(Carregando Índice)... (Carregando Índice)... |
Última revisão: 29/10/2015
Comentários de assinantes: 0
Kristine Phillips, MD, PhD
Professora Assistente, Programa de Esclerodermia, Divisão de Reumatologia, Universidade de Michigan, Ann Arbor, MI
Artigo original: Phillips K, MD, PhD. Scleroderma and Related Disorders. ACP Medicine. 2012.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Paulo Henrique Machado
Revisão técnica: Dr. Lucas Santos Zambon
As doenças do espectro da esclerodermia formam um grupo heterogêneo de distúrbios que são diferenciados por anormalidades nos tecidos conjuntivos da pele e, em alguns casos, por anormalidades em outros órgãos. Cada distúrbio se caracteriza pelo grau de envolvimento cutâneo e interno, assim como pelas características histológicas das biópsias da pele. As doenças do espectro da esclerodermia incluem esclerodermia sistêmica (ES), esclerodermia localizada e fasceíte eosinofílica (FE).
As evidências de esclerodermia sistêmica são espessamento e endurecimento da pele que não se confinam em áreas específicas. As características extracutâneas são observadas com frequência, embora um pequeno subgrupo de pacientes apresente algum envolvimento interno mínimo.
A esclerodermia sistêmica pode se apresentar em um padrão cutâneo difuso, cutâneo limitado e sem padrão cutâneo (esclerose sistêmica sem esclerodermia) ou em padrão sobreposto. A ES cutânea difusa envolve a parede torácica, o abdome, a parte superior dos braços e das coxas. A esclerose sistêmica cutânea limitada se caracteriza pela presença de esclerodermia envolvendo as mãos, os antebraços, os pés ou a face, sem envolvimento do tronco. Um subgrupo de padrão cutâneo limitado de esclerodermia era conhecido anteriormente por síndrome CREST (do inglês calcinosis, Raynaud phenomenon, esophageal dismotility, sclerodactyly, telangiectasias) que se caracterizava pela presença de calcinose, fenômeno de Raynaud, dismotilidade esofágica, esclerodactilia e telangiectasias. O termo CREST deixou de ser utilizado, em parte porque não faz referência ao envolvimento de órgãos internos. A esclerose sistêmica sem esclerodermia se caracteriza pela fibrose de órgãos internos consistente com esclerodermia e autoanticorpos, porém sem fibrose cutânea. Além das evidências de artrite reumatoide, lúpus eritematoso sistêmico, polimiosite ou da síndrome de Sjögren, os pacientes com esclerodermia apresentam uma síndrome de sobreposição. Esclerodermia é uma doença pleomórfica no sentido em que poderá ocorrer em órgãos diferentes, em tipos de processos diferentes e em estágios diferentes da doença em um mesmo paciente em um determinado ponto do curso da doença. A tabela 1 apresenta a classificação de subtipos de esclerodermia.
|
Tabela 1: Classificação das Esclerodermias
| |
|
Forma |
Síndrome
|
|
Esclerodermia (esclerose sistêmica) |
Envolvimento difuso da pele. Envolvimento limitado da pele (síndrome CREST). Características de sobreposição de doença mista nos tecidos conjuntivos.
|
|
Esclerodermia localizada |
Morfeia: placas simples ou múltiplas ou lesões generalizadas. Esclerodermia linear
|
CREST = calcinosis, Raynaud phenomenon, esophageal dismotility, sclerodactyly and telangiectasias.
As informações financeiras estão no final deste capítulo, antes das referências.
A prevalência da esclerose sistêmica é baixa, com 240 pacientes por 1 milhão de pessoas na população norte-americana e 31 pacientes por 1 milhão de pessoas na população do Reino Unido.1 O índice geral de sobrevida é pior em pacientes com esclerodermia difusa em comparação com os pacientes com esclerodermia limitada. No entanto, determinados pacientes com esclerodermia limitada apresentam taxas elevadas de mortalidade. Aproximadamente 10% de pacientes com esclerodermia limitada desenvolvem hipertensão pulmonar, que está associada a uma mortalidade de 60% no período de cinco anos.2 Da mesma forma, os prognósticos para pacientes com esclerodermia limitada que desenvolvem insuficiência renal aguda ou fibrose pulmonar são muito piores. A associação entre exposições ocupacionais ou a solventes e esclerodermia sugere a existência de uma relação causal, embora esses tipos de exposição sejam responsáveis por menos de 10% dos casos de esclerodermia.
Os estudos do sangue periférico de pacientes com esclerodermia mostram que há anormalidades nos fatores de crescimento vascular, como o fator de crescimento do endotélio vascular (VEGF), os fatores de crescimento de fibroblastos, os fatores de crescimento placentário, o fator de crescimento derivado de plaquetas e o fator de crescimento de hepatócitos (HGF). Observa-se um aumento no VEGF nos estágios iniciais da doença e relação com a ausência de ulceração digital na ponta dos dedos. Além disso, as moléculas angiostáticas também aumentam, incluindo moléculas como a endostatina, a trombospondina, a angiostatina e a endoglina solúvel.3,4
As citocinas profibróticas, como o fator de crescimento transformador ß (TGF- ß) e o fator de crescimento do tecido conjuntivo (CTGF), são anormais na esclerodermia. O CTGF ou seus produtos de clivagem é elevado no sangue periférico de pacientes com esclerodermia, sendo que este fato se correlaciona com fibrose cutânea e pulmonar, principalmente nos estágios iniciais da doença.5,6 Os níveis de várias quimiocinas, incluindo o ligante 18 (CCL18) da quimiocina (C-C motif) e o ligante 10 (CXC10) da quimiocina (C-X-C motif), são elevados e possivelmente sejam marcadores de doença pulmonar intersticial (DPI) nos casos de esclerodermia.7,8 Nos alvéolos pulmonares, o fator de necrose tumoral alfa (TGF-a), a interleucina-4 (IL-4) e a IL-5 secretados por células T CD8+ provavelmente desempenhem algum tipo de papel.
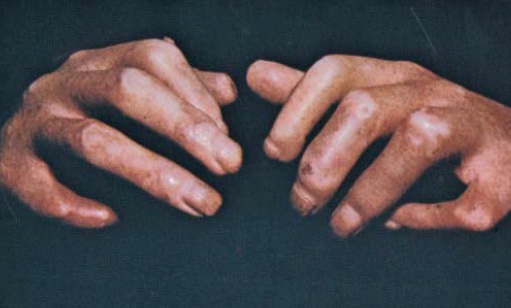
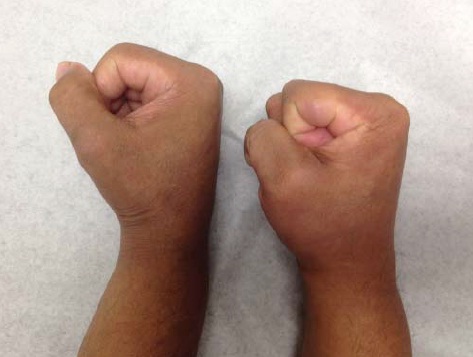
Pele. Muitos pacientes com esclerodermia desenvolvem edema cutâneo nos dedos (descrito pelos pacientes como “dedos fofos”) que é a primeira manifestação de esclerodermia não Raynaud. Logo após esta fase inicial da doença, há uma progressão para o agravamento do endurecimento e, alguns anos mais tarde, para atrofia no estágio pós-fibrótico [ver a Figura 1]. A extensão do endurecimento inicial da pele varia em cada paciente. Outras manifestações cutâneas de esclerodermia incluem contração ou fixação da pele, alteração na pigmentação, calcinose, telangiectasia, formação de fendas orais e fenômeno de Raynaud.

Figura 1: Mãos de um paciente com contraturas secundárias à esclerodermia de longa duração.
Tipicamente, o fenômeno de Raynaud se caracteriza por uma alteração episódica de três fases na cor das mãos em resposta à exposição ao frio ou a um estresse emocional [ver a Figura 2]. Um ou vários dedos desenvolve um tipo de palidez (cor branca), seguida de cianose (cor azul) e finalmente hiperemia reativa causada pela reperfusão (cor vermelha). Observa-se também a ocorrência desse fenômeno em locais como ponta do nariz, língua, orelhas ou dedos dos pés. Os capilares das dobras ungueais de pacientes com fenômeno de Raynaud devem ser avaliados por oftalmoscopia ou com um microscópio capilar de campo amplo. Quaisquer anormalidades nos capilares das dobras ungueais são preditoras do desenvolvimento de esclerodermia.9 Tortuosidade e dilatação dos microvasos são presenças comuns, sendo que a queda dos capilares das dobras ungueais está associada ao desenvolvimento de doença reumática sistêmica.8 Em pacientes que se apresentam com o fenômeno de Raynaud, altos títulos de anticorpos nucleares ou de anticorpos antinucleares (ANA ou mais conhecido em nosso meio como FAN), levantam a possibilidade de doença precoce nos tecidos conjuntivos e indicam a necessidade de um acompanhamento rigoroso. É importante ter sempre em mente que o fenômeno de Raynaud ocorre também em combinação com outros distúrbios reumáticos ou pode seguir um curso idiopático benigno (fenômeno de Raynaud primário).
Figura 2: Fenômeno de Raynaud.
De maneira geral, a síndrome de Raynaud associada à esclerodermia precede o desenvolvimento de endurecimento cutâneo em um período de meses ou mesmo de alguns anos. O intervalo de tempo desde o início do fenômeno de Raynaud até o endurecimento da pele é altamente variável, embora quase todos os pacientes (> 95%) apresentem esta condição em algum momento. Embora, às vezes, siga ou coincida com o início de esclerodermia difusa, o fenômeno Raynaud precede a esclerodermia limitada em vários anos.10 A espessura e a rigidez da pele dos dedos e das mãos aumenta e progride gradualmente a partir dos dedos – estendendo-se para as mãos – até a área do antebraço proximal. Na medida em que começa a ocorrer o envolvimento dos tendões, recomenda-se palpar ou auscultar o atrito entre os tendões, que geralmente coincide com o pico da atividade da doença.
Na fase inicial da esclerodermia, os sintomas de Raynaud podem ser facilmente revertidos com aquecimento local e vasodilatadores. No curso final da esclerodermia, condições como agravamento do endurecimento, fibrose e oclusão da microvasculatura podem contribuir para os episódios de Raynaud que se intensificam em termos de duração ou gravidade, sendo mais difícil fazer a reversão com intervenções padrão . A exposição repetida a condições como isquemia e reperfusão poderá resultar em hipóxia ou na formação de espécies reativas de oxigênio que danificam os tecidos. Nas situações em que a isquemia se agravar, ao longo do tempo poderão surgir ulcerações pequenas nas pontas dos dedos e cicatrizes digitais. Isquemia grave poderá resultar em gangrena digital e perda normal da integridade da ponta dos dedos (atrofia da extremidade digital). Esclerodactilia é a esclerose simétrica dos dedos associada à rigidez, espessamento e fixação da pele, principalmente entre a articulação interfalangiana proximal e a articulação interfalangiana distal. A inflamação dérmica é causada por um infiltrado celular inflamatório misto. A rigidez da pele pode progredir até limitar a mobilidade, com a probabilidade de desenvolver contraturas por flexão em todos os dedos e causar incapacidade profunda.
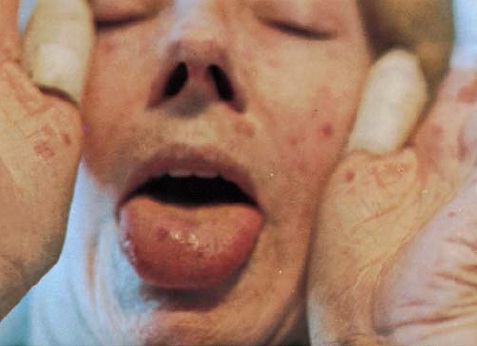
As telangiectasias são ocorrências comuns em muitos pacientes em locais como a face, as palmas das mãos ou a mucosa bucal [ver a Figura 3]. Elas podem ocorrer em grandes quantidades na face e, ocasionalmente, causar preocupações cosméticas. Embora os tratamentos a laser minimizem a aparência, os pacientes devem ser alertados sobre o fato de que muitas companhias de seguro não dão cobertura a esse tipo de tratamento, podendo haver recorrências das telangiectasias. As medicações para tratamento do fenômeno de Raynaud ou para hipertensão arterial pulmonar (HAP) que causam vasodilatação poderão também ressaltar ainda mais a aparência das telangiectasias.
Figura 3: Telangiectasias facial, palmar e bucal em um paciente com esclerodermia.
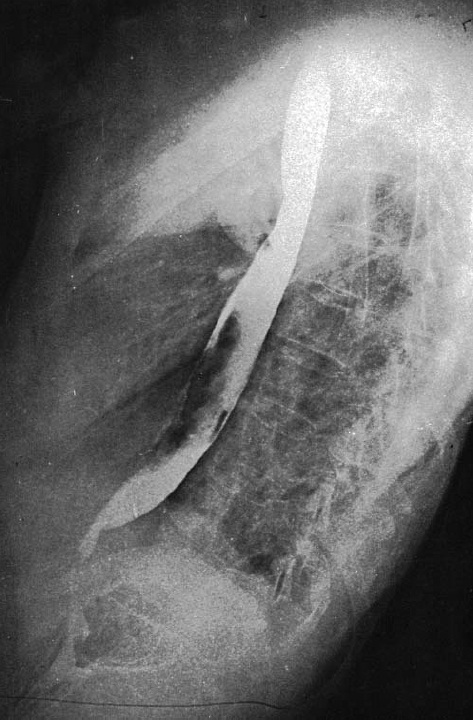
Calcinose é a deposição de cristais de hidroxiapatita de cálcio na pele [ver a Figura 4]. Trata-se de uma complicação muito temida da esclerodermia e ocorre em aproximadamente 22% de pacientes. Observa-se a calcinose com maior frequência nas áreas de atrito (i.e., ao redor da bolsa subcutânea do olécrano), limitando-se apenas a uma área. Acredita-se que a deposição de cálcio seja o resultado de fatores inflamatórios locais. A formação de cristais de hidroxiapatita de cálcio tem a tendência de ser amorfa, sendo que os cristais poderão ser liberados pela pele, e podem ocorrer também em outras áreas de tecidos moles ou de músculos. Não há medicações parenterais para prevenir ou tratar os casos de calcinose. A ocorrência de infecções bacterianas sobrepostas é comum e exige tratamento com antibióticos e desbridamento cirúrgico. De maneira geral não se recomenda o desbridamento cirúrgico em áreas não infectadas, sendo que essa hipótese deve ser considerada com muita cautela devido à possibilidade de má cicatrização. Os pacientes devem ser alertados que, mesmo com o desbridamento cirúrgico, poderá ocorrer recorrência da calcinose.
Figura 4: Radiografia mostrando a presença de calcinose no cotovelo.
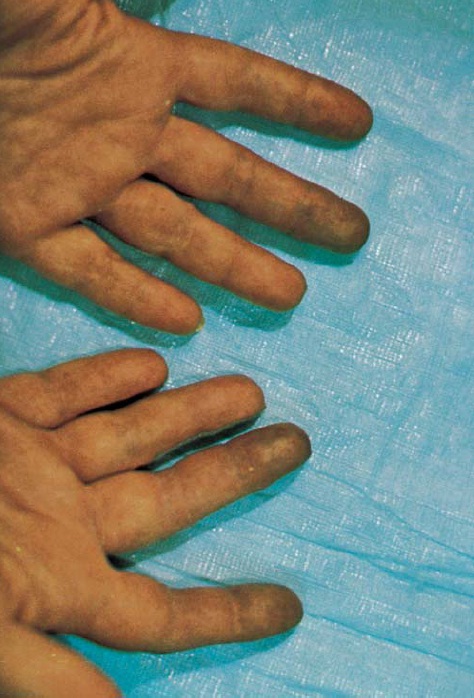
Hipopigmentação e hiperpigmentação são ocorrências prováveis nas áreas com envolvimento da pele [ver a Figura 5]. Observam-se áreas de hipopigmentação sobre as proeminências ósseas, como as clavículas e a área estiloide ulnar do punho, embora possam ocorrer em outros locais. As áreas difusas de hiperpigmentação se estendem sobre áreas de esclerose cutânea. Tanto a hipopigmentação, como a hiperpigmentação, desaparecem espontaneamente, porém, mesmo assim, a melhora total pode levar vários anos e, possivelmente, alguns pacientes nunca recuperem a pigmentação normal.
Figura 5: Mãos de irmãos gêmeos idênticos, uma com esclerodermia (à esquerda) e a outra sem esclerodermia. As características da esclerodermia na mão à esquerda incluem hipopigmentação e hiperpigmentação na pele da região dorsal da mão, esclerodactilia branda, perda da polpa ungueal normal nas extremidades dos dedos e palidez relativa secundária ao fenômeno de Raynaud à temperatura ambiente.
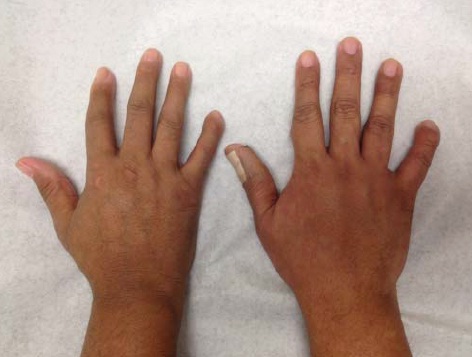
Sistema musculoesquelético. Condições como artrite inflamatória, contraturas por flexão e osteólise ocorrem com frequência nos casos de esclerodermia. Tipicamente, a artrite inflamatória associada à esclerodermia é simétrica e branda, com incidência de menos de 20% em pacientes esclerodérmicos. Com frequência, ocorrem contraturas por flexão nos dedos, assim como em outras articulações; geralmente essas contraturas são consequências da fibrose nos tendões e nas cápsulas articulares e podem diminuir a amplitude de movimento e dificultar o uso do punho [ver a Figura 6].
Figura 6: Punhos de irmãos gêmeos idênticos, um com esclerodermia (à esquerda) e o outro sem esclerodermia. Os pacientes com envolvimento das mãos em casos de esclerodermia apresentam características sutis, incluindo contraturas leves dos dedos e incapacidade para fechar totalmente o punho.
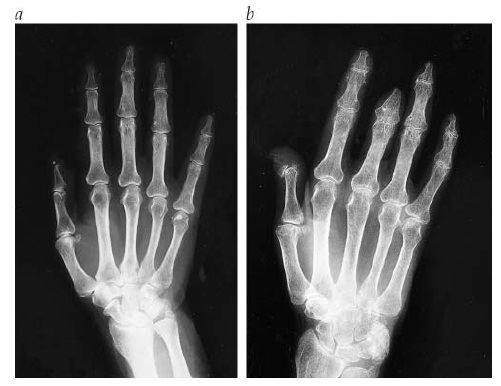
Osteólise acral é a marca registrada da esclerodermia e se caracteriza pela reabsorção das falanges terminais e hipoplasia nos tecidos moles adjacentes, resultando no encurtamento dos dedos. Em geral, observa-se esta condição nas radiografias das mãos com envolvimento de todos os dedos [ver a Figura 7].
Figura 7: (a) Radiografia da mão (filme inicial). (b) Radiografia da mão depois de oito anos mostrando a presença de osteólise acral e encurtamento dos dedos.
De maneira geral, embora apresentem um baixo grau de elevação nas enzimas musculares, os pacientes com esclerodermia não desenvolvem sintomas clínicos de miopatia. Tipicamente, nesses casos, a biópsia muscular não mostra evidências de inflamação. O prognóstico é bom para esses pacientes no que diz respeito à doença muscular, sendo que, na maioria dos casos, não é necessário fazer nenhum tipo de intervenção. Por outro lado, os pacientes com esclerodermia sobreposta e miopatia desenvolvem miosite inflamatória e poderão se beneficiar com imunossupressão específica para o tratamento de fraqueza nos músculos proximais.
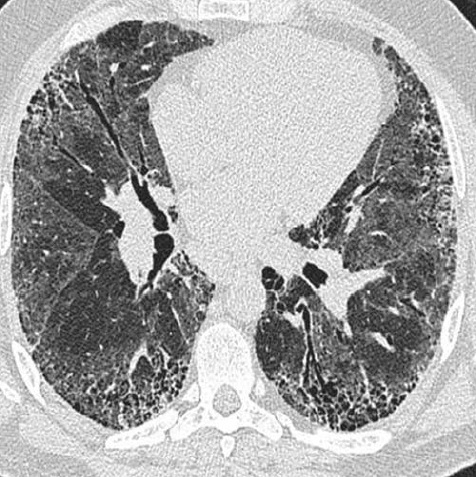
Pulmões. Doença pulmonar intersticial (DPI) é uma das causas principais de morte entre pacientes com esclerodermia sistêmica (ES) pelo fato de que alguns indivíduos com esclerodermia desenvolvem doença pulmonar restritiva grave e progressiva [ver a Figura 8]. Aproximadamente 40% dos pacientes com ES apresentam evidências de DPI através da avaliação da função pulmonar com capacidade vital forçada (CVF) inferior à previsão de 75%. Embora a fisiopatologia exata ainda não tenha sido determinada, a ativação do sistema imune possivelmente contribua para a ocorrência de danos microvasculares precoces. As lesões estruturais resultantes do desbaste microvascular e da redução no parênquima pulmonar funcional poderão produzir hipoxemia e possivelmente contribuir para a ocorrência de danos futuros por meio da formação de espécies reativas de oxigênio. As inflamações causadas por fibroblastos ativados resultam na produção excessiva de matriz extracelular, com bronquiectasia por tração, opacidades do tipo vidro fosco e fibroses que são evidentes nas imagens radiográficas, sendo que os testes da função pulmonar revelam a presença de alteração no desempenho respiratório.
Figura 8: Imagem por tomografia computadorizada de alta resolução em inspiração volumétrica e expiração incremental (posição em prono) de um paciente com esclerodermia cutânea difusa mostrando características, como opacidade do tipo vidro fosco, espessamento septal interlobular e bronquiectasia por tração. Os cistos cheios de ar representam cistos do tipo favo de mel. As descobertas representam doença pulmonar precoce em um paciente com esclerodermia.
Embora alguns especialistas defendam a importância do papel da doença por refluxo gastroesofágico (DRGE) na patogênese de doença pulmonar intersticial (DPI) nos casos de esclerodermia sistêmica, essa hipótese está cercada de controvérsias.11-13 Existem muitas formas diferentes de DPI e, nos casos de esclerodermia, o padrão histológico pulmonar predominante é a pneumonia intersticial inespecífica (PII), com pneumonite intersticial usual em uma minoria de pacientes.14
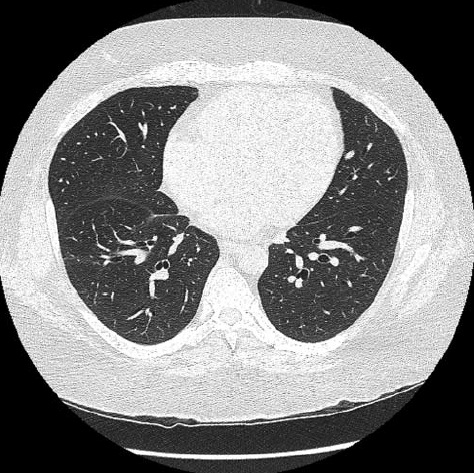
Coração. O envolvimento cardíaco é uma presença provável embora seja assintomático e tenha sido observado em 30% de pacientes em uma série de autópsias15 [ver a Figura 9]. A patogênese não é muito bem compreendida, porém pode resultar em fibrose miocárdica difusa com disfunção ventricular sistólica e diastólica ou anormalidades de condução.16,17
Figura 9: Varredura do tórax por tomografia computadorizada de alta resolução em um paciente com esclerodermia e hipertensão pulmonar grave. Observa-se um aumento no diâmetro da artéria pulmonar e uma falta de homogeneidade generalizada no parênquima pulmonar que reflete uma perfusão mosaica associada à hipertensão pulmonar.
As pressões vasculares do pulmão podem ser afetadas por DPI extensiva, disfunção cardíaca diastólica ou vasculopatia na vasculatura pulmonar de pacientes com esclerodermia. A hipertensão pulmonar resultante poderá produzir morbidade e mortalidade significativas em pacientes esclerodérmicos, além de ser uma causa importante de morte nessa população de pacientes.
A hipertensão arterial pulmonar (HAP) ocorre em um subgrupo de pacientes com hipertensão pulmonar e em pacientes com esclerodermia e é definida como níveis da pressão arterial média acima de 25 mmHg, com evidências de pressão normal de oclusão nos capilares do pulmão ou de pressão ventricular esquerda no final da diástole abaixo de 15 mmHg e resistência vascular pulmonar elevada de três unidades de Wood ou 240 dinas-8/cm2. O grupo 1 da HAP da Organização Mundial da Saúde se caracteriza pela presença de vasculopatia com remodelagem e de obstrução nos arteríolos pulmonares, sendo que a hipertensão arterial pulmonar relacionada à esclerodermia é típica nesse grupo.
Trato gastrintestinal. O envolvimento gastrintestinal nos casos de esclerodermia inclui doença por refluxo gastroesofágico (DRGE), ectasia vascular antral gástrica (EVAG) e hipomotilidade de qualquer segmento intestinal.
Muitos pacientes com esclerodermia sistêmica (ES) e hipomotilidade esofágica são assintomáticos, embora seja importante lembrar que a hipomotilidade esofágica ocorre na grande maioria de pacientes esclerodérmicos.18 A dismotilidade esofágica pode ser confirmada com um esofagrama em pacientes com alterações cutâneas sutis, nos quais pode-se considerar a probabilidade de diagnóstico de esclerodermia [ver a Figura 10]. Ondas peristálticas normais na parte inferior do esôfago e incompetência do esfíncter esofágico inferior resultam em esofagite por refluxo que, ao longo do tempo, poderá progredir para a estrutura esofágica, esôfago de Barrett ou carcinoma esofágico. Observa-se também a presença de esofagite herpética ou por cândida em pacientes imunossuprimidos.
Figura 10: Esofagrama demonstrando a presença de hipomotilidade.
O envolvimento do trato gastrintestinal possivelmente esteja associado à disfunção do nervo autonômico nos estágios iniciais que ao longo do tempo poderá resultar na atrofia dos músculos lisos e em fibrose nos músculos intestinais. Conforme se observa no esofagrama, a hipomotilidade do estômago poderá produzir sintomas de gastroparesia ou provavelmente resulte em pseudodestruição no intestino delgado. O envolvimento do cólon poderá resultar na impactação colônica ou na formação de divertículos de boca larga no cólon, que são patognomônicos de esclerodermia. O crescimento bacteriano excessivo pode resultar na ausência de movimentos peristálticos normais no intestino.
Rins. Pacientes com esclerodermia podem desenvolver proteinúria leve crônica e hipertensão, embora geralmente essas condições não exijam nenhum tipo de intervenção. Antes da década de 1970, a crise renal era a causa principal de morte em pacientes com esclerodermia, porém os resultados melhoraram substancialmente após o desenvolvimento dos inibidores da enzima conversora da angiotensina (iECAs). A taxa de mortalidade em um ano associada à crise renal caiu de 76% para menos de 15%.19 A crise renal esclerodérmica inclui o desenvolvimento rápido de hipertensão, anemia hemolítica microangiopática e insuficiência renal oligúrica. A sobrevida de cinco anos de pacientes com crise renal associada à esclerodermia é de apenas 65%, mesmo com tratamento anti-hipertensivo agressivo.20,21 Aproximadamente 90% dos pacientes com crise renal se apresentam com pressão arterial acima de 150/90 mmHg. A presença de condições como encefalopatia hipertensiva, insuficiência cardíaca congestiva ou arritmia é uma possibilidade.
Cabe lembrar que a esclerodermia sistêmica pode se desenvolver numa idade mais avançada. Tipicamente, a esclerodermia de início tardio é diagnosticada precocemente sendo que a prevalência de esclerodermia sistêmica cutânea limitada é maior nesse grupo etário. As úlceras digitais (UDs) são menos comuns. A hipertensão pulmonar foi observada com mais frequência nessa coorte de pacientes. Provavelmente este fato seja influenciado pela comorbidade cardiovascular subjacente, sendo que a disfunção diastólica contribui para a presença de hipertensão pulmonar.22
Exames de sangue. Diagnóstico. A maior dos pacientes com esclerodermia apresenta resultados positivos no teste de anticorpos antinucleares (FAN), com variações na titularidade. A titulação não se correlaciona com a atividade da doença propriamente dita. Nas situações em que for possível, o teste FAN deve ser feito por ensaio de imunofluorescência , levando-se em consideração que os ensaios de “grânulos” podem ignorar uma grande proporção de pacientes esclerodérmicos com anticorpos nucleolares. Atualmente, o ensaio da polimerase III anti-RNA encontra-se disponível no mercado. O anticorpo da antifibrilarina foi demonstrado em pacientes afro-americanos com esclerodermia.23 A Tabela 2 apresenta uma descrição de anticorpos antinucleares em casos de esclerodermia.
Tabela 2: Anticorpos Antinucleares em Esclerodermia Padrão Imunofluorescente da Coloração de Anticorpos Antinucleares Antígenos Padrão Clínico Frequência Aproximada (%) Especificidade Nucleolar Ribonucleoproteínas nucleolares (nRNPs) Esclerodermia difusa ou limitada 50 Moderada Polimerases RNA I, II e III Esclerodermia difusa 23 Alta Proteínas nucleares PM-1 (PM-Scl) e Ku Sobreposição entre esclerodermia e polimiosite <5 Alta para esclerodermia e polimiosite Fibrilarina (U3 RNP) Esclerodermia difusa 12 Alta Centromérico (manchas grandes)* Proteínas centroméricas (CENP-A, CENP-B e, CENP-C,) Usualmente em esclerodermia limitada (síndrome CREST) 50 (de pacientes com a síndrome CREST) Alta Difuso (manchas finas) DNA topoisomerase tipo 1 Esclerodermia difusa 20 a 33 Alta
*Exige uma linha de células epiteliais de carcinoma humano (HEp-2)
Os anticorpos anticentrômeros (AACs) estão associados à esclerodermia sistêmica cutânea limitada, embora também sejam observados na doença difusa. Os anticorpos anti-isopolimerase são observados em 30% de pacientes com esclerodermia difusa. Os anticorpos anti-U3RNP estão associados à esclerodermia cutânea difusa e à proteína ribonuclear Th ou à proteína ribonuclear To nos casos de doença cutânea.24 Embora os anticorpos anti-RNA polimerase I/III estejam classicamente associados à esclerodermia cutânea difusa, e possam ser muito úteis para identificar pacientes com risco de crise renal, esse tipo de teste não possui sensibilidade e especificidade. Aproximadamente 30% dos pacientes com resultados positivos para o teste de anticorpos desenvolvem crise renal, porém a metade dos pacientes com crise renal não produz esse tipo de anticorpo.22 Existe também a hipótese de uma relação temporal próxima entre o início de algumas malignidades, como câncer no pulmão e esclerodermia concomitante, em pacientes com esses anticorpos.25
O peptídeo natriurético cerebral (BNP) e seu fragmento biologicamente ativo pró-hormônio N-terminal (NT-proBNP) são liberados por miócitos ventriculares em resposta ao alongamento ou hipóxia. O nível de NT-proBNP é mais elevado em pacientes com HAP e pode ser preditor do desenvolvimento de hipertensão arterial pulmonar.26
Monitoramento. A ectasia vascular antral gástrica (EVAG) pode se apresentar com anemia assintomática, de modo que é importante considerar a hipótese de fazer um hemograma completo e um teste de sangue oculto nas fezes. A avaliação do perfil metabólico para incluir a creatinina também é uma opção válida.
A má absorção é uma ocorrência provável mesmo se não houver perda de peso. Nos casos de suspeita de má absorção, os seguintes testes devem ser considerados para avaliação futura: níveis séricos do ácido metilmalônico (elevado nos casos de má absorção), zinco (nível baixo com má absorção), ferro, vitamina B12, 25-hidroxivitamina D, nível de vitamina K ou tempo de protrombina.
Testes de outros líquidos corporais. A biópsia pulmonar e a lavagem broncoalveolar (LBA) são testes invasivos e de utilidade limitada e, consequentemente, não são feitos rotineiramente no contexto de esclerodermia sistêmica e de doença pulmonar intersticial. Embora possivelmente se correlacione com a dimensão da doença na linha de base, aparentemente a LBA não é preditora do curso futuro da esclerodermia sistêmica e da doença pulmonar intersticial.27,28
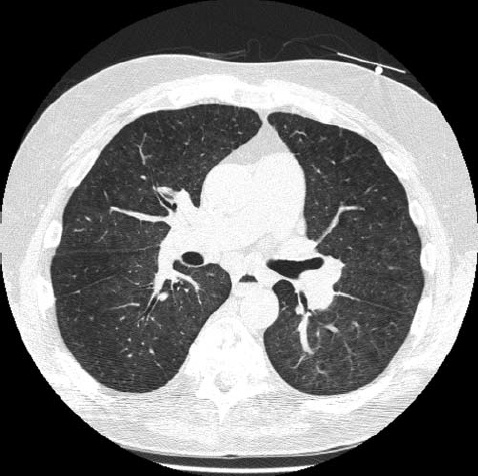
As varreduras por tomografia computadorizada de alta resolução (TCAR) produzem evidências sobre a natureza e a extensão da fibrose pulmonar29,30 [ver a Figura 11]. Provavelmente as varreduras tomográficas padronizadas ou as radiografias torácicas que não mostram os estágios iniciais da doença pulmonar intersticial não sejam as técnicas preferidas para a avaliação de doenças pulmonares nos casos de esclerodermia sistêmica. As anormalidades pericárdicas também podem ser detectadas nas varreduras por TCAR.31
Figura 11: Varredura por tomografia computadorizada de alta resolução em um paciente com esclerodermia cutânea limitada. Fibrose grave com opacidades do tipo vidro fosco com aspecto de favo de mel nas bases.
Crescimento bacteriano excessivo. O teste de respiração 14C-xilose e o teste respiratório com hidrogênio são opções para avaliar o possível crescimento bacteriano excessivo em pacientes com sintomas gastrointestinais. Alguns provedores de serviços de assistência médica disponibilizam mais rapidamente o teste respiratório com hidrogênio.
Testes da função pulmonar. A capacidade de difusão pulmonar de monóxido de carbono (DLCO) é um indicador prognóstico fraco em pacientes com esclerodermia sistêmica.32,33 Embora se reconheça que o declínio na DLCO esteja associado ao agravamento da fibrose no pulmão, este declínio pode também ser o prenúncio do desenvolvimento de hipertensão arterial pulmonar.26,34,35
Eletrocardiografia. Os testes eletrocardiográficos (ECG) podem mostrar atrasos eletromecânicos intra ou interatriais que, reconhecidamente, estão associados ao tempo de duração da doença e ao diâmetro do átrio esquerdo. A dispersão de ondas P (Pd), que possivelmente esteja associada à fibrilação atrial, pode também ser observada na ECG superficial padrão.36
Teste da caminhada de seis minutos. Embora o teste da caminhada de seis minutos seja utilizado com frequência como uma forma de medir resultados nos casos de hipertensão pulmonar, possivelmente esse tipo de teste tenha menor utilidade em esclerodermia, tendo em vista que a artrite e as contraturas da pele podem causar impactos negativos nos resultados.
Biópsia da pele. De maneira geral, a biópsia da pele não é necessária para confirmar o diagnóstico. Todavia, em alguns casos, a biópsia da pele pode ser útil para diferenciar esclerose sistêmica de outras síndromes semelhantes, tais como esclerodermia ou escleromixedema.
Biópsia dos rins. A biópsia dos rins se aplica aos casos de pacientes para os quais o diagnóstico diferencial de insuficiência renal aguda seja muito amplo. No entanto, a biópsia renal não é necessária para confirmar o diagnóstico de crise renal esclerodérmica que se apresentar na forma clássica. Observam-se também infartos e hemorragias subescapulares grosseiramente visíveis. O espessamento da íntima, com proliferação celular organizada de forma concêntrica sem células inflamatórias, é a lesão patológica típica, conhecida como lesão da pele de cebola. Necrose fibrinoide é uma ocorrência provável nas paredes arteriais sem outros sinais de vasculite. A urinálise provavelmente revele a presença de proteinúria, sendo que quaisquer evidências de hematúria não glomerular deverão ser avaliadas por um urologista.
Biópsia do pulmão. Embora, tipicamente, não faça parte dos cuidados rotineiros, a biópsia do pulmão é uma opção a ser utilizada nos casos em que os diagnósticos forem obscuros ou nas avaliações antes de transplantes de pulmão. Observam-se as características da pneumonia intersticial inespecífica (PII) com mais frequência do que as características da pneumonite intersticial usual.
Várias doenças podem se apresentar com pele clinicamente áspera ou com fibrose tecidual, e poderão ser confundidas com esclerodermia sistêmica. Históricos completos e avaliações clínicas ajudam a fazer a distinção entre essas condições e esclerodermia.
Doença do enxerto versus hospedeiro. As manifestações da doença do enxerto versus hospedeiro podem se assemelhar à esclerodermia cutânea localizada ou difusa e acredita-se que essas entidades possivelmente compartilhem uma patogênese comum. Durante a infiltração precoce de células inflamatórias (p.ex., os linfócitos) a liberação de citocinas como a IL-13 provavelmente dispare a ativação de fibroblastos resultando em fibrose.
Lesões associadas a exposições tóxicas. Existem relatos que fazem referência a lesões cutâneas que se assemelham à esclerodermia que são causadas por várias formas de toxinas ou de quimioterapias, e acredita-se que as atividades imunomoduladoras desses agentes possam desencadear a cascata imune que ativa os fibroblastos. A síndrome do óleo tóxico e o uso do L-triptofano na síndrome da mialgia eosinofílica não são comuns nos dias atuais e seu interesse é apenas histórico. As exposições ao óleo de canola desnaturado com anilina foram associadas às características semelhantes à esclerodermia, assim como às exposições ao cloreto de polivinila.
Lesões associadas a medicações. Assim como as exposições tóxicas, vários agentes quimioterápicos estão associados a reações fibróticas localizadas. Reconhecidamente, a bleomicina está associada à fibrose e foi utilizada como um dos componentes de vários modelos animais de doença fibrótica.37 Medicamentos como o paclitaxel, isoladamente, o carbidopa, isoladamente, ou o paclitaxel com carboplatina usados no tratamento de cânceres ginecológicos, como o carcinoma do endométrio ou do ovário, foram implicados nesses tipos de lesão.38 O desenvolvimento pleno desses efeitos poderá levar algumas semanas ou alguns meses e pode ser localizado ou generalizado. Tipicamente, na maior parte dos casos de esclerose cutânea associados à quimioterapia não se observam os efeitos fibróticos viscerais e o fenômeno de Raynaud característico.
A enfuvirtida é um medicamento antirretroviral novo cuja finalidade é o tratamento de HIV-1. A enfuvirtida se liga à proteína necessária para a fusão do HIV a uma célula T CD4+. A administração é pela via subcutânea e as reações no local da injeção são comuns, resultando em um infiltrado celular inflamatório, no aumento de colágeno e em uma aparência semelhante à da esclerodermia.
Síndromes genéticas. Os distúrbios progeroides (progeria) podem se apresentar na infância com manifestações cutâneas que imitam algumas características da esclerodermia difusa. A síndrome da pele rígida é causada por mutações na fibrilina-1, que faz a mediação da ligação da integrina.
Fibrose sistêmica nefrogênica (dermopatia fibrosante nefrogênica). Não foi observado nenhum pacientes com este diagnóstico antes do ano de 1997, quando alguns casos foram descritos no estado da Califórnia.39 Os pacientes se apresentam com endurecimento cutâneo fibrótico rápido associado a alterações na pigmentação e, ocasionalmente, com contraturas por flexão nas extremidades, principalmente com envolvimento simétrico das extremidades inferiores. O período de desenvolvimento das lesões varia de alguns dias a algumas semanas e se tornam crônicas. Os testes sorológicos para doença autoimune são negativos e os capilares das dobras das unhas são normais. A histopatologia revela proliferação de fibroblastos e deposição de mucina, a exemplo do que ocorre nos casos de escleromixedema. O escleromixedema está associado ao espessamento da pele com uma aparência característica de seixos, resultante da deposição de mucina na derme. A maior parte dos casos de escleromixedema está associada à gamopatia monoclonal, especificamente a imunoglobulina IgG-A. Todos os pacientes têm histórico de insuficiência renal e de tratamento com hemodiálise. Não há evidências que indiquem uma associação com o contraste intensificado com gadolínio utilizado nas imagens por ressonância magnética (IRM), embora ainda não tenha sido comprovada a existência de um papel patogenético direto. Atualmente não existe nenhum tratamento eficaz disponível.
Não há tratamentos modificadores de doença para esclerodermia, sendo que o objetivo do gerenciamento são as manifestações em órgãos específicos. Cabe lembrar que, embora o gerenciamento seja desafiador, várias intervenções podem melhorar a morbidade e a mortalidade [ver a Tabela 3].
Doença vascular pulmonar. Prostaciclinas. A prostaciclina é um vasodilatador pulmonar eficaz com propriedades antiplaquetárias e antiproliferativas. Na forma de epoprostenol (Flolan) esse medicamento comprovadamente melhora a capacidade para fazer exercícios e a hemodinâmica cardiopulmonar em pacientes com esclerodermia sistêmica e hipertensão arterial pulmonar. Entretanto, não foi observado nenhum efeito na sobrevida.
|
Tabela 3: Avaliações e Intervenções Principais no Gerenciamento de Esclerodermia
| ||
|
Sistema de Órgãos |
Teste Diagnóstico |
Intervenções Potenciais |
|
Renal |
Automonitoramento semanal da pressão arterial. |
Inibidor da ECA ou bloqueador do receptor da angiotensina.
|
|
Cardíaco e pulmonar |
ECG na linha de base, radiografia do tórax, ecocardiografia Doppler, varreduras por TC de alta resolução e testes da função pulmonar incluindo espirometria e capacidade de difusão para o monóxido de carbono.
Ecocardiografia Doppler anual para avaliar a pressão arterial pulmonar.
Testes da função pulmonar em intervalos de quatro a seis meses se a doença for sintomática ou progressiva.
TC de alta resolução se houver agravamento dos TFPs.
|
Encaminhamento para avaliação e tratamento de hipertensão arterial pulmonar ou fibrose intersticial.
Sildenafil, bosentan ou análogos da prostaciclina para hipertensão pulmonar significativa.
Ciclofosfamida e corticosteroides para fibrose pulmonar progressiva ou grave.
Micofenolato de mofetila como alternativa para a ciclofosfamida. |
|
Gastrointestinal |
Esofagofrafia com bário e endoscopia GI superior.
Avaliação para EVAG no caso de anemia inexplicável e de sangue oculto positivo.
|
Inibidor da bomba de prótons como o omeprazol.
Fotocoagulação de lesões causadas pela EVAG. |
|
Cutâneo e vascular |
Exame serial para úlceras na pele ou isquemia grave. |
Evitar o tabagismo. Terapia antiprurítica. Bloqueadores do canal de cálcio para promover vasodilatação.
|
|
Renal |
Automonitoramento semanal da pressão arterial. |
Inibidor da ECA ou bloqueador do receptor da angiotensina. |
ECA = enzima conversora da angiotensina; TC = tomografia computadorizada; ECG = ecocardiograma; EVAG =ectasia vascular antral gástrica; GI = gastrintestinal; TFP = teste da função pulmonar.
O treprostinil (Remodulin) é um análogo da prostaciclina que ainda não foi avaliado para aplicação específica em casos de esclerodermia sistêmica e hipertensão arterial pulmonar. O iloprost (Ventavis) é outro análogo da prostaciclina que também ainda não foi avaliado para aplicação específica em casos de esclerodermia sistêmica e hipertensão arterial pulmonar. Usualmente, costuma-se prescrever a terapia com administração intravenosa de prostaglandina para pacientes com esclerodermia sistêmica e hipertensão arterial pulmonar e doença funcional classe IV da New York Heart Association ou doença funcional classe III instável ou progressiva.
Antagonistas do receptor da endotelina. Os efeitos benéficos foram comprovados para o bosentan ou ambrisentam nos sintomas de FC (redução de FC) da New York Heart Association, no teste de caminhada de seis minutos (6MWT), no tempo de agravamento clínico e na hemodinâmica em pacientes com hipertensão arterial pulmonar.40 Um subgrupo desses pacientes se apresentou com hipertensão arterial pulmonar associada a doenças nos tecidos conjuntivos, embora essas terapias não tenham sido estudadas para aplicação específica nos casos de esclerodermia sistêmica e hipertensão arterial pulmonar.
Inibidores da fosfodiesterase (PDE). O sildenafil é um inibidor da fosfodiesterase que intensifica os efeitos do óxido nítrico através da inibição da decomposição do monofosfato cíclico de guanosina. Embora esse tipo de terapia ainda não tenha sido estudado para aplicação nos casos de esclerodermia sistêmica e hipertensão arterial pulmonar, os relatos anedóticos sugerem que possivelmente haja algum benefício, assim como sugere uma análise post hoc de 84 pacientes com hipertensão arterial pulmonar associada a doenças nos tecidos conjuntivos.
Doença pulmonar parenquimatosa. A terapia à base de ciclofosfamida é o tratamento mais largamente estudado para doença pulmonar intersticial (DPI) relacionada à esclerodermia sistêmica. A ciclofosfamida é um agente alquilante com efeitos imunossupressivos e vem sendo utilizada no tratamento de doenças neoplásicas, vasculite e lúpus eritematoso. O Scleroderma Lung Study (SLS) foi um teste multicentro randomizado controlado que reuniu 158 pacientes para avaliar a eficácia e a segurança no uso da ciclofosfamida por via oral durante um ano em pacientes com DPI em esclerodermia sistêmica.41 A ciclofosfamida apresentou efeitos significativos, porém modestos, no alívio da dispneia e nos índices de qualidade de vida relatados pelos pacientes. Embora os efeitos da ciclofosfamida tenham sido imediatamente visíveis por vários meses após a interrupção do tratamento, não foi observada nenhuma diferença entre os pacientes tratados e os pacientes não tratados depois de 24 meses.42 Outros estudos sugeriram que o tratamento com ciclofosfamida nos casos de esclerodermia sistêmica não melhora a qualidade de vida ou a função pulmonar.43,44 O Fibrosing Alveolitis Scleroderma Trial, um teste multicentro randomizado controlado que reuniu 45 pacientes, avaliou o desempenho dos corticosteroides e da ciclofosfamida intravenosa seguida de azatioprina em pacientes com DPI em esclerodermia sistêmica.45 Este regime melhorou a capacidade vital forçada (CVF) sem aumentar significativamente o número de eventos adversos sérios.
O micofenolato de mofetila, derivado de um antibiótico fúngico, foi usado para impedir a rejeição de órgãos transplantados e no tratamento de nefrite lúpica. O medicamento foi bem tolerado e seguro para administração em pacientes com doença pulmonar intersticial e esclerodermia sistêmica, sendo que alguns estudos de pequeno porte sugerem que o micofenolato de mofetila pode melhorar a função pulmonar.46,47
O rituximab, um anticorpo monoclonal quimérico, foi usado no tratamento de linfoma e artrite reumatoide. O antígeno CD20 na superfície dos linfócitos B é o alvo principal desse medicamento. Alguns estudos de casos sugerem que pode haver um provável benefício em pacientes com doença pulmonar intersticial e esclerodermia sistêmica que não respondem à prednisolona e à ciclofosfamida.48,49
Os pacientes esclerodérmicos devem ser imunizados contra influenza e organismos pneumocócicos. Além disso, esses pacientes devem ser orientados a evitar o tabagismo. A terapia de suplementação de oxigênio possivelmente seja indicada para pacientes com hipoxemia.
Doença renal e crise renal. Os inibidores da enzima conversora da angiotensina (iECAs) são os agentes de primeira linha para o tratamento de crise renal esclerodérmica, sendo que a dose deverá ser aumentada rapidamente aos primeiros sinais da crise. Doses mais elevadas possivelmente sejam recomendadas nos casos em que a meta principal do tratamento for o controle imediato da pressão arterial. Para atingir esta meta, nos casos em que os pacientes já estiverem tomando a dose máxima de iECAs, é necessário administrar uma terapia anti-hipertensiva adicional sob a forma de bloqueadores do canal de cálcio (BCCs), nitratos ou outros vasodilatadores. O uso de corticosteroides é contraindicado para o tratamento de crise renal esclerodérmica, sendo que ainda não se chegou a estabelecer os efeitos benéficos das medicações imunossupressivas ou das trocas plasmáticas. Embora sejam menos eficazes, os bloqueadores dos receptores da angiotensina foram usados no lugar dos inibidores da enzima conversora da angiotensina.10
Aproximadamente 50% dos pacientes com crise renal esclerodérmica provavelmente precisem fazer diálise, mesmo os que estiverem fazendo os tratamentos atuais. Entretanto, talvez isso deva ser feito apenas temporariamente, tendo em vista que até 50% desses pacientes diminuem gradualmente o uso de diálise dentro de dois anos após o início da crise renal esclerodérmica.50
Doenças musculoesqueléticas. Provavelmente condições como inflamações articulares e sinovite limitem a mobilidade dos pacientes com esclerodermia. A primeira tentativa mais adequada para o tratamento dos sintomas é a administração de acetaminofeno. Os medicamentos anti-inflamatórios não esteroidais (AINEs) comercializados sem prescrição médica são opções a serem utilizadas em doses padrões nas situações em que não houver problemas de gastrite, ectasia vascular antral gástrica (EVAG) ou doença renal. A administração de baixas doses de prednisona (5 mg por dia) é uma opção válida nos casos de agravamento dos sintomas. Nas situações em que for possível, recomenda-se evitar doses de prednisona acima de 10 mg por dia por causa do elevado potencial de risco de doença renal, principalmente em pacientes com esclerodermia sistêmica cutânea difusa. A administração de hidroxicloroquina ou de metotrexato é uma hipótese a ser considerada nos casos de pacientes com sobreposição de alguma doença ou com artrite inflamatória associada.
Doenças gastrointestinais. Os sintomas de alimento “entalado” talvez sejam uma indicação de estreitamento esofágico e justificam a endoscopia superior. A doença ativa por refluxo gastroesofágico (DRGE) deve ser tratada com um inibidor da bomba de prótons, geralmente em doses mais elevadas do que as doses normais.
A ectasia vascular antral gástrica (EVAG) é uma complicação do envolvimento gastrintestinal na esclerodermia, com grande potencial de risco de vida, que poderá ser tratada por gastroenterologistas experientes. Com bastante frequência a EVAG passa despercebida e, consequentemente, não é tratada até o desenvolvimento de anemia significativa. A aplicação de endoscopia com fotocoagulação é imprescindível nos casos de hemorragia ativa ou intermitente, sendo necessários vários cursos de tratamento antes de se conseguir uma hemostasia consistente.
O tratamento mais eficaz para o crescimento bacteriano excessivo, resultando em diarreias prolongadas, é a terapia com antibióticos orais como a rifaximina em doses de 400 mg três vezes ao dia durante dez dias. Outros antibióticos incluem 250 mg de cefalexina quatro vezes ao dia, juntamente com 250 mg de metronidazol três vezes ao dia ou 500 mg de ciprofloxacina duas vezes ao dia por um período de dez dias. No caso de doença gastrointestinal significativa associada a perdas de peso, possivelmente as melhores opções sejam encaminhar o paciente para um nutricionista ou utilizar um agente pró-motilidade logo no início do curso da doença para evitar má nutrição.51 O uso de agentes pró-motilidade como a eritromicina (100 a 150 mg quatro vezes ao dia), metoclopramida (10 a 15 mg quatro vezes ao dia) e domperidona (10 a 20 mg quatro vezes ao dia), pode ser muito útil, embora os dados sobre a eficácia sejam muito limitados. A terapia à base de octreotida (50 µg), com administração subcutânea antes de deitar, é uma alternativa a ser considerada nos casos com sintomas refratários, por causa do envolvimento do intestino delgado.52,53 As varreduras para má nutrição nos pacientes em situação de risco incluem contagem de calorias e níveis pré-albumínicos. Os níveis pré-albumínicos isoladamente não são suficientemente sensíveis para detectar má nutrição nos estágios iniciais. O teste de nutrição parenteral total (NPT) é uma opção nas situações em que houver agravamento da má nutrição.
Úlceras digitais ou de Raynaud. As abordagens terapêuticas para os casos de úlceras digitais (UDs) causadas por esclerodermia apresentam vários desafios. As UDs produzidas por isquemia vascular podem ser confundidas com UDs causadas por traumatismos ou calcinose. Os pacientes com úlceras digitais devem ser orientados a manter o corpo todo aquecido, não apenas as mãos, e evitar a ocorrência de traumatismos diretos nas articulações interfalangianas proximais e nas pontas dos dedos. Recomenda-se manter as unhas sempre aparadas e não permitir a formação de onicocriptose (unhas encravadas). As pessoas devem ser incentivadas a abandonar o tabagismo. O uso de todos os outros vasconstritores deve ser interrompido e, nas situações em que for possível, recomenda-se evitar o uso de simpatomiméticos e de beta bloqueadores.
Caso não sejam contraindicados, os agentes antiplaquetários são opções a serem consideradas. É uma prática comum orientar os pacientes com histórico recente de úlceras digitais a tomar doses baixas de aspirina (81 mg) diariamente. O clopidrogrel também já foi utilizado no tratamento de UDs, porém não há dados publicados que confirmem a eficácia ou a segurança deste medicamento.
A terapia vasoativa inclui terapia de suporte no tratamento do fenômeno de Raynaud, como os bloqueadores do canal de cálcio (BCCs). Os BCCs diminuem em 35% a gravidade dos ataques de Raynaud.54 Um teste randomizado com duração de 16 semanas comprovou que os BCCs diminuem efetivamente a carga ulcerosa (número médio de úlceras)54, que ainda é a terapia de primeira linha para aplicação em pacientes com o fenômeno de Raynaud ou com úlceras digitais. A nifedipina de ação prolongada também é usada com frequência, embora alguns pacientes tenham mais tolerância à anlodipina em comparação com a terapia crônica.
Inibidores da fosfodiesterase tipo 5. Um estudo transversal randomizado controlado55 demonstrou que os inibidores da PDE5 apresentaram resultados benéficos. Relatos de casos e séries de casos sugerem que o sildenafil produz resultados benéficos nos casos de esclerodermia sistêmica e úlceras digitais, que melhoraram de acordo com os resultados de um teste transversal feito com o tadalafil.56
Antagonistas do receptor da endotelina. Pacientes com esclerodermia e úlceras digitais apresentam níveis elevados de endotelina-1, em grande parte por causa das lesões endoteliais associadas a esse tipo de complicação. Os receptores tipo A da endotelina se localizam nas células dos músculos lisos vasculares e os receptores tipo B são encontrados nas células endoteliais e nas células dos músculos lisos vasculares. O bosentan é um antagonista com atividade em ambos os tipos de receptores, sendo que algumas séries de casos e alguns relatos anedóticos sugerem que há um possível benefício em pacientes com esclerodermia sistêmica e úlceras digitais. Alguns testes randomizados e controlados por placebo mostram que há um possível benefício em relação à carga ulcerosa em pacientes com mais de quatro úlceras, embora esses receptores não tenham produzido nenhum efeito no fenômeno de Raynaud.57
Análogos da prostaciclina. Os pacientes tratados com epoprostenol apresentaram um número de casos novos de UDs 50% mais baixo em comparação com os pacientes que haviam sido tratados com as terapias convencionais. No entanto, não se observou nenhum efeito consistente na cicatrização das úlceras digitais existentes.58
O sistema modificado de pontuação cutânea de Rodnan é um método consistente de medição de resultados.59 O questionário respiratório do Hospital St. George foi validado para uso nesta população de pacientes.60 Atualmente encontram-se em fase de desenvolvimento vários índices de respostas combinadas.
A esclerodermia localizada se divide em esclerodermia linear com um subtipo localizado em golpe de sabre (coup de sabre) e morfeia generalizada.
A esclerodermia linear é mais comum na infância e está associada a anormalidades na pele e nos tecidos subcutâneos. Na fase inicial da doença as áreas envolvidas são pequenas e localizadas e, com frequência, segue uma distribuição dermatomal e pode ser unilateral. A esclerodermia em golpe de sabre é um tipo linear e este termo se refere à semelhança com as lesões produzidas por golpes de sabre na cabeça ou no rosto. A lesão cutânea é acompanhada por anormalidades nos tecidos subjacentes derivados do mesênquima.
A morfeia se divide em doença circunscrita (localizada), que é o tipo mais comum, ou doença generalizada, que pode se assemelhar a outras formas de esclerodermia. A morfeia circunscrita se caracteriza pela presença de manchas em áreas escleróticas da pele com variações no diâmetro e na localização. Um dos subtipos é a morfeia gutata em que, de maneira geral, há envolvimento da parte superior do tronco e dos ombros com pequenas pápulas em forma de gota (gutata) hipo ou hiperpigmentadas. Embora sejam muito raras, existem relatos de doença bolhosa. As lesões cutâneas nos casos de morfeia generalizada podem se disseminar e, em geral, há envolvimento simétrico dos membros ou do tronco. É importante fazer a distinção entre morfeia generalizada grave e esclerodermia difusa. Esse tipo de morfeia não está associado a doenças vasculares ou a fibroses em órgãos internos, condições que são comuns nos casos de esclerodermia cutânea difusa. A morfeia generalizada grave exige a administração de regimes imunossupressivos para controlar doenças mais agressivas ou mais generalizadas. Morfeia profunda é uma variante da morfeia que se caracteriza pela presença de bordas mal definidas e de um componente inflamatório significativo.
Existem relatos de esclerodermia localizada depois de tratamentos radioterápicos, mesmo em pacientes sem histórico de doença nos tecidos conjuntivos.61
Alguns pacientes com esclerodermia localizada apresentam resultados positivos nos testes de autoanticorpos.62 Esses autoanticorpos não são preditores do envolvimento de órgãos internos e sua significância não é muito clara. Os autoanticorpos são encontrados frequentemente em pacientes com morfeia generalizada ou linear, principalmente os anticorpos da DNA topoisomerase II, porém atualmente este tipo de teste não está comercialmente disponível no mercado.63
A biópsia de pele é uma técnica importante para fazer a distinção entre morfeia generalizada e fasceíte eosinofílica (FE) e esclerodermia difusa. A melhor abordagem é a obtenção de uma biópsia de espessura total da fáscia considerando que as punções ou as biópsias limitadas não conseguem capturar o envolvimento subcutâneo.
O diagnóstico diferencial inclui fasceíte eosinofílica no caso de morfeia generalizada e alterações esclerodérmicas associadas à radiação no caso de morfeia localizada.
Nas situações em que for administrada por profissionais experientes, a terapia com raios ultravioleta é considerada eficaz para esclerodermia localizada. A utilidade das injeções localizadas de corticosteroides nas lesões não é uniforme, embora tenham sido aplicadas em alguns casos na fase inicial da doença. O uso concomitante de metotrexato e esteroides possivelmente seja uma opção útil nos casos da ampliação de áreas ativas logo no início do curso da doença, fazendo-se o monitoramento da toxicidade pelo metotrexato com testes laboratoriais padronizados.
A fasceíte eosinofílica (também conhecida por doença de Schulman) se apresenta com alterações cutâneas que se assemelham a esclerodermia e enrijecimento doloroso nos tecidos subcutâneos, geralmente envolvendo as extremidades. Observa-se, com frequência, contagem eosinofílica periférica elevada, sendo que a FE pode estar associada à hipergamaglobulinemia. Tipicamente, as biópsias dos tecidos profundos (biópsias de espessura total) mostram que a fasceíte é difusa. Observa-se a ausência do fenômeno de Raynaud e da esclerodactilia.
A influência real desta doença permanece desconhecida. A literatura médica registrou quase 300 casos, sendo que as primeiras descrições clínicas foram feitas por Schulman em meados da década de 1970.64 A fasceíte eosinofílica tende a ser mais frequente em homens do que em mulheres (2:1), embora na população pediátrica a prevalência seja maior em crianças do sexo feminino. Aparentemente a prevalência é maior em brancos, embora haja registros de casos em pacientes afro-americanos e asiáticos. Em relatos anedóticos e em séries pequenas há registros de casos com histórico recente de exercícios vigorosos ou de traumas.65
A fasceíte eosinofílica está associada a uma eosinofilia periférica sanguínea e tecidual e a marcadores inflamatórios elevados. Infiltrados inflamatórios de células T CD8+ e macrófagos poderão se estender desde os septos adiposos subcutâneos até a fáscia superficial e o perimísio. Observam-se níveis elevados de eosinófilos nos tecidos afetados, embora isso não seja tão visível nos casos em que os pacientes tenham iniciado terapia com corticosteroides. Fenótipos citotóxicos ativados66 e citocinas como a IL-5 e outras moléculas profibróticas, como o fator de crescimento transformador (TGF), são encontrados em pacientes com doença ativa.67,68 Outros estudos mostraram que há níveis séricos elevados de manganês superóxido dismutase e do inibidor tecidual da metaloproteinase 1 (TIMP-1).69,70
Provavelmente haja alguma associação entre fasceíte eosinofílica e outras manifestações autoimunes como as citopenias imunomediadas ou as sorologias autoimunes.
A fasceíte eosinofílica é aguda na fase inicial com dor e inchaço nas extremidades que se caracterizam pelo enrijecimento e pela progressão para fibrose cutânea. Os pacientes costumam se queixar de fadiga, dor e coceira nas áreas afetadas, sendo comum a perda de peso. A presença de dispneia e de problemas de deglutição é visível nas situações em que houver fibrose significativa no pescoço ou no tronco, embora, tipicamente, não ocorra envolvimento visceral significativo.
O exame físico revela a presença de condições como contraturas articulares, artrite, neuropatia ou miosite inflamatória. De maneira geral há envolvimento do antebraço, que poderá apresentar enrijecimento, alterações do tipo casca de laranja ou sinais de sulcos. Sinais de sulcos são ranhuras lineares na pele que acompanham o percurso das veias e resultam da perda de tecido subcutâneo nessas áreas. A síndrome do túnel do carpo foi observada em um número substancial de pacientes. Aproximadamente 25% dos pacientes apresentam alguma lesão concomitante de morfeia localizada.
Exames de sangue. A grande maioria, porém nem todos, dos pacientes se apresenta com eosinofilia periférica. Os pacientes podem apresentar também hipergamablobulinemia policlonal ou níveis ligeiramente elevados de enzimas musculares (creatina quinase e aldolase). Observa-se um aumento no número de marcadores inflamatórios (taxa de sedimentação eritrocitária e proteína C reativa) nos estágios iniciais da doença, que poderá ter alguma utilidade no acompanhamento do curso clínico. Não é comum a presença de anticorpos antinucleares (FAN), fator reumatoide ou anticorpos nucleares extraíveis. Em até 10% de pacientes com eosinofilia periférica poderão ocorrer condições como anemia imunomediada, trombocitopenia imunomediada, aplasia de eritrócitos puros, anemia aplásica ou síndromes mielodisplásicas.
Estudos de imagens. As imagens por ressonância magnética (IRM) facilitam a confirmação do diagnóstico e a determinação da extensão da doença. As imagens ponderadas em T2 (sensíveis aos líquidos) provavelmente mostrem a presença de edema muscular e o grau de espessamento fascial e a extensão do edema. As imagens poderão ser instrumentos úteis para recomendar a obtenção de biópsias de espessura total, sendo que as imagens usadas no acompanhamento poderão ajudar no monitoramento das respostas às terapias ou na avaliação da possibilidade de recidiva da doença.71
Biópsias. Eventualmente, para confirmar o diagnóstico, talvez seja necessário obter biópsias cutâneas incisionais de espessura total (biópsias em cunha) da superfície da fáscia ou dos músculos. Embora a pele possa parecer normal e a epiderme seja preservada, a fáscia profunda poderá apresentar infiltração de linfócitos, de células plasmáticas e, com frequência, de eosinófilos.
É imprescindível fazer a distinção entre fasceíte eosinofílica e esclerodermia cutânea difusa e morfeia generalizada. Alguns médicos acreditam que a fasceíte eosinofílica seja um tipo de morfeia, outros não concordam com essa hipótese. Há um grande interesse histórico em torno do fato de que duas entidades clínicas resultantes da ingestão de contaminantes tóxicos sejam semelhantes à fasceíte eosinofílica. Essas entidades incluem o óleo de granola desnaturado com anilina (síndrome do óleo tóxico), síndrome descrita na Espanha em 1981, e a síndrome da mialgia eosinofílica associada ao L-triptofano descrita nos Estados Unidos em 1989.72,73 Os dois distúrbios apresentaram agrupamentos e se caracterizavam pela presença de eosinofilia, fibrose cutânea e evidências de fasceíte. Essas duas síndromes apresentavam características como febre, curso mais agudo e prognósticos piores.
A fisioterapia pode ser importante durante o curso da doença para limitar as contraturas por flexão ou a atrofia muscular e a subsequente incapacitação.
Os corticosteroides são eficazes como tratamento de primeira linha para os casos de fasceíte eosinofílica em mais de 70% de pacientes. Outras terapias foram testadas com graus variados de eficácia, sendo que não há nenhum estudo de larga escala que permita orientar a terapia. A resolução espontânea foi observada em alguns casos.
O início imediato do tratamento com prednisona (ou um corticosteroide equivalente) em doses de 1 mg/kg/dia possivelmente reduza a inflamação e os sintomas. Usualmente, mantém-se essa dose durante o período de algumas semanas a um mês e, em seguida, faz-se a retirada gradual e lenta do medicamento. Pode-se iniciar a administração de uma medicação imunossupressiva de segunda linha, como o metotrexato ou o micofenolato de mofetila, como agente poupador de esteroides ou nas situações em que a doença for extensiva.
De maneira geral, o prognóstico para pacientes com fasceíte eosinofílica é bom. A maior parte dos pacientes que recebe tratamento logo no início do curso da doença consegue a remissão da enfermidade. Existem também relatos de remissões espontâneas. As variáveis clínicas associadas aos piores resultados incluem idade jovem no início, presença de lesões por morfeia e envolvimento do tronco.74
A autora não mantém nenhuma relação comercial com os fabricantes dos produtos ou com os fornecedores dos serviços mencionados neste capítulo.
1.Fathi N, Furst DE, Clements PJ. Management of interstitial lung disease in systemic sclerosis: lessons from SLS and FAST. Curr Rheumatol Rep 2007; 9:144–50.
2.Stupi AM, Steen VD, Owens GR, et al. Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum 1986; 29:515–24.
3.Distler O, Del Rosso A, Giacomelli R, et al. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of ?ngertip ulcers. Arthritis Res 2002;4:R11.
4.Hummers LK, Hall A, Wigley FM, Simons M. Abnormalities in the regulators of angiogenesis in patients with scleroderma. J Rheumatol 2009; 36:576–82.
5.Sato S, Nagaoka T, Hasegawa M, et al. Serum levels of connective tissue growth factor are elevated in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary ?brosis. J Rheumatol 2000; 27:149– 54.
6.Dziadzio M, Smith RE, Abraham DJ, et al. Circulating levels of active transforming growth factor beta1 are reduced in diffuse cutaneous systemic sclerosis and correlate inversely with the modi?ed Rodnan skin score. Rheumatology (Oxford) 2005; 44:1518–24.
7.Luzina IG, Atamas SP, Wise R, et al. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol 2002; 26:549–57.
8.Prasse A, Pechkovsky DV, Toews GB, et al. CCL18 as an indicator of pulmonary ?brotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum 2007;56:1685–93.
9.Koenig M, Joyal F, Fritzler MJ, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: atwenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum2008; 58:3902–12.
10.Denton CP, Black CM. Scleroderma—clinical and patho- logical advances. Best Pract Res Clin Rheumatol 2004;18: 271–90.
11.Christmann RB, Wells AU, Capelozzi VL, Silver RM. Gastroesophageal re?ux incites interstitial lung disease in systemic sclerosis: clinical, radiologic, histopathologic, and treatment evidence. Semin Arthritis Rheum 2010; 40:241–9.
12.Carlo-Stella N, Belloli L, Barbera R, et al. Gastroesophageal re?ux and lung disease in systemic sclerosis. Am J Respir Crit Care Med 2009; 179:1167; author reply 1168.
13.Gilson M, Zerkak D, Wipff J, et al. Prognostic factors for lung function in systemic sclerosis: prospective study of 105 cases. Eur Respir J 2010; 35:112–7.
14.Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of ?brosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med2002; 165 :1581–6.
15.D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of?fty-eight autopsy cases and ?fty-eight matched controls. Am J Med 1969; 46:428–40.
16.Botstein GR, LeRoy EC. Primary heart disease in systemic sclerosis (scleroderma): advances in clinical and pathologic features, pathogenesis, and new therapeutic approaches. Am Heart J1981; 102 :913–9.
17.Bulkley BH, Ridol? RL, Salyer WR, Hutchins GM. Myocardial lesions of progressive systemic sclerosis. A cause of cardiac dysfunction. Circulation 1976;53:483–90.
18.Sallam H, McNearney TA, Chen JD. Systematic review: pathophysiology and management of gastrointestinal dysmotility in systemic sclerosis (scleroderma). Aliment Pharmacol Ther2006; 23:691–712.
19.Steen VD, Costantino JP, Shapiro AP, Medsger TA Jr. Out- come of renal crisis in systemic sclerosis: relation to avail- ability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med1990; 113:352–7.
20.Teixeira L, Mouthon L, Mahr A, et al. Mortality and risk factors of scleroderma renal crisis: a French retrospective study of 50 patients. Ann Rheum Dis 2008; 67:110–6.
21.Walker JG, Ahern MJ, Smith MD, et al. Scleroderma renal crisis: poor outcome despite aggressive antihypertensive treatment. Intern Med J 2003; 33:216–20.
22.Hugle T, Schuetz P, Daikeler T, et al. Late-onset systemicsclerosis—a systematic survey of the EULAR scleroderma trials and research group database. Rheumatology (Oxford) 2011; 50:161–5.
23.Sharif R, Fritzler MJ, Mayes MD, et al. Anti-?brillarin anti- body in African American patients with systemic sclerosis:
immunogenetics, clinical features, and survival analysis. J Rheumatol 2011; 38:1622–30.
24.Mitri GM, Lucas M, Fertig N, et al. A comparison betweenanti-Th/To- and anticentromere antibody-positive systemic sclerosis patients with limited cutaneous involvement. Arthritis Rheum 2003; 48:203–9.
25.Shah AA, Rosen A, Hummers L, et al. Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies. Arthritis Rheum 2010; 62: 2787–95.
26.Allanore Y, Borderie D, Avouac J, et al. High N-terminal pro-brainnatriuretic peptide levels and low diffusing capacity for carbon monoxide as independent predictors of the occurrence of precapillary pulmonary arterial hypertension in patients with systemic sclerosis. Arthritis Rheum 2008; 58:284–91.
27.Strange C, Bolster MB, Roth MD, et al. Bronchoalveolar lavage and response to cyclophosphamide in scleroderma interstitial lung disease. Am J Respir Crit Care Med 2008; 177: 91–8.
28.Goh NS, Veeraraghavan S, Desai SR, et al. Bronchoalveolar lavage cellular pro?les in patients with systemic sclerosis- associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum 2007; 56:2005–12.
29.Strollo D, Goldin J. Imaging lung disease in systemic sclerosis. Curr Rheumatol Rep 2010; 12:156–61.
30.Goldin JG, Lynch DA, Strollo DC, et al. High-resolution CT scan ?ndings in patients with symptomatic scleroderma- related interstitial lung disease. Chest 2008; 134:358–67.
31.Fischer A, Misumi S, Curran-Everett D, et al. Pericardial abnormalities predict the presence of echocardiographically de?ned pulmonary arterial hypertension in systemic sclero- sis-relatedinterstitial lung disease. Chest 2007; 131:988–92.
32.Peters-Golden M, Wise RA, Hochberg MC, et al. Carbon monoxide diffusing capacity as predictor of outcome in systemic sclerosis. Am J Med 1984; 77:1027–34.
33.Morgan C, Knight C, Lunt M, et al. Predictors of end stage lung disease in a cohort of patients with scleroderma. Ann Rheum Dis2003; 62:146–50.
34.Steen V, Medsger TA Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum 2003; 48:516–22.
35.Hachulla E, de Groote P, Gressin V, et al. The three-year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitu- dinal study in France. Arthritis Rheum 2009; 60:1831–9.
36.Aktoz M, Yilmaztepe M, Tatli E, et al. Assessment of ventricular and left atrial mechanical functions, atrial electromechanical delay and P wave dispersion in patients with scleroderma. Cardiol J 2011; 18:261–9.
37.Finch WR, Rodnan GP, Buckingham RB, et al. Bleomycin- induced scleroderma. J Rheumatol 1980; 7:651–9.
38.Clowse ME, Wigley FM. Digital necrosis related to carboplatin and gemcitabine therapy in systemic sclerosis. J Rheumatol2003; 30:1341–3.
39.Cowper S, LeBoit P, Su L, et al. Fibrosing skin condition among patients with renal disease – United States and Europe, 1997–2002.MMWR Morb Mortal Wkly Rep 2002;51: 25–6.
40.Launay D, Sitbon O, Le Pavec J, et al. Long-term outcome of systemic sclerosis-associated pulmonary arterial hyper- tension treated with bosentan as ?rst-line monotherapy followed or not by the addition of prostanoids or sildena?l. Rheumatology (Oxford) 2010; 49:490–500.
41.Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophospha- mide versus placebo in scleroderma lung disease. N Engl J Med2006;354:2655–66.
42.Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-yeartreatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med2007;176:1026–34.
43.Khanna D, Furst DE, Clements PJ, et al. Oral cyclophosphamide for active scleroderma lung disease: a decision analysis. Med Decis Making 2008; 28: 926–37.
44.Nannini C, West CP, Erwin PJ, Matteson EL. Effects of cyclophosphamide on pulmonary function in patients with scleroderma and interstitial lung disease: a systematic review andmeta-analysis of randomized controlled trials and observational prospective cohort studies. Arthritis Res Ther 2008; 10:R124.
45.Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, pro- spective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary ?brosis in scleroderma. Arthritis Rheum 2006; 54: 3962– 70.
46.Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest 2008;133:455–60.
47.Zamora AC, Wolters PJ, Collard HR, et al. Use of mycophe- nolate mofetil to treat scleroderma-associated interstitial lung disease. Respir Med 2008; 102:150–5.
48.McGonagle D, Tan AL, Madden J, et al. Successful treat- ment of resistant scleroderma-associated interstitial lung disease with rituximab. Rheumatology (Oxford) 2008;47: 552–3.
49.Lafyatis R, Kissin E, York M, et al. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum 2009; 60:578–83.
50.Steen VD, Medsger TA Jr. Long-term outcomes of sclero- derma renal crisis. Ann Intern Med 2000; 133:600–3.
51.Baron M, Bernier P, Cote LF, et al. Screening and therapy for malnutrition and related gastro-intestinal disorders in systemic sclerosis: recommendations of a North American expert panel. Clin Exp Rheumatol 2010; 28(2 Suppl 58): S42–6.
52.Perlemuter G, Cacoub P, Chaussade S, et al. Octreotide treatment of chronic intestinal pseudoobstruction secondary to connective tissue diseases. Arthritis Rheum 1999; 42: 1545–9.
53.Soudah HC, Hasler WL, Owyang C. Effect of octreotide on intestinal motility and bacterial overgrowth in scleroderma. N Engl J Med 1991; 325:1461–7.
54.Thompson AE, Shea B, Welch V, et al. Calcium-channel blockers for Raynaud’s phenomenon in systemic sclerosis. Arthritis Rheum2001;44:1841–7.
55.Gore J, Silver R. Oral sildena?l for the treatment of Rayn- aud’s phenomenon and digital ulcers secondary to systemic sclerosis. Ann Rheum Dis. 2005; 64(9):1387.
56.Shenoy P, Agarwal V. Phosphodiesterase inhibitors in the management of autoimmune disease. Autoimmun Rev2010; 9:511–5.
57.Chung L, Fiorentino D. Digital ulcers in patients with systemic sclerosis. Autoimmun Rev 2006; 5:125–8.
58.Badesch DB. Continuous intravenous Epoprostenol for pulmonary hypertension due to the scleroderma spectrum disease. Ann Intern Med 2000; 132: 425–434.
59.Kondo H, Rabin BS, Rodnan GP. Stimulation of lymphocyte reactivity by a low molecular weight cutaneous antigen in patients with progressive systemic sclerosis (scleroderma). J Rheumatol 1979; 6:30–7.
60.Beretta L, Santaniello A, Lemos A, et al. Validity of the Saint George’s Respiratory Questionnaire in the evaluation of thehealth-related quality of life in patients with interstitial lung disease secondary to systemic sclerosis. Rheumatology (Oxford)2007;46:296–301.
61.Colver GB, Rodger A, Mortimer PS, et al. Post-irradiationmorphoea. Br J Dermatol 1989; 120:831–5.
62.Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford) 2006; 45: 614–20.
63.Hayakawa I, Hasegawa M, Takehara K, Sato S. Anti-DNAtopoisomerase IIalpha autoantibodies in localized scleroderma. Arthritis Rheum 2004; 50:227–32.
64.Shulman LE. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia: a new syndrome? J Rheumatol 1974;1:46.
65.Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum 1988; 17:221–31.
66.Toquet C, Hamidou MA, Renaudin K, et al. In situ immunophenotype of the in?ammatory in?ltrate in eosinophilic fasciitis. J Rheumatol 2003; 30: 1811–5.
67.Viallard JF, Taupin JL, Ranchin V, et al. Analysis of leukemia inhibitory factor, type 1 and type 2 cytokine production in patients with eosinophilic fasciitis. J Rheumatol 2001; 28: 75–80.
68.Dziadzio L, Kelly EA, Panzer SE, et al. Cytokine abnormalities in a patient with eosinophilic fasciitis. Ann Allergy Asthma Immunol2003; 90:452–5.
69.Jinnin M, Ihn H, Yamane K, et al. Serum levels of tissue inhibitor of metalloproteinase-1 and 2 in patients with eosinophilic fasciitis. Br J Dermatol 2004; 151:407–12.
70.Jinnin M, Ihn H, Yazawa N, et al. Elevated serum levels of manganese superoxide dismutase in patients with eosinophilic fasciitis. Clin Rheumatol 2003; 22:505.
71.Baumann F, Bruhlmann P, Andreisek G, et al. MRI for diagnosis and monitoring of patients with eosinophilic fasciitis. AJR Am J Roentgenol 2005; 184:169–74.
72.Sánchez-Porro Valadés P, Posada de la Paz M, de Andrés Copa P, et al. Toxic oil syndrome: survival in the whole cohort between 1981 and 1995. J Clin Epidemiol 2003; 56:701– 8.
73.Hertzman PA, Blevins WL, Mayer J, et al. Association of theeosinophilia-myalgia syndrome with the ingestion of tryptophan. N Engl J Med 1990; 322:869–73.
74.Endo Y, Tamura A, Matsushima Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol2007; 26: 1445–51.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.