(Carregando Índice)... (Carregando Índice)... |
Última revisão: 08/05/2017
Comentários de assinantes: 0
|
Artigo original: Schwartz, BL. MD, PhD. Hajjar, J. MD. Allergic Response, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Joud Hajjar, MD
Membro da Equipe de Alergia e Imunologia do Departamento de Medicina Interna da Virginia Commonwealth University (Richmond, VA).
Lawrence B. Schwartz, MD, PhD
Professor de Medicina da Charles & Evelyn Thomas do Departamento de Medicina Interna da Virginia Commonwealth University (Richmond, VA).
Este artigo inicia com uma definição de resposta alérgica e com uma discussão sobre epidemiologia de distúrbios atópicos, bem como sobre o desenvolvimento de hipersensibilidade mediada pela imunoglobulina E (IgE). Os temas abordados na seção sobre mecanismos humorais e celulares das sensibilizações alérgicas incluem células que apresentam antígenos e sensibilização, células T, IgE e receptores de IgE. As discussões sobre mastócitos e basófilos incluem informações sobre mediadores, biomarcadores, eosinófilos e mediadores de eosinófilos. Há também alguns comentários sobre terapia de distúrbios atópicos. As figuras descrevem mecanismos inflamatórios nos casos de inflamações alérgicas, microscopia de um mastócito antes e depois da introdução de um antígeno, e mediadores liberados por mastócitos humanos ativados. Os quadros apresentam descrições de citocinas e quimiocinas selecionadas envolvidas nas inflamações alérgicas mediadas por IgE, níveis séricos de triptase, exemplos de padrões de elevações e interpretações nos níveis séricos de triptase e um resumo das intervenções terapêuticas para tratamento de doenças alérgicas.
ALERGIA & IMUNOLOGIA
Definição de resposta alérgica
O termo “hipersensibilidade” se refere à doença causada por uma resposta imune, independentemente se a resposta é contra patógenos, não patógenos ou individuais, e se é direcionada por anticorpos, linfócitos ou por rotas inatas. O termo “anafilaxia” foi criado em 1902 por Charles Richet, que recebeu o Prêmio Nobel de Medicina em 1913.
Atualmente, esse tipo de resposta alérgica é considerada uma reação imediata de hipersensibilidade deflagrada por alérgenos liberados para hospedeiros com IgE específico para esse tipo de substância de origem natural, produzindo, consequentemente, respostas imunológicas mediadas pela IgE e ativando mastócitos e basófilos para a secreção de mediadores bioativos.
Em 2005, o National Institutes of Health organizou uma conferência de consenso para desenvolver uma definição de trabalho para anafilaxia que pudesse ser usada por médicos ao lado do leito como uma reação alérgica séria cujo início é muito rápido, em geral desencadeando várias combinações de manifestações cutâneas, cardiovasculares, respiratórias e gastrintestinais, podendo até causar a morte.1,2
Esse consenso facilitou o tratamento imediato desses pacientes com epinefrina. Há uma grande confusão sobre a aplicação incorreta do termo alergia ou hipersensibilidade para descrever qualquer reação imprevista aos alimentos, aos medicamentos ou às exposições ambientais. Além disso, podem até ocorrer formas não mediadas pela IgE e pela ativação local e sistêmica de mastócitos ou basófilos, produzindo sinais e sintomas que se assemelham às formas mediadas pela IgE.
Nos casos de situações não atópicas, a exposição de indivíduos desinformados sob o ponto de vista imunológico a um alérgeno resulta na tolerância imunológica ou na persistência da ignorância imunológica, embora, em indivíduos atópicos, a exposição resulte em sensibilização.
Novas exposições de indivíduos sensibilizados aos alérgenos produzem respostas mediadas por meios imunológicos que afetam os órgãos-alvo expostos a esse alérgeno e, a seguir, os mediadores gerados durante a resposta alérgica. Este artigo tem como foco abordar a hipersensibilidade mediada pela IgE.
Epidemiologia dos distúrbios atópicos
As doenças alérgicas são muito comuns. As descobertas feitas por um estudo de 2 anos envolvendo 38.480 crianças (desde a primeira infância até os 18 anos de idade) revelaram que 8% tinham alergia a alimentos.3 Em 2009, entre crianças norte-americanas com idades iguais ou inferiores a 17 anos, 13% sofriam de alergias na pele e 10% de rinoconjuntivite alérgica.4
De acordo com o Centers for Disease Control and Prevention, 14% (aproximadamente, 10 milhões) de crianças norte-americanas com idades iguais ou inferiores a 17 anos foram diagnosticadas com asma e 10% (7 milhões) apresentavam asma persistente. Em termos globais, nos dias atuais, as taxas de sensibilização aplicáveis a um ou mais alérgenos comuns entre crianças em idade escolar se aproximam de 40 a 50%.5
Além do mais, um estudo recente conhecido por Anaphylaxis in America constatou que a prevalência de anafilaxia ao longo da vida é de, pelo menos, 1,8%, ou seja, é muito mais comum do que se reconhecia anteriormente.6
|
*Os autores e os editores agradecem a contribuição de Gregorio Gomez, PhD, autor da edição anterior deste artigo, pelo desenvolvimento e pela redação deste texto. |
Desenvolvimento de hipersensibilidade mediada pela IgE
Fatores de risco genéticos e ambientais para atopia
A “hipótese higiênica” foi proposta no final da década de 1980 e sugeria que a redução na exposição aos micróbios e aos produtos microbianos, assim como a redução na incidência e no tempo de duração das infecções durante o desenvolvimento intrauterino e neonatal, se relaciona ao aumento na incidência e na prevalência de doenças atópicas nos países em desenvolvimento.7
Por exemplo, uma das propostas apresentadas é que o consumo de leite não pasteurizado poderia diminuir o risco de incidência de alergias, possivelmente devido à associação com alterações na expressão genética dos receptores inatos de imunidade nos estágios iniciais da vida.8 Entretanto, chegou-se à conclusão de que o suporte direto à hipótese higiênica é um equívoco.
Embora os fatos indiquem que a exposição aos animais nas fazendas proteja contra asma, as exposições nos bairros urbanos mais pobres tendem a aumentar a prevalência da doença, sendo que, nos países da América Latina, onde as taxas de infecção são elevadas, as taxas de prevalência de asma também são elevadas. Para finalizar, embora a hipótese higiênica se relacione sobretudo às exposições na fase inicial da vida, as exposições ao longo da vida também são determinantes.9
As infecções causadas pelo rinovírus no início da vida, em especial nos casos de histórico de asma materna, estão associadas a uma elevação no risco do desenvolvimento de asma.10-12 A redução na exposição aos raios solares, resultando na diminuição nos níveis de vitamina D, é outro fator de risco potencial para o desenvolvimento de doenças atópicas. A vitamina D afeta a imunidade celular,13 sendo que níveis séricos mais baixos de vitamina D foram associados a uma função pulmonar mais baixa e ao controle precário da asma.14
Entretanto, ainda não se demonstrou se a suplementação de vitamina D melhora a responsividade aos corticosteroides em pacientes asmáticos.15 O tabagismo materno e o consumo de acetaminofeno durante a fase inicial da vida17 são possíveis fatores de risco adicionais para o desenvolvimento de asma em lactentes. São necessários estudos adicionais antes de fazer recomendações finais para a população em geral.
Os antecedentes genéticos desempenham um papel importante na suscetibilidade para desenvolver doenças atópicas, em alguns casos pela influência na produção de IgE. Por exemplo, as mutações genéticas na interleucina-13 (IL-13), IL-4 e no receptor da interleucina-4 (IL-4R) estão associadas à asma e ao nível sérico total de IgE.18
Outras mutações genéticas são específicas de órgãos. A filagrina, por exemplo, é uma proteína produzida por queratinócitos que agrega filamentos de queratina em citoesqueletos, facilitando o processo de cornificação. A perda de filagrina nas mutações funcionais danifica a barreira cutânea e, por conseguinte, aumenta a perda hídrica, prejudica a defesa inata na superfície cutânea, facilita a sensibilização alérgica e aumenta o risco de dermatite atópica de início precoce, a sensibilização aos alimentos e à asma.19
Mecanismos humorais e celulares de sensibilização alérgica
Células que apresentam antígenos e sensibilização
A grande parte dos alérgenos é formada por proteínas encontradas em grãos de pólen, ácaros da poeira doméstica, veneno de insetos, alimentos e fungos, embora não se saiba exatamente o que torna essas proteínas alergênicas. É provável que algumas delas sejam alergênicas por causa de sua atividade proteolítica, e outras, por causa de sua capacidade adjuvante inerente.
Algum carboidrato estranho também pode ser um alérgeno. Por exemplo, a galactose-a-1,3-galactose é expressa em não primatas, mas não em seres humanos. A IgE humana produzida contra esse oligossacarídeo animal foi associada a duas formas distintas de anafilaxia: anafilaxia de início imediato na primeira exposição parenteral a um agente biológico20 e anafilaxia de início tardio que começa dentro de 3 a 6 horas após a ingestão de carne animal (por exemplo, carnes bovina e suína).21
Por exemplo, no tratamento com cetuximabe, e possivelmente com outros produtos recombinantes expressos nas linhas de células murinas, mesmo que a sequência de aminoácidos primários seja “humanizada” e não imunogênica, os grupos de açúcares são adicionados de acordo com o repertório de enzimas das espécies que produzem a célula utilizada para expressar a proteína recombinante. As células animais poderão adicionar metades de carboidratos que não sejam produzidas por seres humanos.
Os alérgenos sofrem endocitose por células que apresentam antígenos (em inglês, antigen-presenting cells [APCs]) profissionais, incluindo células dendríticas, macrófagos e células B, sendo que todas elas expressam o complexo de histocompatibilidade principal (em inglês, major histocompatibility complex [MHC]) II de forma constitutiva.22 Entretanto, apenas as células dendríticas e os macrófagos são capazes de incubar células T virgens e, por conseguinte, são responsáveis pela fase inicial de sensibilização.
As células em que não houver expressão das moléculas do MHC II, a menos que sejam induzidas a expressá-las, incluindo monócitos e células epiteliais, amplificam as respostas secundárias ou amnésicas. As células B ligam-se aos alérgenos e os absorvem através de imunoglobulinas alergênicas específicas que se localizam em sua superfície, ao contrário da absorção endocítica inespecífica usada por outras células apresentadoras de antígenos.
A internalização antigênica resulta na ativação da célula apresentadora de antígenos (suprarregulação do MHC II e das moléculas acessórias) e na fusão de endossomas por lisossomas para formar vesículas especializadas no processamento de antígenos, onde são hidrolisadas em peptídeos. Esses peptídeos lineares se incorporam no sulco de ligação antigênica das moléculas do MHC II durante o transporte para a superfície celular.
Células T
A resposta celular das células T auxiliares (Ths) (da Th0 virgem à Th1 versus Th2 versus Th7) é influenciada por uma grande variedade de fatores, incluindo o tipo e a quantidade de alérgenos, a duração e a via de exposição, e o tipo de citocina produzido durante a diferenciação linfocítica. Por exemplo, as sequências bacterianas de DNA possuem regiões imunoestimuladoras contendo repetições da desoxicitidina-monofosfato-desoxiguanosina (CpG) desmetilada.
As repetições de CpG são reconhecidas como estranhas por um receptor de reconhecimento padrão denominado receptor toll-like (TLR-9). Tipicamente, a estimulação de macrófagos e de células dendríticas por essas repetições de CpG contendo DNA ocorre nas situações em que o oligonucleotídeo se liga ao TLR-9, induzindo a secreção de citocinas Th1, como a IL-12 e IL-18, que induzem a produção de interferon ? (IFN-?) pelas células Th1, que estimulam a mudança de isótipos de células B para IgG1 e IgG4, ao invés de IgE.23
O desenvolvimento da célula Th17 ocorre sob a influência do fator de crescimento transformador ß (em inglês, transformating growth factor-ß [TGF-ß]) e da IL-23.24 As células Th17 são importantes na proteção contra infecções bacterianas e fúngicas e no desenvolvimento de autoimunidade.25
O Quadro 1 apresenta as citocinas e as quimiocinas selecionadas envolvidas em inflamações alérgicas mediadas pela IgE.
Quadro 1
|
CITOCINAS E QUIMIOCINAS SELECIONADAS ENVOLVIDAS EM INFLAMAÇÕES ALÉRGICAS MEDIADAS PELA IgE | ||
|
Citocina |
Fonte |
Função
|
|
IL-4 |
Células Th2, basófilos. |
Diferenciação de células Th0 em Th2 e mudança do isótipo de IgE das células B.
|
|
IL-5 |
Células Th2, mastócitos, eosinófilos. |
Desenvolvimento, ativação, sobrevivência e emergência de eosinófilos a partir da medula óssea.
|
|
IL-13 |
Células Th2, mastócitos. |
Mudança de isótipo da IgE das células B, ativação de eosinófilos, inflamação tecidual.
|
|
GM-CSF |
Células Th2, mastócitos, células epiteliais, macrófagos, células endoteliais.
|
Maturação de granulócitos e macrófagos, ativação e sobrevivência de eosinófilos. |
|
TNF-a |
Monócitos/macrófagos, mastócitos. |
Quimiotaxia e ativação de leucócitos e do endotélio vascular.
|
|
IL-9, -19, -25, -33, TSLP |
Vários tipos de células. |
Desenvolvimento e sobrevivência de células Th-2, aumentando indiretamente a mudança de classe de IgE e o recrutamento de eosinófilos.
|
|
FCT |
Células estromais, células endoteliais, fibroblastos, células epiteliais. |
Desenvolvimento e sobrevivência de mastócitos, melanócitos e das células intersticiais de Cajal; menos importante para outras linhagens hematopoiéticas.
|
|
Eotaxinas 1-3, Rantes |
Células inflamatórias e outros tipos de células. |
Recrutamento e impressão/ativação de eosinófilos e basófilos.
|
|
FCT: fator da célula-tronco; GM-CSF: fator estimulador das colônias de granulócitos e macrófagos; IL: interleucina; Rantes: célula T expressa e secretada normalmente, regulada pela ativação; Th: auxiliar T; TNF: fator de necrose tumoral; TSLP: linfopoietina estromal tímica. |
Por outro lado, a resposta alérgica de Th2 está associada com a produção de IL-4, IL-5 e IL-13, que favorece o envolvimento da IgE e de eosinófilos. As IL-4 e IL-13 compartilham inúmeras funções porque ambas emitem sinais através dos heterodímeros IL-4Ra/IL-13Ra1, enquanto que apenas a IL-4 emite sinais através do heterodímero IL-4Ra/?C.
Entretanto, apenas a IL-4, produzida em abundância por basófilos ativados, é capaz de induzir a diferenciação de células Th0 em células Th2 e de antagonizar a diferenciação de células Th0 em células Th1 ou em células Th17, resultando em inflamações alérgicas mediadas pela IgE.
Indivíduos atópicos que apresentarem fenótipo assimétrico de Th2 devem ter um ambiente permissivo de Th2 durante a sensibilização, incluindo a presença de IL-4. As células T reguladoras (Tregs) se opõem ao desenvolvimento de distúrbios autoimunes e inflamatórios. As Tregs CD4+ expressam níveis elevados de CD25 e do fator de transcrição regulador mestre, Foxp3, e se desenvolvem sob a influência sem oposição do TGF-ß.25
Ige E Receptores Da Ige
Logo após o processamento de alérgenos pelas células que apresentam antígenos e de sua apresentação às células Th2, as células T podem educar as células B a mudarem da produção de IgM ou de IgC para a produção de IgE, sem alterar a especificidade antigênica.
Essa mudança de isótipo pelas células B exige a presença de dois sinais.26O primeiro é disparado pela IL-4 ou pela IL-13, iniciando a transcrição a partir do sítio do promotor da linha germinativa da porção constante da cadeia pesada de IgE. O segundo é liberado por CD154 (CD40L), que é o ligante para o receptor da classificação determinante do antígeno 40 (CD40) nas células B, ativando a recombinase de DNA.
Esses dois sinais levam a uma reorganização dos genes da cadeia pesada, por meio da deleção das regiões constantes da cadeia pesada de IgC e IgM, deixando o gene constante da cadeia pesada da IgE com capacidade para transcrever juntamente com os genes que codificam a porção variável da ligação antigênica da molécula de imunoglobulina.
A estimulação das células B através de CD40 também estimula o crescimento, a diferenciação e a sobrevivência. A CD154 não se expressa nas células T, embora também seja induzível em mastócitos e basófilos.27 É importante ressaltar que essas células segregam IL-13 e IL-4, respectivamente; portanto, podem promover a produção de IgE.
Embora as células Th2 sejam as principais responsáveis pelo início da mudança para IgF específica de antígenos, outras células que produzem IL-4 e IL-13, incluindo mastócitos, basófilos e células T exterminadoras naturais, podem contribuir para o processo durante a resposta primária, antes que as células Th tenham sido treinadas e durante a resposta secundária em sítios periféricos de produção de IgE e de inflamação alérgica, tais como olhos, nariz, pulmões pele e esôfago.
Os níveis totais de IgE variam de 1 a 100IU/mL (2,4 a 240ng/mL), que são bem inferiores aos níveis totais de IgM, IgA e IgC. A vida da IgE também é mais curta na circulação, com meia-vida de 2 a 3 dias, cerca de 10 vezes menos que a vida da IgC. A detecção de níveis antigênicos específicos de IgE, por meio de testes cutâneos de alergia ou de testes séricos in vitro, é uma parte importante do exame completo para verificar a presença de sinais e sintomas relacionados à alergia.
A IgE e seus receptores são partes fundamentais das RESPOSTAS ALÉRGICAS.28 O FCeRI, receptor da IgE de alta afinidade, se expressa de duas formas: um tetrâmero (aß?2), de forma constitutiva nos mastócitos e nos basófilos, e um trímero (a?2) que é induzível em outros tipos de células, incluindo monócitos, eosinófilos e plaquetas.2,29
A cadeia de FCeRIa liga-se à IgE; as cadeias de FCeRia?, com ligação covalente aos elos de dissulfeto, transmitem sinais intracelulares depois da ligação cruzada de FCeRI, e a cadeia de FCeRiß age como uma amplificadora de sinais. É interessante observar que a própria IgE intensifica a expressão de FCeRI nas superfícies de basófilos e de mastócitos, aumentando, consequentemente, a possibilidade de ativação dos mastócitos e dos basófilos contra os alérgenos.30,31
O FCeRII (CD23), um receptor de IgE de baixa afinidade, se expressa nas células B e T, nos monócitos, nos eosinófilos e nas plaquetas.32 O FCeRII possui os principais grupos lipídicos de lecitina tipo C e pedículos helicoidais-a, formando aparadores de forma espontânea, que se soltam da superfície celular pela protease Adam-10.33 A avidez resultante da ligação cooperativa da IgE dos principais grupos lipídicos individuais viabiliza a ligação da IgE livre.
A intensificação na expressão superficial de CD23, pela elevação dos níveis de IgE livre ou pela IL-4, atenua a produção de IgE pelas células B.28,32 A forma expelida ou solúvel de CD23 (sCD23) diminui os níveis de IgE livre, reduzindo, portanto, os níveis do complexo IgE-CD23 na superfície celular e trocando a classe mediada por FCeRII. Ainda resta observar se a modulação farmacológica de FCeRII consegue regular a produção de IgE nos pacientes.
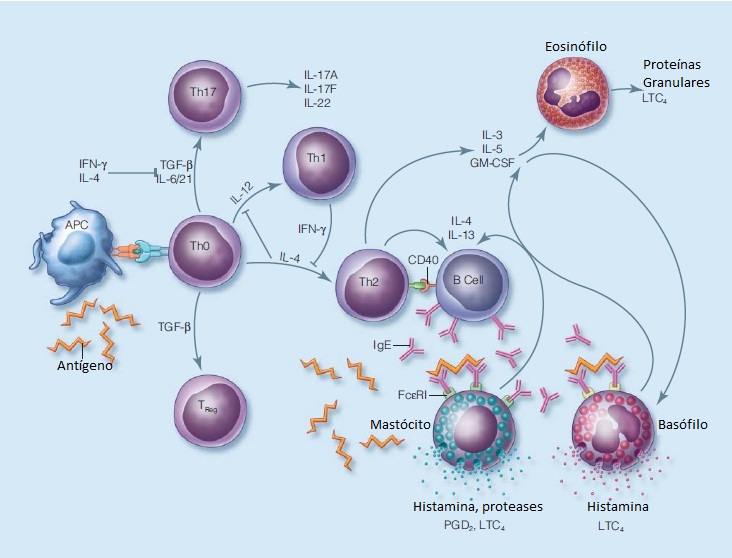
A Figura 1 mostra as rotas inflamatórias em inflamação alérgica.
APCs: células apresentadoras de antígenos; CD40: classificação determinante do antígeno 40; FAP: fator de ativação plaquetária; FCeRIs: receptores de alta afinidade; GM-CSF: fator estimulador das colônias de granulócitos e macrófagos; IFN-?: interferon ?; IL: interleucina; LTC4: leucotrieno C4; PGD2: prostaglandina D2; TGF-ß: fator de crescimento transformador ß; Th: célula T auxiliar; Treg: célula T reguladora.

Figura 1 - Rotas inflamatórias em inflamação alérgica. O antígeno é absorvido pelas APCs e, após o seu processamento, é apresentado para as células Th CD4+. As APCs e as citocinas orientam a resposta da célula Th: (IL-12) a Th1, IL-4 a Th2, TGF-ß, juntamente com a IL-6 em camundongos ou com a IL-21 em seres humanos para Th17, e TGF-ß para as células T reguladoras. A produção de IFN-? durante a resposta de Th1 suprime uma resposta de Th2, enquanto que a produção de IL-4 pelas células Th2 suprime uma resposta de Th1. A IL-4 e o IFN-? suprimem a formação de células Th-17. As IL-3 e IL-4 também estimulam as células B a mudarem a produção de IgM ou IgC para a produção de IgE. A sinalização através da CD40 na célula B também é imprescindível para a mudança de classe da IgE. Outras citocinas de Th2, como a IL-3, IL-4 e o GM-CSF, resultam no desenvolvimento, na sobrevivência e na ativação de eosinófilos e basófilos. A IgE liga-se aos FCeRIs nos mastócitos e nos basófilos; fazendo ligação cruzada por meio de alérgenos multivalentes, a IgE inicia a liberação do mediador.
DÍSTICOS DA FIGURA 1
|
Th17 |
= |
Th17
|
|
IL-17A IL-17F IL-22 |
= |
IL-17A IL-17F IL-22
|
|
Eosinophil |
= |
Eosinófilo
|
|
Granule proteins LTC4 |
= |
Proteínas granuladas LTC4
|
|
IFN-? IL-4 |
= |
IFN-? IL-4
|
|
TGF-ß IL-6/21 |
= |
TGF-ß IL-6/21
|
|
IL-12 |
= |
IL-12
|
|
Th1 |
= |
Th1
|
|
IL-3 IL-5 GM-CSF |
= |
IL-3 IL-5 GM-CSF
|
|
APC |
= |
APC
|
|
Th0 |
= |
Th0
|
|
IFN-? IL-4 |
= |
IFN-? IL-4
|
|
Th2 |
= |
Th2
|
|
CD40 |
= |
CD40
|
|
IL-4 IL-13 |
= |
IL-4 IL-13
|
|
B cell |
= |
Célula B
|
|
TGF-ß |
= |
TGF-ß
|
|
Antigen |
= |
Antígeno
|
|
IgE |
= |
IgE
|
|
FCeRI |
= |
FCeRI
|
|
TReg |
= |
TReg
|
|
Mast cell |
= |
Mastócito
|
|
Basophil |
= |
Basófilo |
|
Histamines, proteases PGD2, LTC4 |
= |
Histaminas, proteases PGD2, LTC4
|
|
Histamine LTC4 |
= |
Histamina LTC4
|
Nas situações em que pessoas sensibilizadas se expõem a algum tipo de alérgeno, o alérgeno se liga ao IgE inserido no FCeRI nos mastócitos e nos basófilos. Se for multivalente, o alérgeno se agregará aos complexos IgE: FCeRI na superfície celular, estimulando a ativação das células, a secreção de mediadores e a produção de sintomas típicos de RESPOSTAS ALÉRGICAS na fase inicial.
O medicamento omalizumabe, um anticorpo monoclonal de IgG humanizado recombinante, direcionado contra a porção Fc da IgE que se liga ao FCeRI, foi aprovado pela Food and Drug Administration para uso no tratamento de asma atópica mau controlada.34 O omalizumabe não consegue ligar-se à IgE que já tenha sido ligada a um FCeRI e, por conseguinte, não ativa nem mastócitos nem basófilos.
Além do mais, o omalizumabe não ativa complementos e tem meia-vida mais longa do que a IgE. O tratamento in vivo com omalizumabe diminui os níveis de IgE livre (porém, não o nível total de IgE) em circulação e infrarregula os níveis de FCeRI nos basófilos35 e mastócitos.30 Estudos in vitro realizados com mastócitos humanos demonstram que o omalizumabe reverte a intensificação da expressão superficial de FCeRI mediada pela IgE e o aumento na sensibilidade à resposta à estimulação de FCeRI.30
Além disso, alguns estudos clínicos confirmaram a eficácia do omalizumabe no tratamento de condições como asma,36 urticária crônica,37,38 aspergilose broncopulmonar alérgica (em inglês, allergic bronchopulmonary aspergillosis [ABPA]),39 e alergia a alimentos.40
Há algumas reações de hipersensibilidade que não são mediadas pela IgE, as quais ocorrem pela ativação direta de mastócitos e/ou basófilos. Por exemplo, a doença respiratória exacerbada por ácido acetilsalicílico (Drea) é desencadeada pelo ácido acetilsalicílico e pelos anti-inflamatórios não esteroides (Aines) inibidores da cicloxigenase-1 (COX-1) que ativam os mastócitos e outros tipos de células em, aproximadamente, 5% de indivíduos asmáticos, sobretudo aqueles com pólipos nasais e com sinusite recorrente (tríade de Samter).
Os pacientes com Drea são tolerantes aos inibidores seletivos da COX-2.41 Embora não se conheça o mecanismo da Drea, tudo indica que ele não envolve a IgE. A dessensibilização ao ácido acetilsalicílico, seguida por terapia diária com esse medicamento, perpetua o estado de tolerância ao ácido acetilsalicílico em pacientes com Drea, melhorando o controle da asma e da polipose nasal, além de possibilitar o tratamento de outras condições, como doenças cardíacas e reumáticas, com inibidores da cicloxigenase.
Outros pacientes desenvolvem urticária e angioedema com os inibidores da cicloxigenase, envolvendo, eventualmente, também os sistemas gastrintestinal e cardiovascular, com a probabilidade de não tolerarem inibidores específicos da COX-2, além de serem mais refratários à dessensibilização em comparação com outras manifestações respiratórias.
Alguns pacientes podem desenvolver reações mediadas pela IgE, tipicamente em relação à cadeia lateral de um determinado tipo de anti-inflamatório não esteroide, porém com tolerância a outros Aines, ou seja, não apresentando nenhuma reatividade cruzada com outros Aines.42 Outros exemplos comuns incluem a síndrome do homem vermelho induzida por vancomicina, coceira, urticária e hipotensão induzida por opiáceos como a morfina; os dois exemplos se referem à ativação direta de mastócitos, sem envolvimento da IgE e de seus receptores.
Para finalizar, a ativação de mastócitos e de basófilos ocorre nas situações em que os anticorpos são gerados contra a FCeRI, agregando esses receptores como alérgenos multivalentes, como se observa em pacientes autoimunes contra urticária.43
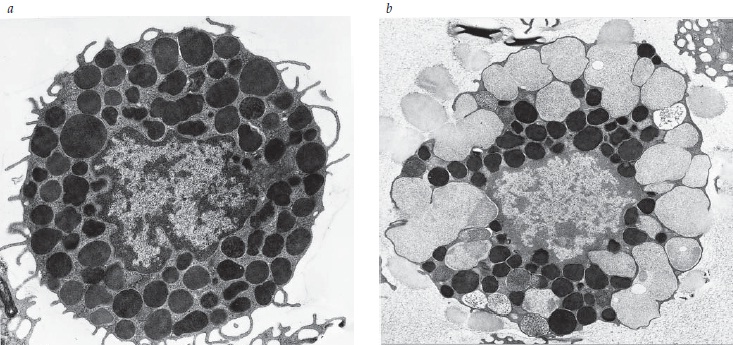
A Figura 2 mostra a macroestrutura de mastócitos em estado de repouso e ativados.
Figura 2 - Macroestrutura de mastócitos em estado de repouso e ativados: (A) antes da introdução de um antígeno, os mastócitos sensibilizados contêm muitos grânulos citoplasmáticos secretores; (B) sessenta segundos depois do tratamento com antígeno, os grânulos periféricos aumentaram de volume e os grânulos adjacentes fundiram entre si e com a membrana plasmática para externalizar os respectivos conteúdos.
Mastócitos e basófilos
Os mastócitos e os basófilos são células efetoras críticas nas doenças alérgicas.44 Essas células expressam níveis elevados de FCeRI que, quando tiverem ligação cruzada por complexos IgE alérgenos, emitem sinais para essas células para se desgranularem e, então, liberam histamina e os outros mediadores nos grânulos secretores, assim como produzem e segregam lipídeos de geração recente como a prostaglandina D2 (PGD2) ? mastócitos; leucotrieno C4 (LTC4) ? mastócitos e basófilos; citocinas e quimiocinas.
A microscopia de mastócitos e de basófilos mostra a presença de grânulos metacromáticos com coloração intensa. De maneira geral, os basófilos completam a maturação na medula óssea influenciada pela IL-3 e circulam na corrente sanguínea e, em seguida, são recrutados para sítios de inflamação, ao passo que os mastócitos maturam nos tecidos periféricos, influenciados sobretudo pelo fator da célula-tronco (FCT), onde residem por vários meses.
Os mastócitos se distribuem estrategicamente nos tecidos ou nas superfícies mucosas que fazem interface com o ambiente externo; os mastócitos se localizam também nas proximidades dos vasos sanguíneos e dos nervos. Em seres humanos saudáveis, dois tipos de mastócitos foram identificados com base nas composições secretoras da protease granular e nos marcadores superficiais: mastócitos triptase-positivos e quimase-negativos (células MCT), mastócitos triptase-positivos e quimase-positivos (células MCTC).45
As células MCT são, em geral, o tipo predominante de mastócitos encontrados na mucosa do intestino delgado, na parede alveolar e no epitélio do trato respiratório. As células MCT são identificadas morfologicamente por uma ultraestrutura granular rica em rolagem; essa ultraestrutura contém triptase ? mas não contém quimase ? catepsina G ou carboxipeptidase de mastócitos.
Há um aumento nos números de células MCTC no epitélio respiratório nos casos de inflamações alérgicas nas vias respiratórias, o que as tornam mais acessíveis aos alérgenos inalatórios. Por outro lado, as células MCTC são o tipo dominante de mastócitos na derme, na conjuntiva, no coração, nas paredes dos vasos sanguíneos e na submucosa do intestino delgado. As células MCT apresentam uma estrutura granular de rede/retículo de baixa rolagem.
Além da triptase, as células MCTC contêm quimase, catepsina G e carboxipeptidase de mastócitos. A expressão do receptor para o complemento 5a (C5aR, CD88) somente no tipo MCTC de mastócitos, pelo menos nos pulmões e na pele, faz a distinção entre esses dois subgrupos.46 As células MCTC e MCT precisam do FCT, o ligante Kit, para seu desenvolvimento e sobrevivência.47
As mutações somáticas do ligante Kit que resultam na ativação constitutiva independente do ligante dessa tirosina quinase estão associadas a condições como mastocitose (um distúrbio clonal) e distúrbios na ativação de mastócitos.2,47,48
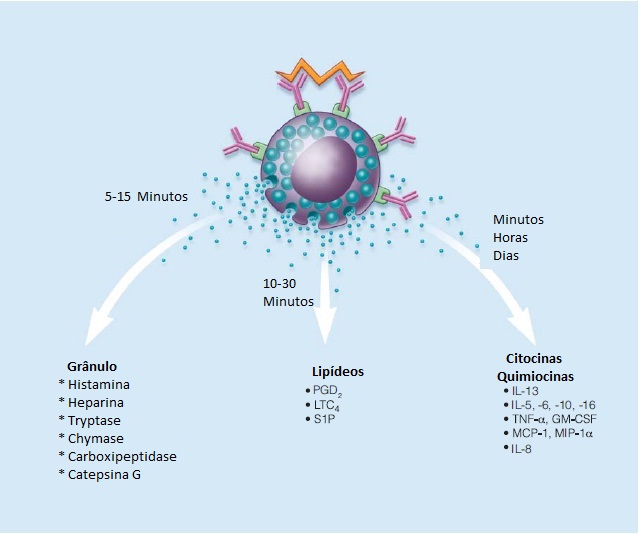
A Figura 3 apresenta os mediadores liberados por mastócitos humanos ativados, incluindo os tipos de mastócito triptase positivo e quimase negativo (MCT) e de mastócito triptase positivo e quimase positivo (MCTC).
GM-CSF: fator estimulador das colônias de granulócitos e macrófagos; IL: interleucina; LTC4: leucotrieno C4; MCP-1: proteína quimiotática dos monócitos (CCL2, ligante de quimiocina [motivo C-C] 2); MIP-1a: proteína inflamatória dos macrófagos 1-a (CCL3); PGD2: prostaglandina D2; S1P: esfongosina-1-fosfato; TNF: fator de necrose tumoral.
*Somente células MCTC.
Figura 3 - Mediadores liberados por mastócitos humanos ativados, incluindo os tipos de mastócito triptase positivo e quimase negativo (MCT) e de mastócito triptase positivo e quimase positivo (MCTC). Ao contrário dos mastócitos, os basófilos (não aparecem na figura) liberam quantidades comparáveis de histamina e LTC4 e quantidades bem menores de triptase e heparina; diferentemente dos mastócitos, eles produzem IL-4 e uma forma intermediária de proteína básica importante, e não produzem quimase, carboxipeptidase A3, catepsina G ou PGD2.
DÍSTICOS DA FIGURA 3
|
5-15 minutes |
= |
5?15min
|
|
Minutes Hours Days |
= |
Minutos Horas Dias
|
|
10-30 minutes |
= |
10?30min
|
|
Granule Histamine Heparin Tryptase Chymase* Carboxypeptidase A3* Cathepsin G3* |
= |
Grânulos Histamina Heparina Triptase Quimase* Carboxipeptidase A3* Catepsina G3*
|
|
Lipids PGD2 LTC4 S1P |
|
Lipídeos PGD2 LTC4 S1P
|
|
Cytokines Chemokines IL-13 IL-5, -6, -10, -15 TNF-a, GM-CSF MCP-1, MIP-1a IL-8 |
= |
Citocinas Quimiocinas IL-13 IL-5, -6, -10, -15 TNF-a, GM-CSF MCP-1, MIP-1a IL-8 |
Os papéis desempenhados pelos mastócitos e pelos basófilos, além de seus envolvimentos óbvios na hipersensibilidade imediata em seres humanos, foram estudados in vivo em modelos murinos. Por exemplo, ao que tudo indica, os basófilos estão envolvidos no desenvolvimento de inflamações alérgicas crônicas independentes dos mastócitos, cujo tempo de duração varia de alguns dias a algumas semanas, depois de um único desafio alergênico cutâneo.49
Foram usados camundongos geneticamente deficientes em mastócitos, cuja deficiência possa ser corrigida por meio da reposição de mastócitos, para estudar a biologia dos mastócitos. Da mesma forma, os mastócitos suprarregulam inflamações, produção de muco e hiperatividade das vias respiratórias, além de remodelarem os pulmões de camundongos que tenham sido sensibilizados e provocados com alérgenos.
Os mastócitos também infrarregulam inflamações, remodelagens e lesões cutâneas nos sítios de hipersensibilidade por contato e de exposição aos raios ultravioleta B, aparentemente através da produção de IL-10,51 e são estimulados diretamente por produtos bacterianos, permitindo que eles direcionem respostas imunes inatas para conter a infecção, enquanto se desenvolve a imunidade adaptativa.44
Em camundongos, as proteases dos mastócitos melhoram a resistência à morbidade e à mortalidade causadas por venenos de cobras, de abelhas, de escorpiões e dos monstros-de-gila (lagartos venenosos), assim como aos efeitos de peptídeos intestinais vasoativos e ao fator de necrose tumoral-a (TNF-a) que dependem das proteases dos mastócitos.52-54
As quimases e as triptases têm potencial para desempenhar diversos papéis nas inflamações, nas defesas de hospedeiros e na homeostase tecidual,55 sugerindo que o antagonismo farmacológico dessas proteases pode apresentar resultados imprevisíveis.
Mediadores de mastócitos e basófilos
Os mastócitos e basófilos expressam a enzima histidina descarboxilase, que converte histidina em histamina, o principal mediador vasoativo pré-formado; quantidades comparáveis são armazenadas nos grânulos secretores desses dois tipos de células, que são liberadas a partir do momento em que as células são estimuladas para promover a desgranulação.
Ao se ligar aos receptores específicos presentes nas várias células, a histamina induz a contração dos músculos lisos das vias respiratórias e a secreção de muco, a vasodilatação, a vasopermeabilidade e o prurido, além de recrutar outras células inflamatórias. O sulfato de condroitina e os proteoglicanos heparínicos também são armazenados nos grânulos dos basófilos e dos mastócitos, sendo que os mastócitos são o sítio principal de armazenamento de heparina.
Ambos os proteoglicanos desempenham algum tipo de papel no empacotamento da heparina e das proteases nos grânulos secretores. A heparina também está envolvida no processamento, na estabilização e nas atividades proteolíticas da quimase e da triptase. A neutralização do pH ácido dos grânulos secretores durante o processo de desgranulação facilita a dissociação da histamina do complexo macromolecular protease-proteoglicanos e sua difusão rápida a partir desse sítio de liberação.
Os mastócitos e os basófilos também secretam mediadores lipídicos de sintetização recente, sendo que ambos os tipos células secretam LTC4 e, possivelmente, o fator de ativação plaquetária (FAP), ao passo que somente os mastócitos secretam a PGD2.
O LTC4 secretado, do qual derivam o leucotrieno D4 (LTD4) e o leucotrieno E4 (LTE4), contrai os músculos lisos das vias respiratórias, estimula a produção de muco, aumenta a permeabilidade dos vasos, vasodilata e promove a quimiotaxia dos eosinófilos. O FAP ativa as plaquetas, os neutrófilos e as células dos músculos lisos e, além disso, pode ser um mediador importante da anafilaxia induzida por alimentos, que foi associada a níveis séricos mais baixos de acetildrolase no FAP, enzima que desativa o fator de ativação plaquetária.56,57
Os basófilos humanos produzem quantidades abundantes de IL-4, enquanto que os mastócitos produzem IL-3; portanto, esses dois tipos de células estimulam a mudança de classe de linfócitos B para IgE. Além disso, os mastócitos produzem um grupo diversificado de citocinas, incluindo o fator estimulador das colônias de granulócitos e macrófagos (GM-CSF), TNFa, IL-5 e IL-8.
Por conseguinte, os mastócitos parecem desempenhar um papel importante no ciclo de realimentação imunorreguladora positiva que perpetua a inflamação. Em parte, o TNFa pode ser armazenado nos grânulos secretores dos mastócitos, e poderá ser disponibilizado para secreção na fase inicial da resposta alérgica.
Os basófilos humanos foram identificados em tecidos com anticorpos monoclonais específicos para esse tipo de célula, como o 2D7, que reconhecem uma forma intermediária de proteína eosinofílica básica principal (em inglês, major basic protein [MBP]) que se acumula seletivamente nos basófilos.58 In vivo, após um desafio alergênico nas vias respiratórias, os basófilos são as células produtoras de IL-4 predominantes.59
Biomarcadores de mastócitos e basófilos
A histamina surge no plasma logo após o início de anafilaxia sistêmica provocada por alérgenos como o veneno da picada de insetos, atingindo um pico dentro de, aproximadamente, 5 minutos após o início dos sinais e sintomas e, em seguida, retornando para a linha de base em um período de tempo que varia de 15 a 30 minutos, o que reflete a ativação de mastócitos ou de basófilos, ou de ambos os tipos de células.
Levando-se em consideração que as elevações nos níveis plasmáticos de histamina são de curta duração, o valor prático com biomarcador clínico de anafilaxia é bastante limitado. Por outro lado, níveis urinários de histamina ou metabólitos histamínicos como a N-metil histamina ou a prostaglandina e os metabólitos de leucotrieno (LT) podem ter maior praticidade.60,61
Entretanto, níveis elevados de metabólitos histamínicos na urina, obtidos durante algum episódio sintomático, rubor ou síncope, não conseguem fazer a distinção entre anafilaxia e mastocitose sistêmica (um distúrbio de hiperplasia clonal dos mastócitos), sendo que, nesse caso, a comparação do nível agudo em relação a um nível na linha de base provavelmente tenha alguma utilidade.
A triptase se difunde para dentro e fora da circulação em velocidades mais lentas do que a histamina. Os níveis séricos da triptase atingem o pico na circulação dentro de um período de tempo que poderá variar de 15 minutos a 1 hora após o início da anafilaxia sistêmica induzida por picada de inseto e declina logo em seguida, com meia-vida de, aproximadamente, 2 horas. Os níveis máximos de triptase nos casos de anafilaxia induzida pela picada de insetos se correlacionam intimamente com o declínio na pressão arterial média.
A triptase se apresenta em formas maduras e imaturas. Com base nisso, são feitas as seguintes considerações:
Em parte, a secreção da forma imatura, a protriptase, por mastócitos não estimulados é espontânea, sendo responsável pela vasta maioria da triptase detectada no soro não anafilático;
A triptase madura, derivada da protriptase, é armazenada no interior dos grânulos secretores como um tetrâmero ativo sob o ponto de vista enzimático ligado ao proteoglicano heparínico, sendo que os mastócitos apresentam níveis 500 vezes mais elevados em comparação com os basófilos, e sua secreção ocorre a partir do desencadeamento da desgranulação celular;
Um imunoensaio comercial (Thermo Fisher Scientific, Waltham, MA) mede as formas maduras e imaturas da triptase (triptase total), enquanto que a disponibilidade de outro imunoensaio aplicável somente à triptase madura é bastante limitada.
Os níveis séricos da triptase madura na linha de base normalmente não são detectáveis (<1ng/mL) em indivíduos saudáveis que não tenham sofrido anafilaxia nas últimas 24 horas. Os níveis séricos totais de triptase na linha de base em indivíduos saudáveis têm uma média de, aproximadamente, 4ng/mL, variando de 1 a 11,4ng/mL.
O Quadro 2 mostra os níveis séricos da triptase.
Quadro 2
|
NÍVEIS SÉRICOS DA TRIPTASE | |||
|
Condição clínica |
Triptase Sérica (ng/mL) |
Proporção entre triptase total e madura | |
|
Total |
Madura | ||
|
Normal |
1?11,4 |
<1 |
NA
|
|
Anafilaxia sistêmica (aguda)
|
>(2 + 1,2 x sBT)* |
>1† |
<10 |
|
Mastocitose sistêmica (não aguda)
|
>20? |
<1 |
>20 |
NA: não aplicável.
*Nível de triptase total aguda acima de 2 + (1,2 x nível de triptase sérica total na linha de base [sBT]).
†Nível relacionado à gravidade clínica (hipotensão), momento da coleta da amostra em relação ao início dos sinais e sintomas, e natureza do estímulo anafilático.
?Reflete sobretudo a carga corporal total de mastócitos nas situações em que o indivíduo está em um estado não anafilático. Um sBT acima de 11,4ng/mL aumenta a possibilidade de um distúrbio subjacente de mastócitos clonais, particularmente em indivíduos que tenham sofrido anafilaxia sistêmica à picada de algum inseto.
Os níveis séricos de triptase madura e de triptase total elevam durante a maior parte das formas de anafilaxia sistêmica, sendo que a elevação nos níveis totais de triptase acima da linha de base é tão sensível quanto os níveis elevados de triptase madura para detectar a ativação de mastócitos.
Na realidade, a comparação entre níveis agudos e na linha de base da triptase total, assim como para histamina, metabólitos histamínicos e metabólitos da PGL2, ajuda a fazer a distinção entre ativação de mastócitos (anafilaxia) e aumento na carga de mastócitos (mastocitose).
Apresentou-se a sugestão de que a elevação mínima no nível total de triptase aguda considerada significativa sob o ponto de vista clínico seja de 2 + 1,2 x os níveis de triptase na linha de base. Além disso, tipicamente, a proporção entre a triptase total e a triptase madura é inferior a 10 e se aproxima da unidade em níveis mais elevados de triptase madura.
O Quadro 3 apresenta exemplos de padrões de níveis de triptase na linha de base e de triptase aguda em vários cenários clínicos.
Quadro 3
|
EXEMPLOS DE PADRÕES DE ELEVAÇÕES NOS NÍVEIS SÉRICOS TOTAIS DA TRIPTASE E INTERPRETAÇÕES | ||
|
Triptase total (ng/mL) |
Interpretação (consistente com) | |
|
Aguda |
Na linha de base | |
|
4 |
1 |
Anafilaxia |
|
4 |
2 |
Normal |
|
10 |
5 |
Anafilaxia |
|
10 |
9 |
Normal |
|
15 |
5 |
Anafilaxia |
|
15 |
12 |
Normal |
|
25 |
5 |
Anafilaxia |
|
25 |
22 |
Mastocitose sistêmica |
|
75 |
10 |
Anafilaxia |
|
75 |
50 |
Anafilaxia + mastocitose sistêmica |
Obs.: As medições da triptase total consideradas isoladamente dão suporte ao diagnóstico clínico de anafilaxia se ambas as amostras, aguda e na linha de base (coletadas antes do evento agudo ou >24 horas após a interrupção de todos os sinais e sintomas de anafilaxia), estiverem disponíveis. Níveis elevados de triptase na linha de base aumentam o risco de anafilaxia mais grave e podem estar associados a distúrbios clonais de mastócitos em níveis superiores a 11,4ng/mL.
Os níveis elevados de triptase total na linha de base, sobretudo nos indivíduos sensíveis ao veneno de insetos, aumentam o risco de anafilaxia mais grave nos campos de picadas de insetos e de imunoterapia contra venenos. Além do mais, os pacientes com histórico de anafilaxia sistêmica às picadas de insetos e com níveis de triptase acima de 11,4ng/mL na linha de base têm prevalência mais alta de doença subjacente de mastócitos clonais (mastocitose sistêmica ou síndrome da ativação de mastócitos).62
A mastocitose sistêmica pode ser diagnosticada pela presença de um critério maior (agregados densos de mastócitos nos tecidos, tipicamente na medula óssea) e por quatro critérios menores (mastócitos CD25- ou CD2-positivo; ativação da mutação c-kit; mastócitos anormais sob o ponto de vista morfológico; nível sérico total de triptase =20ng/mL) ou por três dos critérios menores, sendo que todos os indivíduos com esse distúrbio correm grande risco de apresentar eventos anafiláticos dependentes da IgE e independentes da IgE.2,63
Eosinófilos
Na medula óssea, os eosinófilos se diferenciam de progenitores mieloides multipotenciais CD34+ e dão origem aos progenitores comprometidos com eosinófilos que também expressam Gata-1 e IL-5Ra64 ? que, por sua vez, se desenvolvem em citocinas de resposta, incluindo IL-3, GM-CSF, IL-3365 ou IL-5, sendo que a IL-5 é mais potente e específica para eosinófilos.
A IgA também prolonga a sobrevida dos eosinófilos, que podem ser relevantes em sítios de mucosas.66 O desenvolvimento de basófilos pode depender basicamente da IL-3. Outras citocinas também podem inibir eosinófilos e, possivelmente, o desenvolvimento de basófilos.
Por exemplo, o IFN-a inibe o desenvolvimento de eosinófilos e pode ser usado para tratar pacientes com a síndrome hipereosinofílica (SHE),67 um grupo heterogêneo de distúrbios em que os níveis de eosinófilos são altamente elevados na circulação e desempenham papel importante na patogênese da doença, danificando órgãos como a pele, o coração, os pulmões e o fígado.
Os eosinófilos residem basicamente nos tecidos, em especial na mucosa do intestino e do colo, assim como no timo, no útero e nas glândulas mamárias. Os eosinófilos são recrutados em outros sítios em associação com alguma doença inflamatória causada por fatores quimiotáticos como as eotaxinas 1, 2 e 3, em que sua sobrevivência e seu estado de ativação são melhorados por citocinas como GM-CSF, IL-3 e IL-5. Por outro lado, os eosinófilos em circulação têm meia-vida curta e, em geral, representam apenas 1% do número total de eosinófilos no corpo.
Os eosinófilos são muito úteis na defesa contra organismos parasitas, embora sejam danosos nos casos de doenças não infecciosas nas vias respiratórias como asma e rinite alérgica, que se caracterizam pela presença de inflamação causada por eosinófilos.68 Há várias descrições intrigantes de atividades imunomoduladoras para eosinófilos na apresentação de antígenos, polarização de células T, preparação (priming) de células B, recrutamento e ativação de células dendríticas e de mastócitos, que ainda não foram compreendidas com precisão nas respostas imunes totais.68
A explosão de eosinófilos por várias citocinas provoca alterações na expressão de receptores superficiais e na liberação de mediadores, juntamente com alterações no grau de granulação e na densidade total da célula. Os eosinófilos estimulados e preparados, como aqueles observados na SHE, são, em geral, hipogranulados e de baixa densidade, em comparação com os eosinófilos em estado de repouso.
Embora o FCeRI esteja ausente em eosinófilos não estimulados, existem relatos de sua presença como receptor trimérico a?2 em pacientes com SHE ou eosinofilia secundária a doenças parasitárias ou alérgicas, sugerindo que poderiam ser ativados diretamente por alérgenos. Os eosinófilos de isolamento recente expressam receptores para IgG e IgA.29
A presença de IgA secretora, juntamente com eosinófilos nas superfícies mucosas, sugere a possibilidade de ativação dependente de IgA nesses sítios. Além disso, os receptores para complemento (C3a e C5a) e a IgG indicam a capacidade para responder aos complexos imunes de imunoglobulina G.
As quimiocinas da família C-C são quimiotáticas para os eosinófilos. O receptor de quimiocinas, CCR3, é abundante nos eosinófilos e se liga em, pelo menos, quatro quimiocinas C-C (eotaxinas 1 a 3 e Rantes), além de ser crucial para abrigar eosinófilos para tecidos epiteliais e prepará-los para a liberação de mediadores.
Por exemplo, a eotaxina-3 foi implicada no recrutamento de eosinófilos para o esôfago em pacientes com esofagite eosinofílica, ao passo que as eotaxinas 1, 2 e 3 foram associadas à eosinofilia pulmonar.69,70 Os eosinófilos expressam também uma grande quantidade de selectinas e integrinas que abrigam essas células em sítios inflamatórios.
Mediadores eosinofílicos
Os mediadores inflamatórios pré-formados e armazenados nos grânulos são liberados rapidamente com a ativação dos eosinófilos. Os grânulos eosinofílicos contêm quatro proteínas catiônicas principais: MBP; proteína catiônica eosinofílica (em inglês, eosinophil cationic protein [ECP]), uma ribonuclease fraca; neurotoxina derivada de eosinófilos (em inglês, eosinophil-derived neurotoxina [EDN]), uma ribonuclease potente; peroxidade eosinofílica (em inglês, eosinophil peroxidasei [EPO]).70,71
Grandes quantidades de MBP residem nos grânulos eosinofílicos e agem como biomarcadores de eosinófilos humanos, sobretudo quando observados nos tecidos onde não é possível detectar eosinófilos intactos, como, por exemplo, após o tratamento de pacientes com glicocorticosteroides, que produzem apoptose eosinofílica.72
A MBP também se expressa por basófilos, embora menos que para eosinófilos. Os eosinófilos, quando estimulados por bactérias, C5a ou ligantes de CCR3, liberam DNA mitocondrial com atividade antimicrobiana.73 Os eosinófilos se recuperam, ao contrário dos neutrófilos que liberam DNA nuclear e nunca se recuperam.
A atividade antimicrobiana eosinofílica está associada à geração de radicais de oxigênio reativo e aos complexos de DNA contendo proteínas básicas como ECP e MBP, estendendo o papel antimicrobiano dos eosinófilos dos parasitas para incluir bactérias. As mesmas proteínas básicas que possuem função antimicrobiana podem danificar também os tecidos hospedeiros em pacientes portadores de doenças associadas à eosinofilia, causando danos epiteliais, endoteliais e parenquimatosos em distúrbios eosinofílicos nos pulmões, no coração, no cérebro, no fígado e nos vasos sanguíneos.
A geração de mediadores lipídicos pelos eosinófilos é mais rápida após a estimulação.70 A secreção do FAP ativa plaquetas, neutrófilos e células de músculos lisos. Os principais produtos eicosanoides secretados pelos eosinófilos são o leucotrieno B4 (LTB4), um fator quimiotático para neutrófilos e outros leucócitos, e o LTC4.
Inúmeras citocinas também foram identificadas como produtos eosinofílicos potenciais. Algumas podem agir de forma autócrina ou parácrina para ativar os eosinófilos mais importantes, enquanto que outras podem melhorar o desenvolvimento e a sobrevivência dos eosinófilos. Além disso, os eosinófilos produzem citocinas que regulam as respostas imunes. Por exemplo, os eosinófilos dos tecidos dos pólipos nasais produzem TGF-ß e, por conseguinte, podem contribuir para a patologia estrutural.
Manter a IL-5 (ou IL-5R) como alvo é uma abordagem interessante para o tratamento de pacientes com todos os tipos de SHE, levando-se em conta a atividade não redundante dessa rota de citocinas na linhagem eosinofílica e o pressuposto de que os danos teciduais em pacientes com SHE se relacionam diretamente com a presença de eosinófilos ativados. Os anticorpos monoclonais anti-IL-5 se direcionam para os eosinófilos por meio da neutralização de IL-5, ou seja, impedindo sua ligação com a IL-5R e provocando a morte eosinofílica.
Foram desenvolvidos dois anticorpos anti-IL-5 humanizados diferentes, o mepolizumabe e o reslizumabe, sendo que ambos foram eficazes em testes clínicos para o controle de asma e da SHE.74-79 Cabe observar que, no caso de SHE, os glicocorticosteroides induzem a remissão porque matam os eosinófilos por meio da indução de apoptose, reduzindo os danos teciduais, em comparação com a morte dos eosinófilos por necrose, que poderá causar insuficiência cardíaca aguda a partir do momento em que os eosinófilos invadirem os tecidos do coração e forem necrosados naquele sítio.
Terapia de distúrbios atópicos
As estratégias terapêuticas mais recentes têm como alvo a neutralização das reações alérgicas, ainda que a prevenção e a cura dos distúrbios atópicos sejam os principais objetivos. Os agentes anti-histamínicos bloqueiam a ligação da histamina com seus receptores, o ácido acetilsalicílico e os Aines bloqueiam a síntese da PGD2, e os antagonistas do LT bloqueiam a ligação dos cisteinil-LTs com seus receptores.
Entretanto, a vasta série e redundância de mediadores potentes liberados pelos mastócitos, basófilos e outras células envolvidas nessas reações pode limitar o impacto dos inibidores dos mediadores na atividade da doença.
O Quadro 4 apresenta o resumo de intervenções terapêuticas para doenças alérgicas.
Quadro 4
|
RESUMO DE INTERVENÇÕES TERAPÊUTICAS PARA DOENÇAS ALÉRGICAS | |||
|
| |||
|
Intervenção terapêutica ou medicação
|
Mecanismo de ação |
Indicações |
Indicações potenciais |
|
Anti-histaminas H1 |
Antagonista de receptores da histamina H1. |
Rinoconjuntivite alérgica, urticária. |
Mastocitose, síndromes de ativação de mastócitos.
|
|
Inibidores do LT: montelucaste, zafirlucaste; zileutona |
Inibição da atividade da 5-lipoxigenase. |
Asma, rinite alérgica, broncoconstrição induzida por exercícios físicos.
|
Mastocitose, síndromes de ativação de mastócitos.
|
|
Broncodilatador |
Relaxamento dos músculos lisos dos brônquios por meio da ação sobre os receptores adrenérgicos ß2; pouco efeito sobre a frequência cardíaca.
|
Broncoespasmo em pacientes com doença obstrutiva reversível nas vias respiratórias. |
|
|
Corticosteroides |
Redução da inflamação por meio da supressão da mitigação de leucócitos e sobrevivência de eosinófilos, basófilos e determinados tipos de linfócitos. |
Dermatite atópica, reações de hipersensibilidade aos medicamentos, rinite alérgica, asma alérgica, urticária crônica, distúrbios eosinofílicos.
|
|
|
Omalizumabe |
Anticorpo monoclonal humanizado da IgG; inibe a ligação entre a IgE e o receptor de IgE de alta afinidade nos mastócitos e basófilos.
|
Tratamento de asma alérgica mal controlada. |
Urticária crônica; ABPA. |
|
Anti-IL5 (reslizumabe, mepolizumabe) |
Anticorpos monoclonais humanizados de IgG contra IL-5.
|
|
SHE, asma eosinofílica. |
ABPA: aspergilose broncopulmonar alérgica; IL: interleucina; LT: leucotrieno; SHE: síndrome hipereosinofílica.
Os efeitos dos esteroides tópicos e sistêmicos são limitados sobre os mastócitos, embora impeçam muitos eventos inflamatórios no sentido do fluxo associados a reações alérgicas, em parte por meio do bloqueio do recrutamento ou da sobrevivência de eosinófilos, basófilos e células T. Outros medicamentos são utilizados no tratamento de respostas a esses mediadores específicas de órgãos.
Por exemplo, os agonistas ß-adrenérgicos inalatórios relaxam os músculos lisos dos brônquios e os agentes anticolinérgicos diminuem a produção de muco em pacientes asmáticos, ao passo que a administração intramuscular de epinefrina ajuda a recuperar a integridade vascular durante a anafilaxia sistêmica.
A imunoterapia alergênica (administração de incrementos crescentes de alérgeno[s] proteico[s], como, por exemplo, de pólen ou de veneno de insetos ao longo dos anos), pela forma injetável, sublingual ou oral, é usada para induzir tolerância clínica de longa duração em pacientes sensibilizados de uma maneira antigênica específica, em geral durante vários anos após a descontinuação da imunoterapia.
Diversos estudos bem estruturados demonstraram que a imunoterapia alergênica é eficaz no tratamento de rinite alérgica, conjuntivite alérgica, asma alérgica e hipersensibilidade à picada de insetos. Alguns estudos randomizados controlados também mostraram que a imunoterapia alergênica impede o desenvolvimento de asma em alguns indivíduos com rinite alérgica. A imunoterapia pode ser eficaz nos casos de dermatite atópica nas situações em que essa condição está associada à sensibilidade aeroalergênica.80
A dessensibilização, outro tipo de tolerância clínica, é diferente da tolerância imunológica resultante de aeroalérgenos ou de imunoterapia contra picada de insetos. Os procedimentos de dessensibilização, tipicamente usados em pacientes com hipersensibilidade aos medicamentos mediados pela IgE, parecem tornar as células efetoras (mastócitos e basófilos) menos reativas ou não reativas ao medicamento; portanto, são administradas doses incrementais de alguma substância alergênica durante o período de algumas horas.
A tolerância clínica a essa medicação persiste durante o tempo de administração. Todavia, a sensibilização recorre logo após a descontinuação do uso do medicamento, em geral dentro de alguns dias ou de algumas semanas.
Alguns estudos em andamento sobre imunoterapia sublingual e oral contra alergia por alimentos são promissores, mas ainda não foram recomendados para uso rotineiro. Os protocolos atuais têm capacidade para induzir dessensibilização na maioria dos indivíduos com sensibilidade ao amendoim, aos ovos ou ao leite, reduzindo o risco de reações anafiláticas à ingestão acidental de pequenas quantidades desses alimentos, embora possam ocorrer reações causadas pela imunoterapia alimentar, sobretudo durante a fase de desenvolvimento da doença.
A tolerância no longo prazo, após a descontinuação da imunoterapia, ocorre apenas em uma pequena minoria de pacientes.81-83 Levando-se em consideração a provável importância dos eosinófilos para a alergia e outros distúrbios associados, a IL-5 foi sugerida como alvo molecular potencial no tratamento dessas doenças.
As terapias com antagonistas da IL-5 no desenvolvimento atual incluem dois anticorpos monoclonais anti-IL-5 (mepolizumabe, reslizumabe), um anticorpo monoclonal direcionado para o receptor de IL-5 (benralizumab) e a terapia à base de oligonucleotídeos anti-sentido (TPI ASM8).84 A terapia com embasamento no anticorpo anti-IL-5 foi mais extensivamente estudada, tendo sido realizados vários testes em pacientes com asma brônquica, polipose nasal, dermatite atópica, esofagite eosinofílica, SHE e síndrome de Churg-Strauss.
As estratégias com foco na inibição da IL-5 podem ser bastante promissoras no tratamento de doenças ou de manifestações específicas de uma doença para as quais os eosinófilos são as células efetoras principais.
Ainda é necessário desenvolver terapias melhores para curar ou prevenir o desenvolvimento de fenótipos atópicos. Para prevenir o desenvolvimento desses fenótipos, é imprescindível aprimorar os conhecimentos dos fatores ambientais subjacentes à elevação da prevalência de doenças mediadas pela IgE nas últimas décadas, o que poderia levar a novas estratégias intervencionistas para atrasar o relógio de novas reações alérgicas.
|
Informações financeiras: Joud Hajjar, MD, não tem relações financeiras relevantes a declarar. Lawrence B. Schwartz, MD, PhD, tem um acordo de licenciamento com a Virginia Commonwealth University para o ensaio com a triptase com a Thermo Fisher Scientific; realizou testes clínicos e recebeu honorários da GlaxoSmithKline, atuou como consultor para a Sanofi-Aventis US LLC e recebeu apoio e atuou como consultor e palestrante para a Novartis Corporation e Genentech, membros do grupo Roche.
|
Referências
1. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network Symposium. J Allergy Clin Immunol 2006;117:391–7.
2. Valent P, Akin C, Arock M, et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol 2012;157:215–25.
3. Gupta RS, Springston EE, Warrier MR, et al. The prevalence, severity, and distribution of childhood food allergy in the United States. Pediatrics 2011;128:e9–17.
4. Bloom B, Cohen RA, Freeman G. Summary health statistics for U.S. children: National Health Interview Survey, 2010. Vital Health Stat 10 2011;(250):1–80.
5. Pawankar R, Canonica GW, Holgate ST, Lockey RF. Introduction and executive summary: allergic diseases as a global public health issue. In: Pawankar R, Canonica GW, Holgate ST, Lockey RF, editors. World Allergy Organization white book of allergy. Milwaukee (WI): World Allergy Organization; 2011. p. 11.
6. Boyle JA, Camargo CA Jr, Lieberman PL, et al. Anaphylaxis in America – results from a national telephone survey [abstract]. J Allergy Clin Immunol 2012;129(2 Suppl):AB132.
7. Vercelli D. Mechanisms of the hygiene hypothesis—molecular and otherwise. Curr Opin Immunol 2006;18:733–7.
8. Loss G, Bitter S, Wohlgensinger J, et al. Prenatal and early-life exposures alter expression of innate immunity genes: the PASTURE cohort study. J Allergy Clin Immunol 2012;130:523–30.
9. Brooks C, Pearce N, Douwes J. The hygiene hypothesis in allergy and asthma: an update. Curr Opin Allergy Clin Immunol 2013;13:70–7.
10. Carroll KN, Gebretsadik T, Minton P, et al. Influence of maternal asthma on the cause and severity of infant acute respiratory tract infections. J Allergy Clin Immunol 2012;129:1236–42.
11. Sykes A, Edwards MR, Macintyre J, et al. Rhinovirus 16-induced IFN-alpha and IFN-beta are deficient in bronchoalveolar lavage cells in asthmatic patients. J Allergy Clin Immunol 2012;129:1506–14.
12. Szefler SJ. Advances in pediatric asthma in 2012: moving toward asthma prevention. J Allergy Clin Immunol 2013;131:36–46.
13. Du R, Litonjua AA, Tantisira KG, et al. Genome-wide association study reveals class I MHC-restricted T cell-associated molecule gene (CRTAM) variants interact with vitamin D levels to affect asthma exacerbations. J Allergy Clin Immunol 2012;129:368–73.
14. Gupta A, Sjoukes A, Richards D, et al. Relationship between serum vitamin D, disease severity, and airway remodeling in children with asthma. Am J Respir Crit Care Med 2011;184:1342–9.
15. Goleva E, Searing DA, Jackson LP, et al. Steroid requirements and immune associations with vitamin D are stronger in children than adults with asthma. J Allergy Clin Immunol 2012;129:1243–51.
16. Oh SS, Tcheurekdjian H, Roth LA, et al. Effect of secondhand smoke on asthma control among black and Latino children. J Allergy Clin Immunol 2012;129:1478–83.
17. Kreiner-Moller E, Sevelsted A, Vissing NH, et al. Infant acetaminophen use associates with early asthmatic symptoms independently of respiratory tract infections: the Copenhagen Prospective Study on Asthma in Childhood 2000 (COPSAC(2000)) cohort. J Allergy Clin Immunol 2012;130:1434–6.
18. Spycher BD, Henderson J, Granell R, et al. Genome-wide prediction of childhood asthma and related phenotypes in a longitudinal birth cohort. J Allergy Clin Immunol 2012;130:503–9.
19. Margolis DJ, Apter AJ, Gupta J, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol 2012;130:912–7.
20. Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008;358:1109–17.
21. Commins SP, Platts-Mills TA. Delayed anaphylaxis to red meat in patients with IgE specific for galactose alpha-1,3-galactose (alpha-gal). Curr Allergy Asthma Rep 2013;13:72–7.
22. Klein J, Sato A. The HLA system. First of two parts. N Engl J Med 2000;343:702–9.
23. Kay AB. Allergy and allergic diseases. First of two parts. N Engl J Med 2001;344:30–7.
24. Zuniga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev 2013;252:78–88.
25. Yamane H, Paul WE. Cytokines of the gamma(c) family control CD4+ T cell differentiation and function. Nat Immunol 2012;13:1037–44.
26. Chaudhuri J, Basu U, Zarrin A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 2007;94:157–214.
27. Gauchat JF, Henchoz S, Mazzei G, et al. Induction of human IgE synthesis in B cells by mast cells and basophils. Nature 1993;365:340–3.
28. Dullaers M, De Bruyne R, Ramadani F, et al. The who, where, and when of IgE in allergic airway disease. J Allergy Clin Immunol 2012;129:635–45.
29. Kraft S, Kinet JP. New developments in FcepsilonRI regulation, function and inhibition. Nat Rev Immunol 2007;7:365–78.
30. Gomez G, Jogie-Brahim S, Shima M, Schwartz LB. Omalizumab reverses the phenotypic and functional effects of IgE-enhanced Fc epsilonRI on human skin mast cells. J Immunol 2007;179:1353–61.
31. MacGlashan D Jr. IgE and Fc{epsilon}RI regulation. Ann N Y Acad Sci 2005;1050:73–88.
32. Conrad DH, Ford JW, Sturgill JL, Gibb DR. CD23: an overlooked regulator of allergic disease. Curr Allergy Asthma Rep 2007;7:331–7.
33. Weskamp G, Ford JW, Sturgill J, et al. ADAM10 is a principal ‘sheddase’ of the low-affinity immunoglobulin E receptor CD23. Nat Immunol 2006;7:1293–8.
34. Hamelmann E, Wahn U. Anti-IgE therapy. Clin Allergy Immunol 2008;21:415–27.
35. MacGlashan DW Jr, Bochner BS, Adelman DC, et al. Down-regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997;158:1438–45.
36. Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med 2006;354:2689–95.
37. Magerl M, Staubach P, Altrichter S, et al. Effective treatment of therapy-resistant chronic spontaneous urticaria with omalizumab. J Allergy Clin Immunol 2010;126:665–6.
38. Saini S, Rosen KE, Hsieh HJ, et al. A randomized, placebo-controlled, dose-ranging study of single-dose omalizumab in patients with H(1)-antihistamine-refractory chronic idiopathic urticaria. J Allergy Clin Immunol 2011;128:567–73.
39. Elmallah MK, Hendeles L, Hamilton RG, et al. Management of patients with cystic fibrosis and allergic bronchopulmonary aspergillosis using anti-immunoglobulin E therapy (omalizumab). J Pediatr Pharmacol Ther 2012;17:88–92.
40. Lieberman JA, Chehade M. Use of omalizumab in the treatment of food allergy and anaphylaxis. Curr Allergy Asthma Rep 2013;13:78–84.
41. Gyllfors P, Bochenek G, Overholt J, et al. Biochemical and clinical evidence that aspirin-intolerant asthmatic subjects tolerate the cyclooxygenase 2-selective analgetic drug celecoxib. J Allergy Clin Immunol 2003;111:1116–21.
42. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol 2010;105:259–73.
43. Hide M, Francis DM, Grattan CE, et al. Autoantibodies against the high-affinity IgE receptor as a cause of histamine release in chronic urticaria. N Engl J Med 1993;328:1599–604.
44. Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol 2010;40:1843–51.
45. Schwartz LB. Analysis of MC(T) and MC(TC) mast cells in tissue. Methods Mol Biol 2006;315:53–62.
46. Oskeritzian CA, Zhao W, Min HK, et al. Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. J Allergy Clin Immunol 2005;115:1162–8.
47. Akin C, Metcalfe DD. The biology of Kit in disease and the application of pharmacogenetics. J Allergy Clin Immunol 2004;114:13–9.
48. Alvarez-Twose I, Bonadonna P, Matito A, et al. Systemic mastocytosis as a risk factor for severe Hymenoptera sting-induced anaphylaxis. J Allergy Clin Immunol 2013;131:614–5.
49. Mukai K, Matsuoka K, Taya C, et al. Basophils play a critical role in the development of IgE-mediated chronic allergic inflammation independently of T cells and mast cells. Immunity 2005;23:191–202.
50. Yu M, Tsai M, Tam SY, et al. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest 2006;116:1633–41.
51. Grimbaldeston MA, Nakae S, Kalesnikoff J, et al. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol 2007;8:1095–104.
52. Akahoshi M, Song CH, Piliponsky AM, et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J Clin Invest 2011;121:4180–91.
53. Metz M, Piliponsky AM, Chen CC, et al. Mast cells can enhance resistance to snake and honeybee venoms. Science 2006;313:526–30.
54. Piliponsky AM, Chen CC, Rios EJ, et al. The chymase mouse mast cell protease 4 degrades TNF, limits inflammation, and promotes survival in a model of sepsis. Am J Pathol 2012;181:875–86.
55. Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev 2007;217:141–54.
56. Vadas P, Gold M, Perelman B, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med 2008;358:28–35.
57. Vadas P, Perelman B, Liss G. Platelet-activating factor, histamine, and tryptase levels in human anaphylaxis. J Allergy Clin Immunol 2013;131:144–9.
58. Plager DA, Weiss EA, Kephart GM, et al. Identification of basophils by a mAb directed against pro-major basic protein 1. J Allergy Clin Immunol 2006;117:626–34.
59. Nouri-Aria KT, Irani AM, Jacobson MR, et al. Basophil recruitment and IL-4 production during human allergen-induced late asthma. J Allergy Clin Immunol 2001;108:205–11.
60. Oranje AP, Mulder PG, Heide R, et al. Urinary N-methylhistamine as an indicator of bone marrow involvement in mastocytosis. Clin Exp Dermatol 2002;27:502–6.
61. Raithel M, Zopf Y, Kimpel S, et al. The measurement of leukotrienes in urine as diagnostic option in systemic mastocytosis. J Physiol Pharmacol 2011;62:469–72.
62. Schwartz LB. Laboratory tests to support the clinical diagnosis of anaphylaxis. Available at: http://www.uptodate.com/contents/laboratory-tests-to-support-the-clinical-diagnosis-of-anaphylaxis?detectedLanguage=en&source=search_result&search=tryptase&selectedTitle=1~66&provider=noProvider (accessed September 26, 2013).
63. Valent P, Horny H-P, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leukemia Res 2001;25:603–25.
64. Mori Y, Iwasaki H, Kohno K, et al. Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med 2009;206:183–93.
65. Cherry WB, Yoon J, Bartemes KR, et al. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol 2008;121:1484–90.
66. Bartemes KR, Cooper KM, Drain KL, Kita H. Secretory IgA induces antigen-independent eosinophil survival and cytokine production without inducing effector functions. J Allergy Clin Immunol 2005;116:827–35.
67. Butterfield JH, Weiler CR. Treatment of hypereosinophilic syndromes—the first 100 years. Semin Hematol 2012;49:182–91.
68. Spencer LA, Weller PF. Eosinophils and Th2 immunity: contemporary insights. Immunol Cell Biol 2010;88:250–6.
69. Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006;116:536–47.
70. Blanchard C, Rothenberg ME. Biology of the eosinophil. Adv Immunol 2009;101:81–121.
71. Plager DA, Loegering DA, Checkel JL, et al. Major basic protein homolog (MBP2): a specific human eosinophil marker. J Immunol 2006;177:7340–5.
72. Wright BL, Leiferman KM, Gleich GJ. Eosinophil granule protein localization in eosinophilic endomyocardial disease. N Engl J Med 2011;365:187–8.
73. Yousefi S, Gold JA, Andina N, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 2008;14:949–53.
74. Castro M, Mathur S, Hargreave F, et al. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med 2011;184:1125–32.
75. Corren J. Inhibition of interleukin-5 for the treatment of eosinophilic diseases. Discov Med 2012;13:305–12.
76. Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med 2009;360:985–93.
77. Rothenberg ME, Klion AD, Roufosse FE, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med 2008;358:1215–28.
78. Roufosse FE, Kahn JE, Gleich GJ, et al. Long-term safety of mepolizumab for the treatment of hypereosinophilic syndromes. J Allergy Clin Immunol 2013;131:461–7.
79. Walsh GM. Profile of reslizumab in eosinophilic disease and its potential in the treatment of poorly controlled eosinophilic asthma. Biologics 2013;7:7–11.
80. Schneider L, Tilles S, Lio P, et al. Atopic dermatitis: a practice parameter update 2012. J Allergy Clin Immunol 2013;131:295–9.
81. Burks AW, Jones SM, Wood RA, et al. Oral immunotherapy for treatment of egg allergy in children. N Engl J Med 2012;367:233–43.
82. Fleischer DM, Burks AW, Vickery BP, et al. Sublingual immunotherapy for peanut allergy: a randomized, double-blind, placebo-controlled multicenter trial. J Allergy Clin Immunol 2013;131:119–27.
83. Keet CA, Frischmeyer-Guerrerio PA, Thyagarajan A, et al. The safety and efficacy of sublingual and oral immunotherapy for milk allergy. J Allergy Clin Immunol 2012;129:448–55, 455.
84. Wechsler ME, Fulkerson PC, Bochner BS, et al. Novel targeted therapies for eosinophilic disorders. J Allergy Clin Immunol 2012;130:563–71.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.