(Carregando Índice)... (Carregando Índice)... |
Autor:
Tarso Adoni
Neurologista pelo Hospital da Clínicas da Faculdade de Medicina da USP
Médico Colaborador do Serviço de Neurologia do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 21/12/2008
Comentários de assinantes: 0
Esclerose múltipla (EM) é uma doença inflamatória crônica, imunomediada, que acomete a mielina (substância branca) do sistema nervoso central (SNC). É a principal causa não-traumática de incapacidade em adultos jovens, segundo dados norte-americanos.
Recentes avanços no entendimento da fisiopatologia da doença passaram a incluir a EM também no grupo das doenças degenerativas, e não somente no espectro das doenças inflamatórias crônicas. Isso aconteceu pela evidência de acometimento patológico além da mielina, alcançando os próprios axônios.
EM é uma doença de adultos jovens, com pico de incidência por volta dos 25 anos de idade, mais comum em mulheres (cerca de duas mulheres para cada homem acometido). Raramente inicia-se antes dos 15 ou após os 50 anos de idade. É mais comum em populações de origem caucasiana e mais rara em negros e asiáticos.
Estudos de prevalência dividem o mundo em zonas de baixa, média e alta prevalência (Tabela 1). De modo geral, pode-se dizer que existe um gradiente de latitude: quanto mais distante da linha do Equador, maior a prevalência de EM (quanto maior a latitude, maior a prevalência). Tal achado suscita a especulativa participação de agentes ambientais no aparecimento da EM.
Tabela 1: Zonas de prevalência de EM, segundo Kurtzke
|
Prevalência |
Localidades |
|
Baixa (< 5 casos/100 mil hab.) |
América do Sul, México, maior parte da Ásia e África |
|
Média (5 a 25 casos/100 mil hab.) |
Austrália, sudeste dos Estados Unidos, países do Mediterrâneo (exceto Itália), partes da América do Sul e da Ásia |
|
Alta (> 30 casos/100 mil hab.) |
Europa, sudeste do Canadá, nordeste dos Estados Unidos, Nova Zelândia, sudoeste da Austrália |
Acredita-se que a auto-imunidade desempenha papel relevante na EM. Durante a formação do sistema imunológico, existe a depleção clonal de células com potencial de reagir contra os próprios antígenos. Em todos os indivíduos normais, algumas dessas células escapam da depleção e são mantidas sob vigilância, o que se denomina tolerância. A perda da tolerância imunológica permite que células com potencial de reagir contra antígenos próprios exerçam sua ação auto-imune deletéria.
A causa da perda da tolerância imunológica na EM ainda é assunto sem resposta. Especula-se que o mecanismo de mimetismo molecular possa desempenhar algum papel, pela exposição do sistema imunológico a antígenos (componentes virais) que compartilhem semelhanças estruturais com proteínas da bainha de mielina do SNC.
Outra maneira possível de perda da tolerância é por infecção do SNC, o que causaria lesão tissular e liberação de antígenos sequestrados no SNC para a circulação periférica, desencadeando a ação de células T auto-reativas.
Apesar das diversas tentativas de se isolar um possível agente infeccioso como deflagrador (trigger) da EM, ainda permanece especulativa qualquer associação entre infecção e EM. Os principais agentes suspeitos nos últimos anos, sem qualquer comprovação, são herpes vírus do tipo 6, vírus Epstein-Barr e a bactéria Chlamydia pneumoniae.
Do ponto de vista genético, existem alguns genes candidatos para a suscetibilidade de EM, como o antígeno leucocitário humano (HLA) e o gene que codifica o receptor da célula T, entre outros. Estudo recente realizado pelo International Multiple Sclerosis Genetics Consortium, que se baseou no estudo de polimorfismos gênicos em alelos sabidamente envolvidos com maior suscetibilidade para a EM, encontrou marcadores de predisposição genética nos genes que codificam a subunidade alfa dos receptores da interleucina 2 e da interleucina 7.
Assim, parece que a EM depende, para o seu aparecimento, da ocorrência de interação entre fatores ambientais e a presença de um hospedeiro geneticamente suscetível.
O protótipo do paciente com EM é uma mulher jovem, com idade entre 20 e 30 anos, de pele branca, com história de sintomas neurológicos que aparecem e desaparecem ao longo do tempo.
Os principais sinais e sintomas da EM são déficits motores (piramidais, como fraqueza em um hemicorpo, sinal de Babinski e reflexos profundos exaltados), sensitivos (hipoestesia superficial ou profunda, parestesias), cerebelares (ataxia, desequilíbrio), visuais (diminuição da acuidade visual, diplopia) e de tronco encefálico (oftalmoplegia internuclear – paresia do olho adutor no olhar conjugado lateral com nistagmo horizontal do olho abdutor) e, após algum tempo de doença, neurovegetativos (principalmente urgência urinária, com ou sem incontinência). Alguns sinais e sintomas são bastante sugestivos de EM, como neurite óptica, sinal de Lhermitte (flexão do pescoço leva à sensação de eletricidade percorrendo a coluna), oftalmoplegia internuclear e a piora dos sintomas com elevação da temperatura corpórea (fenômeno de Uhthoff), principalmente após exposição ao sol, após tomar um banho quente, ficar em frente a um forno aceso ou em ambientes fechados muito aquecidos (Tabela 2).
Tabela 2: Características clínicas sugestivas de EM
|
Instalação entre 15 e 50 anos de idade |
|
Evolução em recorrência e remissão |
|
neurite óptica |
|
oftalmoplegia internuclear |
|
fenômeno de Uhthoff |
A definição de episódio clínico típico ou “surto” de EM inclui a comprovação objetiva de novo acometimento neurológico, com duração de pelo menos 24 horas, não atribuível a infecção ou distúrbios metabólicos.
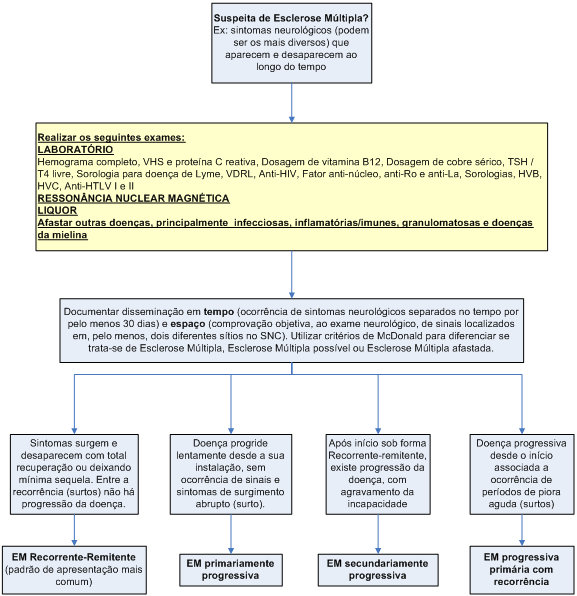
A EM é uma doença que pode se apresentar sob quatro padrões:
1. EM recorrente-remitente;
2. EM primariamente progressiva;
3. EM secundariamente progressiva;
4. EM progressiva primária com recorrências.
Como o próprio nome sugere, os sintomas surgem e desaparecem com total recuperação ou deixando mínima seqüela. Entre a recorrência (surto) dos sintomas, não há qualquer progressão da doença. Esse é o padrão de apresentação mais comum da EM, acometendo 85% dos portadores.
Doença progride lentamente desde a sua instalação, sem a ocorrência de sinais e sintomas de surgimento abrupto (surto). Essa forma representa 15 a 20% dos casos de EM e, diferentemente da forma recorrente-remitente, tende a acometer indivíduos com mais de 40 anos de idade e é mais comum em homens. O quadro clínico clássico é o de paraparesia espástica progressiva associada a sintomas neurovegetativos (bexiga neurogênica com urgência/urge-incontinência ou, mais raramente, retenção urinária).
Após o início da doença sob curso recorrente-remitente, existe progressão da doença, com agravamento da incapacidade. Nesse período, pode ou não haver a ocorrência de surtos. Esse padrão acometerá cerca de 50% dos pacientes após 10 anos do início da doença.
Doença progressiva desde o início associada à ocorrência de períodos agudos de piora (surto), com total recuperação ou permanência de sequelas; há progressão da doença entre os surtos. É a forma menos frequente de apresentação.
A EM é uma doença de exclusão e devem ser afastados outros diagnósticos. Assim, diante de qualquer paciente com EM, é obrigatória a solicitação dos seguintes exames complementares.
Além dos exames de rotina, devem ser pesquisadas alterações reumatológicas e infecciosas (Tabela 3).
Tabela 3: Exames laboratoriais para a investigação de EM
|
Hemograma completo |
|
VHS e proteína C reativa |
|
Dosagem de vitamina B12* |
|
TSH/T4 livre |
|
Sorologia para doença de Lyme |
|
VDRL |
|
Anti-HIV |
|
Fator antinúcleo, anti-Ro e anti-La |
|
Sorologias para HVA, HVB, HVC e Borrelia burgdorferi |
|
Anti-HTLV I e II |
HVA: hepatite A; HVB: hepatite B; HVC: hepatite C; VZV: varicela-zóster vírus.
*Especialmente na forma primariamente progressiva.
O objetivo da RM é descartar outras possibilidades diagnósticas e evidenciar alterações sugestivas de processo desmielinizante. Há critérios radiológicos estabelecidos por Barkoff et al. (Tabela 4) que facilitam o diagnóstico radiológico, desde que excluídas outras causas para os achados.
Tabela 4: Critérios radiológicos de Barkoff (ressonância magnética) para EM
|
Devem ser preenchidos pelo menos 3 dos 4 critérios |
|
Nove lesões hiperintensas em T2 ou 1 lesão que capte gadolíneo em T1 |
|
Pelo menos uma lesão justacortical |
|
Pelo menos uma lesão infratentorial |
|
Pelo menos três lesões periventriculares |
O exame de LCR lombar na EM pode revelar celularidade normal ou aumento discreto do número de células (pleocitose abaixo de 50 células/mm3), perfil linfomonocitário, proteinorraquia (até 40 mg/dL) e glicorraquia normais. Já a eletroforese de proteínas do LCR por método de isoeletrofocalização pode revelar a presença de bandas oligoclonais (que não são patognomônicas de EM e podem ser encontradas em outras doenças do SNC, como panencefalite esclerosante subaguda, neuroesquistossomose, paraparesia espástica tropical, meningites crônicas por fungos, dentre outras) e aumento no índice de IgG.
É de fundamental importância que se tenha em mente o seguinte conceito: “Esclerose múltipla é uma doença que exige, para o seu diagnóstico, a comprovação objetiva de disseminação no tempo e no espaço, desde que não haja melhor explicação para os achados”.
Chama-se disseminação no tempo a ocorrência de sinais neurológicos separados no tempo por pelo menos 30 dias. Chama-se disseminação no espaço a comprovação objetiva, ao exame neurológico, de sinais localizados em, pelo menos, dois diferentes sítios no SNC: papila óptica pálida ao exame de fundo de olho (lesão de nervo óptico) e oftalmoplegia internuclear (lesão localizada no tronco encefálico), por exemplo.
Ainda assim, diante de um paciente com disseminação temporal e espacial, é necessário que se excluam outras doenças que possam se apresentar com o mesmo quadro clínico, realizando os exames citados no item anterior e reavaliando clinicamente. A Tabela 5 mostra alguma dessas doenças.
Tabela 5: Diagnóstico diferencial da EM
|
Doenças inflamatórias |
Lúpus eritematoso sistêmico, doença de Sjögren, doença de Behçet, poliarterite nodosa, encefalomielite disseminada aguda |
|
Doenças infecciosas |
Neuroborreliose de Lyme, leucoencefalopatia multifocal progressiva, neurossífilis, mielopatia do HIV, paraparesia espástica tropical (HTLV) |
|
Doenças granulomatosas |
Sarcoidose, doença de Wegener, granulomatose linfomatóide |
|
Doenças da mielina |
Leucodistrofia metacromática, adrenomieloleucodistrofia |
|
Miscelânea |
Ataxias espinocerebelares, malformação de Arnold-Chiari, degeneração combinada subaguda da medula por deficiência de vitamina B12 |
Para facilitar o diagnóstico de EM têm sido propostos critérios diagnósticos. Os primeiros critérios diagnósticos de EM datam de 1965, feitos por Schumacher. Após esse período, houve a incorporação do auxílio laboratorial no diagnóstico mediante os critérios de Poser, de 1983.
Atualmente, utilizam-se os critérios de McDonald, de 2001 (Tabela 6). Esse último critério considera os avanços proporcionados pela RM e mantém o auxílio do laboratório proposto por Poser.
Tabela 6: Critérios diagnósticos de McDonald*
|
Apresentação clínica |
Dados adicionais para diagnóstico de EM |
|
2 ou mais surtos**; evidência clínica de 2 ou mais lesões |
Nenhum necessário. No entanto, se testes forem realizados e forem negativos (por exemplo, RM e LCR normais), é preciso muita cautela antes de se diagnosticar EM, devendo ser considerados diagnósticos alternativos. Não deve haver explicação melhor para os sintomas e deve haver alguma evidência objetiva que suporte o diagnóstico de EM |
|
2 ou mais ataques; evidência clínica de 1 lesão |
Documentar disseminação no espaço, demonstrável por: alterações típicas na RM que preencham os critérios de Barkhof (Tabela 4); ou 2 ou mais lesões sugestivas de EM associadas a LCR sugestivo; ou novo surto posterior acometendo local diferente |
|
1 surto, com evidência clínica de 2 ou mais lesões |
Documentar disseminação no tempo, demonstrável por: alterações típicas de novos surtos em RM realizadas posteriormente; novo surto posterior |
|
1 surto, com evidência clínica de 1 lesão |
Documentar disseminação no espaço, demonstrável por: alterações típicas na RM que preencham os critérios de Barkhof (Tabela 4); ou 2 ou mais lesões sugestivas de EM associadas a LCR sugestivo; ou novo surto posterior acometendo local diferente Documentar também: Disseminação no tempo, demonstrável por: alterações típicas de novos surtos em RM realizadas posteriormente; novo surto posterior |
|
Progressão neurológica insidiosa sugestiva de EM |
Um ano de progressão de doença (determinada retrospectiva e prospectivamente) associado a 2 ou mais dos seguintes critérios: RM característica de EM (9 ou mais lesões em T2); RM característica em medula espinal (2 lesões focais em T2); LCR sugestivo |
* Se os critérios forem preenchidos e não houver outra explicação para a apresentação clínica, o diagnóstico será de EM. Se houver suspeita clínica de EM, mas os critérios não forem preenchidos, o diagnóstico será de “EM possível”; Se outro diagnóstico surgir durante a avaliação clínica que explique melhor a sintomatologia, o diagnóstico não será de EM.
** Um surto é definido como um episódio de alterações neurológicas provavelmente de origem inflamatória e desmielinizante. Deve haver alterações objetivas (com suporte de achados objetivos) ou observação objetiva de que o evento durou no mínimo 24 horas.
Não existe cura, até o momento, para a EM. O tratamento da EM pode ser dividido em duas partes: fase aguda e fase de manutenção.
Refere-se aos “surtos” (exacerbações ou recorrências), definidos pela ocorrência de sinal ou sintoma neurológico com duração superior a 24 horas. De acordo com o Consenso Expandido do BCTRIMS para o tratamento da EM, com o objetivo de acelerar a melhora funcional (remissão dos sinais ou sintomas), a droga de escolha é a metilprednisolona, 1 g IV, por 3 dias consecutivos (pulsoterapia).
Envolve o uso de drogas imunomoduladoras. Essas drogas apresentam diferentes características biológicas, vias e frequência de administração e efeitos adversos. As drogas atualmente disponíveis são o interferon-beta 1a, interferon-beta 1b e o acetato de glatirâmer. Acredita-se que os efeitos do interferon se devem a menor produção de interferon-gama, aumento da produção de interleucina 10, diminuição da relação Th1/Th2, redução na permeabilidade da barreira hematoencefálica por ação sobre moléculas de adesão endotelial e metaloproteases e efeito sobre as células gliais. O acetato de glatirâmer apresenta efeito terapêutico por expansão de células Th2-específicas com propriedades imunorreguladoras.
A Tabela 7 mostra as características das drogas disponíveis para o tratamento de manutenção da EM.
Tabela 7: Características das drogas disponíveis para o tratamento da EM
|
|
IFN-beta 1b betaferon |
IFN-beta |
IFN-beta |
Acetato de glatirâmer copaxone |
|
Origem |
Eschericia coli |
Ovário de hamster |
Ovário de hamster |
Polipeptídeo sintético |
|
Dose |
8 MUI (250 mcg) |
30 mcg (6 MUI) |
22 mcg (MUI) 44 mcg (12 MUI) |
20 mg |
|
Via de administração |
Subcutânea |
Intramuscular |
Subcutânea |
Subcutânea |
|
Frequência de administração |
Dias alternados |
1 vez/semana |
3 vezes/semana |
Diária |
|
Indicação |
EMRR EMSP com surtos |
EMRR |
EMRR |
EMRR |
|
Contra-indicações |
Gravidez Depressão grave Idéias suicidas Epilepsia não controlada Insuficiência hepática Hipersensibilidade à droga ou à albumina humana |
Idem |
Idem |
Hipersensibilidade à droga |
|
Efeitos adversos |
Reações locais Síndrome gripal Aumento de espasticidade Linfopenia Depressão Idéias suicidas |
Idem |
Idem |
Reações locais |
MIU: milhões de unidades internacionais; EMRR: esclerose múltipla recorrente-remitente; EMSP: esclerose múltipla secundariamente progressiva.
*Secretaria de Saúde do Estado de Minas Gerais.
Existe grande variabilidade individual em relação ao prognóstico da doença. De maneira geral, pode-se dizer que mulheres apresentam um curso mais benigno que homens; quanto maior a idade ao diagnóstico, pior o prognóstico; a forma recorrente-remitente é de melhor prognóstico que a forma primariamente progressiva; o início da doença por neurite óptica ou alterações sensitivas é de melhor prognóstico que o início por sintomas motores ou cerebelares.
Em geral, os portadores de EM morrem com EM, e não de EM, embora haja alguns dados conflitantes na literatura.
A EM é uma doença crônica imunomediada que acomete a mielina do SNC. Afeta principalmente adultos jovens, com pico de incidência por volta dos 25 anos de idade, e é mais comum em mulheres e populações de origem caucasiana.
A EM apresenta-se sob a forma de recorrências (surtos) e remissões de sintomas neurológicos.
Alguns sinais e sintomas são muito sugestivos de EM, como neurite óptica, sinal de Lhermitte (flexão do pescoço leva à sensação de eletricidade percorrendo a coluna), oftalmoplegia internuclear e piora dos sintomas com elevação da temperatura corpórea.
A forma mais comum de EM é a recorrente-remitente, na qual os sintomas surgem e desaparecem com total recuperação ou deixando mínima sequela e sem progressão da doença entre os surtos. Outras formas de evolução são EM primariamente progressiva, EM secundariamente progressiva e EM progressiva primária com recorrências.
A EM é um diagnóstico de exclusão e exige extensa avaliação na busca de diagnósticos alternativos.
Embora nem sempre disponível, a RM é de grande importância no diagnóstico e acompanhamento de pacientes com EM.
EM é uma doença que exige, para o seu diagnóstico, a comprovação objetiva de disseminação no tempo e no espaço, desde que não haja melhor explicação para os achados.
Disseminação no tempo é a ocorrência de sinais neurológicos separados no tempo por pelo menos 30 dias. Disseminação no espaço é a comprovação objetiva, ao exame neurológico, de sinais localizados em, pelo menos, dois diferentes sítios no SNC.
Os surtos de EM são tratados com pulsoterapia de corticóides. Na fase crônica, são usados os imunomoduladores. As drogas atualmente disponíveis são o interferon-beta 1a, interferon-beta 1b e o acetato de glatirâmer.
Algoritmo 1: Avaliação diagnóstica da EM

1. Compston A, Confavreux C, Lassmann H. McAlpine’s multiple sclerosis. 4. ed. London: Churchill Livingstone, Elsevier, 2006.
2. Fleming JO. Diagnosis and management of multiple sclerosis. New York: Professional Communications Inc., 2002.
3. Hafler DA, Compston A, Sawcer S. The International Multiple Sclerosis Genetics Consortium. Risk allelles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357;851-62.
4. Irani S, Lang B. Auto-antibody mediated disorders of the central nervous system. Autoimmunity 2008; 41:55-65.
5. Kurtzke JF, Beebe GW, Norman Jr. JE. Epidemiology of multiple sclerosis in US veterans. 1. Race, sex, and geographic distribution. Neurology 1979; 29:1228-35.
6. McDonald WI, Compston A, Edan G et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the Diagnosis of Multiple Sclerosis. Ann Neurol 2001; 50:121-7.
7. Moreira MA, Lana-Peixoto MA, Callegaro D et al. Consenso Expandido do BCTRIMS para o tratamento da esclerose múltipla. Arq Neuropsiquiatr 2002; 60(3-B):875-80.
8. www.nationalmssociety.org.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.