(Carregando Índice)... (Carregando Índice)... |
Autores:
Francisco Tellechea Rotta
Médico neurologista. Coordenador do Ambulatório de Doenças Neuromusculares da Santa Casa de Misericórdia de Porto Alegre. Especialista em Neurologia e Doenças Neuromusculares pela University of Miami School of Medicine. Título de Especialista pela SBNC.
Daniele Fricke
Médica neurologista e neurofisiologista do Complexo Hospitalar Santa Casa de Porto
Alegre e do Hospital Moinhos de Vento. Título de Especialista pela Academia Brasileira de Neurologia e pela Sociedade Brasileira de Neurofisiologia Clínica (SBNC). Mestre em Clínica Médica pela UFRGS.
Roberta Diehl Rodriguez
Médica neurologista.
Última revisão: 05/03/2014
Comentários de assinantes: 0
Uma paciente do sexo feminino, 18 anos, branca, apresentou, aos 9 anos, crise convulsiva tônico-clônica generalizada ao despertar. Como os resultados dos exames físico geral, neurológico e complementares (eletrencefalograma e ressonância magnética nuclear) foram normais, não se iniciou a administração de nenhuma droga antiepilética. Após três meses da consulta, a paciente apresentou nova crise. Foi prescrito fenobarbital, 100 mg ao dia, porém não houve controle das crises.
Ao retornar para a consulta, quando questionada, relatou a ocorrência de abalos musculares tipo “sustos”, quase diários, que ocorriam logo ao acordar. Os familiares e inclusive na escola também estavam preocupados com alguns episódios em que a paciente pareceu “desligar”.
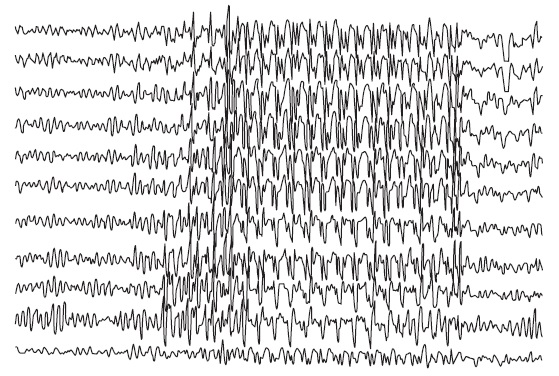
Foi realizado, então, novo eletroencefalograma que evidenciou complexo multiespícula-onda, ponta-onda, com frequência de 3 Hz ou superior, generalizados e simétricos (Fig. 92.1), e iniciado tratamento com valproato de sódio, havendo resolução quase total das crises. Revisando a história médica familiar da paciente, não foram verificados casos de epilepsia ou outra anormalidade neurológica.
As crises epiléticas, convulsivas ou não convulsivas, são manifestações clínicas neurológicas transitórias (p. ex., manifestações motoras, sensitivas, sensoriais, autonômicas, cognitivas, afetivas, etc.) decorrentes de descargas excessivas e hipersincrônicas de uma população de neurônios do encéfalo.
Havendo comprometimento motor (abalos ou contração muscular), essas crises são denominadas convulsivas. Há vários tipos de crises, cada uma com manifestações clínicas características. As crises epiléticas são sintomas comuns de um distúrbio da função cerebral e, por isso, ocorrem frequentemente em condições neurológicas e/ou médicas agudas (p. ex., meningites, hipoxia, coma hiperglicêmico não cetótico). Essas crises que ocorrem apenas quando há uma doença neurológica ou sistêmica aguda e que não recorrem posteriormente não caracterizam a epilepsia.

Figura 92.1
Eletrencefalograma em vigília.
A epilepsia é uma condição crônica caracterizada por crises convulsivas ou não convulsivas que recorrem de forma imprevisível, independentemente de qualquer fator externo. Contudo, isso não significa que algumas crises epiléticas não apresentem mais sensibilidade a determinados fatores desencadeantes externos, como a estimulação luminosa intermitente, mas que elas podem ocorrer independentemente desses fatores. Em geral, vários tipos de crises coexistem em pessoas com epilepsia, e os tipos de epilepsia diferem de acordo com a idade, a causa provável, a história familiar e o prognóstico.
A síndrome epilética constitui-se em epilepsias bem caracterizadas quanto ao tipo de crises, etiologia, prognóstico, idade de início, história familiar, frequência das crises, estudo de imagens, fatores precipitantes e eletrencefalograma.
A epilepsia é uma das doenças primárias do sistema nervoso central (SNC) mais comuns, afetando 40 milhões de pessoas no mundo. De acordo com a Organização Mundial de Saúde (OMS),1 a epilepsia corresponde a 1% da carga global de doenças (global burden of disease), taxa equivalente à de câncer de pulmão nos homens e à de câncer de mama nas mulheres. Anualmente, nos Estados Unidos, cerca de 150 mil adultos apresentam a primeira crise convulsiva e cerca de 40 a 50% desses episódios recorrem e são classificados como epilepsia.
A incidência cumulativa de epilepsia em países desenvolvidos é estimada em 1% aos 15 anos de idade, aumentando para 3% aos 74 anos de idade e para 4,4% aos 85 anos. Esta é significativamente menor do que a incidência cumulativa de apresentar qualquer crise convulsiva, que é de 10% aos 85 anos de idade.
Os homens parecem apresentar uma taxa de incidência de epilepsia um pouco maior do que a das mulheres, mas essa diferença provavelmente decorre da alta prevalência de traumatismo craniano em homens em relação às mulheres. Há uma maior prevalência de epilepsia em indivíduos em idades extremas (nos mais jovens e nos mais velhos).
Os países subdesenvolvidos apresentam maior incidência de epilepsia em relação aos desenvolvidos. Acredita-se que vários fatores contribuam para esse fato, como prevalência maior de doenças parasitárias, lesões ao nascimento, acidentes e violência.
As crises são causadas devido a um desvio do equilíbrio normal entre excitação e inibição dentro do SNC. Pelo fato de existirem diversas propriedades que controlam a excitabilidade neuronal, não é surpresa que existam muitas maneiras diferentes de perturbar esse equilíbrio normal e, por isso, podem apresentar etiologias diversas (Quadro 92.1).
Os sinais e sintomas apresentados por pacientes com epilepsia variam conforme o subtipo da doença.
Crises convulsivas, epilepsias e síndromes epilépticas são classificadas de acordo com um esquema proposto pela International League Against Epilepsy (ILAE): The International Classification of Epileptic Seizures, Epileptic Syndromes and Epilepsy.3 O Quadro 92.2 apresenta uma versão resumida desses sistemas de classificação. A classificação das crises epiléticas divide-as em duas categorias principais: crises parciais (ou focais), cuja atividade é restrita a áreas discretas do córtex cerebral, e crises generalizadas, em que há envolvimento difuso simultâneo de grandes áreas cerebrais em um padrão bilateral simétrico. As crises parciais são subclassificadas conforme o nível de consciência: se mantido, a crise é parcial simples, se alterado, complexa. As crises primariamente generalizadas são subclassificadas pela existência ou não de aspectos característicos de movimento convulsivo.
As epilepsias e as síndromes epiléticas são classificadas, quanto à localização, em generalizadas e localizadas (parciais, focais) e, quanto à etiologia, em sintomáticas, idiopáticas ou criptogênicas. As síndromes sintomáticas indicam epilepsia de etiologia conhecida ou suspeita, criptogênicas de (etiologia desconhecida) e idiopáticas (as sem causa conhecida, mas com forte história familiar e provavelmente com base genética).
O conhecimento dos conceitos discutidos anteriormente, bem como a correta classificação, são essenciais para o manejo de pacientes com epilepsia, já que a escolha do tratamento está baseada no tipo de crise e principalmente na síndrome epilética (associada à história natural e ao prognóstico conhecidos).
Em pacientes com uma primeira crise epilética, deve-se primeiramente caracterizar o evento, determinar se foi de fato uma crise epilética e se foi mesmo a primeira ocorrência. A realização de uma anamnese completa e cuidadosa, e de exame físico e neurológico são muito importantes em pacientes que apresentaram uma crise epilética. O relato de uma pessoa que tenha observado a crise é de extrema valia, principalmente porque auxilia nos diagnósticos diferenciais. Ao conduzir de forma adequada história, exame físico e neurológico, é possível muitas vezes, que o diagnóstico seja realizado sem a necessidade de exames complementares.
Alguns distúrbios episódicos podem se apresentar de forma similar a crises convulsivas ou epilepsia, como síncope, enxaqueca, intoxicações por drogas ou transtornos mentais, como as crises psicogênicas. Não há um único teste, sintoma ou achado clínico que seja eficaz para diferenciar crises epiléticas de eventos não epiléticos.
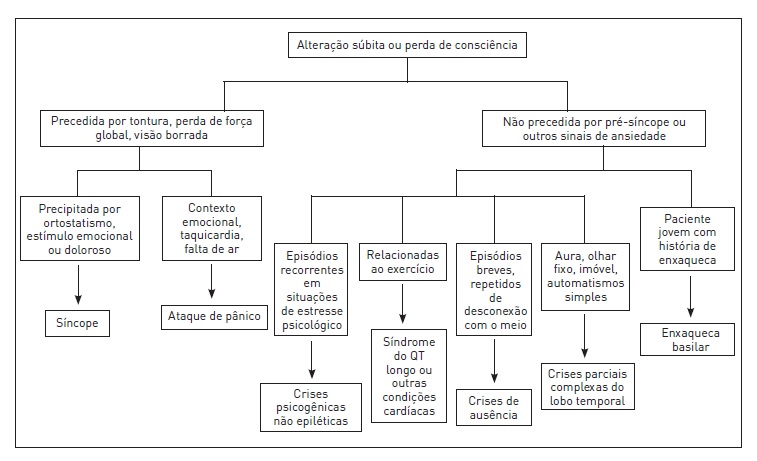
A próxima etapa na avaliação desses pacientes é determinar a causa da crise, pois algumas alterações que podem ocorrer concomitantemente a crises epiléticas requerem tratamento imediato, e outras influenciam muito para decidir sobre o início da terapia antiepilética (Fig. 92.2).
Esse exame deve ser considerado como parte da avaliação neurodiagnóstica de pacientes que apresentaram uma primeira crise epilética provavelmente não provocada, pois indica o risco de recorrência das crises (nível B). Após uma única crise não provocada, a evidência de atividade epileptiforme no EEG aumenta o risco de recorrência para 60%, independentemente se a crise foi focal ou generalizada. Um EEG normal não descarta a possibilidade de uma alteração epilética ter ocorrido, pois cerca de 50% das pessoas clinicamente diagnosticadas com crise epilética apresentam um EEG normal.
Figura 92.2
Diagnóstico diferencial de perda ou alteração súbita de consciência.
Fonte: Adaptada de Engel.5
Recomenda-se que um exame de imagem do encéfalo, tomografia computadorizada (TC) ou ressonância magnética nuclear (RM), seja considerado como parte do exame neurodiagnóstico em adultos que apresentam a primeira crise epilética não provocada (nível B). Para a escolha do exame a ser realizado, deve-se levar em conta de forma individual as circunstâncias clínicas. A TC apresenta a vantagem de um resultado mais rápido, sendo escolhida em situações de emergência, mas a RM evidencia mais sensibilidade, optando-se por ela em situações eletivas.
Eletrólitos, glicose, hemograma e leucograma podem ser úteis em circunstâncias específicas, mas não existem dados suficientes para recomendar a realização deles rotineiramente (nível U).
Esses exames devem ser solicitados de acordo com as circunstâncias apresentadas, pois não há evidências para recomendar a realização deles rotineiramente (nível U).
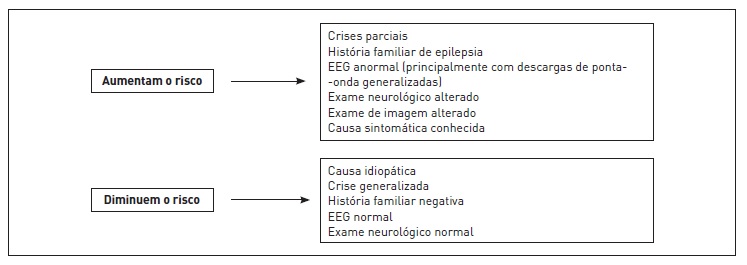
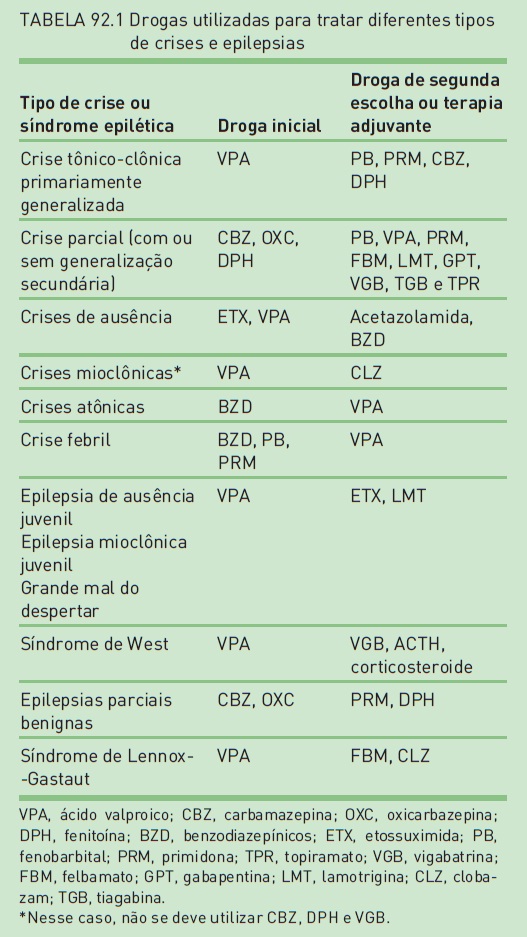
A utilização de drogas antiepiléticas após uma primeira crise não provocada é questionada, devendo-se considerar o risco de recorrência, as condições relacionadas às atividades dos pacientes e a relação entre risco e benefício. A tendência atual é iniciar o tratamento após a ocorrência da segunda crise não provocada (Fig. 92.3 e Tab. 92.1)
Figura 92.3
Fatores de risco para recorrência de crises epilépticas.
Fonte: Adaptada de Sirven.
O estado de mal epilético é definido pela ocorrência de crise epilética prolongada ou crises repetidas sem que haja sinal clínico de interrupção entre os ataques. Não há um consenso sobre o limite de tempo. Estudos experimentais observam que há dano neuronal irreversível após aproximadamente 30 minutos de atividade epilética contínua, e, por esse motivo, esse intervalo de tempo tem sido adotado pela maioria dos profissionais.7
Os estados de mal epilético generalizado convulsivo e não convulsivo são condições neurológicas potencialmente associadas a significativos índices de morbidade e mortalidade; por isso, o tratamento efetivo e imediato deve ser instituído.
O tratamento de escolha para casos de estado de mal epilético generalizado convulsivo é a administração intravenosa de diazepam, 10 mg, ou lorazepam, 4 mg, seguida por 15 a 18 mg/kg de fenitoína. Se as crises continuarem após 10 minutos da primeira injeção, outra dose de diazepam, 10 mg, intravenosa, é recomendada. O estado de mal epilético generalizado convulsivo refratário é tratado com infusão contínua de midazolam, propofol ou barbitúricos.
As características clínicas apresentadas pela paciente, com crises epiléticas tipo mioclonias e ausências, associadas ou não a crises tônico-clônicas generalizadas, padrão eletroencefalográfico com complexos espícula-onda ritmados em torno de 3 Hz ou mais, de forma bilateral e síncrona com início e final abruptos, inexistência de alterações no exame de imagem e idade de início, são compatíveis com o diagnóstico de epilepsia mioclônica juvenil. É administrado valproato de sódio para a paciente, droga de primeira escolha nas epilepsias generalizadas, havendo regressão total das crises.
1.World Health Organization. Atlas: epilepsy care in the world [Internet]. Genova: WHO; 2005 [capturado em 15 set. 2012]. Disponível em: http://www.who.int/mental_health/neurology/Epilepsy_atlas_r1.pdf.
2.Lowenstein DH. Seizures and epilepsy. In: Fauci A, Braunwald E, Isselbacher K, Wilson J, Martin J, Kasper D, et al., editors. Harrison’s principles of internal medicine. 14th ed. New York: McGraw-Hill; 1998. p. 2311-25.
3.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. epilepsia. 2010;51(4):676-85.
4.Scheuer ML, Pedley TA. The evaluation and treatment of seizures. N Engl J Med. 1990;323(21):1468-74.
5.Engel J. Epilepsy global issues for the practicing neurologist. New York: Demos; 2005.
6.Sirven, JL. Antiepileptic drug therapy for adults: when to initiate and how to choose. Mayo Clin Proc. 2002;77(12):1367-75.
7.Meierkord H, Boon P, Engelsen B, Göcke K, Shorvon S, Tinuper P, et al. EFNS guideline on the management of status epilepticus. Eur J Neurol. 2006;13(5):445-50.
Faught E. Epilepsy case studies. Neurol Clin. 2006;24(2):291-307.
Krumholz A, Wiebe S, Gronseth G, Shinnar S, Levisohn P, Ting T, et al. Practice Parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2007;69(21):1996-2007.
Serrano-Castro PJ, Sánchez-Alvarez JC, Cañadillas-Hidalgo FM, Galán-Barranco JM, Moreno-Alegre V, Mercadé-Cerdá JM. Consensus clinical practice guidelines of the Sociedad Andaluza de epilepsia for the diagnosis and treatment of patients with their first epileptic seizure in emergencies. Rev Neurol. 2009;48(1):39-50.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.