(Carregando Índice)... (Carregando Índice)... |
Autores:
Adriana Gelmetti
Doutora em Nefrologia pela Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Aline Lázara Resende
Nefrologista formada pelo Serviço de Nefrologia da Faculdade Federal de Goiás.
Giordano Floripe Ginani
Nefrologista formado pelo Serviço de Nefrologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP).
Última revisão: 06/06/2010
Comentários de assinantes: 0
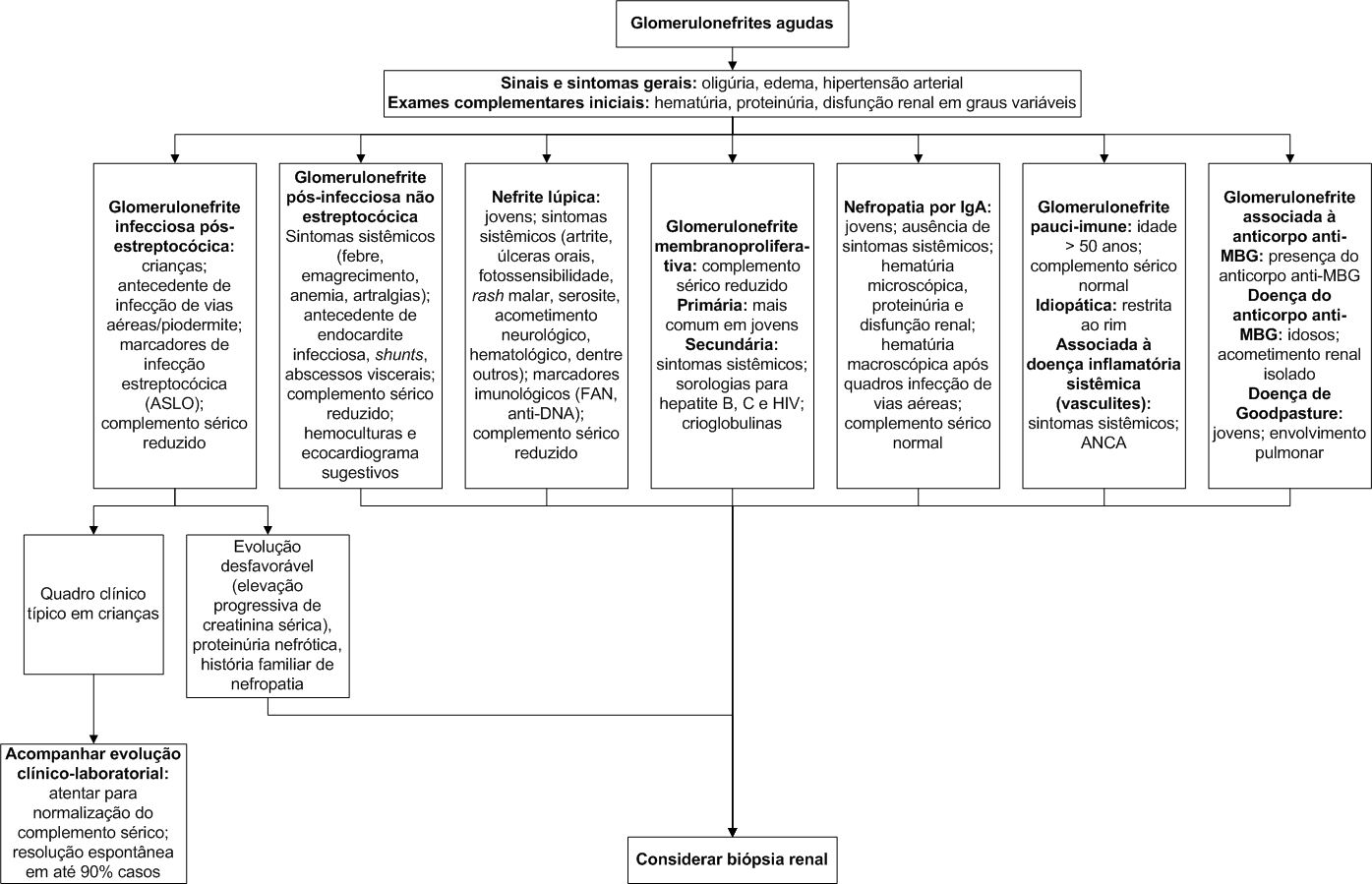
As glomerulonefrites agudas constituem um grupo de doenças caracterizadas clinicamente pelo início abrupto de edema, hipertensão, hematúria e proteinúria.
Histologicamente, caracterizam-se por uma reação inflamatória intraglomerular e proliferação celular, podendo levar à disfunção renal em graus variáveis. Um intenso comprometimento de função renal que evolui num período de dias a semanas caracteriza a glomerulonefrite rapidamente progressiva.
Glomerulonefrites agudas de etiologias diferentes podem evoluir com perda crônica e progressiva da função renal, condição denominada glomerulonefrite crônica.
O diagnóstico preciso e precoce é de extrema importância, uma vez que o tratamento e o prognóstico dependem fundamentalmente da etiologia envolvida.
As glomerulonefrites agudas podem ser classificadas clinicamente de acordo com o sítio e a extensão da lesão em:
primárias: patologias restritas ao rim;
secundárias: quando associadas a doenças sistêmicas.
Quanto à etiologia específica, as glomerulonefrites podem ser classificadas em infecciosas e não infecciosas (Tabela 1).
Tabela 1: Classificação das glomerulonefrites agudas quanto à etiologia
|
Infecciosas |
Não infecciosas |
|
Glomerulonefrite aguda pós-estreptocócica (grupo A, beta-hemolítico) |
Glomerulonefrites primárias: nefropatia da IgA, glomerulonefrite proliferativa mesangial, glomerulonefrite membranoproliferativa, glomerulopatia por IgM, glomerulonefrite rapidamente progressiva idiopática. |
|
Glomerulonefrites não estreptocócicas: endocardite infecciosa, bacteriemia estafilocócica, pneumonia pneumocócica, meningococcemia, febre tifoide, sífilis secundária, infecção aguda viral (citomegalovírus, varicela, Epstein-Barr, coxsackie), micoplasma, toxoplasmose, malária, abscesso visceral, nefrite do shunt, hepatite B ou C com vasculite e/ou crioglobulinemia. |
Glomerulonefrites secundárias a doenças sistêmicas: lúpus eritematoso sistêmico, púrpura de Henoch-Schönlein, vasculite necrotizante, síndrome de Goodpasture, granulomatose de Wegener. |
Apesar da patogênese das glomerulonefrites agudas não ser totalmente esclarecida, existe um número considerável de dados clínicos, imunopatológicos e experimentais que suportam a hipótese de que mecanismos imunológicos sejam comuns às diferentes etiologias da síndrome. Acredita-se que tanto o sistema imune celular quanto o humoral estejam envolvidos.
A lesão humoral no glomérulo pode ser mediada por diversos mecanismos:
1. Formação in situ do complexo antígeno-anticorpo. Neste caso, os anticorpos podem estar direcionados contra as seguintes estruturas:
constituintes normais do glomérulo; p.ex., doença de Goodpasture (o antígeno é um elemento do colágeno que faz parte da estrutura da membrana basal glomerular).
2. Antígenos “plantados” no glomérulo:
exógenos: glomerulonefrite pós-estreptocócica;
endógenos: nefrite lúpica (complexo histona-DNA).
3. Deposição de complexo antígeno-anticorpo circulante.
A extensão da lesão tecidual depende de alguns fatores, como os sítios de deposição, a quantidade e as propriedades biológicas das imunoglobulinas formadoras dos depósitos.
Depósitos mesangiais causam proliferação das células mesangiais e expansão de matriz mesangial. Depósitos subendoteliais, por sua vez, estão em contato direto com a superfície dos capilares glomerulares e têm maior habilidade para recrutar células inflamatórias (neutrófilos e macrófagos) circulantes.
Quanto às características biológicas, sabe-se que algumas imunoglobulinas (como IgG) têm maior capacidade de fixar complemento do que outras (IgA) e, deste modo, apresentam maior potencial nefritogênico.
A deposição de imunocomplexos no rim promove a ativação de frações do complemento (C3a e C5b) e subsequente quimiotaxia de células inflamatórias (neutrófilos e monócitos). A resposta imune celular contribui para a infiltração dos glomérulos por células inflamatórias circulantes. Linfócitos T são encontrados no infiltrado inflamatório, contribuindo para a ativação de macrófagos. Macrófagos ativados liberam citocinas e moléculas de adesão. A ativação de células residentes, como as células mesangiais, também contribui para a produção de citocinas. Os macrófagos e as células mesangiais também são capazes de produzir proteases e oxidantes, responsáveis pela morte celular. Fatores de coagulação ativados podem levar à deposição de fibrina.
O infiltrado de neutrófilos causa obstrução do capilar glomerular e liberação de leucotrienos e tromboxanos, induzindo a vasoconstrição das arteríolas aferentes e queda da filtração glomerular. O aumento na reabsorção distal de sódio, secundário à diminuição na taxa de filtração glomerular, pode estar associado a hipervolemia, edema e hipertensão arterial.
Processos que inicialmente têm componente inflamatório potencialmente reversível podem evoluir com alterações hemodinâmicas (hipertensão e hiperfiltração glomerulares), esclerose glomerular e fibrose tubulointersticial, ocasionando sequelas e perda crônica de função renal.
Os fatores implicados na fisiopatologia estão diretamente associados aos achados da biópsia renal, sendo, portanto, de fundamental importância para o diagnóstico e o tratamento das glomerulonefrites agudas.
O quadro clínico das glomerulonefrites agudas é variável e depende fundamentalmente da etiologia envolvida e do grau de comprometimento histológico. O espectro de manifestações clínicas é caracterizado por alguns achados gerais, como hematúria, oligúria, edema e hipertensão arterial (Tabela 2). Além disso, podem ocorrer proteinúria e perda de função renal.
Tabela 2: Achados clínicos gerais na glomerulonefrite aguda
|
Hipertensão arterial |
Oligúria |
|
Edema |
Hematúria micro ou macroscópica |
A extensão do comprometimento histológico é determinante para as manifestações clínico-laboratoriais e permite classificar as glomerulonefrites em:
1. Glomerulonefrite focal: acometimento inflamatório de menos de 50% dos glomérulos. Estes pacientes habitualmente apresentam hematúria assintomática, proteinúria leve (usualmente < 1,5 g/dia) e, eventualmente, cilindros celulares. Exemplos: nefropatia da IgA, doença de membrana fina, nefrites hereditárias.
2. Glomerulonefrite difusa: envolvimento de mais de 50% dos glomérulos. Classicamente, os pacientes apresentam início abrupto de hematúria micro ou macroscópica, oligúria, edema, hipertensão e queda na taxa de filtração glomerular. Os níveis de proteinúria tendem a ser mais elevados, podendo atingir níveis nefróticos. Exemplos: glomerulonefrite pós-infecciosa, nefrite lúpica, glomerulonefrite membranoproliferativa.
A seguir, serão brevemente descritos os aspectos clínico-laboratoriais mais relevantes para a glomerulonefrite pós-estreptocócica, tida como a causa mais comum e o protótipo das glomerulonefrites difusas agudas.
A glomerulonefrite pós-estreptocócica (GNPE) acomete preferencialmente crianças (pico de incidência entre 6 e 10 anos de idade). Adultos também podem ser acometidos (raramente com mais de 40 anos de idade). Ocorre caracteristicamente após
O quadro clínico clássico da GNPE caracteriza-se por hematúria micro ou macroscópica, edema e hipertensão arterial, mas o espectro de apresentação inclui desde quadros clínicos frustros até insuficiência renal grave. A maioria dos pacientes (80%) apresenta elevação em marcadores imunológicos de infecção estreptocócica, como o anticorpo antiestreptolisina O (ASLO). A fase aguda cursa com hipocomplementemia, habitualmente à custa de redução do componente C3 do complemento, com normalização após 8 semanas. Caso a hipocomplementemia seja persistente, deve-se considerar outras possibilidades diagnósticas, como a glomerulonefrite membranoproliferativa ou a nefrite lúpica. Culturas de orofaringe ou pele não são necessárias.
Frente a um quadro clínico típico de GNPE em crianças, a biópsia renal não é necessária. No entanto, em casos que cursem com elevação progressiva de creatinina sérica, proteinúria nefrótica e história familiar de nefropatia, a biópsia renal pode ser indicada para possibilitar o diagnóstico definitivo. A imunofluorescência evidencia depósitos de IgG e C3 no mesângio e em alças capilares (padrão granular difuso).
A resolução espontânea da GNPE é comum, com taxas de cura de até 90% em crianças. Nos adultos, esta taxa situa-se entre 60 e 70%. A remissão das manifestações clínico-laboratoriais tem correlação com a resolução do processo fisiopatológico renal. Depósitos imunes subendoteliais responsáveis pela ativação de complemento, e consequente influxo local de células inflamatórias, são rapidamente clareados, contribuindo para resolução da disfunção renal (3 a 4 semanas) e da hematúria (3 a 6 meses). Depósitos subepiteliais (humps) são responsáveis por danos às células epiteliais manifestados pela proteinúria. Estes depósitos ficam separados das células circulantes pela membrana basal glomerular, o que limita sua remoção e contribui para a persistência da proteinúria. A proteinúria em níveis nefróticos pode persistir por mais de 6 meses. Proteinúria leve pode ser encontrada em 15% dos pacientes após 3 anos e em 2% dos pacientes após 10 anos.
Diante de um quadro clínico de glomerulonefrite aguda, os principais exames laboratoriais a serem solicitados são:
urina I: permite detectar hematúria, leucocitúria estéril (a ser confirmada com urocultura), cilindros (celulares ou hemáticos) e proteinúria;
proteinúria de 24 horas;
função renal (ureia e creatinina séricas);
ultrassonografia de aparelho urinário: fornece alguns parâmetros de viabilidade renal, além de permitir a identificação de eventuais patologias pós-renais.
Uma vez feito o diagnóstico de glomerulonefrite aguda, outros exames laboratoriais são solicitados de acordo com a suspeita clínica, no intuito de determinar a etiologia da doença:
dosagem de complemento: as glomerulonefrites podem ser classificadas em normo ou hipocomplementêmicas (Tabela 3);
pesquisa de anticorpos antiestreptocócicos (suspeita de glomerulonefrite pós-estreptocócica);
FAN e anti-DNA para investigação de lúpus eritematoso sistêmico;
ANCA (especialmente em casos de glomerulonefrite normocomplementêmica);
pesquisa de anticorpo anti-MBG (principalmente quando a imunofluorescência apresentar um padrão linear de depósito);
pesquisa de crioglobulinas;
sorologias para HIV, hepatites B e C;
hemoculturas e ecocardiograma (suspeita de endocardite ou nefropatia do shunt).
Tabela 3: Glomerulonefrites aguda normo e hipocomplementêmicas
|
Complemento sérico reduzido |
Complemento sérico normal |
|
Patologias renais primárias: glomerulonefrite membranoproliferativa tipos I (50 a 80%) e II (90%). |
Patologias renais primárias: nefropatia da IgA, glomerulonefrite rapidamente progressiva idiopática, doença do anticorpo antimembrana basal. |
|
Patologias renais secundárias: glomerulonefrite pós-estreptocócica (90%), lúpus eritematoso sistêmico (classe III, 75% e classe IV, 90%), crioglobulinemia (85%), endocardite bacteriana (90%), nefropatia do shunt (90%). |
Patologias renais secundárias: poliangeíte microscópica, granulomatose de Wegener, púrpura de Henoch-Schönlein, doença de Goodpasture, abscesso visceral. |
As porcentagens indicam as frequências aproximadas de C3 consumido.
No contexto de uma glomerulonefrite aguda, a biópsia renal nem sempre está indicada. Quadros sugestivos de glomerulonefrite pós-estreptocócica, por exemplo, apresentam um prognóstico habitualmente favorável e podem ser conduzidas mediante acompanhamento clínico-laboratorial. Um parâmetro particularmente importante nestes casos é a normalização do complemento sérico, que deve ocorrer em até 8 semanas.
Pacientes que cursam com alteração de função renal devem ser submetidos à biópsia renal, visando ao diagnóstico definitivo. É importante salientar que a biópsia não deve retardar a instituição do tratamento, especialmente nas suspeitas de glomerulonefrite rapidamente progressiva.
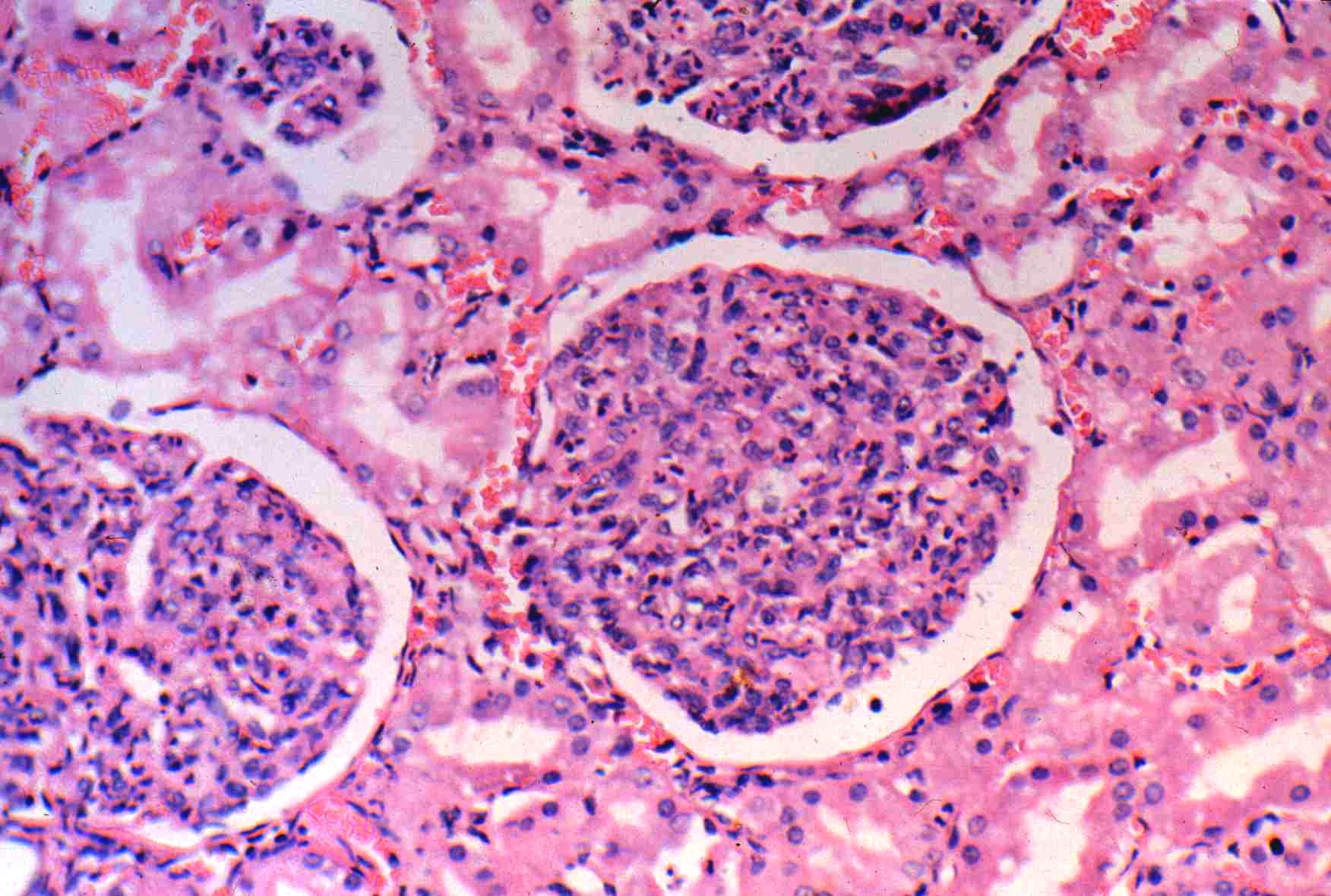
O padrão histopatológico das glomerulonefrites agudas à microscopia óptica é pouco específico. Caracteriza-se pela presença de glomérulos difusamente hipercelulares, com proliferação de células mesangiais e endoteliais, associado a infiltrado de neutrófilos e monócitos (Figura 1).
Figura 1: Glomerulonefrite difusa aguda pós-estreptocócica.

Cortesia de Dra. Denise Malheiros, HC-FMUSP.
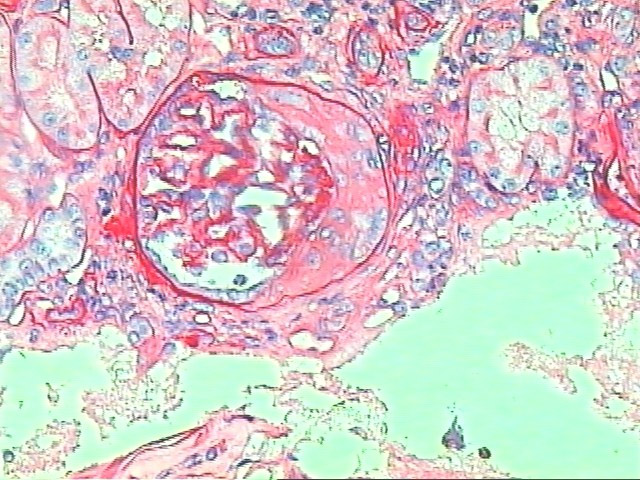
Além disso, a microscopia óptica pode evidenciar a presença de crescentes (Figura 2). Crescentes são achados histológicos que correspondem à presença de pelo menos 2 camadas de células epiteliais glomerulares e fagócitos mononucleares ocupando parcial ou totalmente o espaço de Bowman. Constituem uma resposta inespecífica à rotura do capilar glomerular e extravasamento de fibrina e elementos inflamatórios para o espaço de Bowman.
A presença de crescentes associada a doenças renais primárias ou secundárias está relacionada a maior alteração da função renal com prognóstico potencialmente pior.
Figura 2: Glomerulonefrite crescêntica.
Cortesia de Dra. Denise Malheiros, HC-FMUSP.
A presença ou ausência de componentes do complemento e imunoglobulinas ao exame de imunofluorescência, assim como sua distribuição e padrão, são de fundamental importância para o diagnóstico etiológico:
depósito de IgG em padrão linear caracteriza a glomerulonefrite por anticorpo antimembrana basal glomerular e a síndrome de Goodpasture (acometimento simultâneo de pulmões e rins);
o padrão granular está presente na maioria das glomerulonefrites. Atenção especial ao predomínio de IgA (encontrado na nefropatia por IgA) e no padrão full house (imunofluorescência rica com presença de IgA, IgG, IgM, C3 e C1q), característico da nefrite lúpica;
padrão pauci-imune: imunofluorescência negativa ou pobre, presente nas vasculites sistêmicas e patologias relacionadas ao ANCA.
Os dados de história clínica, exame físico, exames complementares e biópsia renal são fundamentais para a elucidação da etiologia da glomerulonefrite. Seguem alguns tópicos relevantes que devem ser considerados para suspeita clínica de determinadas etiologias.
Contexto clínico de endocardite bacteriana (atenção especial a portadores de próteses valvares e usuários de drogas ilícitas), pacientes com shunts ou abscessos viscerais;
presença de sintomas sistêmicos, como febre, emagrecimento, artralgias e anemia;
geralmente há consumo dos componentes do complemento C1q, C3 e C4;
podem ser detectados no sangue vários autoanticorpos como ANCA (antineutrophil cytoplasmic antibody), FAN (fator antinúcleo), fator reumatoide e crioglobulinas, devido à estimulação policlonal de linfócitos B.
Pico de incidência na 2ª e 3ª décadas de vida;
apresentação clássica: hematúria macroscópica que se manifesta
também pode se manifestar de forma insidiosa, com hematúria microscópica, proteinúria não nefrótica, associados ou não a hipertensão arterial e perda de função renal, ou ainda por meio de síndrome nefrótica clássica (10% dos casos);
aumento na concentração sérica de IgA em 50% dos pacientes;
imunofluorescência revela predomínio de depósitos de IgA no mesângio, podendo haver, em menor intensidade, deposição de IgG, IgM ou componentes do complemento.
Forma sistêmica da nefropatia da IgA, caracterizada por uma vasculite de pequenos vasos com depósitos predominantes de IgA;
púrpura, principalmente em membros inferiores, artralgias e dor abdominal;
mais comum em crianças.
Caracterizado por proliferação mesangial e espessamento com duplicação da membrana basal do capilar glomerular;
pode ser primária/idiopática ou secundária;
a apresentação clínica mais comum é a síndrome nefrótica (em
de 30 a 50% dos pacientes apresentam níveis reduzidos de C3 e CH50;
a forma idiopática é mais frequentemente encontrada em adultos jovens, enquanto a secundária é mais comum em adultos;
a GNMP secundária, encontrada em 50% dos pacientes, pode se associar a doenças infecciosas (vírus da hepatite C e B, HIV, esquistossomose, endocardite, abscessos viscerais, shunts, malária, micoplasma e Epstein-Barr), autoimunes (lúpus eritematoso sistêmico, crioglobulinemia, artrite reumatoide e síndrome de Sjögren), paraproteinemias (nefropatia de cadeia leve, mieloma múltiplo, macroglobulinemia de Waldenström) e microangiopatia trombótica (síndrome hemolítico-urêmica, síndrome anticorpo antifosfolípide, anemia falciforme, nefrite por radiação);
a investigação de quadros sistêmicos é obrigatória e alguns exames laboratoriais são necessários, como pesquisa de anti-DNA, crioglobulina, sorologias para hepatite B e C, enzimas hepáticas e, em casos selecionados, avaliação de neoplasias, incluindo imunoeletroforese de proteínas sérica e urinária.
Mais comum em mulheres, na faixa etária entre 20 e 30 anos;
presença de sinais e sintomas sistêmicos, como artrite, rash malar, febre, úlceras orais, alterações hematológicas (plaquetopenia, anemia hemolítica, linfopenia), serosite e sintomas neurológicos;
a imunofluorescência é habitualmente rica, com presença de IgG, IgA, IgM e frações do complemento (C3 e C1q);
títulos elevados de FAN, presença de anticorpo anti-DNA e hipocomplementemia (
Caracterizada histologicamente pela presença de crescentes;
os pacientes apresentam frequentemente achados clássicos de síndrome nefrítica (hematúria, oligúria e hipertensão) acompanhados de rápida perda de função renal;
atentar para sintomas sistêmicos (como febre e perda de peso), além de aspectos individuais de cada doença;
quanto ao mecanismo fisiopatológico envolvido, podem ser classificadas em nos tipos descritos a seguir.
Responsável por 50% dos casos. Trata-se de uma vasculite de pequenos vasos com pico e incidência na 6ª década de vida. A doença pode ser limitada ao rim (glomerulonefrite rapidamente progressiva idiopática) ou decorrer de doença inflamatória sistêmica (granulomatose de Wegener, doença de Churg-Strauss ou poliangeíte microscópica). Cerca de 90% das vasculites pauci-imunes apresentam ANCA positivo, sendo que o ANCA-c (antiproteinase 3) é mais comum em granulomatose de Wegener e o ANCA-p (antimieloperoxidase), na poliangeíte microscópica e na doença de Churg-Strauss. Imunofluorescência negativa ou pobre é característico.
Corresponde a
Responsável por
O tratamento de suporte é preconizado para todas as glomerulonefrites agudas e visa ao controle dos sinais e sintomas clínicos, como segue.
Dieta hipossódica e restrição hídrica;
se necessário, usar diuréticos (de alça ou em associação), com controle diário de peso;
tratamento da hipertensão com hipotensores;
se o paciente persistir com congestão pulmonar não responsiva às medidas clínicas, indicar hemodiálise ou ultrafiltração.
Dieta hipoproteica;
Tratamento dialítico mediante indicações habituais.
A decisão a respeito do tratamento específico deve ser tomada mediante a avaliação de diversos fatores:
estado clínico do paciente;
etiologia da glomerulonefrite;
manifestações clínico-laboratoriais;
probabilidade de remissão da doença;
efeitos colaterais do tratamento proposto.
A análise da biópsia renal é de fundamental importância, uma vez que pode auxiliar no diagnóstico etiológico e fornece dados de prognóstico, como crescentes, esclerose glomerular, acometimento túbulo-intersticial.
Seguem algumas considerações a respeito do tratamento específico de acordo com a etiologia da glomerulonefrite aguda. Uma revisão detalhada de aspectos referentes ao tratamento está alem dos objetivos deste texto.
Não estão indicados antibióticos, a não ser que haja um processo infeccioso ativo no momento;
o uso de antibiótico profilático também não é recomendado, uma vez que a recidiva é extremamente rara;
em casos de insuficiência renal grave (glomerulonefrite rapidamente progressiva), o tratamento com pulsos de metilprednisolona (seguido de corticoide oral) associado a imunossupressores pode ser indicado. Os casos devem ser avaliados cautelosa e individualmente, uma vez que ainda há controvérsias na literatura quanto aos riscos e benefícios deste tratamento.
O tratamento baseia-se no controle da infecção. Recomenda-se a retirada de shunts e a drenagem de abscessos viscerais, além da cobertura prolongada de antibioticoterapia;
há relatos de boa resposta ao uso de plasmaférese em pacientes já tratados com antibioticoterapia adequada e não responsivos à imunossupressão;
nos casos de glomerulonefrite crescêntica, deve-se aguardar o controle adequado da infecção, antes de se considerar o uso de drogas citostáticas ou pulso com metilprednisolona.
Em função da variabilidade de apresentação clínico-laboratorial, os esquemas terapêuticos devem ser discutidos individualmente;
o uso de inibidores da enzima conversora da angiotensina (iECA) e bloqueadores do receptor AT1 da angiotensina II (BRA) parecem ter benefício na redução da proteinúria;
a presença de proteinúria acima de 1 g/dia, a despeito do tratamento com iECA/BRA, habitualmente é indicação de tratamento com uso de corticoide;
nos casos de glomerulonefrite crescêntica, seguir o esquema descrito em glomerulonefrite rapidamente progressiva por imunocomplexos, com o uso de corticoide e imunossupressores.
Ainda não existe tratamento eficaz para a forma idiopática desta glomerulonefrite;
aspirina e dipiridamol têm sido prescritos na tentativa de limitar a ativação plaquetária secundária à proliferação celular, porém com resultados pouco animadores;
o uso de corticoide pode ser realizado em crianças;
quando se apresenta sob a forma de glomerulonefrite crescêntica, preconiza-se o regime proposto para as crescênticas por imunocomplexos;
nos casos de GNMP secundária, a abordagem terapêutica visa controlar o fator etiológico. Assim, nos casos de associação com vírus C, o mais importante é diminuir a carga viral. Quando a lesão é muito agressiva, admite-se o uso de corticoide e imunossupressores, apesar do risco de aumento da carga viral. Outros recursos, como plasmaférese e anticorpo monoclonal contra molécula CD20 (rituximabe), também têm sido testados, com alguns resultados positivos.
Trata-se de um grupo heterogêneo de patologias;
diagnóstico e tratamento precoces são importantes para permitir a viabilidade renal;
o tratamento das patologias que levam à GNRP geralmente requer uma fase de indução com imunossupressão mais agressiva (visando à remissão da doença) e uma fase de manutenção, na qual o intuito é prevenir recorrências com a menor toxicidade possível;
de uma maneira geral, a indução é feita por meio da pulsoterapia com metilprednisolona (
durante a fase de manutenção, a dose de corticoide é reduzida e a ciclofosfamida é substituída por azatioprina;
na presença de sintomas pulmonares, a plasmaférese está sempre indicada;
a resposta renal à plasmaférese é melhor em pacientes não dialíticos.
1. Massry SG, Glassock RJ. Glomerular diseases In: Textbook of nephrology. 3.ed. Williams & Wilkins. p.681-.;
2. Soares V, Alves MAR, Barros RT. Glomerulonefrites agudas: etiopatogenia, quadro clínico e diagnóstico diferencial. In: Glomerulopatias. Sarvier. p.39-43.
3. Madaio MP, Harrington JT. The diagnosis of glomerular diseases. Arch Intern Med. 2001;161:25-33.
4. Tezar V, Ríhova Z, Jancová E, Rysavá R, Merta M. Current treatment strategies in ANCA-positive renal vasculitis – lesson from European randomized trials. Nephrol Dial Transplant. 2003;18(5):v2-v4.
5. Little MA, Pusey CD. Rapidly progressive glomerulonephritis: current and evolving treatment strategies. J Nephrol. 2004;17:10-19.
6. Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347(10):738-46.
7. Bruchfeld A, Lindahl K, Sthle L, Soderberg M, Schvarcz R. Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and insufficiency. Nephrol Dial Transplant. 2003;18:1573-80.
8. Alric L, Plaisier E, Thebault S, Peron JM, Rostaing L, Pourrat J, et al. Influence of antiviral therapy in hepatitis C virus-associated cryoglobulinemic MPGN. Am J Kidney Dis. 2004;43(4):617-23.
9. Laville M, Alamartine E. Treatment options for IgA nephropathy in adults: a proposal for evidence-based strategy. Nephrol Dial Transplant. 2004;19(8):1947-51.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.