(Carregando Índice)... (Carregando Índice)... |
Última revisão: 20/02/2015
Comentários de assinantes: 0
Annette Esper, MD
Greg S. Martin, MD, MSc, FACP
Gerald W. Staton Jr, MD, FACP
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2014 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr.Lucas Santos Zambon
Entre suas numerosas observações prescientes, em 1821, René Laennec descreveu a aparência do edema em amostras de pulmão obtidas após a morte.1 Contando apenas com seus sentidos naturais, ele concluiu que havia duas formas fisiopatologicamente distintas de edema pulmonar. Uma forma estava associada à patologia cardíaca e ele, então, concluiu que era devido à insuficiência cardíaca. A outra forma aparecia na ausência de patologia cardíaca e foi chamada por Laennec de edema pulmonar “primário ou idiopático”. Estas observações foram provavelmente as primeiras descrições das características patológicas macroscópicas do edema pulmonar hidrostático e do edema pulmonar por permeabilidade—com este último sendo a síndrome clínica identificada há mais de um século como SARA. As contribuições niciais de Laennec para o conhecimento sobre edema pulmonar limitaram-se a observações patológicas macroscópicas, enquanto uma compreensão mais ampla necessitou de avanços técnicos para estudo da função e estrutura pulmonar, bem como do interesse entusiástico de fisiologistas experientes acerca do movimento de líquidos e solutos entre os compartimentos anatômicos.
A nova observação de Laennec de que o edema pulmonar é inevitavelmente resultante de outro processo persiste até hoje, em nossas tentativas de diferenciar clinicamente as causas de edema pulmonar, em grande parte porque o tratamento difere conforme a causa. O edema pulmonar agudo pode ser dividido em duas categorias: edema causado por pressão capilar aumentada (edema hidrostático ou cardiogênico) e edema causado por permeabilidade capilar aumentada (edema pulmonar não cardiogênico ou SARA). Estas duas categorias incluem a maioria dos casos e, em alguns casos de edema pulmonar, mais de um mecanismo pode estar envolvido.
A troca de líquidos e solutos nos pulmões ocorre ao nível da membrana alvéolo-capilar. O lado capilar da membrana é relativamente permeável, possibilitando a filtração constante de líquido e proteínas a partir do espaço vascular para dentro do espaço intersticial. Estima-se que, a cada hora, vários mililitros (talvez, até 50 mL) de líquido normalmente rico em proteínas (cerca de 2/3 da concentração plasmática total de proteínas) sejam filtrados para dentro do interstício.2 A barreira capilar é semipermeável a macromoléculas, por isso existe uma pressão oncótica transmembrana que está diretamente relacionada à diferença de concentração de proteínas entre o plasma e o líquido intersticial.3 O volume do lado alveolar da membrana alvéolo-capilar é constituído por células epiteliais de tipo I, que estão unidas pelas tight junctions (zônulas de oclusão), as quais restringem rigorosamente o fluxo de líquido e solutos sob condições normais.4,5 Desta forma, a barreira existente entre o espaço intersticial e o espaço aéreo é altamente impermeável às macromoléculas.
Em adição à membrana alvéolo-capilar, os linfáticos do pulmão exercem papel importante na regulação de líquidos. O pulmão é rico em linfáticos, cuja origem periférica é o espaço intersticial ao nível dos bronquíolos terminais. Os vasos linfáticos então coalescem ao seguirem na direção do hilo e, eventualmente, se fundem para entrar predominantemente no ducto linfático direito, que então é esvaziado no sistema venoso ao nível da junção da veia jugular interna direita com a veia cava superior. Como consequência de sua ação peristáltica e da presença de uma série de valvas unidirecionais, os linfáticos atuam literalmente como uma bomba de reservatório que regula o volume do espaço intersticial pulmonar distal. Como o espaço intersticial ao nível alveolar deve ser mantido com um volume mínimo, de modo a não interferir nas trocas gasosas, o papel do sistema linfático é essencial para o órgão.
A patogênese do edema pulmonar pode ser entendida analisando a equação de Starling, que define os determinantes do fluxo de líquidos através da membrana capilar pulmonar:
Q = K[(Pcap – Pint) s (pcap – pint)]
Onde Q é o fluxo de líquido; K é o coeficiente de filtração, que é diretamente proporcional à área de superfície endotelial e inversamente proporcional à espessura da parede do capilar alveolar; Pcap é a pressão hidrostática intravascular (capilar); Pint é a pressão hidrostática intersticial; s é o coeficiente de deflexão para proteína (i.e., o grau de permeabilidade a macromoléculas); pcap é a pressão oncótica no plasma; e pint, é a pressão oncótica intersticial. Nesta equação, (Pcap – Pint) representa o gradiente de força hidrostática, enquanto (pcap – pint ) representa o gradiente de força coloidosmótica.6,7
Uma alteração de qualquer um dos fatores da equação de Starling poderia teoricamente levar ao aumento do fluxo de líquido transvascular. Entretanto, na prática clínica, apenas dois destes fatores comumente levam ao edema pulmonar: (1) aumento da pressão capilar, que leva ao desenvolvimento de edema pulmonar cardiogênico ou hidrostático; e (2) diminuição do coeficiente de deflexão, que leva ao edema pulmonar não cardiogênico ou por alteração da permeabilidade.
As respostas fisiológicas à aumentada filtração de líquidos transvascular nos pulmões que aliviam a formação de edema incluem o aumento do fluxo linfático e a diminuição da pressão oncótica intersticial. A defesa primária contra o edema pulmonar é conferida pelo sistema linfático. Normalmente, o líquido filtrado através da membrana capilar é removido pelos vasos linfáticos.8 A reserva linfática é tal que até mesmo um aumento de 4-10 vezes no fluxo de líquido transcapilar pode ser tolerado sem que haja aumento do volume de água nos pulmões.2,9,10
A membrana alvéolo-capilar semipermeável filtra as proteínas com base no tamanho e esta filtração depende da taxa de filtração de líquidos.9,11,12 Portanto, se a membrana de filtração permanecer intacta, o fluxo de líquidos resultará em um filtrado com menor concentração de proteínas. Em consequência, a pressão oncótica intersticial diminui, compensando parcialmente a pressão capilar aumentada que está forçando a filtração—um segundo fator de edema. Todavia, quando a permeabilidade capilar aumenta, este mecanismo protetor é comprometido dependendo do grau de permeabilidade aumentada.
Quando a reserva de remoção de líquidos fica sobrecarregada, o volume de água nos pulmões aumenta, primeiramente no interstício ao redor das vias aéreas, em seguida no interstício em torno dos alvéolos e, por fim, nos alvéolos. As evidências clínicas ou radiográficas de edema pulmonar, portanto, implicam em aumento significativo do fluxo de líquido transcapilar.
Devido às diferentes abordagens terapêuticas de pacientes com edema pulmonar cardiogênico e não cardiogênico, é importante distinguir estas duas condições, ainda que às vezes seja difícil e alguns paciente apresentem componentes de ambas. A história do paciente fornece indícios importantes [ver Tabela 1]. Pacientes com edema pulmonar cardiogênico costumam ter história de cardiopatia ou hipertensão, enquanto os pacientes com edema não cardiogênico podem apresentar condições preexistentes que aumentam o risco de infecção. O paciente com edema pulmonar cardiogênico pode manifestar sintomas de um novo evento cardíaco (dor torácica isquêmica) ou emergência hipertensiva. O paciente com edema pulmonar não cardiogênico quase sempre terá passado por um evento precipitante nítido, como sepse, traumatismo, aspiração de conteúdo gástrico, transfusões múltiplas ou pneumonia.
Os sinais e sintomas são similares nas duas formas de edema pulmonar, com algumas exceções importantes [ver Tabela 1]. Embora ambos os grupos de pacientes desenvolvam dispneia, os pacientes com edema pulmonar cardiogênico geralmente exibem pele fria e diaforética com evidências de disfunção ventricular esquerda (distensão venosa jugular, galope). Em contraste, a maioria dos pacientes com edema pulmonar não cardiogênico exibe pele quente e mostra evidências de circulação hiperdinâmica.
Os exames laboratoriais podem dar indícios do tipo de edema pulmonar. O edema cardiogênico pode ser indicado por um eletrocardiograma anormal (p. ex., evidenciando infarto ou isquemia do miocárdio) ou pela elevação dos níveis séricos de troponina. O ensaio mais moderno para diferenciação dos tipos cardiogênico e não cardiogênico de edema pulmonar é a determinação dos níveis plasmáticos de peptídeo natriurético cerebral (BNP). Níveis de BNP acima de 500 pg/mL sugerem uma possível insuficiência cardíaca, enquanto níveis de BNP abaixo de 100 pg/mL tornam improvável a hipótese de insuficiência cardíaca. Níveis de BNP entre 100 e 500 pg/mL são inadequados para fins de discriminação. Como os níveis de BNP estão aumentados em pacientes com insuficiência renal, a utilidade da quantificação do peptídeo nestes pacientes é menor.
O edema pulmonar, seja qual for o tipo, caracteristicamente se manifesta como opacidades alveolares bilaterais simétricas envolvendo os quatro quadrantes, observadas em radiografia torácica anteroposterior padrão [ver Tabela 1].13 Uma distribuição predominantemente peri-hilar é comum e, ocasionalmente, existe uma demarcação bastante precisa entre a área central de edema pulmonar e a periferia do pulmão, produzindo um padrão característico em “asa de morcego” ou “borboleta”. Este padrão é mais típico do edema pulmonar cardiogênico que do edema pulmonar não cardiogênico [ver Figura 1 e Figura 2]. Esta linha de demarcação não corresponde a nenhuma fronteira anatômica, mas pode ser produzida por gradientes fisiológicos de ventilação, perfusão e fluxo linfático do centro para as partes periféricas do pulmão. De forma menos frequente, o edema é acentuadamente assimétrico ou totalmente unilateral. O edema pulmonar assimétrico pode ocorrer em um paciente que permanece deitado sobre o lado envolvido, à medida que o edema se desenvolve, ou pode resultar de atenuação (enfisema) nas regiões mais radiotransparentes do pulmão. Por outro lado, é impossível determinar o que leva à assimetria em alguns casos e à unilateralidade em outros.
Os aspectos auxiliares que podem ser visualizados de modo rotineiro em uma radiografia torácica anteroposterior obtida com aparelho de raio x portátil podem ajudar a diferenciar o edema pulmonar cardiogênico do não cardiogênico. Um pedículo vascular ampliado e um aumento da proporção cardiotorácica sugerem uma pressão capilar pulmonar aumentada.14 Broncogramas aéreos são mais comuns com edema pulmonar não cardiogênico. A presença ou ausência de efusões pleurais tem menos valor para o estabelecimento do diagnóstico diferencial, embora a presença de efusões pleurais seja mais comum no edema pulmonar cardiogênico.15 Infelizmente, os edemas pulmonares cardiogênico e não cardiogênico muitas vezes não podem ser diferenciados com segurança por meio de radiografias à beira-leito, porque a exposição anteroposterior e a ausência de inspiração total ampliam a silhueta cardíaca. A ecocardiografia de beira de leito pode ser bastante útil para diferenciar as formas cardiogênica e não cardiogênica de edema pulmonar [ver Tabela 1].6 Em pacientes com edema pulmonar cardiogênico, a ecocardiografia consegue detectar e quantificar a função ventricular esquerda anormal e pode ser usada para diagnosticar muitas causas (p. ex., disfunção valvular, disfunção diastólica, miocardiopatia e anomalias focais de movimentação da parede). Em pacientes com edema pulmonar não cardiogênico, observa-se um ventrículo esquerdo normal ou hiperdinâmico e este achado às vezes é acompanhado de dilatação ventricular direita e evidência de hipertensão pulmonar.
|
Tabela 1 edema pulmonar cardiogênico versus não cardiogênico | ||
|
Achados clínicos |
edema pulmonar cardiogênico |
edema pulmonar não cardiogênico |
|
Contexto clínico |
IM prévio, hipertensão, miocardiopatia, IM, emergência hipertensiva, indiscrição dietética, outros. |
Alcoolismo, imunossupressão, sepse, traumatismo, aspiração, pneumonia, outros. |
|
Sintomas |
Dispneia, ortopneia, tosse produtiva com escarro rosado. |
Dispneia, tosse. |
|
Sinais |
Taquipneia, taquicardia, pulsação fraca, hipertensão, pele fria, diaforese, cianose central e/ou periférica, sibilos, estertores, distensão venosa jugular, palpitação LE, galope. |
Hipoxemia grave, taquipneia, febre, hipotermia, pulsação saltante e forte que desaparece rápido, pele quente, cianose, precórdio hemodinâmico. |
|
Achados laboratoriais |
ECG anormal, troponina alta, elevação de BNP > 500 pg/mL. |
BNP < 100 pg/mL. |
|
Achados de radiografia torácica |
Cefalização venosa, pedículo vascular amplo, proporção cardiotorácica aumentada, opacidades peri-hilares, linhas de B de Kerley. |
Opacidades difusas, broncogramas aéreos. |
|
Achados de ecocardiografia |
Fração de ejeção VE diminuída, disfunção diastólica, doença valvar, anormalidades de movimento de parede segmentares, ampliação de VE e AE. |
VE hiperdinâmico. |
|
Achados de cateterismo arterial pulmonar |
POAP aumentada, DC normal ou diminuído, pressão arterial pulmonar aumentada com RVP normal, RVS aumentada, onda v. |
POAP normal ou baixa, pressão arterial pulmonar aumentada com RVP aumentada em presença de sepse, DC aumentado, RVS diminuída. |
BNP = peptídeo natriurético cerebral; DC = débito cardíaco; ECG = eletrocardiograma; AE = átrio esquerdo; VE = ventrículo esquerdo; IM = infarto do miocárdio; POAP = pressão de oclusão da artéria pulmonar; RVP = resistência vascular pulmonar; VD = ventrículo direito; RVS = resistência vascular sistêmica.

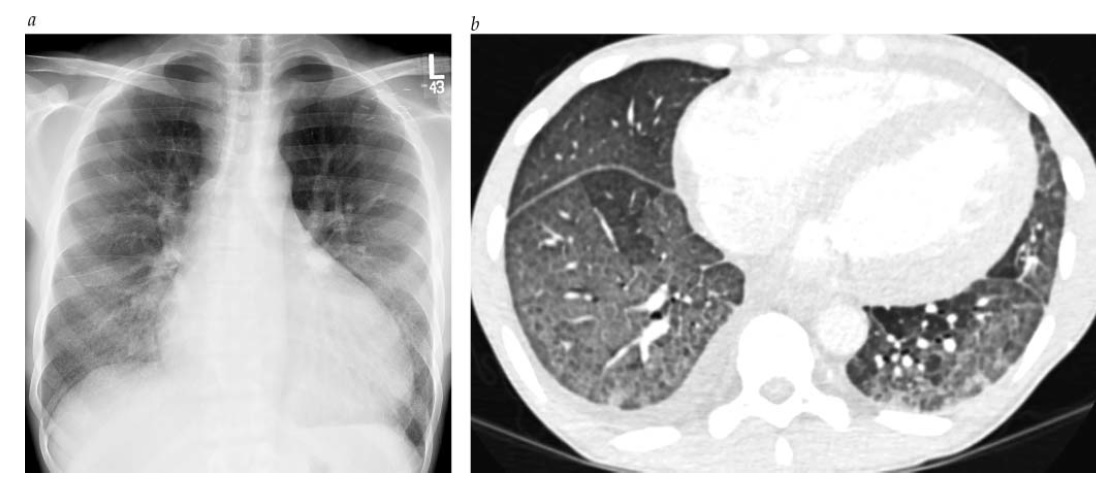
Figura 1 (a) Vista torácica anteroposterior em posição ereta, mostrando uma silhueta cardíaca ampliada com cefalização da vasculatura pulmonar e edema pulmonar intersticial. (b) Imagem torácica axial de tomografia computadorizada, obtida ao nível dos lobos inferiores exibidos nas janelas pulmonares, mostrando opacidades em vidro fosco bilaterais com espessamento septal liso intermisturado, representando edema pulmonar. Há uma pequena camada simples de derrame pleural à direita e cardiomegalia. Cortesia de Eugene Berkowitz, MD.
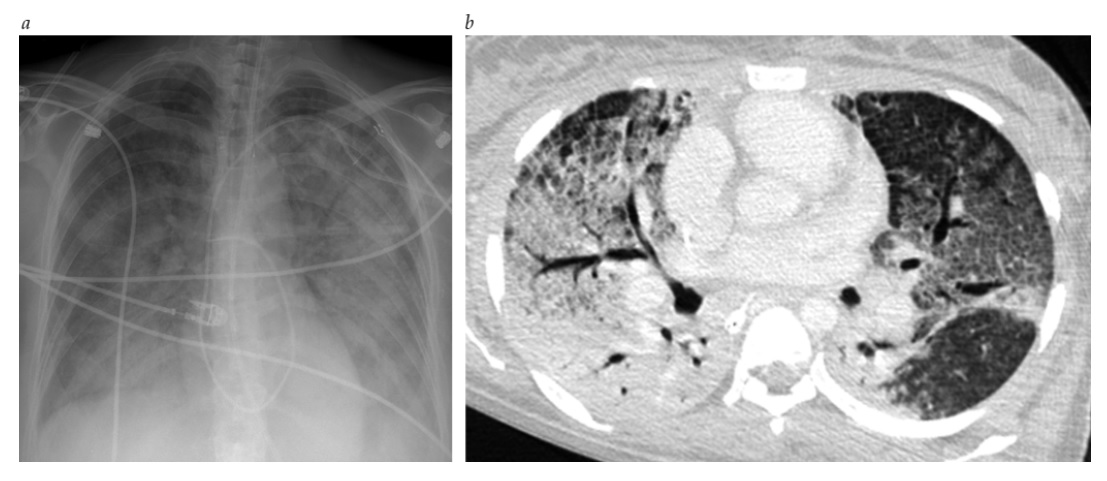
Figura 2 edema pulmonar não cardiogênico. (a) Vista anteroposterior frontal do tórax de uma paciente de 51 anos de idade, com hemorragia subaracnoidea, exibindo opacidades alveolares e intersticiais bilaterais sem derrame pleural significativa – achados consistentes com edema pulmonar não cardiogênico. O coração não está ampliado. A ponta de um cateter de Swan-Ganz é inserida na artéria pulmonar principal direita. (b) Imagem torácica axial de tomografia computadorizada ao nível dos lobos inferiores exibidos nas janelas pulmonares, mostrando opacidades bilaterais intersticiais e alveolares (consolidação) na ausência de efusões pleurais. A biópsia de pulmão em cunha mostrou a ocorrência de dano alveolar difuso. Cortesia de Eugene Berkowitz, MD.
Diante da impossibilidade de diferenciar o edema pulmonar cardiogênico do edema não cardiogênico por meio da avaliação não invasiva destacada anteriormente, a colocação de um cateter arterial pulmonar pode ser considerada. A pressão de oclusão da artéria pulmonar (POAP) é a medida mais útil, contudo outras medidas podem sustentar o diagnóstico e ajudar a tratar o paciente [ver Tabela 1].6
Apesar do apelo lógico do uso de cateterismo arterial pulmonar, nenhum efeito benéfico sobre o resultado final foi atribuído a essa intervenção. Vários estudos sugeriram que o uso de cateteres arteriais pulmonares não melhora a mortalidade e está associado a um risco aumentado de complicações, além de gastos financeiros maiores, em comparação a não adoção dessa intervenção.16
Os achados clínicos do edema pulmonar cardiogênico podem se sobrepor aos achados do edema não cardiogênico. Entretanto, o contexto em que o edema se desenvolve provavelmente será bastante diverso. O edema pulmonar cardiogênico tipicamente ocorre em pacientes com cardiopatia comprovada ou em pacientes que tenham sofrido algum evento cardíaco agudo. Os pacientes podem apresentar sinais clínicos de congestão, incluindo dispneia, edema, estertores e distensão venosa jugular.
O edema pulmonar decorrente de pressão capilar aumentada pode resultar de disfunção sistólica ou diastólica do ventrículo esquerdo, doença da valva mitral, hipervolemia associada à função cardíaca esquerda normal (como poderia ocorrer em um paciente com insuficiência renal) ou obstrução venosa pulmonar.17 A causa mais comum de edema pulmonar cardiogênico é a disfunção ventricular esquerda. Na miocardiopatia congestiva, o desempenho sistólico do ventrículo esquerdo é comprometido, o ventrículo está dilatado e a pressão diastólica final ventricular esquerda (PDFVE) está aumentada. A elevação da PDFVE leva ao aumento da pressão capilar pulmonar e também podem ocorrer elevações dinâmicas da regurgitação mitral.18 Outros tipos de cardiopatia também podem elevar a PDFVE, apesar da função sistólica normal e da euvolemia, diminuindo a complacência ventricular esquerda e produzindo disfunção diastólica.19 A complacência reduzida pode ser persistente (demonstrada pela hipertrofia ventricular esquerda ou miocardiopatia restritiva a partir de cardiopatia infiltrativa) ou transiente (por isquemia miocárdica). A pressão capilar aumentada mesmo com uma PDFVE normal é incomum, mas ocorre com a estenose mitral ou como resultado de obstrução de fluxo venoso pulmonar (doença veno-oclusiva pulmonar).
Os achados clínicos salientes associados ao edema pulmonar cardiogênico são a dispneia intensa, a hipoxemia, a taquipneia e os sinais de atividade simpática aumentada, como taquicardia, hipertensão e diaforese [ver Tabela 1 e Abordagem geral do paciente com suspeita de edema pulmonar, anteriormente]. A hipoxemia é devida ao edema intersticial, que resulta em incompatibilidade de ventilação-perfusão, e ao inundamento alveolar, que resulta em desvio de sangue da direita para esquerda. A hipotensão é incomum, mas pode ocorrer se houver edema pulmonar resultante de um infarto de miocárdio extenso. A falta de ar raramente é aliviada pela correção da hipoxemia, sugerindo que a causa pode ser a ativação de receptores de estiramento intrapulmonares, em vez de hipoxemia. No edema pulmonar cardiogênico, é possível que ocorra cianose central se houver hipoxemia arterial profunda. A cianose é mais frequentemente periférica e resulta de vasoconstrição cutânea intensa e débito cardíaco diminuído. O uso de músculos respiratórios acessórios é comum, devido ao aumento acentuado do trabalho respiratório. No início do curso agudo, pode haver sibilos causados por edema de vias aéreas e líquido intraluminal. Posteriormente, estertores difusos e ásperos são ouvidos.
O oxigênio suplementar deve ser fornecido por máscara ou cânula nasal. Se a hipoxemia não puder ser corrigida com o estabelecimento de taxas de fluxo de oxigênio máximas e com o uso de bolsas-reservatório, a ventilação mecânica com máscara20 ou a intubação endotraqueal serão necessárias. As pressões intratorácicas positivas criadas pelo ventilador abrem os alvéolos em colapso e impedem o retorno venoso. A pressão de ventilação positiva não invasiva (PVPNI) é igualmente útil para o tratamento da hipoxemia e diminui o trabalho respiratório. A PVPNI é benéfica no tratamento do edema pulmonar agudo cardiogênico e pode melhorar a mortalidade.21–23 Embora a hipoxemia esteja sendo tratada, recomenda-se que uma ou mais das várias alternativas terapêuticas sejam empregadas ao mesmo tempo, dependendo dos processos fisiopatológicos subjacentes [ver Tabela 2]. A combinação apropriada de medidas terapêuticas depende da fisiopatologia. Exemplificando, quando o edema pulmonar resulta de uma complicação de hipertensão maligna, os vasodilatadores e diuréticos podem ser suficientes. No caso de uma taquiarritmia causal ou fortemente contribuitiva, a terapia antiarrítmica pode ser a intervenção essencial. As intervenções específicas para condições cardíacas causais são descritas em outra seção [ver Tratamento do edema pulmonar cardiogênico].
Pacientes com edema pulmonar agudo cardiogênico frequentemente são idosos e apresentam múltiplos problemas médicos, entre os quais cardiopatia isquêmica, diabetes e cardiopatia valvular. Em consequência, entre estes pacientes, a mortalidade varia de 6 a 30%.24 Em pacientes internados que sofreram um primeiro episódio de insuficiência cardíaca, a mortalidade hospitalar gira em torno de 8%, com a fração de ejeção diminuída independentemente associada à morte intra-hospitalar.25 A PVPNI, em especial a pressão positiva contínua de vias aéreas (PPCVA), diminui a mortalidade em pacientes com edema pulmonar agudo cardiogênico.22,23 Dentre os pacientes que sobrevivem até a alta, cerca de 70% sobreviverão por 1 ano e 50% apresentarão um estado funcional relativamente satisfatório por períodos mais longos.26
|
Tabela 2 Tratamento do edema pulmonar cardiogênico | |
|
Efeitos almejados |
Abordagem terapêutica |
|
Retorno venoso diminuído |
Venodilatador, pressão positiva de vias aéreas |
|
Minimizar o obstáculo à sístole ventricular |
Dilatador arteriolar, pressão expiratória final positiva |
|
Diminuição do volume intravascular |
Agente diurético |
|
Estimular o miocárdio |
Agente inotrópico |
|
Corrigir arritmias |
Agente antiarrítmico, marca-passo |
|
Aliviar a isquemia |
Intervenções cardíacas percutâneas, agente trombolítico, desvio coronariano |
|
Prevenir a propagação de coágulos |
Aspirina, outros agentes antiplaquetários, anticoagulantes |
A SARA é caracterizada por uma lesão endotelial pulmonar difusa que leva ao desenvolvimento de edema pulmonar, como resultado do aumento da permeabilidade capilar à água, solutos e macromoléculas.6,7 O edema pulmonar visto em pacientes com SARA é caracterizado por uma concentração de proteína no líquido do edema maior do que aquela observada em pacientes com edema pulmonar cardiogênico. Em pacientes com SARA, esta concentração frequentemente corresponde a 80-90% da concentração plasmática de proteínas. Em adição, nos pacientes com SARA, a resposta inflamatória subjacente produz altos níveis de neutrófilos e seus respectivos produtos secretórios no lavado broncoalveolar (LBA). Esta característica distingue o edema não cardiogênico do edema cardiogênico.
Muitos distúrbios clínicos estão associados ao desenvolvimento de SARA [ver Tabela 3], podendo surgir a partir de uma lesão pulmonar direta ou de processos extrapulmonares que causam lesão pulmonar indiretamente.27–29 O risco de desenvolvimento de SARA de um paciente varia conforme o distúrbio predisponente, sendo que o risco aumenta com o aumento do número de distúrbios predisponentes.26 O risco de SARA pode estar aumentado em pacientes com história de consumo abusivo de álcool30 ou tabagismo,31 ou pode ser maior devido à presença de um baixo pH sérico ou de hipoproteinemia32 no momento da agressão. Os fatores genéticos também podem afetar a predisposição.33
Pacientes com SARA podem exibir diversas manifestações, dependendo do fator de risco identificado como deflagrador do desenvolvimento da lesão pulmonar aguda.
A patogênese da SARA é complexa e envolve: (1) lesão do epitélio e endotélio pulmonar, resultando em acúmulo de edema pulmonar rico em proteína; e (2) comprometimento da remoção do edema pulmonar e das células inflamatórias.34 O termo “dano alveolar difuso” descreve a sequência inespecífica e variável de alterações caracteristicamente observadas em pacientes com SARA.7,35,36 O agente ou processo causal geralmente não pode ser determinado com base no padrão histopatológico. Adicionalmente, pode haver resolução das anormalidades em qualquer ponto do curso clínico.
O aspecto histológico do dano alveolar difuso varia durante o período entre o evento precipitante e a realização da biópsia/autópsia, progredindo na seguinte sequência de três fases: fase exsudativa, fase organizacional ou proliferativa subaguda e fase crônica. Uma biópsia obtida em qualquer estágio do desenvolvimento da SARA pode mostrar achados associados a múltiplas fases da evolução do padrão tecidual.
|
Tabela 3 Causas da síndrome de angústia respiratória aguda26 | |
|
Condição |
Exemplos |
|
Lesão pulmonar direta |
|
|
Infecção pulmonar difusa |
Pneumonia bacteriana, viral ou fúngica; pneumonia por Pneumocystis jiroveci |
|
Pneumonite química causada por aspiração |
Aspiração de conteúdo gástrico ou de água (por quase afogamento). |
|
Lesão inalatória |
Inalação de fumaça, cloro ou óxido nítrico |
|
Traumatismo pulmonar direto |
Acidente automobilístico |
|
Lesão pulmonar indireta |
|
|
Reação sistêmica à infecção não pulmonar |
Bacteremia, sepse não bacterêmica, síndrome do choque tóxico |
|
Reação sistêmica à lesão ou inflamação de tecido não pulmonar |
Pancreatite, traumatismo, embolia gordurosa, embolia por líquido amniótico |
|
Reação transfusional |
Transfusão de hemácias |
|
Toxicidade farmacológica |
Salicilatos, agentes citotóxicos |
|
Lesão por reperfusão |
Bypass cardiopulmonar, pós-transplante de pulmão |
|
Outras |
Corrida de maratona, DPSIHT |
DPSIHT = Desvio Porto-sistêmico Intra-hepático Transjugular.
A parte mais inicial da fase exsudativa aguda é caracterizada por edema intersticial e edema intra-alveolar, infiltração de neutrófilos, hemorragia e deposição de fibrina. Uma mistura de fibrina e debris celulares é depositada no espaço alveolar, para formar as chamadas membranas hialinas, que são proeminentes logo após a aquisição da lesão. A mudança das células do revestimento alveolar deixa uma membrana basal descoberta que exerce papel importante no processo subsequente de reparo ou fibrose. Um infiltrado intersticial de células inflamatórias posteriormente se torna mais pronunciado e persiste no decorrer de toda a fase proliferativa.
O desnudamento da membrana basal causa proliferação de pneumócitos de tipo II, produzindo um padrão de hiperplasia nas células do revestimento alveolar. Em pacientes nos quais há resolução da síndrome, estas células proliferantes por fim se diferenciam em pneumócitos de tipo I, restaurando o lado epitelial da parede capilar alveolar e normalizando as trocas gasosas.
A fase proliferativa da SARA é caracterizada por inflamação e proliferação de fibroblastos, a princípio no interstício. Os fibroblastos invadem os espaços alveolares através dos defeitos existentes na membrana basal, em um processo que produz regiões de fibrose intra-alveolar. Durante esta fase, as membranas hialinas desaparecem como resultado de fagocitose ou da organização envolvendo a incorporação do exsudato aos tampões intra-alveolares de fibroblastos proliferantes.
A fase crônica da SARA é caracterizada por regiões de fibrose intensa, regiões focais de expansão excessiva e obliteração vascular pulmonar. Histologicamente, esta fase da doença pode ser similar à fibrose pulmonar idiopática. Contrastando com esta, todavia, a fase crônica da SARA pode melhorar com o tempo.
Investigações extensivas têm levado a um conhecimento mais aprimorado dos mecanismos que levam à SARA 7,34–36 [ver Figura 3].
Os principais sintomas clínicos de edema pulmonar não cardiogênico se sobrepõem aos do edema pulmonar cardiogênico [ver Tabela 1 e Abordagem geral do paciente com suspeita de edema pulmonar, anteriormente]. No edema pulmonar não cardiogênico, contudo, a atividade simpática frequentemente é menor.37 A cianose, quando presente, muitas vezes é decorrente de hipoxemia arterial. A pele é sempre quente (em vez de fria, úmida e pálida) e o pulso pode estar acelerado. A hipoxemia grave é um aspecto importante do edema pulmonar não cardiogênico, devido à lesão pulmonar aguda, sendo usada para definir a gravidade da lesão pulmonar. A hipoxemia geralmente é mais grave na SARA do que no edema pulmonar cardiogênico porque a hipóxia da inundação alveolar, que ocorre antecipadamente na SARA, costuma ser mais grave do que no edema intersticial.15
Tipicamente, a radiografia torácica anteroposterior portátil revela um processo de enchimento alveolar homogêneo e difuso.13 Entretanto, quando examinado por tomografia computadorizada (TC), o padrão de enchimento de espaço aéreo frequentemente parece ser menos homogêneo.38 As radiografias obtidas com o paciente em decúbito dorsal tipicamente mostram um grau maior de consolidação nas zonas pulmonares posteriores do que nas zonas pulmonares anteriores. Esta distribuição, porém, pode ser revertida se o paciente permanecer posicionado em pronação por algumas horas. Com o paciente em pronação, as regiões anteriores se tornam mais consolidadas devido à influência da gravidade, enquanto as regiões posteriores do pulmão apresentam aeração melhorada.
Na SARA, a troca gasosa é caracterizada inicialmente por uma hipoxemia refratária às concentrações crescentes de oxigênio inspirado, implicando a ocorrência de desvio intrapulmonar aumentado. O desvio intrapulmonar é primariamente uma consequência do enchimento alveolar difuso, colapso ou ambos, aliados à microatelectasia. Acredita-se que o colapso alveolar difuso seja causado por anomalias do surfactante, uma substância que normalmente ajuda a manter a distensão alveolar diminuindo a tensão superficial na interface ar-líquido do pulmão.
Inicialmente, a tensão de dióxido de carbono arterial (PaCO2) é baixa ou está dentro da faixa normal, com apenas um aumento apenas modesto da ventilação minuto. Entretanto, a proporção de espaço morto/volume corrente (EM/VC) tende a aumentar com o passar do tempo, de modo que quantidades crescentes de ventilação minuto são requeridas para alcançar uma PaCO2 normal.39 Em alguns casos, o aumento da EM/VC é tão extremo que a hipercapnia não pode ser evitada mesmo que a ventilação minuto seja aumentada até o nível máximo alcançável. Este aumento do espaço morto fisiológico resulta do dano ao leito capilar pulmonar, que cria regiões de alta ventilação em relação à perfusão. Uma causa secundária da necessidade de ventilação minuto aumentada na SARA é a presença de produção aumentada de dióxido de carbono a partir do hipermetabolismo. A alta proporção EM/VC encontrada na SARA é lentamente resolvida e isto, aliado à diminuição da complacência pulmonar (que impõe uma carga sobre a respiração espontânea), contribui em parte para a necessidade frequente de suporte ventilatório mecânico por períodos prolongados. Este tipo de suporte muitas vezes precisa ser mantido, mesmo quando a hipoxemia melhora o suficiente para tornar necessário apenas um pequeno aumento da concentração de oxigênio inspirado.
Na SARA, a mecânica pulmonar é caracterizada primariamente por uma diminuição da complacência pulmonar (elasticidade pulmonar aumentada). Desta forma, elevadas pressões transpulmonares são requeridas para alcançar uma ventilação corrente normal. No início da SARA, quando o edema predomina, uma grande parte da pressão distensora necessária para inflar o pulmão é gasta com a abertura dos alvéolos em colapso. De fato, a complacência pulmonar (i.e., inclinação da curva de pressão-volume) pode estar dentro da faixa normal, se for medida depois que os alvéolos em colapso tiverem sido abertos. Entretanto, ocorre uma diminuição significativa da complacência à medida que o processo patológico evolui ou quando a fibrose alveolar se torna proeminente. Esta diminuição evidente pode ser causada pelo espessamento difuso da membrana alvéolo-capilar, como resultado de fibrose ou pela perda de um alto percentual de unidades alvéolo-capilares disponíveis para ventilação. Foi sugerido que, em pacientes com SARA, a mecânica pulmonar é melhor conceitualizada considerando o pulmão como sendo pequeno e não rígido, porque apenas algumas de suas partes estão envolvidas na troca gasosa.38 Embora os modelos experimentais que comparam a mecânica pulmonar no edema pulmonar não cardiogênico versus edema pulmonar cardiogênico mostrem que ambos os tipos de edema estão associados a aumentos do fluxo linfático e diminuição da complacência, o edema cardiogênico resulta em alterações mais significativas de resistência ao fluxo de ar, em comparação ao edema não cardiogênico [ver Figura 4].3
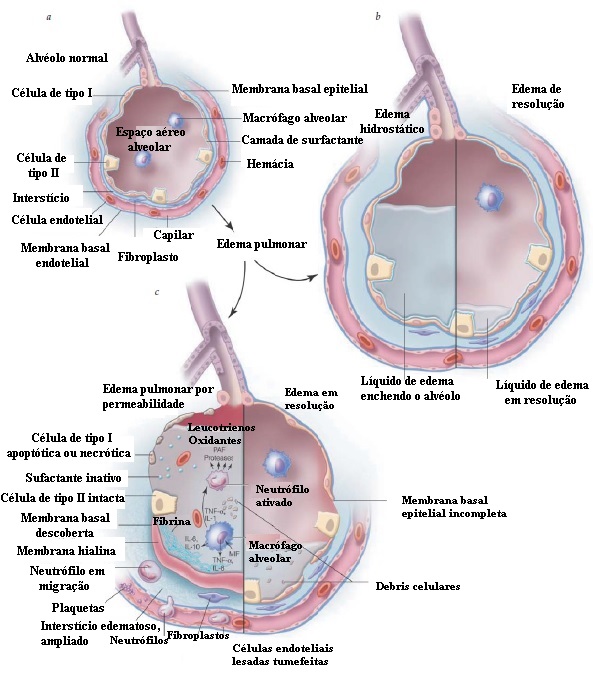
Figura 3 Diferenças entre edema cardiogênico e edema não cardiogênico. (a) Alvéolo normal com membrana alvéolo-capilar intacta e interstício normal. (b) Edema hidrostático com enchimento do interstício e do alvéolo com líquido de edema. A membrana alvéolo-capilar permanece intacta. (c) edema pulmonar por permeabilidade com lesão ao endotélio dos capilares pulmonares, resultando em permeabilidade aumentada da barreira capilar alveolar e influxo de líquido de edema rico em proteínas. A lesão epitelial alveolar resulta em uma membrana basal descoberta e na formação de membranas hialinas. A lesão é adicionalmente mediada pelos neutrófilos que aderem ao endotélio capilar lesado e pela ativação do macrófago alveolar, resultando na secreção de citocinas pró- e anti-inflamatórias. IL = interleucina; MIF = fator inibidor de macrófago; PAF = fator ativador de plaquetas; TNF = fator de necrose tumoral.
Em adição ao efeito sobre EM/VC, as alterações vasculares pulmonares que ocorrem na SARA resultam em aumento da resistência vascular pulmonar e hipertensão pulmonar.40 De fato, a pressão pulmonar arterial está elevada em quase todos os pacientes com SARA moderada a grave. A etiologia da hipertensão pulmonar na SARA tende a ser multifatorial. Entretanto, uma das principais causas subjacentes parece ser a presença de pequenos trombos arteriais pulmonares. A patogênese destes trombos encontrados na SARA é desconhecida, mas é provável que sua formação ocorra in situ.
Muitos pacientes com SARA apresentam certos marcadores de coagulação intravascular acelerada.41 No entanto, apesar da importância evidente da trombose para a produção de hipertensão pulmonar, os benefícios da anticoagulação tradicional para os pacientes com SARA ainda são indeterminados.
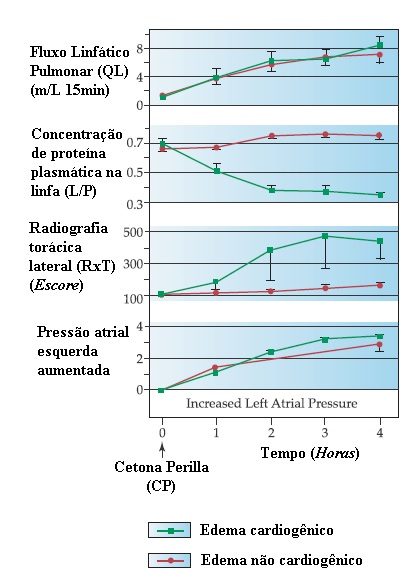
Figura 4 Comparação da mecânica pulmonar e outras variáveis em modelos experimentais de edema pulmonar cardiogênico e edema não cardiogênico. O edema cardiogênico foi induzido aumentando a pressão atrial esquerda, enquanto o edema não cardiogênico foi induzido por infusão de cetona perilla (CP). Ambas as formas de edema mostraram aumento comparável de fluxo linfático (QL) e escores de radiografia torácica (RxT). O edema cardiogênico foi associado a uma proporção diminuída de proteína plasmática na linfa (L/P) e a elevações maiores da resistência ao fluxo de ar (RL), em comparação ao edema pulmonar não cardiogênico.
O tratamento da SARA inclui a abordagem da causa precipitante da síndrome, suporte de cuidados intensivos gerais, prevenção de complicações na unidade de terapia intensiva (UTI) (p. ex., profilaxia para úlceras por estresse e trombose venosa profunda) e tratamento do edema pulmonar [ver Tabela 4].42 Os aspectos ventilatórios e cardiorrespiratórios do tratamento da insuficiência respiratória em pacientes com SARA (p. ex., limitação do volume corrente)43 são discutidos em detalhes em outra seção [consultar no ACP Medicine informação sobre insuficiência respiratória].
O tratamento da causa da SARA, quando viável, deve ser instituído o quanto antes. Exemplificando, no caso de pacientes com SARA associada à sepse, a administração de antibióticos apropriados deve ser iniciada imediatamente, assim como a fonte de infecção deve ser identificada e tratada (p. ex., os abscessos devem ser drenados).
O manejo inicial na UTI deve incluir várias medidas gerais. A prevenção de ulceras de estresse e do sangramento gastrintestinal associado, bem como de trombose venosa profunda e embolia pulmonar é indicada. Resultados favoráveis têm sido alcançados com as tentativas de minimização da infecção nosocomial, particularmente da pneumonia associada à ventilação, com o uso de antibióticos tópicos e sistêmicos.44 Entretanto, a preocupação quanto à indução de organismos resistentes tem tornado esse tipo de terapia controversa. A alimentação entérica é indicada quando o trato gastrintestinal está funcionando, caso contrário deve ser fornecida a nutrição parenteral. Os achados de um estudo recente indicaram que o uso de uma formulação entérica especial, enriquecida com ácido eicosapentaenoico, ácido c-linolênico e antioxidantes, estava associado à diminuição da mortalidade. A fórmula também conferiu vários benefícios clínicos, incluindo melhora da oxigenação, aumento do número de dias livres de ventilação artificial e do número de dias fora da UTI, além de menos eventos novos de disfunção orgânica.45
O tratamento da SARA enfoca a diminuição do edema pulmonar existente, prevenção de edema adicional e modificação da evolução da doença. Quando o paciente se torna hemodinamicamente estável, a adoção de uma estratégia de restrição de líquidos comprovadamente melhora a função pulmonar e encurta a duração da ventilação mecânica e do tratamento intensivo sem aumentar a incidência de insuficiência orgânica extrapulmonar.46 Adicionalmente, a administração de albumina e furosemida pode ser benéfica para pacientes com hipoproteinemia apresentando lesão pulmonar aguda.47 Numerosas tentativas foram empreendidas no sentido de diminuir a resposta inflamatória observada na SARA. Exceto no caso de pacientes com insuficiência suprarrenal absoluta ou relativa, está amplamente demonstrado que o uso de corticosteroides não proporciona nenhum benefício tanto na fase inicial como na fase avançada da SARA.48,49 Numerosos agentes diferentes não modificaram o processo patológico nem melhoraram o resultado [ver Tabela 4].42,50–52
|
Tabela 4 Tratamento para SARA sem envolvimento de ventilação47,86 | |||
|
Tratamento |
Propósito |
Grau de evidência |
Este tratamento é recomendado? |
|
Profilaxia contra úlceras de estresse |
Prevenir úlceras de estresse e sangramento GI. |
A (estudado em pacientes de UTI) |
Sim; neutralização de ácido gástrico permissível. |
|
Profilaxia contra trombose venosa profunda e embolia pulmonar |
Prevenir trombose venosa profunda e embolia pulmonar. |
A (estudado em pacientes de UTI) |
Sim. |
|
Corticosteroides, doses de reposição |
Insuficiência suprarrenal relativa ou absoluta. |
B |
Sim; para pacientes com insuficiência suprarrenal comprovada e choque séptico refratário. |
|
Antibióticos tópicos + sistêmicos profiláticos |
Prevenir infecções nosocomiais. |
A (estudado em pacientes de UTI) |
Controverso; evidências favoráveis. |
|
Nutrição entérica |
Prevenir desnutrição. |
ND |
Sim; desde que a função GI seja satisfatória. |
|
Nutrição parenteral, com baixo teor de carboidrato e alto teor de gordura |
Prevenir desnutrição. |
ND |
Sim; quando a função GI for insatisfatória. |
|
Restrição de líquido e/ou diurese, possivelmente com infusão de albumina |
Prevenir ou minimizar o edema. |
A |
Sim; desde que hemodinamicamente estável e com função renal intacta. |
SARA = Síndrome da Angústia Respiratória Aguda; GI = Gastrintestinal; UTI = Unidade de Terapia Intensiva; ND = Não Determinado.
A incidência da SARA foi estimada em cerca de 190.000 casos anuais e está associada a um impacto significativo sobre a saúde pública.53,54 Embora o prognóstico dos pacientes com SARA esteja relacionado ao grau de lesão pulmonar, outros parâmetros predizem o resultado com maior precisão. Isto não causa surpresa, uma vez que a SARA frequentemente faz parte de uma síndrome de resposta inflamatória sistêmica. A causa dessa síndrome muitas vezes é desconhecida, mas algumas possibilidades são broncopneumonia, translocação de produtos bacterianos ao longo do intestino e liberação persistente de mediadores endógenos na ausência de infecção em curso. A sepse, que costuma estar associada a uma vasodilatação irresponsiva a agentes vasoconstritores, é a causa mais comum de morte durante o curso da doença. Como resultado da adoção de técnicas de suporte ventilatório modernas, a insuficiência respiratória tem sido a causa da morte em menos de 20% dos casos — um fato que destaca a importância da disfunção de outros sistemas orgânicos (p. ex., insuficiência hemodinâmica com choque refratário ou insuficiência renal progressiva) na causa de morbidade e mortalidade. Dados sugerem que os resultados gerais alcançados por pacientes com SARA podem estar melhorando, possivelmente devido aos avanços terapêuticos.55
Uma forma de prever a mortalidade é por meio do número de sistemas orgânicos com insuficiência. A mortalidade aumenta com o número de órgãos com insuficiência e com o número de dias de insuficiência. Há um aumento adicional da mortalidade entre pacientes com mais de 65 anos de idade. A SARA está associada a uma mortalidade significativa, entretanto, com o passar dos anos, tem sido relatada a diminuição ou manutenção da mortalidade.56,57
Se os pacientes sobrevivem à doença aguda que causa a SARA, o prognóstico de retorno da função pulmonar é bom. Os fatores associados a um resultado funcional pulmonar precário são a gravidade da SARA, complacência medida mais baixa e duração da pressão positiva de ventilação.27 A função pulmonar melhora rápido no decorrer das primeiras semanas e, então, mais lentamente ao longo de um período de dois anos. Os sinais e sintomas comuns incluem dispneia ao esforço, tosse, sibilo e estertores persistentes. Os testes de função pulmonar podem demonstrar a presença de fisiologia restritiva; fisiologia obstrutiva, muitas vezes com aumento da reatividade das vias aéreas; ou capacidade de difusão diminuída. A TC pode mostrar um padrão reticular persistente em 85% dos pacientes. Após um ano ou mais do início da SARA, mais de 75% dos pacientes apresentam função respiratória normal58 ou apenas sofrem de algum comprometimento leve. Por outro lado, muitos apresentarão sequelas neuropsicológicas, como comprometimento da memória, da atenção e da concentração; diminuição da velocidade do processamento mental; ou todos estes quatro efeitos ao mesmo tempo.59 Os pacientes também apresentam diminuição da qualidade de vida, devido ao comprometimento funcional físico resultante do desgaste e enfraquecimento muscular.37,54,60,61 Decorridos 5 anos do aparecimento da doença grave, os sobreviventes da SARA com comorbidades apresentam qualidade de vida diminuída e limitações do exercício, enquanto a função pulmonar está relativamente normal.62
O edema pulmonar pode ser resultante de várias agressões ao sistema nervoso central (SNC), incluindo convulsões de “grande mal”, traumatismo craniano, hemorragia subaracnoidea, hemorragia intracerebral e hematoma subdural. Sua causa mais comum é a hemorragia subaracnoidea.63 O denominador comum dessas agressões ao SNC é o fato de serem graves e agudas. Mais frequentemente, o edema pulmonar é agudo, ocorrendo em questão de minutos a horas após o evento envolvendo o SNC. Ocasionalmente, o aparecimento da condição pode ser mais tardio e a progressão é mais gradual, ocorrendo ao longo de vários dias. Os sítios cerebrais considerados zonas deflagradoras de edema pulmonar neurogênico são o hipotálamo e a medula oblonga. A patogênese do edema pulmonar neurogênico é pouco conhecida. A forma aguda, que é mais comum, pode resultar em grande parte de uma intensa atividade simpática associada com hipertensão sistêmica e pooling central do volume sanguíneo. Isto é referido como teoria da “lesão por explosão” (síndrome explosiva). Esta combinação de fatores pode levar a elevações extraordinárias, ainda que transientes, da pressão capilar pulmonar e a um padrão predominantemente cardiogênico de edema pulmonar. A permeabilidade aumentada decorrente da lesão mecânica induzida por pressão no capilar pulmonar ou, possivelmente, do controle do SNC sobre a permeabilidade capilar pulmonar também parece exercer algum papel no desenvolvimento do edema pulmonar neurogênico.
Clinicamente, o edema pulmonar neurogênico costuma ser diagnosticado por meio de sua associação com uma drástica agressão prévia ao SNC. O principal diagnóstico diferencial é a lesão pulmonar por aspiração. Diferente do edema pulmonar neurogênico, a pneumonite química resultante de aspiração frequentemente persiste por mais alguns dias do que o habitual, e muitas vezes é agravada por infecção bacteriana secundária. Se o processo pulmonar cessar rapidamente (i.e., em poucos dias), o provável diagnóstico é o de edema pulmonar neurogênico. Os fatores de risco importantes para o desenvolvimento de edema pulmonar neurogênico incluem a gravidade clínica e radiológica da hemorragia subaracnoidea, bem como a origem na artéria vertebral.64
O tratamento do edema pulmonar neurogênico é basicamente de suporte e similar ao tratamento da SARA (ver anteriormente). Dependendo da gravidade da agressão neurológica, o edema pulmonar neurogênico pode se resolver em 48-72 horas. O uso de diuréticos não é recomendado na ausência de hipervolemia, devido ao risco de hipotensão hipovolêmica, que poderia agravar a lesão ao SNC no contexto de pressão intracraniana (PIC) aumentada.
Foi relatado que o edema pulmonar de alta altitude (EPAA) ocorre em regiões situadas a 2.400-5.000 m de altitude, com uma incidência de 0,5-15%. Um risco maior está associado à juventude, ao sexo masculino, à subida mais rápida, ao esforço pesado e ao ambiente frio.65,66 A condição raramente é encontrada em locais situados a menos de 2.500-3.000 m de altitude e após um período de aclimatização de uma semana a uma altitude específica. A EPAA também pode ocorrer em indivíduos que residem a altas altitudes e retornam para casa após passarem alguns dias em locais de altitude mais baixa. Indivíduos com história de episódios prévios de EPAA apresentam uma probabilidade de recorrência de 60%.
Os sinais e sintomas incluem tosse, que às vezes é produtiva e com escarro rosado ou sanguinolento, dispneia ao esforço, hipoxemia e febre. O aparecimento dos sintomas frequentemente é gradual, mas ocorre tipicamente em 48-96 horas de exposição a altitudes elevadas.65,66 O edema pulmonar fulminante pode ser precedido de sintomas menos graves de mal das montanhas (também conhecida como doença das alturas ou mal agudo de montanha). O edema pode ser difuso, ainda que irregular, ou bastante assimétrico.
Nos estágios iniciais da EPAA, o líquido do LBA revela um líquido de edema contendo hemácias e rico em proteínas. Em estágios avançados, o líquido do LBA é caracterizado pela presença de mais mediadores pró-inflamatórios e granulócitos.67 Com base em medidas hemodinâmicas, a EPAA é caracterizada pelo desenvolvimento de hipertensão pulmonar em algumas horas após a exposição rápida a altitudes elevadas.
A anormalidade fisiopatológica primária subjacente da EPAA é a permeabilidade capilar aumentada. Foi sugerido que a vasoconstrição pulmonar induzida por hipóxia pronunciada pode levar à hiperperfusão de partes menos obstruídas do leito bascular e subsequente lesão endotelial que resulta em vazamento de líquido.65,66 As evidências da ocorrência desse mecanismo advêm, em parte, da observação de que os pacientes que sofreram EPAA apresentam uma vasoconstrição pulmonar hipóxica mais exagerada do que os indivíduos que nunca tiveram a condição.66 Vários mecanismos foram investigados como fatores contribuidores neste processo. A liberação excessiva de vasoconstritores (p. ex., endotelina-1) ou a produção comprometida de vasodilatadores (p. ex., óxido nítrico) exercem algum papel.65 Os mediadores endematogênicos liberados por células endoteliais ou inflamatórias também podem estar envolvidos.65,66 Somando-se a isso, a atividade comprometida da ATPase de Na+/K+ alveolar pode contribuir para a depuração retardada do líquido alveolar.65,66
Todos estes fatores levam a um efeito comum: hipertensão pulmonar hipóxica exagerada. O risco de EPAA pode ser diminuído com a ascenção lenta e estável para altitudes elevadas. Limitar a velocidade de subida a no máximo 300 m/dia é uma medida efetiva até mesmo para indivíduos com tendência à EPAA. O exercício vigoro deve ser evitado durante os primeiros dias de exposição a altitudes elevadas. A prevenção da EPAA também deve envolver a prevenção à elevação excessiva da pressão arterial pulmonar. Agentes como nifedipina, tadala?l e dexametasona comprovadamente previnem a EPAA em indivíduos suscetíveis.65,66,68 A nefedipina de liberação estendida é a mais bem estudada das medicações destinadas à prevenção da EPAA em indivíduos suscetíveis à condição. Os agonistas ?-adrenérgicos inalatórios também previnem a EPAA. Todavia, seu uso rotineiro não é recomendado.69 Descer para altitudes mais baixas quando há manifestação de sintomas da doença das montanhas aguda também deve diminuir o risco de edema pulmonar. Uma vez que o indivíduo tenha desenvolvido edema pulmonar pleno, a administração de oxigênio, PPCVA, nefedipina, tratamento hiperbárico e uma descida rápida são medidas terapêuticas úteis.64,65 Até mesmo a descida de apenas algumas centenas de metros pode ser benéfica. Foi demonstrado que a inalação de óxido nítrico melhora a oxigenação arterial e pode ser útil para pacientes impossibilitados de serem evacuados para altitudes mais baixas.70 Os diuréticos devem ser evitados. Indivíduos que sofreram EPAA apresentam risco aumentado de recorrência e devem ser aconselhados a evitar altitudes elevadas. No caso dos pacientes sem história prévia de problemas com altitude, a subida acima de 3.000 m de altitude não deve ser problemática, desde que haja tempo para aclimatização.71
A rápida expansão de um pulmão que sofreu colapso pode acarretar edema pulmonar ipsilateral ou, ocasionalmente, bilateral.72 A incidência de edema pulmonar por reexpansão (EPR) é considerada baixíssima. O risco de EPR após a drenagem de um pneumotórax ou derrame pleural está relacionada à quantidade de ar ou líquido no espaço pleural, à duração do colapso, à rapidez a reexpansão e às pressões negativas necessárias à reexpansão do pulmão. O desenvolvimento de uma pressão pleural altamente negativa durante a remoção do ar ou do líquido pleural, com consequente diminuição acentuada da pressão hidrostática intersticial, pode ser importante na patogênese da EPR. A alta concentração proteica no líquido do edema sugere uma permeabilidade de membrana aumentada. Esta permeabilidade aumentada poderia ser causada, em parte, pelo estiramento mecânico e deformação dos poros endoteliais, ou pela geração de radicais de oxigênio tóxicos durante a reperfusão do pulmão rapidamente expandido. A depleção de surfactante no pulmão em colapso pode exercer algum papel na gênese da diminuição da pressão hidrostática intersticial durante a reexpansão. O risco de EPR é muito baixo durante a drenagem de um pneumotórax que tenha surgido há menos de três dias.73 Para um pneumotórax considerado mais antigo, a drenagem sob selo d’água, em vez de por aplicação de pressão negativa, pode diminuir o risco de edema. O esvaziamento de líquido pleural não costuma acarretar edema pulmonar, a menos que mais de 1,0-1,5 L de líquido sejam removidos de forma rápida. Foi sugerido que qualquer quantidade de líquido pleural pode ser removida com segurança, enquanto a pressão pleural for mantida em um nível acima de 20 cm de H2O. Por outro lado, ainda é incerto se esta abordagem sempre prevenirá o EPR. Por este motivo, é aconselhável remover os derrames pleurais muito volumosos de forma gradual, no decorrer de várias horas, sempre que possível. A manifestação clínica do EPR é caracterizada pelo rápido aparecimento de dispneia e taquipneia que tipicamente se desenvolvem dentro de 1 hora da reexpansão do pulmão que sofreu colapso. A tosse pode preceder o desenvolvimento de edema pulmonar. O tratamento do EPR é de suporte, não há evidências de que o tratamento com diuréticos seja benéfico. Foi relatado que a taxa de mortalidade associada ao EPR varia de 0 a 20%.74
O transplante de pulmão pode ser complicado pela lesão isquêmica ao aloenxerto, resultando em edema pulmonar não cardiogênico. O desenvolvimento de edema pulmonar no período pós-operatório imediato é referido como resposta de reimplantação pulmonar (RRP).75 A lesão vascular ao aloenxerto resulta em aumento da permeabilidade a proteínas na microcirculação pulmonar, após a reperfusão. Do ponto de vista clínico, a RRP se manifesta como aparecimento de infiltrados pulmonares no aloenxerto dentro de 24 horas após o transplante, e constitui tipicamente um diagnóstico de exclusão. A RRP também pode estar associada à hipotensão sistêmica e a um débito cárdico diminuído. Considera-se que a patogênese envolve citocinas e radicais livres de oxigênio gerados durante a reperfusão do aloenxerto. O tempo de isquemia prolongado e o uso de bypass cardiopulmonar constituem fatores de risco de desenvolvimento de RRP. A hipertensão pulmonar pré-operatória, o tipo de transplante pulmonar, a condição pulmonar subjacente, e a idade e sexo do receptor não são fatores de risco.75
O edema pulmonar pós-obstrutivo (EPPO) também é referido como edema pulmonar de pressão negativa e constitui uma complicação incomum da obstrução das vias respiratórias superiores. Foram descritas duas classes de EPPO. O tipo I está associado a um esforço inspiratório forçado no contexto de obstrução aguda de via respiratória, e pode ser devido ao laringoespasmo, epiglotite, crupe, choque decorrente de corpo estranho, estrangulamento, enforcamento, obstrução de tubo endotraqueal, tumor de laringe, gota, mononucleose, paralisia de cordas vocais no pós-operatório e quase afogamento.76 O tipo II ocorre após o alívio de uma obstrução parcial de via respiratória crônica,76 como adenoidectomia/tonsilectomia, resseção de massa laríngea, correção de estenose coanal ou diminuição de úvula redundante hipertrófica. Em adultos, a causa mais comum de EPPO é o laringoespasmo e os tumores de vias aéreas superiores. A patogênese do tipo I é considerada relacionada ao desenvolvimento de uma pressão intrapleural altamente negativa (50-100 cm H2O) produzida por esforços inspiratórios vigorosos contra uma via respiratória obstruída (manobra de Müller). A pressão intrapleural altamente negativa diminui a pressão hidrostática intersticial, aumenta o retorno venoso e impõe uma pós-carga ao ventrículo esquerdo. Em adição, esta pressão pode levar a uma intensa ativação simpática, hipertensão sistêmica e acúmulo central de volume sanguíneo. Estes fatores, juntos, podem levar ao desenvolvimento de edema pulmonar agudo via aumento do gradiente de pressão transcapilar (i.e., a diferença entre pressão capilar e pressão hidrostática intersticial). A fisiopatologia do EPPO de tipo II envolve inspiração contra uma via respiratória obstruída, similarmente à manobra de Valsalva, resultando em pressões alveolar e pleural positivas. A condição se resolve rapidamente com a remoção da obstrução. O tratamento envolve cuidados de suporte, incluindo a manutenção de uma via aérea livre e oxigenação adequada.
O edema pulmonar agudo não cardiogênico pode ocorrer após a administração de alguns fármacos [ver Tabela 5]. Uma lista mais completa dos fármacos que podem causar edema pulmonar agudo é disponibilizada na Internet (http://www.pneumotox. com). O edema pulmonar pode ocorrer após a injeção intravenosa de heroína ou outros narcóticos.77 Como o líquido de edema exibe uma alta concentração de proteína, foi sugerido que um defeito de permeabilidade poderia atuar como fator patogênico, mas este achado poderia resultar de uma elevação extrema e transiente da pressão capilar, similarmente ao observado no edema pulmonar neurogênico. O início usualmente ocorre em algumas horas após o uso de narcótico, mas ocasionalmente pode ser retardado por até 24 horas. Além dos achados clínicos e radiográficos associados ao edema pulmonar, são observados sinais típicos de intoxicação por narcótico, como constrição pupilar, diminuição da respiração e alteração da atividade mental. Febre e leucocitose não necessariamente indicam a presença de infecção. Assim como no edema pulmonar neurogênico, a consideração primária do diagnóstico diferencial é a aspiração, devido ao nível de consciência alterado.
O tratamento do edema pulmonar induzido por narcótico é suportivo e, em geral, deve incluir intubação com ventilação mecânica, tanto para garantir o suporte ventilatório adequado como para proteger a via respiratória contra aspiração. O papel da naloxona é incerto. Sem dúvida, a naloxona deve ser administrada em qualquer paciente que tenha entrado em overdose de narcóticos e esteja apresentando bradicardia ou hipotensão potencialmente fatal. Do mesmo modo, se a naloxona for administrada em um paciente irresponsivo e hipopneico, que não necessariamente esteja precisando de ventilação mecânica para edema pulmonar, esse paciente poderá ser poupado da intubação. Em contraste, para um paciente intubado em caráter emergencial devido a um edema pulmonar agudo e que se torne clinicamente estável sem comprometimento hemodinâmico, o melhor tratamento pode ser permitir que a intoxicação por narcótico seja revertida de forma gradual e não precipitada. Não há evidência de que a naloxona ajuda a acelerar a resolução do edema pulmonar induzido por narcótico. De fato, foi relatado que a naloxona causa edema pulmonar.78,79 Adicionalmente, a reversão aguda da intoxicação por narcótico em um indivíduo viciado há muito tempo resulta em agitação, com acentuada ativação simpática e um curso clínico menos estável.
|
Tabela 5 Fármacos associados com edema pulmonar agudo |
|
Narcóticos Cocaína Salicilatos Fármacos anti-inflamatórios não hormonais Naloxona Amiodarona (após anestesia geral) Diuréticos tiazídicos Agentes tocolíticos Protamina Citarabina Metotrexato intratecal Bleomicina Dextrana Meio de contraste Etclorvinol Fenotiazinas Colchicina Interleucina-2 Insulina Ácido all-trans-retinoico87 Talco (intrapleural) |
A cocaína causa edema pulmonar agudo, usualmente quando consumida na forma de cocaína base livre.76 A fisiopatologia é incerta. Assim como a heroína, a cocaína leva a um edema pulmonar rico em proteína que sugere lesão celular endotelial e permeabilidade capilar aumentada. Todavia, assim como sugerido para a heroína, a cocaína poderia levar a uma ativação simpática extrema acompanhada de um aumento abrupto e extremado da pressão capilar, que poderia produzir elevação transiente do vazamento de proteínas através da membrana capilar. A cocaína também causa vasoconstrição coronariana, acompanhada de infarto ou isquemia miocárdica aguda, resultando em edema pulmonar.
O edema pulmonar pode ocorrer como complicação subsequente à resseção de pulmão, especialmente após uma pneumonectomia, sendo referido como edema pulmonar pós-pneumonectomia (EPPP). O quadro clínico é consistente com lesão pulmonar aguda ou SARA e ocorre em 7,9% das pneumonectomias, 3,0% das lobectomias e 0,9% das resseções sublobares. Nestes casos, a mortalidade gira em torno de 40%, mas pode ser maior.80,81
A síndrome de EPPP pode estar associada com taquipneia, temperaturas baixas, edema pulmonar, hipoxemia refratária e hipercapnia. Entre os pacientes que desenvolvem EPPP, 35% manifestarão sinais dentro de 24 horas após a cirurgia, enquanto os demais pacientes exibirão sinais ao redor do 3º dia de pós-operatório.82 Os achados radiográficos podem surgir bem depois do aparecimento dos sinais clínicos.
A patogênese do edema pulmonar pós-resseção de pulmão é multifatorial. É provável que haja envolvimento de sobrecarga de líquido no perioperatório, comprometimento da drenagem linfática decorrente de dissecção de linfonodo, bem como de isquemia e lesão por reperfusão.83 O lado e a extensão da resseção pulmonar podem alterar o risco de EPPP, com as resseções de lado direito associadas a um risco maior. O método usado para drenagem perioperatória do espaço da pneumonectomia, a idade do paciente e o uso de quimioterapia neoadjuvante também podem atuar como fatores de risco de desenvolvimento de EPPP.
O edema pulmonar pós-resseção de pulmão pode ser prevenido com a administração de ß-agonistas, provavelmente por regulação positiva dos mecanismos de depuração de líquido ativos.84 O manejo do edema pulmonar pós-resseção de pulmão inclui ventilação mecânica, dando atenção especial às pressões nas vias respiratórias, além de restrição de líquidos e produtos do sangue. O óxido nítrico inalatório pode ser útil em casos graves de EPPP.85 Há relatos do uso de corticosteroides no tratamento de pacientes com edema pulmonar resultante de resseção de pulmão, mas nenhum estudo controlado randomizado foi conduzido até o momento.80
Referências
1.Laennec, RTH. Treatise on the diseases of the chest and on mediate auscultation. London: T. and G. Underwood; 1821. p.98–99.
2.Brigham KL, Woolverton WC, Blake LH, Staub NC. Increased sheep lung vascular permeability caused by Pseudomonas bacteremia. J Clin Invest 1974;54:792–804.
3.Gorin AB, Stewart PA. Differential permeability of endothelial and epithelial barriers to albumin ?ux. J Appl Physiol1979;47:1315–24.
4.Boitano S, Safdar Z, Welsh DG, et al. Cell-cell interactions in regulating lung function. Am J Physiol Lung Cell Mol Physiol2004;287:L455–9.
5.Taylor AE, Gaar KA Jr. Estimation of equivalent pore radii of pulmonary capillary and alveolar membranes. Am J Physiol1970;218:1133–40.
6.Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med 2005;353:2788–96.
7.Piantadosi CA, Schwartz DA. The acute respiratory distress syndrome. Ann Intern Med 2004;141:460–70.
8.Taylor AE. The lymphatic edema safety factor: the role of edema dependent lymphatic factors (EDLF). Lymphology1990;23:111–23.
9.Parker RE, Roselli RJ, Harris TR, Brigham KL. Effects of graded increases in pulmonary vascular pressures on lung ?uid balance in unanesthetized sheep. Circ Res 1981;49:1164– 72.
10.Schraufnagel DE, Agaram NP, Faruqui A, et al. Pulmonary lymphatics and edema accumulation after brief lung injury. Am J Physiol Lung Cell Mol Physiol 2003;284:L891–7.
11.Brigham KL, Harris TR, Bowers RE, Roselli RJ. Lung vascular permeability: inferences from measurements of plasma to lung lymph protein transport. Lymphology 1979;12: 177–90.
12.Gorin AB, Stewart PA. Differential permeability of endothe- lial and epithelial barriers to albumin ?ux. J Appl Physiol1979;47:1315–24.
13.Gluecker T, Capasso P, Schnyder P, et al. Clinical and radio- logic features of pulmonary edema. Radiographics 1999;19:1507–31.
14.Ely EW, Haponik EF. Using the chest radiograph to determine intravascular volume status: the role of vascular pedicle width. Chest 2002;121:942–50.
15.Murray JF. Pulmonary edema: pathophysiology and diagnosis. Int J Tuberc Lung Dis 2011;15:155–60, i.
16.Shure D. Pulmonary-artery catheters—peace at last? N Engl J Med 2006;354:2273–4.
17.Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest 2004;125:669–82.
18.Pierard LA, Lancellotti P. The role of ischemic mitral regurgitation in the pathogenesis of acute pulmonary edema. N Engl J Med 2004;351:1627–34.
19.Andrew P. Diastolic heart failure demysti?ed. Chest2003;124:744–53.
20.Masip J, Roque M, Sanchez B, et al. Noninvasive ventilation in acute cardiogenic pulmonary edema: systematic review andmeta-analysis. JAMA 2005;294:3124–30.
21.Gray A, Goodacre S, Newby DE, et al. Noninvasive ventilation in acute cardiogenic pulmonary edema. N Engl J Med2008;359:142–51.
22.Vital FM, Saconato H, Ladeira MT, et al. Non-invasive positive pressure ventilation (CPAP or bilevel NPPV) for cardiogenic pulmonary edema. Cochrane Database Syst Rev 2008;(3):CD005351.
23.Weng CL, Zhao YT, Liu QH, et al. Meta-analysis: noninvasive ventilation in acute cardiogenic pulmonary edema. Ann Intern Med 2010;152:590–600.
24.Edoute Y, Roguin A, Behar D, Reisner SA. Prospective eval- uation of pulmonary edema. Crit Care Med 2000;28:330–5.
25.Tribouilloy C, Rusinaru D, Leborgne L, et al. In-hospital
mortality and prognostic factors in patients admitted fornew-onset heart failure with preserved or reduced ejection fraction: a prospective observational study. Arch Cardiovasc Dis2008;101:226–34.
26.Adnet F, Le TP, Leberre A, et al. In-hospital and long-termprognosis of elderly patients requiring endotracheal intubation for life-threatening presentation of cardiogenic pulmonary edema. Crit Care Med 2001;29:891–5.
27.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care 2005;11:43–9.
28.Atabai K, Matthay MA. The pulmonary physician in critical care. 5: Acute lung injury and the acute respiratory distress syndrome: de?nitions and epidemiology. Thorax 2002;57:452–8.
29.Rocco PR, Zin WA. Pulmonary and extrapulmonary acute respiratory distress syndrome: are they different? Curr Opin Crit Care 2005;11:10–7.
30.Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit Care Med 2003;31(4 Suppl):S207–12.
31.Iribarren C, Jacobs DR Jr, Sidney S, et al. Cigarette smoking, alcohol consumption, and risk of ARDS: a 15-year cohort study in a managed care setting. Chest 2000;117:163–8.
32.Mangialardi RJ, Martin GS, Bernard GR, et al. Hypoproteinemia predicts acute respiratory distress syndrome develop- ment, weight gain, and death in patients with sepsis. Ibuprofen in Sepsis Study Group. Crit Care Med 2000;28: 3137–45.
33.Marshall RP, Webb S, Bellingan GJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med 2002;166:646–50.
34.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol2011;6:147–63.
35.Marini JJ. Advances in the understanding of acute respiratory distress syndrome: summarizing a decade of progress. Curr Opin Crit Care 2004;10:265–71.
36.Mendez JL, Hubmayr RD. New insights into the pathology of acute respiratory failure. Curr Opin Crit Care 2005;11: 29–36.
37.Steinberg KP, Hudson LD. Acute lung injury and acute respiratory distress syndrome. The clinical syndrome. Clin Chest Med 2000;21:401–17, vii.
38.Rouby JJ, Puybasset L, Nieszkowska A, Lu Q. Acute respiratory distress syndrome: lessons from computed tomography of the whole lung. Crit Care Med 2003;31(4 Suppl): S285–95.
39.Nuckton TJ, Alonso JA, Kallet RH, et al. Pulmonary dead-spacefraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med 2002;346: 1281–6.
40.Moloney ED, Evans TW. Pathophysiology and pharmaco- logical treatment of pulmonary hypertension in acute respiratory distress syndrome. Eur Respir J 2003;21:720–7.
41.Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS. Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia—a review. Crit Care Med 2006; 34:871–7.
42.Jain R, DalNogare A. Pharmacological therapy for acute respiratory distress syndrome. Mayo Clin Proc 2006;81:205–12.
43.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000;342: 1301–8.
44.Liberati A, D’Amico R, Pifferi S, et al. Antibiotic prophylaxis to reduce respiratory tract infections and mortality in adults receiving intensive care. Cochrane Database Syst Rev 2009;(4):CD000022.
45.Pontes-Arruda A, Aragao AM, Albuquerque JD. Effects of enteral feeding with eicosapentaenoic acid, gammalinolenic acid, and antioxidants in mechanically ventilated patients with severe sepsis and septic shock. Crit Care Med 2006;34:2325–33.
46.Wiedemann HP, Wheeler AP, Bernard GR, et al. Compari- son of two ?uid-management strategies in acute lung injury. N Engl J Med 2006;354:2564–75.
47.Calfee CS, Matthay MA. Nonventilatory treatments for acute lung injury and ARDS. Chest 2007;131:913–20.
48.Steinberg KP, Hudson LD, Goodman RB, et al. Ef?cacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 2006;354:1671–84.
49.Matthay MA, Idell S. Update on acute lung injury and critical care medicine 2009. Am J Respir Crit Care Med 2010;181:1027–32.
50.Frank AJ, Thompson BT. Pharmacological treatments for acute respiratory distress syndrome. Curr Opin Crit Care2010;16:62–8.
51.Raoof S, Goulet K, Esan A, et al. Severe hypoxemic respiratory failure: part 2—nonventilatory strategies. Chest 2010;137:1437–48.
52.Papazian L, Forel JM, Gacouin A, et al. Neuromuscular
blockers in early acute respiratory distress syndrome. N Engl J Med 2010;363:1107–16.
53.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353:1685– 93.
54.Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest 2007;131:554–62.
55.Stapleton RD, Wang BM, Hudson LD, et al. Causes and timing of death in patients with ARDS. Chest 2005;128: 525–32.
56.Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest2008;133:1120–7.
57.Phua J, Badia JR, Adhikari NK, et al. Has mortality from acute respiratory distress syndrome decreased over time?
A systematic review. Am J Respir Crit Care Med 2009;179:220–7.
58.Desai SR, Wells AU, Rubens MB, et al. Acute respiratory distress syndrome: CT abnormalities at long-term follow- up. Radiology1999;210:29–35.
59.Hopkins RO, Weaver LK, Pope D, et al. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med1999;160:50–6.
60.Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003;348:683–93.
61.Orme J Jr, Romney JS, Hopkins RO, et al. Pulmonary function and health-related quality of life in survivors of acute respiratory distress syndrome. Am J Respir Crit Care Med 2003;167:690–4.
62.Herridge MS, Tansey CM, Matte A, et al. Functional dis- ability 5 years after acute respiratory distress syndrome. N Engl J Med2011;364:1293–304.
63.Fontes RB, Aguiar PH, Zanetti MV, et al. Acute neurogenic
pulmonary edema: case reports and literature review. J Neurosurg Anesthesiol 2003;15:144–50.
64.Baumann A, Audibert G, McDonnell J, Mertes PM. Neuro- genic pulmonary edema. Acta Anaesthesiol Scand 2007;51: 447–55.
65.West JB. The physiologic basis of high-altitude diseases. Ann Intern Med 2004;141:789–800.
66.Bartsch P, Mairbaurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol 2005;98:1101–10.
67.Maggiorini M. Prevention and treatment of high-altitudepulmonary edema. Prog Cardiovasc Dis 2010;52:500–6.
68.Maggiorini M, Brunner-La Rocca HP, Peth S, et al. Both tadala?l and dexamethasone may reduce the incidence ofhigh-altitude pulmonary edema: a randomized trial. Ann Intern Med 2006;145:497–506.
69.Sartori C, Allemann Y, Duplain H, et al. Salmeterol for the prevention of high-altitude pulmonary edema. N Engl J Med2002;346:1631–6.
70.Anand IS, Prasad BA, Chugh SS, et al. Effects of inhaled nitric oxide and oxygen in high-altitude pulmonary edema. Circulation1998;98:2441–5.
71.Schoene RB. Illnesses at high altitude. Chest 2008;134: 402–16.
72.Sherman SC. Reexpansion pulmonary edema: a case report and review of the current literature. J Emerg Med 2003;24: 23–7.
73.Sohara Y. Reexpansion pulmonary edema. Ann Thorac Cardiovasc Surg 2008;14:205–9.
74.Neustein SM. Reexpansion pulmonary edema. J Cardiotho- rac Vasc Anesth 2007;21:887–91.
75.Khan SU, Salloum J, O’Donovan PB, et al. Acute pulmonary edema after lung transplantation: the pulmonary reimplantation response. Chest 1999;116:187–94.
76.Udeshi A, Cantie SM, Pierre E. Postobstructive pulmonary edema. J Crit Care 2010;25:508–5.
77.Wilson KC, Saukkonen JJ. Acute respiratory failure from abused substances. J Intensive Care Med 2004;19:183–93.
78.Clarke SF, Dargan PI, Jones AL. Naloxone in opioid poisoning: walking the tightrope. Emerg Med J 2005;22:612–6.
79.Schwartz JA, Koenigsberg MD. Naloxone-induced pulmo- nary edema. Ann Emerg Med 1987;16:1294–6.
80.Grichnik KP, D’Amico TA. Acute lung injury and acute respiratory distress syndrome after pulmonary resection. Semin Cardiothorac Vasc Anesth 2004;8:317–34.
81.Dulu A, Pastores SM, Park B, et al. Prevalence and mortal- ity of acute lung injury and ARDS after lung resection. Chest2006;130:73–8.
82.Villeneuve PJ, Sundaresan S. Complications of pulmonary resection: postpneumonectomy pulmonary edema and post- pneumonectomy syndrome. Thorac Surg Clin 2006;16: 223–34.
83.Jordan S, Mitchell JA, Quinlan GJ, et al. The pathogenesis of lung injury following pulmonary resection. Eur Respir J2000;15:790–9.
84.Licker M, Tschopp JM, Robert J, et al. Aerosolized salbutamol accelerates the resolution of pulmonary edema after lung resection. Chest 2008;133:845–52.
85.Rabkin DG, Sladen RN, DeMango A, et al. Nitric oxide for the treatment of postpneumonectomy pulmonary edema. Ann Thorac Surg 2001;72:272–4.
86.Jain R, DalNogare A. Pharmacological therapy for acute respiratory distress syndrome. Mayo Clin Proc 2006;81:205–12.
87.Jung JI, Choi JE, Hahn ST, et al. Radiologic features of all-trans-retinoic acid syndrome. AJR Am J Roentgenol 2002; 178:475.
Agradecimento
Figura 3 – Christine Kennedy
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.