(Carregando Índice)... (Carregando Índice)... |
Última revisão: 25/11/2016
Comentários de assinantes: 0
Epilepsia e transtornos relacionados
BARBARA DWORETZKY, MD
JONG WOO LEE, MD, PHD*
|
Artigo original: Dworetzky, B, MD. Lee, JW, PHD. Epilepsy and related disorders. SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
NEUROLOGIA
|
Epilepsia e transtornos relacionados |
Barbara Dworetzky, MD, e Jong Woo Lee, MD, PhD*
|
*Os autores e os editores agradecem as contribuições dos autores da edição anterior, Giridhar P. Kalamangalam, MD, DPhil, e Jeremy D. Slater, MD, pelos vídeos e pelo desenvolvimento e redação de seções deste artigo. |
|
As informações financeiras são apresentadas ao final deste artigo, antes das referências. |
Definição da doença
Epilepsia é um transtorno cerebral crônico que se caracteriza pela presença de convulsões recorrentes não provocadas. Convulsão é uma mudança rápida de comportamento acompanhada por descargas elétricas no cérebro. Muitos pacientes que se apresentam com uma primeira convulsão se surpreendem ao perceber que se trata de um evento bastante comum.
Qualquer agente desencadeador de uma convulsão reversível ou evitável como o álcool, por exemplo, é um argumento contra a epilepsia subjacente e, por conseguinte, não justifica a administração de tratamento farmacológico.
Epidemiologia
Em torno de 65 milhões de pessoas em todo o mundo sofrem de epilepsia. Nos EUA e nos países em desenvolvimento, a incidência anual é de, aproximadamente, 50 a cada 100 mil indivíduos por ano, sendo que a prevalência é 10 vezes mais elevada. Essas estimativas são ainda maiores nos países cuja renda é mais baixa.
Há algumas evidências de taxas de incidência mais elevadas na população negra,2 embora não existam dados sobre as diferenças raciais e de gênero dos portadores da doença. A elevação nas taxas se relaciona ao fato de que a epilepsia não é uma doença única, mas, sim, um sintoma de disfunção cerebral produzida por uma grande variedade de causas que diferem de acordo com a idade dos pacientes. Entretanto, em dois terços dos casos, não se consegue encontrar uma causa específica.
A prevalência e a incidência cumulativa de epilepsia aumentam com a idade, sendo que os picos máximos ocorrem em idosos e em pessoas muito jovens. As convulsões são ainda mais comuns, ocorrendo em até 10% da população, na maior parte das vezes devido a convulsões febris ou à abstinência de álcool, o que demonstra, claramente, que a maioria das pessoas com novo início de convulsões não evolui para epilepsia.
Etiologia
Os esquemas atuais de classificação discriminam os tipos de convulsão e as síndromes epiléticas. O primeiro sistema amplamente adotado para classificar convulsões epiléticas foi publicado em 1970 por Gastaut.3 Desde então, foram feitas diversas revisões, obtendo-se outra compreensão, bem como novas técnicas para estudar os fenômenos convulsivos.
A classificação atual de convulsões epiléticas foi aprovada em 1981 pela International League Against Epilepsy (ILAE),4 e se fundamenta nas características clínicas da convulsão e nas anormalidades eletrencefalográficas ictais e interictais. Essa classificação foi revisada em 1985,5 com modificações posteriores em 19896 e, mais recentemente, em 2010.7
Classificação das convulsões epiléticas
As convulsões epiléticas são classificadas, de forma ampla, em dois grupos:
convulsões focais (parciais), isto é, aquelas com início clínico ou eletrencefalográfico limitado a uma parte do cérebro;
convulsões generalizadas, isto é, aquelas sem nenhum foco discernível no início.
Observa-se que há, ainda, uma terceira categoria que não se enquadra em nenhuma dessas duas. O Quadro 1 apresenta, com mais detalhes, os tipos de convulsões epiléticas.
Quadro 1
|
CLASSIFICAÇÃO INTERNACIONAL DE CONVULSÕES EPILÉTICAS | ||
|
Convulsões parciais (focais ou locais) |
Convulsões parciais simples (sem alteração na consciência) |
Sinais motores Sintomas somatossensoriais ou sensoriais especiais Sintomas ou sinais autonômicos Sintomas psíquicos
|
|
Convulsões parciais complexas (com alteração na consciência) |
Início parcial simples seguido de alterações na consciência. Alteração na consciência no início. Convulsões parciais simples que evoluem para convulsões generalizadas. Convulsões parciais complexas que evoluem para convulsões generalizadas. Convulsões parciais simples que evoluem para convulsões parciais complexas que, por sua vez, evoluem para convulsões generalizadas.
| |
|
Convulsões generalizadas (convulsivas ou não convulsivas) |
Convulsões de ausência
|
Típicas |
|
Atípicas | ||
|
Convulsões mioclônicas
| ||
|
Convulsões clônicas
| ||
|
Convulsões tônicas
| ||
|
Convulsões tônico-clônicas
| ||
|
Convulsões atônicas
| ||
|
Convulsões epiléticas não classificadas |
| |
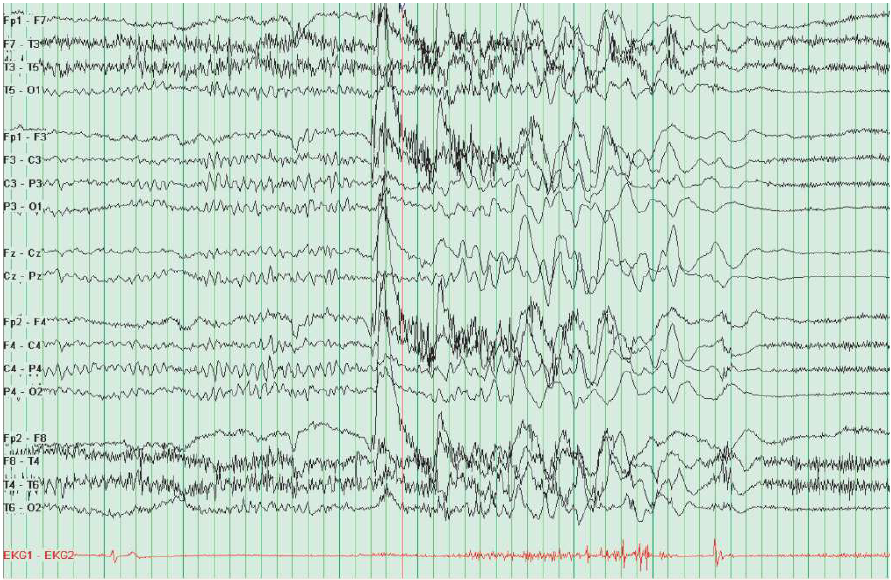
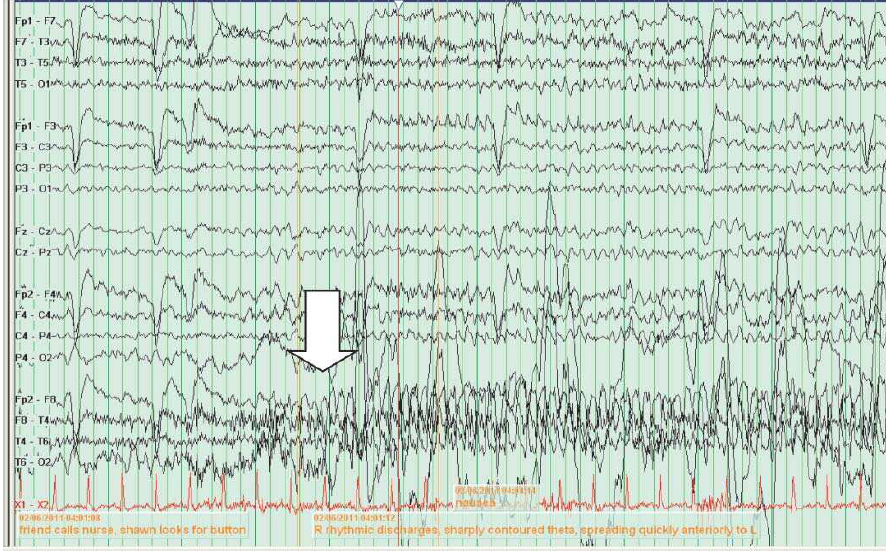
As convulsões focais ou parciais se subdividem em crises parciais simples (CPS), se não houver nenhuma alteração na consciência, e crises parciais complexas (CPC) se a consciência for afetada. Nas convulsões parciais, o eletrencefalograma (EEG) interictal mostra uma descarga focal sobre a região afetada, conforme ilustra a Figura 1.

Figura 1 – Observa-se uma descarga epilética temporal média esquerda durante o estágio II do sono nos EEGs de rotina.
EEGs: eletrencefalogramas.
Os sintomas de CPS dependem do sítio do córtex que for afetado. Por exemplo, as convulsões com origem no córtex occipital podem estar associadas a distúrbios visuais. Da mesma forma, aquelas com origem no córtex sensorial podem produzir parestesias na região correspondente do corpo.
Alucinações gustativas, sensações abdominais e sintomas autonômicos (náusea, vômito, palidez, rubor, diaforese e dilatação das pupilas) ocorrem nas situações em que há envolvimento das estruturas límbicas ou da ínsula. Se o envolvimento abrange apenas uma pequena quantidade do córtex, é possível que o EEG seja normal durante uma CPS, embora se observem, com alguma frequência, alterações rítmicas localizadas (ver o Vídeo 1 [disponível on-line ou em CD]).
Ainda que permaneçam focais, essas convulsões poderão se disseminar para áreas contíguas do córtex, em uma espécie de “marcha”, ou poderão evoluir para CPC. Nessas circunstâncias, um alerta ou aura precede o início da alteração na consciência. Por outro lado, a consciência poderá se alterar a partir do início de uma convulsão. Embora o início ocorra em um hemisfério, as evidências indicam que as CPCs envolvem os dois hemisférios cerebrais.
Outras características clínicas das CPC incluem automatismos ou movimentos involuntários, que se classificam como automatismo oroalimentar, automatismo ambulatório ou automatismo gestural/manual. Os automatismos manuais se caracterizam por movimentos desajeitados, como tentar apanhar qualquer objeto que estiver ao alcance (ver o Vídeo 2 [disponível on-line ou em CD]).
Nos casos de CPC, que se originam nas regiões mesial-frontal ou órbito-frontal, observam-se movimentos de pedaladas ou quaisquer outros movimentos hipermotores. Nesse caso, o EEG pode ser normal e obscurecido pelo artefato em movimento (ver o Vídeo 3 [disponível on-line ou em CD]).
As convulsões generalizadas se subdividem em convulsões de ausência, mioclônicas, clônicas, tônicas e atônicas. As convulsões de ausência (anteriormente conhecidas por “pequeno mal”) se caracterizam pela suspensão súbita da atividade e da função mental, com duração de 5 a 15s, olhar fixo e, às vezes, movimentos involuntários das pálpebras (ver o Vídeo 4 [disponível on-line ou em CD]).
A anormalidade eletrencefalográfica é um padrão generalizado típico de picos e ondas de 3Hz. O indivíduo, normalmente, recupera a atividade que havia sido interrompida ao final da convulsão e não se lembra de nada. A atividade atípica, no EEG, das convulsões de ausência denomina-se “onda de picos baixos”, em geral de 2,5 a 3Hz (ver o Vídeo 5 [disponível on-line ou em CD]).
As convulsões mioclônicas se distinguem por contrações musculares rápidas, involuntárias, que se assemelham a um choque. Comumente, elas se limitam a determinados segmentos musculares, como os músculos flexores das extremidades superiores. A consciência é preservada e o EEG do couro cabeludo mostra uma descarga epileptiforme de alta amplitude, com pico máximo nas regiões frontocentrais (ver o Vídeo 6 [disponível on-line ou em CD]).
As convulsões atônicas se caracterizam pela perda súbita de tônus muscular, o que provoca quedas inesperadas. A perda de consciência é breve ou não há perda de consciência. A eletromiografia (EMG) concorrente confirma o diagnóstico com uma queda transitória de atividade (ver o Vídeo 7 [disponível on-line ou em CD]).
As convulsões tônico-clônicas generalizadas (CTCG), também conhecidas como “grande mal”, são os tipos mais comuns de convulsões generalizadas, e consistem de uma sequência ordenada de convulsões tônicas e clônicas. Quando precedidas por uma CPS ou CPC, são conhecidas por convulsões generalizadas secundárias.
A fase tônica, que é a primeira, se distingue por uma contração sustentada nas extremidades ou nos músculos axiais. Com frequência, nas situações em que a exalação de ar ocorre através da glote fechada, o paciente parece estar “gritando” e apresenta um aspecto apneico. As mordidas laterais na língua costumam ser comuns durante essa fase.
A fase seguinte é a clônica, a qual se caracteriza por uma série de contrações e relaxamentos musculares repetitivos com duração de 1min. A incontinência urinária é uma ocorrência provável durante essa fase. Após a convulsão, há um período de ausência profunda de resposta, cuja duração varia de 10 a 30min, em que o EEG praticamente desaparece, sendo bastante lento (ver o Vídeo 8 [disponível on-line ou em CD]).
Classificação das síndromes epiléticas
A síndrome epilética é um “transtorno epilético que se caracteriza pela presença de um grupo de sinais e sintomas que em geral ocorrem concomitantemente,”4 conforme se pode ver no Quadro 2.
Quadro 2
|
CLASSIFICAÇÃO INTERNACIONAL DE EPILEPSIAS, SÍNDROMES EPILÉTICAS E TRANSTORNOS CONVULSIVOS RELACIONADOS | ||
|
Epilepsias relacionadas à localização (focais ou parciais) |
Idiopáticas
|
Epilepsia infantil benigna com picos centrotemporais |
|
Epilepsia infantil com paroxismos occipitais | ||
|
Epilepsia primária da leitura | ||
|
Sintomáticas |
Epilepsia no lobo temporal | |
|
Epilepsia no lobo frontal | ||
|
Epilepsia no lobo parietal | ||
|
Epilepsia no lobo occipital | ||
|
Epilepsia crônica progressiva parcial contínua | ||
|
Criptogênicas (presume-se que sejam sintomáticas, embora a causa seja desconhecida) |
Epilepsia no lobo temporal | |
|
Epilepsia no lobo frontal | ||
|
Epilepsia no lobo parietal | ||
|
Epilepsia no lobo occipital | ||
|
Epilepsia crônica progressiva parcial contínua | ||
|
Epilepsias generalizadas |
Idiopáticas |
Convulsões neonatais benignas (familiares e não familiares) |
|
Epilepsia mioclônica benigna da infância | ||
|
Epilepsia de ausência na infância | ||
|
Epilepsia mioclônica juvenil | ||
|
Epilepsia com convulsões tônico-clônicas generalizadas ao despertar | ||
|
Sintomáticas |
Etiologia inespecífica: encefalopatia mioclônica precoce, encefalopatia epilética infantil precoce com surtos supressivos | |
|
Outras epilepsias generalizadas sintomáticas | ||
|
Criptogênicas (espasmos infantis) |
Síndrome de Lennox-Gastaut | |
|
Epilepsia com convulsões mioclônico-astáticas | ||
|
Epilepsia com convulsões mioclônico-astáticas | ||
|
Epilepsia com ausências mioclônicas | ||
|
Síndromes específicas (estados de doença em que as convulsões são típicas ou predominantes na apresentação) | ||
|
Epilepsias indeterminadas (características generalizadas ou focais) |
Convulsões neonatais |
|
|
Epilepsia mioclônica grave da infância |
| |
|
Epilepsia com picos contínuos de onda durante o sono com ondas lentas |
| |
|
Afasia epilética adquirida (SLK) |
| |
|
Outras epilepsias indeterminadas sem características generalizadas ou focais inequívocas |
| |
|
Síndromes especiais |
Convulsões relacionadas a situações |
Convulsões febris |
|
Convulsões isoladas ou EE | ||
|
Convulsões causadas por algum evento agudo ou tóxico |
| |
EE: estado epilético; SLK: síndrome de Landau-Kleffner.
Observa-se que, para definir as síndromes epiléticas, são levados em conta diversos fatores como manifestações clínicas, classificações dos tipos de convulsão, EEG e descobertas em imagens neurológicas.
Nas epilepsias relacionadas à localização, as convulsões possuem origem definida e localizada, tendo origem em inúmeros focos. As epilepsias generalizadas são definidas como transtornos epiléticos com convulsões generalizadas. Cada categoria se subdivide nos subgrupos idiopáticos, sintomáticos e criptogênicos.
As epilepsias idiopáticas estão associadas a convulsões epiléticas sem quaisquer outras anormalidades neurológicas ou estruturais no cérebro. Esse tipo de epilepsia tende a apresentar predisposição genética e início relacionado à idade, sendo, em geral, benigna.
As epilepsias sintomáticas são secundárias a alguma anormalidade cerebral específica, de natureza genética (por exemplo, esclerose tuberosa) ou adquirida (por exemplo, trauma, acidente vascular cerebral [AVC], infecção). As epilepsias sintomáticas relacionadas à localização mais comuns talvez sejam localizadas anatomicamente pelo EEG ou pelas técnicas de geração de imagens. Esse tipo inclui epilepsias nos lobos temporal, frontal, parietal e occipital.
Uma terceira categoria, o grupo criptogênico, se refere a epilepsias não idiopáticas, nas quais não é possível detectar a condição subjacente causadora da epilepsia sintomática.
Classificação revisada
Em 2010, a ILAE apresentou uma proposta de revisão na classificação de convulsões e de epilepsias.7 A alteração mais significativa foi a eliminação da distinção entre CPS e CPC. A ILAE incentivou a descrição com embasamento no grau de comprometimento. A seguir, são descritas algumas propostas de alteração nos termos:
os termos “causas idiopáticas”, “causas sintomáticas” e “causas criptogênicas”, que vinham sendo utilizados para descrever a etiologia subjacente das epilepsias, foram substituídos por “causas genéticas”, “causas estruturais ou metabólicas” e “causas desconhecidas”;
os termos referentes a síndromes epiléticas “relacionadas à localização” e “generalizadas” foram substituídos por descrições que refletem melhor a fisiopatologia subjacente: “síndromes eletrolíticas”, que se fundamentam em sinais ou sintomas clínicos, em EEGs e em outras características, definem um transtorno diferenciador e reconhecível; “constelações” referem-se a outros transtornos que não se caracterizam como síndromes, mas representam, clinicamente, transtornos diferenciadores embasados em lesões específicas ou em outras causas – “estruturais ou metabólicas”; “angioma” e “causas desconhecidas”.
Até o momento, não ficou claro o suficiente se essa classificação foi adotada amplamente sob o ponto de vista clínico.
Principais síndromes epiléticas
Síndromes neonatais
Entre as síndromes neonatais, tratadas neste artigo, estão: convulsão neonatal familiar benigna; convulsão neonatal benigna; encefalopatia epilética infantil precoce; encefalopatia mioclônica precoce.
Convulsão neonatal familiar benigna
Esse tipo de síndrome é um diagnóstico de exclusão em neonatos com histórico familiar conhecido. Em geral, o nascimento ocorre dentro do período normal de gestação, e os bebês são neurologicamente normais, sendo que as convulsões costumam ocorrer no segundo ou terceiro dia de vida, embora possam se estender até o terceiro mês.
O EEG interictal é normal, não apresentando anormalidade metabólica. Ainda que a remissão da maior parte dessas convulsões ocorra em torno de 6 semanas, é indicado o uso de fenobarbital ou ácido valproico. A taxa de risco de novos episódios epiléticos varia de 10 a 15%.
Convulsão neonatal benigna
Esse tipo de síndrome, também conhecida como “ataque do quinto dia”, se distingue pela ocorrência de convulsões clônicas ou apneicas em torno do quinto dia de vida em lactentes normais sob o ponto de vista neurológico. O EEG interictal pode ser anormal, com picos focais ou multifocais. Ainda não há evidências quanto à necessidade de se aplicar algum tratamento, e não há risco de episódios epiléticos futuros.
Encefalopatia epilética infantil precoce
Essa condição, também conhecida como síndrome de Ohtahara, faz parte do espectro de encefalopatias epiléticas progressivas dependentes da idade, que poderá incluir a síndrome de West ou a síndrome de Lennox-Gastaut. De maneira geral, não se encontra nenhuma etiologia específica, embora seja comum a presença de lesões cerebrais como porencefalia ou heminegalencefalia. A encefalopatia epilética infantil precoce se caracteriza por convulsões clônicas durante o primeiro mês de vida.
O EEG pode revelar a presença de um padrão de supressão de surtos, com interveniência de surtos de alta tensão. Essa condição poderá progredir para a síndrome de West durante a primeira infância. Embora as convulsões sejam altamente resistentes e os prognósticos neurológicos não sejam muito bons, o tratamento à base de hormônio adrenocorticotrófico (ACTH) mostra-se parcialmente eficaz.
Encefalopatia mioclônica precoce
Embora sua causa seja desconhecida, essa síndrome está associada a erros metabólicos inatos, na maioria das vezes hiperglicemia não cetótica, acidúria proptônica ou acidemia D-glicérica. A maior parte dos infantes com esse tipo de transtorno tem convulsões durante o primeiro mês de vida. A mais comum delas é a mioclonia fragmentar errática, podendo ocorrer também outros tipos.
O EEG mostra um padrão de supressão de surto que poderá evoluir para hipsarritmia, a qual se caracteriza por alta tensão desorganizada que lentifica para descargas epiléticas intercaladas. As imagens neurológicas são comuns. Em geral, as respostas aos medicamentos antiepiléticos (em inglês, antiepileptic drugs [AEDs]), são baixas, sendo que muitos lactentes morrem na primeira infância ou na fase inicial da infância.
Síndromes da primeira infância e do início da infância
Entre as síndromes da primeira infância e do início da infância, tratadas neste artigo, estão: epilepsia parcial benigna da infância; epilepsia mioclônica benigna da infância; síndrome de West; epilepsia mioclônica grave da infância; epilepsia mioclônica grave da infância; convulsões controladas pela piridoxina.
Epilepsia parcial benigna da infância
Esse tipo de transtorno distingue-se pelas CPC durante o primeiro ano de vida em lactentes que seriam intactos sob o ponto de vista neurológico. Os estudos metabólicos e de imagens neurológicas são comuns. O EEG revela a presença de descargas ictais focais, porém é normal sob a ótica interictal. As convulsões são facilmente controláveis com medicamentos como carbamazepina, fenobarbital ou valproato.
Epilepsia mioclônica benigna da infância
As convulsões, nesse caso, consistem de reflexos mioclônicos maciços em pacientes que seriam intactos entre as idades de 5 meses a 5 anos. O EEG mostra descargas generalizadas, apresentando picos e ondas, quase sempre durante o sono. As convulsões são facilmente controláveis com valproato, sendo que as remissões são muito comuns no longo prazo.
Síndrome de West
A síndrome de West é um transtorno devastador que consiste da tríade formada por espasmos infantis, hipsarritmia e retardo mental. A maior parte dos pacientes apresenta algum transtorno estrutural subjacente como, por exemplo, esclerose tuberosa causada por algum transtorno neurocutâneo. Nesse tipo de condição, é também muito comum a presença de outras malformações corticais, infecções neonatais e distúrbios metabólicos, sendo que 15 a 30% dos casos são considerados de origem criptogênica.
As convulsões consistem de espasmos flexores simétricos ou assimétricos, com frequência em agrupamentos, durante a metade do primeiro ano de vida. O EEG se caracteriza pela presença de hipsarritmia: descargas assíncronas com picos e ondas lentas, contínuas e de alta amplitude, com um pano de fundo desorganizado.
Os tratamentos de escolha incluem o uso de ACTH ou vigabatrina. A dependência de piridoxina é uma causa rara dessa síndrome, embora seja recomendável fazer um teste dessa vitamina nas situações em que a etiologia não for muito clara. Em geral, o prognóstico, que depende da etiologia e da capacidade para controlar as convulsões, não é bom.
Epilepsia mioclônica grave da infância
Também conhecida por síndrome de Dravet, é um transtorno que se apresenta com convulsões clônicas unilaterais ou generalizadas ou com estado epilético (EE) durante o primeiro ano de vida, sendo, tipicamente, desencadeado por febre, a partir da qual o paciente costuma apresentar mioclonia errática ou generalizada, ausências atípicas e CPS ou CPC.
Nos momentos iniciais, o EEG interictal e o desenvolvimento neurológico poderão ser normais, porém, de maneira geral, deterioram progressivamente. As imagens neurológicas são comuns. Mesmo que as respostas aos AED sejam desapontadoras, o topiramato e o estiripentol foram eficazes em alguns pacientes. O uso de lamotrigina pode agravar a condição; portanto, deve ser evitado esse medicamento. Mutações sem sentido (nonsense) do gene SCN1a, que codifica a subunidade a do canal de sódio controlado por voltagem, foram identificadas em até 70% dos indivíduos.8
Convulsões controladas pela piridoxina
As convulsões controladas pela piridoxina são raras, apesar de serem reversíveis e associadas ao defeito no gene da glutamato carboxilase. As convulsões parciais de longa duração se apresentam na idade de 3 meses e, na realidade, podem ser avaliadas no útero. O lactente se torna irritável, agitado ou com vômitos antes das convulsões.
O tratamento à base de piridoxina interrompe, de forma imediata, as convulsões e é responsável pela melhora da atividade epileptiforme no EEG, embora seja comum a ocorrência de retardo mental e de leucodistrofia ou atrofia cerebral nas imagens por ressonância nuclear magnética (RNM). Apesar disso, não há necessidade de administrar agentes antiepiléticos convencionais.
Síndromes da fase final da infância e da adolescência
Entre as síndromes da fase final da infância e da adolescência, tratadas neste artigo, estão: epilepsia benigna da infância com picos centrotemporais; epilepsia occipital benigna da infância; epilepsia generalizada com convulsões febris adicionais; epilepsia de ausência; epilepsia mioclônica juvenil; epilepsia com convulsões do grande mal (grand mal) ao despertar; síndrome de Lennox-Gastaut; síndrome de Landau-Kleffner e picos e ondas contínuas lentas durante o sono; síndrome de Rasmussen.
Epilepsia benigna da infância com picos centrotemporais
Tipo mais comum de epilepsia que ocorre na infância, trata-se de um transtorno relacionado à localização, com forte predisposição familiar para convulsões febris ou afebris. As convulsões iniciam no período entre a primeira infância e antes da puberdade (mais comum entre as idades de 5 e 10 anos), e ocorrem sobretudo durante o sono. Essas convulsões parciais se caracterizam por fatores como parestesias faciais, sequestro da fala, sialorreia e sensação de asfixia, seguidos de convulsões tônicas ou clônicas que poderão se tornar generalizadas.
O desenvolvimento neurológico e as neuroimagens são normais. O EEG revela a presença de picos interictais típicos rolândicos/mesotemporais, os quais podem ser unilaterais ou bilaterais. Não há evidências sobre a real necessidade de se aplicar algum tipo de tratamento, sendo, portanto, indicada a prescrição de baixas doses de oxcarbazepina.
Epilepsia occipital benigna da infância
Conhecida como síndrome de Panayiotopoulos, ocorre entre as idades de 2 e 8 anos (tipicamente, na idade de 5 anos), embora existam também evidências de início tardio. As convulsões se caracterizam pelo desvio tônico do olhar, náusea/vômito e manifestações autonômicas (síncope, pupilas dilatadas), assim como sintomas visuais em uma minoria de indivíduos.
O EEG revela a presença de picos occipitais de alta amplitude. A carbamazepina é indicada nas situações em que as convulsões são frequentes. Em geral, a remissão ocorre dentro de um período de tempo que pode variar de 1 a 2 anos.
Epilepsia generalizada com convulsões febris adicionais
Nesse transtorno, diverso sob o ponto de vista fenotípico, os pacientes se apresentam com múltiplas convulsões febris após os 6 anos de idade, e com convulsões de ausência, mioclonia ou convulsões mioclônico-astáticas.
Não existe nenhum padrão eletrencefalográfico específico associado a esse caso. A herança é autossômica, tendo sido associada a vários genes. O tratamento é direcionado para o tipo de convulsão individual; os pacientes são neurologicamente intactos ou apresentam danos leves. De maneira geral, o prognóstico costuma ser bom.
Epilepsia de ausência
Quatro síndromes epiléticas importantes foram associadas às convulsões de ausência típicas: ausência infantil, ausência juvenil, ausência mioclônica e epilepsia mioclônica juvenil (EMJ). A epilepsia de ausência infantil (EAI) é a mais comum dessas condições e tem início entre os 4 e 8 anos de idade. As ausências duram entre 5 e 15s; associadas a movimentos involuntários dos olhos, podem ocorrer com uma frequência de centenas de vezes por dia. Esse tipo de ausência é precipitado pela hiperventilação.
O EEG mostra a presença de descargas pico-onda típicas de 3Hz. Embora a criança seja normal sob o ponto de vista neurológico, o mau desempenho escolar é muito comum antes do diagnóstico. O uso de etossuximida é o tratamento de escolha. Ainda que a remissão das convulsões seja uma grande expectativa, até um terço de pacientes continua tendo convulsões.
O início da epilepsia de ausência juvenil ocorre mais tarde, ou seja, mais perto da puberdade. A frequência das convulsões é mais baixa e o tempo de duração das ausências é mais longo. A maior parte dos pacientes tem episódios convulsivos generalizados, além das ausências, sendo que 15 a 25% poderão sofrer convulsões mioclônicas. Para o tratamento, é indicado o uso de valproato.
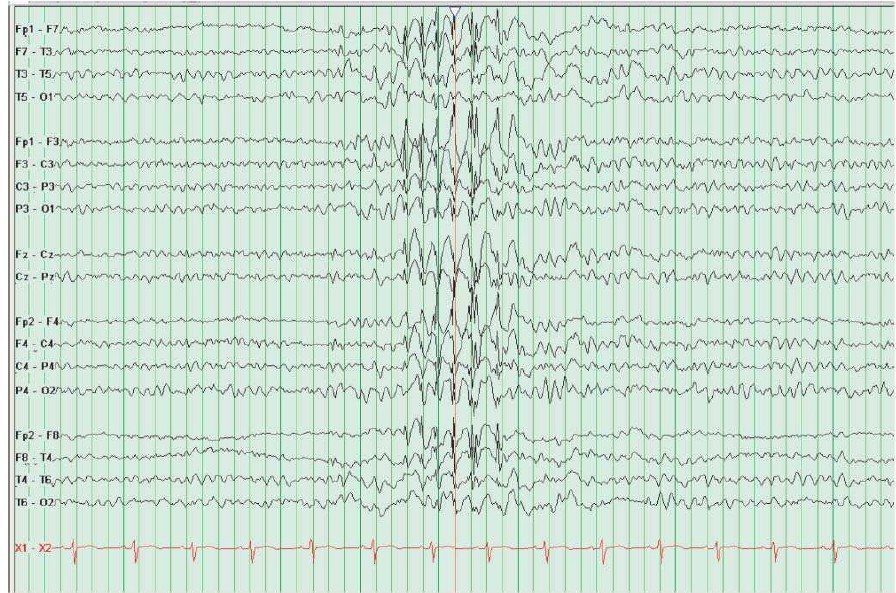
A epilepsia de ausência mioclônica é mais rara do que os transtornos supramencionados. As convulsões, e não as ausências, estão presentes na maior parte dos pacientes. Aproximadamente 50% deles apresentam retardo evolutivo; as convulsões são resistentes ao tratamento e progridem para a síndrome de Gastaut, conforme se pode ver na Figura 2.
Figura 2 – Observa-se uma explosão de picos de 3Hz e uma atividade de onda por 1,5s durante a sonolência (ao centro).
Epilepsia mioclônica juvenil
A EMJ é uma das síndromes epiléticas mais comuns. A idade de início ocorre entre os 12 e 18 anos, com início médio aos 14 anos de idade. As convulsões consistem de reflexos mioclônicos ao despertar, envolvendo principalmente os braços. Quase todos os pacientes têm CTCG, sendo que 30% deles apresentam o fenômeno de ausência. Costumam, também, ser extremamente sensíveis à privação do sono, ao álcool e à fotoestimulação.
Acredita-se que o nível de inteligência seja normal, embora os transtornos comportamentais e de personalidade sejam comuns. Estudos imagiológicos recentes revelam a presença de atrofia frontal e talâmica nas imagens neurológicas. O EEG revela padrões “rápidos” interictais de ondas com múltiplos picos de 5 a 6Hz. O uso de valproato é eficaz na maior parte dos casos. Além disso, recomenda-se um controle do estilo de vida para que os pacientes se livrem das convulsões.
Epilepsia com convulsões do grande mal (grand mal) ao despertar
Esse tipo de transtorno surge na adolescência e na faixa dos 20 anos; consiste de CTCGs que ocorrem logo após o despertar, podendo também se manifestar durante a noite. Além das convulsões generalizadas, os pacientes podem também ter convulsões de ausência e mioclônicas.
A qualidade do sono, nesse caso, é instável. O ECG mostra um pano de fundo interictal anormalmente lento com atividade pico-onda generalizada. O uso de valproato é o tratamento de escolha, e sua aplicação deve ser feita indefinidamente.
Síndrome de Lennox-Gastaut
A síndrome de Lennox-Gastaut é uma encefalopatia devastadora que ocorre na infância com múltiplos tipos de convulsões.
Apesar de haver uma etiologia identificada, na maior parte dos pacientes, como AVC perinatal, infecção, lesão na cabeça e síndrome de Down (SD), a síndrome de Lennox-Gastaut é criptogênica em 25% de casos.
Em geral, esse tipo de transtorno inicia entre as idades de 2 e 8 anos. Os tipos mais comuns são as convulsões tônicas, sobretudo na musculatura axial. Muitos especialistas consideram também a presença universal de ausências atípicas. Algumas vezes, os pacientes têm mioclonia ou convulsões atônicas.
O EEG revela um pano de fundo difusamente lento e desorganizado com anormalidades paroxísmicas que se intensificam durante o sono. As convulsões tônicas ictais consistem de um achatamento atípico no EEG ou de ritmos bilaterais rápidos. De maneira geral, os resultados dos tratamentos são desapontadores. Inicialmente, acreditava-se que o felbamato era eficaz em alguns pacientes.
O uso de rufinamida e de clobazam é uma indicação específica para esse tipo de transtorno. Alternativas como uso de lamotrigina, de valproato, calosotomia (corte do corpo caloso), estimulação do nervo vago (ENV), entre outras, são válidas; entretanto, o prognóstico não é bom, e muitos pacientes apresentam retardo evolutivo.
Síndrome de Landau-Kleffner e picos e ondas contínuas lentas durante o sono
A Síndrome de Landau-Kleffner (SLK) é uma aplasia epilética adquirida. Comumente, os pacientes portadores da doença se desenvolvem de forma normal antes de apresentarem uma regressão na linguagem receptiva por volta dos 3 aos 8 anos de idade. As convulsões ocorrem em 70% de pacientes e, em geral, trata-se de convulsões motoras parciais simples.
As descobertas eletrencefalográficas incluem picos bilaterais temporais independentes ou temporoparietais e ondas lentas, assim como ondas e picos generalizados que são ativados pelo sono. Ao final, a maior parte dos pacientes apresenta descargas bilaterais com picos e ondas na maior parte do sono e com movimentos oculares não rápidos (em inglês, non rapid eye movement [NREM]), consistente com SE eletrográfico de sono de ondas lentas. Os exames imagiológicos costumam ser comuns.
O controle das convulsões é relativamente fácil, e elas costumam remitir durante a vida adulta, em comparação com a regressão da linguagem, que é mais refratária aos tratamentos. O valproato é o medicamento de escolha. Terapias à base de clobazam, vigabatrina, felbamato, corticosteroides e tratamento cirúrgico com múltiplas transecções subpiais podem ser eficazes. Já os carbamazepínicos podem agravar as convulsões.
A presença contínua de picos e de ondas durante o sono lento é um transtorno relacionado que pode fazer parte do mesmo espectro da SLK; o início é mais tardio; o tratamento das convulsões é mais difícil; há perturbações eletrencefalográficas paroxísmicas graves com descargas e picos e ondas em 85% dos gráficos de sono; o foco é frontal e ocorrem deteriorações comportamentais e cognitivas.
Ao contrário de outros tipos de epilepsias no lobo frontal, não ocorrem convulsões tônicas. Diferentemente da SLK, as neuroimagens são, em geral, anormais; observa-se a presença de uma grande variedade de lesões evolutivas estruturais.
Síndrome de Rasmussen
Essa é uma doença hemisférica progressiva devastadora. Sua etiologia é extremamente controversa: as sugestões variam de distúrbio autoimune a etiologia viral. O início desse tipo de síndrome ocorre entre as idades de 1 e 10 anos. O sintoma inicial é uma convulsão parcial, observando-se a presença de epilepsia parcial contínua (EE motor focal simples) em 50% dos pacientes.
A incidência de fatores como hemiatrofia cerebral unilateral progressiva, hemiparesia espástica e retardo mental é bastante comum. As respostas das convulsões são muito baixas, e os tratamentos à base de agentes imunes (corticosteroides, globulina imune intravenosa [IVIg]) não têm eficácia. A hemisferectomia é o tratamento de escolha para impedir que doença progrida para o hemisfério contralateral.
Fisiopatologia
Os mecanismos celulares da hiperexcitabilidade neuronal resultam de uma interação complexa entre os canais iônicos controlados por voltagem, que determinam a excitabilidade das membranas neuronais, e os neurotransmissores que administram a transmissão sináptica excitatória e inibitória através dos canais iônicos controlados por ligantes ou receptores metabotrópicos mediados pela proteína G.
Os principais canais iônicos implicados na epilepsia incluem os canais de sódio, de potássio e de cálcio; cada um desses canais é alvo das terapias farmacológicas, sendo que as mutações em cada um deles influenciam as síndromes epiléticas. O glutamato é o principal neurotransmissor excitatório, cuja ação é mediada através dos receptores ionotrópicos: ácido a-amino-3-hidroxi-5-metil-4-isoxazol propiônico (AMPA), cainato e N-metil-D-aspartato (NMDA) – e através dos receptores do glutamato metabotrópico (mGluRs).
O ácido ?-aminobutírico (em inglês, ?-aminobutyric acid [Gaba]) é o principal neurotransmissor inibitório, que se liga aos receptores ionotrópicos Gabaa e Gabac e ao receptor do Gabab metabotrópico. Outros neurotransmissores que têm alguma implicação na epilepsia incluem a acetilcolina e a adenosina.
As alterações patológicas na epilepsia humana ocorrem, em geral, na formação hipocampal. O hipocampo é formado pelo intertravamento do corno de Ammon com o giro dentado. No interior do corno de Ammon, o hipocampo se subdivide em quatro regiões (CA1 a CA4). A esclerose hipocampal, característica patológica típica da epilepsia no lobo temporal mesial (ELTM), se caracteriza pela perda de neurônios, gliose e atrofia no subcampo CA1, bem como preservação relativa de CA2 e perda celular variável em CA3/4.9
Os substratos patológicos para epilepsia também foram associados a malformações corticais, as quais se classificam em malformações produzidas pela proliferação anormal ou apoptose, migração e organização cortical.10,11 A proliferação anormal poderá resultar em microcefalia, megaencefalia, hamartomas de esclerose tuberosa, tumores neuroepiteliais disembrioplásicos e gangliomas.
A migração anormal poderá causar lisencefalia e heterotopias. A lisencefalia é a manifestação de um córtex de quatro camadas anormalmente espesso e liso. As heterotopias consistem na presença de substância cinzenta em locais inapropriados. Com frequência, observam-se essas condições no revestimento das paredes ventriculares na heterotopia nodular periventricular. Com determinadas mutações genéticas, tem-se uma segunda camada cortical na substância branca (heterotopia da banda subcortical).
A organização anormal está associada a condições como polimicrogiria e displasias corticais. A polimicrogiria se caracteriza pela presença de múltiplos giros pequenos que, na maior parte dos casos, se localizam na região perisilviana.
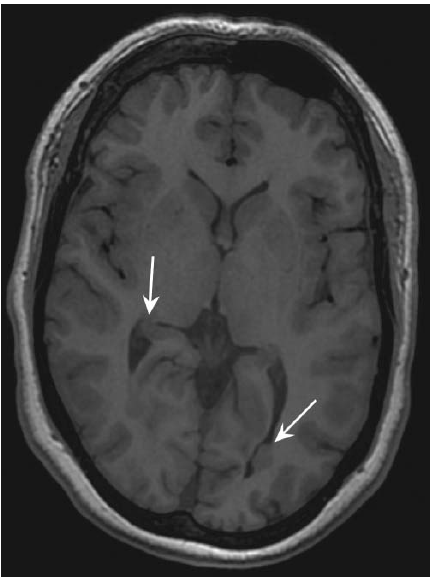
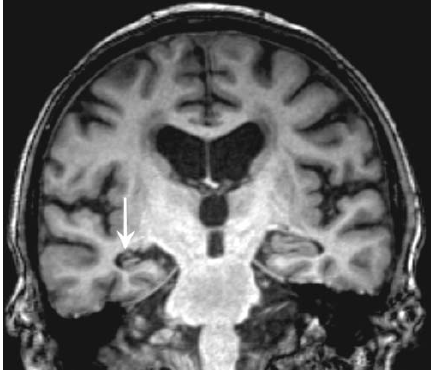
As displasias corticais são os fundamentos patológicos mais comuns da epilepsia focal, conforme mostra a Figura 3. Sob o ponto de vista histológico, as descobertas típicas das displasias corticais incluem células em forma de balão, que se caracterizam por um citoplasma eosinofílico e núcleos com localização excêntrica, bem como neurônios dismórficos anormalmente ampliados.
Figura 3 – As imagens por RNM revelam a presença de pequenas displasias corticais focais (setas) ou de heterotopias de substância cinzenta, responsável pela epilepsia parcial refratária.
RNM: ressonância nuclear magnética.
A epilepsia ainda é um transtorno altamente hereditário, seja em associação com outras anormalidades evolutivas, seja como um transtorno primário isolado. Com a primeira identificação do gene que serviu de base para uma epilepsia idiopática (epilepsia noturna autossômica dominante no lobo frontal), em 1995,12 identificou-se um número cada vez maior de mutações. Não chega a causar nenhuma surpresa o fato de que a maior parte das epilepsias idiopáticas seja formada por canalopatias.
A genética da epilepsia ainda é bastante complexa, excetuando-se as mutações mendelianas com um único gene, as quais são responsáveis por uma pequena minoria de eventos epiléticos. É possível que as mutações mendelianas com um único gene produzam heterogeneidades fenotípicas, ao passo que múltiplos genes poderão produzir o mesmo fenótipo clínico.13
Foram encontradas, por exemplo, três mutações genéticas do canal de sódio e duas mutações do receptor do Gaba para epilepsia com convulsões febris adicionais.14 A variação no número de cópias, refletindo a deleção ou a duplicação de vários alongamentos de DNA, comprovou ser um fator de risco para a epilepsia idiopática.15 Embora, até o momento, tenham sido obtidas poucas melhorias específicas com os cuidados clínicos, é provável que os avanços genéticos resultem em novos mecanismos para os alvos dos tratamentos.
O uso mais amplo dos testes genéticos aplicáveis é o rastreamento de marcadores do antígeno leucocitário humano (em inglês, human leukocyte antigen [HLA]) para hipersensibilidade aos medicamentos. Descobriu-se que, na população do sudeste asiático, a síndrome de Stevens-Johnson ou necrólise epidérmica tóxica ocorre quase que exclusivamente em pacientes portadores do alelo HLA-B*1502. O rastreamento é bastante eficaz para evitar a incidência dessa complicação potencialmente letal.16
Da mesma forma, embora não sejam estatisticamente representativas, algumas descobertas mostraram a presença do alelo HLA-A*3101 e da reação de hipersensibilidade induzida pela carbamazepina em europeus.17 O rastreamento do alelo HLA-B*1502 deve ser feito em todos os pacientes asiáticos antes de se iniciar a administração de carbamazepina.
Os relatórios iniciais indicavam que havia associação de um polimorfismo no gene transportador de medicamentos ABCB1.18 Entretanto, estudos posteriores que utilizaram grupos populacionais não conseguiram replicar essas descobertas.19 O papel dos genes na resistência aos medicamentos ainda não está totalmente esclarecido.
Diagnóstico
O diagnóstico de epilepsia pode se tornar muito difícil considerando que há uma grande variedade de sintomas que são facilmente confundidos com outras condições com apresentação paroxísmica. O primeiro passo é determinar se algum episódio se caracterizou como convulsão definitiva. Históricos cuidadosos são extremamente importantes para se fazer o diagnóstico de convulsão ou de epilepsia, e requerem a reunião de múltiplas fontes de dados sobre o evento.
As convulsões são eventos estereotipados repentinos que duram entre 15s e 3min. Todavia, de um modo geral, os pacientes não se lembram do que aconteceu no decurso desse tempo; apesar disso, devem ser questionados sobre a experiência que tiveram imediatamente antes do episódio, se estavam dormindo ou acordados quando a convulsão teve início e o que estavam fazendo antes do episódio.
Ser acometido por incontinência urinária e morder a língua são sinais úteis de convulsão nas situações em que o evento não tenha sido testemunhado, embora esses fatores não sejam definitivos, estejam eles presentes ou ausentes. Possíveis agentes desencadeadores como febre, privação do sono e abstinência de medicamentos ou de álcool são fundamentais para o diagnóstico, levando-se em consideração que as convulsões provocadas se desviam do curso do tratamento.
É imprescindível conversar com alguma testemunha que possa complementar o máximo possível as informações faltantes, possibilitando a diferenciação de qualquer imitação não convulsiva. Fatos como nível de consciência, tempo de duração do evento e quaisquer sintomas motores ou sensoriais ou autonômicos associados, assim como confusão pós-evento, devem ser questionados.
Os relatos menos úteis de testemunhas incluem declarações como “os olhos estavam virados para cima”, “ele desmaiou”, ou “era uma convulsão”. Qualquer perda de consciência, independentemente do motivo, poderá envolver todos esses sinais, sendo considerada, de forma errônea, como convulsão epilética.
O diagnóstico de epilepsia pode ocorrer no momento da primeira convulsão, considerando que ela permite reconhecer que devaneios recorrentes prévios não diagnosticados na realidade eram convulsões parciais. É fundamental questionar sobre quaisquer eventos recorrentes estereotipados anteriores, incluindo reflexos mioclônicos, levando-se em conta que, em geral, os pacientes não revelam esse tipo de informação de forma voluntária.
De todo modo, os pacientes ou os membros da família reconhecem apenas as convulsões generalizadas como convulsões reais. Mesmo após a obtenção de históricos detalhados, ainda não fica suficientemente claro se o paciente teve uma convulsão epilética. A existência de um irmão epilético na família aumenta a probabilidade de que o evento seja uma convulsão.
Além disso, históricos médicos de retardo evolutivo, traumatismo craniano (com perda de consciência), tumor no cérebro, AVC precedente (em especial, do tipo hemorrágico) e infecção na infância, como meningite ou encefalite, são fatores de risco fortemente associados ao desenvolvimento de epilepsia e, por conseguinte, poderão antecipar o tratamento após a primeira convulsão se nenhum outro fator desencadeador for descoberto nos históricos detalhados.20
O exame neurológico também é imprescindível para o diagnóstico. Os exames neurológicos focais aumentam a probabilidade de que o evento seja uma convulsão; entretanto, na maior parte das vezes, os exames físicos e neurológicos são normais. As descobertas feitas nos exames gerais, tais como erupção cutânea, febre ou rigidez no pescoço, sugerem a presença de uma etiologia infecciosa.
Mais importante ainda é o fato de que outros distúrbios no diagnóstico diferencial poderão ser excluídos ou incluídos através de exames com focos mais específicos, como a pressão arterial (PA) ortostática ou a manobra de Dix-Hallpike, os quais devem ser incluídos no histórico particular.
A avaliação da primeira convulsão deve incluir estudos metabólicos (eletrólitos, cálcio, magnésio, glicose e testes da função renal e hepática), rastreamento toxicológico do soro e da urina e um hemograma completo (em inglês, complete blood count [CBC]). A contagem de leucócitos (em inglês, white blood cells [WBC]) pode ser elevada após uma convulsão; como resultado, é necessário interpretá-la dentro de um contexto clínico.
A Academia Americana de Neurologia (AAN) publica parâmetros práticos para o gerenciamento de condições neurológicas. Esses parâmetros são resultado da opinião consensual de Quality Standards Subcommittee (QSS) específicos, englobando grupos de especialistas que divulgam recomendações gerais com embasamento na metanálise de evidências publicadas mais recentemente.
As recomendações são estratificadas nas categorias A, B, C e U, em ordem decrescente da força de cada evidência. As recomendações de nível A são respaldadas por mais de um teste randomizado com base na população, as de nível U se referem às evidências conflitantes ou à falta de evidências, enquanto as de níveis B e C são intermediárias. A ocorrência de convulsões provocadas depende da administração de muitos medicamentos. Na presença de sintomas e sinais de alguma infecção no sistema nervoso central, é importante fazer punção lombar em adultos imunocompetentes, em geral depois de algum procedimento imagiológico (Nível U).21
No caso de indivíduos imunocomprometidos, neonatos e pessoas mais velhas, os sinais típicos de infecção (como febre ou torcicolo) podem estar ausentes e, nessa hipótese, deve ser considerada a aplicação de punção lombar depois dos estudos de imagens nas situações em que a nova convulsão e as novas descobertas forem focais. Em crianças com idades entre 6 meses e 5 anos com alguma fonte identificável de febre e convulsões generalizadas sem complicações, pode ocorrer a síndrome de convulsões febris benignas, não havendo, portanto, necessidade de exames completos adicionais.
No departamento de emergências, o uso de tomografia computadorizada (TC) do cérebro, nos contextos de exames neurológicos focais, de convulsões com início focal ou de históricos predisponentes (Nível B), facilita a exclusão de hemorragia ou da necessidade aguda de intervenção.21 Entretanto, nos casos de adultos que tenham se recuperado completamente e cujos exames sejam normais, a TC pode ser substituída pela RNM, que é mais eficaz na descoberta da lesão responsável pela convulsão.
As visões coronais e os segmentos finos na RNM ajudam a identificar lesões temporais mediais, sendo que a maior parte dos centros que atendem aos pacientes epiléticos segue protocolos de RNM que aumentam a possibilidade de detectar lesões mais sutis; de maneira geral, não é necessário usar gadolínio nas situações em que a RNM sem contraste é negativa.
Embora estejam disponíveis em alguns centros, os estudos de resolução mais elevada como 3T e 7T não aumentam substancialmente a produtividade acima de 1,5T. A TC com contraste é uma opção nas situações em que a RNM permanecer indisponível por alguns dias ou mais. A EEG deverá ser aplicada o mais rápido possível após a ocorrência de convulsões em adultos (evidência de Nível B), nas situações em que houver grande chance de alguma anormalidade diagnóstica, embora os EEGs de rotina sejam normais em 50% de pacientes com diagnóstico definitivo de epilepsia.
Nos casos em que o EEG inicial for negativo, os testes encefalográficos adicionais poderão revelar a presença de alguma anormalidade epilética definitiva, caso ela exista, sem nenhuma produtividade logo após quatro estudos. Entretanto, alguns centros costumam programar EEGs ambulatoriais de 24 horas, após um primeiro EEG negativo, nas situações em que algum evento levantar suspeitas de convulsão. As descargas epiléticas definitivas em EEGs de adultos, todavia, são altamente específicas para o risco de convulsões, embora, em crianças, sejam menos específicas.
Os parâmetros das práticas atuais publicados pela AAN para investigações sobre as primeiras convulsões atribuem recomendação de Nível B para os EEGs e para as imagens neurológicas (TC ou RNM) e recomendação de Nível U para todos os outros tipos de investigação.22
Se as convulsões ocorrerem em pessoas com epilepsia conhecida, pode haver modificações na avaliação. A verificação da adesão ao regime de tratamento é o primeiro passo nas situações em que o paciente tenha retornado à linha de base sem nenhuma descoberta neurológica, sobretudo ao se observar o nível de um medicamento ou contar as pílulas no frasco ou na caixa de comprimidos – já que a não utilização adequada das doses prescritas é a principal razão das recorrências, o que não costuma ser lembrado no momento da convulsão.
Outras causas também deverão ser investigadas, mesmo nos casos de epilepsia conhecida, considerando que os fatores reversíveis poderiam ter sido mascarados por alterações nas doses dos medicamentos ou no tratamento. Nesse caso, a repetição de imagens do cérebro é uma hipótese a ser considerada quando há alterações inesperadas no tipo, frequência ou gravidade das convulsões sem uma causa óbvia ou nos casos em que os pacientes não tenham retornado à linha de base dentro do período de tempo usual.
Além disso, nas situações em que a cirurgia de ressecção do cérebro for o tratamento recomendado para a epilepsia, ou nos casos em que as varreduras iniciais do cérebro forem normais, não diagnósticas ou não estiverem à disposição para a equipe responsável pelo planejamento procedimental, torna-se essencial repetir o grupo de varreduras com resolução mais alta para qualquer planejamento cirúrgico.
Diagnóstico diferencial
Sob o ponto de vista paroxísmico, podem suceder diversos tipos de eventos transitórios que produzem alterações na consciência, na sensação e/ou outras funções neurológicas. O diagnóstico diferencial de convulsão epilética é bastante amplo, embora inclua síncope, enxaqueca, transtorno psiquiátrico (isto é, transtorno de conversão, ataque de pânico [AP], perturbação dissociativa), transtorno do movimento, transtorno do sono ou distúrbios metabólicos.
Sem dúvida, as imitações mais comuns são episódios de síncope, sobretudo eventos associados a movimentos convulsivos, enxaquecas e transtornos de conversão como as “convulsões” psicogênicas não epiléticas (em inglês, psycogenic nonepileptic seizures [PNESs]), anteriormente conhecidas por “pseudoconvulsões”.
O Quadro 3 mostra, de forma esquemática, o diagnóstico diferencial de convulsão.
Quadro 3
|
DIAGNÓSTICO DIFERENCIAL DE CONVULSÃO | |
|
Condições psiquiátricas |
Convulsão psicogênica não epilética |
|
Estado de fuga ou transtorno dissociativo | |
|
Ataque de pânico | |
|
Transtorno do déficit de atenção | |
|
Eventos cardiovasculares |
Arritmia |
|
Síncope/síncope convulsiva | |
|
Ataque isquêmico transitório/AVC | |
|
Enxaqueca |
|
|
Vertigem paroxísmica |
|
|
Transtorno motor |
Distonia cinesiogênica paroxísmica |
|
Tique ou mioclonia | |
|
Transtornos do sono |
Comportamento REM |
|
Terror noturno ou sonambulismo | |
|
Enurese | |
|
Narcolepsia | |
|
Despertar confusional | |
|
Movimentos periódicos dos membros durante o sono | |
|
Distúrbios metabólicos |
Intoxicação por medicamentos |
|
Hipoglicemia | |
|
Simulação de doença |
|
AVC: acidente vascular cerebral; REM: movimento rápido dos olhos.
Síncope
Os ataques de síncope são imitações comuns de convulsões epiléticas, os quais se manifestam particularmente em pacientes mais idosos. Em indivíduos hipertensos, fatores como doença arterial coronariana (DAC), dor torácica e histórico familiar de morte súbita devem ser avaliados para verificar a possibilidade de doença cardíaca e o risco de morte súbita.
A perda súbita de fluxo sanguíneo e de oxigênio para o cérebro é o mecanismo causativo da perda de tônus postural e chega a provocar “desmaios”. Normalmente, as pessoas recebem um alerta de sensação de desfalecimento ou de obscurecimento auditivo ou visual, e permanecem desligadas durante alguns segundos, com recuperação rápida se tiverem condições de deitar e permitir a perfusão no cérebro. Esses indivíduos poderão apresentar diaforese ou uma aparência pálida antes do ocorrido.
Os eventos ocorridos após um tempo prolongado na posição de pé ou sentado, depois de exercícios físicos ou em ambientes muito quentes, depois de tossir ou no quadro de dor nociva que se assemelha a um estímulo favorecem fortemente o diagnóstico de síncope, ao passo que, se forem seguidos de mordida na língua, incontinência urinária, cianose, giro da cabeça ou déjà-vu, favorecem o diagnóstico de convulsão epilética.
Com frequência, os eventos relacionados à síncope são acompanhados de movimentos “convulsivos” ou mioclônicos conhecidos por síncope “convulsiva”, ainda que não tenham origem epilética. Nos casos de síncope recorrente, os exames completos devem incluir ECG, monitoramento de Holter, radiografia do tórax, ecocardiografia e encaminhamento para um cardiologista.
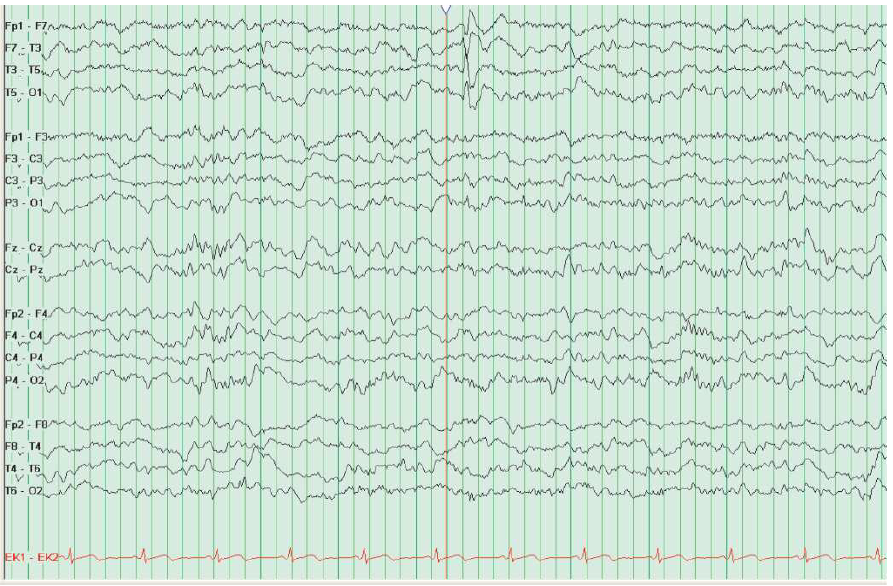
Os eventos de síncope capturados no EEG têm a aparência clássica com a cessação do exame, apresentando, em seguida, uma lentificação delta de alta voltagem seguida de supressão generalizada, conforme se pode ver na Figura 4.
Figura 4 – Este EEG foi feito em um paciente que havia tido síncope convulsiva após a colocação de uma linha intravenosa. Observa-se o complexo QRS simples no EEG, antes das ondas lentas de alta amplitude e do achatamento do gráfico. Não se trata de convulsão.
EEG: eletrencefalograma.
Enxaqueca
Enxaqueca é uma imitação comum de convulsão parcial. Observa-se, por exemplo, que, em geral, as convulsões occipitais são diagnosticadas incorretamente como enxaqueca. Além disso, o diagnóstico de enxaqueca pode ser confuso, em especial na ausência de cefaleia, nos casos de enxaqueca acefálgica ou com aura.
Sob a perspectiva do diagnóstico, as enxaquecas apresentam desenvolvimento mais lento, duram mais tempo e podem não se desenvolverem sob o ponto de vista anatômico. Condições como cefaleia, náusea, vômito e sensibilidade leve poderão ocorrer nos episódios convulsivos, embora tenham características mais marcantes de início e de distribuição que a enxaqueca, o que é muito útil para distinguir os sintomas. O EEG pode ser normal durante as convulsões parciais, desde que não ocorra alteração na consciência, de modo que não é precisamente um teste definidor.
Históricos pessoais e familiares de enxaquecas, assim como desencadeadores específicos como o consumo de chocolate, tiramina e abstinência de cafeína facilitam o diagnóstico, ainda que haja uma quantidade razoável de sobreposição dos dois distúrbios: enxaqueca e convulsões parciais.
Convulsões psicogênicas não epiléticas
As PNESs se assemelham às convulsões diagnosticadas em 20 a 30% dos pacientes encaminhados para centros epiléticos para tratamento de epilepsia incontrolável.23 Aproximadamente três quartos dos pacientes são mulheres, sendo que muitos desses indivíduos sofrem do transtorno do estresse pós-traumático (TEPT), incluindo abuso físico ou sexual na infância.
Normalmente, as PNESs são um transtorno de conversão ou um acesso dissociativo, e não simulação de doença. A distinção entre PNESs e convulsões epiléticas é determinante para um tratamento adequado, embora, em muitos casos, seja muito difícil fazer qualquer tipo de diferenciação, sobretudo nos casos em que a pessoa é também epilética.
A captura de eventos típicos através de EEG com auxílio de vídeo, assim como a avaliação por uma equipe multidisciplinar são primordiais para identificar fatores que contribuem para a perpetuação de eventos. De acordo com relatos, casos de convulsões não epiléticas prolongadas ou frequentes têm sido tratados com administração excessiva e perigosa de benzodiazepínicos, entre outros, sendo o custo do tratamento desses pacientes demasiado elevado.24
O EEG é normal ou obscurecido durante esses eventos, e normal mesmo durante a falta aparente de respostas. É possível que irregularidades menores, ou mesmo variantes normais observadas no EEG, tenham sido registradas como anormais, levando à perpetuação do uso de anticonvulsivantes. A forma mais eficaz de evitar a ocorrência de danos iatrogênicos é descontinuar o uso de todos os AED logo após o diagnóstico definitivo.25
O Quadro 4 mostra a distinção entre convulsões epiléticas e não epiléticas.
Quadro 4
|
DISTINÇÃO ENTRE CONVULSÕES EPILÉTICAS E NÃO EPILÉTICAS
| ||
|
Característica da convulsão |
Epilética |
Não epilética
|
|
Duração |
30s-3min |
Variável, mas sempre prolongada.
|
|
Cabeça |
Fixa |
Lado a lado
|
|
Olhos |
Abertos |
Fechados
|
|
Mordida na língua |
Lateral |
Ponta da língua
|
|
Choro |
Sem choro |
Provável
|
|
Movimentos do corpo |
Sincronizados |
Assíncronos, convulsivos
|
|
Movimentos dos quadris |
Jacksoniano |
Impulso pélvico
|
|
Gagueira/sussurro |
Não |
Provável
|
|
Posictal |
Cianose |
|
Estudos de caso I
(i) A paciente S.G., 36 anos de idade, com diagnóstico de epilepsia na infância, a qual havia sido bem controlada por cerca de 20 anos, relatou que, 2 semanas após um pequeno acidente de carro, desenvolveu um novo tipo de convulsão. Esses novos eventos começaram a ocorrer duas a três vezes por semana, interferindo na sua vida profissional. Um questionamento posterior revelou que ela vinha passando por um estresse significativo, o que teria ocasionado três abortamentos espontâneos. S.G. estava preocupada com a hipótese de perder o emprego devido aos descuidos que cometia. Os novos ataques ocorreram após ter discutido com o marido sobre a hipótese de tentar engravidar novamente. Seu corpo tinha movimentos convulsivos ao mudar de lado no leito, quando estava com os olhos fechados, o que a fazia chorar constantemente. Ela conseguia ouvir o que se passava durante o evento, porém não conseguia responder.(ii) A paciente A.J., 18 anos de idade, vinha tendo episódios de distúrbios visuais como luzes ou linhas brilhantes, no lado direito, seguidos de cefaleia grave, que ocorria mais ou menos uma vez por mês, ao se aproximar o período menstrual. Conforme relatou, a mãe da paciente tinha histórico de cefaleia causada por enxaqueca. Além disso, sentia formigamentos no lado direito da face. A.J. decidiu comparecer a um consultório médico para fazer uma avaliação depois de um episódio de escurecimento da visão que ocorreu de forma súbita e teve duração de 2 minutos.
(iii) O paciente B.L., 59 anos de idade, se apresentou após um episódio de desmaio na igreja. Ele se lembrava de ter sentido o alerta de sensação de desfalecimento e sudorese, bem como da dificuldade em ouvir os sons do coro da igreja. Teria despertado com a congregação ao seu redor, e as testemunhas disseram que havia tido trações corporais súbitas e que “seus olhos rolavam para cima”. B.L. retornou para a linha de base dentro de 10 segundos.
Os três casos relatados não sugerem epilepsia.
A história de S.G. sugere convulsão psicogênica não epilética por causa do novo tipo de ataque, da frequência dos eventos no início e dos agentes desencadeadores supramencionados; entretanto, a captura de um evento em um EEG com vídeo poderia determinar o diagnóstico definitivo, em especial com histórico de epilepsia remota.
O episódio de A.J. se trata de sintomas visuais unilaterais, sugerindo a presença de enxaqueca com perda de visão (sintoma negativo) e de cefaleia, e não convulsão. A RNM do cérebro poderia excluir a hipótese de uma lesão focal, levando-se em conta os sintomas neurológicos unilaterais, embora o histórico familiar positivo seja tranquilizador.
O caso de B.L. se assemelha a uma síncope vasovagal causada pelo pródromo, devido à probabilidade de que o paciente estivesse de pé, em um ambiente quente e repleto de pessoas, e pelo breve estado “posictal”. Movimentos convulsivos e “olhos rolando para cima” são relatos comuns daqueles que testemunham o fenômeno de Bell com perda de consciência. Esse fato não poderia impedir um diagnóstico sólido de síncope convulsiva. O monitoramento de Holter e o EEG são ferramentas muito úteis nesse caso.
Tratamento
O uso de medicações é o grande pilar do tratamento de epilepsia. Há controvérsias sobre a decisão de iniciar o tratamento após uma única convulsão não provocada inequívoca. Uma metanálise revelou que a possibilidade de recorrência depois de uma primeira convulsão não provocada é de 42% nos dois anos seguintes.26 De maneira geral, a recorrência surge dentro dos primeiros 6 meses após a convulsão.
A maior parte dos especialistas não inicia o tratamento após a primeira convulsão não provocada, a não ser que haja outras evidências sugerindo uma maior possibilidade de recorrência. Isso inclui uma lesão cerebral pré-existente (isto é, tumor, AVC), EE no início, descargas epiléticas definitivas no EEG, ou outros fatores como o fato de o paciente ter um irmão com epilepsia ou ter déficits neurológicos focais (paresia de Todd).27
O risco de recorrência é de 70% depois de duas convulsões não provocadas,28 o que justifica o início do tratamento. Ainda há controvérsias a respeito de as duas convulsões não provocadas serem ou não consideradas um evento único dentro de um período de tempo de 24 horas.29,30 O estudo Research Council Multicentre Study of Early Epilepsy and Single Seizures (MESS) apresenta evidências mais completas de vantagens e desvantagens do tratamento precoce.31
Pacientes com uma única convulsão ou com epilepsia precoce –, considerando que havia uma proporção entre as opiniões dos médicos entre prosseguir ou não o tratamento – foram randomizados entre tratamento precoce versus tratamento tardio. Os benefícios dos tratamentos imediatos incluíam incidência de uma quantidade menor de convulsões durante o acompanhamento, com benefícios sustentados por 2 anos. Entretanto, depois de 5 anos, ambos os grupos apresentaram a mesma taxa de remissão de 2 anos. Um número maior de pacientes que recebeu tratamento imediato em vez de tratamento tardio estava tomando AED (60% versus 40%) e, além disso, houve mais eventos adversos com o tratamento imediato.
Está suficientemente claro que o tratamento imediato não impede a ocorrência de epilepsia crônica. Por conseguinte, a decisão de se iniciar, ou não, a administração de AED depende, em parte, da preferência dos pacientes – esses preferem evitar convulsões, mesmo que isso implique no uso de um AED que não tenha sido indicado, ou evitar o uso de medicações? Essa questão deverá sempre ser levantada nas situações em que os dados disponíveis não justificarem enfaticamente o uso de AED.
Logo após a decisão de prosseguir o tratamento, a escolha das inúmeras alternativas é uma tarefa bastante árdua. De maneira geral, os AED se dividem em medicamentos “antigos”, lançados no mercado antes de 1993, incluindo o fenobarbital, ácido valproico, carbamazepínicos e fenitoína, e medicamentos mais recentes.
A era dos AEDs “novos” iniciou com a introdução do felbamato. A partir de então, um grande número de AEDs foram lançados no mercado farmacêutico. A AAN publicou algumas orientações para o uso desses fármacos em casos de epilepsia de novo início e de epilepsia refratária.32,33
Para os casos de epilepsia de novo início, acreditava-se que os AEDs “novos” eram potencialmente mais bem tolerados, embora não tenham sido encontradas evidências da superioridade de qualquer medicamento novo ou antigo. O teste Standard and New Antiepileptic Drugs (Sanad) procurou abordar algumas dessas questões. Em um teste randomizado aberto, observou-se que, no caso de pacientes com epilepsia relacionada à localização, a lamotrigina foi mais bem tolerada que medicamentos como carbamazepina, topiramato ou gabapentina.
A remissão é melhor com a carbamazepina em comparação com a gabapentina e, possivelmente, em comparação com lamotrigina, topiramato ou oxcarbazepinas.34 O valproato atingiu melhor remissão que a lamotrigina, e foi mais bem tolerado que o topiramato nos casos de epilepsias generalizadas ou não classificadas.35 Esses estudos são alentadores, já que confirmam suspeitas clínicas sobre eficácia e tolerância.
Em última análise, a decisão clínica sobre qual medicamento antiepilético é o ideal para iniciar um tratamento depende de diversos fatores, os quais incluem eficácia, preferência dos pacientes, familiaridade dos médicos com determinado medicamento, condições médicas concorrentes, interação medicamentosa potencial, estimativa de tolerância ou complacência, facilidade de uso, rapidez de início, considerações reprodutivas, e considerações financeiras ou de seguro.36
O fator mais decisivo na determinação do medicamento de escolha é avaliar se o paciente apresenta uma síndrome epilética relacionada à localização (parcial) ou uma síndrome epilética generalizada, tendo em vista que, em geral, isso determina a eficácia do medicamento. A apresentação de episódios convulsivos generalizados não ajuda a fazer a distinção entre esses dois tipos de síndrome.
Outros dados como presença de convulsões mioclônicas ou de convulsões de ausência, históricos familiares consideráveis, exames imagiológicos e neurológicos normais, alta sensibilidade à privação do sono e início apropriado para a idade são indicações de que o paciente é portador de uma síndrome epilética generalizada primária.
Basicamente, a presença de descargas generalizadas com picos e ondas no EEG é diagnóstica. O valproato deve ser usado como terapia de primeira linha em homens sempre que houver alguma suspeita ou confirmação de que o paciente tenha epilepsia generalizada. É mais difícil usar o valproato em mulheres por causa do baixo índice de tolerância (ganho de peso é a razão mais comum para interromper o uso do medicamento) e dos efeitos teratogênicos fetais em mulheres em idade de concepção.
Outros AEDs de espectro amplo, reconhecidamente eficazes contra epilepsias generalizadas, são o levetiracetam, topiramato e zonisamida, sendo que a lamotrigina é o mais eficaz para tratamento de convulsões de ausência e poderá ser usada com segurança nas situações em que as ausências forem o componente principal; todavia, não é eficaz o bastante contra episódios convulsivos e, em consequência disso, costuma ser usada em combinação com outro AED.
A lamotrigina pode agravar as convulsões mioclônicas e, por isso, deve ser usada com muita cautela em pacientes com EMJ. Ainda não se conhece a eficácia da lacosamida ou da ezogabine. Alguns AEDs, como a carbamazepina ou fenitoína, não são costumam ser recomendados como terapias de primeira linha, ainda que sejam eficazes contra convulsões generalizadas e não o sejam contra convulsões mioclônicas ou convulsões de ausência.
Muitos pacientes são bem controlados com esses medicamentos, apesar de terem iniciado o seu uso inadvertidamente. Fármacos como gabapentina, pregabalina, fenitoína, ácido valproico ou carbamazepina podem agravar as convulsões de ausência, ou mesmo induzir estado de ausência; portanto, recomenda-se evitar o seu uso nessa categoria de pacientes.
Nas situações em que os pacientes têm grande probabilidade de ter epilepsia focal, praticamente todos os AEDs, com exceção da etossuximida e rufinamida, estão à disposição dos médicos. Com base no estudo Sanad, pode-se deduzir que a carbamazepina, caso seja tolerada, é potencialmente mais eficaz. Entretanto, tendo em vista os efeitos colaterais potenciais no longo prazo, em geral os AEDs mais recentes são os medicamentos de escolha para tratamentos de primeira linha.
Há implicações de custos, tanto para os pacientes, como para a sociedade, iniciar o tratamento com um dos AEDs, em especial se as formulações genéricas não estiverem disponíveis. Cabe ressaltar que as taxas de remissão nos países em desenvolvimento, onde o fenobarbital e a carbamazepina são usadas quase que exclusivamente, se comparam às taxas de remissão nos EUA.
Se a rapidez inicial for um fator importante, o uso dos seguintes AEDs poderá ser adotado, com doses essencialmente terapêuticas: levetiracetam (1000mg, 2x/dia), oxcarbazepina (300mg, 2x/dia), gabapentina (300mg, 2x/dia), pregabalina (75mg, 2x/dia) e alguns dos medicamentos mais antigos (valproato).
O Quadro 5 apresenta, de forma detalhada, os AEDs, contendo a denominação, o mecanismo, a indicação, a dose, os efeitos colaterais e comentários sobre cada um deles.
Quadro 5
|
MEDICAMENTOS ANTIEPILÉTICOS
| |||||
|
Denominação |
Mecanismo |
Indicação |
Dose |
Efeitos Colaterais
|
Comentários |
|
Carbamazepina |
Liga-se aos canais de sódio neuronais para reduzir a deflagração repetitiva sustentada de alta frequência de potenciais de ação. |
Convulsões generalizadas parciais e secundárias. |
Iniciar com 400mg/dia, em 2 doses divididas; dose diária máxima de 1.600mg. |
Estão relacionados à dose e incluem sonolência, tontura, ataxia, discinesias e perturbações visuais. Embora sejam raros, há relatos de reações hematológicas idiossincráticas graves de agranulocitose e anemia aplásica. Induz a isoenzima CYP-450.
|
Considerar o rastreamento do alelo HLA-B*1502 em indivíduos de origem asiática, tendo em vista a associação com o aumento acentuado nas erupções cutâneas. Há, no mercado, diversas formas com ação prolongada. |
|
Etossuximida |
Diminui a explosão de neurônios talamocorticais responsáveis pela geração de descargas de picos e ondas de 3Hz através da redução no limiar baixo das correntes de canal de cálcio (tipo-T).
|
Convulsões de ausência. |
Administrar 500mg, em 2 doses divididas; aumentar para 1.500mg/dia em caso de necessidade. |
Comuns: náusea, vômito, perda de peso, diarreia, dor abdominal, constipação. Sérias: discrasias no sangue. |
|
|
Fosfenitoína |
Ver Fenitoína. |
Ver Fenitoína. |
Administrar dose de carga de 15-20mg EF/kg; pode-se administrar 100-150mg EF/min.
|
Risco mais baixo de hipotensão e de disfunção cardíaca em comparação com a fenitoína. |
|
|
Fenobarbital |
É responsável pelo reforço na transmissão sináptica inibidora mediada pelo Gaba; efeitos nos canais iônicos de cálcio controlados por voltagem; diminuição da excitação mediada por glutamato. |
Todas as convulsões, excetuando-se a convulsão de ausência. |
Administrar 1,5-4mg/kg/dia em doses diárias. |
Comuns: sedação, sonolência, hiperatividade, irritabilidade e insônia. Mais raros: danos cognitivos, depressão, transtornos motores, anemia megaloblástica, osteoporose, distúrbios nos tecidos conjuntivos, teratogenicidade e exacerbação da porfiria.
|
Induz a isoenzima CYP-450, resultando em interações medicamentosas. A formulação IV está disponível no mercado. |
|
Fenitoína |
Provoca inibição dos canais de sódio dependente da taxa (provável mecanismo primário); efeitos sobre os sistemas da calmodulina e do segundo mensageiro do nucleotídeo cíclico, e inibição da liberação de neurotransmissores controlada pela voltagem na sinapse. |
Convulsões de início parcial e convulsões tônico-clônicas generalizadas. Eficácia limitada nas convulsões de ausência, mioclônicas ou atônicas. |
Iniciar com 200-300mg, em 3 doses divididas; aumentar até a dose de manutenção ser atingida (usualmente 300-400mg/dia). Pode-se administrar a dose de carga de 1.000mg em 3 doses divididas, em intervalos de 2h ou por linha IV.
|
Comuns: nistagmo, visão turva, ataxia, fala titubeante, hipertrofia gengival, hirsutismo. Uso de longo prazo: osteoporose, neuropatia periférica, atrofia cerebelar, hipertrofia gengival. Sério: síndrome da luva roxa. |
Metabolização pelo sistema da isoenzima CYP-450, excreção através da urina e das fezes; conversão de cinética de primeira ordem para cinética de ordem zero em níveis na faixa intermediária da linha terapêutica média; indução da isoenzima CYP-450, resultando em múltiplas interações com outras medicações. A formulação IV está disponível no mercado.
|
|
MEDICAMENTOS ANTIEPILÉTICOS
| |||||
|
Denominação |
Mecanismo |
Indicação |
Dose |
Efeitos Colaterais
|
Comentários |
|
Valproato |
Provoca aumento na velocidade de renovação do Gaba e redução na excitação mediada pelo receptor do NMDA glutamato. Inibe a estimulação repetitiva sustentada de neurônios através do bloqueio dos canais de sódio controlados por voltagem.
|
Convulsões parciais complexas, convulsões generalizadas e de ausência. |
Administrar 15mg/kg/dia, em doses divididas, com aumentos semanais de 5-10mg/kg na dose. |
Comuns: sonolência, tremor, náusea, vômito, perda de cabelo, ganho de peso, tremor. Sérios: hepatotoxicidade, pancreatite hemorrágica. |
Eficácia em caso de cefaleia e para estabilização do humor. Formulações IV e de ação prolongada estão disponíveis no mercado. |
|
Clonazepam |
É agonista do receptor do Gaba-A, abre os canais iônicos de cloreto. |
Todas as convulsões. |
Iniciar com, no máximo, 1,5mg em doses divididas; no máximo, 20mg/dia.
|
Sonolência, letargia, possibilidade de dependência. |
|
|
Ezogabine |
Abre o canal de cálcio KCNQ/Kv7, aumenta a corrente K+ tipo-M, hiperpolariza neurônios e reduz a velocidade de estimulação. |
Adjuvante para convulsões parciais. |
Administrar 300mg em doses divididas a cada 8h; dose de manutenção de 600-1.200mg/dia. |
Comuns: sonolência relacionada à dose, tontura, fala titubeante; dificuldade para urinar; pequeno aumento no volume residual após a eliminação da urina. Pode prolongar o segmento QT e produzir efeitos colaterais neuropsiquiátricos atípicos de confusão, psicose ou alucinação.
|
|
|
Felbamato |
Promove interação com os receptores de NMDA, resultando na redução da neurotransmissão aminoácida excitatória. |
Convulsões parciais com ou sem generalização secundária. |
Administrar 1.200mg/dia em doses divididas a cada 8h; aumentar lentamente para a faixa de 2.400-3.600mg/dia. |
Comuns: náusea, cefaleia, insônia. Possibilidade de ocorrer perda de peso significativa com o uso no longo prazo. Sérios: anemia aplásica, insuficiência hepática. Trata-se do AED teratogênico mais sério; devem ser evitados em mulheres na idade de concepção caso seja possível. |
Este medicamento foi desenvolvido originalmente para tratamento da síndrome de Lennox-Gastaut. Recomenda-se fazer um hemograma completo e testes das enzimas hepáticas a cada 2 semanas durante os primeiros 6 meses. Usar apenas em pacientes gravemente refratários.
|
|
Gabapentina |
Provoca redução na liberação de glutamato nas sinapses corticais, redução na liberação dependente de atividade através da interação com canais de cálcio controlados por voltagem tipo P/Q.
|
Convulsões parciais. |
Administrar 300mg/dia com escalonamento rápido para uma dose alvo inicial de 1.800mg/dia em 3 doses divididas; no máximo, 3.600mg/dia. |
Comuns: sonolência, tontura, ataxia, ganho de peso e fadiga. |
Este medicamento não é metabolizado no fígado; poucas interações medicamentosas. |
|
Lacosamida |
Modifica os canais de sódio; desativa estimulações neuronais de alta frequência. |
Convulsões parciais. |
Administrar 100mg, em 2 doses divididas; aumentar para 200-400mg/dia. |
Comuns: tontura (sobretudo em combinação com outros moduladores do canal de sódio), náusea, cefaleia, diplopia. Pode causar prolongamento no intervalo PR no ECG. |
Pequena interação medicamentosa. A formulação IV está disponível no mercado. |
|
MEDICAMENTOS ANTIEPILÉTICOS
| |||||
|
Denominação |
Mecanismo |
Indicação |
Dose |
Efeitos Colaterais
|
Comentários |
|
Lamotrigina |
Bloqueia os canais iônicos de sódio excitatórios de uma forma dependente da voltagem e do uso. |
Convulsões parciais e generalizadas, incluindo convulsões generalizadas primárias. |
Administrar 25mg todos os dias; a dose poderá ser titulada até 25mg a cada 2 semanas, até atingir 100mg/dia, dose de manutenção de 200-400mg/dia. No caso da administração de medicação de indução enzimática: iniciar com 50mg/dia e titular até 300-500mg. No caso da administração de valproato: iniciar com 25mg em dias alternados; titular para 100-200mg/dia.
|
Comuns: tontura, ataxia, sonolência, cefaleia, visão turva, náusea, vômito; insônia (possibilidade). Sérios: erupção cutânea com escalonamento rápido ou coadministração de ácido valproico. Há risco de vida. |
Efeitos significativos na estabilização do humor. A forma com ação prolongada está disponível no mercado. |
|
Levetiracetam |
Modifica a proteína sináptica SV2A; os efeitos no sentido do fluxo não são claros. |
Epilepsia parcial e generalizada. |
Administrar 1.000mg/dia, em 2 doses divididas; aumentar a dose até atingir o alvo de 1.000-3.000mg/dia. |
Comuns: sonolência, irritabilidade, ataxia. |
Não há interações medicamentosas; a eliminação ocorre por via renal. A formulação IV está disponível no mercado. A forma de ação prolongada também está disponível.
|
|
Oxcarbazepina |
Bloqueia os canais de sódio, N/P- e os canais de cálcio tipo R. |
Convulsões parciais. |
Iniciar com 600mg/dia; aumentar 300mg/semana até atingir a dose alvo de 1.200mg/dia; dose máxima de 2.400mg/dia. |
Comuns: tontura, diplopia, fadiga, náusea e vômito, ataxia, dor abdominal, tremor, dispepsia, marcha anormal. Potencialmente sério: hiponatremia.
|
|
|
Pregabalina |
O mesmo que gabapentina; potência mais elevada. |
Convulsões parciais. |
Administrar 75mg, 2x/dia; a dose poderá ser escalonada para 300mg, 2x/dia.
|
O mesmo que gabapentina; pode ser mais grave. |
O mesmo que gabapentina. |
|
Rufinamida |
Provoca limitação da estimulação do potencial de ação dependente do sódio, limitando a estimulação repetitiva sustentada. |
Adjuvante para a síndrome de Lennox-Gastaut. |
Administrar 400-800mg/dia, em 2 doses divididas. Aumentar 400-800mg/dia, a cada 2 dias, até atingir a dose máxima de 3.200mg/dia.
|
Sonolência, vômito, cefaleias, tontura. |
|
|
Tiagabina |
É inibidor potente e específico da absorção do Gaba. |
Convulsões parciais. |
Iniciar com 4mg/dia; aumentar 4 a 8mg/dia semanalmente, até atingir a dosagem de 56mg/dia, em 2 a 4 doses divididas. |
Tontura, astenia, sonolência, náusea, nervosismo, tremor, dificuldade de concentração e atenção, vômito, perturbações comportamentais e aumento de apetite.
|
|
|
Topiramato |
Tem mecanismos múltiplos de ação: efeitos moduladores sobre os canais de sódio, receptores de Gabaa, AMPA/cainato. |
Convulsões parciais com ou sem generalização secundária. Epilepsias generalizadas primárias. |
Administrar 50mg/dia, em 2 doses divididas; alvo: 200-400mg/dia. |
Comuns: sonolência, tontura, ataxia, nervosismo, fadiga, transtornos da fala, lentidão psicomotora, visão anormal, dificuldades de memória, parestesias, perda de peso, cálculos renais. |
Eficácia para profilaxia de enxaqueca. |
|
MEDICAMENTOS ANTIEPILÉTICOS
| |||||
|
Denominação |
Mecanismo |
Indicação |
Dose |
Efeitos Colaterais
|
Comentários |
|
Vigabatrina |
É inibidor seletivo irreversível do Gaba transaminase. |
Convulsões parciais. |
Administrar 1.000mg, em 2 doses divididas; aumentar para até 3.000mg/dia. |
Comuns: sonolência e fadiga, exceto em crianças, nas quais a excitação ou agitação é mais frequente. Outros efeitos no SNC: tontura, nervosismo, irritabilidade, depressão, cefaleia. Sérios: constrição concêntrica bilateral permanente do campo visual.
|
|
|
Zonisamida |
Bloqueia o canal de sódio dependente da dose; bloqueia o canal de cálcio tipo-T. |
Convulsões parciais e convulsões generalizadas primárias. |
Iniciar com 100mg/dia; aumentar para até 500mg/dia. |
Comuns: sonolência, fadiga, ataxia, anorexia, dificuldade de concentração, dificuldades de memória, náusea, vômito, perda de peso. Sérios: hipersensibilidade às sulfonamidas, cálculos renais, oligohidrose. |
Este medicamento poderá ser administrado em uma única dose diária. |
AED: medicamento antiepilético; AMPA: a-amino-3-hidroxi-5-metil-4-isoxazol propiônico; ECG: eletrencefalograma; EF: equivalente de fenitoína; Gaba: ácido ?-aminobutírico; HLA: antígeno leucocitário humano; IV: intravenoso; NMDA: N-metil-D-aspartato; SNC: sistema nervoso central.
A formulação intravenosa com dose equivalente está à disposição no mercado para os medicamentos levetiracetam, fenitoína, lacosamida e valproato. Mesmo com essas medicações, recomenda-se iniciar o tratamento com baixas doses e titular para cima de forma lenta para minimizar a ocorrência de efeitos colaterais.
Nas situações em que a aderência ao regime medicamentoso e o cumprimento às recomendações médicas em relação às dosagens forem preocupantes, é indicada a prescrição de AEDs com dosagem 1x/dia. Esses medicamentos incluem levetiracetam, lamotrigina, zonisamida, valproato, fenitoína e fenobarbital.
O fenobarbital pode parecer útil em determinados casos, considerando que deixar de tomar dose diária pode produzir menos oscilações séricas que outros AEDs com dosagem diária. O descumprimento às instruções do médico é o principal motivo para o progresso das convulsões, o que deverá ser investigado.
Fatores como falta de prescrições médicas por razões financeiras ou de acesso, efeitos colaterais excessivos dos medicamentos e má compreensão das instruções estão entre as razões principais que levam os pacientes a não seguirem os regimes. O uso de uma caixa de comprimidos ou de qualquer outra técnica para simplificar regimes multidoses complicados pode ajudar os pacientes a recuperar as doses faltantes tão logo sejam percebidas.
A facilidade de uso é um dos benefícios da maior parte dos AEDs. Os mais antigos como a fenitoína têm índice terapêutico estreito e sua titulação é muito difícil por causa da necessidade de verificar os níveis séricos. Não é preciso averiguar os níveis dos AEDs modernos, mesmo que, eventualmente, isso tenha de ser feito para se confirmar a adesão ao regime.
Os AEDs diferem entre si na propensão para produzir reações alérgicas. O uso da lamotrigina passou a ser observado com mais atenção pelo fato de causar erupção cutânea, o que pode colocar a vida do paciente em risco com a progressão para a síndrome de Stevens-Johnson. Entretanto, com baixa titulação, a propensão para a ocorrência de erupções cutâneas é menor e não é diferente da propensão quando do uso de carbamazepina ou fenitoína. Nas situações em que os pacientes apresentarem reações alérgicas, valproato, levetiracetam ou gabapentina são opções a serem consideradas porque têm menor probabilidade de produzir erupções cutâneas.37
O início da administração de AEDs no contexto hospitalar em casos de enfermidades críticas representa um grande desafio. Os pacientes se caracterizam pela presença de hemodinâmica instável, absorção não parenteral alterada, enfermidades médicas concorrentes e necessidade de usar muitas outras medicações concorrentes.
A necessidade de tratamento de convulsões de novo início deve ser considerada com muita cautela, tendo em vista que as convulsões provocadas não precisam ser tratadas se a causa subjacente é reversível. Recomenda-se evitar o uso reflexivo de cargas de fenitoína. Diversos estudos demonstraram que há um aumento na tolerância ao levetiracetam sem perda de eficácia, em comparação com a fenitoína.38,39
Ao iniciar o regime de tratamento com um AED, os médicos devem alertar os pacientes sobre os efeitos colaterais. Recomenda-se que as discussões sobre esse tipo de medicamento incluam fatores como efeitos colaterais comuns, os quais, com frequência, desaparecem dentro de algumas semanas, e efeitos colaterais raros, que exigem a descontinuação imediata no uso do AED. Esses esclarecimentos criam expectativas mais realistas e, em última análise, aumentam a satisfação dos pacientes, reduzindo, inclusive, as constantes comunicações com os médicos para sanar dúvidas.
Politerapia
A prática atual é usar os medicamentos como monoterapia. Aumenta-se a dose de forma progressiva até que as convulsões e os efeitos colaterais sejam controlados. O medicamento é considerado um fracasso nas situações em que não é possível controlar as convulsões sem efeitos adversos significativos e, nessas circunstâncias, deve-se escolher um segundo fármaco e repetir o processo. Se o segundo também falhar, a melhor opção é testar uma terceira monoterapia ou tentar uma terapia de combinação.
A terapia de combinação é particularmente útil nos casos em que o paciente tem vários tipos de convulsão (por exemplo, episódios convulsivos generalizados e convulsões de ausência) e os medicamentos tiverem como alvo determinados tipos de convulsão (por exemplo, carbamazepina para convulsões e etossuximida para ausências).
A aplicação da politerapia racional ainda é muito difícil sob os pontos de vista teórico e prático, já que ainda não foi comprovado que os mecanismos farmacológicos são orientações suficientes. Estudos em animais que utilizaram técnicas isobolográficas mostraram que há inúmeras combinações favoráveis e desfavoráveis. A dificuldade para replicar esses resultados de uma forma sistemática em seres humanos resultou em um número bem menor de combinações conhecidas de sinergia ou antagonismo reais.
Comumente, as recomendações se originam mais em séries de casos do que em testes clínicos. As combinações favoráveis associadas à sinergia incluem lamotrigina e valproato, fenitoína e fenobarbital, e levetiracetam e oxcarbazepina. Entre todas elas, a combinação de lamotrigina e valproato é particularmente potente; embora o valproato aumente os níveis de lamotrigina, vários estudos demonstraram que ocorre uma verdadeira sinergia farmacodinâmica.40,41 Por conseguinte, nos casos de pacientes com convulsões intratáveis, recomenda-se colocá-los nesse regime de combinação, embora seja extremamente difícil atingir a combinação ideal devido à interação entre o valproato e a lamotrigina.
A combinação de carbamazepina e lamotrigina é desfavorável, tendo em vista que o perfil de efeitos colaterais é muito pior do que se poderia esperar da combinação linear entre esses dois medicamentos. Acredita-se que a lamotrigina aumente o subproduto da epóxido-carbamazepina, ainda que esse fato não tenha sido efetivamente comprovado como a causa do efeito clínico.42 Apesar disso, o uso dessa combinação é, em geral, bastante eficaz, levando-se em consideração a popularidade relativa individual desses dois medicamentos. Mais recentemente, levantou-se a suspeita de que combinações de lacosamida com outros AEDs, com modulação através do canal de sódio, aumentam os efeitos colaterais da lacosamida; não obstante, essas combinações ainda são eficazes.
Como regra geral, nenhuma combinação de medicamentos deveria ser evitada. Os princípios da politerapia são os mesmos que os da monoterapia; é imprescindível definir, de forma cuidadosa, a síndrome epilética; devem-se utilizar doses mais baixas e que sejam eficazes; devem ser consideradas com muita cautela as interações medicamentosas, a farmacocinética e a condição médica concorrente.
As complicações que surgem com o uso de AEDs, incluindo a dificuldade em seguir as instruções médicas, o custo das medicações, os erros da medicação e os excessos terapêuticos, são ampliadas na politerapia. Deve-se dar atenção especial aos excessos terapêuticos potenciais; ao iniciar o uso de uma segunda medicação, recomenda-se tentar diminuir a primeira caso seja possível.
Em última análise, o insucesso da primeira tentativa de usar um AED é preocupante; 47% dos pacientes respondem ao primeiro AED, 14% respondem ao segundo ou ao terceiro medicamento (respondem ainda menos nas situações em que o insucesso do primeiro AED tenha sido a baixa eficácia em vez dos efeitos colaterais); e 3% respondem ao tratamento com uma combinação de medicamentos.43
Não obstante, em pacientes refratários é válido tentar algum AED alternativo enquanto são avaliadas outras opções de tratamento – como a cirurgia, por exemplo –, tendo em vista que são poucas as chances de os pacientes responderem a testes seriais com AEDs.44
Medicamentos de marcas comerciais versus medicamentos genéricos
Atualmente, os medicamentos genéricos estão disponíveis para quase todos os AEDs prescritos com mais frequência nos EUA. Os medicamentos genéricos e os de marcas comerciais são semelhantes, considerando que têm o mesmo ingrediente ativo, a mesma dose e a mesma via de administração. As únicas diferenças são o processo de fabricação, a substância inativa, a forma, a cor e o sabor.
Os medicamentos genéricos aprovados atendem à norma de bioequivalência da Food and Drug Administration (FDA): o intervalo de confiança (IC) de 90% da média geométrica com transformação logarítmica varia entre 80 e 125% entre os medicamentos genéricos e os medicamentos de referência para as seguintes medições: concentração sérica máxima e área em baixo da curva dos gráficos relacionando tempo e concentração.
Esse fato, em geral, leva a interpretações incorretas, significando que poderá ocorrer uma diferença de 20 a 25% entre a média dos dois produtos. Não se exige equivalência de tempo em relação à concentração máxima. De maneira geral, isso resulta em uma equivalência muito próxima das duas medições dos medicamentos genéricos e dos medicamentos de referência.
O potencial de redução de custos é bastante expressivo. Estima-se uma redução de 11% nos custos das prescrições se todos os medicamentos disponíveis como genéricos forem efetivamente vendidos como genéricos.45 Entretanto, ainda não está claro se essa redução de custos é real; vários estudos demonstraram taxas elevadas de troca medicamentos genéricos por medicamentos com marcas comerciais, aumentando os custos totais de assistência médica.46-48
Além disso, constatou-se que houve um número maior de eventos epiléticos durante a mudança para medicamentos genéricos.47-49,50 Outras variáveis também devem ser levadas em consideração; a janela terapêutica para AEDs é mais estreita em comparação com a maior parte dos medicamentos usados no contexto ambulatorial. A variabilidade entre os lotes de genéricos entre os fabricantes pode ser significativa, mesmo que a variabilidade em cada fabricante seja pequena. Os padrões de fabricação podem variar de forma substancial.
É provável que as normas da FDA para bioequivalência não sejam tão rigorosas; na Dinamarca, o IC de 90% deve ser atingido entre 90 e 111% em vez de 80 e 125%. As consequências das crises epiléticas em pacientes que haviam sido bem controlados constituem um fato bastante significativo e que não pode ser ignorado. Em 2007, a AAN atualizou suas recomendações para a substituição genérica de AEDs, afirmando que a “Academia se opõe à substituição genérica de medicamentos anticonvulsivantes para tratamento de epilepsia sem aprovação médica” e que “FDA aprovou diferenças significativas entre medicamentos genéricos e medicamentos comerciais de referência”.51
A AAN alertou que, no caso dos AEDs, pequenas variações na concentração entre os medicamentos comerciais e seus equivalentes genéricos podem produzir efeitos tóxicos e/ou convulsões. Ao contrário de outras doenças, uma única crise epilética poderá ter consequências devastadoras para o paciente, incluindo perda da carteira de motorista, bem como ocorrência de lesões ou mesmo a morte.
Como resultado, o position paper (carta ou documento de posicionamento) se opôs a toda a legislação estadual ou federal que pudesse impedir a capacidade de um médico para determinar qual AED deveria ser prescrito para epilepsia, assim como às políticas que resultassem da substituição arbitrária de AEDs, incluindo a substituição genérica de AEDs nos pontos de venda sem consentimento prévio do médico e do paciente. Recomenda-se fazer a distinção entre o uso de AEDs para tratamento de epilepsia e o uso de medicamentos para tratamento de outros tipos de transtorno.
Os AEDs genéricos, entretanto, são aparentemente seguros e eficazes para a grande maioria de pacientes. Testes randomizados feitos com fármacos mais antigos não conseguiram demonstrar a inferioridade dos medicamentos genéricos, embora ainda não tenha sido feito nenhum estudo sobre os AEDs mais recentes.52
É bem provável que a causa da maior parte dos eventos relacionados à epilepsia seja a ocorrência de algum evento epilético durante a mudança de tipo de medicamento, o momento da mudança de medicamento – se a mudança é de um medicamento com marca comercial para um medicamento genérico ou de um medicamento genérico para um medicamento com marca comercial.
Os pacientes devem ser tranquilizados quanto a isso, visto que muitos deles acham que é economicamente inviável permanecer no AED com marca comercial devido às restrições do seguro de saúde. Levando-se em consideração as consequências potenciais, é imprescindível intensificar a vigilância (por exemplo, monitoramento sérico do medicamento, manutenção de um diário de convulsões) nas situações em que for necessário mudar o produto farmacológico em pacientes de alto risco. Os pacientes, bem como as farmácias, devem ser orientados a não mudar com frequência os medicamentos genéricos.
Cirurgia epilética
A primeira cirurgia para tratamento de epilepsia foi feita por Victor Horsley, na Inglaterra, na década de 1880. Wilder Penfield e Herbert Jasper desenvolveram a eletrocorticografia na década de 1950 com a finalidade de identificar as regiões epiléticas e de mapear o córtex eloquente com vistas a melhorar os resultados. A partir de então, a cirurgia se tornou menos popular, porém ressurgiu na década de 1980 com técnicas cirúrgicas e de neuroimagens mais aprimoradas.
Em 1987, criou-se a National Association of Epilepsy Centers (NAEC), a qual, atualmente, engloba cerca de 100 centros que servem de pontos de encaminhamento para gerenciamento de pacientes refratários. Nos dois primeiros testes feitos com medicamentos, e que falharam devido à falta de eficácia, os pacientes deveriam ter sido encaminhados para um centro de epilepsia para verificar a necessidade de fazer cirurgia epilética.
A estratégia da cirurgia epilética é identificar o foco das convulsões dos pacientes e fazer ressecções com segurança, sem produzir déficits neurológicos inaceitáveis. As investigações pré-cirúrgicas básicas feitas com RNM cerebral de alta resolução e telemetria com EEG orientada por vídeo (conforme a Figura 5) para identificar áreas epiletogênicas do cérebro aumentam as chances de sucesso cirúrgico.
Figura 5 – Ao despertar, a atividade rítmica teta emerge da cadeia temporal direita de eletrodos (seta), sendo diagnóstica de uma convulsão no lobo temporal direito.
Em particular, a identificação de escleroses hipocampais unilaterais (como mostra a Figura 6) é um fator independente associado a bons resultados. As RNMs de alto campo (3T), com protocolos específicos para epilepsia, são usadas para detectar anormalidades sutis na maior parte dos centros. A telemetria com EEG orientada por vídeo é usada, em geral, no ambiente hospitalar e permite reduzir, de forma gradual, o uso de AEDs.
Figura 6 – A visão coronal de uma imagem por RNM do cérebro demonstra a presença de uma esclerose hipocampal direita grave (seta), sendo o provável início de uma epilepsia que pode ser solucionada por meios cirúrgicos.
RNM: ressonância nuclear magnética.
O EEG concorrente permite correlacionar a semiologia da convulsão com o início do EEG. Técnicas como tomografia computadorizada por emissão de pósitrons (PET) interictal, tomografia computadorizada por emissão de fóton único (SPECT) e magnetoencefalografia (MEG) são modalidades imagiológicas suplementares que, às vezes, são necessárias nas situações em que os focos não são visíveis de imediato (conforme a Figura 7). Os testes neuropsicológicos para avaliar as funções da linguagem e da memória são vitais e ajudam a localizar a área epileptogênica, determinando o tipo e o grau de disfunção cognitiva, e a prever os efeitos cognitivos prováveis da ressecção cortical proposta.
Figura 7 – Varredura tomográfica por emissão de pósitrons do cérebro em um paciente com epilepsia no lobo temporal, demonstrando hipometabolismo temporal esquerdo.
A avaliação psicossocial é muito útil para explorar circunstâncias mais amplas da vida de um paciente, o grau e a qualidade da conectividade social, a preparação para a cirurgia e a vida pós-cirúrgica. O procedimento intracarotídeo de sódio amobarbital (teste de Wada) é uma técnica em que se injeta um barbitúrico de ação curta para determinar a lateralização da fala e da memória. A utilidade dessa técnica invasiva se encontra em fase de declínio, embora ela ainda seja aplicada em muitos centros para tratamento de epilepsia.53
Esse tipo de cirurgia ressectiva contrasta com a radiocirurgia, que é uma alternativa não invasiva feita com radiação orientada a partir de uma faca ? . Nos dias atuais, a abordagem radiocirúrgica se restringe a poucas séries publicadas e aos centros que processam esse tipo de tecnologia. O efeito da radiocirurgia é demorado (>6 meses) e, aparentemente, esse tipo de técnica oferece poucas vantagens nas convulsões ou nos resultados cognitivos. Atualmente, não é uma alternativa realística aplicada de forma ampla em substituição à cirurgia convencional.
Ocasionalmente, aplica-se uma abordagem mais intensiva com EEG invasivo e eletrodos intracerebrais para identificar, de forma mais precisa, a zona onde ocorreu o início da convulsão. Nesses casos, costuma-se fazer o mapeamento do córtex eloquente com estimulação elétrica para identificar as áreas cuja remoção poderia resultar em perda do processamento sensorial e motor ou da capacidade linguística.
Por conseguinte, a cirurgia epilética é um procedimento multidisciplinar que exige a participação de profissionais dedicados e especializados. As taxas de sucesso de resultados favoráveis variam de acordo com a síndrome epilética, assim como a tecnologia e a qualidade dos recursos humanos. Uma revisão de resultados obtidos por vários centros e envolvendo todas as síndromes epiléticas registrou uma taxa de remissão de 66% no período de 2 anos.54
Na população cirúrgica, a epilepsia no lobo temporal com esclerose hipocampal é a causa mais comum de epilepsia focal refratária em adultos. Além disso, trata-se da condição mais sensível ao tratamento cirúrgico, independentemente das convulsões avaliadas com base nos “critérios de Engel”, cujos melhores resultados (Engel 1) variam de 70 a 90%.55 Resultados favoráveis semelhantes podem ser esperados na maior parte dos tipos de epilepsia que resultam de lesões locais, desde que seja possível demonstrar, de forma inequívoca, que a lesão não tenha sido responsável pelas convulsões dos pacientes.
A eficácia do tratamento cirúrgico foi demonstrada de forma conclusiva em um teste clínico randomizado que envolveu pacientes com epilepsia no lobo temporal. Os pacientes foram randomizados para ressecção no lobo temporal versus gerenciamento médico. Depois de 1 ano, os pacientes do grupo cirúrgico tiveram menos convulsões e apresentaram melhor qualidade de vida que os pacientes que haviam recebido terapia médica ideal.56
É provável que a cirurgia tenha sido subutilizada apesar de os parâmetros práticos da AAN recomendarem encaminhamento para um centro de epilepsia após o insucesso no uso de medicamentos de primeira linha. Entretanto, esse fato parece ter pouco impacto sobre os padrões de encaminhamento.57
Outras lesões cerebrais associadas à epilepsia focal intratável em adultos são displasia cortical, tumores de baixo grau, tumor neuroepitelial disembrioplásico, tumor neuroepitelial, encefalomalacia (por exemplo, resultante de AVC prévio) e malformações vasculares. De maneira geral, as displasias corticais são pequenas, difíceis de detectar e extremamente epileptogênicas.
A visualização dessas displasias é muito difícil, mesmo com suspeitas clínicas. A epilepsia não lesional autêntica apresenta grande dificuldade e representa o maior desafio atual para a cirurgia epilética curativa. Calosotomia do corpo caloso e transecções subpiais múltiplas (em inglês, multiple subpial transections [MSTs]) são procedimentos cirúrgicos paliativos que podem ser apropriados em determinadas circunstâncias.
O princípio da calosotomia é dividir o corpo caloso (comumente, os dois terços anteriores, mantendo o esplênio intacto) para impedir a propagação da convulsão entre os hemisférios. Esse tipo de procedimento é, em geral, utilizado para tratar episódios de queda, situação em que é bastante eficaz.
A MST é um procedimento controverso, sendo adotado regularmente apenas em alguns poucos centros de epilepsia. A MST é usada nas situações em que a área de início da convulsão estiver nas proximidades, ou em áreas limítrofes, do córtex eloquente. Esse método se caracteriza por um corte inferior nas fibras em U que ligam a área de início da convulsão do córtex adjacente, deixando intactas as vias descendentes de substância branca.
Os resultados dos desfechos das convulsões e a preservação da função eloquente são variáveis. De maneira geral, o uso de calosotomia e de MST é considerado nas situações em que a cirurgia ressectiva definitiva não é viável. Hemisferectomia é uma técnica reservada apenas para pacientes pediátricos com hemiplegia pré-existente e convulsões parciais excessivamente refratárias envolvendo quase todo o hemisfério, como, por exemplo, a encefalite de Rasmussen ou outras encefalites progressivas.
Terapia de estimulação
A ENV foi aprovada pela FDA em 1997 como terapia adjuvante para convulsões parciais refratárias às medicações anticonvulsivantes. Essa modalidade é uma alternativa válida para o uso em pacientes com epilepsia refratária sob o ponto de vista médico que, por alguma razão, não são candidatos cirúrgicos. O mecanismo exato de ação da ENV ainda permanece desconhecido, embora seja um efeito de projeções corticais e subcorticais difusas dos núcleos do tronco cefálico que se tornam alvos diretos de descargas vagais aferentes.58
As complicações da ENV, além dos riscos cirúrgicos e infecções, incluem alteração na voz, rouquidão, tosse, dispneia e exacerbação potencial da apneia do sono. Os pacientes não conseguirão mais fazer RNM no corpo; a RNM do cérebro ainda será possível em centros médicos terciários com determinados tipos de espirais para uso na cabeça (tipo de transmissão e recepção a 1,5T).
Além da eficácia, um dos benefícios da ENV é que os pacientes não estão sujeitos aos efeitos colaterais típicos dos AEDs (isto é, interações medicamentosas, erros na dosagem, cumprimento das orientações médicas ou efeitos neurocognitivos/neuropsiquiátricos); na realidade, alguns estudos sugeriram a presença de um efeito antidepressivo no dispositivo. Ao contrário dos AEDs, que podem maximizar a eficácia no início do tratamento e se tornar menos eficientes ao longo do tempo, a ENV não está associada à tolerância e parece ganhar eficácia com o passar do tempo.59
Cabe ressaltar que a ENV é eficaz em pacientes com a síndrome de Lennox-Gastaut, tendo em vista que diminui a frequência das convulsões tônicas e atônicas. Para esse tipo de pacientes, recomenda-se avaliar a hipótese de se aplicar ENV antes da calosotomia.
O implante de um dispositivo de ENV é um procedimento rápido que poderá ser executado normalmente em um dia cirúrgico. Faz-se a instalação subcutânea do gerador no lado esquerdo superior do tórax. Os fios condutores devem ser colocados no nervo vago cervical esquerdo e sintonizados com o gerador, visto que contêm um número bem maior de fibras aferentes que o nervo vago direito.
O estimulador possui configurações programáveis; a intensidade e a duração dos pulsos são compatíveis com a condição de cada paciente, e são configuradas para um período de várias semanas. Logo após a programação, o dispositivo começa a operar de forma automática. Além disso, os pacientes poderão ativar o dispositivo por meio de um ímã manual que, após ser ativado, inicia um pulso extra cujos parâmetros são programados de forma separada.
O dispositivo poderá, também, ser desligado temporariamente, mantendo-se o ímã estacionário sobre o estimulador. A vida útil da bateria varia entre 7 e 10 anos; a expectativa é de que esse prazo possa ser melhorado no futuro.
Estudos que analisaram alguns resultados demonstraram que entre 43 e 64% de pacientes apresentam melhoras superiores a 50%,60,61 ou seja, um grau de melhora comparável ao dos AEDs. As taxas de complicações dos implantes de ENVs são baixas e os efeitos colaterais (rouquidão, tosse ou dispneia) são mínimos e melhoram ao longo do tempo. Inúmeros estudos mostraram que houve melhoras na qualidade de vida após ENVs que não dependem do controle convulsivo, incluindo melhoras no humor, no estado de alerta durante o dia e no tempo de recuperação posictal.
A seleção de pacientes para ENV deve ser feita com o máximo rigor possível, da mesma forma que para outros tratamentos cirúrgicos de epilepsia. A cirurgia ressectiva oferece melhores expectativas de cura e, por conseguinte, é uma opção a ser levada em conta antes da decisão de aplicar a técnica de ENV.
Na orientação aos pacientes sobre os riscos e benefícios do implante de um dispositivo de ENV, é muito importante enfatizar que a aplicação desse método raramente consegue resolver as convulsões. Não se deve esperar a ocorrência de benefícios imediatos, como, às vezes, se consegue com os AEDs convencionais.
Uma grande variedade de técnicas de estimulação foi explorada para os casos de epilepsia intratável desde a década de 1970, embora somente a ENV tenha evidências de testes controlados. Os alvos intracranianos incluíam o nervo trigêmeo, uma variedade de estruturas subcorticais (tálamo, subtálamo, caudado) e o próprio foco epilético.
A mais recente dessas técnicas se caracteriza pela estimulação cortical direcionada através de um neuroestimulador responsivo que libera corrente elétrica durante a detecção de convulsões (Neuropace). O fato curioso é que a eficácia de todos esses métodos se assemelha à da ENV. Nenhum desses métodos chegou a ser aprovado pela FDA. A estimulação magnética transcraniana (EMT) e outros novos métodos de estimulação encontram-se, atualmente, em fase de desenvolvimento.
Tratamentos dietéticos
Introduziu-se a dieta cetogênica como tratamento não farmacológico adjuvante para epilepsia após as observações de que a inanição resultava em melhorias nas convulsões. A dieta engloba alta ingestão de gorduras e proteínas e restrição de carboidratos. A consequência metabólica é um estado cetoacidótico que imita a inanição. Já se conhece a eficácia terapêutica desse tipo de tratamento, embora os mecanismos moleculares precisos da dieta ainda não tenham sido elucidados.
Tradicionalmente, a dieta era oferecida apenas para crianças e, em face das graves restrições, sua aplicação se justifica somente por um período limitado de tempo (2 anos ou menos). Entretanto, evidências recentes sugerem que a dieta modificada de Atkins, que é menos restritiva, apresenta eficácia equivalente, inclusive em adultos, podendo ser tão eficaz como em crianças.62
Atualmente, a modificação dietética de acordo com o protocolo modificado de Atkins oferece uma modalidade real e sustentável de tratamento de epilepsia refratária para todas as idades e, portanto, deve ser considerada em combinação com qualquer terapia concorrente. Mais recentemente, observou-se que as dietas com baixo índice glicêmico se aproximam da eficiência das dietas cetogênicas, além de serem muito mais toleráveis e mais simples de colocar em prática.63
O envolvimento imediato de um nutricionista é fundamental para os pacientes e para os médicos. Após o início da terapia, a maior parte dos pacientes aprende rapidamente a administrar suas próprias dietas e a monitorar a cetose com auxílio de tiras reagentes para teste de urina. A suplementação diária de vitaminas e minerais é fundamental.
Recomenda-se monitorar por 3 meses o perfil lipídico no jejum e a excreção de cálcio urinário de 24 horas. A hiperlipidemia é uma consequência rotineira da dieta, sendo que alguns médicos costumam usar as estatinas nos pacientes que persistirem nas dietas por prazos longos. O papel dos bifosfonatos para minimizar a perda óssea causada pela hipercalciúria ainda foi suficientemente esclarecido.
Por fim, é indicada uma hidratação adequada para evitar a ocorrência de nefrolitíase, sendo prudente evitar o uso de dieta em pacientes que estiverem usando AEDs que produzem acidose (isto é, topiramato, zonisamida ou acetazolamida).
Estudos de caso II
Tratamento de síndromes epiléticas comuns
Epilepsia de ausência na infância
(i) O paciente Y.R., 8 anos de idade, de acordo com relato dos pais e informação fornecida pelo professor, estaria “desatento” na escola. O pediatra não observou nada de errado no exame físico e solicitou uma RNM do cérebro, cujo resultado também foi normal. A seguir, a família levou Y.R. ao neurologista, que pediu a ele que soprasse um cata-vento e, então, observou uma falha de atividade ao piscar os olhos. O médico providenciou um EEG de 20 minutos.
Este é um cenário clínico clássico, ou seja, um alarme disparado pelo alerta de um professor. O EEG de Y.R. mostrou uma atividade pico-onda de 3Hz típica de EAI, que se trata de um transtorno autossômico dominante com penetração incompleta. A idade de início varia entre 4 e 8 anos. Esses pacientes apresentam desenvolvimento normal e o resultado do exame neurológico também costuma ser normal.
Esse tipo de síndrome consiste de convulsões de ausência clássicas, que, em geral, ocorrem diversas vezes por dia; 50% dos pacientes também se apresentam com CTCG. Um estudo recente confirmou que a etossuximida é o melhor fármaco de escolha para tratamento de EAI; o valproato (também eficaz, porém menos bem tolerado) e a lamotrigina são medicamentos alternativos.14 O prognóstico é bom, com remissão espontânea depois de 2 a 3 anos na maioria dos pacientes.
Epilepsia benigna da infância com picos centrotemporais
(ii) A família do paciente H.S., 9 anos de idade, estava em uma longa viagem pelo Estado quando percebeu o primeiro sinal. Os pais foram alertados repentinamente por um som de ânsia de vômito. H.S. estava tendo uma convulsão; permaneceu inconsciente e babando, e seu rosto estava sendo empurrado para um lado e repuxando. O episódio cessou depois de 30 segundos. A família parou na clínica de emergência mais próxima, sendo que, naquele momento, H.S. estava desperto e conversando normalmente, embora estivesse um pouco atordoado.
O tipo de convulsão de H.S. (envolvimento bucofacial proeminente) é típico de convulsão benigna da infância com picos centrotemporais. Também conhecida por epilepsia rolândica benigna, esse tipo de doença é comum na infância. A área centrotemporal é a preferida pelas epilepsias benignas da infância, porém o envolvimento occipital também é um subtipo reconhecido, ainda que não tão comum. Raramente, os pacientes se apresentam com foco frontal.
A epilepsia benigna da infância com picos centrotemporais é um transtorno autossômico dominante com penetração dependente da idade. A idade de início varia de 3 a 13 anos. A maior parte dos pacientes apresenta apenas as peculiaridades do EEG, sem convulsões. Nos casos de pacientes que têm episódios convulsivos, as crises ocorrem com pouca frequência e consistem de eventos parciais simples e breves, somatossensoriais ou motores, com generalização secundária ocasional. As convulsões se restringem ao sono em 70 a 80% de casos, e ocorrem dentro de um período de tempo de 20 minutos a 3 horas depois de deitar.
Tipicamente, o EEG revela a presença de picos centrotemporais que se deslocam entre os hemisférios; os picos aumentam durante o sono. Os pacientes respondem bem a quase todos os anticonvulsivantes, sendo que, às vezes, é razoável decidir não fazer nenhum tipo de tratamento. O prognóstico é excelente e a maior parte dos pacientes apresenta remissão antes dos 16 anos de idade.
Espasmos infantis
(iii) O paciente K.G., 3 meses de idade, com nascimento e desenvolvimento normais, desenvolveu episódios posturais curtos. Seu corpo flexionava para frente de forma repentina, com os braços estendidos, apresentando, em seguida, um relaxamento mais lento. Os episódios tendiam a se agrupar causando irritação na criança. Em um período de 6 meses após o início, os pais de K.G. perceberam que houve alguma regressão dos marcos referenciais atingidos anteriormente. Havia um histórico de esclerose tuberosa na família: o pai de K.G. possuía lesões cutâneas e foi confirmado, geneticamente, que tinha a condição, embora nunca tenha sofrido uma convulsão.
Os episódios de K.G. representam espasmos infantis. A condição, conhecida por síndrome de West, envolve uma combinação de espasmos infantis – também denominados ataques em canivete ou crises de Salaam, consistindo de uma flexão repentina (mioclônica) do membro truncal e proximal seguida de um relaxamento lento (tônico) – e um EEG interictal mostrando hipsarritmia (ondas polimórficas irregulares difusas e lentas de alta voltagem e um arranjo desorganizado de picos).
Tipicamente, um enfraquecimento elétrico difuso no EEG acompanha o ataque clínico. Existem duas formas de síndrome de West: criptogênica e sintomática. O início da forma criptogênica ocorre no período de 3 a 7 meses de idade. O retardo mental é frequente, embora o desenvolvimento de alguns lactentes seja normal. Os pacientes respondem de maneira favorável ao ACTH.
A forma sintomática inicia mais cedo (antes dos 3 meses de idade), sendo que se observa um atraso evolutivo antes ou logo após o início da condição. As causas incluem anormalidades genéticas (geralmente, o complexo da esclerose tuberosa), lesão perinatal, malformações cerebrais e doença metabólica. A terapia inclui o uso de medicamentos como ACTH, vigabatrina (sobretudo para uso em pacientes com esclerose tuberosa) ou topiramato; de maneira geral, a resposta é fraca. Pode ocorrer uma progressão para a síndrome de Lennox-Gastaut.
Epilepsia mioclônica juvenil
(iv) O paciente N.M., 15 anos de idade, teve a primeira convulsão no chuveiro. O exame neurológico foi normal. Após ter sido questionado, confirmou ao médico do departamento de emergências que havia sofrido contrações corporais pela manhã durante alguns anos, mas que nunca havia pensado seriamente sobre o assunto.
A RNM do cérebro foi normal, embora o EEG tenha mostrado explosões frequentes de atividades pico-onda rápidas e generalizadas, que é uma forte sugestão de EMJ – síndrome heterogênea sob o ponto de vista genético. Tipicamente, essa síndrome inicia no período dos 12 aos 18 anos de idade, com múltiplos tipos de convulsão, incluindo convulsões mioclônicas, de ausência e CTCG.
Em geral, a mioclonia ocorre pela manhã, ao despertar. Os reflexos súbitos podem se apresentar de uma forma aglomerada, às vezes em um crescendo antes de convulsões generalizadas (embora, esse padrão não seja frequente). Os deflagradores convulsivos incluem privação do sono, consumo de bebidas alcoólicas, estresse emocional e estimulação fótica.
Os pacientes respondem muito bem ao valproato; outras indicações são topiramato e levetiracetam. A lamotrigina poderá agravar a mioclonia. A remissão espontânea é rara, sendo que a maior parte dos pacientes fazem tratamento por toda a vida.
Epilepsia sintomática generalizada
(v) O paciente V.B., 6 anos de idade, tem paralisia cerebral e convulsões desde a primeira infância. As convulsões são de diversos tipos e ocorrem praticamente todos os dias em combinações variadas: episódios de fixação ocular e de interrupção de atividade, reflexos corporais repetitivos e repentinos, episódios de enrijecimento, quedas inesperadas e convulsões totais. V.B. está tomando três anticonvulsivantes diferentes e apresenta um retardo moderado no desenvolvimento: consegue andar sem ajuda, mas necessita de assistência em outras atividades da vida cotidiana. O exame revelou que V.B. é um indivíduo comum, capaz de seguir comandos de um estágio, e com aumento no tônus nos quatro membros.
A situação de V.B. é típica de epilepsia generalizada sintomática, mais conhecida por síndrome de Lennox-Gastaut. O início desse tipo de transtorno ocorre durante o período de 1 a 8 anos de idade, com incidência de diversos tipos de convulsão: atônica, axial tônica, de ausência atípica e CTCG. Episódios de EE refratário são comuns.
As descobertas eletrencefalográficas típicas são lentificação do contexto e padrão de surto-supressão no sono; a atividade epileptiforme consiste de descargas generalizadas de picos e de ondas lentas e de explosões generalizadas de múltiplos picos durante o sono. Sob o ponto de vista clínico, há um atraso evolutivo.
As opções de tratamento incluem valproato, lamotrigina, felbamato e rufinamida, embora, de um modo geral, a resposta terapêutica seja fraca. Levando-se em conta a associação frequente com quedas, esses pacientes são orientados a usar capacete para evitar a ocorrência de lesões no cérebro. Outras intervenções potenciais incluem dieta cetogênica e calosotomia no corpo caloso.
Complicações da Epilepsia e transtornos relacionados
A epilepsia, incluindo os transtornos relacionados, pode apresentar algumas complicações para o paciente portador da doença. Dentre elas, serão abordadas, neste artigo, as seguintes: epilepsia refratária, estado epilético, morte inesperada súbita em pacientes epiléticos, suicídio, declínio cognitivo, efeitos colaterais das medicações, qualidade de vida inferior, restrições para dirigir e estigma.
Epilepsia refratária
Embora seja o tratamento básico para epilepsia, o uso de medicações é eficaz em apenas dois terços de indivíduos epiléticos. Isso significa que um terço das pessoas com epilepsia permanece sem controle da doença, apesar da adesão completa aos regimes prescritos.43 A mortalidade e a morbidade são substancialmente elevadas nesses pacientes sem controle ou refratários, sobretudo se os episódios se caracterizarem pela presença de convulsões generalizadas ou pela alteração no estado de consciência.
Os pacientes com epilepsia refratária correm um risco muito maior de morte causada por EE, de morte inesperada súbita em pacientes epiléticos (em inglês, sudden unexpected death in epileptic patients [Sudep]), acidentes ou suicídio.64 Os pacientes com convulsões incontroláveis sofrem de depressão ou de ansiedade, considerando que a natureza imprevisível desses eventos aumenta o estado de ansiedade, principalmente devido à perda de independência (restrição a dirigir carros), à perda de emprego e ao estigma percebido ou real.
A memória e outras disfunções cognitivas são queixas comuns e fontes de incapacitação. O rastreamento do risco de suicídio e de depressão é imprescindível. Por essas razões, os pacientes epiléticos com resistência aos medicamentos devem ser encaminhados para algum centro com experiência em gerenciamentos mais agressivos, incluindo politerapia ou cirurgia.
Estado epilético
EE é uma emergência médica com risco de vida, definida formalmente como uma “convulsão que persiste por um período de tempo suficiente ou que se repete com frequência suficiente de modo que não ocorre nenhuma recuperação entre ataques”.65
A definição oficial do tempo de duração de uma convulsão é de 30min, com base em estudos que demonstraram o período de tempo necessário para início de uma lesão neuronal. Para a maioria dos especialistas, a definição operacional é de 5min. A incidência cumulativa de EE varia de 10 a 61 em 100.000 indivíduos/ano nos EUA, e provavelmente seja maior nos países em desenvolvimento.
A incidência é mais elevada em pessoas muito jovens e em idosos.66 A causa mais comum de EE é a falta de adesão à terapia com AEDs.67 A mortalidade aumenta com fatores como avanço da idade, duração da convulsão e nível deprimido de consciência na apresentação.67,68 Embora o EE convulsivo generalizado seja a forma reconhecida mais comum, o EE pode ser parcial complexo, de ausência, parcial simples ou não convulsivo.
Em geral, o EE de ausência se deve a alguma agravação, possivelmente pela seleção do AED inapropriado em pacientes com epilepsias generalizadas conhecidas idiopáticas ou sintomáticas. O EE parcial simples se manifesta com convulsões motoras (epilepsia parcial contínua), o qual é classicamente associado à encefalite de Rasmussen; entretanto, o EE parcial também foi observado em associação com condições como doença de Creutzfeldt-Jakob (DCJ), vírus da imunodeficiência adquirida (HIV), tumores e AVC.
Os episódios não convulsivos foram observados com frequência cada vez maior em pacientes portadores de alguma enfermidade neurológica crítica, na medida em que o monitoramento contínuo com EEG passou a ser disponibilizado mais prontamente; nas UTIs para atendimento de pacientes neurológicos, os episódios não convulsivos ocorrem em 18 a 37% de pacientes monitorados, sendo que a maior parte desses indivíduos tem EE não convulsivo.69-72 Isso exige um elevado índice de suspeita.
O monitoramento com EEG por períodos de tempo superiores a 24 horas facilita a detecção dessas convulsões, considerando que apenas 50 a 60% de casos são percebidos na primeira hora.72 O gerenciamento dessas convulsões é particularmente desafiador, tendo em vista que é difícil interpretar muitos padrões eletrencefalográficos nessa população de pacientes e, com frequência, não fica claro se os episódios não convulsivos são meramente epifenômenos da lesão cerebral subjacente. Esse fato é demonstrado com maior clareza em pacientes com EE causado por lesão cerebral anóxica grave, em que o tratamento de convulsões pode se tornar fútil, a não ser que seja feito por razões cosméticas.
A fase inicial do EE convulsivo é hiperdinâmica; ocorre uma liberação maciça de catecolaminas, leucocitose, edema pulmonar e hiperglicemia, resultando em condições como hipertensão, hipertermia e taquicardia. A autorregulação cerebrovascular é mantida. A última fase do EE (>30min) é exaustiva e se caracteriza por fatores como ausência de autorregulação, hipotensão, hipoglicemia e arritmia cardíaca.73 Nos estágios iniciais, esses efeitos sistêmicos do EE devem ser gerenciados de forma agressiva.
Nas situações em que as convulsões persistem, os receptores de Gabaa são internalizados e permanecem fora do alcance dos neurotransmissores. Por outro lado, a subunidades de receptores de NMDA são mobilizadas para a membrana sináptica.74 Isso tem implicações abrangentes no tratamento pelas seguintes razões: o tratamento se torna mais eficaz se for administrado logo no início; os benzodiazepínicos perdem a eficácia à medida que o EE progride.
O gerenciamento deve iniciar com diazepam retal (0,2mg/kg), lorazepam sublingual (1mg) ou outros benzodiazepínicos, antes mesmo da chegada ao hospital. Esses tratamentos imediatos fora do ambiente hospitalar são seguros e eficazes.75 No hospital, o protocolo padrão de EE deve permanecer à disposição. Os medicamentos de primeira linha devem incluir uma benzodiazepina e um AED convencional.
Grande parte das instituições utiliza fenitoína ou fosfenitoína; entretanto, a maioria dos especialistas usa o levetiracetam, pois é mais bem tolerado e há evidências de sua eficácia, mas não de Classe I.76 O valproato e o fenobarbital são usados com mais frequência como agentes de segunda linha, embora ainda existam evidências de eficácia de primeira linha.
Nas situações em que as medidas não tiverem êxito, deve-se iniciar a administração de um segundo AED convencional; é imprescindível providenciar o monitoramento eletrencefalográfico e a transferência para a UTI (ou para um centro terciário de tratamento). O coma anestésico é uma opção para EE refratário. Os fármacos propofol e midazolam estão associados a menos efeitos colaterais relacionados ao tratamento, embora o pentobarbital tenha maior probabilidade de colocar o paciente em supressão de surto mais profunda.
O agente deverá ser titulado para supressão de surto por um período de 12 a 48h e, a seguir, a titulação deverá ser reduzida lentamente. Durante esse período, deve-se manter uma alta dose de AEDs convencionais. Se essas medidas não tiverem efeito, pode-se tentar outras menos convencionais, tais como cetamina, isoflurano, lidocaína, IVg/plasmaférese/esteroides, eletroconvulsoterapia (ECT), hipotermia ou cirurgia. Todas elas apresentam um risco significativo de efeitos colaterais iatrogênicos.
Morte inesperada súbita em pacientes epiléticos
Uma das consequências mais temerárias das convulsões incontroláveis é a morte súbita, que é definida como morte repentina e inesperada, com ou sem testemunhas, não traumática e sem afogamento em pessoas epiléticas, com ou sem documentação de convulsão, excluindo-se os casos de EE nos quais o exame post-mortem não tenha revelado a presença de causas estruturais ou toxicológicas.77
O risco de Sudep aumenta consideravelmente com os seguintes fatores: aumento na frequência das convulsões, uso de politerapia, paciente do sexo masculino, retardo no desenvolvimento, posição em prono na cama e idade entre 20 e 40 anos. Não se conhece com detalhes o fator responsável por uma proporção substancial de mortes em pacientes com epilepsia incontrolável e convulsões recentes, de modo que se enfatiza a necessidade de adesão ao tratamento.
O Quadro 6 apresenta o Protocolo para o Tratamento de EE.
Quadro 6
|
PROTOCOLO PARA O TRATAMENTO DE ESTADO EPILÉTICO | |
|
Tempo (min)/Estágio |
Ação |
|
0-4 |
Fazer o diagnóstico. |
|
Estabelecer acesso IV. | |
|
Administrar oxigênio com máscara. | |
|
Monitorar o EEG e a oximetria de pulso. | |
|
Coletar sangue para CBC, estudos de coagulação, eletrólitos, cálcio, magnésio, fosfato, níveis de AED, gases no sangue, rastreamento toxicológico. | |
|
Administrar antipiréticos em caso de necessidade. | |
|
Proteger as vias respiratórias, fazer sucção frequente, intubar em caso de necessidade. | |
|
5-10 (I) |
Administrar 100mg IV de tiamina, em 50mL de dextrose, a 50% se o paciente for hipoglicêmico. |
|
Administrar 4mg IV de lorazepam por 2min; repetir dentro de 5min se as convulsões persistirem. | |
|
10-20 (IIa) |
Administrar 20mg PE/kg IV de fosfenitoína a 150mg/min. |
|
20-40 (IIIb) |
Transferir para tratamento intensivo. |
|
Administrar 20mg/kg IV de fenobarbital (intubar em caso de necessidade). | |
|
Considerar a adição de 40mg IV de valproato; bolo adicional de 10mg PE/kg de fosfenitoína. | |
|
>40 (III; EE refratário) |
Administrar um bolo de 5-10mg/kg IV de pentobarbital ou um bolo de 0,2mg/kg IV de midazolam ou um bolo de 1-2mg/kg de propofol. |
|
Acompanhar o bolo do medicamento selecionado com uma infusão IV de manutenção com monitoramento contínuo com EEG. | |
AED: medicamento antiepilético; CBC: hemograma completo; EE: estado epilético; EEG: eletrencefalograma; IV: intravenoso; PE: equivalentes de fenitoína.
Suicídio
Alguns estudos mostraram que algumas condições psiquiátricas, como depressão e ansiedade, são mais comuns nos casos de epilepsia refratária em comparação com outros grupos. Aparentemente, a depressão também é um “fator de risco” para epilepsia79 e, sem dúvida, para o suicídio.
O risco de suicídio é mais elevado em pacientes epiléticos, sendo responsável por 5 a 10% de mortes, em comparação com 1,4% na população em geral.80 A epilepsia de novo início aumenta o risco, sobretudo em indivíduos com enfermidade psiquiátrica comórbida.7 Em 2008, a FDA publicou um alerta sobre o risco de suicídio com o uso de AEDs. A análise de 199 estudos de AEDs controlados por placebo, envolvendo 11 AEDs, mostrou que o risco de pensamentos suicidas era 3 vezes mais elevado em pacientes no braço de tratamento, em comparação com o braço de placebo.
Estudos subsequentes confirmaram o risco de suicídio em pacientes que usam AEDs, embora condições comórbidas tenham sido determinantes significativos na ideação suicida relacionada ao uso de AEDs.81 Os resultados de alguns estudos são conflitantes sobre quais medicamentos colocam os pacientes no nível mais elevado de risco.82,83
Todos os pacientes devem ser monitorados com muita cautela sobre a ideação suicida sempre que iniciarem o uso de um novo AED. Cabe ressaltar que o risco de não tomar nenhum AED é muito maior que a preocupação com a ideação suicida atribuível aos AEDs.
Declínio cognitivo
As queixas cognitivas, em especial de memória fraca, estão entre as mais comuns entre os epiléticos. Na realidade, muitos pacientes e suas famílias entendem que as comorbidades cognitivas e comportamentais são muito mais preocupantes que as convulsões. Essas questões são observadas mais claramente em indivíduos com uma frequência maior de crises, com convulsões de maior duração e com epilepsia de novo início, o que leva a crer que as convulsões sejam parte do problema.
Há evidências cada vez mais conclusivas de que mesmo as descargas epiléticas estejam associadas a perturbações cognitivas84 e de que ocorram transtornos nas redes cerebrais totais, conforme demonstram as imagens de RNMs funcionais.85 Entretanto, em muitos pacientes as dificuldades cognitivas podem preceder a primeira convulsão, indicando que existem muitos fatores contribuintes.
Está bastante claro que o uso de medicações, a perturbação do sono e a depressão também podem ser incluídas nas queixas cognitivas por meio da perturbação dos sistemas de vigília. Depois de cirurgias no lobo temporal para tratamento de epilepsia, observou-se que indivíduos com crises continuadas apresentavam maior declínio nas funções cognitivas do que aqueles que não tinham episódios convulsivos.86
Efeitos colaterais das medicações
Os AEDs produzem vários efeitos adversos, e sua tolerância é bastante difícil. Os efeitos adversos mais comuns são produzidos pela dose inicial e incluem sedação, tontura e desconforto gastrintestinal, que poderão ser melhorados com a titulação mais lenta do medicamento. Os efeitos limitadores de dosagem, tais como visão dupla, cefaleia, náusea, lentidão cognitiva e ataxia, poderão ser revertidos interrompendo-se o uso do medicamento; normalmente, esses efeitos são atenuados pela redução na dose.
Os efeitos idiossincráticos raros, porém mais sérios, de determinados tipos de AED como a síndrome de Stevens-Johnson ou pancreatite não podem ser previstos de imediato e deverão ser rigorosamente monitorados. Para finalizar, existem efeitos de longo prazo produzidos pelo uso de AEDs.
A redução na densidade mineral óssea causada pelo uso crônico de agentes indutores da enzima P-450 como a fenitoína, fenobarbital e carbamazepina vem recebendo uma atenção especial. O preditor mais sensível de fratura é a densidade mineral óssea, em combinação com fratura anterior ou com histórico familiar de osteoporose. Nesses indivíduos, a absorciometria radiológica de dupla energia é um método razoável para rastrear o tratamento, em vez da reposição usual de cálcio e de vitamina D.87
Qualidade de vida inferior
Os desfechos para a saúde foram medidos em pessoas com epilepsia. Os dados gerados pelos pacientes sobre a qualidade de vida têm grande potencial para alterar as decisões sobre o tratamento, além das considerações tradicionais de contagens de convulsões e dos efeitos colaterais de medicamentos específicos.
A meta do tratamento de epilepsia, assim como de qualquer outra condição crônica, é ajudar os pacientes a maximizarem seu potencial para um tipo de vida pleno e produtivo e, na medida do possível, a se sentirem normais. Portanto, é imprescindível avaliar a qualidade de vida e os efeitos colaterais dos medicamentos, levando em conta que muitos pacientes poderão ter poucas crises e, mesmo assim, ter baixa qualidade de vida.
As dificuldades sociais também constituem um problema significativo para indivíduos epiléticos, e contribuem para fatores como incidência mais baixa de casamentos, taxa mais baixa de emprego e qualidade de vida inferior. Alguns fatores biológicos que se correlacionam com aumento na incidência de enfermidades comórbidas nos casos de epilepsia são idade mais jovem no começo das convulsões, histórico de danos cognitivos, início da epilepsia no lobo frontal ou no lobo temporal, status econômico mais baixo e função familiar precária.
Muitos esforços foram envidados para aumentar a consciência precoce dessas questões comórbidas não relacionadas às convulsões na tentativa de melhorar a qualidade de vida geral dos pacientes acometidos por essa doença.88
Restrições para dirigir
A capacidade para dirigir tem amplas implicações no trabalho, na sociabilidade, impulsionando, inclusive, as visitas ao médico,89 em especial nas regiões dos EUA em que a sociedade foi estruturada com base em uma cultura que se baseia em dirigir o próprio carro para fins de transporte. O acesso ao transporte público em áreas metropolitanas é bastante precário.
Aproximadamente, 700 mil pacientes epiléticos possuem autorização para dirigir.90 As restrições para dirigir para pacientes epiléticos são reguladas por cada um dos Estados; os requisitos variam de 3 a 18 meses sem ter crises. Os outros seis Estados (Califórnia, Delaware, Nevada, New Jersey, Oregon e Pensilvânia) exigem, obrigatoriamente, um relatório médico.
De maneira geral, essas medidas são contestadas pelos neurologistas, que se preocupam com eventuais sonegações de informações por parte dos pacientes para continuarem dirigindo. Entretanto, mesmo nos Estados em que não seja exigido, o relatório médico torna-se necessário nas situações em que pacientes com epilepsia incontrolável insistirem em continuar dirigindo contra a orientação médica.
Alguns Estados têm regulamentações mais flexíveis que levam em consideração fatores individuais. Esses fatores incluem se as convulsões do paciente são estritamente noturnas, se as convulsões foram desencadeadas por alterações na medicação inicialmente prescrita pelo médico responsável pelo tratamento ou se as convulsões são estritamente parciais simples. O período de tempo ideal de duração é desconhecido, sendo que intervalos mais curtos sem nenhuma convulsão não aumentam, necessariamente, o número de acidentes de carro relacionados a esse tipo de condição.91
No que diz respeito às comorbidades, aparentemente os pacientes epiléticos não causam mais acidentes de trânsito que os pacientes não epiléticos;92 o risco relativo é mais baixo em comparação com pacientes portadores de outras condições médicas, tais como alcoolismo, diabetes ou doença mental.
A responsabilidade legal é um tema emergente em alguns Estados em que os médicos são obrigados a determinar as restrições para dirigir, porém não são providos de imunidade explícita.90 Os médicos devem conhecer as leis de trânsito de seus Estados, bem como orientar adequadamente os pacientes com epilepsia ativa a documentar a orientação no relatório médico.
Estigma
Muitas pessoas com epilepsia podem ter vidas bem-sucedidas e produtivas e continuar trabalhando em regime de tempo integral. Embora as atitudes do público em relação aos portadores de epilepsia tenham melhorado devido à atenção dada ao assunto pela mídia, o estigma social ainda é um grande problema, mesmo que as convulsões sejam controladas. As crianças epiléticas têm taxas de dificuldades sociais mais elevadas, as quais carregam consigo até a vida adulta.93
O conhecimento sobre o tema e a redução nas atitudes públicas negativas devem ser incentivados. O reconhecimento legal, por meio do Americans with Disabilities Act, de que a epilepsia é um tipo de incapacitação dá proteção legal às pessoas epiléticas nos locais de trabalho, de modo que é possível providenciar uma acomodação razoável para as pessoas que não conseguem executar determinadas atividades em seu trabalho. Apesar disso, muitos indivíduos preferem não revelar serem portadores da doença, causando “surpresas” aos demais, o que é uma situação bastante séria. Isso pode ser deveras problemático para os adolescentes.
Prognóstico
Aproximadamente, dois terços de pacientes epiléticos conseguem controlar de modo completo, ou quase inteiramente, as crises com o uso dos medicamentos apropriados. Depois de muitos anos sem convulsões, alguns indivíduos podem, inclusive, interromper o uso de medicamentos sem risco de recorrência.
Após um determinado período de tempo sem convulsões, os pacientes questionam se a descontinuação do uso de AEDs é segura. Esse tema é bastante complexo. As vantagens da interrupção no uso de AEDs são óbvias. A redução de custos é um benefício considerável. Na maior parte dos casos, os pacientes se livram de efeitos colaterais como fadiga, por exemplo, fato que se torna aparente somente após a descontinuação dos medicamentos.
Além disso, inúmeros AEDs estão associados a efeitos colaterais de longo prazo, sobretudo em relação à saúde dos ossos. Deve-se considerar, também, que ainda não são conhecidos os efeitos colaterais crônicos dos AEDs mais recentes, os quais são usados como terapia de primeira linha.
Lembra-se que há um benefício psicológico no sentido de que os pacientes deixam de desempenhar o papel de indivíduos doentes, e há uma melhora da qualidade de vida social e profissional. Os riscos potenciais da descontinuação no uso de AEDs são evidentes. A recorrência de convulsões pode resultar em lesões inesperadas, em limitações ocupacionais e em restrições para dirigir carros. Os benefícios secundários dos AEDs (por exemplo, estabilização do humor com a lamotrigina, alívio da cefaleia com o topiramato) poderão se perder. Para finalizar, a ocorrência de crises depois de um longo período de controle adequado pode se tornar um retrocesso psicológico.
Portanto, a estimativa precisa de recorrência de convulsões após a descontinuação do uso de AEDs é fundamental – tema que foi abordado pelo Medical Research Council AED Withdrawal Study. Este estudo referencial randomizou pacientes para continuação ou retirada lenta de AEDs depois de 2 anos sem convulsões. Nos 2 anos seguintes, 22% dos pacientes que continuaram usando AEDs e 41% dos pacientes que interromperam o uso do medicamento tiveram convulsões.
Longos períodos de tempo sem crises, antes da descontinuação do medicamento, reduziram o risco de episódios convulsivos. Nesse estudo, os riscos que mais aumentaram a probabilidade da recorrência de convulsões incluíram o uso de diversos tipos de AED, ocorrência de crises após o início da terapia com AEDs, idade acima de 16 anos e episódios convulsivos generalizados associados a descargas de picos e ondas no EEG.
Além disso, foram encontrados outros riscos menores. Análises posteriores, feitas a partir desse estudo, resultaram em modelos estatísticos que permitem prever a taxa de recorrência estimada das convulsões de um único paciente.94 Nos casos de pacientes típicos que seriam eleitos para descontinuação de AEDs, sobretudo aqueles que não tiveram crises com uso de apenas um único AED por um período de 2 a 5 anos, com EEG e exame neurológico normais, a probabilidade de recorrência em 2 anos após a interrupção no uso do AED é de 20%; esse é o dobro do risco de recorrência (10%) em comparação com a continuidade no uso do medicamento.
Para esse tipo de candidato, é imprescindível avaliar a relação entre benefícios e riscos do uso de AEDs. Não existe nenhuma orientação para deixar de dirigir carros durante o período de abstinência do medicamento, embora seja prudente considerar um prazo mínimo de 3 meses. Os EEG após a interrupção da medicação ajudam a prever recorrências.
Há determinadas síndromes bem definidas, como a epilepsia rolândica benigna ou a epilepsia de ausência, em que os eventos epiléticos são, geralmente, “fatos superados”. No caso dos pacientes que não se enquadram nessas categorias, os preditores do sucesso da descontinuação de medicamentos incluem estado neurológico normal, ausência de convulsões por, pelo menos, 2 anos (quanto maior o período de tempo, melhor o resultado), um tipo de convulsão simples que tenha sido bem controlada logo após o diagnóstico e, em muitos estudos, EEGs normais.
Mesmo nas situações em que esses critérios são atendidos, o risco de recidiva é de, aproximadamente, 20 a 30%, sendo que o maior risco ocorre no primeiro ano. Não há evidências de que o tratamento precoce, ou qualquer tratamento, altere o histórico natural do transtorno. De acordo com vários preditores, há indivíduos que, provavelmente, nunca se livrarão das convulsões.
Fatores como epilepsia parcial, EEG epileptiforme, ausência de resposta à primeira medicação, menos de 6 meses entre a primeira e a segunda convulsão (ou múltiplas convulsões antes do início do tratamento), anormalidades persistentes no EEG e politerapia são preditores negativos e levam os especialistas a orientar seus pacientes a prosseguir com a medicação.
Os EEGs podem ser muito úteis para fazer a distinção entre epilepsia parcial e epilepsia generalizada, assim como para o prognóstico sobre o risco de recorrência depois de um evento epilético definitivo. Nos casos em que o EEG é epileptiforme, há probabilidade de mais de 80% de outra convulsão, embora, nos casos de EEGs normais, o risco de recorrência varie de 25 a 30%.
Os EEGs são menos úteis nas situações em que o histórico clínico é muito vago, impossibilitando a obtenção do diagnóstico. Os EEGs normais (ou, pelo menos, não epileptiformes) são observados em mais de 50% de pacientes depois de um evento epilético definitivo, embora a sensibilidade aumente para 92% após quatro EEGs.95 Alguns, mas nem todos, os estudos chegaram a comprovar a utilidade dos EEGs para responder perguntas sobre a necessidade de prosseguir com o uso de medicamentos anticonvulsivantes em indivíduos que passaram vários anos sem ter uma convulsão.
As hipóteses em que se deve considerar o encaminhamento para um especialista estão elencadas no Quadro 7, a seguir.
Quadro 7
|
HIPÓTESES DE ENCAMINHAMENTO PARA UM ESPECIALISTA
|
|
incerteza diagnóstica novo diagnóstico medicações que não fazem efeito ou convulsões que não são totalmente controladas efeitos colaterais complicados de uma medicação populações especiais (i.e., mulheres, idosos) gravidez interrupção no uso de um medicamento cirurgia para epilepsia |
Tópicos especiais
As consultas a neurologistas especializados em epilepsia podem ser úteis em muitas situações. Populações especiais como mulheres e idosos poderão ter algum benefício com esse tipo de consulta, tendo em vista que, para esses indivíduos, há abordagens exclusivas em relação ao tratamento e à segurança. A seguir, serão abordados tópicos relativos às pacientes do sexo feminino e aos idosos.
Questões femininas em epilepsia
As mulheres epiléticas apresentam um único problema médico que se fundamenta nos hormônios e na capacidade reprodutiva. Muitas pacientes percebem um aumento na relação da doença com o ciclo menstrual, imediatamente antes do sangramento ou no momento da ovulação. Essa condição é conhecida por epilepsia catamenial, a qual se caracteriza pelo aumento de duas vezes nas convulsões relacionadas ao período menstrual e ocorre, basicamente, em um terço das mulheres epiléticas.
Aparentemente, o estrogênio é epileptogênico, e a progesterona, antiepileptogênica em animais e em seres humanos. A suplementação de progesterona vem sendo utilizada com sucesso limitado para fins de tratamento em mulheres com epilepsia catamenial.96 A intensificação no uso de AEDs ou o uso de benzodiazepinas na fase perimenstrual obteve algum sucesso nas convulsões ocorridas durante esse período.97
As mulheres epiléticas apresentam um aumento na incidência de irregularidades no ciclo menstrual, distúrbios endócrinos reprodutivos como a síndrome do ovário policístico (SOP), hipogonadismo hipogonadotrófico e insuficiência ovariana prematura.96 Portanto, não chega a causar nenhuma surpresa o fato de que a infertilidade é mais elevada em mulheres epiléticas.
As gestações acidentais entre mulheres que tomam contraceptivos orais poderão ser minimizadas dando-se atenção especial aos efeitos dos AEDs no metabolismo hormonal. Medicações antiepiléticas que induzem as enzimas P-450, como a fenitoína, fenobarbital e carbamazepina, poderão resultar em falha contraceptiva, em especial nas situações em que forem usadas com baixas doses de contraceptivos hormonais.
Fármacos mais recentes como a oxcarbazepina e o topiramato também interferem na eficácia dos agentes de controle da natalidade, sobretudo em doses mais elevadas. Atenção especial a sintomas como hemorragia de escape e ajuste para doses mais elevadas e/ou uso contínuo de pílulas anticoncepcionais por via oral pode amenizar esse tipo de interação. As evidências mostram que os agentes contraceptivos orais podem diminuir drasticamente os níveis de lamotrigina e de valproato.
Nas visitas médicas, as mulheres epiléticas devem receber orientação apropriada em relação à concepção o mais rápido possível, tendo em vista que mais de 50% das gestações não são planejadas. Essas pacientes devem receber informações precisas sobre os riscos, embora a probabilidade de terem uma gravidez descomplicada, bem como de conceberem crianças normais, seja de 90%.
Existem vários órgãos que controlam o registro das gestações. Nos EUA, o encaminhamento para o North American AED Pregnancy Registry é muito importante para todas as mulheres epiléticas com gravidez recente (1-888-AED-AED4;
O uso de monoterapia com um AED durante o primeiro trimestre de gravidez pode estar associado à taxa basal de 1,5 a 3% das malformações congênitas mais comuns na população em geral. Entre os medicamentos mais antigos, o valproato representa um risco mais elevado, estando o fenobarbital em segundo lugar, a despeito de a literatura inicial afirmar o contrário.
Os dados mais recentes sugerem a existência de taxas mais elevadas de malformações congênitas maternas (MCMs) com o uso de topiramato.98 O valproato e a carbamazepina estão associados a um aumento na incidência de defeitos no tubo neural, e a suplementação com ácido fólico também não diminui esse risco. Entretanto, a suplementação com ácido fólico tem sido benéfica para reduzir a incidência de MCMs na população em geral.99
Em um grande número de gestações, a lamotrigina apresentou a incidência mais baixa de malformações, semelhante ao risco basal em mulheres. A carbamazepina também apresenta um risco menor do que se pensava anteriormente.100 Não há informações suficientes à disposição sobre muitos dos AEDs mais recentes, embora os dados cognitivos e teratogênicos sobre o levetiracetam sejam animadores.101
O risco de malformações congênitas maternas aumenta com a politerapia, sobretudo na hipótese de o valproato ser um dos medicamentos. Além disso, o valproato tem efeitos cognitivos adversos nas crianças que são expostas no útero, em comparação com a lamotrigina, a carbamazepina e a fenitoína.
Considerando que as CTCG (incluindo as secundariamente generalizadas) apresentam risco de aborto espontâneo e de lesões maternas e fetais, as mulheres com epilepsia ativa devem continuar usando o AED mais eficaz para seu tipo de convulsão durante todo o período de gestação, desde que não coloquem em risco a segurança reprodutiva.
A descontinuação ou a transição para outros medicamentos são fatores a serem considerados bem antes da concepção. Recomenda-se tratar com ácido fólico (nível C) todas as mulheres em idade concepcional que usam AEDs, com, no mínimo, 0,4mg/dia até o máximo de 5mg/dia. As evidências existentes não são suficientes para se recomendar o tratamento de mães com vitamina K antes do parto.102
A frequência de convulsões durante o período de gestação costuma permanecer a mesma dos períodos normais, podendo, eventualmente, aumentar. No caso de mulheres que tenham sido bem controladas por 9 meses antes da gravidez, é bastante provável que elas continuem se sentindo bem durante a gestação.103
É comum o aumento nas convulsões ocorrer no segundo e terceiro trimestres, quando os níveis sanguíneos apresentam tendência de queda, particularmente com o uso de lamotrigina e levetiracetam; o monitoramento sanguíneo mensal pode ser uma orientação bastante útil para aumentos de doses nesse contexto, sendo recomendado para medicamentos com tendência de queda.
A medição de níveis livres também tem alguma utilidade no caso de AEDs com alta ligação proteica (valproato e fenitoína), tendo em vista que, tipicamente, a ligação proteica diminui durante a gestação. A administração de benzodiazepinas de ação curta é uma opção caso ocorram convulsões no trabalho de parto e no parto.
Imediatamente após o parto, as mulheres epiléticas devem se conscientizar de que todos os AEDs penetram no leite materno, em especial no leite que não tiver uma alta ligação proteica. Os fármacos que apresentam ligação proteica mais elevada, como a fenitoína e o valproato, têm uma presença menor no leite materno. Esse fato é bastante significativo no caso de medicamentos sedativos como o fenobarbital e a primidona.
Entretanto, não há nenhuma contraindicação absoluta à amamentação, sendo que os benefícios superam os riscos,104 especialmente se o lactente tiver sido exposto durante todo o período de gestação. As mães com convulsões incontroláveis devem tomar precauções especiais sobre determinados aspectos dos cuidados dos lactentes e adotar medidas apropriadas como, por exemplo, trocar o bebê no assoalho, em vez de utilizar o trocador de fraldas, e não dar banho nos bebês sem supervisão.
Epilepsia em idosos
Os idosos (definidos como idade >65 anos) formam o segmento populacional que mais se expande nos EUA.105 A incidência de epilepsia aumenta de forma vertiginosa com o avançar da idade, superando a incidência no período neonatal na idade de 70 anos, e aumenta para mais de 200/10.000 pessoas com mais de 80 anos de idade.106
O diagnóstico e o gerenciamento da epilepsia em idosos podem se transformar em grandes desafios, e exigem alguma sensibilidade para as circunstâncias típicas desse grupo etário. A etiologia mais comum de epilepsia em idosos é o AVC. Aproximadamente, 30 a 40% de todas as convulsões em pessoas idosas se referem a pacientes que haviam tido AVCs anteriores.107
Outras causas importantes de crises incluem tumores cerebrais, lesões na cabeça e doença de Alzheimer (DA). O diagnóstico pode ser particularmente desafiador, tendo em vista que alguns pacientes se apresentam com episódios convulsivos generalizados. As crises parciais são facilmente confundidas com ataques isquêmicos transitórios.
De maneira geral, os pacientes com tumor cerebral ou DA se apresentam com oscilação no estado mental, que pode não ser distinguível de CPC. Fatores como sintomas neurológicos causados por distúrbios metabólicos, disfunção cardíaca ou efeitos colaterais de medicamentos são mais comuns em pessoas idosas. As causas provocativas de convulsões devem, uma vez mais, ser excluídas antes do diagnóstico de epilepsia. Não se encontra uma etiologia definitiva em 30 a 50% de pacientes. O surgimento de epilepsia generalizada idiopática autêntica na velhice é raro.
Com frequência, os resultados dos estudos de imagens em idosos são anormais e incluem atrofia ou doença microvascular, que, provavelmente, não façam parte dos sintomas. A interpretação do EEG transmite o mesmo aviso de alerta. Por conseguinte, pode-se subdiagnosticar ou superdiagnosticar facilmente a epilepsia, sendo que vale a pena encaminhar os pacientes para um centro especializado para monitoramento por EEG assistido com vídeo.
O tratamento medicamentoso da epilepsia em idosos deve ser considerado com muita cautela, levando-se em conta o estado metabólico dos pacientes e a presença de prescrições concomitantes ou de comorbidades. Diversas alterações farmacocinéticas se tornam significativas em pacientes biologicamente idosos, e poderão levar a níveis séricos imprevisíveis em um único indivíduo ou entre indivíduos: absorção errática, níveis séricos reduzidos de albumina resultando em alterações nas ligações dos medicamentos, fluxo sanguíneo hepático reduzido e massa hepática, taxa de filtração glomerular reduzida e capacidade indutiva reduzida do sistema de enzimas microssômicas.
Um estudo sobre o uso de fenitoína em residentes de casas de repouso, por exemplo, encontrou uma diferença de duas ou três vezes nas concentrações séricas longitudinais sem alterações na dose, sem uso de novas medicações ou sem enfermidades intercorrentes.108 Portanto, a introdução no uso de AEDs deve ser feita com muita cautela, usando-se programas de titulação lenta, dose mínima de manutenção, e dedicando atenção especial aos níveis séricos e às respostas dos pacientes.
As interações medicamentosas se tornam significativas em pacientes idosos que usam diversas outras medicações. É provável que a melhor prática seja a política de escolher um entre os AEDs mais recentes com interações medicamentosas mínimas, nenhuma ligação proteica e eliminação por via renal (por exemplo, levetiracetam, pregabalina, gabapentina, lacosamida).
Para finalizar, tem-se que as comorbidades podem sugerir escolhas naturais (pregabalina ou gabapentina em pacientes com dor neuropática; primidona em pacientes com tremor) ou agentes que devem ser evitados (barbitúricos em pacientes com demência). As evidências atuais sugerem que a lamotrigina é mais bem tolerada e, se possível, deve ser a primeira tentativa.
Entretanto, os anseios dos idosos são os mesmos dos pacientes jovens com epilepsia, incluindo a possibilidade ou não de dirigir, as questões profissionais, o embaraço social e a segurança, entre outros. Com relação aos efeitos colaterais dos medicamentos,109 como era de se esperar, as preocupações são ainda maiores.
Medições da qualidade
Existe uma população significativa de pacientes que não recebe o tratamento ideal para epilepsia. Esse hiato nos tratamentos é marcado sobretudo por disparidades raciais, étnicas ou de outros tipos.110 Na tentativa de garantir tratamentos mais aprimorados e uniformes, a AAN publicou, em 2011, um conjunto de medições da qualidade para o tratamento de epilepsia.
Essas medições consistiam de um conjunto de oito métricas de processo desenvolvidas como formas de medir o desempenho, e com o objetivo de serem implementadas na assistência médica primária e especializada.111 É provável que, com a documentação sobre essas medições, melhore o atendimento de pacientes epiléticos. As medições da qualidade em epilepsia estão elencadas no Quadro 8.
Quadro 8
|
MEDIÇÕES DA QUALIDADE EM EPILEPSIA111
|
|
Avaliar tipo e frequência da convulsão.* Estudar a etiologia da convulsão.* Observar o resultado do EEG. Obter RNM/TC anualmente. Verificar os efeitos colaterais dos AEDs a cada visita. Atentar para os temas relacionados à segurança, como, p.ex., dirigir. Fornecer orientação sobre temas referentes às mulheres.* Discutir anualmente a hipótese de intervenção cirúrgica. |
AEDs: medicamentos antiepiléticos; EEG: eletrencefalograma; RNM: ressonância nuclear magnética; TC: tomografia computadorizada.
*Essas medidas foram recomendadas para o ano de 2012 pelo Physician Quality Reporting System (PQRS).
Pode-se dizer, para finalizar, que há uma demanda pública crescente por dados relacionados à qualidade do atendimento médico. Não se tem certeza absoluta se essas medições serão aplicadas, embora, no futuro, possam vir a ser utilizadas para medir o desempenho; o Centers for Medicare & Medicaid Services (CMS) introduziu três dessas medições em 2012. Para saber mais, consultar o arquivo sidebar para Internet Sources for Epilepsy. http://www.aesnet.org/practice/practice-tools/epilepsy-and-women/epilepsy-pregnancy-registries).
O Quadro 9 contém algumas fontes na Internet para pesquisa sobre a epilepsia.
Quadro 9
|
FONTES PARA PESQUISA SOBRE A EPILEPSIA111
|
|
Leis sobre epiléticos e a obtenção de carteira de motorista: www.epilepsyfoundation.org/resources/drivingandtravel.cfm
Primeiros socorros: http://www.epilepsyfoundation.org/aboutepilepsy/firstaid/index.cfm
Depressão e ansiedade: http://www.epilepsyfoundation.org/aboutepilepsy/relatedconditions
Temas relacionados às mulheres e à gravidez: http://www.aesnet.org/practice/practice-tools/epilepsy-and-women/epilepsy-pregnancy-registries http://www.aesnet.org/go/practice/practice-tools/epilepsy-and-women/checklist-for-women
Temas relacionados ao local de trabalho:
|
|
Obs.: Os autores não mantêm relações comerciais com os fabricantes dos produtos e com os fornecedores de serviços mencionados neste artigo. |
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.