(Carregando Índice)... (Carregando Índice)... |
Última revisão: 28/03/2014
Comentários de assinantes: 1
A) Uma paciente do sexo feminino, 32 anos, branca, comparece ao serviço de emergência devido a cefaleia difusa, fadiga, sonolência e sangramento gengival com início há dois dias. A paciente não apresenta história médica prévia. Ela relata não ter nenhuma alergia medicamentosa significativa e afirma utilizar anticoncepcional oral. Ao realizar exame, verificam-se sinais vitais normais, assim como os exames cardiorrespiratório e abdominal. Observam-se petéquias nos membros inferiores.
A partir da investigação preliminar, foram obtidos os seguintes resultados dos exames: hemoglobina de 7,2 g/dL (esquizócitos presentes no esfregaço de sangue periférico), plaquetas de 18.000/ L, INR de 1,1, tempo de tromboplastina parcial ativada (TTPa) de 32 s, D-dímero negativo, desidrogenase lática (LDH) de 2.568 U/L, creatinina de 0,98 mg/dL.
B) Uma paciente do sexo feminino, 18 anos, branca, comparece ao serviço de emergência com sangramento excessivo após extração dentária oito horas antes da chegada ao hospital. A paciente não apresenta história médica prévia. Ela relata história prévia de menorragia e afirma não utilizar nenhuma medicação, incluindo aspirina. No exame físico, não são evidenciados achados exceto por sangramento no local do procedimento relatado. A partir da investigação preliminar, obtiveram-se os seguintes resultados dos exames: hemoglobina de 10 g/dL, plaquetas de 185.000/ L, desidrogenase lática (LDH) de 195 U/L, INR de 1,1, TTPa de 42 s (normaliza com o teste de mistura), D-dímero negativo, tempo de sangramento de 11 minutos, atividade do fator VIII de 58%.
A hemostasia refere-se ao mecanismo fisiológico que previne a perda de sangue que ocorre após alguma lesão vascular no organismo. Geralmente, predominam na corrente sanguínea os componentes anticoagulantes, de modo que o sangue não coagula quando está circulando pelos vasos sanguíneos. Entretanto, quando há ruptura de um vaso, os pró-coagulantes tornam-se ativados na área de lesão tecidual e sobrepujam os anticoagulantes. É possível distinguir dois componentes:
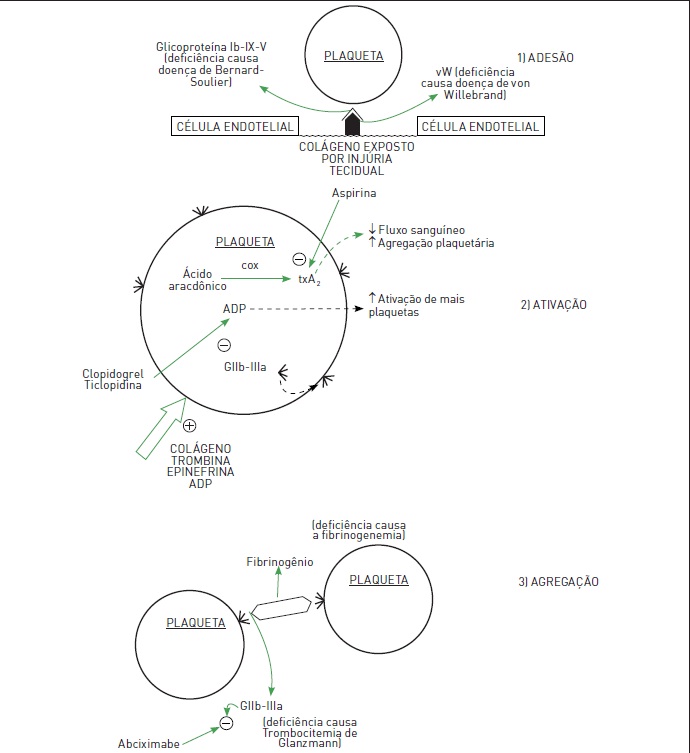
•Hemostasia primária (responsável pelo pronto estancamento do sangramento por meio da constrição vascular e da formação do trombo ou do tampão plaquetário) (Fig. 54.1).
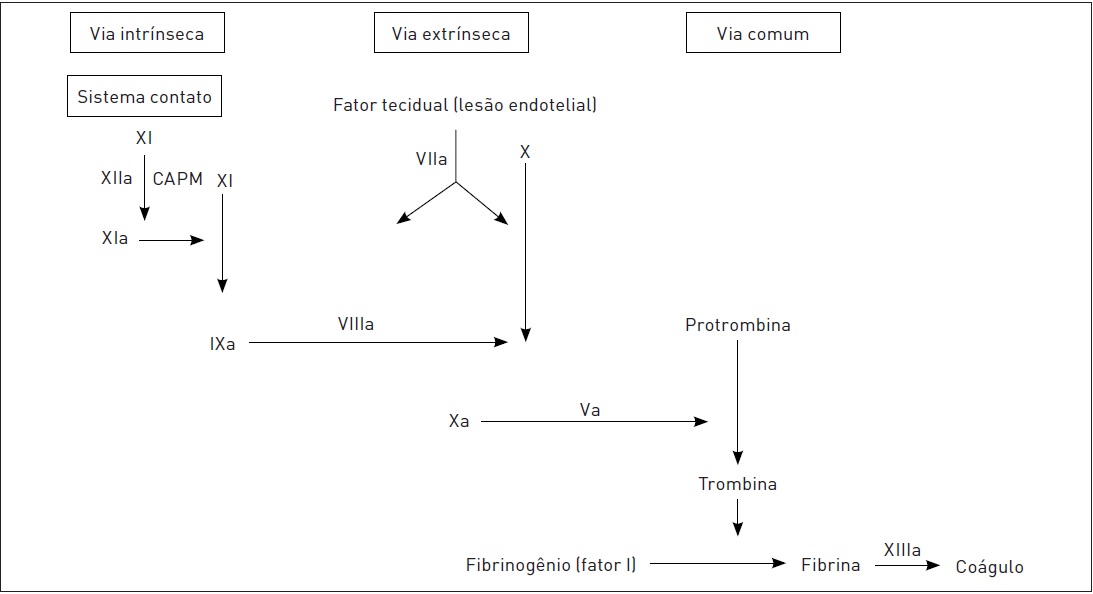
•Hemostasia secundária (implicada na prevenção do ressangramento por meio da estabilização do trombo com a formação da rede de fibrina, formando o coágulo) (Fig. 54.2).
Sistema fibrinolítico: ao mesmo tempo em que é formado o trombo para o controle do sangramento, é iniciado um processo de controle da trombose excessiva. Esse processo visa a dissolver o coágulo e restaurar a integridade vascular. Inicialmente, o endotélio libera o ativador do plasminogênio tecidual (tPA). Este converte o plasminogênio circulante em plasmina, a qual apresenta a capacidade de degradar os polímeros de fibrina, originando os D-dímeros. O endotélio libera o inibidor do ativador do plasminogênio (PAI-1), que degrada o tPA. Os anticoagulantes endógenos fisiológicos são compostos por antitrombina III, proteína C, proteína S e TFPI.
As discrasias hemorrágicas podem ser classificadas, grosso modo, de acordo com o sistema envolvido em sua causa: distúrbios das plaquetas e distúrbios da coagulação. Os defeitos na hemostasia primária (plaquetas) são geralmente caracterizados por sangramento superficial envolvendo a pele ou as membranas mucosas. Sinais como petéquias, sangramento em mucosas e hematomas apresentam-se comumente. Sangramentos em pacientes com alteração da hemostasia primária em geral ocorrem imediatamente após o trauma e não recorrem após parada. Ao contrário, sangramentos que são causados devido a defeitos na hemostasia secundária, como em um paciente com hemofilia, geralmente envolvem tecidos profundos e podem resultar na formação de hematoma intramuscular, hemartrose, hemorragias retroperitoneal, gastrintestinal, geniturinária e cerebral. As hemartroses manifestam-se quase exclusivamente em casos de hemofilia e von Willebrand tipo III (deficiência grave). O sangramento nesses indivíduos pode ocorrer algum tempo após o trauma, evidenciando a inabilidade de o tampão plaquetário ser estabilizado de forma correta. Eventualmente, uma doença congênita pode ser percebida apenas na idade adulta devido à falta de eventos que exigissem mais da hemostasia (p. ex., von Willebrand diagnosticado devido a menorragia na adolescência sem história de extração dentária) e à inexistência de história familiar da doença, embora isso não descarte a possibilidade de ocorrência (cerca de 30% dos casos de hemofilia são causados por mutações novas, não herdadas).
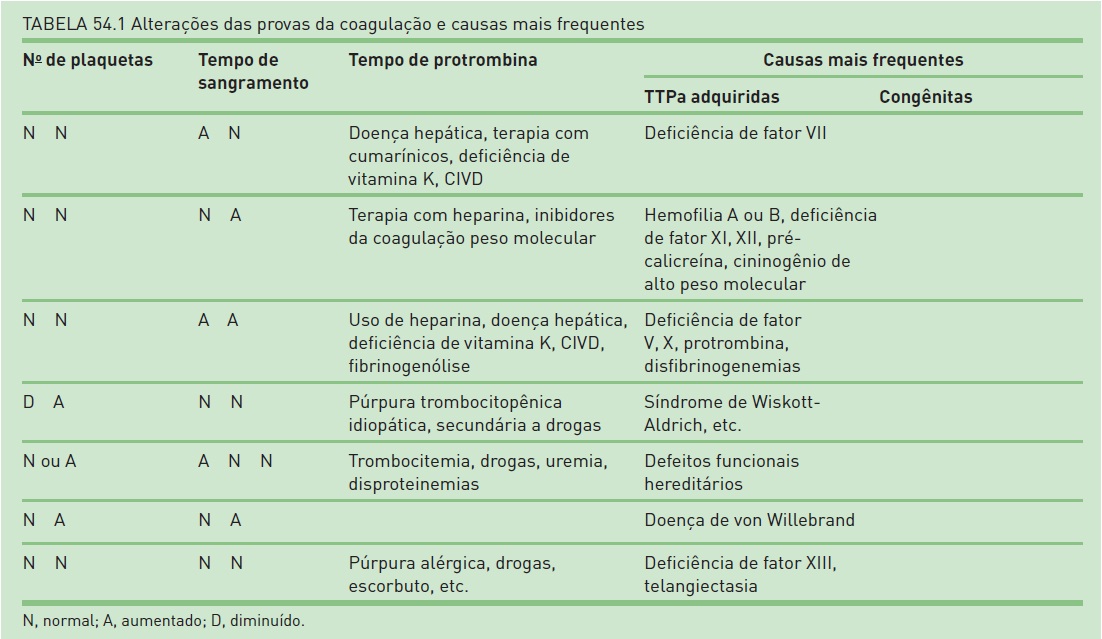
Tempodeprotrombina (TP). O TP avalia os fatores V, VII e X (via extrínseca ou tecidual), a protrombina (fator II) e o fibrinogênio (fator I). Os valores desse teste não se apresentam alterados em caso de deficiência de fator VIII, IX, XI e XII. Doenças hepáticas, assim como defi ciência de vitamina K, alteram seus valores. O resultado atualmente é realizado por meio de um índice normatizado internacional, o INR (international normatized ratio), visando à comparação dos valores obtidos entre laboratórios distintos, devido a diferenças na tromboplastina utilizada. Esse teste é utilizado para monitorar o uso dos anticoagulantes cumarínicos (varfarina).

Figura 54.1
Hemostasia primária: as plaquetas são os componentes com destaque nesse momento. De início, o vaso lesado sofre constrição mediada principalmente por espasmo miogênico local, desencadeado pela lesão direta da parede vascular. 1) Adesão: as plaquetas circulantes aderem ao subendotélio devido à exposição de fibras de colágeno no sítio de lesão vascular. Os receptores da membrana plaquetária, glicoproteína Ib-IX-V, ligam-se ao colágeno. Essa ligação é estabilizada pelo fator de von Willebrand (vW), uma proteína circulante produzida pelas células endoteliais. Após a adesão, as plaquetas sofrem ativação. 2) Ativação: com esse processo, essas células degranulam, emitem pseudópodos e ganham a capacidade de se agregar umas às outras. As substâncias que acarretam ativação plaquetária são o colágeno, a trombina, o ADP e a epinefrina. As plaquetas ativadas liberam, ainda, tromboxano A2 (txA2) (derivado do ácido aracdônico a partir da ação da enzima cicloxigenase) e ADP, que apresentam a propriedade de amplificar esse processo. A próxima etapa é a agregação plaquetária. 3)Agregação: as plaquetas ligam-se umas às outras através das glicoproteínas IIb/IIIa, que utilizam como ponte o fibrinogênio circulante. A hemostasia primária é sufi ciente para estancar o sangramento se a lesão vascular for pequena.
Figura 54.2
Hemostasia secundária: entra em ação a cascata da coagulação. O objetivo é estabilizar o trombo plaquetário. Para isso, é necessário transformar o fibrinogênio plasmático em fibrina. A cascata da coagulação corresponde a um sistema com diversas formas inativas de enzimas proteolíticas que são convertidas em suas formas ativadas e, com isso, desencadeiam reações sucessivas. O último fator, a protrombina (fator II), ativa a trombina (fator IIa), a qual é responsável pela conversão do fibrinogênio plasmático (fator I) em fibrina, formando o coágulo. Todos os fatores de coagulação são sintetizados nos hepatócitos, entre os quais são dependentes de vitamina K (inibidos pela varfarina) os seguintes: II, VII, IX e X. O fator VIII (anti-hemofílico), para não ser degradado na circulação, tem de ser estabilizado pelo fator de von Willebrand. Há dois mecanismos que desencadeiam a cascata da coagulação, provavelmente os dois ocorram ao mesmo tempo: a via intrínseca e a via extrínseca (in vivo esta última parece ser a principal); a partir de um certo ponto, elas seguem uma via comum. O processo da primeira inicia no sistema intravascular e é mais lento, enquanto o da segunda ocorre nos tecidos adjacentes. O estímulo para iniciar o mecanismo da via extrínseca é o fator tecidual ou a tromboplastina tecidual existente nas células do tecido subendotelial, e o da via intrínseca é a exposição do colágeno.
Tempo de tromboplastina parcial ativada (TTPa ou KTTp). Esse teste avalia a maioria dos fatores de coagulação (via intrínseca e comum), exceto os fatores VII e XIII. Não está indicado como screening pré-operatório,exceto se houver suspeita clínica. Ele encontra-se prolongado em caso de deficiência de fator I (fibrinogênio), II (protrombina), V, VIII (hemofilia A), IX (hemofilia B), X, XI e XII, podendo estar prolongado também quando há anticoagulante lúpico (sem distúrbio hemorrágico associado). Os valores do TTPa não são alterados com o uso da heparina de baixo peso molecular. Esse teste é indicado para monitorar a terapia com heparina não fracionada.
Tempo de sangramento. A partir do tempo de sangramento, é possível medir a formação do tampão plaquetário, primeira etapa da coagulação. Os valores normais são de 4 a 7 minutos. Encontra-se prolongado em casos de doenças como von Willebrand, deficiência de fator V e fibrinogênio, defeitos funcionais das plaquetas, como uremia e uso de drogas (principalmente o ácido acetilsalicílico). Contagens de plaquetas menores do que 100.000/ l invalidam o teste. Ele não tem valor como screening pré-operatório. Esse teste apresenta alta variabilidade devido à falta de padronização da técnica na maioria dos laboratórios.
Teste de mistura. Um TTPa prolongado pode ser decorrente da deficiência de um fator de coagulação ou da presença, no plasma, de um inibidor de um fator de coagulação específico (p. ex., anticoagulante lúpico). Mistura-se um volume de plasma do paciente com um mesmo volume de um doador saudável. Se o TTPa for corrigido após o procedimento, o prolongamento previamente observado foi ocasionado devido a deficiência de um fator que pode agora a ser elucidada. Entretanto, se o TTPa não for corrigido (permanecer prolongado), o prolongamento foi causado pela presença de um inibidor.
A púrpura trombocitopênica idiopática (PTI) ou autoimune é uma condição primária adquirida em que autoanticorpos são formados agindo contra as plaquetas. Assim, as plaquetas opsonizadas são reconhecidas pelos macrófagos, no baço, e destruídas. A única manifestação é a trombocitopenia isolada, e a consequência desta é o aparecimento de petéquias e púrpuras.
Essa doença, em adultos, apresenta pico de incidência entre 20 e 50 anos. Há um predomínio de ocorrência em mulheres (2:1). Em crianças, a PTI tem pico de incidência aos 5 anos, não havendo predomínio de incidência conforme o sexo.
Como citado anteriormente, são formados autoanticorpos da classe IgG direcionados contra as plaquetas, cujo antígeno mais envolvido é a glicoproteína de membrana GPIIb/IIIa. No baço e, em menor extensão, no fígado, os macrófagos com receptores Fc do sistema reticuloendotelial reconhecem as plaquetas opsonizadas e as destroem. A meia-vida dessas plaquetas, que era de 7 a 10 dias, passa a ser, então, de apenas algumas horas.
Clinicamente, é possível distinguir duas formas de apresentação: PTI aguda ou infantil e PTI crônica ou do adulto. A PTI aguda ocorre frequentemente duas a três semanas após uma infecção viral e é autolimitada, ou seja, remite em 90% dos casos em um período de até seis meses. A forma do adulto é geralmente de uma doença crônica sem relação frequente com infecções virais, cuja remissão ocorre em menos de 10% dos casos. Essa forma geralmente apresenta um início insidioso e um curso cíclico. Os sintomas relatados pelos pacientes afetados são sangramento de mucosas e pele. Os tipos comuns de sangramento são epistaxe, sangramento oral, menorragia, púrpura e petéquia. Tipicamente, a púrpura e a petéquia em caso de PTI são assintomáticas e não palpáveis. Essas são características que diferenciam essa púrpura das que surgem nas vasculites, que são palpáveis. Os sangramentos graves ocorrem quando as plaquetas estão em níveis menores do que 10.000 células/mm3. Há sangramento fatal quando os níveis das plaquetas estão menores do que 5.000/mm3, mesmo assim raramente ocorre sangramento cerebral. Não há sangramento visceral e de tecidos profundos, com a formação de hematoma, como em casos de distúrbios de coagulação – por exemplo, nas hemofilias. O diagnóstico de PTI é realizado por exclusão. As formas secundárias da doença devem ser descartadas, tais como lúpus eritematoso sistêmico, síndrome antifosfolipídeo, estados de imunodeficiência (deficiência de IgA), doenças linfoproliferativas (p. ex., leucemia linfocítica crônica, linfoma, etc.), infecção pelo vírus da hepatite C e pelo HIV e terapia com drogas, como heparina e quinidina.
Hemograma n O principal achado laboratorial no hemograma é a trombocitopenia (redução do número absoluto de plaquetas no plasma). Valores baixos, como menos de 10.000 células/mm³, podem ocorrer. As outras linhagens hematopoiéticas apresentam-se normais, e o esfregaço do sangue periférico evidencia morfologia normal, exceto pelo aumento do tamanho das plaquetas (megatrombócitos). Esse achado é compatível com plaquetas jovens e representa uma compensação da medula óssea, que aumenta a produção de megacariócitos em resposta à destruição periférica das plaquetas.
Exame da medula óssea n A medula óssea apresenta-se normal. O único achado possível é o aumento do número de megacariócitos. Não é necessário realizar o exame da medula em todos os indivíduos, mas ele é muito recomendado para aqueles com mais de 60 anos a fim de descartar diagnóstico de síndromes mielodisplásicas.
Provas de coagulação n O tempo de trombina e o tempo de tromboplastina parcial ativado encontram-se normais.
Outros exames n Devem ser descartadas possibilidades de outras causas secundárias de púrpura trombocitopênica em adultos com PTI. Dessa forma, além do hemograma, das provas de coagulação e do exame da medula óssea, esses pacientes devem realizar idealmente testes sorológicos para o vírus da hepatite B, o citomegalovírus, o vírus Epstein-Barr, o toxoplasma e o HIV.
A forma que afeta as crianças geralmente é a de uma doença autolimitada e que apresenta remissão espontânea na maioria das vezes; por isso o tratamento raramente é necessário. Já a doença, nos adultos, quase invariavelmente necessita de tratamento. O tratamento está indicado para os pacientes com sintomas e/ou contagem de plaquetas menores do que 20.000 a 30.000 células/mm3. Inicialmente, administra-se prednisona de 1 a 2 mg/kg/dia, por 4 a 6 semanas. Esse fármaco age diminuindo a afinidade dos macrófagos do baço pelas plaquetas recobertas com anticorpos. A contagem de plaquetas começa a subir na primeira semana, e a normalização ocorre em geral até a terceira semana em aproximadamente 80% dos casos. Após 4 a 6 semanas, a dose deve ser ajustada e mantida para a menor possível a fim de manter a contagem de plaquetas um pouco acima de 50.000 células/mm3. Na maioria dos indivíduos, há remissão da trombocitopenia se o tratamento com prednisona for interrompido. Para os pacientes que não respondem inicialmente à administração de corticoide (plaquetas < 30.000/mm3 após 4 a 6 semanas) ou para os que reagem, mas apenas devido a doses de manutenção elevadas, a esplenectomia é o tratamento mais efetivo. Aproximadamente 80% dos pacientes apresentam remissão parcial ou completa.
Para as emergências com sangramento potencialmente fatal ou no pré-operatório, as medidas disponíveis são transfusão de plaquetas (não é realizada rotineiramente para não aumentar a produção de anticorpos), altas doses de dexametasona e administração de imunoglobulina intravenosa (IgIV). Entre essas, a medida que mais rapidamente aumenta o número de plaquetas é a administração de altas doses de IgIV (1 g/kg, por 1 a 2 dias). Esse tratamento, no entanto, é caro, e os seus efeitos duram por, no máximo, 1 a 2 semanas.
A púrpura trombocitopênica trombótica (PTT) e a síndrome hemolítico-urêmica (SHU) são duas condições incomuns, não distintas, potencialmente fatais e com grande sobreposição fisiopatológica e clínica. Ambas são caracterizadas por anemia hemolítica microangiopática e trombocitopenia. Outras manifestações de ocorrência menos comum incluem febre, sintomas neurológicos e perda de função renal. A PTT desenvolve-se mais comumente com sintomas neurológicos, e a SHU, com insuficiência renal. A SHU é uma das principais causas de insuficiência renal aguda em crianças.
Na PTT, os indivíduos mais afetados são os adultos jovens, entre 20 e 50 anos de idade. Há pouca predominância de incidência em mulheres. A SHU ocorre mais comumente do primeiro ano de vida até os 10 anos.
Mecanismos imunes parecem estar envolvidos na PTT. Supostamente é produzido um anticorpo dirigido contra a metaloprotease ADAMST13 (responsável pela quebra do fator de von Willebrand). Não havendo uma adequada degradação dessa protease, acumulam-se multímeros de fator de von Willebrand de ultra-alto peso molecular, os quais resultam em agregação plaquetária e adesão ao endotélio. O dano endotelial é o marco desses distúrbios. Não se sabe sobre o fator inicial que causa a produção desses anticorpos. Entretanto, essa síndrome pode ser precipitada pelo uso de estrógenos, pela gestação, por quimioterápicos em altas doses, por câncer metastático e por certos medicamentos (p. ex., ticlopidina, quinino, ciclosporina, tacrolimus) ou infecções (HIV). Em crianças com SHU, é frequente a doença secundária a infecções, como diarreia por Escherichia coli, cepa O157:H7, que produz a toxina shiga-like.Geralmente, há um período de sete dias entre a diarreia e o início dos sintomas da SHU. Os achados patológicos são trombos hialinos nos capilares e nas pequenas artérias e vênulas, sem inflamação da parede vascular. Estes ocasionam oclusão da microcirculação. Além disso, há incorporação desses trombos à parede dos capilares, representando o achado diagnóstico dessas condições. Diferentes leitos vasculares estão envolvidos na PTT e na SHU.
A doença manifesta-se de forma subaguda, uma vez que se desenvolve durante dias ou semanas. Os primeiros sintomas são mal-estar e fraqueza. Na SHU, é possível identificar história precedente de diarreia, dor abdominal, náuseas e vômitos. Na realização do exame físico, o paciente apresenta-se enfermo, febril e pálido. Podem existir petéquias e equimoses. Os sintomas neurológicos incluem cefaleia, confusão e alteração do nível de consciência, que varia de letargia até coma.
O achado mais comum no hemograma é a anemia, que pode ser grave, com níveis de hemoglobina de até 5 g/dL. As hemácias são destruídas na própria circulação e o seu conteúdo liberado no plasma (hemólise intravascular – anemia microangiopática). Por isso, o dado encontrado mais frequentemente nesse tipo de anemia são as hemácias fragmentadas no sangue periférico (esquizócitos). Sem hemácias fragmentadas não é possível estabelecer o diagnóstico de PTT ou SHU. Os aumentos de bilirrubina indireta são causados por hemólise e isquemia, respectivamente. A medula óssea reage aumentando a produção da linhagem eritrocitária, e, dessa forma, a reticulocitose é marcante. Trombocitopenia (em geral < 50.000/mm3) também é uma alteração muito frequente, que se estabelece conforme a gravidade da anemia e que pode ser grave, principalmente na PTT. Os testes de coagulação, tais como TP, TTPa, fibrinogênio e D-dímeros, apresentam-se também normais, auxiliando no diagnóstico diferencial com coagulação intravascular disseminada. Quanto às provas de função renal, a ureia e a creatinina podem estar alteradas, como na SHU. A biópsia de pele ou de outros locais não é frequentemente necessária para o diagnóstico, mas pode ser um exame importante para o diagnóstico diferencial com vasculites e glomerulonefrites agudas. Quando for necessária, os depósitos hialinos subendoteliais, evidenciados na biópsia, possibilitam o diagnóstico de PTT ou SHU.
Essas duas condições devem ser tratadas rapidamente com plasmaferese, exceto em crianças, que geralmente apresentam uma doença autolimitada. A terapia remove as formas de ultra-alto peso molecular de fator de von Willebrand e o inibidor da metaloprotease ADAMST13 e repõe a enzima deficiente. Deve-se remover grande volume de plasma (60 a 80 mL/kg) e substituí-lo por plasma fresco congelado, o que deve ser realizado até a completa remissão da doença. Essa medida reduz drasticamente a mortalidade desses pacientes, e, com ela, a remissão completa da doença ocorre em 80 a 90% dos casos. Na maioria dos pacientes, a doença não recorre. Os sintomas neurológicos em geral desaparecem sem deixar sequelas. Há recuperação da função renal na maioria dos pacientes.
A doença de von Willebrand é o distúrbio congênito mais comum da hemostasia, afetando 1 a cada 100 a 500 indivíduos. A transmissão é realizada de modo autossômico dominante na maioria dos casos.
O fator de von Willebrand é uma glicoproteína fundamental para a adequada adesão plaquetária ao subendotélio em locais de lesão vascular. Já o processo de agregação plaquetária é alterado nesse distúrbio. O fator é sintetizado pelos megacariócitos e pelas células endoteliais e circula, no plasma, na forma de multímeros de variados pesos moleculares. Os níveis plasmáticos normais são de aproximadamente 1 mg/dL. Apenas os multímeros de alto peso molecular são funcionais e, portanto, são estes que medeiam a adesão plaquetária. Esse fator, independentemente do peso molecular, tem outras importantes funções: ele liga-se ao fator VIII da coagulação (que se encontra deficiente na hemofilia A) e o transporta, de forma a protegê-lo da degradação pelas proteases. Em caso de doença de von Willebrand, a redução dos níveis de fator VIII devido ao aumento de sua degradação não é suficiente para causar um distúrbio secundário da hemostasia.
Há três fenótipos da doença de von Willebrand. No tipo 1 (60 a 80% dos casos), há uma diminuição quantitativa leve a moderada do fator de von Willebrand (em cerca de 50%). No tipo 2 (10 a 30% dos casos), há uma anormalidade qualitativa nessa proteína que impede a formação dos multímeros (2a) ou que causa uma rápida degradação dos multímeros de grande tamanho (2b). O tipo 3 (5 a 10% dos casos) é a forma mais grave, rara e distinta da doença de von Willebrand. Nela, o fator praticamente inexiste, e o modo de herança é autossômico recessivo. Esses subtipos apresentam tratamentos diversos e, por isso, a determinação adequada é importante para o manejo de cada paciente.
A maioria dos casos de doença de von Willebrand é leve. Os sangramentos ocorrem principalmente nas mucosas (nasal e gengival). Um quadro clássico é o de paciente que apresenta hemorragia incisional imediata em cirurgias, traumas ou extração dentária. Pode haver menorragia e epistaxe espontânea. A menorragia é relatada por 60 a 95% das mulheres, e 5 a 20% das mulheres com essa queixa são afetadas pela doença de von Willebrand. Sangramentos graves, como na hemofilia em que ocorre hemartrose, somente ocorrem no tipo 3 da doença.
A contagem de plaquetas apresenta um valor normal. Em caso de suspeita de doença de von Willebrand, o exame de tempo de sangramento deve ser realizado. Este, na maioria das vezes, está prolongado; quando normal, torna-se prolongado com o uso de ácido acetilsalicílico, que aumenta bastante a tendência de sangramento nesse distúrbio. O TTPa encontra-se alargado devido à deficiência parcial secundária do fator VIII. Os níveis de fator de von Willebrand, que estão frequentemente reduzidos, podem ser medidos por meio do antígeno do fator VIII (eletroimunoensaio, Elisa) ou pelo teste do cofator da ristocetina, que mede as propriedades funcionais do fator de von Willebrand em mediar a adesão plaquetária.
Como a tendência para desenvolver sangramento é leve nessa doença, não é necessário nenhum tratamento de manutenção. Deve-se, no entanto, evitar o uso de ácido acetilsalicílico. Se o tempo de sangramento for bastante prolongado, é prudente realizar profilaxia antes de procedimentos cirúrgicos ou dentários. Para o tipo 1 da doença de von Willebrand, a desmopressina (DDAVP) é o medicamento utilizado com esse propósito, sendo administrado poucas horas antes do procedimento. A DDAVP parece agir liberando os estoques de fator de von Willebrand a partir das células endoteliais. Quando for necessário realizar reposição do fator deficiente nos casos mais graves da doença ou após grandes cirurgias ou traumas, tem-se disponível o concentrado do fator VIII (humate-P). Este contém fator de von Willebrand, é isento do risco de transmissão de agentes infecciosos, como o HIV, e pode ser utilizado em qualquer tipo da doença.
As doenças da rota intrínseca da coagulação são caracterizadas por um TTPa prolongado e um TP normal. As formas herdadas incluem deficiência de fatores VIII e IX (hemofilia A e B), pré-calicreína, cininogênio de alto peso molecular, fator IX ou fator XII. Deficiências de fator XII (fator de Hageman), pré-calicreína e cininogênio de alto peso molecular podem ser descartadas facilmente, pois não estão associadas a sangramento clínico excessivo. Para realizar distinção entre as deficiências dos fatores VIII, IX e XI, devem ser efetuados testes específicos. Testes da correção com plasma normal também devem ser realizados para que se tenha certeza de que não há anticorpos contra o fator em estudo. Caso o inibidor do fator VIII seja identificado, o título deve ser quantificado. Entre as doenças adquiridas que provocam alteração na rota intrínseca da coagulação, tem-se o anticoagulante lúpico e os anticorpos contra o fator VIII.
O prolongamento do TTPa e do TP em um paciente com doença herdada indica deficiência de um dos fatores em comum na rota: fator X, V, protrombina ou fibrinogênio. A ocorrência dessas deficiências isoladamente é extremamente rara. Já a deficiência de um ou mais desses fatores está associada a anormalidades adicionais nas rotas intrínseca e extrínseca em muitas das doenças comuns adquiridas da coagulação, como na deficiência de vitamina K, na doença hepática e na CIVD. A constatação de um TP prolongado geralmente sugere doença adquirida.
A verificação de um TP prolongado e um TTPa normal é indício de uma deficiência isolada de fator VII, que ocorre raramente e pode ser uma alteração tanto herdada quanto adquirida. Também pode haver inibidores do fator VII. Além disso, certos casos de CIVD ou disfibrinogenemia podem ocorrer havendo TP prolongado isolado.
A coagulação intravascular disseminada (CIVD) é uma síndrome secundária a uma doença subjacente grave. Essa doença é caracterizada pela ativação excessiva e descontrolada da coagulação. Os níveis intravasculares de trombina estão em tal proporção elevados que excedem os inibidores fisiológicos fibrinolíticos. A partir dessa situação, decorre a deposição de fibrina nos pequenos vasos e, então, a oclusão destes pelo trombo formado. Há um significativo aumento no risco de falência múltipla de órgãos. Esse processo ocasiona, ao final, um grande consumo dos fatores de coagulação, causando, então, a predisposição ao sangramento, principal manifestação clínica da doença. O prognóstico da CIVD é variável e está relacionado à doença de base, que, em geral, é muito grave.
Os diversos agentes etiológicos envolvidos com o desenvolvimento de CIVD causam, em última instância, um estado pró-coagulação por meio da expressão de citocinas. A liberação ou exposição do fator tecidual (via intrínseca) é o principal fator na formação dessa síndrome. A partir desta, ocorre a produção de trombina, a qual fragmenta o fibrinogêneo em fibrina, estimula a agregação plaquetária, ativa os fatores V e VIII e libera o ativador de plasminogênio, que gera plasmina. Formam-se, então, microtrombos na circulação sistêmica, um fenômeno que em geral ocorre por local. O resultado do excesso da atividade da trombina é a hipofibrinogenemia, a trombocitopenia, a depleção dos fatores de coagulação e a fibrinólise. A plasmina, por sua vez, quebra a fibrina, gerando os produtos da degradação da fibrina (PDF), e, além disso, inativa os fatores V e VIII. O sistema fibrinolítico é ativado na tentativa de deter o processo trombótico. Este, no entanto, não é suficiente. Os níveis de antitrombina III, o principal inibidor da coagulação, estão reduzidos, pois são consumidos rapidamente, e a síntese apresenta-se diminuída. Outro importante anticoagulante endógeno, a proteína C, também será reduzida.
395
A CIVD pode ser causada por diversas doenças graves (Quadro 54.1). Atualmente, a CIVD desenvolve-se mais comumente em quadros de sepse – os agentes mais corriqueiros são as bactérias. Classicamente, 30 a 50% dos casos de sepse por bactérias gram-negativas evoluem com CIVD – prevalência semelhante tem sido relatada com bactérias gram-positivas.
Síndrome de Waterhouse-Friedrichsen: infarto hemorrágico da glândula suprarrenal em caso de infecção meningocócica, cujo mecanismo envolve a CIVD.
É possível verificar duas apresentações clínicas distintas para a CIVD: a forma aguda e a forma crônica. Na forma aguda, o quadro clínico é marcado mais comumente por sangramentos. Entretanto, os sintomas trombóticos (isquemia de dígitos, nariz e genitália; acrocianose periférica) podem prevalecer nesse quadro se o estímulo para a coagulação for muito maior do que para a fibrinólise. Os sangramentos podem ocorrer em qualquer local, mas se destacam os localizados em sítios de punção venosa ou onde há cateter e em incisões cirúrgicas. Hemorragia pulmonar, gastrintestinal e cerebral podem ocorrer e são potencialmente fatais. Secundariamente, a CIVD pode ocasionar, em 25% dos casos, anemia hemolítica microangiopática, decorrente do aprisionamento e da lesão das células dentro dos trombos de fibrina. A forma crônica da CIVD afeta mais comumente pacientes com neoplasias sólidas secretoras de fator tecidual e manifesta-se por trombose venosa superficial e profunda (síndrome de Trousseau).
A CIVD apresenta um quadro laboratorial típico: hipofibrinogenemia (< 70 a 100 mg/dL), PDF elevados (o mais sensível destes são os D-dímeros), trombocitopenia (< 100.000 células/mm3) e prolongamento do TP. O TTPa pode ou não estar prolongado. Havendo anemia hemolítica microangiopática, no esfregaço do sangue periférico, observam-se hemácias fragmentadas (esquizócitos). A antitrombina III, um inibidor da coagulação, está reduzida no plasma.
A principal medida terapêutica para pacientes com CIVD deve ser o tratamento e o controle da doença que a desencadeou. No que diz respeito ao sangramento, deve-se realizar a reposição dos produtos em deficiência: transfusão de plaquetas para manter a contagem plasmática maior do que 30.000 a 50.000/mm3, administração de crioprecipitado para elevar o fibrinogênio a valores maiores do que 100 mg/dL, administração de plasma fresco congelado para a reposição de fatores de coagulação deficientes.
As hemofilias são distúrbios hereditários da coagulação. O padrão de herança é ligado ao X, e, por isso, os homens são praticamente os únicos afetados. Fisiopatologicamente, determinados fatores de coagulação apresentam-se deficientes. As principais manifestações das hemofilias são os sangramentos, principalmente em cavidades articulares (hemartrose). A hemofilia A (ou clássica) é a causa mais comum de sangramento grave por distúrbios da coagulação.
A ocorrência da hemofilia A é muito mais comum do que a da B (1:10.000 nascidos do sexo masculino na primeira e 1:100.000 na segunda). Há história familiar da doença em até 65% dos casos. As mulheres podem ser heterozigotas para o gene da hemofilia em um dos cromossomos X. Com isso, a chance de filhos do sexo masculino apresentarem hemofilia é de 50%. Atualmente estão disponíveis testes para identificar mulheres com esse traço.
Na hemofilia A (ou hemofilia clássica), os sangramentos ocorrem devido à deficiência de fator VIII. Na hemofilia B, é o fator IX que está reduzido. Esses fatores são sintetizados no fígado. Os genes que codificam esses fatores estão localizados no braço longo do cromossomo X. Na maioria dos casos, esses fatores de coagulação apresentam-se quantitativamente reduzidos, mas, em alguns poucos casos, eles estão presentes, porém não são funcionalmente efetivos.
A tendência ao sangramento é uma condição dependente dos níveis dos fatores deficientes. Não existem diferenças entre a apresentação clínica da hemofilia A e a da B. Os sítios em que mais comumente ocorrem sangramentos são as articulações que sustentam peso (p. ex., joelho, cotovelo, fêmur, tornozelo), dentro dos músculos, no trato gastrintestinal e no trato geniturinário. A hemartrose é um indicador tão característico da doença que sua ocorrência praticamente confirma o diagnóstico. O quadro é de uma monoartrite de grande articulação: há edema, dor intensa e imobilidade da articulação envolvida. As hemorragias mais temidas são a do sistema nervoso central, que ocorre em 10% dos casos, e a de orofaringe, que pode causar asfixia. Algumas horas após um trauma, pode haver sangramento, que, se não tratado, pode durar dias. Entre os pacientes com hemofilia A, 70% apresentam a forma grave da doença; os com hemofilia B, até 20 a 45%.
As hemofilias são classificadas, conforme a gravidade, da seguinte forma:
•Grave – os níveis do fator são menores do que 1% se comparados aos dos valores normais. Em geral esses pacientes sangram espontaneamente. O diagnóstico é realizado durante a infância, devido a cefaloematoma ou à hemorragia na circuncisão.
•Moderada – os níveis do fator são de 1 a 5%. Os sangramentos ocorrem em pacientes que são submetidos a cirurgias ou sofrem traumas leves/moderados, ou também em crianças de 2 a 4 anos de idade, quando começam a deambular sem apoio. Na doença moderada, deve-se efetuar o diagnóstico diferencial com doença de von Willebrand.
•Leve – os níveis do fator são maiores do que 5%. Há sangramentos apenas quando os pacientes são submetidos a grandes cirurgias ou apresentam politrauma.
Hoje em dia, a maioria dos indivíduos hemofílicos são portadores do HIV. Eles infectaram-se na época em que o tratamento era realizado com concentrado de fator VIII, o qual era preparado a partir do sangue de milhares de doadores, e os testes para HIV e HCV não estavam ainda disponíveis. Atualmente, a Aids é a principal causa de morte nos hemofílicos.
Os achados laboratoriais incluem alargamento do TTPa. Outras medidas da coagulação, como TP, tempo de sangramento, plaquetas e fibrinogênio, apresentam-se normais. O diagnóstico é realizado por meio de ensaios específicos para identificar quais os fatores de coagulação estão deficientes. A medida do fator de von Willebrand evidencia normalidade em indivíduos com hemofilias.
A diferenciação entre os dois tipos de hemofilia é fundamental para a escolha do tratamento adequado. Em todas as formas da doença, deve-se evitar o uso de ácido acetilsalicílico e de outros agentes anticoagulantes.
Hemofilia A n O tratamento atualmente é realizado com fator VIII purificado (obtido a partir de 10.000 doadores e processado para inativação de vírus) ou com o fator VIII recombinante (obtido a partir de engenharia genética). Este está indicado para preparo pré e perioperatório. O preparo pré-operatório para procedimentos cirúrgicos menores pode ser feito com DDAVP em indivíduos com hemofilia moderada. O tempo de meia-vida do fator VIII é de 8 a 12 horas, e, por isso, é necessário realizar infusão continuamente ou, pelo menos, duas vezes ao dia. O DDAVP aumenta a liberação de fator VIII e pode elevar duas a três vezes os seus níveis séricos durante algumas horas.
Hemofilia B n Nesse caso, utilizam-se preferencialmente fator IX purificado e fator IX recombinante. Está disponível também um concentrado do complexo protrombínico parcial que contém os seguintes fatores de coagulação: II, IX e X. Podem ocorrer eventos tromboembólicos devido à presença de alguns fatores ativados nesse preparado.
A) A paciente apresenta sangramento de pequena escala (gengival) e petéquias nos membros inferiores, indicando mais um processo “plaquetário” do que envolvendo a via da coagulação.
Nos exames laboratoriais, a paciente evidencia sinais de anemia hemolítica microangiopática (hemoglobina de 7,2 g/dL com LDH alto, indicando hemólise, e esquizócitos), trombocitopenia (com testes de coagulação normais) e sintomas que afetam o sistema nervoso central, a tríade clássica da PTT.
B)Nesse caso, a paciente apresenta história de menorragia com sangramento excessivo após um procedimento dentário. Esse caso é clássico de doença de von Willebrand, mais especificamente do tipo 1. A paciente evidencia hemorragia incisional imediata após extração dentária e relata menorragia prévia.
397
Os resultados dos exames laboratoriais são os seguintes: anemia com hemoglobina de 10 g/dL, plaquetas de 185.000/ L (normal), LDH de 195 (normal, descartando existência de hemólise), INR de 1,1 (via extrínseca, sem alterações), TTPa prolongado de 42 s (normalizado com o teste de mistura, o que indica a deficiência de um fator da via intrínseca), tempo de sangramento de 11 minutos (evidenciando anormalidade da hemostasia primária, plaquetas) e atividade do fator VIII reduzida (58%).
Os achados laboratoriais que sustentam o diagnóstico incluem o defeito qualitativo das plaquetas (número absoluto dentro da normalidade, mas com tempo de sangramento elevado) e a prolongação do TTPa, corrigida após o teste de mistura. Isso indica a deficiência de um fator, nesse caso, do fator VIII, que é transportado pelo fator de von Willebrand (que está defeituoso ou diminuído).
Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. Annu Rev Med. 2010;61:77-90.
Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357(6):580-7.
Bolton-Maggs PHB, Pasi KJ. Haemophilias A and B. Lancet.2003;361(9371):1801-9.
Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Eng J Med. 2002;346(12):995-1008.
George J. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354(18):1927-35.
Godeau B, Provan D, Bussel J. Immune thrombocytopenic purpura in adults. Curr Opin Hematol. 2007;14(5):535-56.
Kujovich JL. von Willebrand’s disease and menorrhagia: prevalence, diagnosis, and management. Am J Hematol. 2005;79(3):220-8.
Levi M, Ten Cate H. Disseminated intravascular coagulation. N Eng J Med. 1999;341(8):586-2.
Levi M. Disseminated intravascular coagulation. Crit Care Med.2007;35(9):2191-5.
Mannucci PM. Treatment of von Willebrand´s disease. N Engl J Med.2004;351(7):683-94.
McPhee SJ, Papadakis MA, Tierney LM, editors. Current: medical diag- nosis and treatment. 47th ed. New York: McGraw-Hill; 2008.
Nangaku M, Nishi H, Fujita T. Pathogenesis and prognosis of thrombotic microangiopathy. Clin Exp Nephrol. 2007;11(2):107-14.
Psaila B, Bussel JB. Immune thrombocitopenic purpura. Hematol Oncol Clin North Am. 2007;21(4):743-59.
Razzaq S. Hemolytic uremic syndrome: an emerging health risk. Am Fam Physician. 2006;74(6):991-6.
Toh CH, Michael D. Disseminated intravascular coagulation: old disease, new hope. BMJ. 2003;327(7421):974-7.
"Ótima revisão... Parabéns!!"
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.