(Carregando Índice)... (Carregando Índice)... |
Autores:
Andréia Biolo
Médica cardiologista do Serviço de Cardiologia do HCPA. Professora do Programa de Pós-graduação em Ciências da Saúde: Cardiologia e Ciências Cardiovasculares da UFRGS. Doutora em Cardiologia pela UFRGS.
Jordana de Fraga Guimarães
Médica internista.
Última revisão: 31/10/2013
Comentários de assinantes: 0
|
Versão original publicada na obra Fochesatto Filho L, Barros E. Medicina Interna na Prática Clínica. Porto Alegre: Artmed; 2013. |
Um paciente do sexo masculino, 33 anos, procura atendimento ambulatorial queixando-se de dispneia e desconforto precordial. Relata que há dois a três anos apresenta dificuldade progressiva para realização das suas atividades diárias devido à falta de fôlego e à fadiga. O desconforto torácico não está relacionado aos esforços. Nos últimos meses, é frequente a ocorrência de edema de membros inferiores no fim do dia. Ao realizar exame, o paciente evidencia crepitantes finos discretos em campos pulmonares inferiores; ictus propulsivo no quinto espaço intercostal com a linha axilar anterior, B1 hipofonéticas, sopro pansistólico 2+/6 em área de ventrículo esquerdo e com irradiação para a axila; fígado palpável no rebordo intercostal direito de contornos regulares; membros inferiores com pulsos palpáveis, simétricos, edema de 2+/4. Afirma não ser tabagista, nem alcoolista e não apresentar outras doenças.
As miocardiopatias consistem em um grupo heterogêneo de doenças, do ponto de vista fisiopatológico, que afetam primeiramente o miocárdio e não resultam de alterações de outras estruturas do sistema cardiovascular.
De acordo com a etiologia, todas as três formas podem apresentar origem desconhecida (idiopática) ou resultar de fatores genéticos; estas são asmiocardiopatias primárias. Em casos em que há decorrência de doença de causa conhecida ou associação com doenças em outros sistemas, elas são denominadas miocardiopatias secundárias.
A doença das valvas cardíacas, a hipertensão arterial sistêmica, a doença cardíaca congênita e a doença arterosclerótica podem resultar em alterações significativas no músculo cardíaco, as quais, entretanto, são secundárias a essas condições, não sendo consideradas miocardiopatias primárias.
De acordo com os padrões funcionais, as miocardiopatias podem ser classificadas da seguinte maneira:
•miocardiopatia dilatada;
•miocardiopatia hipertrófica;
•miocardiopatia restritiva.
A seguir, essas três condições são abordadas separadamente, pois apresentam peculiaridades. É importante, no entanto, ressaltar um ponto em comum entre elas: o desenvolvimento de insuficiência cardíaca (IC) é frequente e constitui a manifestação principal das miocardiopatias.
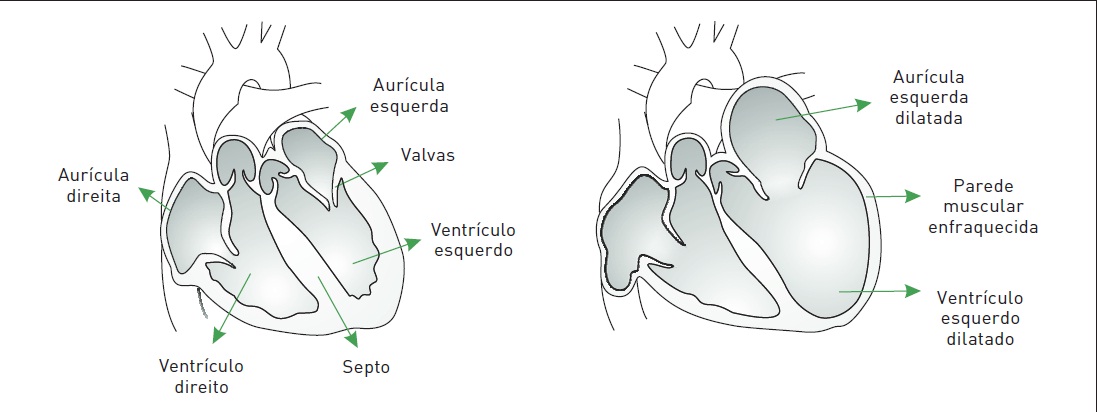
A miocardiopatia dilatada (MCD) caracteriza-se por dilatação das câmaras cardíacas – especialmente do ventrículo esquerdo (VE) – e disfunção sistólica progressiva, havendo redução na fração de ejeção. As paredes livres podem estar adelgaçadas, com espessura normal ou, mais comumente, hipertrofiadas (Fig. 6.1).

Figura 6.1
À esquerda, um coração normal e, à direita, com miocardiopatia dilatada.
A MCD é a mais comum das miocardiopatias e a segunda causa mais frequente de insuficiência cardíaca congestiva (ICC), não superando apenas a doença arterosclerótica das artérias coronárias. A faixa etária mais comumente afetada é entre 20 e 50 anos.
Na maioria das vezes, não é possível identificar uma causa específica dessa doença (MCD idiopática).
Aproximadamente 25 a 35% dos pacientes afetados apresentam uma forma familial de herança autossômica dominante (mutações nos genes da distrofina e desmina, que codificam proteínas do citoesqueleto).
Já uma parcela significativa de 15% pode evidenciar uma sequela tardia de miocardite aguda.
Muitas causas secundárias podem resultar em MCD, e elas devem ser descartadas antes de se estabelecer o diagnóstico de MCD primária ou idiopática (Quadro 6.1).
Entre as causas apresentadas no Brasil, uma comum de MCD secundária é a doença de Chagas – endêmica em estados nordestinos, Minas Gerais e região sudoeste do Rio Grande do Sul.
O quadro clínico é variável e depende do estágio de evolução da doença. Durante muitos anos, devido aos mecanismos compensatórios – lei de Frank-Starling –, os pacientes permanecem assintomáticos. Pode haver dor torácica em até 30 a 40% dos indivíduos, mas a angina típica é incomum.
A lei de Frank-Starling é o aumento do volume diastólico final no VE que aumenta o débito cardíaco.
Com a evolução e a falência desses mecanismos, ocorrem as manifestações de insuficiência cardíaca congestiva.
O comprometimento da função ventricular esquerda resulta em congestão e baixo débito cardíaco. A congestão pulmonar manifesta-se por dispneia, ortopneia, dispneia paroxística noturna e crepitantes finos na ausculta pulmonar. A sobrecarga do ventrículo direito (VD) causa falência dessa câmara, e instala-se, então, o quadro de congestão sistêmica (hepatomegalia, turgência jugular, edema de membros inferiores, ascite) associado ao de congestão pulmonar. Os sintomas de baixo débito incluem limitação para os esforços, fadiga e astenia.
Exame do tórax e dos vasos periféricos
Em pacientes com MCD, verificam-se as seguintes manifestações em exames do tórax e dos vasos periféricos:
•Ictus cordis desviado para a esquerda e para baixo, hipocinético e aumentado (> 3 a 4 cm de extensão).
•B1 hipofonética; presença de B3 e B4; desdobramento paradoxal de B2 (se houver bloqueio de ramo esquerdo).
•Sopro de regurgitação mitral (pansistólico) secundária à dilatação do VE.
•Crepitantes finos nos terços pulmonares inferiores.
•Pulso filiforme ou pulso alternante.
No Quadro 6.2, constam os principais exames complementares que podem ser realizados em pacientes com MCD e suas respectivas alterações.
As complicações apresentadas por pacientes com MCD são as seguintes:
•Arritmias: ocorrem com frequência extrassístoles supraventriculares ou ventriculares, fibrilação atrial ou flutter atrial, podendo ser significativos fatores de descompensação. Decorrem do estiramento das fibras cardíacas pela dilatação das câmaras.
•Trombose intracavitária: a ocorrência de significativa disfunção sistólica e fibrilação atrial são fatores de risco para a formação de trombos, especialmente no ápice atrial. Também é uma causa de descompensação clínica nesses pacientes.
Determina-se o prognóstico dos pacientes com MCD idiopática de longa data por estabilidade da função ventricular e da capacidade de compensação hemodinâmica. Em geral, correlaciona-se com a classe funcional em que se mantêm, havendo uma mortalidade, em um ano, de menos de 10% para aqueles em classe I, de 10 a 15% para os em classe II, de 20 a 25% para os em classe III e de mais de 50% para os que permanecem em classe IV apesar da terapia agressiva para alívio da congestão.
O tratamento da MCD consiste principalmente no manejo da ICC e das suas complicações. Neste capítulo,resume-se essa abordagem. Mais detalhes sobre esse assunto são apresentados no Capítulo Insuficiência cardíaca. O tratamento das causas específicas é abordado em capítulos específicos.
•Fármacos utilizados em casos de IC (ver Capítulo Insuficiência cardíaca): inibidores da enzima
conversora da angiotensina, diuréticos, espironolactona, digitais e betabloqueadores.
•Arritmias: digitais para o controle da frequência cardíaca na fibrilação atrial. Indica-se a utilização
de cardiodesfibrilador implantável para pacientes com alto risco de desenvolver arritmias
potencialmente fatais.
•Anticoagulação: administra-se varfarina com o objetivo de manter um índice normatizado
internacional (INR) entre 2 e 3 em casos selecionados.
•Transplante cardíaco: indica-se esse procedimento em casos de doença avançada refratária ao
tratamento clínico e cuja causa não seja uma doença sistêmica não tratável.
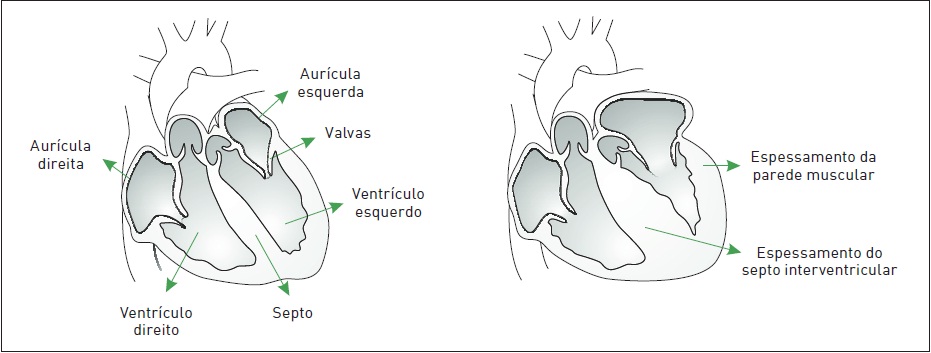
A miocardiopatia hipertrófica (MCH) caracteriza-se por hipertrofia concêntrica do miocárdio, principalmente do VE, e enchimento diastólico anormal (disfunção diastólica), sem uma causa identificada – não associada à estenose aórtica e à hipertensão arterial sistêmica (Fig. 6.2). A função sistólica está normal ou elevada, com fração de ejeção que pode apresentar índices altos de até 80%. A MCH é uma das causas mais comuns de morte súbita, principalmente em atletas jovens.
Aproximadamente 50% dos casos de MCH são familiais, e o padrão de transmissão mais frequente é o autossômico dominante, com expressão variável. Os demais casos são esporádicos. Não foram identificadas causas secundárias para a MCH.
As mutações mais comuns relacionam-se às proteínas do sarcômero, sendo identificadas mais de 100 mutações e explicando, dessa forma, a acentuada heterogeneidade da doença. As mutações afetam mais comumente a cadeia pesada da betamiosina no cromossomo 14, seguidas das mutações que afetam a proteína C que se liga à miosina e à troponina T. Entende-se que a hipertrofia cardíaca que ocorre em pacientes com MCH é um mecanismo compensatório à contração deficiente dos miócitos.
A hipertrofia do VE em geral não é simétrica, afetando principalmente o septo interventricular. Indivíduos com significativa hipertrofia do septo interventricular, aproximadamente 25% dos casos de MCH, apresentam uma forma particular dessa doença: a miocardiopatia obstrutiva (MCO), antigamente denominada estenose subaórtica hipertrófica. Em casos de MCO, há um gradiente de pressão (obstrução) dinâmico da via de saída do VE, relacionado ao estreitamento da área subaórtica devido à posição da cúspide anterior da valva mitral contra o septo interventricular hipertrofiado, isto é, movimento anterior sistólico (MAS). Esse gradiente é dinâmico, porque pode surgir ou desaparecer de acordo com o estado cardiocirculatório.
Figura 6.2
À esquerda, um coração normal e, à direita, com miocardiopatia hipertrófica.
A MCH também cursa com isquemia miocárdica por meio dos seguintes mecanismos:
•Maior demanda metabólica por aumento da massa de VE e estado hipercontrátil.
•Redução da perfusão subendocárdica na diástole por aumento das pressões de enchimento do VE.
•Espessamento das arteríolas coronárias intramurais.
Histopatologicamente é possível observar acentuada hipertrofia (aumento do tamanho celular) dos miócitos (incomum em outras causas de hipertrofia cardíaca), desarranjo dos feixes miocárdicos e até mesmo dos elementos contráteis dos sarcômeros e fibrose intersticial.
A apresentação clínica dos pacientes com MCH é variável.
Muitos permanecem assintomáticos, sendo identificados em avaliações de rotina. Outros apresentam sintomas como dispneia e angina de peito.
Consequente ao aumento das pressões de enchimento (devido à disfunção diastólica pela redução da capacidade de relaxamento), a pressão no átrio esquerdo se transmite ao leito venoso pulmonar. Nesse momento, instala-se os sintomas de congestão pulmonar, como dispneia. Esse fenômeno agrava-se na realização de esforços, e a dispneia é o sintoma mais comum.
Já o segundo sintoma mais comum, a angina do peito, é um indício de isquemia miocárdica e também manifesta-se em geral durante a realização de esforços.
Outros sintomas incluem pré-síncope e síncope, os quais estão relacionados à redução súbita do débito cardíaco.
Nas crianças, a ocorrência de síncope é um fator de prognóstico ruim.
A principal causa de morte em casos de MCH é a morte súbita, a qual ocorre com mais frequência em crianças e adultos jovens. Os mecanismos exatos não são totalmente conhecidos, mas se acredita que a isquemia durante exercício físico intenso desencadeie arritmias ventriculares fatais.
Cerca de 10 a 15% dos pacientes desenvolvem adelgaçamento e dilatação do miocárdio e disfunção sistólica, apresentando prognóstico muito desfavorável.
Exame do tórax e dos vasos periféricos
Em pacientes com MCH, verificam-se as seguintes manifestações em exames do tórax e dos vasos periféricos:
•Ictus cordis de localização habitual, propulsivo, sustentado.
•B4 é bastante comum e representa a contração atrial contra um ventrículo não complacente.
•Sopro sistólico de ejeção em borda esternal esquerda e área aórtica. Esse sopro pode representar dois fenômenos diferentes:
•Hipercontratilidade do VE
•Obstrução do trato de saída de VE
•Pulso bisferiens (dois picos sistólicos) é característico.
No Quadro 6.3, constam os principais exames complementares que podem ser realizados em pacientes com MCH e suas respectivas alterações.
Os dois principais diagnósticos diferenciais são com a cardiopatia hipertensiva e com a estenose aórtica, também causas de hipertrofia concêntrica de VE. Nesses casos, porém, esta geralmente é simétrica.
O tratamento dessa condição cardíaca envolve orientações quanto à realização de atividades físicas, ao uso de drogas inotrópicas e cronotrópicas negativas e aos procedimentos para a redução do septo interventricular. Os pacientes devem evitar a prática de exercícios físicos extenuantes e a participação em atividades competitivas. As drogas utilizadas para o controle dos sintomas incluem os betabloqueadores e os antagonistas dos canais de cálcio (p. ex., verapamil). Indica-se a realização de miomectomia septal para pacientes muito sintomáticos a despeito de uma terapia medicamentosa otimizada. Em centros com experiência nessa cirurgia, as taxas de mortalidade cirúrgica são de aproximadamente 1%. A alcoolização septal é uma alternativa percutânea para redução dos gradientes de obstrução do VE.
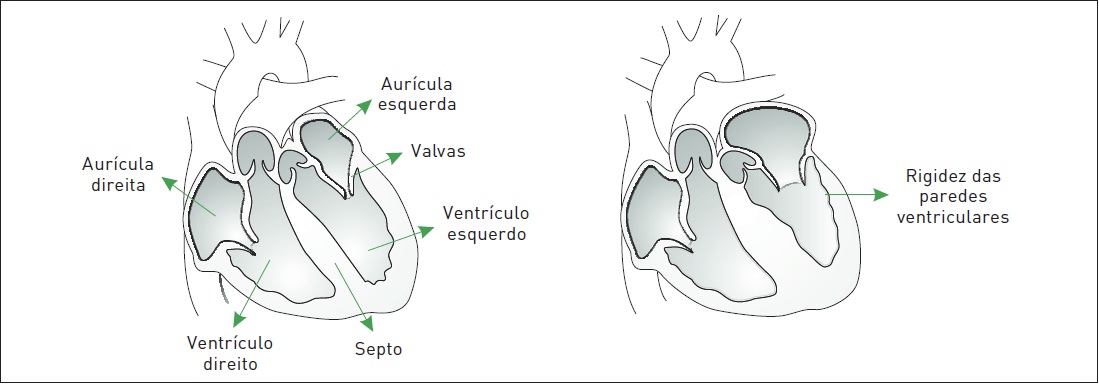
A miocardiopatia restritiva (MCR) caracteriza-se por redução da complacência ventricular (paredes excessivamente rígidas), resultando em enchimento ventricular deficiente durante a diástole (disfunção diastólica), com função sistólica normal ou discretamente reduzida. As câmaras ventriculares podem estar um pouco diminuídas, com tamanho normal ou discretamente aumentadas. É comum observar aumento de tamanho atrial (Fig. 6.3).
Entre as miocardiopatias, a MCR é o tipo que ocorre menos frequentemente, representando menos de 1% de todos os casos de ICC.
Figura 6.3
À esquerda, um coração normal e, à direita, com miocardiopatia restritiva.
Muitas vezes, não é possível identificar uma causa específica (MCR idiopática). Em outros casos, a história familiar é positiva (MCR familiar principalmente com padrão autossômico dominante). Nessas duas situações, predomina o componente fibrótico sobre o infiltrativo.
Muitas causas secundárias de MCR foram identificadas, como as seguintes:
•Infiltrativas: amiloidose, sarcoidose, hemocromatose, doença de Fabry, glicogenoses, eosinofilias (endomiocardiopatia eosinofílica de Löeffler), infiltração neoplásica.
•Fibróticas: fibrose endomiocárdica, rejeição ao trans- plante cardíaco, irradiação do mediastino.
Nos estágios mais precoces da doença, os pacientes apresentam-se assintomáticos. Com a evolução, ocorrem as manifestações de ICC.
O comprometimento da função diastólica determina aumento das pressões de enchimento no átrio esquerdo e congestão pulmonar (dispneia, ortopneia, dispneia paroxística noturna, crepitantes finos na ausculta pulmonar). A sobrecarga do VD causa falência dessa câmara, e, nesse momento, instala-se o quadro de congestão sistêmica (hepatomegalia, turgência jugular, edema de membros inferiores, ascite) associado ao de congestão pulmonar. A despeito de fração de ejeção normal (50 a 65%), pode haver sintomas de baixo débito, como fadiga e astenia, aos esforços.
Exame do tórax e dos vasos periféricos
Em pacientes com MRC, observam-se as seguintes manifestações em exames do tórax e dos vasos periféricos:
•Ictus cordis normal.
•B3 e B4 são bastante comuns.
•Sopro de regurgitação (pansistólico) de insuficiência mitral secundária à dilatação das câmaras.
•Crepitantes finos nos terços pulmonares inferiores.
•Achados de congestão (distensão venosa jugular, hepatomegalia dolorosa, ascite, edema de membros inferiores).
No Quadro 6.4, constam os principais exames complementares que podem ser realizados em pacientes com MCR e suas respectivas alterações.
É importante realizar o diagnóstico diferencial com pericardite constritiva, a qual apresenta manifestações clínicas parecidas com as da MCR, mas é potencialmente curável por meio de cirurgia.
A verificação de espessura aumentada das paredes ventriculares na ecocardiografia pode causar confusão com o diagnóstico de miocardiopatia hipertrófica. No entanto, nesse caso, o espessamento geralmente é assimétrico, maior, e o ventrículo apresenta elevada fração de ejeção.
As complicações apresentadas por pacientes com MCR são as seguintes:
•Arritmias: ocorrem com frequência taquiarritmias atriais e/ou ventriculares, fibrilação atrial ou flutteratrial e bloqueios no sistema de condução cardíaca. Decorrem da infiltração ou fibrose, as quais se estendem também para o tecido de condução cardíaco.
•Trombose intracavitária: a ocorrência de significativo aumento das cavidades atriais e fibrilação atrial crônica ou intermitente são fatores de risco para a formação de trombos, especialmente no ápice atrial.
O manejo da MCR envolve o tratamento da ICC diastólica e das suas complicações.
•Fármacos utilizados para ICC diastólica: diuréticos e nitratos, os quais reduzem a pré-carga e, com isso, combatem os sintomas congestivos.
•Anticoagulação oral: administra-se varfarina com o objetivo de manter um INR entre 2 e 3, principalmente se houver baixo débito cardíaco e fibrilação atrial intermitente ou crônica.
• Transplante cardíaco: indica-se esse procedimento para casos de doença avançada refratária ao tratamento clínico e cuja causa não seja uma doença sistêmica não tratável.
A miocardite é um processo inflamatório, geralmente resultante de uma infecção. Observa-se histopatologicamente um infiltrado inflamatório (linfócitos, neutrófilos ou eosinófilos) adjacente à área de necrose e degeneração dos miócitos. Morfologicamente o coração pode apresentar-se com tamanho normal ou dilatado e pode haver um certo grau de hipertrofia. Relata-se a evolução de miocardite viral aguda para MCD.
A causa mais comum de miocardite continua sendo o Trypanosoma cruzi (doença de Chagas). Outros agentes incluem o vírus Coxsackie B, adenovírus,influenza A, citomegalovírus, parvovírus B19,HHV-6, HSV-1, HIV, VRS.
As causas não infecciosas da doença são uso de álcool, antraciclinas, ocorrência de sarcoidose, esclerodermia e lúpus eritematoso sistêmico.
A apresentação clínica varia desde subclínica até um quadro súbito de ICC, arritmias graves e morte súbita. Alguns pacientes relatam história clínica de sintomas de infecção de vias aéreas superiores. Homens jovens são em geral mais afetados.
Os sintomas são os de uma infecção viral, havendo febre, prostação, mialgias, desconforto torácico. No exame físico, as bulhas apresentam-se abafadas, e a existência de B3 e sopro de insuficiência mitral é frequente.
O diagnóstico da miocardite é sugerido pela combinação de sinais e sintomas apresentados, mas os exames complementares são de extrema importância para sua confirmação e para a exclusão de outras causas de IC.
No Quadro 6.5, há os principais exames complementares que podem ser realizados em pacientes com miocardite e suas respectivas alterações.
O tratamento envolve as medidas de suporte e a terapia tradicional para a IC. Devido à doença avançada e ao bloqueio atrioventricular, pode ser necessário o implante de marca-passo. O uso de imunossupressores e a realização de terapias antivirais ainda não são indicados e dependem de estudos controlados que evidenciem seu benefício.
O caso apresentado sugere um quadro típico de ICC. No entanto, a idade do paciente e a inexistência de fatores de risco para doença arterial coronariana afastam a principal etiologia (doença coronariana aterosclerótica) como causa para a síndrome apresentada e apontam para algum acometimento primário do miocárdio.
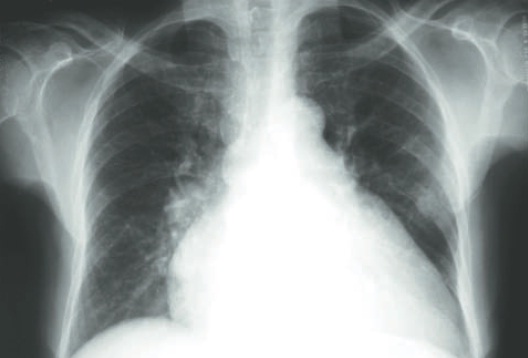
O paciente é avaliado inicialmente com raio X de tórax e eletrocardiograma (ECG) em repouso. O primeiro exame (Fig. 6.4) evidencia relação diâmetro cardíaco/ diâmetro torácico de 0,6, duplo contorno na sombra cardíaca direita e perda da concavidade e retificação do bordo cardíaco esquerdo. O ECG apresenta bloqueio atrioventricular de primeiro grau, sobrecarga de VE e alargamento difuso e não específico do QRS. No eco-cardiograma transtorácico verificam-se aumento dos diâmetros intracavitários de VE e átrio esquerdo, fração de ejeção de 34%, com espessura normal das paredes livres e disfunção sistólica global. Nesse momento da investigação, o diagnóstico altamente provável é o de miocardiopatia dilatada.
A etapa seguinte é a explicação de sua etiologia. O paciente afirma não apresentar casos semelhantes na família, não havendo também alcoolismo e outros sintomas que sugerissem uma condição secundária para a sua doença. Investiga-se, então, a possibilidade de doença de Chagas. Solicita-se sorologia da fase crônica (Elisa), a qual evidencia altos títulos para esse protozoário. Dessa forma, confirma-se o diagnóstico de miocardiopatia dilatada secundária à doença de Chagas e insuficiência cardíaca congestiva.
Figura 6.4
Raio X do caso clínico.
Elliot P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet.2004;363(9424):1881-91.
Ellis CR, Salvo T. Myocarditis: basic and clinical aspects. Cardiol Rev.2007;15(4):170-7.
Goldman L, Ausiello D. Cecil’s textbook of medicine. 22nd ed. Phila- delphia: Saunders; 2004.
Hughes SE, McKenna WJ. New insights into the pathology of inherited cardiomyopathy. Heart. 2005;91(2):257-64.
Kasper D, Fauci A, Longo DL, Braunwald E, Hauser SL, Jameson JL, editores. Harrison: medicina interna. 16. ed. Rio de Janeiro: McGraw- -Hill;2006.
Kumar V, Abbas A, Fausto N. Patologia: bases patológicas das doenças. 7. ed. Rio de Janeiro: Elsevier; 2005.
Lantieri LC, Bertoletti JC, organizadores. Interpretação eletrocardiográfica adulta e pediátrica. Porto Alegre: Artmed; 2006.
Sherrid MV. Pathophysiology and treatment of hypertrophic cardiomyopathy. Prog Cardiovasc Dis. 2006;49(2):123-51.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.