(Carregando Índice)... (Carregando Índice)... |
Autores:
Renata Areza Fegyveres
Médica Neurologista. Pesquisadora Colaboradora do Grupo de Neurologia
Cognitiva e do Comportamento (GNCC) e do Centro de Referência de Distúrbios Cognitivos (CEREDEIC) do Hospital das Clínicas da Faculdade de Medicina da USP. Doutoranda do Departamento de Neurologia da Faculdade de Medicina da USP.
Paulo Caramelli
Professor Adjunto e coordenador do Grupo de Neurologia Cognitiva e do Comportamento
do Departamento de Clínica Médica da UFGM.
Última revisão: 23/09/2014
Comentários de assinantes: 0
O aumento da população de idosos, tanto do ponto de vista relativo como do absoluto, é uma das alterações demográficas mais marcantes do último século. Essa alteração faz parte do processo denominado transição demográfica, que ocorre secundariamente à redução da taxa de natalidade e ao aumento da expectativa de vida.
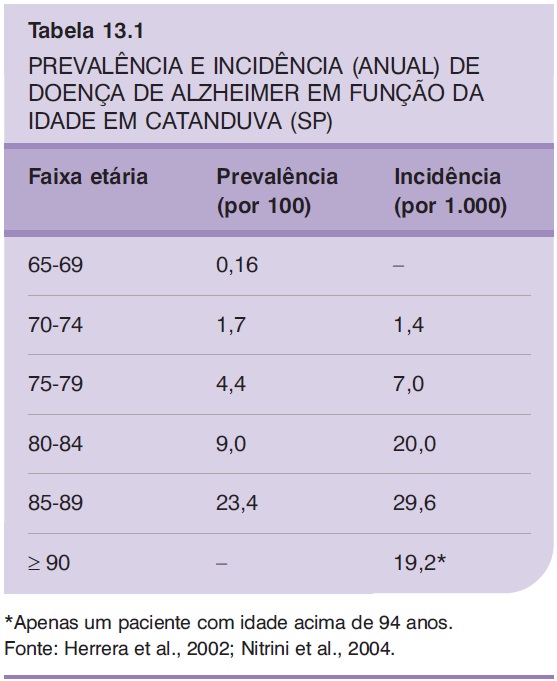
Em estudo epidemiológico brasileiro de base populacional (Herrera et al., 2002) (Tabela 13.1), em que foram avaliados 1.656 sujeitos com idade superior ou igual a 65 anos, foi encontrada prevalência de demência de 7,1% (correspondendo a 118 casos), sendo que, destes, 55,1% receberam o diagnóstico de doença de Alzheimer (DA). A prevalência aumentou com a idade e foi maior entre as mulheres, além de correlacionar-se inversamente com a escolaridade. Outros estudos estrangeiros confirmam prevalência semelhante; no entanto, a comparação entre os estudos é difícil porque são utilizados diferentes critérios e métodos diagnósticos, além de haver variação na seleção e no tamanho da amostra (Lopes; Bottino, 2002).
Em relação à incidência da doença no Brasil, Nitrini e colaboradores (2004), reavaliando após 39 meses a coorte do estudo de prevalência mencionado anteriormente (Herrera et al., 2002), encontraram taxa anual de 7,7 por 100.000 indivíduos com idade superior ou igual a 65 anos. Não houve diferença na incidência de DA em relação ao gênero, embora as mulheres tenham apresentado maior taxa de incidência em idades mais avançadas.
Existem quatro fatores de risco bem definidos para DA (Quadro 13.1). O processo de envelhecimento acarreta a redução do número de neurônios, da arborização dendrítica e da densidade sináptica no córtex e em regiões subcorticais (Cummings et al., 1998), o que se correlaciona com o aumento da incidência de DA com o avançar da idade (Jorm; Jolley, 1998). Outro fator é a presença do alelo e4 da apolipoproteína E (apoE), proteína plasmática relacionada com o transporte de colesterol, que apresenta três alelos (e2, e3, e4) e seis fenótipos. A distribuição da apoE é variável nas diversas etnias, e estudos epidemiológicos demonstram que, em indivíduos com DA, a frequência do alelo e4 é desproporcionalmente alta (Farrer et al., 1997), inclusive na população brasileira (Souza et al., 2003). Os outros dois fatores de risco são história familiar e síndrome de Down.

Na literatura mundial, existe uma grande variedade de termos para denominar o comprometimento cognitivo e a síndrome demencial causados pela doença cerebrovascular (DCV). Alguns são relacionados à síndrome clínica ou aos substratos neuropatológicos, outros estão relacionados aos achados de neuroimagem, e poucos são, realmente, bem definidos, com critérios estabelecidos. Alguns exemplos dessa terminologia são os termos demência vascular, demência aterosclerótica, comprometimento cognitivo vascular, demência por múltiplos infartos, encefalopatia de Binswanger, leucoaraiose e demência vascular subcortical. Neste capítulo, o conjunto das demências causadas pela DCV será chamado de demência vascular (DV).
No estudo de Herrera e colaboradores (2002), 9,3% dos pacientes com demência foram diagnosticados como tendo DV. Posteriormente, no estudo de incidência de demência realizado nessa mesma coorte de idosos, 18% dos 50 casos incidentes preencheram critérios diagnósticos para DV (Nitrini et al., 2004). A DV foi a segunda (no estudo de incidência) ou a terceira (no estudo de prevalência) causa de demência na população idosa nessa comunidade.
Em outros estudos brasileiros, realizados em ambulatórios especializados, foram encontradas frequências mais altas de DV, refletindo características das populações estudadas e critérios de seleção diferentes. Silva e Damasceno (2002) encontraram 24,9% de casos de DV de um total de 261 pacientes com demência, enquanto no estudo de Vale e Miranda (2002) 19,3% de 186 pacientes receberam diagnóstico de DV.
Os dados de prevalência e de incidência obtidos em outros países são bastante variáveis, refletindo diferenças étnicas e sócio-demográficas e critérios de seleção distintos (Yoshitake et al., 1995; Di Carlo et al., 2002; Fitzpatrick et al., 2004; Zhang et al., 2005; Shaji; Bose; Verghese, 2005). Em relação aos estudos de incidência, por exemplo, foram observadas taxas anuais variando de 3,3 (Di Carlo et al., 2002) a 14,6 por 1.000 pessoas (Fitzpatrick et al., 2004). Na maior parte desses estudos, no entanto, a DV corresponde à segunda causa mais frequente de demência, após a DA.
Atualmente, o termo demência mista refere-se a uma situação bastante comum na prática clínica: um paciente que apresenta história e exame físico compatíveis com DA, mas que, no entanto, apresenta lesões vasculares significativas em exame de neuroimagem estrutural, que podem estar contribuindo para o quadro cognitivo. Esse indivíduo também não preenche critérios para serem enquadrados no diagnóstico de DV.
Estudo mencionado anteriormente (Herrera et al., 2002; Nitrini et al., 2004; Vale; Miranda, 2002; Silva; Damasceno, 2002) mostram que a prevalência desse tipo de demência varia de 1,9 a 14,5%, assumindo taxas mais expressivas em idades mais avançadas, especialmente após os 80 anos.
A grande maioria dos casos de DA é de ocorrência esporádica. Formas familiares, em que há padrão de herança autossômica dominante, também são descritas, perfazendo, no entanto, menos de 2% do total de casos da doença. Nesses indivíduos, já foram identificadas mutações nos cromossomos 1, 14 e 21. O gene que codifica a proteína precursora do amiloide encontra-se no cromossomo 21, enquanto os cromossomos 14 e 1 relacionam-se, respectivamente, com as proteínas pré-senilinas 1 e 2. Deve haver participação de outros genes ainda não identificados em uma parcela de casos familiares da doença(Ertekin-Taner, 2007).
Dentre os casos de DA familiar em que anormalidades genéticas são identificadas, a maior parte (cerca de 50% dos casos) decorre de mutações no gene da pré-senilina 1, no cromossomo 14. Todos os casos de DA familiar têm como característica a idade de início dos sintomas antes dos 65 anos, mais frequentemente antes dos 60 anos. Embora as anormalidades genéticas sejam responsáveis por uma absoluta minoria dos casos, elas oferecem fortes evidências da participação dos fatores genéticos na patogênese da DA, além de permitirem melhor compreensão dos mecanismos moleculares relacionados à fisiopatologia da doença (Erte-kin-Taner, 2007).
A DA apresenta dois marcadores histopatológicos fundamentais: as placas senis extracelulares, cujo constituinte molecular principal é o peptídeo ß-amiloide,e os emaranhados neurofibrilares intracelulares, formados pela proteína tau hiperfosforilada. A presença dessas duas lesões em quantidades determinadas é obrigatória para o diagnóstico anatomopatológico da DA (Consensus..., 1997).
A proteína tau está associada aos microtúbulos, participando da formação estrutural do citoesqueleto neuronal e conferindo estabilidade ao sistema. Essa proteína é codificada por um gene localizado no cromossomo 17 e, por razões ainda não totalmente conhecidas, sofre processo de hiperfosforilação na DA. A proteína tau hiperfosforilada deixa de se ligar aos microtúbulos e forma filamentos helicoidais pareados insolúveis, que se agregam formando os emaranhados neurofibrilares. O número de emaranhados aumenta com o processo de envelhecimento, em indivíduos com ou sem demência. Entretanto, há diferenças quantitativas, e a densidade de emaranhados neurofibrilares é maior nos pacientes com DA do que em indivíduos normais da mesma idade, sendo que, nesses últimos, as alterações são restritas às estruturas límbicas (Ballatore; Lee; Trojanowski, 2007).
O córtex entorrinal e a formação hipocampal são as primeiras áreas cerebrais acometidas pelos emaranhados neurofibrilares. Posteriormente, há comprometimento de núcleos colinérgicos localizados no prosencéfalo basal (particularmente do núcleo basal de Meynert) e de áreas neocorticais associativas, com relativa preservação de áreas corticais primárias (Braak; Braak, 1991). A expressão clínica determinante da síndrome demencial, com um espectro de alterações cognitivas e comportamentais, ocorre justamente nesse estágio neuropatológico (Braak; Braak, 1998).
A placa senil é outra lesão neuropatológica característica da DA. As placas são constituídas em grande parte pelo peptídeo ß-amilóide, que possui 40 a 42 aminoácidos e é formado a partir do processo de clivagem anormal de uma proteína precursora (proteína precursora do peptídeo ß-amiloide). Essa proteína precursora é codificada por um gene localizado no cromossomo 21 e pode ser degradada por três enzimas: a, ß e ?-secretases. A ação da a-secretase corresponde à clivagem da proteína precursora que ocorre em situação fisiológica. Na DA, por indução de mutação genética (mais raramente) ou por outros mecanismos ainda não totalmente estabelecidos, predominam as ações da ? e daß-secretases, formando fragmentos que se depositam no espaço extracelular, inicialmente sob forma de oligômeros solúveis de amiloide e, posteriormente, de placas senis difusas que contêm formas insolúveis do peptídeo (Eckman; Eckman, 2007; Ferreira; Vieira; De Felice, 2007).
Sabe-se que o peptídeo ß-amiloide, especialmente o fragmento contendo 42 aminoácidos, e mesmo os oligômeros solúveis que se agregam e vão formá-lo têm efeito neurotóxico e promovem uma cascata de eventos (inclusive com a participação de mediadores inflamatórios) no espaço extracelular, levando à formação das placas senis neuríticas. Tais placas representam lesões maduras em cuja região central se acumulam densos depósitos do peptídeo ß-amilóide,circundados por terminações nervosas (axônios e dendritos) degeneradas.
As placas senis neuríticas localizam-se principalmente em áreas neocorticais, sem obedecer ao mesmo padrão têmporo-espacial de distribuição topográfica descrito para os emaranhados neurofibrilares (Braak; Braak, 1991).
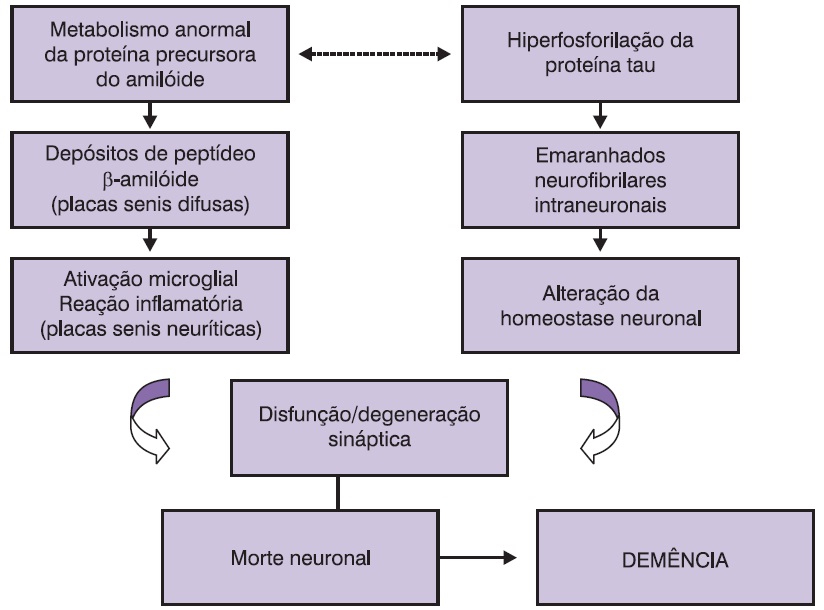
Há muitas controvérsias na literatura científica sobre o papel relativo de cada uma dessas lesões na patogenia da DA, bem como sobre qual evento fisiopatológico (cascata do amiloide ou hiperfosforilação da proteína tau) ocorre primeiro (Arriagada et al., 1992; Caramelli et al., 1998). O que é bem estabelecido, no entanto, é que esses processos culminam com disfunção sináptica e subsequente morte neuronal, que se traduzem, do ponto de vista macroscópico, por significativa atrofia cerebral e, clinicamente, pela síndrome demencial. A Figura 13.1 apresenta um diagrama esquemático da fisiopatologia da DA.
O comprometimento neuropatológico descrito afeta de maneira progressiva diversas vias de neurotransmissão. Déficits glutamatérgicos, noradrenérgicos e serotoninérgicos, entre outros, são bem reconhecidos. Entretanto, o déficit colinérgico é o que ocorre de forma mais consistente, com importantes implicações no tratamento farmacológico disponível atualmente. Tal déficit é decorrente, em grande parte, do acúmulo relativamente precoce de emaranhados neurofibrilares no núcleo basal de Meynert, que constitui importante fonte de inervação colinérgica para o córtex cerebral. Sabe-se que essas vias de projeção colinérgica a partir do prosencéfalo basal têm um importante papel no aprendizado e na memória.
O diagnóstico anátomo-patológico da DA depende da distribuição e da quantidade das lesões neuropatológicas descritas. O aspecto quantitativo é fundamental, uma vez que pacientes idosos sem demência também podem apresentar tais lesões. De acordo com os critérios histopatológicos propostos pelo National Institute on Aging e pelo Reagan Institute Working Group on Diagnostic Criteria for Neuropathological Assesment for Alzheimer’s Disease (Consensus ..., 1997), a probabilidade do diagnóstico anátomo-patológico de DA é dividida em: 1) alta: numerosos emaranhados neurofibrilares e placas senis neuríticas presentes no hipocampo, no córtex entorrinal e em áreas neocorticais (20 a 30 por campo, no aumento de 10 vezes); 2) intermediária: densidade moderada de placas senis neuríticas neocorticais e emaranhados neurofibrilares em áreas límbicas (5 a 10 por campo, no aumento de 10 vezes); e 3) baixa: pequena densidade dos dois tipos de lesões, confinadas a estruturas límbicas.
Figura 13.1
Sequência de eventos fisiopatológicos na DA.
Quatro mecanismos fisiopatológicos básicos estão relacionados à ocorrência da DV: doença de grandes vasos, doença de pequenos vasos, hipoperfusão ou mecanismos hipóxico-isquêmicos e hemorragia intracraniana (Brun, 1994).
Aterosclerose e doença de pequenos vasos são as causas principais de infarto cerebral. Infartos lacunares ou múltiplos microinfartos de núcleos da base, tálamo, tronco encefálico e substância branca estão associados a mais da metade dos casos de DV do tipo subcortical (Kalaria et al., 2004). Na microscopia, observam-se degeneração fibrinóide e lipo-hialinose nas artérias perfurantes pequenas. A diferença entre estado lacunar e leucoaraiose difusa é questão de intensidade. Na última, há lesões confluentes na substância branca profunda e desmielinização, predominando nas regiões periventriculares e subcorticais posteriores.
Na DV subcortical, ocorre isquemia dos núcleos colinérgicos localizados no prosencéfalo basal (que são irrigados pelas artérias penetrantes, altamente suscetíveis à hipertensão arterial) ou isquemia dos núcleos da base e da substância branca (que se associam ao restante do córtex por meio de projeções colinérgicas corticais). Esse padrão lesional acarreta redução na concentração cerebral dos níveis de acetilcolina (Tomimoto et al., 2005). Embora a redução das aferências colinérgicas na DV subcortical seja menor do que aquela observada na DA e na demência com corpos de Lewy, ela constitui a base neuroquímica para a utilização de drogas inibidoras da colinesterase no tratamento da DV subcortical, o que será discutido adiante. Há indícios também de que a estimulação colinérgica produz aumento no fluxo cerebral cortical, o que também pode ser relevante nas implicações terapêuticas da DV (Román, 2005).
Sua fisiopatologia conta tanto com aspectos relacionados às alterações do metabolismo presentes na DA quanto com déficits que ocorrem na DV, como descrito anteriormente. Em casos individuais, no entanto, é praticamente impossível avaliar a parcela efetiva de contribuição de cada um desses dois processos patológicos para o quadro clínico final.
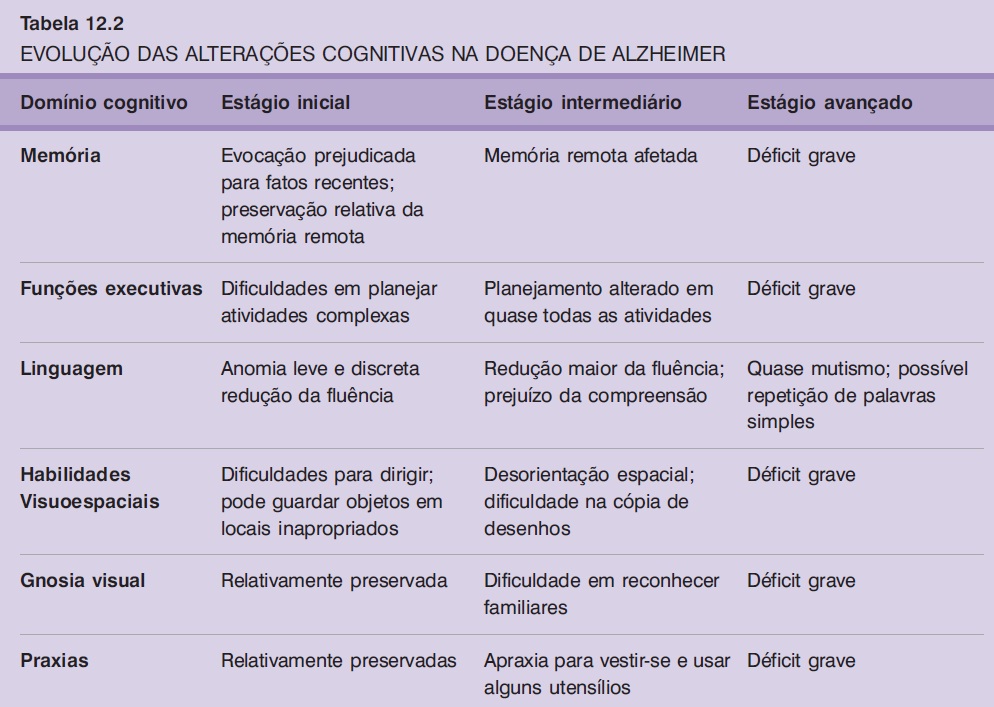
A DA progride em três estágios de uma maneira relativamente previsível e consistente (Cummings; Benson, 1992) (Tabela 13.2). No primeiro estágio, o achado clínico predominante é a perda de memória para fatos recentes. A linguagem também pode estar alterada, com discurso vazio, poucos substantivos e pobreza de ideias, além de anomia e dificuldade de geração de uma lista de palavras (p. ex., em um minuto, produzir uma lista dos animais que conhece). Habilidades visuoespaciais também podem estar alteradas, e o paciente pode perder-se nos arredores, se deixado sozinho. A fala e outras funções motoras estão normais.
No segundo estágio, todos os domínios intelectuais continuam a deteriorar. A alteração da linguagem é caracterizada por discurso fluente parafásico, compreensão alterada e repetição relativamente preservada (Cummings et al., 1985). Ambas as memórias recente e remota estão bastante acometidas. Progressivamente, as habilidades visuoespaciais vão sendo prejudicadas, e os pacientes perdem-se dentro da própria casa. Cálculo e abstração também estão bastante comprometidos.
No estágio final, todas as funções cognitivas estão gravemente comprometidas. A fluência verbal se reduz a ecolalia, palilalia ou mutismo. Ocorre incontinência esfincteriana, e o paciente assume uma postura de flexão dos quatro membros, com rigidez generalizada. O óbito geralmente ocorre por pneumonia aspirativa ou infecção do trato urinário com sepse.
Alguns sinais de alerta podem ajudar médicos e familiares a detectar a DA em fase inicial (Quadro 13.2). Alterações de comportamento, desde apatia até delírios, alucinações, agitação e agressividade, são comuns no curso da doença, ocorrendo em cerca de 80% dos casos (Tatsch et al., 2006).
Os quadros clínicos dos pacientes com demência no contexto de DCV podem ser divididos em três grandes grupos (Quadro 13.3): demência por múltiplos infartos (DMI), demência subcortical isquêmica (DSI) e demência por infarto único localizado em área estratégica para o funcionamento cognitivo (DIE).
O paciente com DMI apresenta, usualmente, história de doença tromboembólica, hipertensão arterial e outras evidências de doença aterosclerótica, como angina e arteriopatia periférica, entre outras. O quadro clínico depende do sítio, do número e da extensão das lesões, mas geralmente os pacientes apresentam combinação de sintomas e sinais característicos de lesão cortical e de lesão subcortical. No entanto, os últimos predominam. Classicamente, a progressão é “em degraus”, com um platô na evolução. Alterações esfincterianas (urinárias) e de marcha ocorrem precocemente no curso da doença, às vezes precedendo as alterações cognitivas. Achados de paralisia pseudobulbar (p. ex., disfagia, disartria, choro patológico) são característicos. Flutuações no desempenho cognitivo e confusão noturna são muito comuns e podem gerar dificuldade para se diferenciar a DV da demência com corpos de Lewy. Podem ser observados, também, labilidade emocional levando à incontinência emocional, irritabilidade e apatia, além de sintomas psicóticos.
A DSI inclui o estado lacunar e a encefalopatia de Binswanger (leucoaraiose difusa). Geralmente, ocorre em pacientes hipertensos de longa data, que apresentam declínio cognitivo insidioso, às vezes mesclado com episódios de piora cognitiva mais acentuada, devido às lacunas ou, mais raramente, a eventos tromboembólicos maiores. Além do quadro demencial, um conjunto de achados piramidais e extrapiramidais pode estar presente. Entretanto, alguns casos não estão ligados a sinais neurológicos focais, apesar de apresentarem alterações grosseiras de substância branca na ressonância magnética (RM) de crânio (Smid et al., 2001). Muitos pacientes com DCV de substância branca procuram atendimento médico devido a queixas cognitivas, porém não preenchem critérios para diagnóstico de demência; apresentam déficits sutis e têm pontuações normais no MiniExame do Estado Mental (Folstein; Folstein; McHugh, 1975). O termo “comprometimento cognitivo vascular” foi proposto para designar estes casos (Rockwood et al., 1999; Román, 2004). A síndrome da DV subcortical se distingue da DA por acometer predominantemente as funções executivas e por o comprometimento de memória episódica não ser tão acentuado. Alterações de humor, especialmente depressão (e também apatia), são muito comuns.
A DIE ocorre quando uma lesão única, em área considerada crítica para o funcionamento cognitivo, resulta em demência. Evidentemente, um paciente que sofre um acidente vascular isquêmico de artéria cerebral média à esquerda apresentará algum grau de afasia, associada a alexia e/ou agrafia, e também algum grau de alteração atencional e de memória, assim preenchendo critérios para demência. No entanto, o termo “infarto estratégico” é reservado para infartos pequenos e em localizações que resultem em comprometimento cognitivo maior do que o esperado, geralmente sem os sinais neurológicos focais clássicos. Por exemplo, no infarto da porção medial do tálamo (ramos paramedianos da artéria cerebral posterior), pode ocorrer confusão mental, seguida de amnésia persistente e grave, o que pode se assemelhar à síndrome de Korsakoff. Apatia e paralisia do olhar vertical e/ou apraxia palpebral são comuns nesses casos. Nos infartos do giro angular, há início súbito de afasia fluente, com alexia e agrafia, alteração de memória, desorientação espacial e apraxia de construção.
O quadro clínico da demência mista apresenta componentes das duas enfermidades descritas anteriormente. Caracteriza-se frequentemente por história clínica típica da DA, porém com lesões significativas no estudo de neuroimagem estrutural; sendo assim, não preenche critérios clínicos para nenhuma das duas doenças exclusivamente.
O diagnóstico de DA é feito por exclusão de outras possíveis causas de demência mais facilmente diagnosticadas. Exame neurológico inespecífico, ou seja, ausência de, por exemplo, sinais piramidais, movimentos involuntários, alteração no exame de motricidade ocular extrínseca e exames laboratoriais de sangue que não sugerem nenhuma outra causa de demência são características que corroboram o diagnóstico de DA. A exclusão de outras causas de demência por exame de neuroimagem também se faz necessária.
Os critérios diagnósticos do Manual de Diagnóstico e Estatístico de Transtornos Mentais da Associação Americana de Psiquiatria (DSM-IV) são os mais comumente utilizados e preconizam a presença de síndrome demencial insidiosa e gradual com comprometimento de memória obrigatório, além de mais um dos seguintes distúrbios: afasia, apraxia, agnosia ou distúrbio de funções executivas, em associação com prejuízo de funcionamento das atividades diárias. Os critérios propostos pelo National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) em conjunto com Alzheimer Disease and Related Disorders Association (ADRDA) (McKhann et al., 1984) são uma alternativa aos critérios do DSM-IV (Quadro 13.4). Dividem a probabilidade do diagnóstico em DA definida, provável e possível e enfatizam a confirmação do padrão de declínio cognitivo por avaliação neuropsicológica completa, após testes de rastreio cognitivo como o miniexame do estado mental (MEEM) (Folstein; Folstein; McHugh, 1975) e a escala de demência de Blessed (Blessed; Tomlinson; Roth, 1968).
O diagnóstico clínico de DA teve sua sensibilidade e especificidade aumentadas nas últimas décadas devido à aplicação de critérios diagnósticos padronizados (Victoroff et al., 1995). Idealmente, os déficits cognitivos devem ser comprovados por avaliação neuropsicológica especializada. Outros achados que corroboram o diagnóstico de DA são diminuição do volume hipocampal com respectivo aumento do volume do corno temporal dos ventrículos laterais na RM de crânio (Frisoni et al., 1996) e demonstração de hipometabolismo fronto-têmporo-parietal bilateral na tomografia computadorizada com emissão fotônica única (SPECT), que corresponde ao chamado padrão B, típico achado nessa doença (Bergman et al., 1997). Esse último aspecto, no entanto, pode não estar presente em muitos casos de DA, além de ser aparentemente mais frequente nos casos de DA pré-senil (início dos sintomas antes dos 65 anos de idade) (Nitrini et al., 2000).
A avaliação neuropsicológica é um passo fundamental para caracterizar a presença da síndrome demencial e determinar quais são os domínios cognitivos acometidos e o quanto estes estão comprometidos. No caso da DA, predomina o déficit de memória nos estágios iniciais e intermediários da doença. Existem inúmeros testes neuropsicológicos com esses objetivos: alguns mais simples, que avaliam superficialmente todas as funções, e outros mais complexos, que avaliam uma função específica com maior profundidade. O MEEM (Folstein; Folstein; McHugh, 1975), que tem versão brasileira com sugestões de utilização (Brucki et al., 2003) e pontuações de corte para diferentes níveis de escolaridade (Bertolucci et al., 1994), o teste de fluência verbal para categorias (animais) (Brucki et al., 1997; Caramelli et al., 2007) e o teste de memória de figuras (Nitrini et al., 1994; 2004), com boa sensibilidade para indivíduos analfabetos, são exemplos de testes mais simples de aplicação rápida. Há também baterias de avaliação neuropsicológica mais completas e trabalhosas, já traduzidas para o português, como o
Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) (Bertolucci et al., 1998; 2001) e a escala de demência de Mattis (Porto et al., 2003).
Outro instrumento muito útil e que possui versão para uso no Brasil é a Clinical Dementia Rating (CDR), uma entrevista semiestruturada que avalia o funcionamento cognitivo e o desempenho funcional e que permite não apenas o diagnóstico de DA como também seu estadiamento (Chaves et al., 2007; Hughes et al., 1982). Instrumentos diagnósticos para avaliação das manifestações neuropsiquiátricas das demências também estão disponíveis; destes, o mais utilizado é o inventário neuropsiquiátrico (Cummings, 1997; Camozzato et al., 2008).
Os critérios diagnósticos mais utilizados atualmente para DV são os do National Institute of Neurological Disorders and Stroke em conjunto com a Association Internationale pour la Recherche et l’Enseigne ment en Neurosciences (NINDS-AIREN) (Quadro 13.5). O diagnóstico de DV é realizado levando-se em conta a história clínica, os achados de exame, a avaliação neuropsicológica e os dados de neuroimagem.
A avaliação neuropsicológica geralmente evidencia comprometimento de memória episódica menos intenso do que na DA, enquanto a atenção e as funções executivas e motoras estão mais gravemente acometidas. Na linguagem, há algumas diferenças em relação à DA, como, por exemplo, pior desempenho de fluência verbal fonêmica nos pacientes com DV (Lafosse et al., 1997; Tierney et al., 2001; Desmond, 2004). O padrão de alteração cognitiva na DV é consistente com disfunção frontal e subcortical.
A neuroimagem estrutural faz parte dos critérios diagnósticos de DV. Os achados da DMI e da DSI muitas vezes se confundem. A DMI é caracterizada por lesões isquêmicas nos territórios de artérias maiores, enquanto a DSI envolve pequenos vasos. Devido ao fato de que, freqüentemente, há coexistência de condições degenerativas, como DA com DCV, sugere-se considerar DV somente os casos em que há alterações relevantes na tomografia computadorizada (TC) ou na RM. Tanto a extensão quanto a localização das lesões devem ser levadas em consideração. Múltiplos infartos e focos com aumento de sinal nos núcleos da base e no tálamo são sugestivos do diagnóstico de DV. A ausência de lesões vasculares na RM exclui o diagnóstico de DV.
Há inúmeras doenças que fazem parte do diagnóstico diferencial das três formas de demência abordadas neste capítulo (Quadro 13.6). Ao avaliar o paciente, deve-se sempre levar em conta o contexto. O quadro clínico pode ser bastante variável, com acometimento predominante de determinado domínio cognitivo ou com alteração global de todas as funções. Por exemplo, a síndrome de Korsakoff cursa com déficit intenso de memória recente, ou seja, de aprendizagem de novas informações (síndrome amnéstica), além de ocorrerem confabulações. A demência alcoólica apresenta-se com um quadro mais gradual e progressivo, com acometimento de todas as funções cognitivas, além de alterações comportamentais e de humor. Nas disfunções endócrinas, principalmente nas tireoidianas, podem ocorrer lentidão do pensamento, alterações repentinas do estado mental, delírios e alucinações. Já o espectro de manifestações da deficiência de vitamina B12 é bastante amplo. Além do comprometimento cognitivo, podem estar presentes alterações hematológicas (anemia megaloblástica é típica), neuropatia periférica, mielopatias e alterações de humor.
Alguns pontos que devem ser valorizados para o estabelecimento do diagnóstico são cronologia dos fatos, velocidade de progressão do quadro, uso de medicações, antecedentes pessoais e familiares de doenças clínicas e alterações do humor. É importante estimar o impacto do déficit cognitivo nos domínios pessoal, social e profissional.
Quando as causas de síndrome demencial são rastreadas, é necessário realizar uma série de exames complementares com o objetivo principal de descartar causas tratáveis de demência ou mesmo cofatores que contribuem para a piora do quadro cognitivo. É importante lembrar que duas causas de demência podem coexistir, assim como ocorre na demência mista (p. ex., DA e hidrocefalia de pressão normal).
O tratamento farmacológico da DA pode ser dividido em tratamento dos sintomas cognitivos e dos sintomas neuropsiquiátricos e uso de neuroprotetores, ou drogas que visam a reduzir a velocidade de progressão da doença.
O reconhecimento da ocorrência de déficit colinérgico na DA, secundário ao acometimento intenso e precoce de núcleos colinérgicos localizados no prosencéfalo basal, como o núcleo basal de Meynert, justificou inúmeras tentativas de manipulação farmacológica da sinapse colinérgica, com o emprego de agentes com diferentes mecanismos de ação. Os inibidores da colinesterase (IChE) são as drogas que se mostraram eficazes nesse sentido, e três compostos dessa classe estão atualmente disponíveis para emprego na prática clínica: donepezil, galantamina e rivastigmina (NE I, GR A). A tacrina foi a primeira droga lançada comercialmente, mas, embora reconhecidamente eficaz, seu uso foi abandonado devido à toxicidade hepática (elevação significativa de transaminases hepáticas em cerca de 25% dos casos tratados), que não é observada com os outros três agentes (Doody et al., 2001).
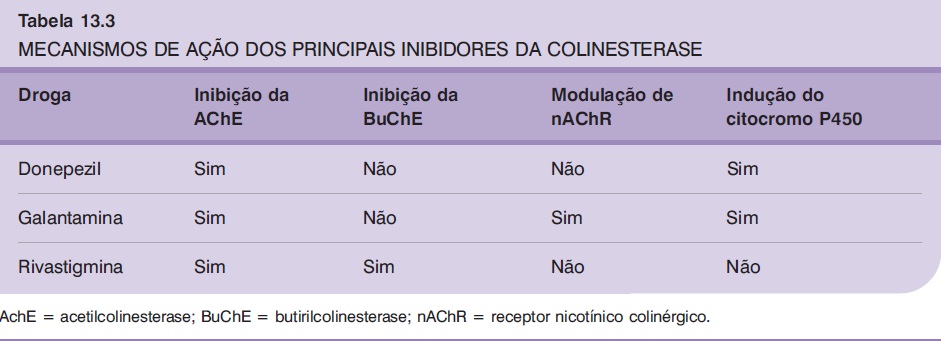
Essas drogas inibem a acetilcolinesterase (e, eventualmente, a butirilcolinesterase), enzimas responsáveis pela hidrólise da acetilcolina na fenda sináptica, levando ao aumento da disponibilidade do neurotransmissor. Embora o mecanismo básico de ação seja comum, diferenças na forma de inibição da enzima ou em mecanismos de ação adicionais conferem particularidades a cada uma das drogas. No entanto, não há até o momento evidências concretas de superioridade terapêutica de um agente sobre outro. Por esse motivo, a escolha da droga deve se basear na experiência clínica, no perfil de interações medicamentosas, na tolerabilidade e na posologia. A disponibilidade atual de três drogas permite beneficiar um número maior de pacientes, uma vez que aqueles que não toleram determinado IChE podem passar a receber outra medicação. Além disso, alguns pacientes podem não apresentar resposta a uma das drogas, mas responder a outra medicação.
A Tabela 13.3 apresenta as características principais do mecanismo de ação dos três IChEs mais empregados atualmente no tratamento da DA.
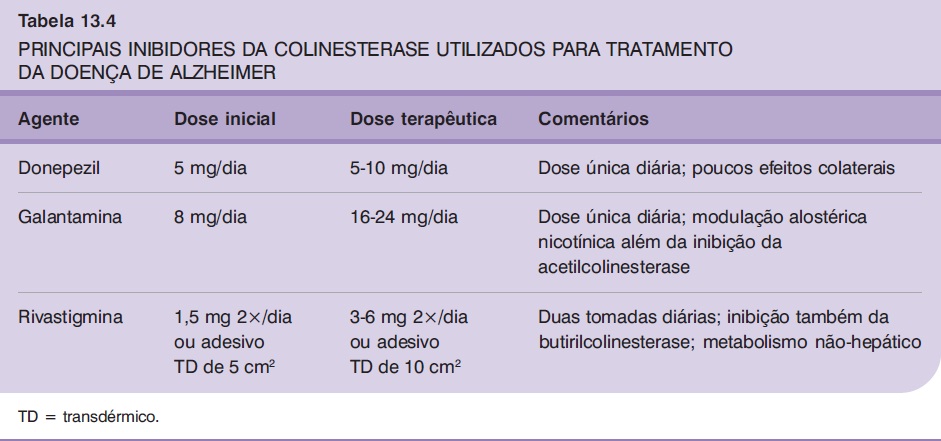
Os principais eventos adversos dos IChEs são náuseas, vômitos e diarreia, decorrentes de inibição da acetilcolinesterase periférica. A administração lenta da dose é a medida mais efetiva na redução desses sintomas, embora, em alguns casos, isso não seja suficiente para proporcionar boa tolerabilidade e o tratamento deva ser interrompido. A recomendação é de que as doses sejam aumentadas a cada quatro semanas, ou mesmo após períodos maiores, dependendo da ocorrência de eventos adversos. No caso do donepezil, a dose inicial recomendada é de 5 mg/dia (dose única), de preferência à noite (embora também possa ser administrado pela manhã nos casos de pacientes que apresentem insônia), podendo-se aumentar a dose para 10 mg/dia após quatro semanas de tratamento. A galantamina, por sua vez, deve ser iniciada na dose de 8 mg uma vez ao dia (após o café da manhã) durante quatro semanas, passando-se em seguida para 16 mg/dia. A dose pode ser aumentada para 24 mg/dia após novo período de quatro semanas. A rivastigmina deve ser prescrita na dose inicial de 1,5 mg duas vezes ao dia, com aumentos para 3 mg e, posteriormente, 4,5 mg duas vezes ao dia após intervalos de pelo menos quatro semanas. A dose máxima recomendada é de 12 mg/dia, divididos em duas tomadas diárias de 6 mg. Alternativamente, a rivastigmina pode ser administrada sob forma de adesivo transdérmico, iniciando-se com o de 5 cm2 de diâmetro (aplicação de uma vez ao dia, correspondendo à dose de 4,6 mg/dia), que, após quatro semanas, pode ser substituído pelo de 10 cm2 (correspondendo à dose de 9,5 mg/dia) (Tabela 13.4).
Ensaios clínicos controlados com placebo revelam que os IChEs têm efeito terapêutico sobre sintomas tanto cognitivos quanto comportamentais, além de promoverem melhora do desempenho funcional (atividades de vida diária). Os ensaios clínicos caracteristicamente incluíram pacientes com demência de intensidade leve a moderada, tratados durante cerca de seis meses (Doody et al., 2001). Em alguns dos trabalhos, foi feita extensão do período de avaliação por até cinco anos, embora em regime aberto (sem controle com placebo), e, nesses casos, as comparações foram feitas com dados sobre a evolução natural da doença a partir de outros estudos(grupo-placebo histórico).
Nos ensaios clínicos, a avaliação da eficácia sobre os sintomas cognitivos é feita com a escala ADAS-Cog (Alzheimer’s Disease Assesment Scale cognitive subscale), que avalia diferentes funções, como memória, linguagem, orientação e habilidades construtivas. A pontuação dessa escala varia de 0 a 70 pontos, sendo que escores mais altos indicam maior comprometimento. Ao final do período de observação (usualmente seis meses), as diferenças de pontuação na ADAS-Cog entre o grupo tratado com droga ativa e o grupoplacebo variam de 1,6 a 4,4 pontos nos diferentes ensaios clínicos. Esse dado indica que, embora os IChEs sejam superiores ao placebo, a magnitude de seu efeito é modesta (Lanctôt et al., 2003). Uma das funções cognitivas que mais parece se beneficiar do tratamento com IChE é a atenção (Caramelli et al., 2004).
Os benefícios do tratamento com IChE podem ser observados a curto prazo ou simplesmente por redução da velocidade de progressão da doença.
Existem padrões diferentes de resposta individual: alguns pacientes exibem resposta clinicamente relevante, enquanto outros não exibem esse mesmo perfil. Não foram identificados até o momento fatores relacionados a melhor resposta terapêutica, e, portanto, o tratamento com IChE deve ser considerado em todo paciente com DA com sintomatologia cognitiva de intensidade leve a moderada (Doody et al., 2001; Lanctôt et al., 2003).
Nas fases mais avançadas da doença (DA moderada a grave), a memantina – droga que atua como antagonista não competitivo dos receptores NMDA (N-metil-D-aspartato) do glutamato – mostrou-se eficaz, levando à melhora cognitiva e funcional e à redução do grau de dependência (Reisberg et al., 2003; Bakchine; Loft, 2008; Wilkinson; Andersen, 2007; Calabrese; Essner; Forstl, 2007; van Dyck et al., 2007). Em condições normais, a memantina permite a neurotransmissão glutamatérgica; porém, em condições de estimulação crônica, essa droga inibe a excitotoxicidade causada pelo glutamato.A posologia inicial recomendada é de 5 mg/dia, com aumento de 5 mg/dia a cada semana. A posologia final recomendada é de 10 mg a cada 12 horas, e a tolerabilidade é muito boa.
Mais recentemente, foi demonstrado que a associação da memantina a um IChE (no caso, donepezil ou rivastigmina) em pacientes com DA moderada a grave promove benefício clínico e funcional superior ao efeito do IChE isoladamente, sem maior incidência de eventos adversos (Dantoine et al., 2006; Tariot et al., 2004; Weycker et al., 2007), o que justifica a instituição do tratamento combinado nesses casos. Da mesma forma, o donepezil demonstrou eficácia também em pacientes com DA grave, embora esta não seja ainda uma indicação terapêutica aprovada no Brasil (Winblad et al., 2006; Black et al., 2007).
Conforme mencionado na seção anterior, diversos ensaios clínicos demonstraram efeito terapêutico dos IChEs sobre sintomas comportamentais (como apatia, delírios, alucinações e agressividade, entre outros) na DA. O benefício terapêutico observado nesses estudos ocorre tanto no controle de sintomas comportamentais já existentes quanto no retardo do aparecimento de novos sintomas. Dessa forma, é lícito considerar o tratamento com IChE como a primeira opção no tratamento farmacológico dos sintomas neuropsiquiátricos na DA (Doody et al., 2001; Sink; Holden; Yaffe, 2005).
Nos pacientes que não respondem ao tratamento com IChE, a prescrição de neurolépticos é bastante frequente na prática clínica. No entanto, é importante ressaltar que há ainda relativa escassez de ensaios clínicos controlados com o uso de neurolépticos em pacientes com demência. Os primeiros estudos, realizados previamente à década de 1980, avaliaram neurolépticos clássicos (clorpromazina, tiotixene e haloperidol) em pacientes com demência de distintas etiologias. Posteriormente, surgiram ensaios clínicos com haloperidol, carbamazepina, loxapina, tioridazina e, mais recentemente, com os neuroléticos atípicos, em especial risperidona, olanzapina, quetiapina e aripiprazol (Kindermann et al., 2002; Sink; Holden; Yaffe, 2005; Mintzer et al., 2007; Ismail et al., 2007).
Em geral, os ensaios clínicos revelaram eficácia terapêutica dos diferentes compostos em comparação com o placebo, com perfil de eventos adversos (sobretudo extrapiramidais) mais favorável para a classe dos neurolépticos atípicos. Embora agências reguladoras, como a ANVISA no Brasil e o FDA nos Estados Unidos, não tivessem aprovado nenhuma droga dessa classe farmacológica para o tratamento de sintomas neuropsiquiátricos em pacientes com demência, os resultados desses ensaios clínicos justificaram indicações “extrabula” para a prescrição desses medicamentos em casos selecionados, utilizando-se doses mais baixas do que as empregadas em pacientes mais jovens com transtornos psicóticos e pelo menor tempo possível. A recomendação consiste em sempre iniciar o tratamento com doses mais baixas, que podem ser ajustadas conforme a resposta clínica, sem, no entanto, ultrapassar as doses máximas recomendadas. Nos casos em que não há resposta às doses máximas preconizadas, é sugerido que a medicação seja substituída por outra. Os regimes posológicos seriam, para a olanzapina, de 5 a 10 mg/dia; para a risperidona, de 0,5 a 1,5 mg/dia; para a quetiapina, de 12,5 a 100 mg/dia; e para o aripiprazol, de 7,5 a 15 mg/ dia (Kindermann et al., 2002).
Entretanto, um estudo de metanálise publicado em abril de 2005 pelo FDA, nos Estados Unidos, revelou aumento de mortalidade (sobretudo cardiovascular) relacionado ao uso dos neurolépticos atípicos em pacientes idosos com demência (Food and Drugs Administration, 2005). Recomenda-se, portanto, que o uso dessa classe de antipsicóticos seja revisto em pacientes com demência até que novos dados relativos a essa questão sejam publicados. Alternativamente, o emprego dessa classe de drogas poderia ser indicado, com extrema cautela e pelo menor tempo possível, a pacientes idosos com demência que não apresentem fatores de risco vascular (Bullock, 2005).
Outras alternativas terapêuticas (também “extrabula”) para o tratamento de quadros de agitação e agressividade, embora avaliadas de forma menos sistemática, são alguns anticonvulsivantes com efeito estabilizador do humor (especialmente carbamazepina e ácido valpróico) e o antidepressivo trazodona (Kindermann et al., 2002; Martinon-Torres; Fioravanti; Grimley, 2004; Sink; Holden; Yaffe, 2005; Herrmann; Lanctôt, 2007).
Sintomas depressivos, e mesmo depressão maior, também podem ocorrer em pacientes com DA. Nos casos em que ocorrem tais sintomas, tratamento farmacológico pode ser indicado. Ensaios clínicos com antidepressivos tricíclicos (amitriptilina, clomipramina e imipramina) e com inibidores seletivos da recaptação de serotonina (fluoxetina, citalopram, paroxetina e sertralina) demonstraram eficácia dos diferentes agentes. Poucos desses estudos compararam duas ou mais drogas antidepressivas, e a preferência é dada aos inibidores seletivos da recaptação de serotonina, devido à sua melhor tolerabilidade em pacientes idosos e ao maior risco de piora cognitiva com o uso de tricíclicos (em função dos efeitos anticolinérgicos). Dentre os inibidores seletivos, o mais bem avaliado na DA é a sertralina, que se mostrou eficaz em um ensaio clínico controlado em que a melhora da sintomatologia depressiva foi acompanhada de melhora dos sintomas comportamentais e do desempenho em atividades de vida diária (Lyketsos et al., 2003; Lebert, 2003).
O impacto do tratamento farmacológico sobre a evolução da DA ainda é objeto de discussão. Em um ensaio clínico controlado em que pacientes com DA foram aleatoriamente distribuídos em quatro grupos de tratamento (vitamina E na dose de 2.000 UI/dia, selegilina na dose de 10 mg/dia, tratamento combinado dos dois compostos e placebo), observou-se que, ao final de dois anos, os pacientes dos três grupos de tratamento tiveram evolução mais lenta da doença em relação ao grupo-placebo, sem diferenças significativas entre os grupos (Sano et al., 1997). O efeito terapêutico seria baseado na ação antioxidante da vitamina E e da selegilina. Não há, até o momento, outro estudo semelhante na literatura, que tenha buscado não apenas replicar os resultados obtidos, como ainda verificar a eficácia de doses menores de vitamina E. Recentemente, entretanto, em um estudo de metanálise (Miller et al., 2005), foi observado inesperado aumento de mortalidade associada ao emprego de vitamina E em doses iguais ou superiores a 400 UI/dia, o que mais uma vez obriga a rever o uso da suplementação vitamínica na dose previamente recomendada.
Outras medidas terapêuticas, como terapia de reposição estrogênica (em mulheres) e uso de antiinflamatórios, embora com evidências de efeito neuroprotetor em estudos epidemiológicos, não demonstraram eficácia clínica em ensaios clínicos controlados. Por essa razão, não são recomendadas no tratamento farmacológico da DA.
O manejo terapêutico da DV passa necessariamente pelas estratégias de prevenção – tanto primária quanto secundária – de eventos cerebrovasculares, com o objetivo de identificar e controlar os fatores de risco, como hipertensão arterial sistêmica, diabete melito, dislipidemia, fibrilação atrial e tabagismo.
A ocorrência de déficit de acetilcolina também está presente na DV, secundariamente ao comprometimento vascular de núcleos colinérgicos do prosencéfalo basal. Esses núcleos são irrigados por artérias perfurantes, que são comumente acometidas em pacientes hipertensos. Paralelamente, as vias de projeção colinérgica para o neocórtex trafegam pela substância branca e podem ser interrompidas por lesões vasculares subcorticais (Román, 2004).
A observação do déficit colinérgico na DV motivou a realização de ensaios clínicos com IChE. Diversos ensaios clínicos controlados com placebo têm demonstrado a eficácia dos três principais IChEs (donepezil, galantamina e rivastigmina) em pacientes com DV de intensidade leve a moderada (NE I, GR A) (Erkinjuntti; Román; Gauthier, 2004; Auchus et al., 2007). Esses estudos também costumam utilizar a escala ADAS-cog para mensurar a intensidade do comprometimento cognitivo antes e depois do tratamento. O efeito é modesto (com diferença média de dois pontos em relação ao grupo-placebo), embora significativo do ponto de vista estatístico. Há também evidências de que o tratamento com IChE produz efeitos positivos sobre sintomas neuropsiquiátricos e melhora funcional e reduz estresse (sobrecarga) do cuidador. As posologias dos IChEs na DV são as mesmas preconizadas para o tratamento da DA, com recomendações semelhantes de administração das doses (Erkinjuntti; Román; Gauthier, 2004; Róman et al., 2005; Craig; Birks, 2006).
A memantina também se mostrou eficaz em pacientes com DV, tanto em casos leves quanto em casos moderados a graves (Winblad; Poritis, 1999; Wilcock; Möbius; Stöffler, 2002; Smith; Wells; Borrie, 2006). No entanto, pacientes com comprometimento mais intenso parecem apresentar maior benefício com a medicação do que pacientes em casos mais leves. Há também sugestão de que os pacientes com DV isquêmica subcortical seriam aqueles que mais se beneficiariam do tratamento com memantina (Möbius; Stoffler, 2002).
Os ensaios clínicos controlados revelaram benefícios do tratamento com memantina em doses de 10 a 20 mg/dia, tanto sobre os sintomas cognitivos quanto em relação ao desempenho funcional, além de o tratamento reduzir a necessidade diária de cuidados dos pacientes.
Até o momento, poucos estudos avaliaram a eficácia dos IChEs e da memantina na demência mista (Langa; Foster; Larson, 2004; Potkin et al., 2006). Há evidências, no entanto, de superioridade terapêutica sobre o placebo, especialmente dos IChEs. Destes, o mais bem estudado foi a galantamina (Erkinjuntti et al., 2002), o que resultou em sua aprovação como indicação terapêutica para o tratamento da DA com DCV.
O tratamento dos fatores de risco cardiovascular, especialmente hipertensão e hiperlipidemia, bem como intervenções a fim de proteger acidentes vasculares futuros, constituem a maneira mais efetiva para proteger as funções cerebrais como prevenção primária, secundária e terciária ou diminuir a progressão da doença já estabelecida. Estudos adicionais necessitam ser realizados para definir melhor a medicação apropriada para atingir esses objetivos.
Uma perspectiva interessante para o tratamento farmacológico da DA são as estatinas. Alguns estudos epidemiológicos já haviam demonstrado associação inversa entre o uso de estatinas e o diagnóstico da doença. Recentemente, foram publicados alguns ensaios clínicos controlados que demonstraram melhora clínica na cognição de pacientes com DA (Sparks et al., 2005; Li et al., 2007; Hinerfeld et al., 2007). Inúmeros outros compostos encontram-se em fase de estudos pré-clínicos e clínicos na DA, incluindo drogas que agem sobre mecanismos fisiopatológicos mais específicos (p. ex., inibidores de secretases ou imunização antiamiloide), o que deverá resultar em formas bem mais eficazes de tratamento da doença no futuro (Machado; Caramelli, 2006).
1. Auchus AP, Brashear HR, Salloway S, Korczyn AD, De Deyn PP, Gassmann-Mayer C, et al. Galantamine treatment of vascular dementia: a randomized trial. Neurology. 2007 Jul 31;69(5):448-58.
2. Bakchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results of a randomised, double-blind, placebo-controlled 6-monthstudy. J Alzheimers Dis. 2007 Jul;11(4):471-9.Corrected and republished in: J Alzheimers Dis. 2008 Feb;13(1):97107.
3. Ballatore C, Lee VM, Trojanowski JQ. Tau-mediatedneurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007 Sep;8(9):663-72.
4. Bergman H, Chertkow H, Wolfson C, Stern J, Rush C, Whitehead V, et al. HM-PAO (CERETEC) SPECT brain scanning in the diagnosis of Alzheimer’s disease. J Am Geriatr Soc. 1997 Jan;45(1):15-20.
5. Bertolucci PH, Brucki SMD, Campacci SR, Juliano Y. Omini-exame do estado mental em uma população geral: impacto da escolaridade. Arq Neuropsiquiatr. 1994;52:1-7.
6. Bertolucci PH, Okamoto IH, Brucki SM, Siviero MO, Toniolo Neto J, Ramos LR. Applicability of the CERAD neuropsychological battery to Brazilian elderly. Arq Neuropsiquiatr. 2001 Sep;59(3-A):532-6.
7. Bertolucci PHF, Okamoto I, Neto JT, Ramos LR, Brucki SMD. Desempenho da população brasileira na bateria neuropsicológica do Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Rev de Psiq Clin. 1998 Mar/ Abr;25(2 Parte 1):80-3.
8. Black SE, Doody R, Li H, McRae T, Jambor KM, Xu Y, et al. Donepezil preserves cognition and global function in patients with severe Alzheimer disease. Neurology. 2007 Jul 31;69(5):459-69.
9. Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968 Jul;114(512):797-811.
10. Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer’s disease. J Neural Transm Suppl.1998;53:127-40.
11. Braak H, Braak E. Neuropathological stageing ofAlzheimer-related changes. Acta Neuropathol.1991;82(4):239-59.
12. Brucki SM, Nitrini R, Caramelli P, Bertolucci PH, Okamoto IH. Suggestions for utilization of themini-mental state examination in Brazil. Arq Neuropsiquiatr. 2003 Sep;61(3B):777-81.
13. Brucki SMD, Malheiros SMF, Okamoto IH, Bertolucci PHF. Dados normativos para o teste de fluência verbal categoria animais em nosso meio. Arq Neuropsiquiatr. 1997;55:5661.
14. Brun A. Pathology and pathophysiology of cerebrovascular dementia: pure subgroups of obstructive and hypoperfusive etiology. Dementia. 1994 May-Aug;5(3-4):145-7.
15. Bullock R. Treatment of behavioural and psychiatric symptoms in dementia: implications of recent safety warnings. Curr Med Res Opin. 2005 Jan;21(1):1-10.
16. Calabrese P, Essner U, Forstl H. Memantine (Ebixa) in clinical practice: results of an observational study. Dement Geriatr Cogn Disord. 2007;24(2):111-7.
17. Camozzato AL, Kochhann R, Simeoni C, Konrath CA, Pedro Franz A, Carvalho A, et al. Reliability of the Brazilian Portuguese version of the Neuropsychiatric Inventory (NPI) for patients with Alzheimer’s disease and their caregivers. Int Psychogeriatr. 2008 Apr;20(2):383-93.
18. Caramelli P, Carthery-Goulart MT, Porto CS, CharchatFichman H, Nitrini R. Category fluency as a screening test for Alzheimer disease in illiterate and literate patients. Alzheimer Dis Assoc Disord. 2007 Jan-Mar;21(1):65-7.
19. Caramelli P, Chaves ML, Engelhardt E, Machado JC, Schultz RR, Vale FA, et al. Effects of galantamine on attention and memory in Alzheimer’s disease measured by computerized neuropsychological tests: results of the Brazilian MultiCenter Galantamine Study (GAL-BRA-01). Arq Neuropsiquiatr. 2004Jun;62(2B):379-84.
20. Chaves ML, Camozzato AL, Godinho C, Kochhann R, Schuh A, de Almeida VL, et al. Validity of the clinical dementia rating scale for the detection and staging of dementia in Brazilian patients. Alzheimer Dis Assoc Disord. 2007 JulSep;21(3):210-7.
21. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997 Jul-Aug;18(4 Suppl):S1-2.
22. Craig D, Birks J. Galantamine for vascular cognitive impairment. Cochrane Database Syst Rev. 2006 Jan 25;(1):CD004746.
23. Cummings JL, Benson DF, Hill MA, Read S. Aphasia in dementia of Alzheimer type. Neurology 1985;35: 394-7.
24. Cummings JL, Benson DF. Dementia: a clinical approach. 2nd ed. Boston: Butterworths; 1992.
25. Cummings JL, Vinters HV, Cole GM, Khachaturian ZS. Alzheimer’s disease: etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology. 1998 Jul;51(1 Suppl 1):S2-17; discussion S65-7.
26. Cummings JL. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology. 1997 May;48(5 Suppl 6):S10-6.
27. Dantoine T, Auriacombe S, Sarazin M, Becker H, Pere JJ, Bourdeix I. Rivastigmine monotherapy and combination therapy with memantine in patients with moderately severe Alzheimer’s disease who failed to benefit from previous cholinesterase inhibitor treatment. Int J Clin Pract. 2006 Jan;60(1):110-8.
28. Desmond DW. The neuropsychology of vascular cognitive impairment: is there a specific cognitive deficit? J Neurol Sci. 2004 Nov 15;226(1-2):3-7.
29. Di Carlo A, Baldereschi M, Amaducci L, Lepore V, Bracco L, Maggi S, et al. Incidence of dementia, Alzheimer’s disease, and vascular dementia in Italy. The ILSA Study. J Am Geriatr Soc. 2002 Jan;50(1):41-8.
30. Doody RS, Stevens JC, Beck C, Dubinsky RM, Kaye JA, Gwyther L, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001 May 8;56(9):1154-66.
31. Eckman CB, Eckman EA. An update on the amyloid hypothesis. Neurol Clin. 2007 Aug;25(3):669-82.
32. Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet. 2002 Apr 13;359(9314):1283-90.
33. Erkinjuntti T, Román G, Gauthier S. Treatment of vasculardementia—evidence from clinical trials with cholinesterase inhibitors. J Neurol Sci. 2004 Nov 15;226(1-2):63-6.
34. Ertekin-Taner N. Genetics of Alzheimer’s disease: a centennial review. Neurol Clin. 2007 Aug;25(3):611-67.
35. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997 Oct 2229;278(16):1349-56.
36. FerreiraST,VieiraMN,DeFeliceFG.Solubleproteinoligomers asemergingtoxinsinAlzheimer’sandotheramyloiddiseases. IUBMB Life. 2007 Apr-May;59(4-5):332-45.
37. Fitzpatrick AL, Kuller LH, Ives DG, Lopez OL, Jagust W, Breitner JC, et al. Incidence and prevalence of dementia in the Cardiovascular Health Study. J Am Geriatr Soc. 2004 Feb;52(2):195-204.
38. Folstein MF, Folstein SE, McHugh PR. “Mini-mentalstate”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975 Nov;12(3):189-98.
39. Food and Drugs Administration; Public Health Advisory. Deaths with antipsychotics in elderly patients with behavioral disturbances [on-line]. Maryland, USA; 2005. [acesso 08 jul 2008] Disponível em: http://www.fda.gov/ cder/drug/advisory/antipsychotics.htm.
40. Frisoni GB, Beltramello A, Weiss C, Geroldi C, Bianchetti A, Trabucchi M. Linear measures of atrophy in mild Alzheimer disease. AJNR Am J Neuroradiol. 1996 May;17(5):913-23.
41. Herrera E Jr, Caramelli P, Silveira AS, Nitrini R. Epidemiologic survey of dementia in a communitydwelling Brazilian population. Alzheimer Dis Assoc Disord. 2002 Apr-Jun;16(2):103-8.
42. Herrmann N, Lanctôt KL. Pharmacologic management of neuropsychiatric symptoms of Alzheimer disease. Can J Psychiatry. 2007 Oct;52(10):630-46.
43. Hinerfeld DA, Moonis M, Swearer JM, Baker SP, Caselli RJ, Rogaeva E, et al. Statins differentially affect amyloid precursor protein metabolism in presymptomatic PS1 andnon-PS1 subjects. Arch Neurol. 2007 Nov;64(11):1672-3.
44. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982 Jun;140:566-72.
45. Ismail MS, Dagerman K, Tariot PN, Abbott S, Kavanagh S, Schneider LS. National Institute of Mental Health Clinical Antipsychotic Trials of Intervention EffectivenessAlzheimer’s Disease (CATIE-AD): baseline characteristics. Curr Alzheimer Res. 2007 Jul;4(3):325-35.
46. Jorm AF, Jolley D. The incidence of dementia: a metaanalysis. Neurology. 1998 Sep;51(3):728-33.
47. Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T. Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci. 2004 Nov 15;226(1-2):75-80.
48. Kindermann SS, Dolder CR, Bailey A, Katz IR, Jeste DV. Pharmacological treatment of psychosis and agitation in elderly patients with dementia: four decades of experience. Drugs Aging. 2002;19(4):257-76.
49. Lafosse JM, Reed BR, Mungas D, Sterling SB, Wahbeh H, Jagust WJ. Fluency and memory differences between ischemic vascular dementia and Alzheimer’s disease. Neuropsychology. 1997 Oct;11(4):514-22.
50. Lanctôt KL, Herrmann N, Yau KK, Khan LR, Liu BA, LouLou MM, et al. Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: ameta-analysis. CMAJ. 2003 Sep 16;169(6):557-64.
51. Langa KM, Foster NL, Larson EB. Mixed dementia: emerging concepts and therapeutic implications. JAMA. 2004 Dec 15;292(23):2901-8.
52. Lebert F. Serotonin reuptake inhibitors in depression of Alzheimer’s disease and other dementias. Presse Med. 2003 Jul 26;32(25):1181-6.
53. Li G, Larson EB, Sonnen JA, Shofer JB, Petrie EC, Schantz A, et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology. 2007 Aug 28;69(9):878-85.
54. Lopes MA, Bottino CMC. Prevalência de demência em diversas regiões do mundo: análise dos estudos epidemiológicos de 1994 a 2000. Arq Neuropsiquiatr.2002;60:61-9.
55. Lyketsos CG, DelCampo L, Steinberg M, Miles Q, Steele CD, Munro C, et al. Treating depression in Alzheimer disease: efficacy and safety of sertraline therapy, and the benefits of depression reduction: the DIADS. Arch Gen Psychiatry. 2003 Jul;60(7):737-46.
56. Machado JC, Caramelli P. Treatment of dementia: anything new? Curr Opin Psychiatry. 2006 Nov;19(6):575-80.
57. Martinon-Torres G, Fioravanti M, Grimley EJ. Trazodone for agitation in dementia. Cochrane Database Syst Rev. 2004 Oct 18;(4):CD004990.
58. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984 Jul;34(7):939-44.
59. Miller ER 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosagevitamin E supplementation may increase all-causemortality. Ann Intern Med. 2005 Jan 4;142(1):37-46.
60. Mintzer JE, Tune LE, Breder CD, Swanink R, Marcus RN, McQuade RD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind,placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007 Nov;15(11):918-31.
61. Möbius HJ, Stöffler A. New approaches to clinical trials in vascular dementia: memantine in small vessel disease. Cerebrovasc Dis. 2002;13 Suppl 2:61-6.
62. Nitrini R, Buchpiguel CA, Caramelli P, Bahia VS, Mathias SC, Nascimento CM, et al. SPECT in Alzheimer’s disease: features associated with bilateral parietotemporal hypoperfusion. Acta Neurol Scand. 2000 Mar;101(3):172-6.
63. Nitrini R, Caramelli P, Herrera E Jr, Bahia VS, Caixeta LF, Radanovic M, et al. Incidence of dementia in a communitydwelling Brazilian population. Alzheimer Dis Assoc Disord. 2004 Oct-Dec;18(4):241-6.
64. Nitrini R, Lefèvre BH, Mathias SC, Caramelli P, Carrilho PE, Sauaia N, et al. Neuropsychological tests of simple application for diagnosing dementia. Arq Neuropsiquiatr. 1994 Dec;52(4):457-65.
65. Porto CS, Fichman HC, Caramelli P, Bahia VS, Nitrini R. Brazilian version of the Mattis dementia rating scale: diagnosis of mild dementia in Alzheimer’s disease. Arq Neuropsiquiatr. 2003 Jun;61(2B):339-45.
66. Potkin SG, Alva G, Gunay I, Koumaras B, Chen M, Mirski D. A pilot study evaluating the efficacy and safety of rivastigmine in patients with mixed dementia. Drugs Aging. 2006;23(3):241-9.
67. Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003 Apr 3;348(14):1333-41.
68. Rockwood K, Bowler J, Erkinjuntti T, Hachinski V, Wallin A. Subtypes of vascular dementia. Alzheimer Dis Assoc Disord. 1999 Oct-Dec;13 Suppl 3:S59-65.
69. Román GC, Wilkinson DG, Doody RS, Black SE, Salloway SP, Schindler RJ. Donepezil in vascular dementia: combined analysis of two large-scale clinical trials. Dement Geriatr Cogn Disord. 2005;20(6):338-44.
70. Román GC. Cholinergic dysfunction in vascular dementia. Curr Psychiatry Rep. 2005 Mar;7(1):18-26.
71. Román GC. Vascular dementia. Advances in nosology, diagnosis, treatment and prevention. Panminerva Med. 2004 Dec;46(4):207-15.
72. Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al. A controlled trial of selegiline, alphatocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med. 1997 Apr 24;336(17):1216-22.
73. Shaji S, Bose S, Verghese A. Prevalence of dementia in an urban population in Kerala, India. Br J Psychiatry. 2005 Feb;186:136-40.
74. Silva DW, Damasceno PB. Demência na população de pacientes do Hospital das Clínicas da Unicamp. Arq Neuropsiquiatr. 2002;60:996-9.
75. Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA. 2005 Feb 2;293(5):596-608.
76. Smid J, Nitrini R, Bahia VS, Caramelli P. Clinical characterization of vascular dementia: retrospective evaluation of an outpatient sample. Arq Neuropsiquiatr. 2001;59: 390-3.
77. Smith M, Wells J, Borrie M. Treatment effect size of memantine therapy in Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 2006 Jul-Sep;20(3):133-7.
78. Souza DR, de Godoy MR, Hotta J, Tajara EH, Brandão AC, Pinheiro Júnior S, et al. Association of apolipoprotein E polymorphism in late-onset Alzheimer’s disease and vascular dementia in Brazilians. Braz J Med Biol Res. 2003 Jul;36(7):919-23.
79. Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Browne P, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005 May;62(5):753-7.
80. Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004 Jan 21;291(3):317-24.
81. Tatsch MF, Bottino CM, Azevedo D, Hototian SR, Moscoso MA, Folquitto JC, et al. Neuropsychiatric symptoms in Alzheimer disease and cognitively impaired, nondemented elderly from a community-based sample in Brazil: prevalence and relationship with dementia severity. Am J Geriatr Psychiatry. 2006 May;14(5):438-45.
82. Tierney MC, Black SE, Szalai JP, Snow WG, Fisher RH, Nadon G, et al. Recognition memory and verbal fluency differentiate probable Alzheimer disease from subcortical ischemic vascular dementia. Arch Neurol. 2001 Oct;58(10):1654-9.
83. Tomimoto H, Ohtani R, Shibata M, Nakamura N, Ihara M. Loss of cholinergic pathways in vascular dementia of the Binswanger type. Dement Geriatr Cogn Disord. 2005;19(56):282-8.
84. Vale FA, Miranda SJ. Clinical and demographic features of patients with dementia attended in a tertiary outpatient clinic. Arq Neuropsiquiatr. 2002 Sep;60(3-A):548-52.
85. van Dyck CH, Tariot PN, Meyers B, Malca Resnick E; for the Memantine MEM-MD-01 Study Group. A 24-weekrandomized, controlled trial of memantine in patients with moderate-to-severe Alzheimer disease. Alzheimer Dis Assoc Disord. 2007 Apr-Jun;21(2):136-43.
86. Wilcock G,Möbius HJ, StöfflerA;MMM500group. Adoubleblind,placebo-controlled multicentre study of memantine in mild to moderate vascular dementia (MMM500). Int Clin Psychopharmacol. 2002 Nov;17(6):297-305.
87. Wilkinson D, Andersen HF. Analysis of the effect of memantine in reducing the worsening of clinical symptoms in patients with moderate to severe Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;24(2):138-45.
88. Winblad B, Kilander L, Eriksson S, Minthon L, Båtsman S, Wetterholm AL, et al. Jansson-Blixt C, Haglund A; Severe Alzheimer’s Disease Study Group. Donepezil in patients with severe Alzheimer’s disease: double-blind, parallelgroup,placebo-controlled study. Lancet. 2006 Apr 1;367(9516):1057-65.Erratum in: Lancet. 2006 Jun 17;367(9527):1980. Lancet. 2006 Nov 11;368(9548):1650.
89. Winblad B, Poritis N. Memantine in severe dementia: results of the 9M-Best Study (Benefit and efficacy in severely demented patients during treatment with memantine). Int J Geriatr Psychiatry. 1999 Feb;14(2):135-46.
90. Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, et al. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama Study. Neurology. 1995 Jun;45(6):1161-8.
91. Zhang ZX, Zahner GE, Román GC, Liu J, Hong Z, Qu QM, et al. Dementia subtypes in China: prevalence in Beijing, Xian, Shanghai, and Chengdu. Arch Neurol. 2005 Mar;62(3):447-53.
www.abneuro.org.br
www.abraz.com.br
www.alz.org
www.anvisa.gov.br
www.apsen.com.br
www.cochrane.org
www.janssen-cilag.com.br
www.ludbeck.com
www.novartis.com.br
www.saude.gov.br
www.scielo.org.br
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.