(Carregando Índice)... (Carregando Índice)... |
Autor:
João Evangelista Bezerra Neto
Hematologita pelo Hospital das Clínicas da Faculdade de Medicina da USP
Oncologista pelo Hospital Israelita Albert Einstein
Ex médico preceptor e assistente da Diciplina de Hematologia do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 26/10/2008
Comentários de assinantes: 0
Leucemias crônicas são neoplasias originadas dos precursores hemotopoiéticos granulocíticos ou linfocíticos da medula óssea. A característica comum a todos os tipos de leucemia crônica é a capacidade mantida de diferenciação do clone neoplásico, resultando no acúmulo de células maduras na medula óssea e no sangue periférico. Geralmente, apresentam uma evolução lenta, quando comparadas às leucemias agudas. Porém, são resistentes à ação da quimioterapia, o que torna essas doenças raramente curáveis.
Muitos avanços têm ocorrido no entendimento dessas doenças, com o direcionamento do tratamento sendo baseado cada vez mais nas características individuais de cada paciente. Dentre as Leucemias crônicas do tecido linfóide, a leucemia linfocítica crônica
Dentre as Leucemias crônicas originadas das células linfóides, a LLC é a mais comum. Outras são a leucemia de células pilosas (LCP) e a leucemia prolinfocítica (LPL).
A LLC é uma neoplasia clonal de linfócitos B maduros, porém não competentes. O clone maligno que origina a doença apresenta uma proliferação lenta e mantida de linfócitos B pequenos, maduros e resistentes à morte celular. Esses linfócitos irão se acumular infiltrando progressivamente a medula óssea e outros órgãos do sistema imunológico, como o baço e os linfonodos.
A LLC corresponde a aproximadamente 30% de todos os casos de leucemia em adultos no Ocidente. É predominantemente uma doença de pacientes idosos, sendo a idade média ao diagnóstico de 65-70 anos. Entretanto, aproximadamente 20% a 30% dos pacientes apresentam menos de 60 anos, com 5% a 10% dos casos ocorrendo antes dos 50 anos de idade. Cada vez mais, a LLC é diagnosticada acidentalmente a partir de linfocitose observada no hemograma, aumentando progressivamente o número de casos em acompanhamento ainda em estágios iniciais e a proporção de pacientes jovens.
Os fatores de risco tradicionais para neoplasias hematológicas, como radiação, benzeno e exposição a quimioterápicos, não têm o mesmo papel na LLC. Entretanto, aproximadamente 10% dos pacientes com LLC têm uma história clara de neoplasias linfóides de células B na família, sugerindo a presença de um fator genético familial. Do mesmo modo, a LLC é rara em países como China, Coréia e Japão, com sua incidência permanecendo constante entre descendentes de imigrantes no Ocidente, excluindo fatores ambientais como predisponentes para a doença.
A LPL, originalmente classificada como uma variante da LLC, é composta por linfócitos maduros, mas com características morfológicas e imunofenotípicas distintas da LLC. Na grande maioria dos casos, as células são originárias de linfócitos B, mas até 20% expressam perfil de linfócitos T. É bem menos freqüente que a LLC, respondendo por 10% das leucemias do adulto. Já a LCP, constituída por linfócitos B maduros e ativados, é uma leucemia rara, correspondendo a cerca de 2% das leucemias.
A LLC e a LPL têm apresentação clínica semelhantes. Dos pacientes com LLC, aproximadamente 25% são assintomáticos no momento do diagnóstico. Linfocitose isolada observada durante hemograma de rotina é muitas vezes o motivo para iniciar a investigação. Fadiga é o principal motivo de procura por assistência médica em ambos os casos. A presença de sintomas B indica doença mais avançada. Linfadenomegalia móvel, fibroelástica e indolor é a alteração mais comumente detectada ao exame físico, sendo freqüentemente acometidas as cadeias cervicais, supraclaviculares e axilares (tabela 1).
Tabela 1: Sinais e sintomas em leucemia linfocítica crônica
|
Assintomático |
25% |
|
Sintomas B* |
20% |
|
Linfadenomegalia |
50-90% |
|
Esplenomegalia |
25-50% |
|
Hepatomegalia |
20-25% |
|
Reação alterada à picada de insetos |
5% |
*Definição de sintomas B: perda de peso > 10% do peso habitual nos últimos 6 meses, febre > 38ºC por duas ou mais semanas sem evidência de infecção, sudorese noturna sem evidência de infecção, fadiga importante, limitando as atividades habituais do paciente
O crescimento rápido e isolado de um único linfondo ou grupamento linfonodal pode indicar transformação da doença para uma forma mais agressiva, em geral um linfoma de grandes células B, mas podendo também ser uma leucemia aguda, uma LPL, um linfoma Hodgkin ou um linfoma T (síndrome de Richter). Esplenomegalia e hepatomegalia discretas também são comuns. Alguns doentes apresentam aumentos maciços do baço, semelhantes ao visto em doentes com LMC e leishmaniose visceral
A LCP apresenta uma evolução muito lenta. Os primeiros sintomas são relacionados à pancitopenia resultante da infiltração medular maciça no momento do diagnóstico. Aproximadamente 90% dos pacientes apresentam esplenomegalia e 35% apresentam hepatomegalia. Adenomegalia periférica é rara e, quando ocorre, os linfonodos são pequenos. Entretanto, o comprometimento de gânglios abdominais é mais freqüente e indica um mau prognóstico.
O diagnóstico das Leucemias crônicas é feito com base em exames laboratoriais. O esfregaço de sangue periférico permite o diagnóstico de casos típicos. A característica marcante da LLC é a presença de linfocitose isolada constituída por linfócitos pequenos e de aparência madura, caracterizado por uma peculiar fragilidade da membrana celular, o que leva freqüentemente à ruptura das células durante a preparação do esfregaço e formação de restos celulares conhecidos como “sombras de Gumprecht”. Uma contagem de linfócitos no sangue periférico maior que 5.000 cels/ml mantido por mais de um mês é um dos critérios estabelecidos em consensos para a conclusão do diagnóstico, embora com o uso de imunofenotipagem à presença de componente monoclonal possa ser comprovada com contagens menores. Muitas vezes, há a presença de prolinfócitos, células maiores com núcleo irregular e nucléolo central evidente, mas constituindo menos de 10% das células neoplásicas. Uma variante mais agressiva, conhecida como LLC/LPL, apresenta uma concentração maior de prolinfócitos, que pode chegar a 55% das células neoplásicas (tabela 2). Na LPL clássica, os prolinfócitos devem corresponder a, pelo menos, 55% das células neoplásicas. No caso da LCP, células típicas da doença são encontradas na maioria dos pacientes, embora algumas vezes sejam pouco freqüentes no sangue periférico.
Tabela 2: Alterações laboratoriais da leucemia linfocítica crônica
|
Anemia (Hb < 11,0 g/dl) |
5-20% |
Pior prognóstico |
|
Trombocitopenia (plaquetas < 100.000/mcl) |
5%-16% |
Pior prognóstico |
|
Anemia hemolítica auto-imune |
10% |
Cerca de 35% tem teste de antiglobulina direta positivo; apenas 11% desenvolve anemia, em geral tardia |
|
Trombocitopenia auto-imune |
5% |
Raramente leva a sangramentos relevantes |
|
Agranulocitose |
0,5% |
Em geral tem percentil de neutrófilos baixo, porém número absoluto normal (>1.500/mcl) |
|
Hipogamaglobulinemia |
8% |
Com a evolução da doença acomete até dois terços dos doentes. Predispõe à infecção bacteriana |
|
Pico monoclonal de gamaglobulina |
8% |
Em geral IgM ou IgD |
|
Aplasia eritrocítica pura |
1% |
Investigação com mielograma e contagem de reticulócitos |
|
Elevação de DHL e b2-microglobulina |
60% |
Não são alterações específicas de LLC. Indicam pior prognóstico |
A análise da medula óssea sempre demonstra infiltração. Na LLC, a realização de mielograma ou biópsia de medula óssea revela infiltração geralmente como nódulos intertrabeculares. Tanto na forma variante mais agressiva (LLC/LPL) quanto na LPL clássica, a infiltração tende a ser difusa. A biópsia de medula é o teste definitivo para o diagnóstico da LCP, já que a punção aspirativa tende a ser seca.
O perfil característico pela imunofenotipagem ajuda a completar o diagnóstico (tabela 3). Os linfócitos da LLC expressam o CD19, típico de linfócitos B, em associação com o CD5, presentes normalmente em linfócitos T. A presença de CD23 e uma baixa expressão de imunoglobulinas de superfície complementam o perfil e permitem o diagnóstico diferencial com outras neoplasias linfóides. A linfócitos da LPL-B expressam fortemente imunoglobulina em sua superfície, sendo positivos para FMC7 e CD79b. Diferentemente da LLC, geralmente são negativos para CD23 e CD5. As células da LPL apresentam marcadores de linfócitos B maduros, porém, além de não expressar CD5, apresentam positividade para FMC7, CD11c, CD103 e HC2 – incomuns nas outras doenças linfoproliferativas.
A realização de biópsia linfonodal não é um procedimento necessário para o diagnostico de Leucemias crônicas. Está indicado somente quando há o crescimento rápido e desproporcional de um ou mais linfonodos, a fim de diagnosticar uma possível transformação para um linfoma de alto grau.
Tabela 3: Perfil imunofenotípico das linfoproliferações crônicas B
|
Marcadores |
LLC |
LPL-B |
LCP |
LNH |
|
SmIg |
+/- |
++ |
++ |
++ |
|
CD23 |
+ |
-/+ |
-/+ |
-/+ |
|
CD5 |
+ |
-/+ |
- |
- |
|
FMC7 |
- |
++ |
++ |
++ |
|
CD79b |
- |
++ |
++ |
++ |
|
CD19/20 |
++ |
++ |
++ |
++ |
|
CD25 |
-/+ |
-/+ |
++ |
-/+ |
|
HC2/CD103 |
- |
- |
++ |
- |
Diagnóstico Diferencial
A LLC se relaciona fortemente com um subtipo de linfoma não-Hodgkin de grau indolente – o linfoma linfocítico pequeno. Trata-se, na verdade, de apresentações clinicas distintas de uma mesma doença, visto que no linfoma linfocítico pequeno não houve disseminação das células para o sangue periférico.
O principal diagnóstico diferencial das leucemias linfóides crônicas ocorre com linfomas não-Hodgkin indolentes em suas formas leucêmicas. O linfoma esplênico da zona marginal, linfoma linfoplasmocítico, linfoma de células do manto e o linfoma folicular podem ser diferenciados das Leucemias crônicas em até 85% das vezes com base apenas na apresentação clínica, morfologia e imunofenotipagem. Em alguns casos, a histologia e/ou testes moleculares são necessários.
Tratamento
Como, até o momento, as Leucemias crônicas do tecido linfóide são incuráveis, os pacientes recebem uma abordagem tratamento paliativo.
Na LLC, em alguns casos, o acompanhamento clínico isolado é uma conduta aceitável. Em virtude da idade avançada ao diagnóstico e da evolução lenta de grande parte dos casos, com sobrevida global alcançando 15 anos, muitos pacientes não necessitarão de qualquer tratamento. Nessa população, o início precoce do tratamento não demonstrou aumentar a sobrevida global, sendo algumas vezes até prejudicial.
Dois critérios para estadiamento semelhantes são utilizados correntemente para estratificação dos pacientes com LLC: o sistema de Binet e o de Rai (tabela 4). Para pacientes em estádios iniciais e intermediários, é aconselhável um período de observação, com intervalos de três a seis meses, a fim de definir se a doença é estável ou progressiva. O tratamento quimioterápico somente deve ser iniciado em doenças progressivas ou quando houver sintomas que prejudiquem a qualidade de vida. Entretanto, alguns pacientes em estádios precoces precisarão de tratamento pouco tempo após o diagnóstico e alcançarão uma sobrevida máxima de 7-8 anos. Isso faz com que 25% dos pacientes diagnosticados em estádio precoce venham a falecer por efeito direto da LLC, e uma proporção semelhante tenha complicações sérias relacionadas à LLC ou ao seu tratamento. A identificação desses pacientes de maior risco e o início precoce de tratamento nesse grupo é uma abordagem que vem sendo testada em estudos clínicos. Na prática diária, no entanto, o início da quimioterapia ainda deve se basear em critérios estabelecidos em consensos especializados (tabela 5).
Tabela 4: Estádio clínico de Rai para leucemia linfocítica crônica
|
Estádio |
Órgão afetado |
Hb (g/dl) |
Plaquetas/mm3 |
Sobrevida |
Risco |
|
0 |
– |
> 11 |
> 100.000 |
12 |
Baixo |
|
1 |
Linfonodos |
> 11 |
> 100.000 |
7,5 |
Intermediário |
|
2 |
Fígado/baço |
> 11 |
> 100.000 |
7,5 |
Intermediário |
|
3 |
Indiferente |
< 11 |
> 100.000 |
1,5 |
Alto |
|
4 |
Indiferente |
< 11 |
< 100.000 |
1,5 |
Alto |
Tabela 5: Indicações de tratamento na LLC (estádio precoce)
|
Anemia e/ou trombocitopenia (estádio de Rai III-IV) |
|
Hepatoesplenomegalia ou adenomegalia sintomática |
|
Presença de sintomas B (fadiga, febre, perda de peso) |
|
Fenômenos imunológicos à anemia hemolítica auto-imune, trombocitopenia imunológica |
|
Tempo de duplicação de linfócitos < 6 meses |
|
Infecções recorrentes |
Tradicionalmente, o tratamento da LLC é baseado no uso de agentes alquilantes. O clorambucil é a droga mais utilizada dessa classe. Embora produza resposta em 60%-80% dos casos, os agentes alquilantes não são capazes de aumentar a sobrevida dos pacientes. Associações baseadas em agentes alquilantes aumentam a taxa de resposta, mas nenhum benefício de sobrevida é obtido. Os análogos de purina são capazes de gerar resposta mais intensa e duradoura quando comparados aos agentes alquilantes. A fludarabina é a droga mais usada desse grupo. Como agente único de primeira linha para tratamento, a fludarabina produz resposta em 80% dos casos, sendo 38% de resposta completa. A combinação da fludarabina com o agente alquilante ciclofosfamida aumenta ainda mais a taxa de resposta e a duração da remissão, mas ganho na sobrevida ainda não é alcançado.
Na LLC, o rituximab, anticorpo monoclonal anti-CD20, aumenta as taxas de resposta completa e da duração da resposta quando associado à quimioterapia em primeira linha. A comparação com resultados históricos sugere que essa associação de rituximab à quimioterapia tradicional pode aumentar a sobrevida global. Estudos fase III estão em andamento para testar essa hipótese. O alemtuzumab, anticorpo monoclonal anti-CD52, é indicado no tratamento de pacientes com doença recidivada e refratária à fludarabina. É também o único agente com ação efetiva em doenças que apresentam deleção do braço curto do cromossomo
A LPL tende a ser resistente ao tratamento com esquemas baseados em agentes alquilantes. Tanto esquemas mais agressivos como o CHOP ou o uso de fludarabina produzem resposta em cerca de 50% dos doentes, mas de curta duração. A alemtuzumab também não é capaz de gerar respostas satisfatórias nesses pacientes. Novas modalidades terapêuticas vêm sendo estudadas.
A LCP, por sua evolução indolente, pode ser acompanhada sem tratamento em pacientes que estejam assintomáticos. Nos casos indicados, os análogos de purina são as drogas de primeira escolha. A cladribina, agente mais utilizado dessa classe, produz taxa de remissão completa em 70%-90% dos pacientes, com resposta mantida por cinco anos em mais de 70% destes. O interferon-alfa, menos efetivo que os análogos de purina, é atualmente utilizado somente em pacientes que apresentam intensa citopenia com depressão imunológica associada, em que o uso de análogos de purina poderiam aumentar muito o risco de infecção. Nesses casos, o tratamento com interferon-alfa antes do uso de análogos de purina é capaz de melhorar as citopenias e diminuir os riscos de infecções.
A LMC é uma neoplasia hematológica do grupo das síndromes mielo-proliferativas. Conseqüência de uma alteração na célula progenitora hematopoiética, a LMC resulta na proliferação excessiva principalmente da série granulocítica, mas outras linhagens podem ser afetadas. Tanto a diferenciação quanto a função dessas células é mantida durante as fases iniciais da doença. A LMC é uma doença que segue um curso bifásico ou trifásico, tendo inicialmente uma fase crônica e indolente, que acaba evoluindo para uma fase acelerada, e uma fase blástica, geralmente fatal.
Responsável por aproximadamente 15%-20% das leucemias em adultos no Ocidente, com uma incidência de 1-2 casos a cada 100 mil pessoas, a maior parte dos casos ocorre na quinta década de vida e o único fator de risco conhecido é a exposição à radiação ionizante.
A LMC foi responsável por grandes avanços para o entendimento das neoplasias. Foi a primeira doença descrita com uma alteração cromossômica recorrente, o cromossomo Filadélfia. Também foi a primeira doença na qual se descobriu ser esta alteração cromossômica responsável diretamente pela sua fisiopatologia, e, atualmente, com o surgimento do mesilato de imatinibe, é também a primeira doença na qual um agente terapêutico foi desenvolvido especificamente para atingir o defeito molecular existente.
A LMC é caracterizada pela presença de uma alteração citogenética recorrente – o cromossomo Filadélfia – resultado da translocação do protooncogene ABL
No momento do diagnóstico, o cromossomo Filadélfia pode ser encontrado em 90%-95% dos casos de LMC por meio da análise citogenética das células da medula óssea. Nos 5%-10% restantes ocorrem translocações alternativas, gerando, porém, o mesmo gene anômalo. Também é possível se detectar o gene BCR-ABL por métodos moleculares como o PCR. A ausência de cromossomo Filadélfia e do gene BCR-ABL exclui o diagnóstico de LMC. A entidade conhecida previamente como LMC-Filadelfia negativo e denominada atualmente “LMC atípica” pela Organização Mundial de Saúde é uma doença com características mielo-proliferativas e mielo-displásicas, com quadro clínico e prognóstico diferente da LMC clássica.
O cromossomo Filadélfia, entretanto, não é patognomônico da LMC, podendo ser encontrado em 20%-25% dos adultos com leucemia linfóide aguda e raramente em pacientes com leucemia mielóide aguda.
A característica mais importante da LMC é a proliferação de leucócitos da linhagem granulocítica. A doença geralmente segue um curso trifásico:
Tabela 6: Critérios para diagnóstico de fase avançada na leucemia mielóide crônica
|
Fase acelerada |
Fase blástica |
|
Blastos > 15% |
Blastos > 30% |
|
Blastos + promielócitos > 30% |
Doença extramedular |
|
Basófilos > 20% |
|
|
Plaquetas < 100.000mm3 sem relação com tratamento |
|
|
Evolução clonal |
|
Critérios do MD Anderson Cancer Center
Dos doentes diagnosticados na fase crônica, aproximadamente metade é assintomática, sendo a doença detectada por um exame de rotina. Quando presentes, os sintomas mais freqüentes estão relacionados à anemia, esplenomegalia e à replicação celular aumentada (tabela 7). Com a evolução para uma fase acelerada/blástica, há o surgimento de novos sintomas e piora dos já existentes, como febre alta, dor óssea, aumento da esplenomegalia e, eventualmente, sinais de falência medular, tal qual ocorre numa leucemia aguda.
Tabela 7: Sinais e sintomas relacionados à leucemia mielóide crônica
|
Sinais e sintomas |
Causas e repercussões clínicas |
|
Sintomas sistêmicos (febre, perda de peso, sudorese noturna) |
Em virtude do hipermetabolismo gerado pela hiperleucocitose |
|
Esplenomegalia |
Presente em 48%-76% dos pacientes, com um aumento maciço e importante do baço, em geral muitos centímetros abaixo do rebordo costal esquerdo, podendo causar desconforto abdominal e saciedade precoce |
|
Hepatomegalia |
Ocorre em até 50% dos casos |
|
Gota |
Em virtude do aumento dos níveis de ácido úrico pelo alto catabolismo celular |
|
Dor óssea |
Em virtude da expansão da medula óssea, acometendo principalmente o em esterno |
|
Leucostase |
sintomas por hiperleucocitose são eventos raros nas LMCs, mesmo com contagens leucocitárias extremamente elevadas, em virtude da maior deformidade das células maduras em comparação com os blastos |
|
Eventos trombóticos |
Como as outras síndromes mieloproliferativas, fenômenos trombóticos, como síndrome de Budd-Chiari (trombose das veias supra-hepáticas), podem estar presentes |
A elevação na contagem leucocitária é característica da LMC. O esfregaço de sangue periférico é característico e demonstra um desvio escalonado da série granulocítica, com predomínio de formas maduras. Os blastos correspondem a menos de 5% dos granulócitos. Com a progressão da doença para fases mais avançadas, o número de blastos aumenta no sangue periférico. Basofilia e eosinofilia estão muitas vezes presentes.
Anemia leve, normocítica e normocrômica está presente em 45% dos casos. Quando há progressão da doença, a anemia tende a piorar. Até 30% dos doentes apresentam trombocitose, em alguns casos com contagem > 1.000.000. Os testes de função plaquetária são anormais, e o paciente pode ter eventos trombóticos ou hemorrágicos.
Uma característica marcante na LMC são os níveis de fosfatase alcalina nos leucócitos. Essa enzima é diminuída (< 10) em pacientes com LMC, sendo aumentada em outras síndromes mielo-proliferativas e em leucocitoses reacionais. É, portanto, de grande valia no diagnóstico diferencial da LMC.
Com o aumento expressivo na contagem de granulócitos, os níveis de vitamina B12 tendem a estar aumentados na LMC. Esse achado é secundário ao aumento da concentração de transcobalamina do tipo 1, naturalmente sintetizada pelos neutrófilos. Do mesmo modo, os níveis de ácido úrico podem estar aumentados, com hiperuricemia e hiperuricosuria, predispondo o paciente a crises de gota.
O mielograma e a biópsia de medula óssea mostram hipercelularidade da linhagem granulocítica, com maturação preservada. Pode haver leve grau de fibrose. No entanto, o mielograma é inespecífico para o diagnóstico de LMC, e a demonstração do cromossomo Filadélfia ou do gene BCR-ABL é necessária para o diagnóstico.
No cariótipo das células da medula, no qual geralmente são analisadas cerca de 20-30 metáfases, é possível encontrar o cromossomo Filadélfia na grande maioria dos casos. Com o uso de sondas específicas dirigidas aos genes envolvidos na translocação, pela técnica de FISH
O diagnóstico diferencial da LMC se dá principalmente com reações leucemóides e com outras síndromes mieloproliferativas (tabelas 8 e 9).
Tabela 8: Diagnóstico diferencial da leucemia mielóide crônica
|
Diagnóstico diferencial |
Causas |
Comentários |
|
Reação leucemóide |
Leucocitose reacional a algum processo infeccioso ou inflamatório |
Leucograma em geral < 50.000 cels/mm3, sem esplenomegalia FAL aumentada, sem cromossomo Filadélfia |
|
Síndromes mielo-proliferativas |
Policitemia vera, mielofibrose idiopática crônica, trombocitemia essencial |
Diagnóstico diferencial feito por hematócrito, esfregaço de sangue periférico, mielograma e biópsia de medula óssea FAL (aumentadas nas outras mielo-proliferativas) e ausência de cromossomo Filadélfia |
Tabela 9: Características das síndromes mielo-proliferativas
|
|
Hematócrito |
Leucócitos |
Plaquetas |
FAL* |
Clínica |
leucemia** |
|
LMC |
? |
?? |
? ou ? |
? |
Infiltração leucêmica |
80% |
|
PV |
?? |
? ou ? |
? ou ? |
? |
Hiperviscosidade |
5%-25% |
|
MIC |
? ou ? |
? ou ? ou ? |
? ou ? ou ? |
? |
Leucoeritroblastose |
5%-10% |
|
TE |
? |
? ou ? |
?? |
? |
Trombose e hemorragia |
Raro |
LMC – leucemia mielóide crônica; PV – Policitemia vera; MIC – Mielofibrose idiopática crônica; TE – Trombocitemia essencial; * Fosfatase alcalina leucocitária; **Porcentagem que evolui para leucemia aguda; ? Normal; ? Aumentado; ? Diminuído
Com o surgimento do imatinibe, inibidor da atividade tirosina-quinase da proteína BCR-ABL, houve uma mudança importante na condução dos pacientes com LMC. Considerado atualmente o tratamento-padrão, o imatinibe vem sendo utilizado cada vez mais em situações em que antes seriam realizados tratamentos mais agressivos, como o transplante de medula óssea.
Após o diagnóstico, o controle da contagem de leucócitos é o passo inicial do tratamento. Durante a fase crônica, o uso da hidroxiuréia controla a contagem de leucócitos com relativa facilidade. Entretanto, o uso de quimioterápicos orais não é capaz de reduzir a quantidade de células com cromossomo Filadélfia na medula óssea, não impedindo a evolução da doença para a crise blástica, que tende a ocorrer após quatro a seis anos do diagnóstico. O segundo passo, então, é o uso de terapêuticas que levem ao controle citogenético, fato associado a uma maior sobrevida.
O transplante de medula óssea alogênico
O interferon-alfa
O imatinibe é hoje o tratamento-padrão para pacientes recém-diagnosticados com LMC. Utilizado na dose de 400 mg/d em pacientes na fase crônica, o imatinibe gera resposta citogenética em mais de 80% dos pacientes. Essa resposta é mantida com o uso contínuo da droga, com menos de 3% dos pacientes abandonando o tratamento por causa dos efeitos colaterais. Alguns pacientes alcançam resposta molecular completa, ou seja, a ausência de detecção do produto do gene BCR-ABL pelo PCR, mas praticamente todos apresentam recorrência da doença se houver a interrupção do tratamento. A sobrevida estimada com imatinibe alcançou 83% em cinco anos no estudo IRIS, estudo fase III, comparando imatinibe com IFN-a. Alguns pacientes, entretanto, apresentam resistência ao imatinibe, geralmente secundária à ocorrência de mutações no gene BCR-ABL. Com o aumento da dose do imatinibe para 600 mg ou 800 mg/dia, uma nova resposta é alcançada na maioria dos pacientes, mas essa é geralmente fugaz. Novas drogas inibidoras de tirosino-quinase, como o dasatinibe e o nilotinibe, são capazes de resgatar parte desses pacientes, gerando resposta citogenética em até 45% dos casos.
As respostas a todos os tipos de tratamento tendem a ser piores à medida que a LMC progride para a fase acelerada e blástica. Inibidores da atividade tirosino-quinase do BCR-ABL geram resposta em cerca de 20%-40% dos pacientes nessa situação, mas muitos voltam a progredir mesmo após o tratamento. O transplante de medula óssea, apesar do potencial curativo, é associado a elevado índice de mortalidade, tanto por toxicidade quanto por progressão.
A escolha do melhor tratamento a ser oferecido para pacientes com LMC é uma questão ainda em debate. Com o surgimento de terapia medicamentosa eficaz, cada vez mais pacientes declinam do transplante de medula óssea. Entretanto, em alguns casos, o tratamento com imatinibe gera respostas apenas parciais ou transitórias. Não há, até o momento, escore de risco baseado no uso de imatinibe. A escolha do tratamento, então, é baseado em escores de risco para transplante de medula óssea – de acordo com o Grupo Europeu de Transplante de Sangue e Medula Óssea
Tabela 10: Escore de risco com transplante de medula óssea do EBMT
|
Número de fatores de risco |
Probabilidade de evento em 5 anos | ||
|
|
Sobrevida global |
Mortalidade pelo transplante |
Recidiva |
|
0 - 1 |
80 |
17 |
16 |
|
2 - 4 |
60 |
32 |
22 |
|
> 4 |
38 |
41 |
31 |
Fatores de risco:
Idade: < 20 anos = 0;
Fase da doença: fase crônica = 0; fase acelerada =1; fase blástica = 2
Tempo entre diagnóstico e transplante: < 12 meses = 0; > 12 meses = 1
Tipo de doador: aparentado = 0; não-aparentado = 1
Combinação de doador: feminino com receptor masculino = 1; outras combinações = 0
Gratwohl et al., 1998
Tabela 11: Chance de resposta a tratamento medicamentoso baseado no escore de Sokal
|
Fatores de risco |
Sobrevida livre de progressão com imatinibe | |
|
Idade Tamanho do baço Contagem de plaquetas Contagem de basófilos % de blastos na MO |
Baixo |
91% |
|
Intermediário |
84% | |
|
Alto |
69% | |
Cálculo do escore de risco disponível no site da Leukemia-net: <http://eln.zms-hosting.com/sites/leukemia-net/content/e58/e495/e1733>
Nas Leucemias crônicas a capacidade de diferenciação do clone neoplásico é mantida, resultando no acúmulo de células maduras na medula óssea e no sangue periférico.
Geralmente, apresentam uma evolução lenta, quando comparadas às leucemias agudas, porém são mais resistentes a quimioterapia.
A LLC é a leucemia crônica originária das células linfóides mais comum.
A LLC é uma neoplasia maligna em que há a proliferação de linfócitos B maduros, porém imunoincompetentes, e que acomete principalmente idosos. A doença costuma ter uma evolução indolente, muitos pacientes não necessitando de tratamento.
Na LLC, o tratamento quimioterápico deve ser iniciado em pacientes com doença progressiva ou estádio avançado. A escolha do esquema deve ser baseado em fatores como idade, presença de comorbidades e presença de marcadores indicativos de pior prognóstico.
A LPL tem evolução mais agressiva, sendo resistente aos esquemas quimioterápicos usuais. Novas formas de tratamento vêm sendo pesquisadas.
A LCP apresenta uma alta taxa de resposta a análogos de purida, como a cladribina. O interferon-alfa pode ser utilizado temporariamente em pacientes com risco muito aumentado de infecção.
O tratamento da LMC evoluiu bastante nos últimos anos com a introdução dos inibidores de tirosino-quinase, como o imatinibe.
O imatinibe na dose de 400 mg/dia é o tratamento-padrão para pacientes com LMC. O transplante de medula óssea alogênico pode ser indicado em pacientes com risco elevado de progressão com uso de inibidores tirosino-quinase ou se for a vontade do paciente.
Pacientes com falha terapêutica com imatinibe podem ser resgatados com o manejo dos inibidores de tirosino-quinase (imatinibe em altas doses, dasatinibe, nilotinibe) ou com transplante de medula óssea. Pacientes com respostas subótimas não têm indicação de tratamento definida, mas o manejo com drogas inibidoras da atividade tirosino-quinase do BCR-ABL é o mais recomendado.
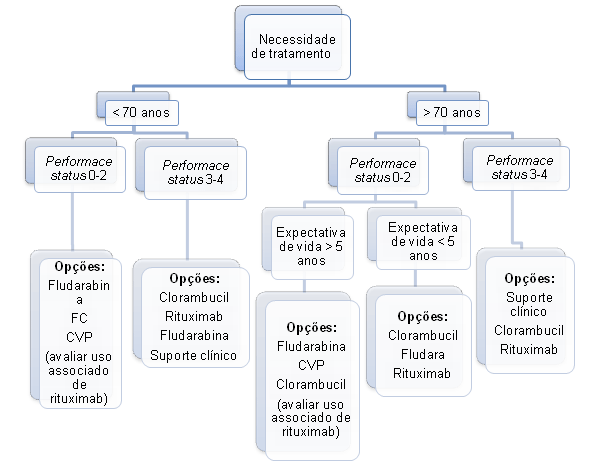
Algoritmo 1: Orientação do tratamento da leucemia linfocítica crônica

CVP – ciclofosfamida, vincristina, prednisona
FC – fludarabina, ciclofosfamida
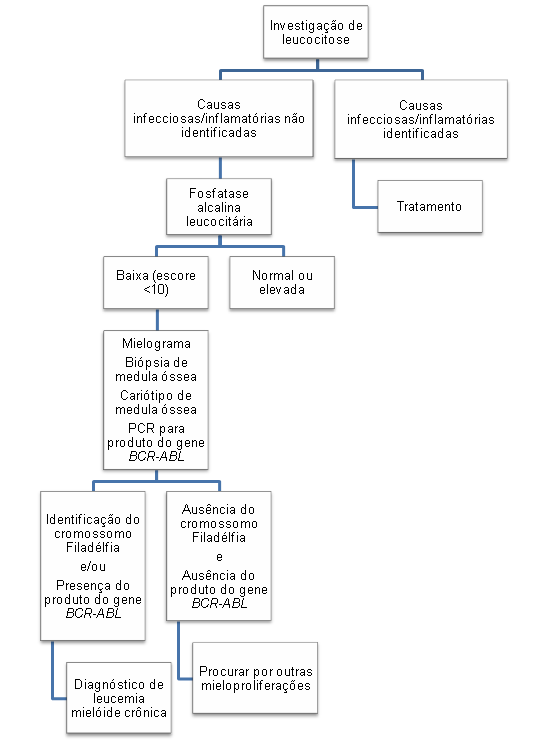
Algoritmo 2: Investigação diagnóstica para suspeita de leucemia mielóide crônica
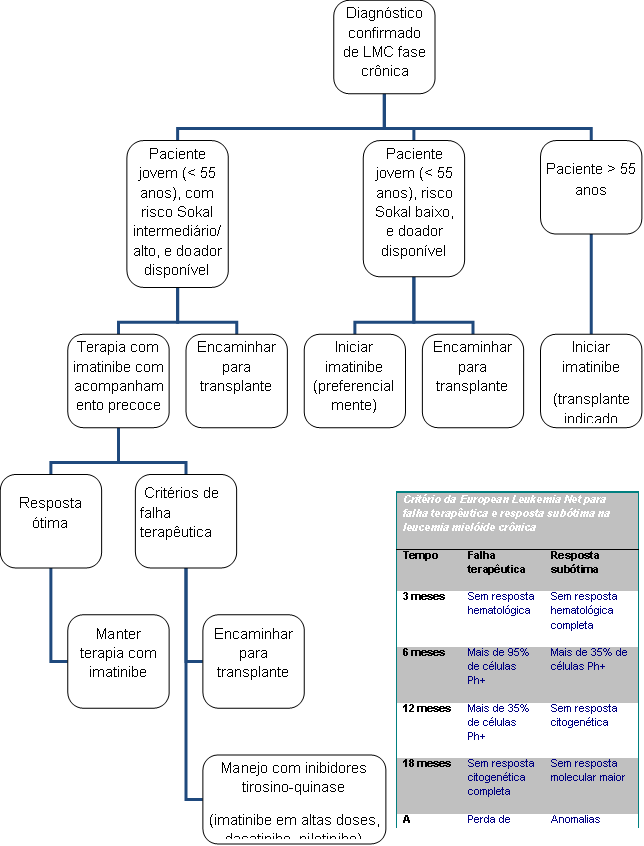
Algoritmo 3: Tratamento da leucemia mielóide crônica
1. Druker BJ, Guilhot F, O’Brian SG et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355(23): 2408-17.
2. Falcão, RP. leucemia linfóide crônica. In: Zago MA, Falcão RP, Pasquini R, Hematologia – Fundamentos e prática. 2 ed. Rio de Janeiro: Atheneu; 2001.
3. Gratwohl A, Hermans J, Goldman JM, Arcese W, Carreras E, Devergie A et al, for the Chronic Leukemia Working Party of the European Group for Blood and Bone Marrow Transplantation. Risk assessment for patient with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Lancet 1998;352:108792.
4. Hoffman R, Benz Jr. EJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE et al. Hematology – Basic principles and practice. 3. ed. New York: Churchill Livingstone; 2000.
5. Manuais de Condutas da Sociedade Brasileira de Oncologia Clínica.
6. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, 2008. Disponível em: <www.nccn.org>.
7. Osier D, Fegan C, Hillemen P et al. Guidelines on the diagnosis and management of chronic lymphocytic leukaemia. Br J Haematol 2004;125(3): 294-317.
8. Pasquini R. leucemia mielóide crônica, variantes da leucemia mielóide crônica. In: Zago MA, Falcão RP, Pasquini R. Hematologia – Fundamentos e prática. 2. ed. Rio de Janeiro: Atheneu; 2001.
9. Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999;340:1330-40.
10. Shanafelt TD, Call TG. Current approach to diagnosis and management of chronic lymphocytic leukemia. Mayo Clin Proc 2004;79(3):388-98.
11. Wiernik PH, Goldman, JM, Dutcher JP, Kyle RA. Neoplastic diseases of the blood. 4. ed. Cambridge: Cambridge University Press; 2003.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.