(Carregando Índice)... (Carregando Índice)... |
Última revisão: 05/04/2019
Comentários de assinantes: 0
|
Artigo original: Cochrane, T I, MD, MBA. Amato, A A, MD. Myopathies, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon.
|
Anthony A. Amato, MD
Chefe da Divisão de Doenças Neuromusculares no Departamento de Neurologia do Brigham and Women’s Hospital. Professor de Neurologia na Harvard Medical School (Boston, MA).
Thomas I. Cochrane, MD, MBA
Neurologista Associado na Divisão de Doenças Neuromusculares do Departamento de Neurologia do Brigham and Women’s Hospital. Professor Assistente de Neurologia na Harvard Medical School (Boston, MA).
Resumo
As doenças musculares (miopatia) podem ser adquiridas ou hereditárias. Os sintomas incluem fraqueza nos músculos esqueléticos, atrofias, cãibras musculares ou mialgias e problemas funcionais nos músculos respiratórios, faríngeos, faciais ou oculares. Cabe aos médicos identificar as miopatias tratáveis e iniciar as terapias antes que ocorram fraquezas permanentes. Nos casos de pacientes com distúrbios que não possam ser tratados, é extremamente importante tomar providências como tratamento de suporte apropriado, reabilitação, orientação genética e suporte psicológico. Este artigo apresenta informações sobre as miopatias comuns, incluindo distrofias musculares, hipertermia maligna, miopatias metabólicas, miopatias mitocondriais e encefalopatias; canelopatias iônicas, paralisias periódicas e miotomias não distróficas; e miopatias induzidas pelo uso de medicamentos. São enfatizados também temas como apresentação clínica, diagnóstico, patogênese e terapia. Os quadros apresentam descrições sobre a classificação genética de cíngulo límbico e de distrofias musculares distais; proteínas envolvidas nos casos de miopatia miofibrilar (MFM); outras miopatias distais; e miopatia induzida por medicamentos antirreumáticos, anti-inflamatórios e imunossupressores. As figuras mostram a membrana sarcolêmica e as proteínas enzimáticas associadas às distrofias musculares, as proteínas sarcoméricas e nucleares associadas às distrofias musculares e as vias metabólicas principais utilizadas pelos músculos.
|
miopatias |
As doenças musculares (miopatia) podem ser adquiridas ou hereditárias. Os sintomas incluem fraqueza nos músculos esqueléticos, atrofias, cãibras musculares ou mialgias e problemas funcionais nos músculos respiratórios, faríngeos, faciais ou oculares. Normalmente, os músculos proximais são afetados com maior gravidade em comparação com os músculos distais. Algumas miopatias hereditárias estão associadas a fraquezas distais proeminentes, embora a maior parte delas produza mais fraqueza proximal do que distal.
A fraqueza nos músculos oculares (por exemplo, ptose, diplopia, oftalmoparesia) sugere a presença de determinados tipos de miopatias congênitas, hereditárias ou adquiridas. As fraquezas faciais, faríngeas, cervicais ou axiais resultam na redução da expressão facial, disfonia, disfagia, queda da cabeça ou camptocormia. Em geral, nos estágios avançados, os músculos respiratórios podem ser afetados.
A fraqueza crônica está associada ao desgaste muscular. De um modo geral, os reflexos tendinosos profundos são preservados, embora possam se tornar ausentes nos casos de músculos gravemente enfraquecidos ou atrofiados. A maior parte das miopatias afeta apenas os músculos esqueléticos, ainda que, em alguns casos, ocorra também o envolvimento dos músculos cardíacos e dos músculos lisos. Comumente, os pacientes com miopatia não apresentam perturbações sensoriais ou disfunção autonômica, sendo que a maioria dos casos de miopatia não é dolorida.
A principal função do exame físico é localizar descobertas relacionadas aos músculos e excluir doenças que possam imitar sintomas e sinais miopáticos, tais como síndromes nos neurônios motores inferiores, neuropatias motoras, distúrbios de transmissão neuromuscular ou fraqueza psicogênica. Os históricos familiares completos ajudam a avaliar a probabilidade de alguma doença hereditária.
O histórico evolutivo, às vezes, revela a existência de sintomas não reconhecidos na primeira infância ou na infância (por exemplo, alimentação difícil, retardo no desenvolvimento das atividades de sentar e caminhar, assim como uma grande dificuldade para manter um bom condicionamento físico), que poderiam apontar na direção de miopatia hereditária.
Cabe aos médicos identificar as miopatias tratáveis e iniciar as terapias antes da ocorrência de fraquezas permanentes. Nos casos de pacientes com distúrbios intratáveis, providências como tratamento de suporte apropriado, reabilitação, orientação genética e suporte psicológico são extremamente importantes. Neste artigo, descreveremos as miopatias mais comuns, com ênfase em temas como apresentação clínica, diagnóstico, patogênese e terapia.
Distrofias Musculares
As distrofias musculares são miopatias hereditárias que se caracterizam pela presença de necrose nos tecidos musculares cuja reposição é feita com tecidos conjuntivos e adiposos. Tradicionalmente, as distrofias são classificadas conforme o fenótipo clínico. Cada vez mais, as classificações se baseiam na mutação genética causativa e na(s) proteína(s) anormal(is) resultante(s). A proteína defeituosa pode ser um componente do sarcolema, núcleo, sarcômero, matriz extracelular ou de uma enzima.
Distrofias Musculares Recessivas Ligadas ao X
Distrofinopatias
As distrofinopatias são causadas pela deficiência de distrofina, uma proteína citoesquelética 427 kDa em forma de bacilo. A distrofina corresponde a 5% de todas as proteínas citoesqueléticas sarcolêmicas e fixa a actina filamentosa (actina-F) ao sarcolema.2?7 A distrofina reforça a membrana plasmática mantendo uma ligação mecânica entre o citoesqueleto e a matriz extracelular.4?7
A ausência ou deficiência de distrofina enfraquece e rompe a membrana sarcolêmica, resultando na penetração de cálcio e em necrose das fibras musculares. As duas distrofinopatias mais importantes são a distrofia muscular de Duchenne (DMD) e a distrofia muscular de Becker (DMB).
As distrofinopatias são distúrbios recessivos ligados ao X produzidos por mutações no gene da distrofina no braço curto do cromossomo X na posição Xp21. A distrofina contém mais de 2.000kb de DNA e codificam 79 exons. Provavelmente por causa do tamanho da distrofina, as mutações são responsáveis por um terço de casos. Aproximadamente, dois terços de casos são causados por grandes deleções, isto é, até cerca de 1 milhão de pares de bases.
Entre 5 a 10% dos casos de DMD resultam de mutações pontuais que produzem códons de parada prematuros.8 As mutações de fase de leitura que causam perda quase total de distrofina geralmente resultam nos fenótipos mais graves de DMD, ao passo que as mutações inseridas no quadro de leitura produzem distrofina semifuncional e, na maioria das vezes, produzem fenótipo menos grave da doença.2,6?9
Distrofia muscular de Duchenne. A DMD afeta um entre 3 mil nascidos vivos do sexo masculino. Os garotos afetados se tornam sintomáticos depois que começam a andar, comumente na faixa etária entre 2 e 3 anos. Muito raramente, a DMD afeta os bebês do sexo feminino. As manifestações precoces de DMD são marcha desequilibrada, postura lordótica, pseudo-hipertrofia da panturrilha, contraturas articulares e caminhar nas pontas dos pés.
Essas manifestações são acompanhadas de fraqueza e desgaste muscular progressivo; ações como levantar-se do chão ou de uma cadeira baixa, subir escadas e erguer os braços se tornam muito difíceis. O esvaziamento gástrico tardio poderá causar episódios súbitos de vômito e dor abdominal. Esse tipo de doença é implacavelmente progressivo. Aos 12 anos de idade, as crianças afetadas têm que usar cadeiras de rodas; a maioria morre aos 25 anos, aproximadamente, devido às complicações associadas à insuficiência respiratória.
Com frequência, o músculo cardíaco também chega a ser afetado. Insuficiência cardíaca congestiva (ICC) e arritmia ocorrem em um momento mais tarde no curso da doença.10 Possivelmente, ocorra um envolvimento leve do sistema nervoso central (SNC), que, comumente, se manifesta como irritabilidade, hiperatividade ou disfunção cognitiva.
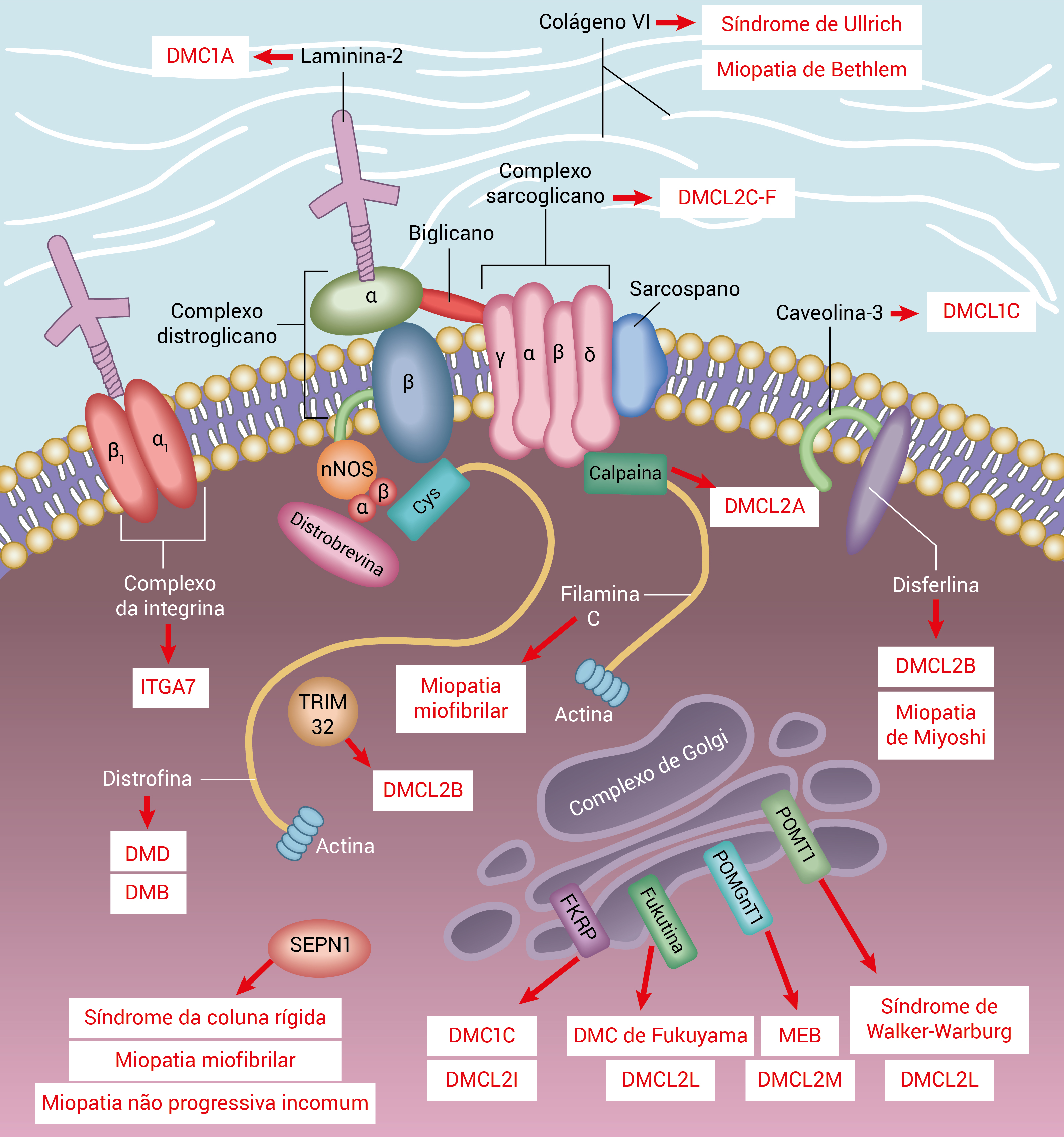
A Figura 1 mostra a membrana sarcolêmica e as proteínas enzimáticas com distrofias musculares.
DMB: distrofia muscular de Becker; DMD: distrofia muscular de Duchenne; DMC: distrofia muscular congênita; DMCL: distrofia muscular cíngulo-límbica; DMVC: doença muscular-visual-cerebral; SEPN1: selenoproteína N1.

Figura 1 - Membrana sarcolêmica e proteínas enzimáticas com distrofias musculares. As caixas indicam as doenças que são causadas por mutações proteicas individuais. A distrofina fixa o citoesqueleto formado por filamentos de actina à matriz extracelular por meio do complexo distroglicano. A distrofina interage intracelularmente com a distrobrevina e as sintrofinas (a e ß). O complexo sarcoglicano e os complexos distroglicanos se conectam através de um biglicano, que fixa ambos ao colágeno extracelular, e os sarcoglicanos d e ? interagem com a filamina C. As proteínas do complexo de Golgi afetam o estado de glicosilação do distroglicano-a e fazem a mediação de sua ligação à matriz extracelular. POMT1, POMGnT1 e LARGE também são mediadores da glicosilação, e acredita-se que se localizam no complexo de Golgi.
Os níveis séricos de creatinoquinase (CK) nos casos de DMD geralmente são elevados e chegam a atingir até 20.000IU/L, embora diminuam com a perda de massa muscular. As biópsias musculares revelam a presença de miopatia grave; os macrófagos produzem fagocitose nas fibras nervosas; com frequência, a presença de células T CD8+ é muito comum, e elas invadem as fibras musculares; ocorre um aumento nos tecidos conjuntivos; e as fibras musculares hipercontraídas são comuns.
O diagnóstico poderá ser confirmado pela ausência de distrofina nos testes imuno-histoquímicos ou pela técnica de immunoblot.9,11 Entretanto, os testes genéticos substituíram as biópsias nos casos de distrofinopatias, tendo em vista que são menos invasivos e estão se tornando gradativamente mais acessíveis sob a ótica de custo.
O tratamento inclui terapia respiratória e fisioterapia, assim como suporte psicológico, para o paciente e sua família. Recomenda-se a orientação genética para todos os pacientes. A terapia com glicocorticoides (por exemplo, 0,75 mg/kg/dia de prednisona) comprovadamente melhora a resistência e a função muscular no curto prazo (6 meses a 2 anos) e lentifica a velocidade de deterioração.12
O medicamento deflazacorte, um esteroide sintético, tem efeito terapêutico semelhante e talvez esteja associado aos efeitos colaterais de febre. Esse medicamento é comercializado na Europa, na América Central e na América do Sul, porém não está disponível no mercado norte-americano.
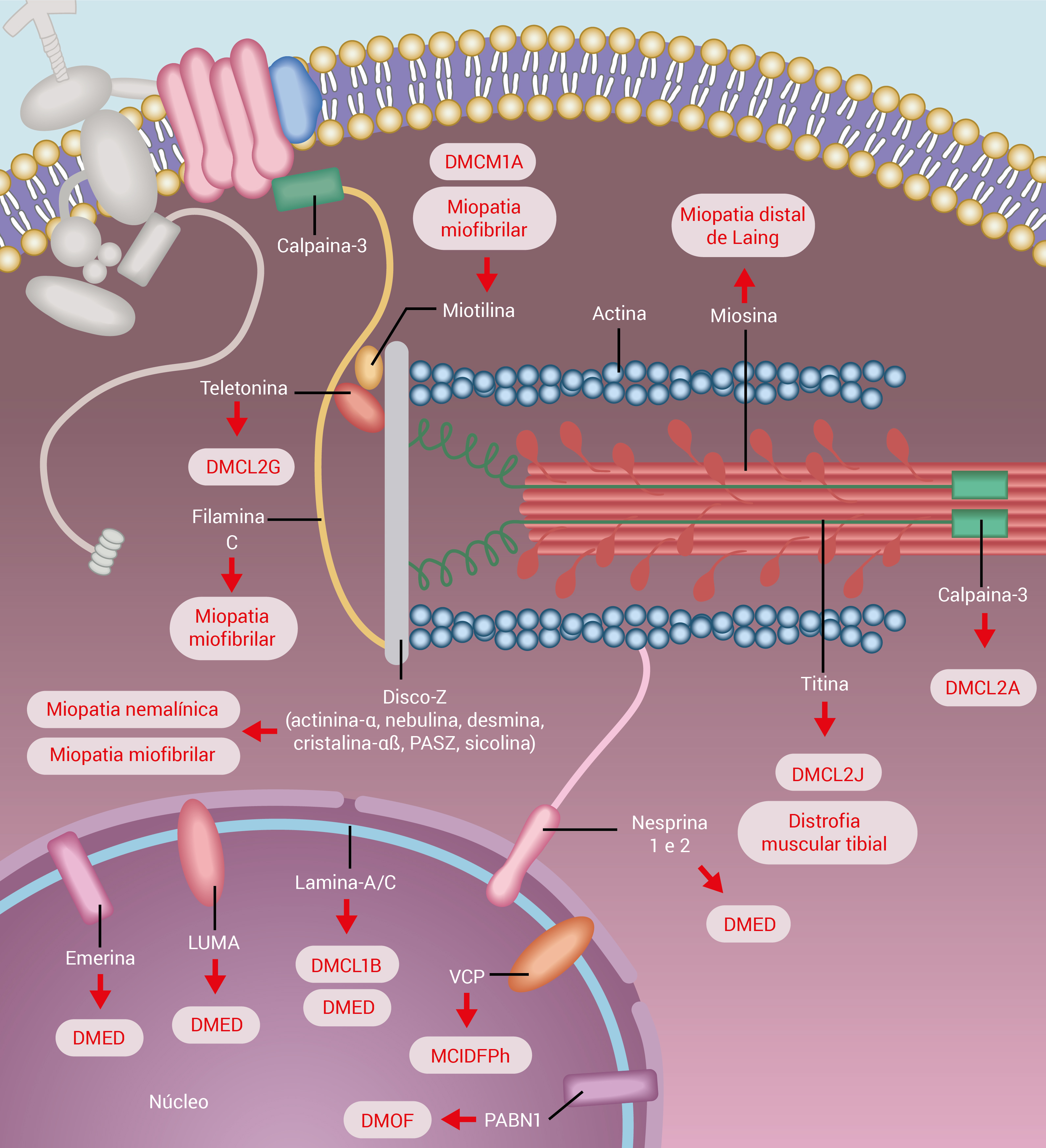
A Figura 2 mostra as proteínas sarcoméricas e nucleares associadas a distrofias musculares.
DMCL: distrofia muscular cíngulo-límbica; DMED: distrofia muscular de Emery-Dreifuss; DMOF: distrofia muscular oculofaríngea; MCIDFPh: miosite com corpúsculos de inclusão com demência frontal de Paget hereditária; PASZ: proteína associada ao disco-Z.
Figura 2 - Proteínas sarcoméricas e nucleares associadas a distrofias musculares. As doenças associadas às mutações nessas proteínas estão identificadas nas caixas da figura.
Distrofia muscular de Becker. A DMB e a DMD são distúrbios alélicos, embora a DMB geralmente inicie mais tarde e seu progresso seja lento. A idade de início da DMB é variável. Alguns casos são identificados na idade de 3 anos ou aos 70 anos; a idade média de início é de 12 anos. O espectro fenotípico da DMB é bastante amplo.
As formas leves se manifestam apenas como cãibras musculares, intolerância aos exercícios, mioglobinúria, elevação assintomática dos níveis séricos de CK, fraqueza muscular leve ou miopatia no quadríceps.2?6 Com frequência, a dor na panturrilha com esforço físico é um dos sintomas, sendo que é comum o aumento de volume na panturrilha.
A maior parte dos pacientes perde a ambulação por volta dos 40 anos de idade (faixa de 10 a 70 anos). A idade para a morte também varia de 23 a 89 anos (idade média de 42 anos). As manifestações cardíacas são comuns e a cardiomiopatia pode ser grave. Entretanto, não há nenhuma relação entre a gravidade dos sintomas cardíacos e a da miopatia.
Aproximadamente, 65% dos pacientes com DMB apresentam deleções inseridas no quadro de leitura do gene da distrofina, produzindo distrofina truncada e/ou semifuncional.2?7,9 As descobertas das biópsias se assemelham às de pacientes com DMD, embora não sejam tão graves, sendo que a coloração da distrofina é atenuada, mas não ausente. Não existe nenhuma terapia eficaz para tratamento de DMB, embora algumas séries menores tenham sugerido que há possíveis benefícios com o uso de corticosteroides.13
Transmissores femininos e distrofinopatia em mulheres. Com frequência, os transmissores femininos de mutações da distrofina são sintomáticos, embora possivelmente tenham fraqueza muscular, cãibras musculares, hipertrofia isolada na panturrilha, fadiga e níveis séricos elevados de CK.3?7,11,14 Em situações raras, os casos de DMD plenamente desenvolvida ocorrem em heterozigotos femininos nas situações em que o cromossomo X paterno, que abriga um gene normal da distrofina, é desativado em uma grande proporção de células embrionárias (hipótese de Lyon).
O Quadro 1 apresenta uma lista das distrofias musculares mais comuns.1
Quadro 1
|
Classificação Genética de Distrofia Muscular Cíngulo-Límbica | |||
|
Doença |
Herança |
Cromossomo |
Proteína afetada
|
|
Distrofias de Duchenne/Becker ligadas ao X |
XR |
Xp21 |
Distrofina |
|
DMEDX1 |
XR |
Xq28 |
Emerina |
|
DMEDX2/escapuloperoneal |
XR |
Xq27.2 |
FHL1 |
|
Distrofia muscular cíngulo-límbica DMCL1A |
AD |
5q22.3-31.3 |
Miotilina |
|
DMCL1B |
AD |
1q11-21 |
Laminas A e C |
|
DMCL1C |
AD |
3p25 |
Caveolina-3 |
|
DMCL1D |
AD |
6q23 |
DNAJB6 |
|
DML1E |
AD |
2q35 |
Desmina |
|
DMCL1F |
AD |
7q32.1 |
TNPO3 |
|
DMCL2A |
AR |
15q15.1-21.1 |
Calpaina-3 |
|
DMCL2B |
AR |
2p13 |
Disferina |
|
DMCL2C |
AR |
13q12 |
Sarcoglicano-? |
|
DMCL2D |
AR |
17q12-21.3 |
Sarcoglicano-a |
|
DMCL2E |
AR |
4q12 |
Sarcoglicano-ß |
|
DMCL2F |
AR |
5q33-34 |
Sarcoglicano-d |
|
DMCL2G |
AR |
17q11-12 |
Teletonina |
|
DMCL2H |
AR |
9q31-33 |
Ubiquitina-ligase-E3 (TRIM32) |
|
DMCL2i |
AR |
19q13 |
Proteína relacionada à fukutina (FKRP) |
|
DMCL2J |
AR |
2q31 |
Titina |
|
DMCL2K |
AR |
9q31 |
POMT1 |
|
DMCL2L |
AR |
11p14.3 |
Anoctamina-5 (ANO-5) |
|
DMCL2M |
AR |
9q31-33 |
Fukutina |
|
DMCL2N |
AR |
14q24 |
POMT2 |
|
DMCL2O |
AR |
1p32 |
POMGnT1 |
|
DMCL2P |
AR |
31p21 |
Distroglicano-a (DAG1) |
|
DMCL2Q |
AR |
8q34 |
Plectina |
|
DMCL2R |
AR |
2q35 |
Desmina |
|
DMCL2S |
AR |
4q35 |
TRAPC11 |
|
miopatias miofibrilares |
AD |
5q22.3-31.3 |
Miotilina |
|
AD |
10q22.2-23.2 |
ZASP | |
|
AD |
7q32.1 |
Filamina-c | |
|
AD |
11q21-23 |
Cristalina ad | |
|
AD/AR |
2q35 |
Desmina | |
|
AR |
2q31 |
Titina | |
|
AD |
6q23 |
DNAJB6 | |
|
AD |
7q32.1 |
TNPO3 | |
|
MCI hereditária MCI hereditária-AR |
AR |
- |
GNE |
|
MCI-H com DFT e doença de Paget |
AD |
- |
VCP |
|
MCI-H 3 |
AD |
- |
MyHC Ha |
|
Distrofias/miopatias distais Welander |
AD |
2p13 |
TIA |
|
Udd |
AD |
2q31 |
Titina |
|
Quadro 1: Continuação
| |||
|
Doença |
Herança |
Cromossomo |
Proteína afetada
|
|
Markesbery-Griggs |
AD |
10q22.3-23.2 |
PASZ |
|
Nonaka |
AR |
9q1-p1 |
GNE |
|
Miyoshi |
AR |
2p13 |
Disferlina |
|
Laing |
AD |
14q11 |
MyHC7 |
|
Williams |
AD |
7q32.1 |
Filamina C |
|
Miopatia nebulínica |
AR |
2q21.2-q22 |
Nebulina |
|
Miopatia com início precoce e mutação 9 semelhante à Kelch (Drosofilia) |
AD |
9p22 |
9 semelhante à Kelch |
|
Outras distrofias DMED3 |
AD |
6q24 |
Nesprina-1 |
|
DMED4 |
AD |
14q23 |
Nesprina-2 |
|
DMED5 |
AD |
3p25.1 |
LUMA |
|
Distrofia escapuloperoneal |
AD |
2q35 |
Desmin |
|
DFEU1 |
AD |
4q35 |
DUX4 |
|
DFEU2 |
AD |
18p11 |
SMCHD1 |
AD: autossômica dominante; AR: autossômica recessiva; DFEU:distrofia facioescapuloumeral; DFT: demência frontotemporal; DMED: distrofia muscular de Emery-Dreifuss; GNE: UDP-N-acetilglicosamina 2-epimerase/N-acetilmanosamina quinase; MCI-H: miosite com corpúsculos de inclusão hereditária; MyHC: cadeia pesada de miosina; PASZ: proteína associada ao disco-Z; PDZ: proteína contendo motivo; POMGNT1: 0-manose ß-1,2-N-acetilglicosaminila transferase; POMT1: gene da 0-manosiltransferase; VCP: proteína contendo valosina; XR: recessiva ligada ao X.
Cardiomiopatia dilatada ligada ao X. Cardiomiopatia dilatada ligada ao X é resultado da deficiência de distrofina nos músculos cardíacos, mas não nos músculos esqueléticos. Os pacientes se apresentam com algum distúrbio cardíaco progressivo; ICC ocorre na segunda ou terceira décadas de vida.
Os portadores femininos que manifestam a doença têm cardiomiopatia com início lento que se apresenta ao redor da quinta década de vida. Acredita-se que as deleções que ocorrem nas proximidades do exon 1 do gene da distrofrina, que afetam a expressão ou a função da distrofina no músculo cardíaco, sejam as causadoras da doença.4?7
Distrofia Muscular de Emery-Dreifuss
A Distrofia Muscular de Emery-Dreifuss (DMED) tem várias formas genéticas: duas mutações recessivas ligadas ao X, a emerina e FHL1 (DMEDX1 e DMEDX2) e, pelo menos, quatro formas autossômicas dominantes (DMED-AD). Essas formas de DMED são clinicamente indistinguíveis. A DMED-XL é causada por mutações na emerina, uma proteína com membrana nuclear15; a DMED-AD, geralmente, é causada por mutações no gene da lamina A/C, que codifica duas proteínas das laminas nucleares, A e C.16
Comumente, as mutações da lamina A/C causam cardiomiopatia e defeitos de condução.15 As proteínas com membranas nucleares integrais interagem com as lâminas nucleares, que são proteínas com filamentos intermediários no lado nucleico (interno) da membrana nuclear. A emerina se liga à lamina A, um dos produtos do gene da lamina A/C. Outras proteínas envolvidas na DMED-AD incluem a nesprina 1 e 2 e a LUMA e todas as proteínas associadas à estabilização da membrana nuclear.
A DMED causa:
fraqueza humeroperoneal e desgaste no bíceps, no tríceps, no músculo tibial anterior e no músculo peroneal, que progride lentamente para um padrão escapulopelvicoperoneal envolvendo o músculo peitoral e o músculo pélvico;
desenvolvimento precoce de contrações nos cotovelos, tendão de Aquiles, pescoço e coluna, que poderá ocorrer antes que haja uma fraqueza significativa;
envolvimento cardíaco, que se apresenta como defeitos de condução, com bradicardia e intervalo PR prolongado.
A presença de paralisia atrial isolada é uma forte sugestão de DMED. De um modo geral, as portadoras femininas de DMED-XL não têm fraqueza, embora possivelmente desenvolvam bloqueio cardíaco.
A DMED inicia no decorrer das primeiras duas décadas de vida. O diagnóstico é feito com base na apresentação clínica, níveis ligeiramente elevados de CK e biópsias musculares, sendo que a confirmação diagnóstica é feita por análises mutacionais. As biópsias musculares revelam a presença de características miopáticas inespecíficas, embora a ausência de emerina dê suporte ao diagnóstico de DMED-XL.
Os testes genéticos atualmente disponíveis identificam os portadores que possivelmente tenham defeitos de condução cardíaca. O reconhecimento imediato e a colocação de marca-passo cardíaco evitam a ocorrência de morte súbita ou de ataques de síncope.
Embora não haja nenhum tratamento médico específico, os pacientes precisam fazer avaliações cardíacas e, com frequência, é necessário fazer implante profilático de marca-passo e de desfibriladores intracardíacos, levando-se em consideração a grande propensão para arritmias cardíacas. A fisioterapia provavelmente lentifique o desenvolvimento de contrações.
Distrofias Musculares Cíngulo-Límbicas
As distrofias musculares cíngulo-límbicas (DMCLs) são causadas por um grande número de mutações e se classificam como autossômicas dominantes (DMCL1A-F) ou autossômicas recessivas (DMCL2A-J).1 O termo cíngulo-límbica se refere à fraqueza nos músculos proximais da cintura escapular e da cintura pélvica. Entretanto, as diversas mutações associadas ao fenótipo clássico de DMCL também poderão causar fraqueza distal.
Distrofias Musculares Cíngulo-Límbicas Autossômicas Dominantes
Essas distrofias correspondem a menos de 10% de todas as DMCLs. Elas se apresentam como fraqueza muscular proximal e distal lentamente progressiva e níveis séricos elevados de CK. As DMCLs tendem a ser mais leves que a DMCL autossômica recessiva (DMCL2). Não há nenhum tratamento específico para esse grupo de distúrbios.
DMCL1A. Esse tipo de distúrbio é causado por mutações na miotilina, que é uma proteína imprescindível para a formação e a manutenção normal de sarcômeros.17 As mutações na miotilina produzem fraqueza nos músculos proximais e distais e podem causar disartria, hipofonia e voz anasalada. Esse distúrbio é alélico em relação à miopatia miofibrilar (MFM) causada por mutações na miotilina.17
DMCL1B. Esse tipo de distúrbio é causado por mutações na lamina A/C; o fenótipo desse distúrbio se assemelha ao da DMED-AD. Além de apresentarem sintomas de DMCL, alguns pacientes têm lipodistrofia familiar parcial que se caracteriza por uma redução nas gorduras subcutâneas, resistência à insulina, elevação nos níveis de triglicérides, níveis baixos de lipoproteínas de alta densidade (HDLs), diabetes melito e aumento no risco de doença vascular aterosclerótica.6,16
DMCL1D. Esse tipo de distúrbio é causado por mutações no gene da caveolina-3.15,18?21 A caveolina é uma proteína sarcolêmica associada à renovação da membrana. Os pacientes poderão apresentar fenótipos clínicos diferentes; talvez eles se apresentem também com DMCL, hiperckemia isolada, doença nos músculos ondulados ou miopatia distal.
DMCL1D. É causado por mutações no gene da desmina. O início ocorre na segunda década de vida ou um pouco mais tarde e os pacientes se apresentam com arritmia cardíaca ou cardiomiopatia dilatada. As mutações na desmina resultam de alterações miofibrilares e são também classificadas como a MFM tipo 1.22,23
DMCL1E. Esse tipo de distúrbio é causado por mutações no DNAJB6. A fraqueza proximal nas pernas é proeminente, sendo que o início da doença varia entre os 20 e 60 anos de idade. Em geral, os pacientes perdem a ambulação algumas décadas após o início da doença.
DMCL1F. É causado por mutações no gene da transportina 3 (TNP03), que codifica um receptor de importação nuclear para os fatores de junção do RNA mensageiro precursor. As biópsias musculares revelam a presença de características de MFM.24
Distrofias Musculares Cíngulo-Límbicas Autossômicas Recessivas (DMCL2)
Foram identificadas as seguintes formas dessas distrofias:15,25
DMCL2A. Essa é a forma mais comum de DMCL causada por mutações no gene da calpaína-3 que se localiza no cromossomo 15q15.1-q15.3.25 A calpaína-3 é uma protease ativada por cálcio que desempenha papel importante na diferenciação muscular.
O início da doença ocorre entre 8 e 30 anos de idade, sendo que os pacientes se apresentam com fraqueza nos músculos da cintura pélvica, em especial os músculos glúteos, preservando os abdutores do quadril. Observa-se com frequência a presença de escápulas aladas, assim como o envolvimento da parte posterior da coxa. Os níveis séricos de CK geralmente são elevados, sendo que, às vezes, chegam a atingir mais de 9.000 IU/L. O ensaio Western Blot ajuda a confirmar o diagnóstico.
DMCL2B. Esse tipo de distúrbio é causado por mutações no gene da disferlina.26,27 Embora seja uma proteína associada à membrana, a disferlina não faz parte do complexo da distrofina-glicoproteína. A disferlina interage com a caveolina e possivelmente esteja envolvida no reparo da membrana.
Dois fenótipos foram descritos nas mutações da disferlina: miopatia de Miyoshi, que se caracteriza por fraqueza distal com níveis séricos muito elevados de CK e envolvimento precoce do músculo gastrocnêmio, sendo que a DMCL2B se apresenta como fraqueza nos músculos distais e proximais em pacientes na fase final da adolescência ou no início dos 20 anos.
Mutações idênticas podem causar a miopatia de Miyoshi e a DMCL2B.26,27 A ausência de disferlina talvez produza perturbações na nova selagem da membrana e interferências nos reparos das fibras musculares danificadas, possivelmente como resultado de defeitos no tráfego vesicular no interior das fibras musculares.6,15,26,27
DMCL2C, 2D, 2E, 2F. Esses quatro distúrbios são sarcoglicanopatias causadas por mutações em genes que codificam os membros do complexo sarcoglicano: DMCL2C (gene do sarcoglicano-? no cromossomo 5q33), DMCL2D (gene do sarcoglicano-? no 17q12), DMCL2E (sarcoglicano-ß no cromossomo 4) e DMCL2F (sarcoglicano-d, mapeado no 13q12).15,28,29 Em todos esses quatro distúrbios, os três outros componentes do complexo sarcoglicano se perdem ou são parcialmente ausentes, embora o complexo distroglicano seja normal.4,6,15,21,28
As mutações nos sarcoglicanos rompem a ligação entre os sarcolemas e a matriz extracelular. As sarcoglicanopatias são responsáveis por, aproximadamente, 10% de todos os casos de DMCL.29 As sarcoglicanopatias produzem fraqueza, variando de leve a grave, nos músculos proximais (especialmente nos músculos das pernas), elevações nos níveis séricos de CK para algo em torno de 3.000IU/L, alterações distróficas nos músculos e, com frequência, hipertrofia na panturrilha.
A idade de início é variável. Técnicas como imunocitoquímica e análise immunoblot de espécimes de biópsias musculares demonstram a ausência ou deficiência grave de sarcoglicanos. Aparentemente, a gravidade da fraqueza muscular depende do grau da expressão residual de sarcoglicanos. Cardiomiopatia dilatada poderá ocorrer de forma isolada ou em combinação com miopatia esquelética.
DMCL2G. Esse distúrbio é causado por mutações no gene que codifica a teletonina, uma proteína sarcomérica que se localiza no disco Z dos músculos esqueléticos e provoca o rompimento da estrutura sarcomérica.30 A DMCL2G é um distúrbio raro e relativamente leve que ocorre na infância, geralmente com envolvimento dos músculos distais.30 As biópsias dos músculos mostram a presença de vacúolos no interior das fibras musculares. Os níveis séricos de CK são entre 10 a 30 vezes mais elevados.
DMCL2H. Esse distúrbio é uma doença muito rara que afeta a população de huteritas canadenses da região de Manitoba e é causada por mutações no gene TRIM32 que codifica a E3-ubiquitina ligase.31,32 A DMCL2H é alélica em relação à miopatia sarcotubular e está associada a dilatações no retículo sarcoplasmático (RS), possivelmente devido à desregulação na renovação da proteína miofibrilar.33
DMCL2I. É causado por mutações no gene relacionadas à proteína fukutina (FKRP).31,34 A DMCL2I e a DMC1C são alélicas. Esses dois distúrbios estão relacionados a alterações na expressão do distroglicano-a (a-DG) resultantes de defeitos na glicosilação. O fenótipo da DMCL2I é bastante comum, ao passo que o fenótipo da DMC1C é raro. Em alguns países (por exemplo, no Reino Unido),34 a DMCL2I é uma das formas mais comuns de DMCL.
O início da doença ocorre na fase inicial da infância ou na vida adulta. Os pacientes se apresentam com fraqueza muscular proximal e aumento no tamanho das panturrilhas, nível sérico elevado de CK, às vezes com aumento no volume da língua e, com frequência, com envolvimento respiratório e cardíaco. O nível de inteligência é normal.
DMCL2J. Esse distúrbio é causado por mutações no gene da maior proteína conhecida, a titina. Além da DMCL2J, que se apresenta com fraqueza proximal com início na infância, as mutações na titina podem produzir miopatia distal autossômica dominante (miopatia distal tipo Udd) e uma miopatia hereditária com insuficiência respiratória precoce (MHIRP) autossômica dominante.1
DMCL2K, DMCL2M, DMCL2N e DMCL2O. Às vezes, esses distúrbios são conhecidos por distroglicanopatias-a secundárias. Normalmente, eles se apresentam na infância, sendo que, nessa hipótese, se classificam como distrofia muscular congênita (DMC). Nas situações em que se apresentam em um momento mais tarde na infância ou na fase inicial da vida adulta, esses distúrbios produzem um fenótipo de DMCL. A DMCL2K é o resultado de mutações na proteína O-manosiltransferase 1 (POMT1) e, comumente, está associada à DMC de Walker-Warburg.
A DMCL2M é causada por mutações na fukutina, que foram originalmente descritas em pacientes com o tipo mais grave (e mais comum) da DMC de Fukuyama. As mutações na proteína O-manosiltransferase 2 (POMT2) podem produzir DMCL2N, que, por sua vez, pode causar também a síndrome de Walker-Warburg. A DMCL2O é o resultado de mutações na proteína O-manose-ß-1,2-N-acetilglicosaminil transferase (POMGTn1).
DMCL2L. Esse distúrbio é o resultado de mutações na proteína transmembrânica anoctamina-5 (ANO-5), que é um canal de cloreto ativado com cálcio. A DMCL2L está associada à fraqueza no cíngulo límbico com atrofia no quadríceps e mialgias, embora alguns pacientes apresentem fraqueza distal com fraqueza proeminente na panturrilha (semelhante à miopatia de Miyoshi).1,35?37 De um modo geral, não há envolvimento cardíaco ou respiratório.
DMCL2P. Esse distúrbio, também conhecido por distroglicanopatia-a, é causado por mutações no gene que codifica a a-DG. A DMCL2P se apresenta na primeira década de vida com danos cognitivos graves.
DMCL2Q. É o resultado de mutações no gene que codifica a plectina-1. A deficiência de plectina-1 está associada à epidermiólise bolhosa (formação de bolhas na pele e nas membranas mucosas com início na primeira infância ou no início da infância); além disso, é também considerada uma forma de miastenia congênita. Além da hipotonia congênita ou neonatal, com fraqueza proximal progressiva, condições como ptose e oftalmoplegia também são ocorrências comuns.
DMCL2R. Esse distúrbio é o resultado de mutações na desmina. A maior parte dos casos de desminopatia primária se refere a heranças autossômicas dominantes e se classifica como DMCL1E ou como MFM. Essa mutação recessiva na desmina se apresenta no período entre a infância e a vida adulta, com fraqueza proximal lentamente progressiva e insuficiência ventilatória.38,39
DMCL2S. Esse distúrbio é o resultado de mutações na subunidade 11 do complexo de partículas de proteínas de transporte (TRAPPC11). Há um relato recente de DMCL2S nas populações síria e huterita que se apresenta com coreoatetoide infantil ou com movimentos distônicos, convulsões, ataxia no tronco e retardo mental. A presença de fraqueza proximal com escoliose e displasia no quadril é comum. A TRAPPC11 é importante no tráfego de proteínas entre o retículo endoplasmático e o complexo de Golgi.
miopatias Miofibrilares
Os filamentos intermediários fazem a integração mecânica das miofibrilas e protegem as fibras musculares contra o estresse mecânico. As miopatias miofibrilares (MFMs) afetam os filamentos musculares intermediários, provocando a desintegração miofibrilar que envolve o disco Z do sarcômero. Esse processo resulta no rompimento miofibrilar que se observa na microscopia eletrônica (ME) e no acúmulo excessivo de desmina e de outras proteínas miofibrilares.1,40?47
Embora se apresentem comumente entre as idades de 25 e 45 anos, as MFMs podem ocorrer em qualquer idade. Elas se apresentam com defeitos de condução cardíaca ou com uma miopatia esquelética com início distal que progride até envolver os músculos proximais, faciais ou respiratórios.43
A suspeita diagnóstica ocorre a partir do momento em que se acumulam produtos miofibrilares nas biópsias musculares; a confirmação do diagnóstico é feita pela análise das mutações. Não existe nenhum tratamento específico para esses distúrbios, embora o reconhecimento imediato seja muito importante para evitar a ocorrência de morte súbita causada por arritmias.
Distrofias Musculares Congênitas
As DMCs são distúrbios autossômicos recessivos que se caracterizam pela presença de hipotonia e fraqueza perinatal e de biópsias musculares com aparência distrófica. Com frequência, esses distúrbios estão associados a anormalidades nos olhos, malformações no cérebro e anormalidades cognitivas.48?50
No período neonatal, o nível sérico de CK é bastante elevado, em geral na casa dos milhares. Um esquema recente de classificação apresentou a proposta de classificar as DMCs de acordo com a localização das proteínas defeituosas e a patogênese da distrofia.51?53
As principais categorias de DCMs são as seguintes:
defeitos em genes que codificam as proteínas estruturais da lâmina basal, a matriz extracelular ou as proteínas sarcolêmicas que ligam a lâmina basal;
defeitos que causam danos na glisosilação da a-DG;
DCM causada por mutações nas selenoproteínas.
Dcm Causada por Defeitos na Lâmina Basal, Matriz Extracelular Ou Proteínas De Ligação Da Lâmina Basal
DCM1A. É responsável por mais de 40% dos casos; ela é conhecida também por DCM clássica ou deficiência de merosina (laminina a2). Essa doença está associada ao cromossomo 6q22 e a defeitos na merosina, a espinha dorsal da base da membrana.
Ao contrário das outras DCMs, em geral não há malformações no cérebro ou retardo mental, embora seja elevada a incidência de epilepsia (12 a 30%); essa doença está associada às alterações na substância branca identificadas nas imagens por ressonância magnética (IRMs). Esse tipo de doença pode se manifestar mais tarde no período inicial da vida adulta.4,6,50
Alguns pacientes apresentam deficiência parcial de merosina, resultante de alguma mutação leve ou de causas secundárias, geralmente mutações nos genes que codificam a fukutina ou a proteína relacionada com a fukutina (FKRP).
Classicamente, os pacientes se apresentam com hipotonia neonatal e fraqueza, níveis séricos elevados de CK, efeitos motores tardios importantes, neuropatia axonal e fraqueza nos músculos respiratórios.4,6,50 Alguns pacientes com DCM apresentam descobertas neuropáticas, levando-se em consideração que a a2-laminina, ß1-laminina e ?1-laminina também estão presentes nas células de Schwann.
O Quadro 2 apresenta as proteínas envolvidas na MFM.
Quadro 2
|
Proteínas Envolvidas na Miopatia Miofibrilar | |
|
Proteína |
Características |
|
Desmina |
Proteína filamentosa intermediária tipo III; miofibrilas com ligação cruzada ao nível do disco Z que as conecta a outras organelas celulares e, consequentemente, escoram o citoesqueleto. No coração, a desmina é intensificada em discos intercalados e é o componente principal das fibras de Purkinje. |
|
Cristalina-aß |
Essa proteína pertence à pequena família de proteínas de choque térmico e serve de acompanhante para a desmina. |
|
Miotilina |
Proteína sarcomérica no disco Z com forte expressão nos músculos esqueléticos e com fraca expressão no músculo cardíaco; essa proteína desempenha um papel importante no conjunto de sarcômeros. |
|
ZASP |
Componente principal do disco Z; essa proteína mantém sua integridade estrutural durante a contração. |
|
Filamina C |
A filamina C faz ligação cruzada ao nível do disco Z e se liga a outras proteínas do disco Z e interage com o complexo distrofina-distroglicano no sarcolema. |
|
BAG3 |
A BAG3 participa das rotas antiapoptóticas e tem forte expressão nos músculos esqueléticos e cardíacos. |
|
DNAJB6 |
As mutações podem se apresentar com um fenótipo de MFM (com predominância de fraqueza distal que progride para proximal) ou com fraqueza proximal cíngulo-límbica, sendo que, nesse caso, seria classificada como DMCL1E. |
|
TNPO3 |
As mutações podem se apresentar com um fenótipo de MFM ou como DMCL1F. |
|
DMCL: distrofia muscular cíngulo-límbica. |
DCM1B. Esse distúrbio se refere à DMC com um fenótipo que é essencialmente o mesmo da DCM1A (DCM clássica), embora seja um pouco mais leve e somente com deficiência parcial de merosina. A DCM1B foi mapeada nos sítios do cromossomo 1q42, porém o gene afetado ainda não foi identificado. A DCM positiva para a merosina também foi associada em três casos às mutações no gene da integrina-a7, que se localiza no cromossomo 12q13, um dos quais incluía retardo mental com IRM normal.54,55
Doença de Ulrich e Bethlem. Existem três formas de DCM que se caracterizam por contrações articulares; os pacientes com esses distúrbios não apresentam defeito na glicosilação nem retardo mental. Esses distúrbios são a DMC com síndrome da coluna rígida (RSMD), a miopatia de Ulrich e a miopatia de Bethlem. Não há nenhuma terapia disponível para o tratamento desses distúrbios.
A miopatia de Ulrich é causada por mutações em uma das cadeias de polipeptídeos que forma o colágeno VI, que é imprescindível na interação com a matriz extracelular.56 Esses pacientes apresentam mobilidade distal excessiva em combinação com contraturas proximais, espinha rígida ao nascer e fraqueza muscular. As biópsias musculares mostram ausência de colágeno VI. As miopatias de Ulrich e de Bethlem são distúrbios alélicos.
A miopatia de Bethlem, uma doença autossômica dominante, é causada por mutações em subunidades de proteínas da matriz extracelular no colágeno VI a1, a2 e a3.53 O início desse tipo doença ocorre na infância ou na adolescência. As manifestações clínicas incluem fraqueza muscular leve e contraturas em diversas articulações, cujas presenças ocorrem mesmo no nascimento.
DCM causada por problemas na glicosilação de a-DG
DCM1C. É uma doença rara causada por mutações no gene FKRP, que se localiza no cromossomo 19q13.3. A DCM1C e a DMCL2I são distúrbios alélicos. Nos casos de DCM1C, a fraqueza se manifesta na primeira semana após o nascimento. Os pacientes não conseguem atingir ambulação independente. Esse tipo de doença é marcado por envolvimento respiratório; a inteligência pode ser normal. Suspeita-se da presença de DCM1C pela redução variável ou pela ausência de a-DG nos músculos ou pela redução no tamanho de a-DG nas análises immunoblot.48,49
DCM1D. É causado por uma mutação no gene LARGE. Trata-se de um distúrbio raro que poderá resultar em retardo mental grave e distrofia. Confirma-se o diagnóstico pela ausência de a-DG nas biópsias musculares.48,49
DCM de Fukuyama (DCMF). Esse tipo de distúrbio é a DCM mais comum em indivíduos de descendência japonesa. A DCMF é causada por uma mutação no gene da fukutina, resultando na deficiência de fukutina e glicosilação reduzida em a-DG, detectada pela coloração reduzida de a-DG no músculo. Os pacientes portadores desse distúrbio apresentam malformações no cérebro, retardo mental profundo e anormalidades oftalmológicas. A doença inicia antes da idade de 7 meses, e os pacientes nunca aprendem a andar. Em geral, esses pacientes morrem em torno da idade de 20 anos.48,49
Doença muscular-visual-cerebral. A doença muscular-visual-cerebral (MVC) é causada por uma mutação no gene POMGnT da glicosiltransferase, resultando na deficiência de a-DG confirmada por imunocitoquímica. Os pacientes se apresentam com hipotonia, fraqueza, hidrocefalia variando de leve a moderada, hipoplasia cortical ou cerebelar e anormalidades visuais (por exemplo, miopia, microftalmia e hipoplasia no nervo ótico).48?50,54
Sindrome de Walker-Warburg. A síndrome de Walker-Warburg resulta de mutações no gene POMGGnT1 da O-manosil transferase. Essa síndrome é a forma mais grave de DCM e compartilha as mesmas características clínicas de FMDC e da doença muscular-visual-cerebral. Em geral, os pacientes morrem em torno da idade de 3 anos.48?50,54
DCM causada por mutações na selenoproteína N1
Síndrome da coluna rígida. A síndrome da coluna rígida, ou distrofia muscular com coluna rígida (DMCR), é o resultado de mutações na selenoproteína N1 (SEPN1) que se localiza no cromossomo 1q35-36.36?38 Os pacientes se apresentam com fraqueza muscular e rigidez espinal distintiva que resulta em incapacidade de flexionar o pescoço, escoliose e dificuldades respiratórias.
miopatias causadas pela nemalina devido a mutações nas proteínas com filamentos finos
As miopatias causadas pela nemalina são agrupadas com base na descoberta histopatológica de bacilos nas biópsias musculares. De um modo geral, essas miopatias se apresentam como miopatias congênitas, com fraqueza generalizada no nascimento, ou mesmo com acinesia fetal, embora seja comum a presença de formas mais leves na vida adulta, sendo que também foram descritas formas distais e nas extremidades inferiores.
Aparentemente, sete defeitos genéticos autossômicos são responsáveis pela miopatia causada pela nemalina. Em seis desses defeitos, a função do produto genético está associada aos filamentos sarcoméricos finos.57 Os genes implicados nesse processo estão envolvidos na manutenção de uma estrutura normal do disco Z. Esses genes incluem a a-tropomiosina (TPM3), a ß-tropomiosina (TPM2), a nebulina (NEM2), a troponina T (TnT1) e a a-actina (ACTA1).58
miopatias DISTAIS
Essas miopatias afetam, predominantemente, as mãos e/ou os pés no início de seu curso, ao contrário da maioria das miopatias, que afetam mais gravemente os músculos proximais. Algumas miopatias distais apresentam um fenótipo distinto que poderá ajudar a direcionar os testes genéticos. Esses distúrbios se sobrepõem claramente a alguns tipos de distrofia muscular cíngulo-límbica e a alguns tipos de MFM, conforme mostra o Quadro 3.1,59
Quadro 3
|
Outros Tipos de Miopatia Distal | |
|
Miopatia |
Características
|
|
Miopatia de Welander |
Distúrbio autossômico dominante distal causado por mutações em TIA1 no cromossomo 2. |
|
Miopatia de Markesbery-Griggs |
Distúrbio causado por mutações em ZASP no cromossomo 10. Esse distúrbio é alélico em relação a algumas formas de MFM associadas a mutações em ZASP. |
|
Miopatia de Nonaka |
Distúrbio autossômico recessivo causado por uma mutação no gene GNE que se localiza no cromossomo 9. O início ocorre no começo da fase adulta; esse distúrbio está associado a um fenótipo distinto, também descrito como miopatia de corpo de inclusão hereditária autossômica recessiva (h-IBM). Nesse distúrbio, a fraqueza se apresenta inicialmente nos músculos tibiais anteriores da perna. A fraqueza progride no sentido proximal, preservando o quadríceps. Nas biópsias musculares, observa-se a presença de vacúolos. |
|
Miopatia de Miyoshi |
Os relatos iniciais indicam que esse distúrbio é causado por mutações na disferlina e é alélico em relação em relação à DMCL2B. Mais recentemente, concluiu-se que as mutações na anoctomina-5 causam o fenótipo de Miyoshi, também conhecido por miopatia de Miyoshi tipo 2, que é alélica com DMCL2L. A miopatia de Miyoshi está associada à fraqueza distal na perna, afetando preferencialmente o compartimento posterior (panturrilhas). Embora seja miopática, em geral a biópsia não revela a presença de vacúolos. |
|
Miopaia de Laing |
É causado por mutações na cadeia pesada 7 da miosina (MyHC7); ele causa um tipo de miopatia distal (queda progressiva dos pés, dedos e fraqueza no pescoço) e, ocasionalmente, cardiomiopatia. |
|
Miopatia de William |
É causado por mutações na filamina C e se manifesta como queda progressiva nos pés; é alélico a algumas formas de miopatia miofibrilar. |
MFM: miopatia miofibrilar.
Distrofias Autossômicas Dominantes Com Um Único Fenótipo
Distrofia Miotônica
A distrofia miotônica (DM) é a distrofia muscular mais comum em adultos, cuja incidência é de um entre 8 mil, e com prevalência de, aproximadamente, cinco em cada 100 mil indivíduos. Existem também dois tipos desse tipo de distrofia: DM1 (DM clássica, doença de Steinert) e DM2 (miopatia miotônica proximal [MMPRO]). Ambas são síndromes multiorgânicas autossômicas dominantes; elas têm muitas semelhanças nas manifestações clínicas.60,61
A DM1 tem fenótipos distintos:
ptose, sem envolvimento dos músculos extraoculares;
atrofia dos músculos masseter e temporal, resultando em uma configuração facial estreita exclusiva;
atrofia no músculo esternocleidomastoideo, com preservação relativa do músculo cervical posterior e do músculo da cintura escapular (sinal clínico que diferencia a DM1 de distrofia facioescapuloumeral [DFEU]);
atrofia distal, com ligeiro envolvimento proximal nos estágios iniciais da doença;
envolvimento dos músculos palatais e faríngeos, com provável desenvolvimento de disartria e disfagia.4,6,60,61
A miotonia - relaxamento lento de uma contração muscular normal ? é um sinal clínico importante. Miotonia de preensão é a incapacidade de soltar imediatamente o punho depois de uma contração forçada. A percussão na eminência tenar ou nos músculos extensores dos dedos também mostra o relaxamento lento da miotomia percussiva.
As características sistêmicas incluem defeitos na condução cardíaca, disfunção mental leve (geralmente, com expressões ou comportamentos infantis ou inapropriados), atrofia testicular, calvície frontal, catarata, envolvimento do trato gastrintestinal (com motilidade e esvaziamento tardio), hipersonia e resposta reduzida à hipóxia, levando à má concentração e à apatia.
Os fatores que distinguem DM1 de DM2 são o envolvimento preferencial dos músculos proximais na DM2, incidência reduzida de antecipação em DM2 e a raridade da disfunção cognitiva em DM2. Somente nos casos de DM1, as crianças de mães afetadas correm o risco de adquirir DM congênita: movimentos retais reduzidos e hipotonia neonatal/infantil grave, dificuldades de alimentação, fraqueza facial e desconforto respiratório.
O diagnóstico clínico de DM se baseia em descargas miotônicas na eletromiografia (EMG). A presença de catarata também é um suporte. De um modo geral, na DM1, porém raramente na DM2, a idade de início é progressivamente mais precoce em sucessivas gerações (antecipação genética).
A DM1 é causada por uma repetição na sequência instável dos trinucleotídeos citosina, timina e guanina (CTG) na região de codificação não proteica de um gene da proteinoquinase denominado DMPK, que se localiza no cromossomo 19.60?63 Em pessoas ligeiramente afetadas, uma região de repetição polimórfica de CTG no gene da proteinoquinase expande em 50 a 80 repetições; em pessoas gravemente afetadas, provavelmente ocorram mais de 2 mil repetições.
A magnitude da expansão dos trinucleotídeos formados pela CTG aumenta com as gerações e é responsável pela antecipação. As medições da expansão de CTG são utilizadas para confirmar a presença de DM em membros da família para fins de diagnóstico pré-natal ou para a orientação efetiva de pessoas assintomáticas que correm o risco de desenvolver DM.
A DM2 é causada por uma repetição no tetranucleotídeo CCTG expandido em uma região não codificável do gene ZNF9 (proteína 9 zíncica) localizada no cromossomo 3q21 que codifica um fator de transcrição. Nos casos de DM1 e DM2, o produto do RNA do gene mutante é o fator patogênico, e não o produto da proteína genética.
Essa é a primeira doença conhecida que é causada por RNA prejudicial.60 O RNA mutante forma inclusões no núcleo (inclusões ribonucleares). Comprovou-se que a repetição de RNA se liga às proteínas na família de ligação muscular e interfere na função normal de processamento do RNA.63
A terapia é sintomática e preventiva. O suporte emocional e a educação envolvendo as precauções necessárias para evitar quedas e lesões são essenciais. O monitoramento rigoroso do estado cardíaco, em especial durante a administração de anestesia, é extremamente importante.
Medicamentos como quinino, procainamida, mexiletina, fenitoína e bloqueadores-ß ajudam a aliviar a miotonia, embora não tenham nenhuma ação contra a fraqueza. Entre esses agentes, a mexiletina aparentemente é o mais eficaz. A testosterona não obteve sucesso como terapia para DM. O modafinil pode diminuir a sonolência durante o dia e melhorar o humor.
Distrofia Facioescapuloumeral
A DFEU é a terceira forma mais comum de distrofia muscular. De um modo geral, os sintomas se apresentam no decorrer da segunda década de vida. Embora seja uma doença autossômica dominante, algo em torno de 25% dos casos de DFEU resultam de mutações novas.64,65
A heterogeneidade fenotípica é significativa. Alguns pacientes com mutação por deleção não têm as características distintivas de DFEU e se apresentam com miopatia no cíngulo límbico ou mesmo com miopatia distal. Em alguns membros da família, o envolvimento dos músculos faciais é mínimo. Em 10% das famílias, ocorre o mosaicismo do tipo germinal; isso significa que mais de um irmão é afetado em uma geração, sem envolvimento dos pais.64,65
Os pacientes se apresentam com fraqueza nos músculos faciais (em especial, o músculo orbicular do olho), com preservação do músculo extraocular e do masseter. A fraqueza precoce nos músculos escapulares produz escápula alada, dando aos ombros uma aparência de inclinação para frente. A fraqueza no músculo tibial anterior, que resulta em pés caídos, é uma presença quase constante. O progresso da doença é lento, com longos períodos de estabilidade.
A atrofia na língua é comum. A progressão ocorre de uma forma descendente: o envolvimento dos músculos da cintura escapular é seguido pelo envolvimento do bíceps, do tríceps e dos músculos da cintura pélvica. Uma grande maioria de pacientes apresenta anormalidades nos vasos capilares da retina, descolamento retinal e problemas auditivos; essas descobertas são mais frequentes nos casos de DFEU que se apresentam na primeira infância.
Nos casos de DFEU, o nível sérico de CK é ligeiramente elevado, com preservação do músculo cardíaco. As descobertas das biópsias musculares variam bastante e incluem a presença de células inflamatórias. A suspeita diagnóstica se fundamenta em bases clínicas, sendo que a confirmação é feita pela análise do DNA.
Aproximadamente, 95% de casos (DFEU tipo 1) são causados por uma deleção em uma série de repetições de 3,3kb (D4Z4) no cromossomo 4.64 Os 5% de casos remanescentes são de DFEU tipo 2, causada por mutações na manutenção estrutural de domínios cromossômicos articulados flexíveis contendo o gene 1 (SMCHD1) que codifica o D4Z4, um modificador da cromatina. As mutações em DFEU1 e DFEU2 resultam na superexpressão tóxica de DUX4 na região de D4Z4.66
Distrofia Muscular Oculofaríngea
A distrofia muscular oculofaríngea (DMOF) é uma doença autossômica dominante rara que, geralmente, se manifesta entre as idades de 40 e 60 anos. A DMOF causa ptose e disfagia; esses dois distúrbios podem ser muito graves. Fraqueza distal leve é uma ocorrência comum. A DMOF é causada por uma repetição na expansão do trinucleotídeo GCG no gene da proteína de ligação poli-A 2 (PABP2) no cromossomo 14q11.2.67
Miopatia Hereditária com Corpúsculos de Inclusão e Com Doença de Paget e Demência Frontotemporal
A miopatia hereditária com corpúsculos de inclusão e com doença de Paget nos ossos e demência frontotemporal (h-IBMPPD) é um distúrbio autossômico dominante raro causado por mutações no gene que codifica a proteína contendo valosina (VCP).68 Esse tipo de miopatia se caracteriza pela presença de fraqueza proximal e distal e com uma leve assimetria. De um modo geral, o início da doença ocorre na vida adulta; os níveis de CK são normais ou ligeiramente elevados.
Hipertermia Maligna
A hipertermia maligna (HM) ocorre em um entre 50 mil a 100 mil adultos durante as anestesias gerais, sobretudo nos casos em que o halotano é usado isoladamente ou em combinação com relaxantes musculares despolarizantes como a succinilcolina. A HM descreve um aumento rápido no metabolismo, durante o qual a temperatura do corpo poderá ser superior a 43ºC.
A HM se apresenta com taquicardia, rigidez muscular (causada por contraturas musculares que poderão progredir para rigor ou morte), aumento na permeabilidade muscular (resultando em níveis séricos elevados de K+, Ca+, Na+ e CK) e mioglobinúria. Trismo ou espasmo no músculo masseter durante a indução de anestesia podem ser indicações da presença de hipertermia maligna.
Embora a HM ocorra em alguns pacientes sem uma doença muscular conhecida, os indivíduos em situação de risco incluem aqueles que têm anormalidades musculoesqueléticas congênitas múltiplas, deslocamento congênito isolado do quadril ou doença medular central; acredita-se que também ocorra em alguns pacientes com DMD ou DMB. A suscetibilidade é herdada como uma característica autossômica dominante.
A hipertermia maligna é causada pela incapacidade de controlar as concentrações intramusculares de cálcio produzidas por um SR defeituoso. Em até 50% de heredogramas, os alelos mutantes foram encontrados no receptor de rianodina (RyR),69 que faz a ligação entre o SR e o túbulo transverso, ou no gene CACNA1S que codifica a subunidade a1 do canal de cálcio tipo L controlado por voltagem.69,70
A terapia com dantroleno é um tratamento eficaz contra hipertermia maligna; essa terapia diminui a liberação de cálcio do SR sem alterar sua reabsorção. A administração intravenosa de dantroleno (2 a 10mg/kg a cada 5 minutos) deve ser feita na fase inicial do episódio enquanto a perfusão muscular ainda for adequada.
O dantroleno poderá ser administrado na dosagem de 2mg/kg, 10 a 15 minutos antes da aplicação da anestesia nos casos de pacientes reconhecidamente suscetíveis à HM. O uso de agentes anestésicos (por exemplo, óxido nitroso e tiopental) e de relaxantes musculares não despolarizantes é a melhor forma de evitar episódios de HM em pacientes suscetíveis.
miopatias Metabólicas
Princípios da Energia Muscular
As miopatias metabólicas se caracterizam pela presença da deficiência de metabolismo energético nas seguintes rotas:
glicólise/glicogenólise;
metabolismo lipídico;
fosforilação oxidativa em mitocôndrias.
O glicogênio e os ácidos graxos são as duas fontes principais de energia para os músculos. Essas rotas metabólicas convergem para a coenzima A acetílica (acetil CoA), que, a seguir, é oxidada em mitocôndrias por meio do ciclo de Krebs e da corrente respiratória.71?73 No estado de repouso, a energia muscular resulta basicamente da oxidação de ácidos graxos livres (AGLs).
Durante os exercícios aeróbicos de alta intensidade, o glicogênio muscular é a fonte principal de combustível para a fosforilação oxidativa. Os estoques de glicogênio muscular se esgotam depois de 90 minutos de exercícios. Os exercícios prolongados aumentam o uso de AGLs e de glicose sanguínea (suprida pela gliconeogênese hepática).
A disponibilização de AGLs pelos tecidos adiposos permite que as pessoas saudáveis façam exercícios moderados por muitas horas. Em pacientes com miopatias metabólicas, os sintomas se tornam evidentes durante as atividades que exigem aumentos nas demandas metabólicas, como, por exemplo, nos exercícios físicos.
Glicogenoses
A glicogenose se refere aos defeitos enzimáticos que ocorrem ao longo das rotas glicogenolíticas ou glicolíticas. Esses distúrbios se caracterizam pela presença de fatores como intolerância aos exercícios, mialgia, cãibras e mioglobinúria. A fraqueza progressiva (não episódica) pode se desenvolver em alguns pacientes.
Deficiência de Fosforilase Muscular
A deficiência de fosforilase A (doença de McArdle) é o protótipo da glicogenose: a glicogenólise é reduzida, resultando também na redução da disponibilidade de piruvato. Trata-se da segunda causa mais comum de mioglobinúria recorrente.71?73
A doença de McArdle é autossômica recessiva e se apresenta após a idade de 15 anos com intolerância aos exercícios e mioglobinúria. Depois de um breve descanso após a mialgia e rigidez, ambas induzidas pelo esforço físico, os pacientes poderão retomar a atividade com maior resistência (fenômeno do segundo vento) por causa do aumento na mobilização de ácidos graxos livres e da glicogenólise hepática.
A fraqueza muscular fixa poderá ocorrer em um momento mais tarde na vida. De um modo geral, os níveis séricos de CK são elevados. Tradicionalmente, a incapacidade de produzir lactato venoso foi examinada por meio de testes isquêmicos de exercícios com os antebraços. Esse tipo de teste vem sendo utilizado cada vez menos como teste diagnóstico, tendo em vista que produz resultados positivos falsos, é inespecífico, pode ser dolorido e, possivelmente, resulte em rabdomiólise focal.
Os testes não isquêmicos com exercícios têm o mesmo valor diagnóstico e não têm as desvantagens dos testes isquêmicos. As biópsias mostram ausência de fosforilase, vacúolos subsarcolemais e acúmulo de glicogênio. Confirma-se o diagnóstico por meio da análise bioquímica dos músculos e pela análise molecular das células sanguíneas. Esse defeito é causado por mutações no gene PYGM que se localiza no cromossomo 11q13 e que poderão ser detectadas nos leucócitos de mais de 90% de pacientes.
Não há nenhum tratamento disponível para a doença de McArdle, embora o treinamento em exercícios aeróbicos e as dietas com alto teor proteico possam ser bastante úteis. A administração de uma carga de sucrose antes dos exercícios pode melhorar a tolerância à atividade física e proteger contra rabdomiólise induzida por exercícios.74
Deficiência de Fosfofrutoquinase
A deficiência de fosfofrutoquinase (PFK) é uma doença autossômica recessiva causada por mutações distintas na subunidade M que se localiza no cromossomo 1. Trata-se de um defeito glicolítico com consequências funcionais que se assemelham àquelas observadas na doença de McArdle. Suspeita-se da deficiência de PFK em pacientes com intolerância aos exercícios, náusea e mioglobinúria.
Fatores como longo histórico de hemólise compensada, contagem elevada de radiculócitos, nível elevado de bilirrubina e hiperuricemia, também sugerem deficiência de PFK, principalmente em determinados grupos étnicos, como japoneses e judeus asquenazes. O diagnóstico da doença é confirmado por estudos bioquímicos nos músculos e análise molecular nas células sanguíneas.75 Não existe nenhum tratamento específico. Os pacientes devem evitar as dietas com alto teor de carboidratos. Alguns especialistas defendem as dietas cetogênicas.
Deficiência de Fosfoglicerato Quinase
A deficiência de fosfoglicerato quinase (PGK) é um distúrbio recessivo raro ligado ao X que se apresenta com intolerância aos exercícios, episódios de mioglobinúria, anemia hemolítica, miopatia e, ocasionalmente, retardo mental leve.71?73
Deficiência de Fosfoglicerato Mutase
A deficiência de fosfoglicerato mutase (PGAM) é uma doença muito rara que afeta apenas os músculos. Nos EUA, esse tipo de doença foi identificado somente em afro-americanos.
Deficiência De Maltase Ácida
A deficiência de maltase ácida (DMA) é uma doença de armazenamento de glicogênio autossômica recessiva causada pela deficiência de a-glicosidase, uma enzima que degrada o glicogênio lisossômico, que é codificada em um gene que se localiza no cromossomo 17q13.71?74,76 As mutações ou pequenas deleções que causam aderências anormais afetam a expressão da a-glicosidase.76,77 A DMA tem importância diagnóstica especial porque é potencialmente tratável com terapia de reposição enzimática.
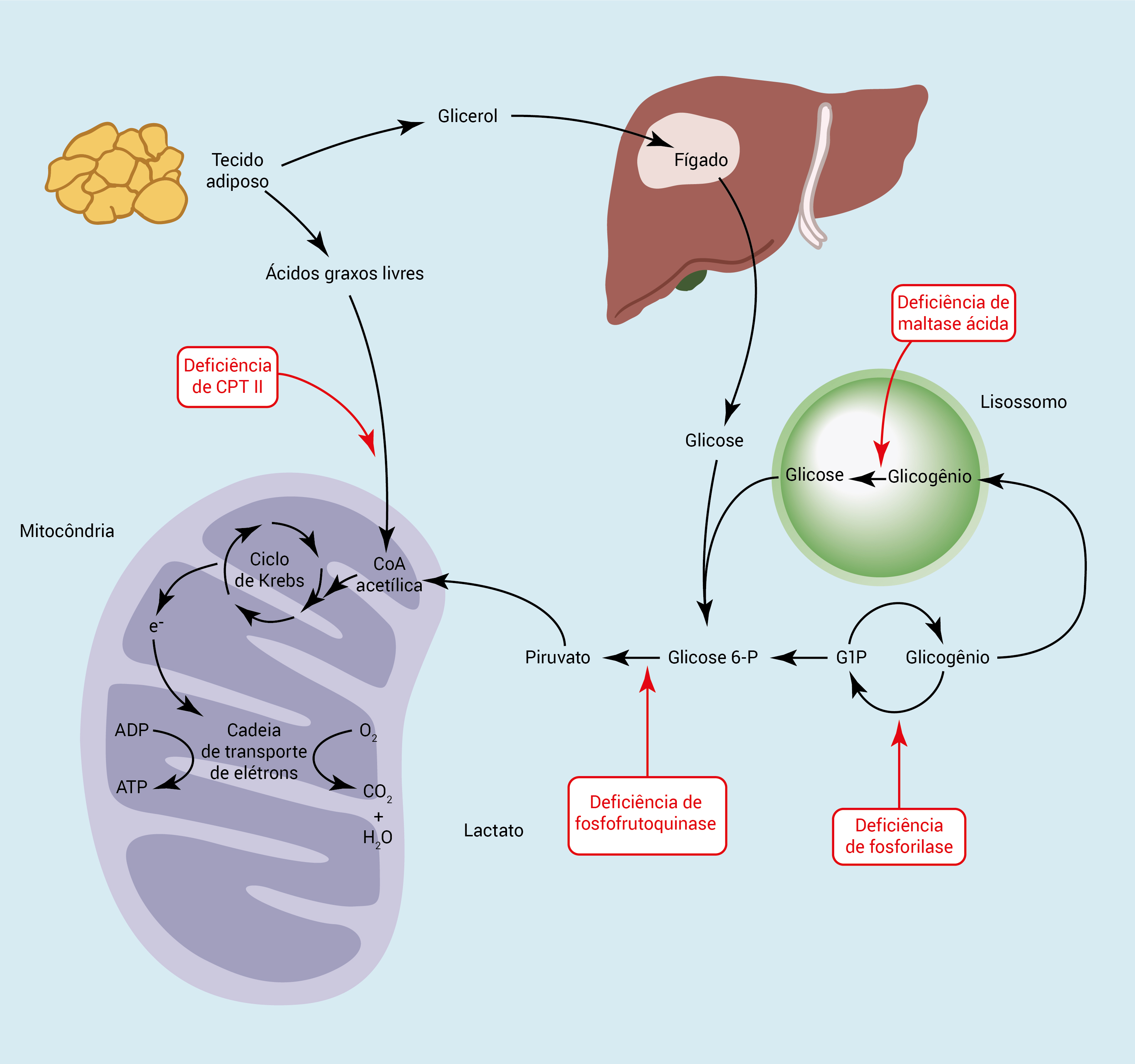
A Figura 3 mostra as rotas metabólicas principais utilizadas pelos músculos.
ADP: difosfato de adenosina; ATP: trifosfato de adenosina; CoA acetílica: coenzima acetílica A; CPT: carnitina palmitoiltransferase.
Figura 3 - Rotas metabólicas principais utilizadas pelos músculos. Nos músculos, o glicogênio é convertido na CoA acetílica. Os ácidos graxos livres são transportados para o interior das mitocôndrias, onde são ß-oxidados para CoA acetílica. Em seguida, a CoA acetílica é usada no ciclo de Krebs, produzindo elétrons livres, para uso na cadeia de transporte eletrônico, ATP, água e dióxido de carbono. Alguns dos defeitos enzimáticos mais comuns que causam a doença aparecem na cor vermelha na figura.
Existem três formas clínicas de DMA: na primeira infância, na infância e na vida adulta.76,77 A forma que prevalece na primeira infância (doença de Pompe) se apresenta nos primeiros meses de vida, como hipotonia, fraqueza e aumento nas dimensões do coração, da língua e do fígado; alterações respiratórias e cardiovasculares levando à morte antes dos 2 anos de idade.
Na forma infantil de DMA, os pacientes se apresentam com eventos motores tardios, fraqueza nos músculos proximais, envolvimento dos músculos respiratórios e aumento na panturrilha. Caso não seja tratado, esse tipo de doença leva à morte na segunda década de vida.
A forma adulta de DMA se manifesta em pessoas com idade acima de 20 anos, como fraqueza nos músculos proximais que se assemelha à poliomiosite ou como distrofia no cíngulo límbico. Fraqueza nos músculos respiratórios pode ser um dos sintomas em um terço de casos de DMA em adultos. O acúmulo de glicogênio ocorre predominantemente nos músculos, ainda que a deficiência enzimática ocorra também nos músculos, no fígado e em culturas de fibroblastos.
Os pacientes apresentam níveis séricos elevados de CK; a EMG revela a presença de descargas mioclônicas proeminentes (sem miotonia clínica), principalmente nos músculos paraespinais; e as biópsias mostram vacúolos lisossômicos cheios de glicogênio. A DMA causa fraqueza fixa, porém não causa intolerâncias aos exercícios e mioglobinúria, considerando que não há comprometimento no uso de glicogênio e glicose. As fibras musculares passam por um processo autofágico devido às anormalidades nos lisossomos.
O rastreamento rápido de DMA pode ser feito com pontos de sangue seco. Se esse tipo de rastreamento revelar que a atividade da a-glicosidase é, realmente, reduzida, então o diagnóstico poderá ser confirmado por meio da análise da genética molecular.78 O diagnóstico é muito importante, tendo em vista a disponibilidade da terapia de reposição enzimática, e está associado a uma grande melhora na distância de ambulação e na estabilização da função pulmonar em um período de 18 meses.79
Outras Glicogenoses Raras
As glicogenoses musculares raras incluem os distúrbios causados pela deficiência de enzimas musculares específicas; esses déficits incluem deficiência de lactato desidrogenase (LDH), deficiência de ß-enolase, deficiência da enzima desestruturadora (debrancher), deficiência da enzima estruturadora (brancher) e deficiência de aldolase.71?73
miopatias Causadas por Armazenamento de Lipídeos
Os ácidos graxos de cadeia longa (AGCLs) são a principal fonte de energia para os músculos durante a execução sustentada de exercícios. Os AGCLs exigem transferência ativa através das membranas mitocondriais. Esse tipo de transporte depende da disponibilidade de L-carnitina e de duas enzimas, as carnitina palmitoiltransferases (CPT I e CPT II), que se localizam nos lados externo e interno das membranas mitocondriais, respectivamente.
No interior das mitocôndrias, a ß-oxidação é facilitada por desidrogenases da enzima colesterol aciltransferase (acil CoA).71?73 As miopatias por armazenamento de lipídeos são causadas por problemas na oxidação de ácidos graxos nas mitocôndrias, como resultado dos déficits em carnitina e CPT, alterando o transporte de ácidos graxos através das membranas mitocondriais, e em enzimas associadas à ß-oxidação.71
Deficiência De Carnitina
A carnitina é derivada basicamente das dietas, embora 25% sejam sintetizadas no fígado. A carnitina é extremamente importante para a oxidação dos AGCLs, e sua deficiência produz disfunção em órgãos como fígado, coração e músculos, todos eles muito dependentes da oxidação dos AGCLs.71
A deficiência primária de carnitina (DPC) é um distúrbio infantil autossômico recessivo incomum causado por mutações no gene SLC22A5, que codifica o transportador catiônico 2 dependente de íons de sódio (OCTN2).
Esse processo reduz o número de receptores funcionais de carnitina com alta afinidade e altera o transporte de carnitina através das membranas.71 Os pacientes com DPC se apresentam com cardiomiopatia progressiva, episódios de hipoglicemia hipocetótica (por causa da disfunção hepática) e miopatia proximal. Os lipídeos se acumulam nos músculos e formam pequenas gotículas lipídicas.
A maior parte das deficiências de carnitina é secundária e resulta de:
distúrbios mitocondriais (por exemplo, defeitos na oxidação-ß, produzindo compostos de acila para ligação com a carnitina);
doença renal, causando excreção excessiva de carnitina;
tratamento com medicamentos, em especial a terapia com zidovudina (AZT) e com valproato.80 Os resultados dos tratamentos com suplementação de carnitina apresentaram variações significativas.
Deficiência de Cpt
A deficiência de CPT II é a miopatia metabólica mais comum, causada por mutações no gene de CPT II, que se localiza no cromossomo 11p11-p13.71?73 A deficiência de CPT II é a causa mais comum de mioglobinúria em adultos jovens. Os pacientes se apresentam com ataques de rigidez muscular, cãibras, mialgia e mioglobinúria logo após a execução prolongada ou sustentada de exercícios físicos, em especial depois de jejum ou nos casos em que a energia muscular depender do uso de AGCLs, e não do uso de glicogênio ou glicose.
Os pacientes com deficiência de CPT II não apresentam condições como tolerância reduzida aos exercícios, fenômeno do segundo vento ou sinais de alerta de mialgia, que os impeça de continuar a fazer exercícios. A resistência muscular e os níveis séricos de CK são normais entre os ataques da doença. O diagnóstico é feito por meio da medição da atividade da enzima CPT II nos músculos ou por testes genéticos. Normalmente, as biópsias musculares não revelam muita coisa.
Ainda não está suficientemente claro porque a deficiência de CPT produz ataques intermitentes de mioglobinúria e por que não há acúmulo de lipídeos nos músculos.71?73 Não há nenhuma terapia para impedir os ataques de mioglobinúria. São recomendadas dietas com alto teor de carboidratos e baixo teor de ácidos graxos, refeições frequentes e ingestão extra de carboidratos antes e durante a execução de exercícios sustentados.
Nos lactentes, a deficiência de CPT I se manifesta como síndrome de Reye, acompanhada de encefalopatia hepática, hipoglicemia hipocetótica e hiperamonemia.
Outros Distúrbios do Metabolismo De Lipídeos
Outros distúrbios menos comuns do metabolismo de lipídeos incluem deficiência múltipla de desidrogenação da coenzima A (MADD), doença de armazenamento de lipídeos neutros com miopatia (DALNM) e doença de armazenamento de lipídeos neutros com ictiose (DALNI).81 Esses distúrbios podem se apresentar acompanhados de fraqueza no período inicial da infância ou na vida adulta.
A MADD é um distúrbio autossômico recessivo causado por mutações que afetam a flavoproteína transportadora de elétrons (FTE) ou a FTE desidrogenase (FTEDH). As suspeitas de MADD devem recair sobre pacientes cujos espécimes de músculos apresentarem aumento na deposição de lipídeos, com aumento nas concentrações séricas e urinárias de ácidos orgânicos com múltiplos comprimentos nas ligações de carbono.
A doença de armazenamento de lipídeos neutros se caracteriza pelo acúmulo sistêmico de triglicérides no citoplasma e inclui dois distúrbios distintos: DALNM e DALNI (também conhecida por síndrome de Chanarin-Dorfman). A DALNM se apresenta como miopatia distal e queda progressiva dos pés; ela é causada por um gene que codifica a lipase triglicerídica adiposa (ATGL). Essa enzima catalisa a etapa inicial da hidrólise triglicerídica.
Uma entre as características laboratoriais mais úteis é a presença de corpos de Jordan, que são as inclusões lipídicas nos leucócitos observadas nos esfregaços de sangue. Como o próprio nome indica, os indivíduos afetados pela DALNI apresentam lesões cutâneas associadas. Esse tipo de distúrbio é causado pelas mutações no gene que codifica o coativador da ATGL.
Deficiência de Mioadenilato Desaminase
Aproximadamente, 1 a 2% da população apresenta uma deficiência da enzima mioadenilato desaminase (AMP desaminase ou AMPDA). A herança da doença é autossômica recessiva. Essa enzima tem um papel importante na manutenção dos níveis do trifosfato de adenosina (ATP) ao agir em combinação com a adenilato quinase.
A adenilato quinase converte duas moléculas de difosfato de adenosina para cada ATP e AMP. A seguir, a adenilato desaminase converte a AMP em monofosfato de inosina, resultando na produção de amônia. O estresse muscular ativa a reação.
Os estudos iniciais sugeriam que os pacientes com deficiência de AMPDA tinham mialgia ou intolerância aos exercícios. Entretanto, muitas pessoas com deficiência de AMPDA são assintomáticas e perfeitamente normais. Os relatos de deficiência enzimática variam de hipotonia congênita a esclerose lateral amiotrófica. Essa fraca correlação entre o defeito enzimático e os sintomas clínicos dificulta a interpretação dessa entidade.
As biópsias musculares não apresentam anormalidades na microscopia ótica; a AMPDA está ausente na imunocoloração. O teste de exercícios com o antebraço é uma técnica de rastreamento bastante útil. Os pacientes com deficiência de AMPDA produzem quantidades normais de lactato, porém pequenas quantidades ou nenhuma quantidade de amônia e hipoxantina, sendo que ambas são subprodutos da reação da AMPDA.
miopatias e Encefalopatias Mitocondriais
Os defeitos no metabolismo mitocondrial afetam não apenas os músculos e o sistema nervoso, mas também outros órgãos. Esses distúrbios se caracterizam pela presença de um defeito primário na produção de energia mitocondrial. O defeitos genéticos nas enzimas que produzem energia mitocondrial podem ser causados por mutações no DNA nuclear ou no DNA mitocondrial (mtDNA), de forma que não são distúrbios herdados exclusivamente das mães.
A principal função das mitocôndrias é gerar ATP por fosforilação oxidativa (OXPHOS), usando cinco complexos enzimáticos (designados como I, II, III, IV [citocromo-c oxidase], e V) que se localizam na membrana mitocondrial interna.61?64 As mitocôndrias contêm seus próprios DNA extracromossômicos (mtDNA), que são distintos do DNA nuclear.
A organização estrutural do mtDNA é altamente compacta; ela não tem introns. Como resultado, as mutações aleatórias no mtDNA, geralmente, atacam uma sequência codificadora e, com frequência, produzem doenças. Além disso, o mtDNA é suscetível aos danos produzidos por radicais de oxigênio devido à proximidade em relação à produção desses radicais pela OXPHOS e por causa de seus mecanismos mínimos de reparo.82?84
O mtDNA é uma herança materna (herança não mendeliana). As doenças causadas por mutações nos genes OXPHOS com codificação nuclear seguem um padrão mendeliano de herança. As mutações no mtDNA são heteroplasmáticas, significando que o mtDNA normal e mutante coexistem em uma única célula. Uma célula, geralmente, contém entre 2 e 10 moléculas de mtDNA que permitem a persistência de mutações letais (isto é, alteração letal de OXPHOS) em organismos viáveis.82?84
As biópsias musculares em pacientes com distúrbios mitocondriais revelam a presença de fibras vermelhas rasgadas na coloração tricrômica, ou de fibras azuis rasgadas na coloração com succinato desidrogenase (SDH), em razão do acúmulo subsarcolêmico de mitocôndrias. As fibras negativas para a citocromo-c oxidase sugerem a presença de alguma deficiência nas enzimas da cadeia respiratória. Na EM, as mitocôndrias apresentam inclusões paracristalinas ou cristas anormais. Mutações, deleções ou depleções específicas poderão ser detectadas por meio de estudos do mtDNA.
O tratamento de todos os distúrbios mitocondriais é de suporte e sintomático. Com frequência, são utilizadas coenzima Q10, creatina, carnitina e vitaminas, porém sem evidências claras de sua eficácia. A recomendação é fazer exercícios moderados, embora exista uma preocupação teórica com a proliferação preferencial de mitocôndrias anormais em resposta aos exercícios.
Síndrome de Kearns-Sayre e Oftalmoplegia Externa Progressiva Crônica
A síndrome de Kearns-Sayre (SKS) se apresenta em pacientes com idade inferior a 20 anos e com oftalmoplegia, ptose, retinite pigmentosa e fraqueza miopática. É muito comum a presença de fatores como baixa estatura, defeitos na condução cardíaca, níveis proteicos elevados no líquido cefalorraquidiano, perda auditiva sensorineural e níveis séricos elevados de lactato.
As biópsias musculares revelam a presença de fibras vermelhas rasgadas. Nas pessoas com idade acima de 20 anos, nas quais a oftalmoplegia seja o fenótipo predominante, a SKS é classificada como oftalmoplegia externa crônica progressiva (OECP).82?84
A SKS e a OECP mais limitada se caracterizam por uma única grande deleção do mDNA que fica entre nove e 50 pares de base. Em geral, a deleção é esporádica e, raramente, a herança é materna. A fosforilação oxidativa (OXPHOS) é defeituosa, e há uma redução nos níveis de atividade nos complexos I e IV.
Encefalomiopatia Neurogastrintestinal Mitocondrial
Uma forma especial autossômica recessiva de OECP, essa doença é uma síndrome multissistêmica conhecida por encefalomiopatia neurogastrintestinal mitocondrial (ENGIM).82?84 Aproximadamente, 60% dos pacientes se apresentam antes dos 20 anos de idade com oftalmoplegia externa progressiva acompanhada de disfunção intestinal, neuropatia periférica e leucoencefalopatia.
De um modo geral, a ENGIM está mais associada às mutações no gene nuclear para a timidina fosforilase (TYMP), resultando em problemas na replicação e manutenção do mDNA. Os pacientes se apresentam com elevação nos níveis séricos de timidina e redução na atividade da timidina fosforilase leucocitária.
Mutações Pontuais No MTDNA Com Herança Materna
Síndrome Melas
A presença de condições como encefalopatia mitocondrial, acidose láctica e episódios que se assemelham a AVC é uma característica da síndrome Melas. Os pacientes poderão apresentar também perda visual, baixa estatura, cardiomiopatia, diabetes melito ou degeneração pigmentar na retina que se assemelha à síndrome de Kearns-Sayre ou oftalmoplegia externa crônica progressiva. As biópsias musculares revelam a presença de fibras vermelhas rasgadas. Até 80% de pacientes apresentam mutações pontuais no mtDNA no gene leucina do RNA transportador (tRNA).
Síndrome MERRF
A síndrome MERRF se caracteriza pela presença de epilepsia mioclônica e miopatia com fibras vermelhas rasgadas, ataxia, demência, surdez e anormalidades cardíacas, ainda que sua expressão seja variável e dependa do grau de heteroplasmia. Aproximadamente, 80% dos pacientes com MERRF têm uma mutação no gene da lisina do tRNA.82?84
Neuropatia Óptica Hereditária de Leber
A neuropatia óptica hereditária de Leber (NOHL) é a causa mais comum de cegueira em adultos jovens (a prevalência é maior em homens em comparação com as mulheres). Os pacientes se apresentam com perda visual indolor, subaguda e bilateral. Em até, pelo menos, 90% das famílias, foram observadas diversas mutações no mtDNA. Alguns pacientes com NOHL têm encefalopatia, surdez, ataxia, mielopatia ou distonia.82?84
Síndromes de Deflexão no MTDNA
Essas síndromes são distúrbios autossômicos recessivos que afetam os músculos, o fígado, os rins e o cérebro. Em geral, mas nem sempre, essas síndromes são fatais depois dos 3 anos de idade devido a condições como encefalopatia ou insuficiência respiratória. Uma grande variedade de mutações nucleares e no mtDNA prejudicam replicações e reparos no mtDNA.82?84
Canelopatias Iônicas, Paralisias Periódicas e Miotonias não Distróficas
As canelopatias iônicas que afetam a excitabilidade das fibras musculares produzem uma ampla gama de distúrbios, tais como paralisia periódica, miotonia e ataxia-mioquimia episódica.85?88 Esses distúrbios formam um grupo raro de doenças que, em geral, iniciam na infância e, tipicamente, se apresentam com ataques de paralisia.
As alterações nos níveis séricos de potássio são muito comuns durante os ataques de paralisia. Algumas formas de miotonia também são comuns. Suspeita-se da presença desses distúrbios nas situações em que houver histórico de ataques semelhantes em membros da família, em especial se os ataques forem provocados por repouso após os exercícios ou por refeições enriquecidas com carboidratos; o diagnóstico poderá ser confirmado por meio da análise genética do DNA no sangue.
Nas células musculares e nervosas, a abertura dos canais de Na+ controlados por voltagem provoca um aumento rápido na permeabilidade do Na+, despolarizando a membrana. Para iniciar a próxima ação potencial, os canais de Na+ deverão ser fechados para permitir a repolarização.85?87,89 A abertura dos canais de Na+ controlados por voltagem permite o efluxo do íon Na+, hiperpolarizando a membrana celular. Os canais de Cl- contribuem para a repolarização através da estabilização do potencial da membrana.
As perturbações na excitabilidade da membrana poderão resultar em miotonia, com ou sem paralisia periódica. A miotonia se manifesta como rigidez indolor depois de períodos de inatividade e melhora com a execução de movimentos contínuos (fenômeno de aquecimento). A miotonia que se desenvolve após a exposição ao frio e se agrava com os exercícios se denomina miotonia paradoxal ou paramiotonia.
Nos casos de miotonias não distróficas, a miotonia é causada por distúrbios no canal de sódio ou de cloreto. A excitabilidade da membrana também poderá resultar em paralisia periódica - ataques de fraqueza associados à hipercaliemia (produzida por mutações no canal de Na+) ou por hipocaliemia (produzida por mutações no canal de Ca+ controlado por voltagem). Os pacientes com paralisia periódica cardiodisrítmica (síndrome de Andersen) ou com ataxia-mioquimia episódica apresentam mutações no canal de potássio.86,87,89
Distúrbios no Canal de Sódio
Nos músculos esqueléticos, os distúrbios no canal de sódio resultam de mutações no gene SCN4A do canal de sódio. As mutações no SCN4A podem produzir paralisia hipercalêmica periódica (hiper-KPP), paramiotonia congênita (PMC), miotonia agravada por potássio (anteriormente denominada miotonia responsiva à acetazolimida ou miotonia oscilante) e paralisia periódica hipocalêmica tipo 2 (hipoKPP2). Os pacientes com esses distúrbios alélicos apresentam diversos graus de miotonia ao fechar os olhos, mastigar, engolir e pegar algo com as mãos.
A hiperKPP e a PMC (também conhecida por miotonia paradoxal) podem coexistir em um mesmo indivíduo ou em uma mesma família, e são doenças herdadas de uma forma autossômica dominante. Os ataques de hiperKPP podem iniciar na primeira infância, ou na infância, e são precipitados pelo repouso depois de exercícios, estresse, administração de K+, exposição ao frio e determinados tipos de alimentos.
A miotonia leve e a paramiotonia nas pálpebras são comuns. A exposição ao frio poderá causar rigidez, principalmente na face, na língua, nas pálpebras e nas mãos. Os sintomas de PMC e de hiperKPP são tratados com inibidores da anidrase carbônica como a acetazolamida e diclorfenamida.86,87,89
Distúrbios no Canal de Potássio
As mutações pontuais no gene KCNA1 do canal de potássio, que se localiza no cromossomo 12, foram associadas à ataxia-mioquimia episódica. Outras mutações nos genes do canal de potássio produzem a síndrome do QT longo. A síndrome de Andersen é causada por mutações KCNJ2 do canal de potássio, que codifica o gene retificador interior Kir21 do canal de K+, que se localiza no cromossomo 17q.
A síndrome de Andersen se caracteriza pela presença de paralisia periódica, que poderá ser acompanhada de condições como fraqueza fixa; síndrome do QT longo com arritmias cardíacas ventriculares; e características craniofaciais dismórficas, tais como micrognatia, orelhas caídas, baixa estatura e sindactilia. De modo geral, a síndrome de Andersen e outros distúrbios no canal de potássio melhoram com a administração de inibidores da anidrase carbônica.86,87,89
Distúrbios no Canal de Cálcio
Os distúrbios no canal de cálcio se apresentam como paralisia hipocalêmica periódica tipo 1 (hipoKPP1). A hipoKPP1 é uma doença autossômica dominante causada por mutações no gene do canal de cálcio tipo L no cromossomo 1q31-q32.86,87,89,90 Esse tipo de doença se apresenta como paralisia episódica que inicia na primeira ou segunda décadas de vida, comumente durante o sono.
A hipoKPP1 é precipitada por fatores como consumo elevado de carboidratos, repouso depois de exercícios e excitação. Durante os ataques, a concentração sérica de K+ diminui e ocorre retenção urinária de Na+ e água. Entre os ataques que ocorrem em um momento mais tarde na vida, muitos pacientes desenvolvem miopatia permanente com progressão lenta.
Esse tipo de doença deve ser distinguido de todas as causas de hipocaliemia secundária que produz fraqueza. Em geral, não ocorre paralisia nos casos de hipocaliemia secundária, a menos que a concentração sérica de K+ caia abaixo de 2,5 a 3mEq. Nos casos de hipoKPP, os ataques poderão ocorrer mesmo nas situações em que a concentração sérica de K+ estiver próxima do nível normal.
A hipoKPP pode estar associada à tirotoxicose, particularmente em indivíduos de descendência asiática. Nesses casos, a hipoKPP responde ao propranolol ou desaparece a partir do momento em que o paciente se torna eutireoideo. A frequência dos ataques, assim como a fraqueza entre ataques, melhora com a administração de inibidores da anidrase carbônica.91,92
Distúrbios no Canal de Cloreto
A miotonia congênita autossômica dominante (doença de Thomsen) ocorre na primeira década de vida e, com frequência, está associada à hipertrofia muscular. A miotonia congênita autossômica recessiva (miotonia do tipo Becker) se manifesta mais tarde na vida, ainda que seja muito mais grave.86,87 Ambas as doenças foram associadas a mutações distintas do gene CLCN1 do canal de cloreto, que se localiza no cromossomo 7q32.85,87
Os pacientes com esses distúrbios não apresentam fraqueza ou paralisia periódica, embora na miotonia do tipo Becker provavelmente ocorra fraqueza transitória que, às vezes, chega a ser grave. A fraqueza melhora com a prática de exercícios. Miotonia percussiva típica e miotonia generalizada, experimentadas pelos pacientes como “rigidez” e hipertrofia nas pernas e nádegas, são características comuns. A rigidez miotônica melhora com a prática de exercícios (fenômeno do aquecimento), e os sintomas miotônicos respondem ao uso de fenitoína, mexiletina ou tocaínida.
miopatias Induzidas por Medicamentos
Define-se miopatia induzida por medicamentos como a manifestação subaguda (raramente aguda) de sintomas miopáticos, tais como fraqueza muscular, fadiga, mialgia, elevação de CK ou mioglobinúria, em pacientes sem doença muscular que se expõem a doses terapêuticas de determinados tipos de medicamentos.93?95
Normalmente, os sinais clínicos ou bioquímicos do envolvimento muscular melhoram após a descontinuação no uso do agente suspeito, dando suporte ao efeito causativo do medicamento miotóxico ofensor. Entretanto, às vezes, a miotoxicidade é irreversível, como se observa em alguns análogos de nucleosídeos que se incorporam na cadeia do mtDNA.96?98
As biópsias musculares podem documentar a miotoxicidade, embora às vezes não tenham nenhuma utilidade informativa, como costuma ocorrer em alguns pacientes com mioglobinúria ou fraqueza muscular leve causada pelo uso de estatinas ou de análogos de nucleosídeos.93?95
Os agentes miotóxicos podem causar miopatia que afeta diretamente as organelas musculares, tais como mitocôndrias, lisossomos, microtúbulos ou filamentos espessos;93?95 alterando os antígenos musculares e, por conseguinte, induzindo reações inflamatórias imunológicas; ou induzindo efeitos sistêmicos secundários, tais como perturbações eletrolíticas ou má absorção nutricional ou vitamínica.
Diversos medicamentos podem produzir efeitos miotóxicos, isoladamente ou em combinação com outros. Além de saber quais medicamentos são miotóxicos, os médicos devem permanecer sempre alertas em relação à miotoxicidade dos fármacos lançados recentemente no mercado.
Medicamentos que Abaixam os Níveis de Colesterol: miopatias Induzidas Por Estatinas
Diversos medicamentos que abaixam os níveis de colesterol produzem um tipo de miopatia que se caracteriza pela presença de fraqueza nos músculos proximais, mialgia, elevação nos níveis séricos de CK e alterações miopáticas na EMG, incluindo potenciais de fibrilação e repetição de descargas miotônicas ou complexas. As biópsias musculares podem revelar a presença de alterações mínimas ou inespecíficas e de fibras necróticas ocasionais ou fibras “vermelhas rasgadas”.94,95,99,100
De modo geral, a recuperação gradual ocorre após a retirada do medicamento ofensor, embora provavelmente ocorra um efeito “de cabotagem” em que a miopatia se agrava por um curto período de tempo após a descontinuação no uso do medicamento. Ocasionalmente, as estatinas poderão desencadear algum tipo de miopatia necrozante com mediação autoimune e suprarregulação do complexo de histocompatibilidade principal (MHC).88,94,101
Levando-se em conta que o colesterol é o componente principal dos esteróis das membranas musculares, a redução na quantidade de colesterol normal disponível para a síntese membranosa poderá aumentar a fluidez das membranas, resultando em condições como sarcolema instável, descargas miotônicas e rabdomiólise nos casos mais avançados.88,94,95,101 Além disso, as estatinas inibem a produção de ubiquinona, interferindo, consequentemente, na ATP mitocondrial e no metabolismo energético no interior dos miócitos.
Os seguintes sintomas ou sinais miopáticos poderão ser observados com o uso de estatinas:
hiperckemia assintomática e oscilante (até 10 vezes) em até 5% de pacientes;
mialgia, com ou sem hiperckemia, em até 9 a 25%;
fraqueza muscular variando de leve a moderada, com elevação nos níveis de CK atribuível à miopatia tóxica ou a uma miopatia necrozante autoimune que responde às terapias imunes, sendo que, em algumas poucas situações, as estatinas revelaram a presença de uma condição miopática pré-existente;
rabdomiólise, definida como uma elevação aguda em CK (>15.000 em relação ao limite superior da normalidade), geralmente acompanhada de mialgia, fraqueza e mioglobinúria.94
A administração concomitante de ciclosporina e lovastatina em pacientes de transplantes com hiperlipidemia aumenta a incidência de miopatia com rabdomiólise. A cerivastatina é a estatina mais frequentemente associada aos sintomas miopáticos; na sequência, a tendência foi acompanhada pelos medicamentos lovastatina, fluvastatina, sinvastatina e pravastatina.88,94,95,101
O risco de incidência de miopatia induzida por estatinas aumenta com o uso de doses mais elevadas das estatinas com ação lipofílica (isso inclui todas as estatinas com exceção da pravastatina e rosuvastatina), assim como com a administração de terapias concomitantes à base de medicamentos como genfibrozil, amiodarona, colchicina ou ciclosporina.88,101
Em geral, os sintomas miopáticos melhoram após a descontinuação dos medicamentos. Nas situações em que os sintomas persistirem ou continuarem a se agravar, é necessário obter biópsias musculares para excluir a eventual presença de miopatia necrozante com mediação imune que exija o uso de imunoterapia com esteroides ou aplicação intravenosa de globulina imune (IVIg), ou de alguma outra condição que não havia sido reconhecida anteriormente. O valor terapêutico da coenzima Q10 no tratamento de sintomas miopáticos ainda permanece incerto.88,101
Miopatia Induzida por Medicamentos Antirreumáticos, Anti-Inflamatórios e Imunossupressores
Miopatia com Zidovudina e Outros Análogos de Nucleotídeos
A zidovudina pode causar fraqueza nos músculos proximais, mialgia (predominantemente nas coxas e nas panturrilhas), fadiga, alterações miopáticas na EMG e níveis séricos elevados de CK.80,95?97 Os sintomas desaparecem em 4 a 6 semanas após a descontinuação da zidovudina.96,97 A histologia mostra a presença de fibras vermelhas rasgadas, acúmulo de lipídeos e inúmeras fibras negativas para a citocromo-c oxidase, sugerindo disfunção mitocondrial.80,96,97
A zidovudina inibe a polimerase complementar do DNA na matriz mitocondrial, resultando em até 78% de depleção do mtDNA muscular.96,97 A estavudina também pode ser miotóxica, entre outros análogos de nucleotídeos usados no tratamento de HIV/AIDS. Condições como síndrome de lipodistrofia, acidose láctica e miopatia foram observadas em terapias antirretrovirais altamente ativas, consistindo de um dos inibidores da protease recentemente introduzidos no mercado em combinação com dois análogos de nucleotídeos, em especial a estavudina (d4T).98
Contaminantes Farmacológicos que Causam Predominantemente Fascite
O l-triptofano contaminado foi responsável pelo surto de uma síndrome de fascite eosinofílica, miosite, espessamento da pele, neuropatia axonal e outras manifestações sistêmicas. Um processo imune contra fibroblastos na matriz extracelular desencadeado pelo agente contaminante foi envolvido como uma das causas dessa síndrome, que passou a ser conhecida por síndrome de eosinofilia-mialgia.103
Uma síndrome semelhante atribuída ao uso de óleo contaminado (síndrome do óleo tóxico) ocorreu na Espanha. A miofascite macrofágica é uma entidade documentada principalmente na França e que foi resultado da aplicação de vacinas contendo alumínio. As biópsias feitas no sítio de injeção da vacina mostraram o acúmulo de macrófagos no epimísio, no perimísio e no endomísio.
Miopatia Causada por Enfermidade Crítica
Em alguns pacientes com enfermidade crítica prolongada, existe a possibilidade de se desenvolver um tipo de miopatia grave, resultando em fraqueza generalizada, incapacidade para retirar gradualmente a ventilação mecânica, atrofia muscular, níveis séricos normais ou moderadamente elevados de CK e alterações miopáticas na EMG.
Em geral, a fraqueza melhora lentamente.95,104?106 A miopatia resultante de enfermidade crítica poderá coexistir com uma neuropatia axonal, embora a paralisia seja causada predominantemente pela miopatia. O diagnóstico diferencial deve incluir defeitos de transmissão neuromuscular, tais como miastenia grave, síndrome de Guillain-Barré, miopatia inflamatória ou necrozante aguda e paralisia periódica.
O Quadro 4 mostra a miopatia induzida por medicamentos antirreumáticos, anti-inflamatórios e imunossupressores.
Quadro 4
|
Miopatia Induzida por Medicamentos Antirreumáticos, Anti-Inflamatórios e Imunossupressores | |
|
Medicamento |
Ação |
|
D-Penicilamina |
Esse medicamento pode desencadear complicações neuromusculares imunomediadas, incluindo polimiosite e miastenia, em até 0,6% de pacientes tratados. |
|
Cloroquina e hidroxicloroquina |
Comumente, com administração de doses elevadas de cloroquina (500mg/dia) no longo prazo. Sob os pontos de vista clínico e histológico, esse tipo de miopatia se assemelha à doença de Pompe e se caracteriza pela presença de fraqueza muscular, nível geralmente normal de CK e múltiplos vacúolos contendo material positivo de fosfatase ácida atribuível ao aumento na atividade da enzima lisossômica.102 A reversão da miopatia é lenta após a descontinuação no uso do medicamento. |
|
Colchicina |
Esse medicamento inibe o crescimento de microtúbulos e produz miopatia vacuolar com acúmulo de lisossomos e de vacúolos autofágicos. Os sintomas incluem fraqueza nos músculos proximais, elevação no nível sérico de CK, envolvimento sensorial distal e arreflexia. Os sintomas desaparecem em um período de 4 a 6 semanas após a descontinuação no uso do medicamento. |
|
Ciclosporina e tacrolimo |
Raramente, causam miopatia por si mesmos, mas estão implicados em miotoxicidade quando usados concomitantemente com as estatinas ou com a colchicina.93?95 |
CK: creatinoquinase.
As biópsias musculares revelam a perda seletiva de filamentos espessos (miosina) e a presença de vários graus de necrose. A coloração com adenosina trifosfatase (ATPase) mostra áreas de palidez central em muitas fibras.95,105,106 A expressão da calpaína é aumentada e sugere que há alteração na homeostase do cálcio e intensificação na proteólise. A microscopia eletrônica revela a perda seletiva e extensiva de miosina em monofilamentos espessos; entretanto, os monofilamentos finos e os discos Z são preservados. A miopatia melhora lentamente com reabilitação intensa.
Miopatia Induzida por Esteroides
O hipercortisolismo crônico causado pela síndrome de Cushing ou pela administração de corticosteroides pode causar miopatia. Comumente, a fraqueza nos membros proximais é branda e preserva os músculos flexores do pescoço.95 O nível sérico de CK é normal, as biópsias mostram apenas atrofia nas fibras tipo II e a EMG, geralmente, é normal.94,95
Toxicidade Causada por Medicamentos Recreativos
As drogas ilícitas e o consumo de álcool são responsáveis pela maioria dos pacientes com mioglobinúria aguda.107 Cocaína, heroína, anfetamina, fenciclidina (PCP) e etanol podem causar miopatia com vários graus de agudeza e gravidade.94,95 O alcoolismo pode causar miopatia aguda ou crônica. Os pacientes intoxicados que permanecem imobilizados por longos períodos de tempo podem desenvolver isquemia e necrose muscular.
A miopatia aguda indolor e reversível poderá ocorrer em pacientes com alcoolismo se a concentração sérica de K+ cair abaixo de 2,5mEq. Má nutrição, inatividade e doença neurológica também contribuem para o estado de fraqueza. Os alcoólatras inveterados poderão apresentar elevação assintomática no nível sérico de CK ? até 20 vezes mais elevado que os níveis normais ?, que poderá ser agravado com a atividade física.
|
Informações Financeiras: Thomas I. Cochrane, MD, MBA, não tem informações financeiras relevantes a declarar. Anthony A. Amato, MD, faz parte do Comitê de Consultoria Médica da empresa Biogen. |
Referências
1. Narayanaswami P, Weiss M, Selcen D, et al. Summary of evidence-based guideline: diagnosis and treatment of limb-girdle and distal muscular dystrophies. Neurology 2014. [In press]
2. Manzur AY, Muntoni F. Diagnosis and new treatments in muscular dystrophies. J Neurol Neurosurg Psychiatry 2009;80:706–14.
3. Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev 2002;12:349–61.
4. Beggs AH, Koenig M, Boyce FM, et al. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990;86:45–8.
5. Mathews KD. Muscular dystrophy overview: genetics and diagnosis. Neurol Clin 2003;21:795–816.
6. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731–40.
7. Mendell JR, Buzin CH, Feng J, et al. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001;57:645–50.
8. Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology 2003;99:1–19.
9. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93.
10. Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology 2003;99:1–19.
11. Flanigan KM, von Niederhausern A, Dunn DM, et al. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet 2003;72:931–9.
12. Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev 2008;(1):CD003725.
13. Backman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord 1995;5:233–41.
14. Prior TW, Bartolo C, Pearl DK, et al. Spectrum of small mutations in the dystrophin coding region. Am J Hum Genet 1995;57:22–33.
15. Nigro V. Molecular bases of autosomal recessive limb-girdle muscular dystrophies. Acta Myol 2003;22:35–42.
16. Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999;341:1715–24.
17. Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology 2004;62:1363–71.
18. Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998;18:365–8.
19. Gazzerro E, Bonetto A, Minetti C. Caveolinopathies: translational implications of caveolin-3 in skeletal and cardiac muscle disorders. Handb Clin Neurol 2011;101:135–42.
20. Zatz M, de Paula F, Starling A, et al. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 2003;13:532–44.
21. Betz RC, Schoser BG, Kasper D, et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet 2001;28:218–9.
22. Greenberg SA, Salajegheh M, Judge DP, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol 2012;71:141–5.
23. Messina DN, Speer MC, Pericak-Vance MA, McNally EM. Linkage of familial dilated cardiomyopathy with conduction defect and muscular dystrophy to chromosome 6q23. Am J Hum Genet 1997;61:909–17.
24. Melià MJ, Kubota A, Ortolano S, et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 2013;136:1508–17.
25. Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995;81:27–40.
26. Liu J, Aoki M, Illa I, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998;20:31–6.
27. Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract Neurol 2009;9:314–23.
28. Duggan DJ, Gorospe JR, Fanin M, et al. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 1997;336:618–24.
29. Angelini C, Fanin M, Freda MP, et al. The clinical spectrum of sarcoglycanopathies. Neurology 1999;52:176–9.
30. Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet 2000;24:163–6.
31. Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003;60:1246–51.
32. Frosk P, Greenberg CR, Tennese AA, et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat 2005;25:38–44.
33. Kramerova I, Kudryashova E, Venkatraman G, et al. Calpain 3 participates in muscle remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet 2007;15:1006.
34. Norwood F, de Visser M, Eymard B, et al. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. EFNS Guideline Task Force. Eur J Neurol 2007;14:1305–12.
35. Bolduc V, Marlow G, Boycott KM, et al. Recessive mutations in the putative calcium-activated chloride channel anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet 2010;86:213–21.
36. Schessl J, Kress W, Schoser B. Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and Miyoshi type of muscular dystrophy. Muscle Nerve 2012;45:740–2.
37. Cetin N, Balci-Hayta B, Gundesli H, et al. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: distinct histopathological outcomes compared with desminopathies. J Med Genet 2013;50:437–43.
38. Henderson M, De Waele L, Hudson J, et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol 2013;125:917–9.
39. Amato AA, Kagan-Hallet K, Jackson CE, et al. The wide spectrum of myofibrillar myopathy suggests a multifactorial etiology and pathogenesis. Neurology 1998;5:1646–55.
40. Dalakas MC, Park KY, Semino-Mora C, et al. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000;342:770–80.
41. Goldfarb LG, Vicart P, Goebel H, Dalakas MC. Desmin myopathy. Brain 2004;127:723–34.
42. Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 2004;127:439–51.
43. Goldfarb JG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009;119:1806–13.
44. Banwell BL, Russel J, Fukudome T, et al. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol 1999;58:832–46.
45. DeBleecker JL, Engel AG, Ertl BB. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Pathol 1996;55:563–77.
46. Nakano S, Engel AG, Akiguschi I, et al. Myofibrillar myopathy. III. Abnormal expression of cyclin-dependent kinases and nuclear proteins. J Neuropathol Exp Neurol 1997;56:850–6.
47. Nakano S, Engel AG, Waclwik AJ, et al. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Pathol 1996;55:549–62.
48. Hewitt JE, Grewal PK. Glycosylation defects in inherited muscle disease. Cell Mol Life Sci 2003;60:251–8.
49. Grewal PK, Hewitt JE. Glycosylation defects: a new mechanism for muscular dystrophy. Hum Mol Genet 2003;12:R259–64.
50. Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006;9:427–43.
51. Muntoni F, Voit T. 133rd ENMC International Workshop on Congenital Muscular Dystrophy (IXth International CMD Workshop) 21–23 January 2005, Naarden, the Netherlands. Neuromuscul Disord 2005;15:794–801.
52. North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord 2014;24:97–116.
53. Hayashi YK, Chou FL, Engvall E, et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 1998;19:94–7.
54. Clement E, Mercuri E, Godfrey C, et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 2008;64:573–82.
55. Wallgren-Petterson CW, Sewry CA, Nowak KJ, Laing NG. Nemaline myopathies. Semin Pediatr Neurol 2011;18:230–8.
56. Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleratonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci U S A 2001;98:7516–21.
57. Wallgren-Petterson C, Laing NG. 138th ENMC Workshop: Nemaline myopathy, 20–22 May 2005, Naarden, the Netherlands. Neuromuscul Disord 2006;16:54–60.
58. Barohn RJ, Amato AA, Griggs RC. Overview of distal myopathies: from the clinical to the molecular. Neuromuscul Disord 1998;8:309–16.
59. Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet 2013;93:744–51.
60. Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol 2007;20:572–6.
61. 61 Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 2003;60:657–64.
62. Kaliman P, Llagostera E. Myotonic dystrophy protein kinase (DMPK) and its role in the pathogenesis of myotonic dystrophy 1. Cell Signal 2008;20:1935–41.
63. Kanandia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science 2003;302:1978–80.
64. Padberg GW, van Engelen BG. Facioscapulohumeral muscular dystrophy. Curr Opin Neurol 2009;22:539–42.
65. Rose MR, Tawil R. Drug treatment for facioscapulohumeral muscular dystrophy. Cochrane Database Syst Rev 2004;(2):CD002276.
66. Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–81.
67. Shanmugam V, Dion P, Rochefort D, et al. PABP2 polyalanine tract expansion causes intranuclear inclusions in oculopharyngeal muscular dystrophy. Ann Neurol 2000;48:798–802.
68. American Association of Neuromuscular and Electrodiagnostic Medicine. Diagnostic critera for late-onset (childhoo¬d and adult) Pompe disease. Muscle Nerve 2009;40:149–60.
69. Stowell KM. Malignant hyperthermia: a pharmacogenetic disorder. Pharmacogenomics 2008;9:1657–72.
70. Benca J, Hogan K. Malignant hyperthermia, coexisting disorders, and enzymopathies: risks and management options. Anesth Analg 2009;109:1049–53.
71. DiMauro S, Lamperti C. Muscle glycogenoses. Muscle Nerve 2001;24:984–99.
72. Hirano M, DiMauro S. Metabolic myopathies. Adv Neurol 2002;88:217–34.
73. Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep 2010;10:118–26.
74. Vissing J, Haller RG. The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. N Engl J Med 2003;349:2503–9.
75. Vasconcelos O, Sivakumar K, Dalakas MC, et al. Nonsense mutation in the human phosphofructokinase muscle subunit gene associated with retention of intron 10 in one of the isolated transcripts in Ashkenazi Jewish patients with Tarui disease. Proc Natl Acad Sci U S A 1995;92:10322–6.
76. Engel AG, Hirschhorn R, Huie ML. Acid maltase deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. p. 1559.
77. van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010;362:1396–406.
78. Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007;68:99–109.
79. Ohkuma A, Noguchi S, Sugie H, et al. Clinical and genetic analysis of lipid storage myopathies. Muscle Nerve 2009;39:333–42.
80. Dalakas MC, Leon-Monzon ME, Bernardini I, et al. The zidovudine-induced mitochondrial myopathy is associated with muscle carnitine deficiency and lipid storage. Ann Neurol 1994;35:482–7.
81. Matthews E, Fialho D, Tan SV, et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 2010;133:9–22.
82. Hassani A, Horvath R, Chinnery PF. Mitochondrial myopathies: developments in treatment. Curr Opin Neurol 2010 Jul 21. [Epub ahead of print]
83. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003;348:2656–68.
84. McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. Lancet Neurol 2002;1:343–51.
85. Meola G, Hanna MG, Fontaine B. Diagnosis and new treatment in muscle channelopathies. J Neurol Neurosurg Psychiatry 2009;80:360–5.
86. Celesia GG. Disorders of membrane channels or channelopathies. Clin Neurophysiol 2001;112:2–18.
87. Kleopa KA, Barchi RL. Genetic disorders of neuromuscular ion channels. Muscle Nerve 2002;26:299–325.
88. Greenberg SA, Amato AA. Statin myopathies. Continuum Muscle Dis 2006;12;169–84.
89. Raja Rayan DL, Hanna MG. Skeletal muscle channelopathies: nondystrophic myotonias and periodic paralysis. Curr Opin Neurol 2010 Jul 14. [Epub ahead of print]
90. Fouad G, Dalakas M, Servidei S, et al. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul Disord 1997;7:33–8.
91. Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve 1983;6:182–6.
92. Tawil R, McDermott MP, Brown R, et al. Randomized trial of dichlorphenamide in the periodic paralyses. Working Group on Periodic Paralysis. Ann Neurol 2000;47:46–53.
93. Sieb JP. Myopathies due to drugs, toxins, and nutritional deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. p. 1693.
94. Dalakas MC. Toxic and drug-induced myopathies. J Neurol Neurosurg Psychiatry 2009;80:832–8.
95. Dalakas MC. Inflammatory and toxic myopathies. Curr Opin Neurol Neurosurg 1992;5:645–54.
96. Dalakas MC, Illa I, Pezeshkpour GH, et al. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med 1990;322:1098–105.
97. Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med 1995;1:417–22.
98. Brinkman K, Smeitink JA, Romijn JA, et al. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 1999;354:1112–5.
99. Rosenson RS. Current overview of statin-induced myopathy. Am J Med 2004;116:408–16.
100. Sieb JP, Gillessen T. Iatrogenic and toxic myopathies. Muscle Nerve 2003;27:142–56.
101. Hodel C. Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicol Lett 2002;128:159–68.
102. Kuncl RW. Colchicine myopathy and neuropathy. N Engl J Med 1987;316:1562–8.
103. Illa I, Dinsmore S, Dalakas MC. Immune-mediated mechanisms and immune activation of fibroblasts in the pathogenesis of eosinophilia-myalgia syndrome induced by L-tryptophan. Hum Pathol 1993;24:702–9.
104. Danon MJ, Carpenter S. Myopathy with thick filament loss following prolonged paralysis with vacuronium during steroid treatment. Muscle Nerve 1991;14:1131–9.
105. Bella I, Chad DA. Neuromuscular disorders and acute respiratory failure. Neurol Clin 1998;16:391–417.
106. Hirano M, Ott B, Raps E, et al. Acute quadriplegic myopathy: a complication of steroids, nondepolarizing blocking agents or both. Neurology 1992;42:2082–7.
107. Melli G, Chaudhry V, Cornblath DR. Rhabdomyolysis: an evaluation of 475 hospitalized patients. Medicine (Baltimore) 2005;84:377–85.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.