(Carregando Índice)... (Carregando Índice)... |
Última revisão: 06/08/2014
Comentários de assinantes: 0
Paciente do sexo masculino, 7 meses e 15 dias de idade, branco, referendado à consulta devido à irritabilidade, febre, diurese abundante e má evolução ponderal. A gestação cursou com polidrâmnio desde o 4º mês e necessitou de cinco amniocenteses. Nasceu de parto cesáreo, com idade gestacional de 32 semanas, devido a sofrimento fetal, com peso de 1.090 g.
Ao ser admitido no serviço médico o paciente apresentava-se desnutrido, desidratado, com distensão abdominal, peso de 5.450 g, comprimento de 60 cm, perímetro cefálico de 42 cm, pressão arterial de 80/40 mmHg, fácies sugestiva (face com forma triangular, olhos grandes e orelha de abano).
O exame bioquímico revelou níveis elevados de renina e aldosterona; potássio sérico de 2 mEq/L; bicarbonato de 18,2 mEq/L; pH urinário de 8,0; fração de excreção de sódio (FENa) de 1,09 mL/dL do filtrado glomerular (FG); fração de excreção de potássio (FEK) de 29,9 mL/dL FG; fração de excreção de claro (FECl) de 3,0 mL/dL FG. A ultrassonografia mostrou a presença de nefrocalcinose.1
As tubulopatias hereditárias são entidades raras e difíceis de se diagnosticar devido ao grande número de alterações eletrolíticas. As alterações genéticas incluem mutações em diversos genes que codificam as proteínas dos diferentes transportadores tubulares. As condições de mutações monogênicas são raras, mas algumas doenças podem envolver alterações genéticas complexas com a participação de diferentes poliforfismos em diversos genes que contribuem para o fenótipo da doença.2-5
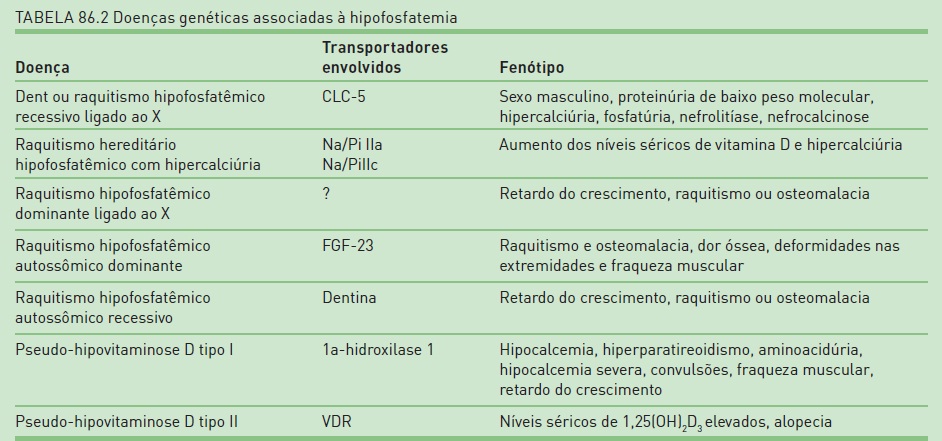
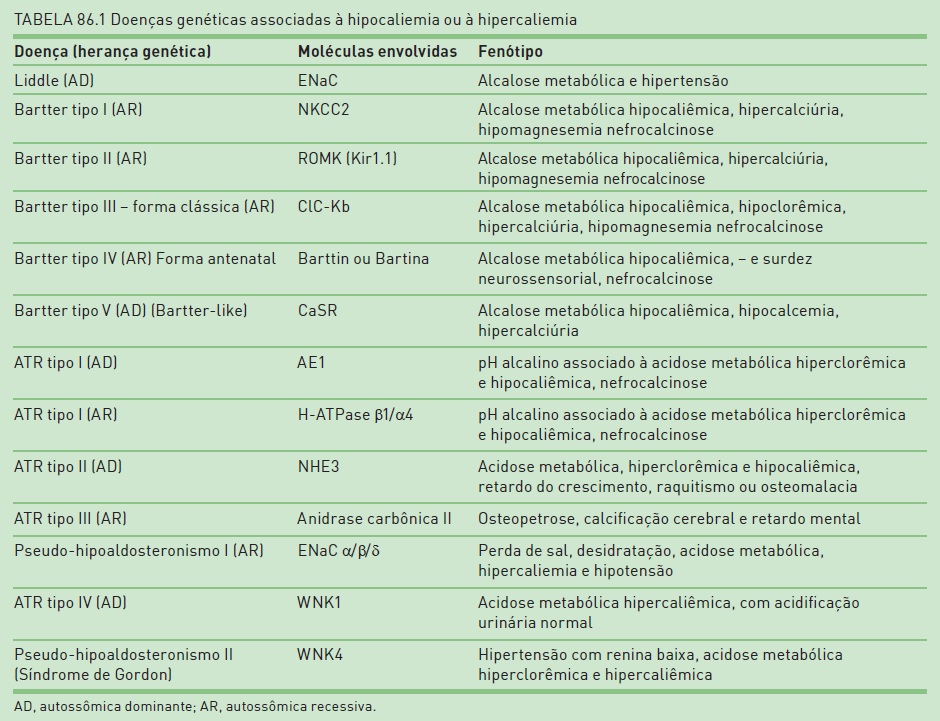
Neste capítulo, serão abordados os distúrbios genéticos que resultam em alterações no transporte tubular de sódio, potássio, fósforo, cálcio e magnésio e como esses distúrbios se apresentam fenotipicamente; assim, o defeito genético será discutido a partir das manifestações clínicas mais relevantes, incluindo as que cursam com hipo ou hipercaliemia (Tab. 86.1), hipofosfatemia (Tab. 86.2), hipo ou hipercalcemia (Tab. 86.3) e hipomagnesemia (Tab. 86.4).
Em 1962, Frederic Bartter publicou seu trabalho descrevendo dois pacientes com alcalose metabólica hipocalêmica, hiperaldosteronismo, pressão normal e hiperplasia do aparelho justaglomerular.4 Em 1966, Gitelman e colaboradores descreveram uma doença familiar caracterizada por hipocaliemia, hipomagnesemia, hipocalciúria, alcalose metabólica e hiperaldosteronismo hiper-reninêmicao.5 Mais estudos mostraram que a síndrome de Gitelman era causada por mutações nos genes que codificavam os transportes nos canais de NaCl e magnésio nos segmentos corticais do néfron distal sensíveis a tiazídicos.
A prevalência estimada da síndrome de Gitelman é de 25 casos por milhão de habitantes, enquanto a prevalência de sujeitos heterozigóticos é de ~ 1% na população branca.5
As síndromes tubulares, em especial a de Gitelman e Bartter, são distúrbios genéticos induzidos por mutações em genes que codificam os transportadores dos canais de Na, Cl, Ca e Mg na membrana apical das células corticais dos túbulos renais.2,4,5
O manuseio tubular de sódio, potássio, cálcio, magnésio e cloro depende de uma adequada atividade molecular em porções específicas de diferentes segmentos dos túbulos renais, em particular: túbulos proximais, alça ascendente espessa de Henle e segmentos distais. Mutações em diferentes genes provocam síndromes associadas a alterações eletrolíticas e acidobásicas com diferentes fenótipos. A patogênese dessas síndromes será mais bem descrita adiante.3-5
Muitos pacientes com síndromes tubulares renais podem ser assintomáticos, mas é frequente a presença de diferentes sinais dependendo do tipo de alteração genética. Por exemplo, na síndrome de Bartter antenatal , podem-se observar:
•Polidrâmio entre 24 e 36 semanas de gestação
•No líquido amniótico: sódio, potássio e prostaglandinas normais
•Cloro elevado no líquido amniótico
•Prematuridade
No paciente adulto, pode-se observar fraqueza, além de:
•Alcalose metabólica
•Hipocaliemia
•Hipotensão
•Parestesias
As diferentes síndromes tubulares apresentam uma variedade de achados clínicos com diagnósticos laboratoriais e tratamentos específicos. Por exemplo, na síndrome de Gitelman, é observada, nos adultos jovens com desenvolvimento normal, uma redução da capacidade de trabalho. Muitos apresentam hipotensão arterial, fraqueza, parestesias e paralisia hipocalêmica. Como são inúmeras as doenças tubulares renais, as características clínicas, o diagnóstico, a patogênese e o tratamento das principais serão apresentados, com mais detalhes, a seguir.
Definição
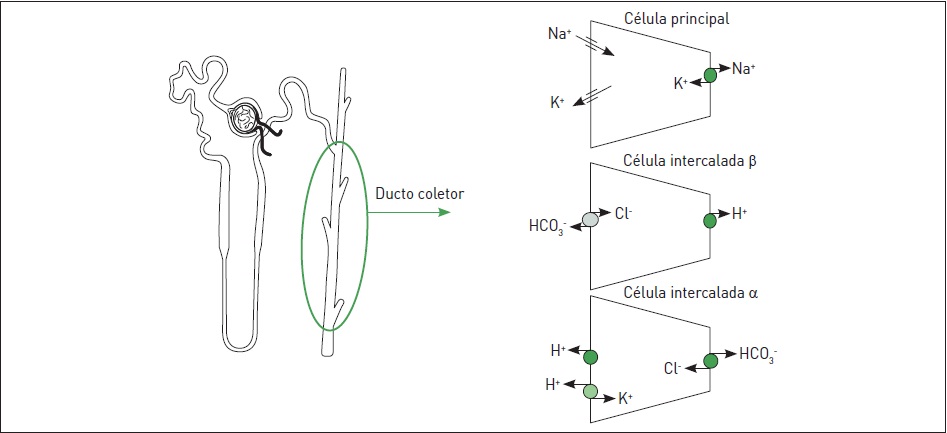
A síndrome de Liddle é definida como um raro distúrbio autossômico dominante caracterizado por um defeito primário na reabsorção de Na+ pelos ductos coletores.2 A Figura 86.1 sumariza as principais vias de transporte do ducto coletor, destacando-se que a reabsorção de sódio, nessa região, ocorre primariamente através do canal epitelial de Na+ (ENaC).
Etiologia
Essa síndrome ocorre por mutações nos genes que codificam as subunidades ß e Y do canal ENaC, resultando na hiperatividade do canal e, portanto, na excessiva reabsorção de Na+ no ducto coletor, criando um gradiente elétrico negativo no lúmen tubular, o que leva as células principais a secretar K+, e as células intercaladas, através da bomba de prótons, a secretar H+.
Sinais e sintomas/diagnóstico
A história familiar de pacientes com hipertensão em idade jovem e hipocaliemia sugere a síndrome de Liddle. Complicações cardiovasculares e cerebrovasculares pela hipertensão são comuns e frequentes causas de morte em pacientes não diagnosticados e não tratados. Os diagnósticos diferenciais incluem hiperplasia congênita de suprarrenal, resistência familiar ao cortisol, tumor de suprarrenal e síndrome do excesso aparente de mineralocorticoide.
O fenótipo dessa doença se caracteriza por alcalose metabólica, hipocaliemia e hipertensão. Ocorre em pacientes jovens, e a hipocaliemia pode não estar presente na apresentação. A retenção de Na+ leva a uma expansão de volume, que inibe a secreção de renina e a secreção de aldosterona e promove hipertensão arterial sistêmica, caracterizando-se por uma rara forma de hipertensão com renina baixa.3
Patogênese
O desenvolvimento de alcalose metabólica, hipocaliemia e hipertensão, típico dessa doença, pode simular um estado de pseudo-hiperaldosteronismo, pois o paciente tem hiporreninemia e baixas taxas de secreção de aldosterona, não respondendo, portanto, à espironolactona.

Tratamento
O tratamento baseia-se na administração de diuréticos poupadores de K+ , que fecham diretamente os canais de Na+, como a amilorida e o triantereno, associados à restrição de sal na dieta, para normalizar a pressão, reverter a perda urinária de K+ e a hipocaliemia.
Definição/patogênese
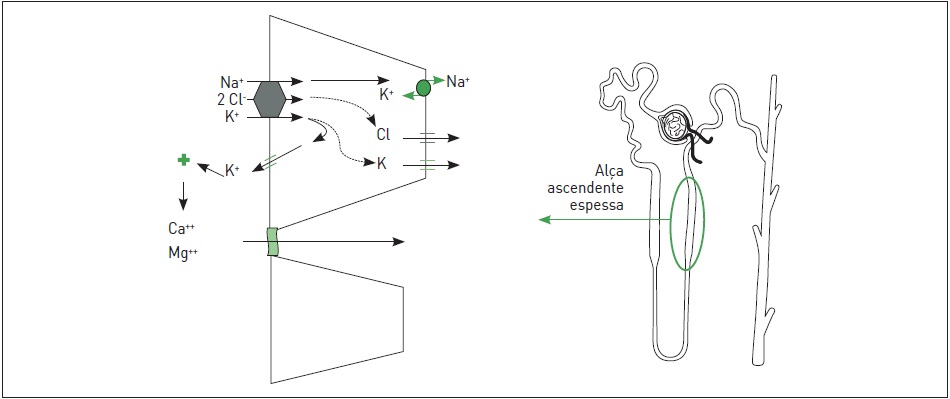
A síndrome de Bartter é uma tubulopatia caracterizada pela redução do transporte de sódio e cloro na porção ascendente espessa da alça de Henle. Conforme esquematizado na Figura 86.2, a reabsorção de sódio e cloro nessa região é feita através do cotransportador Na/K/2Cl (NKCC2), inibido pelos diuréticos de alça. O cloro reabsorvido via NKCC2 é transferido para o interstício através de canais para cloro (ClC-Kb) presentes na membrana basolateral da célula tubular. Um aspecto desse mecanismo de reabsorção é que parte do potássio reabsorvido é devolvido para o lúmen através de canais específicos para potássio (ROMK ou Kir1.1), o que torna a luz tubular positiva em relação à célula, sendo esse mecanismo essencial para a reabsorção passiva de cátions, principalmente Ca++ e Mg++ por via para celular, por simples repulsão de cargas.
Classificação
Esse mecanismo explica uma das características da síndrome de Bartter, que é a hipercalciúria. Mutações em qualquer um desses transportadores resultam em manifestações típicas da síndrome de Bartter, que pode ser classificada em cinco tipos, dependendo da molécula afetada:
•BarttertipoI– mutações no cotransportador Na/K/2Cl.
•Bartter tipo II – mutações no canal ROMK, responsável pela recirculação de K+.
•Bartter tipo III ou Bartter clássica – mutações nos canais de cloreto (ClC-Kb).
•Bartter tipo IV ou Bartter antenatal – também denominada síndrome da hiperprostaglandina E, com mutações na bartina (Barttin), que atua como uma subunidade ß dos canais de cloro CLC-Ka e CLC-Kb (3).
•Bartter tipo V – pacientes com doença familiar hipocalcêmica com hipercalciúria podem apresentar o fenótipo de Bartter, também chamado de Bartter-like.
A redução no transporte tubular na alça ascendente resulta em aumento na carga de sódio e cloro que atinge as porções mais distais do néfron. O aumento de fluido no ducto coletor aumenta a secreção de potássio e hidrogênio, resultando em alcalose metabólica hipocalêmica. Por outro lado, o prejudicado transporte de sódio e cloro na porção espessa da alça de Henle causa um déficit na recirculação do potássio célula-lúmen e, consequentemente, na redução do gradiente elétrico lúmen positivo, que normalmente direciona a reabsorção paracelular de cálcio e magnésio, levando a um aumento na excreção urinária desses cátions1.
647
Figura 86.1
Principais mecanismos de transporte do ducto coletor.
Sinais e sintomas/diagnóstico
O paciente apresenta polidipsia e poliúria. A pressão arterial é normal ou baixa, apesar de um aumento da atividade da renina plasmática, porque há aumento da resistência vascular ao efeito pressórico da angiotensina.5 Bartter tipo IV ou antenatal é a forma mais grave, cursando com polidrâmnio, devido à excessiva poliúria intraútero, prematuridade e surdez neurossensorial. No período perinatal, há febre, vômitos, diarreia, marcada hipercalciúria e nefrocalcinose, podendo evoluir para falência renal.2
A hipercalciúria é uma característica secundária da síndrome de Bartter. O fenótipo é semelhante ao da administração crônica de diuréticos de alça, tipo furosemida.
A síndrome de Bartter clássica (tipo III) geralmente aparece na infância e frequentemente está associada a retardo mental ou a retardo do crescimento. Há alcalose metabólica hipocalêmica, hipoclorêmica, e o magnésio geralmente encontra-se normal ou discretamente reduzido. Por razões não bem esclarecidas, há aumento da produção e da excreção urinária de prostaglandinas. Por outro lado, a consequente perda de sal e água na urina resulta em ativação do sistema renina-angiotensina-aldosterona. A atividade da renina está aumentada, e, no exame anatomopatológico, a hiperplasia do aparelho justaglomerular é um achado típico.
Tratamento
Como o defeito tubular não pode ser corrigido, o tratamento baseia-se em minimizar os efeitos secundários do aumento de prostaglandina e aldosterona. A indometacina tem mostrado bons resultados. Diuréticos poupadores de K+, como espironolactona e amilorida, devem ser associados em doses que podem chegar até 300 mg e 40 mg ao dia respectivamente. A suplementação com cloreto de potássio faz-se necessária.5
Figura 86.2
Principais mecanismos de transporte na porção espessa da alça ascendente de Henle.
Definição
A síndrome de Gitelman, ou variante hipocalciúrica hipomagnesêmica da síndrome de Bartter, considerada como uma forma mais branda, é uma tubulopatia causada por mutações no gene que codifica o cotransportador sódio-cloro, sensível aos diuréticos tiazídicos localizado no túbulo distal.6
Sinais e sintomas/diagnóstico
Geralmente aparece na infância tardia (após os 6 anos) ou até na fase adulta. Caracteriza-se por alcalose metabólica hipocalêmica, hipomagnesêmica e hipocalciúrica. O paciente pode apresentar poliúria, polidipsia, desidratação, hipotensão e, em alguns casos, vômitos. A inibição do cotransportador sódio-cloro no túbulo distal resulta na redução da reabsorção desses íons. Semelhante à síndrome de Bartter, o aumento da demanda de sódio para o ducto coletor resulta em maior secreção de potássio; além disso, o aumento na produção de aldosterona, como resultado da depleção de volume via sistema renina-angiotensina, provoca um aumento adicional da secreção de potássio e também de hidrogênio no ducto coletor (Fig. 86.1), resultando em alcalose metabólica hipocalêmica. Como essa é uma forma mais branda de tubulopatia, essas alterações não são suficientes para estimular a produção sistêmica e renal de prostaglandinas e, portanto, a excreção urinária de prostaglandinas mantém-se normal.
Patogênese
A inibição do cotransportador Na/Cl estimula a reabsorção de cálcio por um mecanismo semelhante ao efeito do tiazídico, resultando em hipocalciúria. Esse mecanismo ainda não está totalmente elucidado, mas parece que o decréscimo da reabsorção de Na+ no túbulo distal estimula o trocador Na/Ca na membrana basolateral, com consequente aumento da reabsorção de cálcio na membrana apical. Além disso, o decréscimo na concentração de Clintracelular aumenta a polaridade da membrana apical, estimulando também a reabsorção de cálcio através de canais de cálcio. Mais recentemente, foi demonstrado que a hipocalciúria também é decorrente da contração de volume extracelular e consequente aumento do transporte de cálcio no túbulo proximal.7 A hipomagnesemia provavelmente seja consequência de redução na reabsorção de magnésio no túbulo distal, o qual é equipado com canais para Mg++, que se encontram reduzidos, tanto na síndrome de Gilteman quanto na vigência de uso de diuréticos tiazídicos.6
Tratamento
O tratamento da síndrome de Gitelman consiste em minimizar a hipomagnesemia com reposição de cloreto de magnésio. O estado acidobásico e a excreção de cálcio tendem a normalizar. Para a correção da hipocaliemia, devem ser administrados sais de potássio ou ainda diuréticos poupadores de potássio, como espironolactona e amilorida
Definição
É uma doença caracterizada pela incapacidade do néfron distal de secretar íons hidrogênio na urina (Fig. 86.1 ).
Etiologia/classificação
A acidose tubular renal (ATR) distal pode ser um distúrbio primário ou secundário. A ATR distal secundária pode ocorrer nas doenças autoimunes, mais frequentemente em mulheres acima dos 40 anos, acompanhando quadros de síndrome de Sjögren, lúpus eritematoso sistêmico, doenças autoimunes hepáticas ou tireoidianas. Pode também ser ocasionada por drogas como anfotericina B, foscarnet, meticilina, ciclosporina, lítio, amilorida, trimetoprima e abuso de analgésicos.8
Patogênese
Nas células intercaladas tipo a do ducto coletor, a disfunção pode estar na bomba H+ ATPase, que secreta prótons, ou no trocador de ânions Cl-/HCO3-, também chamado de AE1 (anion exchanger 1), presente na membrana basolateral. A forma primária familiar pode ser de herança autossômica dominante com mutações no gene que codifica o trocador Cl-/HCO3- (AE1) ou de herança autossômica recessiva com mutações nos genes que codificam respectivamente as subunidades ß1 e a4 da H+ ATPase.9
Sinais e sintomas
A ATR distal, em sua forma clássica, é caracterizada pela presença de um pH inapropriadamente alcalino mediante um quadro de acidose metabólica hiperclorêmica e hipocalêmica. Clinicamente, em alguns casos, a ATR tipo I pode se associar a déficit de crescimento, osteomalacia ou raquitismo, fraqueza muscular, perda auditiva neurossensorial e progressiva falência renal.10 A associação com nefrocalcinose ou nefrolitíase é mais frequente, devido à presença de hipercalciúria e hipocitratúria.11 A ATR distal pode se apresentar de forma incompleta, sem acidemia espontânea, onde o defeito na acidificação urinária só se torna evidente com teste provocativo de sobrecarga ácida, geralmente realizado com sobrecarga de cloreto de amônio.12
Tratamento
O tratamento baseia-se na alcalinização por meio de reposição de bicarbonato de sódio ou citrato de sódio ou potássio, 1 a 2 mEq/kg/dia para adultos e 4 a 8 mEq/kg/dia para crianças. Dá-se preferência ao uso de citrato de potássio, pois, assim, a hipocaliemia é também controlada.8
Definição
A acidose tubular renal proximal é caracterizada pela incapacidade do túbulo proximal de reabsorver HCO3-. Da mesma maneira que a ATR distal, existe a forma primária e a secundária.
Etiologia/classificação/patogênese
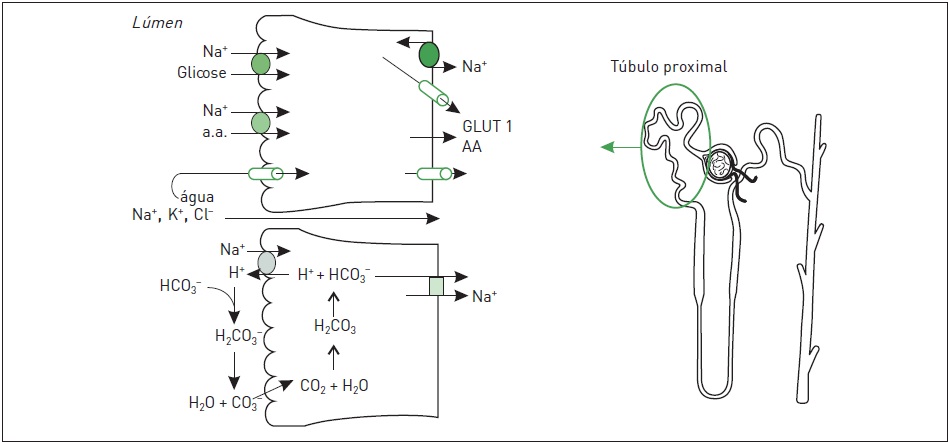
A acidose tubular renal proximal primária pode estar associada a outros defeitos de reabsorção tubular proximal, como a síndrome de Fanconi (menor reabsorção de aminoácidos, glicose e fósforo). Um resumo dos principais mecanismos de transporte no túbulo proximal está representado na Figura 86.3.
As formas secundárias se associam ao uso de drogas (acetazolamida, tetraciclina vencida, aminoglicosídeos, ácido valproico, chumbo, cádmio e mercúrio) ou à presença de outras condições clínicas ou doenças renais ou sistêmicas, tais como deficiência de vitamina D, hiperparatireoidismo, doença medular cística, síndrome de Alport, amiloidose, transplante renal, etc.13 As formas primárias podem ter herança autossômica dominante provavelmente devido a uma alteração no trocador Na-Hna membrana luminal molecularmente cconhecido como NHE-3 (Na/H exchanger 3). A herança também pode ser autossômica recessiva, forma mais comum, com mutação no cotransportador Na-HCO3 (NBC1) na membrana basolateral. O distúrbio pode ocorrer também por uma mutação no gene codificador da anidrase carbônica.
Figura 86.3
Principais mecanismos de transporte no túbulo proximal (a.c., anidrase carbônica).
Sinais e sintomas/diagnóstico
Clinicamente, há retardo do crescimento, raquitismo ou osteomalacia. Mais raramente, na forma autossômica recessiva com alterações oculares, pode haver catarata, glaucoma, ceratopatia da córnea; defeitos dentários; retardo motor e da capacidade intelectual; além de hipotireoidismo e hiperamilasemia em alguns casos.13 O paciente apresenta acidose metabólica, hiperclorêmica e hipocalêmica, pois o aumento de fluxo de Na+ e fluido levam a um aumento da secreção de K+ no néfron distal, com consequente hipocaliemia.
O pH urinário é ácido, apesar do aumento da fração de excreção de HCO3- acima de 15%,11 porque há um reajuste no túbulo, que se torna capaz de reabsorver bicarbonato quando a acidose metabólica torna-se acentuada e a carga de bicarbonato a ser filtrada se reduz.
Tratamento
O tratamento baseia-se na reposição de bicarbonato, e, devido à grande importância do túbulo proximal na reabsorção de bicarbonato, doses maiores são necessárias, 10 a 15 mEq/kg/dia. O uso de citrato de potássio é bastante útil, com a vantagem de repor potássio também.14
Acidose tubular renal tipo III
A combinação das acidoses tubular proximal e distal, também denominada de acidose renal tipo III, é causada pelas mutações no gene que codifica a anidrase carbônica tipo II, levando à menor conversão de ácido carbônico para bicarbonato e vice-versa. Clinicamente, observa-se osteopetrose, calcificação cerebral e retardo mental.13
Definição
A ATR tipo IV é constituída de um grupo heterogêneo de entidades que cursam com acidose metabólica hipercalêmica, com acidificação urinária normal. Pode ser causada pela redução na excreção urinária de NH4+, deficiência de aldosterona ou resistência à aldosterona (pseudo-hipoaldosteronismo). Acompanha vários estados hipercalêmicos, mas mais frequentemente é observada em estados de hipo ou pseudo-hipoaldosteronismo, que podem aparecer isoladamente ou no contexto de uma doença parenquimatosa renal. Nefrocalcinose ou nefrolitíase não são comuns.
Classificação
Pode ser primária, aparecendo transitoriamente na infância, também considerada uma variante do pseudo-hipoaldosteronismo tipo I, onde provavelmente há um distúrbio de maturação no número ou na função dos receptores de aldosterona.14 A forma secundária pode ser por deficiência de aldosterona na ausência de doença renal (Addison, hiperplasia congênita de suprarrenal) ou devido a nefropatias que cursam com hipoaldosteronismo hiporreninêmico (nefropatia diabética, lúpus eritematoso sistêmico, nefropatia do HIV, glomerulonefrites agudas). A ATR tipo IV secundária também pode ser induzida por drogas (inibidores da enzima conversora da angiotensina, heparina, inibidores da cicloxigenase, diuréticos poupadores de potásssio, trimetoprima, pentamidina, ciclosporina, digitálicos, bloqueadores ß-adrenérgicos, agonistas a-adrenérgicos, succinilcolina). Finalmente, a ATR tipo IV pode ser causada por resistência à aldosterona devido aos distúrbios genéticos (pseudo-hipoaldosteronismo tipo I e tipo II) ou ainda por nefrite intersticial crônica ou por uropatia obstrutiva (13). A infecção do trato urinário pode causar resistência transitória à ação da aldosterona.15
Pseudo-hipoaldosteronismo tipo I
É uma entidade caracterizada por perda de sal, desidratação, acidose metabólica, hipercaliemia e hipotensão. Há elevada atividade da renina plasmática e da concentração de aldosterona. Na forma autossômica dominante, a resistência à aldosterona é limitada ao rim e ocorre devido à mutação no gene que codifica o receptor do mineralocorticoide. Na forma autossômica recessiva, a resistência à aldosterona acomete vários órgãos (rim, intestino, pulmão, glândulas sudoríparas e salivares) e ocorre pela mutação nos genes que codificam as subunidades a, ß e Y do ENaC.9
Pseudo-hipoaldosteronismo tipo II
Também é conhecido como síndrome de Gordon e caracteriza-se clinicamente pela presença de hipertensão com renina baixa, acidose metabólica hiperclorêmica e hipercalêmica. A herança é autossômica dominante com mutações nos genes WNK4 e WNK1, que codificam quinases que aumentam a condutância ao cloro. O WNK4 normalmente inibe os canais de K+ (ROMK), inibe o cotransportador tiazídico sensível e aumenta o transporte paracelular de cloro no túbulo distal e no ducto coletor. A mutação do WNK4 leva à exacerbação dessas atividades.
Tratamento
O tratamento da ATR tipo IV depende da etiologia. Drogas retentoras de potássio devem ser suspensas. O hipoaldosteronismo devido à insuficiência suprarrenal responde a corticoide. Nos casos de hipoaldosteronismo hiporreninêmico, devem ser usados corticoide e diuréticos de alça. Para os casos de pseudo-hipoaldosteronismotipo I, a suplementação de sal é suficiente para corrigir a perda de sódio e não há resposta a corticoide. Para opseudo-hipoaldosteronismo tipo II, baixas doses de tiazídico corrigem a hipertensão e a hipercaliemia. Na Tabela 86.1, são apresentados os defeitos genéticos associados às tubulopatias com perda ou retenção de K+.
Definição/quadro clínico
A doença de Dent é uma síndrome ligada ao X, caracterizada por hipercalciúria e nefrolitíase, também conhecida pelas denominações de nefrolitíase recessiva ligada ao X, raquitismo hipofosfatêmico recessivo ligado ao X ou proteinúria de baixo peso molecular com hipercalciúria e nefrocalcinose.
Etiologia/diagnóstico/aspectos genéticos
Todas elas representam a mesma entidade, cujo defeito molecular comum é uma mutação no gene CLCN5 que codifica o canal para cloro CLC-5,16 expresso no túbulo contornado proximal, porção espessa da alça de Henle e células intercaladas dos ductos coletores. Devido à herança ligada ao X, acomete somente o sexo masculino e o fenótipo é variável. Além do raquitismo, há proteinúria de baixo peso molecular, hipercalciúria, fosfatúria, nefrolitíase e nefrocalcinose, podendo evoluir com perda de função renal. As mães dos pacientes afetados geralmente têm níveis intermediários de proteinúria de baixo peso molecular, como a RBP (retinol binding protein) e ß2-microglobulina.17 Em alguns casos, relata-se aumento da excreção urinária de potássio e hipocaliemia. Outros distúrbios do túbulo proximal podem também estar presentes, como aminoacidúria, glicosúria, uricosúria e alteração da acidificação urinária.18
A diferença entre a doença de Dent e a síndrome de Fanconi é que, na segunda, a hipercalciúria, a nefrocalcinose e a nefrolitíase não são comuns.19
Patogênese
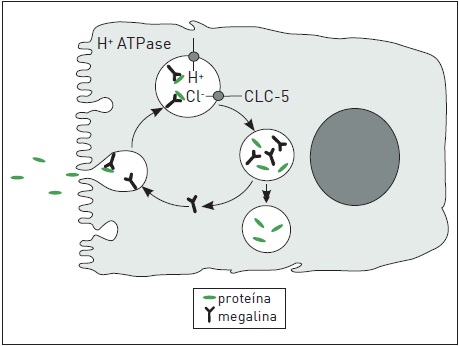
Os canais CLC-5 são cruciais no processo de endocitose, que é o principal mecanismo responsável pela reabsorção de proteínas de baixo peso molecular no túbulo proximal. Conforme esquematizado na Figura 86.4, a endocitose de proteínas é mediada pelo receptor megalina , cuja recirculação membrana-endossomo-membrana depende da acidificação dos endossomos, que, por sua vez, depende da presença dos CLC-5 e da bomba de prótons (H-ATPase) localizados na membrana endossomal.
Mutações no canal CLC-5 impedem ou dificultam a acidificação do endossomo, resultando no déficit de reabsorção de proteínas e na complexa síndrome de Dent. Assim, a reabsorção de 25 (OH)-vitamina D3, de sua proteína ligadora bem como do paratormônio (PTH) estão diminuídas e, consequentemente, todos os mecanismos fisiológicos mediados por esses hormônios ficam debilitados. A perda urinária de vitamina D causa uma variação nos níveis de vitamina D ativa. Esse desequilíbrio poderia explicar a hipercalciúria e a nefrolitíase comumente observados nessa síndrome. A disfunção do CLC-5 na porção espessa da alça de Henle seria outra explicação para a hipercalciúria, já que essa porção do túbulo é o maior sítio de reabsorção de cálcio. Mecanismo semelhante parece ocorrer com a reabsorção de fósforo via transportador Na-Pi, levando à fosfatúria.20
Figura 86.4
Mecanismo de reabsorção de proteínas mediada por receptor (megalina) no túbulo proximal.
Tratamento
O tratamento é feito com suplemento de fósforo. Doses baixas de vitamina D podem ser usadas. A hidroclorotiazida deve ser usada com cautela, já que alguns pacientes têm hipocaliemia na abertura do quadro.
Definição
É um distúrbio autossômico recessivo raro, caracterizado pela diminuição na reabsorção de fosfato devido à mutação nos genes que codificam o cotransportador sódio-fósforo tipo 2a (Na- Pi-IIa) ou tipo 2c (Na-Pi-IIc).
Aspectos clínicos/diagnóstico
Clinicamente, o paciente apresenta retardo do crescimento, raquitismo ou osteomalacia. Há fosfatúria, hipofosfatemia, aumento dos níveis séricos de vitamina D e hipercalciúria. A hipercalciúria, assim como na doença de Dent, pode predispor à nefrocalcinose e à litíase renal. Devido ao aumento da 1,25(OH)2D3, a para tireoide encontra-se suprimida.21
Tratamento
O tratamento feito com suplemento de fósforo, 1 a 2,5 g/ dia, restitui o nível sérico de fósforo, reduzindo o de vitamina D, acelera o crescimento, e as manifestações clínicas de raquitismo ou osteomalacia desaparecem.
Recentes estudos mostraram que existe uma nova classe de fatores reguladores do fosfato, os fatores fosfatúricos, denominados de fosfatoninas e que estão relacionados com doenças hipofosfatêmicas, como o raquitismo hipofosfatêmico ligado ao X (XLH), o raquitismo hipofosfatêmico dominante (ADHR), a osteomalacia oncogênica hipofosfatêmica e a hipofosfatemia autossômica recessiva.22 Esses fatores incluem o fator de crescimento fibroblástico (FGF-23) e o FRP4 (secreted frizzled-relatedprotein), e, além de reduzirem diretamente a reabsorção de fosfato, eles suprimem a 25(OH)-vitamina D a-hidroxilase e, consequentemente, a formação de 1,25(OH)2D3.
O XLH resulta de uma mutação no gene PHEX (phos-phate-regulating gene with homologies to endopeptidases on the X-chromosome),23 gene regulador de fosfato homólogo a endopeptidases no cromossomo X. O PHEX é expresso nos osteoblastos, odontoblastos, ovário, pulmão, paratireoides, cérebro e músculo, mas não é expresso no rim. Na ausência funcional do PHEX, há uma inapropriada expressão e elevação dos níveis circulantes das fosfatoninas. Sugeriu-se, portanto, que o PHEX estaria envolvido na clivagem e inativação das fosfatoninas. O XLH é caracterizado por retardo do crescimento, raquitismo ou osteomalacia. Os níveis de calcitriol estão inapropriadamente baixos ou normais na vigência de hipofosfatemia. O nível sérico de PTH está normal, e o de cálcio pode estar normal ou baixo.24 O tratamento é feito com suplemento oral de fósforo e calcitriol com monitoração dos níveis séricos. Esse tratamento está longe do ideal, já que a resolução completa do raquitismo raramente ocorre.
O ADHR é caracterizado, assim como o XLH, por hipofosfatemia, fosfatúria, nível de vitamina D normal ou baixo, raquitismo e osteomalacia. O paciente pode apresentar ainda dor óssea, deformidades nas extremidades e fraqueza muscular.25 A ADHR está relacionada com mutações do fator de crescimento do fibroblasto, FGF-23 (fibroblast growth factor). O FGF-23 é expresso no cérebro, timo, intestino delgado, coração, fígado, linfonodos, tireoide, paratireoide e medula óssea35. O efeito do FGF-23 no cotransportador Na-P não está claro, mas sabe-se que a administração de FGF-23 recombinante em ratos promove fosfatúria e diminuição dos níveis séricos de fósforo. O número de cotransportadores Na- Pi-IIareduz-se após administração crônica de FGF-23. A expressão por tempo prolongado do FGF-23 aumenta a concentração de PTH e diminui os níveis de 1,25(OH)2D3, em estudos experimentais. Como os níveis de FGF-23 estão aumentados na ADHR e também na ostemalacia oncogênica, foi sugerido que o FGF-23 representaria uma fosfatonina. O tratamento de ADHR é semelhante ao da XLH e consiste na combinação de fósforo e vitamina D.
O ARHR tem parâmetros clínicos, bioquímicos e histomorfométricos semelhantes ao XLH e ao ADHR. No entanto, a análise do DNA de três famílias afetadas sugeriu um tipo de herança autossômica recessiva com a mutação do gene 4q21 que codifica a matriz proteica da dentina (DMP1, dentin matrix protein 1). O nível sérico de FGF-23 apresentou-se elevado nos pacientes homozigotos para a mutação no DMP1, sugerindo que o DMP1 regula a expressão do FGF-23.
É um distúrbio autossômico recessivo causado por uma mutação inativadora do gene que codifica a 1a-hidroxilasevitamina D3, responsável pela síntese da 1,25(OH)2D3, por meio da hidroxilação da 25-hidroxivitamina D3.26 É caracterizado por hipocalcemia, hipofosfatemia, hiperparatireoidismo e aminoacidúria. Aparece em crianças nos primeiros meses de vida, com hipocalcemia severa, convulsões, fraqueza muscular, retardo do crescimento e achados típicos de raquitismo nas imagens radiológicas41. O raquitismo porpseudo-hipovitaminose D difere dos outros tipos de raquitismo hereditário, porque o nível sérico da 1,25(OH)2D3 está muito baixo ou indetectável, e o da 25(OH)D3 está normal ou aumentado, sugerindo um defeito na atividade da 1a-hidroxilase25(OH)D3 causada pela mutação no gene CYP27B1. O tratamento com calcitriol é necessário para a correção do raquitismo.
É uma doença que resulta de mutação no gene que codifica o receptor da vitamina D (VDR). Pacientes com pseudo-hipovitaminose tipo II têm um aumento importante dos níveis séricos de 1,25(OH)2D3 e respondem pobremente às doses de calcitriol usadas no tratamento. Dependendo do tipo de defeito resultante da mutação, o paciente responderá ou não às doses de calcitriol, que é o tratamento de escolha.27 A presença de alopecia está associada à forma severa da doença, mas sua relação com a resistência da vitamina D não está muito clara. Existem também casos descritos com VDR normal causados por uma “rinucleoproteína nuclear” que interfere com a interaçãoVDR-DNA. Esses pacientes não apresentam alopecia. O suplemento de cálcio pode ser usado para os pacientes que não respondem ao tratamento com vitamina D. A seguir, são apresentados, na Tabela 86.2, os defeitos genéticos associados às tubulopatias com perda de fósforo.
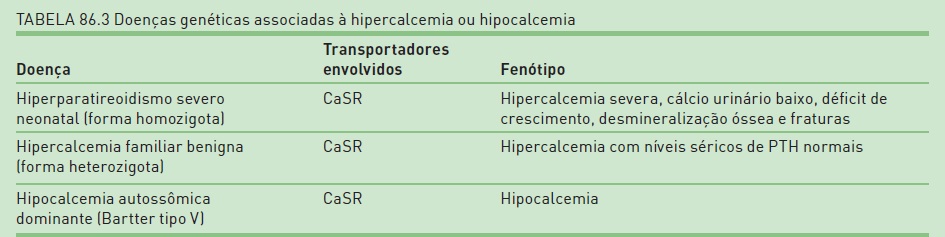
Definição
A doença hipercalcêmica hipocalciúrica familiar benigna (FHH) é uma síndrome de apresentação branda, sendo, na grande maioria dos casos, assintomática, muitas vezes diagnosticada acidentalmente quando o paciente faz exames de rotina.
Etiologia/patogênese
É causada por uma mutação inativadora do gene que codifica o receptor sensor de cálcio (CaSR), o qual é expresso na paratireoide e nos rins, sendo fundamental para a homeostase de cálcio. O CaSR é um sensor de cálcio que, em condições de cálcio sérico elevado, suprime a liberação de PTH pela paratireoide. Quando da inativação do receptor, a ligação do cálcio a este não resulta na supressão da secreção de PTH, que se mantém inapropriadamente elevado.
No rim, o CaSR é expresso principalmente na porção espessa da alça de Henle e no ducto coletor, cuja função fisiológica é inibir a reabsorção de cálcio,28 adequando, assim, a excreção urinária de cálcio aos níveis séricos. Assim, uma mutação inativadora faz com que a hipercalcemia não resulte em hipercalciúria, mas sim em aumento da reabsorção de cálcio (hipocalciúria) e de magnésio.29 Em crianças heterozigotas, há hipercalcemia, hipocalciúria e hipermagnesemia.
Sinais e sintomas/diagnóstico
Clinicamente, o paciente com FHH apresenta hipercalcemia com níveis séricos de PTH normais ou discretamente elevados, sem resposta à paratireoidectomia. O fósforo pode estar normal ou discretamente diminuído. O que diferencia a FHH do hiperparatireoidismo primário é a ausência de osteopenia, hipostenúria ou outros sinais de hipercalcemia, comumente vistos no hiperparatireoidismo primário. Além disso, na FHH, a fosfatase alcalina sérica e a excreção urinária de AMP cíclico estão normais. A FHH é uma doença de curso benigno.
Tratamento
Não está indicado paratireoidectomia parcial ou qualquer outro procedimento agressivo.
O NSHPT é a forma homozigota da mutação do CaSR30 e se caracteriza por uma hipercalcemia muito severa, manifestando se em recém-nascidos ou lactentes até os 6 meses. O cálcio urinário é baixo, há déficit de crescimento, desmineralização óssea e fraturas. É uma condição grave com indicação precoce de paratireoidectomia.
Definição
A ADH é caracterizada pelo aumento da excreção urinária de cálcio com concentrações séricas de cálcio abaixo do nível normal. Também é conhecida como Bartter tipo V. Contrariamente à FHH, é causada por uma mutação ativadora do gene que codifica o CaSR.31
Quadro clínico/diagnóstico
Nessa síndrome, a paratireoide fica hiper-responsiva ao Ca++ extracelular, a ativação do receptor ocorre mesmo com níveis normais ou baixos de cálcio sérico e o cálcio urinário fica elevado. O achado de hipocalcemia com níveis de PTH baixo e marcada redução da excreção de cálcio urinário sugerem ADH, que pode ser confirmada por análise da mutação CaSR. O paciente geralmente é assintomático. É importante diferenciar a ADH do hipoparatireoidismo, porque o tratamento com vitamina D para corrigir a hipocalcemia pode levar a hipercalciúria, nefrocalcinose e perda da função renal. Os pacientes com ADH podem apresentar um defeito na reabsorção tubular de Na+ e Cl- com consequente perda desses eletrólitos, e o balanço negativo de Na+ e Cl- leva consequentemente a um hiperaldosteronismo secundário e à hipocaliemia, simulando uma síndrome de Bartter (síndrome Bartter-like), que alguns autores chamam de Bartter tipo V.
Tratamento
O tratamento deve ser reservado a pacientes sintomáticos, e a vitamina D deve ser dada somente para alívio dos sintomas, e não com o objetivo de normalizar a calcemia, porque, além das complicações já citadas, o paciente com ADH e nível sérico de cálcio normal apresenta poliúria, polidipsia e desidratação, provavelmente pelo aumento da atividade do CaSR mutante no ducto coletor. Em pacientes com hipercalciúria persistente, apesar do tratamento, pode ser usado diurético tiazídico.32
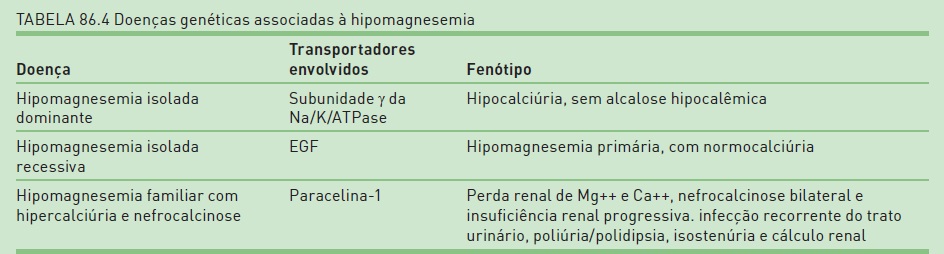
O magnésio, segundo cátion intracelular mais abundante, desempenha várias funções, incluindo a síntese proteica, a estabilidade dos ácidos nucleicos, a excitabilidade neuromuscular e a fosforilação oxidativa e, portanto, variações na concentração extracelular de Mg++ podem resultar em disfunção celular importante. A hipomagnesemia resultante da perda renal de Mg++ está mais frequentemente associada a modificações no transporte de outros íons, sendo, portanto, secundária, conforme descrito nas síndromes de Bartter e, principalmente, de Gilteman. A perda renal primária e isolada de Mg++ é rara e pode aparecer tanto na forma dominante como na forma recessiva.
Tipicamente se associa a hipocalciúria, semelhante aos portadores da síndrome de Gilteman, porém os pacientes com IDH não apresentam outro distúrbio, como alcalose hipocalêmica. O gene envolvido é a subunidade da bomba Na/K/ATPase. Essa subunidade é expressa no túbulo convoluto distal, onde o Mg++ é reabsorvido através do canal para Mg++ presente na membrana luminal (TRPM6). A relação entre o influxo de Mg++ através do canal TRPM6 e a função da subunidade Y da bomba ainda não foi esclarecida, mas há indícios de que seu mau funcionamento resultaria em acúmulo de potássio intracelular e consequente despolarização da célula, o que resultaria na redução do transporte de Mg++.33
655
Indivíduos afetados apresentam hipomagnesemia primária que aparece precocemente durante a infância e se distingue da forma dominante pela ausência de hipocalciúria. Um estudo recente sugere que a origem genética dessa doença está relacionada a mutações no gene que codifica o fator de crescimento epitelial (EGF), o qual exerce um papel estimulante sobre a atividade do canal para Mg++ (TRPM6) expresso no túbulo convoluto distal.34
Caracteriza-se por excessiva perda renal de Mg++ e Ca++, nefrocalcinose bilateral e insuficiência renal progressiva. Manifestações clínicas, como infecção recorrente do trato urinário, poliúria/polidipsia, isostenúria e cálculo renal, permitem distinguir a FHHNC de outras tubulopatias que cursam com perda renal de Mg++.35 Os pacientes afetados apresentam mutações variáveis no gene PCLN-1, que codifica a paracelina-1, um membro da família de proteínas denominadas claudinas, que formam as tight junctions do epitélio tubular renal, típicas da alça ascendente espessa de Henle. Cerca de 70% da carga filtrada de Mg++ é reabsorvida na alça ascendente espessa de Henle por via paracelular, cuja força propulsora inclui a diferença de potencial lúmen positivo em relação ao interstício, resultante da recirculação de K+ que ocorre nessa região do néfron (Fig. 86.2 ). Há evidências de que aparacelina–1 (claudina-16), junto com a claudina-19, regula a permeabilidade paracelular a cátions no ramo espesso da alça de Henle.36 Ausência ou déficit de função daparacelina-1 resulta em diminuição na reabsorção de Mg++ e Ca++ e no fenótipo característico da FHHNC. Os pacientes portadores dessa síndrome não apresentam sinais clínicos de perda de sal na urina ou ativação do sistema renina-angiotensina, e respondem adequadamente aos diuréticos de alça com natriurese, porém sem aumento da excreção urinária de Mg++ ou Ca++.
A síndrome de Bartter neonatal tem suas características clínicas, bioquímicas e genéticas com evolução que pode chegar à insuficiência renal crônica e mimetiza algumas entidades clínicas, como pseudo-Bartter e acidose tubular renal. O paciente em questão apresentou aspectos sugestivos da síndrome de Bartter neonatal com polidrâmnios, parto prematuro, episódios de febre, desidratação, retardo ponderal, hipercalciúria e nefrocalcinose. A dificuldade diagnóstica, nesse caso, deveu-se à presença de acidose metabólica hipocalêmica transitória, que não é parte do diagnóstico da síndrome de Bartter, habitualmente acompanhada de alcalose metabólica.1
1.Amin AB, Cerqueira AT, Casella RG. Síndrome de Bartter neonatal com acidose metabólica: relato de caso. J Bras Nefrol. 2002;24(4):194-8.
2.Scheinman SJ, Guay-Woodford LM, Thakker RV, Warnock DG. Genetic disorders of renal electrolyte transport. N Engl J Med.1999;340(15):1177-87.
3.Warnock DG. Liddle syndrome: an autosomal dominant form of human hypertension. Kidney Int. 1998;53(1):18-24.
4.Amirlak I, Dawson KP. Barterr syndrome: an overview. QJM.2000;93(4):207-15.
5.Graziani G, Fedeli C, Moroni L, Cosmai L, Badalamenti S, Ponticelli C. Gitelman syndrome: pathophysiological and clinical aspects. QJM.2010;103(10):741-8.
6.Chadha V, Alon US. Hereditary renal tubular disorders. Semin Nephrol.2009;29(4):399-411.
7.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115(6):1651-8.
8.Ring T, Frische S, Nielsen S. Clinical review: renal tubular acidosis-aphysicochemical approach. Crit Care. 2005;9(6):573-80.
9.Alper SL. Familial renal tubular acidosis. J Nephrol. 2010;23 Suppl16:S57-76.
10.Khositseth S, Sirikanerat A, Wongbenjarat K, Opastirakul S, Kho- prasert S, Peuksungnern R, et al. Distal renal tubular acidosis associated with anion exchanger 1 mutations in children in Thailand. Am J Kidney Dis. 2007;49(6):841-850.e1.
11.Devuyst O, Pirson Y. Genetics of hypercalciuric stone forming diseases. Kidney Int. 2007;72(9):1065-72.
12.Cheidde L, Vieira TC, Lima PR, Saad ST, Heilberg IP. A novel mutation in the anion exchanger 1 gene is associated with familial distal renal tubular acidosis and nephrocalcinosis. Pediatrics. 2003;112(6 Pt 1):1361-7.
13.Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol.2002;13(8):2171-7.
14.Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-70.
15.Bonny O, Rossier BC. Disturbances of Na/K balance: pseudohypoaldosteronism revisited. J Am Soc Nephrol. 2002;13(9):2399-414.
16.Stechman MJ, Loh NY, Thakker RV. Genetic causes of hypercalciuric nephrolithiasis. Pediatr Nephrol. 2009;24(12):2321-32.
17.Igarashi T, Günther W, Sekine T, Inatomi J, Shiraga H, Takahashi S, et al. Functional characterization of renal chloride channel, CLCN5, mutations associated with Dent’sJapan disease. Kidney Int.1998;54(6):1850-6.
18.Gambaro G, Vezzoli G, Casari G, Rampoldi L, D’Angelo A, Borghi L. Genetics of hypercalciuria and calcium nephrolithiasis: from the rare monogenic to the common polygenic forms. Am J Kidney Dis.2004;44(6):963-86.
19.Vilasi A, Capasso G. Proteomics and tubulopathies. J Nephrol. 2010;23 Suppl 16:S221-7.
20.Devuyst O, Thakker RV. Dent’s disease. Orphanet J Rare Dis. 2010;5:28.
21.Segawa H, Aranami F, Kaneko I, Tomoe Y, Miyamoto K. The roles ofNa/Pi-II transporters in phosphate metabolism. Bone. 2009;45 Suppl1:S2-7.
22.Schiavi SC, Kumar R. The phosphatonin pathway: new insights in phosphate homeostasis. Kidney Int. 2004;65(1):1-14.
23.Carpenter TO, Insogna KL, Zhang JH, Ellis B, Nieman S, Simpson C. Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab. 2010;95(11):E352-7.
24.Vaisbich MH, Koch VH. Hypophosphatemic rickets: results of along-term follow-up. Pediatr Nephrol. 2006;21(2):230-4.
25.Alizadeh Naderi AS, Reilly RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol. 2010;6(11):657-65.
26.Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, et al. Inactivating mutations in the 25-hydroxyvitamin D3 1alphahydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med. 1998;338(10):653-61.
27.Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006;116(8):2062-72.
28.Vezzoli G, Soldati L, Gambaro G. Roles of calcium-sensing receptor (CaSR) in renal mineral ion transport. Curr Pharm Biotechnol.2009;10(3):302-10.
29.Hebert SC. Extracellular calcium-sensing receptor: implications for calcium and magnesium handling in the kidney. Kidney Int.1996;50(6):2129-39.
30.Ward BK, Cameron FJ, Magno AL, McDonnell CM, Stuckey BG, Ratajczak T. A novel homozygous deletion in the calcium-sensing receptorligand-binding domain associated with neonatal severe hyperparathyroidism. J Pediatr Endocrinol Metab. 2006;19(1):93-100.
31.Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med.1996;335(15):1115-22.
32.Sato K, Hasegawa Y, Nakae J, Nanao K, Takahashi I, Tajima T, et al. Hydrochlorothiazide effectively reduces urinary calcium excretion in two Japanese patients with gain-of-function mutations of the calcium-sensingreceptor gene. J Clin Endocrinol Metab. 2002;87(7):3068-73.
33.Sha Q, Pearson W, Burcea LC, Wigfall DA, Schlesinger PH, Nichols CG, et al. Human FXYD2 G41R mutation responsible for renal hypo- magnesemia behaves as an inward-rectifying cation channel. Am J Physiol Renal Physiol. 2008;295(1):F91-9.
34.Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest. 2007;117(8):2260-7.
35.Benigno V, Canonica CS, Bettinelli A, von Vigier RO, Truttmann AC, Bianchetti MG. Hypomagnesaemia-hypercalciuria-nephrocalcinosis: a report of nine cases and a review. Nephrol Dial Transplant.2000;15(5):605-10.
36.Hou J, Renigunta A, Konrad M, Gomes AS, Schneeberger EE, Paul DL, et al. Claudin-16 and claudin-19 interact and form a cation-selectivetight junction complex. J Clin Invest. 2008;118(2):619-28.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.

 647
647