(Carregando Índice)... (Carregando Índice)... |
Autores:
Francisco Tellechea Rotta
Médico neurologista. Coordenador do Ambulatório de Doenças Neuromusculares da Santa Casa de Misericórdia de Porto Alegre. Especialista em Neurologia e Doenças Neuromusculares pela University of Miami School of Medicine. Título de Especialista pela SBNC.
José Otávio Dworzecki Soares
Médico neurologista. Doutorando em Imunologia pela USP.
Última revisão: 20/06/2014
Comentários de assinantes: 0
Uma paciente do sexo feminino, 29 anos, branca, casada, natural e procedente de Porto Alegre, que exerce a profissão de comerciaria, há três anos apresentou visão borrada, de instalação aguda, no olho esquerdo, com leve dor na movimentação ocular. A paciente consultou com um oftalmologista, que determinou exame ocular normal. Ela foi encaminhada para um neurologista que não percebeu alterações e atribuiu os sintomas a depressão e ao estresse, orientando o uso de antidepressivo. O borramento visual regrediu após quinze dias do início dos sintomas. Após doze meses, a paciente desenvolveu parestesia no hemicorpo direito associada a ataxia. Foi novamente examinada por um neurologista e, conforme avaliação complementar e clínica, cogitou-se o diagnóstico de doença desmielinizante – esclerose múltipla.
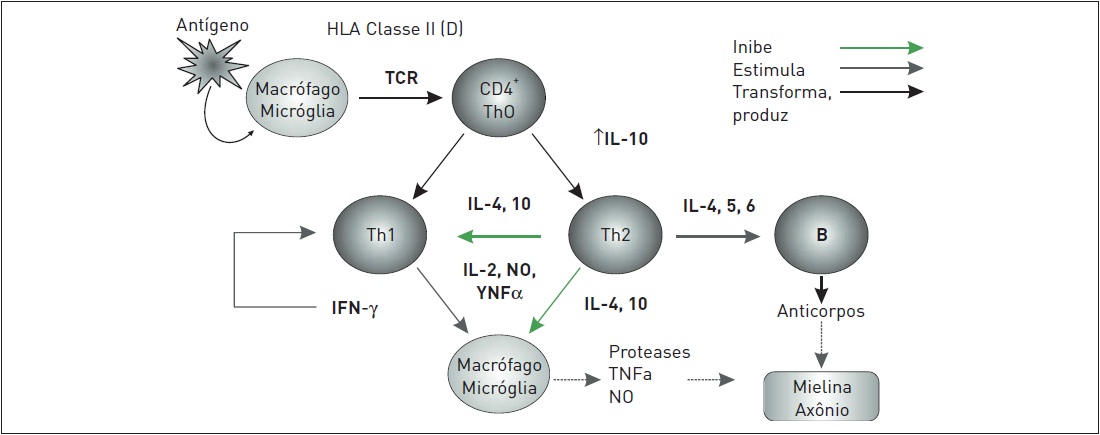
A esclerose múltipla (EM) é uma doença autoimune que causa inflamação, desmielinização, dano axonal, destruição estrutural e atrofia neuronal do sistema nervoso central (SNC). Existe uma predisposição genética para a produção de linfócitos T ativados que expressam antígenos contra as moléculas existentes na bainha de mielina. Esses linfócitos T ativados atravessam a barreira hematencefálica e geram a diferenciação dos linfócitos T CD4 Th0 em T CD4 Th1 e T CD4 Th2.
A ativação e a proliferação das células Th1 desencadeia grande produção de citocinas inflamatórias, resultando em uma sucessão da cascata pró-inflamatória. As células Th2 iniciarão um processo de produção de citocinas anti-inflamatórias, porém em menor intensidade (Fig. 97.1). Dessa forma, esses linfócitos T ativados dentro do SNC, por meio da ativação de mecanismos inflamatórios descritos, irão lesar as células de Schwann, causando desmielinização, atacar a micróglia (oligodendrócitos), bem como causar dano axonal direto aos neurônios desde o início da doença até a sua fase mais tardia. Portanto, a EM trata-se de uma doença multifatorial e complexa que representa o principal distúrbio autoimune do SNC e acarreta um grande prejuízo a indivíduos jovens diagnosticados com essa doença crônica neurodegenerativa.
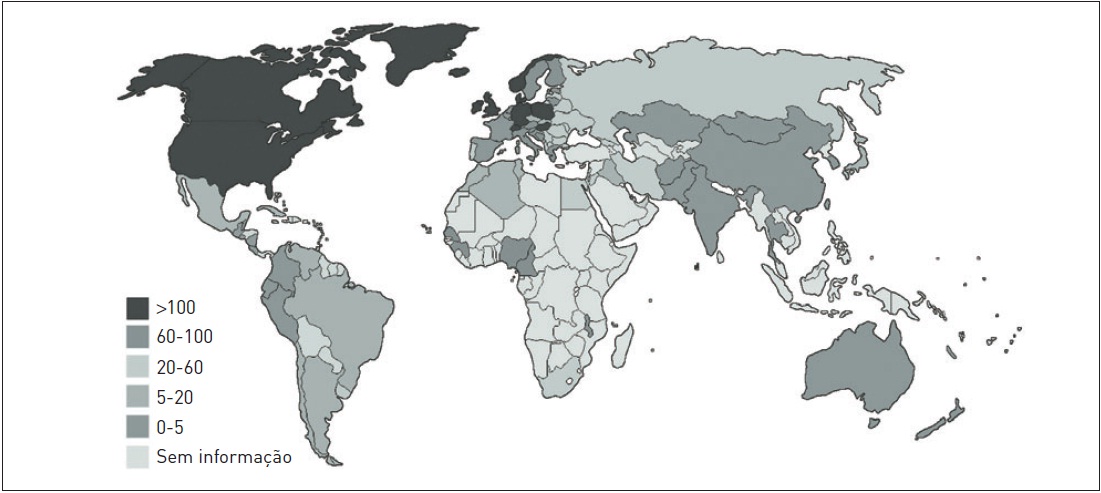
A prevalência da EM varia conforme a localização geográfica, sendo mais frequente nas regiões setentrionais do planeta. Os Estados Unidos, o Canadá e os países da Europa apresentam os maiores índices de ocorrência, conforme consta na Figura 97.2. No Brasil, a prevalência estimada é de 15 a 18/100 mil habitantes. Em relação à raça, os brancos são mais afetados, e existe uma diferença de prevalência entre os sexos, sendo as mulheres mais acometidas (2:1). A EM ocorre com mais frequência em indivíduos na faixa etária de 20 a 40 anos, e parece haver um aumento da incidência em crianças e adolescentes, podendo corresponder a 10% do total de casos de EM. É possível constatar, então, que a EM é uma doença de baixa prevalência, porém de grande impacto socioeconômico, pois compromete adultos jovens.

Figura 97.1
Mecanismo fisiopatológico da ativação dos linfócitos Th0 no sistema nervoso central.
Figura 97.2
Prevalência de esclerose múltipla no mundo (prevalência por 100 mil habitantes).
Nota: 93 países responderam o questionário fornecendo dados para este mapa.
Fonte: Adaptada de World Health Organization.1
A EM não apresenta uma etiologia definida. Essa doença afeta indivíduos com predisposição genética e estímulo ambiental para sua manifestação. Dessa forma, uma resposta autoimune anormal dirigida contra os componentes do SNC evidencia a doença. A teoria de uma mutação genética nos povos escandinavos, foi defendida por Poser, em 1995, como a origem da EM, visto que a Península Escandinava apresenta as taxas mais altas de prevalência da doença e que, de acordo com as migrações populacionais, as regiões do planeta com população mestiça e influências europeias dos vikings vêm identificando loci genéticos similares em suas populações.
Diversos estudos epidemiológicos confirmam que, embora um padrão de hereditariedade não tenha sido observado, existe uma suscetibilidade genética maior, cujo determinante principal está relacionado a alelos do sistema human leukocyte antigen (HLA) no braço curto do cromossomo 6p21-23.
Os estímulos ambientais que estão associados ao surgimento da EM podem ser internos ou externos.
Os estímulos internos são os de origem hormonal. O fato de a doença ser mais prevalente em mulheres está relacionado aos níveis de estrogênio e à expressão desse hormônio na resposta imune nos linfócitos Th1. Além disso, é evidente a acentuação da resposta imunológica no puerpério, outro fator intrínseco que pode auxiliar no desenvolvimento de surtos da doença em indivíduos predispostos.
Quanto aos fatores extrínsecos, os processos infecciosos são apontados como possíveis fatores precipitantes do início da doença. As exposições a agentes virais ou bactérias nas fases iniciais da vida, de forma ainda não compreendida, criariam as condições necessárias para o surgimento de uma resposta crônica autoimune inadequada após alguns anos. Entre esses agentes, pode-se citar herpes simples vírus, HTLV, citomegalovírus, vírus Epstein-Barr, coronavírus, sarampo, caxumba, rubéola, HIV, adenovírus, enterovírus, retrovírus, herpes-zóster, clamídia e borrélia. Entre outros fatores extrínsecos, a exposição ambiental à luz solar, de forma moderada, atuaria como um fator protetor, já que a relação da prevalência da EM é inversamente proporcional à latitude terrestre. Uma explicação para essa teoria é evidenciada por estudos que comprovam o fator protetor da vitamina D em indivíduos com predisposição para a doença, principalmente pela indução de células T regulatórias. Como a principal fonte de vitamina D é proveniente do estímulo da luz solar, esta seria uma teoria plausível. Além disso, a radiação ultravioleta também apresenta efeitos imunossupressores. Portanto, uma combinação de fatores é que determina a deflagração da EM.
O quadro clínico da EM pode ser bastante diversificado. Deve-se atentar, entretanto, para os sintomas que se instalam de forma mais prevalente no início da doença (Tab. 97.1) e para a forma de apresentação, uma vez que isso determina a classificação quanto ao desenvolvimento da doença.
De acordo com a apresentação clínica e o desenvolvimento da doença, pode-se classificar a EM nas quatro formas evolutivas a seguir:
Remitente recorrente: Essa é a forma clínica mais prevalente, correspondendo a 80 a 85% dos casos. Os episódios de exacerbação da doença (surtos) são seguidos por períodos variáveis de estabilização. Nos primeiros surtos, a recuperação neurológica, de um modo geral, é completa. Com a progressão da doença, o paciente pode permanecer com alguma sequela do surto precedente, porém há alguma autonomia funcional nos períodos estáveis. Deve-se salientar os conceitos de surto e remissão (Quadro 97.1), bem como considerar que o intervalo entre os surtos deve ser de 30 dias e que não pode existir progressão dos sintomas entre eles.
Primariamente progressiva: Essa forma de apresentação caracteriza-se pela manifestação de sinais e sintomas neurológicos de forma lenta e progressiva desde o início, com uma progressão mínima de seis meses, sem haver nenhum sinal de remissão. É a classificação menos frequente, correspondendo a 6 a 10% dos casos. Em geral, apresenta-se mais tardiamente, por volta dos 40 anos, e podem ocorrer flutuações clínicas e períodos de estabilização.
Secundariamente progressiva: Essa forma clínica corresponde à progressão da forma remitente recorrente. Assim, quando há piora lenta e gradual entre os surtos, o paciente passa a ser classificado dessa forma. O tempo médio decorrido da forma remitente recorrente para a secundariamente progressiva é de dez anos.
Primariamente progressiva com surtos: Essa é bastante rara. Ocorre evolução lenta e gradual; entretanto há alguns surtos episódicos. Além disso, ressalta-se que alguns pacientes, antes de serem considerados com o diagnóstico de esclerose múltipla clinicamente definida (EMCD), podem ser classificados conforme as seguintes formas clínicas:
Síndrome clínica isolada (clinical isolated syndrome [CIS]): São os casos nos quais o paciente apresenta o primeiro surto de EM, geralmente com resolução espontânea, sem nenhum antecedente prévio de manifestações neurológicas. Os casos mais típicos são de neurite óptica, mielite transversa ou síndromes de tronco ou cerebelar. Entretanto, o paciente não preenche todos os critérios diagnósticos (abordados a seguir). De acordo com diversos aspectos, os casos de CIS podem ser classificados como de alto ou baixo risco para desenvolvimento de EMCD.
Síndrome radiológica isolada (radiological isolated syndrome [RIS]): Esses casos são identificados ao acaso. O paciente que é submetido a um exame de ressonância de encéfalo por algum outro motivo, como traumatismo craniano ou cefaleia, pode evidenciar as imagens características de lesões desmielinizantes, correspondendo aos critérios imagenológicos para EM, sem haver, entretanto, qualquer manifestação clínica. Esses casos devem ser investigados quanto aos possíveis diagnósticos diferenciais e acompanhados neurologicamente.
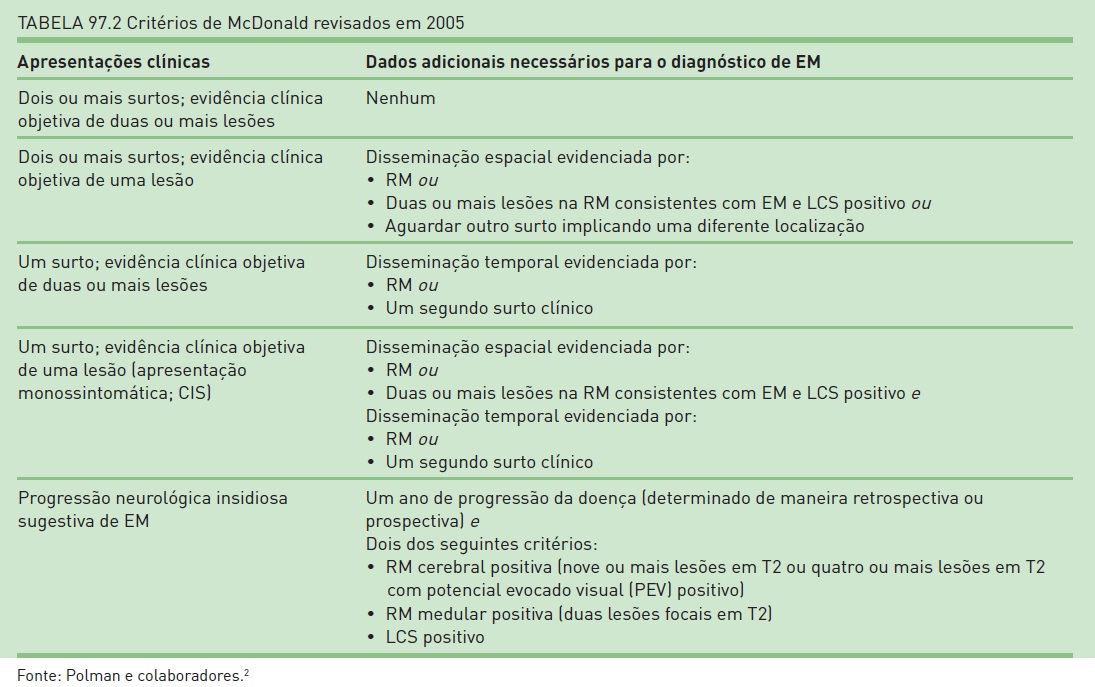
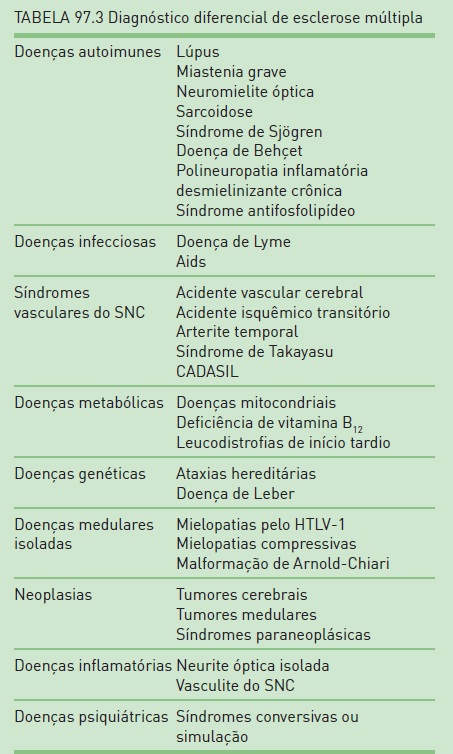
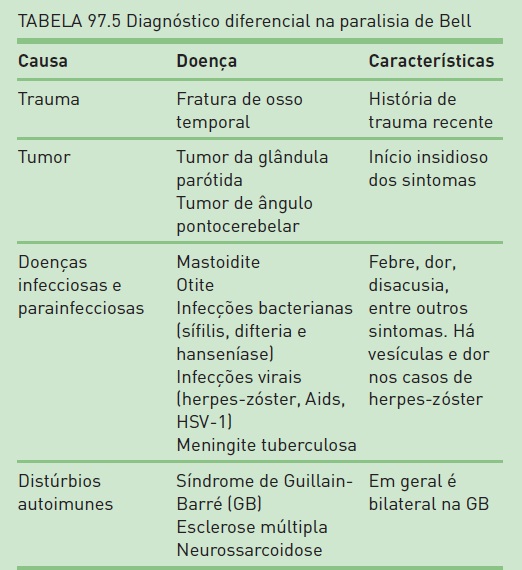
O diagnóstico de EM é baseado em um conjunto de critérios que considera o quadro clínico e os exames complementares (ressonância magnética de encéfalo, líquido cerebrospinal (LCS) e potenciais evocados) para a determinação do diagnóstico – critérios de McDonald, publicados em 2001 e que foram revisados em 20052 (Tab. 97.2). Portanto, de acordo com a combinação dos dados clínicos do paciente e os resultados dos exames paraclínicos, pode-se concluir o diagnóstico ou não. Logo, não há um marcador biológico para o diagnóstico de EM, e o achado de RM de encéfalo apenas não é suficiente para essa determinação (RIS). Além disso, é muito importante que seja realizado o diagnóstico diferencial, uma vez que este é amplo e que, devido a heterogeneidade de apresentações clínicas, grande parte dos pacientes consulta com, pelo menos, outros três médicos antes de saber o diagnóstico correto (Tab. 97.3).
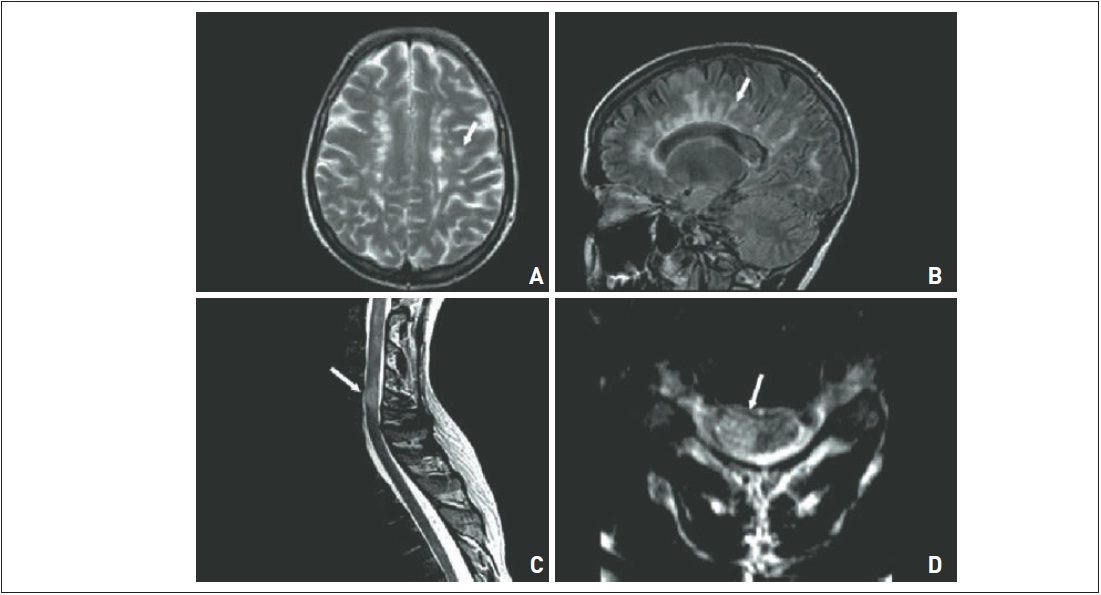
Figura 97.3
Imagens de encéfalo e medula espinal em T2, nos planos axial (A e D) e sagital (B e C). Lesões hiperintensas ovaladas, distribuídas ao longo do corpo caloso (caloso-septal) e de localização periventricular – dedos de Dawson (B). Lesão medular no segmento cervical (C e D).
O acompanhamento do paciente é fundamental na determinação do diagnóstico em casos de suspeita, pois o período de desenvolvimento da doença e a disseminação das lesões cerebrais são fatos incontestáveis. A RM de encéfalo auxilia bastante e com melhor sensibilidade a identificar as lesões da EM no SNC. Com o aumento do poder dos campos de intensidade em telas dos aparelhos de RM, bem como o primoramento de novas técnicas de aquisição das imagens, é cada vez mais evidente e precoce a identificação das lesões, determinando a idade destas e seu substrato histopatológico.
As características típicas da doença são lesões em hipersinal nas sequências ponderadas em T2 e FLAIR e hiposinal em T1. As placas de desmielinização de EM têm uma preponderância pela região periventricular e se apresentam com aspecto ovalado e perpendicular aos ventrículos laterais e ao maior eixo do corpo caloso, configurando um formato digitiforme nos cortes sagitais, denominados dedos de Dawson (Fig. 97.3). As sequências apontadas como mais sensíveis para sua avaliação são aquelas com pulso adicional de transferência de magnetização (MTC), tanto pré como pós-gadolínio, que são fundamentais para a avaliação de qualquer paciente com EM.
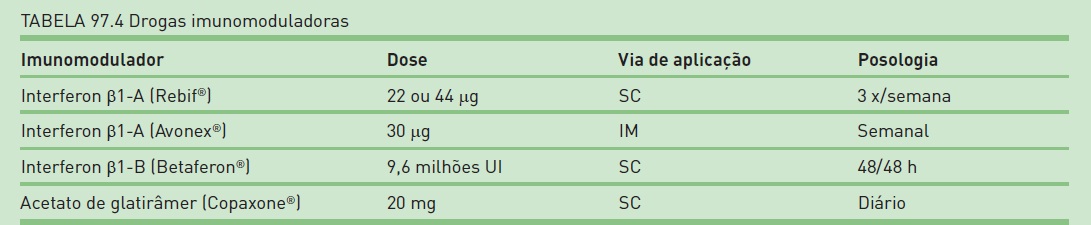
O tratamento de pacientes com EM é focado no controle dos surtos e da progressão da doença. O tratamento dos surtos sempre deve ser instituído com corticosteroides em altas doses, via intravenosa, sendo a pulsoterapia com metilprednisolona a forma mais padronizada em todo o mundo. Quando o objetivo é o tratamento da doença, as drogas imunomoduladoras são a primeira linha de medicações atualmente indicadas. Essas medicações (Tab. 97.4) são indicadas para o tratamento das formas remitente recorrente e dos casos de CIS. Para casos de forma primária progressiva, não há medicação que seja indicada como padrão-ouro para tratamento. Com o desenvolvimento da doença, as drogas imunossupressoras são indicadas, como o mitoxantrona e a ciclofosfamida. Os anticorpos monoclonais apresentam excelentes resultados nos ensaios clínicos para tratamento da EM remitente recorrente que falharam com as medicações de primeira linha.
Entre esses, o natalizumabe já é aprovado em quase todo o mundo, e seu índice de controle de surtos anual é superior a 60%. No momento da publicação deste livro, surge a possibilidade de terapias orais, como fingolimode, teriflunomida, laquinimode, ácido fumárico e cladribina. O transplante autólogo de células-tronco ainda está em análise por um grupo de pesquisa mundial e apresenta a perspectiva de bons resultados no controle e na modificação da história natural da doença. A escolha terapêutica deve ser realizada pelo neurologista com experiência em EM e embasada na progressão da doença do paciente, na forma clínica, no prognóstico e nas demais características particulares de cada indivíduo, bem como no perfil de segurança e na eficácia de cada medicação.
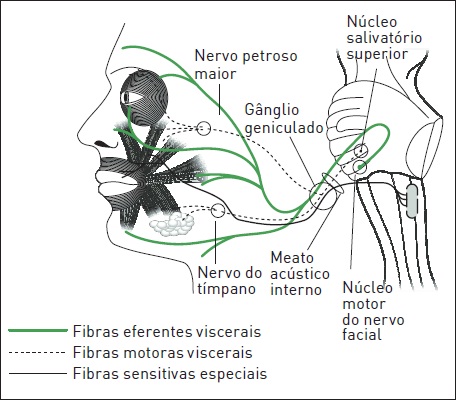
A paralisia facial periférica (PFP) idiopática, também denominada paralisia de Bell (PB), é uma afecção aguda e idiopática que envolve o nervo facial perifericamente. O nervo facial é responsável pela inervação motora dos músculos da face. Além disso, presenta fibras parassimpáticas para as glândulas lacrimais, parótidas, submandibulares e sublinguais, bem como fibras sensitivas para os dois terços anteriores da língua (Fig. 97.4). A PB foi observada, em 1821, por Charles Bell e é ocasionada por uma inflamação do VII nervo craniano em qualquer porção de seu trajeto, desde seu núcleo pontino até as ramificações distais. Em geral, o gânglio geniculado é o sítio mais frequente de lesão.
Não se sabe sobre a etiologia da PB. A inflamação do nervo resulta em edema e compressão do nervo facial no interior de seu canal no osso temporal. A infecção pelo herpes simples tipo 1 (HSV-1) é correlacionada como uma possível causa para a PB, sendo estimado um percentual de 60 a 70% dos casos com essa infecção viral concomitante. Entretanto, estudos não conseguiram isolar o DNA viral em biópsias do nervo, colocando em dúvida essa causalidade.
Figura 97.4
Anatomia do nervo facial.
A PB não ocorre com mais frequência em indivíduos de determinado sexo e nem em algum lado específico da face. Essa doença apresenta incidência anual de 15 a 30/100 mil indivíduos e pode afetar qualquer faixa etária, sendo seu pico de incidência entre 30 e 40 anos. É mais prevalente em gestantes. Em pessoas com doenças crônicas, tais como hipertensão e diabetes melito, há mais risco de ocorrência de PFP idiopática. A PB é responsável por 70% dos casos de PFP. A taxa de risco de recorrência é de cerca de 8% em quase todos os estudos.
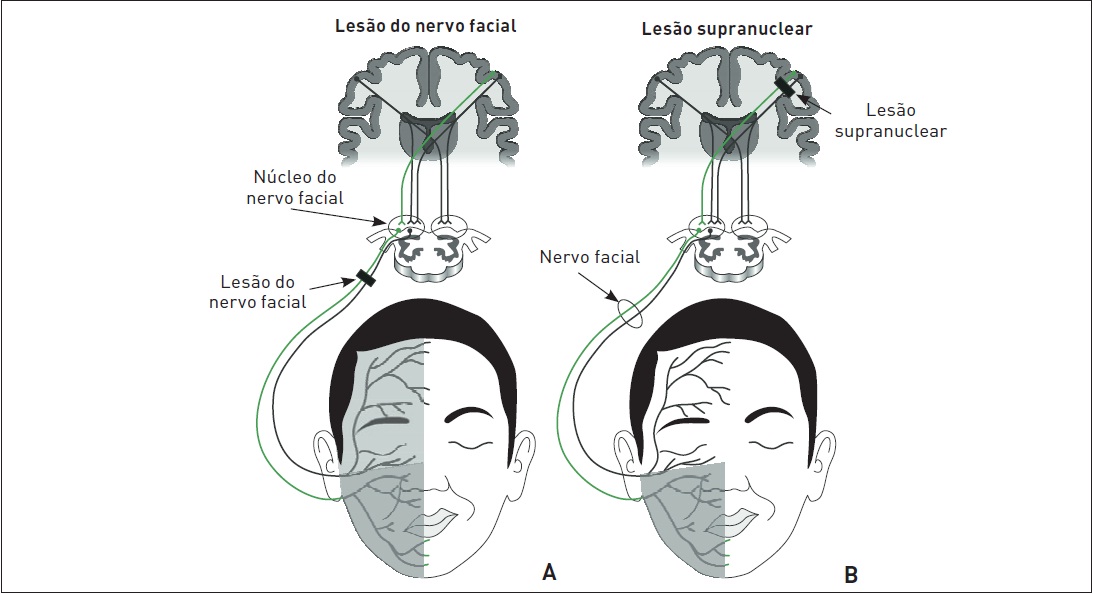
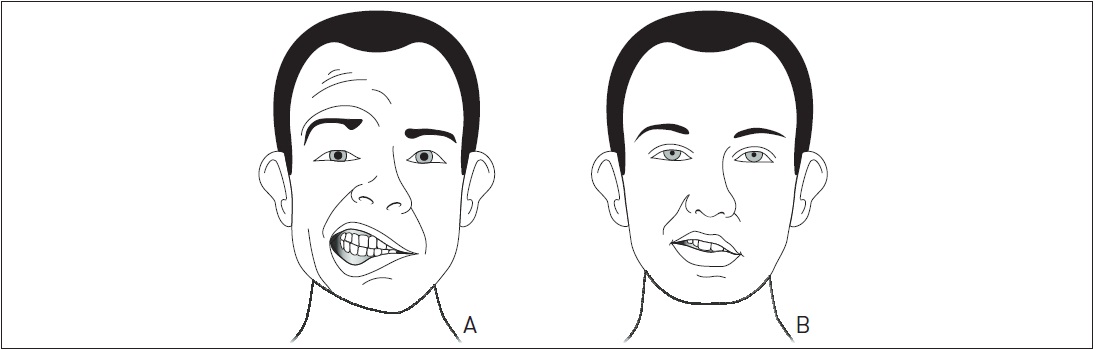
É importante diferenciar a PFP da paralisia facial central. Classicamente, a PFP acomete toda a hemiface, enquanto a paralisia facial central não afeta a metade superior da face. Isso ocorre devido à dupla inervação do andar superior da hemiface, pois há fibras corticonucleares provenientes de ambas as metades do córtex que se dirigem para cada um dos núcleos do nervo facial (Figs. 97.5 A e B e 97.6 A e B).
Figura 97.5
Lesão do nervo facial periférico (A) e lesão supranuclear (B).
Os pacientes com PB tipicamente relatam fraqueza ou paralisia completa de todos os músculos de um lado da face (Fig. 97.6 A e B). As marcas de expressão facial e o sulco nasolabial desaparecem. Na testagem da mímica facial, há alterações de força em uma hemiface, sendo mais evidente o desvio da rima labial para o lado contralateral ao lado paralisado. Ao tentar assobiar, os pacientes não conseguem devido à perda de ação dos músculos bucinador e orbicular dos lábios. A pálpebra não fecha, pois o músculo orbicular não age. Com isso, ocorre um ressecamento ocular pela falta de lubrificação do globo ocular e pela diminuição da produção de lágrimas. Pode-se observar o sinal de Bell, que se caracteriza pelo deslocamento do globo ocular para cima e para fora na tentativa de fechamento com força do olho. Muitos pacientes relatam uma alteração sensitiva na hemiface comprometida, porém a sensibilidade é preservada na PB. Dor leve a moderada no ângulo da mandíbula é descrita em alguns casos. Hiperacusia ocorre devido à paralisia do músculo estapédio (inervado por um ramo do nervo facial).
Os sinais e os sintomas da PB apresentam-se de forma aguda, e essa condição pode piorar em três dias. A recuperação pode ocorrer entre três semanas e alguns meses. Cerca de 80% dos pacientes restabelecem completamente a expressão da mímica facial. Existem alguns fatores de mau prognóstico para a recuperação da PB, descritos no Quadro 97.2.
Figura 97.6
Paralisia facial periférica (A) e paralisia facial central (B).
Fonte: Adaptada de Patten.3
O diagnóstico de pacientes com PB é basicamente clínico. Devido à apresentação dos sintomas e à paralisia periférica do VII nervo craniano unilateral no exame neurológico, pode-se determinar a PFP com todos os dados anteriormente citados. Porém, é muito importante ressaltar que não devem existir indícios de outras alterações no exame clínico e no neurológico. Deve-se atentar para o exame dos demais nervos cranianos, a realização de otoscopia e a palpação das parótidas que, se evidenciarem alterações, podem indicar outras causas para a PFP. Os exames complementares devem ser realizados conforme cada paciente. De uma maneira geral, recomenda-se a solicitação de exames laboratoriais – hemograma completo, velocidade de sedimentação globular, glicemia, provas de função tireoidiana, VDRL, FAN, sorologias para HIV, hepatites e outros que forem oportunos. A coleta de líquido cerebrospinal é restrita aos casos dos quais se suspeita de quadros imunológicos, tumores ou tuberculose. Exames de imagem são indicados quando houver a necessidade de descartar outros diagnósticos diferenciais, como tumores e doenças desmielinizantes. O estudo de neurocondução motora do facial, realizado após sete dias do início do quadro, pode estimar o grau de perda axonal e auxiliar o prognóstico. Os possíveis diagnósticos diferenciais estão citados na Tabela 97.5.
O uso de corticosteroides, via oral, é comumente considerado para o tratamento de pacientes com PB. A prednisona, 60 mg ao dia, tem sido indicada pelo período de 10 dias, com redução gradual da dose. Esse tratamento deve ser instituído quando tiverem passado 72 horas do início dos sintomas, não havendo indicação após esse período. Deve-se indicar que o paciente utilize colírio lubrificante ocular a fim de evitar o ressecamento do globo ocular e proteger a córnea, com posologia de 1 a 2 gotas no globo ocular afetado e frequência de 2 em 2 horas. Pomadas para proteção ocular devem ser indicadas para dormir.
Após a aplicação da pomada, deve-se colocar um tampão ocular no olho afetado.
O uso de drogas antivirais era indicado para todos os casos, porém um estudo recente publicado na Lancet Neurology – de Engström e colaboradores4 – apontou evidências clinicamente significativas de que não há benefício do uso de medicações antivirais associado ao de corticosteroides. Esse estudo randomizado, multicêntrico, duplo-cego, controlado por placebo, alocou 839 pacientes e observou um benefício do uso de prednisolona em relação ao placebo nos índices de recuperação dos sintomas e brevidade da recuperação. Esses benefícios não foram observados com o uso de valaciclovir associado à prednisolona, nem em uso isolado.
Em casos de suspeita de infecção por herpes-zóster associado, está indicado o uso de aciclovir ou valaciclovir durante sete dias. Ressalta-se que a PB é uma doença benigna, apresentando um índice de recuperação espontânea de 57 a 85%. Porém, o uso de corticoterapia reduz os riscos de sequelas permanentes e reduz o período de recuperação dos sintomas, principalmente nos casos de mau prognóstico (Tab. 97.5). Nos casos em que não houver melhora após duas semanas de tratamento, os pacientes devem ser encaminhados para um especialista a fim de que estes avaliem outras causas. Indivíduos que apresentarem quadros de lesões de córnea devem ser indicados para avaliação oftalmológica. Não há provas de um benefício significativo do tratamento fisioterápico nos casos de paralisia de Bell. Para a confirmação da possibilidade de que os exercícios faciais reduzam o período de recuperação e as sequelas, serão necessários ensaios clínicos randomizados.
A arterite de células gigantes (ACG) – ou arterite temporal – é uma vasculite granulomatosa crônica, imunomediada, descrita, pela primeira vez, por Horton e colaboradores, 5 em 1937. É uma doença rara, sendo a cefaleia e a perda visual os sintomas mais conhecidos; entretanto pode se manifestar de forma atípica. O diagnóstico precoce da doença é fundamental para que o comprometimento visual seja evitado.
A ACG afeta predominantemente brancos, sendo muito infrequente em asiáticos e negros. Ela ocorre mais comumente em indivíduos com mais de 50 anos, sendo as mulheres afetadas com prevalência duas a seis vezes superior à dos homens. A incidência anual na população com mais de 50 anos, na Dinamarca, foi de 76/100.000 habitantes e de 30/100.000 em Olmsted (Minesota, Estados Unidos). A incidência média na população acima de 50 anos é de 20/100.000 indivíduos, com um pico entre os 70 e os 80 anos de idade.
A causa dessa doença é um processo inflamatório vascular. Uma inflamação granulomatosa ocorre nos tecidos envolvidos, principalmente em vasos de médio e grande calibre. Existe uma grande associação entre essa doença e a polimialgia reumática, sendo a apresentação concomitante das duas doenças muito frequente. Entretanto, ainda não se sabe sobre as causas para essa comorbidade. Outra possível etiologia envolvida é a associação com infecções. Algumas séries de casos evidenciaram a associação com epidemias de Mycoplasma pneumoniae, parvovírus B19 e Chlamydia pneumoniae. Porém, necessitam de mais dados para serem confirmadas.
A fisiopatologia da ACG está relacionada à entrada de células T na vasa vasorum pelo reconhecimento de um antígeno desconhecido. As células T CD4 ativadas ocasionam a diferenciação e a migração de macrófagos, resultando na formação de granulomas e células gigantes. Os macrófagos produzem citocinas inflamatórias específicas, dependendo em que camada do vaso sanguíneo estão situados: IL-1 e IL-6 na adventícia; metaloproteinases na camada média; e óxido nítrico sintetase-2 na íntima. Os mecanismos destrutivos na parede arterial estão relacionados a mecanismos de reparo devido à secreção de fatores angiogênicos e de crescimento. A última etapa de degradação é a destruição da camada elástica interna e hiperplasia, com oclusão da luz luminal, características típicas das lâminas histológicas.
A cefaleia é o sinal de alerta dessa doença. A dor inicia de forma aguda, com forte intensidade, muitas vezes latejante, uni ou bilateral, de localização frontotemporal na maioria dos casos. Pode ser contínua e incapacitante, não responsiva aos analgésicos comuns, e acabar interferindo nas atividades de vida diária. O sintoma inicial mais sério é a perda visual, que pode ocorrer em um ou ambos os olhos, decorrente de neuropatia óptica isquêmica. As manifestações visuais ocorrem em 25 a 50% dos casos de ACG. Os pacientes relatam a ocorrência de uma sombra que encobre um dos olhos, que pode progredir para amaurose. Dessa forma, essa perda visual pode ser permanente se o paciente não for submetido a tratamento precoce. Se o primeiro dos olhos comprometido não é tratado imediatamente, o segundo olho é afetado em uma ou duas semanas. Essa perda visual ocorre devido ao envolvimento de ramos das artérias oftálmica, ciliar posterior e de arteríolas retinianas. Há amaurose fugaz em 44% dos casos antes da perda visual, sendo um importante sinal de alerta.
Outras complicações do SNC são menos prevalentes, mas devem ser citadas: diplopia, crises convulsivas e neuropatias periféricas. Sintomas sistêmicos, como febre, mal-estar, anorexia e perda de peso, em geral se manifestam em 50% dos casos.
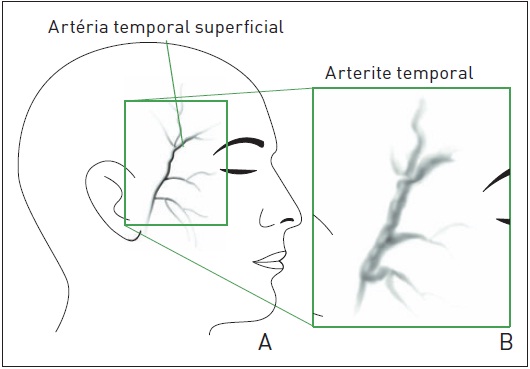
O diagnóstico é realizado por meio de história clínica e exame clínico e neurológico. Neste, pode ser verificada uma artéria temporal espessada, hiperemiada, tortuosa e dolorosa à palpação (Fig. 97.7). O hemograma pode apresentar leucocitose e neutrofilia. A velocidade de hemossedimentação (VHS) está acima de 100 mm em metade dos casos, e o resultado da proteína C-reativa é sempre positivo. O diagnóstico confirmatório é obtido pela biópsia da artéria temporal média, considerada padrão-ouro. O aspecto histológico clássico é um processo inflamatório granulomatoso com células gigantes localizadas entre a junção das camadas íntima e média. A ultrassonografia com Doppler a cores vem sendo estudada como um método auxiliar para o diagnóstico, apresentando grande sensibilidade e especificidade.
Altas doses de corticoterapia devem ser iniciadas no momento em que se suspeita do diagnóstico de ACG. A decisão de tratar não deve ser postergada, pois isso aumenta o risco de perda visual permanente. A biópsia pode ser realizada após administração de corticoide. A dose recomendada é de 1 mg/kg de prednisona ao dia, com uma redução gradual até a dose de manutenção de 10 mg/dia, por 18 a 24 meses.
A pulsoterapia com metilprednisolona pode ser realizada em pacientes que apresentam queixas visuais – 1.000 mg ao dia, durante três dias.
A resposta clínica é a forma mais evidente de monitorar o tratamento, mas a redução dos níveis de VHS também pode ser acompanhada. Agravamentos ocorrem em 30 a 50% dos pacientes, principalmente nos que não realizam um longo curso de uso do corticosteroide.
Figura 97.7
Artéria temporal espessada.
O quadro clínico da paciente é típico de EM remitente recorrente, pois ela apresentou um primeiro surto com remissão completa e espontânea da neurite óptica e posteriormente desenvolveu um novo sintoma neurológico. A observação do período de desenvolvimento da doença, bem como a progressão das lesões encefálicas, é fundamental para a confirmação diagnóstica dessa doença. O acompanhamento e o tratamento, bem como o correto manejo dos surtos, são importantes para a manutenção da qualidade de vida e o controle da progressão da doença.
Assim, a paciente iniciou tratamento imunomodulador e tratamento em Centro de Referência em Tratamento e Pesquisa de EM em sua cidade.
1. World Health Organization [Internet]. Geneva: WHO; c2012 [capturado em 10 set. 2012]. Disponível em: http://www.who.int/en/.
2. Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the McDonald criteria. Ann Neurol. 2005;58(6):840-6.
3. Patten J. Diagnóstico diferencial em neurologia. 2. ed. Rio de Janeiro: Revinter; 2000.
4. Engström M, Berg T, Stjernquist-Desatnik A, Axelsson S, Pitkäranta A, Hultcrantz M, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
5. Horton BT, Magath BT, Brown GE. Arteritis of temporal vessels: report of 7 cases. Proc Staff Meet Mayo Clin. 1937;12:548-53.
Adoni T. Paralisia facial periférica [Internet]. Porto Alegre: MedicinaNET; 2008 [capturado em 15 set. 2012]. Disponível em: http://www.medicinanet.com.br/conteudos/revisoes/1191/paralisia_facial_periferica.
htm.
Azhar SS, Tang RA, Dorotheo EU. Giant cell arteritis: diagnosing and treating inflammatory disease in older adults. Geriatrics. 2005;60(8):26-30.
Cantini F, Niccoli L, Nannini C, Bertoni M, Salvarani C. Diagnosis and treatment of giant cell arteritis. Drugs Aging. 2008;25(4):281-97.
Compston A, Confavreux C, Lassman H, McDonald I, Miller D, Noseworthy J, et al., editors. McAlpine’s multiple sclerosis. 4th ed. Philadelphia: Churchill Livingstone; 2006.
de Araújo Medeiros D, De Miguel E. Doppler color echocardiography in the diagnosis of giant cell arteritis. Acta Reumatol Port. 2009;34(2A):183-9.
International Multiple Sclerosis Genetics Consortium; Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357(9):851-62.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938-52.
Palmer AM. Pharmacotherapy for multiple sclerosis: progress and prospects. Curr Opin Investig Drugs. 2009;10(5):407-17.
Poser C. The dissemination of multiple sclerosis: a Viking saga? A historical essay. Ann Neurol. 1994;36 Suppl 2:S231-43.
Río-Izquierdo J, Montalban X. Natalizumab in multiple sclerosis. Rev Neurol. 2009;49(5):265-9.
Rivera VM. Multiple sclerosis: definitions, epidemiology, genetics and natural history. Barcelona: Viguera; 2009.
Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatic and giantcell arteritis. Lancet. 2008;372(9634):234-45.
Salvarani C, Crowson CS, O’Fallon WM, Hunder GG, Gabriel SE. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum. 2004;51(2):264-8.
Sellebjerg F, Barnes D, Filippini G, Midgard R, Montalban X, Rieckmann P, et al. EFNS guideline on treatment of multiple sclerosis relapses: report of an EFNS task force on treatment of multiple sclerosis relapses. Eur J Neurol. 2005;12(12):939-46.
Teixeira LJ, Soares BG, Vieira VP, Prado GF. Physical therapy for Bell s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev. 2008;(3):CD006283.
Tiemstra JD, Khatkhate N. Bell’s palsy: diagnosis and management. Am Fam Physician. 2007;76(7):997-1002.
Tintoré M, Arrambide G. Early onset multiple sclerosis: the role of gender. J Neurol Sci. 2009;286(1-2):31-4.
Tyler KL. Prednisolone -but not antiviral drugs- improves outcome in patients with Bell’s palsy. Nat Clin Pract Neurol. 2009;5(2):74-5.
Valença MM, Valença LP, Lima MC. Idiopathic facial paralysis (Bell’s palsy): a study of 180 patients. Arq Neuropsiquiatr. 2001;59(3-B):733-9.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.