(Carregando Índice)... (Carregando Índice)... |
Última revisão: 07/03/2018
Comentários de assinantes: 0
Naomi D. L. Fisher, MD
Divisão de Endocrinologia, Diabetes e Hipertensão no Brigham and Women’s Hospital (Boston, MA).
Gail K. Adler, MD, PhD
Divisão de Endocrinologia, Diabetes e Hipertensão no Brigham and Women’s Hospital (Boston, MA).
|
Artigo original: Fisher, N, MD. Adler, G, PhD. Adrenal Hypertension. SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Resumo
As causas secundárias de hipertensão estão associadas à quantidade excessiva dos hormônios principais produzidos pelas glândulas suprarrenais: cortisol, epinefrina e aldosterona. A produção excessiva de aldosterona é conhecida por hiperaldosteronismo primário ou aldosteronismo primário (AP). Todos os indivíduos com AP têm grande risco de incidência de uma grande variedade de distúrbios, tais como fibrilação atrial (FA), doença arterial coronariana (DAC), infarto agudo do miocárdio (IAM) e acidente vascular cerebral (AVC). Feocromocitoma, um tipo raro de tumor (responsável por, ao menos, um em cada 10 mil casos de hipertensão), é marcado por níveis elevados de secreção de catecolaminas, principalmente epinefrina e norepinefrina. A doença de Cushing e a síndrome de Cushing são apresentadas em uma revisão separada.
Definição
As glândulas suprarrenais produzem três hormônios principais: cortisol, epinefrina e aldosterona, sendo que cada um deles está envolvido na manutenção da pressão arterial em níveis normais e, além disso, estão associados à hipertensão quando produzidos em excesso. Os focos principais deste artigo serão as causas secundárias de hipertensão produzidas pela produção excessiva do hormônio adrenal.
Nesta revisão, serão discutidas a hipertensão causada pelo excesso de aldosterona (por exemplo, hiperaldosteronismo primário) e a hipertensão resultante do excesso de catecolaminas (feocromocitoma). Cabe ressaltar a ausência de condições como doença de Cushing e síndrome de Cushing, causadas pelo excesso na produção de glicocorticoides (cortisol) pelas glândulas suprarrenais, que são apresentadas nas revisões dos respectivos tópicos.
Entretanto, levando-se em consideração que a doença de Cushing e a síndrome de Cushing resultam da ativação do cortisol do receptor de mineralocorticoides, os mecanismos discutidos na última seção sobre hiperaldosteronismo são aplicáveis.
Visão Geral
As duas glândulas suprarrenais se localizam no topo de cada rim, sendo que cada uma delas mede, aproximadamente, 5cm na dimensão maior. Essas glândulas são esferas comprimidas que se assemelham a triângulos ou a meias-luas nos estudos de imagens. Às vezes, é possível distinguir a porção central da glândula e os dois segmentos. A título de referência dimensional, a espessura da glândula não deve ser superior ao do segmento ipsilateral do diafragma.
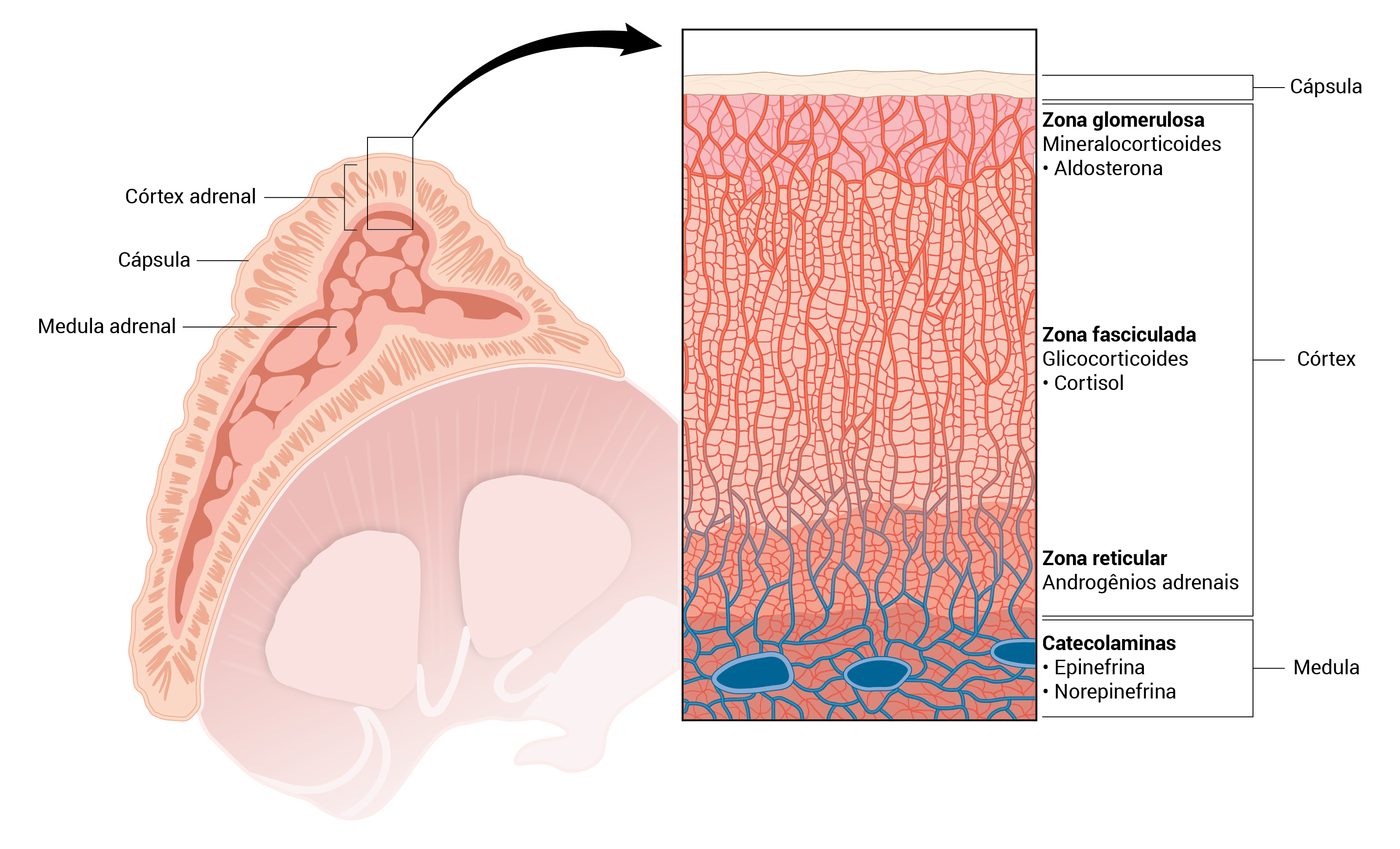
Sob o ponto de vista histológico, a glândula suprarrenal é composta de um córtex e de uma medula. Partindo da cápsula interna, observam-se três zonas: glomerulosa, fasciculada e reticular. A aldosterona é produzida na zona glomerulosa; o cortisol, na zona fasciculada; e os hormônios esteroides sexuais, na zona reticular.
A glândula suprarrenal segrega pequenas quantidades de estrogênio e androgênio, assim como a androstenediona e o sulfato de dehidroepiandrosterona, que são esteroides precursores do androgênio. As catecolaminas são produzidas na medula adrenal, cuja estrutura é análoga à de um glânglio simpático, já que é composta de células de cromatina inervadas por axônios simpáticos pré-sinápticos.
A Figura 1 mostra uma glândula suprarrenal normal.

Figura 1 - Glândula suprarrenal normal. Este diagrama mostra a secção transversal de uma glândula suprarrenal à esquerda, com a anatomia detalhada das zonas que produzem hormônios à direta.
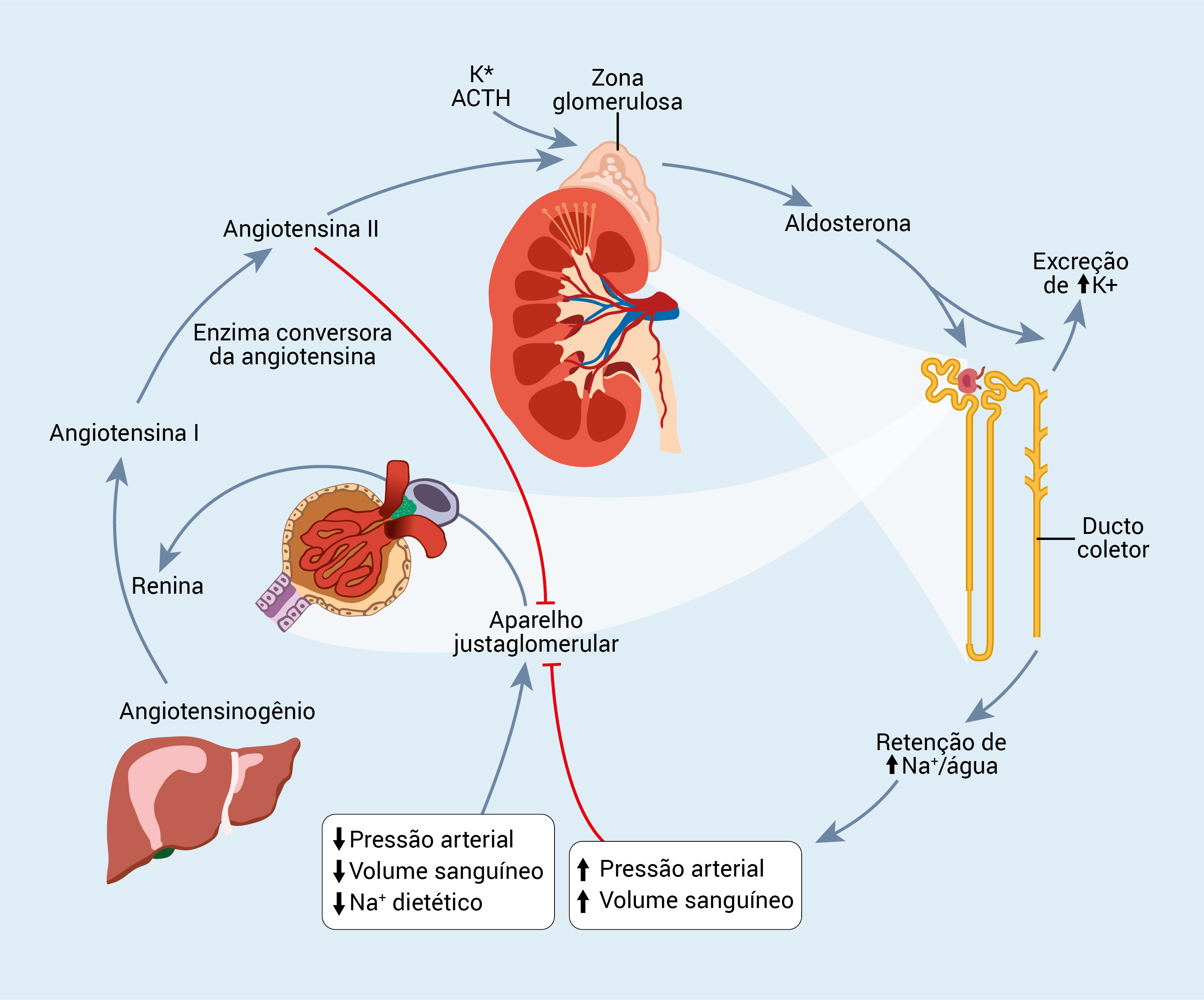
A aldosterona é o principal mediador do sódio e da homeostase volumétrica, além de promover a retenção de sal e água. A concentração da aldosterona é regulada por loops de feedback. A renina, um hormônio polipeptídeo secretado pelas células justaglomerulares renais, hidrolisam o angiotensinogênio em angiotensina I, que, a seguir, é convertida em angiotensina II pela enzima conversora da angiotensina, principalmente nos pulmões.
Em combinação com o hormônio adrenocorticotrófico (em inglês, adrenocorticotropic hormone [ACTH]) e com o potássio, a angiotensina II estimula a síntese e a secreção da aldosterona adrenal. A aldosterona age no ramo espesso ascendente do loop de Henle, reforçando a retenção de sal e expandindo o volume vascular.
A Figura 2 mostra a regulação da produção de aldosterona.
ACTH: hormônio adrenocorticotrófico.
Figura 2 - Regulação da produção de aldosterona. Os principais estimuladores do sistema renina-angiotensina-aldosterona são queda na pressão arterial, volume sanguíneo ou consumo de sódio dietético. Por outro lado, a elevação na pressão arterial suprime o sistema.
A epinefrina, que é uma catecolamina (conhecida também por adrenalina), tem papel importante na resposta de luta ou fuga (os dois termos são idênticos sob o ponto de vista etimológico, sendo que o primeiro é derivado do latim ad renal [para os rins] e o último do grego epi nefros [sobre os rins]). A ação das catecolaminas é constringir a vasculatura e estimular a renina, ativando, consequentemente, o sistema renina-angiotensina.
A hidrocortisona - ou cortisol - é extremamente importante para a vida. As causas adrenais de hipertensão devem ser levadas em conta nas situações em que não houver probabilidade de hipertensão essencial, ou quando sinais, sintomas ou descobertas laboratoriais específicas sugerirem um diagnóstico secundário, conforme mostra o Quadro 1.
Quadro 1
|
HIPÓTESES DE SUSPEITAS DE CAUSAS ENDÓCRINAS DE HIPERTENSÃO |
|
Início antes da idade de 25 anos ou após a idade de 50 anos. Pressão fora de controle, mesmo com uso de três medicamentos, incluindo um diurético (resistente à hipertensão). Sintomas: palpitações, cefaleias, sudorese (feocromocitoma). Sinais: ruído abdominal (estenose na artéria renal); estrias roxas largas e outros estigmas da síndrome de Cushing. Resultados laboratoriais: hipocaliemia (hiperaldosteronismo).
|
Hiperaldosteronismo
Epidemiologia
O hiperaldosteronismo primário, geralmente abreviado para aldosteronismo primário (AP), se caracteriza pelo aumento na produção de aldosterona causado por algum tipo de anormalidade adrenal. A aldosterona age sobre as células epiteliais do túbulo coletor renal, cuja finalidade é promover a reabsorção de sódio e a excreção de potássio e hidrogênio.
Considerando que a reabsorção de sódio resulta na reabsorção de água, com frequência os indivíduos com excesso de aldosterona desenvolvem hipertensão, assim como hipocaliemia e alcalose metabólica. De acordo com algumas estimativas, entre 2 a 10% de pacientes com hipertensão e em até 20% de pacientes com hipertensão resistente têm AP.1-3 O AP ocorre igualmente em homens e mulheres e em todas as idades.
O hiperaldosteronismo secundário ocorre nos casos em que houver ativação do sistema renina-angiotensina, aumentando a produção de aldosterona. As causas de hiperaldosteronismo secundário que produzem hipertensão incluem estenose na artéria renal, doença parenquimatosa renal e, muito raramente, tumores que produzem renina.
O hiperaldosteronismo secundário ocorre com mais frequência em resposta a deficiências volumétricas intravasculares crônicas resultantes de condições como insuficiência cardíaca ou asceíte associada à cirrose hepática. Consequentemente, a epidemiologia do hiperaldosteronismo secundário reflete a epidemiologia do distúrbio subjacente.
Etiologia/Genética
A causa do AP é um grupo de distúrbios esporádicos e familiares. As formas esporádicas de AP incluem adenoma que produz aldosterona (APA) ou hiperplasia adrenal idiopática (HAI)/hiperplasia adrenal bilateral (HAB). Os carcinomas que produzem aldosterona são extremamente raros. Até o presente momento, há três formas hereditárias conhecidas de AP: hiperaldosteronismo familiar (HF) tipos I, II e III.4
O HF-I ou aldosteronismo remediável por glicocorticoides (ARG) é uma síndrome autossômica dominante causada por uma mutação crossover, resultando em um novo gene híbrido que produz quantidades excessivas de aldosterona, como consequência do controle anormal pelo ACTH.
Especificamente, o promotor do gene 11ß-hidroxilase (CYPP11B1), responsivo ao ACTH (gene que codifica a etapa enzimática final na via biossintética do cortisol), aciona a expressão do gene aldosterona sintase (CYP11B2), que codifica a etapa enzimática final na síntese da aldosterona.5 Em HF-I, o tratamento com dexametasona suprime os níveis de ACTH e desativa a expressão do gene híbrido aberrante CYP11B1/CYP11B2, reduzindo os níveis de aldosterona.
Ainda não se sabe qual é o defeito genético que leva ao HF-II. O HF-III é causado por mutações no gene KCNJ5, que codifica o canal 4 de potássio (K+) acoplado ao retificador interno da proteína G ativada.6 Essas mutações KCNJ5 produzem alterações no potencial membranoso das células da zona glomerulosa e, consequentemente, aumentam a produção de aldosterona. Cabe ressaltar que, aproximadamente, 40% dos adenomas que produzem aldosterona têm mutações não germinais esporádicas no gene KCNJ5.
Patogênese
A capacidade da aldosterona para induzir o quadro clássico de hipertensão, hipocaliemia e alcalose metabólica depende da ativação dos receptores renais de mineralocorticoides. Por outro lado, essas condições resultam na ativação do canal de sódio epitelial (em inglês, epithelial sodium channel [ENaC]), que estimula a retenção de sódio e de água e promove a excreção de potássio e hidrogênio.
A restrição na ingestão de sódio neutraliza os efeitos da aldosterona por meio da redução na retenção renal de sódio, reduzindo, consequentemente, a retenção volumétrica e a excreção de potássio e hidrogênio. Além disso, a suscetibilidade de determinados indivíduos aos efeitos totais do excesso de aldosterona varia substancialmente. Por exemplo, alguns indivíduos apresentam elevações na pressão arterial sem hipocaliemia.
Diagnóstico
Manifestações Clínicas
Os pacientes com AP se apresentam com hipertensão e, com frequência, com hipocaliemia e alcalose metabólica. Levando-se em consideração que o hiperaldosteronismo esporádico pode ocorrer em qualquer idade, o novo início de hipocaliemia é uma possibilidade de AP.
O diagnóstico deve também ser considerado em pacientes com hipertensão e nos casos de lesões suprarrenais ou em pacientes com hipertensão resistente. O início de hipertensão em indivíduos mais jovens, assim como um forte histórico familiar de início precoce de hipertensão, é uma grande possibilidade de AP familiar.
O risco de fibrilação atrial (FA), hipertrofia ventricular esquerda, doença arterial coronariana (DAC), infarto agudo do miocárdio (IAM), acidente vascular cerebral (AVC), danos renais e problemas no metabolismo da glicose, é muito maior em indivíduos com AP, em comparação com indivíduos com hipertensão essencial.8?11
A etiologia dessas complicações cardiometabólicas não é atribuída aos efeitos clássicos sobre a homeostase do sal e da água, mas aos outros efeitos cardiometabólicos da aldosterona. A expressão do receptor mineralocorticoide se materializa através de múltiplos tipos de células, sendo que sua ativação resulta na ocorrência de condições como inflamação, estresse oxidativo, danos vasculares, disfunção vascular, lesão cardíaca, remodelagem cardíaca, lesão renal, AVC e distúrbios metabólicos.12?14
As consequências clínicas desses efeitos adversos se tornam imediatamente aparentes em pacientes com AP, o que leva muitos médicos a identificar e tratar indivíduos com esse tipo de distúrbio.
Exame Físico
O exame físico revela a presença de pressão arterial elevada simétrica entre os braços. O restante do exame geralmente não apresenta nenhum fato significativo.
Testes Laboratoriais
Exames de sangue. Nos casos de AP, a elevação no nível de aldosterona resulta na retenção de sódio e água, expansão de volume e supressão na atividade da renina plasmática (ARP). Consequentemente, os testes de rastreamento de AP incluem ARP, níveis séricos ou plasmáticos de aldosterona e níveis de potássio.15 Níveis elevados de aldosterona em combinação com níveis baixos de ARP e, portanto, proporção elevada entre aldosterona e ARP sugerem a presença de AP.
Os valores utilizados com mais frequência são superiores a 15ng/dL para aldosterona e acima de 30 para a razão aldosterona/ARP, com a aldosterona expressa em ng/dL e a ARP em ng/mL/hora.11,16 Entretanto, o ponto de corte recomendado para a razão aldosterona/ARP varia entre valores acima de 20 a valores acima de 40, dependendo do grupo específico de investigação.11,17
Considerando que o potássio estimula a secreção de aldosterona, é extremamente importante manter o nível sérico de potássio na faixa normal para evitar a ocorrência de resultados negativos falsos. Muitas medicações anti-hipertensivas afetam a razão aldosterona/ARP.
Entretanto, de um modo geral, é possível interpretar essa razão sem interromper a terapia anti-hipertensiva excetuando-se os antagonistas do receptor de mineralocorticoides, que devem ser descontinuados por 4 semanas antes da medição da ARP e da aldosterona. Alguns médicos defendem a suspensão no uso de todos os diuréticos antes de iniciar os testes.
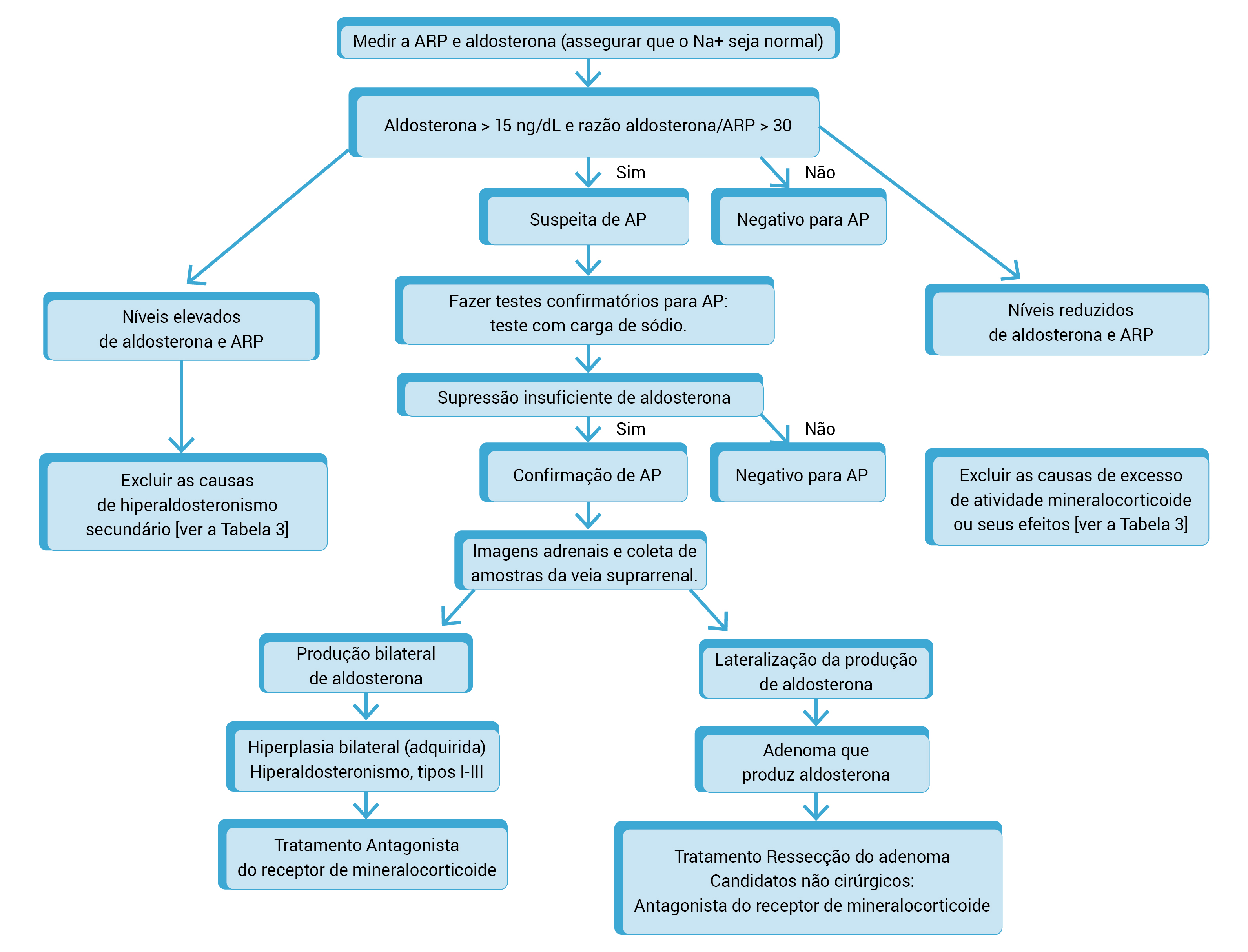
A Figura 3 mostra o exame completo para hiperaldosteronismo primário em um paciente hipertenso.
AP: hiperaldosteronismo primário; ARP: atividade da renina plasmática.
Figura 3 - Exame completo para hiperaldosteronismo primário em um paciente hipertenso. A avaliação inicia com a medição da atividade da renina plasmática e da aldosterona.
Testes fisiológicos. Os pacientes com resultado positivo no teste de rastreamento deverão fazer ao menos um teste confirmatório para possibilitar a determinação do diagnóstico. Esses testes confirmatórios incluem três versões do teste de supressão de sal (supressão salina intravenosa, supressão dietética oral de sal e supressão de fludrocortisona) e um teste de desafio com captopril.
Seja qual for o teste de supressão utilizado, antes que ele seja iniciado, o nível sérico de potássio do paciente deverá ser suplementado até atingir a faixa normal e o nível de potássio precisa ser monitorado, levando-se em consideração que a carga de sal abaixa o nível de potássio em pacientes com AP. Reduções excessivas nos níveis séricos de potássio poderão gerar consequências clínicas adversas e produzir resultados negativos falsos.
O Quadro 2 apresenta o efeito das medicações anti-hipertensivas sobre a atividade da renina plasmática e a aldosterona.
Quadro 2
|
EFEITO DAS MEDICAÇÕES ANTI-HIPERTENSIVAS SOBRE A ATIVIDADE DA RENINA PLASMÁTICA E A ALDOSTERONA
| |
|
Medicações |
Mecanismo
|
|
Medicações que aumentam a razão aldosterona/ARP |
|
|
Bloqueador ß |
Diminui a renina mais do que a aldosterona |
|
Agonista a2 central |
Diminui a renina mais do que a aldosterona |
|
Inibidor da renina |
Diminui a renina pelo ensaio da ARP (eleva a CDR); diminui a aldosterona
|
|
Medicações que diminuem a razão aldosterona/ARP |
|
|
Diuréticos com perda de potássio (loop e tiazida) |
Aumenta a renina mais do que a aldosterona |
|
Diurético com preservação de potássio |
Aumenta a renina mais do que a aldosterona |
|
Ieca/bloqueador do receptor da angiotensina II |
Aumenta a renina, pode levar a uma redução modesta no nível de aldosterona |
|
BCC (especialmente o do tipo dihidropiridina) |
Aumento modesto na renina; efeito mínimo sobre a aldosterona |
|
Antagonista do receptor de mineralocorticoides |
Aumenta a renina mais do que a aldosterona |
ARP: atividade da renina plasmática; BCC: bloqueador do canal de cálcio; CDR: concentração direta da renina; Ieca: inibidor da enzima conversora da angiotensina.
O teste intravenoso (IV) com solução salina e o teste oral com carga de sódio são os testes confirmatórios mais comuns. No teste IV com solução salina, os pacientes incapazes recebem infusões com 2 litros de solução salina normal durante 4 horas. Níveis séricos de aldosterona acima de 10ng/dL, ao final do período de infusão, são consistentes com hiperaldosteronismo, sendo que valores de 5 a 10ng/dL são indeterminados.11
No teste oral de carga de sódio, os pacientes recebem doses adicionais de sal por meio de caldos ou de comprimidos de cloreto de sódio durante 4 dias para elevar o consumo diário de sódio acima de 200mEq/dia. Recomenda-se coletar uma amostra de urina de 24 horas do terceiro ao quarto dia. A presença de AP é confirmada por níveis urinários de aldosterona acima de 12 a 14µg por 24 horas e níveis urinários de sódio acima mEq por 24 horas.15,16
O Quadro 3 apresenta o diagnóstico diferencial para ARP anormal e níveis anormais de aldosterona, incluindo os casos de AP.
Quadro 3
|
DIAGNÓSTICO DIFERENCIAL DE ATIVIDADE ANORMAL DA RENINA PLASMÁTICA E NÍVEIS DE ALDOSTERONA | |||
|
Condição |
Efeito sobre os níveis de aldosterona e ARP |
Causas possíveis | |
|
Aumento no nível de aldosterona e na razão aldosterona/ARP |
Hiperaldosteronismo primário |
Adenoma que produz aldosterona Hiperplasia bilateral idiopática Hiperaldosteronismo familiar, tipos I?III Carcinoma que produz aldosterona (causa muito rara)
| |
|
Redução no nível de aldosterona e na razão aldosterona/ARP |
Excesso de atividade mineralocorticoide |
Síndrome de Cushing Ingestão de alcaçuz Excesso aparente de mineralocorticoides Hiperplasia adrenal congênita (deficiência de 11ß-hidroxilase ou 17a-hidroxilase) Tumores que produzem deoxicorticosterona (causa muito rara)
| |
|
|
Efeito excessivo de mineralocorticoides |
Síndrome de Liddle Pseudo-hipoaldosteronismo tipo 2
| |
|
Aumento no nível de aldosterona e na ARP |
Hiperaldosteronismo secundário |
Causas de hiperaldosteronismo secundário que resultam em hipertensão: |
Estenose na artéria renal Hipertensão essencial (nível elevado de renina) Hiperatividade simpática Tumor que produz renina (causa muito rara)
|
|
|
|
Causas de hiperaldosteronismo secundário que não resultam em hipertensão: |
Depleção volumétrica Hemorragia Insuficiência cardíaca Cirrose hepática Abuso crônico de diuréticos Vômito/diarreia crônico Abuso de laxantes Síndrome de Bartter Pseudo-hipoaldosteronismo tipo 1 |
ARP: atividade da renina plasmática.
O teste de supressão de fludrocortisona por 4 dias não é feito com muita frequência, tendo em vista que sua execução é muito difícil no ambiente ambulatorial. O teste de desafio com captopril aproveita-se da vantagem da capacidade desse medicamento para diminuir a angiotensina II de forma aguda e, consequentemente, o nível de aldosterona nos casos de hipertensão essencial, mas não nos casos de AP. Esse tipo de teste perdeu sua importância porque não reproduz alta especificidade e sensibilidade.
Estudos de imagens. As imagens podem ser obtidas por meio de varreduras na glândula suprarrenal por tomografia computadorizada (TC) ou por imagens por ressonância magnética (IRM), sendo que ambas as técnicas conseguem detectar a presença de hiperplasia adrenal bilateral e adenomas adrenais.
Entretanto, considerando que a ocorrência de adenomas adrenais não funcionais é muito comum, a presença de alguma lesão adrenal não significa necessariamente que ela seja a fonte do excesso de produção de aldosterona. Os adenomas não funcionais e os aldosteronas apresentam as mesmas características de alto teor de gordura nos estudos com imagens. Por esse motivo, recomenda-se coletar amostras das veias suprarrenais na maioria dos pacientes que forem candidatos cirúrgicos.
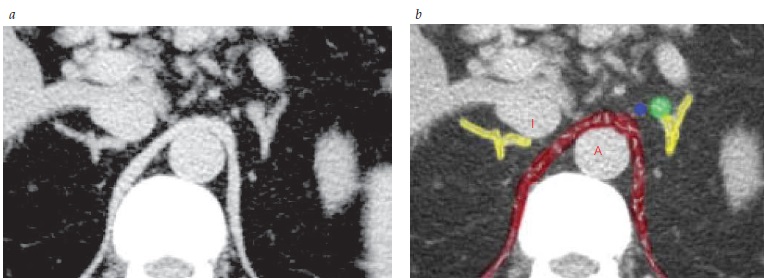
A Figura 4 mostra imagens de hiperaldosteronismo.
TC: tomografia computadorizada.
A: aorta; I: veia cava inferior.
Figura 4 - Imagens de hiperaldosteronismo. (A) Imagens por TC de um adenoma adrenal esquerdo causando hiperaldosteronismo. (B) Sob o ponto de vista cirúrgico, o pequeno nódulo verde com dimensão abaixo de um centímetro localizado no topo da imagem adrenal esquerda é comprovadamente um aldosteronoma. As glândulas suprarrenais aparecem em amarelo; os pilares do diafragma estão sobrepostos na cor vermelha. O ponto azul indica a veia suprarrenal esquerda.
Coleta de amostras na veia suprarrenal. A coleta de amostras na veia suprarrenal ajuda a determinar se uma ou ambas as glândulas suprarrenais são fontes de excesso de aldosterona. Esse tipo de procedimento deverá ser feito somente nos casos em que o paciente for candidato à ressecção cirúrgica de alguma lesão adrenal lateralizante.
A coleta de amostras da veia suprarrenal deverá ser feita por um especialista na técnica e com bastante experiência na colocação precisa de cateteres. O sangue usado para determinar os níveis de aldosterona e de cortisol deve ser obtido na veia cava inferior (ou em uma veia periférica) e em ambas as veias suprarrenais na linha de base e após a administração intravenosa do ACTH.
Níveis de cortisol na veia adrenal pelo menos 2 a 3 vezes superiores aos níveis de cortisol na veia periférica (10 vezes após a administração de ACTH) indicam que o cateter está no interior da veia suprarrenal.11 A seguir, é possível determinar a razão entre aldosterona e cortisol em cada glândula suprarrenal. Recomenda-se a lateralização na produção de aldosterona nas situações em que a razão entre aldosterona e cortisol for superior em uma glândula suprarrenal em relação à outra; de modo geral, utiliza-se uma razão de 4:1 para indicar a lateralização.11
Diagnóstico Diferencial
O AP deve ser diferenciado das síndromes de excesso de atividade mineralocorticoide ou do efeito não mediado pela aldosterona e de hiperaldosteronismo secundário. Nessas síndromes, os pacientes são hipertensos, embora ocorra supressão da ARP e da aldosterona. Por outro lado, nos casos de hiperaldosteronismo secundário, os pacientes poderão ser hipertensos ou não, e os níveis de ARP e de aldosterona são elevados.
Excesso de atividade mineralocorticoide independente da aldosterona
A síndrome do excesso aparente de mineralocorticoides (EAM) é marcada pela presença de hipertensão e hipocaliemia, embora ocorra supressão na ARP e nos níveis de aldosterona. Nesse distúrbio raro, há um aumento na produção de mineralocorticoides, excetuando-se a aldosterona.
A forma familiar de EAM é causada por meio da inativação de mutações na enzima 11ß-hidroxiesteroide desidrogenase tipo 2 (11ßHSD2).18 O cortisol e a aldosterona ativam o receptor de mineralocorticoides. Nos rins e em outros tecidos, a 11ßHSD2 converte o cortisol em cortisona inativa, impedindo a ativação do receptor de mineralocorticoides pelo cortisol.
O cortisol não é desativado nas situações em que a 11ßHSD2 não é funcional. Ele se liga ao receptor de mineralocorticoides causando hipertensão, hipocaliemia e acidose metabólica. Com frequência, as formas genéticas de EAM se apresentam na infância. Nos casos em que a atividade da 11ßHSD2 é normal, os níveis urinários de cortisol de 24 horas são mais baixos que os níveis urinários de cortisona.
Nos casos de EAM, ocorre o inverso; os níveis de cortisona são baixos e a razão entre cortisol e cortisona é elevada. Em crianças, razões urinárias entre cortisol e cortisona acima de 5 sugerem a presença de EAM. O excesso aparente de mineralocorticoides pode ser diagnosticado por meio de testes genéticos.
O uso de antagonistas do receptor de mineralocorticoides é o tratamento de primeira linha. Como opção, pode-se usar a amilorida e o triantereno para reduzir a atividade do canal de sódio. Para finalizar, a administração de um glicocorticoide que não ativar o receptor de mineralocorticoides (por exemplo, dexametasona) levará à supressão da produção do ACTH e também à redução do cortisol endógeno.
Existem duas condições clínicas adquiridas nas quais a enzima 11ßHSD2 está associada ao excesso de atividade mineralocorticoide. Uma das formas adquiridas é causada pela ingestão de alcaçuz ou de outros compostos (por exemplo, carbenoxolona) que inibem a atividade da enzima 11ßHSD2.19
O ácido glicirretínico, um composto que está presente no alcaçuz derivado da raiz da Glycyrrhiza glabra, inibe a enzima 11ßHSD2 (o alcaçuz é usado na fabricação de doces, embora muitos confeitos denominados alcaçuz não contenham extrato da raiz de alcaçuz e, portanto, não podem causar esse tipo de síndrome), assim como o fumo de mascar e a goma de mascar. O tratamento se limita a interromper a ingestão de alcaçuz.
A síndrome de Cushing é um segundo distúrbio que poderá ser considerado uma forma adquirida de excesso aparente de mineralocorticoides e que apresenta níveis extremamente elevados de cortisol, como os níveis que ocorrem nos casos de ACTH ectópico.
Nesse contexto, a capacidade de a enzima 11ßHSD2 converter todo o cortisol em cortisona excede os limites normais e os pacientes desenvolvem hipertensão, hipocaliemia e alcalose metabólica. A produção de mineralocorticoides adrenais, que não seja a aldosterona, pode contribuir para o excesso de atividade mineralocorticoide nos casos de síndrome de Cushing.
O gerenciamento médico desses pacientes envolve tratamentos para limitar a produção de cortisol e para bloquear os receptores de glicocorticoides e de mineralocorticoides. O uso de adrenalectomia emergencial é uma opção a ser considerada, levando-se em consideração os efeitos clínicos devastadores desses níveis muito elevados de cortisol na síndrome de Cushing ectópica.
A produção excessiva de mineralocorticoides adrenais, que não seja a aldosterona, pode levar à síndrome de excesso de mineralocorticoides. A presença de tumores adrenais produz deoxicorticosterona mineralocorticoide.20 Além disso, há duas formas de hiperplasia adrenal congênita nas quais ocorre produção excessiva de mineralocorticoides: deficiência de 11ß-hidroxilase e de 17a-hidroxilase. Nesses tipos de distúrbio, os níveis de ARP e de aldosterona são baixos.
A hiperplasia adrenal congênita é facilmente diferenciada do excesso aparente de mineralocorticoides, tendo em vista que os pacientes se apresentam na infância com hiperplasia adrenal congênita clássica com crise adrenal devido a problemas na produção de cortisol. Nesses casos, os pacientes são tratados com glicocorticoides que abaixam o nível do ACTH e diminuem a produção excessiva de mineralocorticoides.
Efeitos do Excesso de Mineralocorticoides Independentes do Nível de Aldosterona
Nas condições com efeitos do excesso de mineralocorticoides independentes do nível de aldosterona, há defeitos nos alvos posteriores do receptor de mineralocorticoides resultando em condições como hipertensão, hipocaliemia e alcalose metabólica. A aldosterona e a atividade da renina plasmática são suprimidas e não ocorre nenhum aumento em outros ligantes do receptor de mineralocorticoides.
A síndrome de Liddle é causada por um defeito genético no gene SCNN1B ou SCNN1G. Esses genes codificam subunidades do ENaC. O defeito genético prejudica a remoção dos canais de sódio da superfície das células tubulares coletoras, resultando no excesso de atividade no canal de sódio epitelial.22 Os pacientes portadores da síndrome de Liddle se apresentam no início da vida com hipertensão, hipocaliemia e alcalose metabólica.
O teste genético é o método mais confiável para diagnosticar esse tipo de síndrome. O tratamento dessa síndrome é feito com amilorida ou triantereno com o objetivo de bloquear o excesso da atividade no canal de sódio epitelial; os antagonistas do receptor de mineralocorticoides não são eficazes, tendo em vista que o defeito se localiza em um ponto posterior em relação ao receptor de mineralocorticoides.
O pseudo-hipoaldosteronismo tipo 2 é o resultado de mutações em WNK1 e WNK4, que produzem alterações na atividade do canal de sódio epitelial.21 Os pacientes com esse distúrbio genético podem apresentar, em qualquer idade, condições como expansão de volume, hipertensão e supressão na atividade da renina plasmática.
Apesar de serem variáveis, os níveis de aldosterona não são necessariamente suprimidos. Além disso, ao contrário do que ocorre nos casos de AP, o defeito no canal de sódio resulta em hipercaliemia, mas não em hipocaliemia, sendo que a hipercaliemia geralmente precede a hipertensão. Esse tipo de distúrbio não se refere simplesmente ao efeito do excesso de mineralocorticoides, tendo em vista o efeito diferencial sobre o potássio. Os tratamentos incluem hidroclorotiazida e ligantes potenciais do potássio.
Hiperaldosteronismo secundário
O hiperaldosteronismo secundário pode ou não estar associado à hipertensão. Tipicamente, esta doença está associada à hipertensão e resulta de alguma patologia renal subjacente, incluindo estenose na artéria renal, insuficiência renal crônica e, muito raramente, tumores secretores de renina. O tratamento deverá ter como foco a causa subjacente.
O hiperaldosteronismo secundário associado à pressão arterial normal ou à hipotensão geralmente ocorre em distúrbios que se caracterizam pela presença de volume vascular reduzido. As causas renais incluem nefrite crônica, acidose tubular renal e nefropatias com perda de cálcio e magnésio. O abuso crônico de diuréticos também é uma das causas.
As causas gastrintestinais incluem vômito crônico, abuso de laxantes e diarreia crônica de qualquer natureza. Provavelmente, as causas mais comuns sejam insuficiência cardíaca crônica e cirrose hepática com asceíte. O melhor tratamento é aquele com foco no distúrbio subjacente.
Os distúrbios familiares que prejudicam a retenção de sal e água mediada pela aldosterona pelos rins levam à depleção de volume e, secundariamente, à ativação do sistema renina-angiotensina-aldosterona. A síndrome de Bartter é causada por um déficit no transporte de cloreto no ramo espesso ascendente do loop de Henle e está associada a alcalose hipocalêmica, hiperreninemia e hiperaldosteronismo.23
O padrão de anormalidades eletrolíticas imita o padrão observado nos casos de uso abusivo de diuréticos. Pseudo-hipoaldosteronismo tipo 1 é outro distúrbio familiar que se apresenta nos quadros de níveis elevados de ARP e de aldosterona, mas não nos quadros de hipertensão. Nesse tipo de distúrbio, as mutações levam a um receptor não funcional de mineralocorticoides ou a um canal de sódio epitelial não funcional, resultando em depleção volumétrica.24
Gerenciamento
O tratamento médico de AP poderá ser feito com um antagonista do receptor de mineralocorticoides ou com ressecção cirúrgica de um aldosteronoma. Além disso, a restrição ao consumo dietético de sódio minimiza os efeitos do volume e do potássio típicos do excesso de aldosterona.
A meta dos antagonistas do receptor de mineralocorticoides é aumentar a dose da medicação até que os níveis potássicos sejam controlados, sem necessidade de utilizar quaisquer outras medicações controladoras de potássio; a pressão arterial seja mantida sob controle e não ocorra mais a supressão da atividade da renina plasmática.
Os antagonistas do receptor de mineralocorticoides disponíveis no mercado são os medicamentos espironolactona e eplerenona; a eplerenona é mais seletiva para o receptor de mineralocorticoides. A espironolactona também bloqueia os receptores de androgênio e progesterona, que produzem efeitos colaterais, incluindo disfunção sexual, ginecomastia, sensibilidade nos seios e menstruações irregulares.
A adição de amilorida ou de triantereno diminui a atividade do canal de sódio epitelial nos casos em que não for possível controlar a hipocaliemia com o bloqueio do receptor de mineralocorticoides. De modo geral, o gerenciamento da pressão arterial exige a adição de outras medicações anti-hipertensivas, em especial nos casos de hipertensão de longa duração.
Recomenda-se tratamento médico nos casos de hiperplasia adrenal bilateral. Não se recomenda o uso de adrenalectomia bilateral, tendo em vista que ela poderá produzir insuficiência adrenal. Talvez a melhor opção seja o bloqueio do receptor de mineralocorticoides para os casos raros de aldosteronismo remediável por glicocorticoides, embora a terapia à base de glicocorticoides possivelmente seja útil em pacientes específicos.
A ressecção cirúrgica de adenomas que produzem aldosterona é um tratamento bastante eficaz para doença unilateral. Nas situações em que houver um cirurgião adrenal especializado à disposição, a cirurgia, preferencialmente por meio de um procedimento laparoscópico, é a melhor opção para uso em pacientes que sejam bons candidatos cirúrgicos. A hipertensão melhora em quase todos os pacientes tratados por meios cirúrgicos; entretanto, a maioria dos pacientes com hipertensão persistente precisa receber terapia anti-hipertensiva.
A eficácia relativa do gerenciamento médico de longo-prazo com antagonistas do receptor de mineralocorticoides, em comparação com adrenalectomia, ainda é incerta, considerando que não há dados suficientes de estudos de longo prazo que fizeram a comparação dos resultados em pacientes com aldosteronoma unilateral tratado com cirurgia versus os pacientes tratados com bloqueio do receptor de mineralocorticoides.
Complicações
O bloqueio insuficiente do receptor de mineralocorticoides poderá resultar em hipertensão mediada pela aldosterona e hipocaliemia, assim como em complicações associadas ao excesso de aldosterona, incluindo FA, hipertrofia ventricular esquerda, AVC, lesões renais e problemas no metabolismo da glicose. A cirurgia produz suas próprias complicações.
Pode ocorrer supressão na produção de aldosterona por glândulas suprarrenais normais nos casos de AP causado por algum aldosteronoma unilateral. Nessas circunstâncias, poderá haver hipoaldosteronemia transitória após a ressecção do adenoma secretor de aldosterona. Os pacientes poderão ser tratados com aumento na ingestão dietética de sódio e, caso seja necessário, com pequenas doses de fludrocortisona até a recuperação funcional da zona glomerulosa.
Prognóstico
O prognóstico para pacientes com produção excessiva controlada de aldosterona é excelente. Entretanto, geralmente os pacientes com AP ou AP no contexto de hipertensão pré-existente precisam de terapia anti-hipertensiva, além do controle dos efeitos da aldosterona.
Feocromocitoma
Epidemiologia
Feocromocitoma é um tipo muito raro de tumor responsável por, ao menos, 1 a cada 10 mil casos de hipertensão.25,26 Esse tipo de tumor afeta igualmente homens e mulheres, e geralmente se apresenta da terceira à quinta décadas de vida, com incidência de dois a oito casos por milhão por ano.
Etiologia/Genética
As síndromes mendelianas são reconhecidas em um percentual cada vez maior de pacientes com feocromocitoma, incluindo neoplasia endócrina múltipla tipos IIa e IIb, doença de von Hippel-Lindau, neurofibromatose e síndromes de paraganglioma, identificadas por mutações em um número crescente de genes reconhecidos, incluindo as subunidades da succinato desidrogenase.27.
Após a confirmação do diagnóstico de feocromocitoma, recomenda-se que todos os pacientes sejam envolvidos nos debates sobre os testes genéticos com focos específicos. Essa orientação teve origem no percentual cada vez maior de casos de feocromocitoma associados a mutações germinais, assim como no percentual crescente de pacientes com mutações suscetíveis, mesmo sem evidências de histórico familiar. Algumas dessas mutações, como, por exemplo, aquelas envolvendo a succinato desidrogenase B, estão associadas a um risco mais elevado de metástase e a piores prognósticos.28
Patogênese
A maior parte (90%) de todos os tumores de células da crista neural que segregam catecolaminas se localiza na medula adrenal e se denomina feocromocitoma. Os tumores extra-adrenais dos glânglios simpáticos e parassimpáticos se denominam paragangliomas.29 As apresentações são semelhantes.
A medula adrenal corresponde a 10% do peso da glândula suprarrenal. Ela é composta principalmente de células cromafínicas, cuja denominação se deve à combinação de amarelo e castanho após a coloração com sais cromáticos. As células da medula são inervadas diretamente por células nervosas simpáticas pré-gangliônicas. Portanto, essas células secretoras de epinefrina são análogas aos neurônios pós-gangliônicos em outras áreas do sistema nervoso simpático.
No entanto, essas células não são neurônios e não têm dendritos ou axônios. A epinefrina é o produto principal secretado pela medula adrenal, enquanto que o restante do sistema nervoso simpático utiliza a norepinefrina como neurotransmissor. A razão dessa diferença é que o suprimento de sangue para a medula adrenal tem origem no plexo capilar que drena o córtex adrenal.
O sangue capilar é extremamente rico em cortisol, e talvez seja a concentração mais elevada de cortisol no corpo humano, sendo que o cortisol induz a enzima catecol-0-metiltransferase, que converte norepinefrina em epinefrina.
Diagnóstico
Manifestações Clínicas
O diagnóstico de feocromocitoma é confirmado por meio de suspeitas clínicas. Embora esse tipo de tumor seja raro, é imprescindível considerar esse diagnóstico, já que pode ser extremamente letal. Mais de um terço dos feocromocitomas são encontrados incidentalmente durante cirurgias não relacionadas ao tumor ou em autópsias. As manifestações clínicas de feocromocitoma variam amplamente; hipertensão é a descoberta mais consistente.30
A hipertensão paroxísmica marca em torno da metade dos casos; uma proporção significativa de casos tem hipertensão sustentada, e aproximadamente 10% dos pacientes podem ser normotensivos. Os sintomas mais comuns incluem cefaleia, palpitações e sudorese; em um dos estudos, essa tríade apresentou mais de 90% de sensibilidade e especificidade, embora seja importante lembrar a proporção considerável de pacientes cujas autópsias comprovaram a presença de tumores clinicamente fora de suspeita.30
Com frequência, os pacientes se queixam de ansiedade e uma sensação de pânico e de mau pressentimento durante os episódios. Outros sintomas incluem náusea, dor abdominal e perda de peso. Os episódios poderão ser desencadeados por mudança de posição (por exemplo, curvar-se, deitar-se sobre um dos lados), atividade física ou uso de medicamentos que forçam a liberação de catecolaminas (por exemplo, antidepressivos tricíclicos, ß-bloqueadores, nicotina, álcool e queijo).
Exame Físico
A maior parte dos pacientes com feocromocitoma se apresenta com hipertensão. Durante os episódios, é mais provável a presença de palidez do que de rubor; com frequência, essa distinção é muito útil para distinguir feocromocitoma de outros episódios. A hipotensão ortostática acompanha muitos pacientes com feocromocitoma.
Testes Laboratoriais
Em última análise, o diagnóstico de feocromocitoma depende da confirmação da produção excessiva de catecolaminas. O melhor ensaio tem sensibilidade e especificidade mais elevadas, tendo em vista que os resultados negativos falsos e positivos falsos positivos aumentam substancialmente as dificuldades para fazer o diagnóstico.
Os testes diagnósticos clássicos incluem medições plasmáticas e urinárias das catecolaminas, metanefrinas urinárias fracionadas e metanefrinas totais. Recomenda-se usar o ensaio mais recente para medir as concentrações plasmáticas das metanefrinas livres (metanefrinas e não metanefrinas) como o teste de rastreamento melhor e mais conveniente para muitos pacientes. O ganho com múltiplas medições no rastreamento inicial é muito pequeno.
Exames de sangue. Na maior parte dos casos, as medições plasmáticas de metanefrinas livres, tanto a metanefrina como a não metanefrina, são os melhores testes de rastreamento.31?33 Esse tipo de teste evita a coleta de amostras de urina de 24 horas, além de apresentar uma altíssima sensibilidade, variando de 97 a 99%, o que torna os resultados negativos extremamente úteis para a exclusão de feocromocitoma; a especificidade é de cerca de 90%.28
As metanefrinas plasmáticas livres são produzidas continuamente por meio do metabolismo das catecolaminas no interior das células tumorais dos feocromocitomas. Por outro lado, a secreção de catecolaminas por tumores de feocromocitomas é episódica. Além disso, os níveis de metanefrinas plasmáticas livres geralmente não são elevados pela estimulação simpática, ao contrário das catecolaminas, que são liberadas em resposta.
Muitos estudos examinaram as metanefrinas plasmáticas retiradas de um cateter venoso permanente, embora a punção venosa seja mais prática e amplamente usada na maior parte dos processos de rastreamento. Qualquer elevação na metanefrina plasmática fracionada, na metanefrina urinária ou na não metanefrina até quatro vezes acima do limite superior da normalidade geralmente é considerada diagnóstica de resultados positivos falsos.34
Há um número maior de resultados positivos falsos nos casos em que as elevações forem mais modestas; de um modo geral, os resultados positivos falsos são atribuídos ao uso concomitante de medicações, postura sentada e idade mais avançada. Níveis muito elevados podem ser causados pelo uso de antidepressivos tricíclicos, inibidores da monoamina oxidase, simpatomiméticos, cocaína e bloqueio a.28
O acetaminofeno interferiu em estudos mais antigos, mas deixou de ser um problema nos ensaios mais recentes. Nos casos em que os valores forem indeterminados, recomenda-se repetir as medições na posição em supino para eliminar essa causa comum de aumento nos resultados positivos falsos; deve-se interromper o uso de medicações com alto potencial de gerar dúvidas.28
Após a confirmação do diagnóstico de feocromocitoma, recomenda-se que todos os pacientes sejam envolvidos em um debate sobre os testes genéticos com foco nas mutações mais suspeitas e que dependem de características clínicas específicas.
Exames de urina. Os exames tradicionais para diagnóstico de feocromocitoma são medições das catecolaminas urinárias fracionadas e da excreção de metanefrina urinária em coletas de urina de 24 horas. A especificidade é mais elevada para as metanefrinas urinárias. Para fazer esses testes, a urina deve ser coletada em meio ácido (tipicamente, os laboratórios fornecem recipientes adequados) e não precisa ser refrigerada.
A medição da creatinina também é importante porque indica a integralidade da coleta. Alguns ensaios mostram que é mínima a interferência de medicamentos como diuréticos, vasodilatadores, BCC e Ieca nos casos em que for necessário tratar a hipertensão. As medições urinárias de 24 horas das metanefrinas e das catecolaminas produzem menos resultados positivos falsos que as medições da metanefrina plasmática fracionada, que é um atributo preferível para testar pacientes de baixo risco.35
As metanefrinas fracionadas medidas na urina são metabólitos diferentes das metanefrinas livres medidas no plasma e, além disso, são produzidas em partes distintas do corpo por meio de processos metabólicos que não estão diretamente relacionados ao tumor propriamente dito. As metanefrinas urinárias fracionadas e totais são medidas após a etapa de desconjugação e refletem amplamente os níveis de metanefrinas conjugadas produzidas fora do tecido tumoral.
Estudos de imagens. A TC ou as IRMs ajudam a localizar o tumor nas situações em que houver concordância entre o quadro clínico e os testes bioquímicos. A TC tem excelente resolução espacial para detectar massas adrenais, mesmo que elas tenham apenas alguns milímetros, desde que as amostras colhidas sejam finas. As IRMs são particularmente úteis porque esses tumores geralmente brilham nas imagens ponderadas T2.
Nas situações em que a TC e as IRMs não conseguirem revelar a presença de algum tumor adrenal em casos confirmados por meios bioquímicos, a cintilografia usando metaiodobenzilguanidina (MIBG) radiomarcada é uma técnica bastante útil para localização, em especial os tumores fora da glândula suprarrenal, tais como tumores no corpo da carótida, no coração, na bexiga urinária e no órgão de Zuckerkandl.
A MIBG foi desenvolvida por imagens da medula adrenal e de suas doenças, e continua sendo a melhor radiofarmacologia para imagens funcionais; os rastreamentos por TC com emissão de pósitrons talvez sejam uma técnica promissora para detectar feocromocitomas.36,37
Testes fisiológicos. Os testes de provocação têm sido utilizados para confirmar ou excluir a presença de feocromocitomas, embora tenham sido abandonados por causa da baixa sensibilidade e do risco de precipitar hipertensão. Nos dias atuais, os testes de supressão são raramente utilizados para fins diagnósticos.
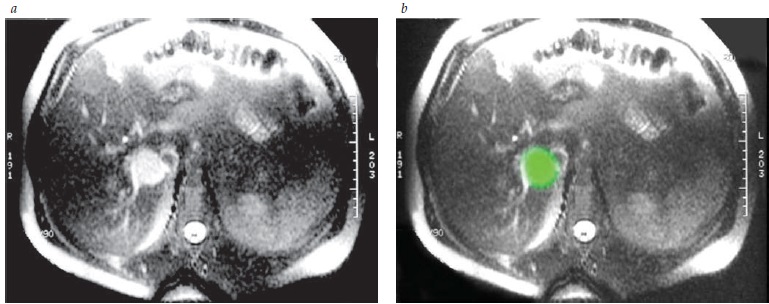
A Figura 5 mostra um feocromocitoma de 4cm na IRM.
IRM: imagem por ressonância nuclear magnética.
Figura 5 - (A) Um feocromocitoma de 4cm brilhando na IAM ponderada em T2. (B) O tumor aparece na cor verde.
Biópsias. Os feocromocitomas são neoplasmas altamente vasculares e, por isso, os procedimentos para biópsia estão associados a fatores como hemorragia com risco de vida, crise hipertensiva, ruptura capsular com implantação tumoral e morte. De um modo geral, as biópsias não são indicadas para confirmar o diagnóstico de feocromocitoma.
Diagnóstico Diferencial
O diagnóstico diferencial de feocromocitoma inclui ansiedade e ataques de pânico, tireotoxicose, uso de anfetaminas, uso de cocaína e uso de medicamentos sem prescrição médica cujos efeitos dependem das catecolaminas, como os atomizadores para congestão nasal.
Gerenciamento
O tratamento de feocromocitomas é cirúrgico, sendo que a ressecção laparoscópica é o tratamento padrão na maior parte dos casos. Esse tipo de cirurgia deve ser feito apenas por equipes experimentadas e especializadas no gerenciamento de feocromocitomas. Os dois componentes mais importantes dos cuidados pré-operatórios incluem bloqueio a-adrenérgico e expansão volumétrica.
O bloqueio a-adrenérgico completo deverá ser induzido antes do procedimento cirúrgico para evitar a ocorrência de crise hipertensiva intraoperatória. A preparação deve iniciar dentro de um período de 7 a 14 horas antes do procedimento planejado; há diversos regimes comprovadamente eficazes.
Anteriormente, a terapia clássica era o uso de uma dose inicial de 10mg de fenoxibenzamina, por via oral, duas vezes ao dia, com aumento diário na dose inicial e, no sétimo dia, o paciente deveria estar tomando ao menos 1mg/kg/dia, em três doses divididas. Os antagonistas a 1 específicos, incluindo a doxamina (2 a 16mg/dia), são comprovadamente eficazes e seguros, sendo que alguns estudos mostraram que há menor incidência de hipotensão pós-operatória com o uso desses medicamentos.38
Bloqueios adequados estão associados à redução na pressão arterial e na hipotensão ortostática com a recuperação do volume vascular. Com frequência, é necessário fazer o ß-bloqueio para tratamento de taquicardia, porém somente após o estabelecimento do bloqueio a.
O Quadro 4 contém o diagnóstico diferencial de feocromocitoma.
Quadro 4
|
DIAGNÓSTICO DIFERENCIAL DE FEOCROMOCITOMA |
|
Ataques de pânico/ansiedade aguda Tireotoxicose Angina Uso de anfetaminas Uso de cocaína Uso de medicações frias sem prescrição médica contendo fenilefrina ou pseudoefedrina Inibidores da monoamina oxidase com interação dietética (p. ex., queijo envelhecido, vinho tinto e cerveja) Hipoglicemia aguda Tumor cerebral Hemorragia subaracnoide Fogachos na menopausa Toxemia na gravidez Interrupção no uso de medicamentos agudos: clonidina, bloqueio ß, álcool. |
Complicações
A mortalidade causada por feocromocitomas resulta do excesso não tratado de catecolaminas produzindo danos cardiovasculares ou de alguma doença maligna.39 Caso não sejam identificados, esses tumores podem causar morbidade cardiovascular significativa levando à morte e à morte súbita durante procedimentos cirúrgicos e procedimentos obstétricos.
A incidência de feocromocitoma durante a gravidez é um grande risco para o feto e para a mãe; com diagnóstico e gerenciamento adequados, a mortalidade diminuiu para, aproximadamente, 5%.39 Em situações raras, os tumores com aumento de volume podem produzir sintomas de efeito de massa ao se estenderem até os tecidos adjacentes.
Prognóstico
De modo geral, o prognóstico é excelente após o reconhecimento precoce e a remoção cirúrgica de feocromocitomas benignos. Os feocromocitomas malignos devem ser tratados com redução cirúrgica do tumor, bloqueio a com fenoxibenzamina e cogerenciamento de um oncologista. A radioterapia é bastante útil nos casos de dor óssea, e obteve-se algum sucesso com a quimioterapia de combinação, incluindo ciclofosfamida, vincristina e dacarbazina.
|
Informações Financeiras: Gail K. Adler, MD, PhD, foi consultora da Pfizer no Japão. Naomi D. L. Fisher, MD, não tem nenhuma informação financeira relevante a declarar. |
Referências
1. Mosso L, Carvajal C, González A, et al. Primary aldosteronism and hypertensive disease. Hypertension 2003;42:161–5.
2. Stowasser M, Gordon RD. The renaissance of primary aldosteronism: what has it taught us? Heart Lung Circ 2013;22:412–20.
3. Clark D, Ahmed MI, Calhoun DA. Resistant hypertension and aldosterone: an update. Can J Cardiol 2012;28:318–25.
4. Mulatero P, Monticone S, Rainey WE, et al. Role of KCNJ5 in familial and sporadic primary aldosteronism. Nat Rev Endocrinol 2013;9:104–12.
5. Lifton RP, Dluhy RG, Powers M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992;355:262–5.
6. Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011;331:768–72.
7. Zennaro MC, Rickard AJ, Boulkroun S. Genetics of mineralocorticoid excess: an update for clinicians. Eur J Endocrinol 2013;169:R15–25.
8. Chen W, Li F, He C, et al. Elevated prevalence of abnormal glucose metabolism in patients with primary aldosteronism: a meta-analysis. Ir J Med Sci 2014;183:283–91.
9. Savard S, Amar L, Plouin PF, Steichen O. Cardiovascular complications associated with primary aldosteronism: a controlled cross-sectional study. Hypertension 2013;62:331–6.
10. Mulatero P, Monticone S, Bertello C, et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J Clin Endocrinol Metab 2013;98:4826–33.
11. Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 2007;66:607–18.
12. Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol 2010;6:261–73.
13. Pojoga LH, Baudrand R, Adler GK. Mineralocorticoid receptor throughout the vessel: a key to vascular dysfunction in obesity. Eur Heart J 2013;34:3475–7.
14. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med 2009;150:776–83.
15. Funder JW, Carey RM, Fardella C, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008;93:3266–81.
16. Stowasser M, Gordon RD, Rutherford JC, et al. Diagnosis and management of primary aldosteronism. J Renin Angiotensin Aldosterone Syst 2001;2:156–69.
17. Rossi GP, Belfiore A, Bernini G, et al. Comparison of the captopril and the saline infusion test for excluding aldosterone-producing adenoma. Hypertension 2007;50:424–31.
18. Monnens L, Levtchenko E. Distinction between Liddle syndrome and apparent mineralocorticoid excess. Pediatr Nephrol 2004;19:118–19.
19. Ferrari P. The role of 11b-hydroxysteroid dehydrogenase type 2 in human hypertension. Biochim Biophys Acta 2010;1802:1178–87.
20. Gupta S, Melendez J, Khanna A. Deoxycorticosterone producing tumor as a cause of resistant hypertension. Case Rep Med 2010;2010:372719.
21. Melcescu E, Phillips J, Moll G, et al. 11Beta-hydroxylase deficiency and other syndromes of mineralocorticoid excess as a rare cause of endocrine hypertension. Horm Metab Res 2012;44:867–78.
22. Kellenberger S, Gautschi I, Rossier BC, Schild L. Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest 1998;101:2741–50.
23. Fremont OT, Chan JC. Understanding Bartter syndrome and Gitelman syndrome. World J Pediatr 2012;8:25–30.
24. Bonny O, Rossier BC. Disturbances of Na/K balance: pseudohypoaldosteronism revisited. J Am Soc Nephrol 2002;13:2399–414.
25. Beard CM, Sheps SG, Kurland LT, et al. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc 1983;58:802–4.
26. Stenström G, Svärdsudd K. Pheochromocytoma in Sweden 1958–1981. An analysis of the National Cancer Registry Data. Acta Med Scand 1986;220:225–32.
27. Fishbein L, Orlowski R, Cohen D. Pheochromocytoma/paraganglioma: Review of perioperative management of blood pressure and update on genetic mutations associated with pheochromocytoma. J Clin Hypertens (Greenwich) 2013;15:428–34.
28. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915–42.
29. Eisenhofer G. Screening for pheochromocytomas and paragangliomas. Curr Hypertens Rep 2012;14:130–7.
30. Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev 2003;24:539–53.
31. Raber W, Raffesberg W, Bischof M, et al. Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med 2000;160:2957–63.
32. Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002;287:1427–34.
33. Václavík J, Stejskal D, Lacnák B, et al. Free plasma metanephrines as a screening test for pheochromocytoma in low-risk patients. J Hypertens 2007;25:1427–31.
34. Kannan S, Purysko A, Faiman C, et al. Biochemical and radiological relationships in patients with pheochromocytoma: lessons from a case control study. Clin Endocrinol (Oxf) 2014;80:790–6.
35. Sawka AM, Jaeschke R, Singh RJ, Young WF. A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003;88:553–8.
36. Pacak K, Eisenhofer G, Ilias I. Diagnosis of pheochromocytoma with special emphasis on MEN2 syndrome. Hormones (Athens) 2009;8:111–16.
37. Jacobson AF, Deng H, Lombard J, et al. 123I-meta-iodobenzylguanidine scintigraphy for the detection of neuroblastoma and pheochromocytoma: results of a meta-analysis. J Clin Endocrinol Metab 2010;95:2596–606.
38. Prys-Roberts C, Farndon JR. Efficacy and safety of doxazosin for perioperative management of patients with pheochromocytoma. World J Surg 2002;26:1037–42.
39. Prejbisz A, Lenders JW, Eisenhofer G, Januszewicz A. Mortality associated with phaeochromocytoma. Horm Metab Res 2013;45:154–8.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.