(Carregando Índice)... (Carregando Índice)... |
Última revisão: 11/11/2015
Comentários de assinantes: 0
Reproduzido de:
Formulário Terapêutico Nacional 2010: Rename 2010 [Link Livre para o Documento Original]
Série B. Textos Básicos de Saúde
MINISTÉRIO DA SAÚDE
Secretaria de Ciência, Tecnologia e Insumos Estratégicos
Departamento de Assistência Farmacêutica e Insumos Estratégicos
Brasília / DF – 2010
Fabiana Wahl Hennigen
Na Rename 2010: seção 16.6
t Xarope 667 mg/mL
t Agente coadjuvante em prevenção e tratamento de encefalopatia porto-sistêmica, incluindo pré-coma e coma hepáticos.
t Hipersensibilidade à lactulose.
t Galactosemia.
t Pacientes em dieta com restrição de galactose.
t Obstrução intestinal.
t Usar com cuidado nos casos de:
– diabete melito e intolerância à lactose.
– diarreia após a dose inicial (reduzir a dose imediatamente e descontinuar se a diarreia persistir).
– uso por mais de 6 meses ou em pacientes predispostos a anormalidades eletrolíticas, como idosos (monitorar as concentrações séricas de eletrólitos).
– uso concomitante com anti-infecciosos orais (monitorar para possível inadequação da resposta à lactulose).
– lactação.
– crianças (não há dados sobre eficácia e segurança para profilaxia e tratamento de encefalopatia hepática).
– uso concomitante com outros laxativos (não associar, pois a evacuação com fezes moles resultante pode falsamente sugerir a obtenção da dose adequada de lactulose).
t Categoria de risco na gravidez (FDA): B.
t Dose inicial de 20 a 30 g (30 a 45 mL), por via oral, a cada 6 ou 8 horas, ajustar a dose a cada 1 ou 2 dias para produzir 2 a 3 evacuações com fezes moles por dia. Dose usual: 60 a 100 g/dia (90 a 150 mL/dia). No manejo de episódios agudos, 20 a 30 g, a cada 1 a 2 horas para induzir rápida diarreia.
t Diluir em água, usualmente 60 a 120 mL, para administração por sonda gástrica.
t Quando usado via retal durante os estágios de pré-coma ou coma hepáticos, diluir 200 g (300 mL) com 700 mL de água ou cloreto de sódio 0,9%. A solução diluída deve ser administrada por meio de sonda retal e retida por 30 a 60 minutos, a cada 4 a 6 horas.
t Pouco absorvido (menos que 3% da dose).
t Tempo para início de ação: 24-48 horas.
t Lactulose não absorvida é metabolizada pelas bactérias do cólon, formando ácido lático e ácido acético.
t Lactulose absorvida não é metabolizada e é excretada inalterada na urina em até 24 horas.
t Comuns: flatulência, diarreia (dose alta), desconforto abdominal, náusea, vômito e cãibras.
t Graves: hipernatremia e hipopotassemia.
t Anticoagulantes cumarínicos orais têm seu efeito aumentado. Monitorar o tempo de protrombina e, se necessário, ajustar a dose do anticoagulante.
t Orientar que a solução pode ser misturada com suco de fruta, água, leite ou alimentos para melhorar o sabor.
t Orientar para ingerir com 250 mL de líquido e, pelo menos, 6 a 8 copos de líquido por dia, para auxiliar o amolecimento das fezes.
t Informar que pode ser necessário aguardar de 1 a 3 dias para obtenção de melhora clínica.
t Conservar sob temperatura ambiente (15 a 30° C) para reduzir a viscosidade.
Proteger do congelamento.
t Descartar a solução se estiver opaca ou muito escura.
Julia Salvan da Rosa
Na Rename 2010: item 5.5.2.1
t Comprimido 150 mg
t Solução oral 10 mg/mL
t Tratamento de infecção por HIV (em combinação com outros fármacos antirretrovirais)
t Prevenção de transmissão materno-fetal do HIV (em combinação com zidovudina)
t Tratamento de infecção crônica por hepatite B
t Hipersensibilidade à lamivudina.
t Usar com cuidado nos casos de:
– obesos, com insuficiência hepática (ver Apêndice C) ou fatores de risco para doença hepática, hepatites B ou C.
– coinfectados com HIV/HCV, recebendo terapia antirretroviral em combinação com interferona 1 alfacona e ribavirina (aumento do risco de hepatotoxicidade).
– pacientes com hepatite B crônica (pode haver recorrência após suspensão de lamivudina).
– crianças com história de pancreatite ou fatores de risco para desenvolvimento da doença.
– insuficiência renal (ver Apêndice D).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Dose de 2 mg/kg (associados a 4 mg/kg de zidovudina), por via oral, a cada 12 horas, durante 7 dias.
t De 3 meses a 12 anos: 4 mg/kg, por via oral, a cada 12 horas. Dose máxima: 300 mg/dia.
t De 2 a 11 anos: 3 mg/kg, por via oral, uma vez ao dia. Dose máxima: 100 mg/dia.
t Com mais de 50 kg: 150 mg, por via oral, a cada 12 horas ou 300 mg, uma vez ao dia.
t Com menos de 50 kg: 2 mg/kg, por via oral, a cada 12 horas.
t Mãe: 150 mg (associados a 600 mg de zidovudina), por via oral, no início do trabalho de parto, seguidos de 150 mg a cada 12 horas (associados a 300 mg de zidovudina, a cada 3 horas) até o parto. Após o parto, lamivudina 150 mg a cada 12 horas durante 7 dias.
t Dose de 100 mg/dia, por via oral, a cada 24 horas.
t Início da resposta: 4 a 8 semanas.
t Meia-vida: 5 a 7 horas, 2 horas (crianças), 2,3 horas (grávidas), 14 horas (neonatos).
t Excreção: renal (70%, em forma inalterada).
t É removida por diálise, porém, não é necessária dose adicional de lamivudina.
t Exantema e/ou prurido (10%), alopecia.
t Cefaleia (35%), fadiga (27%), depressão (9%), insônia/distúrbios do sono (11%), tontura (10%) neuropatia (12%).
t Dispepsia (5%), dor abdominal (9%), anorexia (10%), náusea e vômitos (13%), diarreia (18%), pancreatite em adultos (0,3%) e crianças (14% a 18%).
t Lipodistrofia, hiperglicemia, hepatotoxicidade, acidose lática (com grave hepatomegalia e esteatose).
t Neutropenia, anemia, trombocitopenia.
t Artralgia (5%), mialgia (8%), dor musculoesquelética (12%), rabdomiólise.
t Ototoxicidade.
t Calafrios ou febre (10%)
t Tosse (18%).
t Interferona 1 alfacona e ribavirina: aumentam o risco de acidose lática (potencialmente fatal) e descompensação hepática pela lamivudina. Monitorar os pacientes e avaliar a relação risco-benefício. Se for apropriado, descontinuar a lamivudina, ou reduzir/descontinuar a dose da ribavirina e/ou interferona.
t Sulfametoxazol + trimetoprima: aumentam o efeito/toxicidade da lamivudina, podendo intensificar os efeitos adversos. Monitorar os pacientes que usam esta associação. Alterações nas doses destes fármacos não são indicadas.
t Zalcitabina: diminui o efeito da lamivudina. A administração concomitante não é recomendada.
t Orientar para a possibilidade de administrar o medicamento concomitantemente ou não com alimentos.
t Procurar um serviço de saúde caso tenha dor estomacal repentina, náusea, vômito, febre. Pode ser sinal de pancreatite.
t Reforçar orientações sobre prevenção da transmissão do HIV.
t Manter os comprimidos e solução oral na embalagem original bem fechadas, ao abrigo de ar, luz e umidade e à temperatura ambiente, entre 15 e 30 ºC.
Atenção: como sinonímia para lamivudina (nome que corresponde a denominação comum Brasileira) também é empregada a abreviatura 3TC, entretanto, não se recomenda a prescrição de fármacos por abreviaturas ou siglas.
José Gilberto Pereira
Na Rename 2010: item 13.3
t Comprimido 50 mg + 12,5 mg t Comprimido 100 mg + 25 mg
t Doença de Parkinson.
t Hipersensibilidade conhecida a levodopa ou benserazida.
t Lesões de pele não diagnosticadas, melanoma maligno ou história prévia desta doença.
t Glaucoma de ângulo fechado.
t Uso concomitante de reserpina e inibidores da monoamina oxidase (inclusive duas semanas antes de iniciar levodopa+benserazida).
t Usar com cuidado nos casos de:
– doenças pulmonares.
– úlcera péptica ativa.
– doenças graves hepáticas, renais, cardiovasculares e medulares ósseas.
– diabete melito, hipertireoidismo, feocromocitoma, osteomalácia.
– depressão e outras doenças psiquiátricas graves.
– glaucoma de ângulo aberto.
– retirada do medicamento (deve ser gradual para reduzir risco de síndrome neuroléptica maligna e rabdomiólise).
– anestesia com narcóticos (suspender o medicamento pelo menos oito horas antes da anestesia).
– idosos (introduzir com incrementos graduais das doses).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t De 100 a 200 mg de levodopa e 25 a 50 mg de benserazida (razão 4:1), por via oral, duas vezes ao dia. A dose diária pode ser aumentada gradualmente com 50 a 100 mg de levodopa e 12,5 a 25 mg de benserazida a cada 3 a 7 dias em 3 a 4 doses divididas. Durante a progressão da doença há necessidade de aumento de dose para manutenção da eficácia clínica; não exceder a dose diária de 800 mg de levodopa e 200 mg de benserazida.
t Dose inicial de 50 mg de levodopa e 12,5 mg de benserazida, por via oral, uma ou duas vezes ao dia. Aumentar a dose diária em 50 mg de levodopa e 12,5 mg de benserazida a cada 3 a 4 dias, de acordo com a resposta clínica.
t Absorção: cerca de 60% é rapidamente absorvido pelo trato gastrintestinal; alimento reduz em 15% a absorção.
t Pico de resposta clínica ocorre até o final do primeiro mês de tratamento.
t Metabolismo: predominantemente no trato gastrintestinal e em menor extensão pelo fígado.
t Excreção: renal (90%).
t Meia-vida de eliminação da levodopa é de 1,5 a 2 horas, aumentada em 25% nos idosos. A depuração encontra-se reduzida nesta faixa etária.
t Arritmias cardíacas e hipotensão ortostática (ocasionais); hipertensão, dor no peito e flebites (menor frequência que os primeiros).
t Prurido e exantema; recorrência primária de melanoma maligno.
t Hiperprolactinemia e aumento de TSH.
t Síndrome neuroléptica maligna tem sido reportada durante a retirada abrupta da levodopa+benserazida.
t Elevação do ácido xanturênico foi observada em indivíduos com deficiência de vitamina B6; Náuseas e vômitos (frequentes, especialmente no início do tratamento); sialorreia, disfagia, flatulência, anorexia, disgeusia e diarreia (ocasionais); sangramentos gastrintestinais e dispepsia (raros).
t Leucopenia, trombocitopenia, redução do tempo de tromboplastina, anemia hemolítica e não hemolítica (raros).
t Elevações de bilirrubina e fosfatase alcalina.
t Agitação, ansiedade, distúrbios do sono e depressão (frequentes); o uso prolongado pode levar a desorientação, confusão mental, ilusão e discinesia; convulsões podem ocorrer em indivíduos com déficit renal.
t Indução da dopa-descarboxilase ocorre gradualmente entre o terceiro e quarto mês de tratamento com levodopa+benserazida, podendo ser necessária readequação de dose.
t Perda de resposta clínica após vários anos de tratamento, relacionada à progressão da doença.
t Disfunção sexual, retenção e/ou incontinência urinária (raros).
t Bromperidol, droperidol, fenilalanina, fenitoína e fosfenitoína, kava-kava, sais de ferro, tirosina: podem reduzir a efetividade; monitorar eficácia terapêutica da levodopa+benserazida e aumentar a dose desta se necessário.
t Bupropiona e indinavir aumentam os efeitos adversos; monitorar Efeitos adversos da associação levodopa + benserazida e reduzir a dose desta se necessário.
t Espiramicina reduz a concentração sérica com perda dos efeitos antiparkinsonianos; ajuste da dose de levodopa+benserazida pode ser necessário.
t Inibidores da MAO e linezolida: o uso concomitante é contraindicado.
t Metoclopramida tem seus efeitos extrapiramidais aumentados; o uso concomitante deve ser evitado.
t Orientar para o relato de alergias e uso concomitante de outros medicamentos.
t Orientar para o relato de comorbidades, particularmente diabetes melito, glaucoma, câncer de pele, doenças mentais, doenças dos rins, fígado e pulmão.
t Orientar para tomada do medicamento longe das refeições e, particularmente de alimentos ricos em proteínas; contudo, somente para adaptação ao início do tratamento, pode-se recomendar a tomada com alimentos.
t Recomendar dieta rica em vitamina B6 (bananas, ovos galados, ervilha, carnes, amendoim e cereais integrais).
t Orientar sobre o tempo até que surjam efeitos significativos no controle da doença (cerca de até 30 dias).
t Alertar sobre a necessidade de seguir rigorosamente a dose e os horários de tomada do medicamento; e não interromper abruptamente o seu uso.
t Orientar para o caso de esquecimento da dose, a mesma deverá ser desconsiderada se faltarem duas horas ou menos até a próxima dose, nunca duplicar a dose.
t Orientar para a guarda do medicamento sempre longe do alcance das crianças.
t Orientar os pacientes diabéticos que o medicamento interfere no resultado dos testes de glicose e corpos cetônicos na urina.
t Orientar que o medicamento diminui os reflexos e que ao executar atividades como dirigir ou operar máquinas perigosas poderá expor a riscos de acidentes.
t Orientar que o uso do medicamento poderá dar tonalidade escura à saliva, urina e suor; sabor amargo e sensação de queimação na língua poderão estar presentes.
t Armazenar o medicamento sob temperatura de 15 oC e 30 oC, em embalagens bem fechadas, protegido da luz, calor e umidade. Não colocar em geladeira ou no congelador.
Atenção: no início do tratamento pode ocorrer sedação excessiva e sono de início súbito; alertar o paciente para execução de atividades que requerem atenção e reflexos rápidos, como dirigir e/ou operar máquinas perigosas.
Jose Gilberto Pereira
Na Rename 2010: item 13.3
t comprimido 250 mg + 25 mg t comprimido 100 mg + 25 mg
t comprimido 100 mg + 10 mg
t Doença de Parkinson.
t Outras formas de parkinsonismo não induzidas por fármacos.
t Hipersensibilidade conhecida a levodopa ou carbidopa.
t Lesões de pele não diagnosticadas, melanoma maligno ou história prévia desta doença.
t Glaucoma de ângulo fechado.
t Uso concomitante de reserpina e inibidores da monoamina oxidase (inclusive duas semanas antes de iniciar levodopa + carbidopa).
t Usar com cuidado nos casos de:
– doenças pulmonares.
– úlcera péptica ativa.
– doenças graves hepáticas, renais e cardiovasculares.
– diabete melito, disfunções hormonais hipotalâmicas e hipofisiárias.
– depressão e outras doenças psiquiátricas graves.
– glaucoma de ângulo aberto.
– retirada do medicamento (deve ser gradual para reduzir risco de síndrome neuroléptica maligna e rabdomiólise).
– lactação (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Inicialmente, levodopa 100 mg + carbidopa 25 mg, três vezes ao dia, aumentando diariamente em 50 a 100 mg de levodopa + 12,5 a 25 mg de carbidopa, ou em dias alternados de acordo com a resposta clínica, até o máximo de 800 mg de levodopa + 80 a 100 mg de carbidopa diariamente em doses divididas.
t Ou então, iniciar com levodopa 50 a 100 mg + carbidopa 10 a 12,5 mg, três a quatro vezes ao dia, aumentando diariamente em 50 a 100 mg de levodopa + 12,5 a 25 mg de carbidopa, ou em dias alternados de acordo com a resposta clínica, até o máximo de 800 mg de levodopa + 80 a 100 mg de carbidopa diariamente em doses divididas.
t Ou então, iniciar com levodopa 125 mg + carbidopa 12,5 mg, uma a duas vezes ao dia, aumentando diariamente em 125 mg de levodopa + 12,5 mg de carbidopa, ou em dias alternados de acordo com a resposta clínica.
Nota: dose mínima diária de 70 mg de carbidopa é necessária para a inibição completa da dopa-descarboxilase periférica. A dose de carbidopa deve ser estabelecida de acordo com a presença e intensidade de Efeitos adversos como náuseas e vômitos.
t Absorção: cerca de 90% da levodopa é rapidamente absorvido pelo trato gastrintestinal; alimento reduz em 30% a absorção.
t Distribuição: apenas 10% a 20% da levodopa plasmática alcança o líquido cérebro-espinhal.
t Pico de resposta clínica ocorre até o final de três semanas de tratamento.
t Metabolismo: predominantemente no trato gastrintestinal e em menor extensão pelo fígado; carbidopa reduz o efeito de primeira passagem da levodopa, aumentando a biodisponibilidade desta.
t Excreção: renal (levodopa 70% a 80%, carbidopa 30%).
t Meia-vida de eliminação da levodopa é de 1,5 a 2 horas.
t Arritmias cardíacas e hipotensão ortostática (ocasionais); hipertensão e enfarte agudo do miocárdio (menos frequentes que os primeiros).
t Exantema, alopecia, alterações na coloração da pele, cabelos e unhas, rápido crescimento das unhas e diminuição da secreção sebácea; recorrência primária de melanoma maligno.
t Angioedema, urticária e prurido (raros).
t Artrite gotosa, ocronose cartilaginosa, dores nas costas e nos ombros, cãibras musculares (raros).
t Hiperprolactinemia e aumento de TSH; hiperuricemia (menos frequente).
t Náuseas e vômitos (frequentes, especialmente no início do tratamento); sialorreia, disfagia, flatulência, anorexia, disgeusia e diarreia (ocasionais); sangramentos gastrintestinais e dispepsia (raros).
t Leucopenia, trombocitopenia, redução do tempo de tromboplastina, anemia hemolítica e não hemolítica (raros).
t Elevações de aspartato aminotransferase, bilirrubina e fosfatase alcalina.
t Discinesia, incluindo movimentos coreiformes e distonia (efeitos frequentes, 30% a 80%, graves e dependentes da dose); agitação, ansiedade, distúrbios do sono e depressão (frequentes); o uso prolongado pode levar a desorientação, confusão mental, ilusão e flutuação dos sintomas da doença.
t Síndrome neuroléptica maligna tem sido reportada durante a retirada abrupta da levodopa+carbidopa.
t Visão borrada, diplopia, blefaroespasmo e crises oculogiras (raros).
t Perda de resposta clínica após vários anos de tratamento, relacionada à progressão da doença.
t Disfunção sexual, retenção e/ou incontinência urinária (raros).
t Bromperidol, droperidol, fenilalanina, fenitoína e fosfenitoína, kava-kava, sais de ferro, tirosina: podem reduzir a efetividade; monitorar eficácia terapêutica da levodopa + carbidopa e aumentar a dose desta se necessário.
t Bupropiona e indinavir aumentam os efeitos adversos; monitorar Efeitos adversos da associação levodopa + carbidopa e reduzir a dose desta se necessário.
t Espiramicina reduz a concentração sérica com perda dos efeitos antiparkinsonianos; ajuste da dose de levodopa + carbidopa pode ser necessário.
t Inibidores da MAO e linezolida: o uso concomitante é contraindicado.
t Metoclopramida tem seus efeitos extrapiramidais aumentados; o uso concomitante deve ser evitado.
t Orientar para o relato de alergias e uso concomitante de outros medicamentos.
t Orientar para o relato de comorbidades, particularmente diabetes melito, glaucoma, câncer de pele, doenças mentais, doenças dos rins, fígado e pulmão.
t Orientar para tomada do medicamento longe das refeições e, particularmente de alimentos ricos em proteínas, contudo, somente para adaptação ao início do tratamento, pode-se recomendar a tomada com alimentos.
t Orientar sobre o tempo até que surjam efeitos significativos no controle da doença (cerca de até três semanas).
t Alertar sobre a necessidade de seguir rigorosamente a dose e os horários de tomada do medicamento, e não interromper abruptamente o seu uso.
t Orientar para o caso de esquecimento da dose, a mesma deverá ser desconsiderada se faltarem duas horas ou menos até a próxima dose, nunca duplicar a dose.
t Orientar para manter o medicamento sempre longe do alcance das crianças.
t Orientar os pacientes diabéticos que o medicamento interfere no resultado dos testes de glicose e corpos cetônicos na urina
t Orientar que o medicamento diminui os reflexos e que ao executar atividades como dirigir ou operar máquinas perigosas poderá expor a riscos de acidentes.
t Armazenar o medicamento sob temperatura de 15oC e 30oC, em embalagens bem fechadas, protegido da luz, calor e umidade.
t Solução extemporânea de levodopa+carbidopa pode ser preparada com 10 comprimidos (100/25 mg ou 100/10 mg) e 2 g de ácido ascórbico em pó para 1 litro de água potável, agitar suavemente por 5 minutos e filtrar. A solução permanece estável por 24 horas à temperatura ambiente e por 7 dias sob refrigeração. Descartar a solução se houver descoloração ou escurecimento.
Atenção: no início do tratamento pode ocorrer sedação excessiva e sono de início súbito; alertar o paciente para execução de atividades que requerem atenção e reflexos rápidos, como dirigir e/ou operar máquinas perigosas.
Karen Luise Lang
Na Rename 2010: item 18.4.4
t Comprimido 1,5 mg.
t Contracepção de emergência.
t Hipersensibilidade a qualquer componente da fórmula.
t Sangramento genital de etiologia desconhecida.
t Porfiria.
t Doença arterial grave, distúrbios tromboembólicos
t Gravidez. Fator de risco na gravidez (FDA): X (ver apêndice A).
t Usar com cuidado nos casos de:
– diabetes.
– insuficiência hepática (ver Apêndice C).
– ocorrência de vômito em até 2 horas após a administração (a dose poderá ser repetida; se necessário, administrar antiemético).
t Não é recomendado antes da menarca, mas pode ser usado durante o ciclo menstrual.
t Dose única de 1,5 mg, por via oral, ingerida preferentemente até 72 horas após relação sexual desprotegida. Eficácia ainda se mantém até 120 horas após o intercurso sexual desprotegido.
t Metabolismo: hepático.
t Meia-vida de eliminação: 43 horas.
t Excreção: preponderantemente renal.
t Náusea, vômito, diarreia, dor abdominal, alterações no apetite.
t Cefaleia, vertigens, fadiga.
t Irregularidades menstruais, sensibilidade mamária, cistos ovarianos.
t Fosfenitoína pode induzir o metabolismo de levonorgestrel, com redução da eficácia.
t Tacrina pode ter incidência de Efeitos adversos aumentada pelo levonorgestrel. Avaliar redução da dose da tacrina.
t Orientar para ingerir no máximo até 72 horas após relação sexual desprotegida, para obter melhor efetividade.
t Orientar para ingerir o comprimido com alimento ou leite para evitar desconforto gástrico.
t Orientar para o caso de ocorrência de vômito em até 2 horas após tomar o comprimido; se ocorrer, repetir a dose.
t Alertar para a possibilidade de atraso ou adiantamento da próxima menstruação. Recomendar uso de um método de barreira até a próxima menstruação.
t Alertar sobre a necessidade de notificar dor no baixo ventre, que pode ser sinal de gravidez ectópica.
t Alertar para não adotar o método de proteção contraceptiva de emergência como método regular de controle de natalidade.
t Informar que não protege contra infecção por HIV ou qualquer doença sexualmente transmissível e que não é eficaz para interromper gravidez existente.
t Armazenar sob temperatura ambiente, em recipiente bem fechado.
Cláudia Du Bocage Santos Pinto
Marcela de Andrade Conti
Na Rename 2010: item 18.2
t Comprimidos de 25 microgramas, 50 microgramas e 100 microgramas
t Tratamento de manutenção em hipotireoidismo.
t Supressão da secreção de hormônio estimulante da tireoide (TSH), em situações específicas, como nos carcinomas diferenciados da tireoide.
t Tireotoxicose.
t Hipersensibilidade a hormônios tireoidianos.
t Enfarte do miocárdio recente.
t Angina e hipertensão arterial não tratadas.
t Insuficiência suprarrenal não corrigida.
t Tratamento da obesidade ou emagrecimento.
t Usar com cuidado nos casos de:
– doenças cardiovasculares, como hipertensão, insuficiência cardíaca e enfarte do miocárdio (a dose inicial deve ser reduzida à metade e o ajuste deve ser lento e gradativo).
– pan-hipopituitarismo ou predisposição à insuficiência suprarrenal (iniciar tratamento com corticoides antes de introduzir levotiroxina).
– hipotireoidismo de longa data.
– diabetes insipidus ou melito (provável necessidade de aumentar a dose de insulina ou de antidiabéticos orais).
– idosos (maior sensibilidade aos efeitos dos hormônios tireoidianos; introduzir de forma gradual, em dose 25% mais baixa em relação a adultos jovens).
– mulheres em pré-menopausa (risco de osteoporose; utilizar a menor dose possível).
t Categoria de risco na gravidez (FDA): A (ver Apêndice A).
t Neonatos: dose inicial de 5 a 10 microgramas/kg/dia, via oral. Quando houver risco de falência cardíaca, deve-se considerar o uso de doses abaixo de 25 microgramas/dia.
t Até 12 anos:
t 0 a 3 meses: 10 a 15 microgramas/kg/dia.
t 3 a 6 meses: 8 a 10 microgramas/kg/dia.
t 6 a 12 meses: 6 a 8 microgramas/kg/dia.
t 1 a 5 anos: 5 a 6 microgramas/kg/dia.
t 6 a 12 anos: 4 a 5 microgramas/kg/dia.
t Acima de 12 anos: 2 a 3 microgramas/kg/dia.
t O ajuste de 25 microgramas/dia deve ser feito a cada 2 a 4 semanas. Em maiores, pode-se reduzir a hiperatividade iniciando o tratamento com ¼ da dose recomendada e aumentando a mesma em ¼ a cada semana até atingir a dose almejada. Dose semelhante à de adultos deve ser empregada a partir do completo crescimento e puberdade.
t Administrar pela manhã, com estômago vazio, ou ao menos 30 minutos antes de uma refeição. Os comprimidos podem ser triturados e suspensos em 10 a 15 mililitros de água. A suspensão deve ser imediatamente ingerida.
t iniciar com 50 a 100 microgramas ao dia, por via oral, com acréscimo de 25 a 50 microgramas, a cada 3 a 4 semanas, até normalização do metabolismo. Dose de manutenção: 100 a 200 microgramas ao dia.
t Em casos de doença cardíaca, hipotireoidismo grave e pacientes acima de 50 anos, iniciar com 25 microgramas uma vez ao dia, acrescentando 25 microgramas a cada 4 a 6 semanas, até a normalização do metabolismo. Dose de manutenção de 50 a 200 microgramas ao dia.
t 2 a 6 microgramas/kg/dia, via oral, durante 7 a 10 dias.
t Tempo para pico de efeito sérico: 2 a 4 horas.
t Início da ação: 3 a 5 dias (oral); 6 a 8 horas (intravenosa).
t Pico do efeito: aproximadamente 24 horas (intravenosa).
t Meia-vida de eliminação: 6 a 7 dias (eutiroidismo); hipotireoidismo: 9 a 10 dias; hipertireoidismo: 3 a 4 dias.
t Sintomas de hipertireoidismo: angina, arritmias cardíacas, palpitações, taquicardia, vômitos, diarreia, tremores, excitabilidade, insônia, cefaleia, rubor facial, sudorese, intolerância ao calor, perda de peso, fraqueza muscular, cãibras e febre.
t Menos frequentes: alopecia, alterações no ciclo menstrual, aumento do apetite e irritabilidade.
t Reações de hipersensibilidade: erupção cutânea, prurido e edema. Observação: Normalmente ocorrem em doses excessivas e regridem após redução da dose ou interrupção temporária do tratamento
t Antiácidos: redução da absorção da levotiroxina.
t Anticoagulantes orais: têm seus efeitos potencializados, aumentando o risco de hemorragias.
t Carbonato de cálcio, colesevelam, colestiramina, ferro: diminuição da absorção da levotiroxina.
t Estrogênios, fenitoína, imatinibe, lopinavir, rifampicina, ritonavir, sinvastatina: diminuição do efeito da levotiroxina e piora do hipotireoidismo.
t Sevelâmer, soja: diminuição da absorção da levotiroxina.
t Orientar para ingerir com 250 mL de água e com o estômago vazio, 30 minutos antes ou 2 horas após o café da manhã.
t Alertar para evitar alternância de fabricantes, porque produtos diferentes podem não ter o mesmo efeito.
t Informar que pode ser necessário de 6 a 8 semanas para o medicamento começar a fazer efeito. Não interromper o tratamento sem falar com o médico.
t Conservar sob temperatura ambiente, entre 15 e 30 ºC, em recipientes bem fechados e ao abrigo da luz.
t Se houver necessidade de triturar e misturar o comprimido, utilizar apenas água ou, se necessário, alimentos que não apresentem grandes quantidades de soja, ferro ou fibras.
Atenção: biodisponibilidades diferentes são observadas entre as diversas apresentações comerciais de levotiroxina sódica. Assim, alcançada a estabilização do paciente, a prescrição não deve ser, a priori, alterada.
Rogério Hoefler
Na Rename 2010: item 10
t Emulsão injetável 100 mg/mL (10%)
t Emulsão injetável 200 mg/mL (20%)
t Fonte de calorias e de ácidos graxos essenciais em nutrição parenteral total.
t Hiperlipidemia.
t Nefrose lipídica.
t Pancreatite aguda com hiperlipidemia.
t Usar com cuidado nos casos de:
– anemia, distúrbios da coagulação, risco de embolia gordurosa, doença pulmonar, pancreatite, hipercolesterolemia, hepatopatias.
– insuficiência hepática (ver Apêndice C).
– neonatos prematuros ou com baixo peso (risco aumentado de acúmulo de gordura nos pulmões).
t Uso hospitalar e restrito para prescrição apenas por especialista.
t Verificar história de alergia a ovos ou a algum tipo de óleo.
t A bandagem em torno do cateter deve ser trocada a cada três dias ou quando estiver úmida, suja ou solta.
t Categoria de risco na gravidez (FDA): C.
t Dose inicial de 0,5 g/kg, por infusão intravenosa, em 24 horas. Dose máxima: 3,0 g/kg/dia, sem exceder a velocidade de 1,0 g/kg em 4 horas.
t Dose inicial de 0,01 g/minuto, por infusão intravenosa, durante os primeiros 15 a 30 minutos, aumentando a velocidade de infusão até o limite de 0,1 g/ kg/hora. Dose máxima: 3,0 g/kg/dia e 60% do total diário de necessidade calórica.
t Dose inicial de 0,1 g/minuto, por infusão intravenosa, durante os primeiros 15 a 30 minutos, aumentando a velocidade de infusão para 0,2 g/minuto. Dose máxima no primeiro dia: 500 mL. Dose máxima em dias subsequentes: 2,5 g/kg/dia e até 60% do total diário de necessidade calórica.
Nota: a infusão deve ser realizada por veia periférica ou cateter venoso central. Em lactentes, deve-se utilizar sistema automatizado de infusão para garantir melhor controle da velocidade de administração de pequenos volumes da emulsão.
t Metabolismo: hepático.
t Meia-vida de eliminação: 30 minutos.
t Sepse, caracterizada por calafrios, febre e dor de garganta (mais frequente).
t Reações alérgicas, urticária (raras).
t Trombocitopenia (rara).
t Hepatotoxicidade, icterícia (raras).
t Dispneia (rara).
t Conservar sob temperatura ambiente, entre 15 e 30 ºC. Não congelar.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Incompatível com: albumina, aminofilina, ampicilina, cefalotina, gentamicina, penicilina, tetraciclina, tobramicina, ácido ascórbico, ácido fólico, cloreto de magnésio, cloreto de potássio, sais de cálcio, heparina sódica, cimetidina, metildopa, fenitoína e complexo vitamínico B.
t A mistura com glicose e aminoácidos deve ser efetuada com técnica e cuidados de assepsia adequados. Após preparo, deve ser mantida sob refrigeração e utilizada dentro de 24 horas.
t Se mantido sob refrigeração, deixar chagar à temperatura ambiente antes de administrar.
t Não usar equipos cujos filtros tenham poros menores que 1,2 micra.
Atenção: há risco aumentado de morte em neonatos prematuros ou com baixo peso por apresentarem maior susceptibilidade ao acúmulo de gordura nos pulmões, devido à sua imaturidade hepática e baixa depuração de lipídios.
Vanessa Rocha Machado
Na Rename 2010: item 5.5.2.4
t Comprimido (200 mg + 50 mg)
t Solução oral (80 mg + 20 mg)/mL
t Tratamento de infecção por HIV, em combinação com outros antirretrovirais.
t Hipersensibilidade a lopinavir ou ritonavir.
t Porfiria aguda.
t Usar com cuidado nos casos de:
– distúrbios na condução cardíaca.
– insuficiência hepática (ver Apêndice C), hepatites B e C crônicas, cirrose ou pancreatite recente.
– insuficiência renal (ver Apêndice D).
– hemofilia A e B.
– diabetes melito ou hiperglicemia.
– pancreatite (suspender o tratamento).
t Há risco de resistência cruzada com outros inibidores de protease.
t A solução oral contém propilenoglicol, pelo que deve ser evitada em grávidas e lactantes (ver Apêndice B).
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Dose de (16 + 4) mg/kg de lopinavir + ritonavir ou (300 + 75) mg/m2 de lopinavir + ritonavir, por via oral, 2 vezes ao dia. Neste caso, não é recomendada a combinação com efavirenz, nevirapina, amprenavir ou nelfinavir.
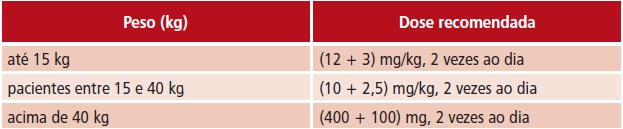
t Sem combinação com efavirenz, nevirapina, amprenavir ou nelfinavir: dose de (230 + 57,5) mg/m2 de lopinavir + ritonavir, por via oral, 2 vezes ao dia ou dose calculada por peso corporal (ver tabela abaixo), até a dose máxima: (400 + 100) mg de lopinavir + ritonavir, 2 vezes ao dia.

t Uso em combinação com efavirenz, nevirapina, amprenavir ou nelfinavir: a dose de lopinavir + ritonavir deve ser aumentada. Recomenda-se dose de (300 + 75) mg/m2 de lopinavir + ritonavir, por via oral, 2 vezes ao dia ou dose calculada por peso corporal (ver tabela abaixo), até a dose máxima: (533 + 133) mg de lopinavir + ritonavir, 2 vezes ao dia.
t Dose de (800 + 200) mg de lopinavir + ritonavir, por via oral, 1 vez ao dia.
t Dose de (400 + 100) mg de lopinavir + ritonavir, por via oral, 2 vezes ao dia. Pacientes com ou sem tratamento anterior com antirretrovirais, em terapia combinada de lopinavir + ritonavir com efavirenz, nevirapina, fosamprenavir, amprenavir ou nelfinavir:
t Dose de (600 + 150) mg de lopinavir + ritonavir, por via oral, 2 vezes ao dia.
t Pico de concentração plasmática: 4 a 6 horas.
t Início de resposta: 3 semanas.
t Meia-vida: 5 a 6 horas.
t Biodisponibilidade: aumenta com alimentos ricos em lipídio(s).
t Metabolismo: hepático.
t Excreção: fecal 20% e renal (2%).
t Não são removidos por diálise.
t Hiperglicemia, dislipidemia, lipodistrofia, síndrome de Cushing, distúrbios eletrolíticos em crianças.
t Hipotireoidismo, disfunção sexual.
t Aumento dos níveis de transaminases e descompensação hepática.
t Náusea, diarreia, vômito, dor abdominal, anorexia, xerostomia, alterações no paladar.
tPancreatite.
t Hematomas espontâneos, sangramento, anemia, neutropenia, trombocitopenia.
t Astenia, parestesia, mialgia, miosite, rabdomiólise.
t Exantema, prurido, síndrome de Stevens-Johnson, acne, alopecia.
t Dor no peito, palpitações, bradicardia, hipertensão, bloqueio atrioventricular.
t Agitação, ansiedade, depressão, insônia, cefaleia, efeitos extrapiramidais.
t Ácido fusídico, posaconazol, quinupristina + dalfopristina: aumento da concentração plasmática e risco de toxicidade pelo ritonavir. Monitorar o sinais e sintomas de toxicidade (diarreia, náuseas, mialgia, febre, anormalidades hepáticas). É recomendado ajuste de dose de ritonavir, quando necessário. Atenção especial para o ácido fusídico devido a hepatoxicidade, sendo que a associação deve ser evitada.
t Ácido valproico, bupropiona, fenitoína, fenorbarbital, lamotrigina, paroxetina, metadona: redução da concentração plasmática dos fármacos que atuam a nível de sistema nervoso central. Monitorar o paciente quanto a perda da eficácia dos mesmos e considerar aumento de dose, se necessário. Monitorar os sinais de crises convulsivas no uso de fenitoína concomitante com ritonavir e precipitação de crise de abstinência quando a combinação é com metadona.
t Alho (Allium sativum): redução na concentração dos inibidores da protease, aumento no risco de resistência antirretroviral e ineficiência do tratamento. Evitar o uso de alho na alimentação e monitorar os sintomas de toxicidade pelos inibidores da protease, ajustando a dose conforme necessário.
t Anfetaminas, aprepitanto, claritromicina, fentanila, rifabutina: aumento da concentração plasmática destes fármacos associados a terapia com lopinavir + ritonavir. Sinais e sintomas de toxicidade: fentanila pode levar a depressão do sistema nervoso central e respiratória; claritomicina tem como sintomas de toxicidade náuseas, diarreia. dispepsia e toxicidade renal; anfetaminas causam agitação, taquicardia, dispneia, hipertensão.
t Anticoagulantes (acenocumarol, varfarina): podem ter sua concentração alterada. Monitorar estreitamente os parâmetros de coagulação. Pode ser necessário ajuste de dose.
t Antidepressivos/ansiolíticos (alprazolam, amitriptilina, clonazepam, desipramina, fluoxetina, imipramina, nefazodona, trazodona): pode haver aumento da concentração plasmática e do risco de toxicidade destes fármacos. Monitorar os sinais e sintomas de toxicidade (xerostomia, sedação, visão borrada, hipotensão, cefaleia, confusão, náuseas, sonolência, retenção urinária, arritmias cardíacas). Reduzir a dose do antidepressivo/ansiolítico, se necessário. Em particular, nefazodona e fluoxetina podem manifestar alterações cardíacas e neurológicas.
t Antineoplásicos/imunossupressores (docetaxel, dutasterida, everolimo, tacrolimo, vimblastina): risco de aumento da concentração plasmática e de toxicidade destes fármacos. Monitorar os níveis plasmáticos e sinais e sintomas de toxicidade (nefrotoxicidade, hiperglicemia, hiperpotassemia, desordens neuropsiquiátricas, obstipação, desordens hematológicas, diarreia). Reduzir a dose do imunossupressor/antineoplásico, se necessário.
t Antirretrovirais (amprenavir, delavirdina, efavirenz, indinavir, maraviroque): pode haver aumento da concentração plasmática destes antirretrovirais associados ao ritonavir. Monitorar o paciente quanto os Efeitos adversos (elevação das transaminases hepáticas, tontura, exantema). Redução da dose pode ser necessária. Associações de ritonavir com delavirdina ou efavirenz podem induzir o aumento da concentração plasmática de ritonavir, sendo necessário monitorar os sinais de toxicidade pelo ritonavir (diarreia, náuseas, mialgia, febre, anormalidades hepáticas) e ajuste na dose do mesmo.
t Bloqueadores de canais de cálcio (anlodipino, bepridil, diltiazem, verapamil): podem ter aumento da sua concentração plasmática e risco de toxicidade. Monitorar sinais e sintomas de toxicidade (tonturas, cefaleia, rubor, edema periférico, hipotensão, arritmias cardíacas). Reduzir a dose do bloqueador de canais de cálcio, se necessário.
t Carbamazepina, clozapina, risperidona: pode haver aumento da concentração plasmática e do risco de toxicidade destes fármacos. Monitorar os sinais e sintomas de toxicidade (efeitos extrapiramidais, sedação, cefaleia, náuseas, fraqueza, convulsão, hipotensão, arritmias cardíacas). Reduzir a dose, se necessário. Atenção especial para a clozapina quando a dose diária ultrapassar 300 mg ou 3,5 mg/kg.
t Cetoconazol, itraconazol: aumento da concentração plasmática dos fármacos antifúngicos. Monitorar os sinais e sintomas de toxicidade (parestesias ou efeitos gastrintestinais persistentes). Redução da dose do antifúngico é recomendada, sendo que doses superiores a 200 mg não são indicadas na terapia combinada com lopinavir + ritonavir.
t Colchicina, dexametasona, fluticasona, prednisona: aumento da concentração plasmática dos anti-inflamatórios. Pode ocorrer toxicidade fatal na combinação de colchicina com ritonavir, estando contra indicada para pacientes com disfunção renal e/ou hepática. Monitorar os sinais e sintomas no uso concomitante de ritonavir com corticosteroides para o risco de síndrome de Cushing (ganho de peso, rubor na face e pescoço, hipertensão e aparecimento de pelos pelo corpo) e redução da dose do corticosteroide.
t Contraceptivos, levotiroxina: redução ou perda da eficácia destes fármacos associados a terapia com lopinavir + ritonavir. Orientar para o uso de outro método contraceptivo e monitorar estreitamente o níveis dos hormônios tireoidianos (levotiroxina).
t Darunavir, nelfinavir, nevirapina, tipranavir: redução da concentração plasmática do lopinavir. Ajuste na dose do lopinavir + ritonavir são necessários: considerar o esquema terapêutico citado nesta monografia para a associação de lopinavir + ritonavir com outros antirretrovirais.
t Didanosina, rifapentina: redução ou perda da atividade antirretroviral. Deve ser avaliada a possibilidade de substituição por outros fármacos com a mesma finalidade terapêutica. Na terapia combinada de ritonavir com didanosina a administração dos fármacos deve ser feita com duas horas e meia de diferença.
t Digoxina, disopiramida, metoprolol, mexiletina: aumento da concentração plasmática dos fármacos cardiovasculares. Monitorar os sinais e sintomas de toxicidade (náuseas, vômitos, arritmias cardíacas, efeitos colinérgicos, hipotensão, tontura, falência cardíaca, sedação, bradicardia). Monitorar a concentração plasmática, principalmente, da digoxina e mexiletina, reduzindo a dose quando necessário.
t Lovastatina, sinvastatina, rosuvastatina: aumento do risco de desenvolver miopatias ou rabdomiólise. Monitorar o paciente quanto aos sinais de mialgia, fragilidade e fraqueza, assim como níveis de creatina quinase.No caso de creatina quinase aumentada ou diagnóstico/suspeita de miopatias ou rabdomiólise, deve-se suspender o uso das estatinas.
t Quinidina, bosentana, pimozida, Hypericum perforatum (erva-de-são-joão), ergotamina e análogos, voriconazol e rifampicina: o uso concomitante é contraindicado.
t Sildenafila: aumento do risco de Efeitos adversos (hipotensão, rubor, cefaleia, mudanças visuais, priapismo). O uso concomitante é contraindicado.
t Tenofovir: pode ter aumento da sua biodisponibilidade. Monitorar marcadores de integridade óssea e concentrações plasmáticas de aminotransferases, além dos sinais de toxicidade hepática e renal.
t Teofilina: redução na concentração plasmática da teofilina. Aumento na dose de teofilina pode ser necessário.
t Orientar para ingerir com alimento.
t Orientar para o uso de outro método contraceptivo devido redução da eficácia dos contraceptivos hormonais à base de estrogênios quando em uso concomitante com lopinavir + ritonavir.
t Informar ao paciente que o lopinavir + ritonavir apresenta inúmeras e significativas interações. Antes de usar qualquer outro medicamento, inclusive plantas medicinais, informar ao médico.
t No caso de terapia combinada com didanosina, orientar para ingerir didanosina sem alimento uma hora antes ou duas horas depois da dose de lopinavir + ritonavir.
t Reforçar orientações sobre prevenção da transmissão do HIV.
t Armazenar a solução oral e cápsulas, preferentemente, sob refrigeração, entre 2 e 8 ºC. Se armazenar à temperatura ambiente, consumir em 2 meses.
t Armazenar o compromido à temperatura ambiente, entre 15 e 30 ºC.
Atenção: a associação lopinavir + ritonavir apresenta um número elevado de importantes Efeitos adversos , por isto deve ser realizada pesquisa específica sobre este aspecto antes de introduzir ou descontinuar a associação de lopinavir + ritonavir ou outros medicamentos no esquema terapêutico do paciente.
Helena Lutéscia Luna Coelho
Na Rename 2010: item 4
t Comprimido 10 mg
t Xarope 1 mg/mL.
t Alívio de sintomas de alergia, febre do feno, rinite alérgica ou vasomotora,
prurido, urticária.
t Hipersensibilidade à loratadina.
t Porfiria.
t Recém-nascidos e bebês prematuros.
t Usar com cuidado nos casos de:
– hipertrofia prostática, retenção urinária, glaucoma, obstrução piloro-duodenal e epilepsia.
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– crianças e idosos (podem desenvolver reação paradoxal de hiperexcitabilidade).
– direção de veículo ou operação de maquinário (risco de acidentes por eventual efeito sedativo da loratadina).
– lactação (ver Apêndice B).
t Fator de risco na gravidez (FDA): B.
t Adultos e crianças a partir de 6 anos: 10 mg, por via oral, uma vez ao dia.
t Adultos e crianças a partir de 6 anos, com falência hepática ou diminuição da função renal (depuração endógena de creatinina < 30 mL/min.): 10 mg, em dias alternados.
t Crianças com 2-5 anos: 5 mg, por via oral, uma vez ao dia.
t Crianças com 2-5 anos, com falência hepática ou diminuição da função renal (depuração endógena de creatinina < 30 mL/min.): 5 mg, em dias alternados.
t Absorção: bem absorvida por via oral. A ingestão concomitante de alimentos pode aumentar a absorção em 40%.
t Início da ação: 1-3 horas
t Duração da ação: dose única de 24 a 48 horas; doses múltiplas de 24 horas a 8 dias.
t Tempo para pico de concentração: 1,3 horas
t Pico de efeito: 8-12 horas; em idosos, a taxa de absorção e o pico plasmático é cerca de 55% maior que em jovens.
t Biotransformação: hepática; metabólito ativo: desloratadina
t Eliminação urinária e fecal, 80% do total da dose administrada
t Meia-vida: 8,4 horas
t Não atravessa a barreira hematencefálica
t Diálise não altera farmacocinética.
t Raros: hipotensão, edema, palpitação, taquicardia, urticária, exantema, mialgia, sudorese, fotossensibilidade, visão borrada, icterícia, necrólise hepática, coloração da urina, dor ao urinar*, anafilaxia, ganho de peso, secura nasal*, faringite, dispneia, congestão nasal, broncoespasmo, zumbido*, astenia, depressão, cefaleia, insônia, confusão*, tremor, tontura*, convulsão.
(* sintomas mais frequentes em idosos)
t Pode ocorrer sedação se a dose recomendada de loratadina for excedida.
t Amiodarona: recomenda-se realização eletrocardiográfica (ECG) antes e após a primeira dose. Se for observado prolongamento do intervalo QT, deve-se interromper o uso da loratadina e monitorar o ritmo cardíaco.
t A administração com alimentos, água ou leite reduz a irritação gástrica.
t Recomenda-se interromper o uso uma semana antes da realização de testes de pele com alergênios, pois podem ocorrer resultados falso-negativos.
t Não usar durante o aleitamento materno.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC. Manter em recipiente bem fechado, longe de calor e luz direta. Não congelar (xarope).
Rosa Martins
Na Rename 2010: itens 14.1, 14.4.6
t Comprimido 50 mg.
t Segunda escolha nos casos de intolerância ao IECA, nas indicações:
– insuficiência cardíaca congestiva (ICC).
– hipertensão arterial sistêmica.
– profilaxia de acidente cerebrovascular em pacientes hipertensos com hipertrofia ventricular esquerda.
– nefropatia diabética em pacientes com diabete melito tipo 2 e história de hipertensão.
t Hipersensibilidade ao fármaco.
t Gravidez. Categoria de risco na gravidez (FDA): C (primeiro trimestre) e D (segundo e terceiro trimestres) (ver Apêndice A).
t Usar com cuidado nos casos de:
– angioedema atual ou história de angioedema.
– depleção de volume (corrigir depleção antes de iniciar o tratamento para prevenir hipotensão).
– insuficiência hepática (ver Apêndice C).
– insuficiência renal (ver Apêndice D).
– hiperpotassemia.
– estenose da artéria renal.
– insuficiência cardíaca congestiva grave
– lactação (ver Apêndice B).
t 0,7 mg/kg, por via oral, a cada 24 horas. Dose máxima: 50 mg/dia.
t Dose inicial: 12,5 mg, por via oral, uma vez ao dia. Dobrar a dose a cada 7 dias, se necessário, até 50 mg/dia.
t Dose inicial 50 mg, por via oral, uma vez ao dia. Dose de manutenção: 25 a 100 mg, por via oral, uma vez ao dia ou dividido a cada 12 horas.
t Dose inicial: 50 mg, por via oral, uma vez ao dia. Dose de manutenção: 100 mg, por via oral, uma vez ao dia. Associado a hidroclorotiazida 12,5 a 25 mg, por via oral, uma vez ao dia.
t Dose inicial: 50 mg, por via oral, uma vez ao dia. Dose de manutenção: 100 mg, por via oral, uma vez ao dia de acordo com controle da pressão arterial.
t Biodisponibilidade: 25% a 35%
t Pico de concentração: 1 a 1,5 horas.
t Duração da ação: 24 horas (em hipertensão)
t Metabolismo hepático 14% via citocromo P450 isoenzimas CYP2C9 e CYP3A4. Metabolito ativo. Sofre metabolismo de primeira passagem.
t Meia-vida de eliminação: 1,5 a 2 horas para o fármaco e 4 a 9 horas para o metabólito ativo.
t Excreção: 13% a 35% renal, depuração renal 42 a 75 mL/min; 50% a 60% biliar.
t Não é dialisável.
t Angina, bloqueio atrioventricular de segundo grau, acidente cerebrovascular, hipotensão (principalmente em paciente com depleção de volume), enfarte do miocárdio, arritmias, síncope (todos menos de 1%). Hipotensão ortostática dose dependente (menor do que 0,5% com dose de 50 mg e 2% com dose de 100 mg).
t Prurido, exantema, alopecia, dermatite, pele seca, eritema, rubor, fotossensibilidade, sudorese, urticária (todos menos de 1%).
t Gota, hiperpotassemia, hiponatremia.
t Diarreia (2%), dispepsia (1%), alteração no paladar, pancreatite.
t Anemia, linfoma maligno, trombocitopenia.
t Hepatotoxicidade
t Dor nas pernas e costas, cãibra muscular e mialgia (todos entre 1% a 1,8%), rabdomiólise.
t Astenia, ataxia, confusão, tontura, hiperestesia (sensibilidade diminuída a estímulos), insônia ou transtorno do sono, comprometimento da memória, enxaqueca, parestesia, síndrome de Parkinson, neuropatia periférica, sonolência, tremor, vertigem (todos menos de 1%).
t Visão borrada, sensação de queimação ocular, conjuntivite, diminuição da acuidade visual, tinido (todos menos de 1%).
t Ansiedade, depressão, sensação de nervosismo, distúrbio do pânico, distúrbio psicótico (todos menos de 1%).
t Nefrotoxicidade.
t Diminuição da libido, impotência (todos menos de 1%).
t Tosse, infecção respiratória superior (8%), congestão nasal (2%), sinusite (1%), alterações no seio frontal (1,5%).
t Angioedema.
t Anti-inflamatórios não-esteroides, fluconazol, rifampicina: podem diminuir a efetividade da losartana. Monitorar pressão arterial.
t Lítio: pode ter a toxicidade (fraqueza, tremor, sede, confusão) aumentada pela losartana. Monitorar para sinais e sintomas específicos.
t Medicamento não recomendado durante a gravidez. Orientar prevenção.
t Aumento de pressão arterial pode não ter sintoma, não deixar de usar o medicamento sem falar com o médico.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar entre 15 e 30 °C, proteger do calor, umidade e luz direta.
t Formulação extemporânea – suspensão oral 2,5 mg/mL: colocar 10 comprimidos de losartana potássica 50 mg num recipiente com 10 mL de água purificada, agitar por 2 minutos, deixar em repouso durante 1 hora e agitar por mais 1 minuto; adicionar esta mistura a 190 mL de Ora-plus e Ora-sweet 50/50 e agitar por mais 1 minuto. Armazenar sob refrigeração. Estável por até 4 semanas. Agitar bem antes de usar.
Atenção: máximo efeito hipotensor é alcançado após 3 a 6 semanas. A incidência de alguns Efeitos adversos varia de acordo com a doença de base (hipertensão e nefropatia diabética) e tendem a ser mais frequentes naqueles com nefropatia diabética.
Consta no documento:
“Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial.”
O objetivo do site MedicinaNet e seus editores é divulgar este importante documento. Esta reprodução permanecerá aberta para não assinantes indefinidamente.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.