(Carregando Índice)... (Carregando Índice)... |
Autor:
Rodrigo Antonio Brandão Neto
Médico Assistente da Disciplina de Emergências Clínicas do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 27/06/2014
Comentários de assinantes: 0
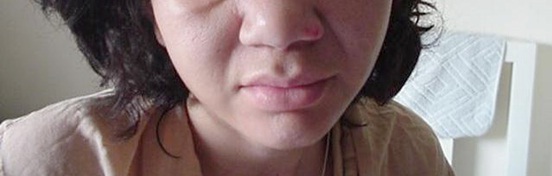
Mulher de 39 anos de idade, com história de aumento de extremidades e alterações de características faciais, também teve diagnóstico recente de HAS. A imagem mostra alterações características de fácies acromegálica com nariz em sela, apresentando mandíbula proeminente.
O diagnóstico de acromegalia foi confirmado pela dosagem de IGF-1 aumentada e ausência de supressão do GH com teste de supressão com glicose, foi diagnosticado macroadenoma hipofisário e programada intervenção cirúrgica.

A acromegalia é uma síndrome causada pelo excesso crônico de hormônio de crescimento ou GH, que em mais de 95% dos casos é causada por um adenoma hipofisário que secreta GH. Se o excesso de GH surgir antes do fechamento das cartilagens de crescimento (infância/adolescência), ocorre ganho excessivo de altura (gigantismo).
A grande maioria dos tumores é de macroadenomas (80-90%). Os adenomas hipofisários podem produzir apenas GH (60%) ou serem compostos de mais de um tipo celular (tumores mistos), produzindo GH em associação a outro hormônio hipofisário (mais comumente, prolactina). Causas mais raras de acromegalia incluem a secreção ectópica de GHRH (5% - por carcinoides brônquicos ou gastrointestinais), o excesso de GHRH causado por doenças hipotalâmicas (1% - hamartoma etc.) e, muito raramente, o carcinoma hipofisário secretor de GH e os tumores extra-hipofisários secretores de GH.
A acromegalia atinge igualmente ambos os sexos e é mais comum entre os 30-50 anos de idade. A doença é associada com aumento de cerca de duas vezes na mortalidade, principalmente por causas cardiovasculares e respiratórias (sobrevida encurtada em 5-10 anos).
As manifestações clínicas se instalam de forma lenta e gradual, o que determina um atraso médio de cerca de 10 anos até o diagnóstico. Os sintomas típicos do excesso de GH são o aumento das extremidades: crescimento das mãos e pés (alargamento dos dedos, anéis muito apertados, mãos “acolchoadas”, aumento do número dos calçados), crescimento grosseiro de protuberâncias faciais (orelhas, nariz, queixo, lábios, crista supraorbitária), afastamento entre os dentes, macroglossia e aparecimento das chamadas “skin tags”. Também pode haver aumento das vísceras: coração, rins, fígado e tireoide. Hiperidrose e oleosidade da pele, bem como aumento das linhas e pregas cutâneas, são comuns. Artralgia é uma queixa muito frequente. O paciente também pode queixar-se de sintomas devidos ao efeito de massa do adenoma hipofisário: cefaleia, perda de visão (hemianopsia temporal) ou hipopituitarismo.
As complicações do excesso de GH incluem hipertensão arterial, insuficiência cardíaca, aterosclerose grave e precoce, osteoartrose, apneia do sono (até 80%), síndrome do túnel do carpo (em um terço dos casos) e diabetes mellitus (em até 50%). Hiperprolactinemia pode ser observada em cerca de 30% dos acromegálicos, com ou sem galactorreia. A acromegalia também aumenta o risco para neoplasia de cólon (pólipos e câncer), e possivelmente para tumores de mama, pele, estômago e tireoide.
O diagnóstico de acromegalia é feito pelo achado de GH e IGF-1 (insulin-like growth factor 1) aumentados. Como a secreção de GH é pulsátil, a coleta de amostras ao acaso tem valor limitado; por isso faz-se necessário o uso de testes de supressão farmacológica do GH para comprovar a presença de secreção autônoma (tumoral) desse hormônio. O padrão-ouro para o diagnóstico é realizado com o teste de supressão com glicose e a dosagem de IGF-1. A hipófise normalmente suprime a secreção de GH frente ao estímulo glicêmico. Usa-se glicose (75g por via oral), com dosagem de GH a cada 30min por duas horas. Em indivíduos normais, deve haver supressão do GH para <1ng/mL, o que não acontece em acromegálicos, mas resultados falso-positivos podem ocorrer em pacientes com diabetes mellitus, doença hepática ou renal, anorexia nervosa e outras condições. Recentemente estão utilizando critérios mais estritos considerando suprimidos apenas valores abaixo de 0,4 ng/ml do GH.
A dosagem de IGF-I é também útil no diagnóstico e no seguimento de pacientes acromegálicos. Trata-se de um fator de crescimento de produção hepática e tecidual, que aumenta em resposta a secreção de GH. Como os níveis de IGF-I não oscilam durante o dia, sua dosagem fornece ideia da secreção integrada de GH nas 24 horas. Os resultados devem ser interpretados levando-se em consideração o sexo e a idade do paciente. Virtualmente todos pacientes acromegálicos apresentam IGF-I aumentado para sexo e idade. A primeira escolha para tratamento é a cirurgia por via transesfenoidal, capaz de curar 40-90% dos microadenomas e 25-50% dos macroadenomas. Outras opções são radioterapia e tratamento medicamentoso com os análogos da somatostatina e pegvisomant que atuam no receptor de GH, além dos agonistas dopaminérgicos.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.