(Carregando Índice)... (Carregando Índice)... |
Autores:
Samuel Katsuyuki Shinjo
Médico Assistente do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da USP
Clínico geral e Reumatologista pelo Hospital das Clínicas da Faculdade de Medicina da USP
Mestre e Doutor em Ciências pela Universidade Federal de São Paulo
Ari Stiel Radu Halpern
Professor Colaborador da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP. Médico Assistente Doutor do Serviço de Reumatologia do Hospital das Clínicas da FMUSP.
Mauricio Levy-Neto
Professor Colaborador da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP. Médico Assistente Doutor do Serviço de Reumatologia do Hospital das Clínicas da FMUSP.
Rosa Maria Rodrigues Pereira
Professora Livre-docente da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP. Médica Assistente Doutora do Serviço de Reumatologia do Hospital das Clínicas da FMUSP.
Última revisão: 22/11/2011
Comentários de assinantes: 0
As vasculites sistêmicas são um grupo de doenças caracterizadas por um processo inflamatório de vasos sanguíneos levando a necrose da parede do vaso e obstrução da luz. O espectro clínico das vasculites sistêmicas é muito variado, dependendo, em grande parte, do tamanho e da localização dos vasos acometidos (Tabela 1).
Quando ocorre de forma isolada, são denominadas vasculites primárias. Outras vezes, porém, surgem na vigência de doenças sistêmicas autoimunes, neoplásicas ou infecciosas, sendo denominadas então de vasculites secundárias.
Várias condições clínicas podem mimetizar quadros de vasculite, devendo ser consideradas no diagnóstico diferencial das vasculites sistêmicas (Tabela 2).
Normalmente as vasculites sistêmicas acometem múltiplos órgãos e tecidos. No entanto, mais raramente as manifestações podem ser limitadas a um único órgão ou sistema, como nas vasculites exclusivamente cutâneas ou nas formas isoladas em sistema nervoso central. Tradicionalmente, as vasculites costumam ser classificadas de acordo com o calibre dos vasos acometidos (Tabela 3). Mais recentemente, a descrição da presença, no soro de portadores de algumas formas de vasculites, de anticorpos contra o citoplasma de neutrófilos (ANCA) trouxe auxílio no diagnóstico, particularmente de portadores de granulomatose de Wegener e poliangiite microscópica.
Algumas vasculites acometem sobretudo crianças (doença de Kawasaki e púrpura de Henoch-Schönlein), enquanto outras acometem adultos (AT, poliarterite nodosa, poliangiite microscópica) ou idosos (arterite de células gigantes).
Tabela 1: Classificação de vasculites de acordo com o calibre do vaso acometido
|
Grande calibre |
Arterite de Takayasu; arterite de células gigantes |
|
Médio calibre |
Poliarterite nodosa; doença de Kawasaki |
|
Pequeno calibre |
ANCA associado: poliangiite microscópica; granulomatose de Wegener; síndrome de Churg-Strauss |
|
Imunocomplexo mediado: doença de Henoch-Schönlein; vasculite crioglobulinêmica; vasculite urticariforme hipocomplementêmica; angiite leucocitoclástica cutânea; doença de Goodpasture |
Tabela 2: Diagnóstico diferencial de vasculites
|
Doença vascular |
Aterosclerose, displasia fibromuscular, amiloidose |
|
Infecções |
Sífilis, tuberculose, doença de Lyme, hepatite viral crônica, meningoencefalite, sepse |
|
Diátese sanguínea |
Púrpura trombocitopênica trombótica, síndrome antifosfolípide, síndrome hemolítico-urêmica, coagulação intravascular disseminada, medicamentosa (heparina, warfarina) |
|
Embolização |
Mixoma, êmbolo de colesterol, endocardite infecciosa |
|
Drogas |
Cocaína, anfetaminas, ergotaminas |
|
Vasoespamos |
Feocromocitoma |
|
Outros |
Neoplasia, síndrome hipereosinofílica, linfoma intravascular, síndrome de hiperviscosidade, doenças do tecido conectivo |
Tabela 3: Manifestação clínica de vasculites de acordo com o tamanho do calibre do vaso acometido
|
Pequenos vasos |
Vênulas pós-capilares |
Púrpura palpável |
|
Arteríolas e capilares |
Livedo reticular | |
|
Capilares glomerulares |
Hematúria, disfunção renal | |
|
Capilares pulmonares |
Hemoptise, dispneia | |
|
Médios vasos |
Cutâneo |
Necrose, úlceras, infartos digitais |
|
Artérias epineural |
Moneurite multiplex | |
|
Artérias do trato gastrintestinal |
Dor abdominal, hemorragia digestiva baixa, úlceras intestinais, infarto visceral | |
|
Artérias renais |
Infarto e disfunção renal | |
|
Artérias coronárias |
Angina, infarto miocárdico | |
|
Grandes vasos |
Aorta e seus ramos |
Diferença de pulso e de pressão, sopros, claudicação vascular |
|
Artéria carótida externa |
Cefaleia temporal, cegueira |
A arterite de Takayasu (AT) é uma vasculite primária sistêmica que acomete vasos de grande calibre, ou seja, a aorta e seus ramos principais, além de artéria pulmonar. É conhecida também por doença sem pulsos, aortite esclerosante, coarctação reversa, síndrome do arco aórtico, arterite primária da aorta, aortite estenosante, tromboarteriopatia oclusiva, síndrome da aortite e aortoarterite medial idiopática.
É uma doença rara predominante na Ásia, mas que ocorre em todo o mundo. A incidência anual nos EUA, por exemplo, é de 2,6/milhões de indivíduos. Já no Japão, é aproximadamente 10 vezes mais frequente que nos EUA. Na América Latina, também parece ser muito mais frequente que na Europa e nos EUA.
O início da doença geralmente ocorre antes dos 40 anos de idade, predominantemente no sexo feminino.
A causa de AT é desconhecida, mas acredita-se que fatores genéticos, hormonais e ambientais estejam envolvidos no desencadeamento da doença. Casos de gêmeos idênticos concordantes para a doença foram descritos, inclusive um par criado separadamente. O grande predomínio em mulheres jovens entre a menarca e a menopausa faz pressupor participação de hormônios femininos no seu desencadeamento. Com relação a fatores ambientais, a doença parece ser mais frequente em países com maior prevalência de tuberculose. No entanto, tratamento empírico para tuberculose não parece modificar o curso da doença.
A vasculite inicia-se na porção da parede arterial entre as camadas média e adventícia e progride envolvendo toda a parede arterial. Infiltrado celular mononuclear invade a parede, aparentemente através dos vasos da vasa vasorum, e ocorre formação de granuloma não caseoso e destruição da parede do vaso. Na maioria dos casos, a doença evolui de forma muito lenta e o processo inflamatório é progressivamente substituído por fibrose, que predomina na camada adventícia e leva lentamente à oclusão arterial. Ocorre também proliferação da camada íntima que predispõe à ocorrência de trombose. Por outro lado, pode ainda ocorrer a formação de aneurismas, bem como a formação de placas de ateroma, que parece ocorrer em maior quantidade nas artérias previamente lesadas. De forma geral, em cerca de 3/4 das artérias acometidas predominam os estreitamentos do calibre, que variam de estenoses leves a oclusão da artéria. Por outro lado, as dilatações arteriais costumam predominar na aorta ascendente e, com frequência, provocam insuficiência aórtica que, por sua vez, é um dos marcadores de mau prognóstico.
O quadro clínico varia conforme o local e o grau de acometimento vascular. A Tabela 4 ilustra a frequência de acometimento vascular em AT.
Na fase inicial da doença, o paciente pode apresentar sintomas constitucionais e/ou manifestações sistêmicas inespecíficas (febre, emagrecimento, indisposição, cefaleia, dor torácica atípica, dor abdominal, eritema nodoso etc.). No entanto, também pode cursar de uma forma totalmente assintomática, quando a lesão vascular ocorrer de uma forma lenta e progressiva e, portanto, há tempo hábil para o desenvolvimento de circulação arterial colateral e o paciente, surpreendentemente, não apresenta qualquer complicação, mesmo após a oclusão da artéria. Neste caso, a doença pode, por exemplo, ser descoberta casualmente em uma consulta médica por outro motivo. O achado clínico que leva a suspeita nestes casos é a diminuição de pulso, a presença de sopro cardíaco ou extracardíaco (carótidas, abdominal etc.) ou a assimetria da medida da pressão arterial nos membros. Por meio de exames de imagens (p. ex., tomografia computadorizada e angiorressonância dos vasos), é possível confirmar tais lesões vasculares.
Por outro lado, as manifestações sistêmicas podem ser extremamente exuberantes como reflexo de intensa atividade inflamatória vascular, levando o paciente a acompanhamento médico e investigação exaustiva que pode durar meses ou anos. Nesta fase, conhecida também por fase pré-estenótica, é muito difícil estabelecer o diagnóstico, pois o processo inflamatório está ocorrendo nas grandes artérias sem que se consiga evidenciá-lo. Após esta fase, o paciente pode ou não apresentar um período assintomático de semanas, meses ou anos até o início da segunda fase, conhecida como fase pós-estenótica. Nesta fase, há predomínio de sintomas de isquemia de tecidos ou órgãos. Isquemias graves e de evolução rápida são raras, mas podem levar o paciente a óbito ou a dano irreversível. No caso de isquemia renal, ocorre em cerca de 1/3 dos pacientes e, na maioria dos casos, provoca hipertensão arterial renovascular de difícil controle medicamentoso ou perda de função renal. Este envolvimento é responsável por mais da metade das indicações cirúrgicas na doença.
Se em vez de estenose houver formação de aneurismas, estes podem se tornar sintomáticos ou complicar com compressões extrínsecas, tromboses com embolias a distância, dissecção ou ruptura. O aneurisma da aorta ascendente costuma provocar dilatação do anel valvar adjacente, causando insuficiência aórtica, com consequente insuficiência cardíaca congestiva.
Às vezes, a AT ocorre de forma associada com várias doenças inflamatórias, entre as quais se destacam as doenças inflamatórias intestinais que ocorrem em até 5% dos pacientes, principalmente a doença de Cröhn. Entre as outras, estão a policondrite recidivante, a granulomatose de Wegener, a espondilite anquilosante, a febre reumática e a glomerulonefrite membranoproliferativa.
Tabela 4: Frequência de vasos sanguíneos envolvidos em arterite de Takayasu
|
Vaso sanguíneo |
Acometimento (%) |
|
Aorta |
65 |
|
Arco da aorta e ramos principais |
35 |
|
Aorta torácica |
17 |
|
Aorta abdominal |
47 |
|
Artérias subclávias |
93 |
|
Artérias carótidas comuns |
58 |
|
Artérias renais |
38 |
|
Artérias vertebrais |
35 |
|
Tronco celíaco |
18 |
|
Artérias ilíacas comuns |
17 |
|
Artérias pulmonares |
5 |
O exame físico de um paciente com AT deve sempre incluir a palpação dos pulsos periféricos à procura de sopros e medida de pressão arterial nos quatro membros. De uma consulta a outra, deve-se estar atento para o surgimento ou a diminuição progressiva da intensidade ou desaparecimento de pulsos, agravamento dos sopros ou frêmitos ou queda da pressão arterial em algum membro. A presença de um sopro novo deve alertar o clínico para a necessidade de repetição de métodos de imagem. Presença de eritema nodoso sugere atividade de doença.
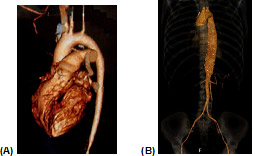
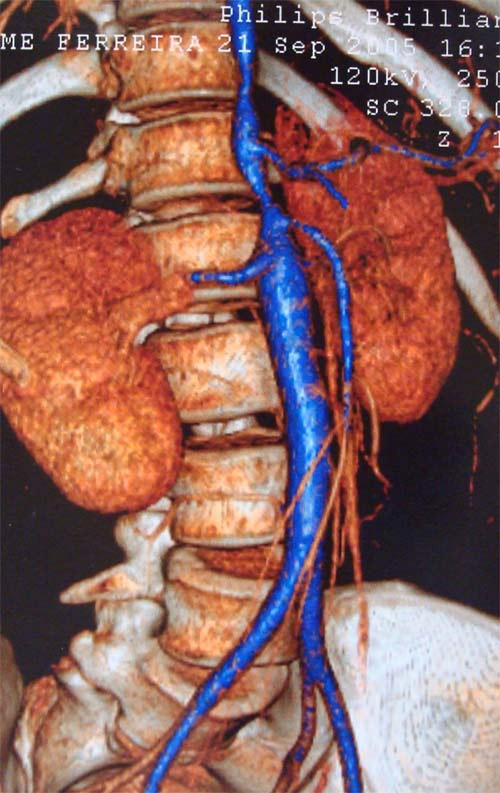
Em virtude do risco envolvido, é impossível indicar biópsia de grandes artérias, a não ser que exista indicação cirúrgica, que só ocorre na evolução, em cerca de 1/3 dos pacientes. Por isso, diferentemente das outras vasculites, o diagnóstico de AT não se baseia na biópsia. Como não existe nenhum exame marcador para esta doença, seu diagnóstico é baseado na clínica e no aspecto das lesões por método de imagem (p. ex., arteriografia digital, angiotomografia ou angiorressonância, sendo este último preferível, dadas a utilização de contraste menos tóxico e a ausência de irradiação). Pelos exames radiológicos, espera-se encontrar imagens sugestivas de AT, como a presença de estenose e/ou aneurismas de aorta e/ou de seus ramos principais. A Figura 1 mostra angiotomografia (reconstrução), demostrando a presença de estenose da artéria carótida e subclávia esquerda (A) e esctasia da aorta descendente (B). Já na Figura 2, há uma estenose da aorta toracoabdominal, ads renais e da mesentérica inferior.
As lesões na AT costumam poupar os óstios, com exceção das artérias coronárias, que podem mostrar estenoses do óstio em razão do envolvimento da aorta ascendente.
Entre as complicações e os sinais de mau prognóstico em AT, temos a presença de aneurismas, hipertensão renovascular, insuficiência aórtica e retinopatia isquêmica. Desse modo, é importante avaliar regularmente a presença ou não de tais sinais, ou seja, além de exames de imagens, o ecocardiograma, Doppler de artérias renais e a avaliação de fundo de olho.
Figura 1: Estenose da artéria carótida e subclávia esquerda (A) e ectasia da aorta descendente (B).

Figura 2: Estenose da aorta toracoabdominal, das artérias renais e da mesentérica inferior.
Entre os diagnósticos diferenciais de AT estão a doença de Ehlers-Danlos tipo IV e a síndrome de Marfan, as quais não costumam apresentar elevações das provas de fase aguda.
Anemia de doença crônica e plaquetose também podem ser utilizadas como parâmetro de atividade. Estes exames em geral são utilizados para monitorar longitudinalmente a atividade da doença e a resposta ao tratamento. Alguns pacientes, entretanto, apresentam provas de fase aguda pouco alterada, mesmo em atividade, e outros, mesmo fora de atividade permanecem sempre com provas inflamatórias de fase aguda elevadas.
Síndrome de Cogan e doença de Behçet muitas vezes envolvem artérias de grande calibre e, nesta circunstância, costumam se apresentar com provas inflamatórias extremamente elevadas, podendo ser confundidas com a AT. Ajuda na diferenciação da primeira a presença de ceratite e do distúrbio vestibular periférico; e da segunda, a presença de úlceras orais e genitais, lesões pustulosas, pseudofoliculite e história de trombose venosa.
Em geral, na fase aguda da doença, é necessário administrar entre 0,5 e 1 mg/kg/dia de prednisona oral para o controle da atividade de doença, desaparecimento dos sintomas sistêmicos e melhora das provas da fase aguda. A maioria dos pacientes entra em remissão clínica e laboratorial com esta medida. Nos casos que não respondem a doses altas de corticosteroides e naqueles em que recidivas de atividade sempre ocorrem ao se tentar redução da dose de prednisona, está indicada a utilização de imunossupressores. Esta indicação se baseia em estudos não controlados, em sua maioria, relatos de poucos casos. O imunossupressor mais usado é o metotrexato, mas existem relatos de bons resultados com ciclofosfamida, azatioprina, ciclosporina, micofenolato mofetil e leflunomida. Mais recentemente, têm-se empregado agentes biológicos como o anti-TNF para casos refratários à associação de corticosteróides com imunossupressores.
Indica-se também a utilização de baixas doses de aspirina ou outros antiagregantes plaquetários, por tempo indeterminado, visando a prevenir fenômenos trombóticos relacionados às lesões das grandes artérias. Outra preocupação é o desenvolvimento de ateromatose sobre as lesões vasculíticas. Para sua prevenção, em geral indica-se controle cuidadoso dos níveis de colesterol com atividade física, dieta e uso de hipolipemiantes, se necessário. Não existem, entretanto, trabalhos convincentes que demonstrem a eficácia destas medidas na AT, e sua indicação baseia-se em estudos fisiológicos.
A arterite de células gigantes (ACG) é uma vasculite que acomete vasos de grande e médio calibre, preferencialmente os ramos extracranianos das carótidas e, em particular, a artéria temporal superficial. Afeta principalmente indivíduos de cor branca, sexo feminino (3:1) e > 50 anos de idade.
É também conhecida como arterite temporal, cefaleia de Horton, doença de Horton, doença de Hutchinson-Horton e arterite cranial.
A ACG é mais frequente na Europa, no Canadá e no norte dos EUA. A incidência anual é estimada em 17/100 mil indivíduos nos EUA. Por outro lado, é menos frequente nos países de latitude menor e incide mais em brancos do que em orientais ou negros.
Inicialmente ocorre a ativação de macrófagos e linfócitos T junto à vasa-vasorum das artérias envolvidas, levando, em última análise, a destruição do vaso acometido. Além disso, há também produção de fatores angiogênicos que auxiliam na destruição da parede e provocam hiperplasia da íntima que predispõe a trombose.
O início da doença pode ser insidioso ou abrupto e a maioria dos doentes apresenta sintomas constitucionais (anorexia, perda de peso, febre baixa, fadiga e depressão). Em alguns casos, febre de origem indeterminada pode ser o quadro predominante.
Cefaléia, que ocorre em cerca de 2/3 dos casos, geralmente é de forte intensidade, localizada apenas em uma área da cabeça, podendo ser o primeiro sintoma da doença. Hipersensibilidade da região temporal, com incômodo ao usar óculos, pentear os cabelos ou apoiar a cabeça no travesseiro à noite é queixa frequente.
Uma das manifestações mais específicas da ACG é a claudicação de mandíbula, com dor em músculo masseter ou temporal relacionada ao esforço de falar ou mastigar, que desaparece com o repouso. Podem ainda ocorrer dores intensas na região retroauricular ou na articulação temporomandibular. Claudicação ou parestesia de língua também pode ocorrer.
Sintomas oculares como borramento visual, diplopia, amaurose fugaz e escotomas ocorrem em
Inflamação da aorta, com dilatação da ascendente levando a insuficiência aórtica ou obstrução de ramos da croça e até mesmo de ramos abdominais, pode ocorrer. Neste caso, os sintomas variam, na dependência do território irrigado pelos ramos.
Em metade dos casos, a ACG pode cursar com polimialgia reumática. A polimialgia reumática, por sua vez, é uma doença inflamatória sistêmica, geralmente autolimitada, que ocorre em indivíduos > 50 anos de idade e é comumente associada com elevados valores de velocidade de hemossedimentação e anemia (50% dos casos). Ela é caracterizada por um início subagudo ou crônico de dores difusas pelo corpo, rigidez matinal e dolorimento na região cervical, ombros e cintura pélvica em pacientes acima dos 50 anos. Os sintomas são geralmente simétricos, mas assimetria da dor pode ocorrer. Alguns pacientes queixam-se de fraqueza, fadiga, anorexia, perda de peso e febre. A polimialgia reumática apresenta uma resposta rápida à corticoterapia em doses baixas.
Entre as manifestações clínicas mais raras em ACG, podemos citar claudicação de língua, perda irreversível da visão, disfagia, claudicação de membros superiores, acidente vascular cerebral, artrites periféricas, distúrbios vestibulares, hipoacusia e neuropatias periféricas.
O espessamento da artéria temporal, com nodulações, rigidez da parede, diminuição ou ausência de pulsos, são os achados mais sugestivos ao exame físico. Sopros devido a estenoses em subclávias ou carótidas e de insuficiência de válvula aórtica podem ocorrer. Diminuição de pulsos periféricos ou diferença de pressão arterial entre membros sugerem envolvimento de ramos da aorta.
Entre os exames laboratoriais, a VHS é quase sempre muito elevada, entre 80 e 100 mm/h. Plaquetose e anemia de doença crônica podem ocorrer, porém são inespecíficas.
A ultrassonografia com Doppler das artérias temporais pode ser útil para evidenciar área de vasculite da artéria temporal, bem como para indicar o local a ser biopsiado. A biópsia de artéria temporal deve ser indicada e o maior segmento possível da artéria (
Entre os diagnósticos diferenciais da ACG, podemos citar, principalmente, a polimialgia reumática. O diagnóstico diferencial de polimialgia reumática, por sua vez, inclui artrite reumatoide, hipotireoidismo, infecções (osteomielite, endocardite infecciosa), fibromialgia, neoplasia (mieloma múltiplo, leucemia, linfoma), polimiosite, bursite/tendinite, Parkinson, drogas (estatinas e corticosteroides), amiloidose, mieloma múltiplo e macroglobulinemia de Waldenströn.
A presença de erosões ósseas periarticulares, presença de fator reumatoide e/ou nódulos reumatoides tornam o diagnóstico de artrite reumatoide mais favorável. Com relação à polimiosite, ocorre mais fraqueza do que dor em região em cintura escapular e pélvica. Além disso, o aumento de creatinofosfoquinase geralmente ocorre em polimiosite. Dores generalizadas associadas à fadiga sugerem fibromialgia.
Outros diagnósticos diferenciais são as vasculites sistêmicas (AT, granulomatose de Wegener, poliarterite nodosa).
Corticosteroides em altas doses é o tratamento de escolha para a ACG. Habitualmente, usa-se prednisona em dose de
O uso de aspirina deve ser indicado para todos os pacientes que não tenham contraindicação, pois está associado à menor risco de complicações isquêmicas tardias. Investigação e consequente profilaxia ou tratamento para osteoporose está indicado em todos os pacientes.
A doença de Kawasaki (DK) é uma vasculite autolimitada e sistêmica que acomete vasos de médio calibre. É conhecida também por síndrome febril linfonodo-mucocutânea, acomete principalmente crianças < 5 anos de idade (
Apesar de ser autolimitada, complicações como aneurisma de artéria coronariana, comprometimento da função miocárdica com insuficiência cardíaca, infarto do miocárdio, arritmia e oclusão arterial periférica podem ocorrer, levando a um aumento da morbidade e da mortalidade.
A causa da DK permanece desconhecida. No entanto, possíveis agentes etiológicos seriam de origem infecciosa, genética (genes HLA como B5, B44, Bw51, DR3 e DRB3*0301 em brancos; B54, Bw15, e Bw35 em japoneses; e Bw51 em israelenses) e imunológica. Neste último caso, lesão dos vasos sanguíneos parece ser resultado de uma resposta imune aberrante, levando a injúria de células endoteliais e lesão da parede dos vasos.
A DK manifesta-se com um quadro febril, conjuntivite não purulenta, eritema dos lábios e mucosa oral, língua “em framboesa”, adenomegalia, eritema palmar com descamação palmoplantar e rash cutâneo, assemelhando-se muito a um processo infeccioso. Estes sinais clínicos básicos podem não estar presentes simultaneamente.
Outras manifestações clínicas, como poliartrite e mais raramente envolvimento do sistema nervoso central com meningite asséptica, comprometimento de par craniano e envolvimento pulmonar, podem ocorrer.
As manifestações cardíacas, como pericardite e miocardite, são comuns na fase aguda. Podem surgir insuficiência cardíaca congestiva, taquicardia e arritmias atriais e ventriculares. A complicação cardíaca mais grave é a arterite coronariana, com detecção de aneurismas geralmente de
O aneurisma da artéria coronária ocorre em
Alguns achados podem auxilar no diagnóstico de DK, como provas de atividade inflamatória elevadas, leucocitose com desvio à esquerda, trombocitose, anemia normocrômica e normocítica, leucocitúria, elevação de transaminases (30% dos casos), alteração do perfil lipídico (elevação de triglicérides e LDL e diminuição do HDL), hiponatremia e hipoalbuminemia.
Os diagnósticos diferenciais da DK são outras causas de exantemas em crianças: sarampo, adenovírus, echovírus; reações a drogas como Steven-Johnson ou doença do soro; artrite idiopática juvenil forma sistêmica.
A recomendação da terapia inicial inclui uso de imunoglobulina intravenosa (IGIV) (2 g/kg), administrada em única infusão por
O tratamento com IGIV diminui a incidência de aneurismas gigantes em mais de 95%.
Poliarterite nodosa (PAN) é uma vasculite sistêmica primária necrosante que compromete artérias de médio calibre. Apresenta espectro de manifestação ampla, desde uma forma leve/limitada à progressiva e fatal, acometendo sistema nervoso central e periférico, rins, músculos, trato gastrintestinal e cutâneo.
A prevalência populacional estimada para PAN é de
A incidência da doença aumenta com a idade, com pico na 6ª década de vida, e existe uma predominância no sexo masculino na razão de 1,5:1.
A maioria dos casos de PAN é idiopática, embora o vírus da hepatite B (HBV) e a leucemia de células cabeludas sejam importantes na patogênese de alguns casos. Outros agentes virais descritos são vírus da hepatite C, vírus da HIV, citomegalovírus e parvovírus B19. Na PAN da infância, observou-se associação com elevados títulos de antiestreptolisina (ASLO), sugerindo infecção prévia por estreptococos do grupo A.
A PAN estabelecida é caracterizada por reação inflamatória com necrose fibrinoide em paredes de artérias. Por outro lado, a PAN não acomete as veias, em contraste com outras formas de vasculites sistêmicas, particularmente as vasculites ANCA associadas (granulomatose de Wegener, poliangiíte microscópica e síndrome de Churg-Strauss). Os infiltrados celulares contêm leucócitos polimorfonucleares e células mononucleares. Fragmentos de leucócitos (leucocitoclasia) podem ser notados. Necrose da parede arterial resulta em uma aparência eosinofílica e homogênea, referida como necrose fibrinoide. Interrupção da lâmina elástica interna e externa é observada, podendo evoluir para o desenvolvimento de dilatação aneurismática. As lesões arteriais são caracteristicamente focais e segmentares, o que pode ser revelado pela coexistência de artérias com lesões em diversos estágios evolutivos (agudo, crônico e fibrótico), além de artérias normais dentro da mesma amostra examinada.
Na fase inicial, sintomas e sinais constitucionais (fadiga, fraqueza, febre, artralgias) podem estar presentes em 70% dos casos. Manifestações em órgãos específicos também podem ocorrer.
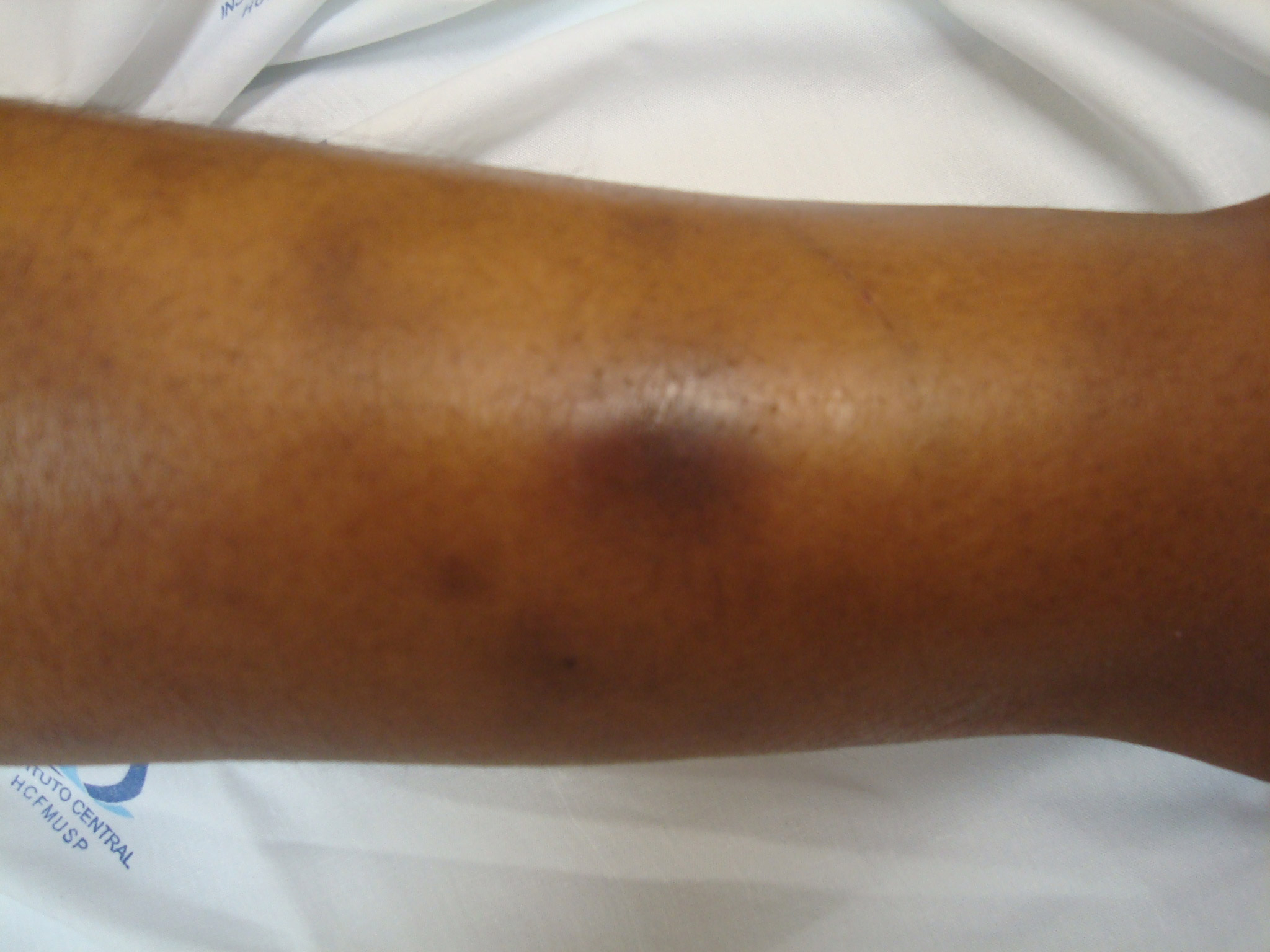
No caso de acometimento cutâneo, pode ocorrer a presença de livedo reticularis, úlcera cutânea, nódulos subcutâneos, eritema nodoso (Figura 3), erupção vesicular ou bolhosa e/ou úlceras Tais lesões podem ser focais ou difusas, e geralmente aparecem em membros inferiores. Envolvimento cutâneo progressivo pode ser grave, incluindo infarto e gangrena de dedos das mãos e dos pés e também em outras áreas, além de ulcerações com extensão até tecido subcutâneo.
Com relação aos rins, há envolvimento destes em 50% dos casos. O acometimento renal leva a hipertensão arterial e diferentes graus de insuficiência renal. Ruptura de aneurismas de artéria renal pode causar hematomas perirrenais. Infartos renais múltiplos também podem ocorrer em indivíduos com extensa vasculite. Estreitamento incompleto do lúmen de artérias inflamadas leva a isquemia glomerular, mas não a inflamação ou necrose. Assim, o sedimento urinário, quando alterado, mostra somente proteinúria subnefrótica e, às vezes, modesta hematúria; no entanto, cilindros hemáticos são geralmente ausentes.
Acometimento do sistema nervoso periférico ocorre em 65% dos casos. A mononeuropatia multiplexa (ou polineuropatia assimétrica) é um dos achados mais comuns. A neuropatia é geralmente assimétrica no início, mas o envolvimento adicional de outros ramos nervosos leva a polineuropatia simétrica distal mais confluente. Nervos cranianos são menos comumente envolvidos.
A dor abdominal é um sintoma intestinal inicial em pacientes com arterite de mesentérica. O desconforto pode ser intermitente ou contínuo, e pode ser referido após as refeições. Perda de peso pode ser secundária a má absorção intestinal. Doença progressiva pode resultar em infarto intestinal com perfuração. Outros sintomas gastrintestinais incluem náuseas, vômitos, melena, diarreia sanguinolenta e extenso sangramento gastrintestinal.
Com relação a acometimento cardíaco, pode ocorrer cardiomiopatia isquêmica como reflexo de vasculite, estreitamento ou oclusão das artérias coronárias.
Envolvimento muscular ocorre em 50% dos casos, com sintomas de mialgias e fraqueza muscular. Enzimas musculares (creatinofosfoquinase) podem estar levemente elevadas.
Se houver dor testicular, este sítio tem sido recomendado como um bom local de biópsia para diagnóstico.
Lesões de pele e a presença de evidência objetiva de comprometimento neurológico motor (mão ou pé caído) devem sempre ser pesquisadas. A presença de hipertensão arterial pode refletir a disfunção renal.
Figura 3: Eritema nodoso na fase anterior da perna.
O diagnóstico clínico de PAN é baseado na presença de sintomas, exame físico e testes laboratoriais compatíveis com esta doença. No entanto, o diagnóstico deve ser confirmado por biópsia sempre que possível, devido à raridade da mesma e ao potencial de eventos adversos relacionados ao tratamento. Na ausência de um sítio óbvio para biópsia tecidual, pode-se indicar angiografia, buscando a presença de microaneurismas de vasos na circulação renal, hepática ou mesentérica.
As alterações laboratoriais são inespecíficas e demonstram apenas a natureza inflamatória da doença ou seu envolvimento visceral. Assim, as provas inflamatórias de fase aguda estão elevadas, o hemograma costuma revelar leucocitose, plaquetose e anemia normocrômica, normocítica. Análise do sedimento urinário, com proteinúria e hematúria, alteração da função renal e sorologias para hepatite (vírus B e C) devem sempre ser pesquisadas quando houver suspeita de PAN.
A eletroneuromiografia geralmente revela neuropatia axonal. Além de mostrar a extensão dos envolvimentos neurológico e muscular, pode ser útil no direcionamento da biópsia nestas localizações, em casos de comprometimento subclínico.
A confirmação diagnóstica pode ser por análise histopatológica, revelando arterite necrosante característica, sendo pele, músculo e nervo os locais preferenciais para a realização de biópsia, por serem menos invasivos. No entanto, a realização de biópsias renal e hepática deve ser considerada somente após a arteriografia mostrar ausência de microaneurismas, pelo risco de ruptura.
Quando a confirmação histopatológica da doença não pode ser obtida, deve-se considerar a arteriografia visceral, nos casos de envolvimento renal ou dor abdominal. A angiografia abdominal pode revelar múltiplos microaneurismas intraparenquimatosos de artérias de médio calibre em
Os diagnósticos diferenciais de poliarterite nodosa são: outras vasculites (granulomatose de Wegener, poliangiíte microscópica, síndrome de Churg-Strauss, crioglobulinemia, vasculite isolada de nervos periféricos); infecções (endocardite, infecção fúngica profunda (histoplasmose, paracoccidioidomicose, blastomicose); colagenoses (lúpus eritematoso sistêmico, doença mista do tecido conectivo, artrite reumatoide (vasculite reumatóide, doença de Still); miscelâneas (sarcoidose, eritema nodoso, displasia fibromuscular, linfoma, doença intestinal inflamatória).
O uso de corticosteroide é a medicação de primeira linha para o tratamento da PAN. Deve-se iniciar o tratamento com altas doses, iniciando com dose única diária ou doses divididas de prednisona, variando de
No seguimento dos pacientes com PAN, o quadro clínico e a VHS devem ser monitorados. Se a clínica do paciente melhorar e a VHS retornar ao normal, deve-se iniciar a diminuição gradativa da dose de corticosteroide. Frequentemente, o paciente é mantido com doses baixas por um período indefinido.
Os imunossupressores devem ser associados à prednisona principalmente nas seguintes situações:
vasculite rapidamente progressiva ou com envolvimento de órgãos viscerais;
quando a prednisona não controla a atividade e progressão da doença;
quando a prednisona não pode ser reduzida devido à recidiva da doença.
Ciclofosfamida é o imunossupressor de escolha para induzir remissão da doença na dose de 2 mg/kg/dia ou em forma de pulsoterapia (0,6 g/m2 superfície corpórea) intravenosa mensal até completar um total de
Efeitos tóxicos secundários a terapêutica devem sempre ser considerados no tratamento a longo prazo dos pacientes com vasculites. Terapêutica apropriada para prevenir a osteoporose induzida por corticosteroide deve ser associada conjuntamente. Efeitos deletérios da ciclofosfamida incluem cistite hemorrágica, neoplasias (hematológica, bexiga), citopenias, infecção e falência ovariana.
Poliangiíte microscópica (PAM) é uma vasculite sistêmica de pequenos vasos (vênulas, capilares, arteríolas) tipicamente levando a uma glomerulonefrite crescêntica associada à vasculite sistêmica generalizada. O quadro de acometimento renal associado à hemorragia alveolar também é conhecido como Síndrome pulmão-rim.Trata-se deuma vasculite necrosante não granulomatosa que, em geral, se acompanha de glomerulonefrite pauci-imune, e faz parte do espectro das vasculites associadas à presença de ANCA. O achado histopatológico é de um infiltrado inflamatório misto bastante semelhante ao encontrado na poliarterite nodosa, porém, ao contrário desta última, pacientes com PAM tipicamente evoluem com glomerulonefrite.
A incidência é de 4 casos/milhão/ano, e é mais comum que a clássica poliarterite nodosa, porém ligeiramente menos comum que a granulomatose de Wegener.
Ocorre tipicamente na 4ª e 5ª décadas de vida, numa razão de 1 homem para 1 mulher.
A exemplo de outras vasculites, ocorre um processo inflamatório na parede vascular, porém de pequenos calibres (arteríolas, capilares e vênulas). No caso da glomerulonefrite, por meio de estudo de imunofluorescência, evidencia-se natureza pauci-imune. Além dos rins, pode ocorrer acometimento pulmonar com quadro de capilarite pulmonar.
Frequentemente são encontrados infiltrados pulmonares migratórios, semelhantes aos da síndrome de Churg-Strauss e hemorragia alveolar, idêntica à que ocorre na granulomatose de Wegener e síndrome de Goodpasture.
As 5 principais manifestações clínicas vistas em PAM são glomerulonefrite (cerca de 80% dos casos), perda de peso (> 70%), mononeurite multiplex (60%), febre (55%) e alterações cutâneas diversas (> 60%).
A Tabela 5 ilustra as principais manifestações clínicas de poliangiíte microscópica.
Tabela 5: Principais manifestações clínicas de poliangiíte microscópica
|
Órgão |
Manifestação clínica |
|
Constitucional |
Emagrecimento, febre, anorexia |
|
Cabeça |
Rinite, úlceras orais, púrpura em palatino, esclerite e uveíte |
|
Pulmões |
Capilarite pulmonar, hemorragia alveolar, infiltrado não específico, fibrose pulmonar, derrame pleural |
|
Trato gastrintestinal |
Vasculite mesentérica, aneurisma mesentérico |
|
Rins |
Glomerulonefrites |
|
Pele |
Púrpura palpável, úlceras, lesões vesicobolhosas |
|
Articulações |
Poli ou oligoartrite migratória, artralgias |
|
Nervos periféricos |
Mononeurites multiplex sensitivas ou motoras |
|
Sistema nervoso central |
Vasculite central |
Oacometimento renal típico na PAM é de uma glomerulonefrite pauci-imune, crescêntica, idêntico ao observado na granulomatose de Wegener ou em certas formas com mainfestação renal isoalada ANCA de padrão perinuclear é detectado em até 70% dos casos.
Deve ser feito com outras vasculites, como a poliarterite nodosa (não é uma vasculite ANCA associada e acomete vasos de médio calibre), granulomatose de Wegener (vasculite ANCA associada, em especial, cANCA, vasculite necrosante granulomatosa), síndrome de Churg-Strauss, crioglobulinemia, púrpura de Henoch-Schönlein e doença de Goodpasture. Além disso, doenças infecciosas (endocardite, HIV, hepatites B e C), pneumopatias (fibrose pulmonar intersticial), doenças autoimunes sistêmicas (lúpus eritematoso sistêmico e artrite reumatoide) e outras (doença inflamatória intestinal, hepatite autoimunes, colangite esclerosante) também devem ser diferenciadas.
O tratamento clássico daa PAM é feito com prednisona (1 mg/kg/dia) e ciclofosfamida (2 mg/kg/dia) oral para indução de remissão da doença. Uma vez atingida à remissão a dose de corticosteroide deve ser gradualmente reduzida até a sua completa suspensão.
Classicamente, a ciclofosfamida era mantida por pelo menos 1 ano após a remissão da doença. Este esquema terapêutico tradicional, embora eficiente seja bastante tóxico, relacionado com uma incidência considerável de eventos adversos graves incluindo citopenias e infecções oportunísticas, cistite e câncer de bexiga. Alternativamente, tem se utilizado esquemas imunossupressores alternativos, particularmente na manutenção da remissão depois de atingido a remissão da doença.
Pulsoterapia com corticosteroide pode ser usada para casos graves e a plasmaférese está indicada em casos de nefrite refratária. Outras modalidades terapêuticas como a gamaglobulina intravenosa e os anticorpos anti-CD20 parecem promissores, embora ainda não suficientemente estudados.
A granulomatose de Wegener (GW) é uma vasculite sistêmica primária. É o protótipo da vasculites ANCA-associada. Sua tríade clínica clássica é representada pela presença de inflamação granulomatosa necrosante das vias aéreas superiores, acometimento pulmonar e uma glomerulonefrite crescêntica. No entanto, graus varioados dee vasculite sistêmica afetando literalmente qualquer órgão ou sistema pode ser observado.
A GW acomete indivíduos de todas as raças e faixas etárias, tem uma maior incidência entre brancos provenientes do norte da Europa e entre adultos da 5ª década de vida. A incidência é de 10/milhão de pessoas/ano.
A GW é caracterizada por processo inflamatório granulomatoso da parede vascular. O quadro renal é idêntico ao observado na PAM, ou seja, uma glomerulonefrite crescêntica, pauci-imune
Manifestações constitucionais como febre, perda de peso e astenia são muito frequentes, particularmente no início da doença.
Manifestações relacionas as vias aéreas superiores estão presente na imensa maioria dos pacientes incluindo rinorréia purulenta, sinusites de repetição, otite, ulceras orais, desabamento de septo e estenose subglótica O envolvimento pulmonar, renal e outras manifestações sistêmicas podem ocorrer logo no início da doença ou surgir posteriormente com a evolução do quadro. A Tabela 6 mostra as principais manifestações clínicas de GW.
Tabela 6: Principais manifestações clínicas de granulomatose de Wegener
|
Órgão |
Manifestações |
|
Nasal |
Rinorreia, epistaxe, obstrução nasal, perfuração do septo nasal, ¨nariz em sela¨ (Figura 4) |
|
Seios nasais |
Sinusite de repetição |
|
Ouvidos |
Hipoacusia |
|
Oral |
Úlceras orais |
|
Olhos |
Pseudotumor orbitário, esclerite, episclerite, ceratite, uveíte anterior |
|
Traqueia |
Estenose subglótica |
|
Pulmões |
Nódulos, lesões cavitárias (Figura 5), infiltrado pulmonar não específico, hemorragia alveolar, lesões brônquicas |
|
Coração |
Lesão valvular, pericardite |
|
Trato gastrintestinal |
Vasculite mesentérica, infarto esplênico |
|
Rins |
Glomerulonefrite |
|
Pele |
Púrpuras palpáveis, nódulos subcutâneos |
|
Articulações |
Oligo ou poliartrite migratória, artralgias |
|
Nervos periféricos |
Mononeurites multiplex motora ou sensitiva |
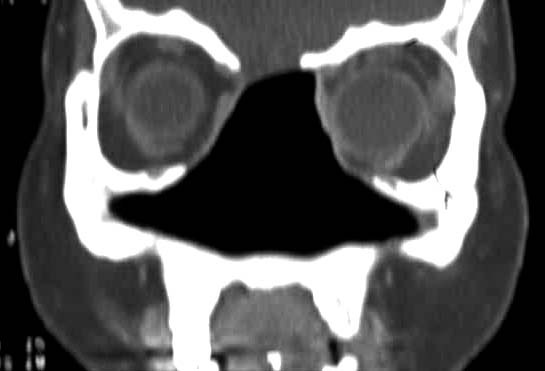
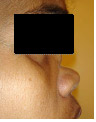
Na ausência de um tratamento adequado a doença é considerada grave. Na sua forma sistêmica ela costuma ser falar em até 5 anos. Porém, além da forma clássica da GW, existem formas limitadas (25% dos casos) que geralmente acometem apenas as vias aéreas superiores, muito embora casos limitados aos pulmões também tenham sido descritos. .As manifestações de GW conferem grande morbidade à doença, seja no aspecto estético facial, seja na dificuldade de tratamento, com recidivas e infecções intercorrentes frequentes. A estenose subglótica, em particular, pode evoluir independentemente da remissão de outros sintomas da doença, para a necessidade de traqueostomia definitiva. A associação entre destruição (Figura 4), levando à desabamento de septo (“nariz em sela”) - Figura 5 - e proptose ocular confere uma fácies bastante característica da doença.
Figura 4: Destruição do septo nasal vista a tomografia computadorizada em corte frontal.
Figura 5: Nariz em “sela”, decorrente da destruição do septo nasal.
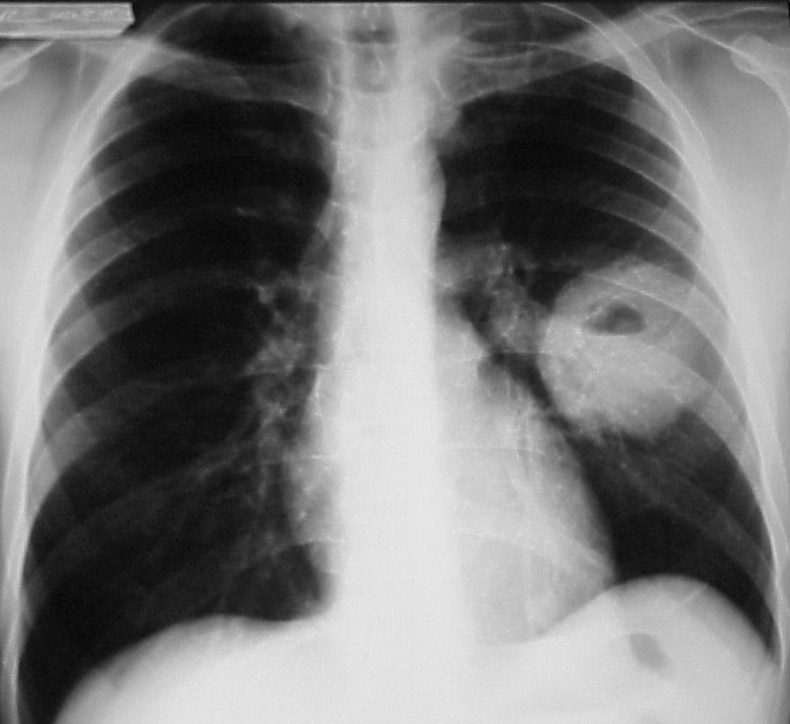
Muitos pacientes com envolvimento pulmonar são assintomáticos no início da doença, evoluindo progressivamente com quadro de tosse, dispneia e escarros hemoptoicos. As imagens radiológicas podem revelar nódulos ou massas cavitadas (Figura 6) ou não, infiltrados intersticiais e infiltrados pleurais. A hemorragia alveolar maciça pode ser uma manifestação inicial e catastrófica da GW.
Figura 6: Presença de uma lesão cavitada em um portador de granulomatose de Wegener.
Raramente a doença apresenta-se de imediato com sinais de insuficiência renal, embora cerca de 50% dos casos apresentem alterações do sedimento urinário ao diagnóstico.
Manifestações oculares ocorrem em cerca de 50% dos casos de GW. Uveíte, conjuntivite e massas retro-orbitárias causando proptose ocular respondem pela maioria dos casos. Além disso, também são observados casos de ulcerações corneanas, episclerite, neuropatia óptica, obstrução de ducto lacrimal e vasculite retiniana.
O uso de cANCA apresenta-se não só no diagnóstico precoce, mas também no acompanhamento terapêutico e na prevenção de recidivas da GW. O diagnóstico deve ser confirmado, sempre que possível, por um exame histológico de um órgão com atividade de doença. No entanto, o patologista que examina o material deve estar familiarizado com o estudo das vasculites sistêmicas e o local de biópsia deve ser escolhido criteriosamente. Os achados característicos sugestivos de GW pela biópsia do tecido acometido são:
vasculite de pequenos vasos;
necrose fibrinoide com infiltrado inflamatório neutrofílico;
presença de granulomas, incluindo granulomas em paliçada;
presença de células gigantes ou granulomas incompletos.
Os diagnósticos diferenciais de GW são:
outras vasculites (poliarterite nodosa, poliangiíte microscópica, síndrome de Churg-Strauss, púrpura de Henoch-Schönlein, crioglobulinemia mista, síndrome de Goodpasture, arterite de células gigantes);
infecções (doenças micobacterianas, infecções fúngicas (histoplasmose, blastomicose, paracoccidioidomicose), pneumonia estreptocócica com glomerulonefrites);
neoplasias (carcinoma nasofaríngeo, linfoma, linfoma angiocêntrico, doença de Castleman);
doença granulomatosa (sarcoidose);
doenças autoimunes sistêmicas (lúpus eritematoso sistêmico, artrite reumatoide, policondrite recidivante).
Deve-se verificar se trata de GW forma sistêmica ou limitada. No primeiro caso, geralmente requer tratamento mais agressivo com uso de ciclofosfamida e altas doses de corticosteroides. A combinação de ciclofosfamida (2 mg/kg/dia) e corticosteroides (prednisona 1 mg/kg/dia), sendo este último precedido geralmente de pulsoterapia de metilprednisolona (1 g/dia, 3 dias, parenteral), promove uma resposta terapêutica inicial satisfatória em > 90% dos casos. Ciclofosfamida diária por via oral promove um período de remissão maior que por via parenteral e intermitente (P. ex.:, mensal).
No caso de forma limitada, pode-se utilizar ciclofosfamida ou outros imunossupressores, como metotrexato, para poupar o uso de corticosteroides.
É aconselhada a utilização de antibiótico profilático para P. jiroveci com dapsona 100 mg/dia ou sulfametoxazol/trimetoprim, durante o tratamento de GW.
A síndrome de Churg-Strauss (SCS) é uma vasculite necrosante sistêmica primária ANCA associada. Histologicamente apresenta infiltrado tissular eosinofílico, formação de granuloma extravascular e vasculite necrosante.
Acomete indivíduos com asma e frequentemente rinite alérgica sendo, por isso, também conhecida como angiíte alérgica e granulomatose alérgica.
A incidência é de
A presença de quadro alérgico é comum a todos os pacientes com SCS. No entanto, a causa é desconhecida. Vários agentes têm sido considerados como desencadeadores, como uso de antagonistas de receptores de leucotrienos, cocaína, acetaminofeno, carbamazepina e macrolídeos.
Sob o ponto de vista fisiopatológico, a lesão tissular é causada por infiltrado eosinofílico e produtos de degranulação neutrofílica. A ativação de linfócitos T parece ser importante para a manutenção da eosinofilia. Além disso, a presença de ANCA sugere modular o fenótipo da doença.
Os marcadores de ativação de linfócitos T estão elevados em pacientes com síndrome SCS em atividade clínica, e se correlacionam com marcadores de lesão endotelial.
Didaticamente, podemos dividir a história natural da doença em 3 fases:
pródromo: caracterizado pela presença de doenças alérgicas (asma, rinite alérgica); esta fase frequentemente dura anos);
infiltrado tissular/eosinofilia: é a fase marcada pela eosinofilia periférica, infiltrado eosinofílico tissular (pulmões, trato gastrintestinal e outros tecidos);
vasculites: fase caracterizada pela presença de vasculites necrosantes em pulmões, coração, nervos e pele.
A presença de asma é um achado em pacientes com SCS. Sintomas alérgicos em vias aéreas superiores, principalmente rinite alérgica e polipose nasal, também ocorrem em cerca de 2/3 dos pacientes. Alguns pacientes, depois da primeira fase em que predominam os fenômenos alérgicos, apresentam uma segunda fase em que são encontrados infiltrados inflamatórios nos tecidos, como pneumonite, hepatite ou enterite eosinofílica.
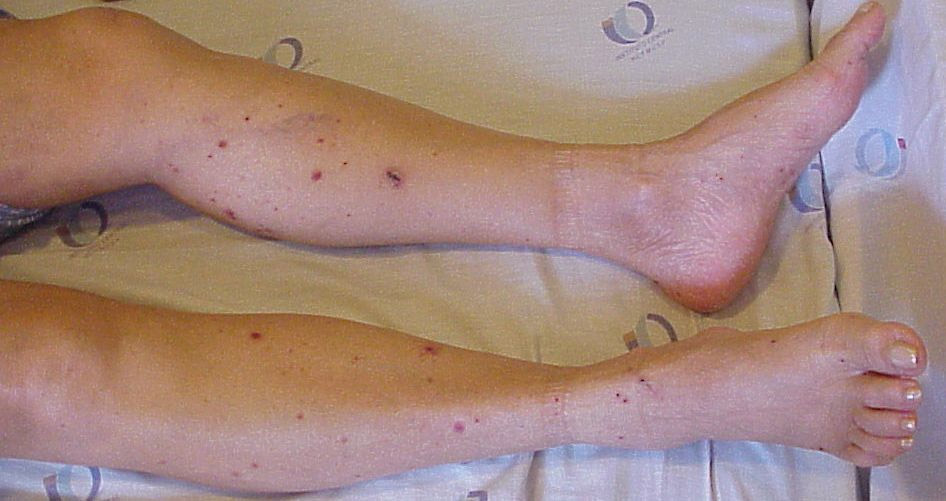
Na terceira fase, inicia a vasculite, que se acompanha em geral de sintomas multissistêmicos. Além da asma, nos pulmões costumam ocorrer pneumonites migratórias, caracterizada por infiltrados localizados, que em geral já foram tratados anteriormente com antibióticos, pensando-se em pneumonia bacteriana, mas que na evolução radiológica de controle mudam de localização. Hemorragia alveolar também pode ocorrer. Na pele podem surgir nódulos, pápulas, púrpuras (Figura 7) ou lesões urticariformes. Neuropatia periférica caracterizada como mononeurite, mononeurite múltipla ou polineuropatia ocorre em cerca de 60% dos pacientes. Cerca de metade dos pacientes apresentam envolvimento cardíaco, principalmente pericardite, miocardite ou isquemia miocárdica. O envolvimento gastrintestinal ocorre também em cerca de metade dos pacientes e caracteriza-se por dor abdominal, diarreia, hemorragia digestiva ou colite. O envolvimento renal também ocorre, porém com uma frequência menor que nas outras vasculites ANCA associadas. Em termos histológicos, é comum encontrar glomerulonefrite segmentar e focal com crescentes.
Figura 7: Presença de púrpuras palpáveis em membros inferiores em uma paciente portadora de síndrome de Churg-Strauss.
Para confirmação do diagnóstico, além da história prévia de alergias e dos achados clínicos, auxiliam os achados de eosinofília periférica, de biópsia de tecido acometido mostrando vasculite com infiltrado granulomatoso ou eosinofílico e a presença do pANCA.
Como no início da doença as manifestações clínicas não são necessariamente simultâneas, é importante estar atento para as manifestações predominantes e avaliá-las dos seus diagnósticos diferenciais. Assim sendo, se predominarem sintomas pulmonares, devemos fazer diagnóstico diferencial com as síndromes pulmonares eosinofílicas (pneumonia eosinofílica crônica, broncopneumonia alérgica relacionada à aspergilose, síndrome hipereosinofílica idiopática, síndrome de Löeffer).
Se ocorrer eosinofilia periférica com acometimento de múltiplos órgãos, devemos levar em consideração a síndrome hipereosinofílica idiopática, variantes mieloproliferativas atípicas.
Se predominar sinais vasculíticos e com acometimento sistêmico, considerar a granulomatose de Wegener, poliangiíte microscópica e arterite nodosa.
O manejo terapêutico de SCS tem sido um desafio. Não há uma guia estabelecida. Muitos dos pacientes podem ser tratados efetivamente somente com o uso de corticosteroides, sem necessidade de imunossupressores.
Por outro lado, o uso de imunossupressores tem sido utilizado para casos mais graves. Assim sendo, p. ex., o uso de ciclofosfamida (2 mg/kg/dia, via oral) tem sido utilizado como no caso de acometimento neuronal (central ou periférico), glomerulonefrites, envolvimento cardíaco, hemorragia alveolar. No caso de pacientes com menor gravidade, pode-se utilizar metotrexato (dose máxima de
A púrpura de Henoch-Schönlein (PHS) é uma vasculite sistêmica primária imunomediada, autolimitada, que acomete principalmente as crianças, entre 4 e 9 anos de idade. Cursa com púrpuras palpáveis, artrite ou artralgia, dor abdominal e envolvimento renal.
A incidência é de 140 casos/milhão/ano. Ocorre uma predominância no sexo masculino (
Em 2/3 dos casos, a doença desenvolve-se aproximadamente 10 dias após uma infecção de via aérea superior. Apesar desta associação, nenhum microorganismo ou antígeno ambiental tem sido isolado como a causa importante de PHS.
O achado histológico característico da PHS é uma vasculite leucocitoclástica, com presença de restos de neutrófilos, acompanhada de depósitos de imunecomplexos contendo IgA nos órgãos afetados. A biópsia de pele das lesões purpúricas demonstra o envolvimento de pequenos vasos na derme papilar. Há um predomínio de neutrófilos e monócitos no infiltrado inflamatório e a imunofluorescência demonstra depósitos de IgA, C3 e fibrina na parede dos vasos envolvidos.
As manifestações clássicas da doença são púrpuras palpáveis (principalmente em membros inferiores), artrite ou artralgia, dor abdominal e envolvimento renal (hematúria). Essas manifestações clínicas podem ocorrer no curso de dias a semanas e podem variar na ordem de surgimento das complicações.
Manifestações cutâneas: púrpuras palpáveis desenvolvem-se em todos os pacientes. No entanto, a clássica lesão de pele da PHS não é a apresentação inicial em 25% das crianças afetadas, sendo difícil fazer o diagnóstico de PHS antes do aparecimento do quadro cutâneo em pacientes com clínica inicial somente de dor abdominal ou artrite. O rash geralmente se inicia com eritema, mácula ou urticária evoluindo como púrpura palpável, equimoses ou petéquias. A distribuição da lesão geralmente é simétrica, localizada em áreas dependentes de gravidade ou pressão, como membros inferiores.
Artrite e artralgia: ocorrem em mais de 80% dos pacientes, sobretudo em grandes articulações como joelhos e tornozelos. São geralmente migratórias ou transitórias, oligoarticulares e não-deformantes. Quando ocorre artrite, a sua natureza é não deformante.
Sintomas gastrintestinais: aparecem em 60% das crianças, sendo que em aproximadamente 30% tem evidência de sangramento pelo trato gastrintestinal. As manifestações podem ser leves, como náuseas, vômitos, dor abdominal em cólica e íleo paralítico transitório, ou comprometimento mais grave (hemorragia gastrintestinal, isquemia ou necrose intestinal, intussuscepção e perfuração intestinal).
Envolvimento renal: ocorre em 40% dos casos e surge quase sempre após a manifestação cutânea. Pode se manifestar como hematúria microscópica transitória até glomerulonefrite rapidamente progressiva e insuficiência renal. Proteinúria com níveis de síndrome nefrótica e hipertensão arterial são raras, podendo haver evolução para a insuficiência renal crônica. O achado histopatológico renal típico é glomerulonefrite focal e segmentar com depósitos de IgA.
Outros órgãos: complicação rara de trato respiratório e sistema nervoso central têm sido descrita. Quando presente, as formas mais frequentes são hemorragia alveolar e convulsões. Envolvimento testicular ocorre em 10% dos pacientes e pode mimetizar torção testicular.
Pode ocorrer leucocitose discreta à moderada. Hipercalemia ocorre em disfunção renal avançada. Exame urinário pode evidenciar hematúria a proteínuria. Provas de atividade inflamatória como velocidade de hemossedimentação está aumentada em 1/3 dos casos. Aproximadamente 60% dos pacientes apresentam aumento elevado de IgA sérico. O nível sérico de complementos (C3 e C4) está normal. Autoanticorpos como FAN, fator reumatoide e ANCA são negativos.
A PHS deve ser diferenciada de outras vasculites (vasculite de hipersensibilidade, poliangiíte microscópica, síndrome de Churg-Strauss, granulomatose de Wegener, crioglobulinemia mista, poliarterite nodosa), doenças autoimunes (lúpus eritematoso sistêmico, artrite reumatoide), doenças infecciosas (infecção viral e bacteriana), neoplasias (leucemia), nefropatias (nefropatia por IgA).
O uso de antiinflamatórios não-hormonais pode aliviar o quadro de artralgias, porém pode agravar sintomas gastrintestinais e piorar a função renal quando esta já está comprometida. O uso de dapsona (100 mg/dia) pode ser efetivo, provavelmente por interferir na interação entre a IgA e neutrófilos. O uso de corticosteroides pode aliviar quadros articulares e sintomas gastrintestinais. No entanto, não parece melhorar o quadro cutâneo (rash) e a sua efetividade em quadro renal é controversa.
Em caso de nefropatia (P. ex.: em síndrome nefrótica e presença de > 50% de crescentes), pulsoterapia com metilprednisolona combinada com azatioprina ou ciclofosfamida pode auxiliar no bom êxito.
Vasculite crioglobulinêmica é uma vasculite de pequenos vasos desencadeada por depósitos de crioglobulinas.
As vasculites crioglobulinêmicas ocorrem na proporção de 3 mulheres para 1 homem, com média de idade de 50 anos ao diagnóstico.
Crioglobulinas são imunoglobulinas com propriedades de serem precipitáveis a <
A crioglobulina é subdividida em 3 subtipos clínicos. Quando constituídas por uma única variedade de imunoglobulina, é denominada crioglobulinemia monoclonal (tipo I), que raramente se apresenta com vasculite importante. Quando se apresenta com mais de uma variedade de imunoglobulinas, denomina-se crioglobulinemia mista (tipos II ou III), conforme mostrada na Tabela 7.
Tabela 7: Classificação dos tipos de crioglobulinemias
|
Subtipo |
Monoclonalidade |
Fator reumatoide |
Doenças |
|
Tipo I |
Sim (IgG ou IgM) |
Ausente |
Macroglobulinemia de Waldenström, linfoma, mieloma múltiplo |
|
Tipo II |
Sim (IgM monoclonal e IgG policlonal) |
Presente |
Doenças associadas ao tipo III com componente monoclonal ou associadas ao tipo I quando o pico monoclonal tiver atividade de fator reumatoide |
|
Tipo III |
Não (IgG e IgM policlonal) |
Presente |
Doenças autoimunes (Sjögren, lúpus eritematoso sistêmico, artrite reumatoide), doenças infecciosas (mononucleose, hepatite viral B ou C, endocardite bacteriana) |
O quadro clínico mais frequente é a tríade constituída por fraqueza e indisposição, lesões purpúricas em membros inferiores e artralgias ou artrites. As principais complicações desta vasculite são neuropatia periférica e glomerulonefrite.
Ao exame físico, a púrpura palpável é o achado mais frequente e é predominantemente em membros inferiores. Pode-se encontrar, ainda, rash em membros inferiores, membros superiores e no tronco. Pode ocorrer a presença de fenômeno de Raynaud.
A presença de artralgias é comum. Os principais locais de acometimento são as articulações interfalangeanas proximais, metacarpofalangeanas e joelhos. A presença de artrite é incomum e, quando presente, não é deformante.
A presença de neuropatia motora é predominante em relação à sensitiva.
O envolvimento renal ocorre em 1/3 dos casos e é tipicamente uma glomerulonefrite membranosa que, por sua vez, pode mostar períodos de exacerbação e remissão. A glomerulonefrite rapidamente progressiva ocorre somente em pequeno número de casos.
Acometimento hepático ocorre em poucos casos. Quando presente, devemos considerar a presença de vírus pela hepatite C.
Sintomas de sistema nervoso central costumam ser consequência de hiperviscosidade sanguínea. É associada principalmente com a vasculite crioglobulinêmica do tipo I. A presença de hiperviscosidade é uma indicação para plasmaférese.
Acometimento do trato gastrintestinal é incomum, mas ocasionalmente pode cursar com quadro de abdome agudo.
A biópsia cutânea com estudo de imunofluorescência pode evidenciar vasculite leucocitoclástica imunocomplexo-mediada, com depósito de IgG, IgM, C3 e outros imunorreagentes dentro e ao redor da parede de vasos de pequenos e médios calibres. Trombose vascular também é evidente na maioria dos casos.
Laboratorialmente, podemos encontrar:
anemia discreta, trombocitopenia (no caso de doença hepática), alteração da função renal e hepática;
aumento de VHS, refletindo a atividade da doença;
FAN presente na maioria dos casos, com fator reumatoide positivo em 80% dos casos;
hipocomplementemia, particularmente com relação a C4;
presença de sorologia para hepatite C em aproximadamente 90% dos casos;
hemocultura negativa.
Como diagnósticos diferenciais, devemos levar em consideração: síndrome de Sjögren, lúpus eritematoso sistêmico, artrite reumatoide, além de outras vasculites como poliarterite nodosa, vasculite de hipersensibilidade, púrpura de Henoch-Schönlein, poliangiíte microscópica e granulomatose de Wegener.
O quadro clínico da crioglobulinemia em geral desaparece com o tratamento da sua causa primária. Caso o paciente apresente neuropatia periférica ou glomerulonefrite na apresentação da doença, além do tratamento da doença pode ser útil a realização de plasmaférese para retirada dos imunocomplexos circulantes, evitando maiores sequelas renais ou neurológicas, enquanto se aguarda o efeito da terapêutica da doença de base.
Quando a crioglobulinemia é secundária a doenças do tecido conectivo, deve-se iniciar o tratamento com corticosteroides e imunossupressores. Se secundária a doenças neoplásicas de células B, deve-se instituir a quimioterapia. Nos casos relacionados a infecções, estas devem ser tratadas. Nas crioglobulinemias essenciais, em geral administra-se corticoterapia sistêmica e imunossupressores em casos refratários ou recorrentes.
|
Critério |
Definição |
|
Idade de início < 40 anos |
Aparecimento de sinais ou sintomas relacionados à AT em idade < 40 anos |
|
Claudicação de extremidades |
Aparecimento e piora de fadiga e desconforto nos músculos de uma ou mais extremidades com o uso, especialmente de extremidades superiores |
|
Diminuição de pulso braquial |
Enfraquecimento da pulsação em uma ou ambas as artérias braquiais |
|
Diferença de pressão arterial > |
Diferença de mais de |
|
Sopro sobre as artérias subclávias ou a aorta |
Sopro audível sobre uma ou ambas as artérias subclávias ou a aorta abdominal |
|
Anormalidade arteriográfica |
Arteriografia com estreitamento ou oclusão da aorta, seus ramos primários ou grandes artérias na região proximal dos membros superiores ou inferiores, não devidos a aterosclerose, displasia fibromuscular ou causas semelhantes; alterações geralmente focais ou segmentares |
|
Para a AT requer pelo menos 3 dos 6 itens. | |
|
Para arterite de células gigantes requer 3 dos 5 itens: |
|
Idade de início > 50 anos |
|
Cefaleia de inicio recente ou novo tipo de cefaleia |
|
Dor à palpação ou diminuição do pulso da artéria temporal |
|
Velocidade de hemossedimentação maior que 50 mm/hora |
|
Evidência histológica de arterite (lesões granulomatosas, geralmente com células gigantes ou infiltrado celular mononuclear) |
|
Para doença de Kawasaki, requer a presença de febre durando pelo menos 5 dias sem qualquer outra explicação combinada, com pelo menos 4 de 5 dos seguintes critérios: |
|
Conjuntivite bilateral não purulenta |
|
Alterações de mucosa oral: lábios hiperemiados ou rachados, hiperemia de faringe ou língua “em framboesa” |
|
Alterações de extremidades: eritema de palma e planta dos pés, edema de mãos e pés (fase aguda) e descamação periungueal (fase convalescente) |
|
Rash polimorfo |
|
Linfoadenomegalia cervical (pelo menos 1 linfonodo > |
|
Para poliarterite nodosa, é necessário pelo menos 3 dos 10 itens seguintes: |
|
Perda de peso, não explicada, superior a |
|
Livedo reticularis |
|
Dor ou dolorimento testicular |
|
Mialgias (excluir cintura escapular e pélvica) |
|
Mononeurite ou polineuropatia |
|
Aparecimento de pressão arterial diastólica superior a |
|
Elevação sérica de úreia (superior a 40 mg/dL) ou creatinina (superior a 1,5 mg/dL) |
|
Infecção pelo vírus da hepatite B (HbsAg ou anti HbsAg) |
|
Anormalidades arteriográficas características (aneurismas ou oclusões) |
|
Biópsia de artéria de pequeno ou médio calibre contendo polimorfonucleares na parede arterial |
|
Para a granulomatose de Wegener, é necessário pelo menos 2 dos 4 itens: |
|
Inflamação oral ou nasal (úlceras orais dolorosas ou não ou descarga nasal purulenta) |
|
Presença de infiltrado nodular, fixo ou cavitário à radiografia simples de tórax |
|
Sedimento urinário com hematúria com ou sem cilindros hemáticos |
|
Infiltrado inflamatório granulomatoso na parede vascular, peri ou extravascular |
|
Para a síndrome de Churg-Strauss, é necessário pelo menos 4 dos 6 itens: |
|
Asma (história de chiado ou achado de sibilos expiratórios difusos) |
|
Hemograma com eosinofilia periférica > 10% na contagem diferencial |
|
Mono (incluindo mononeurite múltipla) ou polineuropatia |
|
Infiltrado pulmonar não fixo à radiografia simples de tórax |
|
Anormalidade dos seios da face à radiografia simples |
|
Biópsia mostrando infiltrado eosinofílico extravascular |
|
Poliangiíte microscópica é caracterizada pela presença de: |
|
Hemorragia alveolar e/ou glomerulonefrite rapidamente progressiva |
|
Demonstração histológica de vasculite de pequenos vasos ou de presença de glomerulonefrite necrosante pauci-imune |
|
Sintomas sugestivos de vasculite envolvimento de pequenos vasos |
|
Púrpura de Henoch-Schönlein é definida pela presença de, pelo menos, 2 dos seguintes critérios: |
|
Púrpura palpável (lesão cutânea hemorrágica não relacionada a trombocitopenia) |
|
Início da doença com < 20 anos de idade |
|
Angina intestinal ou isquemia intestinal manifestada por diarreia sanguinolenta |
|
Biópsia evidenciando vasculite leucocitoclástica |
|
Critérios |
Sorológicos |
Patológicos |
Clínicos |
|
Maiores |
Crioglobulinemia mista |
Vasculite leucocitoclástica |
Púrpura |
|
Menores |
Fator reumatoide + |
Infiltração por clone de linfócitos B (fígado ou medula óssea) |
Hepatite crônica |
|
|
Infecção pelo HCV+ |
|
Glomerulonefrite membranoproliferativa |
|
|
Infecção pelo HBV+ |
|
Neuropatia periférica |
|
|
|
|
Úlceras cutâneas |
Um paciente é considerado como tendo crioglobulinemia mista se pelo menos os seguintes critérios estiverem presentes: os três maiores, ou crioglobulinemia mista + 2 clínicos + 2 sorológicos ou patológicos.
A arterite de Takayasu é uma vasculite primária sistêmica que acomete vasos de grande calibre, ou seja, a aorta e seus ramos principais, além de artéria pulmonar.
Ocorre geralmente antes dos 40 anos de idade e principalmente em sexo feminino.
Ocorre inflamação da parede vascular, levando a estenose ou a dilatação (aneurismas) dos vasos lesados.
Na fase inicial da doença, o paciente pode apresentar sintomas constitucionais e/ou manifestações sistêmicas inespecíficas.
Achado clínico: diminuição de pulso, presença de sopro cardíaco ou extracardíaco ou assimetria da medida da pressão arterial nos membros.
Sinais de mau prognóstico: presença de aneurismas, hipertensão renovascular, insuficiência aórtica e retinopatia isquêmica.
Diagnóstico diferencial: doença de Ehlers-Danlos tipo IV, síndrome de Marfan, síndrome de Cogan, doença de Behçet.
Tratamento: corticoterapia. Se necessário, associar imunossupressores.
Arterite de células gigantes: vasculite de vasos de grande e médio calibre, preferencialmente os ramos extracranianos das carótidas e, em particular, a artéria temporal superficial.
Afeta indivíduos de cor branca, sexo feminino e > 50 anos de idade.
O início da doença pode ser insidioso ou abrupto, e a maioria dos doentes apresenta sintomas constitucionais.
Cefaléia, que ocorre em cerca de 2/3 dos casos, geralmente é de forte intensidade, localizada apenas em uma área da cabeça, podendo ser o primeiro sintoma da doença.
Sintomas oculares, como borramento visual, diplopia, amaurose fugaz e escotomas, ocorrem em
Em metade dos casos, a ACG pode cursar com polimialgia reumática.
Entre os exames laboratoriais, a VHS é quase sempre muito elevada, entre 80 e 100 mm/h.
Na dúvida quanto à hipótese de ACG, a biópsia de artéria temporal deve ser indicada e o maior segmento possível da artéria deve ser retirado e analisado.
Diagnóstico diferencial: polimialgia reumática e AT.
Tratamento: corticosteroides em altas doses é o tratamento de escolha para a arterite de células gigantes. Embora controverso para casos refratários ou com recidivas, está indicada a utilização conjunta de imunossupressores. O uso de aspirina deve ser indicado para todos os pacientes que não tenham contraindicação, pois está associado à menor risco de complicações isquêmicas tardias.
Doença de Kawasaki: vasculite autolimitada e sistêmica que acomete vasos de médio calibre.
Acomete principalmente crianças < 5 anos de idade (
Complicação temível: aneurisma de artéria coronária (
Quadro clínico: quadro febril, conjuntivite não purulenta, eritema dos lábios e mucosa oral, língua “em framboesa”, adenomegalia, eritema palmar com descamação palmoplantar e rash cutâneo, assemelhando-se muito a um processo infeccioso. Estes sinais clínicos básicos podem não estar presentes simultaneamente.
Exames laboratoriais: provas de atividade inflamatória elevadas, leucocitose com desvio à esquerda, trombocitose, anemia normocrômica e normocítica, leucocitúria, elevação de transaminases (30% dos casos), alteração do perfil lipídico (elevação de triglicérides e LDL e diminuição do HDL), hiponatremia e hipoalbuminemia.
Tratamento: imunoglobulina intravenosa. Associar ácido acetilsalicílico. Em caso de aneurisma de coronárias, dose menor de ácido acetilsalicílico pode ser mantida, para evitar trombose.
Tratamento com imunoglobulina intravenosa diminui a incidência de aneurismas gigantes em mais de 95%.
Poliarterite nodosa: vasculite sistêmica primária necrosante não granulomatosa que compromete artérias de médio calibre.
Apresenta espectro de manifestação ampla, desde uma forma leve/limitada a progressiva e fatal, acometendo sistema nervoso central e periférico, rins, músculos, trato gastrintestinal e cutâneo.
Na fase inicial, sintomas e sinais constitucionais podem estar presentes em 70% dos casos.
Manifestações em órgãos específicos também podem ocorrer, com acometimento do sistema geniturinário, nervoso, trato gastrintestinal, cutâneo e musculoesquelético.
Raramente acomete pulmões e baço.
Pode formar microaneurismas.
Em alguns casos, há associação com hepatite tipo B.
Poliangiíte microscópica é uma vasculite sistêmica necrosante não granulomatosa pauci-imune de pequenos vasos, em geral, acompanhada de glomerulonefrite pauci-imune.
Faz parte das vasculites associadas aos ANCA e de síndrome pulmão-rim.
Frequentemente são encontrados infiltrados pulmonares migratórios, semelhantes aos da síndrome de Churg-Strauss e hemorragia alveolar, idêntica à que ocorre na granulomatose de Wegener e síndrome de Goodpasture.
Diagnóstico diferencial: granulomatose de Wegener, síndrome de Churg-Strauss, crioglobulinemia, púrpura de Henoch-Schönlein, doença de Goodpasture e quadro infeccioso (HIV, hepatites B e C).
Tratamento: corticosteroide e imunossupressores (ciclofosfamida, metotrexato, azatioprina, micofenolato mofetil, anti-CD20).
Granulomatose de Wegener é uma vasculite primária sistêmica. Há a forma sistêmica e limitada.
É protótipo de vasculites ANCA associadas, além de vasculite com formação de granuloma necrosante e pauci-imune.
Caracterizam-se por uma tríade clássica de inflamação granulomatosa necrosante das vias aéreas superiores e inferiores, glomerulonefrite crescêntica e vasculite sistêmica afetando predominantemente pequenos vasos.
Acomete indivíduos de todas as raças e faixas etárias, com maior incidência entre brancos provenientes do norte da Europa e entre adultos na 5ª década de vida.
No início da doença, ocorre a presença de sintomas constitucionais, além de história de sinusopatia de repetição.
Manifestações clínicas clássicas: pseudotumor orbitário, nariz “em sela”, nódulos ou cavitações pulmonares, glomerulonefrite rapidamente progressiva pauci-imune.
Presença de cANCA.
A síndrome de Churg-Strauss é uma vasculite necrosante sistêmica primária ANCA associada, com formação de granuloma e infiltração de eosinófilos, tanto na parede vascular como no tecido extravascular.
Acomete indivíduos com asma e frequentemente rinite alérgica.
Etiologia é desconhecida. Vários agentes têm sido considerados como desencadeadores, como uso de antagonistas de receptores de leucotrienos, cocaína, acetaminofeno, carbamazepina, macrolídeos.
A púrpura de Henoch-Schönlein é uma vasculite sistêmica primária imunomediada, autolimitada.
Acomete principalmente crianças entre 4 e 9 anos de idade, com predominância no sexo masculino (
Cursa com quadro de púrpuras palpáveis, artrite ou artralgia, dor abdominal e envolvimento renal.
Em 2/3 dos casos, a doença desenvolve-se aproximadamente 10 dias após uma infecção de via aérea superior. Apesar desta associação, nenhum microorganismo ou antígeno ambiental tem sido isolado como a causa importante de PHS.
Vasculite crioglobulinêmica é uma vasculite de pequenos vasos desencadeada por depósitos de crioglobulinas.
Ocorre na proporção de 3 mulheres para 1 homem, com média de idade de 50 anos ao diagnóstico.
A vasculite crioglobulinêmica é uma associação entre a presença de crioglobulina sérica e de vasculite.
O quadro clínico mais frequente é a tríade constituída por fraqueza e indisposição, lesões purpúricas em membros inferiores e artralgias ou artrites. As principais complicações desta vasculite são neuropatia periférica e glomerulonefrite.
A biópsia cutânea com estudo de imunofluorescência pode evidenciar a presença de vasculite leucocitoclástica imunocomplexo-mediada, com depósito de IgG, IgM, C3.
Diagnósticos diferenciais: síndrome de Sjögren, lúpus eritematoso sistêmico, artrite reumatoide, além de outras vasculites como poliarterite nodosa, vasculite de hipersensibilidade, púrpura de Henoch-Schönlein, poliangiíte microscópica e granulomatose de Wegener.
1. Carlson JA, Chen KR. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopathol 2006;28:486-506.
2. Imboden JB et al. Current - Rheumatology (Diagnosis & treatment). 2 ed. Lange Medical Books/McGraw-Hill, 2006.
3. Gayaraud M et al, Long-term follow up of polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Arthritis Rheum 2001;44: 666-675.
4. Guillevin L et al. Classification of systemic vasculitis. Presse Med 2007;36:845-853.
5. Hoffman GS et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488-498.
6. Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;41:1521.
7. Jennette JC et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37:187-192.
8. Jennette JC, Falk RJ. Nosology of primary vasculitis. Curr Opin Rheumatol 2007;19:10-16.
9. Kallenberg CG. Antineutrophil cytoplasmic autoantibody-associated small-vessel vasculitis. Curr Opin Rheumatol 2007;19:17-24.
10. Nordström DCE et al. Roadmap to vasculitis. Acta Reum Port 2006;31:15-36.
11. Seo P, Stone JH. Large-vessel vasculitis. Arthritis Rheum 2004;51:128-139.
12. Skare TL. Vasculite cutânea. Acta Reum Port 2003;28:10-26.
13. Stone HJ. Classification and diagnostic criteria in systemic vasculitis. Best Pract Res Clin Rheumatol 2005;19:209-221.
14. Tedeschi A et al. Cryoglobulinemia. Blood Rev 2007;21:183-200.
15. Weyand MC, Goronzy JJ. Medium and large-vessel vasculites. N Engl J Med 2003;349:160-169.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.