(Carregando Índice)... (Carregando Índice)... |
Última revisão: 21/05/2013
Comentários de assinantes: 0
Colin H. Chalk, MD, CM
Associate Professor, Department of Neurology and Neurosurgery, McGill University and Director, Division of Neurology, Montreal General Hospital.
Artigo original: Chalk CH. Diseases of the peripheral nervous system. ACP Medicine. 2008;1-20.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2011 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figura 1 – George Kelvin.
Tradução: Soraya Imon de Oliveira
Revisão técnica: Dr. Euclides Furtado de Albuquerque Cavalcanti
O sistema nervoso periférico (SNP) abrange todos os nervos cranianos (exceto o 1º e o 2º), raízes nervosas, gânglios espinais, nervos segmentares, plexos nervosos e nervos de membros. O papel do SNP é simples, porém vital: conectar o sistema nervoso central (SNC) ao ambiente externo e ao milieu intérieur do corpo.
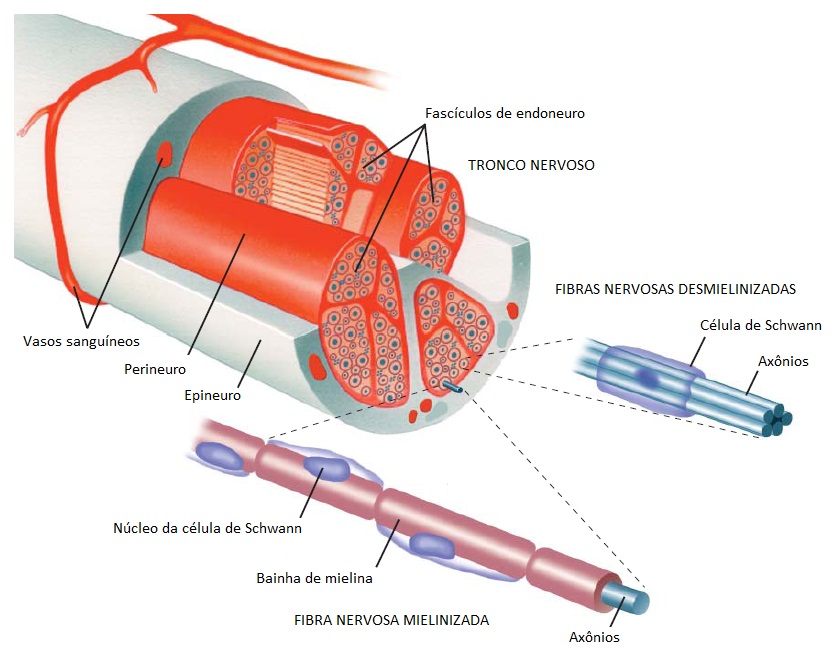
Os nervos periféricos são compostos por fascículos de endoneuro, que contêm vários milhares de axônios e também diversas células de sustentação [Figura 1]. Cada fascículo endoneural está envolto por um envelope de perineuro (várias camadas concêntricas de células fibroblasto-símile). Os troncos nervosos são compostos por vários fascículos endoneurais que são unidos pelo epineuro (uma estrutura frouxa de colágeno e fibroblastos). Existem 2 redes complementares de vasos sanguíneos junto aos nervos: (1) um sistema epineural de arteríolas e vênulas, que são tributárias dos principais vasos dos membros; e (2) uma série de microvasos do tipo capilares junto ao endoneuro. As anastomoses existentes entre estas 2 redes são extensivas e resultam em um robusto sistema de suprimento.

Figura 1. Os nervos periféricos são compostos por vários fascículos de endoneuro, cada um dos quais é envolto pelo perineuro. Todos os fascículos são unidos por um tecido conectivo frouxo, o epineuro. Cada fascículo de endoneuro contém até vários milhares de axônios. O sangue é fornecido ao endoneuro por uma rede de microvasos capilares, derivada de arteríolas e vênulas localizadas no epineuro que, por sua vez, são ramos dos principais vasos dos membros.
Cada axônio é uma extensão longitudinal do corpo celular de um neurônio. O funcionamento e a manutenção dos axônios dependem da maquinaria genética e bioquímica do corpo celular. Um sistema de transporte axonal bidirecional e dependente de energia transporta as moléculas sinalizadoras e estruturais entre o corpo celular e o terminal do axônio. Os axônios motores, sensoriais e autonômicos estão misturados na maioria dos nervos, porém os corpos celulares estão agrupados separadamente na medula espinal e no tronco encefálico (motores), gânglios da raiz dorsal (sensoriais) e gânglios paravertebrais e somáticos (autonômicos).
Grosso modo, 1/4 dos axônios pertencentes ao SNP são mielinizados. A mielina é produzida e mantida pelas células Schwann, que são as células de sustentação mais importantes do endoneuro. Ao longo do desenvolvimento, as células de Schwann elaboram quantidades maiores de membrana citoplasmática para formar uma espiral em torno de alguns axônios. A membrana citoplasmática forma os envoltórios lamelares de mielina. Uma única célula de Schwann mieliniza 0,2 a 1,8 mm de membrana citoplasmática ao longo da extensão de um axônio. Isso significa que a mielina encontrada ao longo de toda a extensão de um único axônio é o produto coletivo das ações de várias centenas de células de Schwann consecutivas. Os axônios não mielinizados, que são mais numerosos, também estão intimamente associados às células de Schwann. O desenvolvimento normal dos axônios e o desenvolvimento normal das células de Schwann são interdependentes. Muitos dos detalhes sobre as complexas interações entre estes processos são conhecidos.1
Para os anatomistas antigos, a divisão do sistema nervoso nas partes central e periférica foi intuitivamente razoável. Esta divisão não é arbitrária. Existem muitas diferenças entre o SNP e o SNC consideradas importantes do ponto de vista clínico. A diferença mais evidente entre ambos reside na simplicidade funcional do SNP. Em essência, o SNP consiste em um sistema de retransmissão simples que transporta sinais motores, sensoriais e autonômicos entre o SNC e as estruturas somáticas. O correlato clínico é o repertório limitado de sinais e sintomas produzidos pelo mau funcionamento do SNP. É possível inferir relativamente pouco acerca da causa de um distúrbio de nervo periférico, partindo apenas dos sinais e sintomas.
Uma segunda característica distintiva do SNP é sua acessibilidade. Diferente do cérebro e da medula espinal, que estão bem protegidos por caixas ósseas, a maioria dos principais nervos periféricos situa-se logo abaixo da pele ao longo de grande parte de seu curso. Esta acessibilidade simplifica o exame direto das características elétricas e histológicas dos nervos periféricos. A avaliação clínica das doenças do SNP tem sido bastante facilitada pelos exames de condução nervosa e biópsias de nervo, os quais podem ser realizados essencialmente sem oferecer nenhum risco ao paciente.
As capacidades regenerativas diferem entre o SNP e o SNC. No SNC maduro, há pouco recrescimento dos axônios após uma lesão. Entretanto, um nervo periférico lesado frequentemente regenera em longas extensões e restabelece as conexões funcionais. Desta forma, muitos distúrbios envolvendo nervos periféricos apresentam potencial de recuperação significativa da função com o tratamento.
Por muito tempo, a diferença de capacidade regenerativa foi considerada relacionada a alguma propriedade intrínseca dos neurônios do SNP. Entretanto, parece que o aspecto crítico é o ambiente ao qual os neurônios são expostos.1 Se um feixe de fibras de nervos do SNC (p. ex., nervo óptico ou medula espinal) é transeccionado e imediatamente reanastomosado, então há pouca renovação axonal. Entretanto, se a terminação proximal da mesma lesão é anastomosada a um enxerto de nervo periférico, surgem brotos axonais que se estendem por muitos milímetros através do enxerto de nervo periférico e podem até estabelecer conexões funcionais com neurônios-alvo do SNC.
As bases celulares e moleculares desta diferença entre os tecidos de sustentação do SNP e do SNC não são totalmente conhecidas. A célula de Schwann (que é exclusiva do SNP) parece ser decisiva, embora outras células (p. ex., fibroblastos e macrófagos) provavelmente também estejam envolvidas. Acredita-se que a célula de Schwann e o milieu extracelular do nervo periférico fornecem um ambiente favorável ao crescimento axonal, atraindo e talvez guiando os brotos axonais. Duas classes principais de moléculas influenciam o crescimento e desenvolvimento dos axônios em brotamento in vitro: fatores de crescimento e moléculas de adesão. Algumas destas moléculas (p. ex., fator de crescimento de nervo, N-caderina e laminina) foram bem caracterizadas.1 Enfim, nosso conhecimento sobre os eventos moleculares que intensificam ou inibem o crescimento axonal após a lesão de um nervo pode resultar em tratamentos que melhorem o reparo e a recuperação dos nervos em seres humanos, no entanto isso ainda não é uma realidade clínica.
Os distúrbios do SNP produzem combinações de sintomas motores, sensoriais e autonômicos. Estes sintomas são essencialmente determinados pela classe de fibras nervosas afetadas (p. ex., fibras motoras ou sensoriais) e pela localização das lesões, em vez de pela etiologia do processo. Os sintomas motores incluem o enfraquecimento (uma queixa comum), atrofia muscular, fasciculações e cãibras. Estes dois últimos sintomas motores podem ser descobertos apenas mediante perguntas específicas dirigidas ao paciente. Os sintomas sensoriais incluem a perda de certos tipos de sensação e a presença de formigamento, picadas, agulhadas e sensação de queimação. A descrição da qualidade e distribuição dos sintomas sensoriais fornecida pelo próprio paciente frequentemente é mais reveladora do que o exame sensorial.
A dor é um sintoma comum em distúrbios de nervos periféricos e muitas vezes está associada a outros sintomas neuropáticos. Quando a dor é o único sintoma, sua causa provavelmente está relacionada a outras estruturas, e não aos nervos periféricos. Os sintomas autonômicos são heterogêneos, e muitas vezes é possível descobri-los apenas por meio de perguntas diretas, pois podem não parecer especialmente neurológicos ao paciente. Os sintomas autonômicos comuns são as tonturas ou desmaios ortostáticos, sudorese excessiva ou diminuída, impotência ou falha ejaculatória, e comprometimento da motilidade gastrintestinal (em particular o retardo do esvaziamento gástrico).
As pesquisas laboratoriais frequentemente são necessárias ao esclarecimento da natureza de um distúrbio de nervo periférico. Além dos exames empregados na avaliação de outros tipos de doenças neurológicas (p. ex., punção lombar), também existem técnicas de exame direto da histologia e fisiologia dos nervos periféricos. Os exames neurofisiológicos (em especial os exames de condução nervosa) podem ser considerados uma parte rotineira da avaliação de qualquer paciente com polineuropatia. A biópsia de nervo, por sua vez, é um procedimento invasivo e está associada ao aparecimento de sintomas sensoriais problemáticos e persistentes em alguns pacientes. Em consequência, recomenda-se que a biópsia de nervo seja reservada para pacientes com sinais e sintomas neurológicos incapacitantes ou para os casos em que poucos diagnósticos específicos estão sendo investigados.
Os exames de condução nervosa podem fornecer suporte para um diagnóstico de neuropatia e constituem uma forma relativamente objetiva de seguir o curso da doença. Estes exames também podem levar o clínico a inferir se a doença está afetando principalmente os axônios ou suas bainhas de mielina – uma distinção importante no diagnóstico diferencial da polineuropatia. Os exames de condução nervosa fornecem uma indicação dos números de grandes axônios sensoriais e motores mielinizados funcionais em vários nervos, bem como a velocidade da transmissão de impulsos nestas fibras. O procedimento consiste na despolarização de um pequeno segmento de nervo com um choque elétrico rápido e o registro do vale de potenciais de ação resultante, à medida que estes potenciais se propagam pelo nervo. Ambas as fibras, motoras e sensoriais, são ativadas por este procedimento. As respostas motoras e sensoriais são distinguidas umas das outras por meio do registro feito a partir de um feixe cutâneo do nervo (sensorial) ou a partir de um músculo inervado (motor). Se os exames de condução nervosa mostrarem a diminuição das amplitudes motoras e sensoriais aliada à preservação das velocidades de condução, é provável que o processo patológico subjacente seja a perda ou destruição dos axônios. Em contraste, se as amplitudes estiverem relativamente preservadas e houver um retardo acentuado da velocidade de condução ou bloqueio de condução, é provável que exista uma anormalidade envolvendo a bainha de mielina.
Eletromiografia com agulha
A eletromiografia com agulha consiste em uma investigação complementar que geralmente é realizada em paralelo aos exames de condução. Em um paciente com neuropatia periférica, a eletromiografia é útil na detecção de pequenos graus de perda axonal que podem não ser detectados pelos exames de condução nervosa. A eletromiografia também pode ser útil para indicar a localização precisa da lesão em uma mononeuropatia. O procedimento requer o registro da atividade elétrica em um músculo, por meio da inserção de um eletrodo com agulha no músculo. Os músculos são examinados durante o repouso e na contração voluntária leve. Quando a atividade elétrica produz um padrão anormal, o eletromicrógrafo consegue determinar se o enfraquecimento apresentado pelo paciente resulta de um músculo adoecido ou de um nervo motor comprometido.
A biópsia de nervo pode ser útil do ponto de vista diagnóstico, em casos de pacientes cuidadosamente selecionados. A biópsia de nervo pode fornecer um diagnóstico específico (p. ex., vasculite necrotizante), contudo mais frequentemente produz informações menos específicas que podem ser úteis quando combinadas a outros dados clínicos. Além das alterações no número ou tamanho dos axônios, a anormalidade mais comumente encontrada no exame de biópsia de nervo é a inflamação, em geral relacionada a uma função imunológica alterada. Em certas ocasiões, uma doença que afeta tanto o SNC como o SNP (p. ex., leucodistrofia metacromática) pode ser facilmente diagnosticada pelo exame de biópsia de nervo. O nervo sural é o preferido, na maioria dos casos, porque a obtenção da biópsia deste nervo resulta em uma área pequena de perda sensorial, em geral bem tolerada, junto à lateral do pé.
O exame patológico adequado de uma amostra de biópsia de nervo requer o uso de técnicas que muitas vezes estão disponíveis apenas em laboratórios especializados. Na falta destes estabelecimentos ao nível local, o paciente deve ser encaminhado a um centro apropriado.
Os sintomas de neuropatia podem ser tratáveis mesmo que a causa da neuropatia seja incurável ou desconhecida. A adoção de medidas simples pode aliviar significativamente os sintomas de muitos pacientes.
A órtese de tornozelo-pé consiste em um dispositivo simples que compensa o pé caído. O principal benefício proporcionado pela órtese de tornozelo-pé reside na maior estabilidade do tornozelo, com consequente melhora do equilíbrio e superação da tendência a prender os dedões dos pés nas bordas dos degraus de escadas, do meio-fio e de tapetes. Um peso leve, devidamente ajustado à órtese de tornozelo-pé e inserido no sapato do paciente, em geral é a opção mais confortável. Os pacientes com enfraquecimento de punho e extensores dos dedos podem ser beneficiados pelo uso de uma braçadeira que mantenha o punho e dedos da mão em posição neutra. Contudo, este tipo de imobilizador tem pouca serventia para os casos em que haja significativo enfraquecimento concomitante da musculatura intrínseca das mãos.
A dor, em particular nos pés, em muitos casos acompanha as perturbações sensoriais associadas à neuropatia. Paradoxalmente, nas polineuropatias acompanhadas de dor proeminente, os déficits neurológicos costumam ser mínimos, e há uma dissociação entre a incapacitação real e o nível de angústia apresentado pelo paciente. Os pacientes podem interpretar a dor como sendo um sinal da existência de um distúrbio grave que ameaça não apenas sua independência como também sua vida. Em alguns casos, a simples tranquilização do paciente, enfatizando o quão mínima foi a perda da função neurológica, ajuda na superação efetiva da dor neuropática.
As ações não farmacológicas podem ser tão efetivas quanto as medicações. É preciso estar atento aos calçados que o paciente usa. Os sapatos recomendados para uso são aqueles que não apertam e têm solado flexível, os quais devem ser calçados com meia grossa. A dor neuropática tende a ser agravada pelos extremos de temperatura – em especial o calor –, e uso de sandálias abertas pode proporcionar alívio. A sustentação de peso por tempo prolongado costuma piorar a dor neuropática. Aos pacientes que trabalham em pé, recomenda-se fazer pausas curtas e frequentes para sentar.
Molhar o pé pode proporcionar um breve alívio a muitos pacientes. Pode ser particularmente útil imergir os pés em água fria (sem gelo) à altura dos tornozelos durante 15 a 20 minutos, antes de ir dormir. Embora este alívio tenha curta duração, pode ser suficiente para permitir que o paciente adormeça e tenha um sono de boa qualidade. Para alguns pacientes, a água morna funciona melhor do que a água fria, enquanto outros preferem alternar água fria e água quente (banhos de contraste) para obter melhor alívio. A inspeção diária dos pés à procura de lesões não detectadas – um hábito importante que precisa ser desenvolvido pelos pacientes com neuropatia – pode ser facilmente combinada à realização dos banhos de pé noturnos.
As medicações podem ser úteis para fins de tratamento da dor neuropática, contudo as metas da terapia devem ser realistas. O alívio completo da dor é improvável. Sendo assim, o objetivo da terapia deve ser tornar a dor mais tolerável sem adicionar os efeitos colaterais intoleráveis da medicação. Dentre as numerosas opções de fármacos que podem ser usados no tratamento da dor neuropática, os tricíclicos (em especial a amitriptilina), a gabapentina e a carbamazepina são empregados com maior frequência. A pregabalina, um derivado da gabapentina, é um agente moderno e efetivo. Vários estudos controlados randomizados envolvendo pacientes com dor neuropática de diversas etiologias demonstraram que cada um destes fármacos é superior ao placebo. Entretanto, faltam estudos que comparem diretamente estes fármacos entre si.2 As dores lancinantes e paroxísmicas podem ser mais propensas a responderem à carbamazepina (200 mg, 3 vezes/dia) ou à gabapentina (300 mg, 3 vezes/dia), enquanto a amitriptilina (10 a 30 mg, na hora de dormir) pode ser mais efetiva para os casos de entorpecimento ardente contínuo. A gabapentina é provavelmente o fármaco menos propenso a causar efeitos colaterais problemáticos. As dosagens efetivas de gabapentina apresentam uma variação considerável. Para a maioria dos pacientes, uma dose inicial satisfatória é 300 mg (3 vezes/dia), que pode ser titulada de modo crescente ao longo de várias semanas para 1.800 a 3.600 mg/dia ou mais, de acordo com a resposta. Os efeitos colaterais anticolinérgicos e hipnóticos podem tornar a amitriptilina intolerável para alguns pacientes, embora o efeito hipnótico possa ser benéfico nas ocasiões em que os sintomas neuropáticos mais incômodos se manifestam durante a noite – uma situação comum.
O tratamento deve ser conduzido por um período mínimo de 1 mês, para que seja possível chegar a quaisquer conclusões acerca da utilidade de um determinado fármaco para o alívio da dor neuropática. Além dos agentes tricíclicos, carbamazepina e gabapentina, os salicilatos e outros analgésicos simples proporcionam alívio a alguns pacientes. Assim como para outros tipos de dor crônica, a prática habitual consiste em evitar o uso de narcóticos, contudo os potenciais benefícios proporcionados pelos opiáceos no tratamento da dor neuropática crônica estão sendo reavaliados.3 Os fármacos de 2ª linha para uso no tratamento da dor neuropática incluem o baclofeno, mexiletina, ácido valproico e topiramato. O uso tópico da pomada de capsaicina promove depleção do neurotransmissor conhecido como substância P, junto ao corno dorsal da medula espinal, e ajuda alguns pacientes. Na maioria dos casos, todavia, as despesas e a inconveniência da capsaicina tópica superam quaisquer benefícios.
Em alguns pacientes, a dor permanece refratária a todas essas ações. Estes pacientes apresentam problemas complicados relacionados ao tratamento. A depressão concomitante pode agravar a situação, e o encaminhamento psiquiátrico pode ser conveniente. O encaminhamento do paciente a um centro multidisciplinar de tratamento da dor também pode ser considerado.
É útil agrupar as doenças do SNP em doenças que afetam nervos isolados (mononeuropatias) e doenças com envolvimento difuso do SNP (polineuropatias). Uma mononeuropatia pode afetar qualquer nervo craniano ou de membro. Em geral, as mononeuropatias ocorrem em indivíduos saudáveis, produzem sintomas problemáticos sem incapacitação significativa e frequentemente melhoram de modo espontâneo, com o passar do tempo.
As neuropatias podem surgir em qualquer nervo craniano individual [Tabela 1]. O VII (facial), V (trigêmeo) e III (oculomotor) nervos cranianos são responsáveis pela maioria das mononeuropatias encontradas na prática clínica.
Tabela 1. Neuropatias cranianas comuns
|
Nervo craniano |
Efeitos da lesão |
Causas |
|
Olfatório (I) |
Perda da percepção do cheiro |
Traumatismo, meningioma da fenda olfatória |
|
Óptico (II) |
Perda da visão (monocular) |
Neurite óptica, tumor da hipófise, neuropatia óptica isquêmica, glioma do nervo óptico, doença de Leber |
|
Oculomotor (III) |
Enfraquecimento da adução, elevação e depressão do olho Ptose Pupila dilatada e não reativa |
Traumatismo, isquemia microvascular, compressão por aneurisma ou massa, tumor ou AVC afetando o tronco encefálico, tumor orbital |
|
Troclear (IV) |
Enfraquecimento da depressão e torção do olho |
Traumatismo, isquemia microvascular, tumor ou AVC afetando o tronco encefálico, tumor orbital |
|
Trigêmeo (V) |
Enfraquecimento do movimento de mastigação e outros movimentos mandibulares Perda da sensibilidade facial e do reflexo corneal |
Neuralgia do trigêmeo (tic douloureux), esclerodermia e outras doenças do tecido conectivo, tumor do osso petroso |
|
Abdutor (VI) |
Enfraquecimento da abdução do olho |
Traumatismo, pressão intracraniana elevada, isquemia microvascular, tumor ou AVC afetando o tronco encefálico, tumor orbital |
|
Facial (VII) |
Enfraquecimento de metade da face Perda do paladar Hiperacusia |
Paralisia de Bell, doença de Lyme, sarcoidose, herpes zóster, tumor no ângulo cerebelopontino, tumor ou AVC afetando o tronco encefálico |
|
Vestibulococlear (VIII) |
Surdez unilateral Vertigem Nistagmo |
Neuronite vestibular, schwanoma acústico |
|
Glossofaríngeo (IX) |
Disfagia Enfraquecimento da elevação do palato Perda do reflexo da ânsia |
Doença neuronal motora, tumor no forame jugular, carcinoma nasofaríngeo, metástases para a base do crânio, neuralgia do glossofaríngeo |
|
Vago (X) |
Disfagia Disartria Enfraquecimento da elevação do palato Perda do reflexo da ânsia |
Doença neuronal motora, tumor no forame jugular, metástases para a base do crânio, carcinoma nasofaríngeo |
|
Acessório (XI) |
Enfraquecimento dos músculos esternocleidomastoideo e trapézio |
Traumatismo (cirúrgico e outros), tumor no forame jugular |
|
Hipoglosso (XII) |
Enfraquecimento da língua |
Doença neuronal motora, tumor na base no crânio, traumatismo ou dissecção da artéria carótida |
A neuropatia facial idiopática (paralisia de Bell) é a mais comum das neuropatias cranianas. Sua incidência anual aproximada é de 25 em cada 100.000 indivíduos. O principal sintoma é o enfraquecimento facial unilateral, que surge de forma abrupta e muitas vezes é precedido ou acompanhado de uma dor atrás da orelha. Alguns pacientes podem ter sofrido uma infecção leve no trato respiratório superior dentro de um período de 2 semanas, antes do aparecimento da condição. O enfraquecimento facial geralmente chega ao pico em 24 horas após a manifestação inicial e é do tipo neuronal motor inferior, afetando as regiões superior e inferior da face. Alguns pacientes apresentam alteração do paladar em metade da língua e hiperacusia. Em muitos casos, os pacientes queixam-se de um entorpecimento facial, porém nenhuma perda sensorial é encontrada.
Quando a história e o exame fornecem dados típicos da paralisia de Bell e a condição é branda e não progressiva, os exames são desnecessários. Os casos severos ou progressivos devem ser encaminhados a um neurologista para confirmação do diagnóstico e decisão acerca da intervenção necessária. Ocasionalmente, a sarcoidose, doença de Lyme e herpes zóster manifestam-se como uma neuropatia facial. A realização de exames para excluir a hipótese destas condições pode ser uma medida apropriada. Quando o exame neurológico revela a existência de algo mais do que uma neuropatia facial unilateral, ou quando o enfraquecimento evolui no decorrer de várias semanas ou meses, deve ser considerado um diagnóstico alternativo, como um tumor do ângulo cerebelopontino ou uma lesão no tronco encefálico.
A história natural de paralisia de Bell não tratada geralmente é favorável: 85% dos pacientes apresentam recuperação total dentro de 1 ano. Entretanto, alguns aspectos clínicos estão associados a um prognóstico ruim, tais como idade avançada, paralisia facial total e a existência de hiperacusia ou alteração do paladar. O espasmo hemifacial (uma síndrome de contração involuntária, breve e indolor dos músculos faciais) ocasionalmente se desenvolve após a paralisia de Bell e outros tipos de neuropatia facial.
Apesar do consenso geral de que a patogênese da paralisia de Bell envolve a inflamação do nervo facial, é discutido se a condição constitui um fenômeno autoimune pós-infecção ou é resultado da infecção viral direta do nervo facial. O vírus do herpes simples de tipo 1 foi implicado pela presença de seu DNA em amostras de nervo facial obtidas de 11 dentre 14 pacientes com paralisia de Bell, mas o mesmo não ocorreu em casos de pacientes com outras causas de neuropatia facial, incluindo o herpes zóster.4
Devido à provável participação de uma resposta inflamatória, a paralisia de Bell muitas vezes é tratada com glicocorticoides. Vários estudos sugeriram que a terapia à base de glicocorticoides acelera a recuperação, embora uma metanálise relatada em 2004 tenha concluído que não foram demonstradas evidências claras da eficácia dos glicocorticoides.5 Contudo, um recente estudo randomizado controlado, que envolveu 500 pacientes, constatou que a administração de 25 mg de prednisolona, 2 vezes/dia, dentro de um período de 72 horas após o aparecimento da condição, resultou na recuperação da função facial de 83% dos pacientes em 3 meses, comparativamente aos 64% de recuperação observados no grupo tratado com placebo.6 Diante das evidências sugestivas da participação do vírus do herpes simples na paralisia de Bell, os agentes antivirais frequentemente são prescritos aliados aos glicocorticoides. Alguns dados clínicos sustentam esta abordagem, porém um amplo estudo conduzido recentemente sobre este assunto constatou que não há vantagens em usar aciclovir, seja isolado ou combinado à prednisona. Em geral, a melhor evidência atualmente disponível favorece a instituição de um curso breve de glicocorticoides para tratamento da paralisia de Bell. O argumento que justifica a adição de um agente antiviral possui um atraente embasamento teórico, entretanto faltam evidências clínicas. O risco de efeitos adversos associados a cursos de curta duração destas classes farmacológicas é extremamente baixo.
A prevenção do ressecamento corneal com o uso de uma compressa ocular é importante para os pacientes incapazes de fechar totalmente o olho. A descompressão do nervo facial era amplamente utilizada, porém este procedimento foi abandonado em grande parte devido à falta de comprovação de sua eficácia. As injeções de toxina botulínica são úteis no tratamento do espasmo hemifacial.
A neuralgia do trigêmeo (ou tic douloureux) consiste em uma síndrome de dor facial que acomete pacientes de meia-idade e idosos. Nesta condição, o paciente apresenta uma dor intensa e paroxísmica que se manifesta várias a dúzias de vezes por dia. A dor frequentemente se assemelha a um choque elétrico intenso. Essa dor tem duração de alguns segundos e é localizada junto à distribuição do trigêmeo, geralmente nos territórios maxilar ou mandibular. Os paroxismos de dor podem ser deflagrados por estímulos táteis mínimos que incidam sobre a lateral afetada da face (p. ex., barbear, lavar o rosto ou mastigação). Entre os paroxismos, os pacientes podem apresentar uma dor de fundo, todavia na ausência de entorpecimento ou parestesia. A neuralgia do trigêmeo pode ser confundida com dor de dente, se os aspectos característicos não forem identificados. A neuralgia glossofaríngea, que é uma síndrome similar e bem menos comum, produz paroxismos de dor de garganta ou faringe.
Frequentemente, é difícil realizar o exame dos nervos cranianos em pacientes com neuralgia do trigêmeo, uma vez que os paroxismos de dor podem ser deflagrados pelo procedimento. Mesmo assim, não há perda sensorial nem outros sinais. Alteração em nervos cranianos e outros sinais neurológicos devem conduzir a uma investigação imediata em busca de lesões estruturais do nervo ou núcleo do trigêmeo ou, ainda, de uma lesão no tronco encefálico. A esclerose múltipla é o diagnóstico mais provável nesta situação, porém este diagnóstico em geral já está bem estabelecido antes da ocorrência dos episódios de neuralgia do trigêmeo.
A causa da neuralgia do trigêmeo é alvo de debates, embora muitos pesquisadores acreditem que o fator responsável seja a compressão ou distorção do nervo trigêmeo decorrente das pulsações oriundas de uma artéria cerebelar superior ectática. Podem ocorrer remissões espontâneas, mas estas raramente são duradouras.
A base do tratamento médico é a carbamazepina (600 a 1.200 mg/dia). As terapias alternativas são o baclofeno (30 a 60 mg/dia) e a gabapentina (300 a 1.200 mg, 3 vezes/dia), apesar da falta de evidências que sustentem a efetividade e superioridade destes fármacos em relação à carbamazepina.7 Se a neuralgia do trigêmeo se torna refratária à medicação, é possível tentar abordagens cirúrgicas que geralmente são bem-sucedidas. O ramo do trigêmeo acometido pode receber uma injeção de álcool ou fenol, ou uma lesão pode ser produzida percutaneamente no gânglio do trigêmeo. Entretanto, com estes procedimentos, o paciente troca a dor por graus variáveis de anestesia, e isto nem sempre é preferível. A descompressão microvascular para alívio dos supostos efeitos oriundos da artéria cerebelar superior parece ser altamente efetiva quando realizada por um cirurgião experiente. Entretanto, este procedimento requer uma osteotomia na fossa craniana posterior. A radiocirurgia com gamma knife está sendo empregada em alguns centros como alternativa à descompressão microvascular. O procedimento parece ser bastante seguro, porém os dados de seguimento a longo prazo obtidos ainda são limitados.8
As paralisias isoladas do 3º nervo craniano são incomuns, sendo difícil estabelecer um diagnóstico diferencial entre causas prejudiciais à vida e causas benignas. O aparecimento da condição é abrupto, e os sintomas usuais são a diplopia e a cefaleia. A diplopia é horizontal e vertical, sendo que a relação entre as 2 imagens muda conforme a direção do olhar fixo. A cefaleia pode ser retrorbital ou difusa. A maioria dos pacientes tem consciência da ptose, e alguns podem notar a dilatação da pupila.
As principais considerações relacionadas a um paciente com paralisia isolada do 3º nervo são a compressão por uma massa em expansão (particularmente um aneurisma da artéria comunicante posterior) e a conhecida lesão microvascular, em que a paralisia oculomotora é considerada isquêmica e decorrente de uma doença envolvendo o suprimento sanguíneo do nervo. Tão logo fique evidente que os únicos sinais apresentados pelo paciente são aqueles associados à paralisia do 3º nervo unilateral, um exame pode ajudar a distinguir uma lesão compressiva de uma lesão microvascular. Havendo paralisia pupilar, a hipótese de lesão compressiva torna-se mais provável, e, para estes casos, indica-se realizar exames de imagem craniana por tomografia computadorizada (TC) ou imagem de ressonância magnética (RM) e angiografia cerebral. Uma paralisia de 3º nervo poupadora da pupila é provavelmente microvascular. Os pacientes afetados são tipicamente idosos, diabéticos e hipertensos. Entretanto, no caso de um paciente jovem não diabético e normotenso, é recomendável realizar um exame de imagem craniana independentemente da preservação pupilar. Para a obtenção de imagens vasculares, a angiografia por ressonância magnética e a angiografia por TC (ambas técnicas não invasivas) estão substituindo cada vez mais a angiografia cerebral convencional.
As mononeuropatias de nervos do membros são comuns. A compressão mecânica do nervo em um local vulnerável é frequentemente considerada a causa da condição, ainda que nem sempre seja comprovada de maneira convincente. Um quadro clínico idêntico pode ser produzido por outras patologias, entre as quais um tumor de nervo. A síndrome autossômica dominante de suscetibilidade hereditária às paralisias por pressão deve ser considerada em casos de pacientes com história de mononeuropatias compressivas recorrentes ou quando o traumatismo precipitador é trivial, se comparado ao déficit resultante.
A STC é causada pela compressão do nervo mediano distal quando este atravessa o túnel localizado no punho, que é formado pelos ossos do carpo ao nível inferior e pelo ligamento do carpo ao nível superior [ver Lombalgia e problemas musculoesqueléticos comuns].
Diagnóstico. Na STC, o paciente queixa-se de um entorpecimento da mão (p. ex., sensações de picada, perda sensorial ou ambas), que é desagradável ou doloroso. O entorpecimento geralmente ocorre junto à distribuição sensorial mediana, porém alguns pacientes relatam envolvimento de todos os dedos da mão ou desconforto no antebraço. Os sintomas são caracteristicamente episódicos e surgem à noite (muitas vezes, chegam a acordar o paciente), bem como durante a prática de atividades em que os punhos são flexionados ou estendidos (p. ex., tricotar ou segurar o volante do carro). Agitar vigorosamente a mão geralmente proporciona alívio imediato, e isto é bastante sugestivo de STC. O entorpecimento pode tornar difícil ou desconfortável para o paciente utilizar a mão apropriadamente, contudo é incomum haver um enfraquecimento verdadeiro. Quando presente, o comprometimento do movimento preênsil de pinçamento relacionado ao enfraquecimento dos músculos tenares pode interferir na execução de certas atividades, como o ato de segurar uma caneta. No caso de alguns pacientes (particularmente os idosos), as queixas sensoriais podem ser mínimas, e os sintomas de enfraquecimento tenar podem predominar.
Na maioria dos pacientes, a STC provavelmente se desenvolve em decorrência de um estreitamento congênito do túnel do carpo. Certas ocupações que exigem a flexão/extensão do punho repetidas vezes (p. ex., marcenaria, costura e usar o teclado do computador) podem predispor o paciente ao desenvolvimento da STC. Esta síndrome é comum durante a gestação (provavelmente devido à retenção de líquido generalizada) e pode ocorrer após lesões como a fratura de Colles. Ocasionalmente, a STC ocorre no contexto de uma doença sistêmica que diminui a margem interna do túnel do carpo, como a artrite reumatoide, hipotireoidismo, amiloidose e acromegalia. Os pacientes com polineuropatias diabéticas e outros tipos de polineuropatia podem apresentar predisposição ao desenvolvimento de STC sobreposta e outras mononeuropatias. Em pacientes com diabetes, a rigidez e o volume aumentados do ligamento do carpo podem ser fatores importantes.
O exame pode revelar a existência de déficits sensoriais ou motores em uma distribuição mediana, mas frequentemente resulta normal, em particular no caso de pacientes jovens que apresentam sintomas intermitentes. A percussão sobre a região do nervo mediano no punho pode produzir uma parestesia transiente nos dedos inervados por este nervo (sinal de Tinel). Segurar o punho em extensão ou flexão forçada durante 1 a 2 minutos pode precipitar os sintomas sensoriais do paciente, que são prontamente aliviados quando o braço é solto e pende junto à lateral do corpo, sem angulação do punho (sinal de Phalen). Nem o sinal de Tinel nem o sinal de Phalen são totalmente específicos ou sensíveis.
Os exames de condução nervosa muitas vezes ajudam a sustentar um diagnóstico clínico de STC. O princípio essencial consiste em demonstrar o retardo da condução no nervo mediano no túnel do carpo em relação à condução nos nervos vizinhos (ulnar ou radial). Os exames de condução nervosa apresentam alta sensibilidade diagnóstica para STC, mas podem resultar normais em casos brandos. Após a cirurgia, os resultados destes exames em geral melhoram sem, contudo, normalizar, mesmo que os sintomas do paciente sejam completamente aliviados.
Tratamento. Para os pacientes que apresentam sintomas leves e não possuem déficits, a tranquilização pode ser suficiente. O uso de um imobilizador que mantenha o punho em posição neutra muitas vezes é efetivo como medida dirigida aos sintomas noturnos. Há pacientes que são beneficiados pela aplicação periódica de injeções de glicocorticoide dentro do ligamento do carpo. O seccionamento do ligamento do carpo, que constitui um procedimento de baixo risco, é amplamente realizado no tratamento da STC e parece proporcionar uma melhora substancial e duradoura para a maioria dos pacientes. A cirurgia é recomendada para os pacientes com sintomas problemáticos que são irresponsivos aos tratamentos não cirúrgicos, bem como para aqueles com déficits sensoriais ou motores progressivos. As técnicas endoscópicas de liberação do túnel do carpo parecem aliviar os sintomas de modo similar às técnicas cirúrgicas abertas convencionais. Os procedimentos endoscópicos podem permitir que os pacientes retomem o trabalho mais cedo, porém são mais onerosos e estão associados a taxas de complicações provavelmente um pouco mais altas.9
A neuropatia ulnar resulta mais frequentemente de uma lesão mecânica no cotovelo, onde o nervo repousa ao nível subcutâneo e sobre o assoalho inflexível do sulco epicondilar. Distalmente ao cotovelo, o nervo pode ser comprimido ao passar sob a aponeurose do flexor ulnar do carpo e entrar no compartimento muscular profundo do antebraço. No punho, o nervo volta a ser relativamente superficial e pode ser comprimido com a sustentação de peso excessivo nas mãos, como ocorre quando longas distâncias são percorridas de bicicleta.
Diagnóstico. Os pacientes com neuropatias ulnares apresentam graus variados de sintomas motores e sensoriais. Os pacientes observadores percebem que o distúrbio sensorial envolve a metade medial do dedo anelar, além do dedo mínimo e da porção medial da mão. O enfraquecimento, quando presente, geralmente permanece confinado na mão. Esta, por sua vez, aparentemente pode apresentar um enfraquecimento geral, ou podem haver queixas específicas, como a dificuldade para segurar uma caneta ou esticar os dedos.
O enfraquecimento e o desgaste são mais facilmente identificados no 1º músculo interósseo dorsal (entre o polegar e o indicador) e nos músculos hipotenares. A perda total da sensação ulnar é incomum, porém a sensação frequentemente é alterada junto à distribuição ulnar. A comparação das faces medial (ulnar) e lateral (mediana) do dedo anelar pode fornecer evidências convincentes da existência de uma neuropatia ulnar. A produção de sensações de entorpecimento ou elétricas na mão a partir da manipulação suave do nervo no cotovelo favorece este como o sítio da patologia. Os exames eletrodiagnósticos são úteis em casos de neuropatia ulnar, tanto para confirmar o diagnóstico como para localizar o sítio preciso da lesão.
Em casos raros, a neuropatia ulnar pode resultar da aplicação de uma compressão na porção superior do braço ou na mão. Devido ao arranjo dos feixes motores e sensoriais no punho e na mão, a compressão ulnar na mão pode envolver apenas os ramos motores. Esta condição resulta no enfraquecimento da mão na ausência de dor e de sintomas sensoriais, podendo ser confundida com uma doença neuronal motora em fase inicial. Este tipo de neuropatia ulnar é causado pela sustentação de peso nas mãos, como pode ocorrer com o uso de andador de 4 apoios ou quando se anda de bicicleta por tempo prolongado.
Tratamento. O tratamento apropriado para a neuropatia ulnar no cotovelo ainda não foi definido. Embora a condição seja comum, as informações existentes sobre sua história natural são surpreendentemente limitadas. É comum haver melhora espontânea, em particular nos pacientes que passam por um episódio nitidamente precipitante de compressão. No caso dos pacientes em que os traumatismos repetidos no nervo são considerados a causa da condição, é comum a neuropatia ulnar melhorar quando o ato de se apoiar sobre os cotovelos é evitado. Em pacientes selecionados que apresentam sinais e sintomas progressivos mesmo quando submetidos à terapia conservadora, é possível considerar um dos vários procedimentos cirúrgicos existentes. Faltam comparações randomizadas entre terapias cirúrgicas e conservadoras, embora um recente estudo comparativo não randomizado tenha sugerido que a probabilidade de obter resultados satisfatórios pode ser maior para os pacientes não submetidos à cirurgia.10
A neuropatia do plexo braquial (também conhecida como amiotrofia neurálgica e síndrome de Parsonage-Turner) é incomum, embora seja provavelmente subdiagnosticada. A história é típica. O paciente apresenta uma dor severa e ardente, similar à dor da bursite, localizada no ombro e sem nenhum fator precipitador evidente. Decorridos vários dias a 2 semanas, a dor desaparece, e o paciente percebe o enfraquecimento do braço. Mais frequentemente, os músculos do cíngulo do membro superior são afetados, contudo pode haver envolvimento da mão ou do antebraço. Em alguns casos, o enfraquecimento é notavelmente focal ou irregular. Pode haver perda sensorial com distribuição em dragona, mas este não constitui um aspecto proeminente. A recuperação dos pacientes é gradual, e em 90% dos casos há pouco ou nenhum déficit após um período de 1 a 2 anos do aparecimento da condição. Em alguns pacientes, ambos os braços são afetados ao mesmo tempo ou de modo sequencial, e alguns pacientes apresentam episódios recorrentes.
A neuropatia do plexo braquial é mais comum em jovens e homens de meia-idade, mas pode afetar indivíduos de ambos os sexos e de todas as faixas etárias. O exame de biópsias do plexo braquial revelou a ocorrência de uma inflamação linfocítica exuberante, e o distúrbio parece ser imunomediado.11 Entretanto, não se sabe se as terapias imunomoduladoras são benéficas. Também existe uma forma autossômica dominante de neuropatia do plexo braquial, que é caracterizada por episódios recorrentes de dor e enfraquecimento do ombro.
A neuropatia peroneal é mais frequentemente o resultado de uma lesão no nervo peroneal, no ponto onde o nervo envolve a cabeça da fíbula, passando da fossa poplítea ao compartimento anterior da perna. Os ramos peroneais distais são superficiais ao nível do tornozelo, onde às vezes são comprimidos por calçados apertados.
Diagnóstico. Os pacientes com neuropatia peroneal no joelho geralmente apresentam pé caído unilateral, em consequência do enfraquecimento dos dorsiflexores do tornozelo. Entretanto, o pé caído pode ser causado por uma radiculopatia de L5, plexopatia lombossacral, neuropatia ciática proximal ou lesão no córtex cerebral motor contralateral. A radiculopatia de L5, sendo o diagnóstico diferencial mais comum, é sugerida pela existência de uma lombalgia que irradia para a perna e de um enfraquecimento dos inversores do tornozelo e abdutores do quadril, além do pé caído. Os exames de eletrodiagnóstico podem ser particularmente úteis para fins de localização da lesão em pacientes com pé caído.
O traumatismo (p. ex., fratura da fíbula) costuma ser responsável pela neuropatia peroneal do joelho. A compressão produzida durante os estados de consciência alterada (p. ex., durante a anestesia e o coma) representa outra causa comum. A herniação de um cisto de Baker pode comprometer o nervo. Em pacientes que não apresentam este tipo de fator precipitador, a neuropatia peroneal no joelho é atribuída, na maiorira das vezes, ao hábito de sentar com as pernas cruzadas – uma explicação possível, ainda que difícil de comprovar.
Tratamento. O tratamento da neuropatia peroneal no joelho geralmente é conservador. Na maioria dos pacientes que sofrem traumatismo no nervo, a melhora ocorre com o passar do tempo, em particular quando o nervo peroneal distal apresenta alguma função residual. No traumatismo severo, o nervo às vezes é totalmente transeccionado, e o resultado costuma ser precário. Neste caso, a anastomose cirúrgica é o procedimento que oferece melhores chances de recuperação. Se não houver nenhuma outra causa evidente da neuropatia peroneal, os pacientes podem ser orientados a evitar sentar com as pernas cruzadas. A melhora pode ocorrer lentamente. Seja qual for a causa, o paciente deve tentar usar uma órtese de tornozelo-pé para compensar o pé caído. A maioria dos pacientes acaba descobrindo que este aparelho simples promove uma melhora significativa da habilidade de caminhar [ver Tratamento de sintomas neuropáticos, anteriormente].
A meralgia parestésica é caracterizada por sintomas sensoriais junto à distribuição do nervo cutâneo lateral da coxa. Os pacientes relatam certo grau de combinação de perda sensorial, parestesia com formigamento e hipersensibilidade sobre a região anterolateral da coxa. Os limites da área anormal tipicamente apresentam uma definição nítida e não estão associados a anomalias motoras ou reflexas. A meralgia parestésica geralmente é atribuída à compressão do nervo cutâneo lateral pelo ligamento inguinal, à medida que este passa do retroperitônio para a região anterior da coxa. A síndrome é mais comum em obesos, talvez devido ao aumento do estresse mecânico sobre o ligamento inguinal. Em casos raros, o nervo cutâneo lateral é lesado durante a cirurgia ou pode ser comprometido pela presença de linfonodos inguinais aumentados ou outras massas. A maioria dos pacientes precisa apenas ser tranquilizada, pois os sintomas costumam desaparecer com o passar do tempo. O tratamento com medicamentos, como a amitriptilina, pode ser experimentado diante da manifestação de sintomas incômodos e persistentes. A exploração cirúrgica da região inguinal é o último recurso.
A plexopatia lombossacral é semelhante à neuropatia do plexo braquial. A história típica de envolvimento do plexo lombossacral começa com o aparecimento de uma dor na região anterior da coxa, geralmente severa e contínua, que dura várias semanas. Conforme a dor vai desaparecendo, surge um enfraquecimento que mais comumente envolve o quadríceps e os flexores do quadril. Este enfraquecimento muitas vezes permanece assintomático até o paciente sofrer uma queda, dando a falsa impressão de evento agudo. Pode haver um desgaste impressionante do quadríceps, e o joelho não apresenta reflexo. Os sinais sensoriais são mínimos. Os sinais e sintomas iniciais às vezes envolvem apenas as partes lombar e sacral do plexo, contudo o processo tende a se disseminar por todo o plexo e envolver o lado contralateral. Uma recuperação bastante gradual é a regra.
A plexopatia lombossacral caracteristicamente afeta homens de meia-idade e idosos. Os pacientes com frequência são diabéticos (neste caso, às vezes são empregados os termos amiotrofia diabética, neuropatia femoral diabética, neuropatia diabética proximal e neuropatia do radioplexo lombossacral diabética), embora uma síndrome idêntica ocorra em indivíduos não diabéticos. A patogênese da plexopatia lombossacral é tema de debate. A base isquêmica há muito é defendida, em particular porque a condição ocorre com frequência em pacientes que apresentam complicações microvasculares do diabetes. Contudo, o contrapeso das evidências fornecidas pelos exames de biópsia agora favorece a participação de um processo inflamatório, que em alguns casos se assemelha a uma forma restrita de vasculite necrotizante.12
Os exames eletrodiagnósticos são úteis para sustentar o diagnóstico de plexopatia lombossacral. Frequentemente, é necessário realizar exames de imagem da pelve para excluir as hipóteses de invasão maligna do plexo lombar, hematoma retroperitoneal e abscesso do psoas.
A imunomodulação tem sido recomendada para o tratamento da plexopatia lombossacral. Os agentes utilizados com esta finalidade incluem os corticosteroides, azatioprina e imunoglobulinas endovenosas, apesar da falta de estudos controlados randomizados. A dor dura mais do que algumas semanas e pode ser aliviada por fármacos como a amitriptilina.
Determinar que um paciente possui uma polineuropatia geralmente não é uma tarefa difícil. Entretanto, como as centenas de causas possíveis de polineuropatia produzem sinais e sintomas semelhantes, pode ser um desafio chegar a um diagnóstico específico. O diagnóstico diferencial pode ser simplificado pela aplicação das seguintes perguntas:
1. Os sinais e sintomas estão ajustados a um padrão diferente daquele associado à polineuropatia sensoriomotora simétrica? Várias síndromes distintas, diferentes da polineuropatia sensoriomotora simétrica, apresentam diagnósticos diferenciais mais restritos. Alguns exemplos são as mononeuropatias múltiplas (também conhecidas como mononeurites múltiplas), neuropatia sensorial atáxica, neuropatia predominantemente autonômica ou pura, e polineuropatia de início agudo [Tabela 2].
2. Os exames de condução nervosa sugerem a ocorrência de desmielinização? Após a obtenção da história e realização do exame físico, os exames de condução nervosa são a próxima etapa lógica. Se for observado um retardo acentuado da velocidade de condução ou diante de outra evidência eletrofisiológica sugestiva de desmielinização, o diagnóstico diferencial é mais restrito [Tabela 3].
3. A polineuropatia é adquirida ou hereditária? Esta pergunta representa um aspecto importante a ser considerado em todos os casos de pacientes com polineuropatia simétrica crônica. A neuropatia hereditária é a causa mais frequentemente negligenciada de polineuropatia não diagnosticada.13 O pé cavo e os dedos do pé em martelo podem ser indícios significativos de um diagnóstico de neuropatia hereditária e devem ser sempre procurados de forma específica. As neuropatias hereditárias frequentemente são brandas, não causam incapacitação e apresentam progressão bastante lenta. Por isso, não costumam se manifestar até a meia-idade ou subsequentemente e com frequência não são identificadas nas famílias [Tabela 4].
Tabela 2. Padrões distintivos de neuropatia com diagnósticos diferenciais limitados
|
Mononeuropatias múltiplas |
|
Vasculite |
|
Diabetes melito* |
|
Sarcoidose |
|
Crioglobulinemia |
|
Suscetibilidade hereditária às paralisias por pressão |
|
Hanseníase |
|
Infecção pelo HIV* |
|
Invasão neoplásica de nervos ou raízes nervosas |
|
Granulomatose linfomatoide |
|
Doença de Lyme |
|
Neuropatia multifocal com bloqueio de condução |
|
Neurofibromatose |
|
Neuropatia sensorial atáxica |
|
Neuropatia sensorial paraneoplásica (câncer de mama ou de pequenas células do pulmão) |
|
Síndrome de Sjögren |
|
Neuropatia associada à proteína monoclonal |
|
Toxicidade da cisplatina |
|
Toxicidade da piridoxina |
|
Deficiência de vitamina B12 |
|
Tabe dorsal |
|
Poligangliopatia sensorial inflamatória não maligna |
|
Ataxia espinocerebelar hereditária |
|
Neuropatia com características autonômicas proeminentes |
|
Diabetes melito |
|
Amiloidose |
|
Neuropatia autonômica e sensorial hereditária |
|
Pandisautonomia aguda |
|
Poligangliopatia autonômica sensorial paraneoplásica |
|
Polineuropatia de início agudo |
|
Síndrome de Guillain-Barré e variantes |
|
Neuropatia vasculítica |
|
Porfiria |
|
Doença de Lyme |
|
Substâncias tóxicas (p. ex., arsênico, chumbo, tálio) |
|
Poliomielite |
|
Difteria |
|
Botulismo |
*Geralmente, sob a forma de polineuropatia simétrica.
Tabela 3. Diagnóstico diferencial da polineuropatia desmielinizante
|
Síndrome de Guillain-Barré (PRDI aguda) |
|
PRDI crônica |
|
Neuropatia associada à proteína monoclonal |
|
Mieloma osteoesclerótico |
|
Difteria |
|
Toxicidade do maleato de perexilina |
|
Neuropatia sensorial e motora hereditária de tipo 1 |
|
Suscetibilidade hereditária às paralisias por pressão |
PRDI = polirradiculoneuropatia desmielinizante inflamatória
Tabela 4. Características básicas das neuropatias hereditárias
|
|
Neuropatia sensorial e motora hereditária |
Neuropatia autonômica e sensorial hereditária |
Neuropatia motora hereditária |
|
Aspectos clínicos |
Desgaste muscular distal, enfraquecimento e perda sensorial |
Perda sensorial distal Disautonomia variável |
Enfraquecimento e desgaste muscular, mais frequentemente proximal |
|
Pé cavo |
Artropatia neurogênica | ||
|
Úlceras plantares | |||
|
Sítio de anormalidade |
Axônio ou célula de Schwann |
Neurônios ganglionares autonômicos e de raiz dorsal |
Células do corno anterior |
|
Herança |
Na maioria dos casos, autossômica dominante |
Autossômica dominante |
Autossômica dominante |
|
Em alguns casos, autossômicos recessivos ou ligados ao X |
Autossômica recessiva |
Autossômica recessiva | |
|
Defeitos genéticos conhecidos |
Doença de Charcot-Marie-Tooth de tipo 1 |
Desconhecidos |
Doença de Werdnig-Hoffmann (e outros tipos de aparecimento na infância) Linkage a 5q Síndrome de Kennedy (atrofia muscular bulboespinal ligada ao X) Defeito no gene codificador do receptor de androgênio (repetição da trinca CAG) |
|
Duplicação ou mutação em ponto do gene PMP22 (cromossomo 17) | |||
|
Mutações em ponto do gene Po (cromossomo 1) | |||
|
Mutações em ponto do gene Cx32 (cromossomo X) | |||
|
Mutações em ponto do gene de resposta de crescimento inicial (cromossomo 10) | |||
|
Doença de Charcot-Marie-Tooth de tipo 2 | |||
|
Linkage a 1p, 3q e 7p |
As investigações adicionais das neuropatias axonais sensoriomotoras simétricas podem ser guiadas pelas informações colaterais fornecidas pela história. Diante da possível existência de uma neuropatia hereditária, os parentes de 1º grau do paciente devem ser examinados. A causa de uma neuropatia adquirida pode ser uma medicação tomada pelo paciente [Tabela 5] ou alguma doença médica concomitante, mais comumente o diabetes melito e a insuficiência renal crônica. Em alguns casos, a neuropatia pode ser a manifestação de uma vasculite sistêmica ou malignidade oculta.
Tabela 5. Fármacos que podem causar polineuropatia
|
Antineoplásicos |
|
Cisplatina (a neuropatia sensorial atáxica é dose-limitante) |
|
Bortezomibe |
|
Suramina |
|
Taxoides (docetaxel, paclitaxel) |
|
Alcaloides da vinca (vincristina, vinblastina; a neuropatia é dose-limitante) |
|
Antirretrovirais |
|
Didanosina (didesoxinosina) |
|
Estavudina (d4T) |
|
Zalcitabina (didesoxicitidina) |
|
Outros antimicrobianos |
|
Cloranfenicol |
|
Dapsona (principalmente a neuropatia motora; doses mais altas do que na terapia da hanseníase) |
|
Isoniazida (evitável pela suplementação com piridoxina) |
|
Metronidazol (cursos prolongados) |
|
Nitrofurantoína |
|
Fármacos utilizados em doenças reumatoides |
|
Cloroquina |
|
Colchicina |
|
Ouro |
|
Talidomida |
|
Diversos |
|
Amiodarona (antiarrítmico) |
|
Dissulfiram (antiabuso) |
|
Perexilina (neuropatia desmielinizante) |
|
Fenitoína (após anos de uso; neuropatia bastante discreta) |
|
Piridoxina (megadoses causam neuropatia sensorial atáxica) |
|
Sinvastatina |
Para os pacientes adultos, os exames laboratoriais de rotina considerados razoáveis podem incluir a determinação da glicemia de jejum, concentração de hemoglobina glicada, níveis séricos de creatinina, hemograma completo, radiografia torácica (para fumantes), velocidade de hemossedimentação e concentração de proteína C reativa, fator reumatoide, anticorpos antinúcleo e – para pacientes com mais de 50 anos de idade – ensaios de imunofixação de proteínas plasmáticas e urinárias. O exame de biópsia de nervo pode ser valioso para a busca de informações específicas. O exame de biópsia não faz parte da rotina e tem utilidade somente em raros casos, como último recurso.
A neuropatia periférica é comum em pacientes com diabetes melito. Dentre os vários tipos de neuropatia diabética, a mais comum é sem dúvida a neuropatia sensoriomotora simétrica distal, comumente referida como polineuropatia diabética. Estimar a incidência e severidade da polineuropatia diabética é uma tarefa difícil por causa dos seguintes fatores: (1) o modo como a condição é definida; (2) as populações estudadas; e (3) os esforços empreendidos para garantir a exclusão de outras possíveis causas de neuropatia. Em um estudo prospectivo populacional sobre norte-americanos, principalmente os descendentes de norte-europeus, a polineuropatia diabética foi encontrada em 54% dos pacientes com diabetes melito de tipo 1 e em 45% dos pacientes com diabetes melito de tipo 2.14 Entretanto, a polineuropatia sintomática foi encontrada em apenas 15% da coorte, e somente 1 paciente apresentou déficits neurológicos incapacitantes. A severidade da polineuropatia apresentou uma correlação mais estreita com o grau de hiperglicemia (hemoglobina glicada média) do que com a duração do diabetes.15 Neste estudo e também em outros estudos mais amplos, a prevalência da polineuropatia diabética aumentou com a duração do diabetes, e houve uma forte correlação entre a presença de polineuropatia diabética, retinopatia e nefropatia. Um corolário prático importante destas observações é o de que o diagnóstico de polineuropatia diabética provavelmente está incorreto quando determinado em um paciente recém-diagnosticado com diabetes e sem outras complicações diabéticas.
Patogênese. A patogênese da polineuropatia diabética continua sendo discutida. Os dados fornecidos pelos exames de eletrofisiologia e biópsia de nervo indicam que a degeneração axonal constitui o principal processo patológico, embora possa haver desmielinização secundária. Os potenciais mecanismos da patogênese incluem um processo isquêmico secundário à microangiopatia, análogo à nefropatia e retinopatia diabética; alterações em proteínas estruturais, em consequência das reações de glicação governadas pela hiperglicemia crônica; ou lesão causada pelas espécies reativas de oxigênio geradas no metabolismo alterado da glicose.16 Estes mecanismos não são mutuamente exclusivos, podendo coexistir e interagir em muitos pacientes.
Diagnóstico. A polineuropatia apresenta a clássica distribuição em “bota e luva” dos sintomas, em geral uma combinação de perda sensorial e sensação desagradável de entorpecimento ou ardência. A perda sensorial nos pés e dedos da mão, bem como o leve enfraquecimento dos pés e tornozelos são típicos. É possível esperar que a polineuropatia diabética piore lentamente, com o passar dos anos,17 ainda que esta progressão possa ser influenciada pelo tratamento do diabetes. Os déficits neurológicos incapacitantes e a piora rápida não são eventos esperados na polineuropatia diabética, mas se forem observados devem levar à investigação imediata de outras possíveis causas.
Tratamento. Os dados a respeito do efeito do controle glicêmico sobre a polineuropatia diabética continuam sendo acumulados. Dentre os pacientes com diabetes de tipo 1 seguidos pelo Diabetes Control and Complications Trial, 5% dos pacientes submetidos à terapia com controle glicêmico rigoroso desenvolveram polineuropatia ao passo que 13% dos pacientes tratados do modo convencional desenvolveram a condição.18 Um estudo de seguimento de uma coorte de receptores de transplantes de pâncreas demonstrou a ocorrência de melhora significativa dos parâmetros de condução nervosa e, talvez, uma modesta melhora clínica. Nos pacientes do grupo controle, tratados com regimes convencionais à base de insulina e não submetidos ao transplante, houve piora das condições no decorrer do mesmo período.19 Em contraste, o United Kingdom Prospective Diabetes Study, que envolveu quase 4.000 pacientes com diabetes de tipo 2, falhou em mostrar a existência de uma diferença na prevalência da neuropatia entre pacientes tratados de maneira convencional e aqueles submetidos a um controle glicêmico rigoroso, durante um período de até 10 anos de seguimento.20 Sendo assim, o controle glicêmico ideal parece diminuir o risco de desenvolvimento de polineuropatia diabética em pacientes com diabetes de tipo 1, mas nenhum efeito protetor foi demonstrado em pacientes com diabetes de tipo 2. Na neuropatia estabelecida, um controle glicêmico rigoroso pode produzir efeitos benéficos, contudo ainda é preciso demonstrar se alguma melhora clinicamente significativa é alcançada após várias décadas. Uma questão relacionada é se os pacientes que têm apenas comprometimento da tolerância à glicose apresentam risco aumentado de desenvolvimento de neuropatia e se esta poderia ser prevenida nestes pacientes. Este aspecto ainda não foi esclarecido, mas as potenciais implicações podem ser amplas, e há estudos adicionais em andamento.21
Os estudos sobre os possíveis mecanismos patogênicos envolvidos no desenvolvimento da polineuropatia diabética conduziram a uma miríade de estudos clínicos sobre agentes com potencial de prevenir ou amenizar a neuropatia. Exemplificando, uma consequência intracelular da hiperglicemia crônica é o aumento da atividade da via de poliol – uma sequência de reações por meio das quais as células conseguem produzir frutose a partir da glicose. Acredita-se que uma atividade aumentada da via de poliol exerce vários efeitos deletérios sobre o metabolismo celular, sendo que a via pode ser bloqueada pela inibição da etapa limitante da velocidade do processo, a qual é catalisada pela aldose redutase. Os estudos sobre os inibidores da aldose redutase em animais diabéticos mostraram-se promissores, contudo os estudos clínicos realizados com seres humanos foram decepcionantes. Uma metanálise recente concluiu que não há evidências da efetividade dos inibidores de aldose redutase no tratamento da neuropatia diabética em seres humanos.22 Outros agentes testados foram: acilcarnitina, aminoguanidina (diminui os produtos finais da glicosilação avançada), óleo de onagra (uma fonte enriquecida de ácido linoleico), ácido alfalipoico (um varredor de radicais livres do oxigênio) e fator de crescimento de nervo. Um amplo estudo randomizado, controlado por placebo, sobre o uso de fator de crescimento de nervo recombinante humano ao longo de 12 meses, falhou em demonstrar a melhora de diversas medidas clínicas e laboratoriais de função, apesar dos resultados promissores obtidos pelos estudos de fase II.23 Um estudo recente sobre o ácido alfa-lipoico encontrou alguns benefícios, em comparação ao grupo tratado com placebo, embora o tempo de seguimento tenha sido de apenas 5 semanas.24
Os pacientes com polineuropatia diabética devem receber aconselhamento sobre cuidados adequados para os pés, incluindo uma inspeção minuciosa diária das superfícies plantares, para que as lesões menores possam ser detectadas ainda em fase inicial e as complicações significativas, como as úlceras plantares ou a osteomielite, possam ser prevenidas. A adição de medidas de tratamento da dor neuropática frequentemente se faz necessária.
As outras variedades de neuropatia diabética geralmente têm a polineuropatia diabética como pano de fundo. Há 2 mononeuropatias (oftalmoplegia diabética e neuropatia diabética proximal) que também podem ocorrer em pacientes diabéticos [ver Neuropatia oculomotora e Plexopatia lombossacral, anteriormente].
Neuropatia autonômica diabética. Graus variáveis de neuropatia autonômica diabética são encontrados na maioria dos pacientes com polineuropatia diabética. Entretanto, os sinais e sintomas autonômicos predominam em alguns indivíduos. A hipotensão ortostática, o comprometimento da motilidade gastrintestinal (incluindo a gastroparesia) e o embotamento dos sintomas de alerta de mediação simpática de hipoglicemia são problemas importantes para o tratamento. Alguns sintomas autonômicos podem ser tratados de maneira efetiva. A gastroparesia, por exemplo, pode ser tratada com metoclopramida, e o ortostatismo pode ser tratado com meias elásticas, expansão de volume e vasopressores (p. ex., midodrina).
Neuropatia truncal diabética. A neuropatia truncal diabética consiste em uma síndrome dolorosa que afeta um ou vários dermátomos torácicos, geralmente de modo unilateral. Do ponto de vista clínico, trata-se de um processo de radiculopatia torácica com desenvolvimento abrupto de hipersensibilidade tátil ou perda sensorial, apresentando distribuição em dermátomos. O envolvimento motor geralmente pode ser demonstrado por eletromiografia, e às vezes há desenvolvimento de uma saliência assimétrica no abdome, devido ao enfraquecimento unilateral da musculatura abdominal. O principal diagnóstico alternativo é a radiculopatia por herpes zóster, que geralmente é identificada pelo aparecimento de erupções cutâneas vesiculares. A radiculopatia diabética truncal é autolimitada, embora os sintomas agravantes possam persistir por várias semanas. A administração de analgésicos, suplementados com amitriptilina, gabapentina ou outros fármacos dirigidos à dor neuropática, constitui a base da abordagem de tratamento.
Até 60% dos pacientes com insuficiência renal crônica desenvolvem polineuropatia. O risco de neuropatia urêmica está relacionado à duração e severidade da insuficiência renal. O retardo da velocidade de condução motora é percebido quando a depuração da creatinina cai para menos de 10% do normal, embora a correlação existente entre o retardo da condução e os sintomas seja apenas grosseira. A neuropatia urêmica tem natureza sensoriomotora, com predominância distal dos sinais e sintomas. Uma vez estabelecida, a neuropatia urêmica tende a piorar lentamente e o sintoma mais problemático consiste em uma desagradável disestesia nos pés. O enfraquecimento motor incapacitante raramente ocorre. O principal achado patológico na neuropatia urêmica é a degeneração axonal, que é mais abundante nas partes mais distais do SNP. Em termos de etiologia, o comprometimento neuronal ou da função axonal provavelmente resulta do acúmulo de uma substância neurotóxica ou de substâncias que normalmente são excretadas pelos rins, contudo os detalhes são desconhecidos. Tanto a diálise como o transplante renal geralmente produzem um efeito benéfico. A diálise pode prevenir, estabilizar ou muitas vezes diminuir a severidade da neuropatia. Os efeitos do transplante renal podem ser notáveis, e até mesmo uma neuropatia urêmica severa pode apresentar chances de melhora nos meses subsequentes ao transplante. A ampla disponibilidade destes tratamentos tornou a neuropatia urêmica sintomática relativamente incomum.
Uma polineuropatia axonal distal branda às vezes ocorre em pacientes com hipotireoidismo, acromegalia ou policitemia. Estas neuropatias não são incapacitantes, porém os sintomas sensoriais podem ser problemáticos. Os pacientes internados em unidades de terapia intensiva (UTI) apresentando síndrome de disfunção de múltiplos órgãos e sepse por vezes desenvolvem uma severa polineuropatia axonal. A patogênese desta condição, denominada polineuropatia do paciente crítico, é desconhecida.25 A suspeita de polineuropatia do paciente crítico em geral surge quando um paciente de UTI não pode ser desmamado do ventilador, mesmo apresentando uma função cardiopulmonar adequada, ou quando um paciente de UTI consciente apresenta enfraquecimento de membro. Por ser difícil examinar adequadamente o SNP em indivíduos severamente doentes e como a polineuropatia do paciente crítico constitui um diagnóstico de exclusão, pode ser difícil estabelecer o diagnóstico com clareza. Um quadro clínico semelhante também pode ser produzido pela miopatia do paciente crítico, sobretudo em pacientes tratados com altas doses de glicocorticoides e bloqueadores neuromusculares de ação prolongada (p. ex., vecurônio). Dados limitados sugerem que a neuropatia do paciente crítico melhora nos pacientes que sobrevivem à condição.26
As polineuropatias hereditárias são comuns e frequentemente negligenciadas, sobretudo em adultos.12 Muitos pacientes permanecem relativamente assintomáticos durante vários anos e, por este motivo, podem vir a procurar a atenção médica somente ao atingirem a meia-idade ou mesmo uma idade mais avançada. Uma progressão bastante lenta dos sintomas, no decorrer de vários anos, é sugestiva de causa hereditária. Os pacientes com polineuropatias hereditárias podem queixar-se de um entorpecimento que, para eles, geralmente significa diminuição da sensibilidade. Os sintomas de formigamento ou parestesia com agulhadas e alfinetadas em um paciente com polineuropatia favorecem uma causa adquirida. Entretanto, a queixa de ardência nos pés é inespecífica, sendo observada nas polineuropatias hereditárias e adquiridas. Como regra, as neuropatias hereditárias surgem e evoluem de modo simétrico. Uma progressão assimétrica sugere uma polineuropatia adquirida.
Diante da suspeita de polineuropatia hereditária, pode ser necessário empreender esforços consideráveis para estabelecer o diagnóstico. O simples interrogatório do paciente para obtenção de uma história familiar de neuropatia raramente é produtivo. As perguntas devem ter caráter mais geral, tais como perguntar se algum familiar apresenta queixas relacionadas ao pé, tem deformidade no pé (em especial no peito do pé) ou necessita de sapatos especiais, usa bengala ou suportes. Uma história familiar negativa não exclui um diagnóstico de neuropatia hereditária. Por causa da cronicidade ou suavidade, ou ainda por ambos, a condição muitas vezes não é percebida na família. Nos casos recessivos, por definição, nenhum dos pais é afetado e é possível que os irmãos(ãs) sejam poupados(as). A paternidade pode não estar tão bem definida quanto parece. A próxima etapa mais efetiva consiste em examinar os parentes de 1º grau, em particular aqueles com qualquer tipo de história de problemas com o pé ou com a habilidade de caminhar. Embora este procedimento seja demorado, apresenta um bom rendimento e é mais custo-efetivo do que uma bateria de exames laboratoriais que podem resultar todos negativos.
O conhecimento sobre os defeitos genéticos específicos relacionados às diversas neuropatias hereditárias está se expandido rapidamente,27 porém a classificação clínica ainda é importante. O termo “doença de Charcot-Marie-Tooth” é aplicado com frequência de maneira indistinta a qualquer tipo de neuropatia hereditária, embora seja mais útil dividir as neuropatias hereditárias em 3 categorias principais, de acordo com as características clínicas: (1) neuropatia sensorial e motora hereditária; (2) neuropatia autonômica e sensorial hereditária; e (3) neuropatia motora hereditária (também denominada atrofia muscular espinal) [Tabela 4]. Até o momento, foram identificados 20 genes responsáveis pela neuropatia hereditária, e os testes para detecção de alguns destes defeitos genéticos hoje são amplamente disponibilizados (p. ex., duplicação do gene PMP22 [neuropatia sensorial e motora hereditária de tipo 1] e mutações no gene da conexina-32 [neuropatia sensorial e motora hereditária ligada ao X]). Uma fonte abrangente e atualizada de informações sobre os testes genéticos atualmente disponíveis pode ser encontrada em www.geneclinics.org. Embora ainda não existam terapias específicas para as neuropatias hereditárias, o diagnóstico correto continua sendo importante para fins de prognóstico, ensino e aconselhamento genético. De uma forma geral, os déficits associados às neuropatias hereditárias apresentam uma evolução bastante lenta, a expectativa de vida é normal, e a maioria dos pacientes preserva a capacidade de andar por toda a vida.
Síndrome de Guillain-Barré (SGB). A síndrome de Guillain-Barré (SGB) – ou polirradiculoneuropatia desmielinizante inflamatória (PRDI) aguda – é a causa mais comum de paralisia generalizada aguda no mundo ocidental. Os hospitais gerais terciários atendem vários pacientes com SGB a cada ano, sendo que o tratamento destes pacientes frequentemente é feito em conjunto com clínicos gerais. A história natural de SGB é favorável na maioria dos casos, porém a obtenção de um resultado satisfatório depende da prestação de uma assistência médica e de enfermagem meticulosa durante o pico da incapacitação neurológica.
Estabelecer o diagnóstico da SGB não é uma tarefa difícil, uma vez que os sinais tenham se manifestado totalmente. Contudo, durante os primeiros dias, os sintomas podem ser vagos, e os sinais, enganosos. É comum um paciente ser liberado da unidade de emergências após receber um diagnóstico de ansiedade e retornar somente depois de 1 a 2 dias, apresentando enfraquecimento de membro progressivo e evidente. O 1º sintoma, na maioria dos casos, é uma parestesia com formigamento que surge nos pés e se dissemina proximalmente a cada hora. O enfraquecimento é notado decorridas algumas horas ou dias. Alguns pacientes manifestam apenas sintomas motores, sem sintomas sensoriais. Classicamente, os sintomas a princípio são simétricos nos membros distais e prosseguem no sentido proximal (paralisia ascendente). Os exames de condução nervosa fornecem evidências da existência de um processo desmielinizante afetando as raízes espinais e os nervos periféricos (polirradiculoneuropatia desmielinizante). As variações desta apresentação clássica são comuns e incluem síndromes nas quais os nervos cranianos (p. ex., síndrome de Miller Fisher) ou o envolvimento autonômico são predominantes, assim como as síndromes essencialmente axonais e não desmielinizantes.28 É evidente que a SGB deve ser considerada dentro de uma categoria ampla de manifestações, embora todas as variantes clínicas compartilhem os principais aspectos de serem distúrbios imunomediados do SNP, apresentarem início agudo e seguirem um curso monofásico autolimitado. Além dos sintomas motores, sensoriais ou autonômicos, muitos pacientes apresentam uma dor que mais frequentemente se manifesta como uma dor profunda nas costas ou nos membros ou, ainda, como uma dor disestésica em um membro.29
A disfunção autonômica pode ser um problema significativo para alguns pacientes com SGB, sobretudo para aqueles com enfraquecimento severo. Uma flutuação acentuada da pressão arterial e uma hipotensão refratária constituem os principais problemas, embora também possa haver hipertermia, paralisia pupilar e arritmias cardíacas.
O enfraquecimento muscular pode aumentar nos primeiros dias, após os quais permanece estável durante vários dias ou semanas. O enfraquecimento pode variar de leve (p. ex., leve enfraquecimento da dorsiflexão do tornozelo) a severo. A quadriparesia flácida acompanhada de paralisia da musculatura respiratória ocorre em até 30% dos pacientes. Durante os primeiros 2 dias é impossível prever a intensidade do enfraquecimento que determinado paciente desenvolverá. Por isso, é recomendável que qualquer paciente com SGB permaneça sob observação no hospital até que o grau de severidade da condição se torne evidente. Como regra geral, o enfraquecimento chega ao máximo dentro de 14 dias. O período de enfraquecimento estável que se segue, antes do início da recuperação, tem duração de dias a meses (duração média de 4 semanas). Existe uma correlação grosseira entre a severidade do enfraquecimento e o intervalo de tempo até o início da recuperação. Uma vez iniciada a fase de recuperação, o paciente geralmente apresenta progressos visíveis a cada semana. Após 1 ano do aparecimento da condição, a maioria dos pacientes já terá alcançado uma recuperação total ou substancial, contudo até 15% dos pacientes continuarão acamados ou dependentes de cadeira de rodas. Os episódios recorrentes de SGB, que por vezes ocorrem ao longo de vários anos, afetam cerca de 3% dos pacientes.
O evento patológico fundamental na SGB consiste na descorticação da mielina dos axônios por ação dos macrófagos, que ocorre de modo irregular em todo o SNP. Acredita-se que uma cascata de eventos envolvendo mecanismos imunológicos celulares e humorais seja ativada, além disso infiltrados inflamatórios linfocíticos são encontrados com frequência no nervo e nas raízes nervosas examinadas por biópsia ou autópsia. Os fatores deflagradores e alvos moleculares específicos do ataque imunológico são desconhecidos. Há muito tempo persiste uma preocupação com o fato de a vacinação contra gripe atuar como fator precipitador da SGB. Estudos epidemiológicos retrospectivos descobriram a existência um pequeno risco relativo de SGB entre os receptores da vacina durante as épocas de gripe do período de 1992 a 1994.30 Estudos sobre a patogênese da SGB enfocaram o potencial papel de uma infecção prévia por Campylobacter jejuni na reatividade cruzada entre antígenos bacterianos e neurais e na produção de autoanticorpos antigangliosídeos – ambos fenômenos observados em um grande número de pacientes com SGB.28
O diagnóstico diferencial de uma polineuropatia sensoriomotora aguda é limitado [Tabela 2]. Antes do desenvolvimento da vacina contra a pólio, o principal diagnóstico diferencial consistia na diferenciação entre SGB e poliomielite. Hoje, porém, a poliomielite é uma condição rara. Quando sinais de doença sistêmica acompanham uma polineuropatia aguda, torna-se necessário considerar as hipóteses de neuropatia vasculítica, infiltração linfomatosa das raízes nervosas, porfiria aguda intermitente, difteria e envenenamento com arsênico. A lesão isquêmica da ponte a princípio pode causar quadriparesia flácida aguda e mimetizar a SGB antes do desenvolvimento de sinais neuronais motores superiores evidentes. Em alguns pacientes com SGB, os sintomas sensoriais não impressionam, e deve-se excluir outras possíveis causas do enfraquecimento de membro agudo, entre as quais a miastenia grave, miopatias inflamatórias ou hipocalemia. No caso das variantes de nervo craniano da SGB (p. ex., síndrome de Miller Fisher), devem ser excluídas as hipóteses de miastenia grave e botulismo.
A SGB é diagnosticada principalmente com base nos aspectos clínicos, no entanto os achados laboratoriais podem ajudar a sustentar o diagnóstico e excluir outras possíveis causas de polineuropatia aguda. Na SGB, o líquido cerebrospinal (LCE) tipicamente apresenta níveis elevados de proteína, níveis normais de glicose e ausência de pleiocitose. Quando a pleiocitose mononuclear é encontrada, torna-se necessário considerar seriamente a possibilidade de infiltração meníngea por um linfoma ou carcinoma. Uma polirradiculopatia aguda similar à SGB, porém com pleiocitose no LCE, pode ocorrer no momento da soroconversão do HIV [ver Polineuropatias causadas por doenças infecciosas, adiante]. Os exames de condução nervosa na SGB geralmente mostram um retardo amplamente disseminado das velocidades de condução ou, na maioria dos casos, um bloqueio de condução proximal e podem fornecer algumas informações prognósticas. Nem o LCE nem as anormalidades de condução nervosa são diagnósticas da SGB, e ambos os exames podem resultar normais, particularmente durante os primeiros dias da doença.
Depois que o diagnóstico de SGB é estabelecido, a 1ª prioridade é monitorar estreitamente a função da musculatura respiratória e prepará-la para uma possível intervenção, em caso de desenvolvimento de insuficiência ventilatória. É importante obter frequentemente medidas de cabeceira da capacidade vital ou da força inspiratória máxima ou, ainda, de ambos os parâmetros, bem como realizar uma avaliação clínica em busca de sinais de fadiga da musculatura respiratória. Um paciente cuja capacidade vital seja inferior a 20 mL/kg está propenso ao desenvolvimento de insuficiência ventilatória franca e, neste caso, a possibilidade de intubação eletiva deve ser fortemente considerada.
Diversos estudos clínicos amplos demonstraram que 2 terapias – plasmaférese e imunoglobulina endovenosa (IVIg) – melhoram a velocidade de recuperação dos pacientes com SGB moderada a severa (p. ex., pacientes incapazes de andar).31,32 Ambos os tratamento parecem modular o processo inflamatório que produz lesão no nervo, porém não são considerados promotores de remielinização nem de regeneração nervosa. É possível demonstrar um efeito benéfico somente quando a plasmaférese ou a administração de IVIg são iniciadas em 2 semanas após o aparecimento dos sintomas neuropáticos. É provavelmente sensato iniciar o tratamento assim que o diagnóstico for estabelecido, quando o enfraquecimento for suficientemente severo ou se houver progressão dos sinais enquanto o paciente estiver sob observação. Geralmente, são instituídos 5 tratamentos ao longo de 5 a 10 dias. Alguns pacientes podem apresentar recidivas e necessitar de tratamento adicional (p. ex., 2 vezes/semana durante 3 semanas). O tratamento combinado (plasmaférese seguida de IVIg) não proporciona nenhuma vantagem nítida em relação à instituição isolada de cada tratamento.33 Os glicocorticoides não proporcionam benefícios ao serem usados de forma isolada e, em alguns casos, chegam a produzir um pequeno efeito negativo.34 Existem dados conflitantes sobre os possíveis benefícios proporcionados pela adição de metilprednisolona endovenosa ao tratamento com IVIg. Se esta adição de fato confere alguma vantagem, parece ser pequena.34 A plasmaférese e a IVIg parecem apresentar a mesma efetividade e serem semelhantes em termos de custos e índices de recidiva. Entretanto, muitos clínicos acreditam que a IVIg é o tratamento de escolha por ser consideravelmente mais fácil de administrar do que a plasmaférese. Como a IVIg é produzida a partir de um pool de sangue de diversos pacientes, é difícil eliminar totalmente as preocupações relacionadas ao risco de infecção, apesar dos múltiplos procedimentos de purificação atualmente empregados e da inexistência de casos comprovados de infecção transmitida por IVIg por mais de uma década. Também existem dúvidas quanto à adequação do suprimento de IVIg para atender à demanda. Uma nova estratégia terapêutica consiste na filtração do LCE. Um estudo controlado pequeno constatou que esta modalidade é no mínimo tão efetiva quanto a plasmaférese, além de estar associada a menos complicações.35
Como muitos pacientes permanecem acamados durante vários meses, uma meticulosa assistência de enfermagem é essencial. É necessário adotar medidas preventivas contra o aparecimento de úlceras de pressão e trombose venosa, sendo que o paciente está exposto ao perigo constante de contrair infecções urinárias e pulmonares. A dor é um dos principais problemas para alguns pacientes e parece estar relacionada em parte à imobilização e em parte à inflamação e disfunção do nervo. A prática regular de exercícios de fisioterapia de amplitude de movimento e o uso judicioso de analgésicos e agentes como a amitriptilina são ações benéficas. Uma tarefa particularmente desafiadora para todos os membros da equipe consiste no suporte psicológico ao paciente. Existem vários relatos eloquentes sobre a perspectiva dos próprios pacientes em relação a SGB.36
Polirradiculoneuropatia desmielinizante inflamatória (PRDI) crônica. Assim como a SGB, a PRDI constitui uma neuropatia imunomediada. O evento patológico fundamental consiste na descorticação da mielina dos axônios por ação dos macrófagos, que retarda ou bloqueia a condução de impulsos nervosos, provocando enfraquecimento e perda sensorial. Clinicamente, a PRDI difere da SGB em diversos aspectos importantes. O aparecimento da PRDI é insidioso, com o desenvolvimento de sinais e sintomas no decorrer de várias semanas a meses, diferentemente do curso inicial rápido da SGB. Em contraste com o curso monofásico e autolimitado da SGB, a história natural da PRDI é variável. O padrão habitual da PRDI é uma piora lenta que ocorre com o passar dos meses e produz uma incapacitação moderada crônica. Alguns pacientes desenvolvem um curso recidivante e remitente, enquanto poucos sofrem remissão gradual e espontânea da condição. Um envolvimento respiratório ou autonômico significativo, comum na SGB, é incomum na PRDI.
Nenhum achado clínico ou laboratorial isolado é patognomônico da PRDI, sendo que o diagnóstico é estabelecido a partir da combinação de uma história adequada, sinais de neuropatia, evidências eletrofisiológicas de desmielinização e um LCE acelular com conteúdo proteico aumentado.37 O exame de biópsia de nervo pode sustentar o diagnóstico, mas em geral é desnecessário. Um tipo semelhante de neuropatia crônica pode ocorrer em pacientes com gamopatia monoclonal, mieloma osteoesclerótico e infecção pelo HIV.
A eficácia de alguns tratamentos para PRDI foi demonstrada em estudos controlados. A plasmaférese38 e a IVIg39 promovem melhora em 2 a 3 semanas, na maioria dos casos. Entretanto, esta melhora dura pouco, e os pacientes necessitam de tratamentos repetidos ou terapias alternativas de longa duração. A administração de altas doses de prednisona também é efetiva,40 porém a resposta obtida geralmente é menos significativa e mais lenta, sendo que o paciente precisa enfrentar as complicações associadas ao uso prolongado de um glicocorticoide. Um estudo cruzado (crossover) duplo-cego sobre o efeito do interferon-beta na PRDI resistente ao tratamento não demonstrou nenhum benefício.41 Entretanto, relatos anedóticos sustentam o uso de outros fármacos imunomoduladores, incluindo a ciclosporina, azatioprina, interferon-alfa, ciclofosfamida e micofenolato de mofetil. O monitoramento atento do comprometimento neurológico e o tratamento da condição por neurologistas com experiência em PRDI são importantes.
Polineuropatia associada à proteína monoclonal. A polineuropatia associada a gamopatia monoclonal podem ocorrer em alguns pacientes com amiloidose, mieloma múltiplo, mieloma osteoesclerótico, macroglobulinemia de Waldenström ou linfoma. Se nenhuma destas condições hematológicas for encontrada, então o paciente é considerado com gamopatia monoclonal de significado indeterminado (GMSI). A relação existente entre GMSI e neuropatia é indefinida, porém evidências epidemiológicas e patológicas sugerem que a GMSI causa neuropatia.
A neuropatia da GMSI apresenta características clínicas e eletrofisiológicas variáveis. Frequentemente, existem evidências eletrofisiológicas e patológicas de uma desmielinização proeminente, enquanto o quadro clínico é bastante semelhante ao da PRDI. Alguns pacientes desenvolvem uma neuropatia axonal dolorosa e apresentam déficits discretos. A neuropatia associada à IgM tende a ser mais severa, com uma ataxia mais significativa e um número maior de evidências de retardo e dispersão da condução nervosa, que apontam a existência de alguma relação entre a classe de imunoglobina e as características exibidas pela neuropatia.42
A neuropatia da GMSI em geral apresenta evolução lenta, embora o grau de incapacitação seja variável. As abordagens terapêuticas são destinadas a suprimir a produção da proteína monoclonal ou removê-la da circulação. Um estudo controlado demonstrou que a plasmaférese é benéfica para os pacientes com gamopatias de IgG e IgA, mas não promove benefícios em casos de gamopatia de IgM.43 Acredita-se amplamente que a IVIg apresente eficácia similar, porém nenhum estudo controlado foi conduzido. Existem relatos pouco confiáveis de respostas ao clorambucil, melfalano e prednisona. O interferon-alfa mostrou-se promissor em casos de neuropatia por IgM, mas um estudo controlado demonstrou a ausência de benefícios em relação ao placebo.44
Neuropatia amiloide. A polineuropatia caracterizada pela deposição de amiloide no nervo ocorre sob 2 formas: (1) como um aspecto desenvolvido por cerca de 15% dos pacientes com amiloidose sistêmica, nos quais a proteína amiloidogênica é uma imunoglobulina; e (2) como distúrbio autossômico dominante denominado polineuropatia amiloide familiar (PNAF), em que o amiloide na maioria das vezes consiste em formas mutadas de transtirretina, uma proteína do soro normal.45 Ambos os tipos são característicos de uma polineuropatia sensoriomotora simétrica, que é distinguida pela presença de aspectos autonômicos predominantes ou significativos. A amiloidose também é uma causa rara de STC.
O diagnóstico da neuropatia amiloide é estabelecido pela biópsia de nervo. A neuropatia amiloide adquirida e a neuropatia amiloide hereditária são similares em termos de histopatologia, mas podem ser distinguidas por análise imuno-histoquímica. Estudos que investigaram o uso de melfalano, glicocorticoides e colchicina demonstraram que estes agentes exercem pouco efeito sobre o resultado apresentado pelos pacientes com amiloidose primária, incluindo qualquer tipo de melhora da neuropatia. Diferente da amiloidose primária, a neuropatia geralmente constitui a principal causa de incapacitação na PNAF, e a maioria dos pacientes sobrevive por uma década ou mais após o diagnóstico da condição. O transplante hepático representa uma opção de tratamento para a PNAF, sobretudo para os pacientes mais jovens. Alguns pesquisadores relatam que o transplante hepático leva à melhora da função nervosa periférica em pacientes com PNFA, ainda que esta não seja uma experiência universal.46
Mieloma osteoesclerótico. O mieloma osteoesclerótico é uma variante do mieloma em que os pacientes apresentam uma ou, em alguns casos, várias lesões ósseas osteoescleróticas. O exame de biópsia revela uma proliferação de plasmócitos maligna. Em geral, estas lesões ósseas fazem parte de uma constelação que inclui polineuropatia, organomegalia, anormalidades endócrinas, gamopatia monoclonal e alterações cutâneas (síndrome POEMS). A polineuropatia é tipicamente desmielinizante e semelhante à PRDI. É importante reconhecer a síndrome POEMS, uma vez que a irradiação da lesão óssea pode proporcionar uma melhora clínica significativa.
Neuropatia paraneoplásica. Várias síndromes neurológicas distintas não metastásicas que se desenvolvem em pacientes com câncer foram descritas desde a década de 1950. Acredita-se que as síndromes paraneoplásicas possuem uma base imunológica e se desenvolvem como consequência da tentativa do hospedeiro de montar uma resposta imune dirigida contra o câncer [ver Distúrbios neoplásicos].
Neuropatia vasculítica. A neuropatia é uma manifestação frequente de certas vasculites sistêmicas. A neuropatia vasculítica é isquêmica e representa uma consequência do envolvimento inflamatório dos vasos fornecedores de nutrientes para os nervos. Devido ao robusto suprimento sanguíneo do nervo e sua relativa resistência à lesão isquêmica, o desenvolvimento de neuropatia na vasculite implica um extensivo envolvimento vascular. A vasculite tende a ser irregular, e a assimetria do envolvimento dos nervos é um aspecto comum, de modo que o envolvimento de nervos individuais é frequente, enquanto os nervos adjacentes são poupados. Esta é a clássica síndrome de mononeuropatias múltiplas. Entretanto, à medida que o envolvimento do suprimento sanguíneo do nervo evolui e mais nervos se tornam envolvidos, pode ficar mais difícil identificar um padrão de mononeuropatias múltiplas. Cerca de 30% dos casos de neuropatia vasculítica são polineuropatias simétricas no momento do diagnóstico inicial, 30% são polineuropatias assimétricas e 40% são mononeuropatias múltiplas.47
Entre os pacientes com neuropatia vasculítica, as principais vasculites sistêmicas associadas são a poliarterite nodosa, vasculite reumatoide, síndrome de Sjögren, granulomatose de Wegener e angiite granulomatosa alérgica (síndrome de Churg-Strauss). A neuropatia é particularmente comum na poliarterite nodosa, sendo encontrada em pelo menos metade de todos os casos. Os aspectos clínicos e neuropatológicos da neuropatia nestes distúrbios são similares, sendo que o diagnóstico específico depende de aspectos não neurológicos sistêmicos. A suspeita de neuropatia vasculítica muitas vezes pode surgir a partir do contexto clínico isolado, porém o diagnóstico definitivo depende do exame de biópsia do nervo. Os anticorpos anticitoplasma de neutrófilo são encontrados com frequência em pacientes com neuropatia vasculítica, contudo os resultados falso-positivos dos exames limitam sua utilidade diagnóstica.48
A neuropatia vasculítica é tratada pelo tratamento da vasculite sistêmica subjacente. Esta terapia geralmente requer altas doses de prednisona, ciclofosfamida ou ambas. Em casos de vasculite, a neuropatia raramente é causa de morte, embora possa acarretar uma incapacitação significativa. Diante da possibilidade de tratar com sucesso os eventos prejudiciais à vida, como a insuficiência renal ou cardiorrespiratória, as perspectivas de melhora neurológica são boas, mesmo que esta melhora demore vários meses para ocorrer.49
Os pacientes que apresentam aspectos clínicos e neuropatológicos de neuropatia vasculítica às vezes não exibem evidências de vasculite sistêmica. Esta síndrome, denominada neuropatia vasculítica não sistêmica, parece estar associada a uma história natural mais benigna do que a vasculite sistêmica. Os corticosteroides são utilizados com frequência nos casos em que o paciente apresenta incapacitação neurológica progressiva ou significativa, embora nenhum estudo controlado tenha sido realizado. A maior série retrospectiva sobre pacientes com neuropatia vasculítica não sistêmica sugeriu que a imunoterapia combinada (corticosteroides + ciclofosfamida) é mais efetiva do que o uso isolado de corticosteroides em termos de indução de remissão e melhora da incapacitação.50
Neuropatias relacionadas a outras doença do tecido conectivo. A neuropatia periférica pode ocorrer em outras doenças do tecido conectivo, como o lúpus eritematoso sistêmico [ver Lúpus eritematoso sistêmico], embora seja difícil determinar se a neuropatia é uma complicação direta da doença ou um efeito secundário de outra complicação (p. ex., secundária ao desenvolvimento de insuficiência renal). Em geral, considera-se que nestes casos a neuropatia possui uma base inflamatória, embora sejam poucos os estudos patológicos que sustentam esta opinião. Uma neuropatia sensorial atáxica, clinicamente bastante similar à síndrome paraneoplásica [ver Distúrbios neoplásicos], foi descrita em pacientes com síndrome de Sjögren e é causada pela infiltração inflamatória dos gânglios da raiz dorsal.51 As neuropatias também ocorrem em pacientes com síndrome sicca, que não apresentam os aspectos extraglandulares da síndrome de Sjögren.52
As substâncias tóxicas aos nervos periféricos incluem uma variedade de compostos químicos industriais, compostos naturais e fármacos. A maioria das neuropatias tóxicas surge distalmente, progride de maneira insidiosa ao longo de semanas a meses e apresenta aspectos eletrofisiológicos de neuropatia axonal. Com algumas exceções, a degeneração axonal constitui a principal característica histológica, e o exame de biópsia de nervo raramente é útil para esclarecer a etiologia.
Fármacos. A neuropatia fármaco-induzida constitui um problema frequente, sobretudo na prática hospitalar. É importante obter uma história farmacológica detalhada, como parte da investigação de qualquer polineuropatia. A neuropatia periférica é um efeito adverso bem estabelecido associado ao uso de numerosos fármacos [Tabela 5]. Em circunstâncias ideais, antes que o um fármaco seja rotulado como potencialmente neurotóxico, o paciente afetado deve estar comprovadamente livre de outras potenciais causas de neuropatia e a suspensão do uso do fármaco deve resultar em alguma melhora clínica. É importante notar que a recuperação pode demorar vários meses. Em algumas neuropatias tóxicas, o paciente passa por um fenômeno conhecido como coasting (cabotagem), em que a neuropatia continua a piorar durante algumas semanas após a cessação da exposição. Para a maioria das toxinas de nervo periférico bem estabelecidas, há evidências experimentais de toxicidade em animais e culturas de células neuronais. Com o uso de alguns antineoplásicos (p. ex., cisplatina), a neuropatia pode ser um efeito colateral dose-limitante. A prevenção da neuropatia induzida pela quimioterapia por meio da administração de neurotropinas foi relatada em alguns modelos de experimentação animal, contudo os estudos envolvendo seres humanos não obtiveram sucesso. Um estudo randomizado sugeriu que a vitamina E diminuiu a incidência de neuropatia entre pacientes tratados com cisplatina ou paclitaxel. Este estudo, no entanto, é pequeno e não cego.53
Compostos químicos industriais. A obtenção da história ocupacional do paciente pode ser importante, porque existem compostos químicos industriais comprovadamente neurotóxicos [Tabela 6]. A neurotoxicidade da maioria destes compostos foi descoberta após o aparecimento de grupos de casos envolvendo trabalhadores de indústrias específicas. Subsequentemente, o alerta para o perigo imposto por estes compostos diminuiu a incidência de casos. A maioria destes compostos químicos produz neuropatias axonais associadas a alterações patológicas inespecíficas. A comprovação de que uma neuropatia resulta da exposição a um composto químico exige um trabalho epidemiológico detalhado, que seja sustentado por estudos com animais ou culturas de tecido.
Tabela 6. Compostos químicos industriais que podem causar polineuropatia
|
Acrilamidas |
|
Cloreto de alila |
|
Dissulfeto de carbono |
|
Dimetilaminopropionitrila |
|
Óxido de etileno |
|
Hexano* |
|
Metilbrometo |
|
Metil-butil-cetona* |
|
Ésteres organofosforados |
|
Bifenis policlorados |
|
Tricloroetileno |
|
Vacor |
*A exposição a estes compostos também ocorre em consequência do vício em cheirar cola e outros solventes de abuso.
Metais. Além dos fármacos contendo ouro e platina, o envenenamento por outros metais pode acarretar neuropatia. A exposição frequentemente resulta de tentativas homicidas ou suicidas, de modo que o valor da história pode ser duvidoso. O diagnóstico deve ser estabelecido a partir dos aspectos clínicos associados. Exemplificando, a neuropatia por chumbo é predominantemente motora, com predileção pelos membros superiores, e está associada à dor abdominal, constipação e anemia. Além de causar neuropatia, o envenenamento com arsênico produz dor abdominal, vômitos, diarreia, alterações na pele e nas unhas, e pancitopenia. A neuropatia causada por tálio é distinguida pelo aparecimento de alopécia e dor abdominal. Os compostos de mercúrio inorgânicos e orgânicos podem causar neuropatia, mas os efeitos sobre o SNC geralmente predominam [ver Tratamento de envenenamento e superdosagem de fármacos].
O diagnóstico de neuropatia causada por metal depende da demonstração do aumento da excreção urinária do metal ou do aumento dos níveis deste metal nos cabelos ou nas unhas do paciente. A solicitação destes exames deve ser feita com urgência se houver suspeita clínica, sendo que este tipo de teste não faz parte da pesquisa de rotina da polineuropatia.
Etanol. Os alcoólatras crônicos comumente apresentam uma neuropatia dolorosa e distal. Os principais sintomas são as dores ardentes e agudas, bem como o entorpecimento dos pés e, em alguns casos, das mãos. A perda sensorial ou a hipersensibilidade dolorosa nos pés, a perda dos reflexos do tornozelo e o leve enfraquecimento distal constituem o quadro típico. Permanece indeterminado se a neuropatia é causada por um efeito tóxico direto do etanol, por desnutrição ou por ambos fatores.
As tentativas de produzir neuropatia com etanol em animais bem nutridos falharam. Contudo, em culturas de células neuronais, foi possível inibir o crescimento celular com o uso de concentrações moderadas a altas de etanol. Um estudo conduzido na Dinamarca, que investigou em detalhes uma coorte de alcoólatras que bebiam cerveja e apresentavam neuropatia, não encontrou diferenças clínicas, eletrofisiológicas nem histológicas entre indivíduos bem nutridos e desnutridos.54 A cerveja dinamarquesa é suplementada com tiamina e piridoxina, por isso é improvável que a deficiência destas vitaminas tenha causado neuropatia nesses pacientes.
Embora a patogênese da neuropatia em alcoólatras seja discutida, observa-se um contexto clínico característico em que outros sinais e sintomas do alcoolismo geralmente estão presentes, incluindo doença hepática crônica, comprometimento da memória e ataxia da marcha. A estimativas da ingesta de etanol são notoriamente pouco confiáveis. Entretanto, no estudo dinamarquês, somente desenvolveram neuropatia os pacientes que consumiram 3 L de cerveja ou 300 mL de destilados por dia, durante um período mínimo de 3 anos. Um corolário importante destas observações está no fato de a neuropatia alcoólica ser incomum em um paciente com neuropatia que consume bebidas alcoólicas de forma moderada, está bem nutrido e não apresenta sinais de doença hepática crônica. Assim, neste tipo de paciente, outras causas de neuropatia devem ser investigadas.
O tratamento da neuropatia alcoólica é teoricamente simples, porém difícil na prática. A ingesta de etanol deve ser suspendida e uma nutrição adequada precisa ser instituída. Se for possível adotar estas medidas, uma melhora gradual pode ser esperada, ainda que demore meses para ocorrer e seja incompleta.
Deficiências nutricionais. A polineuropatia pode ser uma manifestação da inanição. Isto é observado nas vítimas da fome e nos prisioneiros de guerra. Os componentes precisos da dieta responsáveis pelo desenvolvimento de neuropatia na inanição são indefinidos, embora uma ou mais vitaminas do complexo B sejam consideradas essenciais. Algumas deficiências vitamínicas podem causar neuropatia em situações específicas, mas não existe nenhuma base fisiológica para a prescrição rotineira de multivitamínicos para neuropatia em casos de pacientes com dieta normal.
A deficiência de tiamina (vitamina B1) produz beribéri, cujos principais aspectos são a polineuropatia e a insuficiência cardíaca. A neuropatia é distal e axonal, acompanhada de sintomas sensoriais dolorosos. Com a progressão, pode haver desenvolvimento de enfraquecimento distal. O envolvimento de nervos cranianos foi descrito em relatos antigos de beribéri.
A deficiência de piridoxina (vitamina B6) é responsável pela neuropatia causada pela isoniazida, que aumenta a excreção da piridoxina. A administração da vitamina com isoniazida previne o desenvolvimento da neuropatia. Entretanto, doses excessivas de piridoxina (em moda na década de 1970) produzem uma severa neuropatia sensorial.
Uma polineuropatia leve pode fazer parte de uma síndrome neurológica causada pela deficiência de cobalamina (vitamina B12). No entanto, a deficiência desta vitamina provavelmente não se manifesta apenas como polineuropatia. Os principais aspectos clínicos da condição são resultantes de mielopatia (degeneração combinada subaguda).
A deficiência de vitamina E decorrente de má absorção pode acarretar uma síndrome atáxica causada pela degeneração dos processos centrais e periféricos do neurônios ganglionares da raiz dorsal. Alguns pacientes podem apresentar envolvimento cerebelar.
Hanseníase. A hanseníase (lepra), uma doença infecciosa micobacteriana dos nervos periféricos, é provavelmente a causa mais comum de polineuropatia em todo o mundo [ver Infecções causadas por Mycobacterium leprae e micobactéria não tuberculosa]. A maioria dos casos ocorre em regiões tropicais e subtropicais, embora existam focos endêmicos ao longo da costa do golfo da Flórida e na Louisiana, Estados Unidos. A maioria dos pacientes com hanseníase que vivem na América do Norte e na Europa é de imigrantes oriundos de países onde a hanseníase é uma doença comum.
A perda sensorial constitui o sintoma cardinal da hanseníase e muitas vezes é descoberta por causa de uma lesão indolor. Devido aos requerimentos de temperatura de Mycobacterium leprae, os nervos mistos e sensoriais cutâneos localizados em partes do corpo que apresentam baixa temperatura ambiente são mais propensos a serem afetados. O resultado é uma distribuição de sinais diferente daquela observada em outras polineuropatias, com perda sensorial na área localizada sobre as orelhas externas, nos arcos zigomáticos e nas superfícies extensoras das articulações. Os principais nervos tendem mais a ser afetados nos pontos em que seguem próximos à superfície (p. ex., o nervo ulnar na região do cotovelo). O envolvimento dos nervos cutâneos em geral é nitidamente demarcado, em especial na forma tuberculoide da hanseníase, com a epiderme e a derme sobrejacente sendo afetadas e produzindo a clássica mácula anestésica. O enfraquecimento motor não ocorre até que a perda sensorial esteja bem estabelecida. O enfraquecimento geralmente é irregular e assimétrico, podendo sugerir outras causas de mononeuropatias múltiplas. O diagnóstico da hanseníase é estabelecido com base nos achados do exame de biópsia de nervo ou pele, utilizando-se o método de Fite para corar e identificar M. leprae.
A hanseníase é uma doença curável. O grau de recuperação pode ser limitado nos casos de doença em estágio avançado. Por isso, é importante que a doença seja diagnosticada e tratada antes que o paciente desenvolva déficits neuropáticos significativos [ver Infecções causadas por Mycobacterium leprae e micobactéria não tuberculosa].
Infecção pelo HIV. Vários distúrbios do SNP ocorrem em pacientes infectados pelo HIV, em alguns casos durante os estágios iniciais da infecção e em outros, somente após a progressão para Aids [ver HIV e Aids]. É bastante comum a ocorrência de neuropatia dolorosa e distal em pacientes com Aids. O principal sintoma é uma ardência incômoda contínua, principalmente nos pés, que evidencia certo grau de perda sensorial. O envolvimento motor geralmente é mínimo, embora os pacientes muitas vezes estejam debilitados em decorrência de infecções concomitantes e perda de peso. Em alguns casos, a causa é identificável – exemplificando, a deficiência de vitamina B12 ou o tratamento com um agente antirretroviral comprovadamente neurotóxico (p. ex., 2,3-didesoxicitidina ou zalcitabina)55 –, mas em alguns pacientes pode permanecer indefinida. O genoma do HIV foi detectado nos neurônios ganglionares da raiz dorsal e nas células satélite de pacientes com Aids e neuropatia.56 Além disso, a expressão do genoma do HIV em camundongos transgênicos promove doença nervosa periférica.57 Ainda é necessário comprovar se a neuropatia apresentada por indivíduos infectados pelo HIV é de fato resultante da infecção direta dos nervos pelo vírus.
A neuropatia é um efeito colateral dose-limitante associado ao uso dos agentes antirretrovirais análogos de nucleosídeo (p. ex., zalcitabina). Felizmente, porém, nem a zidovudina (AZT) nem os inibidores de protease parecem causar neuropatia. Em um estudo de fase II, o fator de crescimento de nervo, quando comparado ao placebo, amenizou a dor neuropática de pacientes infectados pelo HIV que apresentavam neuropatia sensorial distal, embora a dor no sítio de injeção tenha acarretado um mascaramento em uma parte significativa da população de pacientes tratados.58
Alguns pacientes infectados pelo HIV desenvolvem SGB e PRDI, tipicamente ainda no início do curso da infecção pelo vírus. Estes pacientes podem apresentar aspectos associados à infecção pelo HIV, sendo necessário considerar a realização de testes de detecção do vírus em pacientes com ambas as condições. O quadro neurológico e a resposta ao tratamento dos pacientes infectados pelo HIV são similares ao observado em pacientes não infectados pelo HIV, exceto quanto à ocorrência habitual de fagocitose linfocítica no LCE.
Os pacientes com positividade para HIV podem desenvolver uma síndrome de mononeuropatias múltiplas. A biópsia de nervo pode mostrar a presença de infiltrados inflamatórios perivasculares, vasculite necrotizante ou inclusões de citomegalovírus (CMV). Os pacientes com evidências de infecção pelo CMV podem ser responsivos ao ganciclovir59 [ver Infecções por herpes vírus]. Os pacientes com infecção por HIV agravada por uma síndrome de linfocitose infiltrativa difusa podem apresentar uma neuropatia axonal subaguda. Os exames de biópsias de nervo oriundas destes pacientes têm demonstrado a presença de infiltrados perivasculares notáveis de linfócitos CD8+, sendo que a neuropatia parece ceder com a administração de corticosteroides ou zidovudina.60
A polirradiculopatia lombossacral aguda é uma síndrome devastadora de enfraquecimento da perna, perda sensorial peroneal e na perna e retenção urinária, que se desenvolve ao longo de 1 a 2 semanas, em geral em pacientes com Aids em estágio avançado. O LCE mostra uma pleiocitose polimorfonuclear distintiva. Na maioria dos casos, há evidências de uma infecção concomitante por CMV e o quadro neurológico é produzido pela invasão das raízes nervosas lombossacrais pelo CMV. O diagnóstico e tratamento imediato com ganciclovir pode resultar em melhora [ver Infecções por herpes vírus]. Um quadro similar pode ser produzido por um linfoma.61
Doença de Lyme. A doença de Lyme é causada pela espiroqueta Borrelia burgdorferi, que é transmitida aos seres humanos por carrapatos ixodídeos. A doença ocorre em todo o mundo, contudo é mais comum no nordeste dos Estados Unidos e no norte da Europa [ver Doença de Lyme e outras zoonoses causadas por espiroquetas]. A neuropatia periférica pode ocorrer no início ou na fase tardia da doença de Lyme. Não está claro se as manifestações neurológicas são causadas diretamente pela invasão do tecido nervoso pelas espiroquetas ou pela resposta imune do hospedeiro ao organismo.
Os principais aspectos neurológicos iniciais são as neuropatias cranianas, as radiculopatias espinais ou ambas.62 A cefaleia e a rigidez cervical podem acompanhar os sintomas nervosos periféricos, refletindo a inflamação meníngea. A neuropatia facial é a mais comum das neuropatias cranianas e frequentemente é diagnosticada de modo equivocado como paralisia de Bell. Entretanto, diferente da paralisia de Bell idiopática, a neuropatia facial bilateral é comum na doença de Lyme. As demais neuropatias cranianas são menos frequentes.
O envolvimento da raiz nervosa espinal tipicamente começa com uma dor de distribuição radicular, seguida de enfraquecimento em 1 a 4 semanas. O enfraquecimento muitas vezes é assimétrico e irregular, semelhante às mononeuropatias múltiplas. Em adição aos sinais radiculares predominantes, é comum haver evidências eletrofisiológicas de uma polineuropatia amplamente disseminada e branda. Tanto as neuropatias cranianas como as espinais, na doença de Lyme em estágio inicial, estão associadas a uma história natural favorável e com a recuperação ocorrendo em algumas semanas a meses. O tratamento antibiótico pode acelerar a recuperação.
Na doença de Lyme em estágio tardio, os pacientes podem apresentar uma polineuropatia distal leve ou uma dor radicular com sinais sensoriais. Muitos destes pacientes apresentam sintomas prévios de doença localizada ou inicialmente disseminada. Diferente da neuropatia craniana e da radiculoneuropatia da doença em estágio inicial, a neuropatia periférica da doença tardia tende a não ser resolvida sem tratamento.
O diagnóstico da doença de Lyme baseia-se na história de uma possível exposição ao carrapato, sinais e sintomas compatíveis, bem como exames sorológicos positivos para B. burgdorferi. Na doença em estágio inicial, o envolvimento neurológico é quase sempre acompanhado de pleiocitose linfocítica no LCE. Na doença tardia, contudo, a pleiocitose é rara, e o LCE pode ser normal. A biópsia do nervo sural, obtida durante o estágio inicial ou tardio da doença, revela a ocorrência de degeneração axonal e infiltração inflamatória perivascular, que são achados inespecíficos. Particularmente durante a doença em estágio tardio, a resposta a um curso de antibióticos pode fornecer evidências mais convincentes do diagnóstico [ver Doença de Lyme e outras zoonoses causadas por espiroquetas].
Infecção pelo vírus varicela zóster (VZV). O vírus varicela zóster (VZV), integrante da família dos herpes vírus humanos, é o patógeno viral mais comum no SNP [ver Infecções por herpes vírus]. Após a infecção inicial pelo VZV, que geralmente ocorre durante a infância sob a forma de catapora, alguns vírions do VZV podem entrar nos axônios sensoriais cutâneos e serem levados por transporte axonal retrógrado até os corpos celulares neuronais dos gânglios cranianos ou da raiz dorsal, onde permanecem na forma inativa.
Anos depois, o vírus pode voltar ao estado ativo e proliferante, em decorrência de alterações não totalmente esclarecidas que ocorrem na função imune. Os efeitos citopáticos da replicação do VZV e a resposta imune que se segue produzem uma intensa ganglionite necrótica inflamatória, com consequente alteração sensorial, dor e erupções cutâneas vesiculares nos dermátomos conhecidas como herpes zóster.
O herpes zóster é mais comum em indivíduos com idade acima de 60 anos, embora também ocorra em indivíduos mais jovens. A incidência desta condição é significativamente maior entre pacientes imunocomprometidos, que também apresentam risco de infecção disseminada pelo VZV associada a uma mortalidade considerável. O herpes zóster geralmente é unilateral, envolvendo 1 a 3 dermátomos adjacentes. Os dermátomos do tórax e do trigêmeo (em especial o oftálmico) são os mais frequentemente afetados.
O sintoma inicial é a dor no dermátomo. Decorridos 3 a 7 dias, surgem as erupções vesiculares. É difícil demonstrar a perda sensorial antes de as lesões cutâneas começarem a cicatrizar. O enfraquecimento motor é relatado por até 30% dos pacientes, mas pode não ser notado pelo paciente distraído pela dor. As lesões cutâneas persistem por 7 a 10 dias e então são resolvidas, frequentemente deixando áreas despigmentadas e com cicatriz. É possível esperar que o enfraquecimento motor melhore espontaneamente na maioria dos pacientes. A dor também melhora lentamente, exceto em indivíduos que desenvolvem neuralgia pós-herpética (NPH).
A NPH é definida em geral por uma dor que persiste por mais de 4 a 8 semanas após a cicatrização das lesões cutâneas. O risco de NPH após um episódio de herpes zóster aumenta com o avanço da idade e chega ao máximo de 45% em pacientes com mais de 65 anos. Os pacientes com NPH apresentam uma dor ardente e contínua nos dermátomos, à qual podem se sobrepor dores lancinantes de curta duração. Em muitos casos, o dermátomo afetado também apresenta alodinia, e, deste modo, o toque da roupa ou até do cabelo pode causar uma dor torturante. Alguns pacientes com NPH apresentam melhora gradual, que ocorre no decorrer de várias semanas a meses, contudo a persistência dos sintomas por vários anos não é incomum. A prevenção da NPH é provavelmente a principal justificativa para o uso de agentes antivirais por um paciente imunocompetente. Um amplo estudo controlado com placebo sobre o uso do famciclovir demonstrou a ocorrência de uma diminuição nítida da incidência da NPH, em especial entre os idosos.63 Considera-se, mesmo sem comprovação, que o aciclovir proporciona um benefício semelhante. Em um estudo, a adição de prednisolona a cursos de aciclovir com duração de 7 ou 21 dias não alterou a frequência de NPH,64 embora os glicocorticoides possam diminuir a dor de fase aguda. Um amplo estudo controlado com placebo, envolvendo adultos imunocompetentes com mais de 60 anos de idade, constatou que uma vacina com VZV vivo e atenuado produziu uma diminuição significativa da incidência de NPH durante um período de seguimento de 3 anos.65
A NPH representa um formidável desafio terapêutico. A amitriptilina ou outros medicamentos tricíclicos têm sido usados como agentes de 1ª linha, porém a gabapentina está sendo cada vez mais empregada e se mostrou superior ao placebo em um estudo controlado.66 A dor lancinante pode ser amenizada pela carbamazepina ou outros agentes anticonvulsivos. A aplicação tópica de anestésicos locais e o uso prolongado de opiáceos são defendidos por alguns clínicos.67
O autor não possui relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
O autor atua como conselheiro ou consultor junto à Talecris Biotherapeutics.
1. Chen Z, Yu W, Strickland S. Peripheral regeneration. Annu Rev Neurosci 2007;30:209.
2. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No.: CD005454. DOI: 10.1002/14651858.CD005454. pub2.
3. Foley KM. Opioids and chronic neuropathic pain. N Engl J Med 2003;348:1279.
4. Murakami S, Mizobuchi M, Nakashiro Y, et al. Bell palsy and herpes simplex virus: identifi cation of viral DNA in endoneurial fluid and muscle. Ann Intern Med 1996;124: 27.
5. Salinas RA, Alvarez G, Ferreira J. Corticosteroids for Bell’s palsy (idiopathic facial paralysis). Cochrane Database of Systematic Reviews 2004, Issue 4. Art. No.: CD001942. DOI: 10.1002/14651858.CD001942.pub2.
6. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007;357:1598.
7. He L, Wu B, Zhou M. Non-antiepileptic drugs for trigeminal neuralgia. Cochrane Database of Systematic Reviews 2006, Issue 3. Art. No.: CD004029. DOI: 10.1002/14651858. CD004029.pub2.
8. Gorgulho A, De Salles AAF. Impact of radiosurgery on the surgical treatment of trigeminal neuralgia. Surg Neurol 2006;66:350.
9. Scholten RJPM, Mink van der Molen A, Uitdehaag BMJ, et al. Surgical treatment options for carpal tunnel syndrome. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No.: CD003905. DOI: 10.1002/14651858.CD003905. pub3.
10. Beekman R, Wokke JH, Schoemaker MC, et al. Ulnar neuropathy at the elbow: follow-up and prognostic factors determining outcome. Neurology 2004;63:1675.
11. Suarez GA, Giannini C, Bosch EP, et al. Immune brachial plexus neuropathy: suggestive evidence for an infl ammatory-immune pathogenesis. Neurology 1996;46: 559.
12. Dyck PJB, Windebank AJ. Diabetic and nondiabetic lumbosacral radiculoplexus neuropathies: new insights into pathophysiology and treatment. Muscle Nerve 2002;25:477.
13. Dyck PJ, Oviatt KF, Lambert EH. Intensive evaluation of referred unclassifi ed neuropathies yields improved diagnosis. Ann Neurol 1981;10:222.
14. Dyck PJ, Kratz KM, Karnes JL, et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology 1993; 43:817.
15. Dyck PJ, Davies JL, Wilson DM, et al. Risk factors for severity of diabetic polyneuropathy: intensive longitudinal assessment of the Rochester Diabetic Neuropathy Study cohort. Diabetes Care 1999;22:1479.
16. Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Pract Neurol 2006; 2:620–8.
17. Dyck PJ, Davies JL, Litchy WJ, et al. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study cohort. Neurology 1997;49:229.
18. The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group. Ann Intern Med 1995;122:561.
19. Navarro X, Sutherland DER, Kennedy WR. Long-term effects of pancreatic transplantation on diabetic neuropathy. Ann Neurol 1997;42:727.
20. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. UK Prospective Diabetes Study Group. Lancet 1998;352:837.
21. Dyck PJ, Dyck PJB, Klein CJ, Weigand SD. Does impaired glucose metabolism cause polyneuropathy? Review of previous studies and design of a prospective controlled population-based study. Muscle Nerve 2007;36:536.
22. Chalk C, Benstead TJ, Moore F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No.: CD004572. DOI: 10.1002/14651858.CD004572.pub2.
23. Apfel SC, Schwartz S, Adornato BF, et al. Effi cacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy. JAMA 2000;284:2215.
24. Ziegler D, Ametov A, Barinov A, et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care 2006;29:2365.
25. Zochodne DW, Bolton CF, Wells GA, et al. Critical illness polyneuropathy: a complication of sepsis and multiple organ failure. Brain 1987;110:819.
26. De Jonghe B, Sharshar T, Lefaucheur J-P, et al. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA 2002;288:2859.
27. Parman Y. Hereditary neuropathies. Curr Opin Neurology 2007;20:542.
28. Hughes RAC, Cornblath D. Guillain-Barré syndrome Lancet 2005;366:1653.
29. Moulin DE, Hagen N, Feasby TE, et al. Pain in GuillainBarré syndrome. Neurology 1997;48:328.
30. Lasky T, Terracciano GJ, Magder L, et al. The Guillain-Barré syndrome and the 1992–1993 and 1993–1994 infl uenza vaccines. N Engl J Med 1998;339:1797.
31. Raphaël JC, Chevret S, Hughes RAC, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database of Systematic Reviews 2002, Issue 2. Art. No.: CD001798. DOI: 10.1002/14651858.CD001798.
32. Hughes RAC, Raphaël J-C, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database of Systematic Reviews 2006, Issue 1. Art. No.: CD002063. DOI: 10.1002/14651858.CD002063. pub3.
33. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Lancet 1997;349:225.
34. Hughes RAC, Swan AV, van Koningsveld R, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database of Systematic Reviews 2006, Issue 2. Art. No.: CD001446. DOI: 10.1002/14651858.CD001446.pub2.
35. Wollinsky KH, Hülser P-J, Brinkmeier H, et al. CSF fi ltration is an effective treatment of Guillain-Barré syndrome. Neurology 2001;57:774.
36. Bowes D. The doctor as patient: an encounter with Guillain-Barré syndrome. Can Med Assoc J 1984;131:1343.
37. Lewis RA Chronic infl ammatory demyelinating polyneuropathy. Neurol Clin 2007;25:71.
38. Hahn AF, Bolton CF, Pillay N, et al. Plasma exchange therapy in chronic infl ammatory demyelinating polyneuropathy: a double-blind, sham-controlled, cross-over study. Brain 1996;119:1055.
39. Mendell JR, Barohn RJ, Freimer ML, et al. Randomized controlled trial of IVIg in untreated chronic infl ammatory demyelinating polyradiculoneuropathy. Neurology 2001;56: 445.
40. Dyck PJ, O’Brien PC, Oviatt KF, et al. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann Neurol 1982;11:136.
41. Hadden RDM, Sharrack B, Bensa S, et al. Randomized trial of interferon beta-1a in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 1999;53:57.
42. Gosselin S, Kyle RA, Dyck PJ. Neuropathy associated with monoclonal gammopathies of undetermined signif cance. Ann Neurol 1991;30:54.
43. Dyck PJ, Low PA, Windebank AJ, et al. Plasma exchange in polyneuropathy associated with monoclonal gammopathy of undetermined signif cance. N Engl J Med 1991;325:1482.
44. Kwan Ji Paraproteinemic neuropathy. Neurol Clin 2007; 25:47.
45. Plante-Bordeneuve V, Lalu T, Misrahi M, et al. Genotypic-phenotypic variations in a series of 65 patients with familial amyloid polyneuropathy. Neurology 1998;51:708.
46. Stangou AJ, Hawkins PN. Liver transplantation in transthyretin-related familial amyloid polyneuropathy. Curr Opin Neurol 2004;17:615.
47. Burns TM, Schaublin GA, Dyck PJB. Vasculitic neuropathies. Neurol Clin 2007;25:89.
48. Chalk CH, Homburger HA, Dyck PJ. Anti-neutrophil cytoplasmic antibodies in vasculitic peripheral neuropathy. Neurology 1993;43:1826.
49. Chang RW, Bell CL, Hallett M. Clinical characteristics and prognosis of vasculitic mononeuropathy multiplex. Arch Neurol 1984;41:618.
50. Collins MP, Periquet, MI, Mendell JR, et al. Nonsystemic vasculitic neuropathy: insights from a clinical cohort. Neurology 2003;61:623.
51. Griff n JW, Cornblath DR, Alexander E, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjögren’s syndrome. Ann Neurol 1990;27:304.
52. Grant IA, Hunder GG, Homburger HA, et al. Peripheral neuropathy associated with sicca complex. Neurology 1997;48:855.
53. Argyriou AA, Chroni E, Koutras A, et al. Vitamin E for prophylaxis against chemotherapy-induced neuropathy: a randomized controlled trial. Neurology 2005;64:26.
54. Behse F, Buchthal F. Alcoholic neuropathy: clinical, electrophysiological, and biopsy f ndings. Ann Neurol 1977; 2:95.
55. Simpson DM, Tagliati M. Nucleoside analogue-associated peripheral neuropathy in human immunodef ciency virus infection. J Acquir Immune Def c Syndr 1995;9:153.
56. Brannagan TH, Nuovo GJ, Hays AP, et al. Human immunodef ciency virus infection of dorsal root ganglion neurons detected by polymerase chain reaction in situ hybridization. Ann Neurol 1997;42:368.
57. Thomas FP, Chalk C, Lalonde R, et al. Expression of human immunodef ciency virus type 1 in the nervous system of transgenic mice leads to neurological disease. J Virol 1994; 68:7099.
58. McArthur JC, Yiannoutsos C, Simpson DM, et al. A phase II trial of nerve growth factor for sensory neuropathy associated with HIV infection. Neurology 2000;54:1080.
59. Roullet E, Assuerus V, Gozlan J, et al. Cytomegalovirus multifocal neuropathy in AIDS: analysis of 15 consecutive cases. Neurology 1994;44:2174.
60. Berger JR, Simpson DM. The pathogenesis of diffuse inf ltrative lymphocytosis syndrome: an AIDS-related peripheral neuropathy. Neurology 1998;50:855.
61. So YT, Olney RK. Acute lumbosacral polyradiculopathy in acquired immunodef ciency syndrome: experience in 23 patients. Ann Neurol 1994;35:53.
62. Logigian EL. Peripheral nervous system Lyme borreliosis. Semin Neurol 1997;17:25.
63. Tyring S, Barbarash RA, Nahlik JE, et al. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia. Ann Intern Med 1995;123:89.
64. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med 1994;330:896.
65. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271.
66. Rowbotham M, Harden N, Stacey B, et al. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA 1998;280:1837.
67. Rowbotham M. Managing post-herpetic neuralgia with opioids and local anaesthetics. Ann Neurol 1994;35:S46.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.