(Carregando Índice)... (Carregando Índice)... |
Autores:
Juliana Keller Brenner
Médica residente do Serviço de Medicina Interna do HCPA.
Alexandre Sauer da Silva
Acadêmico da Faculdade de Medicina da UFRGS. Bolsista Probic/Fapergs/ UFRGS do Serviço de Endocrinologia do HCPA.
Artur Boschi
Médico internista. Médico residente do Serviço de Endocrinologia do HCPA.
Sandra Pinho Silveiro
Médica endocrinologista. Professora associada do Departamento de Medicina Interna da UFRGS. Preceptora do Programa de Residência
Médica em Endocrinologia do HCPA. Professora
do Programa de Pós-graduação em Ciências Médicas: Endocrinologia da UFRGS. Doutora em Medicina: Ciências Médicas pela UFRGS.
Última revisão: 09/06/2014
Comentários de assinantes: 0
Uma paciente do sexo feminino, 50 anos, branca, procura o serviço de emergência devido a emagrecimento de 10 kg em um ano, cansaço, fraqueza muscular proximal e desânimo há dois anos, com piora progressiva. Ela relatou escurecimento difuso da pele e episódios de diarreia há três dias, associado a dor abdominal. A paciente também apresenta hipotireoidismo primário em tratamento com levotiroxina.
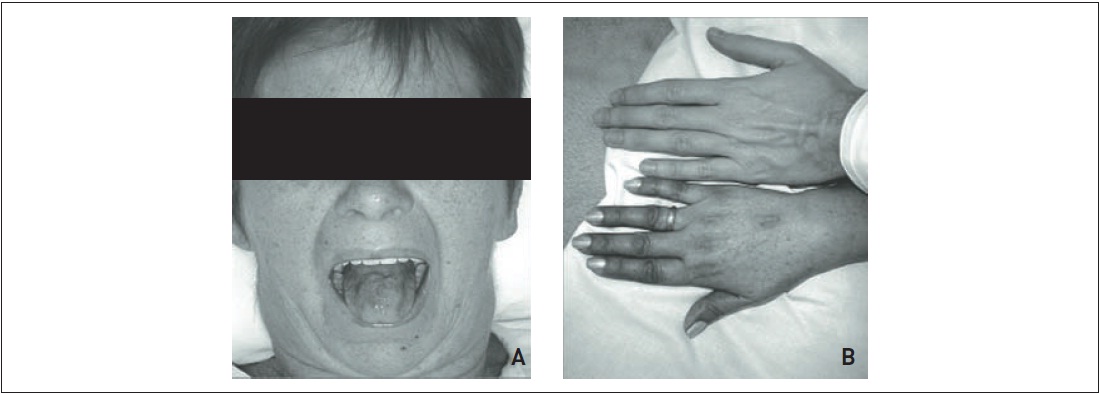
Ao realizar exame físico, constata-se hipotensão postural, mucosas desidratadas e escurecimento de pele em áreas fotoexpostas (Fig. 31.1).
Os exames laboratoriais evidenciam leucograma com 9.170 leucócitos (4.440 neutrófilos/420 eosinófilos/3.480 linfócitos), sódio de 130 mEq/L (135-150), potássio de 8,2 mEq/L (3,5-5,0), creatinina de 1,71mg/dL (até 1,2), hormônio estimulador da tireoide (TSH) de 8,9 UI/mL (0,4-4,5), anticorpo antitireoperoxidase (anti-TPO) de 319UI/L (até 35), cortisolde 2,9 g/dL (4,3-22), hormônio adrenocorticotrófico (ACTH) de 2.166 pg/mL (10-52), renina maior do que 50 ng/mL/h (0,51-2,64), aldosterona menor do que 2,2 ng/dL (4-31), sulfato de de idroepiandrosterona (SDHEA) de 26,2 g/dL (18,9-205) e, na gasometria, consta acidose metabólica leve em compensação. O nível de anticorpo anti 21 hidroxilase é de 121 (1,0).
Pesquisa sérica de paracoccidioidomicose e histoplasmose forneceram resultados negativos, e verifica-se Mantoux não reator.
Na tomografia computadorizada (TC) de abdome superior, constatam-se glândulas suprarrenais normais.

Figura 31.1
Paciente com quadro de insuficiência suprarrenal primária crônica, apresentando (A) hiperpigmentação da pele da face e (B) das mãos (comparar com mão do indivíduo saudável) e da mucosa oral.
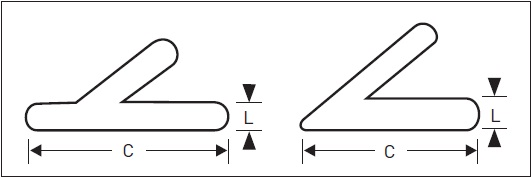
As glândulas suprarrenais estão localizadas nos polos superiores dos rins no retroperitônio, pesando em média 2 g e medindo de 4 a 6 cm de comprimento (C) e de 2 a 3 cm de largura (L) (Fig. 31.2). Elas podem ser divididas em duas regiões, uma mais externa, o córtex, e outra interna, a medula, as quais diferem quanto à origem embriológica, ao tipo de hormônio produzido e ao mecanismo regulatório por que respondem.
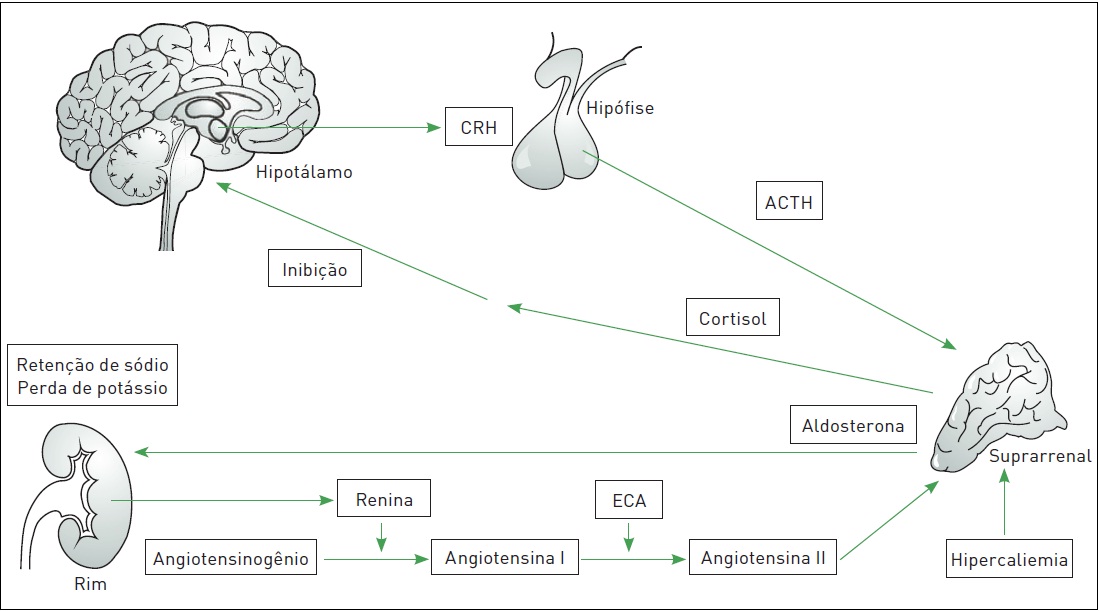
O córtex, responsável pela síntese de hormônios esteroides, é constituído de três zonas distintas: a glomerulosa, mais externa, produtora de mineralocorticoides (principalmente aldosterona) que promovem retenção de sal; a fasciculada, envolvida na síntese de glicocorticoides (principalmente cortisol) com efeitos significativos sobre o metabolismo intermediário e a função imune; e a reticular, mais interna, encarregada dos hormônios com atividade androgênica (SDHEA). A regulação, a síntese e a secreção dos hormônios do córtex da suprarrenal têm origem no eixo hipotálamo-hipófise-suprarrenal (HHS), com exceção da aldosterona, que responde primariamente ao eixorenina-angiotensina (Fig. 31.3). Quando a secreção de hormônios do córtex da suprarrenal é inferior à adequada, seja em estado basal ou devido a estímulos estressores, decorrentes de destruição ou supressão do eixo HHS, há um quadro de insuficiência suprarrenal (IS).
Figura 31.2
Tamanho e possíveis formas da glândula suprarrenal: peso médio de 2 g, 4 a 6 cm de comprimento (C) e 2 a 3 cm de largura (L).
A insuficiência suprarrenal pode ser classificada como primária, secundária ou terciária. A primária, também denominada doença de Addison, em homenagem à Thomas Addison, que primeiramente a descreveu em 1855, ocorre quando a suprarrenal é incapaz de secretar os corticosteroides e os mineralocorticoides por uma destruição total do córtex suprarrenal.
A IS secundária ocorre quando há uma deficiência de hormônio adrenocorticotrófico (ACTH) hipofisário, e a terciária, quando a deficiência é de hormônio liberador de corticotrofina (CRH) hipotalâmico. Nesses dois últimos tipos de IS, também chamadas de insuficiência suprarrenal central, há perda do estímulo trófico das suprarrenais após algum tempo, mas somente a produção de corticosteroides estará comprometida, pois o eixorenina-angiotensina-aldosterona (RAA) mantém-se intacto, não afetando a produção de mineralocorticoide, que praticamente independe do ACTH.
Figura 31.3
Regulação da produção de cortisol pelo eixo hipotálamo-hipófise-suprarrenal e da aldosterona pelo eixo renina-angiotensina-aldosterona.
CRH, hormônio liberador de corticotrofi na; ACTH, hormônio adrenocorticotrófico; ECA, enzima conversora da angiotensina.
Formas adquiridas de IS primária são relativamente raras, com uma prevalência de casos de 12/100.000 habitantes, e podem ocorrer em qualquer idade, com pico aos 40 anos (20 a 40 anos), sendo as mulheres mais frequentemente afetadas do que os homens (2,6:1). Em países desenvolvidos, a causa mais corriqueira de IS primária é a suprarrenalite autoimune. Já a IS central acaba sendo de incidência mais comum, devido ao frequente uso de esteroides em diversas modalidades terapêuticas, o que acarreta supressão do eixo HHS. Ela apresenta uma prevalência estimada de casos de 20/100.000 habitantes, afetando mais frequentemente as mulheres do que os ho- mens. O diagnóstico é realizado principalmente por volta dos 60 anos, e a principal causa é a suspensão abrupta da terapia com glicocorticoides.
As causas de IS estão listadas no Quadro 31.1.
Os sinais e sintomas da IS manifestam-se conforme o grau de acometimento da função suprarrenal, o envolvimento ou não da função mineralocorticoide (afetando o balanço de água e sódio) e o grau de estresse existente (Quadro 31.2). O aparecimento dos sintomas ocorre geralmente de forma gradual e pode não ser percebido até que haja uma crise suprarrenal. A principal característica clínica que diferencia a IS primária da central é a pigmentação cutânea existente praticamente sempre na primeira, exceto quando a deficiência hormonal é de início recente, quando ainda não houve tempo para ocorrer esse evento. Há essa hiperpigmentação devido à redução do feedback negativo do cortisol, causando um aumento hipofisário da produção e clivagem da POMC (pró-ópio-melanocortina), um precursor de diversos peptídeos, entre eles o ACTH e o hormônio estimulador dos melanócitos. Observa-se hiperpigmentação em áreas fotoexpostas, axilas, mamilos, áreas de cicatrizes recentes, palma das mãos e mucosas (gengival, vaginal, anal). Outra diferença fundamental entre a IS primária e a secundária é o fato de que, nesta, não há envolvimento significativo do sistema RAA, pois esse sistema é regulado essencialmente pelos níveis séricos de potássio e pelo volume circulante efetivo. Dessa forma, praticamente não há alterações relacionadas aos distúrbios hidreletrolíticos e à volemia, como hipercaliemia e hipotensão postural grave. No entanto, pode haver hiponatremia dilucional causada por aumento da secreção da vasopressina, decorrente da deficiência de cortisol, e também hipotensão leve.
A crise suprarrenal é mais frequente na IS primária do que na secundária ou na terciária, pois há depleção de volume e hipotensão (consequência do defeito no sistema RAA) concomitante à perda de tônus vascular (falta de corticosteroides). É caracterizada por hipercaliemia, hiponatremia; perda urinária de sódio e depleção do volume plasmático com aumento da ureia.
A confirmação da IS inclui um processo dos três estágios a seguir:
1) Existe insuficiência suprarrenal? Para essa confirmação, é necessário demonstrar níveis de cortisol inapropriadamente baixos.
2) A insuficiência suprarrenal é primária ou secundária? Para a diferenciação, deve-se determinar se a deficiência de cortisol é causada pela deficiência de ACTH/ CRH ou por hipofunção primária da glândula (avaliar função mineralocorticoide).
3) Qual a etiologia da insuficiência suprarrenal? Nesse último estágio, procura-se a causa básica da doença.
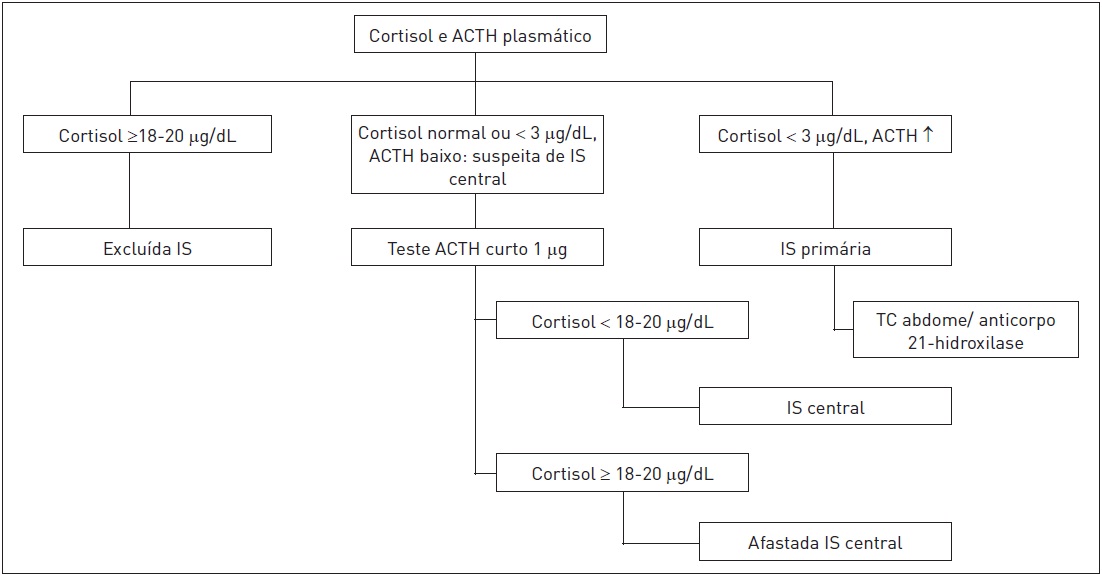
Um fluxograma para a investigação de IS é apresentado na figura 31.4.
Cortisol sérico. Um valor de cortisol sérico menor do que 3 g/dL é altamente sugestivo de IS com uma especificidade de 100%, mas com uma baixa sensibilidade (36%). Dessa forma, a dosagem isolada do cortisol matinal sérico não é um bom preditor de alteração na função suprarrenal e deve ser realizada junto a dosagem do ACTH. Valores basais de cortisol maiores do que 20 g/dL praticamente afastam o diagnóstico, assim como valores maiores do que 25 g/dL em vigência de estresse tornam o diagnóstico de IS improvável. Devem ser consideradas as possíveis interferências na dosagem de cortisol decorrentes de alteração na globulina carreadora de cortisol (CBG), como, por exemplo, a elevação da CBG devido ao uso de estrogênio oral, que provoca aumento dos níveis do cortisol total embora a fração livre permaneça inalterada.
Dosagem plasmática de ACTH. O nível de ACTH deve ser medido junto com o do cortisol sérico na avaliação inicial. Se o índice de cortisol for inapropriadamente baixo e o de ACTH for alto, diagnostica-se IS primária. Se ambos estiverem baixos, o paciente apresenta IS secundária ou terciária. Eventualmente, o resultado da dosagem de ACTH é demorado, e o paciente precisa realizar o diagnóstico de IS com mais brevidade. Nesses casos,realiza-se o teste de estimulação com ACTH intravenoso (cortrosina aquosa) na dose de 250 g. Deve-se ter o cuidado de coletar e centrifugar a amostra para ACTH em temperatura baixa para preservação do hormônio (tubo gelado).
Avaliação mineralocorticoide. Essa avaliação é realizada por meio da medida de potássio, sódio, renina e aldosterona plasmática. Na IS primária, a atividade ou concentração de renina está aumentada, e a aldosterona está reduzida. O nível de potássio pode estar elevado, e o de sódio, reduzido. Na IS secundária ou terciária, a renina e a aldosterona apresentam níveis normais, bem como o potássio sérico.
Anticorpos. A presença de anticorpos contra a 21-hi-droxilase, responsável pela primeira hidroxilação na formação de cortisol, pode ocorrer nos casos de suprar renalite autoimune. Confirmado o diagnóstico, deve-se estar atento para a presença de outras doenças autoimunes associadas, como tireoidite de Hashimoto, falência gonadal, diabetes melito tipo 1, anemia perniciosa e hipoparatireoidismo. O TSH pode estar elevado de forma moderada devido à deficiência do cortisol e é normalizado com a reposição do corticoide, não indicando necessariamente a presença de hipotireoidismo.
Figura 31.4
Investigação de insuficiência suprarrenal.
Teste do ACTH curto/teste da cortrosina aquosa: Esse teste deve ser realizado nos pacientes em que o diagnóstico de IS está sendo considerado e naquelas situações em que não se dispõe de resultado rápido da dosagem de cortisol e ACTH plasmático. Não é necessária a realização desse teste se o paciente apresenta um cortisol matinal no limite superior da normalidade (> 20 g/dL). O teste é realizado por meio de injeção intravenosa de ACTH sintético de duas formas: com baixa dose de 1 g (dose mais fisiológica para suspeita de IS secundária), dosando-se o cortisol após 20 ou 30 minutos da injeção, ou com alta dose de 250 g (suspeita de IS primária), dosando-se o cortisol após 30 a 60 minutos. Ausência de resposta (cortisol < 18 g/dL) no teste com 250 g confirma IS primária, e essa ausência de resposta no teste de 1 g de ACTH sugere IS secundária (que poderia responder com teste de dose mais alta).
Teste da hipoglicemia insulínica: Na IS secundária ou terciária de início recente (p. ex., uma a duas semanas após cirurgia hipofisária), os testes de escolha são o de ACTH de baixa dosagem ou hipoglicemia insulínica. Nesses casos, as suprarrenais ainda não estão completamente atrofiadas, podendo reagir caso o estímulo seja intenso (p. ex., 250 g de ACTH). O teste com hipoglicemia induzida pela administração de insulina IV baseia-se no princípio de que a hipoglicemia estimula a secreção de ACTH, causando um aumento nos seus níveis e nos níveis de cortisol (resposta normal > 18 g/dL) apenas em indivíduos sem lesão hipofisária ou hipotalâmica. O cortisol é medido antes da administração de insulina (0,1 U/kg) e após 30, 60, 90 e 120 minutos, e a glicemia deve atingir o nível de 40 mg/dL ou menos. Esse teste é útil nos casos em que se quer observar concomitância com deficiência de GH, mas apresenta os riscos inerentes à hipoglicemia e é contraindicado em pacientes com cardiopatia isquêmica ou história de convulsões.
A tomografia computadorizada (TC) de abdome deve ser realizada nos casos de IS primária. O achado de calcificações ou aumento bilateral das suprarrenais elimina o diagnóstico de suprarrenalite autoimune e sugere o de doenças infecciosas, hemorrágicas ou neoplásicas (Fig. 31.5). Um raio X de tórax deve ser solicitado para avaliar a presença de tuberculose ou doenças fúngicas. A punção aspirativa de suprarrenal guiada por TC pode auxiliar na determinação da causa da IS. Nos casos de IS secundária ou terciária, a ressonância magnética (RM) de sela túrcica deve ser realizada.
Figura 31.5
TC de abdome em paciente com hemorragia suprarrenal bilateral (setas) após uso de enoxaparina.
Orientaçãoaopaciente. O paciente e seus familiares devem ser orientados quanto à importância do tratamento e de portar consigo uma pulseira ou documento que informe o diagnóstico de IS, bem como uma dose de glicocorticoide injetável nos casos de emergência (trauma grave, como queimaduras ou atropelamentos) (Tab. 31.1).
Crise suprarrenal. Não se deve esperar o diagnóstico definitivo para iniciar o tratamento em pacientes com doença aguda. Pode-se coletar uma amostra de sangue para exames futuros e imediatamente iniciar o tratamento com reposição volêmica (1 a 3 L de soro fisiológico 0,9% nas primeiras 12 a 24 horas), controlando diurese e status volêmico. Em paciente sem o diagnóstico prévio de IS, administração de dexametasona intravenosa (bólus com 4 mg) é preferível por não interferir na medida de cortisol sérico. Já nos pacientes que estavam em tratamento com glicocorticoides, qualquer preparação intravenosa pode ser utilizada (dexametasona, 4 mg, ou hidrocortisona, 100 mg), sendo preferível hidrocortisona para aqueles com potássio elevado. Tal medida rapidamente reduz a produção inapropriada de vasopressina, aumentando a excreção de água livre e corrigindo a hiponatremia. O corticoide intravenoso pode ser reduzido em 1 a 3 dias com substituição para dose de manutenção por via oral. Não é necessária a reposição aguda de mineralocorticoide devido à demora no início de seu efeito. Esta pode ser iniciada quando a infusão venosa salina não mais for necessária.
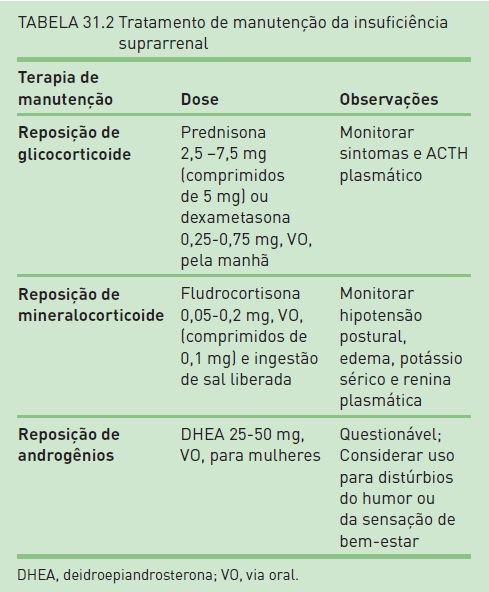
Tratamento de manutenção. O tratamento de manutenção é baseado na educação do paciente sobre sua doença, na reposição de corticosteroides, de mineralocorticoides e possivelmente de androgênios em algumas mulheres (Tab. 31.2).
A paciente desse caso apresenta IS primária, tendo como causa a suprarrenalite autoimune. A presença de hiponatremia e hipercaliemia com as manifestações clínicas apresentadas gerou suspeita de IS de etiologia primária, uma vez que há envolvimento do eixo mineralocorticoide (presença dos distúrbios hidreletrolíticos e alteração de volemia). A dosagem baixa de cortisol e elevada de ACTH confirma a origem primária. A etiologia da IS primária é a suprarrenalite autoimune, confirmada por meio de dosagem no anticorpo contra a 21-hidroxilase. A coexistência com doença tireoidiana autoimune (presença de altos títulos de anti-TPO) corrobora a etiologia, sugerindo a presença da síndrome poliglandular autoimune.
Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab. 2009;94(4):1059-67.
Arlt W, Allolio E. Adrenal insufficiency. Lancet. 2003;361(9372):1881-93.
Bornstein RS. Predisposing factors for adrenal insufficiency. N Engl J Med.2009;360(22):2328-39.
Bouillon R. Acute adrenal insufficiency. Endocrinol Metab Clin North Am.2006;35(4):767-75.
Gurnell EM, Hunt PJ, Curran SE, Conway CL, Pullenayegum EM, Huppert FA, et al. Long-term DHEA replacement in primary adrenal insufficiency: a randomized, controlled trial. J Clin Endocrinol Metab.2008;93(2):400-9.
Kazlauskaite R, Evans AT, Villabona CV, Abdu TA, Ambrosi B, Atkinson AB, et al. Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. 2008;93(11):4245-53.
Kronenberg HM, Melmed S, Polonsky KS, Larsen PR. Williams textbook of endocrinology. 11th ed. Philadelphia: W.B. Saunders; 2008.
Magnotti M, Shimshi M. Diagnosing adrenal insufficiency: which test is best--the 1-microg or the 250-microg cosyntropin stimulation test? Endocr Pract. 2008;14(2):233-8.
Perry R, Kecha O, Paquette J, Huot C, Van Vliet G, Deal C. Primary adrenal insufficiency in children: twenty years experience at the Sainte-Justine Hospital, Montreal. J Clin Endocrinol Metab. 2005;90(6):3243-50.
Reisch N, Arlt W. Fine tuning for quality of life: 21st century approach to treatment of Addison’s disease. Endocrinol Metab Clin North Am.2009;38(2):407-18.
Wade M, Baid S, Calis K, Raff H, Sinaii N, Nieman L. Technical details influence the diagnostic accuracy of the 1 µACTH stimulation test. Eur J Endocrinol. 2010;162(1):109-13.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.