(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Hematologia
acp-medicine Hematologia
Última revisão: 01/08/2018
Comentários de assinantes: 0
|
Artigo original: Seldin D, MD, PhD. Sloan M, MD. Approach to the Patient with Benign Hematologic Disorders, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
David C. Seldin, MD, PhD
Chefe da Seção de Hematologia-Oncologia e Professor de Medicina e Microbiologia na Boston University School of Medicine (Boston, MA).
J. Mark Sloan, MD
Professor Assistente de Medicina na Boston University School of Medicine (Boston, MA).
Resumo
A hematologia se refere basicamente à função e aos distúrbios dos elementos formados a partir do sangue - eritrócitos (RBCs), leucócitos (WBCs) e plaquetas -, assim como os fatores que controlam a hemóstase. Os hematologistas têm tido um papel importante nas pesquisas biomédicas e translacionais básicas. O trabalho desses especialistas, impulsionado em parte pela facilidade de coletar sangue e medula óssea para fins de estudos, possibilitou o conhecimento dos distúrbios no sangue e de níveis moleculares fundamentais. As técnicas desenvolvidas para o estudo de hematologia geralmente são adotadas por outras disciplinas. Este artigo apresenta algumas discussões sobre anatomia do sistema hematopoiético, hematopoiese e medula óssea, exame físico de pacientes hematológicos, avaliação do hematoma completo (CBC) e esfregaço do sangue periférico, e coagulação. Os quadros apresentam uma descrição dos parâmetros do CBC, descobertas feitas nos esfregaços de sangue periférico, descrições, índices e significância dos RBCs; descobertas laboratoriais em eritrocitose; doenças geralmente associadas a eosinofilia e exames completos úteis; medicações comuns fortemente associadas à trombocitopenia; e pontuações dos 4Ts para determinar a probabilidade pré-teste de trombocitopenia induzida pelo uso de heparina. As figuras mostram as três frações de sangue centrifugado, o linfonodo, as células-tronco hematopoiéticas, aspirado da medula óssea e procedimento para biópsia, arquitetura do microambiente da medula óssea, petéquias, tipos de WBVC encontrados nos esfregaços de sangue periférico, teste direto de antiglobulina, células mieloides e sistema de coagulação.
|
Abordagem aos Pacientes com Distúrbios Hematológicos Benignos |
O sangue total pode ser facilmente separado por gravidade ou por uma leve centrifugação em três frações: eritrócitos (RBCs), uma camada contendo leucócitos e plaquetas, e plasma. A subdivisão adicional dessas frações básicas revela a incrível complexidade do sangue; por exemplo, somente o plasma contém mais de 1.000 proteínas exclusivas, cuja composição proteônica se altera nos estados de doença.1
A hematologia se refere principalmente à função e aos distúrbios dos elementos sanguíneos formados - eritrócitos (RBCs), leucócitos (WBCs) e plaquetas -, assim como aos fatores que controlam a homeostase.
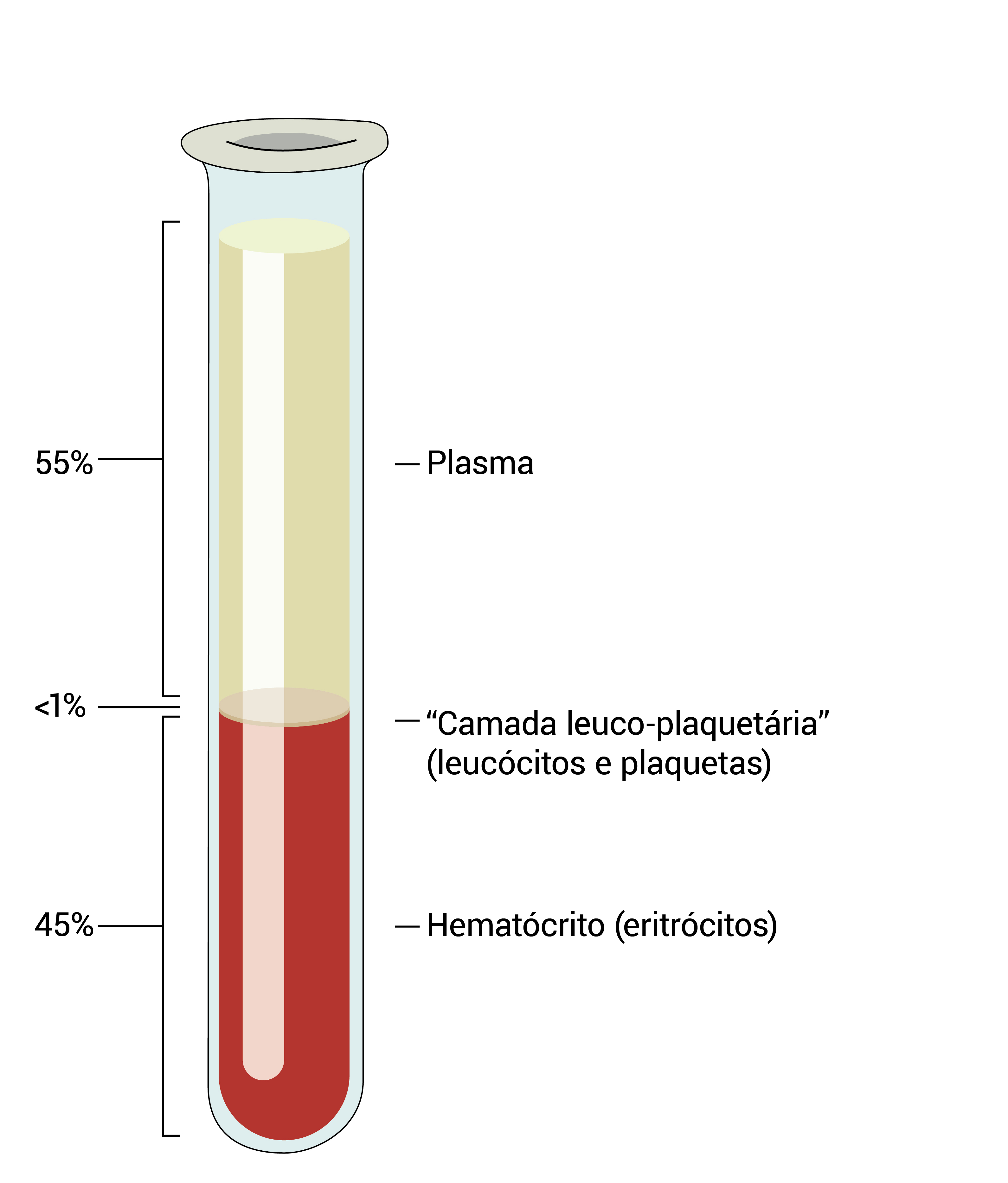
A Figura 1 mostra o sangue estabelecido ou centrifugado, que se separa em três frações: eritrócitos, plasma e camada leucoplaquetária (buffy coat) contendo leucócitos e plaquetas.

Figura 1 - O sangue estabelecido ou centrifugado se separa em três frações: eritrócitos, plasma e camada leucoplaquetária (buffy coat) contendo leucócitos e plaquetas. O percentual de sangue composto por eritrócitos é conhecido por hematócrito (45%, neste exemplo).
Os hematologistas têm tido um papel fundamental nas pesquisas biomédicas e translacionais básicas. O trabalho desses profissionais, impulsionado parcialmente pela facilidade na coleta de sangue e de medula óssea para estudos, viabilizou a compreensão de muitos distúrbios do sangue em níveis moleculares fundamentais.
As técnicas desenvolvidas para os estudos de hematologia geralmente são adotadas por outras disciplinas. Os exemplos incluem identificação de mutações pontuais responsáveis pela anemia causada por células falciformes, a primeira doença a ser tipificada sob o ponto de vista molecular,2 assim como identificação da translocação equilibrada dos cromossomos 9 e 22 nos casos de leucemia mielocítica crônica (LMC), a primeira translocação cromossômica recorrente identificada em uma malignidade.3
Anatomia do Sistema Hematopoiético
O sistema hematopoiético se compõe de sangue periférico, medula óssea e sistema linfático. O sangue dá sustentação à vida por meio do transporte de oxigênio e dos nutrientes essenciais, da remoção de resíduos e da liberação dos fatores humorais e celulares necessários para a defesa dos hospedeiros. As plaquetas e os fatores de coagulação, trabalhando em combinação com as células endoteliais vasculares, mantêm a integridade do sistema.
Em indivíduos adultos, todos os componentes celulares do sangue são produzidos na medula óssea. A quantidade de medula óssea celular diminui com o avanço da idade; em adultos, o volume de medula óssea funcional se localiza na pelve, nas vértebras e nos ossos longos proximais. Em alguns estados patológicos, como a doença conhecida por mielofibrose primária, a medula óssea deixa de ser acolhedora e o corpo tenta produzir sangue em algum outro local, tipicamente no baço e no fígado. Esse processo é conhecido por hematopoiese extramedular.
Embora todas as células do sistema imune tenham origem na medula óssea, os linfócitos (células responsáveis pela imunidade adaptativa) migram para os órgãos linfoides periféricos (linfonodos, baço e timo) e circulam entre o sangue e o sistema linfático. O sistema linfático periférico recircula o líquido extracelular; o líquido extracelular atravessa os linfonodos e os canais linfáticos até, finalmente, retornar para o sangue.
Nos linfonodos, os linfócitos geralmente são expostos aos antígenos provenientes de patógenos infecciosos. Os linfócitos respondem por meio da proliferação e do refino dos receptores de antígenos nas situações em que essa exposição ocorre no contexto apropriado de células apresentadoras de antígenos coestimuladas. A arquitetura intrincada dos linfonodos reflete o processo elaborado através do qual o sistema imune adaptativo identifica e responde aos patógenos.
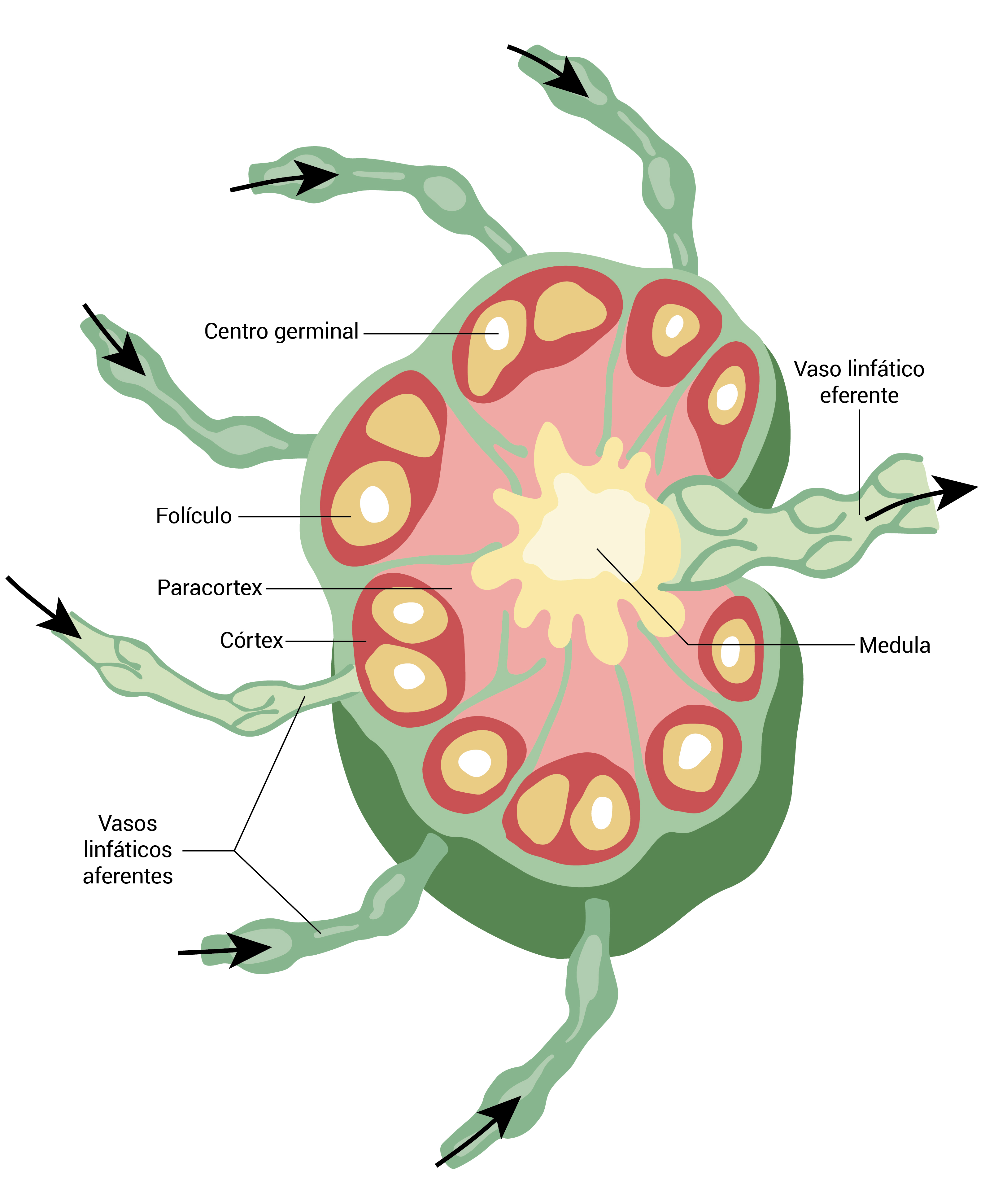
O córtex dos linfonodos contém sobretudo células B organizadas em folículos linfoides; com frequência, esses folículos têm áreas de proliferação intensa de células B conhecidas por centros germinais. Nos centros de proliferação, os linfócitos já foram expostos a um antígeno e trabalham para se multiplicar e aumentar sua afinidade com aquele antígeno específico. De um modo geral, o folículo é circundado por uma zona do manto contendo grupos de células B heterogêneas sob o ponto de vista funcional. As áreas difusas do córtex e do paracórtex se compõem basicamente de linfócitos T, como mostra a Figura 2.
Figura 2 - A organização dos linfonodos otimiza a exposição e a resposta do sistema imune aos patógenos infecciosos. As respostas imunes adaptativas são geradas e refinadas no córtex e nas áreas paracorticais. Os folículos contendo áreas centrais de proliferação de células B (centros germinais) são conhecidos por folículos secundários e indicam a presença de linfonodos “ativos”. O rompimento da arquitetura nodal ocorre nos casos de malignidades linfonodais.
Hematopoiese e Medula Óssea
A fabricação dos componentes celulares do sangue periférico e do sistema imune é um processo conhecido por hematopoiese. As células-tronco hematopoiéticas se diferenciam em qualquer uma das células do sangue periférico e, além disso, passam por um processo de autorenovação.4 De um modo geral, na medida em que as células hematopoiéticas amadurecem, a via de diferenciação delas é conhecida por dendrograma.
A primeira etapa na diferenciação de células-tronco primitivas é a formação de linfoides comprometidos ou de progenitores mieloides. Os progenitores mieloides produzem células B, células T e células exterminadoras naturais. Os progenitores mieloides comuns precisam de várias etapas adicionais e passam por ciclos de ampliação antes de sua prole formar eritrócitos, megacariócitos (que florescem para formar plaquetas) ou células mieloides maduras (principalmente granulócitos e monócitos).
Os patologistas costumam usar a frase “hematopoiese com linhagem tripla normal” nas situações em que examinam a medula óssea para descrever medulas que produzem, de forma apropriada, megacariócitos, RBCs e células mieloides maduras.
O processo hematopoiético é rigorosamente regulado por um sistema intrincado de fatores de crescimento e de citocinas. Atualmente, muitos desses fatores de crescimento são usados na prática clínica. A administração do fator estimulador de colônias de granulócitos (GM-CSF) agiliza a recuperação de neutrófilos depois da quimioterapia (Qt) mielossupressiva.5
Análogos da eritropoietina são os medicamentos usados com mais frequência em pacientes com insuficiência renal com a finalidade de substituir a baixa produção de eritropoietina endógena.6 Os miméticos da trombopoietina aumentam a contagem de plaquetas em pacientes com trombocitopenia imune (TPI) por meio da expansão da megacariopoiese na medula óssea em funcionamento.7
A medula óssea precisa trabalhar incessantemente para manter o estado de equilíbrio das células no sangue e para responder a demandas específicas. O tempo de vida total dos neutrófilos é de apenas alguns dias, dos quais apenas 6 horas são utilizadas na circulação. A maior parte da medula óssea dedica-se à produção de neutrófilos, mesmo na ausência de infecção.
Embora a longevidade dos RBCs seja maior (um RBC normal dura aproximadamente 120 dias), a medula óssea deve fabricar constantemente novos eritrócitos para substituir os RBCs que se perderam através de senescência, sangramento ou hemólise. As plaquetas têm meia-vida intermediária e, geralmente, circulam por um período de 7 a 10 dias. Elas são substituídas através da germinação citoplasmática de megacariócitos - células multinucleares que se localizam na medula.
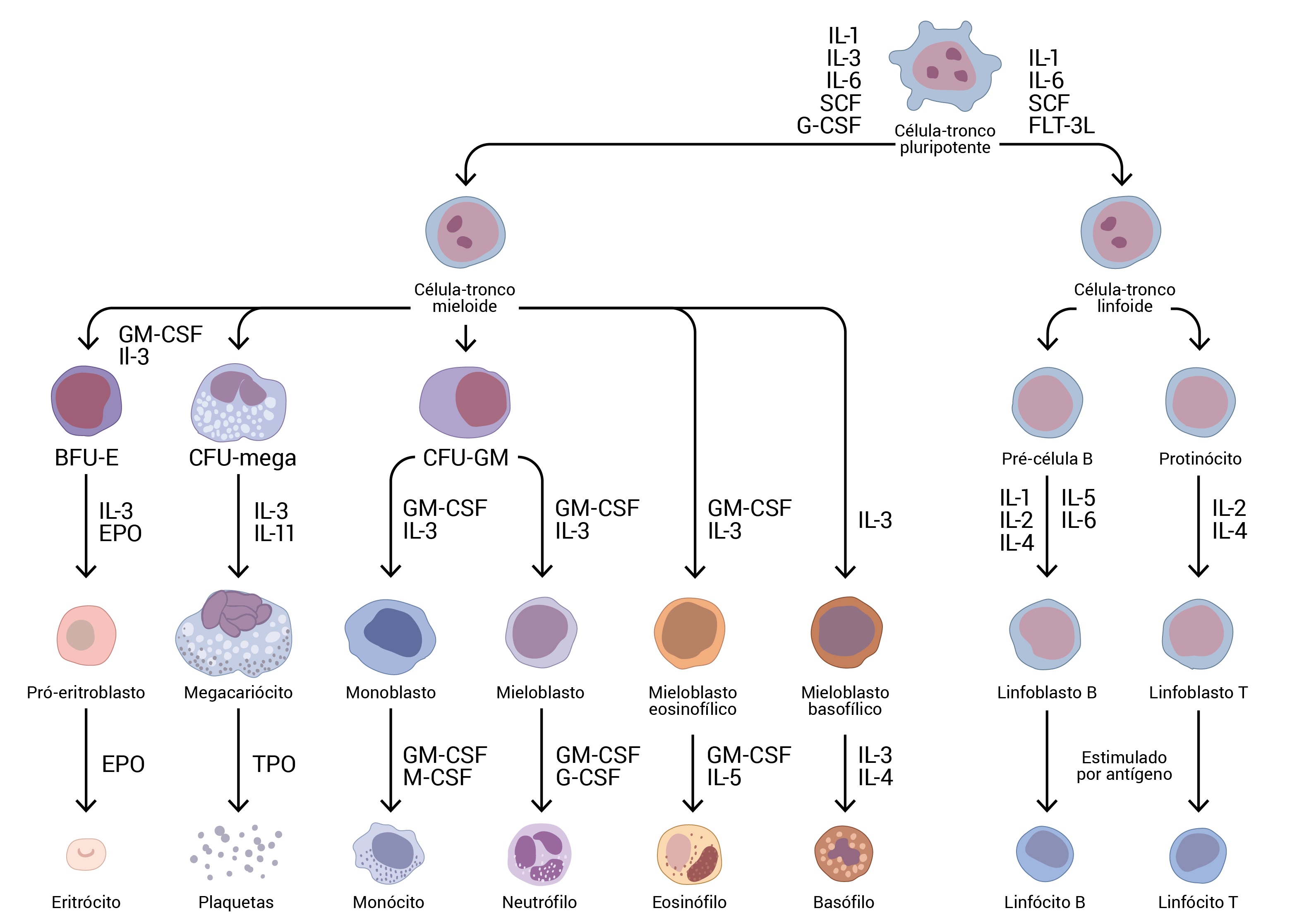
A Figura 3 mostra células-tronco hematopoiéticas da medula óssea.
BFU-E: unidades formadoras de grupos de células eritroides; CFU-GM: unidades formadoras de colônias de granulócitos e macrófagos; CFU-mega: unidades formadoras de colônias de megacariócitos; EPO: eritropoietina; EPOR: componente superficial do receptor de eritropoietina; FLT-3L: ligante da tirosina quinase-3 semelhante ao FMS; GM-CSF: fator estimulante da colônia de granulócitos e macrófagos; IL: interleucina; M-CSF: fator estimulante da colônia de macrófagos; SCF: fator da célula-tronco; TPO: trombopoietina.
Figura 3 - As células-tronco hematopoiéticas da medula óssea se diferenciam em qualquer célula no sangue periférico. Várias citocinas ajudam a orientar a diferenciação na direção que o corpo exigir naquele momento. Algumas dessas citocinas, como, por exemplo, a EPO, a TPO e o GM-CSF, são usados clinicamente para manipular a saída da medula óssea.
Qualquer número de fatores pode causar impacto na expectativa de longevidade celular in vivo. Por exemplo, o estresse oxidativo diminui substancialmente a vida dos RBCs em pacientes com deficiência na glicose-6-fosfato-desidrogenase (G6PD). Em estado de equilíbrio, aproximadamente dois terços da medula hematopoiética são dedicados à produção de células mieloides (sobretudo neutrófilos), enquanto que um terço produz células sanguíneas. Os hematologistas denominam esse fenômeno de razão entre mieloide e eritroide (M:E); a razão M:E normal é de 3:1.
A medula óssea tem a característica incrível de expandir sua capacidade produtiva de qualquer linhagem de células por meio do aumento da celularidade e da alteração na proporção de células. Por exemplo, em pacientes com perda sanguínea significativa, a medula celular se expande para encher a maior parte da medula óssea, invertendo a razão M:E, de modo a haver a predominância de eritroides. A hipercelularidade também é típica de malignidades hematológicas como leucemia ou mielodisplasia, que enche a medula por meio da expansão clonal de células neoplásicas.
Reticulócitos são RBCs que deixaram de ter núcleo, mas que ainda têm o RNA mensageiro (mRNA) que fora transcrito quando ainda havia núcleo. Levando-se em consideração que, nessas células, o mRNA se decompõe em algumas horas e não é substituído, sua presença no RBC é um excelente marcador para eritrócitos jovens.
Os reticulócitos podem ser percebidos em esfregaços de sangue a partir de uma coloração azulada sobreposta em sua cor rosa-salmão normal. Os reticulócitos podem ser quantificados com mais precisão por meio de uma coloração supravital para ácido nucleico, em comparação com a visualização através de microscópios ou de citômetros de fluxo. O número de reticulócitos em 100 RBCs geralmente é expresso em percentual.
Esse percentual sempre é elevado no quadro de anemia por causa da redução no denominador do total de RBCs. Portanto, para avaliar com precisão a produção da medula óssea, é imprescindível calcular a contagem “ajustada” de reticulócitos, isto é, a contagem medida de reticulócitos x [Hgb]/normal.
Por exemplo, um homem com contagem de reticulócitos de 6% aparentemente tem resposta medular apropriada à anemia, mas se a hemoglobina for de 5mg/L, em comparação com o nível normal de cerca de 15mg/L, a contagem ajustada de reticulócitos será de apenas 2%, que não é suficiente para compensar anemias profundas. O aumento na presença de reticulócitos indica que, provavelmente, a causa da anemia seja a perda de RBCs por meio de sangramentos ou pelo aumento na renovação através de hemólise ao invés de produção inadequada.
Anemias hipoproliferativas com baixa contagem de reticulócitos implicam em algum grau de disfunção medular, supressão, deficiência de eritropoietina ou insuficiência de matéria-prima para a eritropoiese (por exemplo, ferro, ácido fólico). Embora alguns marcadores de “plaquetas reticuladas” tenham sido desenvolvidos no esforço de fazer uma distinção semelhante para as plaquetas, nenhum deles chegou a ser utilizado na prática rotineira.
Com frequência, o exame da medula óssea é importante para avaliar a saúde do sistema hematopoiético, para o diagnóstico de estados hipoproliferativos ou para excluir ou definir os estágios de malignidades hematológicas. Trata-se de um procedimento de rotina que geralmente é bem tolerado e, portanto, não deve ser adiado desnecessariamente. Os pacientes devem permanecer na posição prona ou de lado para que a pele sobre a crista ilíaca póstero-superior possa ser esterilizada e anestesiada, da mesma forma que o tecido periósteo do íleo.
Na maior parte das vezes, não é necessário aplicar anestesia geral em adultos, tendo em vista que o tempo de desconforto para os pacientes é muito curto. As primeiras amostras líquidas da medula óssea são removidas por aspiração. Com frequência, os pacientes sentem a pressão negativa na cavidade medular como uma dor aguda produzida pela “aspiração”.
Na maior parte dos casos, divide-se o aspirado em três porções para que sejam analisadas em três laboratórios distintos; uma porção vai para o laboratório de histologia; uma para o laboratório de citometria de fluxo para verificar a eventual presença de populações de células anormais; e outra, para o laboratório de citogenética para analisar os cariótipos e a genética molecular por meio da reação em cadeia da polimerase (PCR) ou da hibridização in situ por fluorescência (FISH).
Os atributos morfológicos das células são mais bem analisados nos aspirados, que permitem obter um diferencial de células. A identificação das células em aspirados medulares é mais complicada, em comparação com o sangue periférico, tendo em vista que são células de todas as fases do desenvolvimento, e muitas linhas de células compartilham estágios imaturos. Conforme já mencionado, um diferencial medular normal tem razão M:E de 3:1, e dois terços das células mieloides devem ser maduras.
Normalmente, obtém-se uma pequena biópsia central (1 a 2cm) juntamente com o aspirado. Ao contrário da dor aguda associada ao aspirado, o paciente percebe a perfuração do núcleo como pressão, que normalmente não é dolorida. Após a descalcificação e o seccionamento, é possível examinar a arquitetura e a celularidade do núcleo. A quantidade de medula celular diminui com o avanço da idade, e a expectativa é que o percentual de medula celular contida em uma biópsia possa ser prevista por meio da seguinte equação:
% de celularidade = 100 – idade
Colorações imuno-histoquímicas (IHCs) específicas permitem identificar e quantificar populações de células específicas para avaliar a patologia. Por exemplo, uma coloração IHC para CD138 identifica as células plasmáticas; espera-se que haja uma grande abundância dessas células nos mielomas múltiplos. A hibridização in situ para mRNA também identifica populações de células anormais na medula óssea, como, por exemplo, demonstrando a predominância clonal de células plasmáticas nas discrasias dessas células.
Para fins diagnósticos, as informações produzidas pelo aspirado e pela biópsia são complementares e, inicialmente, recomenda-se fazer ambos os testes. Entretanto, às vezes, a medula óssea está tão “repleta” de células leucêmicas ou com tanta infiltração de reticulina ou fibrose que é impossível obter células por aspiração; este fenômeno é conhecido por “punção seca”.
Nesse contexto, um procedimento adjuvante bastante útil é tocar suavemente o núcleo da medula formando uma lamela para coloração. Essas “preparações obtidas pelo toque” poderão ser coloridas, preservando-se a citologia antes da fixação como se fossem um aspirado.
Exame Físico de Pacientes Hematológicos
De um modo geral, os problemas hematológicos são diagnosticados por meio de exames laboratoriais ou de investigações patológicas. Entretanto, há um conjunto de descobertas indispensáveis feitas nos exames físicos. Com frequência, anemia significativa sob o ponto de vista clínico poderá ser detectada pela alteração na palidez dos pacientes; em pessoas de pele escura, esse fato poderá ser observado na conjuntiva.
Os pacientes gravemente anêmicos em geral se erguem lentamente e apresentam taquicardia exagerada com exercícios. Na maior parte dos indivíduos, os casos graves de esplenomegalia podem ser determinados por meio do exame físico. Para tanto, o paciente deve permanecer na posição em supino e relaxado, apoiando as pernas na mesa de exame em um ângulo de 45º.
Enquanto estiver examinando o lado direito do paciente, com a mão espalmada ou ligeiramente encurvada, o examinador deve palpar primeiramente o quadrante inferior esquerdo e, a seguir, deslocar as mãos para cima na direção da margem costal esquerda. Em seguida, deve instruir o paciente a respirar lenta e profundamente para verificar se o diafragma empurra o baço para baixo até atingir uma posição palpável.
Ocasionalmente, a percussão pode ser útil. O baço da maior parte das pessoas não é palpável ou a ponta do baço é palpável apenas na inspiração máxima. Às vezes, é necessário obter imagens por ultrassonografia ou por tomografia computadorizada (TC) em pacientes obesos ou muito musculosos para determinar o tamanho do baço.
Embora a cirrose com hipertensão portal seja a causa mais comum de aumento nas dimensões do baço, a esplenomegalia é uma característica marcante de várias doenças hematológicas como mielofibrose, leucemia linfocítica crônica (LLC), LMC, linfomas esplênicos, doença de Gaucher e, em menor extensão, anemia hemolítica autoimune (AHAI).
O exame da pele e da boca também é muito importante. Tipicamente, a presença de mucosite ou de candidíase oral indica algum defeito imune significativo. Equimoses em áreas não traumatizadas podem ser sinais de coagulopatia significativa sob o ponto de vista clínico ou de trombocitopenia grave.
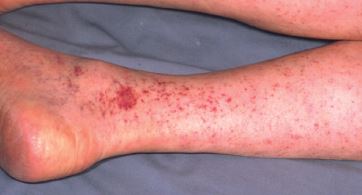
A trombocitopenia pode produzir petéquias. As petéquias são causadas por sangramentos momentâneos nos vasos capilares. Sob a perspectiva clínica, elas surgem como manchas planas arredondadas pequenas que não perdem a cor quando são pressionadas. De um modo geral, as petéquias são mais proeminentes em áreas dependentes, em especial na parte inferior das pernas e no palato.
A gravidade da trombocitopenia não é o único determinante de petéquias, tendo em vista que os pacientes com púrpura trombocitopênica idiopática (PTI) desenvolvem mais petéquias em comparação com pessoas com trombocitopenia por outras causas na mesma contagem de plaquetas.
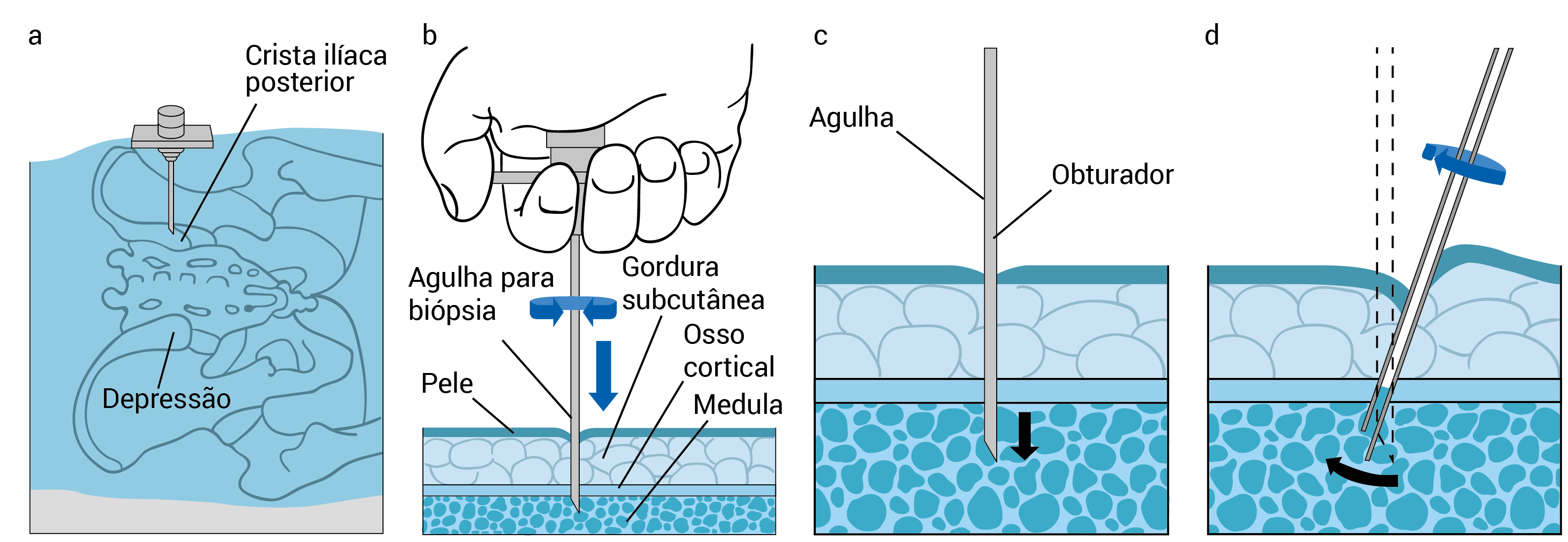
A Figura 4 mostra um procedimento para aspirado e biópsia na medula óssea.
Figura 4 - Procedimento para aspirado e biópsia na medula óssea. (A) A crista ilíaca posterior é o sítio para coleta de amostras. (B) A agulha deve atravessar a pele até atingir o espaço medular. (C) Aspiração da amostra da medula. (D) Remove-se cuidadosamente a amostra para biópsia.
Avaliação do Hemograma Completo e do Esfregaço de Sangue Periférico
O hemograma completo (CBC) automatizado é um dos testes requisitados com maior frequência em toda a medicina. A maioria dos instrumentos automatizados modernos determina a contagem, o volume e a estrutura interna das células por meio da análise da luz espalhada por um lêiser. O contador automático gera também relatórios sobre o cálculo de hematócritos (proporção de sangue ocupado pelos eritrócitos), além de fazer medições diretas de hemoglobina.
Tipicamente, outros índices de RBC também são registrados, incluindo volume corpuscular médio (VCM; tamanho médio do RBC), índice de anisocitose (RDW; medição da variação no tamanho do RBC), hemoglobina corpuscular média (HCM; correlaciona com VCM) e concentração de hemoglobina corpuscular média (CHCM).
Entre todos esses índices de RBC, o VCM e o RDW são os mais úteis para determinar a etiologia de anemia. Por exemplo, em geral a anemia ocasionada pela deficiência de ferro tem VCM baixo (microcitose) e RDW elevado, enquanto que a anemia perniciosa está associada com mais frequência a um VCM elevado (macrocitose) devido à má absorção de vitamina B12.
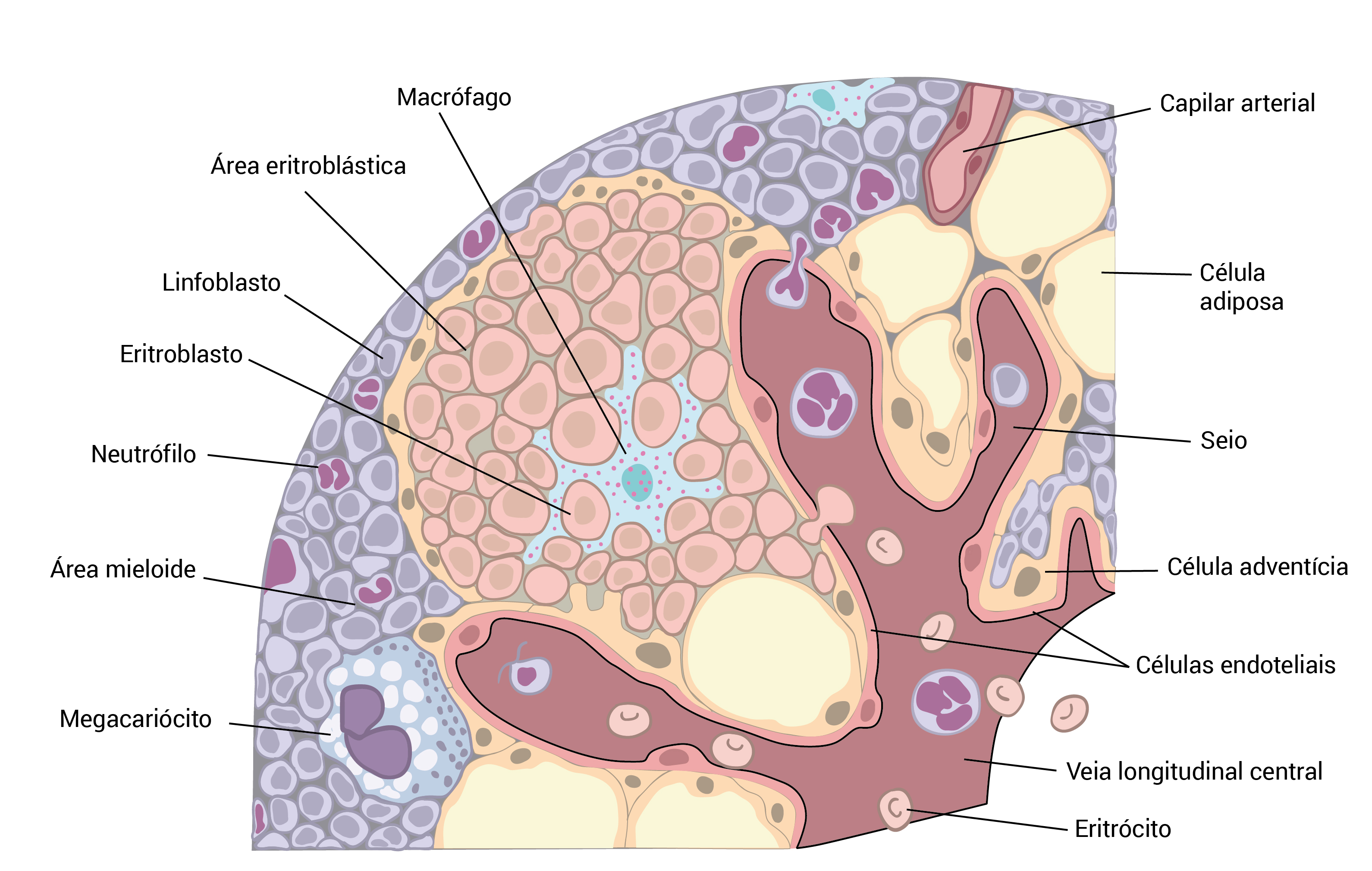
A Figura 5 mostra a arquitetura do microambiente da medula óssea mostrando o tecido hematopoiético, e a Figura 6, as petéquias.
Figura 5 - Arquitetura do microambiente da medula óssea mostrando o tecido hematopoiético. As células mieloides imaturas permanecem perto das trabéculas ósseas, enquanto que os megacariócitos normalmente se localizam nas proximidades dos sinusoides.
Figura 6 - Petéquias são pequenos pontos arredondados produzidos por micro hemorragias. Geralmente, elas estão associadas à trombocitopenia grave e são observadas com mais frequência nos casos de trombocitopenia imune (PTI) em áreas dependentes ou no palato. As áreas coalescentes de petéquias denominam-se púrpura.
Embora o CBC automatizado e o diferencial de leucócitos sejam indicações importantes para o diagnóstico de hematopoiese anormal, as informações mais importantes somente poderão ser obtidas por meio do exame microscópico do sangue. Determinados diagnósticos com risco de vida, variando de leucemia a púrpura trombocitopênica trombótica (PTT), ainda dependem da habilidade dos médicos e tecnólogos para identificar e interpretar as anormalidades observadas nos exames microscópicos do sangue.
A avaliação dos esfregaços periféricos tem custo baixo e está amplamente disponível no mercado. Portanto, o exame de esfregaços de sangue periférico é uma habilidade essencial para qualquer internista. Observadores experientes conseguem estimar os índices de RBC com alta precisão apenas por meio da inspeção de esfregaços de sangue periférico. Todo índice de RBC anormal tem um correlato visual. Por exemplo, anisocitose é um termo que descreve RBCs de tamanhos distintos, que são registrados pelos instrumentos automatizados como RDW elevado.
O Quadro 1 apresenta valores de CBC e índices normais de RBC.
Quadro 1
|
Parâmetros para Hemogramas Completos com Faixas Normais |
|
Leucócitos, 4,0?11,0 x 103/µL Células polimorfonucleares (neutrófilos), 40?75% Linfócitos, 15?54% Monócitos, 4?13% Eosinófilos, 0?7% Basófilos, 0?1% Células polimorfonucleares absolutas, 1,8?7,0 x 103/µL Linfócitos absolutos, 1,1?3,5 x 103/µL Monócitos absolutos, 0,2?0,9 x 103/µL Eosinófilos absolutos, 0,0?0,6 x 103/µL Basófilos absolutos, 0,0?0,1 x 103/µL Hb, 11,8?16,0g/dL Hct, 36,0?47,0% Volume celular médio (MCV), 80?97fL Hemoglobina corpuscular média (HCM), 27,0?33,0pg Concentração da hemoglobina corpuscular média, 32,0?36,0g/dL RDW, 12, ?14,5% Plt, 150?400 x 103/µL |
Hb: hemoglobina; Hct: hematócrito; Plt: plaquetas; RDW: índice de anisocitose.
Além disso, a capacidade para executar diferenciais manuais de leucócitos é extremamente importante. Esses diferenciais têm o objetivo de determinar o número e a proporção de cada tipo de leucócito, assim como de observar determinadas anormalidades no tamanho, no núcleo ou no citoplasma dos leucócitos e identificar qualquer população anormal de células.
As diferenças na proporção e na maturidade da composição de WBCs são extremamente úteis. Por exemplo, qualquer aumento nos neutrófilos geralmente é conhecido como reação leucemoide, que é a reação apropriada a infecções ou ao estresse sistêmico. Entretanto, a presença de células mieloides imaturas e o aumento nos basófilos e eosinófilos sugere doença mieloproliferativa. A linfocitose sustentada é típica de LLC.
Além da composição do diferencial de WBC e dos índices e morfologia dos RBCs, recomenda-se dar atenção especial a determinadas células ou padrões de anormalidades no sangue, tendo em vista que poderão significar a presença de alguma doença subjacente grave. O primeiro caso é a presença de células muito imaturas (também conhecidas por blastos).
Os blastos são células de grande porte com proporções elevadas entre núcleo e citoplasma; tipicamente, o núcleo contém cromatina rendilhada e núcleos proeminentes. A presença de blastos é suficiente para fazer o exame completo e avaliar a eventual presença de anemia aguda. O termo leucoeritroblastose se refere à presença de RBCs nucleados e de células mieloides imaturas no sangue periférico, normalmente em combinação com células em lágrima (também conhecidas por dacrócitos).
Com frequência, observa-se o quadro leucoeritroblástico nos estados em que a medula tenha se tornado fibrosa e sofre infiltração de alguma infecção ou malignidade. Para finalizar, a presença de esquistócitos, que são RBCs lacerados durante a passagem em uma microvasculatura ocluída ou rompida, pincipalmente em combinação com trombocitopenia, é a marca registrada de anemia microangiopática trombótica (AMT). Isso possivelmente seja o sinal de um diagnóstico com risco de vida como PTT ou coagulação intravascular disseminada (CID).
Nos casos de CBC anormal, o mais lógico seria avaliar a adequação de cada uma dessas três “linhagens” (RBCs, células mieloides e plaquetas) de forma independente. Muitos distúrbios podem afetar uma única linhagem. Entretanto, a chance de um paciente apresentar um distúrbio primário na medula óssea aumenta significativamente na medida em que ocorre o envolvimento de mais linhas de células.
Em outras palavras, uma anormalidade em mais de uma linhagem é mais preocupante para qualquer problema subjacente na medula óssea, em comparação com uma anormalidade em uma única linhagem, como, por exemplo, a anemia isolada. Nessas circunstâncias, a aspiração e biópsia da medula óssea são os passos iniciais para fazer o diagnóstico.
No entanto, é importante ter sempre em mente que há algumas condições que ocasionam citopenias múltiplas e não são produzidas por disfunção medular. Os exemplos incluem a síndrome de Evans, uma combinação de anemia hemolítica autoimune (Ahai) e púrpura trombocitopênica imune (PTI), ou PTT, que produz trombocitopenia e anemia hemolítica.
De um modo geral, os hematologistas fazem a distinção entre distúrbios primários e secundários. Nos distúrbios secundários, a medula responde apropriadamente aos estímulos provenientes de qualquer parte do corpo. Os distúrbios medulares primários agem de forma autônoma para produzir muitos ou alguns componentes celulares.
Por exemplo, os hematologistas se referem ao número elevado de plaquetas como uma reação ao baixo nível de ferro secundário à trombocitose, enquanto que se considera que qualquer paciente com distúrbio mieloproliferativo (por exemplo, trombocitemia essencial [TE]) e contagens elevadas de plaquetas tenha distúrbio medular primário.
Significância da Anemia
Os médicos que trabalham em diversas especialidades se defrontam com o desafio de determinar a significância e a etiologia da anemia. Anemia, particularmente em pacientes hospitalizados, é uma ocorrência muito comum e poderá se desenvolver tão insidiosamente que graus leves ou mesmo moderados da doença poderão passar despercebidos.
As transfusões poderão ser feitas, sem qualquer tentativa para determinar se ou porque elas são necessárias. O sangue é um recurso escasso e, portanto, é imprescindível considerar em primeiro lugar a razão pela qual um paciente é anêmico e, em segundo lugar, qual é a melhor forma de abordar o processo subjacente à anemia.
O Quadro 2 apresenta os correlatos de outros descritores de esfregaços periféricos com índices de RBC.
Quadro 2
|
Descobertas dos Esfregaços Periféricos, Descrições e Índices e Significância dos Eritrócitos | ||
|
Descrição do esfregaço periférico |
Correlato automático |
Significância |
|
Anisocitose (células de tamanhos diferentes). |
RDW elevado |
Geralmente observada nos casos de deficiência de ferro, talassemia, depois de transfusões ou como sinal de saúde precária da medula óssea.
|
|
Hipocromia (RBCs pálidas). |
CHCM reduzida |
Deficiência de ferro
|
|
Policromasia (células com cor azulada). |
Contagem elevada de reticulócitos
|
Perda de sangue ou hemólise. |
|
Poiquilocitose (células com formas diferentes). |
|
Descrição inespecífica de células de formas diferentes.
|
|
Esferocitose (células sem palidez central). |
CHCM elevada |
Esferocitose hereditária, anemia hemolítica, hemoglobinopatia ou aglutinina fria.
|
CHCM: concentração de hemoglobina corpuscular média; RBC: eritrócitos; RDW: índice de anisocitose.
É extremamente importante os médicos se conscientizarem de que nenhum tipo de anemia é normal; anemia de qualquer grau significa que há alguma patologia subjacente. Na esmagadora maioria de casos, o diagnóstico pode ser feito por meio de exames completos diretos e de baixo custo.
Embora a anemia se torne mais prevalente em pessoas mais velhas, ignorar a anemia em pacientes idosos sem uma investigação apropriada é uma grande imprudência. Ainda que a “anemia do envelhecimento” tenha sido associada à resistência à eritropoietina ou às formas mais sutis de mielodisplasia, trata-se de um diagnóstico de exclusão.8
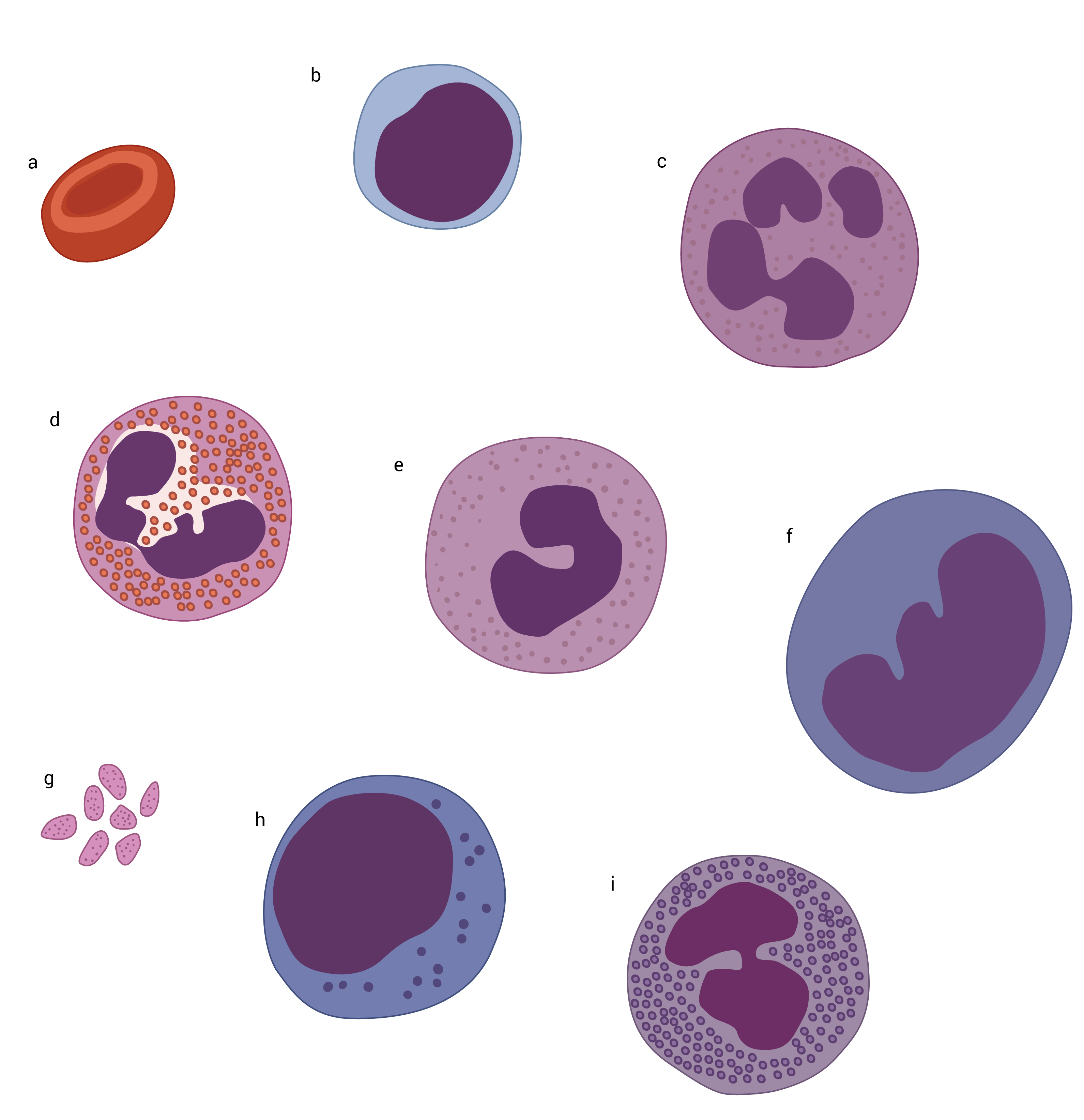
A Figura 7 apresenta os tipos de WBC encontrados nos esfregaços do sangue periférico de indivíduos normais.
Figura 7 - (A) Eritrócito. (B) Linfócito. (C) Neutrófilo. (D) Eosinófilo. (E) Banda neutrofílica. (F) Monócito. (G) Plaquetas. (H) Linfócito granular grande. (I) Basófilo.
A significância clínica da anemia depende de muitos outros fatores além da gravidade da doença. Embora seja sensível às alterações agudas na hemoglobina, o corpo humano tem a capacidade de se adaptar às alterações crônicas; alguns pacientes com anemia provavelmente não apresentem nenhum sintoma, que é sinal da existência de algum problema permanente.
O nível de atividade dos pacientes também é um contribuinte importante para a sintomatologia. No mesmo nível de hemoglobina, os pacientes sedentários apresentam menos sintomas que os pacientes ativos. Com frequência, os sintomas de anemia são inespecíficos e sutis e se sobrepõem de forma significativa aos problemas cardiopulmonares.
Os pacientes com anemia leve geralmente se queixam de fadiga, capacidade física reduzida para fazer exercícios e dificuldade para se concentrar. A deficiência de ferro, mesmo na ausência de anemia, pode causar síndrome de fadiga e desempenho cardíaco subótimo, que poderá melhorar com a reposição de ferro.
Na medida em que a anemia se torna mais grave e o volume intravascular se contrai, possivelmente os pacientes se queixem de sintomas ortostáticos como, por exemplo, sensação de desfalecimento ao ficar de pé. A anemia grave impede mesmo as atividades mais leves e, em alguns pacientes, poderá causar dispneia em repouso ou angina. De um modo geral, as mortes atribuíveis à anemia subaguda não ocorrem até a hemoglobina atingir menos que 5g/dL, embora muitos pacientes consigam sobreviver em níveis mais baixos.9
Avaliação da Anemia
Define-se anemia como uma anormalidade que se caracteriza por níveis baixos de hematócrito ou baixas concentrações de hemoglobina no sangue. Níveis normais de hemoglobina variam entre 13,5 e 17,5g/dL do sangue total. Tipicamente, o nível de hematócrito é três vezes o nível de hemoglobina, cuja faixa normal varia de 40 a 51%, exceto nos estados de aglomeração de RBCs, em que poderá ocorrer algum erro no cálculo do hematócrito.
Mulheres na idade concepcional e pessoas idosas geralmente têm níveis de hemoglobina na faixa inferior da normalidade. A anemia é muito comum em pacientes hospitalizados. Isso é tipicamente multifatorial e a causa possivelmente seja alguma enfermidade subjacente que exija hospitalização, flebotomia, uso de medicações e diluição (geralmente causada pela administração de cristaloides ou por mobilização de edema com o paciente na posição em supino).
A busca da causa subjacente inicia logo após o reconhecimento da anemia. Existem muitas abordagens à classificação de anemia. A mais intuitiva é separar a anemia em algum defeito de produção (anemia hipoproliferativa) ou baixa longevidade dos eritrócitos (perda periférica ou destruição). Com frequência, isso pode ser feito por meio da avaliação de esfregaços periféricos ou por medições das contagens de reticulócitos.
Nas situações em que a anemia for causada apenas por sangramentos ou pela baixa longevidade dos eritrócitos, a medula deve responder aumentando-se a eritropoiese, e a doença poderá ser detectada por níveis elevados de reticulócitos no sangue periférico. Provavelmente, sejam necessários vários dias para produzir uma resposta dos reticulócitos à anemia. A medula óssea de adultos funcionais consegue sustentar indefinidamente reticulocitoses de 10% por dia.
Os sangramentos significativos geralmente são óbvios, embora os pacientes e os médicos não consigam detectar baixos níveis de sangramento no trato gastrintestinal, principalmente no intestino delgado. A hemorragia retroperitoneal, que ocasionalmente é espontânea, também produz perda substancial de sangue antes de produzir sintomas.
A flebotomia frequente em pacientes hospitalizados provavelmente resulte em volumes expressivos de perda de sangue. A prática de coletar diversos conjuntos de culturas de sangue para excluir principalmente a hipótese de bacteremia, embora seja justificada na maior parte das vezes, exige quantidades significativas de amostras de sangue.
Anemias Hemolíticas
A maneira usual de confirmar a hemólise é medir a lactato desidrogenase (LDH), uma enzima liberada por RBCs hemolizados, e a haptoglobina, que elimina a hemoglobina livre no sangue. Portanto, os episódios agudos devem aumentar a LDH e diminuir a haptoglobina. Nenhum desses valores é específico para hemólise; mais especificamente, observam-se níveis elevados de LDH em muitos estados diferentes de doença (por exemplo, pneumonia, linfoma). No entanto, a haptoglobina não detectável quase sempre está associada à hemólise significativa sob o ponto de vista clínico.
A LDH pode ser um marcador bastante útil para acompanhar o processo hemolítico ao longo do tempo. Níveis elevados de bilirrubina indireta implicam no catabolismo acelerado da hematina e são encontrados nos casos de hemólise significativa. A hemólise produz muitos sintomas e condições além da anemia. A hemólise intravascular aguda pode precipitar insuficiência renal. A hemólise significativa geralmente está associada à trombose e, caso persista, poderá causar hipertensão pulmonar e outras morbidades sérias, mesmo se a anemia for bem compensada pelo aumento na eritropoiese.
De um modo geral, os médicos costumam fazer a distinção entre hemólise intravascular e extravascular. Hemólise intravascular implica na destruição de RBCs no interior do espaço vascular causada por danos estruturais graves. Essa ocorrência pode ser súbita e grave, como a hemólise que ocorre nos casos de reação aguda às transfusões hemolíticas devido à administração de sangue do tipo ABO incompatível.
A cor do plasma da hemoglobina livre de pacientes com hemólise intravascular é castanho-avermelhada. A hemólise extravascular é uma aceleração dos meios tradicionais do corpo humano para decompor RBCs defeituosos no baço e no fígado. Esse fato ocorre nas situações em que os RBCs forem danificados na circulação sem sofrer lise.
Logo após a suspeita ou confirmação de hemólise, deve-se fazer o TDA, também conhecido por teste direto de Coombs. O TDA permite verificar se há algum anticorpo ou revestimento de complemento na superfície dos eritrócitos. Resultados positivos do TDA estabelecem se as causas da hemólise são imunológicas. Ahai por anticorpos quentes é uma causa comum de hemólise com TDA positivo.
Reações agudas ou tardias a transfusões hemolíticas também produzem TDAs positivos. A anemia hemolítica por aglutinina fria também produz hemólise com TDAs positivos (geralmente intravascular), embora, tipicamente, apenas o complemento reveste a superfície eritrocitária. É importante ressaltar que muitos pacientes com anemias imuno-hemolíticas apresentarão algum defeito subjacente de tolerância imune, como vírus da imunodeficiência adquirida (HIV) ou alguma malignidade linfoide, predispondo-os a essa condição.
A anemia hemolítica com TDA negativo tem um amplo diagnóstico diferencial, embora geralmente seja causada por condições hereditárias que resultam na redução da longevidade dos RBCs ou no aumento na suscetibilidade a danos. De um modo geral, o diagnóstico exato exige investigações laboratoriais. A suspeita de problemas eritrocitários estruturais, como esferocitose hereditária, geralmente é levantada pela morfologia dos esfregaços periféricos ou pelos índices de RBC (isto é, CHCM elevada).
Hemoglobinopatias, como anemia da célula falciforme, são suspeitas clinicamente e confirmadas pela eletroforese da hemoglobina. A G6PD produz anemia hemolítica com TDA negativo em pacientes suscetíveis quando forem expostos a estresse oxidativo, geralmente na forma de uma medicação. A hemoglobinúria paroxística noturna (HPN), uma condição adquirida em que os RBCs são anormalmente suscetíveis ao complemento, é uma causa tratável rara, porém séria, de hemólise com TDA negativo.
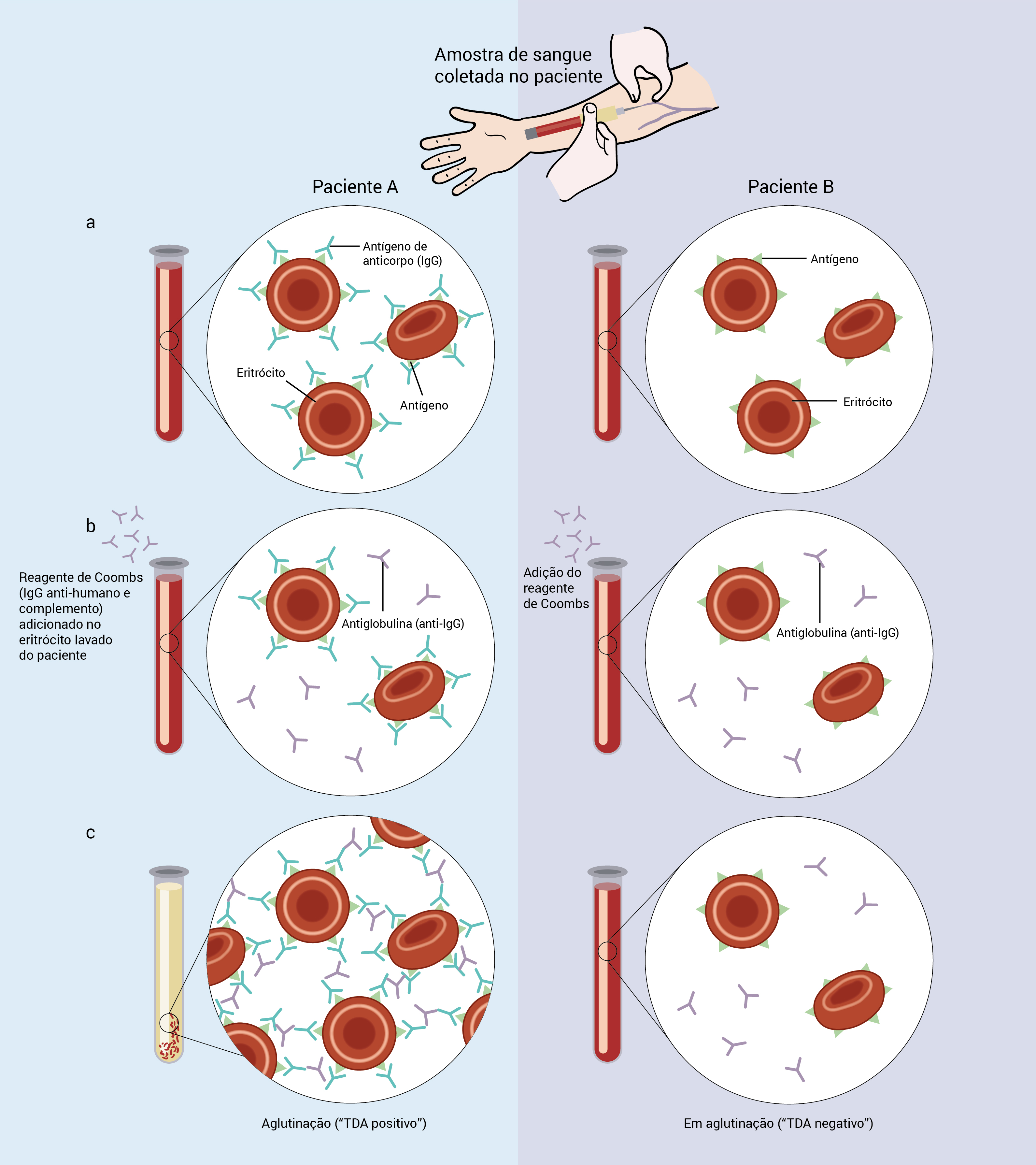
A Figura 8 mostra o TDA.
Ahai: anemia hemolítica autoimune; TDA: teste direto da antiglobulina.
Figura 8 - O TDA, também conhecido por teste direto de Coombs, determina se os eritrócitos de um paciente estão revestidos com anticorpo ou complemento. (A) O paciente A tem um anticorpo IgG ligado aos eritrócitos, da mesma forma como nos casos de Ahai. O paciente B é normal. (B) O reagente de Coombs (IgG anti-humano e complemento) foi adicionado aos eritrócitos lavados de ambos os pacientes. (C) O reagente de Coombs se liga e faz ligação cruzada com o anticorpo ou complemento ligado aos eritrócitos (Paciente A), ocasionando aglutinação. Essa aglutinação indica a presença de uma resposta imune inapropriada aos eritrócitos do paciente. O paciente B não tem anticorpo para os eritrócitos e, consequentemente, não ocorre nenhuma aglutinação (TAD negativo).
Anemias Hipoproliferativas
A etapa única na avaliação de anemias hipoproliferativas é a classificação que se fundamenta no VCM: as anemias normocíticas têm VCM na faixa normal; as microcíticas têm VCM baixo; as macrocíticas têm VCM elevado. A identificação de linfócitos em esfregaços como padrão interno é extremamente útil para classificar o tamanho dos eritrócitos (RBCs): RBCs normais devem ter o mesmo tamanho que o núcleo de um linfócito em repouso.
Anemias macrocíticas. Diagnósticos específicos podem ser feitos em quase todos os pacientes com macrocitose sustentada, principalmente em indivíduos com VCM sustentado acima de fL. Fatores como histórico detalhado, exame físico e revisão de esfregaços periféricos geralmente são suficientes para sugerir o diagnóstico. Observa-se a presença de macrocitose em qualquer condição que cause replicação anormal do DNA.
A macrocitose poderá ser iatrogênica, pois a síntese do DNA sofre impactos de muitas medicações citorredutoras ou antivirais (por exemplo, hidroxiureia ou zidovudina [AZT]). A síntese do DNA também pode sofrer impactos das deficiências de vitamina B12 (cobalamina) ou de folato. De um modo geral, a macrocitose com anormalidades de maturação nuclear é conhecida por alteração megaloblástica e pode ser demonstrada nos esfregaços periféricos pela presença de neutrófilos com segmentação extrema.
Em quase todos os casos, a deficiência de vitamina B12 é resultado da má absorção. Os pacientes com ressecções intestinas ou com desvio intestinal também poderão desenvolver má absorção por deficiência de vitamina B12. Outros pacientes poderão desenvolver um processo autoimune que impede a elaboração de fatores intrínsecos pelas células parietais do estômago.
Os fatores intrínsecos são imprescindíveis para absorção da vitamina B12 no intestino delgado. Esse fato é conhecido historicamente como anemia perniciosa. Os testes sorológicos para células parietais e anticorpos de fatores intrínsecos substituíram o teste tradicional de Schilling para o diagnóstico de anemia perniciosa. Provavelmente, a deficiência de vitamina B12 seja acompanhada de neuropatia e de disfunções neurocognitivas.
A deficiência de folato ocorre com mais frequência em pessoas com consumo abusivo de bebidas alcoólicas e dieta subótima. Embora se baseie fundamentalmente nas medições dos níveis de folato e de vitamina B12, o diagnóstico laboratorial de deficiência de folato ou de vitamina B12 está sujeito a diversos tipos de armadilha.
Os níveis de ácido fólico podem aumentar rapidamente após a reposição por meio de dieta ou de suplementação, de modo que é extremamente importante ter muita cautela ao interpretar um único nível de folato, sobretudo em pacientes hospitalizados. As medições dos níveis de folato no interior dos eritrócitos estão menos sujeitas a oscilações.
Ocasionalmente, observa-se a deficiência fisiologicamente significativa de vitamina B12 em níveis baixos da vitamina. Os melhores indicadores metabólicos da deficiência de folato e de vitamina B12 são a homocisteína e o ácido metilmalônico; ambos os níveis são elevados nos casos de deficiência de vitamina B12, enquanto que a deficiência de folato aumenta apenas o nível de homocisteína.10 Esses dois marcadores são artificialmente elevados nos casos de insuficiência renal.
Outras causas comuns de macrocitose poderão ser excluídas pelo histórico ou pela investigação laboratorial. Essas outras causas incluem consumo abusivo de álcool, disfunção hepática, hipotireoidismo ou envenenamento por metais pesados. Levando-se em consideração que os reticulócitos são maiores que os RBCs normais, a reticulocitose robusta pode aumentar substancialmente o VCM.
Os pacientes, em especial os mais velhos, com anemia macrocítica inexplicável devem ser avaliados com aspiração na medula óssea e biópsia para verificar a eventual presença da síndrome mielodisplásica (SMD), uma condição clonal que se caracteriza por hematopoiese ineficiente.
Anemia microcítica. A anemia microcítica é bastante comum. Tipicamente, a anemia por deficiência de ferro causa microcitose (VMC baixo), anisocitose (RDW elevado) e, em circunstâncias mais graves, hipocromia (CHCM baixa) e morfologia eritrocitária anormal (por exemplo, células em forma de lápis). A presença de trombocitose reativa é comum na deficiência de ferro e, ocasionalmente, pode ser profunda.
Os estudos laboratoriais do ferro são usados para confirmar o diagnóstico. As pessoas com deficiência de ferro clinicamente significativa e sem complicações possivelmente apresentem saturação de transferrina (calculada dividindo-se a capacidade total de ligação do ferro pelo nível de ferro) inferior a 10% e nível de ferritina abaixo de 20ng/mL.
Os níveis de ferro livre e a saturação de ferro poderão ser afetados por ingestão recente ou transfusão de ferro. Como a ferritina é um reagente de fase aguda, os valores duvidosos devem ser interpretados com muita cautela; todavia, níveis de ferritina acima de 100ng/mL excluem definitivamente a hipótese de deficiência de ferro.
Os aspirados da medula óssea possibilitam avaliar diretamente os estoques de ferro, embora a aplicação dessa técnica raramente seja necessária na prática clínica. A deficiência de ferro em homens ou em mulheres que não estiverem menstruando sempre justifica a realização de um exame completo para excluir perda de sangue GI oculto por meio de colonoscopia e endoscopia superior, tendo em vista que poderá ser a forma de apresentação de câncer no colo ou de câncer gástrico.11
A talassemia também produz anemia microcítica. O RDW é tipicamente elevado, porém menos elevado que nos casos de deficiência de ferro. Como a talassemia é uma condição hereditária, os valores do VMC medidos ao longo do tempo são muito úteis para determinar a consistência.
Os resultados dos estudos laboratoriais de ferro são normais ou elevados por causa da associação entre avidez do ferro e talassemia. A eletroforese da hemoglobina define muitas hemoglobinopatias e sugere a presença de talassemia-ß através da elevação da hemoglobina A2, ao passo que a talassemia-a exige diagnósticos genéticos mais sofisticados.
Inflamação crônica da anemia (também conhecida pelo termo menos usual de anemia de doença crônica) é a reação da medula óssea com alguma inflamação com essa origem. Normalmente, é mais profunda em infecções crônicas como osteomielite ou condições reumatológicas. A hepcidina é uma molécula elaborada em ambientes inflamatórios que regulam negativamente a mobilização do ferro a partir do estoque e tem papel importante na fisiopatologia da anemia ocasionada por inflamações crônicas.12
Ao inibir o transportador de ferro ferroportina, a hepcidina impede a absorção de ferro no trato gastrintestinal ou surge a partir do estoque de ferro no sistema reticuloendotelial. A anemia ocasionada por inflamações crônicas pode ser normocítica ou microcítica; os níveis de ferritina (como medida da estocagem de ferro) geralmente aumentam, assim como os níveis de marcadores inflamatórios como a velocidade de hemossedimentação (VHS).
A secreção de eritropoietina aumenta em pessoas com rins saudáveis e agrava a anemia. A capacidade de secretar eritropoietina poderá se perder logo no início do curso da doença renal; os defeitos de secreção de eritropoietina se correlacionam fracamente com o declínio na taxa de filtração glomerular (TFG).
Observa-se claramente que as pessoas com diabetes melito geralmente têm algum defeito de secreção de eritropoietina no início do curso da doença renal.13 A secreção de eritropoietina em resposta à anemia também diminui em vários estados como, por exemplo, no casos de câncer.
Rupturas transitórias ou permanentes na eritropoietina, enquanto outras células são preservadas, é uma condição descrita como aplasia eritrocitária pura. Há formas congênitas (por exemplo, anemia de Diamond-Blackfan) e formas adquiridas de aplasia eritrocitária pura. Observa-se a incidência de aplasia eritrocitária transitória depois de infecções virais, não apenas com o parvovírus B19, mas também com hepatite e HIV.
A significância dos episódios aplásicos de RBC transitória geralmente é muito exagerada em pacientes com RBC de vida curta, como ocorre em pacientes com anemia de células falciformes. A aplasia eritrocitária também foi associada ao uso de determinados tipos de medicamentos, doença autoimune, timoma, gravidez e malignidades (geralmente anemia com grandes linfócitos granulares).14 O exame completo para essas condições subjacentes é extremamente importante, embora muitos casos de aplasia eritrocitária permaneçam idiopáticos.
Os pacientes com anemia hipoproliferativa inexplicável devem ser avaliados para verificar a eventual presença de discrasia de células plasmáticas, considerando que anemia é uma apresentação comum de mielomas múltiplos.15 Isso poderá ser feito por testes no sangue periférico por eletroforese das proteínas séricas (EPS), por imunização sérica por eletroforese (ISE) e pela medição da cadeia leve que circula livremente no soro ou da imunofixação urinária.
Em pacientes mais velhos, a síndrome mielodisplásica (SMD) também é uma hipótese a ser considerada após a exclusão de outras etiologias. Embora a avaliação de esfregaços de sangue periférico possa ser sugestiva, o diagnóstico de SMD quase sempre depende do aspirado e da biópsia da medula óssea com análise citogênica.
Eritrocitose (Policitemia)
Eritrocitose e policitemia são termos que descrevem níveis elevados de hematócrito ou de hemoglobina. A incidência de eritrocitose é menos frequente que a de anemia. As medições de hemoglobina devem ser repetidas em pacientes que não estiverem agudamente enfermos, tendo em vista que poderão ocorrer hemoglobinas transitoriamente elevadas com hemoconcentração (depleção volumétrica), transfusão recente ou coleta/manuseio inapropriado de espécimes.
Após a confirmação, é imprescindível fazer um exame completo para determinar se o problema é primário para a medula óssea ou secundário para o efeito das medicações ou hipoxemia. Determinados tipos de medicamentos podem produzir eritrocitose, principalmente os análogos de eritropoietina e a testosterona, incluindo medicamentos sujeitos a prescrições ou de uso indiscriminado para melhorar o desempenho atlético.
A hipoxemia, assim como a anemia, estimula a secreção de eritropoietina pelo aparelho justaglomerular dos rins que, consequentemente, aumenta a eritropoiese no interior da medula. As causas mais comuns de hipoxemia incluem situações fisiológicas como viver em grandes altitudes e estados patológicos como doença pulmonar obstrutiva crônica (DPOC), apneia obstrutiva do sono, síndrome de hipoventilação da obesidade, exposição ao monóxido de carbono (geralmente nos casos de tabagismo inveterado) e desvio de sangue ou incompatibilidade na ventilação/perfusão no interior do pulmão.
A saturação de oxigênio no estado de repouso é o primeiro passo na avaliação de policitemia secundária, embora não exclua muitas entre as outras causas. Esses processos produzem policitemia secundária. A policitemia primária é conhecida como policitemia vera ou policitemia “autêntica”. Trata-se de um distúrbio clonal na medula óssea produzido por precursores de eritrócitos que proliferam independentemente do estímulo da eritropoietina.
A policitemia primária é um entre os quatro distúrbios mieloproliferativos clássicos, sendo que os outros três são LMC, mielofibrose idiopática (MFI) e TE. Nos casos de policitemia vera, deve-se suprimir completamente a eritropoietina. Mutações na quinase JAK2 são quase universais na policitemia vera (estão presentes em, aproximadamente, 50% de casos de TE e mielofibrose), de modo que o sequenciamento direto (feito no sangue periférico) para a mutação V617F no gene JAK2 é bastante útil para a exclusão de policitemia vera.
Os pacientes com policitemia são predispostos à trombose, sendo que a doença poderá se transformar em mielofibrose ou anemia, ou em pancitopenia ou leucemia mielógena aguda secundária. A policitemia vera pode ser controlada com bastante eficiência por flebotomia ou hidroxiureia, para normalizar os níveis de RBCs, e ácido acetilsalicílico para impedir a ocorrência de trombose.
Leucopenia
O primeiro passo da avaliação de pacientes com leucopenia é determinar qual subgrupo de WBCs está inapropriadamente baixo e identificar se é isolado ou se faz parte de uma bicitopenia ou pancitopenia. O diagnóstico diferencial de linfopenia (contagem baixa de linfócitos) é bastante limitado.
Observa-se com frequência a incidência de linfocitopenia em infecções virais, incluindo o resfriado comum. Ela ocorre também em associação com infecções mais sérias tais como HIV, doença reumatológica, sarcoidose ou linfoma de Hodgkin, ou com uso de fármacos (principalmente corticosteroides ou Qt citotóxica).
A incidência de neutropenia (contagem baixa de neutrófilos) é muito mais comum. A contagem absoluta dos neutrófilos (CAN) é a medição mais apropriada de neutropenia; para calcular a CAN, multiplica-se a contagem total de WBCs pelo percentual de neutrófilos no diferencial. A imunidade inata depende basicamente dos neutrófilos como primeira linha de defesa contra infecções bacterianas e fúngicas.
Embora a suscetibilidade a infecções aumente com a gravidade da neutropenia, a maior parte dos pacientes com CAN acima de 1.000/µL possivelmente não apresente nenhum problema infeccioso. Os pacientes com CAN inferior a 500/µL correm grande risco de incidência de sepse, normalmente ocasionada pela translocação da flora endógena para a corrente sanguínea.
Tipicamente, nesse contexto, a presença de febre é o primeiro sinal de infecção. Febre em pacientes neutropênicos é uma emergência e exige o início imediato da aplicação de antibióticos empíricos de espectro amplo, incluindo a cobertura gram-negativa de antipseudomonas.
Os pacientes com neutropenia grave com mais de uma semana de duração correm grande risco de infecções fúngicas invasivas, como a aspergilose pulmonar, que exija diagnóstico rápido e início imediato da terapia. A Infectious Diseases Society of America (IDSA) publica regularmente guias para orientar os médicos na escolha do tratamento mais apropriado de infecções em hospedeiros neutropênicos.17
O Quadro 3 contém as descobertas laboratoriais em eritrocitose.
Quadro 3
|
Descobertas Laboratoriais em Eritrocitose | ||
|
|
Policitenia vera |
Policitenia secundária (p. ex., hipóxia) |
|
Nível de eritropoietina |
Reduzido |
Normal ou elevado
|
|
JAK2 |
Presença da mutação V617F |
Tipo selvagem
|
Existem diferenças genéticas importantes nas contagens de leucócitos que devem ser consideradas nas avaliações de leucopenia, tendo em vista que algumas pessoas, incluindo as de etnia africana, podem apresentar números baixos de neutrófilos em circulação (ocasionalmente, CAN <1.000/µL), porém sem nenhum problema infeccioso, provavelmente devido às reservas medulares normais ou elevadas. A presença de monócitos também é um fator modificador; a monocitose protege contra infecções generalizadas.
Neutropenia é uma consequência esperada da Qt mielossupressiva, sendo que o grau e a duração da doença poderão ser previstos pelo regime de tratamento utilizado e por fatores específicos dos pacientes. Pacientes idosos e portadores da infecção pelo HIV são mais suscetíveis à prolongação da neutropenia e de infecções sérias e, com frequência, recebem fatores de crescimento mieloide depois da Qt.
Outras causas de neutropenia incluem medicamentos não quimioterápicos que produzem mielossupressão relacionada à dosagem ou a alguma “agranulocitose” idiossincrática como, por exemplo, fenitoína, trimetoprima + sulfametazol (Bactrim), propiltiouracil e carbamazepina.18
Embora algumas condições reumatológicas como lúpus eritematoso sistêmico possam causar neutropenia, outras linhas de células também poderão ser afetadas. A síndrome de Felty se caracteriza pela presença de artrite reumatoide, esplenomegalia e neutropenia. Outras etiologias de neutropenia incluem a radiação, sendo que distúrbios na medula óssea como mielofibrose idiopática, anemia aplásica e mielodisplasia podem ser acompanhados de neutropenia.
Malignidades hematológicas, como leucemia aguda, geralmente se apresentam com neutropenia mesmo que a contagem total de WBCs seja ou não elevada. Além disso, há pacientes com defeitos congênitos no desenvolvimento de células mieloides que têm neutropenia crônica ou cíclica. Recentemente, alguns casos de neutropenia cíclica foram vinculados à herança autossômica dominante de mutações no gene ELA2, que codifica a elastase neutrofílica, assim como a outros genes que regulam o desenvolvimento de células mieloides.
Leucocitose
A leucocitose é um problema comum na hematologia consultiva. Em primeiro lugar, deve-se considerar que tipo de célula é responsável por níveis elevados de leucócitos. O exame do CBC e dos esfregaços de sangue confirma a ausência de blastos e exclui a maioria dos casos de leucemia aguda. Tipicamente, a linfocitose sustentada em adultos indica a presença de LLC, embora a doença possa ocorrer em caráter transitório com infecções virais como o vírus de Epstein-Barr.
As populações de linfócitos granulares atípicos ou grandes geralmente indicam a presença de doença reumatológica. A monocitose é um sinal inespecífico de estresse medular ou de inflamação sistêmica, sendo que populações expressivas de monócitos geralmente acompanham a neutropenia autoimune ou anunciam a recuperação da medula depois de mielossupressão induzida por medicamentos.
Raramente, malignidades hematológicas distintas produzem monocitose. Basofilia quase sempre implica na presença de algum distúrbio mieloproliferativo subjacente, tipicamente LMC. As células imaturas (células mieloides antes do estágio de banda) são raras ou inexistentes no sangue periférico.
A avaliação da medula óssea quase sempre é indicada nos casos em se observa imaturidade muito séria no sangue periférico, a menos que ocorra um estresse sistêmico óbvio e profundo (como, por exemplo, um choque séptico) e as células imaturas desapareçam na medida em que o paciente se recupera. Contagens muito elevadas de WBCs podem aumentar a viscosidade do sangue como decorrência da leucostase ou “sedimentação”.
Na maior parte dos casos, isso se torna um problema nas situações em que há um grande número de blastos em circulação; considerando que são muito grandes, essas células expressam moléculas adesivas aberrantes e se movimentam com dificuldade na microcirculação. A leucostase e a hiperviscosidade se manifestam como dispneia e hipoxemia, com problemas neurológicos não focais ou com sangramentos.
No caso de suspeita, a leucostase causada por leucocitose exige tratamento urgente com citorredução e, eventualmente, com leucoferese. Felizmente, trata-se de um caso raro em que a população de células é formada por células maduras (tipicamente, neutrófilos e linfócitos), mesmo na hipótese de leucocitose extrema.
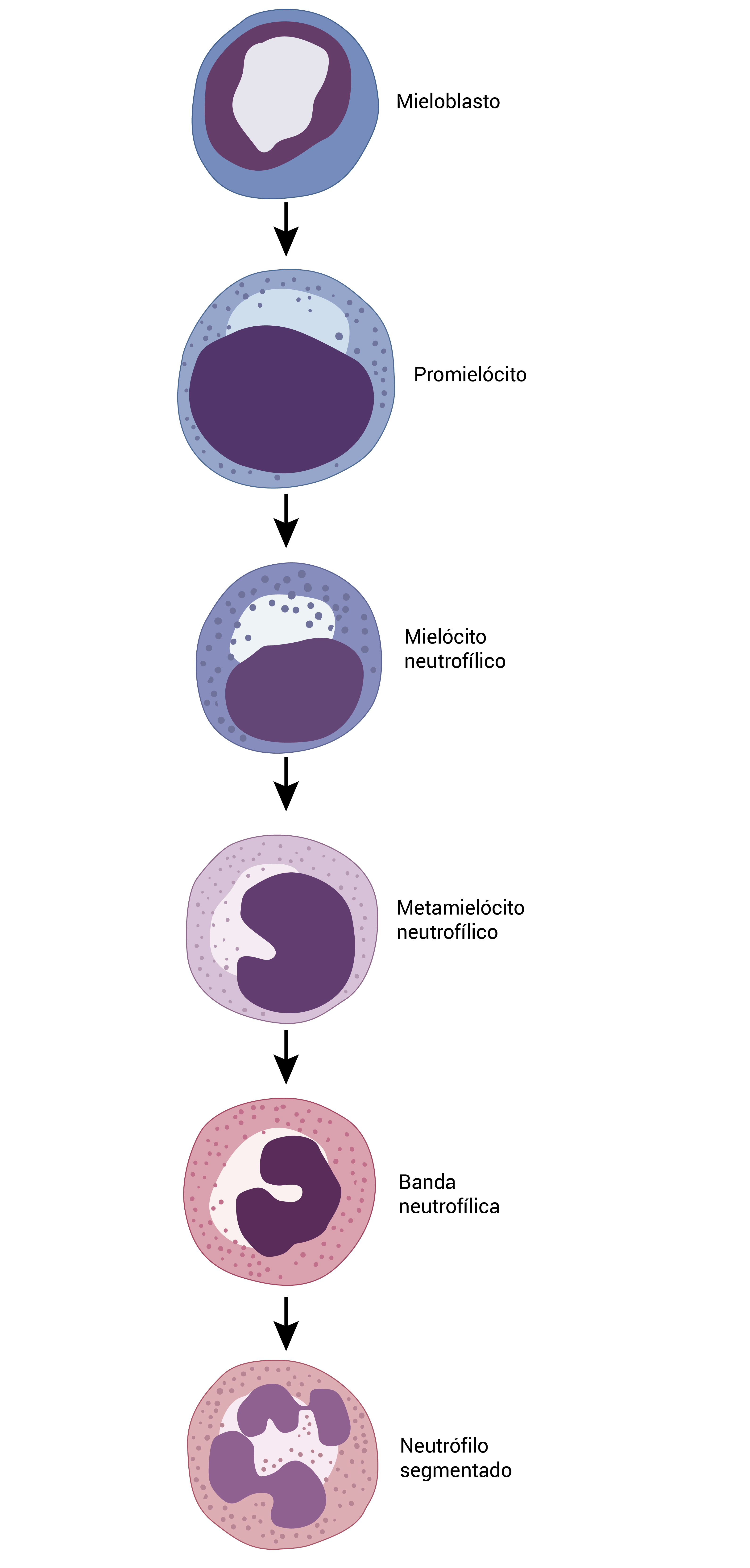
A Figura 9 ilustra as células mieloides e como se dá o processo de maturação.
Figura 9 - As células mieloides sofrem alterações típicas no tamanho, na forma nuclear e nos grânulos citoplasmáticos durante o processo de maturação. A combinação dessas características permite atribuir cada célula precursora a um estágio específico de maturação.
O problema diagnóstico mais comum surge a partir do momento em que os neutrófilos predominam na população de células anormais. Há muitas causas de neutrófilos reativos ou de reações leucemoides. Neutrofilia é uma resposta fisiológica a uma infecção, inflamação ou estresse. A ausência de baço funcional resulta no aumento permanente, porém tipicamente inexpressivo, na contagem de neutrófilos.
Os pacientes asplênicos possivelmente se apresentem com neutrofilia exuberante em resposta ao estresse ou a infecções. Tabaco, lítio e, em especial, esteroides como a prednisona, podem aumentar a contagem de neutrófilos. Na realidade, as respostas neutrofílicas imediatas ao estresse, aos esteroides e mesmo aos fatores de crescimento mieloides, são decorrências da demarcação de leucócitos no sangue em resposta às citocinas, precedendo o aumento na produção de medula óssea e na saída para o sangue.
Na ausência de causas reativas de neutrofilia, uma das opções é considerar a presença de algum distúrbio mieloproliferativo subjacente. Os distúrbios mieloproliferativos, especificamente LMC, produzem elevação lenta, estável e persistente na contagem de leucócitos, embora a contagem oscile em resposta a vários estímulos.
A LMC pode ser diagnosticada pela presença da translocação t(9;22), que forma o cromossomo de Filadélfia. Isso poderá ser feito mais facilmente pelo teste de PCR do sangue periférico para o produto do gene bcr-abl. Embora a pontuação da fosfatase leucocitária alcalina (FLA) tenha sido usada no passado para ajudar a distinguir entre LMC e neutrofilia reativa (a pontuação tendia a ser mais baixa nos casos de LMC), esse tipo de avaliação deixou de ser usada na prática clínica.
Elevações na contagem de leucócitos também podem ocorrer em outros distúrbios mieloproliferativos como TE, mielofibrose ou policitemia vera. Caso seja positivo, o teste da PCR no sangue periférico para detectar a mutação V617F do gene JAK2 poderá ser diagnóstico, tendo em vista que essa mutação é encontrada em, aproximadamente, 50% dessas doenças.16 Ocasionalmente, os médicos solicitam aspirado e biópsia da medula óssea para facilitar a distinção de reação leucemoide versus distúrbio medular primário.
A presença de eosinofilia também é muito comum. Tipicamente, define-se eosinofilia como contagem absoluta de eosinófilos (CAE) superior a 1.500 células/µL e com diagnóstico diferencial bastante amplo. Os efeitos pró-inflamatórios dos eosinófilos podem ser destrutivos nos casos de hipereosinofilia moderada ou grave, com toxicidades em vários órgãos.19
A hipereosinofilia sustentada pode ocasionar danos substanciais nos órgãos, em especial fibrose endomiocárdica, tromboembolismo e complicações respiratórias ou neurológicas. A produção de eosinófilos pela medula óssea é estimulada basicamente pela citocina interleucina-5 (IL-5). Os níveis dessa citocina provavelmente sejam elevados em várias circunstâncias.
As principais associações são infecções parasitárias, alergias ou doenças autoimunes. Ocasionalmente, a eosinofilia produz algumas dicas para ajudar no diagnóstico da doença de Adams ou de diversas vasculites, como, por exemplo, a síndrome de Churg-Strauss. A eosinofilia pode também estar associada a diversos neoplasmas, sendo que o linfoma de Hodgkin é o mais frequente.
A síndrome hipereosinofílica é uma elevação sustentada na contagem absoluta de eosinófilos. Pode ser o marcador de alguma doença subjacente como a síndrome de Churg-Strauss ou o resultado da desregulação no crescimento hematopoiético. Mesmo com exames completos exaustivos e biópsia da medula óssea, às vezes é muito difícil identificar a etiologia de eosinofilia ou fazer a distinção entre subtipos da síndrome hipereosinofílica.
Alguns pacientes com eosinofilia idiopática geralmente apresentam uma população anormal de linfócitos que produz IL-5,20 ao passo que outros apresentam uma reorganização genética envolvendo o receptor do fator de crescimento derivado de plaquetas.21
Ainda que esses casos não sejam comuns, sua identificação é fundamental, considerando que esses pacientes respondem a tratamentos específicos, como a terapia com o inibidor da tirosina quinase imatinib. Os eosinófilos são extraordinariamente sensíveis aos corticosteroides, geralmente desaparecendo algumas horas após a administração de uma dose de esteroides sistêmicos.
Significância Clínica da Trombocitopenia
A importância clínica da trombocitopenia depende da gravidade da doença e da causa subjacente. Os seres humanos têm mais plaquetas do que a quantidade necessária para a homeostase. Embora a contagem normal de plaqueta esteja acima de 150.000/µL, em geral ocorrem pequenos defeitos de ferimentos ou sangramentos identificáveis até a contagem cair para menos de 50.000/µL.
O sangramento aumenta com a ocorrência de traumas (geralmente descritos como “sangramento incômodo”) entre 20.000 e 50.000/µL; em contagens abaixo de 20.000/µL, provavelmente ocorra sangramento espontâneo, cujo risco aumenta nas contagem de plaquetas abaixo de 10.000/µL. Um erro muito comum é expor os pacientes aos efeitos colaterais desnecessários das transfusões ou de outros tratamentos médicos para obter contagens de plaquetas quase normais, levando-se em conta que as contagens seguras (>20.000/µL) são suficientes.
Na ausência de contraindicação para transfusão ou nas circunstâncias em que a transfusão de plaquetas não é eficaz (como nos casos de PTI), grande parte dos médicos procura manter a contagem de plaquetas acima de 10.000 ou 20.000/µL para impedir a ocorrência de sangramentos espontâneos e acima de 50.000/µL nos casos em que há sinais de sangramento ou em que se prevê a administração de algum procedimento invasivo.
Existem outros fatores modificadores, além da quantidade de plaquetas, que influenciam o risco de sangramento, considerando que alguns pacientes se apresentam com sangramento significativo sob o ponto de vista clínico em contagens de plaquetas relativamente elevadas, enquanto em outros pacientes o sangramento é mínimo, a despeito de contagens excessivamente baixas. O uso de medicamentos antiplaquetários, como ácido acetilsalicílico ou clopidogrel, interfere na função das plaquetas e aumenta substancialmente o risco de sangramento no contexto de trombocitopenia.
A trombocitopenia é um problema comum na hematologia consultiva, sobretudo em pacientes agudamente enfermos. Com frequência, ela acompanha infecções ou enfermidades críticas de todos os tipos; de um modo geral, direciona-se o tratamento para a enfermidade subjacente, fazendo-se transfusões nas situações em que forem necessárias.
A trombocitopenia grave em pessoas jovens com CBC normal quase sempre indica a presença de PTI. A transfusão de plaquetas, em geral, é ineficaz ou apenas transitoriamente eficaz nos casos de PTI. A administração de esteroides é o tratamento de primeira linha para PTI; tipicamente, utiliza-se globulina imune intravenosa (IVIg) nos casos que exigirem respostas rápidas.22
Quase todos os medicamentos causam trombocitopenia; alguns ocasionam a doença com bastante frequência. Os medicamentos produzem trombocitopenia por meio da estimulação de mecanismos diferentes, tipicamente por anticorpos induzidos por fármacos ou pela supressão de megacariócitos na medula óssea. Recomenda-se considerar também fatores como o momento de início da trombocitopenia e a prevalência documentada da doença em relação a um determinado medicamento.
Embora, de um modo geral, a trombocitopenia induzida por fármacos inicie em 5 a 7 dias após o início da terapia, pode ser variável e dificultar a detecção imediata da doença. Na ausência de culpados óbvios, uma das opções é a elaboração de um plano de descontinuação gradual de medicamentos desnecessários, o que geralmente consome muito tempo; pode levar uma semana ou mais até que o efeito do medicamento tenha “desaparecido”. Nos casos de trombocitopenia grave, pode ser necessário descontinuar o uso de todas as medicações e substituir outras medicações de acordo com a necessidade.
Com frequência, a trombocitopenia acompanha a disfunção hepática. Isso ocorre por diversas razões. A trombopoietina, hormônio responsável pela megacariopoiese, é fabricada no fígado e seu nível é inapropriadamente baixo nos casos de disfunção hepática. Condições como cirrose e hipertensão portal produzem esplenomegalia, que é uma sequestradora de plaquetas, ocasionando trombocitopenia e respostas insatisfatórias às transfusões. Além disso, as viremias por qualquer causa podem prejudicar a função medular. Em particular, HIV e hepatite C geralmente estão associadas à PTI.24
O exame completo para trombocitopenia deve sempre procurar excluir as etiologias mais sérias. Existem quatro causas de trombocitopenia em que a falha em fazer um diagnóstico imediato poderá gerar consequências graves. Essas causas devem ser consideradas formalmente na avaliação de cada paciente trombocitopênico.
Tipicamente, as leucemias agudas se apresentam com trombocitopenia por causa de falhas na hematopoiese normal ou de CID. O exame cuidadoso dos esfregaços de sangue na busca de blastos (células grandes com razões elevadas entre núcleo e citoplasma, cromatina rendilhada e núcleos proeminentes) exclui a maioria dos casos de leucemia aguda, embora as biópsias da medula óssea sejam imprescindíveis nos casos raros de leucemia aleucêmica e para exames adicionais de todos os casos de suspeita de leucemia.
O Quadro 4 apresenta as doenças geralmente associadas à eosinofilia e os exames úteis.
Quadro 4
|
Doenças Geralmente Associadas à Eosinofilia e Exames Úteis | ||
|
Causa |
Exame útil |
Observações adicionais |
|
Infecções |
Exame parasítico O&P das fezes. |
Causa mais comum de eosinofilia. |
|
Infecções helmínticas (Strongyloides e esquistossomíase) |
Sorologia para Strongyloides e esquistossomíase. |
A supressão corticosteroide da eosinofilia em infecções parasitárias ocultas poderá resultar em infecção extrema. |
|
HIV |
Terapia empírica.
|
|
|
Doença alérgica |
Histórico detalhado das medicações. |
Os anticonvulsivantes e o alopurinol são os vilões farmacológicos mais comuns. |
|
Alergia a medicamentos |
Teste de alergia. |
|
|
Asma atópica, demência |
|
|
|
Rinite alérgica
|
|
|
|
Distúrbios respiratórios |
Níveis de IgG. |
|
|
Pneumonite por hipersensibilidade
|
Imagens torácicas. |
|
|
Doença vascular colagenosa |
Varredura por TC e Anca para síndrome de Churg-Strauss. |
Tipicamente, a síndrome de Churg-Strauss produz asma, infiltrados pulmonares e neuropatia. |
|
Vasculite de Churg-Strauss |
|
|
|
Lúpus |
Marcadores sorológicos reumatológicos para lúpus. |
|
|
Sarcoidose |
Biópsia tecidual para sarcoides.
|
|
|
Anormalidades endócrinas |
Cortisol sérico. |
|
|
Insuficiência adrenal
|
|
|
|
Êmbolos colesterólicos |
|
Cateterização pós-cardíaca comum.
|
|
Distúrbios gastrintestinais |
Endoscopia. |
|
|
Doença intestinal inflamatória |
Colonoscopia. |
|
|
Gastroenterite eosinofílica
|
|
|
|
Hipereosinofilia associada a malignidades |
Varredura por TC no pescoço/tórax/abdome/pelve. |
Causada pela produção de tumor por IL-5, IL-2 ou GM-CSF. |
|
Linfoma (em especial o linfoma de Hodgkin) |
|
|
|
Tumores sólidos avançados
|
|
|
|
Expansão clonal em distúrbios mieloides
|
Biópsia da medula óssea. |
A LMC geralmente é acompanhada por basofilia. |
|
LMC |
|
|
|
LMA
|
|
|
|
SHE |
Citometria de fluxo para populações de células T anormais. |
Provavelmente seja difícil distinguir eosinofilia clonal de processos não clonais. |
|
Variante linfocitária |
|
|
|
Leucemia eosinofílica crônica |
FISH ou fusão FIP1L1-PDGFRA. |
|
|
SHE idiopática |
Biópsia da medula óssea. |
|
Anca: anticorpo citoplasmático antineutrofílico; FISH: hibridização in situ por fluorescência; GM-CSF: fator de estimulação de colônias de granulócitos-macrófagos; HIV: vírus da imunodeficiência adquirida; IgG: imunoglobulina G; IL: interleucina; LMA: leucemia mieloide aguda; LMC: leucemia mielógena crônica; O&P: ovos e parasitas; SHE: síndrome hipereosinofílica; TC: tomografia computadorizada.
A trombocitopenia induzida por heparina (TIP) é uma doença autoimune em que se forma um inibidor contra a proteína denominada ADAMTS13, que é responsável pela clivagem dos multímeros de von Willebrand. O resultado é uma TMA, geralmente acompanhada por alguma disfunção neurológica e renal. Caso não seja tratada, a TIP se torna uma doença fatal, ainda que o tratamento à base de troca de plasma seja bastante eficaz.25
Embora a TIP esteja historicamente associada a uma série de cinco fatores como trombocitopenia, anemia, febre, confusão e insuficiência renal, é imprescindível um nível elevado de suspeita para fazer o diagnóstico em tempo hábil.
O Quadro 5 apresenta uma lista de medicamentos comuns que estão fortemente associados à incidência de trombocitopenia.23
Quadro 5
|
Medicações Comuns Fortemente Associadas á Trombocitopenia |
|
Antimicrobianos (vancomicina, trimetoprima + sulfametazol, linezolida, antibióticos beta lactâmicos, ganciclovir). Heparinas (não fracionada e de baixo peso molecular). Medicações anticonvulsivantes (carbamazepina, fenitoína, ácido valproico). Medicações antiarrítmicas (amiodarona, digoxina). Aines Diuréticos (HCTZ, furosemida, acetazolamida). Quinino |
Aines: medicamentos anti-inflamatórios não esteroides; HCTZ: hidroclorotiazida.
A presença de anemia hemolítica com características microangiopáticas (em especial a presença de esquizócitos nos esfregaços periféricos), febre e danos neurológicos pode ser tão sutil como a cefaleia, sendo que a disfunção renal exige avaliação emergencial para TIP. De um modo geral, o teste para a atividade da proteína denominada ADAMTS13 é bastante útil, embora o tratamento com troca de plasma em casos fortemente suspeitos não possa aguardar os resultados do teste confirmatório.
A CID também produz trombocitopenia e anemia hemolítica microangiopática. Essa doença é causada pela ativação difusa e desregulada da cascata de coagulação, que poderá produzir tanto coagulação quanto hemorragia inapropriada. Tipicamente, é provocada por infecções sérias ou por alguma malignidade subjacente, embora também tenha sido observada em problemas vasculares.26
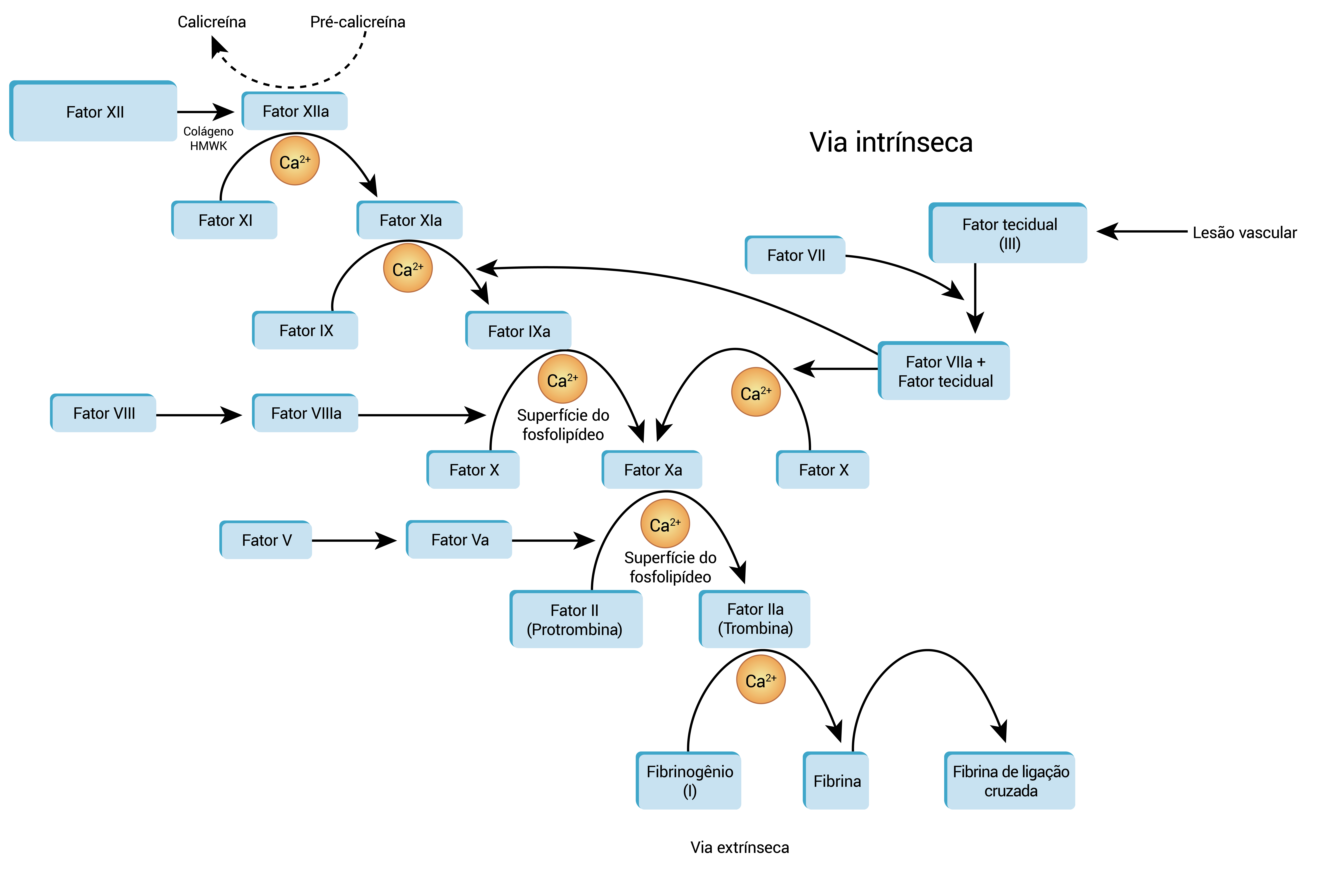
A CID pode ser excluída na avaliação de esfregaços periféricos para esquizócitos, assim como na análise dos parâmetros de coagulação. Nos casos de CID, a razão normalizada internacional (INR)/TP e o TTP tipicamente são elevados pela depleção de fatores. Os níveis de fibrinogênio geralmente são baixos, embora na CID crônica o fígado provavelmente consiga compensar altas renovações.
Os subprodutos da fibrinólise, mais especificamente o dímero-D e os produtos da decomposição da fibrina, apresentam níveis quase sempre elevados. O tratamento é basicamente direcionado para a causa subjacente, embora seja importante suplementar os níveis de fibrinogênio para diminuir o sangramento e, em circunstâncias selecionadas, administrar baixas doses de heparina para inibir a trombina e facilitar a atenuação da CID.
A trombocitopenia induzida pela heparina (TIH) é ocasionada por um anticorpo que age contra o complexo formado por heparina + fator plaquetário 4 (PF4) e estimula a ativação inapropriada das plaquetas. Ainda que, normalmente, a trombocitopenia seja modesta, o perigo de TIH surge a partir de um estado profundo de coagulação com capacidade de formar coágulos na circulação venosa e arterial.
O diagnóstico é importante em pacientes nos quais a heparina tenha sido usada entre 7 a 14 dias antes do início da trombocitopenia, ou mais rapidamente nos casos em que o paciente tenha sido previamente exposto à heparina. A pontuação “4 Ts” prevê a probabilidade pré-teste de TIH com base no grau da trombocitopenia, no momento da exposição à heparina, na presença de trombose e na ausência de outras causas.27
A disposição imediata do enzimaimunoensaio (Elisa) para confirmar a existência de um anticorpo anti-PF4 é altamente sensível, sendo que os resultados negativos quase sempre excluem este diagnóstico. O ensaio funcional da liberação de serotonina é sensível e muito mais específico, porém nem sempre está imediatamente à disposição.
Nos casos de suspeita de TIH, recomenda-se descontinuar o uso de heparina não fracionada e de baixo peso molecular (enoxaparina ou dalteparina) e iniciar um processo de anticoagulação alternativo com um inibidor direto da trombina, mesmo na ausência de coágulos comprovados.28
O Quadro 6 contém a pontuação 4Ts para determinar a probabilidade pré-teste de trombocitopenia induzida pelo uso de heparina.27
Quadro 6
|
Pontuação 4ts para Determinar a Probabilidade Pré-Teste de Trombocitopenia Induzida Pelo Uso De Heparina | ||||
|
Pontos |
Trombocitopenia |
Momento da trombocitopenia |
Trombose ou outras sequelas |
Outras causas |
|
2 |
Queda nas plaquetas >50% ou nadir de 20?100 x 103/µL |
Início nos dias 5 a 10 ou no 1º dia após a reexposição nas situações em que o paciente tenha recebido heparina dentro de 30 dias. |
Confirmação de nova trombose; necrose na pele; reação sistêmica aguda depois da administração intravenosa de heparina.
|
Nenhuma causa aparente. |
|
1 |
Queda de 30 a 40% na contagem de plaquetas ou nadir de 10 a 19 x 103/µL |
Início no 10º dia ou =1 dia antes da exposição à heparina (há 30?100 dias). |
Trombose progressiva ou recorrente em algum sítio; lesões cutâneas não necrozantes; suspeita de trombose (não comprovada).
|
Possível (a critério do médico). |
|
0 |
Queda na contagem de plaquetas <30% ou nadir plaquetário <10 x 103/µL |
Contagem de plaquetas <4 dias sem exposição recente à heparina.
|
Nenhuma. |
Definitivas (a critério do médico). |
Obs.: Determina-se uma pontuação para cada uma das quatro colunas, lendo-se uma contagem total entre 0 e 8. 0 a 3: baixa probabilidade; 4 a 5: probabilidade intermediária; 6 a 8: alta probabilidade.
Trombocitose
Ao contrário da eritrocitose, as contagens elevadas de plaquetas são muito comuns e nem sempre isso significa a presença de alguma patologia subjacente. O aumento na quantidade de plaquetas com inflamações e a ocorrência de trombocitose reativa podem ser observados em qualquer estado inflamatório. Deficiência de ferro é uma causa frequente de trombocitose secundária.29
Levando-se em consideração que essas plaquetas estão funcionando normalmente, os eventos coagulantes (arteriais e venosos) são raros e, de um modo geral, não é necessária nenhuma terapia antiplaquetária agressiva ou tratamento citorredutor. Na ausência de qualquer uma dessas condições, deve-se considerar a eventual presença de trombocitose causada por algum processo mieloproliferativo.
A TE é um distúrbio mieloproliferativo que produz elevações lentas, porém progressivas, na contagem de plaquetas. Ao contrário da maior parte das doenças mieloproliferativas, isso poderá ocorrer em pacientes mais jovens, principalmente nas mulheres, embora seu curso seja indolente nesse contexto. A mutação JAK2 V617F pode ser encontrada no sangue periférico de 50% de pacientes com TE; a busca dessa mutação provavelmente seja uma ferramenta diagnóstica bastante útil.
Com frequência, a TE é uma descoberta incidental, embora os pacientes se queixem também de cefaleia, eritromelalgia (dor abrasadora ou calor nas extremidades) ou sintomas neurológicos. Tipicamente, é possível controlar os sintomas de TE com doses baixas de aspirina; a citorredução com hidroxiureia diminui o risco de formação de coágulos em pacientes de alto risco.30 Outros distúrbios mieloproliferativos (LMC, policitemia vera, mielofibrose idiopática) também podem causar trombocitose, porém, tipicamente, têm outras anormalidades associadas.
Coagulação
O sistema hemostático inclui plaquetas e proteínas plasmáticas cuja função é interromper os sangramentos. Trata-se de um sistema intrincado de fatores com a função de promover e estabilizar os coágulos no sítio da lesão, embora ele limite os coágulos aos sítios de lesões com propagação mínima.
A função inadequada do sistema de coagulação poderá ocasionar hemorragias, ao passo que a regulação inadequada poderá resultar em trombose. Nossos conhecimentos atuais do sistema hemostático, embora incompletos, são suficientes para compreender a maior parte dos distúrbios de sangramento e de coagulação.
As plaquetas em circulação no sangue aderem ao colágeno na membrana basal nas situações em que houver ruptura do sistema vascular. O fator de von Willebrand forma uma ponte entre a membrana basal e a glicoproteína Ib na superfície plaquetária. Após a adesão, as plaquetas são ativadas para recrutar outras plaquetas que se expandirão e reforçarão o tampão. As substâncias liberadas pelas plaquetas para facilitar o recrutamento são o tromboxano e a glicoproteína IIB/IIIA.
Existem vários distúrbios hereditários envolvendo a função das plaquetas cujos diagnósticos são feitos por estudos de agregação de plaquetas. A cascata de coagulação ocorre na superfície de plaquetas que já haviam sido recrutadas para o sítio da lesão. O uso da superfície das plaquetas nessa reação aumenta substancialmente a concentração local das proteínas ativadas pela coagulação.
Trombina é a enzima diretamente responsável pela conversão de fibrinogênio em fibrina, substância que forma o coágulo real. A explosão inicial de trombina é formada pela via extrínseca quando o fator VII ativado é exposto ao fator tecidual. A geração de trombina aumenta substancialmente pela ampliação do feedback em vários pontos da via intrínseca; a via intrínseca gera a quantidade de trombina necessária para os coágulos de fibrina.
O sistema de coagulação é adequado especificamente para respostas rápidas: seus componentes atuam no sangue e ficam imediatamente disponíveis no sítio da lesão. O corpo tem mecanismos igualmente poderosos que permanecem nos respectivos locais para limitar o processo de coagulação. Por exemplo, a proteína C ativada degrada o fator Va, a antitrombina III é um inibidor potente da trombina na presença de heparina e a plasmina degrada a fibrina.
No sistema de coagulação, a compreensão da estrutura básica dessa via é extremamente importante para avaliar pacientes com sangramento ou com problemas de hipercoagulação. A via usual, que inicia no fator Xa e termina na produção de fibrina, é o ponto de convergência das vias intrínseca e extrínseca. A via extrínseca, conforme já mencionado, é a principal responsável pela geração do sinal inicial que se propaga em uma pequena explosão de trombina que, a seguir, se amplia por toda a via intrínseca.
Todos os fatores, excetuando-se os VIII e III (fator tecidual), são produzidos basicamente no fígado. O fator tecidual se localiza em todos os tecidos conjuntivos, e o fator VIII é sintetizado e armazenado principalmente no interior das células endoteliais, em vesículas que se denominam corpos de Weibel-Palade. A função da via extrínseca é medida pelo tempo de protrombina (TP), cuja expressão mais conveniente é como o INR.
A integridade da via intrínseca é medida pelo tempo de tromboplastina parcial (TTP). Para aumentar o TTP acima do nível normal, é imprescindível que ocorram deficiências substanciais em uma única proteína (<35% da normalidade) ou múltiplas anormalidades na cascata de coagulação. Nem todas as proteínas da via intrínseca são enfatizadas igualmente no TTP. Por exemplo, o TTP é relativamente insensível para deficiências no fator IX, ao passo que é mais sensível para a depleção dos fatores VIII e XI.
O INR e o TTP, quando usados em conjunto, são instrumentos poderosos para identificar problemas na cascata de coagulação. Por exemplo, INR elevado com TTP normal sempre indica alguma deficiência no fator VII, considerando que a mesma pessoa não pode ser deficiente no fator tecidual.
TTP normal com INR normal significa que há algum problema em um ou mais fatores na via intrínseca (VIII, IX, XI, XII). Elevações no INR e no TTP indicam a existência de algum problema na via comum ou depressão global nos fatores de coagulação. TTPs baixos são o resultado do enchimento excessivo do tubo durante a flebotomia ou de elevação no fator VIII, que pode ser observado nos quadros inflamatórios.
O desenvolvimento de muitos outros testes ajudou a elucidar diversos problemas dentro da cascata de coagulação. Além do TP/INR e do TTP, as medições mais úteis são as dos níveis de fatores individuais e do tempo de trombina (TT). O TT mede o tempo necessário para gerar um coágulo de fibrina depois da adição de trombina ao plasma. Considerando que se encontra no fundo da cascata de coagulação, pouquíssimas coisas poderão prolongar o TT.
Mais especificamente, os TTs permanecem elevados apenas na presença de um inibidor da trombina (tipicamente a heparina), na insuficiência de fibrinogênio ou (muito mais raramente) se houver defeitos fibrinogênicos. Portanto, um meio sofisticado de comprovar que TTPs elevados são ocasionados pela contaminação por heparina, é definir um nível normal de fibrinogênio e um TT elevado.
O reconhecimento de padrões na depleção de fatores específicos também é muito importante para determinar a etiologia de coagulopatia. A constatação de que os fatores dependentes da vitamina K (II, VII, IX, X) são baixos, enquanto outros fatores são preservados, é uma evidência da deficiência de vitamina K ou da ingestão de antagonistas da vitamina K, a varfarina em especial. A depleção global de todos os fatores de coagulação é uma ocorrência típica para CID, ao passo que os níveis do fator VIII são preservados nos casos de insuficiência hepática.
A doença de von Willebrand é um distúrbio hereditário autossômico dominante da homeostase que ocorre com bastante frequência na população branca, cuja prevalência chega a atingir 1 a 2% na população em geral.31 O fator de von Willebrand tem papel duplo no sistema hemostático. Ele forma a ponte inicial entre colágeno e plaqueta no sítio da lesão e protege o fator VIII contra a degradação na circulação.
A doença de von Willebrand tipo I é a forma mais comum e indica a presença de uma quantidade anormalmente baixa da proteína de von Willebrand que, em outras circunstâncias, seria funcionalmente normal. A doença de von Willebrand tipo II indica a presença de um defeito na função proteica, mesmo em quantidades normais. O tipo III se caracteriza pela ausência do fator de von Willebrand e por sangramento grave, comparável à deficiência grave do fator VIII nos casos de hemofilia A. Há inúmeras formas de aumentar a hemóstase em pacientes com doença de von Willebrand.32
Avaliação de Sangramentos
O primeiro passo na avaliação de pacientes para verificar uma eventual tendência para sangramentos é a obtenção de históricos completos. É imprescindível verificar se o problema do paciente é permanente ou adquirido. A presença ou ausência de ferimentos, epistaxe, menorragia, sangramento pós-cirúrgico, sangramento depois da extração de dentes e contusões causadas por contato no esporte provavelmente seja útil para definir o problema.
Levando-se consideração que muitos problemas relacionados a contusões e a sangramentos são hereditários, é extremamente importante reunir todas as informações sobre sangramentos potenciais ou problemas de coagulação em todos os parentes de primeiro grau.
A Figura 10 apresenta um diagrama do sistema de coagulação.
Figura 10 - A via extrínseca e a via intrínseca convergem para formar a via comum. A conversão de fibrinogênio em fibrina pela trombina forma um coágulo.
O rastreamento de sangramentos inclui contagem de plaquetas, TP/INR-TTP, fibrinogênio, painel de von Willebrand (níveis antigênicos de von Willebrand, níveis do fator VIII e níveis do cofator ristocetina ? é também necessário fazer a análise do multímero do Fator de von Willebrand se qualquer um desses fatores for anormal), e estudos de agregação de plaquetas, para rastrear defeitos qualitativos nas plaquetas.
O tempo de sangramento, envolvendo cortes na pele e a medição do tempo para formação de coágulos não são reproduzíveis e desfigurantes; não têm nenhum papel confiável na avaliação da cascata de coagulação e na função das plaquetas.33 O rastreamento de sangramentos totalmente normais exclui a esmagadora maioria dos problemas diagnosticáveis de sangramento.34
Trombofilia
A coagulação inapropriada é uma causa substancial de morbidade e de mortalidade. Tipicamente, as tromboses se dividem em eventos arteriais ou venosos. Os coágulos arteriais se manifestam como infarto agudo do miocárdio (IAM), acidente vascular cerebral (AVC), isquemia nos membros ou infarto em órgãos. De um modo geral, os coágulos venosos têm origem nas extremidades, embora possam ocorrer também nas veias abdominais.
O termo trombose venosa profunda (TVP) é usado para descrever coágulos nas veias profundas maiores do corpo. Os sintomas de TVP são inespecíficos e incluem dor, vermelhidão ou inchaço na extremidade afetada. É fundamental aperceber-se de que uma proporção elevada de TVPs é assintomática, e o diagnóstico pode ser obtido apenas com imagens.35
O tromboembolismo se refere ao processo em que os coágulos que têm origem em um local poderão se decompor e percorrer na direção do fluxo sanguíneo até se alojarem em algum outro local, bloqueando o êmbolo na circulação distal. Nos casos de embolia pulmonar (EP), um coágulo, ou um fragmento de coágulo, geralmente tem origem nas veias proximais da perna, decompõe-se e percorre o lado direito do coração e se aloja nos pulmões.
Os sintomas e descobertas clássicas da EP incluem dor torácica pleurítica, hipóxia e taquicardia, porém nenhuma dessas condições está presente ou é transitória. Até um terço das embolias pulmonares não apresenta nenhum sintoma simultâneo.36 Embora todas as TVPs precisem de tratamento, a TVP proximal nas pernas é a condição mais séria.
O diâmetro das veias proximais das pernas é suficientemente grande, de modo que um coágulo embolizado proveniente desse local pode ser potencialmente letal. As TVPs nas extremidades superiores geralmente são ocasionadas por cateteres intravasculares ou pela síndrome de Paget-Schroetter, em que o retorno sanguíneo dos braços apresenta danos anatômicos.37
As TVPs nas extremidades superiores raramente são suficientemente grandes para causar EPs com risco de vida, embora possam produzir complicações locais. Embolismos paradoxais podem ocorrer nas situações em que um coágulo no sistema venoso atravessar para o sistema arterial, geralmente através de um forame oval patente, produzindo AVC, infarto ou isquemia nos membros.
Tipicamente, as causas locais de trombose arterial envolvem ruptura de placas ateromatosas, trauma direto ou fluxo sanguíneo turbulento. As causas sistêmicas de trombose arterial geralmente são produzidas por ativação inapropriada de plaquetas.
As causas sistêmicas mais significativas de trombose arterial incluem TIH, anticorpos antifosfolipídicos (especificamente anticorpos da anticardiolipina ou anticoagulante para lúpus), distúrbios mieloproliferativos, CID, hemoglobinúria paroxística noturna ou hiper-homocisteinema.
A reperfusão, caso seja indicada (como no caso de IAM com elevação de ST), deverá ser feita imediatamente por meios mecânicos ou farmacológicos. Tipicamente, o tratamento envolve terapia antiplaquetária, como aspirina ou clopidogrel, normalmente em combinação com outros coagulantes. Caso seja identificada alguma outra causa subjacente, esse tratamento é a melhor opção.
A TVP ou tromboembolismo venoso (TEV) exige anticoagulação imediata e eficaz para evitar embolismos ou a propagação de coágulos. Além de prevenir contra a ocorrência de embolismo, a anticoagulação imediata facilita a reabsorção da trombose pelo corpo. Com frequência, os coágulos novos e desorganizados se dissolvem em alguns dias após a anticoagulação, ao passo que a resolução dos coágulos mais antigos e organizados é mais demorada e, com frequência, eles se tornam crônicos.
Todos as TVPs precisam de, pelo menos, 3 meses de anticoagulação.38 O tempo final de anticoagulação deve ser individualizado para cada paciente e deverá levar em consideração o risco de recorrência da TEV, assim como o risco de sangramento e a inconveniência da anticoagulação de longo prazo. Considera-se um evento provocado as situações em que um coágulo for o culpado plausível por algum fator de risco não permanente.
Por exemplo, a TVP que ocorrer logo após a reposição total do quadril por meios cirúrgicos e imobilização pós-operatória. Nessa hipótese, são necessários apenas de 3 a 6 meses de anticoagulação, porque se presume que não haverá recorrência do coágulo, tendo em vista que o fator de risco para trombose não está mais presente. Outros fatores de risco modificáveis incluem gravidez, contraceptivos orais, outros compostos estrogênicos (como o megesterol), obesidade, malignidade, tabagismo, imobilidade prolongada ou viagens em longas distâncias, em especial nas viagens por via aérea.
As TVPs não provocadas ou idiopáticas são mais preocupantes, considerando que não há nenhum fator de risco que possa ser modificado para impedir a ocorrência de eventos coagulantes adicionais. Observa-se que os pacientes com TVPs não provocadas têm uma predisposição sistêmica para formar coágulos. Muitas dessas predisposições sistêmicas (ou trombofilias) foram identificadas e podem ser hereditárias ou adquiridas.
As causas hereditárias são produzidas por deficiências ou resistência às proteínas ou mutações anticoagulantes que elevam os níveis das proteínas anticoagulantes. A tendência mais pronunciada foi encontrada em famílias com deficiências parciais na antitrombina III, seguidas por deficiências de proteína C e proteína S.
Embora sejam trombofilias menos poderosas, as mutações no fator V (mutação no fator V de Leiden) ou no gene da protrombina (mutação PT20210) são mais comuns.39 A maior parte dos pacientes apresenta apenas um defeito heterozigótico, embora a homozigosidade e a heterozigosidade composta também sejam eventos possíveis. Provavelmente outros fatores de risco hereditários sejam identificados no futuro.
Muitos fatores de risco adquiridos estão associados ao TEV. Esses fatores de risco incluem anticorpos antifosfolipídeos (anticorpos anticardiolipina ou anticoagulante de lúpus), malignidades, hemoglobinúria paroxística noturna, hemólise, gravidez ou uso de medicações. O teste para causas hereditárias ou adquiridas de trombofilia ainda é uma área que está sujeita a muitas controvérsias.40
Embora o rastreamento de pacientes para trombofilias possa ser elucidativo, e ocasionalmente útil, em geral não orienta as decisões de gerenciamento e acredita-se que não tenha muita utilidade para impedir a ocorrência de eventos futuros. Por exemplo, os pacientes com TVP não provocada têm, aproximadamente, a mesma chance de recorrência, tenham ou não uma mutação do fator V de Leiden.
Os pacientes com TVP não provocada têm uma chance significativa de recorrência (algo em torno de 30% nos primeiros 3 anos) nas situações em que a anticoagulação é descontinuada e, com frequência, são candidatos à anticoagulação de longo prazo. Quase sempre a anticoagulação de longo prazo é indicada para episódios recorrentes de TEV. A publicação de orientações consensuais facilita a tomada de decisões.38
|
Informações Financeiras: J. Mark Sloan, MD, e David Seldin, MD, PhD, não têm informações financeiras relevantes a declarar. |
Referências
1. Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002;1:845–67.
2. Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood 2008;112:3927–38. DOI:10.1182/blood-2008-04-078188.
3. Koretzky GA. The legacy of the Philadelphia chromosome. J Clin Invest 2007;117:2030–2. DOI:10.1172/JCI33032.
4. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008;132:631–44. DOI:10.1016/j.cell.2008.01.025.
5. Bohlius J, Herbst C, Reiser M, et al. Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst Rev 2008;(4):CD003189. DOI:10.1002/14651858.CD003189.pub4.
6. Jones M, Ibels L, Schenkel B, Zagari M. Impact of epoetin alfa on clinical end points in patients with chronic renal failure: a meta-analysis. Kidney Int 2004;65:757–67. DOI:10.1111/j.1523-1755.2004.00450.x.
7. Kuter DJ. New thrombopoietic growth factors. Blood 2007;109:4607–16. DOI:10.1182/blood-2006-10-019315.
8. Carmel R. Anemia and aging: an overview of clinical, diagnostic and biological issues. Blood Rev 2001;15:9–18. DOI:10.1054/blre.2001.0146.
9. Viele MK, Weiskopf RB. What can we learn about the need for transfusion from patients who refuse blood? The experience with Jehovah’s Witnesses. Transfusion 1994;34:396–401.
10. Stabler SP. Clinical practice. Vitamin B12 deficiency. N Engl J Med 2013;368:149–60. DOI:10.1056/NEJMcp1113996.
11. Cook JD. Diagnosis and management of iron-deficiency anaemia. Best Pract Res Clin Haematol 2005;18:319–32. DOI:10.1016/j.beha.2004.08.022.
12. Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003;101:2461–3. DOI:10.1182/blood-2002-10-3235.
13. Bosman DR, Winkler AS, Marsden JT, et al. Anemia with erythropoietin deficiency occurs early in diabetic nephropathy. Diabetes Care 2001;24:495–9.
14. Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. Br J Haematol 2008;142:505–14. DOI:10.1111/j.1365-2141.2008.07216.x.
15. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011;364:1046–60. DOI:10.1056/NEJMra1011442.
16. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97. DOI:10.1016/j.ccr.2005.03.023.
17. Freifeld AG, Bow EJ, Sepkowitz KA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 2011;52:e56–93. DOI:10.1093/cid/cir073.
18. Andersohn F, Konzen C, Garbe E. Systematic review: agranulocytosis induced by nonchemotherapy drugs. Ann Intern Med 2007;146:657–65.
19. Simon HU, Rothenberg ME, Bochner BS, et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 2010;126:45–9. DOI:10.1016/j.jaci.2010.03.042.
20. Simon HU, Plotz SG, Dummer R, Blaser K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med 1999;341:1112–20. DOI:10.1056/NEJM199910073411503.
21. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003;348:1201–14. DOI:10.1056/NEJMoa025217.
22. Cines DB, Bussel JB. How I treat idiopathic thrombocytopenic purpura (ITP). Blood 2005;106:2244–51. DOI:10.1182/blood-2004-12-4598.
23. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
24. Chiao EY, Engels EA, Kramer JR, et al. Risk of immune thrombocytopenic purpura and autoimmune hemolytic anemia among 120 908 US veterans with hepatitis C virus infection. Arch Intern Med 2009;169:357–63. DOI:10.1001/archinternmed.2008.576.
25. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010;116:4060–9. DOI:10.1182/blood-2010-07-271445.
26. Perry JH, Lazar HL, Quillen K, Sloan JM. Successful long-term management of aneurysm-associated chronic disseminated intravascular coagulation with low molecular weight heparin. J Card Surg 2012;27:730–5. DOI:10.1111/jocs.12010.
27. Lo GK, Juhl D, Warkentin TE, et al. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost 2006;4:759–65. DOI:10.1111/j.1538-7836.2006.01787.x.
28. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood 2012;119:2209–18. DOI:10.1182/blood-2011-11-376293.
29. Akan H, Guven N, Aydogdu I, et al. Thrombopoietic cytokines in patients with iron deficiency anemia with or without thrombocytosis. Acta Haematol 2000;103:152–6. DOI:10.1159/000041038.
30. Beer PA, Erber WN, Campbell PJ, Green AR. How I treat essential thrombocythemia. Blood 2011;117:1472–82. DOI:10.1182/blood-2010-08-270033.
31. Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
32. Rodeghiero F, Castaman G, Tosetto A. How I treat von Willebrand disease. Blood 2009;114:1158–65. DOI:10.1182/blood-2009-01-153296.
33. Lind SE. The bleeding time does not predict surgical bleeding. Blood 1991;77:2547–52.
34. Laposata M. Laboratory medicine: the diagnosis of disease in the clinical laboratory. New York: McGraw-Hill Medical; 2010.
35. Vaitkus PT, Leizorovicz A, Cohen AT, et al; PREVENT Medical Thromboprophylaxis Study Group. Mortality rates and risk factors for asymptomatic deep vein thrombosis in medical patients. Thromb Haemost 2005;93:76–9. DOI:10.1267/THRO05010076.
36. Agnelli G, Becattini C. Acute pulmonary embolism. N Engl J Med 2010;363:266–74. DOI:10.1056/NEJMra0907731.
37. Lee WA, Hill BB, Harris EJ Jr, et al. Surgical intervention is not required for all patients with subclavian vein thrombosis. J Vasc Surg 2000;32:57–67.
38. Kearon C, Akl EA, Comerota AJ, et al, American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e419S–94S. DOI:10.1378/chest.11-2301.
39. Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001;135:367–73.
40. Baglin T, Gray E, Greaves M, et al, British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20. DOI:10.1111/j.1365-2141.2009.08022.x.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.