(Carregando Índice)... (Carregando Índice)... |
Você está em:
Inicial  acp-medicine Reumatologia
acp-medicine Reumatologia
Última revisão: 14/11/2013
Comentários de assinantes: 0
Alexandra Villa-Forte, MD, MPH
Staff Physician, Center for Vasculitis Care and Research, Orthopedic and Rheumatologic Institute, Cleveland Clinic, Cleveland, OH
Brian F. Mandell, MD, PHD, MACP
Professor and Chairman of Medicine, Cleveland Clinic Lerner College of Medicine at Case Western Reserve University, Center for Vasculitis Care and Research, Orthopedic and Rheumatologic Institute, Cleveland Clinic, Cleveland, OH
Artigo original: Villa-Forte A, Mandell BF. Systemic Vasculitis Syndromes. ACP Medicine. 2008;1-15.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figuras 1 e 2 – Seward Hung. Figura 6 – Gary S. Hoffman.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. José Paulo Ladeira.
O diagnóstico de uma síndrome de vasculite primária depende da comprovação da vasculite e da exclusão da hipótese de outras doenças que podem causar vasculite secundária. O diagnóstico de um distúrbio vaculítico primário depende do padrão de envolvimento de órgãos, da histopatologia e do tamanho dos vasos sanguíneos afetados. O diagnóstico não deve ser estabelecido com base apenas nos exames laboratoriais (p. ex., achados sorológicos de anticorpos anticitoplasma de neutrófilo [ANCA] ou crioglobulinas).
Os principais determinantes do prognóstico e do tipo de terapia incluem o distúrbio vasculítico específico, a severidade e extensão do envolvimento de um órgão significativo, a taxa de progressão da doença e a etiologia (quando identificável). O processo inflamatório muitas vezes está associado a sintomas inespecíficos e anormalidades laboratoriais (p. ex., velocidade de sedimentação eritrocitária alta, anemia e febres) que não diferenciam as doenças vasculíticas de outras doenças inflamatórias, infecciosas ou neoplásicas. A natureza tóxica das terapias disponíveis para vasculite sistêmica impõe a necessidade de se estabelecer um diagnóstico acurado.
O médico não deve relutar em realizar exames invasivos na avaliação diagnóstica de pacientes com doenças multissistêmicas, porém a biópsia de um tecido sem envolvimento clínico e o uso de exames menos específicos devem ser evitados.
A 1ª etapa do diagnóstico da vasculite consiste em obter uma história detalhada e realizar o exame físico para comprovar a existência de envolvimento de um órgão específico. É preciso prestar atenção especialmente à pele, aos olhos, às orelhas, às vias aéreas superiores, às articulações, aos linfonodos, aos nervos periféricos e aos vasos de grande calibre. Os exames laboratoriais [Tabela 1] devem ser seletivamente incluídos na avaliação inicial. Os exames especializados, incluindo as sorologias, são idealmente obtidos e interpretados somente após a formulação do diagnóstico diferencial. Na maioria das vezes, a glomerulonefrite é assintomática. Se o teste de dipstick da urina indicar a presença de sangue, leucócitos ou proteínas, o médico deve examinar imediatamente vários sedimentos urinários frescos. A urina não examinada dentro de algumas horas após a coleta perde a utilidade para identificação de cilindros celulares, os quais se degeneram rapidamente ex vivo. A presença de cilindros hemáticos é quase diagnóstica de glomerulonefrite. Também é possível encontrar cilindros de leucócitos. Com base no padrão de envolvimento de órgãos, é possível estabelecer um diagnóstico diferencial que inclua os tipos específicos de vasculite sistêmica e outros distúrbios, conduzindo à realização de exames dirigidos adicionais.
Tabela 1. Exames laboratoriais selecionados para pacientes com doença multissistêmica e possível vasculite
|
Exame |
Comentários |
|
Contagem de plaquetas |
A trombocitose pode ocorrer em paralelo com a resposta de fase aguda. A trombocitopenia não é esperada nas síndromes de vasculíticas primárias; considerar as hipóteses de LES, infiltração da medula, leucemia de células pilosas ou outras malignidades com vasculite secundária, PTT, CID, hiperesplenismo, SAAF, HIV, crise renal do escleroderma e trombocitopenia heparina-induzida |
|
Contagem de leucócitos |
A leucopenia não é esperada em casos de vasculite primária; considerar as hipóteses de LES, leucemias, hiperesplenismo, sepse, mielodisplasia e HIV. A eosinofilia é comum na SCS, podendo ocorrer na GW, artrite reumatoide, crise renal do escleroderma e embolia de colesterol |
|
VHS |
Uma VHS relativamente baixa é observada na CID, insuficiência hepática e hiperviscosidade. A VHS costuma ser normal na PHS, pode estar normal na AT e está normal em < 20% dos casos de ACG |
|
Aminotransferases |
Níveis aumentados de ALT ou AST na doença hepática, miosite, rabdomiólise, hemólise ou necrose miocárdica |
|
Antimembrana basal glomerular |
Útil para avaliação da hemorragia alveolar, com ou sem glomerulonefrite. É igualmente útil para a avaliação da glomerulonefrite normocomplementêmica. Pode estar associado a uma prognóstico ruim diante da positividade para ANCA e anti-MBG na vasculite que se manifesta como doença severa |
|
Anticorpo antinuclear |
Solicitar quantificação deste anticorpo se houver suspeita clínica de LES (ou em casos inexplicáveis de serosite, glomerulonefrite, hemorragia alveolar, leucopenia ou trombocitopenia), e não como teste de avaliação geral para pacientes doentes; um resultado de teste negativo torna a hipótese de LES improvável |
|
ANCA |
Solicitar quantificação deste anticorpo se houver suspeita clínica de GW ou vasculite fármaco-induzida; o teste deve ser realizado por IF ou ELISA |
|
Painel farmacológico |
Solicitar o painel diante de sintomas inexplicáveis envolvendo o SNC, isquemia miocárdica, espasmo vascular, ataques de pânico com aspectos sistêmicos, taquicardia, perfuração nasal; o painel urinário deve ser obtido |
|
Culturas hematológicas |
Útil em quaisquer casos de paciente com doença febril, multissistêmica ou quadros consumptivos; infiltrados pulmonares; isquemia/infarto focal; ou para pacientes previamente submetidos a procedimentos vasculares ou endovasculares |
|
AAF/PTT/TVVR |
Solicitar os exames diante de casos inexplicáveis de trombocitopenia ou trombose arterial ou venosa |
|
Teste cutâneo com PPD (derivado proteico purificado) (± anergia) |
Realizar o teste em qualquer caso de paciente que possa necessitar de terapia com esteroide ou que apresente hematúria ou piúria estéril inexplicável, inflamação granulomatosa, meningite crônica ou possível exposição à tuberculose |
|
Exame de sedimento urinário fresco |
Realizar o exame em todos os pacientes com doença febril, hipertensiva ou multissistêmica inexplicável ou com níveis inexplicavelmente elevados de creatinina |
|
Exames sorológicos virais: hepatite B e C; possivelmente, CMV e HIV |
Solicitar os exames para avaliar anormalidades de transaminases ou níveis elevados de fosfatase alcalina hepática; hipertensão porta; síndromes de PN ou PAM; ou casos inexplicáveis de crioglobulinemia, poliarterite ou vasculite cutânea |
|
Complemento (C3 e C4) |
Não é um teste de avaliação para vasculite; tem utilidade para fins de diagnóstico diferencial de glomerulonefrite; seus níveis estão baixos na crioglobulinemia e podem estar baixos na endocardite; níveis habitualmente normais na PN, PAM, PHS, GW; seus níveis podem estar baixos na vasculite ou glomerulonefrite relacionada à hepatite viral |
|
Aldolase |
A aldolase não apresenta especificidade orgânica; sua distribuição orgânica é similar à distribuição da desidrogenase láctica |
AAF = anticorpo antifosfolipídio; ACG = arterite de células gigantes; ALT = alanina aminotransferase; ANCA = anticorpo anticitoplasma de neutrófilo; AST = aspartato aminotransferase; AT = arterite de Takayasu; CID = coagulação intravascular disseminada; CMV = citomegalovírus; ELISA = ensaio imunossorvente ligado à enzima; VHS = velocidade de sedimentação eritrocitária; GW = granulomatose de Wegener; IF = imunofluorescência; LES = lúpus eritematoso sistêmico; MBG = membrana basal glomerular; PAM = poliangiite microscópica; PHS = púrpura de Henoch-Schönlein; PN = poliarterite nodosa; PR3 = proteinase 3; TTPa = tempo de tromboplastina parcial ativada ; PTT = púrpura trombocitopênica trombótica; SAAF = síndrome do anticorpo antifosfolipídio; SCS = síndrome de Churg-Strauss; SNC = sistema nervoso central; TVVR = teste do veneno da víbora de Russell.
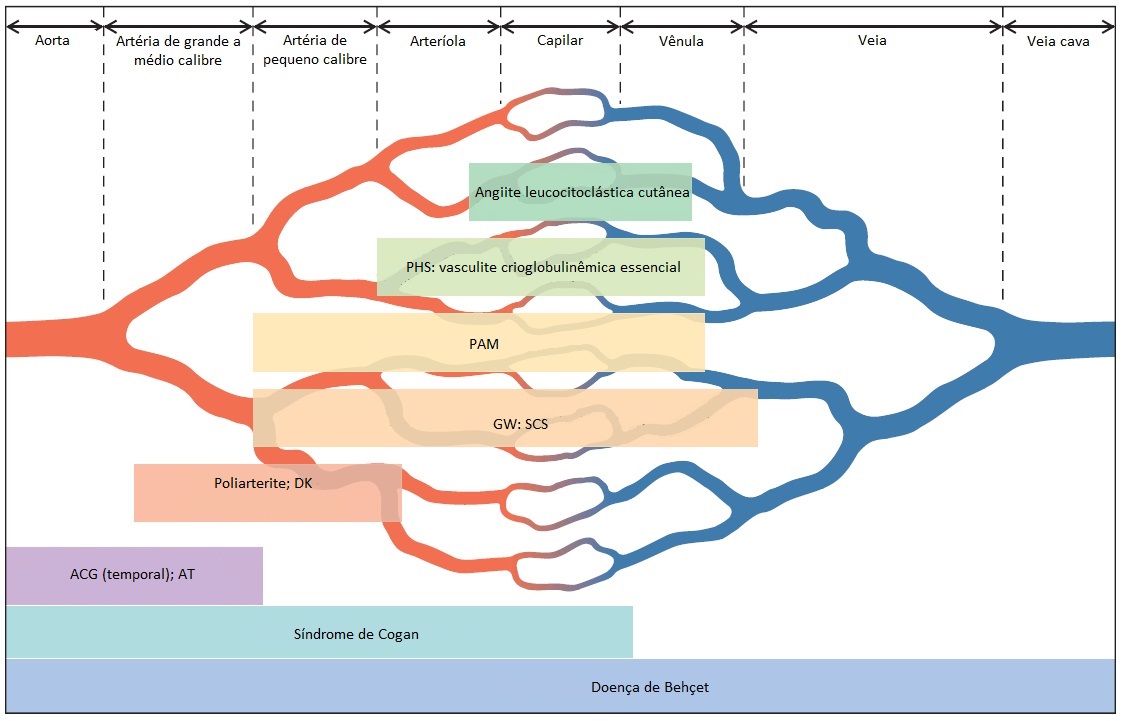
Vários esquemas de classificação foram propostos para organizar os distúrbios vasculíticos sistêmicos em um paradigma consistente. Estas classificações são úteis para distinguir os distúrbios clínicos que apresentam características distintas em termos de prognóstico e resposta ao tratamento.1 Nenhum esquema é perfeito nem universalmente aceito. Todos reiteram as características da doença clássica e enfatizam a especificidade do diagnóstico. Quando um esquema classificatório é estritamente seguido, é comum o paciente que acabou de desenvolver uma doença ainda não totalmente expressa permanecer sem receber um diagnóstico definitivo. Até que as etiologias específicas das síndromes vasculíticas sejam definidas, estas entidades continuam sendo consideradas apenas teóricas, sendo comum haver sobreposição de doenças. Isto não deve ser um impeditivo à instituição da terapia para o paciente que apresenta lesão de progressão rápida em um órgão. Mesmo assim, os sistemas de classificação fornecem definições úteis para fins de comunicação; determinação da terapia inicial; e planejamento dos protocolos de pesquisa [Figura 1]. Os esquemas de classificação mais amplamente utilizados baseiam-se no calibre dos vasos sanguíneos afetados, no padrão de envolvimento orgânico e presença/ausência de inflamação granulomatosa, na ocorrência de deposição significativa de imunocomplexos e nos infiltrados eosinofílicos. Alguns autores propuseram a categoria de vasculite associada ao ANCA, com base na presença ou ausência de ANCA específicos no soro do paciente, em particular dos anticorpos dirigidos contra a proteinase 3 e a mieloperoxidase. No momento, o papel apropriado dos testes para ANCA consiste em sustentar um diagnóstico clínico desenvolvido de forma lógica. Em casos de pacientes que não se ajustam extremamente bem a uma categoria diagnóstica bem-definida, estes exames sorológicos não suplantam a tentativa de se obter um diagnóstico tecidual. A presença do ANCA é insuficiente para estabelecer o diagnóstico de uma síndrome vasculítica primária. O teste para ANCA não é um teste de avaliação.

Figura 1. Classificação das síndromes vasculíticas sistêmicas.
ACG = arterite de células gigantes; AT = arterite de Takayasu; DK = doença de Kawasaki; GW = granulomatose de Wegener; PAM = poliangiite microscópica; PHS = púrpura de Henoch-Schönlein; SCS = síndrome de Churg-Strauss.
Quando os sintomas e achados dominantes (isto é, neuropatia e distúrbio vasculítico) não sugerem a existência de um distúrbio vasculítico específico isolado, os exames sorológicos podem ser úteis. O aspecto mais variável é a confirmação do distúrbio específico por exame de biópsia. O valor da realização de exames indiscriminados para detecção de anticorpos antinucleares, ANCA, fator reumatoide e enzima conversora de angiotensina é discutível. Em contraste, a infecção pelos vírus da hepatite B e hepatite C pode estar associada a uma ampla gama de síndromes vasculíticas. Estas infecções devem ser rotineiramente excluídas em casos de pacientes com vasculite envolvendo vasos de pequeno e médio calibre, quando não há evidências claras da existência de um distúrbio vasculítico clinicamente definido e único, como a granulomatose de Wegener (GW).2
As vasculites sistêmicas são potencialmente prejudiciais à vida e podem necessitar de terapia anti-inflamatória e imunossupressora potente. Os diagnósticos devem ser estabelecidos com o máximo de certeza possível. Entretanto, frequentemente surgem questões relativas aos diagnósticos alternativos ou doenças coexistentes. Mesmo depois que a terapia é iniciada, os médicos devem manter um alto grau de vigilância para detectar problemas médicos não relacionados, complicações da terapia ou ambos. Os sinais e sintomas de infecção não identificada podem ser resolvidos de modo transitório pela terapia com corticosteroides.3 Com o início de uma terapia imunossupressora potente, o paciente fica sujeito a uma suscetibilidade aumentada às infecções oportunistas. Os pacientes que apresentam riscos mais significativos são aqueles com neutropenia marcante ou sob tratamento com doses altas de corticosteroides. Os médicos devem ser particularmente cuidadosos ao atribuírem problemas novos às “exacerbações” de uma doença subjacente sem antes excluir a hipótese de uma infecção nova ou recrudescente. Os pacientes infectados pelo vírus do herpes-zóster podem apresentar febre e dor antes do aparecimento das vesículas. Pneumocystis jiroveci, citomegalovírus (CMV) e infecções fúngicas sistêmicas, bem como a reativação de doenças micobacterianas, são observados com maior frequência em pacientes submetidos à imunossupressão. A imunossupressão produzida por esteroides e outras medicações costuma estar associada ao desenvolvimento de candidíase mucosa que, por sua vez, apresenta uma associação menos frequente com o desenvolvimento de molusco contagioso e sarcoma de Kaposi.
Metotrexato, azatioprina e ciclofosfamida podem causar leucopenia e, com menos frequência, outras citopenias. Os pacientes com função renal diminuída devem usar metotrexato com cautela (se de fato precisarem usá-lo). A dose de ciclofosfamida deve ser reduzida e atentamente monitorada, pois o pró-fármaco (ciclofosfamida) é excretado pelos rins. A disfunção do esvaziamento da bexiga representa uma contraindicação relativamente forte ao uso prolongado da ciclofosfamida, uma vez que a exposição aumentada aos metabólitos tóxicos do fármaco pode predispor o paciente ao desenvolvimento de câncer de bexiga ou cistite. No caso do tratamento dos pacientes que apresentam certas síndromes vasculíticas sistêmicas potencialmente prejudiciais à vida, existe uma tendência à introdução da terapia com um curso de doses altas de corticosteroides de curta duração aliado a um 2º agente imunossupressor, para induzir a remissão e, em seguida, dependendo da doença, reduzir os corticosteroides e manter apenas a terapia imunossupressora com o agente imunossupressor mais efetivo para manter a remissão. O agente não corticosteroide inicial pode ser a ciclofosfamida, que aparentemente é o mais potente dos fármacos imunossupressores não corticosteroides. No entanto, a ciclofosfamida posteriormente é substituída por um agente com perfil de segurança melhor (p. ex., metotrexato ou azatioprina). A terapia com este agente, então, é continuada durante vários meses. Este tipo de abordagem foi mais bem avaliado em pacientes com GW, para os quais foi comprovadamente efetivo.
A vasculite que afeta essencialmente os capilares, vênulas e arteríolas não musculares constitui o padrão mais comum de vasculite e quase sempre envolve a pele. Pode ocorrer em qualquer idade e afeta indivíduos de ambos os sexos com a mesma frequência.
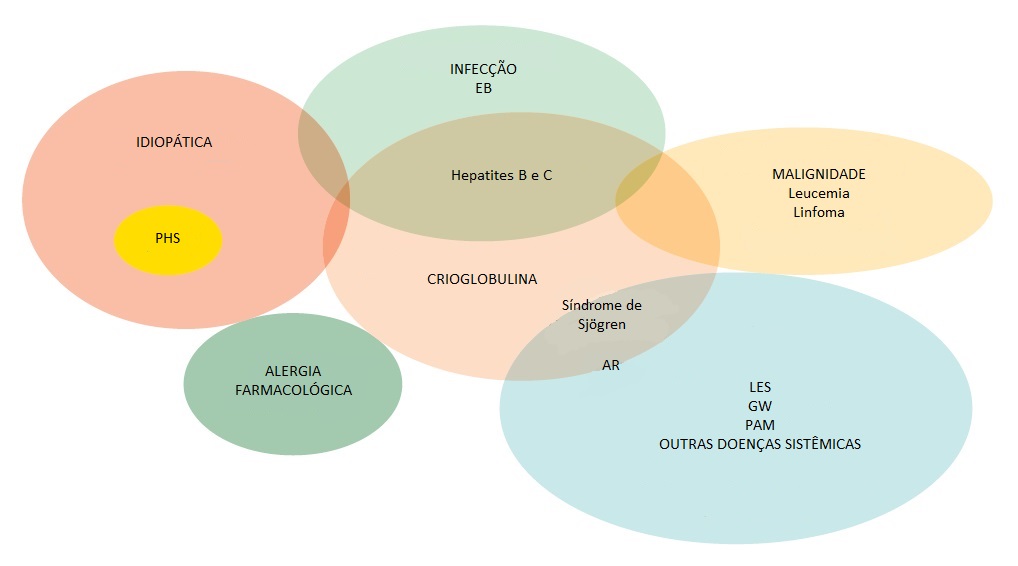
As vasculites de vasos pequenos podem ocorrer como distúrbios idiopáticos (primários), mas frequentemente são secundárias a alergias farmacológicas, endocardite bacteriana e infecções virais, como aquelas causadas pelos vírus das hepatites B e C, Neisseria disseminada ou riquétsias. Pode fazer parte de um distúrbio autoimune sistêmico, como a síndrome de Sjögren, lúpus eritematoso sistêmico (LES) ou artrite reumatoide, ou ocorrer associada a malignidades hematológicas, linfoides e de órgãos sólidos [Figura 2]. As vasculites de pequenos vasos também podem acompanhar doenças comumente associadas ao envolvimento de vasos de grande calibre (p. ex., GW).
Figura 2. Diagrama de Venn ilustrando as relações existentes entre as causas de vasculite de pequenos vasos (“hipersensibilidade”).
AR = artrite reumatoide; EB = endocardite bacteriana; GW = granulomatose de Wegener; LES = lúpus eritematoso sistêmico; PAM = poliangiite microscópica; PSH = púrpura de Henoch-Schönlein.
O envolvimento cutâneo ocorre em muitas síndromes vasculíticas primárias ou secundárias. A obstrução de vasos sanguíneos de pequeno, médio ou grande calibre pode causar livedo, fenômeno de Raynaud ou necrose. As lesões da púrpura que se tornam parcialmente esbranquiçadas sob pressão constituem as manifestações mais comuns de vasculite de pequenos vasos. A vasculite de pequenos vasos, em particular quando associada a infecções, é com frequência acompanhada de deposição de imunocomplexos. Na literatura mais antiga, a vasculite com envolvimento primário das vênulas pós-capilares era denominada vasculite de hipersensibilidade.4 A vasculite de pequenos vasos primária pode ser limitada à pele ou estar associada ao envolvimento visceral, incluindo a hemorragia alveolar, hemorragia ou isquemia intestinal e glomerulonefrite.
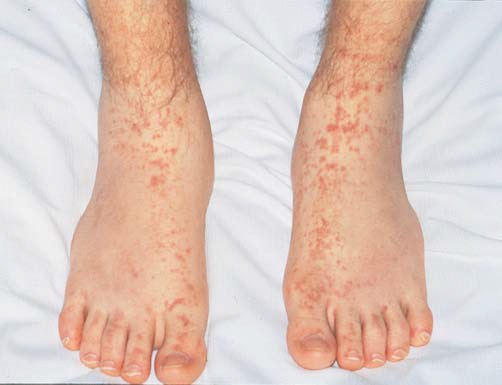
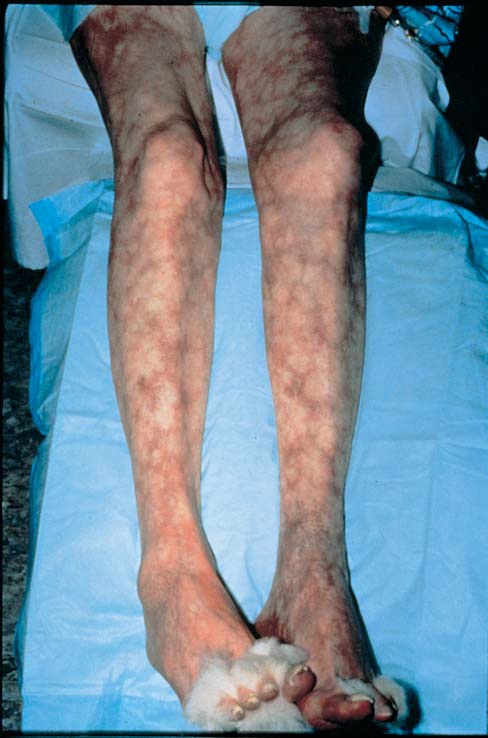
A púrpura tende a ser observada no crescimento recorrente de lesões de idade similar e é mais proeminente nas áreas dependentes de gravidade [Figura 3]. Quando a púrpura não está primariamente localizada nas áreas dependentes de gravidade, devem ser consideradas as hipóteses de doença da aglutinina fria, crioglobulinemia (que pode estar associada a infecções como a hepatite C ou com linfomas), meningocococemia, embolia, doenças infiltrativas e lesão autoinduzida. A vasculite cutânea de qualquer etiologia pode estar associada a um edema notavelmente dependente.
Figura 3. A púrpura palpável de membros distais é a manifestação mais comum da vasculite de pequenos vasos.
Em uma série de casos de vasculite cutânea de pequenos vasos, quase 100% dos pacientes com menos de 20 anos de idade apresentavam doença restrita à pele, enquanto cerca de 40% dos 172 pacientes com idade acima de 20 anos tinham um distúrbio sistêmico associado ou subjacente.4 Um total de 17 pacientes adultos apresentavam vasculite necrotizante sistêmica; 4 tinham malignidade; 4 apresentavam infecção bacteriana causadora de vasculite; 11 tinham crioglobulinemia; e 59 sofriam de púrpura de Henoch-Schönlein. A prevalência da infecção pelo vírus da hepatite C, provavelmente a causa mais comum de crioglobulinemia mista,2 não foi relatada nesta série.
A biópsia é mais útil para a exclusão de causas de púrpura não vasculítica, como amiloidose, leucemia cutânea, sarcoma de Kaposi, linfomas de células T, traumatismo e êmbolos de colesterol ou mixomatosos. A coloração imunofluorescente tecidual é útil para sustentar o diagnóstico de púrpura de Henoch-Schönlein (especificamente, a coloração de IgA), LES ou infecção (o percentual de pacientes com resultados positivos de coloração de imunofluorescência é desconhecido). As células que infiltram e talvez destruam a parede vascular podem ser neutrófilos ou linfócitos, dependendo da doença subjacente. Na maioria dos casos de vasculite de pequenos vasos, a patologia é de angiite leucocitoclástica. A infecção pelo vírus da hepatite C deve ser rotineiramente excluída em casos de pacientes com púrpura inexplicável – um exemplo significativo do fato de a existência de angiite leucocitoclástica não indicar que uma doença apresentada pelo paciente resulta de uma síndrome vasculítica primária. A histopatologia e a presença/ausência de imunoglobulinas detectadas dependem da idade da lesão de onde a biópsia foi extraída.
A angiite leucocitoclástica cutânea é limitada à pele e frequentemente está associada a um fator desencadeante, como os medicamentos. As lesões são quase todas da mesma idade, visto que surgem ao mesmo tempo. Pode haver artralgias, contudo sem envolvimento sistêmico. A causa nem sempre é identificada, e o tratamento inicial inclui os fármacos anti-inflamatórios não hormonais (AINH), a colchicina ou a dapsona. Os casos refratários ou recidivantes podem necessitar de corticosteroides. Outros medicamentos imunossupressores, como a azatioprina ou o metotrexato, têm sido utilizados com resultados variáveis.
A púrpura de Henoch-Schönlein é clinicamente definida como uma síndrome vasculítica de pequenos vasos, em que os aspectos cutâneos em geral são marcantes, enquanto o envolvimento visceral mais significativo é menos frequente. A púrpura de Henoch-Schönlein, que é menos frequentemente observada em adultos do que em crianças e costuma estar associada à deposição vascular e renal de imunocomplexos contendo IgA.5 Entre as manifestações comuns da púrpura de Henoch-Schönlein estão a púrpura, a urticária, a dor abdominal, o sangramento gastrintestinal ou a intuscepção (sobretudo em crianças), as artralgias ou artrite e a glomerulonefrite. Os sintomas viscerais podem preceder as lesões cutâneas apenas em casos raros. A púrpura de Henoch-Schönlein aparentemente pode ser precipitada por medicamentos, bem como infecções estreptocócicas ou virais. Em geral, trata-se de um distúrbio autolimitado. Entretanto, há casos raros (sobretudo em adultos) em que a glomerulonefrite evolui para insuficiência renal. Na ausência de disfunção renal, a púrpura de Henoch-Schönlein frequentemente é uma síndrome autolimitada que pode necessitar apenas de terapia sintomática. As recidivas podem ocorrer em até 1/3 dos pacientes. Como uma parte do envolvimento visceral pode ser significativa, o paciente deve ser monitorado periodicamente até a completa resolução dos sintomas.
A vasculite urticariforme representa um subgrupo peculiar de vasculite de pequenos vasos.6 Sua manifestação clínica consiste no aparecimento de pápulas em forma de vergões ou serpentinas, às vezes acompanhadas de um angioedema circundante ou geograficamente separado. A resolução das lesões individuais é um processo lento, muitas vezes com duração de vários dias. A doença segue um curso mais prolongado, se comparada à urticária típica. Frequentemente, o paciente apresenta um desconforto em queimação e disestésico a partir das lesões. Assim como a púrpura, as lesões da vasculite urticariforme frequentemente estão localizadas em áreas dependentes da gravidade e muitas vezes sua cicatrização é acompanhada de hiperpigmentação da pele ou da formação de uma área de equimose. A maioria dos casos é idiopática, embora tenha sido descrita uma associação com um distúrbio autoimune sistêmico subjacente, como LES, paraproteinemia de IgM ou infecção viral. Em casos raros, a vasculite urticariforme tem sido associada a uma síndrome que inclui hipocomplementemia, doença pulmonar obstrutiva e glomerulonefrite. Esta síndrome difere do angioedema associado à deficiência de C1 esterase, que não causa urticária.
A terapia para vasculite cutânea é voltada essencialmente à eliminação de quaisquer fatores precipitantes subjacentes. As etiologias infecciosas devem ser investigadas e tratadas. Os fármacos potencialmente agressores devem ter o uso suspenso. A associação existente entre mielodisplasia e doença mieloproliferativa deve ser considerada, especialmente diante de evidências de citopenias ou formas celulares anormais presentes no esfregaço de sangue periférico. Na ausência de fatores precipitantes evidentes, pode ser feita uma tentativa de instituir uma terapia de baixo risco com AINH, colchicina, dapsona ou um curso de corticosteroides de curta duração. Estas terapias não são uniformemente efetivas para fins de redução da frequência ou severidade dos ataques. A terapia com corticosteroides deve ser evitada, sempre que possível. O uso de meia-calça ou de meias de sustentação compressoras pode ser útil para limitar o edema significativo que frequentemente acompanha a vasculite cutânea nas pernas.
O envolvimento visceral com disfunção de órgão pode requerer uma abordagem mais agressiva do que aquela empregada na limitação da vasculite cutânea. O uso de doses moderadas de corticosteroides geralmente é efetivo. No contexto das potenciais complicações decorrentes do uso crônico de corticosteroides ou no contexto de um envolvimento visceral grave, a administração de metotrexato, azatioprina, ciclofosfamida ou outros agentes imunossupressores ocasionalmente pode ser necessária [Tabela 2]. A aférese pode ser efetiva no tratamento da vasculite crioglobulinêmica severa. Ao tratar uma doença refratária e crônica de um vaso de pequeno calibre, que não é prejudicial a um órgão nem à vida do paciente, torna-se necessário considerar atentamente a proporção risco-benefício associada às terapias selecionadas.
Tabela 2. Terapias imunossupressoras para vasculite e comentários práticos
|
Fármaco |
Dose |
Avaliação da eficácia |
Comentários |
|
Corticosteroide |
Frequentemente administrado a uma dose inicial de 1 mg/kg/dia (doses divididas na doença severa); reduzido com o objetivo de cessar o tratamento em 6 meses no máximo, se possível; para tanto, outros fármacos são usados. Pode ser usado em pulsos de doses de 1 g/dia durante 3 dias, em casos de doença severa |
Terapia primária para todas as formas de vasculite prejudiciais à vida ou a órgãos; é provavelmente a terapia de ação mais rápida |
De modo ideal, checar a condição basal de PPD; considerar a profilaxia contra Pneumocystis (ao usar doses altas) e osteoporose; monitorar pacientes idosos quanto ao desenvolvimento de glaucoma. As doses altas podem elevar a pressão arterial e os níveis de glicose no soro |
|
Ciclofosfamida |
1 a 3 mg/kg/dia, VO; evitar a neutropenia; o nadir habitualmente ocorre em 9 a 14 dia após o início da terapia ou mudança da dose; diminuir a dose no contexto de insuficiência renal; a dosagem de “pulso” tem sido empregada (0,5 a 1 g/m2), porém com probabilidade maior de neutropenia; administrar as doses de pulso após a diálise |
É a terapia imunossupressora não esteroidal mais potente. O aparecimento de sua ação é indefinido, mas o fármaco deve ser administrado quando a doença severa é identificada, em particular em casos de glomerulonefrite de progressão rápida |
Os efeitos colaterais significativos limitam o uso prolongado deste fármaco: leucopenia, doença mieloproliferativa, dano na bexiga e malignidade. Atualmente, existe uma tendência em induzir remissão na GW e em outra formas severas de vasculite por meio da administração de prednisona e ciclofosfamida, com afunilamento da prednisona e troca da ciclofosfamida por um fármaco menos tóxico (p. ex., azatioprina ou metotrexato) |
|
Azatioprina |
2 a 3 mg/kg/dia, VO. |
Provavelmente, menos potente do que a ciclofosfamida para fins de indução; é útil para manter a remissão durante a redução do corticosteroide |
Não é habitualmente administrada como terapia de indução primária; evitar a leucopenia; pode causar uma reação de hipersensibilidade geradora de confusão, que inclui febre alta, além de presença/ausência de erupção e eosinofilia |
|
Metotrexato |
Administrar 1 x/semana (até cerca de 0,3 mg/kg/dose) aliado à administração diária de ácido fólico (1 mg) |
É menos potente do que a ciclofosfamida no tratamento da doença severa; é útil para manter a remissão durante a redução da dosagem de corticosteroide; diminuir a dose em casos de insuficiência renal branda; evitar o uso em pacientes com níveis de creatinina > 2 mg/dL |
Útil para manter a remissão; pode ser usada como terapia de indução primária com prednisona em casos de pacientes com GW branda; frequência significativa de recidivas entre pacientes com GW mantidos apenas com este fármaco; monitorar a contagem de LEU, creatinina e níveis de transaminase (causa hepatite e pode causar cirrose; evitar a ingesta de etanol); pode ser administrada por via oral ou injeções semanais; o ácido fólico diminui os efeitos colaterais “incômodos” |
GW = granulomatose de Wegener; LEU = leucócitos; PPD = derivado proteico purificado.
A GW é uma doença relativamente incomum e potencialmente letal, caracterizada por inflamação granulomatosa necrotizante e vasculite em vasos de pequeno e médio calibre.7,8 Indivíduos de ambos os sexos e de todas as faixas etárias podem ser afetados.
A GW é caracterizada por necrose parenquimal com um componente vasculítico variável. Múltiplos órgãos frequentemente são envolvidos. A doença apresenta predileção pelos tratos respiratórios superior e inferior, olhos e rins.
Envolvimento do trato respiratório superior. A doença com envolvimento do trato respiratório superior pode ser notável, mas frequentemente é indolente e atribuída a meses ou até anos de doença sinusal rotineira, até a identificação de outras manifestações da GW. Mesmo depois que o diagnóstico é estabelecido e o tratamento imunossupressor é iniciado, a doença sinusal pode ser resistente à terapia. A cronicidade pode ser causada, em parte, por infecção e/ou dano tecidual. O dano anatômico pode incluir perfurações de septo e deformação do nariz em sela. O envolvimento laringotraqueal pode resultar em estenose subglótica que é melhor tratada com injeção local de corticosteroide aliada à terapia de dilatação. É comum haver envolvimento da orelha, particularmente otite média, que pode acarretar perda auditiva. O desenvolvimento de pseudotumores orbitais pode causar proptose, oftalmoplegia, dor intratável e perda da visão. Estas massas inflamatórias e fibrosas podem ser refratárias à terapia anti-inflamatória, terapia imunossupressora e até radioterapia. Conjuntivite, uveíte e esclerite, isoladamente ou em combinação, são comuns.
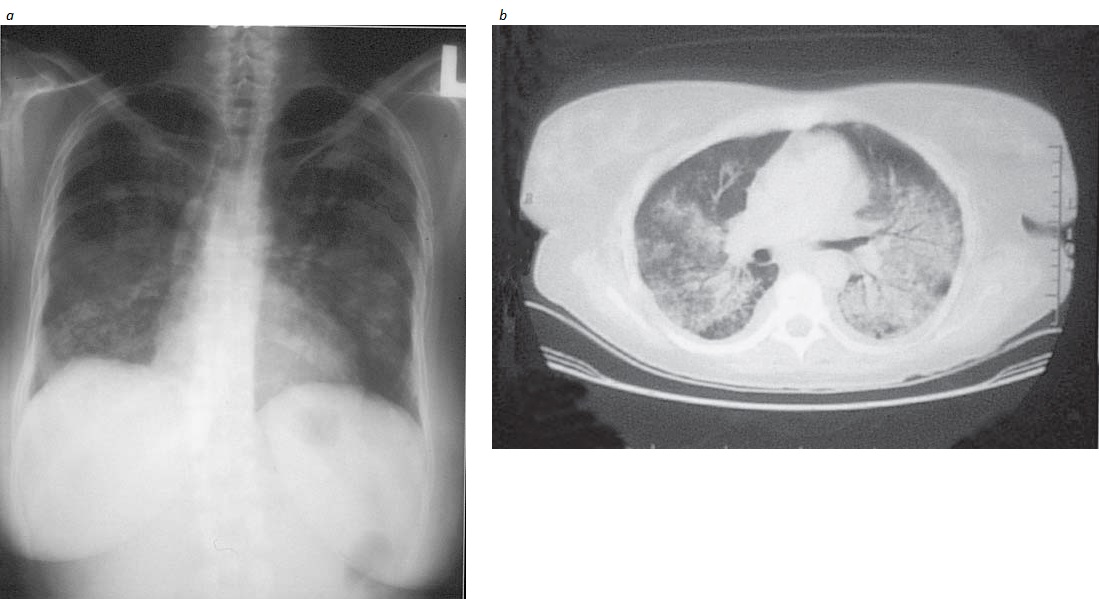
Envolvimento do trato respiratório inferior. O envolvimento pulmonar pode estar ausente no momento do aparecimento da doença, pode ser assintomático ou, ainda, pode estar dramaticamente presente sob a forma de hemorragia alveolar difusa. Um terço das lesões pulmonares observadas nos exames de imagem [Figura 4] são assintomáticas (a tomografia computadorizada é mais sensível do que a radiografia). Os nódulos podem sofrer necrose, com consequente formação de cavidade. O broncoespasmo não é característico do GW. Havendo suspeita de obstrução de vias aéreas, a hipótese de broncoespasmo deve ser considerada para excluir a possibilidade de estenose endobrônquica ou subglótica. Frequentemente, é necessário excluir a hipótese de causa infecciosa da infiltração pulmonar, e a broncoscopia com lavado é útil para esta finalidade. Entretanto, o tecido obtido por biópsia transbrônquica costuma ser insuficiente em quantidade para confirmar o diagnóstico patológico de GW.
Figura 4. Os infiltrados nodulares do pulmão observados na granulomatose de Wegener (GW) aparecem menos extensivamente em uma radiografia-padrão (a) do que na tomografia computadorizada (b).
A biópsia por toracoscopia ou pulmonar aberta frequentemente é o método ideal para demonstração dos achados patológicos típicos da GW e também para excluir as hipóteses de malignidade e infecções atípicas. Os cortes típicos da biópsia pulmonar aberta9 podem conter áreas de necrose (muitas vezes, com um padrão amplo), células gigantes no tecido parenquimal e vasculite. Nem todos os aspectos histopatológicos estarão necessariamente presentes em um mesmo corte de biópsia, sendo que a vasculite pode não ser evidente. Como uma patologia similar à GW pode ser demonstrada em casos de infecções fúngicas e micobacterianas, é essencial usar culturas e colorações especiais para estes agentes.
Glomerulonefrite. A glomerulonefrite é causa comum de morbidade e mortalidade na GW. Esta condição, por sua vez, é muitas vezes referida como sendo uma doença generalizada ou limitada. A existência de glomerulonefrite define a forma generalizada da condição. A glomerulonefrite costuma ser agressiva, mas pode ser relativamente indolente. Também pode ser clínica e patologicamente indistinguível da glomerulonefrite idiopática de evolução rápida e em crescente, além de no geral ser silenciosa do ponto de vista de clínico. A evolução da doença renal subclínica para a doença diálise-dependente pode estender-se por várias semanas. A glomerulonefrite pode estar presente no momento da manifestação da doença ou pode desenvolver-se apenas depois de o paciente ter adoecido, com uma forma aparentemente limitada da doença. A importância das urinálises microscópicas frequentes para as avaliações iniciais e de seguimento dos pacientes com GW não pode ser exagerada. Este monitoramento pode ser feito pelos próprios pacientes, em casa, empregando a análise do dipstik para detectar a hematúria oculta. Sobretudo em casos de pacientes idosos ou debilitados, é possível obter informações valiosas a partir do exame de amostras de urina ocasionalmente coletadas em 24 horas. O exame destas amostras pode fornecer uma estimativa mais acurada da taxa de filtração glomerular do que as estimativas fornecidas pela medida dos níveis séricos de creatinina. A biópsia renal pode revelar a existência de glomerulonefrite focal e segmentar, com alterações glomerulares proliferativas variáveis, formação em crescente e necrose, na ausência de deposição significativa de imunocomplexos (conhecida como glomerulonefrite paucimune). Embora sustentem o diagnóstico de GW, estes achados não são diagnósticos da doença, e a biópsia renal não é o exame preferido para confirmação do diagnóstico específico de GW [ver Exames Laboratoriais, adiante].
Manifestações clínicas adicionais. O envolvimento musculoesquelético é observado em metade dos pacientes com GW. Os sintomas podem ser artralgias ou artrites. Estes sintomas podem ser migratórios, aditivos ou fixos, quanto à distribuição. O fator reumatoide está presente com frequência em pacientes com GW e pode acarretar confusão com o diagnóstico de artrite reumatoide, em que os sintomas articulares são significativos. A doença articular da GW produz erosões somente em casos raros. Os sinais e sintomas neurológicos ocorrem em menos de 50% dos pacientes. A neuropatia periférica é observada em menos de 20% dos casos, enquanto o envolvimento do sistema nervoso central ocorre em menos de 10% dos pacientes. Pode haver desenvolvimento de defeitos oculomotores, devido ao contato com uma massa retro-orbital ou doença sinusal inflamatória. As úlceras e a isquemia gastrintestinal são pouco frequentes, mas podem ser confundidas com uma enteropatia inflamatória, especialmente porque esta última pode estar associada à formação de ANCA (em geral, anticorpo anticitoplasma de neutrófilo perinuclear [p-ANCA]). Até 50% dos pacientes com GW apresentam envolvimento cutâneo com púrpura, paniculite ou ulcerações. Quando presente, a doença cutânea geralmente é paralela à atividade da doença sistêmica. As observações derivadas de estudos clínicos recentes sugerem que os pacientes com GW são predispostos ao desenvolvimento de trombose em veia profunda.10
A inflamação crônica inexplicável do trato respiratório ou do olho, ou, ainda, a presença de glomerulonefrite são consistentes com o diagnóstico de GW. A probabilidade de GW aumenta diante do envolvimento de múltiplos órgãos; a doença das vias aéreas superiores é destrutiva; e os nódulos pulmonares (em especial nas cavidades) são demonstrados por radiografia. Pode haver qualquer combinação de envolvimento de órgãos, todavia a maioria dos pacientes exibe envolvimento das vias aéreas superiores no momento do diagnóstico.
Quando o quadro clínico é totalmente compatível com GW e se os diagnósticos alternativos tiverem sido devidamente excluídos, o achado de anticorpos anticitoplasma de neutrófilo circulantes (c-ANCA) específicos para proteinase 3, por meio do ensaio imunossorvente ligado à enzima (ELISA), é suficiente para estabelecer um diagnóstico provisório e instituir a terapia na ausência de um diagnóstico tecidual. Cerca de 20% dos pacientes com GW podem apresentar p-ANCA com especificidade antimieloperoxidase. Havendo quaisquer aspectos atípicos ou preocupações especiais relacionadas ao início da terapia imunossupressora, ou se o paciente não responder adequadamente à terapia, a confirmação patológica do diagnóstico deve ser agressivamente perseguida. A presença de ANCA não equivale à existência de vasculite. O ANCA pode ser encontrado em outras doenças.
Os níveis de ANCA não são confiáveis como meio de acompanhar a atividade da doença.11-13 Como a GW requer terapia à base de corticosteroides mais um 2º agente para indução de remissão e limitação da probabilidade de recidiva, deve ser distinguida de outros distúrbios inflamatórios, tais como outras síndromes vasculíticas [Tabela 3], que podem ser tratadas de modo efetivo utilizando-se um regime menos tóxico.
Tabela 3. Aspectos clínicos das vasculites
|
Distúrbio |
Órgãos-alvo comuns |
Aspectos patológicos especiais |
Exames laboratoriais especiais |
Comentários |
|
PAM |
Nervo periférico, glomérulo, pulmão (pequenos vasos), trato GI, pele |
Vasculite, GN proliferativa (imunodeposição ausente ou rara*); sem células gigantes |
p-ANCA (antimieloperoxidase) |
Excluir a hipótese de hepatite B e C |
|
PN |
Nervo periférico, trato GI; ausência de GN |
Arterite de artérias musculares médias, sem células gigantes |
Sem ANCA |
Sem envolvimento de pequenos vasos; excluir a hipótese de hepatite B |
|
GW |
Vias aéreas superiores, olho, pulmão (pequenos vasos), glomérulo, nervo periférico, sistema musculoesquelético |
Células gigantes, necrose geográfica, vasculite, eosinofilia leve, GN proliferativa (imunodeposição ausente ou rara) |
c-ANCA (anti-PR3) |
Doença da orelha ou sinusal crônica |
|
SCS |
Nervo periférico, pulmão, coração, pele |
Células gigantes, eosinofilia, vasculite, GN proliferativa (imunodeposição ausente ou rara) |
Eosinofilia ± ANCA (30 a 50% positividade) |
História atópica positiva |
|
DK |
Linfonodos, pele e mucosa oral, coronárias |
Aneurismas inflamatórios em artéria coronária |
|
O tratamento precoce pode prevenir a formação de aneurisma |
|
PHS |
Pele, sistema musculoesquelético, trato GI, glomérulo |
VLC cutânea Deposição tecidual de imunocomplexos de IgA |
|
1/3 das crianças apresentam recidivas |
*A presença de imunodepósitos sugere a possibilidade de infecção pelos vírus das hepatites B ou C.
ANCA = anticorpo anticitoplasma de neutrófilo; c-ANCA = anticorpo anticitoplasma de neutrófilo citoplasmático; DK = doença de Kawasaki; GI = gastrintestinal; GN = glomerulonefrite; GW = granulomatose de Wegener; PAM = poliangiite microscópica; p-ANCA = anticorpo anticitoplasma de neutrófilo perinuclear; PHS = púrpura de Henoch-Schönlein; PN = poliarterite nodosa; PR3 = proteinase 3; SCS = síndrome de Churg-Strauss; VLC = vasculite leucocitoclástica.
O tratamento inicial da GW generalizada justifica a instituição de uma terapia imunossupressora com 2 fármacos. Os corticosteroides podem produzir comprometimento sintomático nas vias aéreas superiores, pulmões, pele e sistema musculoesquelético, no entanto sua redução geralmente resulta na pronta exacerbação da doença, a menos que um 2º agente seja administrado de maneira concomitante. O tratamento da GW deve ser determinado pela severidade da doença. Os pacientes com doença prejudicial à vida ou envolvimento de um órgão significativo devem ser tratados com ciclofosfamida combinada a altas doses de corticosteroide. Depois que o paciente alcança uma melhora ou remissão significativa (3 a 6 meses), a terapia com ciclofosfamida deve ser prontamente substituída pela terapia com metotrexato ou azatioprina como forma de tratamento de manutenção da remissão da doença [Tabela 2]. Esta abordagem atualmente é sustentada por diversos estudos clínicos.14-16
Existem algumas contraindicações relativas fortes ao uso prolongado da ciclofosfamida, incluindo a disfunção da bexiga (risco aumentado de desenvolvimento de câncer de bexiga ou cistite induzida por metabólitos do fármaco) e a leucopenia. Contudo, mesmo na ausência destas contraindicações, a morbidade associada ao uso da ciclofosfamida é significativa.7 Em pacientes com doença mais branda, a administração de doses semanais de metotrexato com ácido fólico (ou leucovorina) pode ser substituída por ciclofosfamida para indução e manutenção da remissão.17 Os pacientes submetidos ao tratamento com agentes imunossupressores devem ser continuamente monitorados quanto à ocorrência de exacerbações da doença, infecções oportunistas e efeitos colaterais da medicação. As exacerbações podem ser mais frequentes em pacientes tratados com metotrexato do que naqueles que recebem cursos mais prolongados de ciclofosfamida, além de ocorrerem frequentemente com a retirada dos corticosteroides.14 Os efeitos colaterais do metotrexato incluem as citopenias e pneumonite fármaco-induzida. O metotrexato pode causar hepatite e, em casos raros, cirrose. Seu uso deve ser evitado no contexto da insuficiência renal e do consumo de bebidas alcoólicas. A azatioprina pode causar uma reação de hipersensibilidade febril e leucopenia. As tentativas de limitar os efeitos colaterais da ciclofosfamida utilizando metotrexato ou azatioprina são justificáveis, porém esta abordagem deve ser acompanhada de um monitoramento atento para se detectar a exacerbação da doença. Alguns autores sugerem o uso de trimetoprima-sulfametoxazol como terapia auxiliar no tratamento da GW, porque existem dados limitados sugerindo que esta terapia pode diminuir a frequência das exacerbações da doença do trato respiratório superior.18 Entretanto, esta abordagem ainda é considerada controversa e não é adotada de modo rotineiro com uma dose integral de metotrexato, pois esta combinação pode resultar em uma toxicidade antifolato adicional. A administração de trimetoprima-sulfametoxazol (3 vezes/semana) é útil para conferir proteção aos pacientes contra a pneumonia por P. jiroveci (antigo P. carinii) durante o curso de terapia imunossupressora intensiva. A higiene sinusal e nasal local, bem como as avaliações otorrinolaringoscópicas periódicas são parte rotineira do tratamento de pacientes com doença no trato respiratório superior. Um estudo randomizado controlado demonstrou que a terapia da necrose antitumoral com etanercepte é inefetiva como terapia auxiliar no tratamento da GW.19 Apesar da alta eficácia na indução da remissão da doença, as terapias convencionais para GW estão associadas a uma substancial toxicidade e a uma elevada taxa de recidivas. Os dados preliminares mostram que a terapia com anti-CD20 (rituximabe) induz remissão em pacientes com GW recidivante, aparentemente de modo independente da positividade para ANCA.20 A eficácia do rituximabe na vasculite associada a ANCA está sendo avaliada em um estudo randomizado controlado (RAVE).
A adoção de medidas profiláticas para prevenção da osteoporose e a realização de densitometria óssea devem ser sempre consideradas diante do uso prolongado de corticosteroides.
A síndrome de Churg-Strauss (SCS), ou angiite granulomatosa alérgica, é uma síndrome rara que afeta artérias e veias de pequeno e médio calibre, associada à asma brônquica e hipereosinofilia.
O componente inflamatório da SCS apresenta similaridades com a GW em termos de envolvimento de órgão e patologia, especialmente em pacientes com doença no trato respiratório superior e inferior ou glomerulonefrite. A SCS difere mais notavelmente da GW no sentido de ocorrer em pacientes com história de atopia, asma ou rinite alérgica frequentemente em curso. A doença é tipicamente descrita como tendo 3 fases: uma fase atópica inicial, em que há rinite alérgica e asma; uma 2ª fase eosinofílica; e uma fase final de vasculite sistêmica. Estas 3 fases nem sempre estão presentes em todos os pacientes e em muitos casos não seguem uma determinada sequência em particular. A eosinofilia é característica e frequentemente acentuada (= 1.000 eosinófilos/mm3). Quando a eosinofilia está presente na GW, em geral é mais modesta (aproximadamente 500 eosinófilos/mm3).
Entre os aspectos órgão-específicos da SCS, estão algumas combinações de infiltrados pulmonares, miocardiopatia, arterite coronariana, polineuropatia (simétrica ou mononeurite múltipla), enteropatia isquêmica, gastrenterite eosinofílica, inflamação ocular, perfurações nasais, glomerulonefrite, nódulos cutâneos e púrpura.21,22
Os infiltrados pulmonares irregulares associados à SCS frequentemente são transitórios e podem estar associados à hemorragia alveolar. Os nódulos pulmonares são incomuns e, diferente do observado na GW, raramente cavitam. As efusões pleurais costumam ser ricas em eosinófilos. Às vezes, é difícil realizar a distinção clínica da condição em relação à pneumonite por hipersensibilidade, aspergilose pulmonar e linfoma pulmonar. Há relatos de vários casos de SCS em pacientes asmáticos que se desenvolveram após a introdução de inibidores de 5-lipoxigenase durante o desmame dos corticosteroides.
Na SCS, a doença cardíaca pode ser severa e constitui a principal causa de mortalidade. A infiltração cardíaca ou o desenvolvimento de arterite coronariana podem acarretar insuficiência cardíaca e síndromes isquêmicas. Os pacientes podem desenvolver uma doença valvular cardíaca que não é tão proeminente nem comum na SCS quanto na síndrome hipereosinofílica idiopática. Há envolvimento neurológico em mais de 60% dos pacientes com SCS. Este envolvimento pode ser severo e geralmente é atribuível à arterite. Pode haver púrpura cutânea, urticária, erupções eritematosas polimórficas e nódulos. O envolvimento gastrintestinal resultante de vasculite isquêmica, a gastrenterite eosinofílica ou ambas as condições podem causar dor, câimbra e diarreia.
A histopatologia tipicamente mostra uma inflamação granulomatosa extravascular com infiltração eosinofílica proeminente, enquanto a presença de vasculite é variável. Os granulomas podem ser encontrados no tecido, em áreas separadas da vasculite demonstrável. Os infiltrados eosinofílicos observados na SCS são mais marcantes do que aqueles encontrados na GW. Eosinófilos abundantes, granulomas e células gigantes estão ausentes na poliarterite nodosa (PN) e na poliangiite microscópica (PAM). A patologia dos nódulos por si só é insuficiente para estabelecer o diagnóstico de SCS, pois uma patologia similar pode ser observada em casos de linfoma e sarcoidose. Na SCS, a glomerulonefrite não costuma ser tão grave quanto na GW e geralmente é focal, segmentar e indistinguível de outras formas de glomerulonefrite paucimune (isto é, a glomerulonefrite com pouca ou nenhuma deposição tecidual de imunocomplexos). Embora os ANCA circulantes sejam observados com menos frequência na SCS (30 a 50% dos pacientes), esta doença é considerada por alguns autores como sendo uma vasculite associada a ANCA.
A SCS costuma ser responsiva à terapia com corticosteroides. Na maioria dos casos, é possível retirar o curso de esteroides. A asma brônquica e a doença sinusal, contudo, podem necessitar da continuidade da terapia mesmo que o componente vasculítico da doença tenha entrado em remissão. Os pacientes com envolvimento de órgão visceral refratário ou severo são tratados de modo empírico com agentes adicionais, tais como a ciclofosfamida, metotrexato ou azatioprina, dependendo da severidade do envolvimento orgânico. Os corticosteroides são reduzidos depois que a remissão é alcançada [Tabela 2].
A PAM é uma vasculite de pequenos vasos predominantemente necrotizante, em que é típico haver envolvimento dos pulmões, rins e sistema nervoso periférico. Uma conferência internacional propôs que a distinção entre PN e PAM fosse baseada na ausência de inflamação granulomatosa em ambas as condições, bem como no envolvimento de arteríolas, capilares, vênulas e capilares glomerulares apenas na PAM.1 O reconhecimento de que a PAM (e não a PN) frequentemente está associada à presença de ANCA (que quase sempre são dirigidos contra a mieloperoxidase) enfatiza ainda mais a distinção entre as 2 entidades. A PAM é similar à GW em muitos aspectos, de modo que os estudos terapêuticos com frequência incluem ambas as doenças.
A PAM, diferente da PN, envolve vasos de menor calibre, que variam de capilares e vênulas a artérias de médio calibre [Figura 1].23 Como envolve os vasos pequenos, a PAM frequentemente se manifesta como glomerulonefrite ou hemorragia alveolar. Do ponto de vista clínico, a PAM pode mimetizar a GW, embora alguns autores definam arbitrariamente a PAM como sendo excludente do envolvimento das vias aéreas superiores. O diagnóstico da PAM na ausência de doença renal pode ser uma tarefa desafiadora, que acaba atrasando o início da terapia.
É preciso tentar confirmar o diagnóstico por meio da demonstração da patologia de vasculite necrotizante na ausência de inflamação granulomatosa. Em oposição à GW, a necrose e a inflamação parenquimal observadas na PAM não são marcantes (à parte das áreas com dano isquêmico). Além disso, as células gigantes não são encontradas na PAM. A biópsia de pulmão obtida em um contexto de hemorragia ou infiltração pulmonar revela a ocorrência de capilarite, um padrão histopatológico que também pode ser observado na GW, LES e doença antimembrana basal glomerular. A biópsia de pulmão obtida de pacientes com suspeita de PAM também é útil para excluir outros diagnósticos pulmonares alternativos. As técnicas toracoscópicas e pulmonares abertas proporcionam um rendimento maior em casos de vasculite, quando comparadas à biópsia transbrônquica. A histopatologia renal revela a existência de glomerulonefrite necrotizante segmentar com pouca ou nenhuma deposição de imunocomplexos, que pode ser indistinguível da glomerulonefite observada na GW. A presença de p-ANCA antimieloperoxidase no soro (detectada em cerca de 60% dos pacientes com PAM) sustenta o diagnóstico clínico de PAM, mas é inespecífica para esta doença.
Como resultado da eficácia comprovada dos regimes terapêuticos vigentes para GW, uma abordagem de tratamento similar é empregada para a PAM. Embora não tenham sido realizados estudos terapêuticos controlados incluindo apenas pacientes com PAM, os dados retrospectivos existentes mostraram que houve diminuição da mortalidade após a adoção desta abordagem.23 Os pacientes com doença severa inicialmente devem ser tratados com uma combinação de ciclofosfamida e altas doses de corticosteroide. A taxa de recidivas pode ser menor na PAM do que na GW.
Os relatos iniciais da PN e PAM não distinguiam adequadamente estas 2 entidades. De uma forma geral, atualmente se admite que a PN é um distúrbio raro ligado à arterite de artérias musculares de calibre médio e sem envolvimento de pequenos vasos. Um aspecto ainda mais importante reside no fato de os pacientes com hepatite viral B ou C não terem sido excluídos dos estudos mais antigos. O reconhecimento da hepatite viral é decisivamente importante, pois a hepatite B ou C crônica2,24 pode induzir uma síndrome vasculítica secundária indistinguível da PN ou da PAM em termos de manifestação, porém diferente quanto ao prognóstico e resposta à terapia.25
A glomerulonefrite e a hemorragia alveolar estão ausentes, por definição, nos pacientes com PN.
A PN afeta as artérias musculares de calibre médio e, assim como a PAM, está associada à neuropatia periférica e isquemia intestinal.26-28 Os pacientes com PN podem apresentar azotemia e hipertensão em decorrência da arterite das artérias renais e da neuropatia isquêmica, mas não devido à glomerulonefrite. Na PN, a formação de microaneurisma em artérias viscerais de médio calibre pode ser notável, e as artérias ocasionalmente podem se romper.
Os sintomas constitutivos, como febre, astenia e mialgias, são comuns. Níveis altos de reagentes de fase aguda, trombocitose, leucocitose e anemia por doença inflamatória são achados comuns, embora não estejam uniformemente presentes e não permitam distinguir a PN da GW ou da PAM.
Diante da suspeita de síndrome clínica de PN ou PAM, torna-se necessário excluir a hipótese de infecção crônica por bactérias (p. ex., endocardite) e vírus (p. ex., hepatites B ou C). A associação com a infecção pelos vírus das hepatites B ou C pode não alterar dramaticamente alguns aspectos das síndromes de PN e PAM. Entretanto, a glomerulonefrite membranosa, crioglobulinemia, glomerulonefrite associada a imunocomplexos, insuficiência hepática e trombocitopenia tendem mais a ocorrer com a vasculite associada à hepatite viral. Não há deposição significativa de imunocomplexos na PN nem na PAM.
A síndrome do anticorpo antifosfolipídio (SAAF) pode mimetizar a PN, manifestando-se como isquemia mesentérica ou insuficiência renal causada por oclusão de vasos mesentéricos e renais.29 Entre os aspectos da SAAF e da arterite que afetam as artérias musculares, está o livedo reticular [Figura 5]. O desenvolvimento de glomerulonefrite, crioglobulinemia, glomerulonefrite associada a imunocomplexos e neuropatia periférica não é esperado na SAAF, a menos que o paciente também tenha LES. A trombocitopenia pode acompanhar a SAAF, mas geralmente está ausente na PN. A embolia de colesterol também deve ser considerada uma causa de livedo, insuficiência renal, eosinofilia e sintomas constitutivos,30 particularmente quando a história clínica inclui um procedimento vascular recente. A biópsia tecidual frequentemente estabelece o diagnóstico.
Figura 5. O livedo reticular é caracterizado pelo aparecimento de manchas vermelho-azuladas nos membros, produzidas pela obstrução de arteríolas dérmicas profundas.
Na situação ideal, o diagnóstico deve ser estabelecido com base na demonstração histopatológica da arterite e do padrão clínico da doença. Uma amostra de biópsia obtida de um tecido com envolvimento clínico e não necrótico, que demonstre a ocorrência de arterite em artérias musculares, constitui o achado de suporte ideal para estabelecer o diagnóstico de arterite de um vaso de médio calibre. No entanto, a obtenção desta amostra de biópsia nem sempre é possível. Os ANCA não são caracteristicamente detectados na PN.
A PN não causa glomerulonefrite, capilarite pulmonar nem doença parenquimatosa pulmonar.
Pode ser difícil demonstrar a existência de uma arterite afetando as artérias musculares de um paciente com PN, sobretudo se houver sintomas constitutivos dominantes e na ausência de tecido afetado facilmente acessível. As tentativas de obtenção de biópsia devem se concentrar no tecido anormal, conforme indicado pelos sintomas ou exames objetivos. A biópsia do nervo sural tornou-se uma opção popular quando se tenta diagnosticar uma arterite que está afetando vasos musculares de médio calibre. O nervo sural é um nervo sensorial puro acessível, e os vasos sanguíneos que o suprem contêm artérias musculares de pequeno e médio calibre. Os exames de condução nervosa podem identificar um nervo sural isquêmico adoecido antes do aparecimento dos achados clínicos.31 Entretanto, múltiplos relatos enfatizaram o baixo rendimento diagnóstico obtido a partir da biópsia de um nervo assintomático e eletricamente normal. Foi relatado que até mesmo os nervos que exibem anormalidades de condução não apresentam patologia diagnóstica em 46% dos casos.32 Existe uma morbidade notável associada à obtenção da biópsia do nervo sural. De um total de 60 pacientes, houve 13 que apresentaram infecções ou atraso da cicatrização da ferida, enquanto outros 3 pacientes apresentaram dor pós-procedimento no nervo sural de onde fora extraída a biópsia.32 A biópsia de um tecido sem envolvimento clínico (isto é, músculo assintomático) fornece um rendimento inferior a 30% para o diagnóstico de PN.
A angiografia abdominal muitas vezes é conduzida durante a avaliação de pacientes que podem apresentar arterite em vasos de médio calibre, nos casos em que a biópsia não foi esclarecedora ou não constitui uma opção. As artérias afetadas pela PN e outros distúrbios de artérias musculares de calibre médio podem desenvolver microaneurismas ou estreitamentos que podem ser visualizados por angiografia. Quando a angiografia é empregada na tentativa de diagnosticar a vasculite necrotizante sistêmica na ausência de evidências patológicas da doença, existem vários alertas a serem considerados. A angiografia possui resolução espacial limitada, e os vasos menores não são nitidamente visualizados. Em pacientes cuja doença afeta principalmente os vasos menores, é improvável que a angiografia seja diagnóstica. Diferentes pesquisadores relataram a presença de aneurismas em 60 a 90% dos pacientes com PN. Os aneurismas demoram para se desenvolver e podem estar ausentes no início do curso da doença. Além de estar associada aos aneurismas, a arterite pode estar associada a estreitamentos que, por sua vez, podem ser mais longos e regulares do que as típicas lesões ateroscleróticas ou obstrução. Para maximizar o rendimento do procedimento, a angiografia deve incluir os vasos celíaco, renal e mesentérico. A falta de envolvimento clinicamente evidente de um órgão (ou seja, ausência de isquemia intestinal) não exclui a possibilidade de se encontrarem vasos anormais na angiografia. Foi sugerido que a visualização dos aneurismas na PN denota a existência de uma doença mais grave. Não está claro se a presença dos aneurismas pode estar alternativamente relacionada à duração real da doença não tratada. Os aneurismas podem ser resolvidos com o tratamento bem-sucedido da arterite associada à hepatite viral ou primária. A presença de microaneurismas viscerais não é diagnóstica de PN e também foi descrita em relatos pouco confiáveis de casos de pacientes com GW e PAM, provavelmente representando o envolvimento de uma artéria muscular de médio calibre nestas doenças. Os microaneurismas também ocorrem em distúrbios não vasculíticos. Relatos de casos isolados descreveram a presença de aneurismas em pacientes com mixoma atrial, endocardite bacteriana, carcinomatose peritoneal e hipertensão arterial severa, bem como o aparecimento dos aneurismas após o uso abusivo de metanfetamina. Existem dados inadequados disponibilizados para avaliação da sensibilidade e especificidade ou determinação do valor preditivo da angiografia abdominal no diagnóstico da arterite necrotizante. Assim como na interpretação do resultado do exame de uma biópsia de paciente com suspeita de vasculite, os exames de imagem devem ser considerados à luz de todo o perfil clínico. A angiografia geralmente é evitada no contexto de uma insuficiência renal significativa ou progressiva.
O tratamento da PN é empírico33 [Tabela 2]. A administração de doses altas de corticosteroides (prednisona ou equivalente, 1 mg/kg/dia) continua sendo a base da terapia para pacientes com doença aguda. O uso isolado de corticosteroides pode ser suficiente em casos de pacientes com PN sem envolvimento orgânico significativo, em que a doença é definida por insuficiência renal (decorrente de isquemia renal), isquemia gastrintestinal, miocardiopatia ou neuropatia periférica. Os pacientes que necessitam de terapia prolongada com corticosteroides para controlar a doença ou os pacientes com marcadores clínicos de doença severa geralmente são tratados com glicocorticoides e um agente imunossupressor adicional, como a ciclofosfamida ou o metotrexato. As indicações para a terapia combinada inicial ainda não foram devidamente estudadas.
Quando o paciente apresenta hepatite B ou C ativa, a instituição de um curso relativamente curto de esteroides deve ser considerada com base na severidade da doença extra-hepática e nos órgãos que apresentam risco agudo de insuficiência, aliada ao início de uma terapia antiviral agressiva. A redução dos esteroides foi associada à exacerbação em casos de necrose hepática.
A doença de Kawasaki (DK) foi descrita em 1967, como uma síndrome de linfonodos mucocutâneos.34 Esta doença tipicamente afeta bebês e crianças pequenas, produzindo manifestações mucosas orais e cutâneas dominantes, febre e arterite coronariana. Em raros casos, pode afetar indivíduos adultos.
A existência de aspectos clínicos característicos permitiu estabelecer os critérios diagnósticos para a DK [Tabela 4]. A vasculite pode envolver vasos de diversos tamanhos, desde vênulas até a aorta. Observa-se uma inflamação proeminente junto às artérias coronarianas maiores, que resulta na formação de aneurisma em cerca de 25% dos pacientes não tratados. As complicações cardíacas prejudiciais à vida imediatas e tardias, aliadas a uma terapia exclusiva (aspirina e gamaglobulina endovenosa), exigem diagnóstico clínico imediato. A biópsia em geral não apenas é desnecessária como provavelmente fornece um diagnóstico inespecífico.
Tabela 4. Critérios diagnósticos da DK
|
Febre persistente (= 5 dias) acrescida de pelo menos 4 das seguintes condições: |
|
1. Conjuntivite bilateral não purulenta |
|
2. Envolvimento da mucosa oral: faringe eritematosa, lábios fissurados ou avermelhados, ou língua do tipo “morango” |
|
3.Anormalidades de tecido mole nas mãos e pés: eritema, edema ou descamação |
|
4. Erupções não vesiculares, polimórficas |
|
5.Adenopatia cervical |
DK = doença de Kawasaki.
Os picos de febre alta podem persistir durante 1 a 2 semanas, caso não sejam tratados. A defervescência rápida costuma ser observada com o início da terapia adequada. Uma conjuntivite não exsudativa surge frequentemente com a febre. É comum haver meningite asséptica (linfocítica). O envolvimento oral inclui eritema, ressecamento e fissura labial, faringite não exsudativa e eritema da língua com papilas bastante proeminentes. As ulcerações mucosas não são características desta condição. Após a febre, os membros distais podem apresentar inchaço, com eritema e sensibilidade não limitados às articulações. A descamação das mãos e pés, frequentemente em lâminas, pode ter início em poucas semanas após o aparecimento da febre. Quando a descamação ocorre no início da DK, pode aparecer ao mesmo tempo que as erupções no tronco e as alterações oculares e labiais. Este tipo de descamação pode mimetizar uma reação farmacológica ou a síndrome de Stevens-Johnson. A erupção em geral é difusa e polimórfica, com componentes urticariantes, morbiliformes, anulares ou de placa, mas não é vesicular. A adenopatia, que está presente em até 75% dos pacientes, é mais evidente na região cervical.
A morbidade e mortalidade (< 3%) da DK estão notavelmente associadas ao desenvolvimento de aneurismas arteriais coronarianos inflamatórios, que são majoritariamente assintomáticos no momento de sua formação. Os aneurismas podem ser detectados por ecocardiografia. Pode haver trombose dentro dos aneurismas, com consequente obstrução direta ou embólica da artéria coronária. Os eventos coronarianos agudos podem ocorrer em algumas semanas ou após muitos anos da doença febril. Um ecocardiograma basal deve ser obtido durante a doença aguda e repetido depois de 2 e 6 semanas. O reconhecimento e tratamento da doença ainda em fase inicial, com administração de imunoglobulina endovenosa e aspirina, diminuíram significativamente a frequência de formação de aneurismas e eventos coronarianos trombóticos.
O tratamento da DK deve ser iniciado com a administração de imunoglobulina endovenosa (uma dose única de 2 g/kg) e aspirina (80 a 100 mg/kg/dia), assim que a doença for seriamente suspeita.35 A aspirina é mais efetiva do que os corticosteroides para fins de prevenção de aneurismas. Na maioria dos casos, a terapia com corticosteroides é desnecessária, e alguns autores a consideram relativamente contraindicada. Os sintomas tendem a responder dentro de alguns dias após a instituição do tratamento com aspirina e imunoglobulina endovenosa. Contudo, nos casos resistentes, os corticosteroides com frequência são adicionados às terapias mencionadas. As crianças com aneurismas múltiplos ou amplos podem necessitar de anticoagulação por tempo prolongado.
A arterite temporal ou de células gigantes (ACG) do idoso e a arterite de Takayasu (AT) são as doenças inflamatórias mais comuns na aorta e em seus principais ramos. Um direcionamento vascular semelhante pode ocorrer na doença de Behçet, síndrome de Cogan e sarcoidose. Estas 2 últimas condições são distinguidas pelo padrão de envolvimento orgânico extra-aórtico. Ainda não foi esclarecido se a AT e a ACG constituem distúrbios distintos ou são o mesmo distúrbio apresentando expressão modificada em grupos de diferentes faixas etárias.
A ACG geralmente afeta indivíduos com mais de 50 anos de idade.36,37 Em muitos pacientes, está associada à síndrome de polimialgia reumática (SPR), a qual é caracterizada por uma dor muscular proximal, que piora à noite e no início da manhã. Pode haver uma sensação subjetiva de enfraquecimento, sem que nenhum enfraquecimento real seja detectado ao exame e sem elevação dos níveis séricos de enzimas musculares. O paciente pode apresentar uma sinovite que frequentemente dificulta a distinção entre ACG e artrite reumatoide de início na idade avançada.
A ACG está variavelmente associada ao aparecimento de febre, sensibilidade no couro cabeludo, cefaleia, claudicação da musculatura da mastigação, aneurismas aórticos inflamatórios e síndromes isquêmicas retinais. Ocasionalmente, os pacientes podem ter artrite oligoarticular, frequentemente em um membro superior, e a síndrome do túnel do carpo. Os sinais e sintomas isquêmicos podem ser clinicamente indistinguíveis daqueles observados na doença arterioesclerótica obstrutiva ou ateroembólica.
O exame de medidas discrepantes da pulsação nos 4 membros, leituras de pressão arterial, aneurismas abdominais e contusões no pescoço, abdome e membros devem fazer parte das consultas de seguimento de rotina dos pacientes com ACG ou SPR. Os achados patológicos de ACG podem ser encontrados nas artérias temporais superficiais de pacientes com SPR, mesmo na ausência de quaisquer sintomas de ACG. Entretanto, a biópsia de rotina das artérias temporais superficiais de pacientes com SPR e na ausência de quaisquer sintomas ou achados de ACG não é uma medida justificável, porque os pacientes que não apresentam achados físicos nem sintomas sugestivos de doença obstrutiva geralmente não desenvolvem perda visual.
Os níveis de reagentes de fase aguda estão aumentados em mais de 80% dos pacientes com SCG, mas não são totalmente confiáveis como marcadores de atividade da doença durante e após a terapia. Um diagnóstico definitivo de ACG geralmente é estabelecido por exame de biópsia da artéria temporal superficial. Na ACG, é comum a patologia revelar a existência de infiltrados de células mononucleares crônicos, obstrução da lâmina elástica interna e presença de células gigantes. Este último achado não é requisito para estabelecer o diagnóstico. A presença de aspectos clínicos característicos, como uma nova cefaleia e a claudicação mandibular, sobretudo acompanhada de SPR concomitante, pode permitir o estabelecimento de um diagnóstico provável na ausência de biópsia ou até mesmo quando o exame de uma biópsia da artéria temporal superficial resulta negativo. Entretanto, como existem outras condições capazes de mimetizar a SCG, incluindo a aterosclerose e a amiloidose, a tentativa de diagnosticar a ACG por biópsia é considerada justificável na maioria dos casos.38 A terapia à base de corticosteroides não produz efeitos rápidos sobre os resultados da biópsia e não deve ser suspendida em casos de pacientes com forte suspeita de ACG que estejam aguardando a realização da biópsia. O exame de uma biópsia bilateral da artéria temporal superficial parece aumentar o rendimento diagnóstico.
Os diagnósticos diferenciais destas doenças obstrutivas inflamatórias de vasos grandes incluem a aterosclerose obstrutiva, dissecação aórtica e dos ramos aórticos, síndrome do anticorpo antifosfolipídio, displasia fibromuscular, sarcoidose, doença de Buerger e outras síndromes vasculíticas primárias menos comuns (p. ex., doença de Behçet).
A AT (doença sem pulsação) é uma doença inflamatória crônica que afeta a aorta e seus principais ramos.39 Diagnosticada geralmente em pacientes jovens do sexo feminino e em idade reprodutiva, a AT também ocorre em crianças pequenas e pacientes idosos de ambos os sexos. A AT está mais comumente associada ao desenvolvimento de estenoses e aneurismas da aorta e dos vasos dos ramos aórticos, do que à ACG.
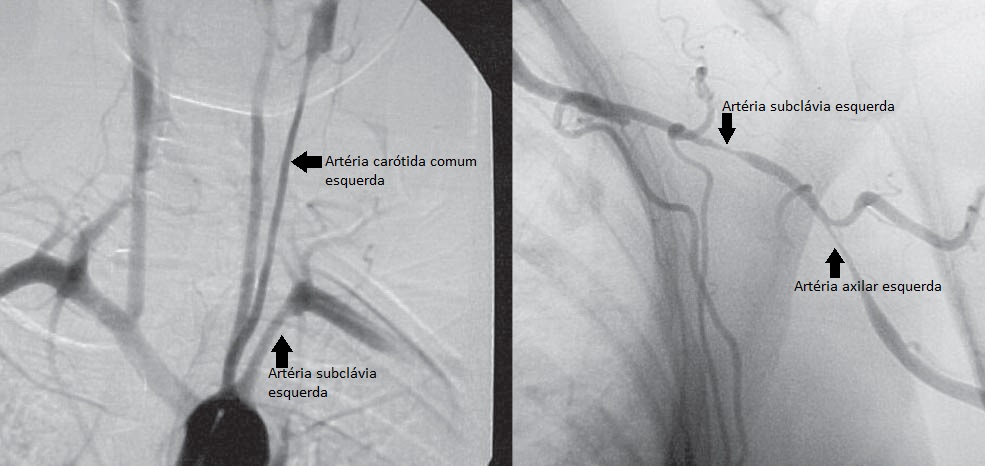
A síndrome clínica manifestada pode incluir uma doença do tipo gripe prolongada, além de um padrão de dor muscular do tipo polimialgia reumática. Muitos pacientes a princípio apresentam sintomas de isquemia cardíaca, cerebral ou nos membros na ausência de quaisquer aspectos constitutivos. Os aspectos característicos da doença refletem a isquemia resultante das estenoses inflamatórias da aorta e seus ramos principais. A isquemia renal pode provocar hipertensão associada a altos níveis de renina. Os sítios predominantes de estenose são os vasos do arco aórtico, em particular as artérias subclávias [Figura 6]. A claudicação do braço com contusões supraclaviculares ou axilares é um achado comum. A dor e sensibilidade junto à artéria superficial (p. ex., carotidinia) podem ser detectadas ao exame, mas não são diagnósticas de AT. A severa hipertensão central produzida pela estenose da artéria renal pode não ser identificada, devido à estenose coexistente da artéria do braço. Desta forma, as leituras da pressão arterial obtidas nos 4 membros devem ser avaliadas inicialmente e monitoradas com frequência. Ocasionalmente, existem estenoses em todos os principais vasos dos membros, e o monitoramento com manguito pode fornecer medidas pouco confiáveis das pressões aórticas centrais. É comum haver acidente vascular cerebral (AVC), muitas vezes relacionado a uma hipertensão central não detectada. É extremamente difícil avaliar a atividade da AT. A presença ou ausência de aspectos constitutivos ou níveis elevados de reagentes de fase aguda são medidas precárias da atividade da doença. Esta impressão é sustentada pelo exame histopatológico vascular realizado durante uma cirurgia de reconstrução. Mais de 40% das amostras vasculares obtidas de pacientes considerados em processo de remissão apresentaram inflamação ativa. Sendo assim, o monitoramento anatômico destes vasos por exame e imagens (imagem de ressonância magnética [IRM] ou angiografia) é obrigatório.
Figura 6. Angiografia de um paciente com arterite de Takayasu (AT) mostrando lesões estenóticas regulares e longas na artéria subclávia esquerda, além do envolvimento de outros ramos dos vasos do arco aórtico.
O diagnóstico de AT geralmente é estabelecido por IRM ou pelas evidências arteriográficas de lesões estenóticas. Os aneurismas são menos comumente observados. Toda a aorta e seus ramos principais devem ser avaliados. A medida rotineira da pressão arterial central no momento da angiografia é essencial, devendo ser comparada às pressões medidas simultaneamente com manguito no braço e na perna. O papel da IRM vascular sequencial e a tomografia por emissão de pósitrons na avaliação e seguimento de pacientes com suspeita de AT atualmente estão sendo investigados. O maior interesse desta investigação é descobrir se as propriedades da imagem da parede vascular podem contribuir para a avaliação da atividade da doença.40 Estas técnicas de imagem podem revelar alterações relacionadas à terapia na espessura da parede do vaso, inflamação, edema e mudanças no diâmetro do lúmen. É difícil obter a documentação patológica na AT, porém a histopatologia da AT, em geral conhecida no momento da cirurgia de revascularização, é semelhante àquela da ACG. A discussão pré-operatória com o cirurgião vascular é obrigatória para garantir a obtenção de amostras adequadas de tecido, quando possível.
Os corticosteroides constituem o tratamento inicial da AT e da ACG. A ACG costuma ser bastante responsiva à terapia com esteroides, embora a dosagem inicial mais apropriada ainda seja controversa. As doses diárias iniciais defendidas são de 20 mg e 1 mg/kg, com redução no decorrer de 8 a 12 meses. A maioria dos autores sente-se confortável em adotar uma dosagem inicial de 40 a 60 mg/dia. Em geral, recomenda-se (sem o suporte de dados fornecidos por estudos controlados) que os paciente com quaisquer sintomas de isquemia ocular sejam inicialmente tratados com altas doses de corticosteroides (pelo menos 1 mg/kg de prednisona ou equivalente, sendo que alguns autores – predominantemente os oftalmologistas – sugerem o uso de metilprednisona em doses de até 1 g/dia durante vários dias). Uma proporção significativa de pacientes com ACG requer vários anos de terapia.
A terapia com altas doses de corticosteroides, especialmente no caso dos idosos, está associada a uma morbidade significativa. A medida dos níveis de reagentes de fase aguda fornece um índice imperfeito de atividade da doença e não deve ser o único fator considerado para guiar o ajuste da dosagem de esteroides. Quando os esteroides produzem efeitos colaterais significativos ou o paciente apresenta recaídas durante a redução, um agente de 2ª linha (p. ex., metotrexato) frequentemente é administrado de modo empírico com a terapia de corticosteroides. Entretanto, o valor dos agentes poupadores de esteroides auxiliares em casos de ACG ainda não está comprovado. Um amplo estudo prospectivo e randomizado falhou em demonstrar um efeito positivo da terapia com metotrexato,41 embora um 2º estudo tenha sugerido um pequeno benefício.42 Em outro estudo recém-concluído, foi demonstrado que a terapia antifator de necrose tumoral não promoveu nenhum benefício adicional à terapia de corticosteroides.43 Foi demonstrado que a adição de aspirina em dose baixa à terapia de corticosteroide diminui a incidência de complicações isquêmicas da arterite temporal.44 Existem apenas relatos pouco confiáveis sobre o sucesso do uso adjunto de azatioprina, metotrexato e micofenolato no tratamento da AT resistente. É preciso atentar especialmente para a prevenção de infecções oportunistas, osteoporose, glaucoma, hiperglicemia e hiperlipidemia.
A cirurgia de reconstrução vascular, angioplastia e colocação de stent são opções terapêuticas auxiliares para alguns pacientes. Entretanto, experiências bastante preliminares sugerem a existência de um alto grau de falha do stent em alguns centros.45 O envolvimento frequente dos vasos subclávios na AT deve ser considerado no momento da escolha do sítio de implante do enxerto para a realização dos procedimentos de desvio coronariano ou carotídeo. A obtenção de enxertos a partir destes vasos deve ser evitada.
A doença de Behçet é uma doença multissistêmica recidivante que tipicamente se manifesta como lesões genitais e orais recorrentes, além de diferentes combinações de várias manifestações clínicas. A histopatologia é caracterizada por uma vasculite que pode afetar artérias ou veias de qualquer tamanho.
A doença de Behçet pode afetar vários sistemas orgânicos: pele, mucosas oral e genital, ocular, musculoesquelético, sistema nervoso central, gastrintestinal e pulmonar. A manifestação clínica mais frequente são as ulcerações orais dolorosas recorrentes, que ocorrem em quase todos os pacientes. As lesões genitais dolorosas estão presentes em pelo menos 75% dos pacientes e, diferente das úlceras, geralmente estão associadas à cicatrização. A doença ocular mais típica é a panuveíte, cujo tratamento pode ser difícil e que pode estar associada a uma inflamação persistente entre os episódios de exacerbação aguda. A lesão cutânea mais frequentemente observada é uma lesão acneiforme com pápulas e pústulas, que pode ser indistinguível da acne comum. Outras lesões incluem o eritema nodoso, nódulos e tromboflebite superficial. As manifestações neurológicas não são comuns e em geral envolvem o sistema nervoso central com lesões cerebrais, meningite asséptica e trombose arterial ou venosa. O envolvimento de artérias de grande calibre pode resultar na formação de aneurisma.
O diagnóstico é baseado nas manifestações clínicas, e não existem exames laboratoriais específicos. Estabelecer o diagnóstico pode ser uma tarefa bastante desafiadora no caso de pacientes em que a expressão clínica da doença não é integral. Embora o teste de patergia possa resultar positivo em casos de pacientes com doença de Behçet, é necessário usar técnica padronizada para evitar a geração de resultados falso-negativos. A patologia pode revelar a existência de vasculite, que é considerada a principal lesão associada à maioria dos aspectos clínicos. Embora a trombose em veia profunda seja observada em até 25% dos pacientes, acredita-se que a tromboembolia pulmonar seja rara na doença de Behçet. A trombose pode envolver veias intra-abdominais de grande calibre. O tratamento dos eventos trombóticos apresentados por estes pacientes é controversa, sendo adotada uma combinação de imunossupressão e anticoagulação.46
O tratamento é ditado pelo envolvimento do sistema orgânico e pela severidade das manifestações clínicas. A doença severa pode requerer o uso de altas doses de glicocorticoides e ciclofosfamida. A azatioprina apresenta certo grau de eficácia no controle da doença ocular inflamatória. Apesar da falta de estudos controlados, o infliximabe tem sido usado com algum sucesso em casos de pacientes com doença ocular severa.47 A colchicina e a pentoxifilina podem promover algum benefício em casos de pacientes com doença ulcerosa mucosa. O interferon e a talidomida apresentam maior eficácia no tratamento da doença ulcerosa mucosa, porém apresentam maior grau de toxicidade associada ao tratamento.
Alexandra Villa-Forte, MD, MPH, não possui relações comerciais com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
Brian F. Mandell, MD, PhD, FACP, recebeu apoio financeiro para fins educacionais da Abbott Laboratories, Aventis Pharmaceuticals, Inc. e da TAP Pharmaceutical Products, Inc. O autor também recebeu apoio financeiro para fins educativos e atuou como consultor ou conselheiro junto às empresas TAP, Sanofi -Aventis e Savient Pharmaceuticals, Inc.
Os fármacos citoxana, corticosteroides, metotrexato, azatioprina, pentoxifilina, colchicina, aspirina e dapsona não foram aprovados pelo Food and Drug Administration para os usos descritos neste capítulo.
1. Jennette C, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994;37:187.
2. Vassilopoulos D, Calabrese L. Hepatitis C virus infection and vasculitis. Arthritis Rheum 2002;46:585.
3. Lawrence EC, Mills J. Bacterial endocarditis mimicking vasculitis with steroid-induced remission. West J Med 1976;124:333.
4. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Cutaneous vasculitis in children and adults: associated disease and etiologic factors in 303 patients. Medicine (Baltimore) 1998;77:403.
5. Gedalia A. Henoch-Schonlein purpura. Curr Rheumatol Rep 2004;6:195.
6. Davis MD, Brewer JD. Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am 2004;24:183.
7. Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488.
8. Rheinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long term outcome in 155 patients. Arthritis Rheum 2000;43:1021.
9. Travis WD, Hoffman GS, Leavitt RY, et al. Surgical pathology of the lung in Wegener’s granulomatosis. Am J Surg 1991;15:315.
10. Merkel PA, Lo GH, Holbrook JT, et al. Brief communication: high incidence of venous thrombotic events among patients with Wegener’s granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis Study. Ann Intern Med 2005;142:620.
11. Hoffman GS. Classifi cation of the systemic vasculitides: antineutrophil cytoplasmic antibodies, consensus and controversy. Clin Exp Rheumatol 1998;16:111.
12. Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies: diagnostic value in systemic vasculitis. Arthritis Rheum 1998;41:1521.
13. Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007;147:611–9.
14. Specks U. Methotrexate for Wegener’s granulomatosis: what is the evidence? Arthritis Rheum 2005;52:2237.
15. Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349:36.
16. Langford CA, Use of a cyclophosphamide-induction methotrexate-maintenance regimen for the treatment of Wegener’s granulomatosis: extended follow-up and rate of relapse. Am J Med 2003;114:463–9.
17. De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52:2461–9.
18. Stegeman CA, Tervaert JW, de Jong PE, et al. Trimethoprimsulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N Engl J Med 1996;335:16.
19. The WGET Research Group. Etanercept in addition to standard therapy in patients with Wegener’s granulomatosis. N Engl J Med 2005;352:351–6.
20. Keogh KA, Ytterberg SR, Fervenza FC, et al. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial. Am J Respir Crit Care Med 2006;173:180–7.
21. Guillevin L, Cohen P, Gayraud M, et al. Churg-Strauss syndrome: clinical study and long-term follow up of 96 patients. Medicine (Baltimore) 1999;78:26.
22. Reid AC, Harrison BW, Watts RA, et al. Churg-Strauss syndrome in a district hospital. Q J Med 1998;91:219.
23. Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory fi ndings in 85 patients. Arthritis Rheum 1999;42:421.
24. Hadziyannis SJ. The spectrum of extrahepatic manifestations in hepatitis C virus infection. J Viral Hepat 1997;4:9.
25. Guillevin L, Lhote F, Cohen P, et al. Polyarteritis nodosa related to hepatitis B virus: a prospective study with long-term observation of 41 patients. Medicine (Baltimore) 1995;74:238.
26. Travers RL, Allison DJ, Brettle RP, et al. Polyarteritis nodosa: a clinical and angiographic analysis of 17 cases. Semin Arthritis Rheum 1979;8:184.
27. Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. Medicine (Baltimore) 1996;75:17.
28. Mandell BF, Hoffman GS. Differentiating the vasculitides. Rheum Dis Clin North Am 1994;20:409.
29. Franchini M, Veneri D. The antiphospholipid syndrome. Hematology 2005;10:265.
30. Om A, Ellahham S, DiScascio G. Cholesterol embolism: an underdiagnosed clinical entity. Am Heart J 1992;124:1321.
31. Wees SJ, Sunwoo IN, Oh SJ. Sural nerve biopsy in systemic necrotizing vasculitis. Am J Med 1981;71:525.
32. Rappaport WD, Valente J, Hunter GC, et al. Clinical utilization and complications of sural nerve biopsy. Am J Surg 1993;166:252.
33. Guillevin L, Lhote F. Treatment of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1998;41:2100.
34. Yeung RS. Pathogenesis and treatment of Kawasaki’s disease. Curr Opin Rheumatol 2005;17:617.
35. Leung DY, Schlievert PM, Meissner HC. The immunopathogenesis and management of Kawasaki syndrome. Arthritis Rheum 1998;41:1538.
36. Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med 2003;139:505.
37. Hoffman GS. Treatment of giant cell arteritis: where we have been and why we must move on (review). Cleve Clin J Med 69 Suppl II:SII117, 2002.
38. Ponge T, Barrier JH, Grolleau JY, et al. The effi cacy of selective unilateral temporal artery biopsy versus bilateral biopsies for diagnosis of giant cell arteritis. J Rheumatol 1988;15:997.
39. Kerr GS, Hallahan CW, Giordano J, et al. Takayasu’s arteritis. Ann Intern Med 1994;120:919.
40. Tso E, Flamm SD, White RD, et al. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum 2002;46:1634.
41. Hoffman GS, Cid MC, Hellmann DB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002;46:1309.
42. Jover JA, Hernandez-Garcia C, Morado IC, et al. Combined treatment of giant-cell arteritis with methotrexate and prednisone, a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001;134:106.
43. Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infl iximabGCA Study Group: Infl iximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007;146:621–30.
44. Nesher G, Berkun Y, Mates M, et al. Risk factors for cranial ischemic complications in giant cell arteritis. Medicine (Baltimore) 2004;83:114.
45. Liang P, Tan-Ong M, Hoffman GS. Takayasu’s arteritis: vascular interventions and outcomes. J Rheumatol 2004;31: 102.
46. Calamia KT, Schirmer M, Melikoglu M, et al. Major vessel involvement in Behcet disease. Curr Opin Rheumatol 2005; 17:1–8.
47. Lindstedt EW, Baarsma GS, Kuijpers RW, et al. Anti-TNFalpha therapy for sight threatening uveitis. Br J Ophthalmol 2005;89:533–6.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.