(Carregando Índice)... (Carregando Índice)... |
Última revisão: 21/03/2016
Comentários de assinantes: 0
Artigo original: Shear NH, Knowles S, Shapiro L. Cutaneous Adverse Drug Reactions. ACP Medicine. 2012.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon
Neil H. Shear, MD, FRCPC
Professor of Medicine, Department of Medicine, Divisions of Dermatology & Clinical Farmacologia, Sunnybrook Health Sciences Centre, University of Toronto, Toronto, Ontario, Canada
Sandra Knowles, BScPhm
Phamacist, Department of Pharmacy, Sunnybrook Health Sciences Centre, University of Toronto, Toronto, Ontario, Canada
Lori Shapiro, MD, FRCPC
Assistant Professor, Department of Medicine, Division of Dermatology, Sunnybrook Health Sciences Centre, University of Toronto, Toronto, Ontario, Canada
Uma reação adversa a medicação (RAM) é definida como qualquer efeito nocivo, não intencional e indesejado de um fármaco, que seja produzido com doses usadas em seres humanos para fins de profilaxia, diagnóstico ou terapia. As RAMs geralmente são classificadas como reações previsíveis ou “no alvo” (com base na farmacologia do medicamento e potencialmente evitável), ou ainda como reações imprevisíveis.1 A maioria das reações cutâneas é considerada imprevisível ou “fora do alvo” e amplamente classificadas como imunológicas (alérgicas) ou idiossincráticas. As RAMs cutâneas podem variar de uma simples erupção exantematosa a síndromes graves (p. ex., síndrome de hipersensibilidade farmacológica [SHF], síndrome de Stevens-Johnson [SSJ], necrólise epidérmica tóxica [NET] e reação do tipo doença do soro). De que forma um fármaco consegue causar reações cutâneas continua sendo um mistério apenas parcialmente compreendido. Os quatro componentes principais de uma reação iniciam com a genética do paciente, a farmacologia e o metabolismo do medicamento, a existência de infecções prévias e a natureza da resposta imune. Muitas RAMs cutâneas graves são desencadeadas, em parte, pela formação de metabólitos oxidativos reativos e, talvez, formação de complexos de anticorpo antifármaco e proteína, proteínas cutâneas ou ambos. A predisposição às erupções fármaco-induzidas pode ser genética e estar relacionada ao metabolismo farmacológico e às respostas imunes. A presença de infecções virais ativas aumenta o risco de algumas RAMs cutâneas, enquanto a presença a longo prazo de reservatórios de vírus pode ter papel na expressão de algumas reações. Este capítulo revisa a fisiopatologia e as manifestações clínicas importantes para o diagnóstico correto e tratamento das RAMs cutâneas.
Estudos epidemiológicos têm mostrado que as RAMs ocorrem em 6,7% de todos os pacientes internados e que cerca de 5% de todos os casos de internação são resultantes de RAMs.2 No Boston Collaborative Drug Surveillance Program, a prevalência de RAMs cutâneas em pacientes hospitalizados foi de 2,2%.3 Os antibióticos foram responsáveis por 75% das reações detectadas. No Harvard Medical Practice Study, cerca de 14% das RAMs em pacientes internados eram de natureza cutânea ou alérgica.4 O custo da morbidade e mortalidade relacionadas com medicamentos tem sido estimado em 30 bilhões de dólares ao ano,5 com as RAMs classificadas como sendo entre a quarta e a sexta causas principais de morte nos Estados Unidos.6
As reações cutâneas a fármacos frequentemente ocorrem em contextos clínicos complicados, que podem incluir a exposição a múltiplos agentes. Fármacos novos cujo curso tenha sido iniciado dentro das últimas seis semanas são agentes causais em potencial, assim como os fármacos usados de modo intermitente, incluindo as preparações vendidas sem prescrição médica, fitoterápicos e compostos naturais.
A morfologia das erupções cutâneas pode ser exantematosa, urticariforme, bolhosa ou pustular. A extensão da reação é variável. Exemplificando, uma vez documentada a morfologia da reação, é possível estabelecer um diagnóstico (p. ex., erupção farmacológica fixa ou pustulose exantematosa generalizada aguda). A reação também pode se manifestar como síndrome (p. ex., reação do tipo doença do soro ou reação de síndrome de hipersensibilidade). A febre está associada com as RAMs cutâneas mais graves [ver Tabela 1].
Os diagnósticos diferenciais podem incluir os exantemas virais (p. ex., mononucleose infecciosa e infecção por parvovírus B19), infecções bacterianas, síndrome de Kawasaki, doenças do colágeno e vasculites, e neoplasias.
O teste cutâneo de penicilina com determinantes maiores e menores é útil para confirmação de reações de hipersensibilidade mediada por IgE contra penicilina. Os testes cutâneos são realizados em seis semanas a seis meses após a cura completa da reação farmacológica cutânea.7 Os testes orais podem ser úteis para o diagnóstico de RAMs, entretanto não devem ser usados em casos de pacientes que tenham apresentado reação grave, como SSJ ou NET. Os testes de contato podem ser úteis no diagnóstico de erupções farmacológicas fixas ou dermatite de contato.8
Erupções exantematosas, também conhecidas como erupções morbiliformes, erupções maculopapulares, exantemas ou erupções escarlatiniformes, são as RAMs cutâneas mais comuns.3 Os exantemas simples consistem em alterações eritematosas que ocorrem na pele sem formação de bolhas nem pústulas.
Muitos fármacos podem causar erupções exantematosas, incluindo as penicilinas, sulfonamidas, barbitúricos, medicações antiepiléticas, inibidores de transcriptase reversa não nucleosídeos (p. ex., nevirapina) e antimaláricos.3 As erupções exantematosas ocorrem em 3-7% dos pacientes que recebem aminopenicilinas, como ampicilina e amoxicilina. Entretanto, estas erupções podem ocorrer em 60-100% dos pacientes sob tratamento com ampicilina ou amoxicilina que, concomitantemente, estejam recebendo terapia de alopurinol ou tenham leucemia linfocítica, mononucleose infecciosa, infecção por citomegalovírus ou hiperuricemia.8
|
Tabela 1 Sinais de alerta de uma erupção farmacológica grave |
|
Sintomas sistêmicos Febre, faringite, mal-estar, artralgia, tosse Linfadenopatia Sintomas cutâneos Eritroderma Envolvimento facial proeminente ± edema Envolvimento de membrana mucosa Sensibilidade cutânea, bolhas ou descamação Púrpura |
Estudos sugerem que algumas erupções exantematosas representam hipersensibilidade mediada por células.9 A etiologia da erupção por ampicilina concomitante com infecção viral é desconhecida, porém a erupção aparentemente não é mediada por IgE e os pacientes conseguem tolerar todos os antibióticos b-lactâmicos, incluindo ampicilina, tão logo o processo infeccioso tenha sido resolvido. Reações similares são observadas em 50% dos pacientes infectados por HIV expostos a antibióticos sulfonamidas.10 As células T fármaco-específicas exercem papel importante nas reações exantematosas, bolhosas e pustulares.11
Os exantemas simples são simétricos e muitas vezes se tornam generalizados. A febre não está associada a erupções exantematosas simples. Estas erupções usualmente ocorrem em uma semana após o início da terapia e em geral se resolvem em 7-14 dias. Uma alteração na cor do exantema, de vermelho brilhante para marrom-avermelhado, indica resolução. A resolução pode ser seguida de escamação ou descamação. Alguns pacientes com erupções exantematosas induzidas por ampicilina ou amoxicilina podem apresentar resultado positivo em teste de contato ou em um teste intradérmico tardio.8 De modo geral, todavia, o teste cutâneo não é considerado útil no diagnóstico de uma erupção exantematosa.
O diagnóstico diferencial de erupção exantematosa induzida por fármaco inclui exantema viral (os pacientes devem ser testados para mononucleose), doença do colágeno e vasculites, infecção bacteriana e infecção por riquétsia. A síndrome de hipersensibilidade deve ser considerada no diagnóstico diferencial.
O tratamento das erupções exantematosas simples geralmente é de suporte. Exemplificando, os anti-histamínicos orais usados em conjunto com banhos calmantes podem ajudar a aliviar a urticária. Os corticosteroides tópicos são indicados para os casos em que os anti-histamínicos não promovem alívio. Os corticosteroides sistêmicos somente são usados em casos graves. A descontinuação do agente agressor é recomendada na maioria dos casos.
A reação de síndrome de hipersensibilidade (também conhecida como SHF ou erupção/reação farmacológica com eosinofilia e sintomas sistêmicos [Síndrome DRESS]) é uma reação farmacológica complexa que afeta vários sistemas de órgãos. Uma tríade de febre, erupção cutânea e envolvimento de órgão interno sinaliza esta síndrome potencialmente fatal. Embora a ocorrência da condição tenha sido relatada em 1 a cada 1.000 e em 1 a cada 10.000 exposições a anticonvulsivantes e antibióticos sulfonamidas, a incidência verdadeira é desconhecida devido à apresentação variável e relatos incertos. Entre os fármacos relatados como causadores desta síndrome, estão os anticonvulsivantes aromáticos (i.e., fenitoína, fenobarbital, oxcarbazepina e carbamazepina), lamotrigina, antibióticos sulfonamidas, dapsona, nitrofurantoina, nevirapina, minociclina e alopurinol. Em uma revisão de 172 casos publicados, o fármaco mais frequentemente associado foi a carbamazepina, seguida do alopurinol, lamotrigina, fenobarbital e sulfasalazina.12
Foram desenvolvidos critérios diagnósticos que incluem exantema, anormalidades hematológicas (p. ex., eosinofilia e/ou presença de linfócitos atípicos), envolvimento sistêmico com aumento de linfonodo ou hepatite (aumento superior a duas vezes nos valores de transaminases séricas), nefrite intersticial ou falência de outro órgão.13
Tem sido sugerido que o metabolismo dos anticonvulsivantes aromáticos pelo citocromo P-450 exerce papel central no desenvolvimento da reação de síndrome de hipersensibilidade com estes fármacos.14 Parece haver uma associação entre a infecção pelo herpesvírus humano tipo 6 (HHV-6) (seja a infecção inicial ou uma reativação) e a síndrome de hipersensibilidade grave.15 A reativação da infecção por HHV-6 é considerada um critério diagnóstico pelo grupo de consenso japonês para SHF.16 As infecções virais podem atuar como/gerar produção de sinais perigosos que levam a respostas imunes danosas aos fármacos, em vez de imunotolerância.17,18
Em um estudo, 75% dos pacientes com reações de síndrome de hipersensibilidade a um anticonvulsivante aromático apresentaram reatividade cruzada in vitro a outros dois anticonvulsivantes aromáticos.14 Adicionalmente, testes in vitro têm mostrado a ocorrência familiar de hipersensibilidade a agentes anticonvulsivantes.14 Embora a lamotrigina não seja um anticonvulsivante aromático, também pode causar reação de síndrome de hipersensibilidade. A lamotrigina e outros anticonvulsivantes também estão associados a reações mais graves (p. ex., SSJ e NET) [ver Erupções Complexas, adiante].
Os antibióticos sulfonamidas podem causar reações de síndrome de hipersensibilidade em indivíduos suscetíveis. A via metabólica primária das sulfonamidas evolve acetilação do fármaco em um metabolito não tóxico e excreção renal. Uma via metabólica alternativa, quantitativamente mais importante em pacientes que são acetiladores lentos, engaja o sistema de oxidase de função mista do citocromo P-450. Estas enzimas transformam o composto parental em metabólitos reativos—a saber, hidroxilaminas e compostos nitrosos, que produzem citotoxicidade independentemente de anticorpo fármaco-específico pré-formado. Na maioria das pessoas, o metabólito é destoxificado. Entretanto, as reações de síndrome de hipersensibilidade podem ocorrer em pacientes incapazes de destoxificar este metabólito (p. ex., indivíduos com deficiência de glutationa).19 Embora o defeito de destoxificação esteja presente em 2% da população, apenas 1 em cada 10.000 indivíduos manifestará uma reação de síndrome de hipersensibilidade em resposta a antibióticos sulfonamidas. Irmãos e outros parentes de primeiro grau de pacientes com problema de destoxificação apresentam risco aumentado (talvez, 1 em 4) de terem defeito similar.
Outras aminas aromáticas, como a procainamida, dapsona e acebutolol, também são metabolizadas a compostos quimicamente reativos. Nós recomendamos que os pacientes que desenvolvem sintomas compatíveis com uma reação de síndrome de hipersensibilidade à sulfonamida evitem estas aminas aromáticas devido ao potencial de reatividade cruzada. Do mesmo modo, foi demonstrado que a sulfassalazina apresenta reatividade cruzada com o sulfametoxazol, de modo que este fármaco também deve ser evitado. Entretanto, não deve haver reatividade cruzada entre sulfonamidas e fármacos que não sejam aminas aromáticas (p. ex., sulfonilureias, diuréticos tiazida, furosemida e acetazolamida).20
O alopurinol está associado ao desenvolvimento de reações farmacológicas graves, incluindo reação de síndrome de hipersensibilidade. Em um total de 60 pacientes com reação de síndrome de hipersensibilidade, o alopurinol foi citado como fármaco responsável em 19 casos (32%).21 Em uma revisão de 13 pacientes apresentando reações adversas ao alopurinol, febre e erupção foram os sintomas mais comumente manifestados. Outras anormalidades associadas foram leucocitose (62%), eosinofilia (54%), comprometimento renal (54%) e disfunção hepática (69%).22 A reativação da infecção por HHV-6 foi relatada em um paciente que desenvolveu reação de síndrome de hipersensibilidade induzida por alopurinol com hepatite, comprovada por análise de reação em cadeia da polimerase mostrando o DNA do HHV-6 em amostra de sangue. O DNA do HHV-6 também foi detectado no líquido cerebrospinal.23 As reações adversas induzidas por alopurinol, incluindo SHF, SSJ e NET, têm sido fortemente associadas à predisposição genética em chineses han. Foi demonstrado que o alelo B*5801 do antígeno leucocitário humano (HLA) é um importante fator de risco genético.24
A suscetibilidade à hipersensibilidade da nevirapina pode ser aumentada pela presença do alelo HLA-DRB1*0101, mas é inibida por contagens baixas de células T CD4+.25 O abacavir também está associado a reações adversas potencialmente fatais em cerca de 8% dos pacientes que recebem este fármaco. Estudos têm mostrado a existência de uma forte associação preditiva entre a reação de síndrome de hipersensibilidade ao abacavir e o HLA-B*5701.26
A reação de síndrome de hipersensibilidade é mais frequente na primeira exposição ao fármaco, com os sintomas iniciais surgindo em 1-6 semanas após a exposição [ver Tabela 2].
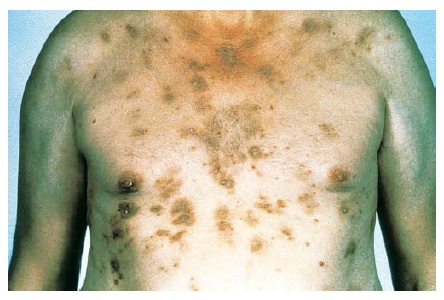
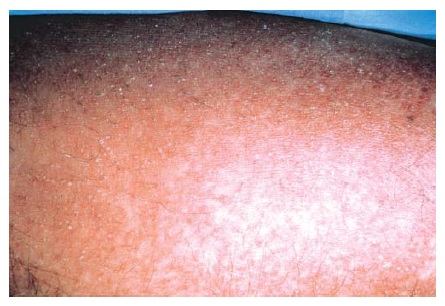
Febre e mal-estar, que podem ser acompanhados de faringite e linfadenopatia cervical, são os sintomas manifestados pela maioria dos pacientes. Estes sintomas frequentemente são seguidos de edema e inchaço da face, sobretudo ao levantar de manhã. Linfocitose atípica com subsequente eosinofilia pode ocorrer durante as fases iniciais da reação, em alguns pacientes. Uma erupção cutânea, que ocorre em cerca de 85% dos pacientes, pode variar de uma erupção exantematosa [ver Figura 1] até as condições mais sérias de SSJ ou NET. O envolvimento hepático é frequente e resulta em hepatite, embora outros órgãos internos possam ser afetados, como o rim (p. ex., nefrite intersticial e vasculite), o sistema nervoso central (p. ex., encefalite e meningite asséptica), o coração (p. ex., miocardite)27 e os pulmões (p. ex., pneumonite intersticial, síndrome da angústia respiratória e vasculite). Um subgrupo de pacientes pode desenvolver hipotireoidismo como parte de uma tireoidite autoimune dentro de dois meses após a iniciação dos sintomas.28 Esta condição é caracterizada por baixos níveis de tiroxina, níveis altos de hormônio estimulador da tireoide e presença de autoanticorpos antitireoide, incluindo anticorpos antimicrossomais.

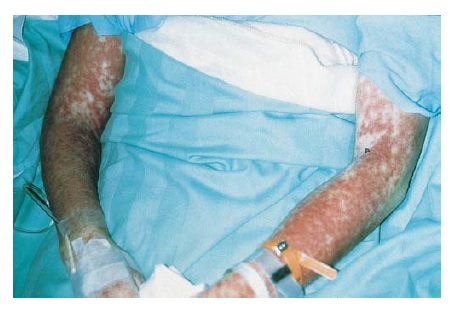
Figura 1 Esta mulher de 35 anos desenvolveu reação de síndrome de hipersensibilidade, caracterizada por febre, erupção e hepatite, decorridos 14 dias do início da terapia com trimetoprima-sulfametoxazol. A erupção é extensa, simétrica, avermelhada e edematosa.
Depois que a ocorrência de uma reação de síndrome de hipersensibilidade é reconhecida a partir do complexo de sintomas de febre, de erupção e de linfadenopatia, alguns exames laboratoriais podem ser usados para avaliar o envolvimento de órgão interno que, por sua vez, pode ser assintomático. Hemograma completo, análise de urina (Urina tipo 1) e determinação dos níveis de transaminases hepáticas e creatinina sérica devem ser realizados. Em adição, o clínico deve ser guiado pelos sintomas que possam sugerir envolvimento de órgão interno específico (p. ex., sintomas respiratórios). A função tireoidiana deve ser avaliada no momento da apresentação da reação de síndrome de hipersensibilidade e, subsequentemente, em 2-3 meses após a manifestação. Uma biópsia de pele pode ajudar a confirmar o diagnóstico quando o paciente apresenta bolhas ou erupção pustular. Infelizmente, não há testes diagnósticos ou confirmatórios prontamente disponíveis. Um teste in vitro empregando um sistema microssomal hepático murino é usado para fins de pesquisa na caracterização de pacientes que desenvolvem reação de síndrome de hipersensibilidade.29 O teste cutâneo também têm sido usado, embora a sensibilidade e especificidade sejam desconhecidas.30 Devido à gravidade da reação, as repetições de desafio oral e dessensibilizações não são recomendadas. De fato, em pacientes com história de reação de síndrome de hipersensibilidade, a reexposição ao agente agressor pode levar ao desenvolvimento de sintomas em um dia.
Embora os sintomas se resolvam na maioria dos pacientes após a descontinuação do fármaco, alguns desenvolvem doença autoimune e/ou produção de autoanticorpos após a resolução da SHF.31 Isto pode incluir o desenvolvimento de diabetes mellitus tipo 1,32 tireoidopatia autoimune32 e lúpus eritematoso.33
|
Tabela 2 Achados clínicos da reação de síndrome de hipersensibilidade e da reação do tipo doença do soro | |||||
|
|
Erupção |
Febre |
Envolvimento de órgão interno |
Artralgia |
Linfadenopatia |
|
Reação de síndrome de hipersensibilidade |
Exantema Dermatite esfoliativa Erupções pustulares Eritema multiforme Síndrome de Stevens-Johnson Necrólise epidérmica tóxica |
Presente |
Presente |
Ausente |
Presente |
|
Reação do tipo doença do soro |
Urticária |
Presente |
Ausente |
Presente |
Presente |
Embora o papel dos corticosteroides seja controverso, a maioria dos clínicos inicia o regime com dosagem de 1-2 mg/kg/dia quando os sintomas são graves, com desmame lento, muitas vezes requerendo semanas a meses. Há relatos de tratamento bem-sucedido com ciclosporina,34 imunoglobulina intravenosa (IVIg)35 e plasmaférese combinada com rituximabe.36 Anti-histamínicos, corticosteroides tópicos ou ambos podem ser usados para aliviar os sintomas. Como o risco de reação de síndrome de hipersensibilidade em parentes de primeiro grau de pacientes que tiveram reações é substancialmente maior do que na população em geral, os familiares devem receber aconselhamento quanto ao risco.
A urticária é caracterizada por pápulas avermelhadas pruriginosas de tamanhos variados, que podem ocorrer com qualquer medicação. Quando os tecidos subcutâneos e dérmicos profundos também estão inchados, a reação é conhecida como angioedema37 [consulte no ACP Medicine informações sobre urticária, angioedema e anafilaxia]. Urticária e angioedema geralmente resultam de uma reação de hipersensibilidade imediata tipo 1. Este mecanismo é caracterizado pela ocorrência de reações imediatas à penicilina e a outros antibióticos. A ligação do fármaco ou seu metabolito à IgE ligada à superfície dos mastócitos cutâneos resulta em ativação, degranulação e liberação de mediadores vasoativos, como histamina, leucotrienos e prostaglandinas. A urticária subsequente à exposição a aspirina ou a fármacos anti-inflamatórios não esteroides (AINEs) é causada pela inibição da ciclo-oxigenase.38 Os analgésicos opiáceos e relaxantes musculares anestésicos também podem causar urticária ao agirem diretamente no mastócito para induzir degranulação.39
As reações urticariformes também podem resultar da ativação não imunológica de mediadores inflamatórios. Fármacos como ácido acetil salicílico e AINEs, meio de contraste e analgésicos narcóticos podem causar diretamente a liberação de histamina dos mastócitos, independentemente de IgE. Os inibidores de enzima conversora de angiotensina (ECA) são causa frequente de angioedema.40 O angioedema induzido por inibidor de ECA pode afetar a língua, a garganta, a face e o intestino delgado.41 O mecanismo dessa reação é desconhecido, mas está relacionado ao acúmulo de bradicinina ou ativação do sistema complemento.
Embora as medicações tendam a causar urticária, angioedema ou ambos, outros agentes causais incluem os alimentos [consulte no ACP Medicine informação sobre alergias alimentares], fatores físicos (p. ex., dermatografismo e urticária colinérgica) [consulte no ACP Medicine informação sobre urticária, angioedema e anafilaxia] e fatores idiopáticos. Certos alimentos contendo proteína podem apresentar reação cruzada com proteínas do látex, tais como banana, kiwi, abacate e castanhas, e causar inchaço e prurido oral, urticária ou sibilos após a ingesta. O risco de alergia ao látex é especialmente alto em crianças com espinha bífida e funcionários da assistência médica.42 A alergia ao látex pode se manifestar como urticária de contato nos sítios de exposição ao látex. Tais reações incluem inchaço labial em indivíduo que tenham enchido um balão de ar ou em criança que tenha chupado chupeta. O contato com os aerossois de pó oriundos das luvas de látex as quais a proteína do látex tenha aderido pode causar sintomas mucosos, como coceira e inchaço nos olhos; corrimento nasal; espirros; ou sibilos. Também pode haver anafilaxia.
Os sinais e sintomas das reações alérgicas mediadas por IgE são tipicamente prurido, urticária, eritema cutâneo, angioedema, náusea, vômito, diarreia, dor abdominal, congestão nasal, rinorreia, edema de laringe e broncoespasmo ou hipotensão, ou ambos. A febre não está associada com reações de urticária nem angioedema. Em geral, as lesões isoladas de urticária duram menos de 24 horas, embora novas lesões possam se desenvolver continuamente. As reações adversas aos inibidores de ECA usualmente surgem dentro de algumas horas após o início do curso do fármaco, mas podem demorar de uma semana a vários meses após o início da terapia. Com o tratamento, o angioedema resultante geralmente se resolve em 48 horas.43
O teste cutâneo pode ser útil em casos de urticária mediada por IgE ou anafilaxia.44 O teste cutâneo de penicilina com determinantes maiores e menores, por exemplo, identifica cerca de 99% dos pacientes que tiveram reação IgE-mediada contra penicilina. Um teste cutâneo para látex é indicador sensível de sensibilização por IgE. As reações positivas do teste cutâneo imediato identificam pacientes que apresentam risco de desenvolvimento de reações IgE-mediadas a partir de agentes de alto peso molecular, como insulina, insulina protamina neutra de Hagedorn (NPH) e vacinas contendo ovo.
A retirada do agente causal é recomendada. Quando ocorre angioedema ou anafilaxia, pode ser necessária a instituição imediata de terapia com adrenalina e esteroides sistêmicos. O alívio sintomático em geral pode ser alcançado com anti-histamínicos (bloqueadores de receptor H1). A segurança dos antagonistas de receptor de angiotensina II para pacientes com história de angioedema associado ao uso de inibidores de ECA ainda é indeterminada, apesar da incidência aproximada de 10% de reatividade cruzada de angioedema entre os pacientes.45
A urticária alérgica deve ser diferenciada da urticária causada por fatores físicos. A urticária fria, por exemplo, é precipitada pela exposição ao frio, ocorrendo em questão de minutos após a imersão das mãos ou do corpo em água fria ou após a exposição ao ar frio. Nos casos graves, os pacientes podem apresentar sintomas sistêmicos, incluindo sibilos e síncope. Uma rara forma familiar de urticária fria, autossômica dominante, foi associada ao cromossomo 1q44.46
A urticária fria pode ser diferenciada de outras formas de urticária se uma reação urticariforme for desencadeada com a aplicação de um cubo de gelo sobre a pele por 5-10 minutos. Outras urticárias físicas também têm causas ou características distintivas. A urticária solar ocorre com alguns minutos de exposição à luz solar e pode ser produzida com a exposição de áreas limitadas de pele à luz solar ou a comprimentos de onda apropriados de luz ultravioleta em uma resposta de fototerapia à pressão física. A urticária colinérgica, que é caracterizada por pequenas pápulas urticariformes, pode ser induzida pela exposição ao calor ou pelo exercício.
Histologicamente, todas as urticárias são caracterizadas por aumento de mastócitos na derme. Edema, alterações vasculares e infiltrados mononucleares são mais acentuados na derme de pacientes com urticária fria. Os infiltrados mononucleares também são mais proeminentes na derme profunda de pacientes com urticária de pressão tardia.
Assim como na urticária induzida por fármaco, a terapia de primeira linha para a maioria das urticárias consiste na administração de anti-histamínicos orais e descontinuar/evitar fatores precipitantes. Psoraleno e luz ultravioleta de comprimento de onda longo (PUVA) é o tratamento que tem sido usado com sucesso em casos de pacientes com urticária solar. O montelucaste tem sido usado com sucesso no tratamento da urticária de pressão tardia,47 e a ciclosporina é promissora para casos de urticária crônica refratária grave.
As reações do tipo doença do soro são definidas por febre, erupção (geralmente urticariforme) e artralgias que ocorrem em 1-3 semanas após a iniciação do fármaco. Outros sintomas, como linfadenopatia e eosinofilia, também podem estar presentes. Contrastando com a doença do soro, as reações do tipo doença do soro não são acompanhadas de imunocomplexos, hipocomplementemia, vasculite nem lesões renais [ver Tabela 2].
Estudos epidemiológicos realizados com crianças sugerem que o risco de desenvolvimento de reações do tipo doença do soro é maior com o uso de cefaclor, em comparação a outros antibióticos, incluindo outras cefalosporinas.48 A incidência geral das reações do tipo doença do soro associada ao cefaclor tem sido estimada em 0,024-0,2% por curso de cefaclor.
Embora a patogênese seja desconhecida, tem sido postulado que em hospedeiros geneticamente suscetíveis o metabolismo do cefaclor produz um metabólito reativo que pode se ligar a proteínas teciduais e desencadear uma resposta inflamatória que se manifesta como reação do tipo doença do soro.48
Outros fármacos implicados no desenvolvimento de reações do tipo doença do soro são cefprozil, bupropiona, minociclina, rituximabe49 e in?iximabe.50 A incidência das reações do tipo doença do soro causadas por estes fármacos é desconhecida.
A descontinuação do fármaco causal e o tratamento sintomático com anti-histamínicos e corticosteroides tópicos são recomendados para pacientes com reações do tipo doença do soro. Um breve curso de corticosteroides orais pode ser necessário para pacientes com sintomas mais graves. O fármaco causador da reação do tipo doença do soro deve ser evitado. No caso do cefaclor e do cefprozil, o risco de reatividade cruzada com antibióticos b-lactâmicos é pequeno e a administração de outra cefalosporina em geral é bem tolerada.51 Entretanto, alguns clínicos recomendam para pacientes que desenvolvem reações do tipo doença do soro, a partir do uso de cefaclor, evitarem todos os fármacos b-lactâmicos. Não há informação disponível sobre reatividade cruzada entre antibióticos derivados de tetraciclina em pacientes com reação do tipo doença do soro induzida por minociclina.
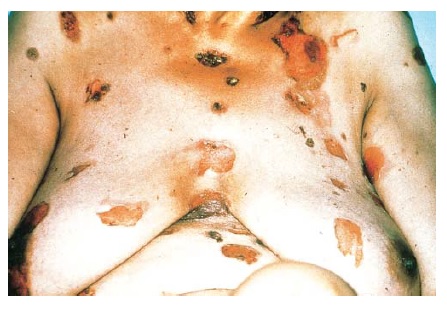
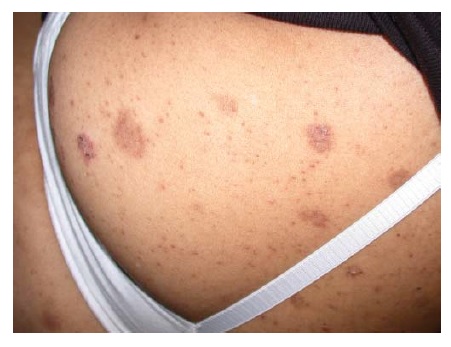
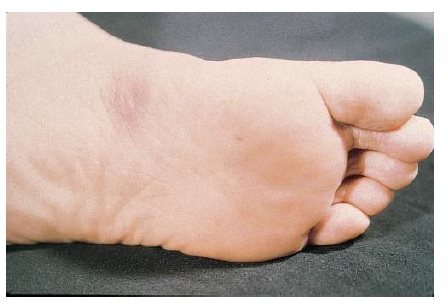
As erupções farmacológicas fixas geralmente aparecem como manchas de cor vermelho brilhante ou vermelho-escuro, eritematosas, pruriginosas e solitárias, que podem evoluir para uma placa edematosa [ver Figura 2]. Em alguns pacientes, pode haver múltiplas lesões. Formação de bolhas e erosão podem ocorrer nas superfícies mucosas. As erupções farmacológicas fixas voltam a ocorrer na mesma área da pele após a readministração da medicação causal.
Figura 2 Este homem de 28 anos usa tetraciclina para tratamento de acne vulgar e desenvolveu erupção farmacológica fixa.
Muitos fármacos têm sido implicados no desenvolvimento de erupções farmacológicas fixas, incluindo fenolftaleína, naproxeno, ibuprofeno, sulfonamidas, tetraciclinas e barbitúricos. A patogênese das erupções farmacológicas fixas está parcialmente elucidada, ainda que as células T CD8+ intraepidérmicas com fenótipo de células efetoras/de memória residentes nas lesões de erupções farmacológicas fixas exerçam papel contribuidor importante.52 Há relatos da existência de um linkage de haplótipo no contexto das erupções farmacológicas fixas induzidas por trimetoprima-sulfametoxazol.53
As erupções farmacológicas fixas são mais comuns nos genitais e na área perianal, embora possam ocorrer em qualquer parte da superfície da pele. O aparecimento de uma erupção farmacológica fixa pode ser abrupto, desenvolvendo-se em 30 minutos a 8-16 horas após a ingesta da medicação. Em pacientes que continuam usando o fármaco agressor, o número de sítios de erupção pode aumentar gradualmente.
Após a fase inicial aguda, que dura dias a semanas, há desenvolvimento de hiperpigmentação residual. Alguns pacientes podem se queixar de ardência ou ferroada nos sítios cutâneos afetados. As manifestações sistêmicas, que estão presentes em cerca de 25% dos casos, podem incluir febre, mal-estar e sintomas abdominais.54
Não há testes diagnósticos conclusivos disponíveis, mas um teste de desafio ou provocação com o fármaco suspeito pode ser útil para confirmar o diagnóstico. O teste de contato no sítio de uma lesão prévia fornece uma resposta positiva em cerca de 40% dos pacientes.55 Foi relatado que os testes cutâneos de Prick e intradérmico fornecem reações positivas em 24 e 67% dos pacientes, respectivamente, mas os resultados variam com diferentes fármacos e padrões de reação.56
O tratamento inclui a descontinuação do agente causal e instituição de terapia sintomática (p. ex., corticosteroides tópicos).
Pseudoporfiria [ver Figura 3] é um distúrbio fototóxico cutâneo que pode ser semelhante à porfiria cutânea tardia (PCT) ou à protoporfiria eritropoiética (PPE). Vários fármacos, incluindo tetraciclina, furosemida, imatinibe e naproxeno, têm sido implicados na PCT- e PPE-pseudoporfiria.57,58 A erupção pode surgir em um dia após a iniciação da terapia ou pode demorar até um ano para se manifestar. A PCT-pseudoporfiria é caracterizada por fragilidade cutânea, formação de bolha e formação de cicatriz em áreas expostas à luz solar; é uma condição que ocorre com o metabolismo normal da porfirina. O segundo padrão clínico mimetiza a PPE e se manifesta como ardência cutânea, eritema, vesiculação, cicatrizes angulares e espessamento ceroso da pele.
Figura 3 Pseudoporfiria subsequente ao uso de naproxeno. Cortesia de Scott Walsh, MD, University of Toronto.
Devido ao risco de formação de cicatriz facial permanente, o fármaco implicado deve ser descontinuado diante da ocorrência de fragilidade cutânea, formação de bolhas ou formação de cicatriz. Em adição, é preciso recomendar ao paciente o uso de protetor solar de amplo espectro e roupas protetoras.
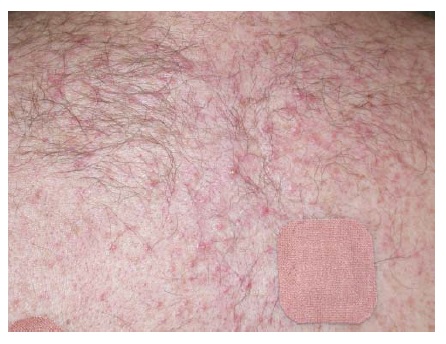
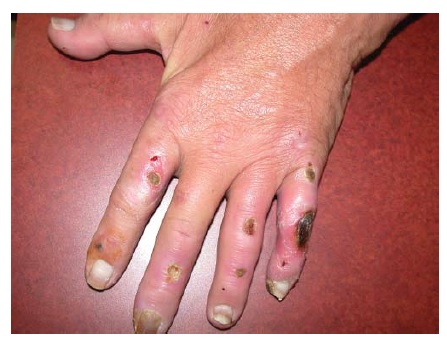
A doença da IgA linear é uma dermatose bolhosa autoimune identificada com base na deposição linear de IgA na zona da membrana basal.59 Esta doença pode ser induzida por fármacos como vancomicina [ver Figura 4], lítio, diclofenaco e amiodarona. A doença fármaco-induzida provavelmente representa uma resposta imunológica ao fármaco agressor.
A doença da IgA linear fármaco-induzida é heterogênea em termos de apresentação clínica. Os casos relatados têm mostrado morfologias semelhantes ao eritema multiforme, penfigoide bolhoso e dermatite herpetiforme. A doença induzida por fármaco não pode ser distinguida da variedade idiopática, seja clínica, histológica ou imunologicamente. Entretanto, os cursos clínicos destas manifestações diferem. Na doença induzida por fármaco, há remissão espontânea tão logo o agente agressor seja suspendido. Na doença da IgA linear idiopática, os imunodepósitos desaparecem da pele quando as lesões se resolvem. Esteroides e dapsona não influenciam o processo de cicatrização, mas são comprovadamente efetivos no tratamento da doença da IgA linear idiopática.60
O pênfigo pode ser induzido ou desencadeado por fármaco (i.e., a doença latente é revelada pela exposição ao fármaco).
Figura 4 Doença da IgA linear induzida por vancomicina. Cortesia de Scott Walsh, MD, University of Toronto.
Os fármacos causadores de pênfigo são penicilina, rifampina, fenilbutazona, propranolol, progesterona, piroxicam, interferon-b, interleucina-2 e levodopa. Um grupo ainda ativo encontrado em fármacos tiol mascarados, como a penicilina e as cefalosporinas, e em fármacos não tiol, como o enalapril, pode contribuir para a patogênese do pênfigo.61 O pênfigo foliáceo [ver Figura 5], causado pela penicilamina e outros fármacos tiol, tende a se resolver de modo espontâneo em 35-50% dos casos. O intervalo médio até o aparecimento da condição é um ano. Anticorpos antinucleares são detectados em 25% dos pacientes afetados.
O pênfigo induzido por fármaco não tiol manifesta aspectos clínicos, histológicos, imunológicos e evolucionários similares aos do pênfigo vulgar idiopático [ver Figura 6]. O pênfigo fármaco-induzido está associado ao envolvimento mucoso. A recuperação espontânea após a retirada do fármaco ocorre em 15% dos pacientes afetados.
O tratamento de pênfigo fármaco-induzido começa com a retirada do fármaco. Os corticosteroides sistêmicos frequentemente são requeridos até que todos os sintomas de doença ativa desapareçam. O seguimento vigilante se faz necessário após a remissão, para detecção de uma recidiva logo no início. O soro do paciente deve ser monitorado regularmente quanto à presença de autoanticorpos.
Figura 5 Pênfigo foliáceo em homem de 64 anos que usa enalapril.
Figura 6 Pênfigo vulgar em mulher de 59 anos que tomava penicilamina para tratamento de artrite reumatoide.
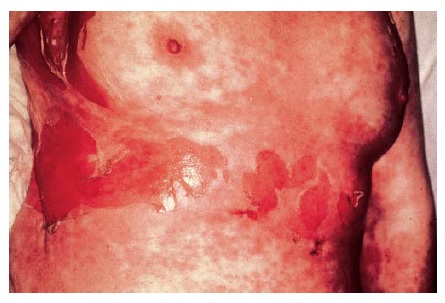
A diferenciação junto ao espectro de SSJ e NET depende da natureza das lesões cutâneas e da extensão do envolvimento da área de superfície corporal. Clinicamente, as reações do espectro de SSJ/NET são caracterizadas pela presença da tríade de erosões da membrana mucosa, lesões-alvo e necrose epidérmica com separação da pele.62 Uma separação inferior a 10% da área de superfície corporal total faz parte da definição de SSJ, enquanto a NET ocorre com o envolvimento de mais de 30% da superfície corporal. Os casos intermediários têm sido chamados sobreposições de SSJ/NET (10-30%). Na SSJ, as lesões-alvo não são tipicamente a lesão de íris trianelar destacada, como se observa no eritema multiforme, sendo esperada uma lesão mais purpúrica ou eritematosa bianelar [ver Figura 7 e Tabela 3]. O envolvimento ocular ocorre em cerca de 75% dos pacientes com SSJ e NET.63
Figura 7 Esta mulher de 50 anos desenvolveu necrólise epidérmica tóxica 17 dias após iniciar a terapia com fenitoína.
|
Tabela 3 Características da síndrome de Stevens-Johnson e da necrólise epidérmica tóxica62 | ||
|
Classificação |
Síndrome de Stevens- Johnson |
Necrólise epidérmica tóxica |
|
Separação epidérmica (% de área de superfície corporal) |
< 10 |
> 30 |
|
Alvos típicos |
Não |
Não |
|
Alvos atípicos |
Plano |
Plano |
|
Manchas |
Sim |
Sim |
|
Envolvimento de membrana mucosa |
Sim |
Sim |
|
Gravidade |
++ |
+++ |
|
Probabilidade de etiologia farmacológica |
++ |
+++ |
Alguns casos de SSJ não estão relacionados a fármacos. Estes distúrbios podem se desenvolver em consequência de vários fatores predisponentes, incluindo infecções, neoplasia e doenças autoimunes. Em geral, quanto mais grave for a reação, maior é a probabilidade de que tenha sido induzida por fármaco. Os fármacos mais frequentemente citados como causa de SSJ e NET são os anticonvulsivantes, antibióticos (p. ex., sulfonamidas), alopurinol e AINEs (p. ex., piroxicam).64 Com o uso dos anticonvulsivantes, o risco parece ser maior durante as primeiras oito semanas de terapia.65 Há relatos de casos graves de RAMs cutâneas à lamotrigina (p. ex., SSJ e NET). Tem sido relatada uma prevalência de RAMs cutâneas graves associadas à lamotrigina de até 1 em cada 1.000 adultos e maior entre crianças. O risco aumenta em presença de ácido valproico. Entre as crianças, as medicações mais frequentemente associadas com SSJ/NET são as sulfonamidas anti-infecciosas, fenobarbital, carbamazepina e lamotrigina.66
A patogênese das RAMs cutâneas graves é desconhecida, embora tenha sido proposta uma base metabólica.67 O envolvimento de células T citotóxicas (CD8) tem sido sugerido.68 A citotoxicidade fármaco-específica mediada por granzima e perforina, restrita ao antígeno de histocompatibilidade principal (MHC)-classe I, pode exercer papel primário no desenvolvimento da NET.69 O envolvimento de Fas-ligante (FasL) solúvel na SSJ/NET também tem sido proposto. Os queratinócitos humanos expressam Fas, um dos conhecidos death receptors (receptores de morte celular), e a interação Fas-FasL induz apoptose em queratinócitos.70 Alternativamente, a fonte de FasL são as células mononucleares do sangue periférico, em vez dos queratinócitos.71 A granulisina é uma proteína citolítica catiônica capaz de matar vários micróbios e células tumorais, além de atuar como fator quimiotático com propriedades pró-inflamatórias, que é super expressa no sangue e no líquido das bolhas da NET. A granulisina pode induzir morte celular por apoptose nos queratinócitos da epiderme.72,73
Vários estudos recentes têm demonstrado que, em indivíduos de etnia chinesa/asiática, o alelo HLA-B*1502 está fortemente associado à SSJ e NET induzida por carbamazepina, mas não à SHF ou às erupções exantematosas induzidas por este fármaco.74,75 Nenhuma correlação entre HLA-B*1502 e SJS/NET induzida por carbamazepina foi encontrada em brancos, até o momento.76 Entretanto, o alelo HLA-A*3101 foi associado a reações de hipersensibilidade induzidas por carbamazepina em indivíduos com descendência norte-europeia.77 Adicionalmente, o alelo HLA-A*3101 apresenta sensibilidade de 60,7% e especificidade de 87,5% ao ser usado como preditor de risco de RAMs cutâneas induzidas por carbamazepina (incluindo SSJ/NET) na população japonesa.77 O HLA-B*5801 pode ser necessário, todavia insuficiente, para a ocorrência de RAM cutânea grave induzida por alopurinol.24 Em um estudo europeu sobre SSJ e NET, o HLA-B*5801 estava presente em 61% dos pacientes com SSJ/NET induzida por alopurinol, incluindo 55% dos pacientes de descendência europeia.76 A triagem para o alelo HLA-B*1502 antes de iniciar a terapia com carbamazepina é recomendada para pacientes de etnia sudeste-asiática.78
O tratamento da SSJ e NET inclui a descontinuação de um fármaco suspeito e a adoção de medidas de suporte como cuidados com a ferida, hidratação e suporte nutricional. Hemogramas completos, quantificação de enzimas hepáticas e radiografias torácicas devem ser realizados para excluir a possibilidade de envolvimento concomitante de órgão interno. O uso de corticosteroides na SSJ e na NET é controverso.79 Alguns clínicos sugerem que os corticosteroides podem ser benéficos quando administrados no início do curso da doença e a uma dosagem relativamente alta. No entanto, outros autores sugerem que os corticosteroides não encurtam o tempo de recuperação e podem aumentar o risco de complicações, inclusive de infecções secundárias e sangramento gastrintestinal.80 A administração de IVIg (0,4-1,0 g/kg/dia durante 2-4 dias), que contém anticorpos bloqueadores de FasL de ocorrência natural, pode conter a progressão da NET, em especial quando a IVIg é iniciada antecipadamente.81 Entretanto, outros estudos falharam em alcançar um resultado melhor em pacientes com NET tratados com IVIg.82 Um amplo estudo retrospectivo sobre o tratamento de SSJ e de NET constatou que nem os corticosteroides nem as IVIgs exerceram qualquer efeito significativo sobre a mortalidade em comparação ao tratamento de suporte isolado.83 Houve um relato de um paciente com NET induzida por etoricoxibe tratado com infliximabe, no qual a progressão da epidermiólise foi detida após algumas horas da administração de única dose.84 Outros fármacos usados no tratamento de pacientes com SSJ/NET incluem a ciclosporina85 e a ciclofosfamida.86 Pacientes que desenvolveram RAM cutânea grave (i.e., eritema multiforme, SSJ ou NET) não devem ser submetidos a um novo teste com o fármaco nem à dessensibilização com a medicação.
Erupções que mimetizam morfologicamente a acne vulgar podem estar associadas com ingesta de fármaco. Iodetos, brometos, hormônio adrenocorticotrópico, corticosteroides, isoniazida, andrógenos, lítio, dactinomicina e fenitoína foram relatados como indutores de lesões do tipo acne.87
A acne induzida por fármaco muitas vezes aparece na face e no dorso [ver Figura 8], mas pode surgir em áreas atípicas, como braços e pernas, sendo geralmente monomórfica. Em geral, os comedões estão ausentes. Não há febre. As erupções acneiformes não afetam crianças em fase pré-puberdade, indicando que o condicionamento hormonal prévio é pré-requisito. A aplicação tópica de tretinoína pode ser útil ante a impossibilidade de suspender o curso do fármaco.
Uma erupção acneiforme ocorre com frequência durante o tratamento com inibidores de receptor de fator de crescimento epidérmico (p. ex., gefitinibe, erlotinibe, cetuximabe). A erupção acneiforme frequentemente é acompanhada de paroníquia, pele seca e fissuras cutâneas. A erupção é dose-dependente, em termos de incidência e gravidade.88 Em uma metanálise e revisão sistêmica envolvendo mais de 1.000 pacientes sob tratamento com cetuximabe como agente único, a incidência de erupção acneiforme foi de 81,6%.89 O tratamento destas reações geralmente envolve medicações tópicas convencionais, como metronidazol, eritromicina e clindamicina.90
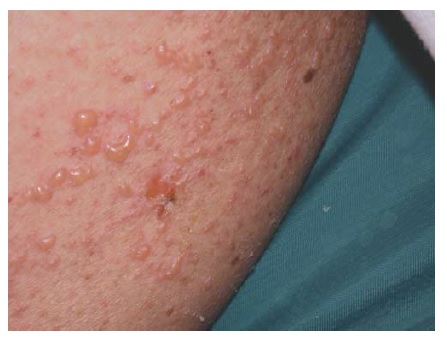
A pustulose exantematosa generalizada aguda é caracterizada por início agudo, febre e erupção cutânea com pústulas estéreis não foliculares em um eritema edematoso, surgindo geralmente com alguns dias de ingesta do fármaco91 [ver Figura 9]. A leucocitose é outro achado comum. Descamação generalizada ocorre após duas semanas. O diagnóstico diferencial inclui psoríase pustular, dermatose pustular subcorneal (doença de Sneddon-Wilkinson ), reação de síndrome de hipersensibilidade com postulação, e erupções pustulares da infância.92 Tem havido vários relatos de pustulose exantematosa generalizada aguda simulando NET.93
A pustulose exantematosa generalizada aguda está mais comumente associada ao uso de antibióticos b-lactâmicos e macrolídeos. Numerosos fármacos diferentes têm sido implicados, todavia, incluindo bloqueadores de canais de cálcio e analgésicos. A incidência estimada é de aproximadamente 1-5 casos por milhão de pacientes ao ano.94 A descontinuação da terapia geralmente é a extensão do tratamento necessário na maioria dos casos, embora alguns pacientes possam requerer o uso de corticosteroides. O teste de contato com o fármaco putativo frequentemente resulta positivo, com uma reação pustular localizada. Resultados positivos de testes de contato e de testes de transformação linfocitária sugerem envolvimento de células T na pustulose exantematosa generalizada aguda.95 CélulasT CD4+ e CD8+ fármaco-específicas têm sido isoladas a partir de sítios de teste de contato e do sangue de pacientes com história do distúrbio.96
Figura 8 Melanoníquia estriada (faixa pigmentada longitudinal) produzida por um nevo melanocítico da matriz ungueal. É muito importante manter um alto índice de suspeita de melanoma subungueal ao identificar uma faixa pigmentada longitudinal na unha.
Figura 9 Pustulose exantematosa generalizada aguda (pequenas pústulas não foliculares sobre uma base avermelhada) em um homem de 70 anos que tomava cloxacilina para tratamento de celulite.
A descontinuação da terapia geralmente é a extensão do tratamento necessário, na maioria dos pacientes. Entretanto, o uso de corticosteroides sistêmicos pode ser indicado para alguns pacientes, dependendo da extensão da reação. A repetição do desafio em geral não é recomendada nas primeiras horas subsequentes à administração do fármaco.
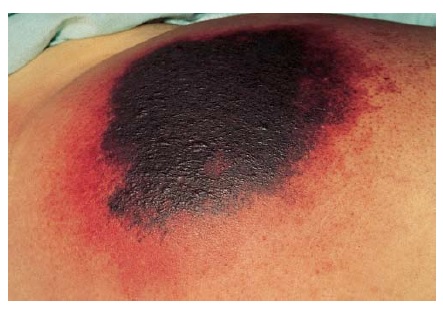
Os fármacos anticoagulantes podem induzir estados hipercoaguláveis com subsequente infarto vascular e necrose cutânea [ver Figura 10]. Cumarínicos e heparina (não fracionada e de baixo peso molecular) podem induzir necrose cutânea. A maioria dos casos tem sido atribuída aos congêneres de cumarínicos (bis-hidroxicoumarin, fenprocoumon, acenocoumarol e varfarina). A necrose cutânea induzida por heparina pode ocorrer associada com síndrome de trombose e trombocitopenia induzida por heparina (Síndrome HITT).97 As dicas clínicas preciosas que podem ajudar a diferenciar estas reações envolvem a localização, momento da ocorrência, contagem de plaquetas e diagnóstico primário [ver Tabela 4].
Figura 10 Necrose cutânea induzida por coumarin em uma mulher de 57 anos que recebeu coumarin como tratamento para fibrilação atrial.
A patogênese da necrose cutânea induzida por cumarínicos é o desenvolvimento paradoxal de trombos oclusivos em vênulas e cutâneas e subcutâneas, causado por um estado hipercoagulável transiente. Esta condição resulta da supressão da proteína C, anticoagulante natural, a uma taxa maior do que os fatores pró-coagulantes naturais. A necrose cutânea induzida por coumarin está associada à deficiência de proteína C e de proteína S, mas não há justificativa para realização de triagem pré-tratamento. Há relatos da existência de uma associação com heterozigosidade para a mutação do fator V de Leiden.98
Estima-se que uma em cada 10.000 pessoas que usam coumarin apresenta risco de desenvolver este evento adverso.97 A prevalência é quatro vezes maior em mulheres do que nos homens. Em ambos os sexos, o pico de incidência ocorre na sexta e sétima décadas da vida. Os pacientes afetados tendem a ser obesos.
A necrose cutânea induzida por cumarínicos surge em 3-5 dias após a iniciação do tratamento. Placas avermelhadas dolorosas se desenvolvem em sítios ricos em tecido adiposo, como mamas, nádegas e quadril. Estas placas podem formar bolhas, ulcerar ou se desenvolver em áreas necróticas. Uma infecção acompanhando, como pneumonia, infecção viral ou erisipela, pode ocorrer em até 25% dos pacientes. A síndrome do dedo do pé roxo pode ocorrer em 3-8 semanas após a iniciação da terapia com cumarínicos.
O tratamento ocasiona a descontinuação do cumarínico, administração de vitamina K e infusão de heparina em doses terapêuticas. Plasma fresco congelado e concentrados de proteína C purificada têm sido usados.99 Recomenda-se a adoção de medidas de suporte para a pele. A cirurgia plástica para conserto se faz necessária para 60% dos pacientes afetados.
As lesões de líquen plano induzidas por fármaco [ver Figura 11] são clínica e histologicamente indistinguíveis das lesões do líquen plano idiopático. Muitos fármacos, incluindo b-bloqueadores, penicilamina, AINEs, ouro e inibidores de ECA, em especial o captopril, têm sido relatados como produtores desta reação. Mais recentemente, erupções líquen plano-símile têm sido relatadas com o uso de antagonistas de fator de necrose tumoral (TNF)-a, como in?iximabe e adalimumabe.100
O período de latência entre o início da administração do fármaco e o aparecimento da erupção é variável. O período de latência médio varia entre dois meses e três anos para penicilamina, cerca de um ano para b-bloqueadores, e 3-6 meses para inibidores de ECA. O período de latência pode ser menor, se o paciente tiver sido previamente exposto ao fármaco.101 Em geral, a resolução costuma ocorrer em 2-4 meses.
Tentativas de repetir o teste com o fármaco agressor tem sido feitas em alguns pacientes, com reativação dos sintomas em 4-15 dias. O teste de contato tem se mostrado inútil na maioria dos casos de líquen plano induzido por fármaco. Entretanto, os resultados dos testes de contato realizados com indutores de erupções farmacológicas de líquen por contato (p. ex., desenvolvedores de cor-filme e materiais restauradores dentais) geralmente são positivos.
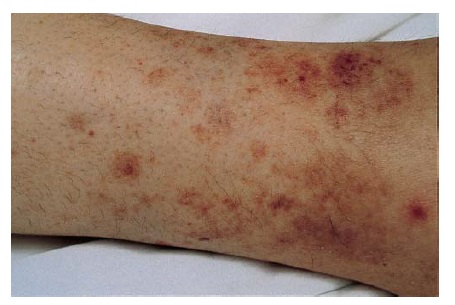
A vasculite induzida por fármaco representa cerca de 10% das vasculites cutâneas agudas e, em geral, afeta vasos pequenos [ver Figura 12]. A vasculite induzida por fármaco deve ser considerada em qualquer caso de paciente com vasculite de pequenos vasos confinada à pele. Os fármacos mais frequentemente associados à vasculite incluem propiltiouracil, hidralazina, fator estimulador de colônia de granulócitos (G-CSF), fator estimulador de colônia de granulócitos-macrófagos (GM-CSF), alopurinol, cefaclor, minociclina, penicilamina, fenitoína e isotretinoína.102 A vasculite leucocitoclástica também tem sido associado ao uso de fármacos anti-TNF , em particular etanercept e in?iximabe.103 O intervalo médio até o aparecimento da vasculite induzida por fármaco é 7-21 dias.104
Figura 11 Erupção farmacológica liquenoide associada ao ramipril. Cortesia de Scott Walsh, MD, University of Toronto.
|
Tabela 4 Dicas clínicas preciosas para identificação de necrose cutânea induzida por anticoagulante | |||
|
|
Intervalo até o aparecimento |
Localização |
Outro |
|
Necrose cutânea induzida por coumarin |
3–5 dias |
Sítios ricos em tecido adiposo |
— |
|
Trombose e trombocitopenia induzida por heparina |
4–14 dias |
Membros |
A trombocitopenia é concomitante |
|
Síndrome do dedo do pé roxo |
3–8 semanas |
Localização acral |
Ocorre com frequência após a angiografia |
A principal característica clínica da vasculite cutânea é a purpura apalpável, encontrada classicamente nos membros inferiores, embora qualquer outro sítio cutâneo possa ser afetado. A urticária pode ser uma manifestação de vasculite de pequenos vasos. Diferente da urticária alérgica não vasculítica, a urticária vasculítica dura mais que um dia, pode evoluir para lesões purpúricas e pode ser acompanhada de hipocomplementemia. Outros achados são bolhas hemorrágicas, urticária, úlceras, nódulos, doença de Raynaud e necrose digital. O mesmo processo vasculítico também pode afetar órgãos internos, como fígado, rim, intestino e sistema nervoso central, além de ser potencialmente fatal.105
Figura 12 Vasculite leucocitoclástica nesta mulher de 47 anos sob tratamento com hidroclorotiazida.
Histologicamente, os pequenos vasos sanguíneos da derme exibem necrose fibrinoide, infiltração polimorfonuclear no interior da parede do vaso sanguíneo, extravasamento de hemácias e poeira nuclear. A imunofluorescência direta pode mostrar depósitos de IgM e C3 nas paredes dos vasos sanguíneos. Sendo assim, estas reações são farmacológicas dependentes de imunocomplexo. Os imunocomplexos podem ser constituídos por anticorpos dirigidos contra haptenos fármaco-relacionados, mas isto ainda precisa ser comprovado.
A vasculite induzida por fármaco pode ser difícil diagnosticar e seu diagnóstico muitas vezes é feito por exclusão. É preciso eliminar as causas alternativas de vasculite cutânea, como infecção ou doença autoimune. A eosinofilia tecidual pode ser um indicador de indução farmacológica na vasculite de pequenos vasos cutâneos.106
O tratamento consiste na retirada do fármaco. A terapia para pacientes com manifestações graves inclui hemodiálise, pulsos de corticosteroide, ciclofosfamida e plasmaférese.
Neil H. Shear, MD, FRCP, tem recebido apoio financeiro para pesquisa clínica ou com fins educacionais, e atuou como consultor para as empresas Roche, Galderma SA, Genesoft Co. Ltd., GlaxoSmithKline, Novartis AG e Fujisawa Healthcare Inc. Sandra Knowles, BScPhm, e Lori Shapiro, MD, FRCPC, não tem relações comerciais com fabricantes de produtos nem prestadores de serviços mencionados neste capítulo.
1.Thong B, Tan T. Epidemiology and risk factors for drug allergy. Br J Clin Pharmacol 2011;71:684–700.
2.Kongkaew C, Noyce P, Ashcroft D. Hospital admissions associated with adverse drug reactions: a systematic review of prospective observational studies. Ann Pharma- cother2008; 42:1017–25.
3.Bigby M. Rates of cutaneous reactions to drugs. Arch Dermatol2001;137:765–70.
4.Leape L, Brennan T, Laird N, et al. The nature of adverse events in hospitalized patients. Results of the Harvard Medical Practice Study II. N Engl J Med 1991;324:377–84.
5.Classen D, Pestotnik S, Evans R, et al. Adverse drug events in hospitalized patients: excess length of stay, extra costs and attributable mortality. JAMA 1997; 277:301–6.
6.Lazarou J, Pomeranz B, Corey P. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–5.
7.Barbaud A, Goncalo M, Bruynzeel D, Bricher A. Guidelines for performing skin tests with drugs in the investigation of cutaneous adverse drug reactions. Contact Dermatitis 2001; 45:321–8.
8.Romano A, Quaratino D, DiFonso M, et al. A diagnostic protocol for evaluating nonimmediate reactions to aminopenicillins. J Allergy Clin Immunol 1999;103:1186–90.
9.Romano A, Quaratino D, Papa G, et al. Aminopenicillin allergy. Arch Dis Child 1997; 76:513–7.
10.Coopman S, Johnson R, Platt R, Stern R. Cutaneous disease and drug reactions in HIV infection. N Engl J Med 1993; 328:1670–4.
11.Hausmann O, Schnyder B, Pichler W. Drug hypersensiti- vity reactions involving skin. Handb Exp Pharmacol 2010; 196:29–55.
12.Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med 2011;124:588– 97.
13.Bocquet H, Bagot M, Roujeau J. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg 1996;15:250–7.
14.Shear N, Spielberg S. In vitro evaluation of a toxic metabolite of sulfadiazine. Can J Physiol Pharmacol 1985;63: 1370–2.
15.Tohyama M, Hashimoto K, Yasukawa M, et al. Association of human herpesvirus 6 reactivation with the ?aring and severity ofdrug-induced hypersensitivity syndrome. Br J Dermatol2007; 157:934–40.
16.Shiohara T, Ijima M, Ikezawa Z, Hashimoto K. The diagnosis of a DRESS syndrome has been suf?ciently established on the basis of typical clinical features and viral reactivation. Br J Dermatol2007; 156:1083–4.
17.Kano Y, Inaoka M, Shiohara T. Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia. Arch Dermatol2004; 140:183–8.
18.Wong G, Shear N. Is a drug alone suf?cient to cause the drug hypersensitivity syndrome? Arch Dermatol 2004; 140: 226–30.
19.Shear N, Spielberg S, Grant D, et al. Differences in metabolism of sulfonamides predisposing to idiosyncratic toxicity. Ann Intern Med 1986; 105:179–84.
20.Johnson K, Green D, Rife J, Limon L. Sulfonamide cross-reactivity: fact or faction? Ann Pharmacother 2005; 39: 290–301.
21.Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol 2010; 146:1373–9.
22.Khoo B, Leow Y. A review of inpatients with adverse drug reactions to allopurinol. Singapore Med J 2000; 41:156–60.
23.Masaki T, Fukunaga A, Tohyama M, et al. Human herpes virus 6 encephalitis in allopurinol-induced hypersensiti- vity syndrome. Acta Derm Venereol 2003;83:128–31.
24.Hung S, Chung W, Liou L, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 2005; 102: 4134–9.
25.Martin A, Nolan D, James I, et al. Predisposition to nevirapine hypersensitivity associated with HLA-DRB1*0101 and abrogated by low CD4 T-cell counts. AIDS 2005; 19: 97–9.
26.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358: 568–79.
27.Bourgeois G, Cafardi J, Groysman V, Hughey L. A review ofDRESS-associated myocarditis. J Am Acad Dermatol 2011 Jun 7. [Epub ahead of print]
28.Gupta A, Eggo M, Uetrecht J, et al. Drug-induced hypo- thyroidism: the thyroid as a target organ in hypersensitiv- ity reactions to anticonvulsants and sulfonamides. Clin Pharmacol Ther 1992; 51:56–67.
29.Elzagallaai A, Knowles S, Rieder M, et al. In vitro testing for the diagnosis of anticonvulsant hypersensitivity syndrome. Mol Diagn Ther 2009; 13:313–30.
30.Elzagallaai A, Knowles S, Rieder M, et al. Patch testing for the diagnosis of anticonvulsant hypersensitivity syndrome: a systematic review. Drug Saf 2009; 32:391–408.
31.Kano Y, Ishida T, Hirahara K, Shiohara T. Visceral involvements and long-term sequelae in drug-induced hypersensitivity syndrome. Med Clin North Am 2010; 94:743–59.
32.Brown R, Rother K, Artman H, et al. Minocycline-induced drug hypersensitivity syndrome followed by multiple autoimmune sequelae. Arch Dermatol 2009; 145:63–6.
33.Aota N, Hirahara K, Kano Y, et al. Systemic lupus erythe- matosus presenting with Kikuchi-Fujimoto’s disease as along-term sequela of drug-induced hypersensitivity syndrome. Dermatology 2009; 218:275–7.
34.Harman K, Morris S, Higgins E. Persistent anticonvulsant hypersensitivity syndrome responding to ciclosporin. Clin Exp Dermatol 2003; 28:364–5.
35.Kano Y, Inaoka M, Sakuma K, Shiohara T. Virus reactivation and intravenous immunoglobulin (IVIG) therapy ofdrug-induced hypersensitivity syndromes. Toxicology2005; 209:165–7.
36.Shaughnessy K, Bouchard S, Morh M, et al. Minocycline- induced drug reaction with esoinophilia and systemic symptoms (DRESS) syndrome with persistent myocardi- tis. J Am Acad Dermatol2010; 62:315–8.
37.Tan E, Grattan C. Drug-induced urticaria. Expert Opin Drug Saf2004;3:471–84.
38.Kowalski M, Makowska J, Blanca M, et al. Hypersensitivity to nonsteroidal anti-in?ammatory drug: classi?cation, diagnosis and amangement. Allergy 2011;66:818–29.
39.Ardern-Jones M, Friedmann P. Skin manifestations of drug allergy. Br J Clin Pharmacol 2011;71:672–83.
40.Dykewicz M. Cough and angioedema from angiotensin- converting enzyme inhibitors: new insights into mechanisms and management. Curr Opin Allergy Clin Immunol 2004;4:267–70.
41.Scheirey C, Scholz F, Shortsleeve M, Katz D. Angiotensin- converting enzyme inhibitor-induced small bowel angio- edema: clinical and imaging ?ndings in 20 patients. AJR Am J Roentgenol 2011; 197:393–8.
42.Taylor J, Erkek E. Latex allergy: diagnosis and management. Dermatol Ther 2004; 17:289.
43.Hoover T, Lippmann M, Grouzmann E, et al. Angiotensin converting enzyme inhibitor induced angioedema: a review of the pathophysiology and risk factors. Clin Exp Allergy2010; 40:50–61.
44.Mirakian R, Ewan P, Durham S, et al. BSACI guidelines for the management of drug allergy. Clin Exp Allergy 2009; 39: 43–61.
45.Beavers C, Dunn S, Macaulay T. The role of angiotensin receptor blockers in patients with angiotensin-converting enzymeinhibitor-induced angioedema. Ann Pharmaco- ther2011; 45:520–4.
46.Hoffman H, Wright F, Broide D, et al. Identi?cation of a locus on chromosome 1q44 for familial cold urticaria. Am J Hum Genet 2000; 66:1693.
47.Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria: a single-blind, placebo-controlled, cross-over clinical study. J Allergy Clin Immunol 2002; 110:484.
48.Kearns G, Wheeler J, Childress S, Letzig L. Serum sickness- like reactions to cefaclor: role of hepatic metabolism and individual susceptibility. J Pediatr 1994;125:805–11.
49.Hellerstedt B, Ahmed A. Delayed-type hypersensitivity reaction or serum sickness after rituximab treatment. Ann Oncol 2003; 14:1792.
50.Gamarra RM, McGraw SD, Drelichman VS, Maas LC. Serumsickness-like reactions in patients receiving intrave- nous in?iximab. J Emerg Med 2006; 30:41–4.
51.Vial T, Pont J, Pham E, et al. Cefaclor-associated serumsickness-like disease: eight cases and review of the literature. Ann Pharmacother 1992; 26:910–4.
52.Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol 2009; 9: 316–21.
53.Ozkaya-Bayazit E, Akar U. Fixed drug eruption induced bytrimethoprim-sulfamethoxazole: evidence for a link to HLA-A30B13 Cw6 haplotype. J Am Acad Dermatol 2001; 45:712–7.
54.Ozkaya E. Fixed drug eruption: state of the art. J Drugs Dermatol2008;6:181–8.
55.Andrade P, Brinca A, Goncalo M. Patch testing in ?xed drug eruptions: a 20-year review. Contact Dermatitis 2011; 65:195–201.
56.Barbaud A, Reichert-Penetrat S, Trechot P, et al. The use of skin testing in the investigation of cutaneous adverse drug reactions. Br J Dermatol 1998; 49:139.
57.Al-Kehnaizan S, Schechter J, Sasseville D. Pseudoporphy- ria induced by propionic acid derivatives. J Cutan Med Surg1999; 3:162–6.
58.Timmer-de Mik L, Kardaun S, Kramer M, et al. Imatinibinduced pseudoporphyria. Clin Exp Dermatol 2009; 34: 705–7.
59.Kuechle M, Stegemeir E, Maynard B, et al. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol 1994; 30:187– 92.
60.Neughebauer B, Negron G, Pelton S, et al. Bullous skin disease: an unusual allergic reaction to vancomycin. Am J Med Sci 2002; 323:273.
61.Brenner S, Bialy-Gohan A, Ruocco V. Drug-induced pemphigus. Clin Dermatol 1998; 16:393.
62.Bastuji-Garin S, Rzany B, Stern R, et al. Clinical classi?cation of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol 1993; 129: 92–6.
63.Gueudry J, Roujeau J, Binaghi M, et al. Risk factors for the development of coular complications of Stevens-Johnsonsyndrome and toxic epidermal necrolysis. Arch Dermatol2009; 145:157–62.
64.Roujeau J, Kelly J, Naldi L, et al. Medication use and the risk ofStevens-Johnson syndrome or toxic epidermal necrolysis. N Engl J Med 1995; 333:1600–7.
65.Rzany B, Mockenhaupt M, Stocker U, et al. Incidence ofStevens-Johnson syndrome and toxic epidermal necrolysis in patients with the acquired immunode?ciency syndrome in Germany. Arch Dermatol 1993; 129:1059.
66.Levi N, Bastuji-Garin S, Mockenhaupt M, et al. Medications as risk factors of Stevens-Johnson syndrome and toxic epidermal necrolysis in children: a pooled analysis. Pediatrics2009; 123:e297–304.
67.Chave T, Mortimer N, Sladden M, et al. Toxic epidermal necrolysis: current evidence, practical management and future directions. Br J Dermatol 2005; 153:241–53.
68.Mockenhaupt M. Severe drug-induced skin reactions: clinical pattern, diagnostics and therapy. J Dtsch Dermatol Ges 2009; 7 :142–60.
69.Nassif A, Bensussan A, Boumsell L, et al. Toxic epidermal necrolysis: effector cells are drug-speci?c cytotoxic T cells. J Allergy Clin Immunol 2004; 114:1209–15.
70.Viard I, Wehrli P, Bullani R, et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998;282:490–3.
71.Abe R. Toxic epidermal necrolysis and Stevens-Johnsonsyndrome: soluble Fas ligand involvement in the pathomechanisms of these diseases. J Dermatol Sci 2008; 52: 151–9.
72.Nickoloff B. Saving the skin from drug-induced detach- ment. Nat Med 2008;14:1311–3.
73.Chung W, Hung S, Yang J, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens- Johnson syndrome and toxic epidermal necrolysis. Nat Med 2008;14:1343–50.
74.Man C, Kwan P, Baum L, et al. Association between HLA- B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese. Epilepsia 2007;48:1015–8.
75.Chen P, Lin J, Lu C, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med2011; 364:1126–33.
76.Lonjou C, Borot N, Sekula P, et al. A European study of HLA-Bin Stevens-Johnson syndrome and toxic epidermal necrolysis related to ?ve high risk drugs. Pharmacogenet Genomics2008; 18:99–107.
77.McCormack M, Al?revic A, Bourgeois S, et al. HLA-A*3101 andcarbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011; 364:1134–43.
78.Fernando S, Broadfoot A. Prevention of severe cutaneous adverse drug reactions: the emerging value of pharmaco-genetic screening. CMAJ 2010; 182:476–80.
79. Patterson R, Miller M, Kaplan M, et al. Effectiveness of early therapy with corticosteroids in Stevens-Johnson syndrome: experience with 41 cases and a hypothesis regarding pathogenesis. Ann Allergy 1994; 73:27–34.
80.Wolf R, Orion E, Marcos B, Matz H. Life-threatening acute verse cutaneous drug reactions. Clin Dermatol 2005; 23: 171–81.
81.French L, Trent J, Kerdel F. Use of intravenous immunoglobulin in toxic epidermal necrolysis and Stevens- Johnson syndrome: our current understanding. Int Immunopharmacol 2006; 6:543–9.
82.Shortt R, Gomez M, Mittmann N, Cartotto R. Intravenous immunoglobulin does not improve outcome in toxic epidermal necrolysis. J Burn Care Rehabil 2004; 25:246–55.
83.Schneck J, Fagot J, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR study. J Am Acad Dermatol2008; 58:33–40.
84.Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with in?iximab. J Dermatol 2010; 37:904–6.
85.Valeyrie-Allanore L, Wolkenstein P, Brochard L, et al. Open trial of ciclosporin treatment for Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 2010; 163:847–53.
86.Trautmann A, Klein C, Kampgen E, et al. Severe bullous drug reactions treated successfully with cyclophospha- mide. Br J Dermatol 1998; 139:1127–8.
87.Momin S, Peterson A, del Rosso J. A status report on drug- associated acne and acneiform eruptions. J Drug Dermatol2010; 9:627–36.
88.Cowen E. Epidermal growth factor receptor inhibitors: a new era of drug reactions in a new era of cancer therapy. J Am Acad Dermatol 2007; 56:514–7.
89.Su X, Lacouture M, Jia Y, Wu S. Risk of high-grade skin rash in cancer patients treated with cetuximab: an anti- body against epidermal growth factor receptor: systemic review andmeta-analysis. Oncology 2009; 77:124–33.
90.Ehmann L, Ruzicka T, Wollenberg A. Cutaneous side- effects of EGFR inhibitors and their management. Skin Ther Lett2011; 16:1–3.
91.Beylot C, Doutre M, Beylot-Barry M. Acute generalized exanthematous pustulosis. Semin Cutan Med Surg 1996; 15:244–9.
92.Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP): results of a multinational case control study (EuroSCAR). Br J Dermatol2007; 157:989–96.
93.Peermohamed S, Haber R. Acute generalized exathema- tous pustulosis simulating toxic epidermal necrolysis. Arch Dermatol2011; 147:697–701.
94.Sidoroff A, Haleevy S, Bainck J, et al. Acute generalized exanthematous pustulosis (AGEP): a clinical reaction pattern. J Cutan Pathol 2001; 28:113–9.
95.Halevy S. Acute generalized exanthematous pustulosis. Curr Opin Allergy Clin Immunol 2009; 9:322–8.
96.Britschgi M, Pichler W. Acute generalized exanthematous pustulosis, a clue to neturophil-mediated in?ammatory processes orchestrated by T cells. Curr Opin Allergy Clin Immunol2002; 2:325–31.
97.Nazarian R, van Cott E, Zembowicz A, Duncan L.Warfarin-induced skin necrosis. J Am Acad Dermatol2009; 61:325–32.
98.Freeman BD, Schmieg RE, McGrath S, et al. Factor V Leiden mutation in a patient with warfarin-associated skin necrosis. Surgery 2000; 127:595–6.
99.Schramm W, Spannagel M, Bauer K, et al. Treatment ofcoumarin-induced skin necrosis with a monoclonal antibody puri?ed protein C concentrate. Arch Dermatol 1993; 19:753–6.
100.Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor- alpha antagonists. J Am Acad Dermatol 2009; 61:104–11.
101.Thompson D, Skaehill P. Drug-induced lichen planus. Pharmacotherapy 1994; 14:561–71.
102.Holder S, Joy M, Falk R. Cutaneous and systemic manifes- tations of drug-induced vasculitis. Ann Pharmacother 2002; 36:130.
103.Moustou AE, Matekovits A, Dessinioti C, et al. Cutaneous side effects of anti-tumor necrosis factor biologic therapy: a clinical review. J Am Acad Dermatol 2009; 61:486–504.
104.Merkel P. Drugs associated with vasculitis. Curr Opin Rheumatol 1998; 10:45.
105.Wiik A. Clinical and laboratory characterisitcs of drug- induced vasculitis syndromes. Arthritis Res Ther 2005; 7: 191.
106.Bahrami S, Malone J, Webb K, Callen J. Tissue eosinophilia as an indicator of drug-induced cutaneous small-vessel vasculitis. Arch Dermatol 2006; 142:155–61.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.

 .
.