(Carregando Índice)... (Carregando Índice)... |
Última revisão: 18/06/2015
Comentários de assinantes: 0
Soma Jyonouchi, MD, e Kathleen E. Sullivan, MD, PhD
Artigo original: Jyonouchi S, Sullivan KE. Deficiencies of Innate and Adaptive Immunity. ACP Medicine. 2013.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Santos Zambon
Os defeitos de função de células B, função de células T e imunidade inata constituem a maioria das imunodeficiências primárias. Cada compartimento tem um conjunto característico de achados arquétipos. Os defeitos funcionais de células B são caracterizados pela produção precária de imunoglobulina que, por sua vez, acarreta em infecções sinopulmonares recorrentes. Os defeitos da função da célula T são caracterizados pela depuração retardada dos vírus, suscetibilidade a infecções oportunistas e alta incidência de doenças autoimunes. Os defeitos da imunidade inata tipicamente estão associados a infecções graves de aparecimento rápido. Em geral, a suspeita de imunodeficiência deve ser levantada quando uma infecção for atípica, acentuadamente mais grave do que o normal ou recorrente. Em alguns casos, patógenos específicos devem alertar o médico para uma potencial imunodeficiência [ver Tabela 1].
Estes distúrbios representam o tipo mais comum de imunodeficiências primárias e são de longe a o principal tipo de imunodeficiência primária em adultos. Nesta categoria, a imunodeficiência comum variável (IDCV) e a deficiência de IgA são os distúrbios mais comuns. A ligação clínica comum que une as deficiências de imunoglobulina são as infecções recorrentes de membranas mucosas. Os distúrbios individuais têm características exclusivas, mas estão todos associados a infecções sinopulmonares e à suscetibilidade a certas infecções gastrintestinais [ver Tabela 2]. O tratamento desses distúrbios enfoca, primariamente, a substituição da imunoglobulina defeituosa e a prevenção de dano a órgãos-alvo decorrentes das infecções. Esta seção descreve os defeitos de imunoglobulina mais comuns e clinicamente relevantes.
Este distúrbio, descrito em 1952, foi uma das primeiras imunodeficiências a ter a etiologia definida.
Genética e patogênese. O gene mutante na agamaglobulinemia ligada ao X (ALX) reside no braço longo do cromossomo X. O gene Btk é uma tirosina quinase necessária à sinalização pelo receptor da célula B.1 Na ausência deste sinal essencial do desenvolvimento, as células B falham em avançar do estágio de célula pró-B para o estágio de célula pré-B. A parada do desenvolvimento neste estágio precursor impede efetivamente as células B maduras de alcançarem o sangue periférico. Em adição, as estruturas linfoides secundárias populadas por células B são bastante hipoplásicas.
Diagnóstico. A manifestação clínica raramente ocorre nas primeiras semanas de vida, porque os anticorpos maternos conferem proteção durante os primeiros meses de vida. Meninos com ALX tipicamente começam a ter infecções no primeiro ano de vida, sendo que as infecções iniciais, frequentemente, envolvem o trato respiratório superior. Pneumonia, artrite, abscessos cutâneos e bronquite também são observados na infância. 2,3 Um achado clínico fundamental é a ausência de tecido tonsilar e adenoidal. Os meninos maiores desenvolvem infecções sinusais recorrentes. As manifestações menos comuns são as infecções cutâneas por Pseudomonas com neutropenia, meningoencefalite indolente por enterovírus e sepse.4,5
Exames laboratoriais. Na ALX, os títulos de IgG, IgA e IgM são extremamente baixos, exceto em pacientes com menos de 3 meses de idade ou que tenham recebido produtos do sangue contendo imunoglobulina recentemente. Há relatos de casos raríssimos de pacientes com níveis normais de IgG que são incapazes de responder a vacinas. As células B do sangue periférico estão praticamente ausentes nesta doença e as respostas a vacinas serão extremamente fracas. Existem várias fenocópias autossômicas recessivas (AR) de ALX e, para distinguir entre esses distúrbios e a ALX, é necessário realizar testes de mutação de Btk ou citometria de fluxo para proteína Btk. Estes testes são comercializados e podem fornecer informação importante para os pais e demais familiares, referente ao risco de recorrência.
Tratamento. A base da terapia é a substituição das imunoglobulinas faltantes. Atualmente, existem várias formas de c-globulina disponíveis, incluindo preparações intravenosas e subcutâneas de imunoglobulina. Seja qual for a estratégia de reposição, a dose típica é 400-800 mg/kg/mês. Para as estratégias de reposição subcutânea semanal, cerca de 1/4 dessa dose é administrada por semana, ou até mesmo doses menores podem ser administradas diariamente. A estratégia de dosagem pode ser titulada para manter níveis mínimos que sejam pelo menos superiores a 500 mg/dL.6 Alguns defendem níveis mínimos mais altos, de pelo menos 800 mg/dL, para um resultado melhor.7,8 Com as infusões subcutâneas, os níveis mínimos tipicamente caem para valores entre 800 e 1.000 mg/dL. Existem vários produtos disponíveis para uso intravenoso e cada um deles difere quanto ao estabilizador e ao processo de purificação. Pacientes individuais podem tolerar um produto intravenoso (imunoglobulina intravenosa [IVIg]) melhor do que outro, mas não há evidências de que um produto qualquer seja mais eficaz do que outro. As infusões subcutâneas parecem produzir menos efeitos colaterais do que as formas intravenosas.9 Enquanto estiverem sob reposição de imunoglobulina, os pacientes não necessitarão de vacinas usuais da infância, porque estarão recebendo proteção comparável do produto de imunoglobulina e não poderão produzir respostas de anticorpo por si próprios. Em geral, é desnecessário adotar medidas preventivas especiais contra infecção. Os pacientes devem estar protegidos contra infecções enterovirais conhecidas. Mesmo com a reposição de imunoglobulina, os pacientes apresentam suscetibilidade aumentada a infecções por Giardia, de modo que sintomas gastrintestinais devem ser prontamente investigados. O monitoramento em longo prazo inclui provas de função pulmonar periódica e avaliações de efeitos colaterais da terapia.
|
Tabela 1 Infecções sentinela | ||
|
Infecção |
Imunodeficiências |
Comentários |
|
Bactéria |
|
|
|
Burkholderia cepacia |
DGC |
Segunda causa mais frequente de morte na DGC. Outras Burkholderia sp. são encontradas. |
|
Chromobacterium violaceum |
DGC |
Secundária à exposição à água salobra |
|
Mycoplasma/Ureaplasma |
Defeitos humorais |
Encontrada com frequência como osteomielite ou artrite |
|
Neisseria meningitidis |
Componentes da via alternativa do complemento e componentes do complemento terminais |
Encontrada com mais frequência em pacientes com sorotipos incomuns ou doença recorrente |
|
Nocardia sp. |
DGC |
As lesões pulmonares frequentemente formam cavidades |
|
Pseudomonas aeruginosa |
Neutropenia |
|
|
Salmonella sp. |
DGC Distúrbios de ativação de macrófago |
Infecções usualmente invasivas com Salmonella sp. de baixa patogenicidade |
|
Serratia marcesens |
DGC |
Com exceção das infecções do trato urinário |
|
Staphylococcus aureus (grave) |
DGC, hiper-IgE |
Infecção visceral mais sugestiva |
|
Sepse estreptocócica |
Deficiência de IRAK4 Deficiência de NEMO Deficiência de MyD88 Asplenia Defeitos do complemento Defeitos de anticorpo |
A deficiência de IRAK4 está associada a manifestações atenuadas de sepse |
|
Micobactéria atípica |
Todos os defeitos afetando a ativação de macrófagos e DGC |
Excluindo a adenopatia cervical por MAI isolada. Infecção pode ser osteomielite. |
|
Vírus |
|
|
|
CMV/EBV |
SLPLX LHF Defeitos graves de célula T |
SLPLX e LHF frequentemente estão associadas à hemofagocitose |
|
Herpes simples |
Deficiências de TBK1, TRAF3, Unc93b e TLR3 (deficiências de STAT1, caspase 8, GATA2 e NEMO) |
Encefalite herpética vista como achado isolado com deficiências de Unc93b e TLR3 (STAT1, caspase 10 e NEMO também estão associadas ao herpes, porém estes defeitos usualmente estão associados a outras infecções) |
|
Influenza (grave) |
Deficiência de TLR3 |
Encontrada em 1 dentre 3 pacientes com encefalite por influenza |
|
Vírus JC |
Hiper-IgM Hiper-IgE GATA2 |
LPM |
|
HHV-8 |
Defeitos graves de célula T e síndrome de Wiskott-Aldrich |
|
|
Varicela |
Defeitos mais significativos de células T e NK |
Pode ser esmagadora ou recorrente |
|
Papilomavírus |
VHIM Epidermodisplasia verruciforme DOCK8, STK4 GATA2, RHOH |
|
|
Infecção grave por vírus respiratório comum |
IDGC ou outros defeitos graves de célula T |
|
|
Fungos |
|
|
|
Aspergillus |
DGC |
|
|
Candida |
DGC e PEACDE |
|
|
Histoplasmose (disseminada,invasiva) |
Defeitos de ativação de macrófago |
|
|
Fungos de baixa patogenicidade |
DGC |
|
|
Parasita |
|
|
|
Criptosporídios |
Hiper-IgM, DOCK8 |
Colangite ascendente na hiper-IgM |
|
Giardia |
Deficiências de anticorpo |
|
|
Pneumocystis jiroveci |
Defeitos graves de célula T Deficiência de NEMO |
|
|
Toxoplasmose |
Defeitos graves de célula T e hiper-IgM |
|
PEACDE = poliendocrinopatia autoimune com candidíase e distrofia ectodérmica; DGC = doença granulomatosa crônica; CMV = citomegalovírus; DOCK8 = dedicador de citocinese 8; EBV = vírus Epstein-Barr; LHF = linfo-histiocitose hemofagocítica familiar; LPM = leucoencefalopatia progressiva multifocal; MAI = Mycobacterium avium intracellulare; NEMO = modulador essencial do fator nuclear-kB; NK = natural killer; IDGC = imunodeficiência grave combinada; TLR = receptor Toll-like ; VHIM = verrugas, hipogamaglobulinemia, imunodeficiência, mielocatexia; SLPLX = síndrome linfoproliferativa ligada ao X.
Tabela 2 Comparação dos defeitos de produção de imunoglobulina Tipo Idade no momento do aparecimento dos sintomas Achados laboratoriais característicos Características clínicas exclusivas Hipogamaglobulinemia transitória da infância 5–6 meses Níveis de IgG, IgA e IgM baixos, com relativa preservação das respostas a vacinas. Os números de célula B estão altos. Tipicamente melhora aos três anos Agamaglobulinemia ligada ao X 5–12 meses Níveis de IgG, IgA, e IgM muito baixos. Sem resposta a vacinas. Os números de células B estão muito baixos ou nulos. Ausência de tonsilas e adenoides Hiper-IgM ligada ao X 5–12 meses Níveis altos ou normais de IgM, com níveis baixíssimos de IgA e IgG. Os números de células B estão normais, mas há ausência de células B transformadas em células de memória e a expressão de CD40L nas células T tipicamente é baixa. Suscetibilidade à pneumonia por Pneumocystis jiroveci Hiper-IgM e CD40 5–12 meses Ausência de células B de memória Rara Hiper-IgM e AID 5–12 meses Alguns pacientes têm IgA detectável e iso-hemaglutininas. As células B de memória são normais. Tecidos linfoides secundários amplos, alta incidência de autoimunidade Hiper-IgM e UNG 5–12 meses Células B de memória normais Rara. Adenopatia é vista. Imunodeficiência variável comum Qualquer idade, com picos em 6 e 20 anos Níveis baixos de IgG, IgA e IgM, embora alguns pacientes apresentem níveis baixos apenas de IgG. As respostas a novas vacinas serão fracas. Outras causas de hipogamaglobulinemia devem ser excluídas. Números variáveis de células B. A autoimunidade pode preceder a alteração clínica do padrão de infecção
Prognóstico. O prognóstico dos pacientes com ALX é considerado excelente. Com exceção de algum evento incomum, a expectativa de vida é normal, assim como o perfil de atividade. Um pequeno subgrupo de pacientes desenvolve condições inflamatórias, como enteropatia inflamatória. Um alerta é o de que a reposição otimizada de imunoglobulina tem sido amplamente disponibilizada há 30 anos e, por este motivo, é possível que as sequelas em longo prazo se tornem evidentes conforme os pacientes com ALX vivam por mais tempo. Tem sido descrito certo grau de declínio da função respiratória, porém sua causa ainda é indeterminada e não está claro se isto representa um aspecto relevante do ponto de vista médico. 10
Existem pelo menos cinco tipos de síndromes de hiper-IgM. Embora compartilhem alguns achados laboratoriais e clínicos, cada tipo também tem achados distintos.
Genética e patogênese. O primeiro tipo de hiper-IgM descrito foi a síndrome hiper-IgM ligada ao X, que se deve a mutações em CD40L.11 Esta proteína presente nas células T é requerida para que a célula T sinalize a troca de isótipo para a célula B. Na falta deste sinal oriundo das células T, as células B produzem IgM mas não fazem a troca de isótipo para IgG ou IgA. Os outros tipos de hiper-IgM são ARs e tipos com mutações conhecidas. Todos afetam as vias da célula B envolvidas na troca de isótipo. Desta forma, todos apresentam achados laboratoriais comuns de níveis séricos normais ou elevados de IgM e de níveis baixos de IgG e IgA. Os três tipos ARs conhecidos são devidos a mutações em CD40 (que codifica o receptor cognato de CD40L), UNG e AID. Estas duas últimas mutações afetam especificamente o processo recombinante de troca de isótipo.
Diagnóstico e exames laboratoriais. O diagnóstico em geral é considerado como suspeita clínica. Embora todos os casos de hiper-IgM estejam associados à ocorrência de infecções sinopulmonares recorrentes, a hiper-IgM ligada ao X está associada a outros dois achados dignos de nota. Pacientes com esta condição são suscetíveis a Pneumocystis jiroveci e podem apresentar neutropenia significativa. As formas ARs não estão associadas a estes achados. Pacientes com defeitos em AID apresentam aumento mais significativo de IgM, de modo geral, em comparação com os outros tipos, e tendem mais a desenvolver adenopatia e autoimunidade.
O diagnóstico pode ser indicado por uma forte suspeita em presença de achados clínicos compatíveis e com o achado de IgM normal ou aumentada, aliado a níveis baixíssimos de IgA e IgG. Os achados de suporte são a ausência de células B de memória à análise de citometria de fluxo. O diagnóstico definitivo tipicamente se baseia na demonstração da anormalidade proteica por citometria de fluxo (CD40 e CD40L) ou análise de mutação. Existem pacientes que parecem ter o fenótipo para o qual a análise de mutação não é reveladora, e estes pacientes são atualmente denominados como indivíduos temporariamente hiper-IgM.
Tratamento. A terapia ideal para todos os pacientes com hiper-IgM não está estabelecida. Atualmente, todos os pacientes são tratados com reposição de imunoglobulina, conforme descrito. Pacientes com hiper-IgM ligada ao X apresentam alta incidência de malignidade na fase adulta. 12,13 Pacientes com hiper-IgM ligada ao X parecem ser beneficiados pelo transplante de células-tronco para prevenção de malignidade, mas faltam dados sobre os resultados alcançados a longo prazo. Pacientes com doador compatível para transplante frequentemente são aconselhados a se submeterem ao transplante de célula-tronco. Na ausência de um doador totalmente compatível, os riscos do transplante aumentam consideravelmente e os benefícios não são tão definidos. Assim, o transplante não costuma ser realizado na ausência de um doador totalmente compatível. Frequentemente, a terapia para os tipos ARs de hiper-IgM é a reposição de imunoglobulina. Pacientes incomuns podem ser candidatos ao transplante de células-tronco hematopoiéticas.
Prognóstico. O prognóstico para os tipos de hiper-IgM ARs é considerado razoavelmente bom. Existem poucos dados em longo prazo, mas os pacientes aparentemente não apresentam problemas potencialmente fatais depois que se submetem à terapia de reposição de imunoglobulina, ainda que a autoimunidade possa ser problemática. 14 O prognóstico para pacientes com a forma ligada ao X é, consideravelmente, pior. Um estudo sobre resultado constatou que, na ausência de transplante, poucos pacientes sobreviveram até os 30 anos de idade. No grupo de faixa etária mais avançada, as mortes foram devidas primariamente à malignidade, com os tumores de ducto biliar sendo os mais comuns. 12 É possível que isto se deva à estimulação por criptosporídios, sendo que estudos de longa duração sobre pacientes protegidos contra criptosporídios estão em andamento.
Este é o mais comum dos estados de deficiência de imunoglobulina tratáveis. Considera-se que sua prevalência gire em torno de 1:30.000, embora esta verificação seja provavelmente imperfeita. A IDCV está associada a vários achados incomuns. Diferentemente de muitas imunodeficiências primárias, não se trata de um distúrbio envolvendo um único gene. Além disso, em contraste com a maioria das imunodeficiências primárias, a IDCV pode se manifestar em qualquer idade, embora ocorram picos de incidência em 6-8 anos e 15-35 anos de idade.
Genética e patogênese. Embora os parentes com defeitos monogênicos sejam bastante raros, considera-se que a vasta maioria dos pacientes apresenta etiologia poligênica complexa com um componente ambiental significativo. Mutações em TACI podem contribuir para a suscetibilidade ou para o fenótipo, contudo o teste de mutação não é clinicamente indicado por ter baixo valor preditivo. Alguns pacientes mostram evidências de defeito de célula B intrínseco, enquanto outros parecem ter um defeito de célula B extrínseco. Um estudo recente identificou altos níveis de variantes de número de cópia em pacientes com IDCV.15 Este estudo, também, sugeriu que os subtipos clínicos de IDCV podem ter base genética.
Diagnóstico. O diagnóstico deve ser suspeito em pacientes que estavam bem e depois desenvolveram um novo padrão de infecções sinopulmonares recorrentes. Também é possível observar infecções incomumente graves e muitos pacientes têm doença autoimune, como as citopenias autoimunes sendo as mais comuns.16 Pacientes em que o aparecimento da condição se deu antes dos 2 anos de idade, geralmente, são excluídos, porque se presume que tenham um processo etiológico distinto.17 Os critérios diagnósticos formais são níveis de IgG e de IgA ou IgM inferiores a 2 desvios padrões (DPs) em relação à média para a idade, além de incapacidade comprovada de responder a vacinas. Todas as outras potenciais causas de perdas secundárias de imunoglobulina devem ser excluídas, porque não existe teste específico para IDCV.
Exames laboratoriais. Não há teste específico para IDCV. O caminho para estabelecer o diagnóstico inclui a realização de exames laboratoriais de suporte e a exclusão de outras potenciais causas. Em geral, os pacientes apresentam diminuição de IgG, IgA e IgM, mas é possível que estes níveis estejam assimetricamente diminuídos. Como esta doença evolui com o passar do tempo, pode haver necessidade de avaliações adicionais. A função precária dos anticorpos tipicamente é demonstrada por meio da avaliação das respostas de anticorpo às vacinas. É importante avaliar as respostas a vacinas recentes. Entre os distúrbios significativos a serem excluídos, estão a hipogamaglobulinemia associada a fármacos, timoma, leucemia linfocítica crônica, síndrome nefrótica, enteropatia perdedora de proteína, HIV e (em crianças) hipogamaglobulinemia transitória da infância (HTI).
Tratamento. Pacientes com IDCV são tratados com reposição de imunoglobulina, conforme descrito. A atenção à doença autoimune e o equilíbrio cuidadoso das medicações imunossupressoras usadas com frequência no tratamento da autoimunidade se fazem necessários. Os pacientes parecem apresentar aumento significativo (ainda que discreto) da frequência de malignidades e os sintomas sugestivos de malignidade devem ser investigados.
Prognóstico. O prognóstico para pacientes com IDCV é variável. Indivíduos com doença autoimune têm expectativa de vida diminuída.18
A deficiência de IgA ocorre em 1:500 a 1:800 brancos.19 É significativamente menos frequente em outras raças. A maioria dos pacientes com deficiência de IgA permanece em bom estado, enquanto alguns têm complicações autoimunes ou infecciosas.
Genética e patogênese. A deficiência de IgA é mais frequentemente esporádica, mas 10-20% dos casos são familiares. Em alguns casos familiares, a deficiência de IgA é vista no mesmo parente com IDCV. Em outros, a deficiência de IgA progride para IDCV. Ao serem examinados, estes múltiplos parentes exibem um padrão genético complexo.
O distúrbio pode ser heterogêneo, mas na maioria dos casos as células B conseguem produzir IgA se forem tratadas com anti-CD40 e interleucina-10 (IL-10), sugerindo que o defeito está localizado no compartimento da célula T.20 Acredita-se que, assim como a IDCV, a deficiência de IgA é um distúrbio poligênico com um deflagrador ambiental. Certas medicações estão associadas a um risco significativo de indução de deficiência de IgA, e a infecção é citada com frequência como potencial deflagrador.
Diagnóstico. A deficiência de IgA está associada a infecções recorrentes, por causa do papel da IgA secretada sobre as membranas mucosas. A maioria das infecções é sinopulmonar e em sua grande maioria bacteriana. A doença autoimune também é mais comum em pacientes com deficiência de IgA e isto pode ser atribuível ao papel da IgA de superfície na proteção do sistema imune sistêmico contra a ativação. A deficiência de IgA está associada ainda a alergias e à asma. Embora esteja claro que muitas séries clínicas de pacientes com doença autoimune ou infecções recorrentes exibam uma frequência maior do que a esperada de indivíduos com deficiência de IgA, um estudo sobre doadores de bancos de sangue constatou que a maioria dos pacientes com deficiência de IgA eram clinicamente sadios.21,22
Os critérios diagnósticos são bastante específicos. Os níveis de IgA devem ser inferiores a 5 mg/dL, com preservação dos níveis de IgG e IgM, bem como das respostas a vacinas. Como a regulação da IgA está altamente associada ao crescimento, a condição somente pode ser diagnosticada após dois anos de idade. Pacientes com níveis baixos de IgA não são considerados deficientes, a menos que os níveis estejam abaixo de 5 mg/dL.
Um aspecto importante do diagnóstico é o estabelecimento da causalidade. A maioria dos indivíduos com deficiência de IgA é sadia, sendo que a ocorrência abrupta de infecções em pacientes de idade avançada comprovadamente deficientes de IgA exige investigação. É necessário procurar fatores adicionais que contribuam para o padrão de infecção.
Exames laboratoriais. Os níveis de imunoglobulina e as respostas a vacinas são informações requeridas para estabelecer o diagnóstico. Alguns laboratórios não estão capacitados para medir os níveis de IgA até o nível requerido para estabelecer o diagnóstico.
Tratamento. Não há intervenção específica para a deficiência de IgA. A IgA não pode ser reposta e é inapropriado administrar IVIg para tratar a deficiência de IgA. No caso dos pacientes com infecções recorrentes, o tratamento dirigido para o alívio das infecções é útil, sendo que a abordagem de alergias concomitantes muitas vezes pode ser benéfica.
Pacientes com deficiência de IgA devem ser alertados quanto à possibilidade de sensibilização com produtos do sangue.23 Qualquer produto do sangue contendo IgA pode servir de imunógeno, de modo que a exposição subsequente pode levar a reações transfusionais não hemolíticas. Isto é incomum, mas os pacientes devem ser alertados quanto ao risco e, quando necessário, podem ser usados produtos do sangue lavados ou livres de IgA.
Prognóstico. Acredita-se que os pacientes com deficiência de IgA, em geral, têm expectativa de vida normal.
Esta condição é razoavelmente comum e considerada uma representação de atraso do desenvolvimento na produção de imunoglobulina.
Genética e patogênese. Apesar dos relatos de ocorrência de HTI em famílias, a condição costuma ser mais esporádica e ainda não está claro se existe uma base genética. A patogênese é completamente desconhecida.
Diagnóstico. A HTI é classicamente encontrada em bebês de 6-18 meses de idade com infecções recorrentes envolvendo os tratos respiratórios superior e inferior. As infecções podem ser uma mistura de infecções bacterianas e virais. Ocasionalmente, são vistos pacientes de idade mais avançada.
Exames laboratoriais. O fenótipo não é tão uniforme quanto inicialmente descrito, mas os critérios mais comuns para o diagnóstico são níveis baixos de IgG (com ou sem níveis baixos de IgA) e de IgM. Um número de células B normal ou alto e uma resposta amplamente intacta a vacinas devem ser observados.24,25 Recentemente, foi relatado que algumas respostas a vacinas podem não ser tão robustas quanto em outras crianças, mas com a administração de uma vacina de reforço, as respostas geralmente normalizam.24 Um critério importante para o diagnóstico é que os níveis de imunoglobulina do paciente se normalizem com o passar do tempo.
Tratamento. Nenhum tratamento se faz necessário, embora os pacientes com hipogamaglobulinemia sustentada possam receber tratamento temporário com reposição de imunoglobulina, caso não seja possível estabelecer o diagnóstico com certeza e/ou o padrão de infecção seja grave.
Prognóstico. Este atraso na produção de imunoglobulinas é considerado uma variante normal. A maioria das crianças tem os níveis de IgG normalizados ao redor dos 2-3 anos de idade. Depois que a criança desenvolve níveis normais de imunoglobulina, considera-se que não há sequelas. Não existem estudos abrangentes sobre esta entidade, mas também não há dados sugerindo que o resultado seja outro que não a normalidade completa. As infecções que surgem durante a fase hipogamaglobulinêmica podem ser significativas. Entretanto, o padrão de infecção melhora ao mesmo tempo ou pouco depois da melhora dos níveis de imunoglobulina.
Os distúrbios de célula T variam de muito graves a brandos e o fenótipo da infecção é correspondentemente amplo. A principal característica dos distúrbios de célula T são as infecções virais prolongadas. Nos defeitos mais graves de célula T, infecções oportunistas são vistas. Quase todos os defeitos de célula T estão associados ao risco aumentado de doença autoimune.
A imunodeficiência grave combinada (IDGC) é assim chamada porque há comprometimento grave tanto do compartimento de célula T como do compartimento de célula B. Na maioria dos casos, não há função detectável de células T ou de células B.
Genética e patogênese. É provável que existam mais de 20 causas diferentes de IDGC ou condições similares à IDGC. Todos os defeitos afetam o desenvolvimento da célula T de modo significativo. Na maioria dos casos, o desenvolvimento da célula T é bloqueado e nenhuma célula T naive é encontrada no sangue periférico. Na falta da ajuda da célula T, as células B não conseguem produzir anticorpo. Em alguns casos, o bloqueio que afeta o desenvolvimento da célula T também afeta o desenvolvimento da célula B e, quando isso ocorre, as células B também estão ausentes no sangue periférico. Caracterizar as populações de linfócitos pode ajudar a identificar o tipo de IDGC e tem algumas implicações para o prognóstico.
Diagnóstico. Muitos estados americanos estão adicionando a IDGC ao programa de triagem de recém-nascidos. Entretanto, até o momento, essa triagem é obrigatória apenas em uma minoria dos estados. A IDGC tipicamente se manifesta entre os 2 e 6 meses de idade.26 As manifestações mais comuns são as infecções persistentes do trato respiratório superior, pneumonia por P. jiroveci (referida como pneumonia por Pneumocystis ou PPC), Candida, diarreia e pneumonia. A suspeita do diagnóstico deve ser levantada em casos de bebês que apresentam achados clínicos compatíveis e uma contagem absoluta de linfócitos baixa.27 As contagens de linfócito são bem mais altas em bebês do que nos adultos, sendo que níveis abaixo de 2.800 células/µL são considerados sugestivos de IDGC, exceto quando há outras explicações para a linfopenia. Duas apresentações variantes devem ser notadas. Os pacientes com IDGC são incapazes de destruir células de enxerto que “pegam” e as células maternas que atravessam a placenta podem estabelecer a doença do enxerto versus hospedeiro.28 Estas crianças desenvolvem eritroderma logo após o parto e podem apresentar outros sinais sugestivos de doença do enxerto versus hospedeiro. As células maternas proliferantes às vezes podem levar a uma contagem de linfócitos normais e podem fazer com que células T sejam detectadas por citometria de fluxo. A segunda variante de apresentação é conhecida como síndrome de Omenn. A síndrome de Omenn está associada ao crescimento oligoclonal de algumas células T que escaparam do bloqueio do desenvolvimento.29 Vários tipos genéticos de IDGC podem estar associados a esta apresentação. Bebês com síndrome de Omenn também apresentam eritroderma de aparecimento precoce e, geralmente, têm populações de linfócitos desviadas no sangue periférico. Estes bebês, frequentemente, apresentam uma drástica hepatoesplenomegalia e adenopatia.
Exames laboratoriais. A maioria dos bebês apresenta contagem absoluta de linfócitos abaixo de 2.800 células/µL. Cada tipo de IDGC está associado a um fenótipo característico de população de linfócitos. Tradicionalmente, a IDGC é classificada de acordo com a presença de células T, células B e células natural killer (NK). Esta classificação costuma ser representada como T-B+NK-, etc. É tradicional documentar a disfunção de célula T e a disfunção de célula B. Entretanto, nos casos com subgrupos nitidamente consistentes, nem sempre é necessário realizar ensaios de proliferação e de imunoglobulina. Em muitos casos, é útil realizar a fenotipagem detalhada da célula T. é importante excluir a hipótese de infecção por HIV. A Tabela 3 detalha os subgrupos de linfócitos característicos encontrados em diferentes tipos de IDGC e de condições similares à IDGC. Quando há suspeita de IDGC e a contagem total de células T se aproxima do normal, é útil analisar os subgrupos de células T naive e de memória. Na síndrome de Omenn ou no enxerto materno, é possível que haja células T presentes alcançando até mesmo números normais. O indício destas variantes de apresentação é que as células T podem ser de apenas um único subgrupo (i.e., CD4 ou CD8) e exibirão uniformemente o fenótipo de memória (i.e., CD45RO). Uma vez estabelecido o diagnóstico de IDGC, a terapia definitiva deve ser iniciada. Pode ser desejável realizar teste genético para os diferentes tipos de IDGC, para obter informação útil sobre o risco de recidiva em irmãos e também para obter informação sobre o prognóstico. No entanto, a terapia não deve esperar a disponibilização dos resultados do teste genético.
Tratamento. A IDGC é uma rara emergência médica imunológica.30 Mesmo uma forte suspeita de IDGC deve conduzir prontamente à iniciação de medidas de isolamento protetor, proteção contra produtos do sangue não irradiados e proteção contra produtos do sangue contendo citomegalovírus (CMV). Em geral, é possível realizar exames de fenotipagem de linfócitos em 1-2 dias. Se houver atraso significativo, a profilaxia para PPC deverá ser instituída. A terapia definitiva é o transplante de célula-tronco, tão logo o diagnóstico tenha sido estabelecido. Durante a espera pelo transplante, o paciente também deve receber IVIg.
|
Tabela 3 Tipos de imunodeficiência grave combinada | |||||
|
Defeito genético |
Defeito de proteína, função |
% de IDGC |
Perfil de linfócitos | ||
|
T |
B |
NK | |||
|
IL2RG |
Cadeia g comum de receptores para IL-2, -4, -7, -9, -15, -21 |
45–50 |
|
+ |
- |
|
ADA |
Adenosina desaminase (metabolismo de purina) |
16 |
- |
± |
- |
|
IL7R |
cadeia a, se receptor de IL-7 |
9 |
- |
+ |
+ |
|
JAK3 |
Janus quinase 3 (ativada por cg) |
6 |
- |
+ |
- |
|
DCLRE1C |
Artemis (recombinação de antígeno de célula T/célula B) |
< 5 |
- |
- |
+ |
|
RAG 1/2 |
Gene ativador de recombinação |
< 5 |
- |
- |
+ |
|
LIG4 |
DNA ligase IV (recombinação célula T/célula B) |
Raro |
- |
+ |
+ |
|
XLF |
Cernunnos (recombinação de célula T/célula B) |
Raro |
- |
- |
+ |
|
PTPRC |
CD45 (requerido para ativação de T, B por antígeno) |
Raro |
-/baixo |
+ |
+/baixo |
|
TCRD, TCRE, TCRZ |
Deficiência de CD3 delta, épsilon e Csi (comprometimento do desenvolvimento da célula T) |
Raro |
-/baixo |
+ |
+ |
|
LCK |
Tirosina quinase de linfócito p56lck (desenvolvimento e ativação de célula T) |
Raro |
-/baixo |
+ |
+ |
|
FOXN1 |
Forkhead box N1 (desenvolvimento do timo e do folículo piloso—ortólogo do camundongo nude) |
Raro |
-/baixo |
+ |
+ |
|
PNP |
Fosforilase purina nucleosídeo (metabolismo de purina) |
Raro |
- |
baixo |
baixo |
|
PTCD |
Coronina 1A (egresso tímico) |
Raro |
- |
+ |
+ |
|
ORAI1/STIM 1 |
Canal de cálcio ativado pela liberação de cálcio |
Raro |
+ |
+ |
+ |
|
Atualmente indeterminado |
Atresia intestinal com IDGC |
Raro |
- |
- |
+ |
|
AK2 |
Disgênese reticular |
Raro |
- |
- |
- |
IL = interleucina; IDGC = imunodeficiência grave combinada.
O transplante de células-tronco para pacientes com IDGC é singular no sentido de que é possível realizar o transplante transpondo as barreiras do antígeno leucocitário humano (HLA), uma vez que os bebês são incapazes de rejeitar o enxerto. Quando disponível, um transplante compatível de um doador irmão ou de sangue de cordão ou medula de doador registrado é ideal. Entretanto, a demora em identificar uma compatibilidade deve ser evitada. Transplantes de qualquer um dos pais também podem ser usados com sucesso. Transplantes fora do contexto de compatibilidade total, em geral, são depletados de células T, para prevenir a doença do enxerto versus hospedeiro. Os transplantes totalmente compatíveis incluem células T maduras e, portanto, reconstituem o paciente mais prontamente do que os enxertos T-depletados. No caso dos enxertos T-depletados, tipicamente demora três meses para aparecerem células T maduras e funcionais no sangue periférico. Quando há disponibilidade de um doador irmão compatível, está é sem dúvida a escolha ideal. A qualidade do enxerto também é influenciada pela escolha do doador, embora a escolha do condicionamento também represente uma influência significativa sobre a qualidade do enxerto. Os transplantes totalmente compatíveis tendem mais a produzir pega real do enxerto de células-tronco e reconstituição de linhagens múltiplas em longo prazo. Assim, a decisão acerca da fonte de transplante deve equilibrar o enxerto ideal (compatibilidade total) com a necessidade de transplante imediato. A maioria dos pacientes com IDGC está doente no momento do transplante e a infecção viral somente poderá ser eliminada depois que o enxerto reconstituir funcionalmente o compartimento de células T. Uma compatibilidade de registro pode oferecer o enxerto ideal. Entretanto, se o doador não puder ser recrutado por muitas semanas, a vantagem do enxerto ideal pode ser anulada pela necessidade de obter uma pega de enxerto que seja rápida o suficiente para combater a infecção.
Prognóstico. A maior parte da mortalidade associada à IDGC é devida ao diagnóstico tardio ou ao atraso em instituir a terapia definitiva. No primeiro mês de vida, o transplante está associado a uma taxa de sobrevida superior a 95%.31 Depois que a criança recebe o transplante, há os riscos de doença do enxerto versus transplante e de perda tardia do enxerto, mas muitos pacientes são verdadeiramente curados, ainda que alguns aparentemente percam o enxerto de células NK e de células B com o passar do tempo. As variáveis que governam os resultados bastante variáveis ainda estão sendo elucidadas, mas incluem a compatibilidade de doador e o condicionamento. Em adição, alguns tipos genéticos de IDGC têm resultados piores, independentemente das outras variáveis.32
A síndrome de DiGeorge e síndrome da deleção cromossômica 22q11.2 não são sinônimos. A síndrome de DiGeorge é uma constelação clinicamente definida de timo hipoplásico, anomalia cardíaca conotruncal e hipoparatireoidismo.33 Muitos destes pacientes terão deleção hemizigota do cromossomo 22q11.2, ainda que nem todos.34
Genética e patogênese. A base genética dos casos de pacientes com deficiência grave de células T com síndrome de DiGeorge é heterogênea. Cerca de 1/3 apresentará deleções hemizigotas do cromossomo 22q11.2 e 1/3 terá mutações em CHD7. Dentre os pacientes que exibem fenótipo mais brando e contagens de célula T diminuídas, ainda que não grave, cerca de 80% terão deleções cromossômicas de 22q11.2. A deleção do cromossomo 22q11.2 leva ao desenvolvimento de hipoplasia tímica decorrente de haplossuficiência de um fator de transcrição conhecido como TBX1.35 Este fator de transcrição regula o desenvolvimento da artéria do arco faríngeo durante a embriogênese. As células migram dos arcos faríngeos para o coração, paratireoides e glândula tímica em desenvolvimento. A haplossuficiência de TBX1 compromete a migração e o crescimento destas células.
Diagnóstico. Os casos mais graves de imunodeficiência são bastante parecidos com a IDGC, com a preocupação adicional das anomalias congênitas. As células T ausentes obrigam a adoção das mesmas precauções que seriam tomadas para pacientes com IDGC. No outro extremo do espectro, cerca de 20% dos pacientes com deleção do cromossomo 22q11.2 têm números normais de células T a partir do nascimento.36 A maioria dos pacientes com deleção do cromossomo 22q11.2 apresenta defeito leve a moderado de produção de células T. A suspeita de diagnóstico de síndrome de DiGeorge ou de síndrome da deleção cromossômica 22q11.2 deve ser considerada em casos de pacientes com anormalidades cardíacas conotruncais, hipocalcemia, fácies dismórficas, hipotonia, retardo de fala, fala nasal ou retardo do desenvolvimento em qualquer combinação.37 Um número crescente de adultos com fenótipos brandos vem sendo identificado. A síndrome de DiGeorge com ausência de células T deve ser distinguida da IDGC, porque será insensível ao tratamento com transplante de célula-tronco. Os aspectos distintivos úteis são a presença de outras anomalias, hipocalcemia e fácies dismórficas. Devem ser realizadas a análise de mutação em CHD7, hibridização in situ fluorescente para deleção do cromossomo 22q11.2 e o ensaio de arranjo de polimorfismo de nucleotídeo único (SNP) para detecção das deleções menos comuns no cromossomo 10.
Exames laboratoriais. Pacientes que exibem o fenótipo clássico de síndrome de DiGeorge com ausência de células T nem sempre precisam ser submetidos a testes genéticos. Entretanto, seus familiares frequentemente desejam realizar o teste para ajudar a determinar os riscos para os futuros descendentes. O teste genético é mais importante em casos ambíguos, porque é importante distinguir entre um distúrbio tratado com transplante de timo e um distúrbio tratado com transplante de célula-tronco. Os testes seriados de número de células T são importantes no início, porque podem mudar de forma bastante drástica, de modo que um paciente inicialmente pode parecer ser candidato ao transplante de timo e, depois, ter número suficiente de células T. Um aspecto importante da análise de células T deve incluir uma determinação das células T naive versus células T de memória. No início da infância, a contagem de células T naive reflete a capacidade tímica. Os testes de citometria de fluxo para emigrações tímicas recentes são disponibilizados em alguns locais, e uma análise de círculos de excisão de recombinação de células T também está começando a ser disponibilizada.
Tratamento. A vasta maioria dos pacientes com síndrome da deleção cromossômica 22q11.2 dispensa intervenção imunológica. Estes pacientes requerem cuidados multidisciplinares e uma abordagem coordenada para suas necessidades de saúde diversificadas e significativas. O sistema imune destes pacientes requer monitoramento para garantir que o compartimento de células T seja suficientemente robusto para lidar com os vírus atenuados contidos nas vacinas com vírus vivos, e para ser capaz de defender o organismo contra os vírus comuns encontrados no contexto escolar. Em casos raros, alguns pacientes podem precisar receber IVIg em decorrência de defeitos de anticorpo secundários.
Pacientes com síndrome de DiGeorge e células T ausentes requerem transplante de timo ou transplante de medula não manipulada de doador irmão totalmente compatível.34 O preparo do enxerto tímico é difícil e deve ser feito apenas em centros especializados. O transplante de medula óssea (ou de sangue periférico coletado) de irmão totalmente compatível fornece mais células T maduras e reconstitui imediatamente o compartimento de células T.
Prognóstico. A expectativa de vida para pacientes com síndrome da deleção cromossômica 22q11.2 é considerada normal. Há casos raros de pacientes que morreram em consequência da anomalia cardíaca, e casos ainda mais raros de pacientes que morreram por infecção ou durante o transplante tímico.38 A qualidade de vida é menos positiva. Um número significativo de adultos sofre com infecções recorrentes, sendo que a doença autoimune é comum. Existem aspectos não relacionados ao sistema imune, ligados ao trabalho e a transtornos psiquiátricos, que podem acarretar ônus significativo ao paciente e seus familiares.
Para pacientes com síndrome de DiGeorge que requerem transplante de timo, o tempo para haver reconstituição de células T é de cerca de 3 meses e, durante este intervalo, os pacientes ficam bastante vulneráveis a infecções. Os pacientes que recebem transplante de medula óssea de irmão totalmente compatível obtêm reconstituição imediata. Em ambos os casos, existem preocupações teóricas relacionadas à durabilidade das novas células T e ao risco de doença autoimune.
A síndrome de Wiskott-Aldrich (SWA) foi uma das primeiras imunodeficiências primárias a ser reconhecida. O defeito afeta muitas células hematopoiéticas, mas é tipicamente caracterizado como um defeito de célula T, porque muitas das manifestações clínicas refletem o comprometimento da função da célula T.
Genética e patogênese. A SWA é um distúrbio ligado ao X que pode se desenvolver em meninas, ainda de que raramente, como resultado da síndrome de Turner ou da inativação assimétrica de X. As mutações no gene WASP são responsáveis por quase todos os casos da tríade clássica de trombocitopenia com plaquetas pequenas, eczema e infecções recorrentes.39 Há algumas evidências da existência de correlação genótipo-fenótipo, contudo pode haver enorme variação da gravidade da doença entre os parentes do indivíduo. A função de WASP é regular e propiciar uma armação para a polimerização de F-actina.40 Esta estrutura de actina serve para polarizar a célula, de modo que o receptor da célula T e suas respectivas moléculas sinalizadoras se alinham na face da célula voltada para a célula apresentadora de antígeno. De modo semelhante, a polarização da actina é essencial para a função dos neutrófilos e macrófagos. Este defeito na actina também está por trás da trombocitopenia, embora ainda não esteja claro se o defeito inteiro é intrínseco à plaqueta ou se alguns aspectos da trombocitopenia são atribuíveis à captação acelerada.
Diagnóstico. A SWA frequentemente é descrita como a tríade clássica de eczema, trombocitopenia e infecção recorrente. O achado mais frequentemente presente é o sangramento e contusão resultantes da trombocitopenia.41 Não raro, os pacientes a princípio são diagnosticados com púrpura trombocitopênica idiopática (PTI) crônica. Um aspecto fundamental para o diagnóstico é o fato de as plaquetas serem pequenas, em vez de grandes, como se observa na PTI. O espectro da infecção e eczema bastante amplo e pode não ser evidente no momento em que a trombocitopenia é identificada. O padrão de herança ligada ao X é visto em cerca de 2/3 dos parentes, enquanto 1/3 apresentam mutações novas. A história familiar materna deve ser especificamente investigada e os familiares que apresentarem evidências de sangramento, infecção, eczema e linfoma deverão ser especificamente identificados.
Exames laboratoriais. A suspeita clínica de SWA pode ser levantada com base na trombocitopenia com plaquetas pequenas. Muitos estabelecimentos não realizam análise de plaquetas devidamente ampla e é útil solicitar a um profissional experiente a confirmação visual das plaquetas pequenas ou determinar o tamanho com auxílio de um instrumento para esta finalidade. O diagnóstico de SWA está associado a um prognóstico desfavorável, portanto a maioria dos casos precisa ser confirmada por teste genético.42 Entre as possíveis exceções, está uma extensa história familiar de SWA e uma situação em que a família não deseja realizar teste genético.
Os exames auxiliares que podem proporcionar benefícios são as avaliações padrão de produção e função de imunoglobulina, e a fenotipagem da célula T. Estes testes são úteis para definir o nível de função imunológica e a necessidade de IVIg.
Tratamento. O tratamento da SWA continua evoluindo. O transplante de células-tronco representa uma opção em potencial, mas, muitas vezes, é reservado para os casos em que há disponibilidade de compatibilidade total.43 Uma consideração adicional é que, neste distúrbio, o êxito do transplante depende da idade e os casos mais bem-sucedidos envolvem pacientes que receberam transplante nas fases iniciais da vida.
Pacientes com fenótipo brando e pacientes que não dispõem da opção de compatibilidade total imediata frequentemente são tratados de modo conservativo, com profilaxia antibiótica e IVIg, conforme a indicação clínica. A esplenectomia tem sido realizada para controlar a trombocitopenia e tem sido bem-sucedida, mas está associada a um risco maior que o usual de sepse pós-esplenectomia e não é recomendada. O tratamento da trombocitopenia é desafiador. Os pacientes tipicamente têm contagem basal de plaquetas da ordem de 50.000-80.000/µL, mas são bastante suscetíveis a episódios agudos de trombocitopenia aumentada, em que as contagens de plaquetas podem cair para 0-10.000/µL. Em alguns casos, a trombocitopenia aguda pode ser tornar crônica.41 IVIg e esteroides representam potenciais intervenções para trombocitopenia aguda, embora não sejam universalmente efetivas.
Prognóstico. A eficácia do transplante de célula-tronco ainda não está totalmente definida, mas os efeitos colaterais aparentemente exclusivos da SWA foram descritos. As citopenias autoimunes pós-transplante são comuns e parecem ocorrer com mais frequência em indivíduos com quimerismo misto. Um enxerto em que ocorre pega durável somente de células T pode proporcionar algum benefício com relação à infecção, mas deixará o paciente suscetível ao desenvolvimento de doença autoimune e linfoma. Poucos estudos relatam os resultados alcançados em longo prazo.
Pacientes não transplantados apresentam risco contínuo de desenvolvimento de doença autoimune e linfoma. Existem pacientes com fenótipo brando que retêm esse fenótipo por toda a vida, assim como há pacientes com fenótipo brando que subsequentemente desenvolvem linfoma na fase adulta. Os pacientes com fenótipo mais clássico apresentam expectativa de vida diminuída e isto leva ao desejo de realizar transplantes nesta população de pacientes.
Os patógenos disparam o sistema imune adaptativo (linfócitos T e B) ligando-se a receptores altamente específicos gerados por recombinação V(D)J dos receptores de antígeno (Ag) das células T e B. A memória imune de longa duração é mantida pelas células de memória T e B, que conseguem mediar uma rápida resposta baseada na lembrança quando da reexposição ao mesmo patógeno. Entretanto, a resposta imune adaptativa precisa de tempo o suficiente (dias a semanas) para se tornar totalmente ativada. Em contraste, o sistema imune inato é capaz de promover respostas imediatas à infecção microbiana e atua como mecanismo de primeira linha de defesa no corpo. Os defeitos das vias de sinalização do receptor Toll-like receptor (TLR) e da IL-12/interferon-g são exemplos de imunodeficiências primárias que destacam a importância do sistema imune inato.
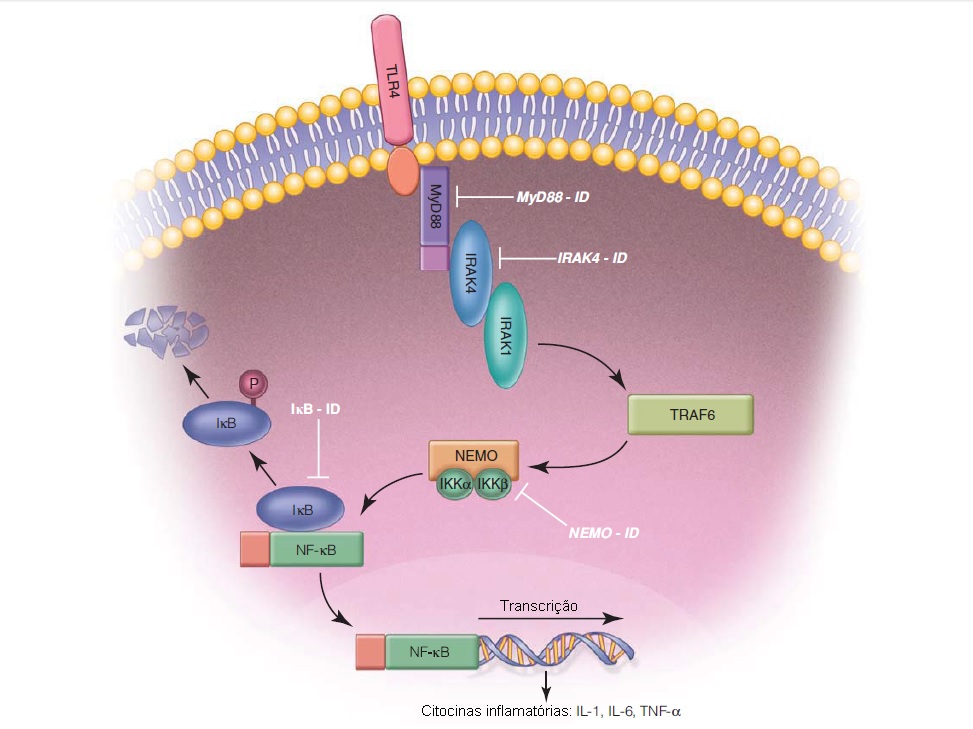
O sistema imune inato usa receptores de reconhecimento de padrão (PRRs) para detectar padrões moleculares associados a patógenos (PAMPs), como endotoxina, lipopolissacarídeo, parede celular bacteriana, flagelos e RNA de fita dupla (dsRNA). Os TLRs são apenas um exemplo destes PRRs. Foram descritos 10 TLRs humanos até o presente, sendo que cada TLR é ativado por um PAMP vital ou bacteriano específico. Após o reconhecimento dos PAMPs pelos TLRs, a sinalização intracelular ocorre através de moléculas adaptadoras MyD88 e quinases associadas ao receptor da IL-1 (IRAK4 e IRAK1), resultando enfim na ativação do fator de transcrição nuclear-kB, (NF-kB), que regula positivamente a produção de citocinas inflamatórias (IL-1, IL-6 e fator de necrose tumoral-a[TNF-a]) [ver Figura 1].44

Figura 1. Representação esquemática da via de sinalização do receptor Toll-like (TLR) 4. A ativação de TLR4 resulta na sinalização via MyD88 e IRAK4, e na ativação do complexo IKK, constituído por NEMO, IKK-a e IKK-b. O complexo IKK fosforila IkB, o inibidor do fator nuclear kB (NF-kB), levando a sua ubiquitilação e degradação. Isto libera o NF-kB para ser translocado para o núcleo e induz a expressão de genes codificadores de citocinas inflamatórias. Com exceção de TLR3, todos os TLRs sinalizam através de uma dependente de MyD88. A sinalização de TLR4 pode ocorrer através de vias dependentes ou independentes de MyD88. Os defeitos envolvendo esta via de sinalização são indicados na ilustração. ID = imunodeficiência; IL = interleucina; TNF-a = fator de necrose tumoral-a.
Genética e patogênese. A deficiência de IRAK4 e MyD88 são defeitos ARs da imunidade inata. MyD88 e IRAK4 são moléculas de sinalização intracelular requeridas para a sinalização mediada por TLR. Em pacientes afetados, a sinalização de TLR em resposta aos PAMPs bacterianos é bastante diminuída. Como a sinalização via TLR3 (que reconhece RNA de fita dupla, um PAMP viral) independe de IRAK4/MyD88, a defesa antiviral parece ser preservada.
Diagnóstico. Pacientes com deficiência de IRAK4 e MyD88 são caracterizados pela suscetibilidade aumentada a infecções por Streptococcus pneumoniae invasivo, Staphylococcus aureus e Pseudomonas. As infecções clostridiais são menos frequentes. Os pacientes aparentemente não apresentam risco de aquisição de doenças fúngicas, virais ou micobacterianas. Uma característica exclusiva desta condição está no fato de muitos pacientes não apresentarem febre nem respostas inflamatórias (elevação mínima da velocidade de sedimentação eritrocitária/proteína C reativa) durante as infecções agudas, devido à capacidade comprometida de regular positivamente das citocinas inflamatórias. Entre todos os pacientes relatados até hoje, tem havido uma mortalidade surpreendente de 40% na primeira década da vida. Entretanto, não relatos de morte após oito anos de idade e nenhuma infecção bacteriana invasiva ocorreu após os 14 anos. Foi postulado que esta melhora é secundária à maturação das respostas imunes adaptativas que ocorre com a idade.45
Os níveis séricos de imunoglobulina, respostas de anticorpo específicas, números de subgrupos de célula T e respostas proliferativas de células T tipicamente permanecem normais. Os testes de função de TLR que medem a produção de citocinas inflamatórias (IL-1, IL-6 e TNF-a) após a estimulação com ligantes de TLR fornecem resultados significativamente reduzidos. O diagnóstico é confirmado por sequenciamento dos genes IRAK4 ou MyD88.45
Tratamento e prognóstico. A iniciação do tratamento é essencial, devido á alta taxa de mortalidade durante a primeira década da vida. As terapias de profilaxia antibiótica e administração de imunoglobulina mensal devem ser mantidas pelo menos até a adolescência, quando as complicações infecciosas se tornam menos comuns. O prognóstico em longo prazo para os pacientes com deficiência de IRAK4 parece ser excelente quando os pacientes sobrevivem além da adolescência.
Genética e patogênese. O NF-kB é um fator de transcrição central, que é ativado subsequentemente a partir da ativação de TLR que induz expressão de um amplo número de genes envolvidos na imunidade e inflamação. Nas células em repouso, o NF-kB está inativo e ligado ao inibidor IkB. quando a célula é estimulada, o IkB é fosforilado pelo complexo IkB quinase, que inclui o NEMO (modulador essencial de NF-kB ou IKKg). Isto permite a dissociação do IkB e translocação de NF-kB para o núcleo para ativação da transcrição genética [ver Figura 1].
Os genes envolvidos na imunidade positivamente regulados pelo NF-kB incluem as citocinas pró-inflamatórias (IL-1, IL-6 e TNF-a), quimiocinas, moléculas de adesão, peptídeos antimicrobianos, genes determinantes de troca de classes de imunoglobulina, óxido nítrico sintetase induzível e fatores de transcrição indutores de interferons tipo I (IRF3, IRF7). Em adição, os genes envolvidos na diferenciação de estruturas ectodérmicas (pele, cabelo e dentes) são ativados pelo NF-kB.46
A deficiência de NEMO resulta em displasia ectodérmica ligada ao X com imunodeficiência e é causada por mutações hipomórficas no gene NEMO (IKBKG) localizado no cromossomo X. Estas mutações resultam no comprometimento da fosforilação e degradação do inibidor de NF-kB, o IkB. Em consequência, o NF-kB fica capturado no citoplasma e não consegue translocar para o núcleo. As mutações null (perda total de função) no gene IKBKG são letais no pré-natal em bebês do sexo masculino, e causam incontinência pigmentar em bebês do sexo feminino.
As mutações de ganho de função envolvendo IkB (gene IKBA ), inibidor do NF-kB, localizadas no cromossomo 14 podem resultar em uma forma autossômica dominante (AD) de displasia ectodérmica com imunodeficiência. Esta mutação resulta no comprometimento da fosforilação de IkB e impede sua degradação, intensificando a função inibitória dessa proteína sobre o NF-kB.
Diagnóstico. Os pacientes com deficiência de NEMO e mutação em IKBA sofrem de infecções bacterianas sinopulmonares por organismos encapsulados, bem como de infecções invasivas (sepse, abscessos cutâneos e meningite) por S. pneumoniae, Haemophilus influenzae, Klebsiella, Salmonella e Pseudomonas. Também têm sido relatadas infecções graves pelo vírus do herpes (vírus do herpes simples [HSV] e CMV). Outras infecções virais relatadas nestes pacientes são a infecção por adenovírus, infecção pelo papilomavírus humano (HPV) e molusco contagioso. Os pacientes também apresentam suscetibilidade aumentada a infecções micobacterianas. Também há relatos de pneumonia por P. jiroveci. Devido ao papel do NF-kB na diferenciação ectodérmica, os pacientes tipicamente apresentam displasia ectodérmica (dentes cônicos, cabelos finos e escassos, e ausência de glândulas sudoríparas). Os achados clínicos adicionais relatados em um subgrupo de pacientes incluem a osteopetrose com linfedema e enteropatia inflamatória.47 Os achados imunológicos encontrados nos pacientes incluem comprometimento da resposta de anticorpo específica, baixa citotoxicidade de célula NK e produção diminuída de citocinas inflamatórias após a estimulação de TLR. Alguns pacientes apresentam níveis elevados de IgM e IgA. Os números dos principais subgrupos de linfócitos e a proliferação de células T frequentemente estão normais.47 O diagnóstico é confirmado com o sequenciamento dos genes IKBKG ou IKBA.
Tratamento e prognóstico. Os pacientes com deficiência de NEMO e mutação de IKBA devem receber terapia de reposição de imunoglobulina para diminuir a frequência de infecções invasivas. A vacinação com a vacina do bacilo Calmette-Guérin (BCG) é contraindicada. Tipicamente, é feita a prescrição de antibióticos profiláticos contra micobactéria e outras infecções. A sobrevida é variável e depende da gravidade dos defeitos. O transplante de células-tronco tem sido realizado em um pequeno número de pacientes com deficiência de NEMO. Entretanto, esta terapia tem sido associada a taxas significativas de morbidade e mortalidade, e seu uso deve ser considerado com cautela.48
As deficiências de TLR3 (AD) e UNC-93B (AR) são dois defeitos inatos que têm sido clinicamente associados a uma estreita suscetibilidade à encefalite por HSV.
Genética e patogênese. TLR3 reconhece o dsRNA produzido por vírus contendo RNA e DNA, durante a replicação. A sinalização do TLR3 se dá por uma via independente de MyD88 e leva à indução de interferons tipo I (a e b). Os interferons tipo I desencadeiam a transcrição de proteínas antivirais, aumentam a apresentação de antígeno viral por moléculas do complexo principal de histocompatibilidade (MHC) de classe I, e ativam células NK.
A UNC-93B é uma proteína do retículo endoplasmático que atua no transporte de TLR7 e TLR9 para os endossomos. A sinalização por estes TLRs leva à indução de interferons antivirais do tipo I.
Diagnóstico. Até o presente, foram relatados três casos de pacientes com deficiência de TLR3. Dois pacientes apresentaram somente meningoencefalite por HSV. Um paciente japonês apresentou encefalopatia associada à influenza. Foram relatados dois pacientes com deficiência de UNC-93B que sofriam meningoencefalite por HSV, mas não desenvolveram outras infecções incomuns. Os exames convencionais para imunidade humoral e imunidade celular resultaram normais para todos os pacientes. Pacientes com deficiência de TLR3 têm menor indução de interferon tipo I após a estimulação com agonista de TLR3. Entretanto, os testes comercialmente disponíveis para função de TLR resultarão normais. Pacientes com deficiência de UNC-93B apresentam menor indução de interferon tipo I após a estimulação por agonistas de TLR7, TLR8 e TLR9. O sequenciamento dos genes TLR3 ou UNC93B pode confirmar o diagnóstico.49–51 Casos adicionais raros de pacientes com outras mutações genéticas têm sido identificados.
Tratamento e prognóstico. A encefalite associada a infecções por HSV deve ser tratada de forma agressiva com aciclovir. A adição de interferon-a pode proporcionar benefício terapêutico no tratamento de infecções por HSV agudas e graves. Devido ao pequeno número de pacientes com deficiência de TLR3 e UNC-93B relatado até hoje, o tratamento ideal e o prognóstico para esses pacientes é indeterminado.
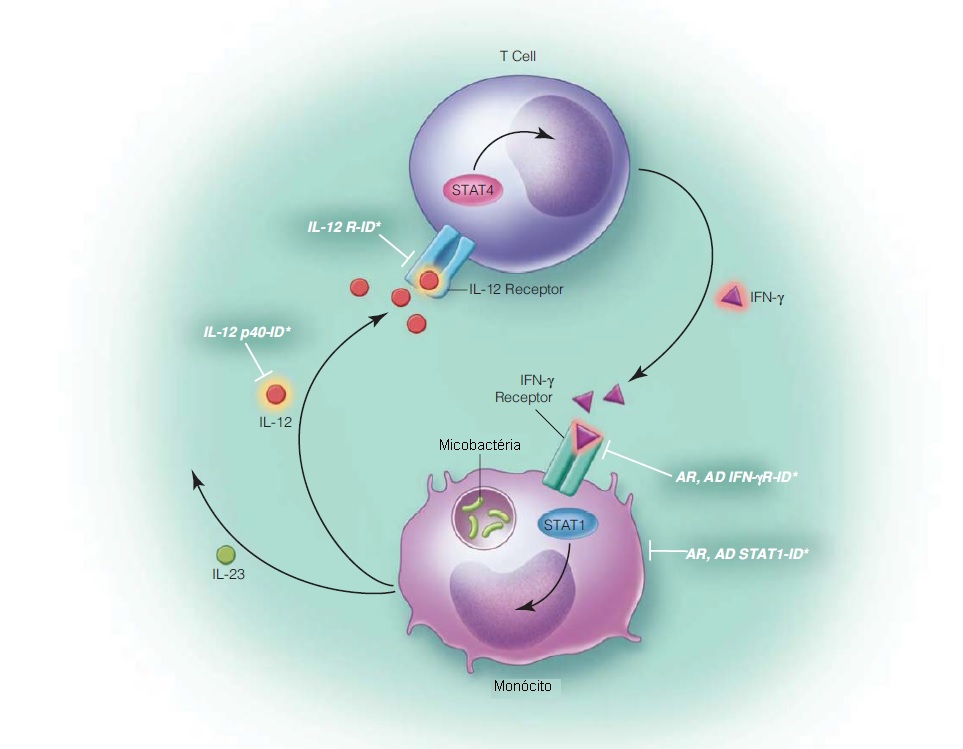
A sinalização pela via IL-12/interferon-g é essencial à ativação dos fagócitos para controlar as infecções causadas por patógenos intracelulares, como micobactéria e salmonela. Macrófagos infectados com micobactéria produzem IL-12, que estimula células T auxiliares de tipo 1 (Th1) e células NK a produzirem interferon-g. O interferon-g, então, ativa os macrófagos para induzir killing das micobactérias e salmonelas (esta transdução de sinal ocorre via STAT1) [ver Figura 2].
Foi relatado que os defeitos envolvendo essa via de sinalização causam suscetibilidade mendeliana a doenças micobacterianas. Estas doenças são caracterizadas pela acentuada suscetibilidade à micobactéria e à salmonela.
Genética e patogênese. Foram descritas ambas as formas, AR e AD, de deficiência de receptor de interferon-g 1 e 2. A deficiência de receptor de interferon-g AR (completa) é causada por mutações envolvendo o domínio extracelular do receptor de interferon-g e resulta na ausência completa de expressão do receptor na superfície celular. A deficiência de receptor de interferon-g AD (parcial) é causada por truncamentos heterozigotos do domínio citoplasmático do receptor de interferon-g 1. Isto resulta no acúmulo de moléculas de receptor não funcional na superfície celular, as quais são incapazes de realizar a transdução de sinal. Também foram relatadas mutações AD envolvendo o receptor de interferon-g 2.52
Diagnóstico. Pacientes com deficiência AR de receptor de interferon-g 1 e 2 apresentam infecções graves por micobactérias e Salmonella, na infância ou início da infância. As infecções tipicamente falham em formar granulomas micobacterianos bem circunscritos. A administração da vacina BCG tipicamente resulta em infecção disseminada.53 Pacientes com deficiência AD de receptor de interferon-g 1 desenvolvem uma forma mais branda da doença e, no final da infância ou na adolescência, apresentam infecção disseminada ou localizada por BCG, infecção por micobactéria pulmonar não tuberculosa (MNT), histoplasmose ou salmonelose. A osteomielite multifocal por MNT é particularmente comum entre estes pacientes e é considerada uma das principais características da doença (79% dos pacientes incluídos em um estudo). Diferente dos pacientes com doença AR, estes pacientes conseguem formar granulomas micobacterianos.
O diagnóstico de deficiência AR de receptor de interferon-g é sugerido pela ausência completa do receptor de interferon-g na superfície dos linfócitos, bem como pelo comprometimento da fosforilação de STAT1 em resposta à sinalização pelo interferon-g. O diagnóstico de deficiência AD de receptor de interferon-g é sugerido por um aumento de 3-5 vezes da expressão do receptor de interferon-g 1 em monócitos. Apesar do número aumentado de receptores de superfície, a fosforilação de STAT1 após a estimulação pelo interferon-g é mínima ou indetectável. O sequenciamento dos genes IFNGR1 e IFNGR2 pode confirmar o diagnóstico.
Tratamento e prognóstico. Os pacientes requerem tratamento agressivo das infecções com agentes antimicobacterianos. Na deficiência AR de receptor de interferon-g, a terapia de reposição com suplementação de interferon-g não traz benefícios, dada a total ausência de receptores de interferon-g na superfície celular. Em contraste, pacientes com deficiência AD de interferon-g conseguem responder a doses maiores de terapia com interferon-g. O transplante de células-tronco hematopoiéticas tem sido realizado com sucesso em um pequeno número de pacientes com deficiência AR de interferon-g, empregando doadores totalmente compatíveis com condicionamento mieloablativo. Entretanto, a doença micobacteriana preexistente estava associada a uma alta mortalidade.54,55 Este tratamento não é indicado para as formas mais brandas ADs da doença.
Genética e patogênese. Foram relatadas mutações ARs no gene do receptor b1 da IL-12. As mutações ocorrem no domínio extracelular do receptor, gerando códons de parada precoces e ausência de expressão da proteína na superfície da célula T. sem a sinalização da IL-12, a secreção de interferon-g pelas células T é bastante comprometida.
Diagnóstico. Os pacientes apresentam infecções disseminadas por micobactérias e Salmonella no início da infância. Os pacientes também desenvolvem infecção disseminada após receberem vacina BCG. Os granulomas são bem contidos e bem organizados. Não há expressão de receptor 1 de IL-12 na superfície das células T e há comprometimento da fosforilação de STAT4 em resposta à estimulação pela IL-12. É possível confirmar o diagnóstico por meio do sequenciamento do gene codificador do receptor b1 da IL-12.
Tratamento e prognóstico. Os pacientes requerem tratamento agressivo das infecções com agentes antimicobacterianos. Como a expressão do receptor de interferon-g e a sinalização subsequente permanecem intactas, a terapia com interferon-g pode ser benéfica para pacientes que falham em melhorar apenas com a terapia antimicobacteriana. Em um estudo internacional envolvendo 141 pacientes com deficiência de receptor b1 de IL-12, observou-se uma mortalidade de 30% associada à média da idade num período de seguimento de 12,7 anos (faixa de 0,5 a 46,4 anos).56
Genética e patogênese. A IL-12 é composta por duas subunidades, p35 e p40, codificadas pelos genes IL-12A e IL-12B, respectivamente. Pacientes com mutações ARs em IL12B têm subunidades p40 de IL-12 indetectáveis.
Diagnóstico. As complicações infecciosas são similares para os pacientes com deficiência de receptor 1 de IL-12 (infecção por BCG, infecções por MNT e sepse por Salmonella). O diagnóstico é sugerido pela ausência de níveis detectáveis de p40 de IL-12.
Tratamento e prognóstico. O espectro clínico pode ser bastante variável, oscilando de infecções micobacterianas fatais no início da vida a indivíduos que apresentam infecções micobacterianas localizadas em fases posteriores da vida. O fenótipo clínico geral é o de uma deficiência mais branda do que completa de receptor de interferon-g.57 O tratamento agressivo das infecções micobacterianas continua sendo a base da terapia. Assim como na deficiência do receptor 1 da IL-12, devido ao fato de a sinalização via receptor de interferon-g permanecer intacta, a terapia com interferon-g pode ser benéfica para pacientes que falham em melhora apenas com a terapia antimicobacteriana.
Figura 2. Representação esquemática da via de sinalização da interleucina-12 (IL-12)/interferon-g (IFN-g). A infecção micobacteriana envolve a secreção de IL-12 por monócitos, que estimula as células T a produzirem interferon-g. O interferon-g se liga ao seu receptor e isto resulta em transdução de sinal por STAT1 e na ativação do macrófago para indução do killing das micobactérias. Os defeitos desta via de sinalização são indicados na ilustração. AD = autossômico dominante; AR = autossômico recessivo; ID = imunodeficiência.
STAT1 é um transdutor de sinal essencial à via do interferon-g (antimicobacteriano) e do interferon-a/b (antiviral). A sinalização do interferon-g resulta em fosforilação e criação de um homodímero de STAT1 conhecido como fator ativador-g (GAF). A sinalização do interferon-a/b leva à formação de um homodímero de STAT1, STAT2 e p48 conhecido como fator gênico estimulado por interferon 3 (ISGF3). Ambas as formas, AD e AR, de deficiência de STAT1 têm sido descritas.
Genética e patogênese. Na deficiência AD (parcial) de STAT1, há comprometimento da dimerização de STAT1 e da formação de GAF que se seguem à sinalização pelo interferon-g, contudo a formação de ISGF3 após a sinalização por interferon-a/b é poupada. Desta forma, a imunidade antimicobacteriana é comprometida, mas a imunidade antiviral permanece intacta. Em contraste, a deficiência AR (completa) de STAT1 resulta em comprometimento significativo de ambas as vias de sinaliza, do interferon-g e do interferon-a/b, sendo observadas infecções micobacterianas e virais. Um terceiro fenótipo, associado a mutações ativadoras e candidíase mucocutânea, não é abordado aqui.
Diagnóstico. A deficiência AD de STAT1 é caracterizada de forma marcante por um fenótipo relativamente brando com suscetibilidade aumentada a infecções micobacterianas (BCG, MNT), consequente ao comprometimento da imunidade mediada pelo interferon-g.58 Em contraste, a deficiência AR de STAT1 causa uma forma grave de doença marcada por infecções micobacterianas e virais (HSV) potencialmente fatais no início da vida, em consequência do comprometimento da imunidade mediada por interferon-g e interferon-a/b.58
Pacientes com deficiência AD de STAT1 têm proteína STAT1 detectável e respostas mediadas por interferon-g diminuídas. Pacientes com deficiência AR de STAT1 não têm expressão detectável da proteína STAT1 e apresentam respostas acentuadamente reduzidas ao interferon-g e ao interferon a/b. O diagnóstico é confirmado pelo sequenciamento do gene STAT1.
Tratamento e prognóstico. Pacientes com deficiência de STAT1, tanto AD como AR, requerem tratamento agressivo das complicações infecciosas agudas. Todos os três casos relatados de deficiência AR de STAT1 têm se mostrado fatais durante a infância. Em contraste, pacientes com deficiência AD de STAT1 têm sobrevivido além da infância.58,59
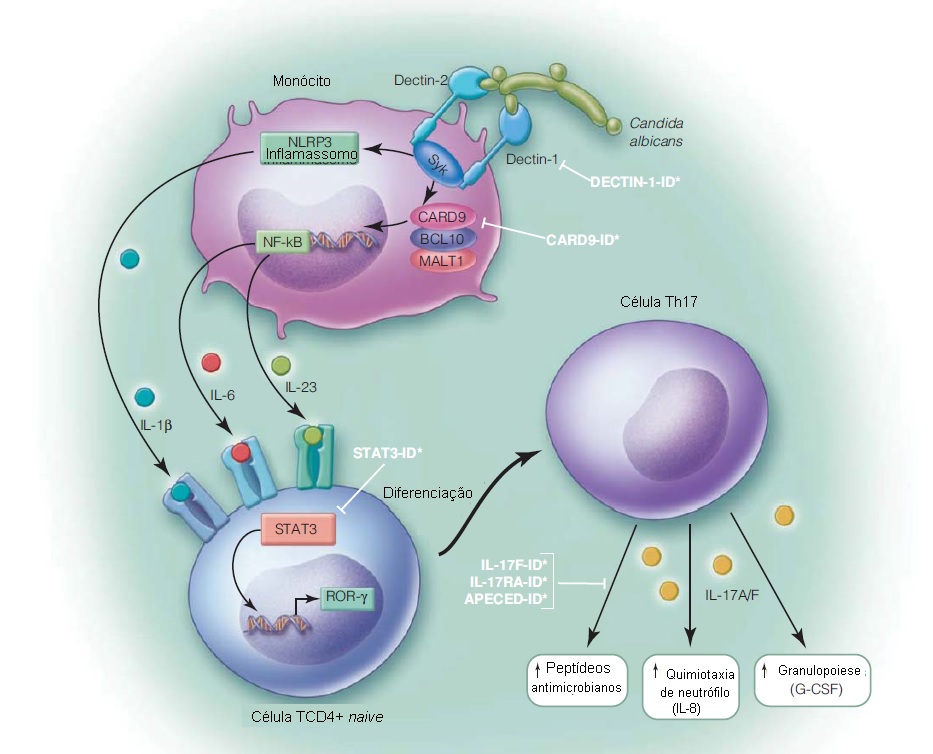
As células Th17 constituem um subgrupo de células T auxiliares secretoras de IL-17. A imunidade mediada por Th17 tem emergido como uma via de sinalização crítica, responsável pelo controle de infecções fúngicas e bacterianas. A diferenciação da célula T naive em células Th17 é estimulada pelas citocinas IL-1b, IL-6 e IL-23, que sinalizam via STAT3.60 A IL-17 secretada pelas células Th17 estimula a granulopoiese via indução de fator estimulador de colônias de granulócitos (G-CSF), recruta neutrófilos para o sítio de infecção via indução de IL-8, e estimula a produção de peptídeos antimicrobianos (p. ex., b-defensinas).61 A importância desta via [ver Figura 3] é destacada pelos defeitos imunes primários, com o comprometimento da imunidade mediada por Th17 caracterizado por infecções fúngicas recorrentes na pele, mucosa e unhas (candidíase mucocutânea crônica [CMC]). Alguns defeitos da imunidade mediada por Th17, como a síndrome da hiper-IgE e a deficiência de IL-17RA, também são caracterizados por infecções bacterianas recorrentes (mais comumente, S. aureus).
Genética e patogênese. Dectina-1 e dectina-2 são PRRs fúngicos expressos em macrófagos, neutrófilos e células dendríticas, que reconhecem b-glucana, um componente da parede celular de Candida albicans. A ligação da b-glucana à dectina-1 e à dectina-2 resulta em sinalização intracelular via proteína adaptadora CARD9, para ativação de NF-kB e do inflamassomo NLRP3, com indução da produção de citocinas pró-inflamatórias (TNF-a, IL-1b, IL-6 e IL-23). IL-1b, IL-6, e IL-23 são requeridas para induzir diferenciação da célula T naive em células Th17, as quais são essenciais à imunidade antifúngica.62
Uma mutação nonsense homozigota em dectina-1 (Y238X) foi descrita em três irmãs de uma família que sofria de CMC, na ausência de outras complicações infecciosas. A mutação resultou na incapacidade do receptor dectina-1 de se ligar à b-glucana e iniciar a produção de citocinas pró-inflamatórias. Os pais eram heterozigotos para a mutação e sofriam de infecções fúngicas mais brandas.63 Entretanto, é preciso notar que indivíduos homozigotos e heterozigotos para dectina-1 Y238X sadios também têm sido descobertos, sugerindo que esta mutação pode ser um fator de risco, em vez de gene causador verdadeiro de CMC.62 Uma mutação nonsense homozigota em CARD9 (Q295X) foi descrita em quatro pacientes de uma família consanguínea que sofria de CMC. A mutação resultou na ausência de expressão de CARD9 e em baixos números de células Th17.64
Diagnóstico. As deficiências de dectina-1 e CARD9 devem ser consideradas em casos de indivíduos com CMC, particularmente se houver padrão de herança AR. O sequenciamento genético pode confirmar o diagnóstico.
Tratamento e prognóstico. Os pacientes requerem tratamento das infecções agudas com agentes antifúngicos apropriados. A profilaxia antifúngica diária pode ser necessária para redução da frequência das infecções.
A deficiência de STAT3 é determinada pela clássica mutação associada ao distúrbio chamado síndrome de Job na literatura antiga.
Genética e patogênese. Mutações no gene STAT3 causam uma forma AD de síndrome da hiper-IgE. A estimulação de células T naive com IL-1b, IL-6 e IL-23 leva à sinalização via STAT3 e indução do fator de transcrição linhagem-específico RORgt, resultando na diferenciação em células Th17.60 A imunidade mediada por Th17 comprometida na deficiência de STAT3 resulta em suscetibilidade aumentada a patógenos fúngicos e bacterianos.
Diagnóstico. Pacientes com deficiência de STAT3 sofrem de candidíase, abscessos cutâneos recorrentes, pneumonia com formação de pneumatocele e dermatite eczematosa. S. aureus é o organismo bacteriano mais comum encontrado pelos pacientes. Na deficiência de STAT3, os achados somáticos incluem aspectos faciais distintivos (nariz amplo, olhos fundos, queixo e testa proeminente), atraso da troca dos dentes primários, fraturas patológicas, escoliose e hipermotilidade articular.
Os achados laboratoriais incluem IgE elevada e eosinofilia. Os níveis totais de imunoglobulina estão normais ou elevados, porém os pacientes podem apresentar respostas precárias às imunizações com proteína e polissacarídeo. O killing do neutrófilo é poupado, mas a quimiotaxia de neutrófilos é comprometida. O número de células Th17 está acentuadamente diminuído. O sequenciamento do gene STAT3 pode confirmar o diagnóstico.
Tratamento e prognóstico. A profilaxia antifúngica pode ser útil para pacientes com candidíase crônica e a profilaxia antibiótica com trimetoprima-sulfametoxazol é útil para prevenir infecções estafilocócicas. Pacientes com deficiência de anticorpo específica podem ser beneficiados pela terapia de reposição de imunoglobulina.
Figura 3. Representação esquemática da resposta imune do tipo T auxiliar 17 (Th17). A ligação de dectina-1 e dectina-2 à Candida albicans resulta em sinalização via Syk e ativação do inflamassomo NLRP, além de secreção interleucina-1b (IL-1b). A sinalização via Syk e o complexo CARD9/BCL10/MALT1 leva à ativação do fator nuclear-kB (NF-kB) e à secreção de IL-6 e IL-23. A ligação das citocinas IL-1b, IL-6 e IL-23 e a sinalização via STAT3 resultam na diferenciação das células T naive em células Th17. A IL-17 secretada pelas células Th17 induz produção de peptídeo antimicrobiano, granulopoiese e quimiotaxia de neutrófilos. Os defeitos nesta via de sinalização estão indicados na ilustração (omitidos na figura: deficiência de DOCK8 e mutações de ganho funcional em STAT1). PEACDE = poliendocrinopatia autoimune com candidíase e distrofia ectodérmica; G-CSF = fator estimulador de colônia de granulócitos; ID = imunodeficiência.
Genética e patogênese. A deficiência de dedicador de citocinese 8 (DOCK8) resulta em uma forma AR de síndrome de hiper-IgE, embora as manifestações infecciosas sejam mais profundas. Similarmente aos pacientes com deficiência de STAT3, há comprometimento da diferenciação de Th17. Entretanto, o mecanismo exato de comprometimento da imunidade mediada por Th17 é indeterminado. DOCK8 é membro da família de fatores de troca de nucleotídeo guanina relacionada ao DOCK180, envolvida na ativação das Rho guanosina trifosfatases e na iniciação da reorganização citoesquelética. Este processo é requerido para o desempenho de algumas funções, incluindo a migração celular, formação de imunossinapse, fagocitose e endocitose/exocitose.65
Diagnóstico. Pacientes com deficiência de DOCK8 sofrem de candidíase, abscessos cutâneos bacterianos e pneumonia. Os pacientes também sofrem de infecções cutâneas virais graves por HSV, molusco contagioso, varicela-zoster e HPV. A doença atópica grave, incluindo asma, eczema, alergias alimentares e alergias ambientais, é comum. Diferentemente dos indivíduos com deficiência de STAT3, os pacientes com deficiência de DOCK8 não apresentam anormalidades somáticas, como aparência facial grosseira, retardo da troca de dentes primários e fraturas patológicas. Esses pacientes apresentam risco aumentado de desenvolvimento de malignidades, como carcinoma de células escamosas e linfoma.65
Entre os achados laboratoriais, estão IgE elevada e eosinofilia. Os níveis totais de IgM e IgA podem estar diminuídos, porém os níveis de IgG estão normais ou aumentados. É possível que os pacientes apresentem resposta precária a antígenos proteicos e polissacarídicos de vacinas. A linfopenia de células T e o comprometimento da ativação da célula T frequentemente estão presentes. O número de células Th17 está significativamente diminuído. O sequenciamento do gene DOCK8 ou a hibridização genômica comparativa podem confirmar o diagnóstico.
Tratamento e prognóstico. A deficiência de DOCK8 é uma imunodeficiência combinada que pode estar associada a taxas significativas de morbidade e mortalidade. A profilaxia antifúngica pode ser útil para pacientes com candidíase, sendo que a profilaxia antibiótica é útil para prevenir infecções bacterianas. Pacientes com deficiência de anticorpo específica podem ser beneficiados pela terapia de reposição de imunoglobulina. O transplante de célula-tronco hematopoiética frequentemente é recomendado para pacientes com deficiência de DOCK8.65
Genética e patogênese. A importância da imunidade mediada por Th17 na defesa antifúngica é destacada pela descoberta de duas imunodeficiências primárias novas resultantes do comprometimento da sinalização da IL-17 (mutações ADs em IL-17F e mutações ARs no receptor de IL-17 [IL-17RA]). Uma mutação heterozigota em IL-17F foi descrita em uma família multicomplexa oriunda da Argentina que apresentava padrão de herança dominante de CMC. Foi constatado que a mutação missense no gene IL17F comprometia a ligação de IL-17F ao receptor da IL-17.66
A deficiência de IL-17RA foi relatada em um paciente cujos pais eram cossanguíneos, como consequência de uma mutação nonsense homozigota no gene IL17RA. A mutação resultou em um códon de parada precoce e isto causou ausência total de expressão do receptor de IL-17 na superfície celular. O paciente sofria de infecções cutâneas por C. albicans e S. aureus.66
Diagnóstico. A deficiência de IL-17F e IL-17RA deve ser considerada em casos de pacientes com CMC e padrão de herança AD e AR, respectivamente. O único paciente relatado com deficiência de IL-17RA ,também, sofria de infecções cutâneas estafilocócicas. O sequenciamento dos genes IL17F ou IL17RA pode confirmar o diagnóstico.
Tratamento e prognóstico. A profilaxia antifúngica pode ser útil para pacientes com fenótipo unicamente de CMC. A profilaxia antibiótica com trimetoprima-sulfametoxazol é útil para prevenção de infecções estafilocócicas.
Genética e patogênese. STAT1 é um transdutor de sinal essencial às vias do interferon-g e do interferon-a/b. As mutações de perda de função em STAT1 podem resultar em suscetibilidade aumentada a infecções micobacterianas e virais. As citocinas essenciais que sinalizam via STAT1 também atuam inibindo a diferenciação em Th17. As mutações AD de ganho de função em STAT1 têm sido associadas à CMC. As mutações missense ocorrem no domínio espiralado do gene STAT1 e resultam em diminuição da desfosforilação de STAT1 ativada. Isto leva ao aumento do acúmulo de STAT1 fosforilada no núcleo e à expressão excessiva de citocinas como interferon a/b e IL-27, que inibem a diferenciação em Th17. Adicionalmente, há afastamento da ativação de STAT3 induzida por IL-6/IL-21 e aproximação da ativação de STAT1 induzida por IL-6/IL-21, comprometendo ainda mais a imunidade mediada por Th17.67,68
Diagnóstico. Este diagnóstico deve ser considerado em casos de pacientes com padrão de herança AD de CMC.
Tratamento e prognóstico. Os pacientes requerem tratamento das infecções agudas com agentes antifúngicos apropriados. A profilaxia antifúngica diária pode ser benéfica para diminuir a frequência de infecções.
Genética e patogênese. A poliendocrinopatia autoimune com candidíase e distrofia ectodérmica (PEACDE) é um distúrbio AR de imunodesregulação. Também é conhecida como síndrome poliendócrina autoimune tipo I (SPA-I).
A PEACDE é causada por mutações no gene AIRE. AIRE está expresso de modo proeminente em células epiteliais tímicas medulares. No timo, AIRE induz expressão ectópica de autoantígenos que, normalmente, seriam restritos aos tecidos periféricos. Isto permite a seleção negativa de células T autorreativas e a indução de tolerância central. A ausência de expressão de AIRE permite que células T autorreativas escapem da seleção tímica negativa. Estas células então persistem e causam autoimunidade em múltiplos órgãos na periferia.69
Foram relatados alguns mecanismos de comprometimento da imunidade antifúngica na PEACDE. Primeiramente, tem sido demonstrado que os pacientes com PEACDE apresentam níveis elevados de anticorpos neutralizadores circulantes dirigidos contra citocinas Th17 (IL-17 e IL-22).70,71 Em segundo lugar, a proteína AIRE aparentemente está envolvida na via de sinalização da dectina-1, formando complexos com CARD9 e Syk. Em pacientes com PEACDE, a ligação da dectina-1 resultou em diminuição da produção de citocinas pró-inflamatórias.72
Diagnóstico. A PEACDE é caracterizada por CMC e poliendocrinopatia (mais frequentemente insuficiência suprarrenal e hipoparatireoidismo). Os pacientes aparentemente não apresentam suscetibilidade aumentada a outros tipos de infecções. Outras possíveis endocrinopatias adicionais incluem insuficiência gonadal, tireoidopatia e diabetes melito dependente de insulina. As manifestações não endócrinas incluem hipoplasia de esmalte, distrofia ungueal, alopécia, má absorção e vitiligo.
O diagnóstico de PEACDE deve ser considerado em pacientes que exibem a tríade de CMC, hipoparatireoidismo e insuficiência suprarrenal. Tipicamente, a CMC surge no início da infância, seguida de hipoparatireoidismo e insuficiência suprarrenal em fases posteriores da vida. O diagnóstico pode ser confirmado por sequenciamento do gene AIRE.
Tratamento e prognóstico. Os pacientes tipicamente requerem terapia antifúngica para CMC, reposição de cortisol para a insuficiência suprarrenal e suplementação de cálcio/vitamina D para o hipoparatireoidismo. Outros distúrbios endócrinos são tratados do mesmo modo como seriam tratados em outros indivíduos quaisquer.
Os autores não mantêm relações comerciais com os fabricantes de produtos ou prestadores de serviços mencionados neste capítulo.
Agradecimentos
Figuras 1-3 – Christine Kenney.
1.Conley ME, Rohrer J. The spectrum of mutations in Btk that cause X-linked agammaglobulinemia. Clin Immunol Immunopathol 1995;76(3 Pt 2):S192–7.
2.Lederman HM, Winkelstein JA. X-linked agammaglobulinemia: an analysis of 96 patients. Medicine (Baltimore) 1985;64:145–56.
3.Winkelstein JA, Marino MC, Lederman HM, et al. X-linkedagammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) 2006;85:193–202.
4.Farrar JE, Rohrer J, Conley ME. Neutropenia in X-linkedagammaglobulinemia. Clin Immunol Immunopathol 1996;81:271–6.
5.Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr 2002;141:566–71.
6.Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 2005;94(5 Suppl 1): S1–63.
7.Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immune deficiency: a meta-analysis of clinical studies. Clin Immunol2010;137:21–30.
8.Quartier P, Debre M, De Blic J, et al. Early and prolonged intravenous immunoglobulin replacement therapy in child-hood agammaglobulinemia: a retrospective survey of 31 patients. J Pediatr 1999;134:589–96.
9.Berger M. Subcutaneous immunoglobulin replacement in primary immunodeficiencies. Clin Immunol 2004;112:1–7.
10.Plebani A, Soresina A, Rondelli R, et al. Clinical, immunological, and molecular analysis in a large cohort of patients withX-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol 2002;104:221–30.
11.Notarangelo LD, Peitsch MC, Abrahamsen TG, et al. CD40lbase: a database of CD40L gene mutations causing X-linkedhyper-IgM syndrome. Immunol Today 1996;17: 511–6.
12.Hayward AR, Levy J, Facchetti F, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys withX-linked immunodeficiency with hyper-IgM. J Immunol1997;158:977–83.
13.Winkelstein JA, Marino MC, Ochs H, et al. The X-linkedhyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82:373–84.
14.Quartier P, Bustamante J, Sanal O, et al. Clinical, immuno- logic and genetic analysis of 29 patients with autosomal recessivehyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol 2004;110: 22–9.
15.Orange JS, Glessner JT, Resnick E, et al. Genome-wideassociation identifies diverse causes of common variable immune deficiency. J Allergy Clin Immunol 2011;127:1360–7.
16.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999;92:34–48.
17.Glocker E, Ehl S, Grimbacher B. Common variable immu- nodeficiency in children. Curr Opin Pediatr 2007;19:685– 92.
18.Chapel H, Lucas M, Lee M, et al. Common variable immu- nodeficiency disorders: division into distinct clinical pheno- types. Blood 2008;112:277–86.
19.Clark JA, Callicoat PA, Brenner NA, et al. Selective IgA deficiency in blood donors. Am J Clin Pathol 1983;80: 210–3.
20.Briere F, Bridon JM, Chevet D, et al. Interleukin 10 induces B lymphocytes from IgA-deficient patients to secrete IgA. J Clin Invest 1994;94:97–104.
21.Wells JV, McNally MP, King MA. Selective IgA deficiency in Australian blood donors. Aust N Z J Med 1980;10:410–3.
22.Ropars C, Muller A, Paint N, et al. Large scale detection of IgA deficient blood donors. J Immunol Methods 1982;54: 183–9.
23.Vyas GN, Perkins HA, Fudenberg HH. Anaphylactoid transfusion reactions associated with anti-IgA. Lancet 1968;2:312–5.
24.Dorsey MJ, Orange JS. Impaired specific antibody repsonse and increased B-cell population in transient hypogamma-globulinemia of infancy. Ann Allergy Asthma Immunol2006;97:590–5.
25.Cano F, Mayo DR, Ballow M. Absent specific viral anti-bodies in patients with transient hypogammaglobulinemia of infancy. J Allergy Clin Immunol 1990;85:510–3.
26.Buckley RH, Schiff RI, Schiff SE, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr 1997;130:378–87.
27.Gossage DL, Buckley RH. Prevalence of lymphocytopenia in severe combined immunodeficiency. N Engl J Med 1990;323:1422–3.
28.Palmer K, Green TD, Roberts JL, et al. Unusual clinical and immunologic manifestations of transplacentally acquired maternal T cells in severe combined immunodeficiency. J Allergy Clin Immunol 2007;120:423–8.
29.Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol 2008;122:1082–6.
30.Rosen FS. Severe combined immunodeficiency: a pediatric emergency. J Pediatr 1997;130:345–6.
31.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood 2002;99:872–8.
32.Bertrand Y, Landais P, Friedrich W, et al. Influence of severe combined immunodeficiency phenotype on the outcome of HLA non-identical, T-cell-depleted bone marrow trans- plantation: a retrospective European survey from the European Group for Bone Marrow Transplantation and the European Society for Immunodeficiency. J Pediatr 1999;134: 740–8.
33.Lischner HW, Huff DS. T-cell deficiency in DiGeorge syndrome. Birth Defects Orig Article Ser 1975;11:16–21.
34.Markert ML, Devlin BH, Alexieff MJ, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood 2007;109:4539–47.
35.Lindsay EA, Vitelli F, Su H, et al. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001;410:97–101.
36.Jawad AF, McDonald-McGinn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr 2001;139:715–23.
37.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet 2007;370:1443–52.
38.Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 1997;34:798–804.
39.Schwartz K. WASPbase: a database of WAS- and XLT- causing mutations. Immunol Today 1996;17:496–502.
40.Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopo- dium formation by a WASP-related actin-depolymerizingprotein N-WASP. Nature 1998;391:93–6.
41.Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 1994;125:876–85.
42.Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrichsyndrome. Curr Opin Hematol 2008;15:30–6.
43.Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al.Long-term outcome following hematopoietic stem-celltransplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood2008;111:439–45.
44.Kumagai Y, Akira S. Identification and functions of patternrecognition receptors. J Allergy Clin Immunol 2010;125:985–92.
45.Picard C, von Bernuth H, Ghandil P, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 2010;89:403–25.
46.Puel A, Picard C, Ku CL, et al. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol2004;16:34–41.
47.Orange JS, Jain A, Ballas ZK, et al. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol2004;113:725–33.
48.Fish JD, Duerst RE, Gelfand EW, et al. Challenges in the use of allogeneic hematopoietic SCT for ectodermal dysplasia with immune deficiency. Bone Marrow Transplant 2009;43: 217–21.
49.Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007;317:1522–7.
50.Hidaka F, Matsuo S, Muta T, et al. A missense mutation of theToll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol 2006;119:188–94.
51.Casrouge A, Zhang SY, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science2006;314:308–12.
52.Al-Muhsen S, Casanova JL. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol 2008;122:1043 –51; quiz 1052–3.
53.Dorman SE, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet 2004;364:2113–21.
54.Roesler J, Horwitz ME, Picard C, et al. Hematopoietic stem cell transplantation for complete IFN-gamma receptor 1 deficiency: a multi-institutional survey. J Pediatr 2004;145: 806–12.
55.Moilanen P, Korppi M, Hovi L, et al. Successful hematopoi- etic stem cell transplantation from an unrelated donor in a child with interferon gamma receptor deficiency. Pediatr Infect Dis J2009;28:658–60.
56.de Beaucoudrey L, Samarina A, Bustamante J, et al. Revisit- ing human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 2010;89:381–402.
57.Picard C, Fieschi C, Altare F, et al. Inherited interleukin-12deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet 2002;70: 336–48.
58.Dupuis S, Dargemont C, Fieschi C, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 2001;293:300–3.
59.Dupuis S, Jouanguy E, Al-Hajjar S, et al. Impaired response tointerferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 2003;33:388–91.
60.McDonald DR. TH17 deficiency in human disease. J Allergy Clin Immunol 2012;129:1429–35.
61.Gaffen SL. An overview of IL-17 function and signaling. Cytokine 2008;43:402–7.
62.Engelhardt KR, Grimbacher B. Mendelian traits causing susceptibility to mucocutaneous fungal infections in human subjects. J Allergy Clin Immunol 2012;129:294–305.
63.Ferwerda B, Ferwerda G, Plantinga TS, et al. Human dectin- 1 deficiency and mucocutaneous fungal infections. N Engl J Med2009;361:1760–7.
64.Glocker EO, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 2009;361:1727–35.
65.Su HC. Dedicator of cytokinesis 8 (DOCK8) deficiency. Curr Opin Allergy Clin Immunol 2010;10:515–20.
66.Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of inter-leukin-17 immunity. Science 2011;332:65–8.
67.van de Veerdonk FL, Plantinga TS, Hoischen A, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 2011;365:54–61.
68.Liu L, Okada S, Kong XF, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011;208: 1635–48.
69.Capalbo D, Giardino G, Martino LD, et al. Genetic basis of altered central tolerance and autoimmune diseases: a lesson from AIRE mutations. Int Rev Immunol 2012;31:344–62.
70.Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 2010;207:299–308.
71.Puel A, Dofinger R, Natividad A, et al. Autoantibodies againstIL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendo- crine syndrome type I. J Exp Med 2010;207:291–7.
72.Pedroza LA, Kumar V, Sanborn KB, et al. Autoimmune regulator (AIRE) contributes to dectin-1-induced TNF- alpha production and complexes with caspase recruitment domain-containing protein 9 (CARD9), spleen tyrosine kinase (syk), and dectin-1. J Allergy Clin Immunol 2012;129: 464–72,472.e1–3.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.