(Carregando Índice)... (Carregando Índice)... |
Autores:
Daniel Soares Freire
Endocrinologista pelo Hospital das Clínicas da Faculdade de Medicina da USP;
Médico Colaborador do Serviço de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP
Marcos Catânia
Médico Assistente da Disciplina de Emergências Clínicas do Hospital das Clínicas da Faculdade de Medicina da USP
Especialista em clínica médica e endocrinologia e metabologia pelo Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 12/06/2009
Comentários de assinantes: 0
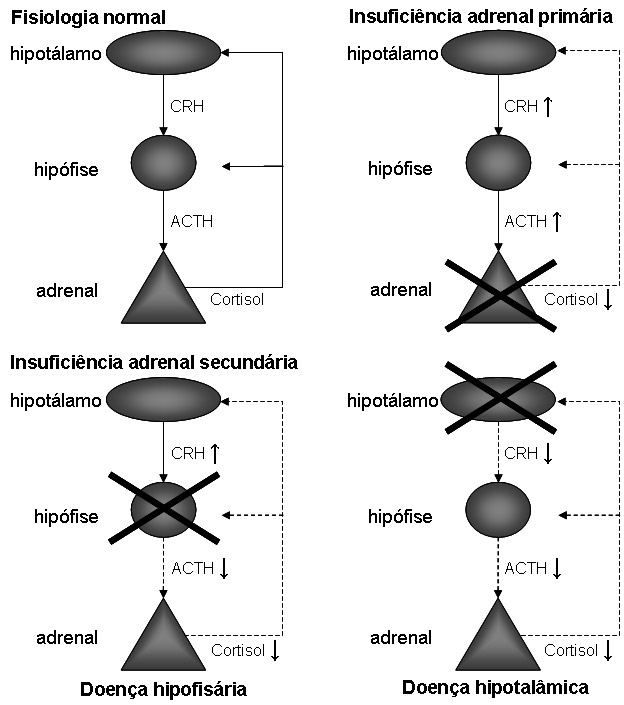
Insuficiência adrenal (IA) é o resultado da deficiência na produção hormonal pelas adrenais, que pode ocorrer por destruição ou disfunção do córtex adrenal (neste caso definida como Insuficiência adrenal primária ou doença de Addison) ou por deficiência na secreção hipofisária do principal fator trófico adrenal, o ACTH (neste caso definida como Insuficiência adrenal secundária). Uma diferença importante entre as duas formas de IA é que a deficiência de mineralocorticóide está presente apenas na IA primária.
A Insuficiência adrenal primária tem prevalência ao redor de 100 casos/milhão e incidência de 4 a 6 por milhão em população caucasiana. O diagnóstico ocorre mais freqüentemente ao redor dos 40 anos afetando mais as mulheres. A Insuficiência adrenal secundária tem prevalência estimada de 150-280 casos/milhão e também afeta mais mulheres que homens, com pico de incidência ao redor dos 60 anos. Este cálculo de incidência e prevalência da doença é entretanto falseado devido ao fato das manifestações da doença serem inespecíficas, não sendo possível estimar o número de pacientes que morrem com a doença sem ter o diagnóstico de Insuficiência adrenal corretamente estabelecido. Pacientes com Insuficiência adrenal nos estados unidos um media passam por 3 a 4 médicos antes do diagnóstico correto ser estabelecido.
As manifestações da Insuficiência adrenal podem ser divididas em três síndromes clínicas: Insuficiência adrenal primária crônica ou doença de Addison, crise adrenal (estado de Insuficiência adrenal aguda que ocorre em pacientes expostos a um estresse agudo como infecção, trauma, cirurgia ou desidratação) e Insuficiência adrenal secundária, com aspectos clínicos variáveis a depender da etiologia específica apresentada dentro de cada síndrome. Descreveremos, para cada síndrome, a etiologia, quadro clínico, estratégia diagnóstica e tratamento, e comentaremos sobre as particularidades no diagnóstico e manuseio da Insuficiência adrenal em pacientes críticos.
Fisiologia da adrenal
Glicocorticóides
São sintetizados na zona fasciculada da adrenal sob o controle do eixo ACTH/CRH, com um ritmo circadiano de concentrações máximas pela manhã e mínimas ao redor da meia-noite. O cortisol, principal glicocorticóide endógeno, circula ligado à CBG (globulina ligadora de corticosteróides, corticosteroid-binding globulin), com menos de 10% na forma livre, que é a fração bioativa. Glicocorticóides são importantes para a manutenção do metabolismo lipídico, protéico e de carboidratos. Modulam o sistema imune e atuam na cicatrização de feridas, na manutenção da integridade cardiovascular e contratilidade cardíaca (o cortisol facilita a produção e a ação das catecolaminas), dentre vários outros efeitos.
Mineralocorticóides
São produzidos na zona glomerulosa principalmente sobre o estímulo do sistema renina-angiotensina ou de hipercalemia. A aldosterona, principal mineralocorticóide endógeno, atua na homeostase do sódio e potássio e na manutenção do volume intravascular.
Andrógenos
DHEA (deidroepiandrosterona) é o principal precursor androgênico produzido pela na zona reticular. Sua síntese também é diurna e agudamente estimulada pelo ACTH, com aumento na adrenarca (6-10 anos) e queda progressiva após os 30 anos.
Fisiopatologia da IA primária
A progressão do déficit funcional adrenal na IA primária evolui de uma glândula adrenal normal, antes da instalação da doença, a um primeiro estágio onde se detecta comprometimento mineralocorticóide inicial verificado pelo aumento da atividade plasmática de renina (APR) e níveis normais ou reduzidos de aldosterona.
Posteriormente, observa-se um estado de menor reserva do setor glicocorticóide avaliado pela diminuição dos níveis de cortisol frente a estímulo com ACTH.
Com a progressão da lesão adrenal, o paciente passa a apresentar elevações do ACTH basal por redução do efeito do cortisol na alça de retroalimentação; nesta situação, o paciente é susceptível à falência adrenocortical aguda ou crise adrenal durante estresse que demande maior nível de cortisol endógeno.
Finalmente, ocorre redução dos níveis basais de cortisol associado a aumento importante dos níveis plasmáticos de ACTH, juntamente com sintomas de Insuficiência adrenal. Tal evolução mostra a progressão do comprometimento das zonas glomerulosa (mineralocorticoide) e fasciculada (glicocorticóide), o que culmina com déficit hormonal basal de cortisol, níveis elevados de ACTH e APR elevada com aldosterona baixa e manifestações clínicas de hipocortisolismo. Cabe ressaltar que deficiência mineralocorticóide ocorre apenas na Insuficiência adrenal primária, causando desidratação e hipovolemia.
É necessária a perda de cerca de 90% do córtex das adrenais para ocorrerem manifestações da Insuficiência adrenal. O quadro clínico será influenciado pela velocidade desta destruição sendo gradual e levando a sintomas de Insuficiência adrenal crônica na maioria das vezes, mas em alguns casos com rápida destruição glandular e apresentações dramáticas, cerca de 25% dos diagnósticos ocorrem em pacientes em crises addisonianas.
Fisiopatologia da IA secundária
A IA secundária decorre de uma falência hipofisária em secretar o principal hormônio estimulador do o córtex adrenal, o ACTH. A deficiência de ACTH pode ser causada por doença hipofisária ou hipotalâmica; neste último caso, ocorre redução da secreção do hormônio liberador de ACTH pelo hipotálamo (CRH).
Como o sistema renina-angiotensina-aldosterona está preservado, a secreção de mineralocorticóide é normal e não se observa hipercalemia. Contudo os pacientes apresentam hiponatremia devido a aumento da liberação de hormônio anti-diurético (ADH) secundário ao déficit de cortisol, na apresentação é freqüente a hiponatremia ser mais pronunciada que em pacientes com Insuficiência adrenal primaria, o que é justificado pelo diagnóstico mais tardio da forma secundária.
Assim como na IA primária, a produção de andrógenos e precursores androgênicos adrenais (como DHEA e DHEAS) está prejudicada na IA secundária.
Figura 1: Fisiopatologia da IA.

Insuficiência adrenal primária – doença de Addison
Adrenalite auto-imune
A principal causa nos países desenvolvidos, correspondendo a mais de 70% dos casos. Em 75% dos casos podem-se identificar anticorpos contra a adrenal, dentro destes o anticorpo anti 21-hidroxilase é o mais prevalente e importante, também o anticorpo anti-17-hidroxilase e anticorpo anti-P450scc são descritos, outros anticorpos anti-adrenal usualmente são associados com outras síndromes. Pode ser isolada (40% dos casos com discreta preponderância do sexo masculino) ou ocorrer como parte de uma síndrome poliglandular auto-imune (60% dos casos com preponderância do sexo feminino), a associação de adrenalite auto-imune com hipoparatireoidismo e candidíase mucocutâne é conhecida como síndrome poliglandular auto-imune do tipo 1, a associação de adrenalite auto-imune e hipotireoidismo auto-imune constitui a síndrome poliglandular auto-imune do tipo 2 também denominada de síndrome de Schmidt, já a associação com diabetes mellitus do tipo 1 caracteriza a síndrome poliglandular do tipo 3 ou síndrome de Carpenter. Por outro lado apenas 1-2% dos pacientes com doença tireoidiana auto-imune ou diabetes mellitus do tipo 1 apresentam Insuficiência adrenal.
Critérios diagnósticos foram propostos para definir o diagnóstico de adrenalite auto-imune, que são os seguintes:
1. Diagnóstico bioquímico de Insuficiência adrenal
2. Adrenal de volume normal ou reduzida em exames de imagens e sem calcificações
3. Presença de anticorpos contra adrenal
4. Exclusão de outras causas de Insuficiência adrenal
5. Associação com outras doenças endócrinas auto-imunes
A presença dos critérios 1 a 4 seriam indicativos de diagnóstico definitivo da doença, enquanto a presença dos critérios 1,2,3 e 5 tornariam este diagnóstico provável.
Infecções
Correspondem à maior parte dos casos nos países em desenvolvimento e no Mundo. A etiologia tuberculosa e fúngica (paracoccidioidomicose, histoplasmose, criptococose e outros) são as mais importantes. Outros agentes infecciosos podem acometer e destruir a adrenal, como o HIV e o citomegalovírus.
A doença de Addison causada por tuberculose aparece pela disseminação hematógena da infecção tuberculosa, nestes casos a doença extra-adrenal costuma estar evidente, inicialmente as adrenais parecem aumentadas com granulomas extensos e caseificação, com o córtex e a medula afetados, durante a evolução a fibrose aparece com a diminuição do tamanho das adrenais, ficando estas normais ou diminuídas, aparecendo calcificações em cerca de 50% dos casos, em raros casos o tratamento da tuberculose leva a melhora da Insuficiência adrenal.
A Insuficiência adrenal pode ocorrer associada à Síndrome da Imunodeficiência adquirida(SIDA) , a prevalência de doença clinicamente significativa nestes pacientes é considerada baixa pela literatura, entretanto quando o teste da cortrosina é realizado cerca de 10% dos pacientes apresentam Insuficiência adrenal, é duvidoso que a infecção pelo HIV apresente papel direto como causa de doença de Addison, mas infecções oportunistas, infiltração de adrenais pelo sarcoma de Kaposi, hemorragias intra-adrenais e o uso de medicações como cetoconazol e rifampicina; esta ultima aumentando o clearence hepático do cortisol; podem levar ao aparecimento da doença.
Infarto de adrenal
Hemorragia intra-adrenal bilateral causada por hemorragia ou trombose da veia adrenal, podem ser causas de Insuficiência adrenal, associa-se principalmente com meningococcemia, CIVD, Síndrome de Anticorpos-antifosfolípides. Infecções por pseudomonas aeroginosa também são associadas com hemorragia de adrenais.
Medicações
Drogas que alteram a biossíntese de cortisol como etomidato, aminoglutemidina, cetoconazol e metirapona. Medicações que aceleram o clearence hepático de cortisol através das enzimas de oxigenação hepática como rifampicina, hidantal e barbituratos podem levar a Insuficiência adrenal em pacientes com limitada reserva adrenal hipofisária ou adrenal. Também o mitotane agente adrenolítico é associado com Insuficiência adrenal.
Adrenoleucodistrofia
Defeito na beta-oxidação de ácidos graxos de cadeia longa resulta em Insuficiência adrenal e doença progressiva desmielinizante. Normalmente afetam meninos de 5-12 anos e em 15% dos casos aparece como Insuficiência adrenal.
O óleo de Lorenzo uma mistura de dois tipos de glicerol têm benefício terapêutico limitado, lovastatina tem sido usada, assim como o fenofibrato podem levar ao aumento da expressão da proteína relacionada a adrenoleucodistrofia, mas como arma terapêutica, permanecem controversos, o transplante da medula óssea é benéfico nos estágios iniciais da doença.
Metástases tumorais
Metástases adrenais,são principalmente achado de necropsia, mas Insuficiência adrenal resultante destas é incomum, pois é necessário a destruição de aproximadamente 90% da glândula adrenal para os pacientes desenvolverem sintomas de hipoadrenalismo. Os principais carcinomas associados com Insuficiência adrenal são o de pulmão e mama.
Hipoplasia adrenal congênita
Condição familiar rara, onde o desenvolvimento do córtex adrenal é interrompido, ocorre em 1 a cada 12.500 nascimentos, a forma ligada ao X é associada com mutações do DAX-1 e é associada a hipogonadismo hipogonadotrófico, mutações no fator esteroidogênico 1(SF-1) também resultam em Insuficiência adrenal por ausência de desenvolvimento da adrenal
Deficiência familiar de glicocorticóide
Causa rara de hipoadrenalismo autossômica recessiva, o eixo renina-angiotensina-aldosterona permanece inalterado ou com alterações discretas respondendo ao estímulo postural e depleção de volume, crianças apresentam-se comumente com hipoglicemia no período neonatal ou posteriormente com hiperpigmentação, freqüentemente com aumento da velocidade de crescimento, estes pacientes apresentam deficiência de glicocorticóide com níveis aumentados de ACTH, ocorrendo por mutações na melanocortina-2 ou receptor de ACTH(MC2R) no cromossomo 18 em alguns dos casos. A variante chamada triplo A ou síndrome de Allgrove´s caracterizada por Insuficiência adrenal, acalasia e alacrimia,com deficiência de mineralocorticóide ocorrendo em 15% dos casos, esta variante ocorre em mutação de outros genes.
Outras causas
Estão descritas na Tabela 1.
Tabela 1: Causas de IA primária
|
Diagnóstico |
Aspectos clínicos associados |
Comentários |
|
Adrenalite auto-imune | ||
|
Isolada |
- |
Associação com HLA-DR3, CTLA-4 |
|
Como parte da síndrome poliglandular auto-imune (SPA): | ||
|
SPA tipo 1 (APEDEC – autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy) |
Hipoparatireoidismo, candidíase mucocutânea crônica, outras doenças auto-imunes |
Mutação do gene AIRE (21q22.3) 15% dos pacientes com adrenalite auto-imune Herança autossômica recessiva |
|
SPA tipo 2 |
Doença tireoideana auto-imune, DM tipo 1, outras doenças auto-imunes (falência gonadal primária, vitiligo, gastrite atrófica, doença celíaca) |
Associação com HLA-DR3, CTLA-4 Herança autossômica dominante com penetrância incompleta. |
|
SPA tipo 4 |
Outras doenças auto-imunes, fora DM tipo 1 e doença tireoideana |
Associação com HLA-DR3, CTLA-4 |
|
Adrenalite Infecciosa | ||
|
Tuberculosa |
Manifestação de tuberculose em outros órgãos |
|
|
AIDS |
Outras manifestações da AIDS |
Patógenos envolvidos: HIV-1, citomegalovírus |
|
Fúngica |
Principalmente em imunodeprimidos |
Criptococose, histoplasmose, paracoccidioidomicose |
|
Doenças genéticas | ||
|
Adrenoleucodistrofia |
Sintomas neurológicos |
Mutação gene ABCD1 Herança recessiva ligada ao X |
|
Hiperplasia Adrenal Congênita | ||
|
Deficiência de 21-hidroxilase |
Genitália ambígua em meninas |
Mutação CYP21; Herança autossômica recessiva. |
|
Deficiência de 11ß-hidroxilase |
Hipertensão e genitália ambígua em meninas |
Mutação CYP11B1 Herança autossômica recessiva |
|
Deficiência 3ß-HSD tipo 2 |
Genitália ambígua em meninos e virilização pós-natal em meninas |
Mutação HSD3B2 |
|
Deficiência de 17a-hidroxilase |
Hipertensão, genitália ambígua em meninos e ausência de puberdade em ambos os sexos |
Mutação CYP17 Herança autossômica recessiva |
|
Hipoplasia adrenal congênita lipóide |
Sexo reverso em pacientes com cariótipo XY |
Mutações gene SIAR e CYP11A |
|
Síndrome Smith-Lemli-Optiz |
Retardo mental, malformações craniofaciais, déficit de crescimento |
Mutação gene DHCR7 |
|
Hipoplasia adrenal congênita | ||
|
Ligada ao X |
Hipogonadismo hipogonadotrófico |
Mutação NROB1 Herança recessiva ligada ao X |
|
Síndrome gene contíguo Xp21 |
Distrofia muscular de Duchenne e deficiência de glicerol quinase (retardo psicomotor) |
Deleção dos genes da distrofia muscular de Duchene, glicerol quinase e NROB1 |
|
Ligada ao SF-1 |
Sexo reverso em pacientes com cariótipo XY |
Mutação NR5A1 Herança autossômica recessiva ou dominante |
|
Síndrome IMAGe (Intrauterine growth retardation, Metaphyseal dysplasia, Adrenal insufficiency, Genital anomalies) |
Retardo de crescimento intrauterino, Displasia metafisária, anomalias genitais |
Mutação desconhecida Herança autossômica recessiva |
|
Síndrome Keams-Sayre |
Oftalmoplegia externa, retinite pigmentosa, defeitos de condução cardíacos, outras endocrinopatias |
Deleções de DNA mitocondrial |
|
Síndromes de insensibilidade ao ACTH (deficiência familiar de glicocorticóide) |
Deficiência glicocorticóide sem alteração da síntese mineralocorticóide |
|
|
Tipo 1 |
Alta estatura |
Mutação no receptor ACTH (MC2R) Herança AR |
|
Tipo 2 |
Sem outros aspectos clínicos associados |
Mutação desconhecida |
|
Síndrome de Allgrove (Síndrome triplo A) |
Alacrimia, Acalasia, sintomas Adicionais (neurológicos, surdez, retardo mental, hiperqueratose) |
Mutações no gene triplo A (AAAS) |
|
Hemorragia adrenal bilateral |
Sintomas da doença causadora |
Choque séptico, meningococcemia (Síndrome Waterhouse-Friederichsen), síndrome antifosfolípide. |
|
Infiltração adrenal |
Sintomas da doença causadora |
Metástases (sítios primários principais: neoplasia pulmonar, de trato gastrointestinal e renal), linfoma primário adrenal, sarcoidose, amiloidose, hemocromatose |
|
Adrenalectomia bilateral |
Sintomas da doença causadora |
Como opção terapêutica para síndrome de Cushing refratária a outras modalidades terapêuticas |
|
Induzida por drogas |
- |
Mitotane, etomidato, cetoconazol, mifepristone, aminoglutemidina, suramin |
HSD = hidroxi-?-5-esteróide desidrogenase; SPA = Síndrome poliglandular auto-imune; 3ß-HSD = 3ß-hidroxi-esteróide desidrogenase.
Insuficiência adrenal secundária
Interrupção de corticóide
A causa mais comum de Insuficiência adrenal secundária é a interrupção abrupta da terapia com glicocorticóides exógenos. Durante a corticoterapia ocorre bloqueio da secreção de ACTH pelos corticotrofos hipofisários com conseqüente atrofia adrenal. Uma vez retirado ou reduzido o aporte de corticóide exógeno, o paciente pode manifestar sintomas de Insuficiência adrenal. De forma semelhante, pode-se observar IA secundária após tratamento da síndrome de Cushing.
Doses menores que 5 mg de prednisona em dose única pela manhã, não parecem causar qualquer espécie de supressão no eixo, também dose de corticóide de curta duração usando em dias alternados, ou qualquer dose de corticóide usada por menos de 3 semanas não parecem causar qualquer espécie de supressão no eixo. Porém em pacientes que usaram dose de 20 mg ou mais de prednisona por período de tempo maior que 3 semanas, ou que pareçam clinicamente cushingóide ou com uso de dose de 7,5 mg ou maior por período de 1 mês ou mais, provavelmete apresentam supressão do eixo. A duração da supressão do eixo é discutível, mas pode durar até 1 ano após a parada do uso de glicocorticóides. Alguns estudos sugerem que pacientes em uso de corticóides em dias alternados não apresentam supressão do eixo.
Tumores hipotálamo-hipofisários
A presença de tumores na região hipotálamo-hipofisária ou o seu tratamento cirúrgico ou radioterápico são importante causa de IA por déficit de ACTH, que habitualmente está associada a outras deficiências hipofisárias ou mesmo pan-hipopituitarismo (é improvável a ocorrência de déficit de ACTH isolado).
Hipofisite linfocítica auto-imune
É outra causa menos freqüente, que afeta principalmente mulheres durante ou logo após a gestação
Outras causas
Estão descritas na Tabela 2.
Tabela 2: Causas de IA secundária
|
Diagnóstico |
Comentários |
|
Hipercortisolismo crônico prévio |
Exógeno (iatrogênico) ou endógeno (síndrome de Cushing) |
|
Tumores hipofisários |
Geralmente acompanhada de outras insuficiências hipofisárias. Pode ocorrer déficit visual por compressão do quiasma pelo tumor (geralmente adenomas de hipófise). |
|
Outros tumores da região hipotálamo-hipofisária |
Craniofaringioma, meningioma, ependimoma, metástases. |
|
Irradiação hipofisária |
O hipopituitarismo se instala geralmente em até 10 anos da irradiação da região selar. |
|
Hipofisite linfocítica auto-imune | |
|
Isolada |
80% relacionadas à gravidez; maior parte manifesta-se por hipopituitarismo, mas pode ocorrer deficiência isolada de ACTH. |
|
Como parte de SPA |
Associada com doença tireoideana auto-imune e, menos freqüentemente, com vitiligo, falência gonadal primária, DM tipo 1, anemia perniciosa. |
|
Deficiência congênita isolada de ACTH |
Defeito enzimático da clivagem da POMC |
|
Síndrome de deficiência da POMC |
Mutação no gene da POMC; tríade clínica de Insuficiência adrenal, obesidade de início precoce e cabelos com pigmentação avermelhada. |
|
Deficiência hormonal hipofisária combinada |
Mutações no gene PROP1 (fator de transcrição hipofisário do Pit1) com desenvolvimento progressivo de pan-hipopituitarismo seguindo a seqüência GH, prolactina, TSH, LH/FSH, ACTH. Mutações no gene HESX1: deficiência hormonal hipofisária combinada associada à hipoplasia de nervo óptico e defeitos encefálicos de linha média (displasia septo-óptica) |
|
Apoplexia hipofisária |
Cefaléia abrupta de forte intensidade, alterações visuais e náusea/vômitos. |
|
Síndrome de Sheehan |
Apoplexia ou necrose hipofisária com início no periparto, decorrente de hipotensão ou perda sangüínea volumosa. |
|
Infiltração hipofisária ou granuloma |
Tuberculose, actinomicose, sarcoidose, histiocitose X, granulomatose de Wegener |
|
Trauma crânio-encefálico |
Lesão de haste hipofisária |
POMC = pró-opiomelanocortina
O quadro clínico da IA depende do tipo de insuficiência (primária ou secundária) e do tempo de instalação (aguda ou crônica). A maior parte dos pacientes diagnosticados tem IA crônica.
A IA aguda, também chamada de crise adrenal, representa uma emergência endocrinológica muitas vezes de difícil diagnóstico, pois o quadro clínico pode não ser tão rico em sinais e sintomas como o observado na IA crônica; de fato, pacientes com crise adrenal grave podem apresentar apenas choque circulatório refratário como quadro clínico.
Insuficiência adrenal crônica
Os sinais e sintomas da IA crônica dependem de quatro mecanismos fisiopatológicos:
Déficit de glicocorticóide
Déficit de mineralocorticóide
Déficit de andrógenos
Aumento de ACTH
Como mostrado na Tabela 3, os quatro mecanismos originam o quadro clínico da IA primária; na IA secundária, não se observa déficit de mineralocorticóide nem aumento de ACTH.
Além disso, algumas características clínicas costumam estar associadas a uma ou outra forma de IA.
Tabela 3: Quadro Clínico da IA Primária e Secundária
|
|
IA primária |
IA secundária |
|
Déficit de glicocorticóide |
+ |
+ |
|
Déficit de mineralocorticóide |
+ |
- |
|
Déficit de andrógenos |
+ |
+ |
|
Aumento de ACTH |
+ |
- |
|
Condições que podem estar associadas |
Vitiligo, hipotireoidismo e outras doenças auto-imunes, infecções sistêmicas (tuberculose, micoses sistêmicas), infecção pelo HIV, neoplasia maligna disseminada (mama e pulmão). |
Uso prévio de glicocorticóide, deficiência de outros setores da hipófise, déficit visual por compressão de vias ópticas por tumor de hipófise, sinais de tumores hipofisários (galactorréia, face acromegalóide etc). |
Tabela 4: Sinais, sintomas e alterações laboratoriais das IAs primária e secundária
|
|
Sinais |
Sintomas |
Alterações laboratoriais |
|
Déficit de glicocorticóide |
• Perda de peso • Febre |
• Fadiga • Indisposição • Fraqueza • Anorexia • Epigastralgia • Náuseas • Vômitos • Mialgias • Artralgias • Tontura |
• Hiponatremia • Anemia • Linfocitose • Eosinofilia • Hipercalcemia • Hipoglicemia • Elevação do TSH* |
|
Déficit de mineralocorticóide |
• Hipotensão |
• Tontura • Avidez por sal |
• Hipercalemia • Azotemia • Acidose metabólica |
|
Déficit de andrógenos |
• Redução de pêlos pubianos e axilares (?) |
• Pele seca (?) • Redução de libido (?) |
|
|
Aumento de ACTH |
• Hiperpigmentação |
|
|
*Elevação de TSH pode decorrer do próprio hipocortisolismo ou de hipotireoidismo primário associado.
Insuficiência adrenal aguda (crise adrenal)
Representa um estado de Insuficiência adrenal aguda que ocorre principalmente em pacientes com IA primária expostos a um estresse agudo como infecção, trauma, cirurgia ou desidratação por diarréia, vômitos ou privação de sal, e que pode levar à morte se não reconhecida e tratada adequadamente.
A crise adrenal também pode acometer pacientes com IA secundária submetidos a estresse agudo, principalmente quando o diagnóstico de IA não é previamente sabido.
O quadro clínico inclui:
Anorexia e vômitos são os sintomas mais importantes, piorando a desidratação e podendo precipitar choque hipovolêmico. Pode ocorrer dor abdominal semelhante a um quadro de abdome agudo.
Fraqueza, apatia e confusão são comuns, assim como febre, que pode estar relacionada a eventual processo infeccioso precipitante da crise como ser devida ao hipoadrenalismo em si.
Hipotensão ocorre em cerca de 90% dos pacientes ocorrendo principalmente ou acentuando-se quando em ortostase, podendo ser acompanhada por síncope em algumas situações, em casos crônicos severos e crises agudas a hipotensão é invariavelmente presente podendo evoluir para choque.
Dentro dos sintomas mais específicos da doença se inclui a “avidez por sal” ocorrendo em até 20% dos casos, perda de peso pode ocorre eventualmente sendo maior do que 15 Kgs em alguns casos.
Se o quadro de IA for recente (como, por exemplo, em caso de hemorragia adrenal aguda), hiperpigmentação e outros sinais de IA crônica não estão presentes.
Hiponatremia, hipercalemia, linfocitose, eosinofilia e hipoglicemia são manifestações adicionais.
A Tabela 5 cita a proporção de sinais, sintomas e alterações laboratoriais em grandes séries de pacientes com Insuficiência adrenal.
Tabela 5: Achados clínicos e laboratoriais na Insuficiência adrenal
|
Sintomas, sinais e achados laboratoriais |
Freqüência(%) |
|
Sintomas: |
|
|
Fraqueza,fadiga, cansaço |
100% |
|
Anorexia |
100% |
|
Sintomas do trato gastrointestinal: |
92% |
|
Náuseas |
86% |
|
Vômitos |
75% |
|
Constipação |
33% |
|
Dor abdominal |
31% |
|
Diarréia |
16% |
|
Avidez por sal |
16% |
|
Sensação de tontura postural |
12% |
|
Dores musculares ou articulares |
6-13% |
|
Sinais: |
|
|
Perda de peso |
100% |
|
Hiperpigmentação |
94% |
|
Hipotensão PAS<110 mmHg |
88-94% |
|
Vitiligo |
10-20% |
|
Calcificação auricular |
5% |
|
Achados laboratoriais |
|
|
Alterações hidroeletrolíticas: |
92% |
|
Hiponatremia |
88% |
|
Hipercalemia |
64% |
|
Hipercalcemia |
6% |
|
Azotemia |
55% |
|
Anemia |
40% |
|
Eosinofilia |
17% |
Hemorragia adrenal bilateral pode se manifestar como crise adrenal. O quadro clínico compreende evidência de sangramento oculto (queda nos níveis de hemoglobina), dor abdominal ou em flanco e manifestações de crise adrenal (hiponatremia e hipercalemia, febre, hipoglicemia, hipovolemia, choque). São fatores de risco para hemorragia adrenal: anticoagulação com heparina ou outras drogas, coagulopatia, doença tromboembólica, estados de hipercoagulabilidade e sepse (infecção por Pseudomonas aeruginosa, meningococcemia – síndrome de Waterhouse-Friederichsen, dentre outros agentes infecciosos).
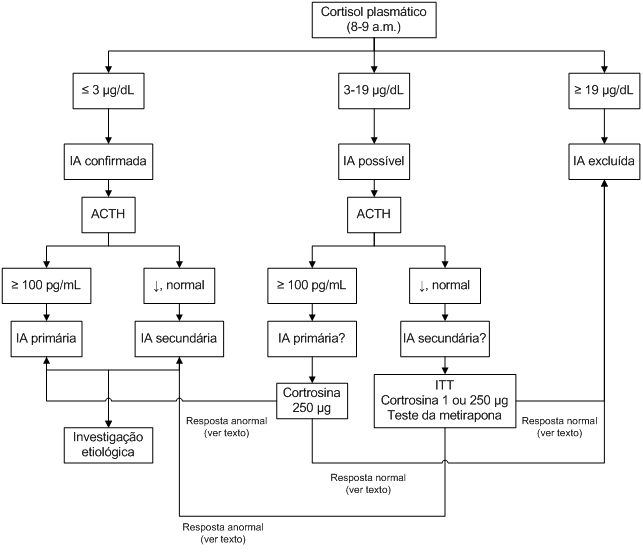
O fluxograma a seguir ilustra como deve ser conduzida a investigação diagnóstica.
Figura 2: Diagnóstico de IA.
Diagnóstico de Insuficiência adrenal
O primeiro teste a ser realizado é a dosagem de cortisol entre 8 e 9 horas da manhã. Valores menores ou iguais a 3 µg/dL são consistentes com o diagnóstico de IA, enquanto valores maiores ou iguais a 19 µg/dL excluem o diagnóstico. Contudo, a maior parte dos pacientes se apresentará com cortisol basal em valores intermediários, entre 3 e 19 µg/dL, e necessitarão de testes adicionais.
O ACTH deve ser dosado pela manhã nos pacientes em que se estabeleceu diagnóstico laboratorial de IA (cortisol = 3 µg/dL) e naqueles em que o diagnóstico é possível mas ainda não foi confirmado (cortisol entre 3 e 19 µg/dL).
Cortisol = 3 µg/dL: níveis de ACTH acima de 100 pg/mL indicam IA primária, enquanto valores baixos ou normais indicam IA secundária.
Cortisol entre 3 e 19 µg/dL: o ACTH será útil para orientar o diagnóstico e sugerir o melhor teste adicional para confirmá-lo ou excluí-lo; níveis de ACTH acima de 100 pg/mL sugerem IA primária, enquanto níveis normais ou baixos podem ser encontrados em pacientes com IA secundária parcial e em pessoas normais. Testes adicionais são necessários para se chegar a um diagnóstico.
Teste da cortrosina com 250 µg
Para avaliar a reserva funcional adrenal, utiliza-se um análogo do ACTH (cortrosina, peptídeo sintético composto pelos primeiros 24 aminoácidos da molécula do ACTH). Neste teste, avalia-se a resposta adrenal através da dosagem de cortisol nos tempos 30 e/ou 60 minutos após estímulo com 250 µg de cortrosina (teste padrão com administração intravenosa ou intramuscular). Indivíduos normais apresentam frente a esse estímulo níveis de cortisol maiores que 18 µg/dL.
Teste de tolerância a insulina (insulin tolerance test, ITT)
É tido como o teste padrão-ouro na avaliação da suspeita de Insuficiência adrenal secundária, uma vez que a hipoglicemia é um potente ativador do eixo hipotálamo-hipófise-adrenal. O teste envolve a administração intra-venosa de insulina regular na dose de 0,1-0,15 UI/kg de peso, com aferição da glicemia e cortisol nos tempos 0, 30, 45, 60, 90 e 120 minutos. Hipoglicemia efetiva (glicemia < 40 mg/dL) com sinais de neuroglicopenia é necessária para que o teste seja adequado. Considera-se normal um pico de cortisol acima de 18 µg/dL. O ITT não deve ser realizado em coronariopatas (sempre realize um ECG antes do teste), pacientes com epilepsia e quando se suspeita de IA grave (cortisol basal < 6,5 µg/dL) e deve ser sempre realizado na presença de um médico e tendo o paciente uma veia canulada para pronta administração de glicose se houver necessidade.
Teste da cortrosina com 1 ou 250 µg
O teste da cortrosina pode ser utilizado como alternativa ao ITT por ser mais simples, não necessitar a presença de um médico para a realização do exame, e por não expor o paciente aos riscos da hipoglicemia. O racional do teste da cortrosina na suspeita de Insuficiência adrenal secundária reside no fato de que ocorre atrofia da glândula e down-regulation dos receptores adrenais de ACTH frente à ausência de estimulação por este hormônio; assim a adrenal perderia a capacidade de responder ao estímulo agudo do ACTH exógeno com incremento na produção de cortisol. Dessa maneira, é um teste inadequado para pacientes com Insuficiência adrenal secundária de instalação recente (como na apoplexia ou no pós-operatório de cirurgia hipofisária), uma vez que a adrenal demora de 2 a 3 semanas para deixar de responder ao ACTH exógeno. A literatura mostra excelente correlação entre o pico de resposta de cortisol atingido com o ITT e com o teste da cortrosina nos pacientes com Insuficiência adrenal secundária. Entretanto, apesar dessa correlação ser alta, alguns pacientes apresentam resposta adequada do cortisol no teste da cortrosina padrão (250 µg) e resposta inadequada no ITT, o que mostra que o teste da cortrosina padrão teria menor sensibilidade que o ITT. Uma consideração importante é a de que a administração de 250 µg de ACTH corresponde a um estímulo supra-fisiológico maciço, muito superior à máxima capacidade de estimulação endógena, o que poderia fazer com que a adrenal respondesse com incremento na secreção de cortisol no teste pela super-estimulação, algo incompatível com a reserva adrenocortical real. Assim, uma opção para melhorar a sensibilidade do teste seria administrar doses menores de ACTH. De fato, alguns trabalhos mostram que o teste com dose baixa de ACTH (teste da cortrosina com 1 µg) tem maior sensibilidade para a detecção de pacientes com Insuficiência adrenal secundária. Assim, na avaliação da suspeita de Insuficiência adrenal secundária, pode-se utilizar o teste com 1 µg de cortrosina (que mesmo assim ainda corresponde a um estímulo supra-fisiológico), pela maior sensibilidade sugerida em alguns estudos, com valor de corte de 18 µg/dL com cortisol colhido 30 e/ou 60 min após estímulo. Ressalve-se que o preparo da dose de 1 µg é algo trabalhoso e sujeito a erros operacionais (envolve a diluição de 1 mL de cotrosina [250 µg/mL] em 249 mL de solução salina, homogeneização e retirada de uma alíquota de 1 mL da solução). Assim, na ausência de equipe de enfermagem treinada, é mais conveniente fazer o teste com 250 µg.
A metirapona (Metapirona®) bloqueia a síntese de cortisol, inibindo a enzima 11ß-hidroxilase, que converte 11-desoxicortisol em cortisol. Pelo bloqueio, ocorre aumento do ACTH e dos níveis de 11-desoxicortisol. Uma resposta normal consiste de um nível de ACTH > 100 pg/mL e um nível de 11-desoxicortisol > 7 µg/dL medidos às 8 horas da manhã após administração de 30 mg/kg de metirapona à meia-noite anterior (dose máxima de 3 g). Na prática, trata-se de um teste pouco utilizado pois a droga não é disponível no nosso meio.
Investigação etiológica
Uma vez feito o diagnóstico de IA primária ou secundária, parte-se para a investigação etiológica. Dados de anamnese e exame físico serão fundamentais para nortear o diagnóstico. Abaixo seguem alguns testes úteis para estabelecer a etiologia da IA.
IA primária
1. Anticorpos contra a adrenal e avaliação de outras doenças auto-imunes
Pacientes com adrenalite auto-imune apresentam auto-anticorpos contra córtex adrenal ou anticorpos anti-21-hidroxilase em mais de 80% dos casos no início do quadro. A determinação desses anticorpos auxilia na definição etiológica principalmente naqueles pacientes com Insuficiência adrenal isolada sem doença auto-imune concomitante e sem história familiar da doença. Como cerca de 60% dos casos de doença de Addison ocorre em associação com outras doenças auto-imunes (configurando uma síndrome polioglandular auto-imune), recomenda-se o rastreamento para hipotireoidismo e diabetes dosando-se TSH e glicemia de jejum.
2. Ácidos graxos de cadeia muito longa (VLCFA)
A adrenoleucodistrofia (ALD) deve ser afastada em pacientes do sexo masculino com Insuficiência adrenal primária isolada, sem evidências de adrenalite auto-imune ou de outras causas primárias. A ALD pode se apresentar inicialmente sem manifestações neurológicas. O diagnóstico é feito com determinação dos níveis plasmáticos de ácidos graxos de cadeia muito longa (cadeia com 24 carbonos ou mais). Entretanto, trata-se de exame pouco disponível na rotina laboratorial.
3. Exames de imagem adrenal
São dispensados nos pacientes com diagnóstico firmado de adrenalite auto-imune ou doença de Addison associada à adrenoleucodistrofia. Se há suspeita de infecção, hemorragia, infiltração ou doença neoplásica deve-se realizar tomografia computadorizada de abdome para avaliação da morfologia das adrenais. Na adrenalite tuberculosa, ocorre aumento das adrenais na fase subaguda, e mais tardiamente se desenvolvem calcificações, que podem ser vistas em radiografia de abdome em cerca de 50% dos casos. Calcificações também podem ocorrer em pacientes com outras doenças invasivas ou hemorrágicas da adrenal.
IA secundária
4. Avaliação de outros setores hipofisários
É importante que os outros hormônios da adeno-hipófise sejam avaliados, pois raramente ocorre disfunção exclusiva do setor corticotrófico. Os testes utilizados para esta finalidade estão descritos no capítulo sobre hipopituitarismo.
5. Exames de imagem hipotálamo-hipofisária
A ressonância nuclear magnética da região hipotálamo-hipofisária é o método de escolha para se estudar a presença de tumores. Caso se evidencie imagem hipofisária compatível com adenoma, é importante ressaltar que apenas os adenomas maiores que 1 cm (macroadenomas) costumam causar déficit hormonal hipofisário. O achado incidental de imagem selar compatível com adenoma (incidentaloma) é relativamente freqüente (pode ocorrer em cerca de 10% dos indivíduos), e não justifica a deficiência investigada. A hipofisite linfocítica inicialmente apresenta aumento da hipófise passível de ser interpretado erroneamente como um tumor hipofisário, e posteriormente leva a atrofia hipofisária, podendo evoluir até uma imagem de sela vazia.
Glicocorticóides
Visando uma reposição hormonal que se aproxime do padrão fisiológico de secreção de cortisol, utilizam-se preferencialmente corticóides de curta duração em 2 ou 3 doses ao dia, com metade a 2/3 da dose total diária administrada pela manhã.
A produção de cortisol de um indivíduo normal varia entre 5 e 10 mg/m2 de superfície corpórea por dia, o que equivale à administração oral de 15-25 mg de hidrocortisona ou 25-37,5 mg de acetato de cortisona (para valores de equivalência de doses entre os fármacos, vide tabela 6). Em geral, em um esquema com duas doses diárias, administra-se a primeira dose ao acordar e a segunda dose cerca de 6 a 8 horas após a primeira.
Corticosteróides de meia-vida mais longa também podem ser utilizados na reposição (p. ex. 5 a 7,5 mg de prednisona ao dia); se por um lado eles podem resultar em níveis inadequadamente altos no período noturno, por outro lado favorecem a posologia através de uma única administração diária, o que pode ser desejável para alguns pacientes com dificuldade de adequação a um esquema com 2 ou 3 doses.
Tabela 6: Equivalência dos glicocorticóides
|
|
Duração da ação terapêutica |
Efeito Glicocorticóide (potência relativa) |
Dose equivalente (mg) |
Efeito mineralocorticóide (retenção relativa de sódio) |
Meia-vida plasmática (minutos) |
Meia-vida biológica (horas) |
|
Hidrocortisona |
Curta |
1 |
20 |
1 |
90 |
8-12 |
|
Cortisona |
Curta |
0,8 |
25 |
0,8 |
30 |
8-12 |
|
prednisona |
Intermediaria |
4 |
5 |
0,2 |
60 |
12-36 |
|
Prednisolona |
Intermediaria |
5 |
4 |
0,2 |
200 |
12-36 |
|
Metilprednisolona |
Intermediaria |
5 |
4 |
0 |
180 |
12-36 |
|
Triamcinolona |
Intermediaria |
5 |
4 |
0 |
300 |
12-36 |
|
Betametasona |
Prolongada |
25 |
0,5 |
0 |
100-300 |
24-72 |
|
Dexametasona |
Prolongada |
25 |
0,75 |
0 |
100-300 |
24-72 |
No seguimento desses pacientes, a adequação da dose de glicocorticóide é baseada essencialmente em critérios clínicos (avaliação da reversão dos sintomas de hipoadrenalismo e aumento na sensação de bem-estar), uma vez que não existe parâmetro laboratorial objetivo adequado para a titulação da dose de glicocorticóide.
O nível plasmático de ACTH, em pacientes com IA primária, é invariavelmente alto antes da dose da manhã e declina após a tomada do corticóide, de tal maneira que a busca de um nível de ACTH normal pela manhã levaria a uma reposição em excesso. Dessa maneira, não se recomenda a dosagem de ACTH no seguimento de pacientes com doença de Addison, a não ser que no acompanhamento o paciente volte a apresentar hiperpigmentação.
O cortisol urinário também não se presta à avaliação da adequação da dose, uma vez que os valores de referência para indivíduos normais não se aplicam a pacientes em reposição (após a tomada, os níveis séricos de cortisol se elevam, saturando a CBG, resultando em aumento da excreção renal). Entretanto, em casos de suspeita de não-aderência à terapêutica, a medida do cortisol urinário pode eventualmente ser útil.
Assim, deve-se buscar níveis de reposição compatíveis com a faixa de produção endógena acima apresentada, visando resolução dos sintomas e melhora na qualidade de vida, levando em conta sinais e sintomas que possam sugerir reposição deficiente (fadiga, falta de energia, astenia, náusea, emagrecimento, hiperpigmentação, hipotensão arterial) ou excessiva (ganho de peso, desenvolvimento de características cushingóides como obesidade central, hipertensão arterial, edema periférico, hipocalemia). Reposição inadequadamente baixa aumenta o risco de crise adrenal e limita a qualidade de vida, enquanto que reposição excessiva pode causar morbidade significativa (intolerância à glicose, diabetes, obesidade, osteoporose).
Em situações de estresse, a dose deve ser aumentada. Pacientes devem ser orientados a adicionar dose equivalente a 5-10 mg de hidrocortisona ao seu esquema habitual pouco antes de realizar atividade física extenuante. Em caso de febre, orienta-se dobrar a dose habitual até a recuperação. Em caso de vômitos ou diarréia, a reposição deve ser administrada por via parenteral. Para cirurgias de grande porte, trauma e doenças críticas, deve-se administrar 100 a 200 mg hidrocortisona por dia, através de infusão contínua ou em doses divididas a cada 6 horas. Para procedimentos cirúrgicos de pequeno a médio porte, doses de até 75 mg de hidrocortisona por dia são suficientes. Em caso de crise adrenal, recomenda-se administração imediata de 100 mg de hidrocortisona seguida de 100-200 mg/24h e reposição volêmica vigorosa visando compensação clínica. Pacientes e familiares devem ser instruídos sobre como proceder frente a situações de estresse ou doença, e o paciente deve levar consigo um cartão informando sobre o seu diagnóstico e sobre como proceder em situações emergenciais. A tabela 7 mostra recomendações de suplementação em situações de estresse.
Tabela 7: Recomendações de suplementação da reposição de glicocorticóide em situações de estresse.
|
Nível de estresse (clínico ou cirúrgico) |
Dose recomendada |
|
Menor (p. ex. cirurgia de hérnia inguinal, colonoscopia, doença febril sem outras repercussões clínicas, gastroenterite, náuseas e vômitos que não levem à desidratação) |
Procedimentos cirúrgicos ou diagnósticos: administrar 25 mg de hidrocortisona no dia do procedimento. Intercorrências clínicas: dobrar a dose habitual de reposição. Considerar reposição parenteral no caso de vômitos e/ou diarréia. |
|
Moderado (p. ex. colecistectomia aberta, hemicolectomia, doença febril com comprometimento do estado geral, pneumonia, gastroenterite severa) |
Procedimentos cirúrgicos: 50-75 mg de hidrocortisona IV no dia do procedimento com retorno à dose usual em 2 dias. Intercorrências clínicas: manter a dose acima até recuperação clínica. |
|
Severo (cirurgias de grande porte – cirurgia cardiovascular, gastroduodenopancreatectomia, hepatectomia, pancreatite aguda) |
Procedimentos cirúrgicos: 100-200 mg de hidrocortisona IV no dia do procedimento com retorno à dose usual em 2 dias. Intercorrências clínicas: manter a dose acima até recuperação clínica. |
|
Crise adrenal |
Administração imediata de 100 mg de hidrocortisona seguida de 100-200 mg/dia |
|
Doença crítica: Insuficiência adrenal relativa e choque séptico (vide texto) |
200 a 300 mg hidrocortisona/dia IV contínuo ou doses divididas por 5 a 7 dias preferencialmente com desmame por 5 a 7 dias. Fludrocortisona 50 µg/dia é opcional*. |
*Pode-se administrar tal esquema a todos os pacientes com choque séptico ou realizar teste com 250 µg de cortrosina antes de se iniciar a reposição, interrompendo a administração de corticóide naqueles que responderam ao teste (vide texto).
Como os hormônios tireoideanos aumentam o clearance de cortisol, pacientes com Insuficiência adrenal com hipertireoidismo descompensado devem ter a dose de reposição dobrada ou triplicada, e para se evitar crise adrenal, reposição de levotiroxina para hipotireoidismo deve ser iniciada após o tratamento da hipofunção adrenal ou após a possibilidade de Insuficiência adrenal ter sido afastada.
Durante a gravidez normal ocorre elevação gradual nos níveis de CBG e, no último trimestre, o cortisol livre também se eleva. Desta forma, recomenda-se elevação de 50% na dose de manutenção no terceiro trimestre. No período periparto, a estratégia de reposição segue a de cirurgia de grande porte, ou seja, 100 mg de hidrocortisona em 24 horas iniciando com o trabalho de parto até 48 horas após o parto, seguido de desmame rápido.
Caso o paciente necessite tratamento com fármacos que aumentem o clearance de cortisol, deve-se aumentar a dose de reposição de glicocorticóide. Para o tratamento de tuberculose com esquema utilizando rifampicina, a dose de reposição deve ser dobrada.
Mineralocorticóides
A reposição com mineralocorticóides é necessária apenas para pacientes com IA primária, através da administração de 50 a 200 µg por dia de fludrocortisona (Florinefe®) em tomada única pela manhã. A monitorização da dose se faz através da medida da pressão arterial, dos níveis de sódio e potássio séricos e visando uma APR entre o valor médio e superior da normalidade. Se o paciente desenvolver hipertensão arterial no seguimento, a dose de mineralocorticóide pode ser gradativamente reduzida, observando-se os níveis de sódio e potássio durante esse processo, a fim de evitar hiponatremia ou hipercalemia. Glicocorticóides também exercem efeito mineralocorticóide (tabela 5), e a depender da dose e do tipo de corticóide utilizado o uso de um mineralocorticóide pode ser dispensado visto que o efeito mineralocorticóide exercido pelo glicocorticóide pode ser suficiente. Exemplificando, 20 mg de hidrocortisona têm uma ação equivalente a 50 µg de fludrocortisona, e habitualmente para doses maiores que 50 mg de hidrocortisona a reposição de fludrocortisona pode ser descontinuada.
Na gestação, a elevação dos níveis de progesterona exerce ação anti-mineralocorticóide, e a reposição de fludrocortisona deve ser ajustada baseada nos níveis pressóricos e de potássio. Como a APR aumenta fisiologicamente durante a gestação, ela não deve ser utilizada com a finalidade de ajuste da dose do mineralocorticóide.
Andrógenos adrenais
Estudos recentes têm sugerido que a reposição de DHEA (25-50 mg/dia) em mulheres com Insuficiência adrenal primária ou secundária poderia ser benéfica em termos de melhora da sensação de bem estar e da libido nessas pacientes. Entretanto, tal conduta é opcional visto que ainda não foi adequadamente apreciada em ensaios clínicos maiores. Não existe uma formulação comercial padronizada disponível para DHEA no Brasil.
A tabela 8 resume a estratégia de reposição hormonal para pacientes com Insuficiência adrenal.
Tabela 8: Resumo das reposições na Insuficiência adrenal
|
Reposição de glicocorticóide* Hidrocortisona 15-25 mg/dia ou acetato de cortisona 25-37,5 mg/dia Administrar em 2 a 3 doses com metade a 2/3 da dose total cedo ao acordar |
|
Reposição mineralocorticóide* (IA primária): Fludrocortisona 50-200 µg/dia em dose única matinal |
|
Reposição com andrógenos adrenais – DHEA em mulheres (opcional): DHEA 25-50 mg/dia em dose única matinal |
*para individualização da dose, vide texto
Para pacientes com Insuficiência adrenal decorrente do uso crônico de corticóides, a recuperação do eixo pode durar de semanas a anos, sendo necessária eventualmente terapia de reposição prolongada. O desmame abrupto, dependendo da circunstância clínica, pode desencadear sintomas de hipoadrenalismo, e eventualmente até crise adrenal. A tabela 9 mostra uma sugestão de esquema de desmame para esses pacientes, levando em conta o tempo e a dose utilizada.
Tabela 9: Sugestão de esquema para realização de desmame de corticóide baseado na dose e tempo de uso
|
Dose (prednisona, em mg/dia) |
Tempo de uso do glicocorticóide | ||
|
Curto (até 2 semanas) |
Intermediário (entre 2 semanas e 2 meses) |
Longo (mais de 2 meses) | |
|
Alta (40-100 mg/dia) |
Reduzir 50% da dose a cada 3-4 dias |
Reduzir 1/3 da dose a cada semana |
Reduzir 1/5 da dose a cada 2 semanas |
|
Média (15-40 mg/dia) |
Não há necessidade de redução gradual; retirada pode ser abrupta |
Reduzir 1/3 da dose a cada semana |
Reduzir ¼ da dose a cada 2 semanas |
|
Baixa (5-15 mg/dia) |
Não há necessidade de redução gradual; retirada pode ser abrupta |
Reduzir 1/3 da dose a cada 3-4 dias |
Reduzir ¼ da dose a cada semana |
Durante a doença crítica, ocorre um aumento na secreção de cortisol proporcional à gravidade da doença, e esse aumento parece ser um mecanismo adaptativo importante na regulação da resposta inflamatória e em outros processos, como sensibilização à ação das catecolaminas. A mortalidade em doentes críticos tem associação com os níveis de cortisol sérico elevados (reflexo da maior gravidade da doença) e baixos (fato coerente com a idéia de que glicocorticóides são importantes para a sobrevivência na doença crítica). Nesse contexto, torna-se difícil determinar qual a resposta adrenal adequada para determinado paciente crítico, que varia de acordo com a gravidade, com os níveis de proteína circulantes (na doença crítica ocorre redução da CBG com maior proporção de cortisol livre que é a fração bioativa, dificultando a interpretação dos níveis do cortisol total; a análise do cortisol livre é disponível apenas experimentalmente) e com o grau de resistência à ação do hormônio nos diferentes tecidos. Nesse contexto, o termo Insuficiência adrenal relativa foi cunhado para se referir àqueles pacientes que, apesar de não preencherem os critérios habituais de hipocortisolismo comentados neste capítulo e de terem eventualmente cortisol elevado em termos absolutos, poderiam se beneficiar da suplementação com corticóide exógeno.
Muitos fatores podem alterar a resposta adrenal durante a doença crítica, como fatores pré-existentes (doenças previamente diagnosticadas ou não que causam Insuficiência adrenal primária ou secundária; uso prévio de corticóides) ou fatores relacionados à doença crítica que reduzem a efetividade do eixo (trauma crânio encefálico, infarto e necrose hipofisária, drogas inibidoras da esteroidogênese usadas em terapia intensiva como etomidato, cetoconazol, fenitoína; hemorragia adrenal, inibição de síntese e aumento da resistência ao cortisol relacionada à sepse), de forma que tal resposta adrenal reduzida pode ser insuficiente e associar-se a um pior prognóstico. Pacientes com AIDS apresentam maior incidência de Insuficiência adrenal em unidades de cuidados intensivos por diversos motivos (infecções da adrenal, fármacos, aumento da resistência periférica à ação do cortisol), e nesses pacientes o limiar para testar o eixo deve ser menor.
Quando os sintomas clássicos de crise adrenal estão presentes, em um paciente com outros comemorativos clínicos de Insuficiência adrenal, como hiperpigmentação, o diagnóstico pode ser realizado mais facilmente, mas em situações menos extremas e com diversas manifestações de Insuficiência adrenal sendo comuns em pacientes críticos (como anorexia, náuseas, vômitos, diarréia, dor abdominal, febre, hipotensão) necessita-se de um maior índice de suspeita. Como hipoglicemia e eosinofilia são incomuns em doentes críticos, sua presença deve alertar para a possibilidade de hipoadrenalismo. A presença de instabilidade hemodinâmica persistente com padrão hiperdinâmico e resistência vascular baixa a despeito de reposição volêmica vigorosa, sem causa que justifique tal cenário, e que não responde às manobras terapêuticas instituídas, deve também alertar para a possibilidade de hipoadrenalismo.
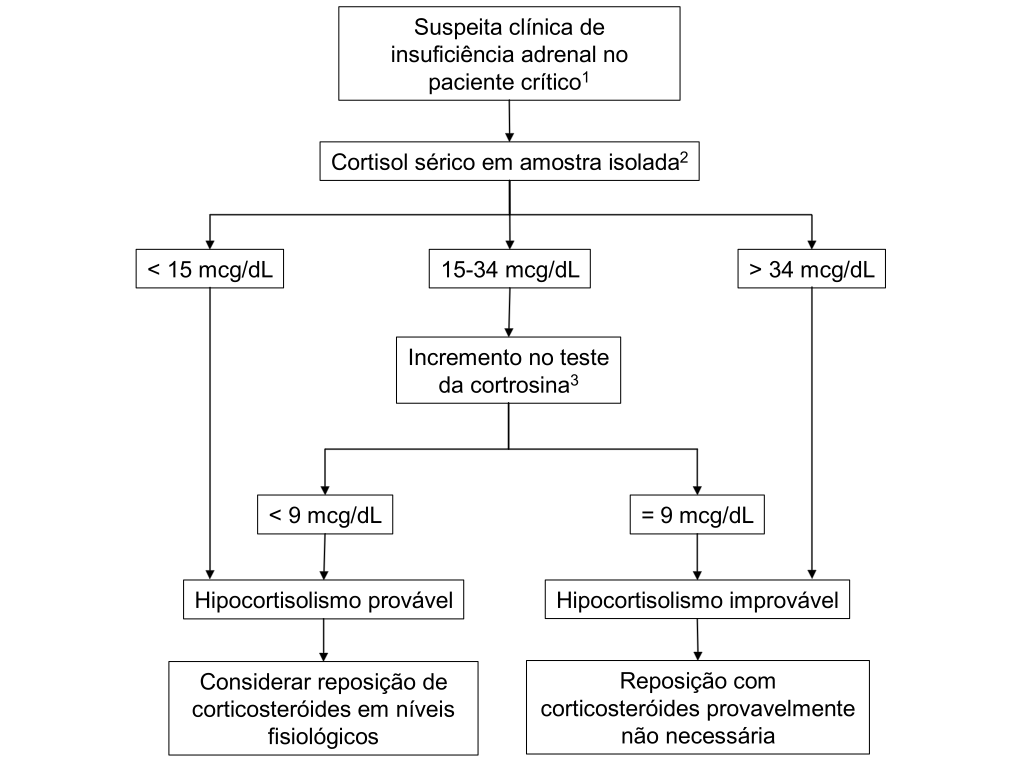
Na avaliação laboratorial, a análise do cortisol em amostra isolada pode auxiliar o diagnóstico, através da utilização de um limiar inferior abaixo do qual é provável a presença de Insuficiência adrenal relativa e um limiar superior acima do qual é improvável. Vários limiares foram propostos para pacientes críticos, e nenhum é plenamente adequado para a finalidade proposta, mas um valor inferior de 15 µg/dL e superior de 34 µg/dL parecem satisfatórios na avaliação daqueles que poderiam se beneficiar da reposição com corticóides.
O ITT não é indicado em pacientes críticos, restando o teste da cortrosina padrão com 250 µg como teste dinâmico de escolha nesses pacientes (o teste com 1 µg não foi validado nessas circunstâncias e não deve ser utilizado). A literatura mostra muita controvérsia a respeito da utilização e da interpretação desse teste em doentes críticos, e uma estratégia possível é a apresentada na Figura 3, onde o teste seria aplicado àqueles pacientes com cortisol basal entre 15 e 34 µg/dL. Um incremento no cortisol menor que 9 µg/dL está associado a pior prognóstico e sugere o diagnóstico de Insuficiência adrenal relativa e a necessidade de reposição. Outra estratégia mais ágil é a de realizar prontamente um teste da cortrosina ao invés de esperar os resultados do cortisol basal para se decidir a necessidade do teste dinâmico. Note que o diagnóstico de Insuficiência adrenal é realizado em termos de resposta incremental apenas em pacientes críticos; para os demais pacientes, utiliza-se o valor absoluto dos níveis de cortisol após o estímulo. Uma limitação importante do teste da cortrosina, já comentada, é que ele não pode ser realizado após insultos agudos à hipófise quando a adrenal ainda pode responder ao ACTH.
Figura 3: Algoritmo de investigação de Insuficiência adrenal em pacientes críticos.
Caso opte-se pela reposição de glicocorticóides, sugere-se uma dose de 200 mg de hidrocortisona por dia, em infusão IV contínua ou em doses divididas de 6 em 6 horas (tabela 6). Em pacientes com choque séptico dependente de drogas vasoativas, o uso de doses fisiológicas de hidrocortisona por 5 a 7 dias (até 300 mg por dia em infusão contínua ou em doses divididas) iniciado até 72 horas do uso de vasopressores, preferencialmente com desmame subseqüente (por 5 a 7 dias), mostrou aumentar a sobrevida e a taxa de reversão do choque. Na literatura há controvérsia se essa reposição deve ser administrada a todos os pacientes com choque séptico, e alguns preconizam que apenas os pacientes com Insuficiência adrenal absoluta ou relativa, diagnosticada pelo incremento de cortisol inferior a 9 µg/dL no teste da cortrosina, se beneficiariam de tal estratégia. Em um estudo administrou-se, junto à hidrocortisona, fludrocortisona na dose de 50 µg/dia, mas outro estudo recente chamado CORTICUS contestou estes resultados iniciais. Não existe aind consenso quanto à reposição de mineralocorticóides para pacientes com choque séptico, pois muitos acreditam que o efeito benéfico verificado deva-se basicamente ao glicocorticóide. Assim, o uso de mineralocorticóides no choque séptico é atualmente conduta opcional.
A Insuficiência adrenal deve sempre fazer parte do diagnóstico diferencial em situações de choque grave ou refratário, e o fato de poder ser causada pelas mais diversas etiologias faz com que todas especialidades médicas devam conhecer e saber diagnosticar e tratar.
A Insuficiência adrenal primária tem prevalência ao redor de 100 casos/milhão e incidência de 4 a 6 por milhão em população caucasiana
Os pacientes apresentam manifestações inespecíficas, passando em media em 3 a 4 médicos antes do diagnóstico correto ser estabelecido
No primeiro estágio da Insuficiência adrenal primaria ocorre comprometimento do setor meniralocorticóide, com aumento da renina plasmática e diminuição da aldosterona, mas raramente nesta fase com manifestações clínicas
É necessária a perda de cerca de 90% do córtex das adrenais para ocorrerem manifestações da Insuficiência adrenal
Cerca de 25% dos pacientes tem seu diagnóstico realizado na circunstância de uma crise addisoniana
Na Insuficiência adrenal secundaria a hiponatremia costuma ser mais pronunciada que na primária
Estes pacientes não costumam apresentar hipercalemia
A principal causa de Insuficiência adrenal nos paises desenvolvidos é a adrenalite auto-imune, com auto-anticorpos contra a adrenal presentes em cerca de 75% dos casos
A tuberculose ainda é uma das principais causas de Insuficiência adrenal nos paises menos desenvolvidos
A principal causa de Insuficiência adrenal secundaria é o uso prévio de glicocorticóides
Os sinais e sintomas nos pacientes com Insuficiência adrenal dependem da velocidade de instalação do quadro
A crise adrenal aguda costuma ocorrer na circunstância de algum insulto grave ao organismo como infecções e cirurgias
Hipotensão e sintomas gastrointestinais com diarréia e náuseas são muito prevalentes
A avidez por sal parece ser um sintoma relativamente específico
Hiperpigmentação sugere causa primaria de Insuficiência adrenal
O primeiro teste diagnóstico na insuficiência adrenalé a dosagem do cortisol, valores menores que 3 µg/dL são confirmativos da doença
O teste da cortrosina é necessário em pacientes em que os valores de cortisol estão entre 3 e 19 µg/dL.
Em pacientes com suspeita de insuficiênca adrenal secundaria, testes com baixa dose de cortrosina, teste da metirapona ou teste de tolerância à insulina podem ser necessários
Anticorpos contra 21-hidroxilase estão presentes no início do quadro em até 80% dos pacientes com adrenalite auto-imune ajudando no diagnóstico etiológico
Calcificações e aumento de tamanho da adrenal podem ocorrer em pacientes com tuberculose e outras doenças invasivas da adrenal
Outras deficiências hormonais hipofisárias são freqüentes em pacientes com Insuficiência adrenal secundária não obviamente associada com uso prévio de glicocorticóides, sendo portanto necessário a dosagem dos hormônios hipofisários
A dose usual de reposição de corticóide na Insuficiência adrenal é de 25-37,5 mg de acetato de cortisona, ou 15-25 mg de hidrocortisona, cortiçoides de maior duração como a prednisona podem ser utilizados em dose de 5-7,5 mg ao dia
A adequação da dose de reposição de glicocorticóides é baseada em critérios clínicos
Na crise adrenal a hidrocortisona pode ser administrada em bolo de 100 mg endovenoso, mantendo dose posteriormente de 100-200 mg ao dia
Alguns estudos mostraram benefício da reposição de glicorticóides em pacientes com choque séptico necessitando de drogas vasoativas
Alguns autores sugerem realizar a reposição apenas nos pacientes com cortisol menor que 15 µg/dL basal ou com resposta menor que 9 µg/dL no teste da cortrosina
1. Stewart, P.M. (2007) The adrenal cortex. In Williams Textbook of Endocrinology eds. H. M. Kronenberg, S. Melmed, K. S. Polonsly & P. R. Larsen). Saunders Elsevier, Philadelphia, PA, pp. 445-503.
2. Aron, D.C., Findling, J.W. & Tyrrell, J.B. (2004) Glucocorticoid & Adrenal Androgens. In Basic & Clinical Endocrinology eds. F. S. Greenspan & D. G. Gardner). McGraw-Hill, New York, NY, pp. 362-413.
3. Minneci, P.C., Deans, K.J., Banks, S.M., Eichacker, P.Q. & Natanson, C. (2004) Meta-Analysis: The Effect of Steroids on Survival and Shock during Sepsis Depends on the Dose. Ann Intern Med 141, 47-56.
4. Hamrahian, A.H., Oseni, T.S. & Arafah, B.M. (2004) Measurements of Serum Free Cortisol in Critically Ill Patients. N Engl J Med 350, 1629-1638.
5. Arlt, W. & Allolio, B. (2003) Adrenal insufficiency. Lancet 361, 1881-1893.
6. Dorin, R.I., Qualls, C.R. & Crapo, L.M. (2003) Diagnosis of Adrenal Insufficiency. Ann Intern Med 139, 194-204.
7. Cooper, M.S. & Stewart, P.M. (2003) Corticosteroid Insufficiency in Acutely Ill Patients. N Engl J Med 348, 727-734.
8. Axelrod, L. (2003) Perioperative management of patients treated with glucocorticoids. Endocrinol Metab Clin North Am 32, 367-383.
9. Coursin, D.B. & Wood, K.E. (2002) Corticosteroid Supplementation for Adrenal Insufficiency. JAMA 287, 236-240.
10. Ten, S., New, M. & Maclaren, N. (2001) Addison's Disease 2001. J Clin Endocrinol Metab 86, 2909-2922.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.