(Carregando Índice)... (Carregando Índice)... |
Última revisão: 11/11/2015
Comentários de assinantes: 0
Reproduzido de:
Formulário Terapêutico Nacional 2010: Rename 2010 [Link Livre para o Documento Original]
Série B. Textos Básicos de Saúde
MINISTÉRIO DA SAÚDE
Secretaria de Ciência, Tecnologia e Insumos Estratégicos
Departamento de Assistência Farmacêutica e Insumos Estratégicos
Brasília / DF – 2010
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: seção 15.4
t Pó para solução injetável 450 UI e 500 UI de concentrado de fator VIII rico em fator de von Willebrand (AE>1.000 UI/ mg).
t Tratamento da doença de von Willebrand.
t Profilaxia de hemorragia em cirurgia de pacientes com doença de von Willebrand.
t História de reação anafilática ou sistêmica grave ao fator VIII de von Willebrand.
t Hipersensibilidade ao fator VIII de von Willebrand ou a qualquer outro componente da formulação.
t Usar com cuidado nos casos de:
– fatores de risco preexistentes para trombose.
– lactação.
t Altos níveis deste fator se associam a trombose, sobretudo em mulheres.
t O produto proveniente de plasma humano pode conter agentes infecciosos, incluindo vírus.
t Em grávidas e pacientes imunocomprometidos há maior risco de transmissão de infecção por parvovírus B19.
t Monitorizar para o desenvolvimento de inibidores de fator VIII.
t Categoria de risco na gravidez (FDA): C.
t Dose inicial 75 UI, por via intravenosa, seguida de 50 a 75 UI, por via intravenosa, a cada 8 a 12 horas, se necessário, até cicatrização completa.
t As doses são definidas de acordo com o tipo da doença e a gravidade da hemorragia:
t Hemorragia leve: epistaxe, sangramento oral, menorragia.
– Hemorragia grave: epistaxe grave ou refratária, sangramento gastrintestinal, trauma do sistema nervoso central, hemartrose ou hemorragia traumática.
t Tipo 1 (doença leve – atividade do fator maior que 30%, com hemorragia grave): dose inicial 40 a 60 UI/kg, por via intravenosa. Dose de manutenção 40 a 50 UI/kg, a cada 8 a 12 horas, por até 7 dias.
t Tipo 1 (doença moderada a grave – atividade do fator menor que 30%, com hemorragia leve): 1 ou 2 doses de 40 a 50 UI/kg, por via intravenosa.
t Tipo 1 (doença moderada a grave – atividade do fator menor que 30%, com hemorragia grave): dose inicial 50 a 75 UI/kg, por via intravenosa. Dose de manutenção 40 a 60 UI/kg, a cada 8 a 12 horas, por até 7 dias.
t Tipo 2 e 3 (hemorragia leve): 1 ou 2 doses de 40 a 50 UI/kg, por via intravenosa.
t Tipo 2 e 3 (hemorragia grave): dose inicial 60 a 80 UI/kg, por via intravenosa. Dose de manutenção 40 a 60 UI/kg, a cada 8 ou 12 horas, por até 7 dias.
t Dose pré-operatória: 60 UI, por via intravenosa, seguidas de 40 a 60 UI, por via intravenosa, a cada 8 a 12 horas, se necessário, até cicatrização completa.
t Em dose única, por via intravenosa, a duração do efeito do fator VIII von Willebrand é de 22 a 26 h.
t Edema facial, rubor, prurido, exantema, urticária.
t Palpitação
t Náusea, alterações no paladar.
t Vertigem, cefaleia, parestesia, sonolência.
t Visão embaçada.
t Anafilaxia: urticária, pressão torácica, prurido, edema, hipotensão, choque.
t Faringite, embolia pulmonar.
t Raros: aumento nos níveis de fibrinogênio, anemia hemolítica, eventos tromboembólicos.
t Alertar sobre a importância de avisar sobre o surgimento de reações alérgicas.
t Estas informações dependem do produtor e do tipo de fator adquirido.
t Manter o pó à temperatura de 2 a 8 ºC, não congelar. Ou à temperatura ambiente durante 2 meses.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Dissolver o concentrado liofilizado, agitando suavemente o frasco, sem sacudi-lo.
t Antes da reconstituição, deixar o frasco à temperatura ambiente. Administrar até 3 horas depois da reconstituição.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 15.4
t Pó para solução injetável 200 UI, 250 UI e 500 UI (concentrado de alta pureza; AE>50 UI/ mg)
t Tratamento e profilaxia de hemorragia na deficiência de fator IX congênito (hemofilia B).
t Coagulação intravenosa disseminada.
t Hipersensibilidade a proteína murina.
t Usar com cuidado nos casos de:
– coagulação intravenosa disseminada, fibrinólise, doença hepática, neonatos e pós-operatório (predisposição a complicações tromboembólicas – menos frequentes nas preparações purificadas).
– desenvolvimento de inibidores de fator IX (risco aumentado de hipersensibilidade, incluindo anafilaxia).
– altas doses (risco aumentado de enfarte do miocárdio, coagulação intravenosa disseminada, trombose venosa e embolia pulmonar).
– lactação.
t Presença de plasma humano no medicamento: risco potencial de transmissão de infecções virais, como hepatite B, HIV, entre outras.
t Indução de tolerância imune com risco potencial para síndrome nefrótica.
t Categoria de risco na gravidez (FDA): C (ver apêndice A).
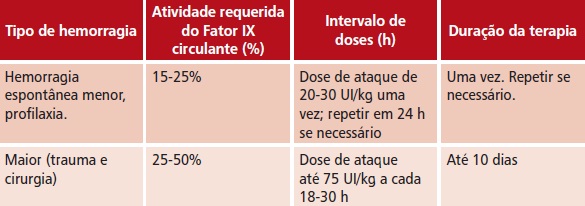
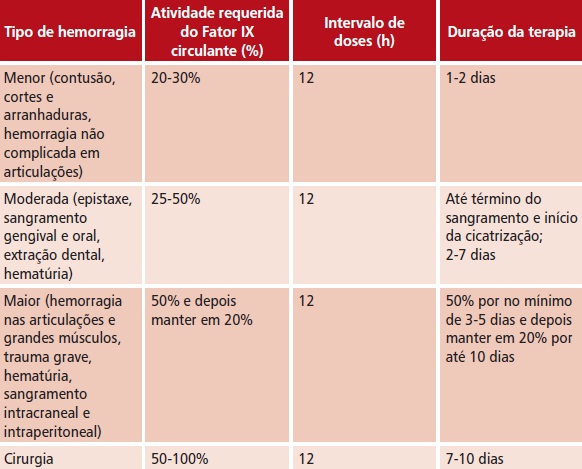
t Via intravenosa. Dose necessária = peso corporal em kg x aumento desejado do Fator IX (porcentagem do normal) x 1 UI/kg
t O quadro abaixo é um guia para dosagem em episódios de sangramento e em cirurgia

t Via intravenosa. Dose necessária = peso corporal em kg x aumento desejado do Fator IX (porcentagem do normal) x 1 UI/kg
t Usar o quadro abaixo como guia para dosagem em pacientes com eventos hemorrágicos e cirurgia em pacientes com hemofilia B, por injeção intravenosa lenta ou infusão intravenosa.
t Reações alérgicas (1,5%), rinite (6,4%), dispneia (3,2%) arrepio e febre (3,1%).
t Náusea (6,2%), redução no sentido da gustação (ageusia), distúrbios gastrintestinais.
t Hipotensão, complicações tromboembólicas.
t Exantema (1,6-7,7%), rubor (3,1%).
t Dor (6,2%) e sensação de queimação no lugar da injeção (1,6-7,7%),
t Vertigem (7,7%), cefaleia (10,8%).
t Orientar pacientes para portar cartão de identificação informando ser portador de hemofilia B.
t Alertar para notificar (ao médico ou farmacêutico) se esquecer de administrar uma dose.
t Orientar a maneira correta de empregar o medicamento em casa, garantindo que todas as instruções foram entendidas.
t Orientar para retirar o medicamento da geladeira antes da hora de usá-lo e injetar quando estiver à temperatura ambiente.
t Alertar para notificar se houver gravidez.
t Estas informações dependem do produtor e do tipo de fator adquirido.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Armazenar o pó e o diluente a temperatura ambiente. Podem ser armazenados entre 4-8oC. Soluções reconstituídas devem ser inspecionadas visualmente para identificar presença de partículas ou descoloração, devem ser filtradas antes da administração e a administração deve ser feita imediatamente à dissolução ou, no máximo, três horas após a reconstituição para evitar contaminação bacteriana. A potência do fator IX é expressa em Unidades Internacionais (UI), cada unidade correspondendo à média da atividade do Fator IX presente em 1 mL de plasma fresco coletado com menos de 1 hora.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 15.4
t Pó para solução injetável 60 kUI (1,2 mg/frasco), 120 kUI (2,4 mg/frasco) e 240 kUI (4,8 mg/frasco) de fator recombinante de coagulação VII ativado
t Hemorragia na deficiência adquirida de Fator VIII e na profilaxia da hemorragia pós-cirúrgica na deficiência de Fator VIII
t Hemorragia na deficiência congênita de Fator VII
t Hemorragia em pacientes com inibidores de Fator VIII ou Fator IX
t Profilaxia da hemorragia pós-cirúrgica na deficiência de Fator VII
t Profilaxia de hemorragia em pacientes com inibidores de Fator VIII e Fator IX (hemofilia A ou B)
t Usar com cuidado nos casos de:
– hipersensibilidade ao fator VII ou a qualquer ingrediente da formulação.
– hipersensibilidade a proteínas bovinas, de camundongos ou hamsters.
– doença aterosclerótica avançada, lesão por impacto, septicemia ou uso concomitante de concentrados de complexo de protrombina ativados (risco aumentado de eventos tromboembólicos ou coagulação intravascular disseminada; pode ser necessário reduzir dose ou interromper tratamento).
– idosos, não-hemofílicos, portadores de hemorragia intracerebral, uso concomitante de complexos concentrados de protrombina não ativada (risco aumentado de eventos adversos arteriais tromboembólicos, incluindo isquemia ou enfarte do miocárdio, isquemia e/ou enfarte cerebral.
– portadores de deficiência congênita de fator VIIa previamente tratados com plasma humano ou fator VII derivado de plasma humano (risco de desenvolvimento de anticorpos; analisar presença de anticorpos se não houver melhora clínica ao tratamento com fator VIIa).
– lactação (ver Apêndice B).
t Monitorizar tempo de protrombina (TP) e atividade coagulante de fator VII, antes e após a administração de fator VIIa recombinante. Se a atividade do fator VIIa não for aumentada adequadamente e o TP não for corrigido, ou se ocorrer sangramento descontrolado após a administração das doses recomendadas, o paciente deve ser avaliado para detecção de desenvolvimento de anticorpos.
t Categoria de risco na gravidez (FDA): C.
t 70 a 90 microgramas/kg em injeção intravenosa direta em 2 a 5 minutos a cada 2 a 3 horas até obtenção da hemostasia.
t 15 a 30 microgramas/kg em injeção intravenosa direta em 2 a 5 minutos, a cada 4 a 6 horas ate obter hemostasia. doses de 10 microgramas/kg são efetivas. Ajustar dose e frequência de administração à resposta individual.
t 90 microgramas/kg em injeção intravenosa direta em 2 a 5 minutos. Repetir doses em intervalos de 2 horas até hemostasia adequada ou até que o tratamento se mostre inadequado, embora a dose e o intervalo de administração possam ser ajustados com base na resposta do paciente e na gravidade do sangramento. Alcançada a hemostasia, continuar a administração do fator VIIa em intervalos de 3 a 6 horas.
t 15 a 30 microgramas/kg em injeção intravenosa direta em 2 a 5 minutos, a cada 4 a 6 horas até obter hemostasia. Doses de 10 microgramas/kg são efetivas. Ajustar dose e frequência de administração à resposta individual.
t 90 microgramas/kg em injeção intravenosa direta em 2 a 5 minutos, imediatamente antes da intervenção. Repetir doses em intervalos de 2 horas enquanto durar a cirurgia. Para dose pós-cirúrgica em cirurgias leves continuar a injeção direta a cada 2 horas pelas primeiras 48 horas e depois a cada 2 a 6 horas até cicatrização. Após cirurgia grave, continuar a cada 2 horas por 5 dias, e então a cada 4 horas até a ocorrência de cicatrização.
t Início: 10 a 20 minutos
t Tempo para máxima concentração plasmática – 15 minutos.
t Depuração endógena – em pacientes com hemofilia o depuração endógena total do fármaco é 30 a 36 mL/kg/h e parece ser independente da dose. Em pacientes pediátricos com hemofilia A ou B, a taxa de depuração endógena é de 67mL/kg/h, valor substancialmente maior que dos pacientes adultos com hemofilia A ou B, que apresentam taxa média de depuração endógena de 32,8 mL/kg/h.
t Meia-vida de eliminação: 2,3 horas. Em pacientes pediátricos com hemofilia A ou B a meia-vida é de 1,3 h.
t Náusea, vômito (1%)
t Oclusão de artéria cerebral, hidrocefalia (55%)
t Angina (0,1%), bradiarritmia (1%), edema (1%), hipertensão (0,8 a 10%), hipotensão (1%), coronariopatia isquêmica aguda, enfarte do miocárdio, taquicardia supraventricular (0,1%)
t Reação no sítio da injeção (1%), prurido (1%), exantema (1%)
t Tromboembolismo venoso, distúrbio da coagulação (1%), redução no fibrinogênio plasmático (2% ou mais), coagulação intravenosa disseminada (1%), epistaxe (0,2%), hemorragia (2%), púrpura (1%), choque, acidente vascular cerebral.
t Hepatotoxicidade (0,1%), com elevação de transaminases e alteração na função hepática.
t Reações de hipersensibilidade, anafilaxia (0,05%), desenvolvimento de anticorpos em pacientes previamente tratados com fator VII (0,06%).
t Artralgia, artropatia (1%), ataxia (0,05%), hemartrose (2% ou mais).
t Oclusão da artéria cerebral, isquemia cerebral, cefaleia (1%), hidrocefalia, hematoma subdural.
t Insuficiência renal aguda (0,05%), piora na função renal (1%).
t Pneumonia (1%), embolia pulmonar.
t Febre (0,3-2%), dor (1%).
t Alertar para:
– desenvolvimento de sintomas de alergia: urticária, pressão no peito, sibilos, hipotensão.
– evitar gravidez
– cuidados em doença renal ou doença da coagulação intravenosa disseminada.
t Estas informações dependem do produtor e do tipo de fator adquirido.
t Antes da reconstituição, deve ser armazenado entre 2 e 25 ºC e protegido da luz solar direta.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t O pó liofilizado deve ser reconstituído. Diluir o pó com água esterilizada. Após a reconstituição a solução pode ser armazenada a temperatura ambiente ou na geladeira. Após a diluição o fármaco deve ser administrado no máximo em três horas.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 15.4
t Pó para solução injetável 250 UI e 500 UI (AE > 100 UI)
t Controle de hemorragia espontânea ou relativa a procedimento cirúrgico em pacientes com hemofilia A.
t Usar com cuidado nos casos de:
– doses altas ou repetidas, sobretudo em pacientes de grupos sanguíneos A, B, ou AB (pode ocorrer anemia progressiva e hemólise intravenosa; menos provável com concentrados altamente purificados).
– pacientes com anticorpos ao fator VIII (risco de ausência de resposta e de aumento nos níveis de inibidor após infusão do fármaco). Empregar fator VIII porcino).
– crianças e idosos (segurança e eficácia não estão bem avaliadas por estudos clínicos; Efeitos adversos semelhantes aos dos adultos).
– idosos (cuidado com a determinação da dose).
– infusão contínua (atenção quanto à estabilidade do fator VIII, contaminação bacteriana, irritação local e tromboflebite e formação de anticorpos).
– lactação.
t Risco de adquirir doenças virais: hepatites A, B, C, Aids e parvovirose.
t Monitorizar para o desenvolvimento de inibidores de fator VIII. t Categoria de risco na gravidez (FDA): C (ver apêndice A).
Pré-operatória em cirurgia maior (hematoma, trauma, hemoptise graves, hematêmese e melena): 50 UI/kg por via intravenosa até alcançar aproximadamente 100% do fator anti-hemofílico normal. Repetir infusões a cada 6 ou 12 horas inicialmente na medida da necessidade até um total de 10 a 14 dias até a cicatrização completa.
t Leve: dose única de 10 UI/kg, via intravenosa, até alcançar aproximadamente 20% do fator VIII anti-hemofílico normal. Repetir se houver evidência posterior de sangramento.
t Moderada: 15 a 25 UI/kg, via intravenosa, até alcançar 30 a 50% do fator VIII normal. Repetir com 10 a 15 UI/kg a cada 8 ou 12 horas se houver evidência posterior de sangramento.
t Grave: iniciar com 40 a 50 UI/kg, via intravenosa, até alcançar 80 a 100% do fator anti-hemofílico normal. Repetir se houver evidência posterior de sangramento.
Dose pré-operatória em procedimentos cirúrgicos menores: iniciar com 15 a 25 UI/kg seguidos de 10 a 15 UI/kg a cada 8 a 12 horas até aumento de 30 a 50% do fator VIII normal.
t Dose pré-operatória em procedimentos cirúrgicos maiores: 50 UI/kg, via intravenosa, até alcançar um aumento de aproximadamente 100% do fator VIII antes da cirurgia. Repetir infusões a cada 6 a 12 horas inicialmente se necessário e até 10 a 14 dias até completar a cicatrização.
t Nota: Dose necessária para crianças e adultos (UI) = peso corporal (kg) x aumento desejado na proporção de fator VIII/(2%/UI/kg)
t Reações alérgicas, incluindo hipotensão, angioedema, calafrios, febre, urticária e anafilaxia.
t Distúrbios gastrintestinais, alteração na percepção de sabores.
t Rubor, palpitação.
t Dispneia, tosse.
t Cefaleia, vertigem, parestesia, sonolência.
t Visão turva.
t Reação no sítio da injeção, exantema.
t Início de ação por via intravenosa: 10 minutos
t Duração por via intravenosa: 12 a 15 horas
t Tempo para alcançar o pico de concentração plasmática: 10 minutos a 2 horas.
t Estas informações dependem do produtor e do tipo de fator adquirido.
t Armazenar o pó entre 2 e 8 ºC, evitar congelar o diluente. Algumas preparações podem ser armazenadas a temperatura ambiente (até 30 ºC), por períodos até 6 meses.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Antes da reconstituição, deixar o diluente a temperatura ambiente, de 15 a 30 ºC, não exceder 37º C.
t Durante a reconstituição, manter o fluxo do diluente em ângulo de 45 graus contra a parede do frasco para minimizar a formação de espuma.
t Administrar por meio de agulha filtradora em 3 horas após reconstituição em velocidade tolerada pelo paciente (em geral tolera-se a dose completa em 5 a 10 minutos)
t Usar apenas seringas plásticas porque a solução tende a aderir a seringas de vidro.
Sheila Silva Monteiro Lodder Lisboa
Na Rename2010: item 15.4
t Pó para solução injetável 500 UI a 600 UI de fator IX (AE > 0,6 UI/ mg)
t Hemorragia por deficiência dos fatores II, VII ou IX – tratamento e profilaxia
t Hemorragia na hemofilia B
t Hemartrose por hemofilia em pacientes com inibidores de fator VIII
t Coagulação intravenosa disseminada.
t Angina.
t Enfarte do miocárdio recente (exceto em hemorragia grave após dose(s) excessiva(s) de anticoagulantes orais e antes de terapia fibrinolítica).
t História de trombocitopenia induzida por heparina.
t Risco de trombose ou coagulação intravenosa disseminada. Provavelmente
menor com as preparações altamente purificadas.
t Usar com cuidado nos casos de:
– história de enfarte do miocárdio ou de coronariopatia.
– insuficiência hepática.
– uso pós-operatório.
t 0,5 UI/kg x peso em kg x aumento desejado (% do normal) a cada 4 ou 6 horas, se necessário. Em cirurgia recomendam-se níveis acima de 25% por pelo menos uma semana após a cirurgia. Para alcançar tais níveis por este período de tempo cada dose, intravenosa, deve ser calculada para aumentar o nível de fator IX em 40 a 60% acima do normal.
t 75 mg/kg de peso, por via intravenosa
t Conforme tipo de hemorragia:
– Menor (hemartrose recente, epistaxe leve, sangramento gengival, hematúria leve): 25 a 35 UI/kg até atingir aproximadamente 20% de fator IX em relação ao normal. Duração da terapêutica: 1 dia
– Moderada (sangramento articular grave, hematoma recente, hemorragia recente, trauma leve, hemoptise leve, hematêmese e melena, hematúria grave): 40 a 55 UI/kg até atingir aproximadamente 40% de fator IX em relação ao normal. Duração da terapêutica: 2 dias ou até cicatrização adequada
– Maior (hematoma, trauma e hemoptise graves, hematêmese e melena): 60 a 70 UI/kg até atingir aproximadamente 60% ou mais de fator IX em relação ao normal. Duração da terapêutica: 2 a 3 dias ou até cicatrização completa.
t Nota: Administração por via intravenosa, como regra geral: 1 UI de fator IX/ kg aumenta o nível plasmático de fator IX em 0,8%. Fórmula para cálculo da dose: número de fator IX necessário = peso corpóreo x aumento desejado do fator IX (% do normal) x 1,2
t Muito raramente náusea, eventos tromboembólicos (enfarte do miocárdio e acidente vascular cerebral), distúrbios de coagulação, tremores e febre, dor, reações alérgicas, inclusive exantema.
t Alertar sobre a importância de avisar sobre o surgimento de reações alérgicas.
t Armazenar sob temperatura de 2 a 8o C em frasco hermeticamente fechado.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Dissolver o concentrado liofilizado, agitando suavemente o frasco, sem sacudi-lo.
t Antes da reconstituição, deixar o frasco à temperatura ambiente. Administrar até 3 horas depois da reconstituição.
Sheila Silva Monteiro Lodder Lisboa
Na Rename2010: item 15.4
t Pó para solução injetável 500 UI
t Hemorragia por deficiência dos fatores II, VII ou IX – tratamento e profilaxia
t Hemorragia na hemofilia B
t Hemartrose por hemofilia em pacientes com inibidores de fator VIII
t Coagulação intravenosa disseminada.
t Angina.
t Enfarte do miocárdio recente (exceto em hemorragia grave após dose(s) excessiva(s) de anticoagulantes orais e antes de terapia fibrinolítica).
t História de trombocitopenia induzida por heparina.
t Risco de trombose ou coagulação intravenosa disseminada. Provavelmente menor com as preparações altamente purificadas.
t Usar com cuidado nos casos de:
– história de enfarte do miocárdio ou de coronariopatia.
– insuficiência hepática.
– uso pós-operatório.
t 0,5 UI/kg x peso em kg x aumento desejado (% do normal) a cada 4 ou 6 horas, se necessário. Em cirurgia recomendam-se níveis acima de 25% por pelo menos uma semana após a cirurgia. Para alcançar tais níveis por este período de tempo cada dose, intravenosa, deve ser calculada para aumentar o nível de fator IX em 40 a 60% acima do normal.
t 75 mg/kg de peso, por via intravenosa
t Conforme tipo de hemorragia:
– Menor (hemartrose recente, epistaxe leve, sangramento gengival, hematúria leve): 25 a 35 UI/kg até atingir aproximadamente 20% de fator IX em relação ao normal. Duração da terapêutica: 1 dia
– Moderada (sangramento articular grave, hematoma recente, hemorragia recente, trauma leve, hemoptise leve, hematêmese e melena, hematúria grave): 40 a 55 UI/kg até atingir aproximadamente 40% de fator IX em relação ao normal. Duração da terapêutica: 2 dias ou até cicatrização adequada
– Maior (hematoma, trauma e hemoptise graves, hematêmese e melena): 60 a 70 UI/kg até atingir aproximadamente 60% ou mais de fator IX em relação ao normal. Duração da terapêutica: 2 a 3 dias ou até cicatrização completa.
t Nota: Administração por via intravenosa, como regra geral: 1 UI de fator IX/ kg aumenta o nível plasmático de fator IX em 0,8%. Fórmula para cálculo da dose: número de fator IX necessário = peso corpóreo x aumento desejado do fator IX (% do normal) x 1,2
t Muito raramente náusea, eventos tromboembólicos (enfarte do miocárdio e acidente vascular cerebral), distúrbios de coagulação, tremores e febre, dor, reações alérgicas, inclusive exantema.
t Alertar sobre a importância de avisar sobre o surgimento de reações alérgicas.
t Armazenar sob temperatura de 2 a 8o C em frasco hermeticamente fechado.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Dissolver o concentrado liofilizado, agitando suavemente o frasco, sem sacudi-lo.
t Antes da reconstituição, deixar o frasco à temperatura ambiente. Administrar até 3 horas depois da reconstituição.
Larissa Niro
Rogério Aparecido Minini dos Santos
Na Rename 2010: item 13.1
t Comprimidos 100 mg
t Solução injetável 50 mg/mL
t Suspensão oral 20 mg/mL
t Convulsões tônico-clônicas generalizadas.
t Convulsões parciais.
t Estado de mal epiléptico.
t Profilaxia e tratamento de convulsões associadas a neurocirurgia ou traumatismo cranioencefálico grave.
t Hipersensibilidade as hidantoínas.
t Porfiria aguda.
t Bradicardia sinusal.
t Bloqueio sinoatrial.
t Bloqueio AV de graus 2 e 3.
t Síndrome de Stokes-Adams (uso parenteral).
t Usar com cuidado nos casos de:
– diabetes melito (hiperglicemia).
– hipotensão.
– insuficiência cardíaca congestiva.
– insuficiência hepática.
– história de doença renal ou hepática (não devem receber o regime de dose oral de ataque).
– se ocorrer exantema ou erupção cutânea (suspender o tratamento).
– uso de álcool (uso agudo: eleva os níveis plasmáticos de fenitoína; uso prolongado: reduz os níveis).
– suspensão do tratamento (pode precipitar estado epiléptico; suspensão deve ser gradual).
– HLA-B * 1502-positivos, mais comuns no Sul da Ásia, incluindo asiáticos índios (aumento do risco de síndrome de Stevens-Johnson e necrólise epidérmica tóxica; evitar utilizar como uma alternativa para carbamazepina em pacientes com teste positivo para esse alelo).
t Monitorar contagem de células sanguíneas.
t Métodos de reanimação devem estar disponíveis.
t Evitar extravasamento (soluções alcalinas são irritantes para os tecidos).
t Categoria de risco na gravidez (ADEC): D (ver apêndice A).
t 5 mg/kg, por via oral, dividido a cada 8 ou 12 horas. Dose de manutenção: 4 a 8 mg/kg/dia. Dose máxima: 300 mg/dia
t Dose inicial 10 a 15 mg/kg, por via intravenosa, até 1 a 3 mg/kg/min. Dose de manutenção: 2,5 a 5 mg/kg a cada 12 horas (neonatos e crianças até 12 anos), ou 100 mg a cada 6 ou 8 horas (12 a 18 anos).
t 5 mg/kg, por via oral, dividido a cada 8 ou 12 horas. Dose de manutenção: 4 a 8 mg/kg/dia. Crianças com idade superior a 6 anos podem necessitar da dose mínima de adultos. Dose máxima: 300 mg/dia.
t 100 mg, por via oral, a cada 8 horas. Dose de manutenção: 100 mg, a cada 6 ou 8 horas. Dose máxima diária: 600 mg.
t Dose inicial 10 a 15 mg/kg, por via intravenosa, até 50 mg/minuto, seguido de dose de manutenção de 100 mg, por via oral ou intravenosa, a cada 6 ou 8 horas.
t 100 a 200 mg, por via intramuscular, a cada 4 horas durante a cirurgia e continuar durante o período pós-operatório. Na administração intramuscular feita durante neurocirurgias, para pacientes previamente controlados por via oral, a dose intramuscular deverá ser 50% maior que a dose oral que vinha sendo utilizada.
t 100 mg, por via oral, a cada 8 horas. Dose de manutenção: 100 mg por via oral, a cada 6 ou 8 horas. Dose máxima: 600 mg/dia.
t Nota: Para pacientes obesos, a dose de ataque deve ser calculada com base no peso corporal ideal mais 1,33 vezes o excesso de sobrepeso; ajuste de dose deve ser considerado em hipoalbuminemia e doença renal.
t Absorção oral: lenta e variável entre as especialidades farmacêuticas.
Insatisfatória em neonatos. Absorção intramuscular: muito lenta, mas completa (92%).
t Pico de concentração plasmática: 1,5 a 3 horas.
t A fenitoína é distribuída no líquido cerebroespinhal, saliva, sêmen, líquidos gastrintestinais, bile e leite materno; também atravessa a placenta, com concentrações séricas fetais iguais às da mãe.
t Metabolismo hepático, especificamente pela família CYP2 de isoenzimas microssomais.
t Excreção renal, aumentada pela alcalinização da urina.
t A meia-vida de eliminação é dependente da dose e concentração plasmática, uma vez que apresenta farmacocinética de saturação.
t Prurido e erupção cutânea, síndrome de Stevens-Johnson, dermatose bolhosa, erupção purpúrea, escarificação, necrólise epidérmica tóxica; lupus eritematoso sistêmico.
t Agranulocitose, leucopenia, trombocitopenia, pancitopenia, transtorno granulocitopênica.
t Obstipação, hiperplasia gengival, náuseas e vômitos.
t Hepatotoxicidade, dano hepático, hepatite tóxica.
t Osteomalácia.
t Confusão mental e nervosismo, ataxia, problemas de coordenação, encefalopatia, cefaleia, insônia, fala emplastada, parestesia, vertigem, coreoatetose.
t Nefrotoxicidade.
t Depressão cardiovascular: bradiarritmias, hipotensão e colapso (particularmente em injeções rápidas).
t Alteração na função respiratória: pneumonia, fibrose pulmonar, insuficiência respiratória e infiltrado pulmonar.
t Beclamida: pode resultar em leucopenia. O perfil hematológico deve ser monitorado regularmente quando doses maiores de fenitoína são dadas concomitantemente a beclamida.
t Delavirdina: pode ter a sua concentração de vale diminuída. O uso concomitante não é recomendado devido à substancial redução da concentração plasmática da delavirdina.
t Erva-de-são-joão (Hypericum perforatum): pode resultar na redução da efetividade da fenitoína, podendo precipitar convulsões. Evitar o uso concomitante.
t Imatinibe: pode ter suas concentrações plasmáticas diminuídas. Se a associação se fizer necessária, a dose de imatinibe deve ser aumentada em pelo menos 50% para manter a eficácia. Ainda assim, a resposta clínica ao imatinibe deve ser sempre monitorada.
t Irinotecano: pode resultar na redução da sua exposição sistêmica com consequente diminuição da eficácia. Considerar o uso de um anticonvulsivante não indutor enzimático, com início de pelo menos 2 semanas antes de instituir a administração do irinotecano.
t Lidocaína: pode ter suas concentrações plasmáticas diminuídas e efeito depressivo cardíaco aditivo. Esta associação deve ser feita com cautela. Monitorar função cardíaca do paciente. Se possível, evitar em pacientes com doenças cardíacas diagnosticadas.
t Lopinavir: pode ter suas concentrações plasmáticas diminuídas. Se a associação se fizer necessária, ajustar a dose do lopinavir para manutenção da efetividade.
t Metotrexato: pode resultar na redução da efetividade da fenitoína e aumento no risco de toxicidade pelo metotrexato. Se a associação se fizer necessária, monitorar níveis plasmáticos de fenitoína e sinais de toxicidade pelo metotrexato.
t Posaconazol: pode resultar na redução das concentrações plasmáticas de posaconazol e aumento da fenitoína. É sugerido que a associação seja evitada. Monitoramento adequado deve ser realizado se o uso concomitante for indispensável.
t Ranolazina: pode ter suas concentrações plasmáticas reduzidas. O uso concomitante com indutores de isoenzima CYP3A, como a fenitoína, é contraindicado.
t Tacrolimo: pode ter sua efetividade diminuída ou aumentar as concentrações plasmáticas de fenitoína. Monitorar as concentrações plasmáticas de tacrolimo, sinais de redução de efetividade e proceder ao ajuste de dose. Monitorar os pacientes para os efeitos da elevação da exposição à fenitoína.
t Voriconazol: pode resultar no aumento das concentrações plasmáticas de fenitoína e redução de voriconazol. Monitorar frequentemente as concentrações plasmáticas e de Efeitos adversos à fenitoína. Se o uso concomitante for necessário, aumentar em 25% a dose intravenosa e em 100% a dose oral de voriconazol.
t Hipersensibilidade a fenitoína e compostos relacionados deve ser investigada antes de iniciar o tratamento.
t A efetividade de contraceptivos hormonais pode ser comprometida. Um método anticoncepcional adicional pode ser necessário. A ocorrência de gravidez deve ser notificada imediatamente ao médico
t Higiene dental/bucal adequada e acompanhamento odontológico são necessários devido à ocorrência de hiperplasia gengival, em especial às crianças. Cuidado nas cirurgias dentárias e tratamento odontológico de emergência.
t Pode ocorrer crescimento anormal e excessivo do cabelo, notado principalmente nas meninas.
t Potencial redução do desempenho escolar pode ocorrer com uso prolongado e em doses altas.
t Pacientes idosos são mais susceptíveis aos Efeitos adversos e toxicidade.
t Devido ao potencial de interação da fenitoína com outros produtos, nenhum medicamento ou substância terapêutica deve ser utilizados sem conhecimento do médico.
t As formas orais devem ser administradas preferentemente com alimentos.
t Os comprimidos de fenitoína não devem ser partidos ou mastigados durante a administração.
t Para a forma líquida um medidor graduado deve ser utilizado para determinação da dose.
t O uso de bebidas alcoólicas não é recomendado durante o tratamento com fenitoína.
t Os pacientes diabéticos que utilizam fenitoína devem monitorar estreitamente os níveis de glicose no sangue.
t Dosagem e posologia devem ser rigorosamente obedecidas. Não interromper o tratamento sem autorização médica.
t Cuidado ao dirigir, usar máquinas ou exercer atividade as quais requerem atenção.
t Armazenar a temperatura ambiente, entre 15 e 30%, e protegidas a luz. Não expor a extremos de temperatura sob risco de perda de estabilidade e deterioração.
t Os comprimidos de fenitoína não devem ser amassados, partidos ou triturados.
t Não está garantida a mesma bioequivalência e biodisponibilidade das especialidades farmacêuticas do comprimido de fenitoína. Isto está relacionado à quantidade de sulfato de cálcio utilizado como adjuvante nas preparações.
t A forma ácida da fenitoína possui 8% a mais de fenitoína base do que a forma sódica.
t A forma sódica de fenitoína contém 0,35 mEq (8 mg) de sódio por 100 mg.
t A suspensão oral de fenitoína possui liberação imediata, portanto não deve ser administrada uma única vez ao dia, mas sim dividida em 2 a 3 doses diárias.
t A suspensão oral deve ser agitada bem antes da medição da dose.
t Armazenar entre 15 e 30 ºC, em recipiente bem fechado, protegido da luz. Não congelar ou expor a temperaturas superiores a 40 ºC.
t Sob baixas temperaturas, a solução de fenitoína pode formar um precipitado que usualmente se dissolve após ser aquecido a temperatura ambiente, entretanto, não utilizar se a solução não estiver límpida.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t O preparo da solução para infusão deve ser feito, imediatamente antes da administração, pela adição da fenitoína em até 50 mL de cloreto de sódio 0,9% injetável e concentração final entre 1 a 10 mg/mL de fenitoína base. O tempo de infusão deve ser no máximo 1 hora. O equipo deve ser enxaguado com cloreto de sódio 0,9% injetável antes e após a administração da solução de fenitoína para minimizar a irritação venosa local. Usar filtro de 0,22 a 0,45 micrômetros no equipo.
t A injeção intravenosa deve ser feita diretamente numa veia larga, com uma agulha de calibre grande ou cateter. A infusão intermitente é preferida à contínua. Evitar esta última.
t A velocidade de infusão máxima para adultos é de 50 mg/min e 1 a 3 mg/ kg/min em neonatos.
t A mistura da fenitoína sódica com outros fármacos para injeção é contraindicada. Mesmo quando adicionada a cloreto de sódio 0,9% injetável, a solução deve ser administrada em no máximo 2 horas após o preparo. Uma coloração levemente amarelada pode surgir, sem, no entanto, alterar a potência da fenitoína.
t Monitorar função cardíaca e os valores de PA durante a administração intravenosa.
t A injeção intramuscular não é recomendada para remissão do estado epiléptico devido à absorção retardada.
t O extravasamento deve ser evitado, pois a injeção de fenitoína é cáustica aos tecidos.
t Transaminases hepáticas, fosfatase alcalina, glicemia e testes da função tireoideana podem ser alterados durante o uso da fenitoína.
t Fenitoína sódica não deve ser adicionada em perfusão intravenosa, devido à falta de solubilidade e precipitação resultante.
Atenção: este medicamento apresenta um número elevado de interações, sobretudo por indução do metabolismo de outros fármacos. Deve ser realizada uma pesquisa específica sobre este aspecto quando se considerar a terapia com fenitoína, bem como ao introduzir ou descontinuar outros medicamentos no esquema terapêutico do paciente.
Maurício Fábio Gomes
Na Rename 2010: item 13.1
t Comprimido 100 mg
t Solução oral 40 mg/mL
t Solução injetável 100 mg/mL
t Controle de crises epilépticas parciais, complexas e tônico-clônicas (segunda escolha).
t Estado de mal epiléptico (para controle após diazepam).
t Convulsões em neonatos e convulsões febris na infância.
t Porfiria.
t Crises de ausência.
t Hipersensibilidade a fenobarbital e a outros barbitúricos.
t Insuficiência hepática grave (ver Apêndice C).
t Insuficiência repiratória.
t Usar com cuidado nos casos de:
– idosos (indução de confusão mental).
– crianças e pacientes enfraquecidos (risco de hipercinesia).
– insuficiência renal (ver Apêndice D).
– insuficiência hepática (ver Apêndice C).
– abuso de álcool e psicotrópicos, depressão.
– tratamento prolongado (podem ocorrer tolerância e dependência física e psíquica).
– suspensão do tratamento após uso prolongado (retirada abrupta pode desencadear estado de mal epiléptico; suspender de forma gradual, com reduções de 10% da dose a cada dia).
– lactação (ver Apêndice B).
t Evitar extravasamento perivascular ou injeção intra-arterial, porque a solução é alcalina.
t Monitorar pressão arterial, respiração e frequência cardíaca durante a administração intravenosa de fenobarbital; equipamento de ressuscitação e ventilação artificial devem estar disponíveis.
t Categoria de risco na gravidez (FDA): D (ver Apêndice A).
t 5 a 10 mg/kg da forma sódica, por via intravenosa, a cada 20 a 30 minutos, até concentração plasmática alvo de 40 mg/L.
t 15 a 20 mg/kg, por via intramuscular ou, na forma sódica, por via intravenosa, seguida por dose de manutenção de 3 mg/kg, por via intramuscular ou oral a cada 8 horas.
t 5 a 8 mg/kg/dia, por via oral, de 1 a 2 doses.
t Crianças e lactentes: administrar de 5 a 10 mg/kg da forma sódica, por via intravenosa (30 mg/minuto). Repetir se necessário, até dose máxima: 40 mg/ kg.
t De 60 a 180 mg, por via oral, à noite.
t Administrar, inicialmente, 10 mg/kg da forma sódica por infusão intravenosa (100 mg/min). Se as convulsões não cessarem, reduzir a taxa para 50 mg/ minuto e adicionar fenitoína. Dose máxima de fenobarbital de 1 a 2 g.
Notas:
t Infundir lentamente (3 a 5 minutos); não exceder 2 mg/kg/minuto em crianças pequenas, 30 mg/minuto em maiores e 60 mg/minuto em adultos.
t Injeções intramusculares devem ser administradas profundamente em grandes músculos, como glúteo máximo ou vasto lateral; existe risco de abscesso estéril nas aplicações superficiais; não administrar mais de 5 mL em cada injeção.
t Absorção gastrintestinal rápida.
t Início de ação: 1 hora (oral), 20-60 minutos (intramuscular), 5 minutos (intravenoso).
t Duração da ação de dose única: 10-12 horas.
t Concentração plasmática terapêutica: 10-40 microgramas/mL.
t Metabolismo: hepático. Fenobarbital é importante indutor de várias isoenzimas microssomais, principalmente CYP3A4 e CYP1A2.
t Meiavida de eliminação: 79 horas (adultos), 21-75 horas (crianças), 110 horas (neonatos).
t Excreção: renal
t Hipotensão, choque.
t Depressão respiratória.
t Obstipação, náusea, vômito.
t Vertigem, sonolência, alucinações, ansiedade, nervosismo, irritabilidade, prejuízo de desempenho cognitivo, dores de cabeça, disartria.
t Eczema esfoliativo (raro), síndrome de Stevens-Johnson (raro), necrólise epidérmica tóxica, urticária, angioedema.
t Agranulocitose (raro), anemia megaloblástica (raro), trombocitopenia (raro), leucopenia.
t Tromboflebite (raro).
t Dano hepático,
t Nefrotoxicidade.
t Osteopenia (raro), raquitismo (raro), hipocalcemia.
t Ácido valproico, cloranfenicol: pode haver inibição do metabolismo de fenobarbital. Considerar redução de doses do fenobarbital.
t Álcool, analgésicos opioides, antidepressivos, benzodiazepínicos: pode haver efeito aditivo de depressão respiratória. Monitorar estreitamente a função respiratória. Considerar redução de doses.
t Contraceptivos orais: redução da concentração de estrógenos com perda da eficácia contraceptiva. Considerar o uso de uma formulação contendo dose maior de estrógenos ou método contraceptivo alternativo.
t Inibidores da MAO, lopinavir, tacrolimo: pode resultar na redução das concentrações plasmáticas destes fármacos. A efetividade do tratamento deve ser monitorada, assim como a concentração plasmática do imunossupressor.
t Inibidores de tirosina cinase: pode resultar na redução das concentrações plasmáticas dos inibidores; aumentar as doses e monitorar efetividade.
t Fenobarbital pode reduzir as concentrações plasmáticas e o efeito de: irinotecano, metoxiflurano, voriconazol e delavirdina (uso concomitante contraindicado); quetiapina e teniposídeo (aumentar as doses destes); anticoagulantes cumarínicos (monitorar tempo de protrombina e, se necessário, ajustar a dose).
t Alertar sobre a importância de informar sobre alergia a fenobarbital ou outro barbitúrico, gravidez, amamentação, bem como a ocorrência de efeitos indesejáveis.
t Alertar para evitar atividades que exijam atenção, como dirigir automóveis e operar máquinas, pelo risco de acidente.
t Orientar para utilizar o medicamento preferentemente com o estômago vazio.
t Alertar para não interromper o tratamento.
t Alertar para não usar bebida alcoólica durante o tratamento.
t A eficácia dos contraceptivos orais pode ser prejudicada, portanto, outro método anticoncepcional adicional deve ser utilizado.
t Armazenar à temperatura ambiente (entre 15 e 30 oC), longe do calor, luz e umidade.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t A solução para administração intravenosa pode ser diluída em igual volume de solução injetável de glicose 5%, Ringer + lactato ou cloreto de sódio 0,9%.
t A forma injetável é incompatível com os seguintes fármacos em misturas intravenosas: anfotericina B, ampicilina, analgésicos opioides, caspofungina, clorpromazina, cimetidina, ciclosporina, benzodiazepínicos, diltiazem, dobutamina, doxorrubicina, doxiciclina, eritromicina, haloperidol, lansoprazol, lidocaína, metildopa, norepinefrina, penicilinas, fentolamina, fenitoína, prometazina, tiamina, vincristina, bloqueadores neuromusculares. Outras incompatibilidades, consultar literatura específica.
Atenção: este medicamento apresenta um número elevado de interações. Considerar a descontinuação de outros medicamentos no esquema terapêutico do paciente quando do uso do citrato de fentanila.
Rosa Martins
Na Rename 2010: itens 14.2, 14.6
t Cápsulas 67 mg e 200 mg (micronizado)
t Dislipidemias associadas a diabetes melito tipo 2, como adjuvante ao tratamento com sinvastatina, se necessário.
t Prevenção primária e secundária de doença arterial coronariana associada a diabetes melito tipo 2.
t Hipersensibilidade ao fenofibrato.
t Doença na vesícula biliar.
t Insuficiência hepática, incluindo cirrose biliar primária e anormalidade da função hepática persistente e inexplicável.
t Insuficiência renal grave (ver Apêndice D).
t Usar com cuidado nos casos de:
– associação com um inibidor da HMG-CoA redutase, diabéticos, idosos, insuficiência renal (ver Apêndice D) e/ou hipotireoidismo (aumento do risco de miopatia incluindo rabdomiólise).
– associação com anticoagulantes (risco durante atividades físicas).
– lactação (ver Apêndice B).
t Aumenta o risco de colelitíase.
t Pode ocorrer miopatia e ser necessário interromper o tratamento.
t Monitorar função hepática, alteração é dose dependente.
t Categoria de risco na gravidez (FDA): C
t Dose inicial: 67 mg, por via oral, a cada 8 horas. Podendo ser diminuída para 2 vezes ao dia ou aumentada para 4 vezes ao dia, de acordo com a resposta. Ou,
t Dose de 200 mg, por via oral, a cada 24 horas.
t Dose de 200 mg, por via oral, a cada 24 horas.
t Dose inicial: 67 mg, por via oral, a cada 24 horas. Podendo aumentar a dose para 200 mg, por via oral, a cada 24 horas, se necessário.
t Biodisponibilidade: 90%, melhora com alimento.
t Início da ação: 6 a 8 semanas.
t Pico de concentração: 4 a 8 horas.
t Metabolismo hepático e renal, via glucoronidação
t Metabolito ativo: acido fenofíbrico.
t Meia-vida de eliminação: 20 a 22 horas.
t Excreção: 60 a 93% renal; 5 a 25% nas fezes.
t Dialisável: não.
t Trombose venosa profunda (1%).
t Exantema (6%), alopecia (0,5%), fotossensibilidade.
t Colelitíase, pancreatite, obstipação (2%), diarreia, dispepsia, flatulência, náusea (2%), desconforto gástrico, dor abdominal (5%).
t Hepatite colestática, hepatite, hepatotoxicidade (3 a 13%).
t Leucopenia, eosinofilia, hiper-homocisteinemia.
t Reação de hipersensibilidade.
t Artralgia, miopatia induzida por medicamento, rabdomiólise, aumento da creatinina fosfatoquinase CPK (3%).
t Cefaleia (3%), fadiga, insônia, tontura.
t Alterações do humor, cognitivas, do sono, da percepção; astenia, letargia, sonolência.
t Nefrotoxicidade,
t Impotência, diminuição da libido.
t Broncoespasmo, embolismo pulmonar.
t Morte por doença cardíaca coronária e total.
t Anticoagulantes orais (acenocumarol, anisindiona, dicumarol, fenindiona, femprocumona e varfarina): podem ter a efetividade/toxicidade aumentada pelo fenofibrato. Monitorar sinais e sintomas específicos de coagulação.
t Colchicina, estatinas (inibidores da HMGCoA redutase): podem ter a efetividade/toxicidade aumentada pelo fenofibrato. Monitorar sinais e sintomas específicos de miosite, miopatia, rabdomiólise (dor, fraqueza ou rigidez muscular) e concentração sérica de creatina cinase.
t Colestipol (sequestradores de ácidos biliares): pode diminuir a efetividade do fenofibrato. Administrar o fenofibrato uma hora antes ou 4 a 6 horas após o colestipol e monitorar sinais e sintomas específicos.
t Ezetimiba, glimepirida: podem ter a efetividade/toxicidade aumentada pelo fenofibrato. Monitorar sinais e sintomas específicos de colelitíase e hipoglicemia respectivamente.
t Orientar a ingestão com alimentos.
t Referir ao médico em caso de dor, fraqueza ou rigidez muscular.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t Armazenar à temperatura ambiente, entre 15 e 30 °C, proteger do calor, umidade e luz direta.
t Uma cápsula de 200 mg de fenofibrato micronizado corresponde ao comprimido de 160 mg
Thais Furtado de Souza
Na Rename 2010: item 6.3
t Solução injetável 300 microgramas/mL.
t Uso restrito em casos de neutropenia grave relacionada a tratamento anti-neoplásico.
t Hipersensibilidade a proteínas derivadas de Escherichia coli ou a filgrastim.
t Quimioterapia mielossupressiva ou radioterapia concomitantes (não administrar no período de 24 horas antes ou após o procedimento).
t Neutropenia congênita grave (síndrome de Kostmann) com citogenética anormal.
t Usar com cuidado nos casos de:
– síndrome respiratória aguda grave em adultos, neutropenia e sepse, leucemia mieloide aguda secundária, anemia falciforme, história de infiltração pulmonar ou pneumonia.
– quimioterapia mielossupressiva (monitorar leucocitose).
– neutropenia congênita (risco de síndrome mielodisplástica ou leucemia mieloide).
– lactação (não há informação sobre concentração no leite materno).
t Monitorar regularmente hemograma do paciente.
t Monitorar aumento do baço (risco de crescimento e ruptura).
t Monitorar densidade óssea (risco de osteoporose). t Categoria de risco na gravidez (FDA): C.
t Iniciar 24 horas após a realização da quimioterapia, com dose de 5 microgramas/kg/dia, por infusão intravenosa em 15 a 30 minutos, ou por infusão contínua nos pacientes recebendo quimoterapia mielossupressiva; a dose pode ser aumentada em 5 microgramas/kg/dia a cada ciclo da quimioterapia, de acordo com a contagem absoluta de neutrófilos. Continuar tratamento até a normalização da contagem de neutrófilos, geralmente por até 14 dias ou mais nos casos de leucemia mieloide aguda.
t Iniciar tratamento pelo menos 24 horas após transplante, com dose de 10 microgramas/kg/dia, por infusão intravenosa com duração de 4 ou 24 horas, ajustando a dose conforme a contagem absoluta de neutrófilos:
t Se a contagem absoluta de neutrófilos for superior a 1000/mm3 por 3 dias consecutivos, a dose deve ser reduzida para 5 microgramas/kg/dia. Suspender tratamento caso a contagem absoluta de neutrófilos permaneça superior a 1000/mm3 por mais 3 dias.
t Se a contagem absoluta de neutrófilos atingir um valor inferior a 1000/mm3 deve-se retornar à dose de 10 microgramas/kg/dia.
t Dose de 6 a 10 microgramas/kg/dia ou de 3 a 5 microgramas/kg, a cada 12 horas, por via subcutânea, administrada ao menos 4 dias antes da primeira leucoferese e continuar até a última. Ajustar dose se a contagem leucocitária periférica for superior 100.000/mm3.
t Início de efeito: 24 horas
t Pico de concentração: 2 a 8 horas após a administração subcutânea.
t Tempo de meia-vida: 3,5 horas.
t Depuração endógena renal: 0,5 a 0,7 mL/min/kg
t Febre (12%).
t Alopecia.
t Náusea (10%) e vômito (7%), obstipação, anorexia, mucosite.
t Esplenomegalia (rara, porém ocorre em 30% dos pacientes com neutropenia crônica grave).
t Dor óssea (33%), osteopenia, artrite reumatoide.
t Retenção de fluido, vasculite cutânea.
t Anemia (65%), anemia falciforme, leucocitose (2%), trombocitopenia (6%).
t Síndrome respiratória aguda, hemorragia pulmonar.
t Dor no lugar da injeção
t Epistasia (9 a 15%).
t Vincristina: pode resultar em neutropenia periférica grave. Restringir a dose de vincristina no primeiro ciclo e monitorar paciente.
t Administrar em diferentes áreas do corpo quando em uso por via subcutânea.
t Em caso de esquecimento de alguma dose contatar o médico ou farmacêutico.
t Notificar o médico sobre sinais e sintomas de dor, transtorno respiratório, infecção, sangramento e demais efeitos adversos.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Antes da diluição, armazenar à temperatura de 2 a 8 °C. Não agitar. Descartar qualquer quantidade após 24 horas à temperatura ambiente. Diluições devem ser preparadas utilizando-se glicose 5%, a concentração final deve estar entre 5 e 15 microgramas/mL. Forma precipitado quando diluído em cloreto de sódio. Plástico e vidro podem adsorver filgrastim. Para minimizar a adsorção no material de armazenagem ou equipamento de infusão deve-se adicionar albumina humana (concentração final 2 mg/mL) em soluções com concentração abaixo de 15 microgramas. As soluções preparadas podem ficar sob temperatura ambiente até o máximo de 24 horas antes da administração, após esse período deverão ser descartadas. Sob temperatura entre 2°C e 8°C a soluções são estáveis por 7 dias. Evitar congelamento, soluções congeladas por mais de 24 horas ou mais de uma vez devem ser descartadas. Em seringas de tuberculina, filgrastim não diluído é estável por 24 horas sob temperatura de 15°C a 30°C e por 2 semanas sob temperatura de 2°C a 8°C. Frascos de filgrastim são estáveis por até 7 dias sob temperatura entre 9°C e 30°C.
Sheila Silva Monteiro Lodder Lisboa
Na Rename 2010: item 15.2
t Solução injetável a 10 mg/mL.
t Reversão do efeito de anticoagulantes.
t Hipoprotrombinemia adquirida ou induzida por varfarina.
t Profilaxia e tratamento de doença hemorrágica de recém-nascido.
t Hipersensibilidade a fitomenadiona ou a qualquer outro ingrediente da formulação.
t Usar com cuidado nos casos de:
– injeções intravenosas (somente em situações emergenciais; administrar lentamente).
– altas doses de fitomenadiona em neonatos, principalmente prematuros (risco de hemólise, hiperbilirrubinemia e icterícia; não exceder dose recomendada).
– hipoprotrombinemia hereditária ou induzida por insuficiência hepática (fitomenadiona pode não ser eficaz nestes casos).
– idosos (reduzir doses).
– insuficiência hepática.
t A administração empírica de fitomenadiona a recém-nascidos para tratamento de doença hemorrágica não deve substituir avaliação clínica e laboratorial pertinentes. Uma pronta resposta à terapêutica com fitomenadiona (encurtamento do tempo de protrombina em 2 a 4 horas), em geral, é diagnóstico de doença hemorrágica no recém-nascido. Falha na resposta terapêutica sugere condição diferente ou distúrbio de coagulação não relacionado à fitomenadiona.
t Determinações periódicas do tempo de protrombina são recomendadas para avaliar a necessidade de terapia adicional com fitomenadiona.
t Categoria de risco na gravidez (FDA): C.
t Dose de 1 mg, por via intramuscular ou subcutânea; doses adicionais podem ser administradas, se necessário, a cada 8 horas.
t Dose de 0,3 mg, por via intramuscular, para recém-nascidos pré-termo com menos de 1 kg de peso; logo após o nascimento.
t Dose de 0,5 mg, por via intramuscular, para recém-nascidos pré-termo com mais de 1 kg de peso, logo após o nascimento.
t Dose de 1 mg, por via intramuscular, para recém-nascidos a termo, logo após o nascimento.
t Inicialmente 2,5 a 10 mg, por via intramuscular ou intravenosa lenta, podendo chegar a 25 mg de acordo com a resposta. A dose inicial pode ser repetida a cada 6 a 8 horas se a resposta for inadequada. Durante o tratamento o tempo de protrombina do paciente deve ser monitorado.
t Sangramento ausente ou de menor importância : 0,5 mg por injeção intravenosa lenta
t Hemorragia moderada: 10 a 20 mg por injeção intramuscular t Hemorragia grave: 5 a 10 mg por injeção intravenosa lenta Aspectos farmacocinéticos clinicamente relevantes 3, 9
t Latência (via intravenosa): ação detectável em 1 a 2 horas, hemorragia controlada em 3 a 6 horas e concentrações normais de protrombina atingidas em 12 a 14 horas (tratamento de hemorragia)
t Reação cutânea por administração intramuscular
t Anafilaxia; reações de hipersensibilidade incluindo rubor, dispneia, broncoespasmo, vertigem, hipotensão e colapso respiratório ou circulatório, os quais podem ser principalmente devido à presença de surfactante de óleo de castor polietoxilado em algumas formulações injetáveis do que devido a fitomenadiona.
t Anemia hemolítica (raro) e trombocitopenia (raro)
t Varfarina: uso concomitante pode causar mudanças ou flutuações do tempo de protrombina.
t Orientar o paciente para restringir a ingestão de vegetais verdes folhosos.
t Alertar para a possibilidade de surgimento de exantema, transtorno respiratório, vertigem, palpitação, paladar estranho na boca, dor, edema ou sensibilidade no lugar da injeção.
t Manter à temperatura ambiente, entre 15 e 30 oC. Não congelar. Proteger da luz.
t Cuidado: a fórmula do medicamento determina a via de administração, verificar informação com o produtor.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Pode ser diluída com cloreto de sódio 0,9%, glicose 5% ou glicose 5% em cloreto de sódio 0,9% para injeção. Todos os diluentes devem ser livres de conservantes.
t As soluções devem ser preparadas imediatamente antes do uso e qualquer porção não usada deve ser descartada.
t Incompatível com amobarbital sódico, cianocobalamina, cloridrato de dobutamina, fenitoína, pentobarbital sódico, secobarbital e secobarbital sódico.
Atenção: fitomenadiona só deve ser administrada por via intramuscular ou intravenosa quando inevitável, pois há riscos de reações adversas graves incluindo fatalidades.
Mirian Parente Monteiro
Na Rename 2010: item 5.3.1
t Cápsula 100 mg e 150 mg.
t Solução injetável 2 mg/mL.
t Pó para suspensão oral 10 mg/mL
t Candidemia
t Candidíase disseminada
t Candidíase de vias urinárias.
t Candidíase esofágica
t Candidíase orofaríngea (tratamento em pacientes com HIV).
t Candidíase vulvovaginal.
t Coccidiomicose (profilaxia e tratamento em pacientes com HIV)
t Criptococose pulmonar (tratamento em pacientes com HIV)
t Meningite criptocócica
t Prevenção de infecções fúngicas em pacientes submetidos a transplante de medula óssea.
t Hipersensibilidade ao fármaco ou a outro componente da fórmula.
t Porfirias agudas.
t Usar com cuidado nos casos de:
– insuficiência renal, especialmente para tratamento prolongado (ver Apêndice D).
– uso de altas doses, tratamentos prolongados ou uso concomitante com outros fármacos hepatotóxicos (monitorar a função hepática).
– aumentos nas concentrações das transaminase séricas (8 vezes ou mais que o limite superior) ou sintomas de doença hepática (risco de necrólise hepática; suspender o tratamento).
– imunocomprometidos que desenvolvem exantema durante a terapia com fluconazol (monitorar rigorosamente e descontinuar o tratamento se as lesões progredirem).
– condições potencialmente pró-arrítmicas por induzirem prolongamento do intervalo QT e torsade de pointes, incluindo o uso de determinados fármacos (aumenta o risco de palpitações, extrassístoles ventriculares e síncope; monitorar eletrocardiograficamente).
– lactação.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t 6 a 12 mg/kg/dia, por via oral ou intravenosa
t 6 mg/kg/dia, por via oral ou intravenosa, no dia 1, então 3 mg/kg/dia, até o máximo de 12 mg/kg/dia, por no mínimo 3 semanas; continuar por mais 2 semanas ou até resolução dos sintomas.
t 6 mg/kg/dia, por via oral, no dia 1, então 3 a 6 mg/kg/dia, até o máximo de 400 mg/dose, por no mínimo 2 a 3 semanas.
t 12 mg/kg/dia, por via oral ou intravenosa, no dia 1, então 6 mg/kg/dia, até o máximo de 12 mg/kg/dia, por 10 a 12 semana após cultura negativa no LCR; para supressão de recorrência a dose recomendada é de 6 mg/kg/dia. Ou então,.
t Indução: 12 mg/kg/dia, por via oral ou intravenosa, no dia 1, então 6 a 12 mg/kg/dia, até o máximo de 800 mg/dia em combinação com flucitosina 25 mg/kg, por via oral, de 6 em 6 horas, por no mínimo 2 semanas.
t Consolidação: 12 mg/kg/dia, por via oral ou intravenosa, no dia 1, então 6 a 12 mg/kg/dia, até o máximo de 800 mg/dia, por no mínimo 8 semanas.
t Manutenção: 6 mg/kg/dia, por via oral, até o máximo de 200 mg/dia; considerar descontinuação quando a contagem de CD4 + se mantiver no mínimo em 200 células/microlitro durante 6 meses, em pacientes assintomáticos com 6 anos e mais de idade e que tenha recebido fluconazol e terapia antirretroviral por pelo menos 6 meses.
t 400 a 600 mg/dia, por via intravenosa
t 200 mg (3 mg/kg)/dia, por via oral ou intravenosa, durante 2 semanas.
t 200 a 400 mg (3 a 6 mg/kg)/dia, por via oral ou intravenosa, por 2 a 3 semanas.
t 100 mg/dia, até o máximo de 400 mg/dia, por via oral ou intravenosa, por 2 a 3 semanas.
t 200 mg, por via intravenosa, no dia 1, seguidos por 100 mg/dia, por no mínimo 2 semanas.
t Não complicada: 150 mg, por via oral em dose única.
t Complicada: 150 mg, por via oral, a cada 72 horas, por 3 doses.
t Recorrente: 150 mg, por via oral, uma vez por semana, por até 6 meses.
t Profilaxia: 400 mg/dia, por via oral; continuar até contagem de CD4+ manter-se no mínimo de 250 células/microlitros durante 6 meses.
t Tratamento: 400 a 800 mg/dia, por via oral ou intravenosa.
t Manutenção: 400 mg/dia, por via oral.
t 200 a 400 mg/dia, por via intravenosa, indefinidamente. Ou então,.
t 400 mg/dia, por via oral ou intravenosa, e flucitosina 100 a 150 mg/kg/dia, por via oral, divididos em 4 doses diárias, durante 10 semanas.
t 400 mg/dia, por via oral ou intravenosa, no dia 1, então 200 mg/dia, até o máximo de 400 mg/dia; tratar por 10 a 12 semana após cultura negativa no LCR.
t 400 mg/dia, por via oral ou intravenosa, no dia 1, então 200 mg/dia, por 10 a 12 semana após cultura negativa de LCR; para supressão de recorrência a dose recomendada é de 200 mg/dia. Ou então,.
t Indução: 400 mg/dia, por via oral ou intravenosa, em associação com anfotericina B 0,7 mg/kg/dia, durante 2 semanas; OU 400 a 800 mg/dia, em associação com flucitosina 25 mg/kg, por via oral, de 6 em 6 horas, durante 4 a 6 semanas.
t Consolidação: 400 mg/dia, por via oral, durante 8 semanas, seguidos de 200 mg/dia indefinidamente ou até contagem de CD4 + se mantiver no mínimo de 250 células/microlitros durante 6 meses sob tratamento antirretroviral.
t 400 mg/dia, por via oral ou intravenosa; pacientes sujeitos a granulocitopenia (menos de 500 neutrófilos/mm3) devem iniciar a prevenção durante vários dias previamente ao início da neutropenia, e continuar por 1 semana após a contagem de neutrófilos alcançar 1000/mm3.
t Absorção: rápida, quase completa e independe da presença de ácidos ou alimentos.
t A terapia intravenosa é geralmente reservada para pacientes que não toleram ou são incapazes de tomar o fármaco pela via oral.
t Biodisponibilidade: acima de 90% (comprimidos e injeção intravenosa)
t Distribui-se por todos os tecidos e fluidos.
t Metabolismo: hepático
t Tempo para o pico de concentração plasmática: 1 a 2 horas. (comprimido, injeção intravenosa). Idosos: 1,3 horas (comprimido, injeção intravenosa).
t Excreção: renal (predominante em forma ativa; 11% como metabólitos).
t Taxa de depuração endógena – 0,27 to 0,63 mL/min/kg. Idosos: 0,124 mL/ min/kg (comprimido, injeção intravenosa). Crianças 5 a 15 anos – 0,4 to 0,66 mL/min/kg (comprimido, injeção intravenosa). Neonatos, idade gestacional de 26 a 29 semanas, 0,18 a 0,333 mL/min/kg (comprimido, injeção intravenosa).
t Excreção Renal (%) – 80% de fármaco inalterado, 11% metabólitos (comprimido, injeção intravenosa). Idosos: 22% do fármaco inalterado (comprimido, injeção intravenosa).
t Meia-vida: 30 horas (triplica em pacientes com DCE inferior a 20 mL/minuto e é muito prolongada em prematuros). Idosos: 46,2 horas (comprimido, injeção intravenosa). Crianças: 15,2 a 25 horas (comprimido, injeção intravenosa). Hemodiálise: 8,7 horas.
t É removido por diálise.
t Anafilaxia
t Síndrome de Stevens-Johnson, agranulocitose, necrólise epidérmica tóxica.
t Hipersensibilidade (febre, calafrios, exantema, prurido)
t Náusea, vômitos, dores abdominais, dispepsia, distúrbio de paladar, flatulência, diarreias em aproximadamente 1,5-8,5% dos pacientes.
t Elevação transitória das enzimas hepáticas (5-11%), necrólise hepática e outros, disfunção hepática.
t Hiperlipidemia, hiperglicemia, leucopenia, trombocitopenia, hipopotassemia.
t Prurido, exantema, incluindo exantema difuso acompanhado de eosinofilia (5%), alopecia, erupção maculo-papular, angioedema.
t Prolongamento do intervalo QT e torsade de pointes.
t Amenorreia.
t Hipopotassemia, hipocortisolismo secundário, efeitos hematológicos e trombocitopenia.
t Tonturas, convulsão, cefaleia (2%), sonolência, delirium/coma, distúrbios psiquiátricos, parestesia de mãos e pés.
Observação: Efeitos adversos do sistema nervoso têm sido relatado em aproximadamente 14-20% das mulheres recebendo dose única de fluconazol para tratamento de candidíase vulvovaginal.
t Acenocumarol, dicumarol, femprocumona, varfarina: aumento do risco de sangramento, por decréscimo do metabolismo do anticoagulante. Em pacientes recebendo terapia anticoagulante, o tempo de protrombina ou a razão internacional normalizada (RNI) deveria ser estreitamente monitorada com a introdução e suspensão do fluconazol, e também durante a terapia simultânea. Ajustes na dose do anticoagulante podem ser necessários a fim de manter o nível desejado de anticoagulação.
t Alfentanila e fentanila: efeitos opioides prolongados ou aumentados (depressão do sistema nervoso central e depressão respiratória). Monitorar cuidadosamente pacientes para sinais e sintomas de toxicidade por opioide.
t Amitriptilina e nortriptilina: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, e parada cardíaca).
t Antagonistas de canais de cálcio diidropiridínicos (anlodipino, felodipino, isradipino, nicardipino, nifedipino): decréscimo do metabolismo do antagonista de canal de cálcio, resultando em aumento das concentrações séricas e toxicidade (tontura, hipotensão, rubor, cefaleia, edema periférico). O uso concomitante não é recomendado (particularmente com isradipino). Se a combinação de agente diidropiridínico com fluconazol for utilizada, monitorar o paciente para sinais de toxicidade e considerar a redução de dose do antagonista de canais de cálcio ou a suspensão de um dos fármacos.
t Astemizol, bepridil, cisaprida, pimozida, tioridazina, ziprasidona: risco aumentado de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes).
t Carbamazepina: risco aumentado de toxicicidade da carbamazepina (ataxia, nistagmo, diplopia, cefaleia, vômito, apneia convulsões e coma).
t Cerivastatina, sinvastatina, rosuvastatina: risco aumentado de miopatia ou rabdomiólise (especialmente com as duas primeiras). Se a administração com fluconazol é necessária, monitorar o paciente para sinais e sintomas de miopatia ou rabdomiólise. Monitorar concentrações de creatinina cinase e descontinuar o uso se houver aumento acentuado deste parâmetro laboratorial, ou em caso de suspeita ou diagnóstico de miopatia ou rabdomiólise.
t Ciclosporina: risco aumentado de toxicidade da ciclosporina (disfunção renal, colestase, parestesia).
t Cimetidina: redução da absorção gastrintestinal, com decréscimo da efetividade do fluconazol.
t Citalopram: risco aumentado de síndrome serotoninérgica. Monitorar sinais e sintomas da síndrome e outros eventos adversos causados pelo citalopram.
t Claritromicina e fluoxetina: aumento do risco de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, e parada cardíaca).
t Colchicina: aumento das concentrações e da toxicidade da colchicina. Se o uso concomitante for necessário, reduzir a dose da colchicina.
t Derivados da ergotamina: risco aumentado de ergotismo (náusea, vômitos, isquemia vasoespástica) devido à inibição do metabolismo pelo citocromo P450 3A4. O uso concomitante com fluconazol é contraindicado.
t Fenitoína e fosfofenitoína: redução do seu metabolismo, com risco aumentado de toxicidade (ataxia, hiperreflexia, nistagmo, tremores). Ao introduzir o fluconazol, monitorar concentrações da fenitoína e sintomas de toxicidade.
t Gemifloxacino, levofloxacino: risco aumentado de cardiotoxicidade (prolongamento do intervalo QT, torsades de pointes, parada cardíaca) por efeitos cardíacos aditivos. A administração simultânea de dois fármacos que prolongam o intervalo QT não é recomendada. Se o uso simultâneo de fluconazol e levofloxacino for considerado necessário, proceder com cautela e monitorar pacientes quanto a dose, ECG e eletrólitos.
t Glimepirida: aumento das concentrações de glimepirida e risco de hipoglicemia, devido à inibição da biotransformação da glimepirida, mediada por isoenzima do citocromo P450 (2C9).
t Losartana: inibição do metabolismo da losartana em seu metabólito ativo. Monitorar pacientes para controle contínuo de sua hipertensão.
t Midazolam: aumento da concentração e toxicidade potencial destes fármacos (sedação excessiva e efeitos hipnóticos prolongados), devido à inibição pelo fluconazol do metabolismo mediado pelo sistema CYP450 3A4 (nefrotoxicidade, hiperglicemia, hiperpotassemia).
t Nevirapina: aumento das concentrações plasmáticas de nevirapina. A coadministração não é recomendada, sendo necessária extrema cautela na administração concomitante, devendo os pacientes ser rigorosamente monitorados em relação aos Efeitos adversos da nevirapina.
t Nitrofurantoína: risco aumentado de toxicidade hepática e pulmonar. Evitar o uso concomitante, mas se este for necessário, monitorar a toxicidade hepática e pulmonar.
t Rifabutina: aumento da concentração sérica e da toxicidade da rifabutina (uveíte, dor ocular, fotofobia, distúrbios visuais). Monitorar estreitamente o paciente para sintomas de toxicidade. Em caso de uveíte, descontinuar a rifabutina e utilizar agentes midriáticos.
t Rifampicina: diminuição das concentrações séricas do fluconazol e da atividade antifúngica.
t Rosuvastatina: exposição aumentada à rosuvastatina e risco aumentado de miopatia ou rabdomiólise.
t Sinvastatina: risco aumentado de miopatia e rabdomiólise. Inibição pelo fluconazol do metabolismo da sinvastatina mediado pelo citocromo P4503A4. Se a administração concomitante com fluconazol for necessária, monitorar o paciente para sinais e sintomas de miopatia ou rabdomiólise. Monitorar os níveis de creatinina cinase e descontinuar o uso se os níveis mostram aumento acentuado, ou em caso de suspeita diagnóstico de miopatia ou rabdomiólise.
t Tacrolimo: concentração aumentada do tacrolimo devido a inibição do seu metabolismo pelo fluconazol, mediado por CYP3A.
t Tipranavir: aumento da exposição ao tipranavir, com aumento do risco de efeitos adversos.
t Trimetrexato: toxicidade aumentada do trimetrexato (supressão da medula óssea, disfunção hepática e renal e ulceração gastrintestinal), por redução do seu metabolismo. Se for clinicamente possível evitar a administração concomitante. Caso esta seja necessária, monitorar os níveis séricos e a toxicidade do trimetrexato (supressão da medula óssea, disfunção hepática e renal e ulceração gastrintestinal).
t Valdecoxibe: aumento da concentração plasmática e dos Efeitos adversos do valdecoxibe (cefaleia, vômitos, náusea e dor abdominal), devido à inibição do metabolimo do valdecoxibe pelo citocromo P4502C9. Monitorar o paciente em relação a efeitos adversos.
t Agitar bem o frasco com a suspensão oral antes de usar. Medir a dose com a colher, copo ou seringa de medida.
t Em caso de perder a hora de tomada do medicamento, tome assim que puder. Se já estiver próximo da hora da dose seguinte, espere até o momento correto da próxima dose. Não use medicamento a mais para compensar a dose que não foi tomada.
t Descartar a sobra da suspensão oral até 14 dias após o término do tratamento.
t Antes de usar o medicamento informar se estiver grávida ou amamentando.
t Usar o medicamento pelo tempo prescrito. Não interromper o tratamento.
t Armazenar a cápsula e o pó para suspensão sob temperatura abaixo de 30ºC.
t Armazenar a suspensão reconstituída entre 30 ºC e 5 ºC. Proteger do congelamento.
t Armazenar soluções injetáveis em refrigerador, não congelar.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t As injeções de fluconazol para infusão intravenosa deverão ser inspecionadas visualmente quanto a possível descoloração e presença de material particulado, antes da administração, sempre que a solução e o frasco permitirem.
t Em crianças a administração por infusão intravenosa, administrar por tempo de 10 -30 minutos, não exceder taxa de infusão de 5-10 mL/minuto.
Atenção: este fármaco apresenta um número muito elevado de Efeitos adversos , sendo necessária uma pesquisa específica sobre este aspecto antes de introduzir ou descontinuar o fluconazol ou outros medicamentos no esquema do paciente.
Elaine Silva Miranda
Rachel Magarinos-Torres
Na Rename 2010: item 8.2
t Solução injetável com 0,1 mg/mL.
t Reversão da sedação por benzodiazepínico.
t Tratamento de sobredose de benzodiazepínico.
t Hipersensibilidade a flumazenil.
t Dependência a benzodiazepínicos ou em pacientes que receberam benzodiazepínicos para tratar condições com risco de morte, tais como estado epiléptico e controle da pressão intracraniana.
t Sinais de intoxicação por antidepressivo tricíclico.
t Pacientes que receberam bloqueador neuromuscular, até que o efeito do bloqueador tenha sido totalmente eliminado.
t Usar com cuidado nos casos de:
– dependência a álcool e/ou psicotrópicos.
– distúrbios do pânico.
– danos cerebrais, cardiopatia
– idosos e crianças.
– insuficiência renal
– lactação.
t A reversão de efeito de benzodiazepínicos pode resultar em convulsões.
t A depressão respiratória induzida por benzodiazepínicos também deve ser tratada com suporte ventilatório.
t A utilização de flumazenil não deve substituir um período de monitoria adequado após o procedimento cirúrgico.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t Dose de 0,01 mg/kg, por via intravenosa (diluído em solução de glicose a 5%, Ringer + lactato ou cloreto de sódio a 9%, lentamente, em veia de grande calibre), em 15 a 30 segundos.
t Caso a resposta não seja obtida dentro dos próximos 45 a 60 segundos, repetir a dose de 0.01 mg/kg a intervalos de 1 minuto.
t Dose máxima total 0,05 mg/kg ou 1 mg, o que for menor.
Nota: A eficácia e segurança na reversão de sedação em menores que 1 ano não foi determinada.
t Dose inicial: 0,2 mg, por via intravenosa (diluído em solução de glicose a 5%, Ringer + lactato ou cloreto de sódio a 9%, lentamente, em veia de grande calibre), em 15 a 30 segundos.
t Caso a resposta não seja obtida dentro dos próximos 30 segundos, administrar nova dose de 0,3 mg em 30 segundos.
t Doses adicionais de 0,5 mg podem ser administradas após intervalo de 60 segundos, até dose máxima acumulada de 2 a 3 mg quando em tratamento intensivo.
t Início de resposta: 1 a 2 minutos. Tempo médio para abertura dos olhos e autoidentificação em torno de 4 minutos.
t Duração de efeito: 30 a 60 minutos.
t Meia-vida de eliminação: 40 a 80 minutos.
t Metabolismo: hepático.
t Excreção: renal.
t Arritmias cardíacas, convulsões generalizadas (especialmente em pacientes epilépticos, usuários crônicos de benzodiazepínicos e com sinais de intoxicação por antidepressivos cíclicos).
t Náusea (11% a 16%), vômito (11%)
t Cefaleia, tontura, sedação, visão borrada, crise de pânico em pacientes com transtorno do pânico.
t Hipotensão, vasodilatação.
t Tromboflebite, exantema, dor no lugar da injeção.
t Após rápida reversão: ansiedade, agitação e medo.
t Oxicodona, tapentadol e zolpidem: o uso concomitante com flumazenil pode resultar em aumento da depressão no sistema nervoso central, podendo aumentar depressão respiratória. Recomenda-se redução de dose inicial da oxicodona a um terço ou à metade da dose; nos outros casos, pode ser necessário reduzir a dose de um ou ambos os medicamentos.
t Orientar para não operar máquinas ou dirigir veículos durante as primeiras 24 horas porque o efeito benzodiazepínico pode reaparecer.
t Armazenar à temperatura entre 15 e 30 ºC.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Compatível com solução de glicose 5%, solução de Ringer + lactato e cloreto de sódio a 0,9%, permanecendo estável por 24 horas.
Atenção: como a meia-vida do flumazenil é menor que a dos benzodiazepínicos, pode ocorrer retorno da sedação após 1 a 2 horas, com possibilidade de depressão respiratória. Doses adicionais poderão ser necessárias. Flumazenil não deve ser utilizado até que o bloqueio neuromuscular associado à anestesia geral seja revertido. A administração deve ser feita por profissional experiente.
Eudiana Vale Francelino
Na Rename 2010: item 21.7
t Colírio 1%.
t Detecção de lesões e corpos estranhos na córnea.
t Auxílio no diagnóstico em angiografia oftálmica. t Auxílio no ajuste de lentes de contato duras.
t Teste de potência lacrimal.
t Tonometria (combinado com lidocaína).
t Hipersensibilidade à fluoresceína ou a outro componente da formulação.
t Uso concomitante com lentes de contato moles.
t Usar com cuidado nos casos de:
– associação com colírio anestésico local – lidocaína (evitar em neonatos devido à imaturidade do sistema enzimático metabolizador desses pacientes).
– direção de veículo ou operação de maquinário (turvação transitória da visão; aguardar restabelecimento para retomar tais atividades).
t Categoria de risco na gravidez (FDA): C.
t Instilar 1 a 2 gotas da solução oftálmica, esperando alguns segundos para que atinja o epitélio corneano. Lavar com água estéril para retirar o excesso, dando sequência ao procedimento.
t Instilar 1 a 2 gotas da solução oftálmica.
– as secreções nasais são examinadas sob luz azul após seis minutos da instilação;
– os canais lacrimais são considerados desobstruídos se traços do corante estiverem presentes nas secreções.
t Raros e locais; ardência passageira é o efeito mais frequente.
t Visão borrada transitória.
t Irritação e erupção cutânea, descoloração amarelada da pele ou olhos.
t Urina de cor amarela brilhante.
t Orientar para remover lentes de contato do tipo macias antes do exame e aguardar pelo menos 1 hora após o exame para recolocá-las. Lavar abundantemente os olhos com cloreto de sódio 0,9% antes da recolocação.
t Alertar para a possível permanência de uma coloração amarela de conjuntiva e pele das pálpebras por 6 a 12 horas.
t Advertir o paciente quanto à cautela para operar máquinas ou dirigir até o restabelecimento da visão.
t Conservar em recipientes fechados, ao abrigo da luz, à temperatura entre 15 e 30 ºC.
Maria Isabel Fischer
Na Rename 2010: item 11
t Solução bucal 2 mg/mL (FN)
t Profilaxia da cárie dental.
t Hipersensibilidade a fluoreto.
t Crianças com menos de 6 meses de idade.
t Áreas onde a água é fluoretada ou onde a quantidade de flúor é naturalmente elevada, acima de 0,6-0,7 ppm.
t Usar com cuidado nos casos de:
– crianças (supervisionar durante a realização dos bochechos).
– uso excessivo e/ou prolongado (pode causar fluorose dos dentes e alterações ósseas em crianças).
– insuficiência renal (ver Apêndice D).
t Não exceder a dose recomendada
t Não deglutir a solução.
t Limpar as superfícies dentárias antes das aplicações para promover maior contato do composto fluoretado com os dentes.
t Bochechos diários, durante 1 minuto, com 5 mL a 10 mL da solução diluída em 1:4 de água (0,05%). Ou então,
t Bochechos semanais, durante 1 minuto, com 10 mL da solução sem diluir.
Nota: para crianças, a dose diária pode variar considerando o conteúdo de fluoreto na água, a dieta utilizada e a idade.
t Não deve haver absorção sistêmica na aplicação tópica por bochechos
t Distúrbios na formação do tecido dentário (fluorose dentária) e alterações ósseas se houver consumo crônico excessivo
t Coloração dos dentes: manchas brancas (doses recomendadas), coloração marrom-amarelada (doses excessivas)
t Exantema.
t Náusea, vômito, diarreia, dor abdominal.
t Leite ou produtos lácteos: pode haver retardo na absorção e diminuição do pico de concentração do fluoreto.
t Sais de cálcio, magnésio e alumínio: pode haver redução na absorção do fluoreto.
t Adotar o período da noite, após escovar os dentes e antes de dormir, para fazer os bochechos. Não deglutir a solução enxaguatória.
t Não comer, beber ou escovar os dentes durante 15 a 30 minutos após a utilização da solução.
t Não utilizar medicamentos contendo sais de cálcio ou de alumínio, ou alimentos ricos em cálcio (leite, queijo, iogurte) no mínimo 2 horas antes e 2 horas após a administração do flúor.
t Armazenar o fluoreto de sódio em recipiente hermético, sob temperatura entre 15 e 30ºC.
t Observar o prazo de validade indicado pelo produtor.
Tatiana Aragão Figueiredo
Elaine Silva Miranda
Na Rename 2010: item 6.1.2
t Solução injetável 50 mg/mL.
t Creme 50 mg/g.
t Carcinomas de mama, pâncreas, estômago, cólon, colorretal, cabeça e pescoço (uso parenteral).
t Carcinomas de células basais superficiais e ceratoses solares (uso tópico).
t Hipersensibilidade ao fármaco e componentes da formulação.
t Mielossupressão grave.
t Deficiência da enzima di-hidropiridina desidrogenase (risco de toxicidade sistêmica com tratamento tópico).
t Desnutrição.
t Gravidez existente ou potencial.
t Lactação (ver Apêndice B).
t Infecções graves.
t Usar com cuidado nos casos de:
– vômito e diarreia intratáveis, queda na contagem de leucócitos e plaquetas, estomatite, hemorragia ou isquemia miocárdica (suspender terapia).
– radioterapia ou quimioterapia prévia.
– insuficiência renal.
– insuficiência hepática (ver Apêndice C).
– histórico de doença cardíaca.
– idosos (são mais Susceptíveis aos efeitos da mielossupressão).
– crianças (eficácia e segurança não estabelecidas).
– metástases envolvendo a medula óssea.
t Categoria de risco na gravidez (ADEC) : D (solução injetável) e (FDA): X (creme para uso tópico) (ver Apêndice A).
t 500 a 600 mg/m2, por injeção intravenosa em bolo, a cada 3 a 4 semanas; ou, 425 mg/m2, por injeção intravenosa em bolo, em dias 1 a 5, a cada 4 semanas. Dose máxima: 800 mg/dia, não excedendo 400 mg/dia em pacientes de alto risco.
t 2.000 mg/m2/dia, por infusão intravenosa contínua, durante 4 a 5 dias, a cada 3 a 4 semanas; ou, 2.300 a 2.600 mg/m2/dia, por infusão intravenosa contínua, no dia 1 de cada semana; ou, 225 mg/m2/dia, por infusão intravenosa contínua, durante 5 a 8 semanas, com radioterapia.
t Aplicar fina camada sobre a lesão, uma ou duas vezes ao dia, até 4 a 6 semanas.
t Duração de efeito: 3 semanas.
t Metabolismo: hepático.
t Excreção: renal e pulmonar.
t Meia-vida: 6 a 22 minutos.
t No uso tópico a absorção é mínima.
t Mielossupressão, anemia, leucopenia, trombocitopenia.
t Hepatite, atrofia do fígado, cirrose, fibrose, necrólise e insuficiência hepáticas.
t Exantema maculopapular, dermatite, alopecia, descoloração da pele, exantema.
t Efeito irritativo local.
t Dor abdominal, estomatite, diarreia, anorexia, náuseas, vômitos, esofagite, úlcera.
t Toxicidade ocular.
t Mais raramente pode ocorrer: irritação ocular, lacrimejamento excessivo acidente vascular cerebral, doença desmielinizante do sistema nervoso central, encefalopatia, cognição prejudicada, neurotoxicidade, neuropatia periférica, palmar-plantar (síndrome pé-mão), angina, cardiomiopatia, arteriosclerose coronária, enfarte do miocárdio, tromboflebite.
t Ácido folínico, metronidazol: o uso concomitante com fluoruracila pode levar à toxicidade por esta última. O uso concomitante de fluoruracila com metronidazol deve ser evitado, se possível, mas se o mesmo for necessário, deve-se monitorar o paciente para sinais de toxicidade por fluoruracila.
t Fenitoína: o uso concomitante com fluoruracila pode resultar em toxicidade aguda por fenitoína (ataxia, hiperreflexia, nistagmo, tremores).
t Hidroclorotiazida: o uso concomitante pode levar a mioelossupressão.
t Levamisol: o uso concomitante pode resultar em hepatotoxicidade.
t Tamoxifeno: o uso concomitante pode aumentar o risco de tromboembolismo. Em pacientes utilizando tamoxifeno na quimioterapia de câncer de mama, o risco da adição de fluoruracila deve ser avaliado em relação aos potenciais benefícios.
t Vacinas com vírus vivos e vacina contra rotavírus: o uso concomitante com agentes quimioterápicos pode resultar em aumento do risco de infecção pela vacina. A vacinação contra rotavírus é contraindicada em pacientes utilizando quimioterápicos. A vacinação com outras vacinas de vírus vivos pode ser realizada decorridos no mínimo três meses do fim da quimioterapia.
t Varfarina: aumento do efeito anticoagulante de varfarina.
t Aumentar ingestão diária de alimentos ricos em tiamina e a ingestão de líquidos durante o tratamento.
t Orientar para não ingerir bebidas alcoólicas enquanto utilizar este medicamento.
t Evitar gravidez.
t Orientar para lavar e secar a área lesionada 10 minutos antes da aplicação.
t Ensinar a proteger as mãos, quando utilizar o creme e a não cobrir com curativo oclusivo. Aplicar o creme preferivelmente com o auxilio de um aplicador não metálico ou utilizando luvas. Evitar aplicar na área de olhos e mucosas.
t Evitar exposição ao sol durante o tratamento.
t Hemograma deve ser pedido com frequência durante o tratamento sistêmico, para contagem de plaquetas.
t Armazenar a solução e o creme à temperatura ambiente, entre 15 e 30 ºC e proteger da luz.
t Leve coloração não significa inativação da solução.
t Se exposta ao frio, a solução precipita, leve aquecimento dissolve novamente o precipitado.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Diluições em solução fisiológica ou glicosada a 5% são estáveis à temperatura ambiente por 72 horas.
t Incompatibilidades: Preparações de fluoruracila são alcalinas, e problemas de compatibilidade podem ocorrer com preparações e fármacos ácidos ou aqueles que são instáveis na presença de álcalis. A fluoruracila é descrita como incompatível com citarabina, diazepam, doxorrubicina (e presumivelmente outras antraciclinas que são instáveis em pH alcalino) e folinato cálcico.
Atenção: recomenda-se que fluoruracila seja administrada somente por médico qualificado ou sob sua supervisão. Devido à possibilidade de reações tóxicas graves, recomenda-se que os pacientes sejam hospitalizados, pelo menos, durante o curso inicial da terapia.
Maurício Fábio Gomes
Na Rename 2010: itens 5.6.2.3, 6.3, 8.2
t Comprimido 15 mg.
t Pó para solução injetável 50 mg.
t Solução injetável 3 mg/mL.
t Resgate em intoxicação causada por antagonistas do ácido fólico.
t Anemia megaloblástica por deficiência de folato (quando terapia oral não for possível).
t Câncer colorretal avançado (tratamento paliativo).
t Administração por injeção intratecal.
t Hipersensibilidade ao folinato de cálcio.
t Anemia perniciosa ou outras anemias megaloblásticas devidas à deficiência de vitamina B12.
t Câncer colorretal em pacientes pediátricos.
t Usar com cuidado nos casos de:
– anormalidades no sistema nervoso central (em combinação com fluoruracila).
– sintomas de toxicidade gastrintestinal de qualquer gravidade (não iniciar ou continuar o uso de folinato de cálcio associado à fluoruracila; aguardar resolução dos sintomas).
– combinação de folinato de cálcio com fluoruracila (maior risco de toxicidade gastrintestinal).
– idosos e pacientes enfraquecidos (em combinação com fluoruracila).
– crianças susceptíveis a convulsões (o folinato pode aumentar a frequência das convulsões).
– história de convulsões (em combinação com fluoruracila).
– insuficiência renal (ver Apêndice D) t Categoria de risco na gravidez (FDA): C.
t Metotrexato em altas doses: administrar 10 mg/m2, por via intramuscular ou intravenosa ou oral, a cada 6 horas, até que as concentrações plasmáticas de metotrexato fiquem abaixo de 0,01 micromolar. Se após 24 horas os níveis de creatinina sérica encontrarem-se 50% acima do normal e as concentrações plasmáticas de metotrexato mantiveram-se acima de 5 micromolar, ou se ao final de 48 horas as concentrações plasmáticas de metotrexato mantiveram-se acima de 0,9 micromolar, a dose de folinato de cálcio deve ser aumentada para 100 mg/m2 e administrada por via intravenosa, a cada 3 horas, até que as concentrações plasmáticas de metotrexato fiquem abaixo de 0,01 micromolar.
t Antagonistas de ácido fólico menos potentes (trimetoprima, pirimetamina); administrar 5 a 15 mg/dia, por via oral, até que a hematopoiese normal seja restaurada.
t Dose de até 1 mg/dia, por via intramuscular ou intravenosa.
t Metotrexato em altas doses: administrar 10 mg/m2, pela via intramuscular ou intravenosa ou oral, a cada 6 horas, até que as concentrações plasmáticas de metotrexato fiquem abaixo de 0,01 micromolar. Se após 24 horas os níveis de creatinina sérica encontrarem-se 50% acima do normal e as concentrações plasmáticas de metotrexato mantiveram-se acima de 5 micromolar, ou se ao final de 48 horas as concentrações plasmáticas de metotrexato mantiveram-se acima de 0,9 micromolar, a dose de folinato de cálcio deve ser aumentada para 100 mg/m2 e administrada por via intravenosa, a cada 3 horas, até que as concentrações plasmáticas de metotrexato fiquem abaixo de 0,01 micromolar.
t Antagonistas de ácido fólico menos potentes (trimetoprima, pirimetamina), administrar 5 a 15 mg/dia, por via oral, até que a hematopoiese normal seja restaurada.
t Dose de até 1 mg/dia, administradas por via intramuscular ou intravenosa, tem-se mostrado efetiva.
t Administrar 200 mg/m2, por via intravenosa, durante 3 minutos, seguidos de 370 mg/m2/dia de fluoruracila, por via intravenosa, durante 5 dias. Repetir a cada 4 semanas por 2 ciclos e seguir a cada 4 a 5 semanas com a dose ajustada de acordo com a tolerância do paciente.
t A absorção oral de folinato de cálcio satura-se em doses únicas maiores que 25 mg.
t Pico de concentração: 28 minutos (intramuscular), 10 minutos (intravenoso), 1,2 horas (via oral)
t Meia-vida de eliminação: 6,2 horas (intravenoso e intramuscular), 5,7 horas (via oral)
t Excreção: renal (80 a 90%) e fecal (5 a 8%).
t Diarreia, náusea, vômitos, estomatites.
t Hipopotassemia (65%).
t Fadiga.
t Alopecia.
t Leucopenia.
t Reações alérgicas.
t Convulsões e/ou sincope (raro), insônia, agitação, depressão (raro).
t Febre após administração parenteral.
t Capecitabina e fluoruracila: o uso concomitante com folinato de cálcio pode resultar no aumento das concentrações de fluoruracila (metabólito da capecitabina) com consequente aumento de toxicidade.
t Fenitoína, fenobarbital e primidona: a concentração plasmática e eficácia destes fármacos pode ser reduzida.
t Se esquecer alguma dose contatar o médico ou farmacêutico para instruções.
t Informar se for necessário utilizar medicamentos contendo sulfa ou anticonvulsivantes.
t Evitar gravidez.
t Manter os comprimidos à temperatura ambiente, entre 15 e 30 oC, ao abrigo de luz direta, calor e umidade.
t Manter solução injetável à temperatura ambiente, entre 15 e 30 oC, proteger de luz direta.
t Observar orientação específica do produtor quanto a reconstituição, diluição, compatibilidade e estabilidade da solução.
t Reconstituir pó com água bacteriostática para injeção para obter concentração de 10 mg/mL; a solução poderá ser utilizada dentro de 7 dias da preparação; concentrações maiores de 10 mg/mL, devem ser administradas imediatamente.
t Compatível com soluções injetáveis de glicose 5%, cloreto de sódio 0,9% e Ringer + lactato, com concentração final de folinato de 1 mg/mL; as mesmas devem ser utilizadas até 24 horas após o preparo.
t Incompatibilidades: droperidol, fluoruracila, foscarnete sódico, trimetrexato, carboplatina, anfotericina B, lansoprazol, cloridrato de epirrubicina, pantoprazol sódico, quinupristina/dalfopristina, bicarbonato de sódio.
Atenção: o folinato de cálcio é um derivado do ácido folínico, entretanto, nos estados unidos é utilizada a denominação genérica leucovirin calcium. A denominação comum brasileira (DCB), oficial no Brasil, é folinato de cálcio, assim, o uso do sinônimo “leucovirina cálcica” deve ser evitado.
Aline Lins Camargo
Na Rename 2010: item 2.2
t Comprimidos 30 mg.
t Dor leve a moderada, aguda ou crônica.
t Depressão respiratória aguda.
t Íleo paralítico.
t Hipersensibilidade à codeína.
t Usar com cuidado nos casos de:
– idosos e enfraquecidos, condições abdominais agudas, doença de Addison, febre, alcoolismo agudo, asma, doença pulmonar obstrutiva crônica, hipotensão, choque, miastenia grave, hipotireoidismo, hipertrofia prostática ou estreitamento uretral, dependência a opioides, pressão intracraniana aumentada, coma, doenças obstrutivas ou inflamatórias do intestino, doenças do trato biliar, convulsões, insuficiência adrenocortical grave.
– insuficiência hepática grave (ver Apêndice C).
– insuficiência renal grave (ver Apêndice D).
– interrupção de tratamento prolongado (a retirada do opioide deve ser gradual).
– lactação (ver Apêndice B).
t A dose pode necessitar de ajuste individual, pois a resposta clínica é amplamente variável.
t Categoria de risco na gravidez (FDA): C (Categoria D para uso prolongado ou em altas doses próximo do parto) (ver Apêndice A).
t De 6 a 12 anos: 0,5 a 1 mg/kg/dose, por via oral, a cada 4 a 6 horas. Até o máximo de 60 mg/dia.
t Dose de 15 a 60 mg, por via oral, a cada 4 horas, quando necessário. Até o máximo de 240 mg/dia.
t Início de efeito: 30 a 60 minutos.
t Pico do efeito: 30 a 120 minutos.
t Duração de efeito: 4 a 6 horas.
t Meia-vida de eliminação: 2,5 a 3,5 horas.
t Metabolismo: hepático – metabólito ativo: morfina.
t Excreção: principalmente renal.
t Frequentes (1->10%)
t Obstipação, náusea, vômito, anorexia e xerostomia.
t Sedação, sonolência, tontura, cefaleia, confusão, dependência física.
t Dificuldade de urinar.
t Fraqueza
t Visão borrada
t Exantema, urticária
t Dispneia
t Convulsões, alucinações, insônia, depressão, pesadelos.
t Taquicardia ou bradicardia, hipotensão.
t Depressão respiratória.
t Álcool: possível aumento dos efeitos sedativos, hipotensores e depressores do sistema nervoso central. Uso concomitante deve ser evitado.
t Benzodiazepínicos e barbitúricos: possível depressão respiratória aditiva. Monitorar paciente para depressão respiratória. Redução da dose de um ou ambos os fármacos pode ser necessária.
t Naltrexona: pode resultar em sintomas de retirada (cólicas abdominais, náuseas, vômitos, lacrimejamento, rinorreia, ansiedade, inquietação, elevação da temperatura ou piloereção) e decréscimo da efetividade da codeína, quando em uso prolongado do opioide. Uso concomitante é contraindicado. Pacientes devem estar sem usar opioides, no mínimo, por 7 a 10 dias antes de iniciar tratamento com naltrexona.
t Agonistas/antagonistas de opioides (por exemplo, naloxona, buprenorfina, nalbufina): pode resultar em sintomas de retirada dos opioides. Antagonistas de opioides devem ser administrados cautelosamente em pessoas com suspeita de dependência física de qualquer agonista de opioide. Se ocorrerem sinais e sintomas de síndrome de retirada, recomenda-se reinstituição da terapia opioide seguida de redução gradual da dose de opioide combinada com suporte sintomático.
t Relaxantes musculares de ação central: possível depressão respiratória aditiva. Monitorar paciente para depressão respiratória. Redução da dose de um ou ambos agentes pode ser necessária.
t Reforçar necessidade de aumentar ingestão de líquido, para evitar obstipação.
t Evitar uso concomitante de hipnóticos, ansiolíticos, medicamentos para alívio sintomático de gripe e resfriados, antialérgicos, pelo risco de tontura ou sonolência. Evitar uso de bebidas alcoólicas durante a utilização do medicamento.
t Evitar realizar atividades que exigem atenção e coordenação motora, como operar máquinas e dirigir.
t Reforçar para não ingerir bebidas alcoólicas enquanto estiver utilizando este medicamento.
t Alertar para não interromper abruptamente o uso deste medicamento, pode ser necessária a diminuição gradual da dose antes da interrupção do tratamento.
t Alertar para a possibilidade de desenvolvimento de dependência.
t Alertar para evitar o uso em excesso do medicamento, devido a sérios Efeitos adversos e potencialidade para levar ao óbito.
t Armazenar à temperatura ambiente, entre 15 e 30 ºC, em embalagens protegidas da luz e bem fechadas.
José Gilberto Pereira
Na Rename 2010: itens 3.2, 6.3
t Solução injetável 4 mg/mL
t Adjuvante do tratamento de meningite tuberculosa.
t Adjuvante do tratamento emergencial de reações anafiláticas.
t Dermatites e dermatoses complicadas e/ou graves.
t Doenças inflamatórias do sistema musculoesquelético.
t Doenças respiratórias graves.
t Edema cerebral associado a tumor cerebral primário ou metastático, craniotomia e traumatismo craniano.
t Exacerbação de doenças inflamatórias intestinais.
t Hipercalcemia associada a câncer.
t Insuficiência andrenocortical primária ou secundária.
t Náusea e vômito induzidos por quimioterapia antineoplásica.
t Púrpura trombocitopênica idiopática.
t Síndrome nefrótica idiopática ou secundária a lúpus eritematoso sistêmico.
t Supressão de curto prazo de inflamação em doenças alérgicas.
t Tratamento paliativo de leucemias, linfomas e outras malignidades.
t Triquinose com envolvimento neurológico e/ou miocárdico.
t Adjuvante do tratamento de meningite bacteriana por H. influenzae ou S. pneumoniae (suspeita ou confirmada).
t Hipersensibilidade a dexametasona ou a outros componentes da formulação.
t Infecções fúngicas, bacterianas ou virais sistêmicas não tratadas com antimicrobiano específico.
t Administração de vacinas com vírus vivos.
t Usar com cuidado nos casos de:
– úlcera péptica e doenças inflamatórias intestinais, diabete melito; hipertensão arterial, insuficiência cardíaca congestiva, recente enfarte agudo do miocárdio; doenças tromboembólicas; malária cerebral, miastenia grave; osteoporose; glaucoma avançado; instabilidade emocional, tendências psicóticas, estresse, epilepsia; psoríase; hipotireoidismo.
– insuficiência hepática.
– insuficiência renal.
– idosos.
– tratamento prolongado em crianças.
– psoríase (pode precipitar psoríase pustular grave na retirada).
– uso concomitante com imunossupressores.
– interrupção após terapia crônica com doses diárias (risco de supressão suprarrenal; a retirada deve ser gradual).
t Monitorar peso, pressão arterial, equilíbrio de fluidos, eletrólitos e glicose sanguínea durante tratamento prolongado.
t Aumenta a susceptibilidade e a gravidade de infecções bacterianas, varicela e sarampo.
t Pode ativar ou exacerbar tuberculose, amebíase e estrongiloidíase.
t Categoria de risco na gravidez (FDA): C (ver Apêndice A).
t 0,2 a 0,4 mg/kg, por bolo ou infusão intravenosa lenta, de acordo com a gravidade da reação.
t acima de 2 meses de idade: 0,15 mg/kg, por via intravenosa, a cada 6 horas, do 2º ao 4º dia de tratamento com antibiótico; coincidir a primeira dose de fosfato dissódico de dexametasona com uma dose do antibiótico.
t Dose inicial: 1 a 2 mg/kg, por via intramuscular ou intravenosa, seguidos de 1 a 1,5 mg/kg/dia, até o máximo de 16 mg/dia, a cada 4 a 6 horas.
t 10 a 20 mg/m2, por via intravenosa antes da quimioterapia; manter com 5 mg/m2 a cada 6 horas, se os sintomas persistirem.
t 0,5 a 24 mg, por bolo ou infusão intravenosa lenta, de acordo com a gravidade da reação.
t 0,15 mg/kg, por via intravenosa, a cada 6 horas, do 2º ao 4º dia de tratamento com antibiótico; coincidir a primeira dose de fosfato dissódico de dexametasona com uma dose do antibiótico.
t Dose de inicial: 10 mg, por via intravenosa, seguidos de 4 mg, por via intramuscular, a cada 6 horas, durante 2 a 4 dias; após, reduzir dose gradualmente e interromper ao observar melhora dos sintomas; no caso de tumor cerebral não ressecável, manter o tratamento com 2 mg, por via intramuscular ou intravenosa, a cada 8 ou 12 horas.
t 20 mg, por via intravenosa, antes da quimioterapia. Dose de manutenção: 8 mg a cada 12 horas, por via oral ou intravenosa, por até 3 dias.
t Ver esquemas terapêuticos na seção “Medicamentos utilizados no manejo das neoplasias”.
t Para uso intra-articular ou intralesional, a dose é baseada no local da aplicação: grandes articulações de 2 a 4 mg; pequenas articulações de 0,8 a 1 mg; bursas de 2 a 3 mg; bainha dos tendões de 0,4 a 1 mg; tecidos moles de 2 a 6 mg; e gânglios de 1 a 2 mg. O intervalo de doses pode variar de 3 a 5 dias até 2 a 3 semanas.
t 0,5 a 9 mg/dia, por via intramuscular ou intravenosa, de acordo com a necessidade, gravidade e resposta clínica.
t Início da resposta nas reações alérgicas agudas: 8 e 24 horas após administração intramuscular, permanecendo por 4 dias.
t Até 65% da dose é excretada na urina em 24 horas.
t Meia-vida de eliminação varia com a idade: 2,3 a 9,5 horas (até 2 anos); 2,8 a 7,5 horas (de 8 a 16 anos); e 3 a 6 horas no adulto.
t Retenção de sódio, edema e hipertensão; arritmias cardíacas e cardiomiopatia podem ocorrer em crianças.
t Acne, hematomas, dermatite, equimose, eritema facial, atrofia, hirsutismo, dificuldade de cicatrização de feridas, sudorese, estrias, telangiectasia, rosácea, dermatite perioral, prurido vulvar; e superinfecção mucocutânea.
t Hiperglicemia grave em pacientes com diabetes melito (1% a 46%), acompanhada de cetoacidose e coma hiperosmolar; ocasionalmente ocorre hipertireoidismo, dislipidemias e porfiria; supressão da suprarrenal pode ocorrer tanto por administração sistêmica quanto tópica; é comum aumento do apetite e ganho de peso.
t Redução da taxa de crescimento das crianças com uso prolongado e em altas doses.
t Náusea; candidíase orofaríngea (33%), úlcera péptica (2%), perfuração e hemorragias gastrintestinais (<1%); raramente pode ocorrer pancreatite.
t Reação leucemoide (leucócitos > 20.000/mm3) tem sido relatada.
t Reações de hipersensibilidade e imunossupressão após uso sistêmico de altas doses (<1%).
t Osteoporose e osteopenia (comuns); osteonecrose asséptica (rara).
t Precipitação de esquizofrenia (>5%); menos frequentemente pode ocorrer euforia, depressão, insônia, mania e alucinações.
t Catarata subcapsular posterior (2,5% a 60%), aumento da pressão intraocular (30%) e dano do nervo óptico; glaucoma de ângulo aberto (após um ano de tratamento sistêmico contínuo).
t Superinfecção generalizada por bactérias, vírus, fungos e parasitas.
t Ácido acetilsalicílico: risco aumentado de toxicidade gastrintestinal e de concentrações séricas subterapêuticas do ácido acetilsalicílico.
t Aminoglutetimida, fenitoína, fenobarbital, fosfenitoína, primidona, rifampicina e rifapentina: podem diminuir a efetividade de dexametasona.
t Antiácidos, antifúngicos azólicos, anti-inflamatórios não-esteroides, bloqueadores dos canais de cálcio, ciclosporina, estrógenos e ritonavir: podem aumentar efetividade e toxicidade de dexametasona.
t Aprepitanto, fosaprepitanto, itraconazol e ritonavir: aumento das concentrações séricas do corticosteroide e do risco de seus efeitos adversos.
t Bloqueadores neuromusculares (alcurônio, atracúrio, brometo de hexaflurônio, galamina, pancurônio, vecurônio): risco aumentado de miopatias; ajustar dose dos bloqueadores.
t Bupropiona: pode haver diminuição do limiar convulsivo; cautela em caso de uso concomitante com corticoides sistêmicos.
t Caspofungina e saquinavir: podem ter sua efetividade diminuída pela dexametasona.
t Fluoroquinolonas (exemplo, ciprofloxacino): o uso concomitante aumenta o risco de ruptura de tendões. Descontinuar fluoroquinolonas na presença de sinais e sintomas.
t Galamina e quetiapina: têm sua efetividade diminuída pela dexametasona.
t Talidomida: aumento do risco de desenvolvimento de necrólise epidérmica tóxica. O uso da combinação com talidomida pode ser útil no tratamento de mieloma refratário, contudo, até que um esquema posológico seguro seja determinado, o uso de dexametasona com talidomida para o tratamento de mielomas recentemente diagnosticados deve ser evitado, a menos que o paciente seja intensivamente monitorado.
t Vacina contra rotavírus: aumento do risco de infecção pela vacina de vírus vivos. A administração da vacina a pacientes imunocomprometidos por corticoides sistêmicos é contraindicada, mas deve prevalecer a avaliação clínica do médico.
t Varfarina: pode aumentar risco de sangramento. Monitorar tempo de protrombina.
t Orientar para relato de história prévia de alergias, hipertensão, diabetes melito, osteoporose, distúrbios gastrintestinais, micoses e outras infecções.
t Orientar para não tomar qualquer tipo de vacina ou vacina sem consulta prévia.
t Alertar para evitar contato com pessoas acometidas de infecções, particularmente as mais comuns na infância.
t Armazenar em recipientes bem fechados e à temperatura ambiente (15 a 30ºC). Manter ao abrigo de luz. Não congelar.
t 1 mg de dexametasona equivale a cerca de 1,3 mg de fosfato dissódico de dexametasona.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t Para infusão intravenosa, diluir em glicose 5% ou cloreto de sódio 0,9%; velocidade de administração intravenosa em crianças: direta (bolo), solução 50 mg/mL em 3 a 5 minutos; injeção intermitente, solução 1 mg/mL, em 20 a 30 minutos.
t A solução injetável, após preparada, deve ser mantida sob refrigeração e utilizada dentro de 24 horas. É necessário cuidado especial para não precipitar a dexametasona durante a preparação.
t Incompatibilidades: cloridrato de difenidramina, doxapram, fenoldopam, nitrato de gálio, idarrubicina e cloridrato de metoclopramida.
Rachel Magarinos-Torres
Na Rename 2010: itens 9, 10.3
t Solução injetável (0,03 g + 0,1567 g)/mL (2 mEq de fosfato/mL)
t Hipofosfatemia grave.
t Nutrição parenteral
t Hiperfosfatemia.
t Hiperpotassemia.
t Insuficiência renal grave (ver Apêndice D).
t Urolitíase.
t Usar com cuidado nos casos de:
– insuficiência renal
– hipoparatireoidismo, rabdomiólise, osteomalácia, raquitismo, pancreatite aguda, problemas da condução cardíaca, hipertensão arterial, desidratação aguda, intolerância a potássio ou fosfato.
t Monitorar níveis plasmáticos de eletrólitos e funções renal e cardíaca durante a infusão intravenosa.
t Categoria de risco na gravidez (FDA): C.
t Dose de 0,15 a 0,33 mmoles/kg/dose, por infusão intravenosa (taxa de 0,2 mmoles/kg/hora), por aproximadamente 6 horas. Repetir as doses até que as concentrações séricas de fósforo mantenham-se acima de 2 mg/dL.
t Dose de 0,5 a 2 mmoles/kg/dia
t Dose de 9 milimoles (aproximadamente 300 mg de fósforo), diluídos em NaCl 0,45%, como infusão intravenosa contínua por 12 horas; repetir em intervalos de 12 horas até a concentração sérica de fósforo alcançar 1 mg/dL.
t Dose equivalente a 12 a 15 milimoles (372 a 465 mg de fósforo) por litro de solução de nutrição parenteral.
Nota: A solução deve ser diluída antes de administrar. A infusão intravenosa deve durar no mínimo 4 horas.
t Bradicardia.
t Hiperpotassemia, hiperfosfatemia.
t Fraqueza.
t Dispneia.
t Edema periférico.
t Antiácidos: o uso concomitante pode reduzir a absorção de fosfatos.
t Conservar à temperatura ambiente, entre 15 e 30 ºC.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t As soluções devem ser límpidas e livres de partículas.
t A adição de fosfato em soluções contendo cálcio ou magnésio pode levar à formação de precipitado.
t É incompatível com soluções de Ringer e Ringer + lactato; ocorre precipitação.
t Cada grama de fosfato de potássio monobásico corresponde a cerca de 7,3 mmol de potássio e de fosfato.
t Cada grama de fosfato de potássio dibásico corresponde a cerca de 11,5 mmol de potássio e 5,7 mmol de fosfato.
Helena Lutéscia Luna Coelho
Na Rename 2010: itens 3.2, 3.3, 4, 5.4, 6.3, 7.1,17.1
t Solução oral 4,02 mg/mL (equivalente a 3 mg de prednisolona/mL)
t Adjuvante no tratamento de pneumonia pneumocística moderada ou grave.
t Artrite reumatoide
t Asma
t Doença de Hodgkin
t Doença pulmonar obstrutiva crônica (exacerbações agudas)
t Doenças alérgicas
t Doenças de pele (pênfigo, dermatite herpetiforme bolhosa, síndrome de Stevens-Johnson, dermatite esfoliativa, micose fungoide, psoríase grave, dermatomiosite sistêmica e dermatite seborreica grave).
t Doenças endócrinas
t Doenças gastrintestinais (enterite e colite ulcerativa)
t Doenças hematopoieticas
t Doenças inflamatórias
t Doenças inflamatórias músculo-esqueléticas
t Doenças neoplásicas
t Doenças oculares (conjuntivite alérgica, úlceras alérgicas marginais da córnea, inflamação do segmento anterior, coriorretinite, uveíte posterior difusa e coroidite, herpes zoster oftálmico, irite e iridociclite, ceratite e neurite óptica).
t Doenças respiratórias (pneumonia pneumocística moderada ou grave, sarcoidose sintomática, tuberculose pulmonar fulminante ou disseminada, síndrome de Loëffler, beriliose e pneumonite aspirativa).
t Esclerose múltipla (exacerbações)
t Linfoma não-Hodgkin t Rejeição de transplantes t Síndrome nefrótica
t Triquinose (com envolvimento neurológico ou miocárdico)
t Tuberculose meníngea
t Hipersensibilidade à prednisolona ou a algum componente da formulação.
t Infecções sistêmicas por fungos, bactérias ou vírus não tratadas com antimicrobiano específico.
t Vacinas com vírus vivos, pois a resposta imune pode ser diminuída pela prednisolona.
t Usar com cuidado nos casos de:
– úlcera péptica, diabete melito (incluindo história familiar), hipertensão arterial, psicose, insuficiência cardíaca congestiva, hipotireoidismo, glaucoma (incluindo história familiar), diverticulite, miastenia grave, herpes simples ocular, osteosporose, tendência psicótica.
– insuficiência hepática.
– insuficiência renal.
– doença inflamatória intestinal.
– crianças e adolescentes (pode ocorrer retardo no crescimento).
– idosos (pode desenvolver osteosporose, principalmente em mulheres na pós-menopausa, ou hipertensão).
– lactação.
t Aumenta a susceptibilidade e a gravidade de infecções como varicela e sarampo.
t Pode ativar ou exacerbar tuberculose, amebíase ou estrongiloidíase.
t Fator de risco na gravidez (FDA): C.
t Iniciar o uso da prednisona o mais cedo possível e dentro de 72 horas do tratamento bacteriano.
t abaixo de 12 anos:
– 1 mg/kg, por via oral, a cada 12 horas, nos dias 1 a 5;
– 0,5 mg/kg, por via oral, a cada 12 horas, nos dias 6 a 10;
– 0,5 mg/kg, por via oral, a cada 24 horas, nos dias 11 a 21.
Asma (exacerbação de moderada a grave)
t 1 mg/kg/dia, por via oral, divididos em 2 doses; até o máximo de 60 mg/dia, durante 3 a 10 dias
t 0,25 a 2 mg/kg/dia, por via oral, em dias alternados
t 0,14 a 2 mg/kg (4 a 60 mg/m2/dia), por via oral, divididos em 3 ou 4 doses.
t 60 mg/m2/dia, por via oral, divididos em 3 doses, durante 4 semanas; Manter com 40 mg/m2, em dias alternados, por mais 4 semanas.
t 40 a 80 mg/dia, por via oral, divididos em 2 doses; até o máximo de 60 mg/ dia, durante 3 a 10 dias
t 7,5 a 60 mg/dia, por via oral, em dias alternados,
t 5 a 60 mg/dia, por via oral, divididos em 3 ou 4 doses.
t 200 mg/dia, por via oral, durante 1 semana; seguidos por 80 mg, em dias alternados, durante 4 semanas
Nota:
t As doses devem ser individualizadas com base na gravidade da doença e na resposta ao tratamento.
t Pacientes com hipertireoidismo requerem doses maiores.
t Absorção: rápida.
t Biodisponibilidade: completa. t Pico de concentração: 1 hora.
t Metabolismo: hepático.
t Eliminação: renal.
t Meia-vida de eliminação: 2 – 4 horas
t Catarata, glaucoma.
t Síndrome de Cushing (obesidade do tronco, face de lua cheia, comprometimento na cicatrização de feridas, estrias, edema, corcova de búfalo), hiperglicemia, insuficiência adrenocortical.
t Tuberculose pulmonar.
t Desequilíbrio de fluidos e eletrólitos.
t Infecções por bactérias, parasitas, fungos e vírus.
t Euforia, depressão, alucinações, insônia, epilepsia.
t Acne, afinamento da pele.
t Telangiectasia.
t Úlcera gastrintestinal, náusea, aumento do apetite, ganho de peso, pancreatite.
t Necrose asséptica óssea, miopatia proximal.
t Reações leucemoides (leucocitose).
t Menstruação irregular.
t Ácido acetilsalicílico: aumento do risco de ulceração gastrintestinal e de concentrações subterapêuticas de ácido acetilsalicílico no sangue.
t Amobarbital, fenitoína, fosfenitoína, primidona e rifampicina: podem reduzir a efetividade da prednisolona.
t Atracúrio, alcurônio, brometo de hexaflurônio, galamina e vecurônio: diminuição da efetividade destes fármacos; prolongada fraqueza muscular e miopatia.
t Contraceptivos em combinação: o uso concomitante com prednisolona pode resultar em aumento do risco de Efeitos adversos dos corticoides (reações neuropsiquiátricas, distúrbios de fluidos e eletrólitos, hipertensão e hiperglicemia).
t Itraconazol: aumento das concentrações séricas do corticosteroide e do risco de seus efeitos adversos.
t Fluoroquinolonas: aumento do risco de ruptura de tendões.
t Quetiapina: diminuição das concentrações sanguíneas de quetiapina. Cautela em caso de uso concomitante.
t Vacina contra rotavírus: aumento do risco de infecção pela vacina com vírus vivos. A vacinação de pacientes em terapia sistêmica prolongada com corticosteroides é contraindicada.
t A administração com alimento pode minimizar a irritação gástrica.
t Não tomar qualquer tipo de vacina ou vacina sem consultar um médico. Evitar contato com qualquer pessoa que tome a vacina oral contra pólio e com pessoas doentes.
t Evitar uso de bebidas alcoólicas.
t Não suspender abruptamente este medicamento após uso prolongado.
t Atletas devem consultar autoridades esportivas, pois pode ter seu uso restrito em alguns esportes.
t Armazenar em recipientes bem fechados, e à temperatura ambiente, de 15 a 30 ºC. Manter ao abrigo de luz, calor e umidade. Não congelar.
Atenção: após uso prolongado, a retirada do fármaco deve ser lenta e gradual, devido ao risco de provocar supressão suprarrenal de reversão demorada.
Beatriz Garcia Mendes
Na Rename 2010: item 5.5.2.3
t Comprimido 300 mg.
t Tratamento de infecção por HIV em conjunto com outros antirretrovirais.
t Tratamento de hepatite B crônica em paciente com doença hepática compensada com evidência de replicação viral e inflamação hepática ativa ou fibrose histologicamente documentadas.
t Reação de hipersensibilidade ao tenofovir ou componentes de sua fórmula.
t Usar com cuidado nos casos de:
– fatores de risco para doenças hepáticas (risco de acidose lática e hepatomegalia com esteatose) (ver Apêndice C).
– fatores de risco de osteopenia ou osteoporose (o tenofovir aumenta o risco de redução da densidade mineral óssea).
– insuficiência renal grave (ver Apêndice D).
– uso concomitante de outros fármacos nefrotóxicos (evitar).
– associação com didanosina em terapia inicial (não é recomendada devido a uma possível falha na resposta terapêutica).
– lactação (ver Apêndice B).
t Há risco de exacerbação da infecção pelo vírus da hepatite B após interrupção do tratamento com tenofovir.
t Terapia tripla com inibidores de transcriptase reversa análogos de nucleosídeos está associada a perda de resposta virológica precoce (não se recomendam as associações tenofovir + lamivudina + abacavir ou tenofovir + lamivudina + didanosina).
t Categoria de risco na gravidez (FDA): B
t Dose de 300 mg, por via oral, uma vez ao dia.
t Dose de 300 mg, por via oral, uma vez ao dia.
t Nota: Para pacientes com dificuldades de deglutição, os comprimidos podem ser dispersos em no mínimo 100 mL de água, suco de laranja ou de uva.
t Biodisponibilidade oral: 25 a 40%.
t Meia-vida: 10 a 14 horas.
t Metabolismo: é rapidamente hidrolisado a tenofovir, sendo depois fosforilado (forma ativa).
t Excreção: renal (70 a 80%).
t Náusea (11 a 16%), vômito (3 a 7%), dor abdominal, diarreia (6 a 11%), flatulência (4%), dispepsia e anorexia (4%),
t Pancreatite, hepatotoxicidade (hepatite, acidose lática e hepatomegalia grave com esteatose).
t Dispneia, tosse.
t Nefrotoxicidade, diabetes insípido nefrogênico, insuficiência renal, efeito nos túbulos proximais renais (incluindo síndrome de Fanconi), proteinúria, poliúria.
t Cefaleia (5 a 10%), depressão (4 a 8%), insônia (3 a 4%), fadiga, tontura (1 a 3%), neuropatia periférica (3 a 5%), convulsões.
t Mialgia, hipertonia, rabdomiólise, astenia (3 a 11%) e artralgia.
t Osteopenia.
t Sudorese, exantema (18%), urticária e febre.
t Lipodistrofia (1%), hipofosfatemia, distúrbios sanguíneos, incluindo anemia, neutropenia e trombocitopenia.
t Síndrome da reconstituição imune.
t Didanosina: aumento das concentrações plasmáticas da didanosina, elevando o risco de toxicidade (neuropatia, diarreia, pancreatite, acidose lática grave). Suspender o uso da didanosina caso sinais ou sintomas de pancreatite, hiperlactatemia sintomática ou acidose lática sejam evidentes. Em adultos pesando 60 kg ou mais com depuração da creatinina endógena (DCE) igual ou superior a 60 mL/min, a dose de didanosina deve ser reduzida a 250 mg uma vez por dia quando é coadministrada com tenofovir. Em adultos com peso inferior a 60 kg, com DCE de 60 mL/min ou mais, é recomendada uma dose de 200 mg didanosina uma vez ao dia.
t Lopinavir: aumento da biodisponibilidade do tenofovir. Acompanhar os pacientes para o aparecimento de sinais e sintomas de toxicidade, incluindo hepática ou renal.
t Ritonavir: aumento da biodisponibilidade do tenofovir. Monitorar os marcadores de integridade óssea bem como as concentrações séricas das aminotransferases. Monitorar os pacientes quanto a sinais e sintomas de toxicidade hepática ou renal.
t Tipranavir: redução da concentração plasmática do tipranavir. Monitorar os pacientes quanto a perda de eficácia e realizar, se possível, o monitoramento das concentrações do tipranavir.
t Pode ser usado com ou sem alimentos, porém a administração com alimentos gordurosos aumenta a concentração plasmática em 14%.
t Informar o médico quanto ao surgimento de reações alérgicas (pruridos, edema na face e nas mãos, edema e parestesia oral e na garganta e transtorno respiratório), fraqueza, perda do apetite, náuseas e vômitos.
t Orientar para o uso durante todo o tempo prescrito, mesmo que haja melhora dos sintomas com as primeiras doses.
t Reforçar orientações sobre prevenção da transmissão do HIV.
t Manter à temperatura ambiente, entre 15 a 30 °C. Proteger do calor, umidade e luz direta.
Atenção: segurança e eficácia não foram estabelecidas em neonatos, crianças e adolescentes menores de 18 anos.
Como sinonímia para fumarato de tenofovir desoproxila (nome que correspondente a Denominação Comum Brasileira) também é empregada a abreviatura TDF, entretanto, não se recomenda a prescrição de fármacos por abreviaturas ou siglas.
Pacientes HIV positivas não devem amamentar devido ao risco de transmissão do HIV ao lactente.
Rosa Martins
Na Rename 2010: itens 14.1 e 14.5
t Comprimido 40 mg.
t Solução injetável 10 mg/mL.
t Solução oral 10 mg/mL
t Edema refratário a outros diuréticos, de diversas causas.
t Edema agudo de pulmão.
t Edema em insuficiência renal crônica
t Insuficiência renal com anúria (ver Apêndice D).
t Estado pré-comatoso associado a cirrose hepática.
t Hipersensibilidade a furosemida e sulfonamidas.
t Usar com cuidado nos casos de:
– indução de diurese em insuficiência renal moderada (pode ser necessário aumentar a dose) (ver Apêndice D).
– hipotensão, hipovolemia e hipopotassemia que precisam ser corrigidas.
– aumento da próstata.
– idosos (são mais sensíveis aos efeitos hipotensores e hidreletrolíticos; pode ser necessária redução da dose ou ajuste do intervalo de administração).
– insuficiência hepática (ver Apêndice C).
– associação com outros fármacos ototóxicos, como vancomicina e aminoglicosídeos (aumento do risco de ototoxicidade).
– lactação.
t Monitorar eletrólitos, particularmente sódio e potássio.
t Categoria de risco na gravidez (FDA): C.
t 1 a 3 mg/kg/dia, por via oral, a cada 24 horas. Dose máxima 40 mg/dia.
t Dose inicial 1 mg/kg, por via intravenosa lenta. Se necessário, aumentar em 1 mg/kg, a intervalo mínimo de 2 horas. Dose máxima 20 mg/dia. Em infusão intravenosa, a velocidade não deve exceder a velocidade de 4 mg/minuto.
t 40 mg/dia, por via oral. Dose manutenção 20 a 40 mg/dia, em dose única matinal. A dose pode ser aumentada para 80 mg diários ou mais, em edemas resistentes. Dose máxima: 600 mg/dia.
t 20 a 40 mg, por via intravenosa lenta. Administrar mais 20 mg a cada 2 horas se necessário. Dose acima de 40 mg administrar por infusão intravenosa lenta (não exceder 4 mg/minuto). Dose máxima 1,5 g/dia.
t Dose inicial 20 a 80 mg, por via oral. Se necessário, repetir a mesma dose inicial ou acrescida em 20 a 40 mg, a cada 6 a 8 horas, até que o efeito diurético seja alcançado. Dose de manutenção: dose que produziu o efeito diurético, a cada 24 horas ou dividida a cada 12 horas. Dose máxima: 600 mg/dia.
t Dose 20 mg/dia, por via oral, aumentando lentamente até obter a resposta desejada.
t Biodisponibilidade 47 a 70%
t Início da resposta diurética: 30 a 60 minutos (oral); 2 a 5 minutos (intravenosa).
t Pico de resposta diurética: 1 a 2 horas (oral); 30 minutos (intravenosa).
t Duração da ação: 4-6 horas (oral), 2 horas (intravenosa).
t Metabolismo hepático, 10%; metabólitos com atividade desconhecida.
t Excreção: renal (60% a 90%).
t Meia-vida de eliminação: 30 a 120 minutos.
t Não dialisável
t Distúrbio hidreletrolítico (24%): hiponatremia, hipopotassemia e hipomagnesemia, alcalose hipoclorêmica, hipocalcemia.
t Hipotensão, hipovolemia/desidratação.
t Náusea, distúrbios gastrintestinais.
t Hiperuricemia (40%) e gota, hiperglicemia, aumento temporário nas concentrações plasmáticas de colesterol (10%) e triglicerídeos.
t Exantema, fotossensibilidade, Síndrome de Stevens-Johnson.
t Depressão medular, agranulocitose, anemia aplástica, anemia hemolítica.
t Pancreatite (com altas doses parenterais).
t Diminuição da densidade mineral óssea
t Ototoxicidade (6%), geralmente com altas doses parenterais, administração rápida e em insuficiência renal.
t Alcaçuz (Glycyrrhiza glabra): aumento do risco de hipopotassemia e/ou redução da efetividade do diurético
t Anti-inflamatórios não-esteroides, ácido acetilsalicílico, colestiramina e colestipol: podem diminuir o efeito farmacológico da furosemida. Evitar o uso concomitante, monitorar sinais e sintomas específicos.
t Anti-inflamatórios esteroides e clofibrato: podem aumentar o efeito farmacológico da furosemida e a concentração das transaminases; clofibrato pode ter seus Efeitos adversos na musculatura esquelética aumentados. Evitar o uso concomitante, monitorar sinais e sintomas específicos.
t Aminoglicosídeos (gentamicina, tobramicina), betabloqueadores (sotalol), digitoxina, digoxina, dofetilida, inibidores da ECA (primeira dose) e lítio: podem ter o efeito farmacológico/tóxico aumentado pela furosemida. Ajustar dose e monitorar sinais e sintomas específicos.
t Bepridil: uso concomitante pode levar a hipopotassemia e subsequente cardiotoxicidade (torsades de pointes). Monitorar potássio e magnésio, considerar substituição por diurético poupador de potássio.
t Bloqueadores neuromusculares (pancurônio, vecurônio): podem ter o efeito aumentado ou diminuído pela furosemida. Ajustar dose e monitorar sinais e sintomas específicos.
t Ginseng: o uso associado aumenta o risco de resistência ao diurético.
t Orientar que pode ser administrada com alimento, se houver desconforto gástrico.
t Orientar quanto ao aparecimento de efeitos indesejáveis.
t Recomendar o aumento da ingestão de alimentos com alto teor de potássio (laranjas, bananas, feijão).
t Recomendar aos diabéticos para monitorar cuidadosamente a glicemia.
t Em caso de esquecimento de uma dose, usar assim que lembrar. Se estiver perto do horário da próxima dose, desconsiderar a dose anterior, esperar e usar no horário. Nunca usar duas doses juntas.
t O armazenamento da formulação oral e parenteral deve ser feito sob temperatura ambiente e controlada, entre 15 e 30 ºC.
t O medicamento deve ser dispensado em embalagem fechada e protegida da luz.
t Observar orientação específica do produtor quanto a diluição, compatibilidade e estabilidade da solução.
t As soluções parenterais são preparadas com a adição de hidróxido de sódio, originando soluções com pH entre 8,0 e 9,3.
t As soluções de furosemida para injeção são alcalinas, não devendo ser misturadas ou diluídas com glicose ou soluções ácidas.
t A injeção de furosemida é visualmente incompatível com injeções de diltiazem, dobutamina, dopamina, midazolam e brometo de vecurônio.
Atenção: este fármaco apresenta um número elevado de Efeitos adversos, sendo necessário realizar pesquisa específica sobre este aspecto, antes de introduzir ou descontinuar furosemida ou outros medicamentos no esquema terapêutico do paciente. Atentar para risco aumentado de ototoxicidade com uso simultâneo de outros fármacos ototóxicos. Sinal de distúrbio hidreletrolítico: cefaleia, hipotensão, cãibra, xerostomia, sede, fraqueza, letargia, tontura, arritmia, oligúria e distúrbio gastrintestinal.
Consta no documento:
“Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial.”
O objetivo do site MedicinaNet e seus editores é divulgar este importante documento. Esta reprodução permanecerá aberta para não assinantes indefinidamente.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.