(Carregando Índice)... (Carregando Índice)... |
Autores:
Marcelo Basso Gazzana
Médico pneumologista, internista e intensivista. Médico do Serviço de Pneumologia do HCPA e do CTI Adulto do Hospital Moinhos de Vento. Mestre em Ciências Pneumológicas pela UFRGS.
Sérgio Saldanha Menna-Barreto
Médico pneumologista. Professor titular do Departamento de Medicina Interna da Faculdade de Medicina da UFRGS. Mestre em Pneumologia pela UFRGS. Doutor em
Cardiologia e Ciências Cardiovasculares pela UFRGS. Pós-doutor em Pneumologia pela Universidade de Toronto, Canadá.
Última revisão: 26/06/2014
Comentários de assinantes: 0
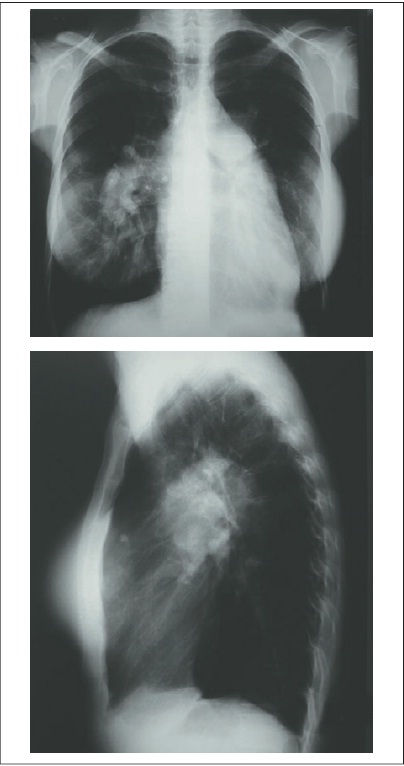
Uma paciente do sexo feminino, 39 anos, casada, tem dois filhos adolescentes, é desportista e apresenta dispneia rapidamente progressiva, estabilizando aos pequenos esforços, com início há cerca de dois anos. Ela relata que, há um ano, teve piora aguda, com internação em unidade de terapia intensiva devido a pneumonia necrosante por bacilos gram-negativos sem identificação da espécie em cultura. Ela tem alta e novamente apresenta quadro de dispneia estável aos médios e pequenos esforços. Não há antecedentes crônicos. Afirma ter realizado cirurgia plástica mamária há cinco anos. Ao realizar exame físico, verificam-se leve cianose central, congestão jugular, frequência respiratória de 20 rpm, pulmões limpos, ausculta cardíaca de 104 bpm e pressão arterial de 112/90 mmHg. A saturação de oxigênio é de 75% em ar ambiente. O eletrocardiograma evidencia ritmo sinusal, sobrecarga de cavidades direitas e bloqueio de ramo direito. A partir de radiografia de tórax, são observados grande aumento da área cardíaca àscustas de cavidades direitas e dilatação dos ramos centrais das artérias pulmonares (Fig. 108.1).
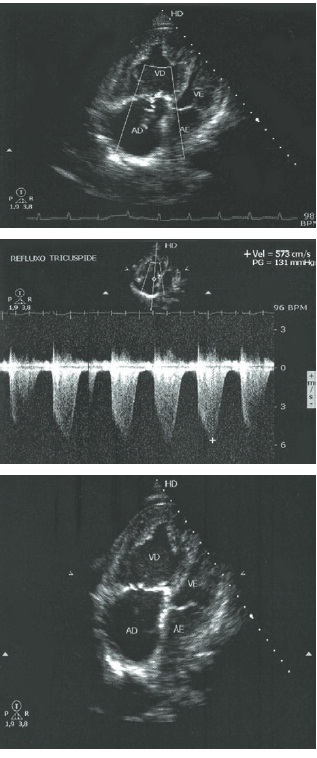
No ecocardiograma Doppler com fluxo a cores, há grande defeito do septo atrial, do tipo seio venoso, com fluxo bidirecional e retorno venoso anômalo: três veias pulmonares direitas drenam no átrio direito e uma veia pulmonar esquerda no átrio esquerdo. A razão entre os fluxos pulmonar e sistêmico (Qp/Qs) é de 3,8.
Diâmetro diastólico de ventrículo direito (VD) é de 4,0 cm, velocidade de regurgitação tricúspide (VRT) de 6,1 m/s, o gradiente transcúspide (GTT) de 146 mmHg, aparelho pulmonar (AP) de 5,4 cm – fluxo lento e formação de contraste espontâneo, veia cava inferior (VCI) dilatada – colapso inspiratório, pressão sistólica de artéria pulmonar (PSAP) de 157 – 160 mmHg. Derrame pericárdico de leve a moderado (Fig. 108.2).
A hipertensão pulmonar (HP) é a elevação persistente da pressão vascular pulmonar, que pode ser causada por aumento isolado do segmento arterial ou por aumento nas pressões dos segmentos venosos e arteriais da circulação pulmonar. A HP não é uma doença específica, mas uma condição fisiopatológica que se manifesta de forma clínica, multidisciplinar e complexa. A classificação da Organização Mundial de Saúde (OMS), revisada em 2008, evidencia a extensão da HP (Quadro 108.1).
A hipertensão arterial pulmonar (HAP) é essencialmente uma condição hemodinâmica caracterizada pelos seguintes parâmetros: pressão arterial pulmonar média maior do que 25 mmHg em repouso, pressão arterial pulmonar em cunha (considerada como pressão capilar) ou pressão do átrio esquerdo, ou pressão diastólica final de ventrículo esquerdo igual ou menor do que 15 mmHg, resistência vascular pulmonar maior do que 3 unidades Wood (> 240 dinas por segundo por cm-5).1
A HP é de ocorrência relativamente frequente, devido à alta prevalência de cardiopatias e pneumopatias, as quais podem cursar com elevação das pressões vasculares pulmonares por mecanismos geralmente identificados. A HAP, entretanto, constitui-se em um grupo de doenças bem menos frequentes, apresentando características fisiopatológicas, histopatológicas e clínicas similares. O principal desafio na abordagem clínica de pacientes com HP é a caracterização diagnóstica e o tratamento da HAP.

Figura 108.1
Radiograma de tórax do paciente mencionado no caso clínico.
A HP é uma anormalidade fisiopatológica que pode complicar várias doenças cardiopulmonares, como insuficiência ventricular e valvopatias esquerdas, doença pulmonar obstrutiva crônica, doenças pulmonares difusas e doença tromboembólica, bem como ser decorrente de outras doenças que possam interferir sobre a função e a estrutura dos vasos pulmonares. Como pode ser observado pela classificação, há cerca de 40 condições clínicas com HP, classificadas em cinco grupos conforme particularidades patológicas, fisiopatológicas, clínicas e terapêuticas.
Não se sabe sobre os dados epidemiológicos populacionais comparativos acerca da prevalência das formas de HP. Por aproximação, um levantamento realizado em 4.579 pacientes examinados em laboratório de ecocardiografia apontou prevalência de HP, definida como pressão sistólica do ventrículo direito/artéria pulmonar maior do que 40 mmHg, de 10,5%. Entre os 483 pacientes com HP, a confirmação diagnóstica evidenciou que 78,7% apresentaram doença do coração esquerdo (grupo 2); 9,7%, pneumopatia ou hipoxemia (grupo 3); 4,2%, HAP (grupo 1); 0,6 %, HPTC (grupo 4); em 6,8% não foi possível definir o diagnóstico.3
O registro do National Institute of Health dos Estados Unidos da América (EUA), cobrindo 32 centros, entre 1/07/1981 e 30/09/1985, proporcionou uma estimativa de incidência de HAP primária (hoje, classificada como idiopática [HAPI]) de 1 a 2 casos por milhão de indivíduos.4 Um registro norte-americano de 578 pacientes adultos (77% mulheres) com HAP, apresentou as seguintes taxas em subgrupos: 48% de HAPI, 4% de HAP hereditária (familial), associadas a doença difusa do tecido conectivo em 30% dos casos, cardiopatia congênita em 11% (70% com defeito atrial septal), hipertensão portal em 7%, anorexígenos em 3% e Aids em 1%.5
O registro nacional francês apontou incidência da HAP, entre os anos de 2002 e 2003, de 2,4 casos por milhão de habitantes adultos por ano, com prevalência anual de 15 casos por milhão da habitantes adultos (30 por milhão na área de Paris), incluindo casos idiopáticos e de etiologia ou associação reconhecidas. Em 674 casos de HAP, as proporções de subtipos foram as seguintes: 39,2% de HAPI, 3,9% de HAP hereditária (familial), 15,3% de doença difusa do tecido conectivo, 11,3% de cardiopatia congênita, 10,4% de hipertensão portal, 9,5% de anorexígenos e 6,2% de aids.6
O registro escocês de morbidades estimou incidência de 7,1 casos por milhão, com prevalência de 52 casos por milhão de indivíduos. Ainda na Escócia, a Scottish Pulmonary Vascular Unit de Glasgow, estudando HAP, apontou, entre 1997 e 2005, incidência de 7,6 casos por milhão e prevalência de 26 casos por milhão de indivíduos.7
Um estudo retrospectivo realizado no Reino Unido, em sete anos de observação, identificou 64 crianças com HAPI, estimando incidência de 0,48 casos por milhão de crianças por ano e prevalência de 2,1 casos por milhão.8
Figura 108.2
Ecocardiograma Doppler com fluxo a cores do paciente mencionado no caso clínico.
A HAP é uma vasculopatia arterial proliferativa.Admite-se que os seus mecanismos patogênicos – tendo como padrão a HAPI – originam-se de disfunção endotelial, que seria decorrente de uma suscetibilidade individual a algum fator ou fatores desencadeadores. Haveria um genótipo permissivo, um fenótipo suscetível e um disparo exógeno. A disfunção endotelial seguiria múltiplas vias moleculares, algumas já reconhecidas, como prostaciclinas, óxido nítrico, endotelina-1, serotonina, produzindo um desequilíbrio entre ações vasoconstritoras e vasodilatadoras e entre apoptose e proliferação. Isso determinaria vasoconstrição arterial e remodelamento da parede vascular, com hipertrofia da camada média muscular, proliferação e fibrose da camada íntima, proliferação da camada adventícia e trombose in situ, com ou sem lesão plexogênica.1,9
A pressão média da artéria pulmonar (PMAP) é determinada por resistência vascular pulmonar (RVP), débito cardíaco (DC) e pressão venocapilar (˜ pressão do átrio esquerdo).9 A elevação de qualquer um desses parâmetros pode produzir aumento da PMAP, sendo o aumento da pressão venocapilar por cardiopatia esquerda (incluindo valvulares) a forma mais frequente de elevação da PMAP. Os valores normais da PMAP em adultos, ao nível do mar, é de até 20 mmHg.9.10 Entre 21 e 24 mmHg, os valores são considerados supranormais e limítrofes para HP. O plano de corte de 25 mmHg para HP é um valor empírico e prevalente entre os pesquisadores.1,9-11
A alteração hemodinâmica básica da HAP é o aumento progressivo da RVP decorrente da arteriopatia e arteriolopatia proliferativas, o que causa evolutivamente elevação da pressão arterial pulmonar e sobrecarga ao ventrículo direito (VD). Essa sobrecarga pode desencadear hipertrofia do VD para compensar o aumento de sua pós-carga,prejudicar seu relaxamento e enchimento, com consequente redução da complacência ventricular e disfunção diastólica de VD, e também resultar em interferência na circulação coronariana. Com essa sobrecarga de pressão à direita, o septo interventricular desvia-se à esquerda e contribui para a limitação de enchimento e do débito do ventrículo esquerdo (interdependência ventricular). A redução do débito cardíaco diminui a pressão aórtica e a circulação pelas artérias coronárias, com isquemia relativa. A pressão capilar pulmonar permanece normal até ocorrer a disfunção tardia do ventrículo esquerdo. Então, o mecanismo hemodinâmico inicial da HAP é o aumento da RVP, e o final é a insuficiência ventricular direita e a falência circulatória.1,9,11
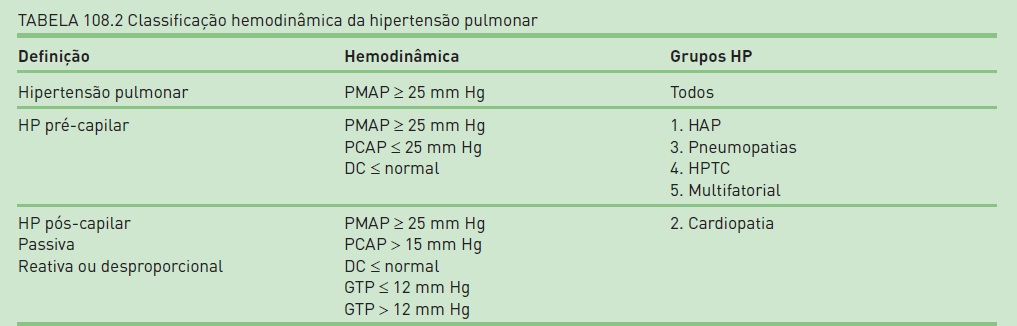
Para os pacientes com PMAP igual ou superior a 25 mmHg, com débito cardíaco normal ou reduzido, deve-se realizar a classificação hemodinâmica com base na pressão venocapilar pulmonar.
Em casos de HAP, a pressão venocapilar é igual ou menor do que 15 mmHg (HP pré-capilar), enquanto nos de HP venosa, a pressão venocapilar é maior do que 15 mmHg (HP pós-capilar). Dessa forma, essa classificação hemodinâmica da HP, imprescindível para o entendimento e o manejo das situações clínicas, baseia-se na relação entre a PMAP e a pressão venocapilar. A HP pré-capilar corresponde aos grupos 1, 3, 4 e 5 da classificação clínica; já a HP pós-capilar, ao grupo 2, correspondente a cardiopatias esquerdas.11,12
Nos pacientes com HP pré-capilar, o gradiente transpulmonar (GTP= PMAP – pressão venocapilar) está elevado, (> 12 mmHg); nos com HP pós-capilar, o GTP é normal (= 12 mmHg), pois a elevação da PMAP é passiva e proporcional à elevação do segmento venocapilar (pressão jusante). Em alguns casos, pode haver ativação de mecanismos reacionais e resultar em remodelamento arterial/arteriolar, com elevação desproporcional da RVP e da PMAP e com aumento do GTP além do aumento primário da pressão venocapilar. A HP considerada desproporcional a alterações funcionais e estruturais pode ocorrer pouco frequentemente em pacientes com pneumopatias ou cardiopatias, caracterizando um quadro fisiopatológico complexo, com componentes pré-capilar e pós-capilar (Tabela 108.2).9,11,12
Os sintomas de HAP são inespecíficos. Dispneia aos esforços é a manifestação inicial e predominante. Inicialmente, a dispneia pode ser confundida com fadiga ou descondicionamento físico. A realização de esforços pode causar pré-síncope ou síncope. Podem ocorrer palpitações, dor torácica anginosa (por isquemia do ventrículo direito), tosse e rouquidão (causados por compressão do nervo laríngeo recorrente: síndrome de Ortner). O fenômeno de Raynaud é observado em poucos pacientes, que geralmente são mulheres. A classificação funcional da New York Heart Association/World Health Organization é bastante utilizada na avaliação clínica (Tabela 108.3).Há sintomas em repouso apenas em casos muito avançados.1,11
O exame físico, na maioria dos pacientes, evidencia impulsão palpável do ventrículo direito, hiperfonese da segunda bulha no foco pulmonar, em geral com desdobramento (às vezes fixo), sopro pansistólico de regurgitação tricúspide e sopro diastólico de insuficiência pulmonar. Terceira e quarta bulhas do ventrículo direito podem estar presentes, assim como distensão jugular, edema em membros inferiores, hepatomegalia, ascite, frialdade das extremidades e cianose de extremidades são observados em casos avançados. Geralmente a ausculta pulmonar é normal. Em casos de abertura de forame oval, pode haver cianose central, o que também ocorre na fisiologia de Eisenmenger (direção direita-esquerda do fluxo comunicante) em casos de cardiopatia congênita com comunicação intracardíaca e hipertensão pulmonar acentuada.1,11
No processo diagnóstico da HP, são necessárias informações sobre antecedentes pessoais e história familiar de HP, doenças difusas do tecido conectivo, tromboembolia venosa, cardiopatia congênita, doenças hepáticas (hipertensão portal), uso de anorexígenos, infecção por HIV-1, doenças da tireoide, anemias hemolíticas, cirurgia com esplenectomia.
Os casos de HAP são apresentados ao médico por meio de sintomas, triagem em pacientes com risco reconhecido ou achado incidental. Os objetivos de diagnóstico consistem em confirmar se há hipertensão arterial pulmonar, determinar as repercussões cardíacas e caracterizar o tipo de HAP. Estabelece-se o diagnóstico de HAPI por exclusão.
O rastreamento de HP é realizado por meio de ecocardiograma com Doppler. O ecocardiograma estima valores de pressão sistólica de ventrículo direito, o que, se não há estenose pulmonar, corresponde à pressão sistólica de artéria pulmonar.Tem-se essa estimativa a partir da determinação da velocidade do jato regurgitante tricúspide em metros por segundo, proporcionalmente ao gradiente transtricúspide em milímetros de mercúrio. Agrega-se a esse valor a estimativa da pressão do átrio direito (cálculos pela equação de Bernoulli: PSAP = 4 [VRT]2 + PAD). A medida fundamental, portanto, é a VRT, cujo valor de corte fisiológico é igual ou menor do que 2,5 m/s. Considera-se a ocorrência de HP se o valor for maior do que 2,7 m/s. Outras variáveis podem reforçar a contribuição do ecocardiograma em casos de HP, como o aumento do diâmetro diastólico do ventrículo direito, da velocidade de regurgitação por meio da valva pulmonar e o encurtamento da aceleração do tempo de ejeção do ventrículo direito.9
Obtém-se a confirmação de HAP por cateterização cardíaca direita e medidas hemodinâmicas plenas da circulação pulmonar. Além das pressões arteriais pulmonares e do débito cardíaco, é essencial a definição segura da pressão venocapilar e da resistência vascular pulmonar. A pressão venocapilar pode ser medida por meio da pressão arterial pulmonar em cunha (admitida como pressão capilar pulmonar, sendo melhor obtida por extrapolação a partir da pressão de oclusão da artéria pulmonar) ou pressão do átrio esquerdo (medida diretamente ou estimada pela pressão de oclusão da artéria pulmonar), ou pressão diastólica final de ventrículo esquerdo (por cateterismo esquerdo ou por comunicação intracardíaca). Se a pressão for igual ou menor do que 15 mmHg, é indício de normalidade e, se for maior do que 15 mmHg, corresponde à hipertensão pós-capilar. Esses dados possibilitam o diagnóstico hemodinâmico correto e o diagnóstico final do tipo de HP. A realização do teste de vasorreatividade com vasodilatadores de ação curta (óxido nítrico inalado, adenosina IV) é indicada em casos de HAP, principalmente nos de HAPI, HAP hereditária (HAPH) e HAP associada a anorexígenos (HAPA). A resposta aceita como positiva para fins de seleção de vaso dilatadores é a redução da PMAP de valores iguais ou superiores a 10 mmHg, atingindo valores inferiores a 40 mmHg, sem queda do índice cardíaco.1,9,11
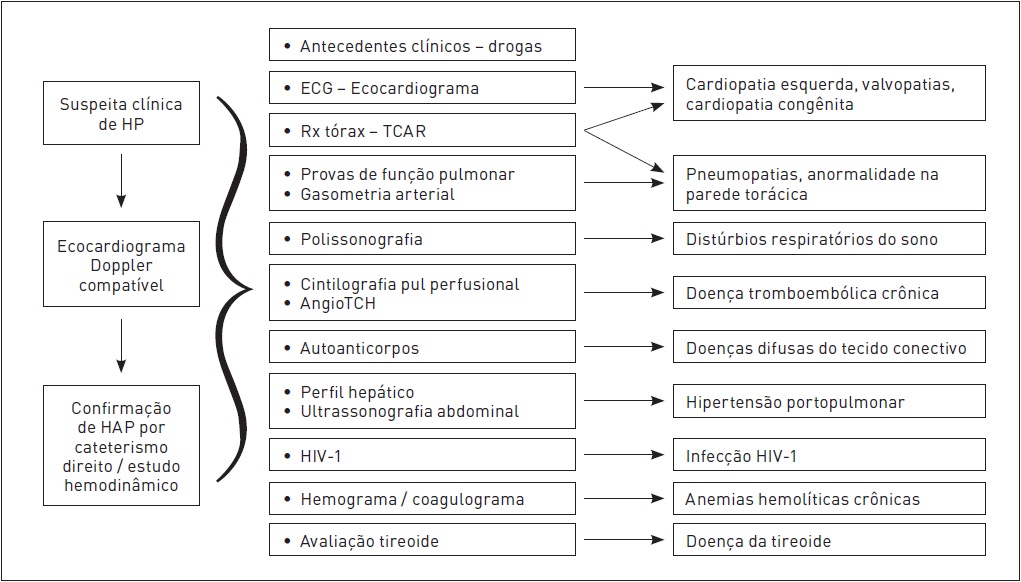
A caracterização diagnóstica para a complementação do diagnóstico implica em investigação que pode contemplar os exames apresentados na Figura 108.3.
Como o processo de investigação de dispneia inexplicada pode requerer testes ergométricos, deve-se considerar que pacientes com hipertensão arterial pulmonar correm risco de síncope se realizarem testes de esforço máximo. A execução de testes de esforço cardiopulmonar (TECP), entretanto, não é contraindicada dependendo do caso.
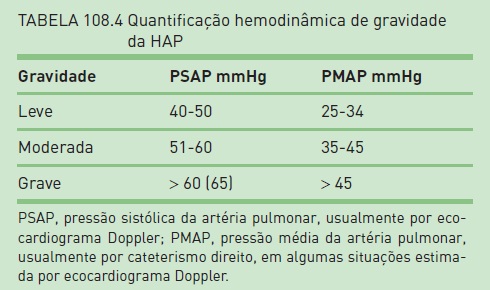
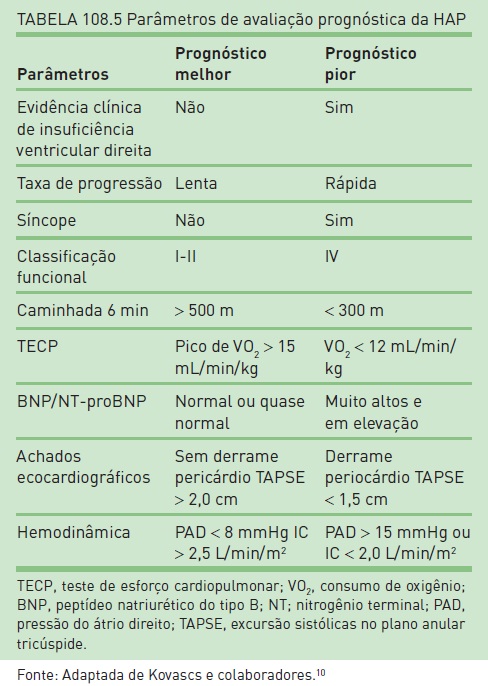
Baseia-se a gravidade da HAP em valores hemodinâmicos (Tabela 108.4), mas também na classificação funcional, sem que haja plena correspondência entre classe funcional e valor de PMAP.1,11 O prognóstico da HAP pode ser estabelecido por outros parâmetros, como os mostrados na Tabela 108.5.
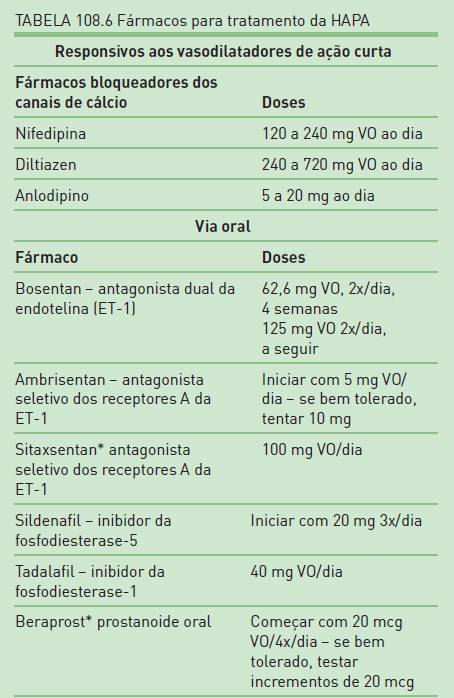
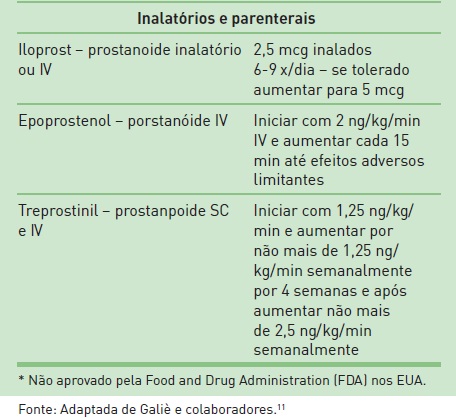
O tratamento, pela disponibilidade de novos fármacos com ações vasodilatadoras e antiprolifetativas, tem melhorado a qualidade de vida, a classe funcional e proporcionado prolongamento da vida em um número substancial de pacientes com HAP. Entretanto, ainda não há esquema terapêutico farmacológico com perspectiva de cura.As estratégias atuais de tratamento clínico da HAP contemplam fármacos mais específicos, com ação vasodilatadora e antiproliferativa, antiadesivos e anticoagulantes, além de medidas gerais e atenção à classificação funcional dos pacientes.12 A relação dos fármacos disponíveis pode ser verificada na Tabela 108.6.
Figura 108.3
Exames para caracterização diagnóstica de hipertensão pulmonar. Podem ser necessários exames adicionais conforme o resultado dos testes apresentados.
Pacientes com HAP sintomática necessitam de medidas gerais de tratamento: evitar gravidez, fazer vacinas contra pneumococo e influenza conforme padronização, evitar excesso de atividade física e realizar reabilitação supervisionada e apoio psicossocial. O uso de diuréticos e de oxigênio depende de avaliação clínica e complementar. A anticoagulação oral com antagonistas da vitamina K não é de prescrição obrigatória; não havendo impedimento ao uso, é indicada em casos de HAPI, HAPH e HAPA. Pacientes com HAP na classificação funcional I devem receber acompanhamento e realizar medidas gerais compatíveis.1,11,13
•A realização de teste de vasorreatividade apresenta recomendação formal para HAPI e relativa para outros tipos de HAP, não sendo indicado para outros grupos de HP.
•O resultado positivo do teste de vasorreatividade possibilita o uso de bloqueadores dos canais de cálcio (BCC), recomendação para HAPI, HAPH e HAPA. Se a resposta for favorável, manter BCC.
Se o teste de vasorreatividade apresentar resultado negativo ou resposta insuficiente no regime de BCC, deve-se realizar tratamento com os fármacos mais específicos, alvodirecionados, conforme classe funcional ou parâmetros de prognósticos: graus de recomendação I, IIa, IIb, III; graus de evidência A, B, C.1,11
•Classe funcional II e melhor prognóstico: I-A: ambrisentan, bosentana, sildenafil, sem ordem de preferência; I- B: tadalafil (B); IIa-C: sitaxsentan.
•Classe funcional III: I-A: ambrisentan, bosentana, sildenafil, sitaxsentan ou iloprost inalado, sem ordem de preferência; I-B: tadalafil, treprostinil subcutâneo (SC) ou inalado; IIa-C: iloprost intravenoso (IV), treprostinil IV; IIb-B: beraprost.
•Classe funcional IV: I-A: epoprostenol IV; IIa-C: ambrisentan, bosentana, iloprost inalado e IV, sildenafil, sitaxsentan, treprostinil SC, IV, inalado – iniciar terapia combinada.
•Combinação de fármacos (bosentana, sildenfil e iloprost ou outro prostanoide disponível), em casos refratários, em incrementos trimestrais.
•Cirurgia em casos refratários ou em franca deterioração – individualizar indicações.
As septoplastia atrial com balão causa um curto-circuito que reduz a pressão do lado direito e permite o enchimento das câmaras esquerdas; a contaminação venosa com consequente dessaturação sanguínea é compensada pelo aumento do débito cardíaco. Isso frequentemente ocorre de forma espontânea pela reabertura do forame oval. Em geral é indicada em casos de síncopes frequentes, e é um procedimento realizado para ganhar tempo em pacientes que aguardam transplante.
Os transplantes pulmonares simples ou duplos, ou de coração-pulmão, em casos com envolvimento do coração esquerdo ou cardiopatias congênitas associadas, têm sido executados em casos sem resposta satisfatória ao tratamento conservador. Relata-se que o pós-operatório imediato é difícil, e a expectativa de vida é menor do que para outros transplantes, como de coração, fígado e rim. No entanto, é um recurso a ser utilizado e valorizado.
A tromboendarterectomia pulmonar é indicada em casos de hipertensão pulmonar tromboembólica crônica com obstrução em vasos centrais e segmentares. Essa cirurgia evidencia resultados satisfatórios e pode ser considerada, em casos selecionados, como a única medida terapêutica de cura para a hipertensão pulmonar. Em alguns casos, os pacientes melhoram, mas necessitam de complementação com tratamento farmacológico.
890
O prognóstico é influenciado pela etiologia/associação subjacente: em se tratando de sobrevida média de pacientes com HP, cerca de 80% dos com cardiopatia congênita sobrevivem por cinco anos, período que é atingido por pouco menos da metade dos pacientes com HAPI; 64% de pacientes com hipertensão portopulmonar sobrevivem por três anos, o que ocorre com menos dos 40% dos pacientes com doenças do tecido conectivo e com cerca de 20% daqueles com infecção porHIV-1.14 A sobrevida média de pacientes com HAPI, que estava entre três e quatro anos do diagnóstico, tem aumentado. Tratamento mais racional e eficaz e cuidados abrangentes têm aumentado a qualidade e a expectativa de vida de muitos pacientes. Principalmente a possibilidade do uso de drogas com ação vasodilatadora e antirremodelante apresenta diferença positiva.15
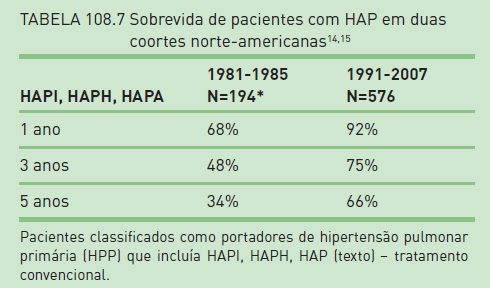
A Tabela 108.7 mostra a diferença de sobrevida em duas coortes norte-americanas de pacientes com HAPI, HAPH e HAPA (classificadas como hipertensão pulmonar primária).
A HAP é uma manifestação fisiopatológica que pode assumir identidade própria, com várias possibilidades etiológicas e um componente idiopático relevante. As fases iniciais da doença são assintomáticas ou pouco sintomáticas. Frequentemente, é por meio da radiografia de tórax ou do ecocardiograma que se cogita a possibilidade da doença. A ecocardiografia com Doppler é um eficiente meio de rastreamento diagnóstico. A confirmação diagnóstica depende de estudo hemodinâmico por cateterismo direito. O teste de vasorreatividade indicado em casos de HAPI pode auxiliar a orientação inicial de tratamento. Novos agentes com ação vasodilatadora e antiproliferativa têm aumentado a qualidade de vida e a sobrevida dos pacientes, mas ainda não apresentam perspectivas de cura. A complexidade na abordagem global do paciente sugere que os centros especializados devam confirmar o diagnóstico e propor um esquema de tratamento que seja acompanhado paralelamente pelo médico assistente.
Na realização de cateterismo cardíaco direito e esquerdo, observou-se: pressão sistólica do ventrículo esquerdo (PSVE): 125 mmHg, pressão diastólica final do ventrículo esquerdo (PD2VE): 10 mmHg, pressão sistólica aórtica (PSAo): 125 mmHg, pressão diastólica aórtica (PDAo): 70 mmHg, pressão média sistêmica (PMS): 100 mmHg, pressão sistólica do ventrículo direito (PSVD): 125 mmHg; pressão diastólica final do ventrículo direito (PD2VD): 12 mmHg, pressão sistólica de artéria pulmonar (PSAP): 125 mmHg, pressão diastólica de artéria pulmonar (PDAP): 57 mmHg, pressão média de artéria pulmonar (PMAP): 80 mmHg, pressão da aurícula direita (PAD): 4 mmHg, Pressão capilar pulmonar ˜PD2VE, débito cardíaco (DC): 6.0 L/min, resistência vascular sistêmica e pulmonar (RVP): 11.66 uW, comunicação interatrial do tipo seio venoso. Cateterização seletiva das três veias pulmonares direitas, todas drenando anomalamente no átrio direito. Injeção de contraste através da CIA na veia pulmonar superior esquerda, que drena corretamente no átrio esquerdo.
A paciente apresenta hipertensão arterial grave, associada a cardiopatia congênita complexa, drenagem anômala parcial, e veias do pulmão direito que drenam no átrio direito e defeito do septo atrial com comunicação interatrial do tipo seio venoso. Há resistência vascular pulmonar pré-capilar elevada, com gradiente transpulmonar muito alto. O débito cardíaco evidencia valor nominalmente normal, mas relativamente reduzido, devido ao tipo de cardiopatia, que é de alto débito. A paciente inicia tratamento com sildenafil, em doses preconizadas e crescentes, objetivando melhora clínica e redução das pressões e da RVP que possibilite a realização de cirurgia corretora. Mesmo sem muita frequência, há casos de sucesso com tal procedimento, que está em processo de avaliação.
1.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573-619.
2.Proceedings of the 4th World Symposium on Pulmonary Hypertension, February 2008, Dana Point, California, USA. J Am Coll Cardiol. 2009;54(1 Suppl):S1-117.
3.Gabbay E, Yeow W, Playford D. Pulmonary arterial hypertension (PAH) is a uncommon cause of pulmonary hypertension (PH) in na unselected population: the Armadale echocardiography study. Am J Respir Crit Care Med. 2007;175:A713.
4.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-23.
5.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-basedregistry for pulmonary arterial hypertension: 1982-2006. Eur Respir J.2007;30(6):1103-10.
6.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France. Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30.
7.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J.2007;30(1):104-9.
8.Moledina S, Hisloop AA, Foster H, Schulze-Neick I,Haworth SG. Childhood idiophatic pulmonary arterial hypertension: a national cohort study. Heart. 2010;96(17):1401-6.
9.Chemla D, Castelain V, Hervé P, Lecarpentier Y, Brimioulle S. Haemodynamic evaluation of pulmonary hypertension. Eur Respir J.2002;20(5):1314-31.
10.Kovascs G, Berghold A, Scheidel S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888-94.
11.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J.2009;30(20):2493-537.
12.Galiè N, Palazzini M, Manes A. Pulmonary hypertension and pulmonary arterial hypertension: a clarification in needed. Eur Respir J.2010;36(5):986-90.
13.Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51(16):1527-38.
14.McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1Suppl):78S-92S.
15.Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-MaitlandM. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35(5):1079-87.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.