(s) excessiva(s)(risco de depressão respiratória).
– Podem ocorrer reações até 24 horas pós-anestésicas: alucinações, delírio, sonhos vividos, comportamento irracional, confusão mental, sendo essas reações menos comuns em pacientes acima de 65 anos e quando administrado por via intramuscular.
t Devido à ocorrência de salivação excessiva e laringoespasmos, não deve ser utilizado como agente único nas intervenções cirúrgicas ou diagnósticas da faringe, laringe ou árvore brônquica (agente anticolinérgico pode ser utilizado para reduzir salivação).
t Categoria de risco na gravidez (ADEC): A (ver Apêndice A).
t 1 a 4,5 mg/kg, por via intravenosa, durante pelo menos 1 minuto.
t 4 a 13 mg/kg, por via intramuscular.
t Dose inicial 0,5 a 2 mg/kg, por infusão intravenosa de solução com concentração de 1 mg/mL, infundida a velocidade máxima de 0,5 mg/kg/minuto.
t Dose de manutenção 0,01 a 0,045 mg/kg/minuto, por infusão intravenosa de solução com concentração de 1 mg/mL, ajustada conforme a resposta.
t A diluição pode ser feita com solução fisiológica, solução glicosada a 5% ou água para injeção.
t Dose inicial 4 mg/kg, por via intramuscular.
t Início da ação: 1 a 2 minutos (anestesia e analgesia por via intravenosa) e 3 a 8 minutos (anestesia por via intramuscular).
t Duração: 5 a 15 minutos (intravenosa), 12 a 25 minutos (intramuscular), e 15 a 30 minutos (analgesia intramuscular).
t Meia-vida: 11 a 17 minutos.
t Metabolismo: hepático.
t Excreção: renal (90%).
t A recuperação ocorre entre 1 e 2 horas, sendo mais demorada após administração intramuscular.
t Hipertensão ou hipotensão arterial, taquicardia ou bradicardia, arritmia cardíaca.
t Movimentos tônicos-clônicos, tremor, alucinações visuais, sonhos vívidos, nistagmo, diplopia.
t Depressão respiratória.
t Anorexia, náusea, vomito, salivação.
t Exantema e hipersensibilidade.
t Bloqueadores neuromusculares não-despolarizantes (atracúrio, pancurônio, tubocurarina): pode causar bloqueio neuromuscular. Caso uso concomitante com cetamina seja necessário, a dose do bloqueador deve ser ajustada e o paciente deve ser monitorado para depressão respiratória.
t Erva-de-são-joão (Hypericum perforatum): pode ocasionar risco aumentado de colapso cardiovascular. O uso da erva-de-são-joão deve ser descontinuado 5 dias antes da utilização do anestésico.
t Halotano: o uso concomitante pode diminuir a pressão arterial. Utilizar com cautela, a pressão arterial deve ser monitorada.
t Alertar para o possível surgimento de tontura, sonolência e confusão.
t Se receber alta dentro do período de 24 horas após o procedimento, deve ser acompanhado até a casa, não deve dirigir, operar máquinas ou praticar qualquer atividade perigosa que necessitar atenção.
t Orientar para evitar o uso de bebida alcoólica e de outros depressores do SNC no período de 24 horas.
t Notificar imediatamente se ocorrer sintomas alérgicos (prurido, edema em mãos, rosto, boca, garganta, distúrbios respiratórios), alteração no batimento cardíaco, síncope e dor, hiperemia ou formação de bolhas no lugar da injeção.
t Notificar a ocorrência de tosse, diplopia, enrijecimento dos músculos, náusea, vômito ou perda de apetite, hiperemia ou suave erupção da pele.
t Manter sob temperatura entre 15 e 30 °C, protegido da luz e calor. Não congelar.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Para administração intravenosa a concentração de 100 mg/mL deve ser diluída antes da aplicação em igual volume de água para injeção, cloreto de sódio 0,9% ou glicose 5%.
t Cetamina e barbitúricos não devem ser preparados na mesma seringa, pois ocorre formação de precipitado.
t Diazepam e cetamina são incompatíveis.
Maria Inês de Toledo e Lívia Luize Marengo
Na Rename 2010: item 5.1.7
t Comprimido 500 mg.
t Solução injetável 2 mg/mL.
t Infecções causadas por bacilos gram-negativos aeróbios sensíveis a ciprofloxacino (infecções urinárias complicadas, geniturinárias, respiratórias, sinusite, cutâneas e de tecidos moles, ósseas e articulares, intra-abdominais – junto com metronidazol).
t Hipersensibilidade ao ciprofloxacino ou a qualquer outra quinolona.
t Histórico de doença nos tendões associada ao uso de quinolonas.
t Gravidez a termo.
t Usar com cuidado nos casos de:
– histórico de epilepsia ou convulsões (diminui o limiar), miastenia grave.
– insuficiência renal (ver Apêndice D).
– crianças e adolescentes (não é antibiótico de primeira escolha; risco de Efeitos adversos sobre as articulações).
– exposição à luz solar (risco de fotossensibilidade).
– alcalinização excessiva da urina (risco de cristalúria).
– uso prolongado (risco de desenvolver colite pseudomembranosa – superinfecção).
– ocorrência de reações psiquiátricas, neurológicas ou de hipersensibilidade (suspender o tratamento).
– lactação (ver Apêndice B).
t Potencial de desenvolver graves reações de hipersensibilidade, inclusive anafiláticas.
t Categoria de risco na gravidez (FDA): C (ver apêndice A).
t 10 a 20 mg/kg, por via oral, a cada 12 horas. Dose máxima diária: 1,5 g.
t 6 a 10 mg/kg, por via intravenosa, a cada 8 horas. Dose máxima diária: 800 mg
t 500 a 750 mg, por via oral, a cada 12 horas.
t 400 mg, por via intravenosa, a cada 8 ou 12 horas.
Nota
t A infusão lenta (60 minutos) da solução diluída em veia de grande calibre diminui o desconforto para o paciente e reduz o risco de irritação venosa.
t Pico de concentração sérica: 1 a 2 horas.
t Meia-vida de eliminação: 3 a 5 horas.
t Metabolismo: hepático (metabólitos ainda ativos).
t Excreção: renal (30% a 50% em forma inalterada) e fecal.
t Náusea, vômito, dispepsia, dor abdominal, flatulência, diarreia, disfagia, anorexia.)
t Pancreatite.
t Cefaleia, tremor, tontura, distúrbios do sono, depressão, confusão, alucinações, convulsões, parestesia, hipostesia, desordens do movimento, astenia.
t Exantema (raramente eritema multiforme e necrólise epidérmica tóxica), prurido, eritema nodoso, petéquias.
t Vasculite.
t Aumento de ureia e creatinina.
t Fotossensibilidade, reações de hipersensibilidade (febre, urticária, angioedema,anafilaxia).
t Artralgia, mialgia, tenossinovite, inflamação e dano no tendão (especialmente em idosos usando corticosteroides).
t Eosinofilia, leucopenia, trombocitopenia, anemia hemolítica.
t Distúrbios de visão, paladar e audição.
t Taquicardia, hipotensão, edema, síncope.
t Insuficiência renal, nefrite intersticial.
t Disfunção hepática (incluindo hepatite e icterícia colestática).
t Probenecida, metoclopramida: aumento do efeito do ciprofloxacino.
t Antiácidos, cátions multivalentes, sucralfato: redução do efeito do ciprofloxacino. Evitar. Se necessário, usar antagonista H2.
t cafeína, teofilina: aumento dos efeitos tóxicos da teofilina. Excitação do SNC. Evitar. Quando não for possível, ajustar dose de teofilina.
t Ciclosporina: aumento da creatinina sérica. Monitorar concentração de ciclosporina e sinais de rejeição do transplante.
t Anticoagulantes orais: aumento do efeito anticoagulante. Monitorar RNI e ajustar a dose.
t Corticosteroides: aumento no risco de ruptura no tendão (durante ou após o tratamento). Descontinuar o ciprofloxacino se houver sintomas.
t Hipoglicemiantes orais, insulina: hiper ou hipoglicemia. Monitorar a glicemia e se necessário suspender a quinolona.
t Sinvastatina: pode aumentar miopatia e rabdomiólise por diminuição do metabolismo da sinvastatina. Monitorar os sintomas de miopatia e níveis de CK. Suspender se houver suspeita de rabdomiólise.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Não tomar com leite e derivados. Não tomar nenhum suco contendo cálcio juntamente com o ciprofloxacino. Se utilizar algum medicamento contendo cálcio, tomar o ciprofloxacino 2 horas antes ou 6 horas depois. Não partir, não quebrar e não mastigar o comprimido.
t Pode causar reações alérgicas graves e fotossensibilidade; utilizar filtro solar para proteger-se da exposição ao sol.
t Comprimidos: Armazenar a temperatura ambiente, 15 a 25 ºC, e proteger da luz.
t Solução injetável: Armazenar em local fresco, 8 a 15 ºC, ou a temperatura ambiente, 15 a 25 ºC, e proteger da luz. Não congelar. Medicamento fotossensível.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Ciprofloxacino injetável é compatível com soro fisiológico 0,9%, soluções de Ringer e Ringer + lactato, soluções de glicose a 5% e 10%, solução de frutose a 10% e solução de glicose a 5% com 0,225% ou 0,45% de cloreto de sódio.
t Administrar logo depois do preparo.
t Incompatível com heparina.
Fernando de Sá Del Fiol e Silvio Barberato Filho
Na Rename 2010: itens 5.1.9; 5.4; 5.6.2.2; 5.6.2.3
t Cloridrato de clindamicina: cápsula de 150 e 300 mg
t Fosfato de clindamicina: solução injetável de 150 mg/mL
t Infecções causadas por bactérias anaeróbias e aeróbias gram positivas.
t Pneumocistose.
t Malária por Plasmodium falciparum (em esquema com derivados da artemisinina ou dicloridrato de quinina).
t Toxoplasmose.
t Profilaxia de endocardite bacteriana em pacientes alérgicos às penicilinas.
t Hipersensibilidade a clindamicina ou lincosamidas.
t Colite pseudomembranosa prévia.
t Colite ulcerativa e enterite.
t Usar com cuidado nos casos de:
– ocorrência de diarreia, cólica abdominal e perda de sangue/muco nas fezes (risco potencial de colite pseudomembranosa; suspender imediatamente o tratamento).
– recém-nascidos, crianças, idosos e pacientes com atopia.
– insuficiência renal.
– insuficiência hepática (ver Apêndice C). t Evitar a administração intravenosa rápida. t Categoria de risco na gravidez (FDA): B.
t 8 a 20 mg/kg/dia, por via oral, divididos a cada 6 ou 8 horas, durante 7 a 10 dias.
t 20 a 40 mg/kg/dia, por via intramuscular profunda ou infusão intravenosa, divididos a cada 6 ou 8 horas, durante 7 a 10 dias.
t De 1 a 6 meses:
t 75 mg, por via oral, a cada 12 horas, durante 5 dias, combinada a sulfato de quinina 125 mg, por via oral, a cada 12 horas, nos 3 primeiros dias.
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, combinada a dicloridrato de quinina 20 mg/kg, por infusão intravenosa durante 4 horas, seguido de 10 mg/kg a cada 8 horas, diluído em 10 mL/kg de solução glicosada a 5% (máximo de 500 mL), ambos durante 7 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t A partir dos 6 meses (malária grave):
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, durante 7 dias, combinada a artesunato de sódio 2,4 mg/kg, por via intravenosa, seguido de 1,2 mg/kg administrado após 12 e 24 horas. Depois, 1,2 mg/kg, a cada 24 horas, durante 6 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, durante 7 dias, combinada a arteméter 3,2 mg/ kg, por via intramuscular, no primeiro dia, seguido de 1,6 mg/kg, a cada 24 horas, por mais 4 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, combinada a dicloridrato de quinina 20 mg/kg, por infusão intravenosa durante 4 horas, seguido de 10 mg/kg a cada 8 horas, diluído em 10 mL/kg de solução glicosada a 5% (máximo de 500 mL), ambos durante 7 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t 5 a 7,5 mg/kg, por via oral, a cada 6 horas, durante 12 meses, combinada a pirimetamina e folinato de cálcio.
t 20 mg/kg, por via oral ou intravenosa, 1 hora ou 30 minutos antes do procedimento, respectivamente.
t 150 a 450 mg, por via oral, a cada 6 horas. Dose máxima diária: 1,8 g.
t 0,6 a 2,7 g, por via intramuscular profunda ou infusão intravenosa, a cada 6 a 12 horas. Doses acima de 600 mg devem ser administradas somente por infusão intravenosa.
t 300 a 450 mg, por via oral, a cada 6 horas, durante 21 dias, combinada a primaquina.
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, durante 7 dias, combinada a artesunato de sódio 2,4 mg/kg, por via intravenosa, seguido de 1,2 mg/kg administrado após 12 e 24 horas. Depois, 1,2 mg/kg, a cada 24 horas, durante 6 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, durante 7 dias, combinada a arteméter 3,2 mg/ kg, por via intramuscular, no primeiro dia, seguido de 1,6 mg/kg, a cada 24 horas, por mais 4 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t 20 mg/kg, por via intravenosa, diluída em 1,5 mL/kg de solução glicosada a 5%, infundida em 1 hora, combinada a dicloridrato de quinina 20 mg/kg, por infusão intravenosa durante 4 horas, seguido de 10 mg/kg a cada 8 horas, diluído em 10 mL/kg de solução glicosada a 5% (máximo de 500 mL de SG 5%), ambos durante 7 dias. Assim que o paciente estiver em condições de deglutir, a dose diária pode ser administrada em comprimidos, por via oral.
t 600 mg, por via oral, a cada 6 horas, durante no mínimo 6 semanas, combinada a pirimetamina e folinato de cálcio.
t 600 mg, por via oral ou intravenosa, 1 hora ou 30 minutos antes do procedimento, respectivamente.
t Nota: Para esquema profilático de toxoplasmose em pessoas com Aids e tratamento de malária por Plasmodium falciparum em grávida, consultar, respectivamente, os guias de tratamento de crianças, adolescentes e adultos infectados pelo HIV e o guia prático de tratamento de malária no Brasil.
t Absorção oral é alta (90%) e não influenciada pela presença de alimentos.
t Pico de concentração em 45 minutos (oral) e 2,5 a 3 horas (intramuscular).
t Meia-vida: 1,5 a 5 horas.
t Metabolismo: predominantemente hepático.
t Excreção: renal (5% a 28%).
t Exantema, dermatite de contato, prurido, xerose cutânea e síndrome de Stevens-Johnson (10%).
t Esofagite, glossite, estomatite, desconforto abdominal, náusea, vomito, dispepsia, gosto metálico na boca (4%), diarreia (10%) e colite pseudomembranosa (0,01% a 1%).
t Hepatotoxicidade.
t Dor local e flebite.
t Neutropenia, eosinofilia, agranulocitose e trombocitopenia
t Atracúrio e tubocurarina: podem prolongar o bloqueio neuromuscular, por efeito aditivo. Monitorar o paciente quanto ao nível de bloqueio e diminuir a dose do relaxante muscular, se necessário.
t Ciclosporina: pode ter sua biodisponibilidade diminuída, por mecanismo não conhecido. Monitorar as concentrações de ciclosporina e aumentar sua dose, se necessário.
t Ingerir com grande quantidade de água.
t Caso surja diarreia, informar ao médico.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Armazenar a cápsula e a solução injetável a temperatura de 20 a 25 oC.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Ao usar pela via intravenosa, a concentração final não deve exceder 18 mg/ mL. Nunca administrar a solução em bolo.
t Solução injetável compatível com solução fisiológica 0,9%, glicose 5% e Ringer + lactato por 8 semanas a 10 oC, 32 dias a 4 oC e 16 dias a 25 oC, quando acondicionada em recipiente de vidro ou PVC.
t Incompatível com: ampicilina, aminofilina, barbitúricos, gliconato de cálcio, ceftriaxona, idarrubicina, sulfato de magnésio, fenitoína, filgrastim, fluconazol, alopurinol e ranitidina.
Tatiana Aragão Figueiredo
Isabella Campagnuci Knust
Na Rename 2010: itens 13.2 e 13.5
t Comprimido 10 e 25 mg.
t Depressão.
t Distúrbios do pânico, associados ou não a agorafobia.
t Transtorno obsessivo-compulsivo.
t Distúrbios da condução cardíaca e enfarte do miocárdio recente.
t Insuficiência hepática.
t Hipersensibilidade ao fármaco ou a outros antidepressivos tricíclicos.
t Fase maníaca do transtorno bipolar.
t Uso de inibidores da monoamina oxidase (IMAO) nos últimos 15 dias (a troca de um IMAO pelo tricíclico ou vice-versa deve guardar o intervalo mínimo de 15 dias).
t Porfiria.
t Crianças com menos de 12 anos.
t Usar com cuidado nos casos de:
– distúrbios obsessivo-compulsivos (a suspensão deve ser gradual; 25% da dose a cada 2 meses).
– cardiopatas, epilépticos, idosos, portadores de hipertrofia prostática, hipertireoidismo, glaucoma de ângulo fechado, asma, alcoolismo, psicoses, em pessoas com ideias suicidas ou distúrbios da cognição, agravo da depressão.
– insuficiência renal.
– eletrochoque concomitante (pode aumentar os riscos da terapia com eletrochoque).
– cirurgia eletiva ou uso de anestesia.
– feocromocitoma.
– lactação (ver Apêndice B).
t Perigo ao dirigir ou realizar outras tarefas que exijam atenção e coordenação motora.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Dose de 5 a 10 mg/dia, por via oral, com aumento de 10 mg a cada três dias, até 50 mg.
t Após, incremento de 25 mg até o máximo de 75 mg/dia.
t A administração costuma ser em dose única diária.
t Dose de 25 mg, por via oral, a cada 24 horas. Aumentar, ao cabo de duas semanas, para 100 a 150 mg/dia. Dose máxima de 250 mg/dia.
t inicialmente dose de 25 mg/dia, por via oral, com aumento gradual conforme necessário para 100 a 250 mg/dia em doses divididas. Dose máxima de 250 mg/dia.
t Iniciar com a dose de 10 mg, por via oral, a cada 24 horas. Aumentar, ao cabo de duas semanas, para 100 a 150 mg/dia.
t Iniciar com a dose de 10 mg, por via oral,a cada 24 horas. Aumentar, ao cabo de duas semanas, para 30 a 75 mg/dia.
t Absorção oral variável, mas não sofre interferência da alimentação.
t Meia-vida: 19 a 37 horas (em média 32 horas).
t Resposta inicial para distúrbio obsessivo-compulsivo é de 4 a 10 semanas e para depressão é de 2 semanas
t Metabolismo hepático, com expressivo efeito de primeira passagem.
t Excreção renal (51 a 60%) e fecal (24 a 32%).
t Hipotensão ortostática (que pode levar a quedas em idosos), alterações eletrocardiográficas, arritmias cardíacas.
t Sedação, tontura, insônia, hipomnésia, fadiga, ansiedade, tremores finos de extremidades, disartria, visão turva, diminuição do limiar convulsivo, discinesias, síndrome parkinsoniana, convulsões, sudorese.
t Aumento do apetite, anorexia, dispepsia, anormalidades da função hepática, diarreia, obstipação, náusea, vômito.
t Retenção urinária, especialmente em idosos com hipertrofia prostática.
t Efeitos anticolinérgicos: xerostomia, midríase, cicloplegia, retenção urinária, diminuição da motilidade gastrintestinal, taquicardia, aumento da pressão intraocular.
t Distúrbios comportamentais, transtornos confusionais, que podem ser acompanhados de ansiedade, alucinações ou delírio (sobretudo em idosos), alteração no sono, cefaleia, mania, tendências suicidas.
t Leucopenia, agranulocitose, eosinofilia, trombocitopenia, púrpura.
t Exantema, dermatite, prurido, fotossensibilidade.
t Ganho de peso ou perda de peso, ginecomastia, galactorreia, aumento testicular, alterações dos níveis glicêmicos, diminuição da libido, queda de cabelo, secreção inapropriada do hormônio antidiurético, hipertermia.
t Ácido valproico, antidepressivos bloqueadores seletivos da recaptação de serotonina, cimetidina, enalapril, inibidores de protease (amprenavir, fosamprenavir, atazanavir), modafinila, suco de toranja ou pomelo (grapefruit): aumento de efeito do antidepressivo. Deve-se monitorar as concentrações plasmáticas do antidepressivo, e, quando necessário, ajustes de doses devem ser efetuados. Acompanhar os sinais e sintomas de toxicidade de antidepressivos tricíclicos (efeitos anticolinérgicos, sedação, confusão, arritmias cardíacas).
t Ademetionina e linezolida: pode levar a síndrome serotoninérgica. O uso concomitante com clomipramina é contraindicado.
t Álcool, anti-histamínicos H1, anticoagulantes cumarínicos: podem ter seus efeitos potencializados. Nos pacientes com terapia anticoagulante oral, o tempo de protrombina deve ser cuidadosamente monitorizado. Conseguir uma dose de anticoagulante capaz de produzir o nível desejado de anticoagulação pode ser difícil e frequentes ajustes da dose dos anticoagulantes podem ser necessárias.
t Anfetaminas, bepridil, cisaprida, epinefrina, fenitoína, fluoxetina, paroxetina, fenitoína, formoterol, gatifloxacino, grepafloxacino, halofantrina, lumefantrina, moxifloxacino, quinidina, vasopressina, venlafaxina: aumentam a toxicidade da clomipramina.
t Carbamazepina, fenitoína, oxibutinina: podem diminuir os efeitos da clomipramina, por aumento do catabolismo. Monitorar as concentrações plasmáticas do antidepressivo para orientar ajustes de doses.
t Clonidina, guanadrel, ioimbina: pode haver diminuição dos efeitos destes fármacos. Monitorar a pressão arterial e, se necessário, ajustar a dose do anti-hipertensivo.
t Diuréticos: aumentam o risco de hipotensão postural.
t Fármacos simpaticomiméticos: o uso concomitante pode levar a hipertensão, arritmia cardíaca e taquicardia. Se estes fármacos são utilizados em associação com clomipramina, deve-se monitorar atentamente e a redução da dose dos simpaticomiméticos pode ser necessária.
t Inibidores da monoamina oxidase (IMAO): a associação pode levar a neurotoxicidade, convulsões, ou síndrome serotoninérgica (hipertensão, hipertermia, mioclônus, alterações do estado mental).
t Lítio: há aumento do risco de toxicidade.
t Maconha (Cannabis sativa): a associação pode levar a delírio e taquicardia.
t Nefopam, olanzapina: utilização concomitante com clomipramina pode levar a convulsões.
t Orientar para evitar uso de bebidas alcoólicas.
t Recomendar a ingestão após a alimentação para prevenir irritação gástrica.
t Alertar em relação à demora para o início da resposta terapêutica.
t Alertar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Orientar para informar se houver mudança na frequência, cardíaca e a levantar-se mais lentamente para evitar hipotensão ortostática.
t Alertar para não suspender o uso de maneira repentina.
t Conservar sob temperatura ambiente, entre 15 e 30 oC, em recipientes bem fechados e ao abrigo da luz.
Atenção: os efeitos terapêuticos podem demorar de 15 a 21 dias para se manifestar. Acompanhamento contínuo de pressão arterial e frequência cardíaca nas semanas iniciais. Não há informação quanto à eficácia e segurança em crianças e adolescentes. Este fármaco apresenta um número muito elevado de interações com medicamentos, sendo necessária uma pesquisa específica sobre este aspecto antes de introduzir ou descontinuar a clomipramina ou outros medicamentos no esquema do paciente.
Paula Pimenta de Souza
Rachel Magarinos-Torres
Na Rename 2010: item 13.4
t Comprimido de 25 mg e 100 mg.
t Solução oral 40 mg/mL.
t Solução injetável 5 mg/mL.
t Esquizofrenia e outros transtornos psicóticos.
t Controle de agitação psicomotora, fase aguda de mania em transtorno bipolar, para estabilizar o paciente até que se observem os benefícios do lítio, e em síndromes demenciais, intoxicações exógenas ou síndromes cerebrais orgânicas.
t Sedação de pacientes clínicos em ventilação mecânica, quando em surtos psicóticos associados a doença grave.
t Psicoses com sintomas negativos.
t Feocromocitoma.
t Depressão medular.
t Depressão do sistema nervoso central.
t Hipersensibilidade a clorpromazina e outras fenotiazinas.
t Coma.
t Histórico de tumores dependentes de prolactina.
t Usar com cuidado nos casos de:
– após o controle da crise psicótica
– suspensão de tratamento prolongado (deve ser gradual).
– uso prolongado de clorpromazina (não associar anestesia espinhal ou epidural).
– idosos (utilizar as menores doses pelo menor tempo possível).
– idosos ou enfraquecidos (risco de hipotensão postural).
– epilepsia (redução do limiar convulsivante).(ver Apêndice B).
– insuficiência hepática (evitar quando grave) (ver Apêndice C).
– renal (evitar quando grave) (ver Apêndice D).
– distúrbios cardiovasculares, cerebrovasculares e doenças respiratórias.
– doença de Parkinson, infecções agudas, leucopenia, hipotireoidismo, miastenia gravis, hipertrofia prostática e glaucoma de ângulo fechado.
– histórico de icterícia, síndrome neuroléptica maligna ou câncer de mama.
– mielografia (descontinuar o uso da clorpromazina no mínimo 48 horas antes).
– exames laboratoriais para detectar fenilcetonúria e presença de salicilato na urina (podem ter resultado falso-positivo).
– testes de gravidez (medida de gonadotropina coriônica na urina pode ser afetada; pode ocorrer resultado falso-positivo ou falso-negativo).
t Exames de lâmpada de fenda e oftalmoscópico para detectar alterações oculares.
t Pode afetar a habilidade de operar máquinas e dirigir.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t de 1 a 5 anos – na crise, dose de 0,5 mg/kg, por via intramuscular, a cada 6 a 8 horas. Dose de manutenção 0,5 mg/kg, por via oral, a cada 4 a 6 horas, ajustado de acordo com a resposta. Dose máxima diária: 40 mg.
t de 6 a 12 anos – na crise, dose de 0,5 mg/kg, por via intramuscular, a cada 6 a 8 horas. Dose de manutenção 25 a 75 mg, por via oral, a cada 8 horas, ajustado de acordo com a resposta. Dose máxima diária: 75 mg.
t Na crise, dose de 25 a 50 mg, por via intramuscular, a cada 6 a 8 horas. Dose de manutenção 75 a 300 mg/dia. Dose máxima diária: 1 g.
t Em crise ou em manutenção, a dose deve ser a metade ou um terço da dose de adulto.
t Boa absorção por via oral. Biodisponibilidade de 32%.
t Metabolismo hepático com metabólitos ativos. Crianças tendem a metabolizar este fármaco mais rapidamente que os adultos.
t Excreção renal (menos de 1% na forma ativa).
t Atravessa barreira hematoencefálica, placenta e pode ser detectado no leite materno.
t Pico plasmático: 2 a 4 horas (oral), 1 a 4 horas (intramuscular).
t Meia-vida plasmática: em torno de 30 horas.
t Sintomas extrapiramidais, como parkinsonismo, acatisia, distonia aguda. Na administração prolongada, discinesia tardia, potencialmente irreversível
t Delírio, síndrome neuroléptica maligna, estado catatônico.
t Sedação, perturbação da regulação da temperatura corporal, apatia, pesadelos, palidez, excitação, insônia, confusão e convulsões
t Sintomas anticolinérgicos, incluindo xerostomia, obstipação, dificuldade de micção, hipotensão, taquicardia, arritmia, midríase, visão borrada e aumento da pressão intraocular.
t Ganho de peso.
t Síndrome semelhante a lúpus eritematoso sistêmico no tratamento prolongado.
t Agranulocitose, leucopenia, leucocitose, anemia hemolítica, trombocitopenia, distúrbios tromboembólicos.
t Aumento do intervalo QT, arritmia cardíaca.
t Icterícia, íleo adinâmico.
t Fotossensibilidade, exantema, pigmentação da pele, córnea e retina, dermatite de contato.
t Amenorreia, galactorreia e ginecomastia devido a hiperprolactemia, disfunção erétil e raramente priapismo.
t Dor e formação de nódulo no local da administração intramuscular.
t Aumento do risco de fraturas de quadril em idosos
t Acetazolamida e amilorida: aumento do risco de hipotensão.
t Amissulprida: aumento de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, enfarte) por efeito aditivo no prolongamento do intervalo QT. A administração concomitante de amissulprida e clorpromazina não é recomendada.
t Amitriptilina e clomipramina: aumento do risco de Efeitos adversos antimuscarínicos.
t Amodiaquina: aumento da concentração plasmática de clorpromazina. Diminuir a dose do antimalárico.
t Anlodipino, isradipino e nifedipino: aumento do risco de hipotensão.
t Anticonvulsivantes (carbamazepina, etossuximida, fenitoína, ácido valproico): aumento do risco de convulsões por diminuição do limiar convulsivante.
t Atenolol, metoprolol, propranolol e timolol: aumento do risco de convulsões e da toxicidade da clorpromazina (sedação, efeitos extrapiramidais e delírio). Monitorar o paciente e reduzir a dose de um, ou ambos fármacos, se necessário.
t Beladona: aumento da mania, agitação e dos efeitos anticolinérgicos, podendo levar à insufuciência respiratória. Descontinuar o uso de beladona quando do excesso de atividade anticolinérgica.
t Benzatropina, biperideno, prociclidina, triexifenidil: diminuição da concentração plasmática e da eficácia da clorpromazina; aumento dos efeitos anticolinérgicos. O uso rotineiro de anticolinérgicos para reduzir os efeitos extrapiramidais de fenotiazinas não é recomendado. Anticolinérgicos devem ser reservados às situações em que os efeitos extrapiramidais ocorrem e em que a dose da clorpromazina não pode ser reduzida. Reavaliar o uso de anticolinérgicos a cada 3 meses.
t Captopril e enalapril: hipotensão por ação anti-hipertensiva sinérgica. Advertir o paciente do risco de hipotensão postural e instruí-lo a levantar-se devagar. Monitorar a pressão arterial.
t Cisaprida: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca). A administração concomitante é contraindicada.
t Esparfloxacino, gatifloxacino, gemifloxacino, grepafloxacino, levofloxacino, moxifloxacino: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca). A administração concomitante é contraindicada.
t Etanol: aumento da sedação por efeito aditivo na depressão do sistema nervoso central. Orientar para não ingerir bebida alcoólica durante o tratamento.
t Fenilalanina: aumento da incidência de discinesia tardia por acúmulo de fenilalanina no cérebro. Monitorar sinais de discinesia tardia.
t Fenobarbital: diminuição da eficácia da clorpromazina por indução do metabolismo hepático. Se a terapia concomitante for necessária, ajustar a dose da clorpromazina para manter ou alcançar seu efeito terapêutico.
t Fentanila, levorfanol, meperidina, metadona, morfina, oxicodona: Aumento da depressão do sistema nervoso central e respiratória por efeitos aditivos. Monitorar sinais de depressão respiratória e do SNC, além de hipotensão.
t Fluconazol, fluoxetina, foscarnete: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca).
t Lítio: fraqueza, discinesia, aumento dos sintomas extrapiramidais, encefalopatia e dano cerebral. Monitorar sinais de toxicidade ou sintomas extrapiramidais. Realizar periodicamente a dosagem plasmática do lítio.
t Mesoridazina, proclorperazina, tioridazina, trifluoperazina: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca). A administração concomitante é contraindicada.
t Metoclopramida: aumento do risco de reações extrapiramidais. Uso concomitante é contraindicado.
t Metrizamida: aumento do risco de convulsão por redução do limiar convulsivante. O uso de clorpromazina deve ser descontinuado ao menos 5 dias antes da administração de metrizamida.
t Paliperidona, pimozida, quetiapina, risperidona, sertindol, sultoprida, ziprasidona: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca). A administração concomitante é contraindicada.
t Procarbazina: uso concomitante pode resultar em depressão do sistema nervoso central.
t Tramadol: o uso concomitante pode aumentar o risco de convulsões.
t Trazodona: aumento do risco de hipotensão. Monitorar a pressão arterial. Advertir o paciente a levantar-se devagar, de modo a evitar hipotensão postural.
t Orientar para evitar o uso de bebidas alcoólicas ou sedativos.
t Orientar para notificar o aparecimento de movimentos involuntários.
t Alertar para a importância de não suspender o tratamento abruptamente.
t Orientar para a exigência de cautela com atividades que exijam atenção, como dirigir e operar máquinas.
t Informar mulheres em idade fértil sobre os riscos e aconselhar a comunicar suspeita de gravidez.
t Conservar sob temperatura ambiente entre 15 e 30 ºC, em recipientes bem fechados e ao abrigo da luz.
t A solução injetável pode ser diluída em cloreto de sódio 0,9%. Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Não administrar a solução injetável de clorpromazina por sistema de infusão intravenosa de plástico (ex.: PVC). Perda por adsorção de aproximadamente 41%. Utilizar tubos de poliolefinas (ex.: polietileno e polipropileno).
t A manipulação requer uso de máscaras e luvas de borracha, pois pode causar dermatite de contato grave em pessoas sensíveis.
Atenção: após injeção intramuscular o paciente deve permanecer deitado e sua pressão arterial deve ser monitorada por 30 minutos.
Este medicamento apresenta interações com um grande número de fármacos, com destaque para as interações que podem resultar em aumento do risco de cardiotoxicidade. Assim, uma consulta à literatura específica deve ser realizada antes de incluir este ou outros medicamentos no esquema terapêutico do paciente.
Larissa Niro
Na Rename 2010: seção 6.1.4
t Pó para solução injetável 20 mg
t Leucemia linfoblástica aguda.
t Leucemia mieloide aguda.
t Hipersensibilidade a daunorrubicina ou a outros componentes da formulação.
t Insuficiência cardíaca congestiva.
t Mielossupressão.
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– doença cardíaca prévia ou insuficiência hepática ou renal (ver Apêndice D).
– radioterapia de tórax ou quimioterapia com altas doses cumulativas de antraciclinas ou ciclofosfamida, ou uso de outros agentes cardiotóxicos.
– manipulação (irritante aos tecidos).
– leucemias secundárias.
– lactentes e crianças (mais Susceptíveis à toxicidade cardíaca).
t Associa-se a toxicidade cardíaca relacionada a dose e a grave mielossupressão mesmo em doses terapêuticas.
t Evitar extravasamento.
t Pode ocorrer hiperuricemia secundária à rápida lise das células leucêmicas.
t Categoria de risco na gravidez (FDA): D.
t Dose de 25 a 45 mg (de daunorrubicina base)/m2, por via intravenosa, uma vez por semana, em combinação com vincristina e prednisona.
t Dose de 30 a 60 mg (de daunorrubicina base)/m2/dia, em infusão intravenosa contínua, nos dias 1 e 3 do ciclo.
t Dose de 45 mg (de daunorrubicina base)/m2, por via intravenosa, nos dias 1, 2 e 3 de um ciclo de 32 dias, em combinação com vincristina, prednisona e asparaginase.
t Dose de 45 mg (de daunorrubicina base)/m2, por via intravenosa, nos dias 1, 2 e 3 do primeiro ciclo e nos dias 1 e 2 do segundo ciclo, em combinação com citarabina.
Nota:
t Em menores de 2 anos ou com menos de 0,5 m2 de superfície corporal, a dose deve ser calculada com base no peso corporal em kg.
t A dose acumulada não pode exceder 300 mg/m2 em crianças com mais de 2 anos.
t A dose acumulada não pode exceder 400 a 600 mg/m2 em adultos.
t Administração somente intravenosa, em injeção durante 1 a 5 minutos ou por infusão (em 100 mL de solução de glicose a 5% ou solução fisiológica) durante 15 a 30 minutos.
t Em caso de extravasamento: aplicar gelo imediatamente por 30 a 60 minutos; após alternar a cada 15 minutos no primeiro dia. Elevar a extremidade por 24 a 48 horas.
t Ajuste de dose para pacientes geriátricos: 30 mg (de daunorrubicina base)/ m2.
t Distribuição: ampla distribuição no corpo especialmente nos rins, fígado, coração e baço. Não atravessa a barreira hematoencefálica.
t Biotransformação: rápida (no prazo de 1 hora) no fígado para produzir o metabólito ativo, daunorrubicinol.
t Metabolismo: hepático e outros tecidos.
t Excreção: fecal: 40%; renal: 25%.
t Meia-vida de eliminação: 14-20 horas.
t Alopecia reversiva, reações alérgicas cutâneas (raras), hipercromia ou hiperemia.
t Náuseas, vômitos (50%), esofagite, estomatite, diarreia, ulceração gástrica.
t Coloração vermelha da urina.
t Cardiotoxicidade (mais frequente em adultos).
t Hiperuricemia.
t Leucopenia, trombocitopenia, mielossupressão (dose dependente).
t Clozapina: aumento de efeito/toxicidade de daunorrubicina. Aumento do risco de agranulocitose. Evitar o uso concomitante.
t Trastuzumabe: aumento de efeito/toxicidade de daunorrubicina.O uso concomitante de trastuzumabe e antraciclinas pode determinar um aumento do risco de disfunção cardíaca. A descontinuação do trastuzumabe deve ser fortemente considerada em pacientes com diminuição da função ventricular esquerda clinicamente significante.
t Vacinas com vírus vivos: o uso concomitante de vacinas com vírus vivos pode resultar em aumento do risco de infecção pelo vírus vacinal. Pacientes recebendo quimioterapia imunossupressiva não devem ser vacinados com vacinas de vírus vivos.
t Vacina oral de rotavírus humano: o uso concomitante de vacina contra rotavírus pode resultar em aumento do risco de infecção pelo rotavírus. A vacina contra o rotavírus é contraindicada em pacientes recebendo quimioterapia imunossupressiva.
t Orientar para aumentar a ingestão de líquidos.
t Orientar pata não interromper o uso do medicamento na presença de náuseas e vômitos.
t Orientar para evitar vacinas, especialmente contra poliovírus, e contato com pessoas próximas que receberam a vacina. Se for imprescindível, usar máscara de proteção.
t Orientar para os devidos cuidados para não se envolver em situações que determinem ferimentos, ou que exponham olhos e mucosas a infecção.
t Orientar para evitar o contato com pessoas com infecção, especialmente durante os períodos de baixas contagens sanguíneas.
t Alertar para antes do início do tratamento identificar história prévia de hipersensibilidade a daunorrubicina, gravidez e lactação.
t Notificar imediatamente se ocorrer febre, tosse ou rouquidão, dores lombares, disúria.
t Embalagem e armazenamento: armazenar à temperatura ambiente, entre 15 e 30 ºC, ao menos que haja outra especificação do produtor. Proteger da luz.
t Todo pó ou solução de daunorrubicina que entrar em contato com a pele ou mucosas deve ser retirado completamente com água ou sabão.
t 21,4 mg de daunorrubicina equivalem a 20 mg de daunorrubicina base.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir o pó no frasco original com 4 mL de água para injeção (5 mg/ mL). Soluções reconstituídas são estáveis por 24 horas sob temperatura ambiente ou 48 horas entre 2 a 8 ºC. Proteger da luz.
t Após reconstituição, retirar a dose desejada para uma seringa estéril contendo 10 a 15 mL de solução de cloreto de sódio 0,9%. Infundir por 2 a 3 minutos por tubo Y (ou equivalente) acoplado a um sistema de infusão rápida contendo glicose 5% ou cloreto de sódio 0,9%.
t É incompatível com heparina sódica e solução de fosfato sódico de dexametasona.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: a solução injetável de daunorrubicina é para uso intravenoso exclusivo. A administração deve ser dentro de um sistema de infusão rápida, apropriado e já instalado. O extravasamento do fármaco provoca necrólise grave nos tecidos locais. Cardiotoxicidade manifesta-se por insuficiência cardíaca congestiva que pode ocorrer durante o tratamento ou meses a anos após a suspensão da terapia e está associada a dose acumulada superior a 400-500 mg/m2 em adultos e 300 mg/m2 em maiores de 2 anos. Mielossupressão grave ocorre em doses terapêuticas, induzindo infecções ou hemorragia.
Marta Maria de França Fonteles
Na Rename 2010: item 14.6
t Solução injetável 12,5 mg/mL
t Segunda escolha em choque cardiogênico e choque séptico, como alternativa a dopamina.
t Hipersensibilidade a dobutamina e aos sulfitos.
t Impedimento de ejeção cardíaca tais como estenose subaórtica hipertrófica idiopática.
t Uso concomitante com isocarboxazida e linezolida.
t Usar com cuidado nos casos de:
– arritmia, enfarte do miocárdio, doença da artéria coronária grave e choque cardiogênico complicado por hipotensão grave.
– hipovolemia (deve ser revertida antes de iniciar o uso da dobutamina).
– uso recente de betabloqueadores (pode comprometer a eficácia da dobutamina).
– lactação (ver Apêndice B).
t Seu uso exige acompanhamento constante de ECG, pressão arterial e potássio sérico.
t Não é recomendado o uso prolongado intermitente.
t Fator de risco na gravidez (FDA): B
t Deve ser diluída em concentrações de 0,25 a 5 mg/mL com glicose 5% + cloreto de sódio 0,45%, glicose 10%, cloreto de sódio 0,9%, glicose 5% + cloreto de sódio 0,9% ou Ringer + lactato.
t 2,5 a 20 microgramas/kg/min, por via intravenosa. Dose máxima: 20 microgramas/kg/minuto.
t 2,5 a 20 microgramas/kg/min, por via intravenosa. Dose máxima: 40 microgramas/kg/minuto.
t Início da ação: 1 a 2 min.
t Duração da ação: 10 min após dose única e até 1 semana após dose múltipla.
t As concentrações plasmáticas de dobutamina no estado estacionário são atingidas cerca de 10 a 12 minutos após o início de uma infusão.
t Meia-vida de eliminação: 1 a 2 min.
t Metabolismo: tecidual e hepático, primariamente por conjugação com ácido glicurônico e metilação pela catecol-O-metiltransferase (COMT).
t Excreção: renal (66%), fezes (20%).
t Angina (1% a 3%), arritmia cardíaca, miocardite eosinofílica (até 7%).
t Trombocitopenia (raro).
t Dor no peito, hipertensão (7.5%), palpitação, taquicardia (aproximadamente 10%), arteriosclerose coronária, fibrilação ou flutter atrial são fatores de risco para desenvolver rápida resposta ventricular.
t Reação no lugar da injeção, possivelmente devido ao sulfito de sódio na formulação, tais como: exantema, eritrema, prurido e flebite; necrólise na pele; broncoespasmo
t Hipopotassemia.
t Náusea (1%-3%), vômito.
t Cefaleia (1% a 3%).
t Incontinência urinária (em doses elevadas por 10 minutos de infusão)
t Desordens na coagulação sanguínea (podem ocorrer com infusões contínuas durante vários dias).
t Parestesia, cãibras nas pernas.
t Dispneia (1%-3%)
t Carvedilol: pode reduzir a eficácia da dobutamina. Se necessário uso concomitante, intensificar a monitoração da pressão arterial e frequência cardíaca. t Isocarboxazida: pode ocorrer crise hipertensiva. Uso concomitante é contraindicado. Caso seja necessário o uso combinado, monitorar a pressão arterial e verificar incidência frequentes de cefaleia ou palpitações, sintomas indicativos de crise hipertensiva. Caso ocorra a crise, descontinuar o inibidor da MAO e administrar 5 mg de fentolamina lentamente por via intravenosa.
t Linezolida: pode resultar em aumento dos efeitos hipertensivos. É contraindicado o uso concomitante sem monitorar eventuais aumentos na pressão arterial. Em pacientes tratados com linezolida, a dose inicial de dobutamina deve ser reduzida e posteriormente ajustada para alcançar a resposta terapêutica desejada.
t Medicamento restrito para uso hospitalar.
t Avisar imediatamente o médico se detectar algum destes efeitos: batimento cardíaco lento, rápido ou irregular; dor no peito, transtorno respiratório, urticária ou erupção cutânea grave.
t Antes da diluição, armazenar a temperatura ambiente (15 a 30 ºC). Não congelar, pode causar cristalização.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Pode ser diluída em solução glicosada a 5%, solução de cloreto de sódio 0,9% ou Ringer + lactato.
t Após sua preparação, a solução para infusão intravenosa se mantém estável por 24 h a temperatura ambiente e 48 h sob refrigeração.
t A solução altamente concentrada (500 mg/50 mL) se mantém estável por 24 h quando protegida da luz.
t A solução concentrada é límpida, incolor a cor palha pálida, mas pode exibir uma coloração rosa que pode ficar mais forte com o passar do tempo, devido a oxidação do princípio ativo. Porém, não significa perda de sua potência até 24h.
t O pH da solução reconstituída é em torno de 2,5 a 5,5.
t Incompatível com solução de bicarbonato de sódio 5% ou qualquer outra solução alcalina.
t Recomenda-se não misturar na mesma solução contendo outros medicamentos. Possui um número elevado de incompatibilidades físicas com fármacos, como aciclovir, bumetamina, gliconato de cálcio, diazepam, digoxina, foscarnete, varfarina, insulina, fenitoína, entre outros.
t Não se recomenda a administração em Y concomitantemente com outros fármacos alcalinos, como: aminofilina, furosemida e tiopental sódico.
Atenção: este fármaco apresenta um número elevado de incompatibilidades, por isso não se recomenda a mistura na mesma solução contendo outros medicamentos, nem a administração em Y concomitantemente a outros fármacos.
Marta Maria de França Fonteles
t Solução injetável 5 mg/mL
t Choque cardiovascular e choque séptico.
t Taquiarritmia.
t Fibrilação ventricular.
t Doença cardíaca isquêmica.
t Feocromocitoma.
t Hipertireoidismo.
t Hipersensibilidade à dopamina.
t Usar com cuidado nos casos de:
– lactação.
– choques decorrentes de enfarte do miocárdio ou história de doença vascular periférica (usar doses baixas para diminuir o risco de isquemia de extremidades).
– angina pectoris.
– arritmias ventriculares.
– uso recente de inibidores da monoamina oxidase (MAO).
– sensibilidade a sulfitos.
– doença vascular oclusiva.
– extravasamento (risco de isquemia periférica, necrólise tecidual e gangrena).
– crianças sob risco de desenvolver hipertensão pulmonar (pode ocorrer aumento da pressão arterial pulmonar em crianças, com o uso de dopamina, após cirurgia cardíaca, e em prematuros com hipotensão).
t Antes de iniciar a terapia com dopamina, corrigir quadros de hipóxia, acidose metabólica e hipovolemia (manter volume sanguíneo adequado durante o tratamento).
t Podem ocorrer arritmias cardíacas.
t Categoria de risco na gravidez (FDA): C (ver apêndice A).
t Dose inicial 2 a 5 microgramas/kg/min, por infusão intravenosa, aumentada gradualmente para 5 a 10 microgramas/kg/min, de acordo com a pressão arterial, débito cardíaco e débito urinário. Em casos graves, até 20 a 50 microgramas/kg/min.
t Durante a descontinuação da infusão pode ser necessária a redução gradual da dose (enquanto expande o volume plasmático com fluidos intravenosos) pois a cessação abrupta pode causar hipotensão grave.
t O fármaco é inativado quando administrado via oral.
t A resposta inicial via intravenosa ocorre em 5 minutos, com duração de 10 minutos após dose única.
t O fármaco possui alto volume aparente de distribuição, atravessando em neonatos a barreira hematoencefálica (não ocorre em adultos) e ultrapassa também a barreira placentária.
t A depuração de dopamina em crianças depende da idade. Crianças menores de 2 anos podem ter uma depuração duas vezes maior que os pacientes pediátricos mais velhos. A maior extensão de depuração de dopamina foi observada em pacientes pediátricos com disfunção hepática e renal
t O tempo de meia-vida de distribuição é de 1,8 minutos e o volume aparente de distribuição é de 1,81 a 2,45 L/kg. A meia-vida de distribuição em crianças (3 meses a 13 anos) é de aproximadamente 1,8 minutos após um intervalo de infusão de 1 a 20 microgramas/kg/min para 1 a 6 dias.
t Metabolismo hepático, renal e sérico: 75% do fármaco é tranformado em ácido homovanílico (metabólito inativo) e 25% em norepinefrina (metabólito ativo)
t Excreção renal: 80%, sendo a maior parte excretada como metabólitos e uma pequena porção é excretada na forma inalterada. A depuração endógena total corporal é de 115 mL/kg/min.
t O tempo de meia-vida plasmática é de 2 minutos, e de eliminação é de aproximadamente 9 minutos (sendo em recém nascidos em torno de 7 min e em crianças em torno de 26 min).
t Angina, bradicardia, hipertensão ou hipotensão, palpitações, taquicardia e arritmias ventriculares (especialmente em altas doses), batimentos ectópicos.
t Dispneia.
t Hipopotassemia.
t Poliúria.
t Gangrena.
t Cefaleia.
t Náusea e vômito.
t Vasoconstrição periférica.
t Hipotensão com tonturas, desmaios, rubor.
t Agitação e irritação
t Ergotamina e análogos (ergotamina, ergonovina): o uso associado a dopamina aumenta risco de vasoconstrição periférica e gangrena em mãos e pés, sendo contraindicado (especialmente para a ergotamina). Se a administração for necessária o paciente deve receber monitoria adequado quanto à vasoconstrição periférica (sensação de frio, pele pálida, dor), especialmente nas extremidades, sendo este o provável mecanismo da interação.
t Fenelzina: uso concomitante com dopamina pode resultar em crise hipertensiva (cefaleia, hiperpirexia, hipertensão), sendo contraindicado.
t Fenitoína e fosfenitoína: uso concomitante com dopamina pode resultar em hipotensão e/ou parada cardíaca.
t Isocarboxazida: uso concomitante com dopamina pode resultar em crise hipertensiva (cefaleia, hiperpirexia, hipertensão), sendo contraindicado. Se for necessário o uso concomitante, controlar a pressão arterial e perguntar ao paciente sobre frequentes cefaleias ou palpitações, uma vez que estes sintomas podem anteceder uma crise hipertensiva. Se ocorrer crise hipertensiva, descontinuar o inibidor da MAO e iniciar terapia hipotensiva (5 mg de fentolamina administrado lentamente por via intravenosa)
t Linezolida: pode haver aumento do efeito hipertensivo. Em pacientes em tratamento com linezolida deve-se iniciar a terapia com dopamina em doses baixas, aumentando-as conforme a resposta do paciente, até atingir os resultados esperados.
t Pargilina: uso concomitante de pargilina e dopamina pode resultar em crise hipertensiva (cefaleia, hiperpirexia, hipertensão), devendo ser evitado. Se estes fármacos forem usados em conjunto, deve-se controlar a pressão arterial e perguntar ao paciente sobre frequentes cefaleias ou palpitações, uma vez que estes sintomas podem anteceder uma crise hipertensiva. Se uma crise hipertensiva ocorrer, descontinuar o inibidor da MAO e instituir terapia para baixar a pressão arterial (5 mg de fentolamina administrado lentamente por via intravenosa).
t Selegilina: risco de aumento do efeito hipertensivo. Dopamina deve ser administrada com precaução em pacientes com terapia crônica com selegilina ou que tenham recebido selegilina nas duas semanas anteriores ao início do tratamento.
t Tranilcipromina: uso concomitante com dopamina pode resultar em crise hipertensiva (cefaleia, hiperpirexia, hipertensão), sendo contraindicado.
t Comunicar o médico se tiver problema de saúde como diabetes, asma, alergias ou problema circulatório ou, ainda, casos de gravidez ou lactação.
t No aparecimento de algum dos seguintes Efeitos adversos deve-se comunicar o médico responsável pelo tratamento: dor no peito, taquicardia ou bradicardia, dispneia, cansaço ou fraqueza incomuns, cefaleia, ansiedade, náusea ou vômito.
t Pode ser necessário diminuir lentamente a dose antes de parar completamente.
t Armazenar sob temperatura ambiente, entre 15 e 30 º C.
t O pH da solução da dopamina está entre 4 e 6,4, não devendo ser adicionada a diluentes alcalinos pois pode ocorrer inativação.
t A solução é sensível à luz e deve ser armazenada com proteção específica e sem contato com ar. Descoloração da solução para amarelo a marrom é um indicativo de decomposição e a mesma não deve ser utilizada.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t O fármaco é inativado em soluções com bicarbonato de sódio 5% e é incompatível com medicamentos alcalinos como furosemida, tiopental sódico, sais de ferro, insulina, ampicilina, anfotericina B, cefepima, indometacina, aciclovir e aminoglicosídeos.
t Os diluentes utilizados para preparação de solução para infusão intravenosa incluem cloreto de sódio 0,9%, glicose 5%, glicose 5% em cloreto de sódio 0,45%, glicose 5% em Ringer com lactato, lactato de sódio. A diluição deve ser feita imediatamente antes da administração e é estável por até 24 horas após a diluição.
t Para preparar a diluição deve-se adicionar de 400 a 800 mg de dopamina a 250 mL de uma solução diluente apropriada. A solução resultante contém de 1.600 a 3.200 microgramas de dopamina por mL.
Atenção: durante a terapia, monitorar: a pressão arterial, eletrocardiograma, o débito e frequência cardíaca, balanço hídrico, a cor e temperatura das extremidades, débito urinário (regimes de doses acima de 20 microgramas/ kg/min) e, diante de um aumento desproporcional de pressão arterial diastólica, reduza o fluxo da infusão. Se houver hipotensão, comunique imediatamente o médico para possibilitar a redução da dose.
Maria Inês de Toledo e Lívia Luize Marengo
t Comprimido 100 mg.
t Infecções causadas por Rickettsia, Chlamidia (psitacose, ornitose, tracoma, doença inflamatória pélvica, uretrite, salpingite, linfogranuloma venéreo, conjuntivite e prostatite) e Mycoplasma
t Tratamento alternativo de sífilis e gonorreia em paciente alérgico à penicilina.
t Peste (Yersinia pestis).
t Granuloma inguinal (Calymatobacterium granulomatis).
t Brucelose (Brucella spp).
t Cólera (Vibrio cholerae).
t Primeira escolha para quimioprofilaxia em viajantes que visitarão regiões de alto risco de transmissão de Plasmodium falciparum na Amazônia Legal, que permanecerão na região por tempo maior que o período de incubação da doença (e com duração inferior a seis meses) e em locais cujo acesso ao diagnóstico e tratamento de malária estejam a mais de 24 horas.
t Tratamento de segunda escolha de malária por Plasmodium falciparum
t Hipersensibilidade a doxiciclina, tetraciclinas ou outros componentes da fórmula.
t Crianças com menos de 8 anos de idade (provoca alterações no crescimento ósseo e durante o desenvolvimento da dentição, com descoloração temporária ou permanente dos dentes e hipoplasia do esmalte).
t Gravidez (ver apêndice A)
t Insuficiência hepática grave (ver Apêndice C).
t Porfiria.
t Lupus eritematoso sistêmico.
t Usar com cuidado nos casos de:
– exposição à luz solar (pode ocorrer fotossensibilidade, manifestada por queimaduras).
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
– insuficiência hepática: evitar altas doses.
t 100 a 200 mg/dia, por via oral, a cada 12 ou 24 horas. A duração de tratamento depende da doença.
t 1,5 mg/kg até 100 mg, por via oral, a cada 24 horas, iniciado 1 dia antes da viagem e mantido até 4 semanas após o retorno
t De 8 a 10 anos (22 a 29 kg): 100 mg, por via oral, dividido a cada 12 horas, durante 5 dias, combinada a sulfato de quinina 750 mg, por via oral, dividido a cada 12 horas, durante os primeiros 3 dias, e primaquina 15 mg, por via oral, em dose única, no 6º dia.
t De 11 a 14 anos (30 a 49 kg): 150 mg, por via oral, dividido a cada 12 horas, durante 5 dias, combinada a sulfato de quinina 1,25 g, por via oral, dividido a cada 12 horas, durante os primeiros 3 dias, e primaquina 30 mg, por via oral, em dose única, no 6º dia.
t Maiores de 15 anos (= 50 kg): 100 mg, por via oral, a cada 12 horas, durante 5 dias, combinada a sulfato de quinina 2 g, por via oral, a cada 12 horas, durante os primeiros 3 dias, e primaquina 45 mg, por via oral, em dose úncia, no 6º dia.
t Não têm absorção prejudicada por alimentos.
t Pico plasmático: 2 horas.
t Meia-vida de eliminação: 15 a 24 horas.
t Metabolismo: parcialmente inativado no trato gastrintestinal por quelação.
t Excreção: urinária (35 a 45%)
t Não é dialisável.
t Alterações dentárias: hipoplasia de esmalte e coloração dos dentes.
t Alteração de crescimento ósseo (10%).
t Insuficiência hepática em grávidas com altas doses.
t Esofagite (1 a 10%), dor epigástrica, anorexia, náusea, vômito, diarreia.
t Fotossensibilidade.
t Leucopenia.
t Reações de hipersensibilidade, zumbidos, cefaleia e distúrbios visuais.
t Contraceptivos combinados: têm sua efetividade reduzida. Se necessário uso concomitante, empregar outro método contraceptivo.
t Fosfenitoína, penicilinas, rifapentina: redução da efetividade da doxiciclina. Monitorar efetividade do tratamento com doxiciclina. Considerar aumento da dose em pacientes que recebem fosfenitoína ou fenitoína. Ajustar dose em pacientes que recebem rifapentina.
t Isotretinoína: pode resultar em pseudotumor cerebral (hipertensão intracraniana benigna). Monitorar pacientes para sinais de pseudotumor cerebral (cefaleia grave, náusea, vômito e transtornos visuais).
t Metotrexato: aumento no risco de toxicidade pelo metotrexato (leucopenia, trombocitopenia, anemia, nefrotoxicidade, ulcerações em mucosas). Monitorar cuidadosamente para evidência de toxicidade do metotrexato, especialmente se for administrado em doses altas. Considerar substituição da doxiciclina.
t Produtos contendo alumínio, cálcio ou magnésio, subsalicilato de bismuto: redução da efetividade das tetraciclinas. Não se recomenda uso concomitante. Se não for possível evitar, administrar tetraciclinas pelo menos 1-2 horas antes de produtos contendo alumínio, cálcio ou magnésio. Monitorar paciente quanto à eficácia do antimicrobiano.
t Rifampicina: pode resultar em redução da concentração plasmática da doxiciclina e potencial perda da eficácia da doxiciclina. Monitorar pacientes quanto à resposta ao tratamento. Considerar administração da doxiciclina em combinação com estreptomicina.
t Sais de ferro: redução da efetividade das tetraciclinas e do ferro. Administrar sais de ferro pelo menos três horas antes ou duas horas após a dose de doxiciclina.
t Orientar para ingerir o medicamento junto às refeições da manhã e da noite com um copo cheio de água.
t Não deitar logo após ingerir o medicamento.
t Alertar para evitar o uso de alimentos ricos em cálcio, antiácidos e suplementos de ferro de 1 a 3 horas antes ou depois de tomar o medicamento.
t Alertar para empregar método alternativo ou adicional para evitar a gravidez se estiver em uso de contraceptivos orais.
t Alertar para usar protetor solar durante exposição solar.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses, sob risco de desenvolvimento de resistência bacteriana.
t Armazenar a temperatura ambiente (de 15 a 30 ºC). Proteger da luz.
Atenção: no Brasil, onde a malária tem baixa incidência e há predomínio de Plasmodium vivax em toda a área endêmica, deve-se lembrar que a eficácia da profilaxia para essa espécie de Plasmodium é baixa, não devendo ser recomendada.
Larissa Niro
t Pó para solução injetável 10 mg e 50 mg
t Leucemia linfoblástica aguda.
t Leucemia mieloide aguda.
t Carcinoma de mama avançado.
t Linfomas de Hodgkin e não-Hodgkin.
t Sarcomas ósseos e não ósseos.
t Neuroblastoma.
t Nefroblastoma avançado.
t Câncer de bexiga, ovário, tireoide, pulmão.
t Estadios avançados de câncer de endométrio, próstata, útero, estômago, pâncreas, fígado.
t Leucemia linfoide crônica.
t Mieloma múltiplo.
t Mesotelioma.
t Tumores de células germinativas de ovário e testículo.
t Carcinoma de cabeça e pescoço.
t Sarcoma de Kaposi relacionado com a Aids.
t Tumor maligno das ilhotas de Langerhans.
t Hipersensibilidade a doxorrubicina ou a outros componentes da formulação.
t Mielossupressão pré-existente.
t Doses cumulativas prévias de doxorrubicina ou outras antraciclinas.
t Insuficiência cardíaca congestiva e arritmias.
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– insuficiência hepática.
– crianças (apresentam maior risco para cardiotoxicidade tardia).
– prévia radioterapia torácica ou quimioterpia com daunorrubicina ou ciclofosfamida (dose total máxima de 450 mg/m2; demais casos, dose total máxima de 550 mg/m2).
– extravasamento.
– associação com citarabina (pode ocorrer colite necrosante).
t Realizar avaliação cardíaca prévia ao tratamento, especialmente em pacientes de alto risco para cardiopatia e em crianças.
t Hiperuricemia secundária pode ocorrer por rápida lise das células leucêmicas.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Cuidado especial com o descarte dos dejetos do paciente.
t Monoterapia: 60 a 75 mg/m2, por via intravenosa, a cada 21 dias. Doses menores são recomendadas nos casos de mielossupressão ou infiltração neoplásica medular.
t Em combinação com outro agente quimioterápico: 40 a 60 mg/m2, por via intravenosa, a cada 21 a 28 dias.
t Monoterapia: 60 a 75 mg/m2, por via intravenosa, a cada 21 dias.
t Em combinação com outro agente quimioterápico: 40 a 60 mg/m2, por via intravenosa, a cada 21 dias ou 28 dias.
t 9 mg/m2, por via intravenosa contínua, a cada 24 horas, dos dias 1 a 4, em combinação com vincristina e dexametasona.
t 60 mg/m2, por via intravenosa, em combinação com ciclofosfamida 600 mg/ m2, por via intravenosa, a cada 21 dias, por 4 ciclos.
t 30 a 50 mg/m2, por via intravenosa, a cada 21 dias ou 28 dias.
t 50 mg, por via intravenosa, nos dias 1 e 22 e repetir o ciclo a cada 42 dias.
t Metabolismo: hepático.
t Excreção: biliar (40%) e renal (5 a 12%).
t Meia-vida de eliminação: 1-3 horas.
t Metabolizada no fígado rapidamente (dentro de 1 hora); o metabolismo da doxorrubicina leva a produção de radicais livres que contribuem para a cardiotoxicidade.
t Meia-vida de distribuição: aproximadamente 5 minutos.
t Não atravessa a barreira hematoencefálica.
t Alterações transitórias eletrocardiográficas, geralmente assintomáticas e autolimitadas.
t Alopecia
t Náuseas; vômitos (21-55%), estomatite, esofagite, mucosite, ulceração do cólon, anorexia.
t Arritmias cardíacas, insuficiência congestiva cardíaca.
t Mielossupressão (75%).
t Coloração vermelha da urina.
t Necrose local ao extravasamento.
t Ciclosporina: pode resultar em aumento da exposição à doxorrubicina e ao seu metabólito ativo doxorrubicinol.
t Cisplatina: o uso concomitante pode resultar em leucemia. Extremo cuidado ao administrar esta combinação altamente tóxica. Avaliar cuidadosamente os riscos e os benefícios.
t Docetaxel: pode resultar em icterícia colestática e colite pseudomembranosa. O médico deve estar ciente dos potenciais riscos desta interação.
t Estavudina: doxorrubicina inibe o efeito de estavudina. A administração concomitante de doxorrubicina e antirretrovirais análogos de nucleosídeos deve ser utilizada com cautela e apenas se os potencias benefícios superarem os riscos.
t Fenitoína: pode haver redução na absorção de fenitoína.
t Glicosamina: pode resultar em redução da efetividade da doxorrubicina. Evitar o uso concomitante.
t Paclitaxel: pode resultar em aumento das concentrações plasmáticas da doxorrubicina e da atividade do seu metabólito, doxorrubicinol. Monitorar concentração plasmática da doxorrubicina.
t Sorafenibe: pode resultar em aumento da exposição à doxorrubicina.
t Trastuzumabe: pode resultar em maior risco de disfunção cardíaca. A descontinuação do tratamento com trastuzumabe deve ser considerada seriamente em pacientes que desenvolvem diminuições significativas na função do ventrículo esquerdo.
t Vacinas com vírus vivos: pode resultar em risco aumentado de infecção pelo vírus da vacina. Pacientes recebendo quimioterapia imunossupressiva não devem ser vacinados com vacina com vírus vivos. Em pacientes com remissão de leucemia, esperar pelo menos 3 meses entre o final da quimioterapia e a vacinação.
t Vacina rotavírus: pode resultar em maior risco de infecção pelo vírus da vacina. A vacina com a vacina de rotavírus é contraindicada em pacientes recebendo quimioterapia imunossupressiva.
t Valspodar: pode aumentar a toxicidade de doxorrubicina. Durante o uso concomitante é indicado a redução de 75% da dose de doxorrubicina em pacientes com câncer.
t Varfarina: o uso concomitante pode aumentar o risco de sangramento.
t Zidovudina: pode resultar em toxicidade hematológica (neutropenia). A administração concomitante deve ser evitada, mas se o uso for necessário, a retirada temporária ou a redução da dose da doxorrubicina ou zidovudina são recomendadas. A contagem de células sanguíneas deve ser monitorada frequentemente incluindo hemoglobina, hematócrito, células brancas e contagem de leucócitos diferencial.
t Orientar para aumentar a ingestão de líquidos.
t Orientar para evitar vacinas, especialmente contra poliovírus, ou contato com pessoas próximas que receberam a vacina. Se for imprescindível, usar máscara de proteção.
t Orientar para evitar o contato com pessoas com infecção, especialmente durante os períodos de baixas contagens sanguíneas.
t Alertar para notificar imediatamente se ocorrer febre, tosse ou rouquidão, dores lombares, disúria.
t Orientar para não interromper o tratamento em caso de náuseas e vômitos.
t Notificar o médico em caso de sangramento incomum ou contusões.
t Cuidar para não se envolver em situações que determinem ferimentos, ou que exponham olhos e mucosas à infecção.
t Identificar antes do início do tratamento história prévia de hipersensibilidade a doxorrubicina, gravidez e lactação.
t Idosos e menores de 2 anos possuem risco maior para cardiotoxicidade.
t Armazenar o frasco com pó à temperatura ambiente, entre 15 e 30 °C, proteger da luz. Não contém conservante.
t Durante a manipulação, atenção para distinguir as diferentes concentrações contidas nos frascos (10 mg e 50 mg).
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir o pó com cloreto de sódio 0,9% injetável na proporção de 5 mL para cada 10 mg, obtendo concentração de 2 mg/mL. Agitar até dissolução. A solução reconstituida é estável por 7 dias à temperatura ambiente, entre 15 e 30 °C, e por 15 dias sob refrigeração entre 2 e 8 °C.
t Após reconstituição, diluir com soro fisiológico 0,9% ou soro glicosado 5%. As soluções de doxorrubicina permanecem estáveis por 48 horas se armazenadas entre 2 e 8 °C e por 24 horas, entre 15 e 30 °C.
t Incompatibilidade: heparina, furosemida, dexametasona, fluoruracila, succinato sódico de hidrocortisona, aminofilina, cefalotina.
t O uso de diluentes bacteriostáticos não é recomendado.
t Embora sensível à luz em baixas concentrações, doxorrubicina não está sujeita a fotodegeneração significativa às concentrações clínicas e precaução especial para proteger as soluções da luz durante a administração não parece ser necessária.
t A administração intravenosa deve ser feita durante 3 a 5 minutos através de tubo Y (ou equivalente), acoplado a sistema de infusão rápida contendo cloreto de sódio 0,9% ou glicose 5%, para evitar eritema local ou rubor facial.
t A infusão contínua deve usar linha central.
t Muito cuidado deve ser tomado para evitar o extravasamento durante a administração intravenosa devido ao risco de ulceração grave e necrólise local. Enrubescimento facial indica que a injeção está muito rápida.
t Em caso de extravasamento: aplicar gelo imediatamente por 30 a 60 minutos; após alternar a cada 15 minutos no primeiro dia. Elevar a extremidade por 24 a 48 horas.
t A doxorrubicina também pode ser administrada por via intra-arterial e por instilação vesical tópica.
t A solução remanescente após utilização deve ser descartada.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: necrose tecidual grave pode ocorrer em caso de extravasamento durante a administração. Não deve ser administrada por vias intramuscular ou subcutânea. Cardiotoxicidade dose dependente manifestada por insuficiência cardíaca congestiva pode ocorrer durante o tratamento ou meses a anos após a suspensão da terapia. A probabilidade de ocorrência deste evento é de 1 a 20%, correspondendo à variação da dose acumulada de doxorrubicina a partir de 300 mg/m2 até 500 mg/m2 independente ou não da presença de fatores de risco associados. Leucemia mieloide aguda secundária associa-se ao uso de doxorrubicina. Doxorrubicina pode determinar mielossupressão grave.
Marta Maria de França Fonteles
t Solução injetável 1 mg/mL
t Anafilaxia (choque anafilático).
t Hipersensibilidade a aminas simpaticomiméticas.
t Dilatação cardíaca e insuficiência coronária.
t Uso concomitante com ciclopropano, anestésicos hidrocarbonetos halogenados ou anestésicos locais em extremidades, como dedos, pés e orelhas (pode desencadear gangrena nessas áreas).
t Uso concomitante com isocarboxazida, fenelzina e di-hidroergotamina devido ao risco de desencadear crise hipertensiva.
t Não usar em casos onde fármacos vasopressores são contraindicados (ex: tireotoxicose, diabetes, grávidas com pressão arterial acima de 130/80 mmHg, hipertensão e outras doenças cardiovasculares), exceto quando diluído em soluções com anestésicos locais para reduzir a absorção e prolongar a ação.
t Trabalho de parto (ver apêndice A).
t Isquemia cerebral.
t Glaucoma.
t Choque não-anafilático.
t Uso de inibidor da monoamina oxidase dentro das 2 semanas anteriores ao início do uso da epinefrina.
t Arritmia cardíaca.
t Usar com cuidado nos casos de:
– doença cardíaca, hipertensão, arritmias, doença cerebrovascular, hipertireoidismo, diabete melito, idosos e doença pulmonar crônica.
– doença de Parkinson e psiconeuroses.
– lactação – avaliar benefício-risco.
– extravasamento (pode ocorrer necrólise e perda tecidual).
– uso prolongado ou dose excessiva de epinefrina (produz aumento da glicemia e da concentração plasmática de ácido lático).
t É recomendado alternar os sítios das injeções, a fim de evitar necrólise devido à vasoconstrição.
t Infusão intravenosa rápida pode causar a morte do paciente, devido à hemorragia cerebrovascular ou arritmias cardíacas.
t Alguns produtos disponíveis no comércio contêm sulfitos como conservantes, substâncias potencialmente alergênicas.
t Inicialmente, a epinefrina administrada por via parenteral pode produzir constrição dos vasos sanguíneos renais e diminuir a formação de urina.
t Fator de risco na gravidez (FDA): C (ver apêndice A).
t Menores de 6 meses: 50 microgramas (0,05 mL de solução 1:1.000), por via intramuscular, repetida se necessário a cada 5 minutos de acordo com a pressão arterial, frequência cardíaca e função respiratória.
t De 6 meses a 6 anos: 120 microgramas (0,12 mL de solução 1:1.000), por via intramuscular, repetida se necessário a cada 5 minutos de acordo com a pressão arterial, frequência cardíaca e função respiratória.
t De 6 a 12 anos: 0,25 mg (0,25 mL de solução 1:1.000), por via intramuscular, repetida se necessário a cada 5 minutos de acordo com a pressão arterial, frequência cardíaca e função respiratória.
t Maiores de 12 anos: 0,5 mg (0,5 mL de solução 1:1.000), por via intramuscular, repetida se necessário a cada 5 minutos de acordo com a pressão arterial, frequência cardíaca e função respiratória.
t Menores de 12 anos: 10 microgramas/kg (0,1 mL/kg de solução 1:10.000), por via intravenosa lenta, na velocidade de 1 mL/minuto, se a circulação estiver inadequada.
t Maiores de 12 anos: 0,5 mg (5 mL de solução 1:10.000), por via intravenosa lenta, na velocidade de 1mL/minuto, se a circulação estiver inadequada.
t 0,5 mg (0,5 mL de solução 1:1.000), por via intramuscular, repetida se necessário a cada 5 minutos de acordo com a pressão arterial, frequência cardíaca e função respiratória.
t 0,5 mg (5 mL de solução 1:10.000), por via intravenosa lenta, na velocidade de 1 mL/minuto, se a circulação estiver inadequada.
t 0,2 a 0,5 mg (0,2 a 0,5 mL de solução 1:1.000), por via subcutânea, a cada 5 minutos conforme necessário.
t Não é absorvida por via oral.
t Início da ação: rápido.
t Duração da ação: curta (1 a 2 minutos).
t Meia-vida de eliminação: 1 minuto.
t Metabolismo: é rapidamente inativada e degradada por enzimas hepáticas e de outros tecidos. É metabolizado pela monoamina oxidase (MAO) e catecol-O-metiltransferase (COMT).
t Excreção: renal.
t A epinefrina é capaz de atravessar a placenta e chegar à circulação fetal.
t Arritmia cardíaca, taquicardia, emergência ou urgência hipertensiva.
t Edema pulmonar.
t Hemorragia cerebral.
t Extremidades frias, palpitação, taquicardia.
t Palidez, sudorese.
t Náusea, vômito.
t Ansiedade, apreensão, nervosismo.
t Astenia, tontura, cefaleia, dor nos olhos, tremor.
t Dispneia.
t Angina, fibrilação ventricular, síncope.
t Convulsão.
t Acidose lática, alteração na glicemia.
t Hipersalivação.
t Trombocitose.
t Nefrotoxicidade, retenção urinária.
t Antidepressivos tricíclicos: uso conjunto pode resultar em taquicardia, arritmias e hipertensão, pela inibição da recaptação da norepinefrina. Evitar uso concomitante. Caso seja necessária administração concomitante, monitorar o paciente cuidadosamente e reduzir a dose da epinefrina.
t Betabloqueadores (carvedilol, labetalol, metipranolol, nadolol, oxprenolol, pindolol, propranolol, tertatolol, timolol): uso concomitante pode causar hipertensão, bradicardia e resistência à epinefrina na anafilaxia. Se possível, evitar o uso concomitante; entretanto, se usado, monitorar pressão arterial cuidadosamente; se um betabloqueador não seletivo causar resistência à epinefrina na anafilaxia, glucagon pode ser efetivo em uma dose de 1 mg ou mais por via intravenosa a cada 5 minutos.
t Di-hidroergotamina e alcaloides do ergot: podem causar aumento extremo da pressão arterial. O uso concomitante é contraindicado.
t Entacapona: uso concomitante pode resultar em risco aumentado de taquicardia, hipertensão e arritmias. Deve-se ter cuidado durante a administração, pacientes devem ser monitorados em relação a efeitos cronotrópicos e arritmogênicos elevados da epinefrina.
t Fenelzina: uso concomitante pode resultar em aumento de efeitos hipertensivos. O uso concomitante é contraindicado.
t Halotano: uso concomitante pode resultar em toxicidade ventricular (arritmia ventricular). Monitorar eventuais arritmias.
t Isocarboxazida: pode resultar em crise hipertensiva. O uso concomitante é contraindicado.
t Linezolida: aumento dos efeitos hipertensivos. O uso concomitante é contraindicado.
t Morfina: pode ter sua concentração plasmática aumentada.
t Epinefrina solução injetável é sensível à luz e deve ser armazenada em recipiente resistente à luz e a temperatura de 15 a 25 ºC. Não congelar.
t Não administrar se a solução estiver rosada, marrom, escurecida ou descolorida; ou se houver precipitado.
t É estável em pH em torno de 5,5.
t A presença de metabissulfito de sódio no produto injetável não deve impedir a administração do mesmo em casos de emergência ou situações alérgicas graves.
t Forma sais na presença de ácidos que são prontamente solúveis em água.
t Soluções aquosas são preparadas com cloridrato de epinefrina, mas a dose é fixada pelo conteúdo de epinefrina equivalente: 1,2 mg de cloridrato de epinefrina equivale aproximadamente a 1 mg de epinefrina.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A epinefrina é estável por 24 horas à temperatura ambiente ou sob refrigeração em Ringer e Ringer + lactato, na concentração de 4 mg/L.
t Em soro glicosado 5%, a estabilidade se mantém na concentração de até 87 mg/L, enquanto as soluções de epinefrina em cloreto de sódio 0,9% apresentam estabilidade na concentração máxima de 1 mg/L com atividade, quando mantidas por 24 horas a 5 ºC.
t Entre os compostos incompatíveis em solução com a epinefrina citam-se aminofilina, ampicilina, cloranfenicol, corticotropina, ciclosporina, hialuronidase, meticilina, penicilina G potássica, procaína, bicarbonato de sódio (soluções alcalinas) e varfarina. Agentes oxidantes podem destruir a epinefrina.
t Este fármaco apresenta um número elevado de incompatibilidades, por isso recomenda-se consultar a literatura antes de misturá-lo na mesma solução contendo outros medicamentos e antes da administração em Y concomitantemente com outros fármacos.
Simone Sena Farina e Fernando de Sá Del Fiol
Na Rename 2010: item 5.2.2
t Comprimido de 400 mg.
t Solução oral 25 mg/mL.
t Tratamento de tuberculose quando há intolerância a rifampicina ou a isoniazida ou a pirazinamida.
t Hipersensibilidade ao etambutol.
t Neurite óptica.
t Pacientes incapazes de relatar alterações visuais, como idosos e menores de 5 anos de idade.
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– idosos.
– lactação.
– crianças abaixo de 13 anos.
t É recomendável realizar exame oftalmológico antes e durante o tratamento.
t Categoria de risco na gravidez (FDA): C.
t 20 a 35 kg: 400 a 800 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a isoniazida, pirazinamida e estreptomicina, seguido de 400 a 800 mg, por via oral, a cada 24 horas, durante 10 meses, combinada a isoniazida.
t 36 a 50 kg: 800 a 1.200 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a isoniazida, pirazinamida e estreptomicina, seguido de 800 a 1.200 mg, por via oral, a cada 24 horas, durante 10 meses, combinada a isoniazida.
t Acima de 50 kg: 1.200 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a isoniazida, pirazinamida e estreptomicina, seguido de 1.200 mg, por via oral, a cada 24 horas, durante 10 meses, combinada a isoniazida.
t 20 a 35 kg: 400 a 800 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a rifampicina, pirazinamida e estreptomicina, seguido de 400 a 800 mg, por via oral, a cada 24 horas, durante 4 meses, combinada a rifampicina.
t 36 a 50 kg: 800 a 1.200 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a rifampicina, pirazinamida e estreptomicina, seguido de 800 a 1.200 mg, por via oral, a cada 24 horas, durante 4 meses, combinada a rifampicina.
t Acima de 50 kg: 1.200 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a rifampicina, pirazinamida e estreptomicina, seguido de 1.200 mg, por via oral, a cada 24 horas, durante 4 meses, combinada a rifampicina.
t 20 a 35 kg: 400 a 800 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a rifampicina e isoniazida, seguido de 7 meses de rifampicina e isoniazida.
t 36 a 50 kg: 800 a 1.200 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a rifampicina e isoniazida, seguido de 7 meses de rifampicina e isoniazida.
t Acima de 50 kg: 1.200 mg, por via oral, a cada 24 horas, durante 2 meses, combinada a rifampicina e isoniazida, seguido de 7 meses de rifampicina e isoniazida.
t Pico de concentração plasmática: 2 a 4 horas.
t Meia-vida: 2,5 a 4 horas, 7 a 15 horas (insuficiência renal).
t Metabolismo: hepático (10 a 20%).
t Excreção: fecal (20% a 22% inalterada) e urinária (50 a 90%).
t Dialisável
t Neurite óptica (1 a 6%), com acuidade visual reduzida e troca entre as cores vermelho e verde (os sintomas recentes são geralmente reversíveis; a pronta retirada pode prevenir o sintoma de troca de cores).
t Neurite periférica, especialmente nas pernas.
t Hiperuricemia e desencadeamento de gota.
t Exantema, prurido, urticária, trombocitopenia
t Antiácidos contendo compostos de alumínio: podem diminuir as concentrações séricas de etambutol. Administrar com intervalo de pelo menos 4 horas.
t Alertar para notificar imediatamente ao perceber qualquer distúrbio visual.
t Orientar que pode ser tomado com alimento para diminuir irritação gástrica. t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses, sob risco de desenvolvimento de resistência bacteriana.
t Deve ser mantido em sua embalagem original, bem fechado, ao abrigo de luz e umidade e à temperatura de 15 a 30 ºC.
Atenção: como sinonímia para etambutol (nome correspondente à Denominação Comum Brasileira) também é empregada a abreviatura E, entretanto, não se recomenda a prescrição de fármacos por abreviaturas ou siglas.
Tatiana Aragão Figueiredo
Elaine Silva Miranda
t Cápsula ou comprimido 20 mg.
t Transtorno depressivo.
t Transtorno obsessivo compulsivo (TOC).
t Hipersensibilidade ao fármaco.
t Uso de inibidores da monoamina oxidase nos últimos 14 dias.
t Fase de mania da doença bipolar.
t Usar com cuidado nos casos de:
– insuficiência hepática (ver Apêndice C).
– lactação.
– epilepsia, doença cardíaca, distúrbios hemorrágicos, diabete melito, susceptibilidade ao glaucoma de ângulo fechado, histórico de mania, transtorno bipolar e pensamentos suicidas.
– eletrochoque concomitante (pode aumentar os riscos da terapia).
– uso concomitante de anti-inflamatórios não-esteroides, ácido acetilsalicílico ou outros fármacos que afetam a coagulação.
t Perigo ao dirigir veículo automotor ou ao realizar outras tarefas que exijam atenção e coordenação motora.
t Categoria de risco na gravidez (FDA): C.
t 20 mg/dia, por via oral, em dose única pela manhã ou à noite, por no mínimo 6 meses. Dose máxima: 80 mg/dia.
t Doses acima de 20 mg/dia devem ser divididas em tomadas matinais e noturnas.
t 40 a 60 mg/dia, por via oral.
t 10 mg/dia, por via oral, com aumento para 20 mg após várias semanas de uso. Dose máxima: 60 mg/dia.
t Absorção oral rápida.
t Latência de efeito antidepressivo: 2-3 semanas.
t Biotransformação no fígado, gerando o metabólito ativo norfluoxetina.
t Meia-vida de eliminação: 1-3 dias após dose única e 4-6 dias após utilização por longo período.
t Dispneia, cefaleias, distúrbios do sono, tonturas, ataxia, tremores, convulsões, alucinações, mania, confusão, agitação, ansiedade, ataques de pânico, ideação suicida, tonturas, ansiedade, arrepios, aumento da sudorese, convulsões, alucinações, confusão, agitação, efeitos extrapiramidais, palpitações.
t Efeitos cardiovasculares como bradiarritmia, insuficiência cardíaca, hipertensão, taquiarritmia.
t Insônia/sonolência, ansiedade.
t Náuseas, vômito, xerostomia, hemorragia gastrintestinal, estomatite, hemorragia digestiva, distúrbios gastrintestinais, anorexia com perda de peso.
t Hipotensão postural, retenção urinária, midríase, distúrbios visuais.
t Disfunção sexual, incluindo ejaculação precoce, anorgasmia, diminuição da libido, galactorreia, hipertrigliceridemia, hipoglicemia, hiponatremia, síndrome da secreção inapropriada de hormônio antidiurético.
t Alopecia, erupção cutânea, urticária, angioedema, fotossensibilidade, artralgia, mialgia.
t Vasculite, distúrbios hemorrágicos incluindo equimoses e púrpura.
t Alprazolam, antidepressivos tricíclicos, antipsicóticos, aripripazol, carbamazepina, clozapina, digitálicos, fenitoína, haloperidol, metoprolol, risperidona, trazodona: têm sua toxicidade aumentada. O uso concomitante não é recomendado.
t Anticoagulantes, antiplaquetários, clozapina, galantamina: têm seus efeitos potencializados. Monitorar sinais de sangramento ao usar fármacos que afetam a coagulação. Monitorar a toxicidade por galantamina, quando usada de forma concomitante: anorexia, náuseas, vômitos, tonturas, arritmias ou hemorragia digestiva.
t Anti-inflamatórios não-esteroides (AINE): aumento do risco de sangramento. Cuidado ao combinar fluoxetina com AINE; monitorar sinais de sangramento.
t Bepridil, ciclobenzaprina, cloroquina, clorpromazina, dolasetrona, droperidol, enflurano, eritromicina, espiramicina, fluconazol, gemifloxacino, halofantrina, halotano, hidroxiquinidina, isoflurano, isradipino, lidoflazina, mefloquina, mesoridazina, octreotida, pirmenol, probucol, proclorperazina, propafenona, quetiapina, quinidina, sematilida, sertindol, sotalol, sulfametoxazol, tedisamila, telitromicina, tioridiazina, trifluoperazina, trimetoprima, venlafaxina, ziprasidona: aumento no risco de cardiotoxicidade. O uso concomitante é contraindicado com bepridil, mesoridazina, tioridiazina, sendo em princípio desaconselhado com os demais, mas se a associação for necessária, deve-se ter cautela no uso.
t Bupropiona, delavirdina e furazolidona: aumento de efeito do antidepressivo. Monitorar as concentrações plasmáticas do antidepressivo para eventuais ajustes de doses. Acompanhar, entre os usuários da associação, os sinais e sintomas de toxicidade da fluoxetina, assim como os sinais de excesso serotoninérgicos (mudanças do estado mental, diaforese, febre, fraqueza, hiperreflexia, incoordenação).
t Ciproeptadina: redução do efeito do antidepressivo.
t Claritromicina: uso concomitante pode resultar em delírio e psicose, devendo ser evitado.
t Ergotamina e análogos: aumento do risco de ergotismo. O uso concomitante é contraindicado.
t Desvenlafaxina, dexfenfluramina, duloxetina, erva-de-são-joão (Hypericum perforatum), fenfluramina, Ginkgo biloba, linezolida, meperidina, milnaciprana, mirtazapina, sibutramina, tramadol, tranilcipromina, trazodona, triptanas: uso concomitante pode produzir síndrome serotoninérgica. O uso concomitante de fluoxetina com linezonida ou tranilcipromina é contraindicado. Após descontinuar o uso da erva-de-são-joão (Hypericum perforatum), esperar duas semanas antes de iniciar terapia com fluoxetina. No caso de uso concomitante, monitorar estreitamente os sintomas da síndrome.
t Flufenazina:riscoaumentadodeparkinsonismo.
t Hipoglicemiantes (como a insulina): redução acentuada dos níveis glicêmicos.
t Inibidores da monoamina oxidase (MAO): o uso concomitante é contraindicado, levando a toxicidade do sistema nervoso central ou síndrome serotoninérgica (hipertensão, hipertermia, mioclônus, alterações do estado mental). Esperar duas semanas após descontinuar o uso de um inibidor da MAO antes de iniciar terapia com fluoxetina. Adicionalmente, esperar no mínimo cinco semanas após a descontinuação da fluoxetina antes de iniciar terapia com inibidor da MAO.
t Lítio: pode resultar em aumento das concentrações de lítio e/ou do risco de síndrome serotoninérgica.
t Metoclopramida: aumento do risco de reações extrapiramidais ou síndrome neuréptica maligna. O uso concomitante é contraindicado.
t Pimozida: risco aumentado de bradicardia, sonolência e cardiotoxicidade. O uso concomitante é contraindicado.
t Ritonavir: pode haver alteração em funções cardíacas ou neurológicas.
t Orientar para não suspender o uso de maneira repentina.
t Alertar que podem ser necessárias quatro semanas ou mais para o início dos efeitos antidepressivos.
t Alertar para não fazer uso de bebidas alcoólicas durante o tratamento.
t Orientar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir.
t Orientar para mudança na frequência cardíaca e a levantar-se mais lentamente para evitar hipotensão ortostática.
t Armazenar em recipiente hermético, à temperatura ambiente, preferentemente entre 15 e 30 ºC. Proteger da luz.
Atenção: este fármaco apresenta um número elevado de Efeitos adversos e deve ser realizada uma pesquisa específica sobre este aspecto ao introduzir ou descontinuar este ou outros medicamentos no esquema terapêutico do paciente. Em pacientes com doença recidivante, a terapia por longos períodos (mínimo de 6 meses) deve ser considerada.
Pacientes devem ser monitorados quando encerrado o tratamento. É recomendada diminuição gradual da dose.
José Gilberto Pereira
t Pó para solução injetável 200 mg e 1 g
t Carcinoma de pâncreas, como primeira linha em carcinoma localmente avançado (estágios II ou III), metastático (estágio IV) ou pacientes previamente tratados com fluoruracila.
t Câncer metastático de mama, como primeira linha em pacientes previamente tratadas com esquema contendo antraciclinas ou pacientes apresentando contraindicações a antraciclinas.
t Câncer de pulmão de não-pequenas-células, como primeira linha em combinação com cisplatina para pacientes com câncer inoperável, localmente avançado (estágios IIIA ou IIIB) ou metastático.
t Câncer avançado de ovário, em combinação com carboplatina em pacientes que tiveram recaída no mínimo 6 meses após terapia com derivado de platina.
t Hipersensibilidade a gencitabina.
t Casos de síndrome hemolítico-urêmica e/ou insuficiência renal foram registrados; disfunção renal aumenta o risco de toxicidade, sendo recomendado monitorar o paciente.
t Pacientes com hepatite, alcoolismo, cirrose ou pacientes com metástase no fígado têm maior risco de toxicidade hepática, devendo ser monitorados.
t Monitorar sinais e sintomas de mielossupressão; redução ou retardo de doses pode ser necessário.
t Descontinuar em caso de toxicidade pulmonar grave.
t Risco de toxicidade aumentada se administrada 7 dias antes ou após radioterapia.
t Infusões de gencitabina com duração maior que 60 minutos aumentam o risco de toxicidade.
t Lactação: (ver Apêndice B).
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t 1 g/m2, por infusão intravenosa por 30 minutos, a cada 7 dias, durante 7 semanas. Interromper por uma semana e repetir a dose por mais 3 semanas. Se necessário, ciclos de 3 semanas podem ser repetidos sempre com intervalos de uma semana sem tratamento entre eles. Em cada ciclo a dose deve ser ajustada para mais (20% a 25%) ou para menos (25% a 50%) de acordo com a ocorrência ou não de granulocitopenia e/ou plaquetopenia.
t 1,25 g/m2, por infusão intravenosa por 30 minutos, nos dias 1 e 8 de cada ciclo de 3 semanas, combinada a paclitaxel 175 mg/m2, por infusão intravenosa por 3 horas, antes de gencitabina, no dia 1 de cada ciclo. Contagem de granulócitos e plaquetas deve preceder o tratamento e o início de cada novo ciclo. Ajuste de dose para menos (25% a 50%) deve ser realizado na ocorrência de granulocitopenia e/ou plaquetopenia.
t 1 g/m2, por infusão intravenosa por 30 minutos, nos dias 1, 8 e 15 de cada ciclo de 4 semanas, combinada a cisplatina 100 mg/m2, por via intravenosa, no dia 1, após gencitabina. Contagem de granulócitos e plaquetas deve preceder o tratamento e o início de cada novo ciclo. Ajuste de dose para menos (25% a 50%) deve ser realizado na ocorrência de granulocitopenia e/ou plaquetopenia.
t Ou então, 1,25 g/m2, por infusão intravenosa por 30 minutos, nos dias 1 e 8 de cada ciclo de 3 semanas, combinada a cisplatina 100 mg/m2, por via intravenosa, no dia 1, após gencitabina.
t 1 g/m2, por infusão intravenosa por 30 minutos, nos dias 1 e 8 de cada ciclo de 3 semanas, combinada a carboplatina com alvo de área sob a curva (AUC) de 4 mg/mL/min no dia 1, após a gencitabina. Nos ciclos subsequentes reduzir a dose de gencitabina para 800 mg/m2. Contagem de granulócitos e plaquetas deve preceder o tratamento e o início de cada novo ciclo. Ajuste de dose para menos (50%) deve ser realizado na ocorrência de granulocitopenia e/ou plaquetopenia.
t 1 g/m2, por infusão intravenosa por 30 a 60 minutos, nos dias 1, 8 e 15 de cada ciclo de 4 semanas, combinada a cisplatina 70 mg/m2, por via intravenosa, no dia 2. Repetir por até 6 ciclos. Contagem de granulócitos e plaquetas deve preceder o tratamento e o início de cada novo ciclo. Ajuste de dose para menos (25% a 50%) deve ser realizado na ocorrência de granulocitopenia e/ou plaquetopenia. Se radioterapia concorrente for requerida, administrar gencitabina 1 semana antes ou depois da radioterapia.
t Tempo para atingir concentração máxima após injeção intravenosa: 30 minutos.
t Área sob a curva (AUC) do metabólito ativo na forma de trifosfato aumenta em 4 vezes quando o tempo de infusão aumenta de 30 para 60 minutos.
t Ligação a proteínas plasmáticas não é significativa.
t Metabolismo intracelular, que produz metabólitos fosfatados ativos com efeito citotóxico.
t Excreção renal (92 a 98%); depuração plasmática é influenciada pelo gênero e idade, sendo até 25% menor nas mulheres jovens, quando comparada aos homens, e diminui até 60%, em ambos os sexos, com o avanço da idade.
t Meia-vida de eliminação é influenciada pelo gênero, idade e duração da infusão, sendo 20% maior nas mulheres idosas, quando comparada aos homens; aumenta até 50%, em ambos os sexos, com o avanço da idade e com o maior tempo de infusão.
t Edema periférico (20%), arritmia cardíaca (3%), raramente insuficiência cardíaca congestiva e enfarte agudo do miocárdio.
t Alopecia (15% a 90%), exantema (10% a 30%), raramente erupções bolhosas e gangrena.
t Hiperglicemia (30%), hipomagnesemia (30%).
t Obstipação (10% a 42%), diarreia (14% a 25%), náusea e vômito (64% a 96%), estomatite.
t Alterações hematológicas desde leves a graves: anemia (65% a 89%), leucopenia (21% a 86%), neutropenia (62% a 90%), trombocitopenia (24% a 85%) .
t Elevação das enzimas hepáticas (10% a 70%), raramente hepatotoxicidade e insuficiência hepática.
t Doenças infecciosas (8 a 28%), raramente anafilaxia e reações de hipersensibilidade.
t Artralgia (24%), mialgia (33%), fadiga (40%), febre (6% a 41%), dor (10% a 48%).
t Parestesias (2% a 38%), neuropatia motora periférica (15% a 35%), neuropatia sensorial (23% a 64%), distúrbio de audição (25%), síndrome leucoencefalopática posterior reversiva.
t Hematúria (13% a 35%), proteinúria (10% a 45%), aumento da creatinina sérica (2% a 38%), síndrome hemolítico-urêmica (0,25%), microangiopatia tromboembólica (0,015% a 0,4%), raramente insuficiência renal.
t Dispneia (1% to 23%), broncoespasmo (até 2%); raramente: síndrome aguda do desconforto respiratório, pneumonia intersticial, edema pulmonar, fibrose pulmonar, insuficiência respiratória.
t Vacina rotavírus: aumento do risco de infecção pelo vírus da vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pelo vírus da vacina. Se a vacina for necessária, administrar a vacina depois de três meses da descontinuação da quimioterapia.
t Varfarina: pode aumentar o risco de sangramento. Se o uso concomitante for necessário monitorar a coagulação sanguínea e proceder ao ajuste de dose da varfarina.
t Evitar vacinas sem conhecimento médico. Evitar contato com pessoas que tenham recebido vacinas com vacina oral contra poliomielite.
t Evitar contato com pessoas que apresentem infecções e alertar sobre a necessidade de evitar infecções, por meio de medidas higiênicas e protetoras, como lavar as mãos com frequência.
t Alertar sobre a ocorrência de Efeitos adversos comuns, como anorexia, diarreia, náuseas e vômito, febre, cefaleia, erosões bucais, urticária e exantema, perda dos cabelos.
t Orientar as pacientes para a utilização de métodos contraceptivos efetivos enquanto fazer uso de gencitabina. Em casos da ocorrência de gravidez, informar o médico.
t Cuidado ao escovar os dentes, passar fio dental ou palito de dentes, barbear-se ou cortar as unhas, evitando sangramentos.
t Evitar esportes de contato ou outras situações as quais pode ocorrer sangramento.
t Armazenar os frascos de gencitabina sob temperaturas entre 20 e 25 °C.
t Seguir protocolo local de manipulação e descarte de quimioterapia antineoplásica.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir, sob agitação, com cloreto de sódio 0,9%, livre de conservantes, até uma proporção de 40 mg/mL.
t Para administração, diluir em cloreto de sódio 0,9%, livre de conservantes, a uma concentração de 0,1 mg/mL. Permanece estável por 24 horas sob temperaturas entre 20 e 25 °C. Não conservar em refrigerador, pois há risco de cristalização.
t Soluções injetáveis de gencitabina são incompatíveis com vários medicamentos em misturas intravenosas. Para maiores detalhes verificar a literatura adequada ou protocolo local.
Rosa Martins
Na Rename 2010: item 14.4.4
t Comprimido 50 mg.
t Solução injetável 20 mg/mL.
t Hipertensão arterial sistêmica grave e refratária.
t Emergência hipertensiva.
t Pré-eclampsia grave e eclampsia.
t Hipersensibilidade a hidralazina.
t Taquicardia grave.
t Insuficiência miocárdica por obstrução mecânica.
t Cor pulmonale.
t Aneurisma aórtico dissecado.
t Doença reumática de valva mitral.
t Porfiria.
t Lupus eritematoso sistêmico.
t Insuficiência cardíaca grave.
t Usar com cuidado nos casos de:
– insuficiência hepática e acetiladores lentos (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– doença coronariana, doença cerebrovascular e doença da valva mitral.
– uso intravenoso (monitorar pressão arterial).
– lactação.
t Categoria de risco na gravidez (FDA): C. (ver Apêndice A).
t Dose inicial 25 mg, por via oral, a cada 12 horas, aumentado até 100 mg, por via oral, a cada 12 horas. Dose máxima diária: 200 mg.
t 5 a 10 mg, por via intravenosa lenta, diluído com 10 mL de soro fisiológico 0,9%, repetido a cada 20 a 30 minutos, se necessário.
t Dose inicial 200 a 300 microgramas/minuto, por infusão intravenosa. Dose de manutenção 50 a 150 microgramas/minuto.
t 12,5 mg, por via intramuscular, repetido a cada 2 horas, se necessário.
t 5 a 10 mg, por via intravenosa lenta, diluído com 10 mL de soro fisiológico 0,9%, repetido a cada 20 a 30 minutos, se necessário.
t 12,5 mg, por via intramuscular, repetido a cada 2 horas, se necessário.
t Biodisponibilidade: 22 a 50%, sendo menor em acetiladores lentos; o efeito de alimentos na biodisponibilidade é controverso.
t Início da ação: 1 hora (oral) e 5 a 15 minutos (intravenosa).
t Pico de concentração: 1 a 2 horas (oral).
t Duração de efeito: 3 a 8 horas (oral) e 1 a 4 horas (intravenosa).
t Metabolismo hepático; metabolismo de primeira passagem significativo, por acetilação. Diferença de resposta entre acetiladores rápidos e acetiladores lentos.
t Excreção: renal (3 a 14%), fezes (3 a 12%).
t Meia-vida de eliminação: 3 a 5 horas (função renal normal), superior a 16 horas (quando DCE for inferior a 20 mL/minuto).
t Efeitos da administração na gravidez: hipotensão materna, partos cesáreos, placenta prévia, baixos escores de Apgar.
t Hipotensão postural, exacerbação de angina, palpitações, taquicardia.
t Anorexia, náusea, vômito, diarreia.
t Cefaleia grave, neuropatia periférica (normalmente reversiva com suplemento de 100 a 200 mg de piridoxina)
t Indução de lúpus eritematoso sistêmico (74%), combinada a doses acima de 100 mg/dia, acetiladores lentos e alteração da função renal.
t Discrasias sanguíneas (raras).
t Exantema (raro).
t Metoprolol, oxprenolol, propranolol: podem ter seus efeitos tóxicos exacerbados, especialmente após administração em jejum. Se o uso concomitante for requerido, preferir betabloqueador de liberação sustentada ou ingesta com alimentos. Monitorar cuidadosamente pressão arterial.
t Alertar que pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora como operar máquinas e dirigir, pelo risco de produzir tontura.
t Alertar para a importância de monitorizar regularmente a pressão arterial.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar os comprimidos e ampolas entre 15 a 30 ºC e protegidos de luz.
Ampolas não devem ser congeladas.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t É estável em veículos como manitol e sorbitol por 21 dias.
t Incompatível com: glicose, frutose, lactose e maltose.
t A adição de fosfato e citrato à solução não tem efeito sobre a estabilidade do veículo.
t Pode haver mudança de cor em solução, o que não indica perda da eficácia.
t A solução injetável deve ser utilizada imediatamente após a preparação.
Luciana dos Santos
Na Rename 2010: itens 11 e 15.1
t Solução injetável de 1 mg/mL.
t Anemia megaloblástica na deficiência de hidroxocobalamina (vitamina B12) (anemia perniciosa).
t Hipersensibilidade ao cobalto, outras cobalaminas e a outros componentes da formulação.
t Usar com cuidado nos casos de:
– hipersensibilidade aos componentes (pode ser necessária uma dose-teste devido ao risco de reações anafiláticas).
– crianças (não recomendado o uso prolongado).
– fotossensibilidade (risco aumentado pelo uso da hidroxocobalamina).
– infusão intravenosa (pode aumentar a pressão arterial).
t Não deve ser administrado antes de diagnóstico estabelecido de deficiência de vitamina B12.
t Monitorar potássio sérico durante as primeiras 48 horas. A reposição do eletrólito, por vezes, é necessária pelo risco de arritmias secundárias.
t Categoria de risco na gravidez (FDA): C.
t 1 mg, por via intramuscular, 3 vezes por semana, durante 2 semanas, seguido por 1 mg, a cada 3 meses.
t 1 mg, por via intramuscular, em dias alternados, até não se observar melhora adicional, seguido por 1 mg a cada 2 meses.
t Dose de 1 mg, por via intramuscular, a cada 2 ou 3 meses.
t Tempo para pico de concentração: 2 horas.
t Duração de efeito: 1-3 meses, após dose única ou 6 meses a 5 anos, após múltiplas doses.
t Meia-vida de eliminação: 26 a 31 horas.
t Metabolismo: fígado (90%), mas também renal e suprarrenal.
t Excreção: renal (50%-70%).
t Aumento transitório na pressão arterial (18% a 28%), angioedema.
t Eritema (94% a 100%), exantema (20% a 44%), prurido e reações no lugar da injeção (6% a 39%), fotossensibilidade.
t Náusea (6% a 11%), diarreia, dispepsia e dor abdominal.
t Linfopenia (8% a 17%)
t Cefaleia (6% a 33%), sonolência.
t Coloração avermelhada na urina (100%).
t Anafilaxia, desenvolvimento de anticorpos
t Hipopotassemia
t Irritação, hiperemia e edema ocular
t Orientar para o uso de protetor solar e óculos. Evitar a exposição solar nas primeiras 2 semanas após o tratamento.
t Evitar o uso de álcool.
t Informar que a urina e as membranas poderão ficar vermelhas após o uso da vitamina.
t Não pode ser administrado por via subcutânea e intravenosa.
t Armazenar o medicamento à temperatura ambiente (entre 15 e 30 °C), proteger da luz.
Maurício Fábio Gomes
Na Rename 2010: itens 6.1.4
t Pó para solução injetável 10 mg
t Leucemia mieloide aguda (em combinação com outros agentes antileucêmicos).
t Hipersensibilidade a idarrubicina ou outras antraciclinas.
t Marcada mielossupressão prévia por quimioterapia ou radioterapia.
t Bilirrubina superior a 5 mg/dL.
t Uso concomitante com a vacina rotavírus humano G1P1[8] (atenuada).
t Usar com cuidado nos casos de:
– pacientes com arritmias, insuficiência cardíaca congestiva, cardiomiopatia induzida por fármacos, hemorragias, doenças infecciosas, mielossupressão.
– idosos e crianças com menos de 2 anos (requerem cuidadoso monitoria pelo risco de ocorrência de efeitos cardíacos).
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– pacientes leucêmicos (pode causar nefropatia urêmica).
– mucosite grave (requer maior espaçamento entre administrações ou redução da dose de idarrubicina em 25%).
– lactação.
t Monitorar a contagem sanguínea total periodicamente, assim como a função cardíaca, hepática e renal.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t Terapia de indução: 12 mg/m2/dia, por via intravenosa lenta (10 a 15 minutos), por 3 dias, associado ou não a citarabina (100 mg/m2/dia, em infusão intravenosa contínua por 7 dias ou 25 mg/m2, em injeção intravenosa em bolo, seguida por 200 mg/m2/dia, em infusão intravenosa contínua, por 5 dias).
t Alternativamente, 8 mg/m2/dia, por via intravenosa lenta (10 a 15 minutos), por 5 dias; monoterapia ou em combinação com outros agentes.
t Terapia de consolidação, em combinação com outros agentes antileucêmicos: 10 a 12 mg/m2/dia, por via intravenosa, por 2 dias.
t Dose de 8 a 10 mg/m2/dia, por via intravenosa, por 3 dias, a cada 3 semanas.
Notas:
t Não deve ser administradas por vias intramuscular ou subcutânea.
t A administração intravenosa deve ser feita lentamente, acoplada a sistema de infusão rápida contendo cloreto de sódio 0,9% ou glicose 5%.
t Evitar extravasamento devido a risco de ulceração grave e necrólise local.
t Metabolismo: hepático.
t Excreção: biliar (8% por via oral e 17% por via intravenosa), renal (16%).
t Meia-vida de eliminação: 22 horas (em monoterapia) e 20 horas (em combinação com outros agentes), 45 horas (metabólito ativo).
t Alterações transitórias eletrocardiográficas, geralmente assintomáticas.
t Alopecia (25-30%), erupções cutâneas (11%), urticária.
t Diarreia (9-22%), náusea (86%), vômitos, estomatite (11%), mucosite, hemorragia gastrintestinal (30%).
t Cefaleia.
t Arritmia cardíaca, dor torácica, insuficiência cardíaca congestiva, enfarte do miocárdio.
t Mielossupressão, anemia (oral 34% e intravenoso 100%), leucopenia (86%).
t Hepatotoxicidade (<5%).
t Nefrotoxicidade (1%), coloração amarelo-escuro da urina.
t Doenças infecciosas.
t Hiperuricemia.
t Atrofia testicular (infertilidade).
t Necrose local ao extravasamento.
t Trastuzumabe: aumento do risco de disfunção cardíaca. É fortemente recomendado que trastuzumabe seja descontinuado durante o uso de idarrubicina.
t Vacina rotavírus humano G1P1[8] (atenuada): aumento do risco de infecção pelo vírus da vacina. O uso concomitante é contraindicado.
t Vacinas com vírus vivos: aumento do risco de infecção pelo vírus da vacina. Se a vacina for necessária, administrar a vacina depois de três meses da descontinuação da quimioterapia.
t Orientar para aumentar a ingestão de líquidos.
t Utilizar contracepção efetiva durante o tratamento, homens e mulheres.
t Orientar para evitar vacinas, especialmente contra poliovírus, e contato com pessoas próximas que recebera a vacina. Se o contato for inevitável, usar máscara de proteção.
t Orientar para evitar contato com pessoas com infecção, especialmente durante os períodos de baixas contagens sanguíneas. Notificar imediatamente se ocorrer sinais ou sintomas de mielossupressão e outras reações adversas.
t Armazenar à temperatura ambiente, entre 15 e 30oC. Proteger da luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir o pó em solução de cloreto de sódio 0,9%, glicose a 5%, solução glicofisiológica 3,3% ou solução de Ringer + lactato. Reconstituir em soluções alcalinas resulta em degradação de idarrubicina. Soluções reconstituídas são estáveis por 7 dias sob refrigeração (2 a 8 oC) e por 72 horas sob temperatura ambiente (15 a 30oC).
t É incompatível com heparina. Verificar literatura específica para outras incompatiblidades.
t Observar protocolos locais para manipulação de substâncias citotóxicas.
Atenção: associa-se a cardiotoxicidade, levando à insuficiência cardíaca congestiva. Este risco é maior em pacientes com cardiopatias preexistentes.
César Augusto Braum
Na Rename 2010: itens 1.2, 14.2, 20.1
t Solução injetável 1% e 2%
t Gel 2%.
t Aerossol 100 mg/mL
t Anestesia local tópica de membranas mucosas.
t Anestesia local infiltrativa.
t Anestesia local regional (bloqueio de nervos periféricos).
t Arritmias.
t Hipersensibilidade à lidocaína ou a qualquer anestésico do tipo amida.
t Infecções cutâneas adjacentes ao sítio de administração.
t Anemia grave.
t Uso concomitante com anticoagulantes.
t Anestesia espinhal ou epidural em pacientes desidratados ou hipovolêmicos.
t Doença cardíaca.
t Síndrome de Adam-Stokes.
t Usar com cuidado nos casos de:
– pacientes com diminuição na condução cardíaca
– idosos.
– hipertensão grave em anestesia epidural caudal e lombar.
– toxicidade no SNC (monitorar sinais e sintomas).
– anestesia espinhal, epidural, caudal ou regional intravenosa (não utilizar soluções contendo preservativos).
– apresentações tópicas (não devem ser utilizadas em grande área corpórea por mais de 2 horas).
– síndrome de Wolff-Parkinson-White.
– lactação.
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
t Categoria de risco na gravidez (FDA): B (ver Apêndice A).
t Corrigir alterações de eletrólitos, principalmente hipopotassemia e hipomagnesemia, antes e durante o uso de lidocaína.
t Corrigir causas subjacentes de arritmia ventricular.
t bloqueio de nervo simpático cervical e lombar, bloqueio periférico braquial, bloqueio periférico intercostal, bloqueio periférico paravertebral, bloqueio periférico pudendal, anestesia local por infiltração, sequência rápida de intubação (pré-indução) em maiores de 3 anos: 3,3 a 4,4 mg/kg.
t Indução da anestesia intravenosa regional: 3 mg/kg de solução a 1%.
t Anestesia local de mucosas (maiores de 3 anos): 3,3 a 4,4 mg/kg de gel 2%, a cada 3 horas, para anestesia de boca e faringe. Máximo de 8 doses ao dia.
t dose inicial 1 mg/kg (máximo de 100 mg), por infusão intravenosa direta. Dose de manutenção: 30 microgramas/kg/minuto (20 a 50 microgramas), por infusão intravenosa contínua.
t Anestesia local percutânea: 0,5 a 30 mL (5 a 300 mg) de solução a 1%, por infiltração.
t Anestesia local para bloqueio regional (bloqueio de nervos periféricos): 10 a 60 mL (50 a 300 mg) de solução a 0,5%, por via intravenosa.
t Bloqueio do nervo braquial: 15 a 20 mL (225 a 300 mg) de solução a 1,5%.
t Bloqueio do nervo intercostal: 3 mL (30 mg) de solução a 1%.
t Bloqueio do nervo pudendo (cada lado) e nervo paracervical (cada lado) para analgesia obstétrica: 10 mL (100 mg) de solução a 1%. No caso de bloqueio do nervo paracervical, a dose não pode ser repetida antes de 90 minutos.
t Bloqueio de nervo simpático cervical: 5 mL (50 mg) de solução a 1%.
t Bloqueio de nervo simpático lombar: 5 a 10 mL (50 a 100 mg) de solução a 1%.
t Bloqueio de nervo paravertebral: 3 a 5 mL (30 a 50 mg) de solução a 1%.
t Sequência rápida de intubação, pré-indução: 1,5 mg/kg, por via intravenosa, 2 a 3 minutos antes da intubação.
t Exame uretral feminino: 60 a 100 mg de gel a 2% dentro da uretra minutos antes do exame.
t Exame uretral masculino: 300 mg de gel a 2% pela uretra. Dose adicional de 300 mg pode ser utilizada para uma anestesia adequada.
t Cateterização uretral em homens: 100 a 200 mg de gel a 2% antes de realizar o procedimento.
t Sondagem e cistoscopia: 600 mg de gel a 2% antes de realizar o procedimento.
t Aftas em imunodeprimidos: 40 a 60 mg de gel a 2%. Dose máxima diária: 300 mg.
t Anestesia de mucosas bucais durante procedimentos: 10 a 50 mg de aerossol a 10%. Não exceder 30 mg de lidocaína por quadrante de gengiva ou mucosa oral no período de 1 hora e meia e de 200 mg em 24 horas.
t Cirurgia de catarata: aplicar o gel a 2%, 3 a 5 vezes, 15 a 20 minutos antes da cirurgia.
t Dose máxima para soluções injetáveis: 4 mg/kg ou 300 mg.
t Dose incial 50 a 100 mg (0,7 a 1,4 mg/kg), por via intravenosa por 2 a 3 minutos, pode-se repetir em 5 minutos até 300 mg num período de 1 hora. Dose de manutenção 1 a 4 mg/minuto, por infusão intravenosa contínua.
t 300 mg, por via intramuscular. Repetir, caso necessário, após 60 a 90 minutos.
t Fibrilação ventricular: dose inicial 1 a 1,5 mg/kg, por via intravenosa. Se necessário, doses adicionais de 0,5 a 0,75 mg/kg, a cada 5 a 10 minutos. Dose máxima: 3 mg/kg.
t Início de efeito: 45 a 90 segundos (intravenosa).
t Pico de concentração plasmática: 10 a 30 minutos (intravenosa), 30 minutos a 2 horas (intramuscular).
t Duração de efeito: 10 a 20 minutos para bloqueio antiarritmico (intravenoso), 100 minutos (bloqueio lombar epidural com solução a 2%), 75 a 135 minutos (bloqueio caudal com soluções a 1% ou 2%), 100 minutos (anestesia espinhal com solução a 2%), 60 a 180 minutos (bloqueio epidural), 30 a 60 minutos (infiltrativa).
t Metabolismo: fígado (aproximadamente 90%)
t Meia-vida: 1 a 2 horas.
t Excreção: renal (10% em forma inalterada).
t Início de efeito: 3-5 minutos (gel a 2%).
t A absorção por mucosas e pele lesada é rápida. Já a absorção por pele íntegra é lenta e incompleta.
t Hipotensão, bloqueio cardíaco, parada cardíaca, bradicardia.
t Náusea, vômitos, incontinência fecal.
t Hipersensibilidade e reações alérgicas (raras).
t Parada respiratória.
t Tontura, distúrbios visuais, tremores, cansaço, convulsões, sonolência, inconsciência, agitação, cefaleia, parestesias, sensação de frio ou calor, paraplegia (acidente raro).
t Retenção urinária.
t Antiarrítmicos (amiodarona, tocainida, disopiramida, mexiletina, procainamida): o uso concomitante pode potencializar Efeitos adversos cardíacos. Monitorar pacientes, especialmente idosos. Ajuste na dose da lidocaina pode ser necessário.
t Betabloqueadores sistêmicos (metoprolol, nadolol, propranolol, pembutolol): Aumento no risco de toxicidade da lidocaína (ansiedade, depressão miocárdica), com pembutolol pode ocorrer aumento do volume de distribuição e aumento do tempo de meia-vida da lidocaína. Monitorar os níveis de lidocaína e ajustar as taxas de infusão.
t Bloqueadores neuromusculares (suxametônio = succinilcolina) pode resultar em toxicidade (depressão respiratória, apneia). Monitorar cuidadosamente.
t Cimetidina: aumento no risco de toxicidade da lidocaina (neurotoxicidade, arritmias cardíacas). Monitorar os níveis de lidocaína e ajustar dose, ou substituir a cimetidina por ranitidina ou famotidina, com menor potencial de alterar o metabolismo da lidocaina.
t Fenitoína: pode resultar em efeito aditivo na depressão cardíaca. Monitorar o paciente e evitar em pacientes com doença cardíaca conhecida.
t Inibidores da protease (amprenavir, atazanavir, ritonavir, indinavir): Aumento da concentração plasmática da lidocaína e potencial toxicidade (hipotensão, arritmias). Monitorar o paciente e os níveis plasmáticos de lidocaína.
t Propofol: pode haver aumento dos efeitos hipnóticos. Pode ser necessário ajustar a dose do propofol.
t Orientar para evitar o contato de anestésico tópico com os olhos. Caso o contato ocorra, lavar os olhos imediatamente com água e proteger até que a sensibilidade retorne.
t Evitar a aplicação tópica de grande quantidade e em grande número de vezes do medicamento.
t Armazenar as soluções sob temperatura ambiente, entre 15 a 30 ºC. O congelamento deve ser evitado.
t Armazenar o gel de lidocaína 2% sob temperatura ambiente, entre 15 e 30 ºC, ao abrigo de luz e umidade.
t Inspecionar visualmente para a presença de qualquer material particulado ou coloração antes da administração.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Lidocaína é incompatível com soluções contendo anfotericina B, sulfadiazina sódica, cefazolina sódica ou fenitoína sódica, soluções alcalinas.
César Augusto Braum
Na Rename 2010: item 1.2
t Solução injetável 5% + 7,5%
t Anestesia espinhal.
t Anestesia espinhal contínua.
t Ver demais contraindicações na monografia da lidocaína.
t Categoria de risco na gravidez (FDA): B.
t Ver demais precauções na monografia da lidocaína.
t até 5 kg: 2,5 mg/kg.
t entre 5 a 15 kg: 2 mg/kg. t mais de 15 kg: 1,5 mg/kg.
t Obstétrica (parto normal): 1 mL (50 mg).
t Obstétrica (cesariana): 1,5 mL (75 mg).
t Cirurgia abdominal: 1,5 a 2 mL (75 a 100 mg).
t Ver monografia do cloridrato de lidocaína.
t Ver monografia do cloridrato de lidocaína.
t Ver monografia do cloridrato de lidocaína.
t Soluções do cloridrato de lidocaína com glicose podem ser autoclavadas uma única vez. A caramelização da glicose pode ocorrer após mais de uma autoclavagem.
t As quantidades não utilizadas da solução devem ser desprezadas devido à ausência de conservantes.
t Ver demais Efeitos adversos na monografia do cloridrato de lidocaína.
César Augusto Braum
Na Rename 2010: item 1.2
t Solução injetável 1% + 1:200.000
t Solução injetável 2% + 1:200.000
t Solução injetável 2% + 1:80.000 (uso odontológico).
t Anestesia infiltrativa.
t Bloqueio nervoso periférico e simpático.
t Anestesia em procedimentos odontológicos.
t Anestesia em sítios com limitada circulação colateral (dedos, orelhas, nariz ou pênis).
t Ver demais contraindicações na monografia da lidocaína (página 565).
t Usar com cuidado nos casos de:
t idosos, hipertensos, diabéticos não-controlados, crianças, pacientes enfraquecidos, pacientes com arritmias graves, hipertireoidismo, doença vascular periférica e com epilepsia (ajustar a dose).
t anestesia epidural ou caudal (não utilizar preparações com preservativo).
t Categoria de risco na gravidez (FDA): C (ver Apêndice A, em lidocaína). t Ver demais precauções na monografia da lidocaína (página 565).
t Até 7 mg/kg de solução a 1% de lidocaína com epinefrina 1:200.000.
t Até 10 anos: de 1 a 1,5 mL (20 a 30 mg) de solução a 2% de lidocaína com epinefrina 1:80.000. Dose máxima: 4 a 5 mg/kg de lidocaina ou 100 a 150 mg em dose única.
t Mais de 10 anos: até 6,6 mg/kg ou 300 mg de lidocaína e 0,003 mg/kg de epinefrina/kg.
t Até 40 mL (até 400 mg) de solução a 1% de lidocaína.
t 1 a 5 mL (20 a 30 mg) de solução a 2% de lidocaína com epinefrina 1:80.000. Dose máxima: 7 mg/kg ou 500 mg.
t Durante a anestesia epidural contínua a presença de epinefrina reduz ligeiramente as concentrações plasmáticas da lidocaína e de um dos metabólitos da lidocaína.
t A redução do pico de concentração da lidocaína pela epinefrina depende do lugar da injeção.
t A presença de epinefrina em anestésicos locais reduz a velocidade de absorção e consequentemente prolonga o tempo de ação.
t Quando a epinefrina for utilizada em anestesia de infiltração pode ocorrer redução do sangramento no campo de operação.
t Ver demais informações na monografia da lidocaína.
t A toxicidade sistêmica da lidocaína é reduzida com a presença da epinefrina.
t Para outros Efeitos adversos ver monografia do cloridrato de lidocaína.
t Amiodarona, atazanavir e ritonavir: aumento da toxicidade da lidocaína.
t Antidepressivos tricíclicos podem resultar em hipertensão, taquicardia e arritmias. Evitar administração concomitante ou reduzir a dose do simpaticomimético.
t Betabloqueadores (propranolol, pindolol, nadolol, timolol, carvedilol etc.): uso concomitante com epinefrina pode resultar em hipertensão, bradicardia, resistência à epinefrina e anafilaxia. Evitar uso concomitante. Se for necessário o uso, monitorar cuidadosamente pressão arterial.
t Di-hidroergotamina, fenelzina, isocarboxazida e linezolida: aumento do risco de hipertensão. O uso concomitante com lidocaína é contraindicado.
t Enflurano ou halotano: aumento do risco de arritmia ventricular devido associação com epinefrina. Monitorar arritmias.
t Entacapona: aumento do risco de taquicardia, hipertensão e arritmias. Monitorar o paciente.
t Sulfato de morfina lipossomal: o uso concomitante com lidocaína + epinefrina (epidural) pode causar aumento do pico de concentração da morfina. Aguardar 15 minutos após administração da lidocaína + epinefrina pela via epidural para administrar o sulfato de morfina lipossomal.
t Suxametônio (succinilcolina): pode haver aumento em seu efeito/toxicidade (depressão respiratória, apneia). Monitorar cuidadosamente para prolongamento do bloqueio muscular e depressão respiratória.
t Para outras interações importantes, ver monografia do cloridrato de lidocaína.
t Ver monografia do cloridrato de lidocaína.
t As soluções contendo lidocaína e epinefrina não podem ser autoclavadas.
t As porções de solução não utilizadas devem ser descartadas.
t Ver demais Efeitos adversos na monografia do cloridrato de lidocaína.
Letícia Figueira Freitas
Elaine Silva Miranda
Na Rename 2010: item 5.6.2.2
t Comprimido 250 mg.
t Segunda escolha para quimioprofilaxia em viajantes que visitarão regiões de alto risco de transmissão de Plasmodium falciparum na Amazônia Legal, que permanecerão na região por tempo maior que o período de incubação da doença (e com duração inferior a seis meses) e em locais cujo acesso ao diagnóstico e tratamento de malária estejam a mais de 24 horas.
t Histórico de doenças neuropsiquiátricas, incluindo depressão, distúrbios de ansiedade e convulsão.
t Problemas mentais ou emocionais.
t Hipersensibilidade à mefloquina, quinina ou quinidina.
t Tratamento recente com quinina.
t Arritmia cardíaca.
t Gestante no primeiro trimestre (ver Apêndice A).
t Usar com cuidado nos casos de:
– distúrbios da condução cardíaca.
– insuficiência hepática grave (ver Apêndice C).
– epilepsia.
– crianças com menos de 6 meses de idade (uso não recomendado)
t 250 mg, por via oral, a cada 7 dias, iniciado 1 semana (preferentemente 2 a 3 semanas) antes da viagem e mantido até 4 semanas após o retorno.
t Biodisponibilidade: 85%, aumentada pela ingestão de água ou alimento antes da administração.
t Pico de concentração plasmática: 2 a 24 horas.
t Ligação às proteínas plasmáticas: 98%. t Volume de distribuição: 13 a 29 L/kg. t Metabolismo: hepático.
t Meia-vida de eliminação: em crianças é mais curta que em adultos, nos quais varia de 10 a 40 dias (21 dias em média).
t Excreção: principalmente fecal, podendo ser também biliar e renal (concentrações subterapêuticas podem persistir no sangue por algumas semanas).
t Náuseas, vômitos, dor abdominal, diarreia, anorexia.
t Sonolência, insônia e sonhos anormais.
t Exantema, prurido, alopecia, urticária, eritema multiforme. Raramente: síndrome de Stevens-Johnson, necrólise epidérmica tóxica, vasculite cutânea.
t Dispneia.
t Anormalidades no ECG; distúrbios circulatórios, taquicardia, bradicardia, hipotensão ortostática, síncope, edema, angina. Raramente: bloqueio atrioventricular.
t Trombocitopenia, leucopenia, leucocitose, agranulocitose, anemia hemolítica.
t Hepatite aguda, esteatose hepática.
t Manifestações neuropsiquiátricas graves (efeito frequente), incluindo cefaleia, vertigem, dificuldade de equilíbrio, encefalopatia, neuropatias motoras e sensoriais, astenia, distúrbios visuais, ansiedade, depressão, ideias suicidas, alucinações, delírio, instabilidade emocional, psicoses, convulsões, confusão, ataques de pânico, agressão, agitação.
t Fraqueza muscular, mialgia, artralgia.
t Alterações em testes de função hepática.
t Ácido valproico: perda de controle de ataque apoplético.
t Anlodipino, felodipino, isradipino, lacidipino, lercanidipino, nicardipino, nifedipino e nimodipino podem aumentar o risco de bradicardia.
t Atomoxetina: aumento do risco de arritmias ventriculares.
t Aurotioglicose: aumento do risco de discrasias sanguíneas/efeito aditivo. Contraindicado o uso concomitante.
t Bepridil, cisaprida, gemifloxacino, halofantrina, isradipino, mesoridazina, pimozida, terfenadina, tioridazina, ziprasidona: pode haver aumento do risco de cardiotoxicidade/efeito aditivo do prolongamento do intervalo QT, torsades de pointes, parada cardíaca. Contraindicado o uso concomitante.
t Betabloqueadores (ver também abaixo, propranolol): aumento do risco de bradicardia.
t Cetoconazol: aumento da concentração plasmática de mefloquina/inibição da isoenzima CYP3A4. Diminuir a dose de mefloquina.
t Digoxina: pode aumentar o risco de bradicardia.
t Etossuximida: antagoniza o efeito anticonvulsivante.
t Ivabradina e moxifloxacino: podem aumentar o risco de arritmias ventriculares.
t Lumefantrina: redução da concentração plasmática de lumefantrina/diminuição da absorção de lumefantrina devido à diminuição da produção de bile. Ingerir o medicamento com alimentos e monitorar redução da eficácia.
t Propranolol: aumento do risco de anormalidades no eletrocardiograma (ECG) e parada cardíaca.
t Quinina: aumento do risco de convulsões, anormalidades no eletrocardiograma, parada cardíaca e diminuição da eficácia da mefloquina. Mefloquina deve ser administrada 12 horas após última dose de quinina.
t Rifampicina: diminuição da concentração plasmática de mefloquina/indução da CYP3A4.
t Orientar o paciente ou cuidador, criteriosamente, sobre a forma de fracionamento de doses, tendo em vista a forma farmacêutica disponível.
t Orientar para tomar água antes da administração do medicamento e ingerí-lo durante ou logo após a refeição, também com bastante água.
t Pode ocorrer vômito após ingestão. Se o vômito ocorrer dentro de 30 minutos após ingestão do medicamento, deve-se readministrar a mesma dose. Se o vômito ocorrer após 30 a 60 minutos, ingerir meia dose e prosseguir com as doses programadas.
t Orientar que o comprimido pode ser triturado e misturado com pequena quantidade de comida, leite e água, para ser administrado para pacientes com dificuldade de deglutição.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC, ao abrigo de luz, calor e umidade.
Atenção: no Brasil, onde a malária tem baixa incidência e há predomínio de Plasmodium vivax em toda a área endêmica, deve-se lembrar que a eficácia da profilaxia para essa espécie de Plasmodium é baixa, não devendo ser recomendada. É importante considerar que o uso isolado de mefloquina propicia o desenvolvimento de resistência bacteriana.
Karen Luise Lang
Na Rename 2010: item 18.3
t Comprimido 500 mg e 850 mg.
t Diabete melito tipo 2 em pacientes obesos.
t Cetoacidose.
t Insuficiência renal (ver Apêndice D).
t Administração concomitante com contrastes radiológicos iodados.
t Anestesia geral.
t Alcoolismo.
t Hipersensibilidade à metformina.
t Diabete gestacional.
t Usar com cuidado nos casos de:
– insuficiência renal (avaliar a função renal antes do início do tratamento e uma a duas vezes durante o ano – ver Apêndice D).
– idosos, especialmente acima de 80 anos (é maior o risco de acidose lática pela redução da função renal).
– ingestão excessiva de álcool, distúrbios hepáticos (ver Apêndice C), hipoxemia, desidratação e septicemia (condições que elevam o risco de acidose lática).
– durante infecções, cirurgias ou traumas (substituir por insulina).
– lactação.
t Pode haver redução da absorção de vitamina B12.
t Categoria de risco na gravidez (FDA): B.
t Dose inicial: 500 mg, duas vezes ao dia, ao desjejum e ao jantar, ou 850 mg, uma vez ao dia. Se necessário, elevar a dose semanalmente, com inclusão de um comprimido, até que se obtenha controle dos níveis de glicose sanguínea ou até que se atinja a dose máxima recomendada de 2.550 mg/dia, fracionada em três administrações (café da manhã, almoço e jantar).
t Biodisponibilidade: 50% a 60%. A absorção é retardada na presença de alimentos no estômago.
t Meia-vida: aproximadamente 6 horas.
t Pico de ação: 1 a 3 horas.
t Efeito máximo: 2 semanas.
t Eliminação: 90% renal.
t Dialisável.
t Sabor metálico, diarreia, flatulência, dor abdominal, indigestão, náuseas, vômitos.
t Anorexia, astenia, fotossensibilidade.
t Acidose lática.
t Hepatotoxicidade.
t Eritema, prurido, urticária.
t Discrasias sanguíneas.
t Bloqueadores beta-adrenérgicos: podem alterar o metabolismo glicêmico causando hiperglicemia, hipoglicemia e hipertensão. Se a associação for necessária, monitorar glicose sanguínea periodicamente. Bloqueadores cardioprevalentes oferecem menor risco de distúrbios glicêmicos e de mascaramento dos sintomas de hipoglicemia.
t Cefalexina e cimetidina: podem elevar as concentrações plasmáticas de metformina pela inibição de sua excreção tubular. Monitorar o surgimento de efeitos adversos associados ao cloridrato de metformina e avaliar redução da dose.
t Ciprofloxacino, outras fluoroquinolonas: alteração do metabolismo da glicose, com hipoglicemia ou hiperglicemia. Se a associação for necessária, monitorar glicose sanguínea periodicamente. Avaliar redução da dose de cloridrato de metformina.
t Contrastes radiológicos iodados: risco de acidose lática e falência renal aguda. O uso simultâneo é contraindicado. Interromper tratamento se houver necessidade de exames radiológicos com administração intravenosa de contrastes radiológicos iodados; restabelecer tratamento após normalização da função renal.
t Enalapril: pode causar acidose lática e hiperpotassemia. Evitar uso simultâneo em pacientes com insuficiência renal.
t Glucomanano: risco de redução da absorção do cloridrato de metformina. Administrar os medicamentos em diferentes períodos do dia.
t Inibidores de monoamina oxidase (IMAO): podem estimular secreção de insulina provocando hipoglicemia, depressão do sistema nervoso central e vertigens. Monitorar glicose sanguínea quando um IMAO for adicionado ou retirado da terapia. Avaliar redução da dose de cloridrato de metformina.
t Plantas como Psyllium (nome utilizado em inglês para designar algumas espécies do gênero Plantago; no Brasil, espécies deste gênero são conhecidas como tansagem), melão-de-são-caetano (Momordica charantia), erva-desão-joão (Hypericum perforatum) e feno-grego (Trigonella foenum-graecum) ou produtos derivados: podem aumentar o risco de hipoglicemia. Se a associação for necessária, monitorar glicose sanguínea periodicamente.
t Topiramato: pode alterar a biotransformação de ambos os fármacos. Monitorar glicose sanguínea quando o topiramato for adicionado ou retirado da terapia. Avaliar redução da dose de cloridrato de metformina.
t Orientar para administrar com alimentos para reduzir os sintomas gastrintestinais. Aumentar a ingestão de água.
t Reforçar a necessidade de evitar a ingestão de bebida alcoólica.
t Ensinar a reconhecer sintomas de acidose lática, como diarreia, hiperventilação, dores ou cãibras musculares, sonolência e cansaço.
t Manter ao abrigo de ar e luz e à temperatura ambiente, de 15 a 30 ºC.
Fabiana Wahl Hennigen
Na Rename 2010: item 16.4
t Comprimido 10 mg
t Solução oral 4 mg/mL
t Solução injetável 5 mg/mL
t Náusea e vômito associados a quimioterapia ou no pós-cirúrgico, doença do refluxo gastresofágico e estase da gastroparesia diabética.
t Hipersensibilidade à metoclopramida.
t Hemorragia, obstrução ou perfuração gastrintestinal.
t Feocromocitoma.
t Epilepsia e outros distúrbios convulsivos.
t Três a quatro dias após cirurgia gastrintestinal.
t Uso concomitante de fármacos com efeitos extrapiramidais, como fenotiazinas.
t Usar com cuidado nos casos de:
– doença de Parkinson, comprometimento da habilidade mental e/ou física.
– idosos (maior risco de parkinsonismo e discinesia tardia).
– crianças e adultos jovens (maior incidência de reações distônicas).
– neonatos (maior risco de metemoglobinemia).
– depressão.
– insuficiência cardíaca congestiva.
– porfiria.
– cirrose.
– lactação (ver Apêndice B).
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D). t Categoria de risco na gravidez (FDA): B.
t Neonatos: 100 microgramas/kg, a cada 6 a 8 horas (vias oral ou intravenosa).
t 1 mês a 1 ano (até 10 kg): 100 microgramas/kg (máximo de 1 mg), 2 vezes ao dia.
t 1 a 3 anos (10 a 14 kg): 1 mg, 2 a 3 vezes/dia.
t 3 a 5 anos (15 a 19 kg): 2 mg, 2 a 3 vezes/dia.
t 5 a 9 anos (20 a 29 kg): 2,5 mg, 3 vezes/dia.
t 9 a 14 anos (30 a 60 kg): 5 mg, 3 vezes/dia.
t 15 a 18 anos (acima de 60 kg): 10 mg, 3 vezes/dia.
t A dose diária não deve exceder a 500 microgramas/kg.
t 1 a 2 mg/kg, por infusão intravenosa, conforme o potencial emético do antineoplásico, 30 minutos antes da administração do antineoplásico, repetindo a cada 2 a 3 horas, por no máximo 5 administrações diárias.
t 10 a 20 mg, por via intramuscular ou intravenosa, próximo ao término da cirurgia, podendo a dose de 10 mg ser repetida a cada 4 a 6 horas, se necessário.
t 10 a 15 mg, por via oral, 30 minutos antes de cada refeição e antes de dormir, até 4 vezes/dia, por até 12 semanas.
t 10 mg, por via oral, 30 minutos antes de cada refeição e antes de dormir, até 4 vezes/dia, por 2 a 8 semanas.
t 10 mg, por via intramuscular ou intravenosa, 30 minutos antes de cada refeição e antes de dormir, até 4 vezes/dia, por até 10 dias.
t 5 mg, por via intramuscular ou intravenosa, próximo ao término da cirurgia. A dose pode ser repetida, se necessário.
t 5 mg, por via oral, 30 minutos antes de cada refeição e antes de dormir, até 4 vezes/dia. A dose pode ser aumentada para 10 mg, 4 vezes/dia, se resposta não for obtida.
t 5 mg, por via oral, 30 minutos antes de cada refeição e antes de dormir, por 2 a 8 semanas. Se necessário, aumentar para 10 mg.
t 5 mg, por via intravenosa, podendo aumentar para 10 mg, se necessário.
Nota:
t doses baixas (até 10 mg) de metoclopramida podem ser administradas via intravenosa direta sem diluição. A injeção intravenosa direta deve ser realizada lentamente por 1 a 2 minutos e a infusão intravenosa por um período não inferior a 15 minutos, pois a administração rápida é associada com ansiedade e agitação transitórias, mas intensas, seguidas por sonolência.
t Absorção: rápida e quase completa.
t Início de ação: oral: 0,5 a 1 hora; intramuscular: 10 a 15 minutos; intravenosa: 1 a 3 minutos.
t Meia-vida de eliminação: 4 a 6 horas.
t Duração da ação: oral: 1 a 2 horas após dose única.
t Arritmia cardíaca reversiva (torsades de pointes), bloqueio atrioventricular, hipertensão ou hipotensão, taquicardia supraventricular, insuficiência cardíaca congestiva, retenção de fluidos.
t Sonolência (10% a 70%), fadiga (10%), inquietação (10%), reações distônicas agudas (menos de 1% a 25%, dose e idade relacionadas), acatisia, confusão, vertigem, ansiedade cefaleia, insônia, discinesia tardia.
t Reações extrapiramidais ocorrem com maior frequência em crianças e adultos com menos de 20 anos e após administração intravenosa de altas doses do fármaco.
t Depressão.
t Mastodínia, hiperprolactinemia, galactorreia.
t Diarreia, náusea.
t Ciclosporina: risco de toxicidade pela ciclosporina (disfunção renal, colestase, parestesia, neurotoxicidade). Evitar o uso concomitante. Monitorar o paciente e os níveis de ciclosporina, ajustando a dose conforme necessário.
t Dexametasona: apresenta sinergismo com o efeito antinauseante e antiemético de metoclopramida, sendo a associação empregada em muitas condições.
t Ciclosporina: risco de toxicidade pela ciclosporina (disfunção renal, colestase, parestesia, neurotoxicidade). Evitar o uso concomitante. Monitorar o paciente e os níveis de ciclosporina, ajustando a dose conforme necessário.
t Didanosina: aumento das concentrações plasmáticas da didanosina. Monitorar o paciente quanto a toxicidade.
t Digoxina: redução dos níveis de digoxina. Monitorar o paciente para redução da resposta terapêutica.
t Levodopa: redução da eficácia da metoclopramida. Aumento da biodisponibilidade da levodopa e da incidência de sintomas extrapiramidais. Evitar o uso concomitante.
t Linezolida: risco de síndrome serotoninérgica (hipertermia, hiperreflexia, mioclônus, disfunção cognitiva). Monitorar o paciente e considerar a descontinuação de um ou ambos os fármacos.
t Mivacúrio/suxametônio (succinilcolina): risco de bloqueio neuromuscular prolongado. Monitorar a função neuromuscular.
t Sertralina/venlafaxina: risco de desenvolvimento de sintomas extrapiramidais. Monitorar o paciente.
t Tacrolimo: aumento da concentração do tacrolimo. Monitorar os níveis plasmáticos e observar sinais de toxicidade (nefrotoxicidade, neurotoxicidade, hiperglicemia, hiperpotassemia). Redução da dose pode ser necessária.
t Tiopental: aumento do efeito hipnótico. Monitorar o grau de sedação do paciente. Redução da dose pode ser necessária.
t Orientar para ingerir 30 minutos antes das refeições e antes de dormir.
t Alertar para a possibilidade de prejudicar a habilidade para realizar atividades que requeiram atenção e coordenação motora.
t Reforçar para a necessidade de evitar o uso de bebida alcoólica e outros depressores do SNC.
t Alertar para a possibilidade de surgirem tremores, rigidez e outros sinais de transtorno extrapiramidal, especialmente em crianças e idosos.
t Armazenar à temperatura entre 20 e 25 °C. É fotossensível e deve ser protegida da luz.
t Para a formulação intravenosa, observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Para infusão intravenosa, o fármaco pode ser diluído em 50 mL de cloreto de sódio 0,9%, glicose 5%, solução glicofisiológica, solução de Ringer ou Ringer + lactato.
t Após diluição, a solução pode ser armazenada por até 48 horas, à temperatura ambiente, protegido da luz, ou por até 24 horas quando não protegida da luz.
Maria Isabel Fischer
Na Rename 2010: itens 2.2 e 8.2
t Solução injetável 0,4 mg/mL
t Depressão respiratória, pós-cirúrgica, por dose excessiva de analgésicos opioides.
t Antídoto em intoxicações por opioides.
t Hipersensibilidade ao fármaco ou a qualquer componente da formulação.
t Usar com cuidado nos casos de:
– pós-operatório em pacientes com doença cardíaca pré-existente ou em uso de fármaco cardiotóxico (risco de edema pulmonar).
– pacientes hipotensos ou que apresentem disfunção na circulação periférica (início de ação pode ser tardio).
– reversão abrupta de depressão pós-operatória por opioide (risco de convulsões, taquicardia ventricular e fibrilação, edema pulmonar, parada cardíaca e potencialmente morte).
– doses excessivas de naloxona podem resultar em significante reversão da analgesia pós-operatória.
– depressão respiratória causada por opioides mistos (agonistas/antagonistas), tais como buprenorfina e pentazocina (pode requerer doses mais altas de naloxona ou a reversão pode ser incompleta; se a resposta for incompleta, indica-se ventilação mecânica).
– durante trabalho de parto em mulheres com hipertensão branda a moderada (risco de episódios hipertensivos graves; monitorar pressão arterial).
– neonatos de mães com suspeita de uso de opioides por longo prazo; não administrar naloxona devido ao risco de convulsões e/ou síndrome aguda de retirada.
– dependência física conhecida ou suspeita a opioides (naloxona precipita sintomas de retirada incluindo dor, hipertensão, suor, agitação, irritabilidade e choro estridente dentro de minutos após a administração e permanece por cerca de 2 horas; observar os pacientes para recorrência de depressão respiratória e outros efeitos narcóticos por pelo menos 2 horas após a última dose de naloxona.
– pacientes em choque séptico; risco de agitação, náusea e vômito, edema pulmonar, hipotensão, arritmias cardíacas e convulsões; uso com cuidado particularmente se o paciente tiver dor, tiver sido tratado previamente com opioide, ou se o paciente tiver desenvolvido tolerância a opioide.
– pacientes hipotensos ou que apresentem disfunção na circulação periférica (início de ação pode ser tardio).
– doença hepática.
– doença renal.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t neonatos (pós-parto): 0,001 a 0,015 mg/kg por via intravenosa ou intramuscular; a dose pode ser repetida se necessário.
t com 1 mês ou mais: 0,001 a 0,015 mg/kg por via intravenosa, intramuscular ou subcutânea; a dose pode ser repetida se necessário.
t neonatos: 0,1 mg/kg, por via intramuscular ou intravenosa; pode ser repetido se necessário.
t com menos de 5 anos ou menos de 20 kg: 0,1 mg/kg por via intravenosa, intramuscular ou subcutânea; pode ser repetida se necessário.
t com 5 anos ou mais ou 20 kg ou mais: 2 mg por via intravenosa, intramuscular ou subcutânea; pode ser repetida se necessário.
t 0,1 a 0,2 mg, por via intravenosa, repetida a cada 2 a 3 minutos, até obter a resposta necessária; repetir a dose, se necessário, em 1 a 2 horas. Ou então, doses subsequentes podem ser dadas por via intramuscular cada 1 a 2 horas até obter a resposta desejada.
t Dose de 0,4 mg a 2 mg, por via intravenosa; se não houver resposta, repetir a dose em intervalos de 2 a 3 minutos, até o máximo de 10 mg. Deterioração da função respiratória pode requerer doses adicionais. Se a função respiratória não melhorar, reavaliar o diagnóstico.
Notas:
t Usar incrementos de 0,1 a 0,2 mg em pacientes dependentes de opioides ou em pós-operatório para evitar grandes alterações cardiovasculares.
t Em adultos, o regime posológico para infusão intravenosa não está bem estabelecido. Administrar 10 mg diluídos em 50 mL de glicose 5% ou soro fisiológico, com velocidade ajustada conforme a resposta. Administrar, preferentemente, utilizando bomba de infusão.
t Por apresentarem início de ação mais lento, as vias intramuscular ou subcutânea devem ser usadas apenas se for inviável a via intravenosa.
t Doses utilizadas em sobredose aguda por opioides podem não ser apropriadas para o manejo de depressão respiratória induzida por opioides e sedação naqueles recebendo cuidado paliativo e em uso prolongado de opioides.
t Início de efeito: menos de 2 minutos (intravenosa), 2 a 5 minutos (intramuscular e subcutânea)
t Duração do efeito: depende da dose e da via de administração, varia entre 20 e 60 minutos
t Metabolismo: hepático.
t Meia-vida de eliminação: 1,2 a 3,5 horas (neonatos); 1 a 1,5 horas (adultos)
t Excreção: renal
t Hiper ou hipotensão, taquicardia ventricular, arritmia ventricular, parada cardíaca, fibrilação ventricular.
t Edema pulmonar.
t Ansiedade, inquietude, convulsão.
t Náusea, vômito, diarreia.
t Alguns Efeitos adversos podem estar associados à retirada do opioide.
t Ioimbina: pode aumentar efeitos adversos. Cautela ao iniciar tratamento.
t Clonidina: pode resultar em hipertensão. Monitorar intensamente a pressão arterial após a administração de naloxona.
t Restrito para uso hospitalar.
t Armazenar a 25 ºC, proteger da luz.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Estável por 24 horas em soro fisiológico 0,9% ou glicose 5% em concentração de 4 microgramas/mL.
t Não misturar com soluções alcalinas.
t Soluções devem ser utilizadas em 24 horas. Após este período porções não utilizadas devem ser descartadas.
Tatiana Aragão Figueiredo
Isabella Campagnuci Knust
Na Rename 2010: item 13.2
t Cápsulas 10 mg, 25 mg, 50 mg e 75 mg.
t Depressão maior.
t Distúrbios da condução cardíaca, após enfarte do miocárdio, arritmias cardíacas, doença hepática grave (ver apêndice C), fase de mania da doença bipolar, porfiria.
t Hipersensibilidade ao fármaco ou a outros antidepressivos tricíclicos.
t Uso de inibidores da monoamina oxidase (IMAO) nos últimos 15 dias (a troca de um IMAO pelo tricíclico ou vice-versa deve guardar o intervalo mínimo de 15 dias).
t Crianças e adolescentes.
t Usar com cuidado nos casos de:
– uso de álcool e outros depressores do SNC (aumento dos efeitos sedativos).
– transtorno bipolar (pode causar reversão maníaca).
– doença cardiovascular, tendência suicida, histórico de etilismo, prostatismo, epilepsia, hipertireoidismo, glaucoma, esquizofrenia e retenção urinária.
– lactação: avaliar potenciais benefícios e riscos.
– idosos (reduzir doses).
– eletrochoque concomitante (pode aumentar os riscos do eletrochoque).
– feocromocitoma.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t 25 a 50 mg, por via oral, a cada 24 horas, à noite.
t Dose inicial de 25 mg, por via oral, a cada 24 horas, à noite, aumentado se necessário até 75 a 100 mg. Dose máxima diária: 150 mg.
t A dose de resposta deve ser mantida por 3 a 4 meses, sendo reduzida à metade.
t O tratamento deve ser mantido por 6 a 12 meses para evitar recidivas.
t Na retirada gradual, diminui-se a dose em 25 mg a cada 2 ou 3 dias. Se os sintomas reaparecerem, retomam-se os níveis iniciais.
t 25 a 50 mg, por via oral, a cada 24 horas, à noite.
t Absorção oral.
t Metabolismo predominantemente hepático.
t Período de latência: 2 a 3 semanas
t Pico sérico: 7 a 8 horas.
t Meia-vida de eliminação: 15 a 39 horas; em idosos pode chegar a 90 horas.
t Eliminação urinária e biliar
t Hipotensão ortostática, taquicardia, arritmia, enfarte do miocárdio, morte súbita.
t Tremor, fraqueza, sonolência, tontura, cefaleia, insônia, alucinações, ataxia, acidente vascular cerebral, crise convulsiva, visão turva.
t Secura na boca, gengivite, aumento do apetite, náusea, anorexia, dispepsia, obstipação, diarreia.
t Diminuição da função hepática, icterícia.
t Agranulocitose, aplasia medular, eosinofilia, trombocitopenia.
t Efeitos anticolinérgicos: xerostomia, midríase, cicloplegia, retenção urinária, diminuição da motilidade gastrintestinal, taquicardia e em altas doses, delírio.
t Ginecomastia, alterações dos níveis glicêmicos, aumento de peso, disfunção sexual, porfiria.
t Urticária, alopecia.
t Sudorese excessiva.
t Ácido valproico, amprenavir, antidepressivos bloqueadores seletivos da recaptação de serotonina, antipsicóticos, bupropiona, cimetidina, fluconazol, fosamprenavir, ritonavir, terbinafina: aumento de efeito do antidepressivo.
Monitorar a concentração plasmática, sinais e sintomas de toxicidade do antidepressivo; ajustes da dose da nortriptilina podem ser necessários.
t Álcool e outros depressores do sistema nervoso central, anticoagulantes cumarínicos, fármacos com efeitos anticolinérgicos (anti-histamínicos H1, antiparkinsonianos e neurolépticos): podem ter seus efeitos potencializados. Em pacientes recebendo anticoagulante oral, o tempo de protrombina deve ser cuidadosamente monitorado e ajustes da dose do anticoagulante podem ser necessários.
t Amiodarona, bepridil, cinacalcete, cisaprida, disopiramida, dofetilida, dolasetrona, droperidol, enflurano, espiramicina, fenitoína, fenotiazinas, fluconazol, haloperidol, halotano, hidrato de cloral, ibutilida, isoflurano, lidoflazina, mesoridazina, octreotida, pentamidina, pimozida, proclorperazina, sulfametoxazol, tioridazina, trimetoprima, vasopressina, venlafaxina, zolmitriptana: pode levar a aumento da toxicidade da nortriptilina. Monitorar a concentração plasmática, sinais e sintomas de toxicidade do antidepressivo; ajustes da dose da nortriptilina podem ser necessários.
t Anfetaminas, antiarrítmicos, antibióticos macrolídeos e quinolonas, anti-histamínicos, antimaláricos, bloqueadores adrenérgicos: aumento da toxicidade da nortriptilina. Monitorar a concentração plasmática, sinais e sintomas de toxicidade do antidepressivo; ajustes da dose da nortriptilina podem ser necessários.
t Carbamazepina e rifapentina: diminuição de efeito da nortriptilina. Monitorar as concentrações plasmáticas da nortriptilina; ajustes de doses podem ser necessários.
t Clonidina, guanetidina, guanadrel, guanfacina: podem ter seus efeitos diminuídos. Monitorar a pressão arterial para possível ajuste de dose dos anti-hipertensivos.
t Inibidores da MAO, linezolida: a associação pode levar a neurotoxicidade, convulsões, ou síndrome serotoninérgica (hipertensão, hipertermia, mioclônus, alterações do estado mental).
t Simpaticomiméticos: o uso concomitante com nortriptilina pode levar a hipertensão, arritmia cardíaca e taquicardia. A vasoconstrição proveniente de fármacos alfa-adrenérgicos e de outros simpaticomiméticos é substancialmente reforçada com a presença de antidepressivos tricíclicos. Se estes fármacos são utilizados em associação com nortriptilina, um acompanhamento atento e uma redução da dose do simpaticomimético é necessária.
t Alertar para evitar uso de bebida alcoólica.
t Orientar que deve ser administrado após a alimentação para prevenir irritação gástrica.
t Informar sobre a demora no início de resposta terapêutica.
t Orientar sobre a mudança de frequência cardíaca e para levantar-se mais lentamente, de modo a evitar hipotensão ortostática.
t Evitar dirigir e operar com máquinas que exijam atenção.
t Não descontinuar o uso de maneira repentina.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas. Caso esqueça uma dose, não use o medicamento pela manhã e espere até a próxima noite.
t Conservar sob temperatura entre 15 e 30 oC, em recipiente bem fechado e ao abrigo da luz.
t Sob forma de cloridrato, apresenta-se como pó branco ou quase branco, solúvel em água. Solução aquosa a 1% tem pH 5,0.
Atenção: este fármaco apresenta um número muito elevado de Efeitos adversos , por isto deve ser feita uma pesquisa específica sobre este aspecto antes de introduzir ou descontinuar nortriptilina ou outros medicamentos no esquema terapêutico do paciente. Os efeitos terapêuticos podem demorar de 15 a 21 dias para se manifestar. Monitorizar pressão arterial e frequência cardíaca nas semanas iniciais.
Fabiana Wahl Hennigen
Na Rename 2010: itens 6.3 e 16.4
t Comprimidos 4 mg e 8 mg.
t Solução injetável 2 mg/mL.
t Profilaxia de náusea e vômito induzidos por antineoplásicos com potencial emetogênico moderado e alto.
t Hipersensibilidade à ondansetrona.
t Usar com cuidado nos casos de:
– sensibilidade cruzada com antagonistas seletivos dos receptores 5-HT3.
– íleo progressivo ou distensão gástrica (podem ser mascarados pelo fármaco).
– pacientes com menos de 4 meses (requerem cuidadoso monitoria).
– uso intravenoso (pode produzir alterações transitórias no eletrocardiograma).
– insuficiência hepática (ver Apêndice C).
t Não estimula o peristaltismo gástrico ou intestinal. Não usar em substituição à sucção nasogástrica.
t Categoria de risco na gravidez (FDA): B (ver Apêndice A).
t 4 a 11 anos: 4 mg, por via oral, 30 minutos antes da quimioterapia, repetir 4 e 8 horas após a primeira dose e prosseguir com 4 mg a cada 8 horas por 1 a 2 dias após o término da quimioterapia.
t 6 meses a 18 anos: 0,15 mg/kg, por via intravenosa, 30 minutos antes da quimioterapia, repetir 4 e 8 horas após a primeira dose. Ou então, 3 a mg/m2, imediatamente antes da quimioterapia (dose única, máxima de 8 mg), repetidos a cada 8 a 12 horas durante a terapia e por pelo menos 24 horas após o término ou seguidos por 4 mg, via oral, a cada 8 a 12 horas, por até 5 dias.
t 8 mg, por via oral, 30 minutos antes da quimioterapia, repetir a dose após 8 horas e, então, a cada 12 horas por 1 a 2 dias após o término da quimioterapia. Ou então, 24 mg em dose única, por via oral, 30 minutos antes do iníco da terapia.
t 0,15 mg/kg, por via intravenosa, 30 minutos antes da quimioterapia, repetir 4 e 8 horas após a primeira dose. Ou então, 0,45 mg/kg, por via intravenosa, 1 vez ao dia.
t Injeção intramuscular: administrar não diluído.
t Injeção intravenosa: administrar por 2 a 5 minutos como solução não diluída.
t Infusão intravenosa diluída em 50 mL de solução de glicose 5% ou cloreto de sódio 0,9%: administrar durante 15 a 30 minutos.
t Boa absorção após administração oral. Alimentos aumentam a absorção.
t Início de ação: 30 minutos.
t Pico de ação: 2 horas.
t Meia-vida de eliminação: 3 a 6 horas.
t Obstipação (6% a 11%), diarreia (2% a 7%), xerostomia (5% a 17%).
t Cefaleia (9% a 27%), mal-estar e fadiga (9% a 13%), sonolência (8%), febre (2% a 8%), tontura (4% a 7%), ansiedade (6%), sensação de frio (2%).
t Prurido (2% a 5%), exantema (1%).
t Distúrbio ginecológico (7%), retenção urinária (5%).
t Reação no sítio de injeção (4%).
t Parestesia (2%).
t Hipóxia (9%).
t Aumento das enzimas hepáticas (1% a 5%).
t Efeitos graves (inferior a 1%): arritmia cardíaca, parada cardíaca, hipotensão, bradicardia, angina, broncoespasmo, laringoespasmo, anafilaxia.
t Apomorfina: aumento do efeito hipotensivo e perda de consciência. O uso concomitante é contraindicado.
t Ciclofosfamida: Redução da exposição sistêmica à ciclofosfamida. Monitorar a resposta terapêutica do paciente e, se necessário, aumentar a dose.
t Mesoridazina, pimozida e tioridazina: alterações transitórias no eletrocardiograma e aumento do risco de cardiotoxicidade. O uso concomitante é contraindicado.
t Orientar para ingerir uma dose oral adicional se ocorrer vômito em até 30 minutos após a administração. Consultar o médico, se o vômito persistir.
t Orientar para administração independentemente do horário das refeições.
t Armazenar entre 2 e 30 °C. Proteger da luz.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A solução injetável é compatível com glicose a 5% ou cloreto de sódio a 0,9%, mantendo-se estável por até 48 horas à temperatura ambiente. Devido ao risco de contaminação microbiana, deve ser usada em até 24 horas.
t Não misturar com soluções alcalinas, pois pode precipitar.
Isabella Campagnuci Knust
Na Rename 2010: item 8.2
t Cápsula 250 mg.
t Antídoto para intoxicação por metais pesados (agente quelante para mercúrio, níquel, chumbo, arsênio e cobre).
t Doença de Wilson.
t Artrite reumatoide grave (alternativa de segunda linha quando não houver resposta ao tratamento convencional).
t Hipersensibilidade a penicilamina.
t Anemia aplástica e/ou agranulocitose. t Insuficiência renal moderada a grave. t Lúpus eritematoso sistêmico.
t Gravidez; exceto na Doença de Wilson (ver Apêndice A)
t Lactação (ver Apêndice B).
t Usar com cuidado nos casos de:
– insuficiência renal (ver Apêndice D).
– hipersensibilidade conhecida a penicilinas (possibilidade de reação cruzada).
– uso concomitante com suplementos contendo sais minerais.
– uso concomitante com medicamentos nefrotóxicos, antimaláricos e citotóxicos.
– idosos (risco de potencializar Efeitos adversos hematológicos). t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t 20 a 40 mg/kg/dia, por via oral, divididos em 3 tomadas.
t Dose usual: 30 a 40 mg/kg/dia, por via oral, divididos em 3 tomadas, 2 horas antes das refeições ou 3 horas depois, durante 1 a 6 meses.
t Dose máxima de 20 mg/kg/dia, por via oral, divididos em 3 tomadas, 2 horas antes das refeições. Dose mínima: 500 mg/dia.
t Dose de manutenção: 15 a 20 mg/kg/dia, por via oral.
t A dose inicial deve ser reduzida ou aumentada em intervalos de 4 semanas, por um período de 3 a 6 meses.
t 1 a 2 g/dia, por via oral, dividido em 3 tomadas, 2 horas antes das refeições ou 3 horas depois.
t O tratamento deve ser continuado por 1 a 2 meses.
t 1,5 a 2 g/dia, por via oral, dividido em 3 tomadas, 2 horas antes das refeições, no 1º ano. A partir do 2º ano, dose de manutenção de 0,75 a 1 g/dia.
t No caso de gravidez, a dose diária deve ser limitada para 1 g. Se o parto for por cesariana, reduzir a dose como consta nas observações.
t 125 a 250 mg/dia, por via oral, aumentando-se a dose mais 125 a 250 mg a cada 1 a 3 meses, até uma resposta satisfatória ou se ocorrer toxicidade.
t Na falta de resposta satisfatória ao tratamento após 3 a 4 meses, o tratamento deve ser descontinuado.
t Muitos pacientes necessitam de dose diária de manutenção de 500 a 750 mg/ dia.
t Após a remissão da doença, o tratamento deve ser continuado por mais de 6 meses, a dose pode ser diminuída em 125 a 250 mg a cada 3 meses. Recorrência pode ocorrer.
t Dose inicial: 125 mg/dia, por via oral. Se a dose for bem tolerada, aumentar 125 mg/dia, no máximo até 750 mg/dia, dividido em 3 tomadas.
t 20 mg/kg/dia, por via oral, dividido em 3 tomadas, 2 horas antes das refeições.
t A dose deve ser ajustada de acordo com a resposta.
t Dose inicial: 125 mg/dia, por via oral, durante 1 mês. Aumentar a dose em 125 mg a cada 4 semanas, até a dose diária máxima de 1g.
Nota:
t em caso de intervenção cirúrgica, reduzir a dose diária para, no máximo, 250 mg, por 6 semanas antes da cirurgia e manter essa dose até a cicatrização completa da ferida operatória.
t Pico de concentração plasmática: 1 a 4 horas.
t Meia-vida de eliminação: 1 a 7,5 horas.
t Metabolismo: hepático.
t Excreção: renal e fecal.
t Náuseas, perda do paladar e anorexia
t Febre
t Reações cutâneas de hipersensibilidade, trombocitopenia, leucopenia, agranulocitose, anemia aplástica, anemia hemolítica.
t Proteinúria, hematúria, síndrome nefrótica.
t Síndrome semelhante a lupo, miastenia grave, síndrome semelhante a artrite reumatoide, polimiosite, dermatomiosite.
t Úlceras na mucosa oral e estomatite, alopecia, pênfigo.
t Alteração da função hepática, hepatite.
t Neurite óptica.
t Bronquiolite, pneumonia.
t Ginecomastia.
t Alimentos, antiácidos, ferro e outros metais: risco de redução do efeito da penicilamina.
t Anti-inflamatórios não-esteroides: aumento do risco de nefrotoxicidade.
t Auranofina, aurotioglicose, aurotiomalato de sódio: aumento do risco de depressão medular.
t Clozapina: pode causar agranulocitose.
t Diazepam (via intravenosa): pode causar flebite.
t Digoxina e zinco: podem ter sua efetividade diminuída pela penicilamina
t Probenecida: pode causar redução do efeito da penicilamina.
t Administrar com o estômago vazio, 1 hora antes da alimentação, ou 2 a 3 horas depois.
t Respeitar intervalo de pelo menos 1 hora para a ingestão de outros medicamentos ou leite.
t Manter a temperatura ambiente, ao abrigo do ar, luz e umidade.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 21.5
t Colírio 2%
t Glaucoma crônico de ângulo aberto.
t Hipertensão ocular.
t Tratamento de emergência de glaucoma agudo de ângulo fechado.
t Irite aguda.
t Uveíte anterior.
t Algumas formas de glaucoma secundário.
t Inflamação aguda do segmento anterior.
t No pós-cirúrgico de glaucoma de ângulo fechado (risco de sinéquia posterior).
t Hipersensibilidade a pilocarpina.
t Usar com cuidado nos casos de:
– doença da retina (risco de descolamento da retina).
– lesão na córnea ou conjuntiva.
– glaucoma de ângulo aberto e tratamento de longa duração (monitorar a pressão intraocular e os campos visuais).
– doença cardíaca, hipertensão.
– asma ou doença pulmonar obstrutiva crônica que requer farmacoterapia.
– úlcera péptica.
– obstrução do trato urinário.
– doença de Parkinson.
– crianças (segurança e eficácia não estão estabelecidas).
– surgimento de sintomas de toxicidade sistêmica (interromper o tratamento).
– lactação: pode ocorrer absorção sistêmica. t Categoria de risco na gravidez (FDA): C.
t Glaucoma crônico de ângulo aberto – 1 gota até 4 vezes ao dia. Para evitar excessiva absorção sistêmica, os pacientes devem pressionar com o dedo o saco lacrimal por 1 a 2 minutos após instilação da solução.
t Pré-operatório em glaucoma agudo de ângulo fechado – 1 gota a cada 10 minutos por 30 a 60 minutos, seguido de 1 gota a cada 1 a 3 horas até redução da pressão intraocular.
t Íris pigmentada pode requerer concentração maior do miótico ou administração mais frequente, correndo o risco de dose excessiva.
t Absorção sistêmica é rara.
t Miose ocorre de 10 a 30 minutos após o uso do colírio e dura de 4 a 8 horas. Redução da pressão intraocular ocorre geralmente em até 75 minutos e persiste por 4 a 14 horas.
t Dor nos olhos, visão turva, espasmo ciliar, lacrimejamento, miopia, cefaleia, congestão vascular conjuntival, queratite superficial, hemorragia vítrea e aumento do bloqueio pupilar.
t Opacidade das lentes após uso prolongado .
t Raramente: efeitos sistêmicos incluindo hipertensão, taquicardia, broncoespasmo, edema pulmonar, salivação, sudorese, náusea, vômito e diarreia.
t Latanoprosta: pode ter sua eficácia reduzida. Administrar a dose noturna de pilocarpina 10 minutos após aplicação da latanoprosta; de preferência, uma hora após latanoprosta.
t Alertar que o colírio de pilocarpina causa dificuldade de adaptação à luz (ofuscamento) e pode causar espasmo de acomodação.
t Alertar para não operar máquinas ou dirigir até que a visão esteja límpida.
t Ensinar a pressionar com o dedo o saco lacrimal por 1 a 2 minutos após instilação da solução, para evitar excessiva absorção sistêmica.
t Orientar para lavar as mãos antes da aplicação.
t Agitar o frasco antes de usar.
t Ensinar cuidado para não contaminar o frasco de colírio, que deve ser de uso exclusivo.
t Dose esquecida: alertar para usar a dose esquecida o mais breve possível. Se estiver perto da hora regular, aplicar a dose normal e ignorar a dose esquecida. Alertar para não aplicar duas doses ao mesmo tempo.
t Manter à temperatura ambiente, longe da luz direta.
Luciana dos Santos
Na Rename 2010: item 12
t Comprimido 50 mg.
t Solução oral 1 mg/mL e 10 mg/mL
t Profilaxia e tratamento de deficiência de piridoxina.
t Prevenção de neurite periférica induzida por fármacos (isoniazida, penicilamina).
t Anemia sideroblástica.
t Hipersensibilidade à piridoxina ou a qualquer componente da formulação.
t Usar com cuidado nos casos de:
– uso prolongado de doses diárias de 50 mg a 2 g (pode ocorrer neurite sensorial).
– adultos recebendo doses diárias acima de 200 mg (podem ocorrer dependência e síndrome de retirada).
– insuficiência renal (ver Apêndice D).
t Categoria de risco na gravidez: A e C (dose superior a dietética recomendada) (ver Apêndice A).
t 5 a 25 mg, por via oral, a cada 24 horas, durante 3 semanas. Após, 1,5 a 5 mg, por via oral, a cada 24 horas.
t Profilaxia: 1 a 2 mg/kg, por via oral, a cada 24 horas.
t Tratamento: 10 a 50 mg, por via oral, a cada 24 horas. Dose máxima: 200 mg por dia.
t Profilaxia na gravidez e lactação: 1,5 a 2,5 mg por via oral a cada 24 horas.
t Tratamento: 25 a 50 mg, via oral, a cada 8 horas.
t Profilaxia: 10 mg, por via oral, a cada 24 horas.
t Tratamento: 50 mg, por via oral, a cada 8 horas.
t 100 a 400 mg/dia, por via oral, em doses divididas.
t Ajuste de dose em hemodiálise: dose suplementar de 10 mg a cada 24 horas.
t Pico de concentração: 1,25 horas.
t Meia-vida de eliminação: 15 a 20 dias.
t Absorção: rapidamente absorvido (jejuno).
t Metabolismo: hepático.
t Excreção: renal (35% a 63%).
t Ocorrem em menos de 1%.
t Acidose, diminuição da lactação
t Ácido fólico sérico diminuído.
t Náusea.
t Reações alérgicas (fotossensibilidade).
t Cefaleia, insônia, epilepsia.
t Enzimas hepáticas aumentadas.
t Parestesia, neuropatia.
t Altretamina: pode ocorrer redução do efeito da altretamina. Evitar o uso concomitante.
t Orientar para adotar na dieta ou aumentar a ingestão de carnes, legumes e cereais, que são ricos em piridoxina.
t Pode ser administrado por via oral sem considerar a presença de alimentos (sem interferência na absorção).
t Em caso de esquecimento de dose, orientar para que o indivíduo a tome assim que lembrar. Não dobrar doses.
t Armazenar à temperatura ambiente, de 15 a 30 º C, em recipientes herméticos e protegido da luz.
Thais Furtado de Souza
Na Rename 2010: item 1.2
t Solução injetável para uso odontológico 3% (30 mg/mL) + 0,03 UI/mL.
t Anestésico local para uso odontológico (anestesia infiltrativa).
t Uso restrito para pacientes que não podem utilizar cloridrato de lidocaína + hemitartarato de epinefrina.
t Hipersensibilidade a um ou a ambos os fármacos.
t Metemoglobinemia.
t Hipovolemia
t Bloqueio cardíaco.
t Usar com cuidado nos casos de:
– hipertensão grave não tratada, doença cardíaca grave, choque, arritmia e bradicardia.
– crianças, idosos e pacientes enfraquecidos.
– doença renal e hepática.
– epilepsia, depressão respiratória, porfiria, miastenia grave e hipóxia.
– doses elevadas (pode ocorrer metemoglobinemia, que pode ser tratada com a administração intravenosa de cloreto de metiltionínio 1%, dose 1 mg/kg).
– lactação: não há estudos conclusivos para determinar o risco durante a amamentação.
t Categoria de risco na gravidez (FDA): B (ver Apêndice A).
Nota:
t dose máxima de prilocaína com felipressina 0,03 UI/mL: 6 a 8 mg/kg, até total de 600 mg.
t 40 mg.
t 40 a 80 mg.
t Início de efeito: 2 minutos.
t Duração de efeito: 2 a 3 horas. t Meia-vida: 10 a 150 minutos.
t Metabolismo: hepático.
t Excreção: renal (1% em forma inalterada).
t Hipotensão, bradicardia, arritmias, metemoglobinemia, convulsão.
t Raro: hipersensibilidade.
t Porfiria.
t Nervosismo, visão borrada, ansiedade, tremor, depressão respiratória, sonolência, perda da sensibilidade, perda da consciência.
t Hialuronidase: aumenta a difusão tópica do anestésico e pode ocasionar aumento da incidência de reação sistêmica. Monitorar a toxicidade da prilocaína.
t Erva-de-são-joão (Hypericum perforatum): pode aumentar risco de colapso cardiovascular ou retardar o efeito da anestesia. O uso da erva-de-são-joão deve ser descontinuado 5 dias antes da utilização do anestésico.
t Óxido nítrico: pode aumentar o risco de metemoglobinemia.
t Pode ocorrer dormência na língua, nos lábios, no interior das bochechas, podendo durar algumas horas após utilização do medicamento.
t Orientar para evitar mastigação de alimentos sólidos até retorno da sensibilidade normal.
t Armazenar sob temperatura entre 15 a 30 °C.
Ana Cláudia de Brito Passos
Na Rename 2010: itens 4 e 6.3
t Solução injetável 25 mg/mL.
t Anafilaxia (adjuvante).
t Terapêutica antineoplásica (adjuvante)
t Hipersensibilidade ou história de reação idiossincrática a prometazina ou a outras fenotiazinas e a sulfitos (ampolas contêm metabissulfito).
t Injeção subcutânea ou intra-arterial.
t Estado comatoso.
t Sintomas do trato respiratório baixo, incluindo asma.
t Crianças com menos de 2 anos apresentam maior risco potencial de depressão respiratória fatal.
t Usar com cuidado nos casos de:
– pacientes com hipertrofia prostática, retenção urinária, glaucoma de ângulo fechado, úlcera péptica estenosante e obstrução piloroduodenal, devido aos efeitos anticolinérgicos.
– durante a indução e a recuperação de anestesia (devido aos efeitos muscarínicos, pode aumentar o risco de regurgitação e aspiração do conteúdo gástrico).
– pacientes com convulsões, depressão da medula óssea, agranulocitose, leucopenia, disfunção hepática (ver Apêndice C) e renal, doença cardiovascular e função respiratória comprometida.
– crianças e idosos (apresentam maior susceptibilidade aos efeitos anticolinérgicos e sobre o sistema nervoso central, podendo ocorrer reação paradoxal).
– pacientes pediátricos com doença aguda associada a desidratação ou com sinais e sintomas sugestivos da síndrome de Reye ou outras doenças hepáticas.
– uso concomitante de outros fármacos que afetam o limiar epileptogênico (exemplo: narcóticos ou anestésicos locais).
– pacientes pediátricos com vômito não complicado (uso não recomendado); limitar o uso em vômito prolongado de etiologia desconhecida.
– síndrome neuroléptica maligna.
– lactação (ver Apêndice B).
– porfiria.
t Monitorar a pressão arterial.
t Reações graves podem ocorrer no lugar da injeção (após injeção única pelas vias intravenosa, subcutânea ou intra-arterial inadvertida); pode ser necessária intervenção cirúrgica como fasciotomia, enxerto cutâneo e amputação; o risco é aumentado com concentrações acima de 25 mg/mL e com taxas de infusão superior a 25 mg/min.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t De 5 a 10 anos: 6,25 a 12,5 mg, por injeção intramuscular profunda, iniciar pela dose mais baixa que produza efeito desejado. Dose máxima: 0,5 mg/ kg/dose.
t 12,5 a 25 mg, em injeção intramuscular profunda ou injeção intravenosa lenta (diluído para 2,5 mg/mL), a intervalos de 4 a 6 horas.
t 25 mg, por via intramuscular ou intravenosa, a intervalos de 2 horas.
t 6,25 a 12,5 mg, por via intravenosa, iniciar pela dose mais baixa que produza efeito desejado, para limitar o risco de dano tecidual.
Nota:
t Não deve ser administrado em concentrações acima de 25 mg/mL e em velocidade superior a 25 mg/minuto. A administração intravenosa deve ser feita em veia calibrosa, nunca em mão ou pulso.
t Início de efeito: 20 minutos (intramuscular), 3 a 5 minutos (intravenosa)
t Pico de concentração plasmática: 2 a 3 horas
t Duração de efeito: 4 a 6 horas, pode persistir por até 12 horas
t Liga-se a proteínas plasmáticas em cerca de 76 a 93%
t Meia-vida: 5 a 15 horas
t Metabolismo: hepático t Excreção: renal e biliar.
t Tontura, lassidão, zumbido, incoordenação, fadiga, insônia, tremores e crises oculogiras.
t Retenção urinária.
t Visão borrada (diplopia).
t Encefalopatia.
t Coreoatetose (movimentos atetoides), distonia.
t Febre e ataque cardíaco.
t Dano tecidual grave no local das injeções, exantema, necrólise tecidual, reações de hipersensibilidade, fotossensibilidade, dermatite e urticária.
t Icterícia, náuseas e vômitos, obstrução intestinal e xerostomia.
t Agranulocitose, leucopenia, trombocitopenia, trombose venosa.
t Bradicardia, taquicardia, hipertensão, hipotensão, doença coronária grave.
t Alucinações, euforia, nervosismo, excitação, estado catatônico, histeria e transtorno psicótico, depressão do SNC, vertigem, sinais extrapiramidais, sedação, sonolência, cefaleia, confusão, debilidade psicomotora, convulsões, síndrome neuroléptica maligna, cognição prejudicada.
t Depressão respiratória, apneia, asma, congestão nasal e broncospasmo.
t Arterioespasmo e gangrena podem ocorrer como resultado de injeção intra-arterial inadvertidamente.
t Alcaloides da beladona: aumento excessivo dos efeitos anticolinérgicos.
t Cisaprida, esparfloxacino, gatifloxacino e grepafloxacino: uso simultâneo com prometazina pode resultar em prolongamento do intervalo QT e/ou torsades de pointes. O uso concomitante é contraindicado.
t Fenilalanina: aumento do risco de discinesia.
t Lítio: em caso de uso concomitante, podem ocorrer fraqueza, discinesias, sintomas extrapiramidais, encefalopatia.
t Meperidina: aumento dos efeitos depressores do sistema nervoso central e respiratório.
t Midodrina: uso concomitante pode aumentar o risco de acatisia.
t Outros fármacos depressores respiratórios: risco de depressão acentuada. Evitar em pacientes pediátricos.
t Procarbazina: risco de depressão do sistema nervoso central.
t Propranolol: pode elevar o risco de aumento da pressão arterial.
t Alertar para notificar imediatamente caso haja sintomas: batimento cardíaco irregular, respiração lenta ou incômodo ao respirar, erupção cutânea, prurido, urticária, pele ou olhos amarelados, dor, ardor ou inchaço no lugar da injeção, visão turva, zumbido nos ouvidos.
t Também notificar em casos de efeitos mais sérios, tais como: sonolência ou tontura, dificuldade para dormir, nervosismo, depressão, boca ressecada, obstipação.
t Orientar para a interrupção do uso 2 dias antes de realizar testes cutâneos de alergia, devido à possibilidade de obtenção de resultados falso-negativos. Este medicamento pode afetar os resultados de alguns exames médicos.
t Pode afetar a capacidade de realizar atividades que exigem atenção e coordenação motora, como operar máquinas e dirigir.
t Recomendar o uso de protetor solar devido ao risco de fotossensibilização.
t Alertar para observar lactentes, devido ao risco de sedação.
t Reforçar a importância de evitar bebidas alcoólicas e outros depressores durante o uso do medicamento.
t Armazenar a temperatura entre 15 e 30 ºC. Não congelar. Manter ao abrigo de umidade e luz, em recipiente bem fechado.
t Não utilizar se estiver com a coloração alterada ou se houver precipitação.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Incompatibilidades em solução: alopurinol, gliconato de cálcio, cetorolaco, substâncias alcalinas, aminofilina, barbitúricos, benzilpenicilina, carbenicilina, cloranfenicol, clorotiazida, cefmetazol, cefoperazona, cefotetana, dimenidrinato, heparina, succinato sódico de hidrocortisona, meticilina, morfina, nalbufina, furosemida, doxorrubicina (formulação lipossomal), e alguns meios de contraste e soluções nutritivas.
Rosa Martins
Na Rename 2010: item 14.2
t Comprimido 150 mg e 300 mg t Solução injetável 3,5 mg/mL
t Fibrilação atrial e flúter.
t Taquicardia supraventricular paroxística (apenas comprimido de liberação imediata).
t Arritmia ventricular (apenas comprimido de liberação imediata).
t Hipersensibilidade ao fármaco.
t Bradicardia sinusal.
t Distúrbios broncoespásticos.
t Choque cardiogênico.
t Desequilíbrio eletrolítico.
t Insuficiência cardíaca congestiva não controlada.
t Hipotensão grave.
t Distúrbios de geração de impulso e/ou de condução sinoatrial, atrioventricular e intraventricular (ex.: bloqueio atrioventricular, síndrome do nó sinoatrial) na ausência de marca-passo artificial.
t Usar com cuidado nos casos de:
– elevada dosagem de anticorpos antinucleares (ANA), como em lúpus eritematoso sistêmico e artrite reumatoide.
– distúrbios hematológicos.
– hipersensibilidade ao propranolol, pela semelhança estrutural.
– insuficiência hepática (ver Apêndice C).
– pós-enfarte do miocárdio com arritmias ventriculares assintomáticas ou pouco sintomática (não utilizar o fármaco pelo risco de aumento da mortalidade).
– lactação – presente no leite materno.
t Pode ocorrer piora ou desencadeamento de insuficiência cardíaca congestiva.
t Pode alterar o limiar do marca passo.
t Eventos pró-arrítmicos (pode ocorrer piora ou desencadeamento de arritmias).
t Comprometimento da espermogênese. t Categoria de risco na gravidez (FDA): C
t Dose inicial: 150 mg, por via oral, a cada 8 horas. Aumentar a dose a cada 3 a 4 dias até 225 a 300 mg, por via oral, a cada 8 horas.
t Dose 2 mg/kg, por via intravenosa direta. Seguido por 0,0078 mg/kg/min, via infusão intravenosa.
t Dose inicial: 150 mg, por via oral, a cada 8 horas. Aumentar a dose a cada 3 a 4 dias até 225 a 300 mg, por via oral, a cada 8 horas.
t Dose de 1 a 2,5 mg/kg, por via intravenosa.
t Dose inicial: 150 mg, por via oral, a cada 8 horas. Aumentar a dose a cada 3 a 4 dias até 225 a 300 mg, por via oral, a cada 8 horas.
t Dose de 2 mg/kg, por via intravenosa direta, seguido de 2 mg/min, por infusão intravenosa.
t Biodisponibilidade: 12%, aumenta com a dose.
t Pico de concentração: 2 a 3 horas.
t Metabolismo hepático. Metabolito ativo. Sofre metabolismo de primeira passagem. Paciente classificado como metabolizador lento, de polimorfismo esparteína/debrisoquina, pode experimentar alta incidência de reação adversa.
t Meia-vida de eliminação: 5 a 8 horas; para os metabolizadores lentos 10 a 32 horas.
t Excreção: 38% renal, depuração endógena renal 1,3 L/kg/h; 53% nas fezes.
t Dialisável: não.
t Angina (5%), dor no peito (2%), fibrilação atrial (1%), bloqueio atrioventricular (0,2 a 2,5%), bradiarritmia (1,5%), insuficiência cardíaca congestiva (2 a 3%), bloqueio de ramo (1,2%), alteração do ritmo funcional do coração, alteração no eletrocardiograma (2%), bloqueio cardíaco, hipotensão (1%), palpitação (3%), disfunção do nodo sinusal, arritmia ventricular (5%).
t Exantema (3%), alopecia (1%), lupus eritematoso cutâneo subagudo.
t Obstipação (7%), diarreia (2,5%), alteração no paladar (7 a 14%), náusea/
vômito (11%), anorexia (2%), desconforto abdominal (2%), dispepsia (3%).
t Equimose (2 a 4%).
t Tontura (12%), fadiga (6%), ataxia (2%), tremor (1%), vertigem/cefaleia
(4%), neuropatia periférica.
t Visão borrada (4%).
t Impotência (menos de 1%).
t Broncoespasmo.
t Bepridil, cisaprida, levometadona, mesoridazina, pimozida, ritonavir, saquinavir, tipranavir, terfenadina, tioridazina, ziprasidona: uso concomitante contraindicado, pelo aumento do risco de cardiotoxicidade devido a efeito aditivo ou inibição do metabolismo.
t Bupropiona, clozapina, fluoxetina, paroxetina, sertralina e tolterodina: podem aumentar o efeito/toxicidade da propafenona. Monitorar sinais e sintomas específicos, principalmente cardiotoxicidade.
t Ciclosporina, digoxina, metoprolol, teofilina e varfarina: podem ter a efetividade/toxicidade aumentada. Monitorar sinais e sintomas específicos.
t Rifampicina e rifapentina: podem reduzir efetividade da propafenona. Monitorar sinais e sintomas específicos.
t Orientar para usar o medicamento mesmo que o paciente esteja sentindo-se bem. Não deixar de usar o medicamento sem falar com o médico.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver a menos de 4 horas do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar os comprimidos e a solução injetável entre 15 e 30 °C, proteger do calor, umidade e luz direta.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Compatível em glicose 5% na concentração de 1 e 2 mg/mL. Mais estável em seringa de polipropileno.
Atenção: terapia intravenosa é superior a dose única de 600 mg por via oral apenas nas primeiras duas horas de administração. Este fármaco apresenta um número elevado de Efeitos adversos , por isso é necessário uma pesquisa específica ao avaliar a terapia com este fármaco. Sinais/ sintomas de cardiotoxicidade: prolongamento do intervalo QT, torsades de pointes, parada cardíaca.
Rosa Martins
Na Rename 2010: itens 2.3, 14.2, 14.3, 14.4.2 e 18.2
t Comprimidos de 10 mg e 40 mg.
t Solução injetável 10 mg/mL.
t Profilaxia da enxaqueca.
t Arritmias cardíacas associadas a tirotoxicose, feocromocitoma, anestesia geral, exercício, emoção e uso de cocaína.
t Tratamento de cardiopatia isquêmica: angina e enfarte agudo do miocárdio.
t Hipertensão arterial sistêmica, em crianças.
t Hipersensibilidade ao propranolol.
t Hipotensão.
t Insuficiência cardíaca descompensada.
t Choque cardiogênico.
t Bradicardia sinusal grave.
t Sindrome do nó sinoatrial
t Bloqueio atrioventricular de 2º e 3º graus.
t Asma ou história de doença pulmonar obstrutiva crônica.
t Acidose metabólica.
t Angina de Prinzmetal
t Doença arterial periférica grave.
t Usar com cuidado em pacientes em uso de anestésicos que diminuam a função do miocárdio.
t Suspender o fármaco no decurso de 1 a 2 semanas. A suspensão súbita pode gerar efeito rebote, com piora de angina de peito, arritmias cardíacas e surgimento de enfarte do miocárdio.
t Deve ser utilizado com cautela em pacientes com história de insuficiência cardíaca congestiva, insuficiência cerebrovascular, doença vascular periférica, miastenia grave, bloqueio atrioventricular de 1º grau, hipertensão portal, reações de hipersensibilidade, hipertireoidismo/tirotoxicose, diabetes melito (pode mascarar sintomas de hipoglicemia).
t Usar com cuidado em pacientes com doença hepática (ver apêndice C) e insuficiência renal (não é necessário ajuste de dose).
t Quando usado por via intravenosa, fazer monitoria eletrocardiográfica e da pressão arterial.
t O risco de Efeitos adversos é aumentado em pacientes idosos. t Categoria de risco na gravidez (FDA): C e D (ver apêndice A).
t Lactação.
t A administração por via intramuscular não é recomendada.
t Abaixo de 35 kg: 10 a 20 mg, por via oral, a cada 8 horas.
t Acima de 35 kg: 20 a 40 mg, por via oral, a cada 8 horas.
t Neonatos: dose inicial 0,25 mg/kg, por via oral, a cada 8 horas, aumentando se necessário até no máximo 2 mg/kg a cada 8 horas.
t Crianças de 1 mês a 12 anos: dose inicial 0,25 a 1 mg/kg, por via oral, a cada 8 horas, aumentando se necessário até no máximo 5 mg/kg/dia, dividido a cada 8 horas.
t Crianças acima de 12 anos: 40 mg, por via oral, a cada 12 horas, aumentado gradualmente até 120 a 240 mg por dia, dividido a cada 8 ou 12 horas. Dose máxima diária: 320 mg.
t Neonatos:
t 0,25 a 0,50 mg/kg, por via oral, a cada 8 horas.
t 0,2 a 0,5 mg/kg, por via intravenosa lenta sob monitoria, a cada 6 ou 8 horas.
t Dose inicial 0,25 a 0,50 mg/kg, por via oral, a cada 6 ou 8 horas, ajustada conforme a resposta, até o máximo de 1 mg/kg, a cada 6 horas. Dose máxima diária: 160 mg.
t 0,25 a 0,5 mg/kg, por via intravenosa lenta sob monitoria, a cada 6 ou 8 horas.
t Dose inicial 40 mg, por via oral, a cada 8 ou 12 horas. Dose de manutenção: 80 a 160 mg/dia. Aumentar a dose no intervalo de uma semana. Dose máxima diária: 240 mg.
t 10 a 40 mg, por via oral, a cada 6 ou 8 horas.
t 1 a 3 mg, por via intravenosa, na velocidade de 1 mg/minuto. Se necessário, a segunda dose pode ser dada após 2 minutos da primeira, dose adicional não deve dada em menos de 4 horas. Dose máxima 10 mg (5 mg em anestesia).
t Dose inicial 40 mg, por via oral, a cada 8 ou 12 horas. Dose de manutenção: 120 a 240 mg/dia.
t Dose inicial 40 mg, por via oral, a cada 6 horas, durante 2 a 3 dias, seguido de 80 mg, por via oral, a cada 12 horas.
t Biodisponibilidade 30 a 70%. A presença de alimento aumenta a biodisponibilidade do propranolol.
t Início da ação: 1 a 2 horas (oral) e 2 a 10 minutos (intravenoso).
t Início da resposta anti-hipertensiva: 2 a 3 semanas.
t Pico de concentração (oral): 1 a 1,5 hora.
t Duração da ação: 6 horas (oral) e 10 a 15 minutos (intravenoso).
t Metabolismo hepático (50 a 70%), extenso metabolismo de primeira passagem; metabólitos inativos.
t Excreção: renal; menos de 1% é excretado em forma inalterada na urina.
t Meia-vida: 4 a 6 horas, podendo ser de 1,1 a 9,9 horas no uso prolongado. Aumento da meia-vida em recém-nascidos e lactentes.
t Não é removido por diálise.
t Distúrbios gastrintestinais
t Insuficiência cardíaca congestiva, hipotensão, bradicardia, transtorno na condução.
t Broncoespasmo, com piora de asma e DPOC.
t Claudicação intermitente, fenômeno de Raynaud.
t Depressão mental, insônia, pesadelos, fadiga (26%), cefaleia.
t Disfunção sexual.
t Aumento do risco de hipoglicemia em diabéticos insulino-dependentes.
t Agentes hipoglicemiantes, bloqueadores alfa-1-adrenérgicos (primeira dose), clonidina (retirada), clorpromazina (fenotiazinas), digitálicos e lidocaína: podem ter efeito/toxicidade aumentado pelo propranolol. Monitorar eletrocardiograma, pressão arterial, bem como sinais e sintomas específicos.
t Agonistas beta-2 adrenérgicos: podem ter o efeito diminuído pelo propranolol. Monitorar sinais e sintomas específicos.
t Antiácidos: podem diminuir o efeito do propranolol. Monitorar sinais e sintomas específicos.
t Amiodarona, bloqueadores de canais de cálcio do tipo di-hidropiridina, di-hidroergotamina, cimetidina, diltiazem, epinefrina, ergotamina, fenilefrina, fentanila, fluvoxamina, haloperidol, mefloquina, propoxifeno, quinidina, sertralina e verapamil: podem aumentar o efeito/toxicidade do propranolol. Monitorar função cardíaca, particularmente em pacientes predispostos a insuficiência cardíaca. Pode ser necessário ajuste de dose.
t Tioridazina: aumenta o risco de cardiotoxicidade. O uso concomitante é contraindicado.
t Orientar para a importância de comunicar ao perceber qualquer sinal de efeito adverso.
t Orientar para não suspender o uso do medicamento.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se o horário da próxima dose for a menos de 4 horas, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t O comprimido deve ser mantido ao abrigo de luz e umidade e à temperatura de 20 a 25 ºC.
t A solução injetável deve ser protegida da luz e é estável em pH 3. Ocorre rápida decomposição em pH alcalino.
t A solução injetável pode ser diluída em solução de cloreto de sódio 0,9% ou solução de glicose a 5%, mas é incompatível com bicarbonato. Verificar instruções do produtor quanto a diluição e administração.
t Formulação extemporânea para uso oral – 1 mg/mL. Preparar a partir de 10 comprimidos de 10 mg de propranolol. Triturar os comprimidos e adicionar lentamente o veículo para suspensão, composto por uma solução contendo etanol 1% e sacarina 0,05% em base aromatizada de polietilenoglicol 8000 (PEG 8000) 33%, em quantidade suficiente para volume final de 100 mL. Rotular: agitar bem antes de usar. Estabilidade: 4 meses à temperatura ambiente ou sob refrigeração.
Atenção: este fármaco apresenta um número elevado de Efeitos adversos, por isto deve ser realizada pesquisa específica sobre este aspecto, antes de introduzir ou descontinuar o propranolol ou outros medicamentos no esquema terapêutico do paciente.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 15.2
t Solução injetável a 10 mg/mL
t Antídoto para dose excessiva de heparina quando há hemorragia ou risco aumentado de hemorragia.
t Neutralização de heparina administrada durante circulação extracorpórea em cirurgias cardíaca e arterial ou procedimentos de diálise.
t Hipersensibilidade à protamina.
t Usar com cuidado nos casos de:
– tratamento prévio com protamina ou insulina-protamina, alergia a peixe e homens inférteis ou vasectomizados (maior risco de reação alérgica).
– após cirurgia cardíaca (monitorar ocorrência de efeito rebote da heparina e colapso circulatório; administrar doses adicionais de protamina).
– insuficiência hepática.
– insuficiência renal.
– cirurgia recente.
– lactação.
– crianças (segurança e eficácia do fármaco não foram estabelecidas).
t A administração muito rápida pode causar hipotensão e anafilaxia graves.
t Monitorar tempo de tromboplastina parcial ativada (TTPA), outros parâmetros de coagulação sanguínea e ajuste sanguínea de protamina para verificação de eficácia e cálculo da dose de protamina.
t Protamina possui efeito anticoagulante quando usada em excesso.
t Categoria de risco na gravidez (FDA): C.
t dose de 1 mg, infundido por via intravenosa durante 10 minutos, neutraliza 80 a 100 UI de heparina, até 15 minutos após administração da mesma.
t Transcorridos 15 a 30 minutos da administração de heparina, deve-se reduzir a dose de protamina à metade uma vez que a heparina já foi parcialmente depurada.
t Transcorridas 2 horas ou mais, a dose de protamina deve ser reduzida a 0,25 a 0,375 mg para cada 100 UI de heparina originalmente administrada.
t Não exceder 50 mg num período de 10 minutos (velocidade de administração de 5 mg/minuto).
t Considera-se que 1 mg neutraliza 100 UI de heparina; administrar 25 a 50 mg, por injeção intravenosa lenta (não excedendo 5 mg/min), seguidos de administração do restante da dose calculada, durante 8 a 16 horas, por infusão intravenosa.
t Não exceder 50 mg num período de 10 minutos (velocidade de administração de 5 mg/minuto).
t Dose de 25 a 50 mg, por via intravenosa, imediatamente após a interrupção da infusão contínua de heparina.
t Não exceder 50 mg num período de 10 minutos (velocidade de administração de 5 mg/minuto).
t Início de ação: 5 minutos (para neutralização de heparina)
t Hipotensão grave, bradicardia.
t Hipertensão pulmonar, dispneia.
t Rubor transitório e sensação de calor se administração rápida, alopecia, necrólise de pele, icterícia, dedos avermelhados.
t Náusea, vômito, pancreatite, diarreia, icterícia, disfunção hepática.
t Redução inexplicável do hematócrito, cansaço.
t Reforçar a importância de não utilizar em caso de reação alérgica prévia à protamina.
t Alertar para notificar sobre o uso de cefalosporinas ou penicilinas injetáveis.
t Manter sob refrigeração, de 2 a 8 °C. Estável por até 2 semanas à temperatura ambiente. Não congelar.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A solução injetável de protamina deve ser usada preferentemente sem diluição adicional, no entanto, se uma diluição adicional for desejada, os diluentes podem ser glicose 5% ou cloreto de sódio 0,9%. Soluções diluídas não devem ser estocadas por não conterem conservantes.
t Incompatibilidade: penicilinas e cefalosporinas.
Ana Cláudia de Brito Passos
Na Rename 2008: item 21.1
t Colírio 0,5%.
t Dor ocular aguda em trauma ocular não penetrante e procedimentos cirúrgicos.
t Anestesia para cirurgia corneana superficial, remoção de corpo estranho e remoção de suturas.
t Tonometria, procedimentos tonográficos e tonoscopia.
t Manipulação do sistema canalicular nasolacrimal.
t Hipersensibilidade à proximetacaína.
t Neonatos prematuros, devido à imaturidade do sistema enzimático metabolizador.
t Não fazer uso prolongado (risco de dano à córnea).
t Categoria de risco na gravidez (FDA): C.
t A segurança, efetividade e esquema de administração em crianças não estão bem estabelecidos, entretanto proximetacaína tem sido usada como anestésico tópico em crianças, pois, dentre os anestésicos locais, é o que causa menos ardor.
t Aplicar 1 ou 2 gotas antes do procedimento.
t Aplicar 1 ou 2 gotas a cada 5 a 10 minutos, até no máximo 3 aplicações, ou aplicar 1 ou 2 gotas 2 a 3 minutos antes do procedimento.
t Aplicar 1 gota a cada 5 a 10 minutos. Repetir a dose 5 a 7 vezes.
t Aplicar 1 ou 2 gotas antes do procedimento.
t Início da anestesia corneana em 20 ou 30 segundos após a aplicação, com duração de efeito de 15 minutos ou mais.
t Irritação e ardor local podem ocorrer várias horas após a aplicação.
t Reação corneana grave e imediata (rara).
t Reação hiperalérgica corneana imediata e aparente (rara).
t Queratite (grave reação hiperalérgica corneal).
t Dermatite de contato alérgica.
t Midríase.
t Exacerbação da Síndrome de Stevens-Johnson.
t Reação de hipersensibilidade retardada.
t Ataque tônico-clônico.
t Hialuronidase: os efeitos tóxicos dos anestésicos locais podem ser aumentados. O uso concomitante é contraindicado.
t Os pacientes devem ser advertidos para não tocar ou esfregar os olhos enquanto persistir anestesia e o olho anestesiado deve ser protegido de poeira e contaminação bacteriana.
t Os pacientes devem ser advertidos quanto ao uso prolongado de anestésicos oculares tópicos, já que tal uso pode causar opacificação corneal permanente e perda da visão.
t Informar ao paciente para não usar este medicamento se tiver tido uma reação alérgica à proximetacaína.
t Alertar aos pacientes para informar ao médico imediatamente se detectar algum destes efeitos secundários: reação alérgica (prurido ou urticária, inchaço em seu rosto ou mãos, inchaço ou formigamento na boca ou garganta, aperto no peito, dificuldade para respirar).
t Alertar aos pacientes para informar ao médico, caso detectar Efeitos adversos menos graves: visão turva, dor ou ardor, prurido, inchaço, hiperemia e lacrimejamento nos olhos.
t Conservar ao abrigo da luz e sob refrigeração (2 a 8 ºC). Soluções com coloração alterada não devem ser usadas.
Fabiana Wahl Hennigen
Na Rename 2010: itens 6.3 e 16.2
t Comprimido 150 mg.
t Solução injetável 25 mg/mL.
t Úlcera péptica de diversas etiologias e outras condições de hipersecreção gástrica, como síndrome de Zollinger-Ellison.
t Doença do refluxo gastresofágico.
t Esofagite erosiva.
t Dispepsia funcional.
t Hipersensibilidade à ranitidina.
t Usar com cuidado nos casos de:
– história de porfiria aguda.
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– crianças (segurança e eficácia não estão estabelecidas para tratamentos longos e para lactentes com menos de um mês).
– uso prolongado (pode causar deficiência de vitamina B12).
– terapia intravenosa prolongada (ocorre aumento de transaminases hepáticas).
– predisposição a distúrbios do ritmo cardíaco (administração intravenosa rápida pode produzir bradicardia).
– fenilcetonúricos (algumas formulações contêm fenilalanina).
– lactação (distribuído no leite, mas não se conhecem efeitos sobre o lactente).
t Pode mascarar sintomas de neoplasia gástrica. t Categoria de risco na gravidez (FDA): B.
t 2 a 4 mg/kg, por via oral, a cada 12 horas, durante 4 a 8 semanas. Dose máxima: 300 mg.
t 2 a 4 mg/kg, por via intravenosa, dividido a cada 6 ou 8 horas. Dose máxima: 200 mg.
t 2,5 a 5 mg/kg, por via oral, a cada 12 horas. Dose máxima: 300 mg/dia.
t 2,5 a 5 mg/kg/dia, por via oral, a cada 12 horas. Dose máxima: 600 mg/dia.
t Dose de 300 mg, por via oral, antes de dormir, durante 4 a 8 semanas.
t Dose de 150 mg, por via oral, a cada 12 horas. Dose máxima diária: 6 g.
t Dose de 50 mg, por via intravenosa ou intramuscular, a cada 6 a 8 horas, ou em infusão intravenosa contínua. Iniciar com 1 mg/kg/hora e aumentar 0,5 mg/kg/hora a cada 4 horas, até no máximo 2,5 mg/kg/hora.
t Dose de 150 mg, por via oral, a cada 12 horas, ou 300 mg antes de dormir, durante pelo menos 6 semanas. Dose de manutenção: 150 mg, por via oral, a cada 12 horas.
t Dose de 150 mg, por via oral, 4 vezes ao dia, durante até 12 semanas. Dose de manutenção: 150 mg, 2 vezes ao dia, por via oral.
t Dose de 150 mg, por via oral, a cada 12 ou 24 horas. Dose máxima diária: 300 mg.
t Injeção intramuscular: não diluir.
t Injeção intravenosa direta: diluir 50 mg em 20 mL de solução compatível, como cloreto de sódio 0,9%, glicose 5% ou Ringer + lactato. A concentração não deve ser superior a 2,5 mg/mL. Administrar por, pelo menos, 5 minutos, não excedendo a velocidade de 4 mL/minuto.
t Infusão intravenosa intermitente: diluir 50 mg em 100 mL de solução compatível. A concentração não deve ser superior a 0,5 mg/mL. Administrar por 15 a 20 minutos, não excedendo a velocidade de 5 a 7 mL/minuto.
t Infusão intravenosa contínua: diluir 150 mg em 250 mL de solução compatível. Administrar com velocidade de 6,25 mg/hora. Para uso em pacientes com síndrome de Zollinger-Ellison ou outras condições hipersecretórias, diluir a uma concentração não superior a 2,5 mg/mL.
t Absorção rápida (50%).
t Tempo para pico de efeito: oral: 2 a 3 horas; intramuscular: 15 minutos.
t Meia-vida de eliminação: oral: 2,5 a 3 horas; intravenosa: 2 a 2,5 horas.
t Duração de ação: oral: 4 a 12 horas (após dose única).
t Diarreia e outros distúrbios gastrintestinais, pancreatite aguda, enterocolite
necrosante no feto ou recém-nascido, insuficiência hepática.
t Bradiarritmia, vasculite.
t Pneumonia.
t Cefaleia, vertigem, alucinações, sonolência, confusão mental, cansaço, depressão.
t Exantema, eritema multiforme.
t Discrasias sanguíneas, incluindo agranulocitose, leucopenia, trombocitopenia, pancitopenia, anemia hemolítica adquirida, anemia aplásica e granulocitopenia.
t Anafilaxia e reações de hipersensibilidade.
t Atazanavir: redução das concentrações plasmáticas do atazanavir, podendo resultar em perda da eficácia ou desenvolvimento de resistência. Pacientes em início de tratamento devem usar atazanavir 400 mg (dose única com alimentos) pelo menos 2 horas antes ou 10 horas após o antagonista dos receptores H2 ou atazanavir 300 mg com ritonavir 100 mg (uma vez ao dia com alimentos) simultaneamente com o antagonista histamínico ou pelo menos 10 horas após a sua administração. Em pacientes com tratamento em andamento, atazanavir 300 mg/ritonavir 100 mg devem ser administrados uma vez ao dia, com alimentos, simultaneamente com o antagonista ou pelo menos 10 horas após.
t Cefpodoxima: redução da sua eficácia. Administrar a cefalosporina com as refeições para otimizar a absorção. Considerar a substituição da terapia antibiótica por outra cefalosporina de terceira geração ou uma de segunda com atividade semelhante. Se possível, substituir a ranitidina (por sucralfato, por exemplo) e usar a cefpodoxima, pelo menos, 2 horas antes. Monitorar o paciente para eficácia antibiótica.
t Dicumarol: aumento ou redução da eficácia anticoagulante. Monitorar o tempo de protrombina e, se necessário, ajustar a dose.
t Didanosina: aumento da concentração plasmática da didanosina e redução da concentração da ranitidina. Monitorar o paciente para sinais e sintomas de Efeitos adversos a didanosina e redução da eficácia da ranitidina.
t Enoxacino: o uso concomitante não é recomendado, pois pode ocorrer redução da eficácia do enoxacino. Ranitidina deve ser administrada, pelo menos, 2 horas após o enoxacino. Monitorar o paciente para eficácia antibiótica.
t Fosamprenavir: redução das concentrações plasmáticas do amprenavir (metabólito ativo) e potencial diminuição da sua eficácia. Ajustar a dose conforme necessário. Considerar o uso de um inibidor da bomba protônica como alternativa à ranitidina.
t Gefitinibe: redução das concentrações plasmáticas do gefitinibe. Monitorar.
t Glipizida: a administração concomitante não é recomendada, pois o paciente pode apresentar hipoglicemia. Monitorar a glicemia e reduzir a dose da glipizida conforme necessário. Considerar a substituição por outro antissecretor (como sucralfato) com menor potencial de interação.
t Itraconazol: em uso concomitante com a ranitidina pode haver redução da absorção do itraconazol, quando este é ingerido com água. Recomenda-se administrar o fármaco com uma bebida à base de cola.
t Risperidona: aumento da biodisponibilidade da risperidona. Monitorar o paciente para aumento de efeitos adversos, como sedação, acatisia, parkinsonismo, dispepsia, taquicardia, obstipação ou xerostomia.
t Tolazolina: redução da eficácia da tolazolina. Aumentar a dose da tolazolina ou descontinuar o uso da ranitidina.
t Varfarina: aumento do risco de sangramento. Monitorar o tempo de protrombina. Considerar o uso de um antagonista H2 com menor potencial de interação, como famotidina ou nizatidina.
t Alertar para a possibilidade de demora de alguns dias para o alívio da dor ulcerosa.
t Alertar para respeitar intervalo de uma a duas horas entre o uso do antiácido e o da ranitidina.
t Informar que alimentos não interferem na absorção do fármaco.
t Administrar o comprimido com um copo cheio de água.
t Reforçar para evitar alimentos, bebidas ou outros medicamentos que possam causar irritação gastrintestinal.
t Reforçar para evitar bebidas alcoólicas.
t Informar para suspender o tabagismo ou, pelo menos, não fumar após a última dose do dia.
t Armazenar os comprimidos a temperaturas entre 15 e 30 °C, proteger da luz e umidade.
t Armazenar a solução injetável entre 4 e 30 °C, proteger da luz e do congelamento. Leve escurecimento da solução não afeta sua eficácia.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t É estável por até 48 horas à temperatura ambiente, quando diluída em cloreto de sódio 0,9%, glicose 5% ou solução de Ringer + lactato.
Silvio Barberato Filho e Simone Sena Farina
Na Rename 2010: itens 5.1.10; 5.2.1 e 21.2
t Pomada oftálmica 1%.
t Tratamento de tracoma.
t Hipersensibilidade à tetraciclina.
Precaução
t Uso prolongado pode promover crescimento de microrganismos resistentes.
t Uma aplicação da pomada em cada olho, 2 vezes ao dia durante 5 dias, ou uma vez ao dia durante 10 dias, por 6 meses consecutivos a cada ano. Repetir o esquema, se necessário.
t Uma aplicação da pomada em cada olho, 2 vezes ao dia, durante 6 semanas, no mínimo.
t Exantema; raramente ardência, queimação local.
t Orientar para o provável aparecimento de visão borrada, que não deve ser persistente.
t Evitar contato com os olhos ou com outras superfícies durante a aplicação e manuseio. Limpar o bico do tubo após aplicação.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC, ao abrigo do ar e da luz. Não congelar.
Luciana dos Santos
Na Rename 2010: item 12
t Cloridrato de tiamina: comprimido de 300 mg e solução oral 10 mg/mL.
t Palmitato de tiamina: solução injetável 100.000 UI/mL (100 mg/mL).
t Prevenção e tratamento de deficiência da tiamina: pelagra, encefalopatia de Wernicke e síndrome de Korsakoff (em alcoólicos), neuropatia periférica (beribéri, grávidas).
t Distúrbios genéticos metabólicos.
t Hipersensibilidade a tiamina ou a qualquer componente da formulação
t Usar com cuidado nos casos de:
– insuficiência renal
– lactação.
t Reservar a via parenteral para casos de coma ou hipotermia de etiologia desconhecida, pois reações anafiláticas podem ocorrer durante ou imediatamente após administração intravenosa ou intramuscular.
t Injeções intravenosas devem ser lentas (em torno de 10 minutos); manter recursos para o tratamento de eventual anafilaxia.
t Realizar teste intradérmico antes da administração parenteral de tiamina em pacientes com suspeita de hipersensibilidade à mesma.
t Lactação: não deve ocorrer em situação de grave deficiência de tiamina.
t Categoria de risco na gravidez: A e C (quando a dose ultrapassar a recomendada) (ver Apêndice A).
t 50 a 200 mg/dia, por via oral ou infusão intravenosa lenta (em 30 minutos).
t Prevenção: 10 a 50 mg/dia, por via oral, durante 2 semanas; seguido de 5 a 10 mg/dia, por via oral, durante 1 mês.
t Tratamento: 10 a 25 mg/dia, por via oral.
t 100 a 300 mg/dia, por via oral ou infusão intravenosa lenta (em 30 minutos).
t 5 a 30 mg/dia, por via intramuscular ou infusão intravenosa lenta (acima de 30 minutos), divididos em 3 doses, por até 2 semanas; seguido de 5 a 30 mg/ dia, por via oral, em dose única ou divididos em 3 doses, por 1 mês.
t 5 a 10 mg/dia, por via intramuscular.
t 100 mg, por via intravenosa lenta (acima de 30 minutos). Manter 50 a 100 mg/dia, por via intramuscular ou infusão intravenosa lenta (acima de 30 minutos), até que se restabeleça a dieta oral. Em alguns casos, nas primeiras 12 horas, a dose poderá variar de 300 a 1000 mg.
t Dose de 10 a 20 mg/dia, por via oral (doses acima de 4 g/dia devem ser divididas).
t Biodisponibilidade oral: 5,3%
t Metabolismo: hepático.
t Excreção: renal.
t Reações de hipersensibilidade à injeção (choque anafilático).
t Angioedema, exantema (menos de 1%).
t Parestesia, sensação de ardor (menos de 1%).
t Ensinar que a tiamina pode ser encontrada em vegetais frescos, carnes e grãos.
t Alertar que bebidas alcoólicas podem diminuir a absorção de tiamina.
t Orientar para ingestão às refeições, de modo a aumentar a absorção.
t Informar que a urina poderá apresentar coloração amarelada.
t Em caso de esquecimento de dose, orientar para tomar a vitamina assim que lembrar. Não dobrar doses.
t Armazenar à temperatura entre 15 a 30 °C, e protegido da luz.
t A forma farmacêutica injetável pode ser administrada por via intravenosa ou intramuscular.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t É instável em soluções neutras ou alcalinas.
t Incompatibilidades: agentes oxidantes e redutores (sais de ferro), bicarbonato, carbonatos, citratos, eritromicina, metoexital, pentobarbital, fenobarbital, secobarbital, tiopental. Em soluções com sulfitos e bissulfitos se torna rapidamente inativo.
t Compatibilidades: solução de glicose 5%, glicose 10%, cloreto de sódio 0,9%, Ringer + lactato.
t A estabilidade das soluções é de aproximadamente 100 dias.
t Coloração azulada indica oxidação, sendo aconselhável o descarte da solução.
Maria Inês de Toledo e Lívia Luize Marengo
Na Rename 2010: item 5.1.8
t Pó para solução injetável 500 mg
t Infecções causadas por Staphylococcus aureus e Staphylococcus epidermidis, resistentes à meticilina (pneumonia, infecções relacionadas a cateter, meningite), e enterococo resistente.
t Hipersensibilidade a vancomicina ou componentes do produto.
t Usar com cuidado nos casos de:
– idosos, pacientes com insuficiência renal (ver Apêndice D) e neonatos (monitorar concentração plasmática da vancomicina; concentrações no pico devem ser mantidas em cerca de 30 microgramas/mL e no vale não podem ultrapassar 5 a 10 microgramas/mL).
– lactação.
t Evitar infusão rápida e alternar os locais de infusão para evitar tromboflebite.
t Monitorar função auditiva.
t Categoria de risco na gravidez (FDA): C.
t Dose inicial de 15 mg/kg, por via intravenosa, seguida de 10 mg/kg a cada 12 horas.
t 10 a 15 mg/kg, por via intravenosa, a cada 8 horas. Dose máxima: 2 g/dia.
t 500 mg, por via intravenosa, a cada 6 horas; ou 1.000 mg a cada 12 horas.
t 500 mg, por via intravenosa, a cada 12 horas; ou 1.000 mg a cada 24 horas.
Observação
t A velocidade de infusão intravenosa não deve exceder a 500 mg em 30 minutos. Isso evita risco de tromboflebite e reação sistêmica.
t Pico de concentração sérica: aproximadamente 63 microgramas/mL imediatamente após dose de 1 g infundida num período de 60 minutos.
t Meia-vida de eliminação: 4 a 11 horas (adultos); 6 a 10 horas (neonatos); 4 horas (recém-nascidos); 2 a 3 horas (crianças).
t Excreção: renal (80% a 90%).
t A vancomicina é dialisável. É pouco removida na hemodiálise convencional, mas ocorre redução significante da concentração em membranas de alto fluxo. Diálise peritoneal reduz a concentração, não significativamente, mas hemoperfusão e hemofiltração efetivamente removem a vancomicina do sangue.
t Comuns: febre, calafrio, flebite no sítio de infusão.
t Reação eritematosa, com prurido e hipotensão, acometendo face, pescoço e tronco, denominada de síndrome do pescoço vermelho (administração intravenosa rápida); exantema (5%), síndrome de Stevens-Johnson, urticária, prurido, necrólise tóxica epidérmica, dermatite exfoliativa.
t Dor e espasmos musculares em dorso e face anterior do tórax.
t Ototoxicidade e nefrotoxicidade (raras).
t Neutropenia, trombocitopenia, eosinofilia, agranulocitose.
t Colitepseudomembranosa(rara).
t Amicacina, anfotericina B, cisplatina, diuréticos de alça e gentamicina: aumento de toxicidade de vancomicina. Monitorar a função renal.
t Anestésicos gerais podem aumentar Efeitos adversos da vancomicina. Se o paciente necessitar anestésicos gerais, completar a infusão de vancomicina antes da indução da anestesia.
t Bloqueadores neuromusculares (suxametônio e vecurônio): podem ter seu efeito aumentado pela vancomicina. Ajustar cuidadosamente a dose do bloqueador neuromuscular e monitorar a função respiratória do paciente.
t Varfarina: aumento do risco de sangramento. Monitorar o tempo de protrombina quando introduzir ou descontinuar o antimicrobiano. Ajuste na dose da varfarina pode ser necessário.
t Informar imediatamente ao médico se houver redução da audição.
t Se o medicamento for injetado muito rápido pode ocorrer: pesadelo, ondas de calor, exantema nas mãos, pescoço e face.
t Manter o pó à temperatura ambiente, entre 15 e 30 ºC.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir o frasco de 500 mg em 10 mL de água estéril para injeção.
t Diluir com glicose a 5% ou cloreto de sódio 0,9% até 5 mg/mL, para infusão intermitente. A solução diluída pode ser guardada em geladeira por até 14 dias.
t Administrar em infusão lenta (60 minutos ou mais para 500 mg e 100 minutos para 1 g).
t Incompatível com soluções de pH alcalino ou com fármacos instáveis em pH baixo.
Rosa Martins
Na Rename 2010: itens 14.2, 14.3 e 14.4.3.
t Comprimidos 80 mg e 120 mg.
t Solução injetável 2,5 mg/mL.
t Taquiarritmias supraventriculares.
t Angina estável, instável e de Prinzmetal.
t Hipertensão arterial sistêmica.
t Hipersensibilidade ao fármaco ou a outros antagonistas do canal de cálcio.
t Hipotensão sintomática.
t Bradicardia.
t Bloqueio atrioventricular de segundo ou terceiro graus.
t Choque cardiogênico
t Insuficiência cardíaca descompensada
t Sindrome do nó sinoatrial
t Disfunção de nós sinusal e atrioventricular.
t Distúrbio de condução infranodal.
t Síndrome de Wolf-Parkinson-White.
t Obstipação crônica.
t Taquicardia supraventricular paroxística em menores de 2 anos.
t Usar com cuidado nos casos de:
– insuficiência ventricular esquerda, cardiomiopatia hipertrófica obstrutiva, bloqueio AV de primeiro grau, isquemia digital, ulceração e gangrena, estenose aórtica.
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– início de tratamento, aumento de dose ou durante retirada de betabloqueador (pode aumentar dor anginosa e/ou risco de enfarte do miocárdio).
– associação com bloqueadores neuromusculares não-despolarizantes (o verapamil retarda a recuperação do efeito desses fármacos).
– descontinuação (a retirada do verapamil deve ser gradual; se for abrupta, pode causar hipertensão de rebote).
– lactação (ver Apêndice B).
– tratamento recente com betabloqueadores adrenérgicos (evitar administração intravenosa em decorrência do risco de hipotensão e assistolia).
– uso intravenoso em crianças.
t Categoria de risco na gravidez (FDA): C.
t Até 1 ano: 0,1 a 0,2 mg/kg, por via intravenosa durante 2 minutos (monitorar com ECG). Se não houver resposta, repetir a dose após 30 minutos.
t De 1 a 15 anos: 0,1 a 0,3 mg/kg (máximo 5 mg), por via intravenosa durante 2 minutos (monitorar com ECG). Se não houver resposta, repetir a dose após 30 minutos (máximo 10 mg).
t 40 a 120 mg, por via oral, a cada 8 horas.
t 5 a 10 mg, por via intravenosa durante 2 minutos (monitorar com ECG). Se não houver resposta, administrar mais10 mg após 30 minutos. Dose máxima: 20 mg.
t 80 a 120 mg, por via oral, a cada 8 horas.
t Dose inicial 80 a 120 mg, por via oral, a cada 8 ou 12 horas. Dose máxima: 480 mg/dia.
t 5 a 10 mg, por via intravenosa durante 3 minutos (monitorar com ECG). Se não houver resposta, administrar mais 10 mg após 30 minutos.
t Dose inicial 40 mg, por via oral, a cada 8 horas.
t Alimento não interfere na absorção
t Biodisponibilidade após administração oral 20 a 35%
t Início de efeito: 30-60 minutos (oral) e 1 a 5 minutos (intravenoso).
t Pico sérico: 1 a 2 horas (oral).
t Duração: 6-8 horas (oral) e 10 a 20 minutos (intravenoso).
t Metabolismo hepático 65 a 80%, via citocromo P450, metabólito ativo. Extenso metabolismo de primeira passagem.
t Excreção: 70% renal, fecal 9 a 16%.
t Meia-vida de eliminação: 2 a 8 horas (dose única); mais de 12 horas (doses repetidas)
t Não dialisável
t Bradicardia (1,4%), depressão da contratilidade miocárdica, hipotensão (1,5-3%), bloqueio atrioventricular (1,2%), edema periférico (1,9%), insuficiência cardíaca congestiva ou edema pulmonar (1,8%).
t Obstipação intestinal (12-42%).
t Hiperplasia gengival (19%).
t Exantema (1,2%), enrubescimento (10 a 20%).
t Dispneia (3%), tosse (5%).
t Ácido acetilsalicílico, álcool, betabloqueadores adrenérgicos, benzodiaze-pínicos, bloqueadores de canais de cálcio, bloqueadores neuromusculares, carbamazepina, ciclosporina, colchicina, digoxina, quinidina, sinvastatina, sirolimo podem ter o efeito/toxicidade aumentado pelo verapamil. Pode ser necessária a redução de dose. Monitorar sinais e sintomas específicos.
t Anestésicos, antifúngicos azólicos, inibidores de protease (indinavir, ritonavir, saquinavir), macrolídeos, tetraciclinas podem aumentar o efeito/toxicidade do verapamil. Monitorar função cardíaca e pressão arterial; retirar ou ajustar a dose dos medicamentos; monitorar sinais e sintomas específicos.
t Dofetilida: pode haver aumento da cardiotoxicidade. O uso concomitante é contraindicado.
t Fenitoína, fenobarbital, nevirapina e rifamicinas podem diminuir o efeito do verapamil. Monitorar sinais e sintomas específicos, aumento da dose pode ser necessário.
t Lítio e oxcarbazepina podem ter o efeito/toxicidade diminuido pelo verapamil. Monitorar sinais e sintomas específicos.
t Orientar para manter boa higiene dental e fazer consultas frequentes ao dentista.
t Orientar para a importância de comunicar ao perceber qualquer sinal de efeito adverso e, ainda, hipotensão.
t Alertar para limitar a ingestão de cafeína e álcool durante o tratamento.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Reforçar que as doses esquecidas não devem ser dobradas.
t Orientar para mudar lentamente de posição durante a terapia para evitar hipotensão postural.
t Ensinar que os comprimidos não devem ser macerados ou mastigados, devendo ser ingeridos inteiros e com alimentos para diminuir a irritação gástrica.
t O armazenamento de comprimidos e da solução injetável deve ser feito sob temperatura entre 15 a 25 ºC e protegido da luz.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Solução é estável a pH entre 3 e 7.
Atenção: este fármaco apresenta um número muito elevado de Efeitos adversos , sendo necessária uma pesquisa específica sobre este aspecto antes de considerar a introdução ou descontinuação do verapamil ou de outros medicamentos no esquema terapêutico do paciente. Sugere-se considerar, além dos medicamentos individualmente, os grupos na avaliação da interação. O efeito adverso edema periférico ocorre dentro de 2 a 3 semanas após o início do tratamento.
Consta no documento:
“Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial.”
O objetivo do site MedicinaNet e seus editores é divulgar este importante documento. Esta reprodução permanecerá aberta para não assinantes indefinidamente.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.