(Carregando Índice)... (Carregando Índice)... |
Última revisão: 20/05/2019
Comentários de assinantes: 0
|
Artigo original: Lang, A.E, MD. Slow E.J, MD, PhD. Parkinsonism and Related Disorders, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão Técnica: Dr. Lucas Santos Zambon.
|
Anthony E. Lang, MD
Professor na Clínica de Distúrbios Motores no Morton and Gloria Shulman e no Edmond J. Safra Program in Parkinson’s Disease do Toronto Western Hospital da University of Toronto (Toronto, ON)
Elizabeth J. Slow, MD, PhD
Professora Assistente na Clínica de Distúrbios Motores no Morton and Gloria Shulman e no Edmond J. Safra Program in Parkinson’s Disease do Toronto Western Hospital da University of Toronto (Toronto, ON)
Resumo
O parkinsonismo descreve os critérios clínicos básicos de tremor, bradicinesia, rigidez e instabilidade postural. Embora o diagnóstico diferencial seja bastante amplo, a doença de Parkinson (DP) é a causa mais comum de parkinsonismo. Este artigo apresenta detalhes sobre temas como epidemiologia, etiologia e genética, patogênese, diagnóstico e diagnóstico diferencial, gerenciamento e prognóstico para DP, demência com corpúsculos de Lewy (DCL), paralisia supranuclear progressiva (PSP), degeneração corticobasal (DCB), parkinsonismo vascular, hidrocefalia com pressão normal e parkinsonismo induzido por medicamentos (PIM). As figuras mostram as características da DP; sumário do diagnóstico; cronograma do curso clínico da DP; gerenciamento e prognóstico da DP; um algoritmo para o tratamento da DP com predominância de tremor; um algoritmo para o tratamento de DP com predominância de bradicinesia e rigidez; alterações em imagens por ressonância nuclear magnética (IRMs) na atrofia de múltiplos sistemas (AMS); alterações em IRMs em casos de PSP; e aumento nos ventrículos associado à hidrocefalia com pressão normal. Os quadros apresentam uma lista de diagnósticos diferenciais de parkinsonismo; dicas de causas alternativas de parkinsonismo não relacionadas à DP (isto é, sinais de alerta de parkinsonismo); como distinguir outras doenças que causam parkinsonismo da DP; tratamento de sintomas motores na DP; efeitos colaterais periféricos e centrais das terapias com agentes dopaminérgicos (agonista da dopamina e levodopa); e tratamento de sintomas não motores na DP.
Definição de Parksonismo
O parksonismo é um termo utilizado para descrever dois ou mais grupos de um subgrupo de sintomas. Esses sintomas incluem:
tremor;
rigidez (uma descoberta feita no exame físico, que, às vezes, é descrita pelo paciente como “enrijecimento”;
lentidão e pobreza de movimentos voluntários (denominadas bradicinesia ou acinesia);
instabilidade postural (evidenciada pela ocorrência de quedas ou identificada no exame físico);
postura encurvada;
presença de congelamento (interrupção breve em um movimento, especialmente evidente ao caminhar).
O método mnemônico TRAP (tremor/tremor; rigidity/rigidez; akinesia/acinesia; postural instability/instabilidade postural) ajuda as pessoas a se lembrarem dos sintomas que descrevem as quatro características principais da doença. A maioria das definições ou dos critérios requer a presença de bradicinesia como uma das características do complexo de sintomas.
Embora o diagnóstico diferencial para parksonismo seja bastante amplo, a causa mais comum da doença em pacientes com mais de 50 anos de idade é a doença de Parkinson (DP), às vezes conhecida por DP “idiopática”. Inúmeros outros distúrbios se apresentam com o parksonismo, incluindo atrofia de múltiplos sistemas (AMS), demência com corpúsculos de Lewy (DCL), paralisia supranuclear progressiva (PSP), degeneração corticobasal (DCB), parksonismo vascular, hidrocefalia normobárica (HNB) e parkinsonismo induzido por medicamentos (PIM).
Doença de Parkinson
Epidemiologia
A DP inicia aos 65 anos de idade,1 porém a faixa etária de incidência é ampla, incluindo DP com início em idades mais jovens (tipicamente, abaixo de 40 anos). A incidência e a prevalência aumentam com o avanço da idade. As estimativas de incidência variam bastante, embora, entre as idades de 55 e 65 anos, a incidência, por 1 mil pessoas, seja de 0,3, aumentando para 4,4 em pacientes com mais de 85 anos de idade.2 Estima-se em 0,3% a prevalência na população total, porém, na população acima de 60 anos, esse número aumenta para 1%. Há uma proporção de 1,5:1 entre homens e mulheres.2
Vários estudos epidemiológicos realizados ao longo dos anos sugerem que há um aumento no risco de desenvolvimento de DP nos casos em que há histórico da doença na família, lesão na cabeça, exposição a pesticidas e constipação, embora, provavelmente, a constipação seja uma manifestação da doença que precede o início das características óbvias (isto é, sintoma “pré-motor” ou “prodromal”).4 Os fatores potenciais de proteção incluíam histórico de tabagismo, consumo de cafeína e hipertensão.4
Etiologia e Genética
A DP é um distúrbio neurodegenerativo, e a perda neuronal óbvia afeta as células da substância negra que produzem dopamina. A perda desses neurônios e a perda subsequente do sistema dopaminérgico nigroestriado são responsáveis por muitos sinais e sintomas de parkinsonismo.
Corpos de Lewy são inclusões compostas de uma agregação proteica anormal encontrada nos neurônios de pacientes com DP, e são a marca registrada patológica da doença. As inclusões se compõem de a-sinucleína (em combinação com muitas outras proteínas) que são encontradas nos neurônios (corpos de Lewy) e nas projeções neuronais (neurites de Lewy).
O Quadro 1 apresenta o diagnóstico diferencial de parkinsonismo e o Quadro 2, as causas alternativas não-DP de parkinsonismo (sinais de alerta de parkinsonismo).
Quadro 1
|
Diagnóstico Diferencial de Parkinsonismo |
|
Neurodegenerativo DP: esporádica e genética* AMS* PSP* DCB* DCL* DH Doença de Alzheimer Doença de Wilson? Calcificação nos gânglios basais (doença de Fahr) Ataxias espinocerebelares (p.ex., SCA-2, SCA-3) Distonia responsiva ao levodopa (não degenerativa) Medicamentos* Agentes bloqueadores do receptor da dopamina (neurolépticos e antieméticos como a metoclopramida), tetrabenazina, lítio, bloqueadores do canal de cálcio (p.ex., flunarizina) Toxinas Manganês, MPTP, monóxido de carbono Vascular* Hidrocefalia com pressão normal* Infeccioso Parkinsonismo pós-encefalítico Doença de Creutzfeldt-Jakob Neoplásico Relacionado a lesões na cabeça |
AMS: atrofia de múltiplos sistemas; DCB: degeneração corticobasal; DCL: demência com corpúsculos de Lewy; DH: doença de Huntington; DP: doença de Parkinson; MPTP: 1-metil-4-fenil-1,2,5,6-tetrahidropiridina; PSP: paralisia supranuclear progressiva.
*A cobertura completa está no texto.
?É extremamente importante considerar e excluir a doença de Wilson em todos os pacientes mais jovens (geralmente < 40 anos, embora ocasionalmente possa ocorrer mais tarde).
Quadro 2
|
Causas Alternativas Não-DP de Parkinsonismo (Sinais de Alerta de Parkinsonismo) |
|
Anormalidades no movimento dos olhos (p.ex., nistagmo, limitação na fixação do olhar vertical, em especial na fixação do olhar para baixo). Disfagia/disartria, precoce e grave. Ataxia: nos membros e na marcha. Sinais nos neurônios motores superiores (p.ex., clônus, reflexos bruscos, espasticidade, reflexos plantares no extensor). Disfunção nos nervos periféricos (p.ex., perda de reflexos). Demência precoce e grave. Quedas/instabilidade postural/congelamento da marcha no início do curso da doença. Parkinsonismo na parte inferior do corpo, com preservação relativa da função dos membros superiores.* Disfunção autonômica, precoce e proeminente. Apraxia/alterações sensoriais corticais/fenômeno do membro alheio. Resposta fraca ao levodopa.? |
DP: doença de Parkinson.
*Não deve ser confundido com a situação comum que se observa no estágio final da DP, em que a função dos membros superiores continua a responder relativamente bem ao levodopa, embora a resposta da “metade inferior” do corpo seja muito mais fraca, o que deixa o paciente com disfunção persistente na marcha e instabilidade postural.
?Às vezes, a presença dessas características clínicas encoraja o uso do termo parkinsonism plus. A ausência de uma resposta ao levodopa é outra dica importante para um diagnóstico alternativo (i.e., parkinsonism minus).
Um trabalho que foi realizado em uma série de autópsias propõe a hipótese de uma disseminação ascendente da patologia de a-sinucleína no interior do cérebro, iniciando na medula ou na parte inferior do tronco cefálico, atingindo a substância negra no estágio intermediário e, finalmente, atingindo as áreas corticais nos estágios finais da doença.
O sistema nervoso autonômico periférico poderá ser afetado mesmo antes do envolvimento do cérebro. Essa hipótese ajuda a explicar as manifestações pré-motoras precoces de DP (incluindo alterações olfativas e transtornos do sono explicados pela ocorrência de alterações na parte inferior do tronco cefálico) e complicações tardias da DP, incluindo demência (explicada por alterações nas regiões corticais).5
O Quadro 3 mostra como distinguir outras doenças que causam parkinsonismo de DP.
Quadro 3
|
Como Distinguir Outras Doenças que Causam Parkinsonismo de Doença de Parkinson | |||
|
Parkinsonismo |
Características diferenciadoras |
Resposta ao levodopa |
Imagens |
|
AMS |
Disautonomia proeminente, disfunção cerebelar (ataxia), sinais no trato piramidal, mioclonia sensível a estímulos, sintomas respiratórios (apneia, estridor), disartria proeminente. |
Até 40% no início; possibilidade de desenvolvimento de distonia craniana causada pelo levodopa. |
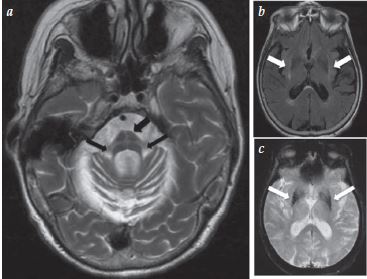
MSA-C: sinal hot cross bun, ponte; MSA-P: alterações estriadas. |
|
PSP |
Quedas precoces, paralisia na fixação do olhar supranuclear vertical, alterações cognitivas e comportamentais. |
Resposta leve em 30%. |
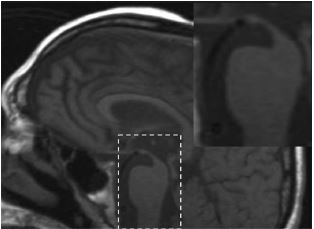
Atrofia no mesencéfalo (p.ex., sinal humminbird).
|
|
DCB |
Disfunção cognitiva, apraxia, membro estranho, perda sensorial cortical; rigidez assimétrica, distonia; mioclonia sensível a estímulos. |
Inexpressiva. |
As IRMs podem mostrar atrofia cortical assimétrica pronunciada. |
|
DCL |
Demência, alucinações visuais, nível oscilante de consciência, sensibilidade para agentes neurolépticos, transtorno do comportamento do sono REM. |
Os sintomas motores podem responder bem. |
- |
|
HNB |
Problemas cognitivos, sintomas urinários, parkinsonismo na parte inferior do corpo (“apraxia da marcha”). |
Geralmente, fraca. |
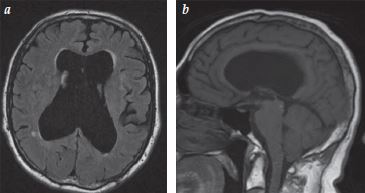
Aumento ventricular desproporcional no grau de atrofia cerebral. |
|
Parkinsonismo vascular |
“Parkinsonismo na parte inferior do corpo”, sinais neurológicos adicionais (p.ex., espasticidade, fraqueza). |
Usualmente fraca, porém com respostas ocasionais. |
As IRMs devem mostrar alterações isquêmicas. |
|
PIM |
Possivelmente, apresente todas as características do parkinsonismo clássico da DP, incluindo tremor em repouso; geralmente, é simétrico; pode ser acompanhado de outros distúrbios motores induzidos por medicamentos (p.ex., discinesia tardia, que se torna evidente apenas após a descontinuação no uso do agente causador). |
Em geral, é baixa, levando-se em consideração que os receptores da dopamina geralmente estão bloqueados. |
- |
AMS: atrofia de múltiplos sistemas; DCB: degeneração corticobasal; DCL: demência com corpúsculos de Lewy; DP: doença de Parkinson; HNB: hidrocefalia normobárica; IRMs: imagens por ressonância magnética; MSA-C: atrofia de múltiplos sistemas com manifestações cerebelares; MSA-P: atrofia de múltiplos sistemas com manifestações parksonianas; PIM: parkinsonismo induzido por medicamentos; PSP: paralisia supranuclear progressiva; REM: movimentos oculares rápidos.
Enquanto 90% dos casos de DP são esporádicos, estima-se que 10% sejam causados por causas genéticas. O provável primeiro gene causativo das formas familiares de DP foi o SNCA, que codifica a proteína a-sinucleína.6 As mutações, duplicações e triplicações desse gene produzem uma forma herdada autossômica dominante de DP.
Com frequência, essa forma familiar tem início em idades mais jovens e, tipicamente, é mais grave que as outras formas herdadas de DP e se caracteriza pelo desenvolvimento de alterações cognitivas e comportamentais; além disso, a progressão da doença é muito mais rápida.7 Outras formas hereditárias de DP com início em idade mais jovem são causadas por mutações nos genes PRKN (codifica a proteína parkina), PINK1 e DJ-1.
As mutações nesses genes produzem formas autossômicas recessivas de DP. De um modo geral, essas três formas familiares de DP têm um curso progressivo mais lento e com menos disfunções cognitivas.7 As evidências indicam que as mutações nesses três genes alteram a função mitocondrial por três vias distintas, o que sugere a participação da disfunção mitocondrial nos casos de DP.
A mutação genética mais comum nos casos de DP descoberta até o presente momento é a do gene LRRK2, que codifica a repetição da quinase rica em leucina. As mutações autossômicas dominantes nesse gene são responsáveis por mais de 1% dos casos de DP esporádica em todo o mundo.8 As mutações em LRRK2 produzem DP com início em idade mais avançada que, no momento, não é possível ser distinguida de DP esporádica.8 As mutações em LRRK2 aumentam a atividade da quinase, implicando na participação das quinases na patogênese de DP.
Embora não sejam diretamente causativos, os maiores fatores de risco genético para o desenvolvimento de DP são as mutações no gene GBA, que codifica a ß-glicocerebrosidase, a enzima lisossômica cuja deficiência se observa na doença de Gaucher.7,9
Patogênese
Inúmeros mecanismos foram envolvidos na patogênese da DP. Hipoteticamente, fatores como neuroinflamação, perda de fatores tróficos e disfunção mitocondrial contribuem para a disfunção e perda neuronal nos casos de DP.7
Acredita-se que as dobras proteicas anormais e a agregação de a-sinucleína desempenhem algum papel no processo, tendo em vista que a a-sinucleína é o principal componente proteico dos corpos de Lewy e das neurites de Lewy, os grandes pilares patológicos da DP. Nos anos mais recentes, surgiu uma nova teoria afirmando que a a-sinucleína poderá se disseminar de uma forma priônica em todo o cérebro.10
Diagnóstico
Manifestações Clínicas
Define-se DP como o início de manifestações motoras, incluindo tremor, rigidez, acinesia/bradicinesia e instabilidade postural. Com frequência, os sintomas iniciam em um lado do corpo, sendo que essa assimetria poderá persistir em todo o curso da doença. A presença de tremor é comum no estado de repouso e desaparece com postura ou ação, tais como usar utensílios domésticos e beber líquidos em um copo.
Na maior parte dos casos, o tremor em repouso se manifesta primeiramente no braço ou na perna, embora ocorra também nas bochechas. Ao longo do tempo, o tremor poderá envolver outras partes do corpo e, possivelmente, se torne evidente mesmo com a postura e ação. De um modo geral, descreve-se a rigidez nos históricos de “enrijecimento”.
Os pacientes poderão se queixar de enrijecimento nos ombros como manifestação inicial e, nessa circunstância, talvez sejam investigados na busca de causas musculoesqueléticas para a dor naquele local. A lentidão motora possivelmente se manifeste com queixas na marcha (lentidão ao caminhar) ou de dificuldade para usar as mãos (por exemplo, abotoar as roupas). Os pacientes se queixam de que levam mais tempo para executar as atividades diárias ou hobbies (por exemplo, tocar piano) do que anteriormente.
A instabilidade postural se torna mais evidente pelo histórico de quedas, embora, na fase inicial do curso da doença, seja descrita como “falta de equilíbrio” ou “instabilidade”. De um modo geral, as quedas no estágio inicial da doença indicam um diagnóstico diferente de DP. Outras manifestações motoras incluem hipomimia ou perda da expressão facial; alteração na voz, com voz suave ou hipofonia e, ocasionalmente, fala arrastada; disfagia; e micrografia, conforme a Figura 1.11

DP: doença de Parkinson.
Figura 1 - Características da DP: (A) hipomimia (face mascarada); (B) micrografia.
De igual importância no histórico é a evidência das manifestações não motoras da DP. A disfunção autonômica, na forma de disfunção urinária, mais frequentemente bexiga superativa, hipotensão ortostática e disfunção erétil, é muito comum nos casos avançados de DP, embora o início precoce desses sintomas seja a indicação de uma causa atípica de parkinsonismo (e não de DP), mais notadamente atrofia de AMS.
As manifestações neuropsiquiátricas, incluindo psicose e disfunção cognitiva, também são muito comuns nos casos avançados de DP, embora sejam também sinais de alerta sugerindo outra causa de parkinsonismo nas manifestações precoces. A presença de depressão, ansiedade e apatia é comum em todos os estágios da doença.
As anormalidades no sono incluem insônia; sonolência excessiva durante o dia (EDS); distúrbio comportamental do sono com movimentos rápidos dos olhos (DCR), uma condição em que o paciente “age” fora dos sonhos; síndrome das pernas inquietas; e movimentos periódicos dos membros durante o sono.12
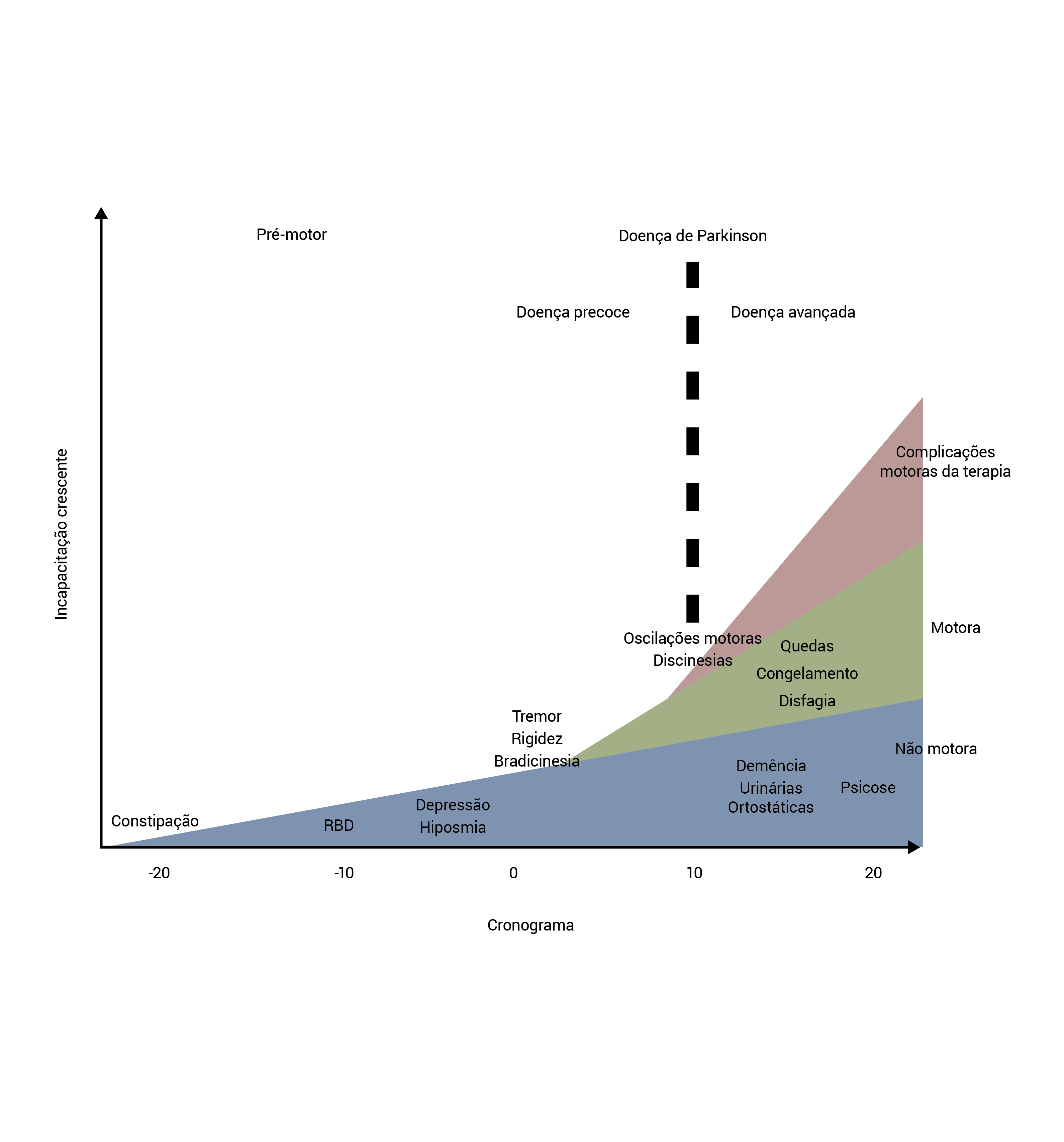
Existem evidências crescentes da presença de manifestações pré-motoras na DP.13 Esses sintomas prenunciam o desenvolvimento das características motoras clássicas de DP, conforme a Figura 2. A deficiência olfativa, comum em mais de 75% de pacientes,14 talvez preceda o diagnóstico de DP em 5 a 10 anos.13 A constipação é uma descoberta comum em todos os estágios da DP e pode preceder o diagnóstico em 15 anos.13 A depressão também pode preceder o diagnóstico de DP. O distúrbio comportamental do sono REM ocorre em 30% dos casos e pode preceder o diagnóstico em 15 anos ou mais.13
DP: doença de Parkinson; RBD: distúrbio comportamental do sono com movimentos rápidos dos olhos.
Figura 2 - Cronograma do curso clínico da DP. Tipicamente, os sintomas não motores de DP iniciam muitos anos antes dos sintomas motores (momento do diagnóstico = momento 0) e aumentam ao longo do tempo. Os sintomas motores aumentam no decorrer do tempo juntamente com as complicações das medicações. O conjunto desses fatores aumenta a incapacitação nos estágios avançados da DP. Esta figura foi modificada com base no original de Kalia LV e Lang AE.7
Exame Físico
Ao observar um paciente com parkinsonismo pela primeira vez, provavelmente o diagnóstico seja aparente de imediato, com alterações óbvias na expressão facial, lentidão motora, postura inclinada e possível tremor em repouso. O exame físico completo é importante para confirmar o diagnóstico e esclarecer a etiologia (isto é, DP ou outra causa de parkinsonismo)
Nos casos de DP, o exame físico geral pode revelar a presença de seborreia em até 60% de pacientes,15 que poderão responder à terapia dopaminérgica. A hipotensão ortostática deve ser examinada registrando-se os sinais vitais depois de 3 minutos nas posições em supino e de pé. Hipotensão ortostática é um sintoma comum de DP e poderá ser exacerbada pelo uso de medicações. A presença de hipotensão ortostática proeminente e grave no início do curso da doença é um sinal de alerta e causa preocupações sobre o diagnóstico de AMS.
A disfunção cognitiva em DP aumenta com o tempo de duração da doença. A Montreal Cognitive Assessment (MoCA) é, atualmente, a melhor ferramenta de rastreamento da disfunção cognitiva.16 É imprescindível examinar os movimentos dos olhos em pacientes com parkinsonismo. Os movimentos dos olhos são relativamente preservados nos casos de DP.
Os pacientes que tiverem dificuldades para fixar novamente o olhar ou limitações para movimentar os olhos (em especial, na fixação descendente do olhar) poderão ter um diagnóstico alternativo, ou seja, de PSP. O mau contato dos olhos é a primeira indicação de anormalidades no movimento dos olhos associadas à PSP.
Os pacientes com parkinsonismo geralmente se apresentam com hipomimia (perda de expressão facial), que poderá ser uma manifestação precoce de DP. A voz de pacientes com DP costuma ser hipofônica, monótona e, às vezes, hesitante. Voz estridente e trêmula, fala arrastada proeminente ou voz anasalada sugerem diagnósticos alternativos (AMS nas duas primeiras situações e PSP na última).
Embora se observem vários tremores nos casos de DP, o mais comum deles é o que ocorre nos membros em estado de repouso absoluto. Com frequência, os tremores em repouso são observados primeiramente nas mãos, mas podem também ocorrer nas pernas. Os tremores nas bochechas também são comuns, porém, comumente, ocorrem mais tarde no curso da doença.
De um modo geral, o tremor é a primeira manifestação motora observada pelos pacientes, mesmo porque é a mais óbvia. Os tremores em repouso costumam ser lentos (4 a 6Hz) e têm a característica de um movimento de “rolar pílulas” com a mão. O tremor desaparece quando o paciente mantém uma determinada postura (por exemplo, braço estendido), porém pode surgir novamente depois de alguns segundos na medida em que a posição atual se torna a nova posição de “repouso”.
Normalmente, o tremor da DP desaparece com alguma ação (por exemplo, estender o braço para pegar uma caneta). Assim como todos os sintomas motores, em geral o tremor começa em um lado do corpo e, ao longo do tempo, pode envolver ambos os lados e diversos membros. Cabe observar que, aproximadamente, 30% de todos os casos de DP nunca manifestam tremor em repouso. Os tremores posturais e cinéticos (ação) de frequência mais elevada também são comuns na DP.
A rigidez é um aumento involuntário no tônus muscular. É mais fácil avaliar o aumento no tônus quando o paciente flexiona e estende passivamente o membro ou o pescoço. A rigidez não depende da velocidade e, ao contrário da espasticidade, se torna mais aparente durante os movimentos lentos e passivos.
Com frequência, a característica conhecida por tremor subjacente em roda dentada pode ser sentida durante essa manobra clínica. O fenômeno do tipo roda dentada deve ser avaliado como uma sensação semelhante à de um mecanismo de catraca que poderá ser observado e palpado com o membro totalmente flexionado e estendido.
Observa-se a presença de bradicinesia ao ver o paciente caminhar para o quarto, sentar-se ou levantar-se de uma cadeira; esse tipo de condição pode também ser observado pedindo ao paciente para executar movimentos alternados rápidos e repetitivos como estalar os dedos, cerrar e soltar os punhos, pronar e supinar o braço ou bater de leve os dedos dos pés por repetidas vezes. A redução na amplitude e/ou a lentificação progressiva de movimentos repetitivos (fadiga) é típica de bradicinesia, assim como a hesitação breve nos movimentos repetitivos.
Alterações posturais e na marcha também podem ser observadas no momento em que o paciente entra no consultório médico. As alterações posturais incluem postura inclinada, ocasionalmente acompanhada de cifose ou escoliose. Outras posturas anormais, que podem ocorrer em estágios posteriores da doença, incluem camptocornia (flexão proeminente na cintura) ou síndrome de Pisa (inclinação do tronco para um lado).
Posturas anormais das mãos e dos pés também se desenvolvem ao longo do tempo (mão ou pé estriatal). O teste de tração produz reflexos posturais anormais; nesse tipo de teste, o examinador permanece de pé diretamente atrás do paciente, puxando-o para trás em cada ombro, criando uma perturbação postural.
A resposta normal é dar um ou dois passos para trás, enquanto que os pacientes com reflexos posturais alterados, geralmente, dão vários passos para recuperar o equilíbrio ou não conseguem parar e podem até cair se não forem segurados pelo examinador (retropulsão); mais tarde, o paciente possivelmente não faça nenhum esforço para recuperar o equilíbrio e poderá cair pesadamente. Na medida em que se agrava a instabilidade postural, os pacientes com DP começam a cair espontaneamente, em geral para trás.
A marcha anormal inclui passos lentos e hesitantes, com passadas mais curtas em uma base mais estreita. Nesses casos, observam-se também passos curtos e progressivamente mais rápidos, conhecidos por marcha festinante, em que o tronco parece ser mais rápido que as pernas. As viradas ao caminhar poderão ser acompanhadas de passos extras (“virar em bloco”).
Outra anormalidade da marcha que se observa com frequência nos estágios finais da DP é o “congelamento da marcha” (em inglês, freezing of gait [FOG]), em que os pés parecem ficar presos ao solo, antes de se liberarem e continuarem a marcha. O fenômeno FOG ocorre com mais frequência no início da marcha, ao dar uma virada (provavelmente, o tipo mais comum), e nos movimentos em espaços muito estreitos (por exemplo, nas portas).
A observação da marcha revela também evidências adicionais de bradicinesia e rigidez, com redução no movimento dos braços ou na retração dos pés. Tipicamente, o ato de caminhar produz também tremor em repouso no membro afetado.
Testes Laboratoriais
Não há testes laboratoriais ou descobertas imagiológicas específicas que permitam fazer a confirmação diagnóstica de DP. As varreduras por tomografia computadorizada por emissão de fótons únicos (SPECT) com o transportador da dopamina (DAT), atualmente, estão disponíveis em muitos países. Tipicamente, essas varreduras são anormais, mesmo nos estágios iniciais da doença, e mostram evidências de depleção estriatal do rastreador do DAT, particularmente no lado oposto dos membros mais afetados.
Embora a varredura do DAT seja bastante útil para fazer o diagnóstico e a distinção entre DP e algumas outras condições, como tremor essencial, não tem nenhuma utilidade para excluir muitas outras causas degenerativas de parkinsonismo que também podem causar perda de células dopaminérgicas na substância negra.
Diagnóstico Diferencial
O diagnóstico definitivo de DP depende de uma avaliação neuropatológica. Nos dias atuais, não existem critérios diagnósticos patológicos padrões para a DP.17 De um modo geral, o diagnóstico depende de perdas neuronais, variando de moderadas a graves, na parte compacta da substância negra (SNc), da presença de corpos de Lewy nos neurônios remanescentes, e da ausência de evidências patológicas de alguma outra doença que possa produzir parkinsonismo.7
O diagnóstico clínico se fundamenta na presença de parkinsonismo, em que a bradicinesia é um critério essencial para o diagnóstico de, pelo menos, algum outro sinal de parkinsonismo. Os critérios de exclusão são utilizados para excluir outras causas potenciais de parkinsonismo.18 As características de suporte ajudam a aumentar a sensibilidade e a especificidade desses critérios e incluem unilateralidade no início, resposta satisfatória ao levodopa (a ser discutida mais adiante) e longa duração da doença.
A especificidade desses critérios diagnósticos varia de 76 a 90% nos estudos clínicos e patológicos.18 Há um movimento para redefinir a DP com o objetivo de incorporar na definição o papel da genética, de subtipos clínicos, de dificuldades cognitivas e de sintomas pré-motores.19
A heterogeneidade dos sintomas motores em pacientes com DP resultou em várias tentativas de classificar subtipos diferentes da doença. Embora ainda não exista nenhuma subclassificação aceitável, uma das abordagens mais comuns é fazer a distinção entre dois subtipos principais: DP com predominância de tremor (com tremor proeminente e, relativamente, poucos outros sintomas motores) e DP sem predominância de tremor (conhecida, alternativamente, como distúrbio da marcha com instabilidade postural [DMIP] ou distúrbio rígido acinético para descrever as características motoras mais proeminentes).20
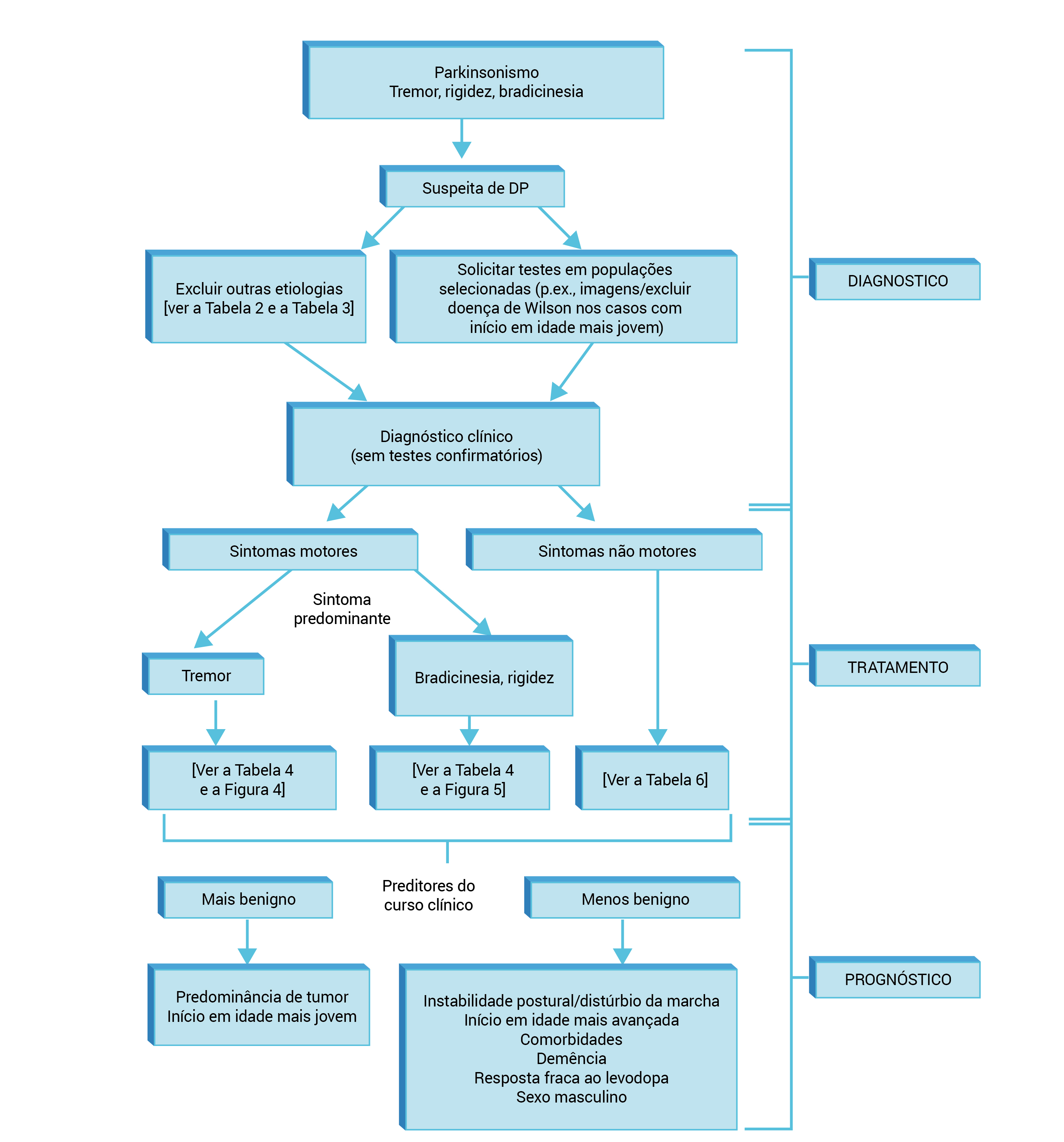
A Figura 3 apresenta o sumário do diagnóstico, o gerenciamento e o prognóstico da DP.
DP: doença de Parkinson.
Figura 3 - Sumário do diagnóstico, do gerenciamento e do prognóstico da DP.
Uma terceira categoria de fenótipo misto ou indeterminado é utilizada para descrever uma síndrome com características motoras equivalentes. Em parte, essas classificações foram descritas em razão de cursos e prognósticos distintos observados em subtipos diferentes. A predominância de tremor de longa duração está associada a um curso com progressão mais lenta e menos incapacitação funcional do que os casos sem predominância de tremor (em especial o DMIP), sugerindo a presença de etiologias subjacentes potencialmente distintas.7
A International Parkinson and Movement Disorder Society criou uma força tarefa para definir os critérios diagnósticos para a DP.19 Na tentativa de abordar subtipos diferentes de DP, essa força tarefa propôs novos critérios que incluem a contribuição genética da doença e as manifestações pré-motoras.
O Quadro 4 contém o tratamento de sintomas motores na DP.
Quadro 4
|
Tratamento de Sintomas Motores na Doença de Parkinson | ||||
|
Medicação |
Dose |
Indicação |
Eventos adversos importantes* |
Comentários |
|
Precursor da dopamina | ||||
|
Levodopa administrado com um inibidor periférico da descarboxilase (carbidopa na proporção de 4:1 ou 10:1 ou benserazida na proporção de 4:1) |
Titular para a dose inicial de 100/25mg, 3x/dia, dose máxima de 375mg/dia ou mais. |
Todos os sintomas motores. |
Periféricos: náusea e vômito, hipotensão ortostática; centrais: discinesias, psicose, oscilações motoras. |
Formulações: liberação imediata para tratamento precoce ou tardio: liberação controlada: acinesia noturna; liberação estendida (Rytary): tratamento tardio de oscilações motoras; LCIG; duodopa: usado com uma bomba para oscilações motoras. |
|
Agonista da dopamina | ||||
|
Pramipexol |
Iniciar com 0,125mg 3x/dia, dose máxima de 4,5mg/dia. |
Todos os sintomas motores. |
Periféricos: náusea, hipotensão ortostática, edema pedal; centrais: sonolência excessiva, incluindo ataques de sono, transtorno do controle de impulsos, psicose. |
Terapia inicial e adjuvante; também disponível na formulação de liberação estendida. |
|
Ropinirol |
Iniciar com 0,25mg, 3x/dia, dose máxima de 24mg/dia. |
Todos os sintomas motores. |
Os mesmos do pramipexol |
Terapia inicial e adjuvante; também disponível como liberação prolongada. |
|
Rotigotina (adesivo) |
Iniciar com 2mg/24h, dose máxima de 16mg/24h. |
Todos os sintomas motores. |
Os mesmos do pramipexol; reações cutâneas locais (dermatite). |
Terapia inicial e adjuvante; terapia tardia para oscilações motoras. |
|
Apomorfina (parenteral) |
Injeção SC de 3?5mg, p.r.n. (ou infusão contínua) |
Todos os sintomas motores. |
Os mesmos do pramipexol; reações cutâneas locais (incluindo nódulos). |
Oscilações motoras no estágio final; exige o uso concomitante de um antiemético (p.ex., domperidona). |
|
Inibidores da monoamina oxidase B (IMAOBs) | ||||
|
Seleginina |
25mg/dia a 5mg, 2x/dia. |
Sintomas leves, oscilações motoras. |
Efeitos colaterais dopaminérgicos de outros medicamentos possivelmente acentuados, insônia, confusão. |
Também disponível como formulação em pastilhas; a última dose deve ser administrada ao meio-dia para evitar insônia. |
|
Rasagilina |
1mg/dia |
Sintomas leves, oscilações motoras. |
Ver acima |
- |
|
Inibidor da catecol-O-metiltransferase (COMT) | ||||
|
Entacapona |
200mg com cada dose de levedopa |
Oscilações motoras. |
Diarreia em 5%; os efeitos do levodopa são acentuados. |
Usar somente como adjuvante. |
|
Tolcapona |
100mg, 3x/dia. |
Oscilações motoras. |
O mesmo que entacapona; hepatotoxicidade. |
O mesmo que entacapona. |
|
Anticolinérgicos | ||||
|
Trihexifenidil ou benzitropina |
1?2mg, 2 a 3x/dia. |
Tratamento inicial de tremor. |
Alucinações, alterações cognitivas, boca seca, visão turva, retenção urinária, constipação. |
Contraindicação relativa para uso em idosos ou em pacientes com alterações cognitivas. |
|
Neurolépticos | ||||
|
Clozapina |
6,25?12,5mg, diariamente ao deitar, dose máxima de 150mg/dia. |
Tremor, discinesias (psicose) [ver o Quadro 6] |
Sedação, hipotensão ortostática, sialorreia, agranulocitose (exige monitoramento regular). |
Baixo risco de agravar o parksonismo. |
|
Outros | ||||
|
Amantadina |
150mg, 1 ou 3x/dia |
Discinesias |
Edema periférico, livedo reticular, alucinações, confusão. |
Possivelmente tenha algum benefício em sintomas motores precoces. |
LCIG: gel intestinal à base de levodopa-carbidopa; SC: subcutâneo.
*[ver o Quadro 5]
Gerenciamento: Manifestações Motoras
Medicações
O gerenciamento médico de manifestações motoras nos casos de DP tem como alvo principal a reposição da dopamina, tendo em vista a perda crítica de neurônios dopaminérgicos na parte compacta da substância negra.
Levodopa. Esse medicamento é reconhecido como padrão de ouro para o tratamento sintomático das manifestações motoras da DP. As respostas adequadas à terapia com levodopa servem também de suporte para o diagnóstico de DP, tendo em vista que muitos parkinsonismos respondem fraca ou transitoriamente a esse tipo de terapia.
A terapia com levodopa poderá ser complicada por efeitos colaterais periféricos (hipotensão ortostática e náusea/vômito) e efeitos colaterais centrais, incluindo oscilações motoras e discinesias, psicose e efeitos colaterais comportamentais (distúrbios de controle de impulsos [DCIs], palpitação e síndrome da desregulação da dopamina).21,22
O levodopa é o precursor imediato do neurotransmissor dopamina e é convertido em dopamina no cérebro e na periferia. O levodopa deve ser administrado com um inibidor periférico da descarboxilase (que impede a conversão de levodopa em dopamina na periferia e, portanto, aumenta a quantidade de levodopa disponível no cérebro) na forma de carbidopa (levodopa/carbidoba, conhecida como Sinemet) ou de benserazida (levodopa/benserazida, conhecida como Prolopa ou Madopar).
A adição de um inibidor periférico da descarboxilase ao levodopa ajuda a impedir a ocorrência de náusea/vômito como efeito colateral devido à formação de dopamina periférica, que age sobre a área postrema (centro de vômito). O levodopa é comercializado nas formulações de liberação imediata e de liberação controlada. O maior problema com o levodopa é a meia vida plasmática curta (˜ 90 minutos).
O Quadro 5 contém os efeitos colaterais periféricos e centrais da terapia dopaminérgica (AD e levodopa).
Quadro 5
|
Efeitos Colaterais Periféricos e Centrais da Terapia Dopaminérgica (AD e Levodopa) | |||
|
Efeito colateral |
Definição |
Tratamento |
Comentários |
|
Periféricos | |||
|
Náusea/vômito |
? |
Adição de carbidopa; adicionar domperidona, 30min antes do levodopa. |
Complicação imediata em até 15%. |
|
Hipotensão ortostática |
? |
Adição de carbidopa; adicionar domperidona, 30min antes do levodopa. |
- |
|
Centrais | |||
|
Motores Oscilações motoras |
Alterações entre “ON” (a medicação dopaminérgica alivia os sintomas motores) e “OFF” (a medicação dopaminérgica perdeu o efeito e os sintomas motores retornam). |
Ver o texto e o Quadro 4. |
Comumente, essas oscilações iniciam depois de 5 anos de terapia com levodopa; a frequência aumenta com a duração da doença. |
|
Discinesias |
Movimentos involuntários, em geral coreiformes, devido ao alto estado da dopamina (pico); muitos pacientes têm discinesias em baixos níveis dopaminérgicos (em geral, distônicas e, às vezes, doloridas). |
Ver o texto e o Quadro 4. |
Comumente, as discinesias iniciam depois de 5 anos de terapia com levodopa; a frequência aumenta com a duração da doença. |
|
Comportamentais | |||
|
Distúrbio do controle de impulsos |
Esse distúrbio inclui jogo patológico, hiperssexualidade, compras compulsivas e compulsão alimentar. |
Redução ou descontinuação no uso do agonista da dopamina. |
O risco aumenta 15% ou mais com uso de ADs; aumento no desenvolvimento de DCIs com históricos de comportamentos aditivos, transtorno obsessivo-compulsivo ou da personalidade impulsiva. |
|
Punding |
Comportamentos estereotipados repetitivos, geralmente sem nenhum propósito (p.ex., manuseio ou separação contínua de objetos) |
Redução na terapia dopaminérgica. |
Ocorre em até 15% de pacientes, usualmente com doença de longo prazo. |
|
Síndrome da desregulação da dopamina |
Uso excessivo compulsivo de terapia dopaminérgica (acima do necessário para tratamento de sintomas motores). |
Redução gradual no uso de levodopa; descontinuar o uso de agonistas de ação curta (p.ex., apomorfina). |
Frequência estimada em 4%; tipicamente, ocorre com medicamentos dopaminérgicos de ação curta como levodopa ou apomorfina. |
|
Psiquiátricos | |||
|
Alucinações |
Tipicamente visual; bem formado, de pessoas ou animais. |
Ver o texto e o Quadro 6. |
Ver o texto e o Quadro 6. |
AD: agonista da dopamina; DCI: distúrbios de controle de impulsos.
A despeito deste fato - as respostas precoces não oscilam de forma muito significativa, embora oscilem com o tempo -, o desenvolvimento de complicações motoras se deve à curta duração da ação do medicamento, sendo que as mais comuns são: desativação das oscilações motoras e movimentos coreicos e distônicos anormais (discinesias). Esses problemas incentivaram o desenvolvimento de formulações diferentes para o levodopa.
A liberação imediata tem ação com duração mais curta e “ON” mais rápido, enquanto que a liberação controlada tem meia-vida plasmática mais longa. Infelizmente, o tempo de início na formulação de liberação controlada é altamente variável, o que torna o gerenciamento dos sintomas muito mais difícil. Uma formulação estendida de levodopa/carbidopa (IPX066, Rytary) se tornou recentemente disponível usando um sistema de liberação de uma única cápsula. Alguns estudos preliminares sugerem que essa formulação produz benefícios nas oscilações motoras nos casos de DP.23
Mais recentemente, o levodopa passou a ser disponibilizado em formulação de gel intestinal conhecida como gel intestinal de levodopa-carbidopa (GILC) ou duodopa. Essa formulação permite a liberação direta de levodopa-carbidopa no intestino delgado através de um tubo de gastrostomia endoscópica percutânea (GEP). Por esse método, é possível atingir níveis plasmáticos mais estáveis de levodopa.
Observou-se que o GILC melhora as oscilações motoras e diminui as discinesias nos casos avançados de DP em comparação com o tratamento oral.24 Esse método de liberação de dopamina tem sido usado na Europa desde 2004, sendo que há dados de longo prazo à disposição que sugerem a eficácia continuada em um período de 8 anos.25 Os efeitos colaterais do GILC se relacionam principalmente ao processo GEP, às oclusões e deslocamentos do tubo de infusão e à polineuropatia.26 As novas formulações futuras com grande potencial incluem a liberação subcutânea e as formas inalatórias de levodopa.26
Agonistas da dopamina. Os agonistas da dopamina (ADs), depois do levodopa, são os medicamentos que produzem mais benefícios no tratamento das manifestações motoras da DP. Os ADs são comercializados na formulação oral (pramipexol, ropinirol), como adesivos transdérmicos (rotigitina) ou como injeções subcutâneas ou sprays intranasais (apomorfina).
Os ADs podem ser usados como monoterapia ou como adjuvantes na terapia com levodopa. Os benefícios potenciais dos ADs em relação ao levodopa incluem chances menores de discinesias. Muito provavelmente, os ADs induzem DCIs; além disso, poderão também causar alucinações visuais/confusão, especialmente em idosos, edema na perna, insônia e ataques de sono.
Inibidores enzimáticos (inibidores da monoamina oxidase B [IMAOBs] e da catecol-O-metiltransferase [COMT]). Os IMAOBs bloqueiam o catabolismo da dopamina no cérebro, aumentando a biodisponibilidade dopaminérgica total. Os IMAOBs incluem a selegilina e a rasagilina. Uma metanálise mostrou que os IMAOBs produzem benefícios leves nos estágios iniciais da DP27 e poderão ser usados como monoterapia ou como adjuvantes à terapia com levodopa.
De um modo geral, os IMAOBs são bem tolerados, embora talvez exacerbem os efeitos colaterais induzidos pelo levodopa nos casos em que forem utilizados com adjuvantes. Essas medicações são administradas uma vez ao dia (rasagilina) ou duas vezes ao dia (selegilina), que poderão ser preferidas pelos pacientes com DP nos estágios iniciais, em comparação com a dosagem de três vezes ao dia de levodopa ou das formulações de AD com liberação imediata.
Levando-se em consideração que esses medicamentos bloqueiam a monoamina oxidase B, o risco da síndrome de serotonina com o uso concomitante de inibidores seletivos de reabsorção da serotonina (ISRSs) é muito baixo, e não se observa o “efeito queijo” com o uso simultâneo de tiamina. Um estudo de longo prazo envolvendo o uso de rasagilina com ISRSs corroborou esse fato.28 Os efeitos modificadores potenciais dos IMAOBs foram estudados ativamente, porém os resultados ainda não foram confirmados.29
A COMT é a enzima responsável pela decomposição do levodopa. O bloqueio dessa enzima aumenta os níveis de levodopa no plasma e no cérebro, assim como a duração da ação do medicamento, aumentando, consequentemente, os níveis de dopamina no cérebro. Os inibidores da COMT (COMTIs) incluem a entacapona, que bloqueia a COMT periférica, e a tolcapona que bloqueia a COMT periférica e central.
Esses medicamentos devem ser usados apenas como adjuvantes do levodopa, tendo em vista que não possuem efeito antiparkinsonismo inerente. Os efeitos colaterais incluem urina de cor escura, diarreia e exacerbação dos efeitos colaterais induzidos pelo levodopa. A hepatotoxidade da tolcapona provocou restrições à disponibilidade clínica desse medicamento.
Terapias não dopaminérgicas. Uma revisão feita por Cochrane demonstrou que o uso de anticolinérgicos foi benéfico em termos de melhoras na função motora na DP,39 embora tenham ocorrido efeitos mistos relacionados ao tremor. A clozapina, um antipsicótico atípico, também melhorou o tremor em pacientes com DP.31 Esse medicamento é utilizado com mais frequência para controlar a psicose associada à DP sem agravar o parkinsonismo, como costuma ocorrer com a maioria dos neurolépticos (com a possível exceção da quetiapina).
Os exames de sangue devem ser monitorados com muito cuidado com o uso de clozapina devido ao grande potencial para o efeito colateral de agranulocitose. Os resultados da administração de amantadina para controlar os sintomas motores da DP são mistos, sendo que uma revisão feita por Cochrane concluiu que as evidências são insuficientes devido à má qualidade dos estudos,32 embora os especialistas da International Parkinson and Movemement Disorder Society tenham concluído que a amantadina talvez seja eficaz como adjuvante ou como monoterapia.31
Esse medicamento é particularmente útil para controlar discinesias induzidas pelo uso de levodopa. Alguns estudos mais antigos de baixa qualidade indicaram que os bloqueadores ß poderiam melhorar o tremor.3
Cirurgia
As opções neurocirúrgicas para o gerenciamento de DP incluem estimulação cerebral profunda (ECP) com implante de eletrodos ou cirurgia lesional/ablativa. A ECP foi introduzida clinicamente em 1987 e, em muitas situações, substituiu as cirurgias lesionais mais antigas por causa da vantagem da reversibilidade, da programabilidade e da capacidade para executar procedimentos bilaterais sem produzir efeitos colaterais permanentes e intoleráveis.
Os alvos da ECP incluem o núcleo subtalâmico (NST) e o segmento interno do globo pálido (GPi), sendo que ambos são eficazes para tratar todas as características principais da DP e, ocasionalmente, o núcleo ventral intermediário (VIM) do tálamo nos casos de tremor refratário ao tratamento médico. Uma metanálise envolvendo o NST e o GPi mostrou que, em ambos os casos, a função motora e as atividades diárias melhoraram sem diferenças significativas nos resultados terapêuticos entre os dois procedimentos.33
A DBS no NST não permitiu uma redução maior no uso de medicamentos dopaminérgicos, ao passo que a DBS no GPi para tratamento de sintomas motores apresentou melhoras mais significativas em resultados psiquiátricos como a depressão. O estudo EARLY-STIM mostrou que a DBS no NST poderá ser benéfica em pacientes com complicações motoras precoces, em comparação com as melhores práticas de gerenciamento médico.34
A seleção apropriada de pacientes é extremamente importante para o sucesso da terapia com ECP. Os melhores candidatos são os pacientes com diagnóstico claro de DP, que continuam a apresentar respostas robustas ao levodopa e que tenham se submetido a testes adequados com outra medicação antiparkinsoniana.
Os pacientes não serão candidatos à cirurgia com ECP se tiverem parkinsonismo atípico, problemas cognitivos ou demência, problemas psiquiátricos ativos significativos, falsas expectativas cirúrgicas ou respostas inadequadas ou não sustentadas ao levodopa. Um princípio clínico básico bastante útil é que a ECP no NST, certamente, não resultará em nenhuma melhora motora mais significativa, em comparação com a melhor resposta do paciente ao levodopa, embora haja mais tempo ON e menos discinesias.
Outras Terapias
Observou-se que outras modalidades terapêuticas também são úteis no gerenciamento dos sintomas motores da DP. Muitos outros estudos avaliaram os benefícios da fisioterapia nos casos de DP. Uma revisão feita por Cochrane demonstrou que houve benefícios na maior parte das medições de equilíbrio e da marcha no curto prazo, embora não tenha havido nenhum benefício na prevenção de quedas.35
Cabe observar que a prática de tai chi chuan foi mais eficaz que os treinamentos em alongamento e resistência na prevenção contra quedas, na redução de alterações no equilíbrio e na melhora da qualidade de vida.36 A hipofonia pode se tornar um problema muito grave na DP e chega a causar isolamento social e uma visão equivocada de deficiência cognitiva. O Lee Silverman Vocal Treatment (LSVT) é um dos tratamentos recomendados para hipofonia.37 O treinamento em marcha sinalizada (cued gait) provavelmente seja eficaz no tratamento de congelamento da marcha.31
Recomendações para Gerenciamento
Doença de Parkinson Precoce (Descomplicada)
Nos dias atuais, não há nenhuma terapia modificadora da doença ou neuroprotetora que seja considerada eficaz. Todas as terapias são sintomáticas; portanto, a decisão de iniciar o tratamento se baseia unicamente na noção sobre o nível de incômodo que os sintomas causam aos pacientes, seja pelos danos funcionais ou pelo embaraço social.
Nos casos de pacientes com DP precoce e sintomas leves de parkinsonismo, observou-se que os IMAOBs (selegilina e rasagilina) trazem alguns benefícios e, por essa razão, são recomendados para o controle sintomático.31,38 O benefício é pequeno, porém o uso desses medicamentos possivelmente adie por vários meses a necessidade de terapia dopaminérgica. Os IMAOBs têm o benefício adicional da dosagem diária menos frequente e de um perfil baixo de efeitos colaterais.
Os anticolinérgicos também apresentaram alguns benefícios e são recomendados para uso nos casos de DP precoce,31 em especial no tratamento de tremor em pacientes mais jovens. Recomenda-se evitar o uso desses agentes em pacientes com mais de 60 anos de idade e em qualquer pessoa com disfunção cognitiva pré-existente. Provavelmente, a amantadina seja eficaz para o controle sintomático inicial do parkinsonismo, ainda que as evidências sejam menos robustas.38
Nas situações em que os pacientes apresentarem um grau maior de problemas funcionais, justifica-se a administração de terapia dopaminérgica com levodopa ou com ADs considerando que essas medicações produzem apenas benefícios leves no parkinsonismo. Entre essas duas medicações, o levodopa é o mais eficaz no controle dos sintomas motores, embora tenha uma chance bem maior de produzir complicações motoras em comparação com os ADs.
Embora também sejam altamente eficazes no tratamento de parkinsonismo precoce e sintomático, os ADs apresentam risco mais elevado de causar distúrbios de controle de impulsos, edema, insônia e alucinações visuais em comparação com o levodopa.
A escolha ideal da primeira medicação se baseia nos sintomas e nas características de cada paciente. A idade é um fator importante na escolha entre levodopa e ADs. Os pacientes mais jovens com DP têm maior chance de desenvolver discinesias precoces e oscilações motoras; portanto, os ADs são a terapia inicial preferida para aplicação em pacientes com idade inferior a 60 anos.
Nos pacientes com mais de 60 anos de idade, o risco de desenvolver complicações motoras precoces e proeminentes, em especial discinesia, é menor e o risco de desenvolver alucinações é maior; portanto, o levodopa talvez seja a medicação preferida para a terapia inicial.3 A introdução do levodopa no tratamento não deve ser adiada em pacientes que tenham receio de desenvolver complicações motoras se as outras medicações não garantirem o controle adequado dos sintomas.
Alguns estudos recentes mostraram que o desenvolvimento de discinesias relacionadas ao uso de levodopa depende menos do tempo de uso do medicamento do que da duração real e da gravidade da DP propriamente dita (isto é, o desenvolvimento de discinesia ocorrerá no mesmo ponto do curso da doença, independentemente do momento em que o paciente iniciou o tratamento com levodopa).39,40 Na realidade, nesses estudos, os pacientes que iniciaram o tratamento com levodopa versus terapias “poupadoras de levodopa” apresentaram resultados semelhantes no longo prazo41 em termos de complicações motoras.
Doença de Parkinson Moderada (Incluindo o Gerenciamento de Complicações Motoras)
De um modo geral, o levodopa e os ADs são as melhores opções para a terapia inicial em pacientes com níveis moderados de incapacitação. De um modo geral, nos pacientes tratados com levodopa por 2 a 5 anos, as complicações motoras, tais como oscilações e discinesias, são indicações para ajustes posteriores no tratamento.
As oscilações motoras podem ser previsíveis (isto é, aquelas relacionadas ao término da ação do levodopa) ou imprevisíveis. As oscilações previsíveis podem ser tratadas inicialmente por: redução no intervalo da dose de levodopa, aumentando consequentemente o número de doses diárias do medicamento; ou adição de AD, um IMAOB ou COMTI.
O uso adjuvante de pramipexol, ropinirol (incluindo as formulações com liberações estendidas e prolongadas) e o tradicional adesivo transdérmico com rotigotina (liberação contínua por 24 horas) reduziram, comprovadamente, em 15%3,38 e de forma significativa o tempo OFF em comparação com placebo.37
A adição de COMTI ou de inibidores da MAOB poderá reduzir o tempo OFF em 1 a 1,5 horas diariamente, sendo que não há nenhuma recomendação clara sobre qual medicamento deve ser o primeiro a ser usado.31 Provavelmente, seja necessário usar diversas medicações de todas as classes para otimizar os benefícios.
As discinesias leves e que não incomodam os pacientes não precisam ser tratadas. Nos casos em que as discinesias incomodarem e forem incapacitantes, uma das opções é tentar reduzir o uso de medicações dopaminérgicas (incluindo levodopa, COMTI, IMAOB ou AD), embora essa abordagem geralmente resulte no agravamento do parkinsonismo e em incapacitação mais séria, incluindo aumento no tempo OFF.
De um modo geral, a amantadina é eficaz para diminuir o tempo de duração e a gravidade das discinesias, e, em geral, é um medicamento bem tolerado.31 A clozapina reduziu as discinesias em um teste duplo cego de pequeno porte.38 Há uma hipótese indicando que, em parte, as oscilações motoras e as discinesias podem ser o resultado da estimulação pulsátil de receptores dopaminérgicos estriatais pós-sinápticos, um fenômeno que ocorre mais tarde no curso da doença, quando as concentrações intracerebrais de levodopa se tornam intimamente ligadas às concentrações plasmáticas do medicamento.7
Portanto, uma das estratégias para tentar reduzir as discinesias e as oscilações motoras é manter um estado estável e mais contínuo de estimulações dopaminérgicas. As várias estratégias para atingir esse estado, além das já descritas, incluem liberação direta do GILC (também conhecido por duodopa) no duodeno24 e infusão subcutânea do agonista da dopamina apomorfina.31 Essas terapias à base de infusões (GILC ou bomba de apomorfina) em combinação com a ECP, em geral, são conhecidas como terapias avançadas, tipicamente reservadas para as oscilações motoras resistentes ao tratamento.
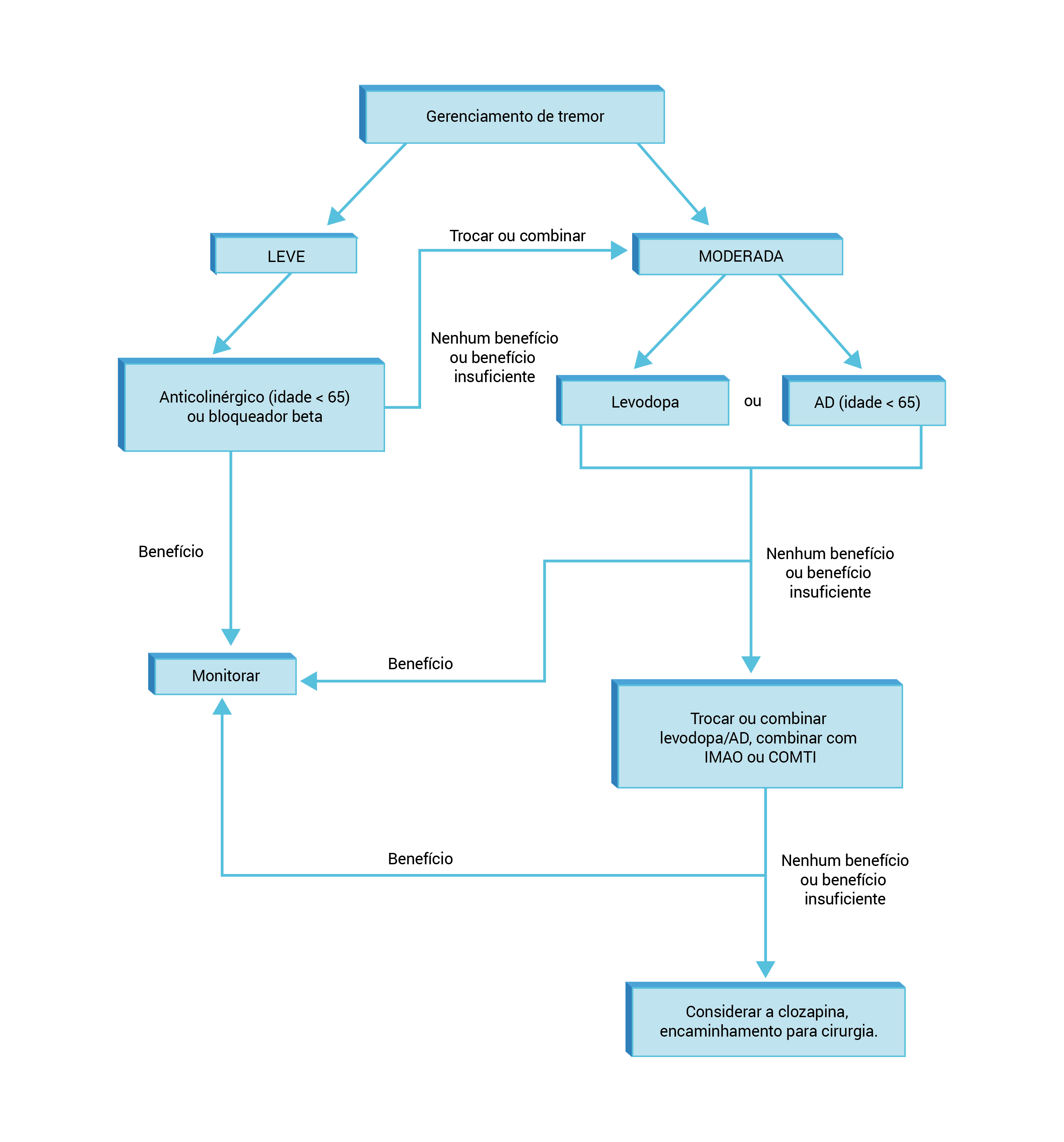
A Figura 4 mostra o algoritmo para tratamento da DP com predominância de tremor.
AD: agonista da dopamina COMTI: inibidor da catecol-O-metiltransferase; DP: doença de Parkinson; IMAO: inibidor da monoaminoxidase.
Figura 4 - Algoritmo para tratamento da DP com predominância de tremor.
O sintoma mais incômodo também poderá ser um fator importante na escolha da medicação. Com frequência, a instabilidade postural e/ou distúrbio da marcha é mais difícil de tratar e tende a ocorrer mais tarde no curso da doença.
Os testes com amantadina ou com um inibidor da colesterase geralmente são feitos nos casos de instabilidade postural/distúrbio da marcha refratários ao levodopa. Nos casos de distúrbio da marcha responsivos ao levodopa, que ocorrem principalmente durante os episódios OFF com oscilações motoras, o uso de ECP também é uma opção a ser considerada.3
Doença de Parkinson avançada (estágio final) e manifestações neuromotoras
Na medida em que a DP progride, os sintomas motores selecionados tendem a se tornar mais responsivos à terapia dopaminérgica (em particular, sintomas axiais como fala, deglutição e distúrbios da marcha) e os sintomas não motores adicionais se tornam problemáticos (embora algumas características não motoras possivelmente se tornem problemáticas, mesmo nos estágios iniciais da doença).
Acredita-se que, em alguns estágios mais avançados, as anormalidades nos neurotransmissores não dopaminérgicos (por exemplo, acetilcolina, glutamato, norepinefrina e serotonina) provavelmente contribuam para as manifestações não motoras da DP. Essas manifestações não motoras da DP incluem dificuldades cognitivas, transtornos psiquiátricos, anormalidades no sono e disfunção autonômica. Embora as opções disponíveis de tratamento sejam relativamente limitadas, há inúmeras recomendações que poderão ser tentadas.
Disfunção neuropsiquiátrica. Demência. De um modo geral, os pacientes com DP desenvolvem comprometimento cognitivo leve (CCL), que, então, poderá evoluir para demência relacionada com DP (DDP). Após o diagnóstico, a probabilidade de ocorrência de CCL ou de demência aumenta ao longo do tempo, e estima-se que mais de 80% de indivíduos tenham DPP depois de 20 anos de duração da doença.42 Geralmente, a DDP difere da demência de Alzheimer que, com frequência, é acompanhada de disfunção executiva precoce e lentificação profunda da cognição.
O uso de inibidores da colinesterase, incluindo a rivastigmina, com descobertas menos robustas para o donepezil e a galantamina, resultou em melhoras na cognição.31 Os benefícios foram modestos com a administração de memantina, um agente bloqueador do receptor do N-metil-D-aspartato (NMDA).31
Psicose. As alucinações visuais são ao mesmo tempo uma manifestação de DP e uma complicação causada pelos medicamentos dopaminérgicos. As alucinações visuais da DP tendem a ser bem formadas em pessoas ou animais.43 Tipicamente, as alucinações começam entre 5 e 10 anos após o início da doença e podem preceder a demência, sendo que a incidência aumenta com o tempo de duração da doença (semelhante à demência); as estimativas indicam que 70% das pessoas são afetadas depois de 20 anos de doença.42
O gerenciamento da psicose nos casos de DP inclui investigação e tratamento das causas reversíveis (por exemplo, infecção, distúrbio metabólico), em especial se a psicose for aguda no início. Os medicamentos também poderão agravar a psicose, sendo, portanto, necessário reduzir ou interromper o uso das terapias farmacológicas.
Os agentes ofensores mais importantes incluem anticolinérgicos (usados no tratamento dos sintomas de DP ou de disfunção urinária), amantadina e AD, embora qualquer medicação usada no tratamento de DP possivelmente seja causativa. A redução no uso de medicações dopaminérgicas aumenta o risco de parkinsonismo.
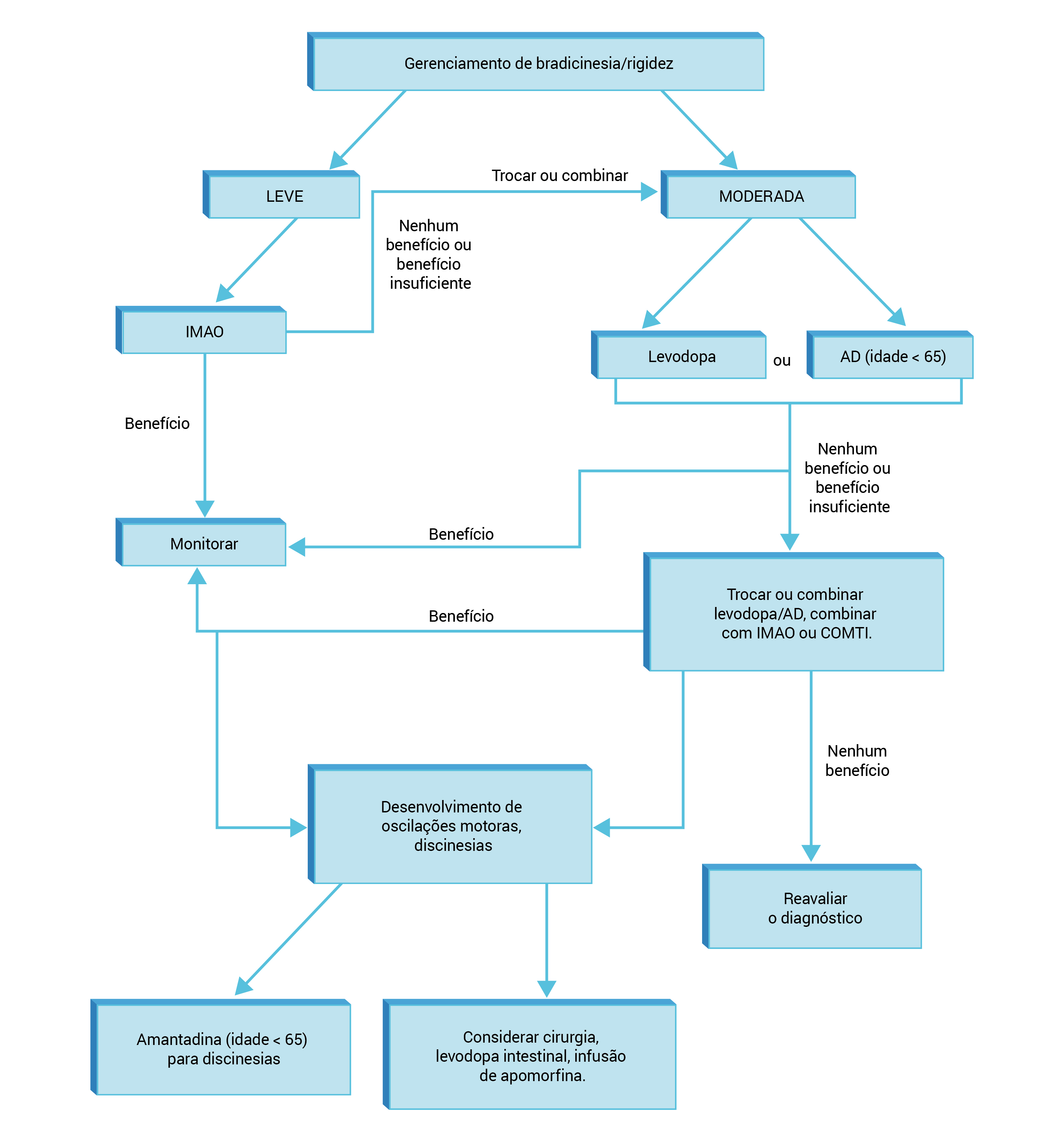
A Figura 5 mostra o algoritmo para tratamento da DP com predominância de bradicardia e rigidez.
AD: agonista da dopamina; COMTI: inibidor da catecol-O-metiltransferase; DP: doença de Parkinson; IMAO: inibidor da monoaminoxidase.
Figura 5 - Algoritmo para tratamento da DP com predominância de bradicardia e rigidez.
Atualmente, a clozapina e a quetiapina são as duas únicas medicações antipsicóticas recomendadas para uso nos casos de psicose causada pela DP, tendo em vista que os antipsicóticos típicos e outros atípicos poderão agravar o parkinsonismo. A quetiapina produz efeito menos robusto sobre a psicose em comparação com a clozapina, embora tenha a vantagem de não precisar de monitoramento sanguíneo.
Observou-se que a clozapina é bastante eficaz no tratamento de psicose causada pela DP44 e produz benefícios adicionais potenciais no tremor e na discinesia, mas com a desvantagem de precisar de monitoramento frequente do sangue. As evidências indicam que os inibidores da acetilcolinesterase diminuem as alucinações, particularmente em pacientes com demência concomitante.
O medicamento pimavanserina é um novo agonista inverso do receptor da serotonina que demonstrou ser eficaz no tratamento de psicose em um teste clínico de fase III (aguardando a aprovação da Food and Drug Administration).45
Depressão. A depressão é um prenúncio do desenvolvimento de sintomas motores e, portanto, do diagnóstico de DP em vários anos. Estima-se que a prevalência de depressão nos casos de DP seja de 40%.31 O uso de antidepressivos em casos de DP apresentou resultados mistos. Os antidepressivos tricíclicos apresentaram resultados mais consistentes em comparação com placebo em diversos estudos.31,46
Os ISRSs e os inibidores da reabsorção da serotonina e norepinefrina (SNRIs) também são muito utilizados para tratamento de depressão nos casos de DP, embora nos estudos que compararam esses medicamentos com placebo os resultados tenham sido menos conclusivos.47 Para finalizar, os ADs também foram eficazes no gerenciamento da depressão.31,46
Técnicas como terapia eletroconvulsivante, estimulação magnética transcraniana repetitiva e psicoterapia são intervenções não farmacológicas que também são utilizadas no tratamento de depressão, embora ainda não existam evidências que deem suporte à sua utilização nos casos de DP.7
Apatia. Define-se apatia como um estado de falta de motivação, que poderá se manifestar como depressão nos comportamentos direcionados para metas e falta de interesse ou de emoções, não necessariamente relacionadas à depressão ou a danos cognitivos.48 A apatia pode preceder o diagnóstico de DP.
A prevalência em diagnósticos recentes de DP varia de 20 a 36%, sendo que a incidência aumenta com o tempo de duração da doença e com o desenvolvimento de dificuldades cognitivas. Existem algumas evidências para o uso de ADs, de inibidores da colinesterase e de metilfenidato no tratamento de apatia nos casos de DP, embora, de um modo geral, o tratamento seja ineficaz.48
Disfunção autonômica. As anormalidades autonômicas, além da constipação (que, com mais frequência, é um fenômeno pré-motor), tipicamente ocorrem entre 5 e 10 anos após o início dos sintomas de DP, cujas frequências variam de 15 a 20% (hipotensão ortostática, disfunção sexual) a 30 a 35% (sialorreia, constipação, sintomas urinários incluindo urgência e noctúria).
O Quadro 6 contém o tratamento de sintomas não motores na DP.
Quadro 6
|
Tratamento de Sintomas não Motores na Doença de Parkinson | |||
|
Sintoma não motor |
Medicação |
Dosagem |
Efeitos adversos |
|
Demência |
Inibidores da acetilcolinesterase: Doneprezil Rivastigmina (oral ou adesivo). |
5?10mg, 1x/dia 3?12mg, diariamente; adesivo: 4,5?9,8mg/24 horas |
Náusea, tontura, cefaleia. |
|
|
Inibidores do NMDA: Memantina |
5?10mg, diariamente |
Confusão, fadiga, tontura, cefaleia. |
|
Alucinações |
Antipsicóticos: Clozapina |
Iniciar com 6,25?12,5mg, diariamente, ao deitar; dose máxima: 150mg/dia |
Sedação, hipotensão ortostática, sialorreia agranulocitose. Sintomas extrapiramidais e sedação. |
|
|
Inibidores da acetilcolinesterase: Rivastigmina |
Ver acima |
Ver acima |
|
Depressão |
ISRSs: Citalopram Fluoxetina Paroxetina Sertralina |
10?20mg, 1x/dia 10?50mg, 1x/dia 20?40mg, 1x/dia 20?200, 1x/dia |
Acatisia, disfunção sexual, anorexia, náusea, sonolência. |
|
|
SNRIs: venlafaxina com liberação estendida |
37,5?225mg, diariamente |
Disfunção sexual, insônia, sonolência, sintomas gastrintestinais. |
|
|
ATCs: Nortriptilina, amitriptilina |
25?150mg, diariamente ou em doses divididas |
Efeitos colaterais anticolinérgicos. |
|
DCR |
Melatonina |
3?15mg, todas as noites |
Sonolência durante o dia, cefaleia, tontura. |
|
|
Clonazepam |
0,25?2mg, todas as noites |
Sedação e confusão. |
|
Sonolência excessiva durante o dia |
Modafinil |
100?400mg |
Psiquiátrico. |
|
|
Metilfenidato |
5?10mg, até 3x/dia |
Palpitações, pediátrico, gastrintestinal, perda de peso. |
|
Hipotensão ortostática |
Fludrocortisona |
0,1?0,3mg, diariamente |
Hipertensão supina, hipocaliemia |
|
|
Midodrina |
2,5mg/dia até, no máximo, 15mg, 3x/dia. |
Hipertensão supina |
|
|
Piridostigmina |
50mg, 3x/dia |
Náusea, sintomas gastrintestinais, aumento na salivação/secreções brônquicas. |
|
|
Yoimbina |
2mg, 3x/dia |
Alucinações, convulsões, insuficiência renal. |
|
|
Indometacina |
50mg, 3x/dia |
Insuficiência renal, sintomas gastrintestinais. |
|
|
Droxidopa |
300mg, 3x/dia |
Hipertensão, taquicardia, náusea/vômito, cefaleia. |
|
Sialorreia |
Glicopirrolato |
1mg, 3x/dia |
Efeitos colaterais anticolinérgicos. |
|
|
Atropina (gotas) |
1?2 gotas de 1% até 4x/dia |
O mesmo que glicopirrolato. |
|
|
Atrovent (spray) |
1?2 sprays até 4x/dia |
O mesmo que glicopirrolato. |
|
|
Toxina botulínica |
A dosagem varia; glândulas parótida e/ou submandibular |
Boca seca, disfagia. |
|
Disfunção sexual |
Sildenafila |
25?100mg |
Eventos cardíacos, priapismo, perda visual. |
ATCs: antidepressivos tricíclicos; DCR: distúrbio comportamental do sono REM; ISRSs: inibidores seletivos de reabsorção da serotonina; NMDA: N-metil-D-aspartato; SNRIs: inibidores de reabsorção da serotonina e norepinefrina.
Hipotensão ortostática. É uma complicação comum do próprio processo da doença ou um efeito colateral de medicações dopaminérgicas.
O gerenciamento inclui medidas como:
evitar os fatores agravantes (por exemplo, refeições exageradas, depleção de volume, anti-hipertensivos, diuréticos, álcool);
fazer intervenções não farmacológicas, como aumento na ingestão de água e sal;
usar meias com alta compressão na cintura e faixas de compressão abdominal;
evitar levantar-se abruptamente da cama durante a noite.31
A melhor evidência para o gerenciamento farmacológico é o uso de midodrina acompanhado pelo uso de fludrocortisona.31 Outras medicações potencialmente úteis com poucas evidências de eficácia incluem piridostigmina, indometacina, yohimbina e droxidopa.3 A domperidona poderá gerar algum benefício nos casos em que a hipotensão ortostática for causada ou agravada pelo efeito periférico do levodopa ou dos ADs.3
Constipação. Com frequência, a constipação é um prenúncio do desenvolvimento de sintomas motores na DP. Levando-se em consideração que há poucos testes terapêuticos sobre o gerenciamento de constipação em casos de DP, o tratamento se assemelha àquele usado na constipação crônica não associada à DP.49 Recomenda-se o uso de polietileno glicol, assim como suplementos de fibras.31
Disfunção urinária. Os sintomas no trato urinário inferior (noctúria, frequência e urgência) são comuns (prevalência de 30%) em homens e mulheres com DP. Com frequência, embora iniciem entre 5 e 10 anos após o início da doença, esses sintomas podem ser uma característica de doença precoce em alguns pacientes. Tipicamente, esse tipo de disfunção está associado à superatividade da bexiga.
A otimização da terapia dopaminérgica noturna poderá gerar algum benefício. Os medicamentos anticolinérgicos também podem ser benéficos, mas podem induzir eventos adversos relacionados à cognição.31,46 Os agentes novos e mais seletivos, como a darifenacina (um antagonista M3) ou o mirabegron (um agonista do receptor ß2), têm menos chances de causar efeitos adversos na cognição.50 A aplicação de injeções de toxina botulínica tipo A no músculo detrusor também é uma opção bastante eficaz.50
Sialorreia. As gotas de atropina poderão ser usadas nos casos de sialorreia na DP, embora possivelmente contribuam ou causem alucinações ou delirium em indivíduos mais velhos, de modo que se recomenda muita cautela para usar essa medicação. Eventualmente, se utiliza o spray de brometo de ipratrópio, ainda que existam poucas evidências de sua eficácia.
O glicopirrolato é um anticolinérgico eficaz e não produz os efeitos colaterais neuropsiquiátricos dos outros agentes anticolinérgicos, incluindo a atropina. Para finalizar, as injeções de toxina botulínica na parótida e/ou nas glândulas submandibulares podem ser eficazes no tratamento de sialorreia.3,46
Transtornos do sono. Os transtornos do sono são prevalentes na DP.51 Observa-se o distúrbio comportamental do sono REM (DCR) em 30% de pacientes e, com frequência, esse distúrbio é um prenúncio do desenvolvimento de sintomas motores. A sonolência excessiva durante o dia (EDS) pode ser, como consequência do efeito colateral de alguma medicação, resultado da doença secundária à disfunção do sono noturno (incluindo apneia obstrutiva do sono), ou provavelmente resulte de uma combinação desses fatores.
A insônia na DP costuma ser uma insônia de manutenção do sono (isto é, o paciente não consegue ficar sonolento) ou insônia no início do sono. As causas potenciais são multifatoriais e incluem fenômeno OFF noturno, noctúria, DCR, pernas inquietas e transtorno do sono associado à DP. Os estudos formais do sono com polissonografia são opções a serem consideradas na avaliação de pacientes com DP e com problemas proeminentes de sono (EDS e insônia).
Distúrbio Comportamental do sono REM (DCR). O uso de clonazepam, um benzodiazepínico, é a terapia de primeira linha para tratamento de DCR. A melatonina também é uma boa opção.3,46
Insônia. O histórico do sono é importante no processo de determinação da causa de insônia. A administração noturna de uma formulação de liberação controlada de levodopa é uma ótima opção para o fenômeno OFF noturno. Os medicamentos eszopidona e melatonina produzem benefícios leves no tratamento de insônia.37,46
Sonolência excessiva durante o dia. As causas potenciais de EDS devem ser revisadas. Nos casos em que a EDS for secundária à insônia ou DCR, esses problemas deverão ser abordados em primeiro lugar. Pode ser necessário reduzir gradualmente a dosagem dos medicamentos sedativos ou mesmo descontinuar gradativamente o seu uso.
Entre as medicações dopaminérgicas, os ADs provavelmente sejam os que mais contribuem para a incidência de EDS, incluindo “ataques de sono” com início repentino, o que talvez justifique alterações nessas medicações. O modafinil e o metilfenidato são recomendados para uso nos casos de EDS em pacientes com DP.31
Prognóstico
Com base em estudos populacionais e em dados obtidos em testes clínicos, observou-se que o prognóstico de DP tem um padrão não linear de alteração ao longo do tempo. Esse padrão consiste de três fases: uma melhora inicial na função motora nos primeiros 2 a 3 anos e um segundo patamar em até 5 a 7 anos conhecido por “período de lua de mel”, sendo que ambos os estágios provavelmente correspondam a uma boa resposta ao levodopa.
Imediatamente após esses dois estágios, há um terceiro período de deterioração de 7 a 9 anos, que se caracteriza pelo agravamento da função motora (em especial, as características motoras axiais resistentes ao levodopa) e pelo desenvolvimento de mais manifestações não motoras incapacitantes.52,53
Os fatores que se correlacionam com taxas mais rápidas de progressão da DP incluem gênero masculino, início em torno da idade de 50 anos, presença de alterações cognitivas no diagnóstico e predominância de um subtipo rígido acinético.52 Conforme já mencionado, determinadas causas genéticas de DP têm um fenótipo mais “benigno” e uma taxa mais lenta de progressão em comparação com a DP esporádica (por exemplo, mutações em Parkin).
Embora a DP esteja associada a um aumento na taxa de mortalidade, as estimativas variam de acordo com o estudo. Em uma revisão sistemática, um grupo de estudos sugeriu uma razão de mortalidade nos casos de DP de 1,5 versus os controles. A mortalidade aumenta com a idade e com a presença de demência.54 Comumente, a morte nos casos de DP é causada pelos efeitos da doença através da imobilidade, aspiração ou quedas, e as lesões subsequentes.
A grande expectativa é de que, no futuro, terapias neuroprotetoras eficazes com foco em diversos mecanismos patogênicos subjacentes, incluindo tentativas para diminuir os níveis de a-sinucleina (por exemplo, anticorpos focados), mudarão o curso desse tipo de doença.56
Demência com Corpúsculos de Lewy
Epidemiologia e Etiologia
A DCL é um distúrbio neurodegenerativo progressivo com características de parkinsonismo e desenvolvimento precoce de alterações cognitivas/demência, alucinações visuais e níveis oscilantes de alerta.56,57 Estima-se que a prevalência seja de 0,7% na população acima de 65 anos de idade, com uma incidência de 3,5 por 100 mil pessoas por ano, que aumenta com o avanço da idade.58
A DCL é a segunda forma mais comum de demência depois da doença de Alzheimer, com uma variação na prevalência estimada de 3 a 26,3% na população de demência com idade acima de 65 anos. A preponderância masculina é de 1,9:1. Os fatores de risco para DCL incluem hipertensão, hiperlipidemia, o alelo ApoE4 e mutações no gene GBA.
A DCL é uma DP semelhante à “sinucleinopatia”, que tem a a-sinucleína agregada nos corpos de Lewy e nas neurites de Lewy como pontos de referência patológicos. A distribuição dessa patologia de Lewy na DCL é mais generalizada e envolve regiões corticais no estágio inicial, explicando o início com demência e alterações comportamentais. A substância negra também é menos envolvida nos estágios iniciais da doença, e o parkinsonismo é mais variável, sendo que alguns pacientes desenvolvem essa característica.
Acredita-se que a DCL seja o extremo de um espectro com DP, sendo que alguns especialistas não veem nenhuma razão para separar as duas condições.19 Na realidade, talvez não seja possível fazer distinção entre a patologia de DCL e de DDP. Entretanto, os cérebros de pacientes com DCL geralmente contêm mais deposição de ß-amiloide e, com frequência, não apresentam outras características da doença de Alzheimer (isto é, placas sem entrelaçamentos).
Diagnóstico
O diagnóstico se baseia em critérios clínicos básicos, incluindo demência, parkinsonismo, nível oscilante de alerta e alucinações visuais que ocorrem na fase inicial do curso da doença.59 A demência nos casos de DCL consiste de alterações na função viso-espacial, na função executiva e na velocidade cognitiva, com preservação relativa da memória nos estágios iniciais.
De um modo geral, o parkinsonismo é considerado mais simétrico no início, com ocorrência de menos tremor em repouso em comparação com a DP, embora se observe também uma apresentação típica de DP. Com frequência, as alucinações visuais espontâneas (isto é, não induzidas por medicamentos como nos casos de DP) geralmente são vívidas e bem formadas, como pessoas ou animais. O distúrbio comportamental do sono REM (DCR) é sugestivo do diagnóstico e, assim como ocorre nos casos de DP, o DCR pode antecipar o diagnóstico por muitos anos. Delírios paranoicos, alucinações não visuais e depressão são características de suporte.
A sensibilidade aos neurolépticos, estimada em 30 a 50% de pacientes com DCL expostos a esses agentes, também é sugestiva de um diagnóstico de demência de corpos de Lewy.60 A presença de disfunção autonômica é comum com hipotensão ortostática, constipação, disfunção urinária e sialorreia. Quedas e síncope são características de suporte.59
Os testes auxiliares são muito úteis para dar suporte ao diagnóstico, incluindo evidências de atividade dopaminérgica nigrostriatal reduzida através de TC por emissão de fótons únicos (SPECT) ou de tomografia computadorizada por emissão de pósitrons (PET) nos lobos occipitais, principalmente com o lobo temporal medial intacto nas imagens (diminuindo a probabilidade de doença de Alzheimer), e lentificando na eletroencefalografia (EEG), embora nenhuma dessas descobertas seja específica para DCL.
Assim como em outros distúrbios neurodegenerativos, o diagnóstico definitivo se baseia na avaliação patológica post mortem.
Gerenciamento
Não há tratamentos específicos para DCL. Os inibidores da colinesterase são opções para o tratamento de alucinações visuais, delírios, apatia, disfunção cognitiva e agitação.61 Na realidade, esses medicamentos possivelmente sejam mais eficazes para DCL e DDP do que para a doença de Alzheimer.
Embora as medicações dopaminérgicas sejam boas opções para o parkinsonismo da DCL, recomenda-se tomar muito cuidado considerando que os pacientes com DCL talvez tenham mais probabilidade de agravar o estado de confusão e a psicose como efeitos colaterais. A seção sobre DP descreve outras características não motoras.
Atrofia de Múltiplos Sistemas
Epidemiologia e Etiologia
A AMS é uma doença neurodegenerativa esporádica progressiva que se caracteriza pela presença de disfunção autonômica no quadro de parkinsonismo, de disfunção cerebelar ou de ambos. O termo AMS substituiu os termos anteriores síndrome de Sky-Drager, degeneração estriatonigra e atrofia olivopontocerebelar, que, sob o ponto de vista patológico, representam a mesma condição. A AMS se subclassifica em AMS-C e AMS-P, isto é, se as manifestações predominantes forem cerebelares ou parkinsonianas.
A prevalência estimada de AMS varia entre 3,4 e 4,9 casos por uma população de 100 mil indivíduos. Tipicamente, o início da doença ocorre na sexta década de vida, sendo que os homens e as mulheres são igualmente afetados. Nesse exato momento, não está suficientemente claro se quaisquer fatores ambientais ou genéticos aumentam o risco de desenvolvimento da doença.
Aparentemente, a genética desempenha um papel importante na determinação do fenótipo; a incidência de AMS-C é mais comum em asiáticos, enquanto que a AMS-P é um fenótipo mais comum na população branca. A neuropatologia mostra diversos graus de atrofia olivopontocerebelar e de degeneração estriatonigra. Inclusão citoplasmática glial é a marca registrada patológica da AMS.
A a-sinucleina é a proteína com dobra anormal encontrada nessas inclusões, o que torna a AMS uma das a-sinucleinopatias que ocorre concomitantemente com DP, DCL e insuficiência autonômica pura.62 A AMS é exclusiva no que diz respeito à deposição oligodendroglial predominante de a-sinucleina agregada, que pode preceder e, de alguma forma, estimular a neurodegeneração.
Diagnóstico
Da mesma forma que a DP, a AMS apresenta manifestações pré-motoras que ocorrem em um período de meses a anos antes do início do parkinsonismo ou dos sinais cerebelares. As manifestações pré-motoras incluem disfunção sexual, hipotensão ortostática, disfunção urinária (retenção ou incontinência de urgência), DCL e estridor respiratório em 20 a 75% de pacientes.63
De um modo geral, os pacientes despertam a atenção neurológica com o início de características motoras de parkinsonismo e/ou ataxia. Parkinsonismo com bradicinesia, rigidez e uma tendência para quedas são características da AMS-P. O tremor em repouso é atípico e, ao invés disso, é comum a presença de tremor postural espasmódico e de tremor de ação em 50% de pacientes.
A AMS-C se caracteriza pela presença de marcha com base ampla, movimentos atáxicos ou descoordenados dos membros e movimentos anormais dos olhos, incluindo nistagmo. Os reflexos nos tendões profundos hiperativos são comuns e, às vezes, são acompanhados de respostas do extensor plantar.
As posturas anormais do pescoço e tronco são observadas com mais frequência nos casos de AMS em comparação com a DP, em que se observa flexão grave do pescoço para frente (antecollis) e flexão (camptocormia) ou inclinação lateral (síndrome de Pisa) do tronco. A insuficiência autonômica se manifesta com disfunção erétil nos homens, incontinência urinária e hipotensão ortostática grave (por exemplo, queda sistólica de 20mmHg ou queda diastólica de 10mmHg depois de permanecer de pé durante 3 minutos).
Outros sintomas e sinais de suporte incluem respiração desordenada na forma de apneia do sono, suspiros inspiratórios ou estridor; mãos e pés frios, que poderão ter cor azulada; hipofonia grave e dificuldade para deglutir; e incontinência emocional (risada ou choro inapropriado). As características que poderiam contestar o diagnóstico incluem início dos sintomas após os 75 anos de idade, presença de alucinações ou demência e histórico familiar de algum distúrbio semelhante.
Assim como na DP, o diagnóstico de AMS é clínico e se divide em definitivo (patologia comprovada), provável e possível, conforme definição dos critérios consensuais.64 Os testes auxiliares poderão produzir alguns benefícios.
As descobertas específicas dos estudos de imagens dão suporte ao diagnóstico de AMS, incluindo atrofia e perda de fibras mielinadas na ponte (conhecidas por sinal de hot cross bun); sinal alto nos pedúnculos cerebelares intermediários na IRM da recuperação de inversão com atenuação do líquido (FLAIR) nos casos de AMS-C; atrofia e evidências de deposição de ferro (sinal diminuído no gradiente da IRM ecocardiográfica65); e gliose no putâmen na AMS-P.
Outros estudos de imagens de suporte incluem hipometabolismo no putâmen na PET com fluorodeoxiglicose. Tipicamente, os estudos SPECT ou PET mostram evidências de desnervação dopaminérgica nigrostriatal pré-sináptica; embora isso seja mais simétrico do que a DP típica, não é um método confiável para diferenciar as duas condições.
Gerenciamento
Nos casos de AMS-P, o parkinsonismo responde ao levodopa em 40% de pacientes nos estágios iniciais da doença.66 Isso poderá ser acompanhado de discinesias induzidas por medicamentos, incluindo distonia orofacial e cervical proeminente. Embora alguns pacientes retenham a resposta ao levodopa pelo tempo de duração da enfermidade, a maioria deles recebe benefício apenas transitório, ou mesmo nenhum benefício, sendo que a ausência de resposta ao levodopa dá suporte ao diagnóstico.
A disfunção autonômica é tratada de uma forma semelhante à disfunção autonômica nos casos de DP. Entretanto, as perturbações autonômicas em geral são mais resistentes e muitos pacientes precisam fazer cateterização nos casos de disfunção na bexiga. As síncopes repetidas, causadas por ortostase em combinação com imobilidade progressiva, finalmente contribuem para o estado de confinamento em cadeira de rodas. Condições como apneia do sono e estridor inspiratório podem ser tratadas com pressão positiva contínua nas vias respiratórias (CPAP).
A Figura 6 mostra as alterações nas IRMs na AMS.
AMS: atrofia de múltiplos sistemas; IRMs: imagens por ressonância nuclear magnética.
Figura 6 - Alterações nas IRMs na AMS. (A) Hiperintensidades pontinas: “sinal de hot cross bun” na ponte (seta grossa negra) e hiperintensidades nos pedúnculos cerebelares intermediários (seta fina negra), na maioria das vezes associadas à AMS com manifestações cerebelares. (B) Hiperintensidades no putâmen bilateral e atrofia (setas brancas). (C) Sinal reduzido no putâmen na sequência de gradiente de eco (setas brancas), sugerindo deposição de ferro. Os itens (B) e (C) geralmente estão associados à AMS com manifestações parkinsonianas.
Prognóstico
A AMS é uma enfermidade implacavelmente progressiva, sendo que 60% dos pacientes são confinados em cadeiras de rodas em um período de 5 anos e uma idade média de 6 a 8 anos para um estado de confinamento no leito.62 A idade média de sobrevida a partir do início dos sintomas varia de 5 a 10 anos, sendo que poucos pacientes vivem até a idade de 15 anos. As causas mais comuns de morte nos casos de AMS incluem infecções (pneumonia por aspiração e urosepse) e morte súbita causada pelo rompimento agudo da estimulação cardiorrespiratória no tronco cefálico.62
Paralisia Supranuclear Progressiva
Epidemiologia e Etiologia
A PSP é uma condição neurodegenerativa esporádica que se caracteriza por parkinsonismo, quedas precoces e anormalidades nos movimentos dos olhos (paralisia supranuclear da fixação vertical do olhar). A PSP também é conhecida por síndrome de Steele-Richardson-Olszewski, em homenagem aos pesquisadores canadenses que descreveram essa condição pela primeira há, aproximadamente, 50 anos.
Acredita-se que a proteína tau com dobras anormais seja a causa subjacente deste distúrbio, ao contrário da a-sinucleina nos casos de DP e AMS. A composição neurofibrilar emaranhada da proteína tau é a marca registrada neuropatológica de PSP.
A patologia da PSP mostra neurodegeneração proeminente no tronco cefálico, juntamente com a neurodegeneração no córtex frontal e em outras estruturas dos gânglios basais.67 A maior parte dos pacientes carrega o haplótipo H1 do gene MAPT (tau).68 Além disso, os fatores de risco ambientais e genéticos associados não estão muito bem definidos.
A prevalência estimada de PSP varia entre 1.5 a 4,9 casos por 100 mil pessoas,69 com uma ligeira preponderância masculina (55%).67 Normalmente, os sintomas iniciam na sétima década de vida, com uma idade mediana de 63 anos.69
Diagnóstico
Sob o ponto de vista clínico, geralmente os pacientes se apresentam com desequilíbrio precoce (instabilidade postural) e quedas, em combinação com a “lentificação” dos movimentos. O parkinsonismo se caracteriza pela predominância de rigidez axial e bradicinesia. Os problemas com a marcha e a estabilidade postural são comuns logo no início da condição, ocasião em que o congelamento da marcha (FOG) pode ser proeminente.
Provavelmente, os membros da família observem alterações sutis na personalidade como apatia, por exemplo. As anormalidades nos movimentos dos olhos são a marca registrada desse tipo de distúrbio e, na ausência delas, o diagnóstico se torna muito difícil. Essas anormalidades poderão ocorrer mais tarde ou não ocorrer em nenhum momento.
As características clínicas mais precoces são lentificação das sacadas verticais (em especial para baixo) e perda das fases rápidas do nistagmo optocinético vertical. Ao longo do tempo, esse processo poderá progredir para a incapacidade total de olhar voluntariamente para cima ou para baixo, o que poderá ser facilmente constatado pelo examinador se ele movimentar rapidamente a cabeça para baixo ou para cima (manobra oculocefálica), que caracteriza a PSP típica.
Os pacientes, em geral, se queixam de visão turva, dificuldade para fixar novamente o olhar, em especial na leitura e na dificuldade com a visão em campos visuais mais baixos (por exemplo, ver que alimentos estão no prato).
Outras anormalidades nos movimentos dos olhos incluem perseguição sacádica proeminente, movimentos dos olhos de pequena amplitude em repouso (reflexos de ondas quadradas) e anormalidade nas pálpebras como velocidade gravemente reduzida ao piscar, blefaroespasmo (ou fechamento forçado dos olhos), ou dificuldade para abrir os olhos conhecida por “apraxia de abertura das pálpebras”.
A aparência típica da face na PSP é uma expressão “preocupada” ou “de surpresa”. A voz anasalada rude é uma característica típica de PSP e a dificuldade para deglutir pode se tornar bastante proeminente. Às vezes, observa-se uma postura estendida do pescoço (espasmo retrocervical). As anormalidades cognitivas são comuns nos casos de PSP e, tipicamente, afetam a velocidade de processamento e a função executiva.
O estado de apatia é comum e talvez seja uma característica precoce da doença. Os sinais de liberação frontal, como o reflexo da preensão palmar (continuar segurando a mão do examinador apesar de ele ter pedido para soltá-la com um toque na palma), podem ser proeminentes, assim como os sinais de desinibição frontal, incluindo o sinal de “aplauso” (incapacidade de parar de bater palmas depois de tentar imitar o examinador bater palmas três vezes). Palilalia ou repetição involuntária de palavras ou frases também é uma ocorrência comum.
O diagnóstico de PSP clássica (às vezes, conhecida por síndrome de Richardson) é clínico e se divide em definitivo (patologia comprovada), provável e possível, de acordo com a definição dos critérios de consenso.70 As características principais são uma síndrome bradicinética gradualmente progressiva, dificuldades para fixação vertical do olhar e instabilidade postural proeminente, com dificuldade para manter o equilíbrio ou para evitar quedas (no primeiro ano).
Os testes auxiliares poderão trazer algum benefício. As descobertas específicas dos estudos de imagens que ajudam a definir o diagnóstico de PSP incluem atrofia no mesencéfalo na IRM, conhecida por sinal do “colibri” ou do “pinguim”, conforme a Figura 7.
IRMs: imagens por ressonância nuclear magnética PSP: paralisia supranuclear progressiva.
Figura 7 - Alterações nas IRMs na PSP. Visão sagital intermediária mostrando atrofia no mesencéfalo, descrita por uma caixa com contorno branco explodido na inserção (geralmente, conhecida por “sinal do colibri”).
Uma área que, atualmente, é objeto de estudos ativos é o desenvolvimento de ligações com PET que possam avaliar com confiabilidade a presença de tau nos cérebros de pacientes com PSP e taupatias relacionadas.71
Estudos neuropatológicos recentes mostraram que a patologia de PSP também pode estar associada a outras apresentações fenotípicas, incluindo parkinsonismo sugestivo de DP (com tremor em repouso e resposta parcial ao levodopa), uma síndrome corticobasal (SCB), um transtorno precoce da fala indicando apraxia da fala ou afasia progressiva primária não fluente, e um distúrbio da marcha, o FOG primário.72
Gerenciamento
Com frequência, o tratamento de PSP inclui o teste com levodopa, que poderá mostrar uma melhora leve e transitória no parkinsonismo em 30% de pacientes.73 Eventualmente, a amantadina poderá produzir algum benefício para o congelamento da marcha e, ocasionalmente, para outros sintomas. As injeções com toxina botulínica são boas opções para blefaroespasmo ou apraxia na abertura das pálpebras. A prevenção de quedas e o gerenciamento da disfagia são extremamente importantes nos casos de PSP para evitar a morbidade e a mortalidade associada a esses sintomas.
Prognóstico
A PSP tem maus prognósticos, sendo que a sobrevida média varia de 5,9 a 9,7 anos, dependendo do estudo.69 Diversos tratamentos com foco na função ou na quantidade de tau encontram-se, atualmente, em fase de desenvolvimento ativo.
Degeneração Corticobasal
Epidemiologia e Etiologia
DCB é uma condição neurodegenerativa esporádica rara que, classicamente, se classifica pela presença de distúrbios motores e de características corticais no cérebro. Os distúrbios motores incluem o parkinsonismo assimétrico que responde fracamente ao levodopa, em geral acompanhado de outros movimentos anormais, incluindo distonia e mioclonia.
As características corticais no cérebro inclui apraxia de membro, disfunção cognitiva, anormalidades comportamentais e transtornos de linguagem, em especial afasia progressiva primária não fluente. A patologia revela perda de neurônios no córtex frontal e parietal, de substância negra e de outras estruturas dos gânglios basais.
Da mesma forma que nos casos de PSP, tau é a proteína agregada anormal encontrada nos neurônios e nas células gliais, o que torna a DCB uma tauopatia. DCB se refere à patologia específica, que possivelmente está associada a uma grande variedade de características clínicas além da apresentação já descrita anteriormente, incluindo uma variante comportamental pura de demência frontotemporal ou uma apresentação que se assemelha à PSP.
O termo síndrome corticobasal (SCB) é usado para descrever a síndrome clínica clássica, que pode ser causada por outros distúrbios, incluindo PSP, doença de Alzheimer, demências frontotemporais (sendo que algumas delas produzem formas familiares de SCB) e mesmo a doença de Creutzfeldt-Jakob (resultando, tipicamente, em uma SCB com progressão rápida).74
Estima-se que a prevalência de DCB seja de 4,9 a 7,3 casos por 100 mil pessoas.75 Tipicamente, a doença se apresenta entre a sexta e oitava décadas de vida, com idade média de início de 63 anos.75 Não há fatores de risco genéticos ou ambientais conhecidos, porém, assim como nos casos de PSP, a maior parte dos pacientes carrega o haplótipo H1 do gene MAPT.68
Diagnóstico
A apresentação clínica se divide em características motoras e disfunção cortical. A característica motora mais comum da DCB é o parkinsonismo assimétrico, cujas características principais são bradicinesia e rigidez em mais 75% de pacientes.76 A presença de instabilidade postural, de quedas e de rigidez axial também é muito comum.
Observa-se a presença de tremor em um terço de pacientes, cuja presença se manifesta ao manter uma postura ou ao executar uma ação, mas raramente ocorre no estado de repouso. Distonia (posturas anormais dos membros) ou mioclonia (movimentos breves que se assemelham a choques) também ocorrem em, aproximadamente, um terço de pacientes.76
Com frequência, a disfunção cortical se caracteriza mais pela presença de alterações cognitivas gerais (70%), sendo que metade dos pacientes apresenta alterações comportamentais. Observa-se também apraxia nos membros (incapacidade para executar comportamentos motores básicos, como, por exemplo, escovar os dentes) em 50% de pacientes, assim como alguma forma de afasia (problemas com produção de linguagem).76
O fenômeno do movimento involuntário dos membros (alien limb) é uma descoberta relativamente específica da DCB; entretanto, a sensibilidade é baixa, considerando que essa condição ocorre em apenas 30% de pacientes. Esse fenômeno se caracteriza por movimentos complexos aparentemente propositais dos membros, porém involuntários, que interferem nas tarefas motoras, ou por uma sensação de que o membro se comporta de uma forma estranha e sem vontade própria.76
Perdas sensoriais corticais, incluindo grafestesia (incapacidade de perceber que está escrevendo na própria pele) e estereognose (incapacidade de reconhecer objetos com base apenas no toque), também ocorrem em 30% de pacientes.76
A DCB pode ser diagnosticada em caráter definitivo apenas pela patologia post mortem. Foram propostos alguns critérios clínicos para DCB dos tipos possível e provável, porém eles ainda não foram validados. A forma SCB de DCB se tipifica pela presença de características motoras (rigidez/acinesia assimétrica em um ou dois membros, distonia, mioclonia) e de perturbações corticais (apraxia em um ou dois membros e perda sensorial cortical).76
As descobertas feitas em estudos de imagens, que dão suporte ao diagnóstico de DCB, incluem atrofia assimétrica no córtex frontal e parietal, embora essas descobertas não sejam sensíveis e nem específicas.75
Gerenciamento
O tratamento de DCB inclui administração de levodopa para parkinsonismo que, ocasionalmente, apresenta benefícios leves a moderados, porém de caráter transitório.76 As injeções com toxina botulínica produzem algum benefício nos casos de distonia e tremor, sendo que a mioclonia responde à terapia sintomática.75 Todavia, ao final, os pacientes acabam se tornando totalmente dependentes.
O gerenciamento da disfagia é essencial nos casos de doença avançada. As terapias com foco na proteína tau, que atualmente estão em desenvolvimento para PSP, também poderão ser aplicadas no tratamento de DCB. Levando-se em consideração a grande dificuldade para fazer um diagnóstico preciso da doença com os pacientes ainda em vida, as varreduras da proteína tau usando a técnica de imagens PET são muito importantes no processo de avaliação das terapias anti-tau nesse tipo de distúrbio.71 Atualmente, os prognósticos são muito fracos e a sobrevida média é de 7,9 anos.75
Parkinsonismo Vascular
Epidemiologia e Etiologia
O parkinsonismo vascular descreve um grupo heterogêneo de distúrbios com características típicas de parkinsonismo, possivelmente devido à presença de doença cerebrovascular. A característica clínica proeminente do parkinsonismo vascular é o distúrbio da marcha ou “parkinsonismo na parte inferior do corpo”. De um modo geral, a idade de início é mais avançada nos casos de parkinsonismo vascular em comparação com a DP, sendo que os homens são mais afetados que as mulheres, possivelmente porque eles têm maior incidência de doenças vasculares.
As estimativas de prevalência variam de 3 a 5% no parkinsonismo considerado de origem vascular.77 Embora existam apenas alguns estudos patológicos, aqueles que estão à disposição sugerem a ocorrência de pequenos ataques (lacunas) nos gânglios basais e de alterações extensivas na substância branca, principalmente nos lobos frontais.77 Ao contrário da DP ou dos distúrbios neurodegenerativos do parkinsonismo atípico descritos anteriormente, o diagnóstico definitivo não se baseia na patologia, tendo em vista que existem critérios diagnósticos aceitáveis para o parkinsonismo vascular.
Diagnóstico
A característica clínica mais clássica de parkinsonismo vascular é o que se costuma chamar de “parkinsonismo na parte inferior do corpo” (ou “metade inferior”) que se caracteriza por um tipo de marcha de passos curtos, festinante ou “magnético”, em geral com uma base ligeiramente ampla, instabilidade postural e quedas. Outras características incluem espasticidade e, ao longo do tempo, a tendência para desenvolver paralisia pseudobulbar, demência, incontinência urinária e reflexos primitivos.78
Com frequência, observa-se a ausência de tremor e as extremidades superiores são muito menos afetadas que as extremidades inferiores. De um modo geral, os pacientes apresentam fatores de risco vascular (por exemplo, hipertensão, diabetes melito, tabagismo) e talvez tenham histórico de acidente(s) vascular(es) cefálico(s). O início da doença pode ser agudo e ter progressão rápida.
As descobertas dos estudos de imagens incluem infartos lacunares múltiplos nos gânglios basais e lesões extensivas na substância branca.79 Embora sejam necessárias para o diagnóstico de parkinsonismo vascular, as alterações nas IRMs não apresentam nenhum padrão específico para essa condição.
Gerenciamento
Uma pequena minoria de pacientes responde ao levodopa ou a outras terapias dopaminérgicas. Acredita-se que o gerenciamento de fatores de risco vascular lentifique a progressão da doença, mas essa hipótese ainda não foi comprovada. A heterogeneidade da apresentação clínica e das descobertas feitas nos estudos de imagens, assim como a ausência de critérios patológicos, transformou o parkinsonismo vascular em um desafio diagnóstico e em uma entidade clínica bastante controversa.80
Hidrocefalia com Pressão Normal
A HNB descreve a síndrome de perturbação na marcha, demência e incontinência urinária, que está associada ao aumento nas dimensões ventriculares nas imagens do cérebro no contexto de pressão normal do líquido cefalorraquidiano (LCR). Os sintomas desenvolvem de forma insidiosa e, tipicamente, iniciam na sétima década de vida ou mais tarde.
O primeiro sintoma mais frequente é uma perturbação na marcha, descrita alternativamente como apraxia “frontal” da marcha ou “marcha magnética” e se compõe de passos curtos, arrastando os pés e com uma base ampla muito semelhante ao que ocorre em pacientes com “parkinsonismo vascular”. Na maior parte dos casos, os pacientes têm histórico de quedas.
Tipicamente, os transtornos cognitivos são “subcorticais”, com raciocínio lento, desatenção e dificuldades nas funções executivas e de planejamento. Os sintomas urinários de bexiga superativa são a terceira característica da tríade e incluem urgência/frequência à incontinência autêntica. A prevalência estimada de HNB varia de 0,1 a 2,9% em pessoas idosas.81
Considerando que há um grande diferencial para os distúrbios individualizados da marcha, distúrbios cognitivos e disfunção urinária, é essencial investigar e excluir outras etiologias potenciais. Considerando que o início típico de HNB ocorre em idades mais avançadas, em geral há etiologias concomitantes que explicam os vários sintomas. Nos estudos de imagens, o aumento ventricular, que poderia se esperar para o grau de atrofia cerebral sem evidências de obstrução, é essencial para o diagnóstico de HNB, conforme mostra a Figura 8.
HNB: hidrocefalia normobárica.
Figura 8 - Aumento nos ventrículos com HNB nas visões axial (A) e sagital (B).
Os investigadores passaram a se interessar pela HNB como uma entre as poucas causas “reversíveis” de demência, cujo gerenciamento definitivo é a colocação de uma derivação (shunt) ventriculoperitoneal (VP) para drenar o líquido cefalorraquidiano. Alguns estudos mostram que a melhora após a cirurgia se aproxima de 80%,82 sendo que é mais óbvia na marcha, ao passo que a disfunção cognitiva tem menor probabilidade de melhorar.83
É muito difícil prever qual condição terá mais benefícios com a cirurgia. De um modo geral, utiliza-se a drenagem de LCR para dar suporte ao diagnóstico e para tentar prever quais pacientes poderão ter benefícios com a cirurgia. A marcha tende a responder melhor à drenagem de LCR e, portanto, deverá ser medida antes e após a drenagem. A cognição também poderá melhorar após a drenagem.
Embora sejam mais fáceis, as punções lombares (PLs) de grandes volumes com remoção de 30 a 50cc de LCR não são sensíveis nem específicas. Nos casos em que os testes de PL de grandes volumes forem negativos, embora a suspeita clínica seja alta, é imprescindível fazer a drenagem lombar externa.84
Esse procedimento implica em um breve período de hospitalização e colocação de um cateter lombar para possibilitar a drenagem contínua (imitando a derivação VT), assim como a avaliação da marcha e da cognição antes, durante e depois do procedimento de drenagem.85
A discussão se relaciona à condição que passou a ser conhecida por “HNB idiopática”. Alguns pacientes com essa síndrome têm algum distúrbio neurodegenerativo subjacente, incluindo PSP ou doença de Alzheimer.86 Nesse caso, tipicamente, a resposta inicial à derivação é acompanhada de um declínio clínico progressivo subsequente.
O termo HNB sintomática foi aplicado à síndrome de HNB em pacientes com histórico prévio de hemorragia subaracnoideia ou meningite nos quais a absorção de LCR possivelmente tenha sido alterada por vilosidades aracnoideias disfuncionais. A HNB sintomática responde muitíssimo bem ao desvio do LCR (derivação VT).
Parkinsonismo Induzido por Medicamentos
O PIM, um efeito colateral relacionado à dosagem de uma grande variedade de medicações, é clinicamente indistinguível de DP com todas as características básicas, incluindo tremor em repouso. As medicações implicadas com mais frequência no desenvolvimento de PIM são os agentes bloqueadores do receptor da dopamina, incluindo os neurolépticos, e os antinauseantes como a metoclopramida, assim como os agentes bloqueadores do canal de cálcio flunarazina e cinarazina, que também bloqueiam os receptores da dopamina.
Os agentes depletores da dopamina reserpina e tetrabenazina também são agentes causativos. Com menos frequência, os ISRSs, o lítio e o valproato de sódio também estão associados ao parkinsonismo. A ocorrência de PIM é mais provável nas mulheres em comparação com os homens (razão de 2:1). As estimativas de incidência variam de acordo com o estudo e o medicamento; um estudo envolvendo pacientes que haviam sido tratados com neurolépticos encontrou uma incidência de 19,4%.87 Os fatores de risco são idade avançada e DP pré-existente.
O tratamento de PIM envolve redução na titulação do agente ofensor, embora os sintomas melhorem somente depois de 1 a 2 meses ou mais. Há poucas evidências que dão suporte à existência de um fenômeno de parkinsonismo tardio (isto é, parkinsonismo induzido pelos medicamentos que ainda estiverem sendo usados após a retirada total do agente ofensor).
Nos casos em que o parkinsonismo persistir, apesar da descontinuação no uso da medicação ofensora, acredita-se que os sintomas persistentes tenham outra etiologia subjacente (por exemplo, DP idiopática concorrente). As imagens obtidas pela técnica SPECT do transportador de dopamina são muito úteis para determinar se o parkinsonismo foi induzido por medicamentos ou se foi causado por algum processo degenerativo persistente.88
Alguns pacientes apresentam resolução do parkinsonismo com a retirada do medicamento ofensor, embora observem, mais tarde, que houve recorrência dos sintomas que não estavam relacionados com o uso posterior do medicamento. Isso sugere que o paciente teve alguma doença subjacente como DP, cujos sintomas não haviam sido mascarados prematuramente pela medicação.
Nos pacientes em que o agente ofensor não possa ser reduzido gradualmente (por exemplo, esquizofrenia que depende de tratamento), o tratamento com anticolinérgicos ou, em alguns casos, com amantadina talvez produza benefícios. Como alternativa, o tratamento neuroléptico poderia ser substituído por agentes menos potentes ou com menor probabilidade de induzir parkinsonismo (por exemplo, clozapina).
Sumário
O parkinsonismo descreve os critérios clínicos básicos de tremor, bradicinesia, rigidez e instabilidade postural. Embora o diagnóstico diferencial seja bastante amplo [ver o Quadro 1], a DP é a causa mais comum de parkinsonismo. O diagnóstico de DP é clínico e se baseia na presença de parkinsonismo e na ausência de outros “sinais de alerta” [ver os Quadros 2 e 3]. Existem muitas opções de tratamento para os sintomas motores [ver o Quadro 4] e não motores [ver o Quadro 5] da DP.
|
Informações financeiras: Elizabeth J. Slow, MD, PhD, não tem nenhuma informação financeira a declarar. Anthony E. Lang, MD, é consultor das empresas Abbvie, Medtronic, Teva e UCB, das quais recebeu honorários. Esta revisão foi feita anteriormente por Bradley Robottom, MD, Jason Hawley, MD, e William Weiner, MD, cujas informações financeiras foram apresentadas por ocasião da publicação inicial. Este texto foi revisado, atualizado e reeditado pelos autores mencionados. |
Referências
1. de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 Suppl 5):S21–3.
2. de Lau LML, Giesbergen PCLM, de Rijk MC, et al. Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology 2004;63:1240–4.
3. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA 2014;311:1670–83.
4. Noyce AJ, Bestwick JP, Silveira-Moriyama L, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol 2012;72:893–901.
5. Braak H, Del Tredici K, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211.
6. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–7.
7. Kalia LV, Lang AE. Parkinson’s disease. Lancet 2015;386:896–912. DOI: 10.1016/S0140-6736(14)61393-3. [Epub 2015 Apr 19]
8. Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 2008;7:583–90.
9. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009;361:1651–61.
10. Visanji NP, Brooks PL, Hazrati L-N, Lang AE. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol Com-mun 2013;1(1):2.
11. Siderowf A, Lang AE. Premotor Parkinson’s disease: concepts and definitions. Mov Disord 2012;27:608–16.
12. Jankovic J, Lang AE. Movement disorders: diagnosis and assessment. In: Bradley WG, editor. Neurology in clinical practice. Vol.1. 5th ed. USA: Elsevier Inc.; 2008. p. 293–325.
13. Goldman JG, Postuma R. Premotor and nonmotor features of Parkinson’s disease. Curr Opin Neurol 2014;27:434–41.
14. Haehner A, Boesveldt S, Berendse HW, et al. Prevalence of smell loss in Parkinson’s disease—a multicenter study. Parkinsonism Relat Disord 2009;15:490–4.
15. Olanaw CW, Stocchi F, Lang AE. The dopaminergic and non-dopaminergic features of Parkinson’s disease. In: Olanaw CW, Stocchi F, Lang AE, editors. Parkinson’s disease: non-motor and non-dopaminergic features. 1st ed. USA: Blackwell Publishing Ltd.; 2011. p. 1–6.
16. Zadikoff C, Fox SH, Tang-Wai DF, et al. A comparison of the Mini Mental State exam to the Montreal Cognitive Assessment in identifying cognitive deficits in Parkinson’s disease. Mov Disord 2008;23:297–9.
17. Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 2009;8:1150–7.
18. Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology 2001;57:1497–9.
19. Berg D, Postuma RB, Bloem B, et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 2014;29:454–62.
20. Marras C. Subtypes of Parkinson’s disease: state of the field and future directions. Curr Opin Neurol 2015;28:382–6.
21. Aquino CC, Fox SH. Clinical spectrum of levodopa-induced complications. Mov Disord 2015;30:80–9.
22. Beaulieu-Boire I, Lang AE. Behavioral effects of levodopa. Mov Disord 2015;30:90–102.
23. Hauser RA, Hsu A, Kell S, et al. Extended-release carbidopa-levodopa (IPX066) compared with immediate-release carbidopa-levodopa in patients with Parkinson’s disease and motor fluctuations: a phase 3 randomised, double-blind trial. Lancet Neurol 2013;12:346–56.
24. Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol 2014;13:141–9.
25. Nyholm D, Klangemo K, Johansson A. Levodopa/carbidopa intestinal gel infusion long-term therapy in advanced Parkinson’s disease. Eur J Neurol 2012;19:1079–85.
26. Poewe W, Antonini A. Novel formulations and modes of delivery of levodopa. Mov Disord 2015;30:114–20.
27. Ives NJ, Stowe RL, Marro J, et al. Monoamine oxidase type B inhibitors in early Parkinson’s disease: meta-analysis of 17 randomised trials involving 3525 patients. BMJ 2004;329:593.
28. Smith KM, Eyal E, Weintraub D, ADAGIO Investigators. Combined rasagiline and antidepressant use in Parkinson disease in the ADA-GIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol 2015;72:88–95.
29. Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009;361:1268–78.
30. Katzenschlager R, Sampaio C, Costa J, Lees A. Anticholinergics for symptomatic management of Parkinson’s disease. Cochrane Database Syst Rev 2003;(2):CD003735.
31. Ferreira JJ, Katzenschlager R, Bloem BR, et al. Summary of the recommendations of the EFNS/MDS-ES review on therapeutic management of Parkinson’s disease. Eur J Neurol 2013;20:5–15.
32. Crosby NJ, Deane KHO, Clarke CE. Amantadine for dyskinesia in Parkinson’s disease. Cochrane Database Syst Rev 2003;(2):CD003467.
33. Liu Y, Li W, Tan C, et al. Meta-analysis comparing deep brain stimulation of the globus pallidus and subthalamic nucleus to treat ad-vanced Parkinson disease. J Neurosurg 2014;121:709–18.
34. Schuepbach WMM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson’s disease with early motor complications. N Engl J Med 2013;368:610–22.
35. Tomlinson CL, Patel S, Meek C, et al. Physiotherapy versus placebo or no intervention in Parkinson’s disease. Cochrane Database Syst Rev 2013;9:CD002817.
36. Li F, Harmer P, Fitzgerald K, et al. Tai chi and postural stability in patients with Parkinson’s disease. N Engl J Med 2012;366:511–9.
37. Grimes D, Gordon J, Snelgrove B, et al; Canadian Neurological Sciences Foundation. Canadian guidelines on Parkinson’s disease. Can J Neurol Sci 2012;39(4 Suppl 4):S1–30.
38. Fox SH, Katzenschlager R, Lim S-Y, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the motor symptoms of Parkinson’s disease. Mov Disord 2011;26 Suppl 3:S2–41.
39. Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain 2014;137(Pt 10):2731–42.
40. Fox SH, Lang AE. ‘Don‘t delay, start today’: delaying levodopa does not delay motor complications. Brain 2014;137(Pt 10):2628–30.
41. PD Med Collaborative Group, Gray R, Ives N, et al. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson’s disease (PD MED): a large, open-label, pragmatic randomised trial. Lancet 2014;384:1196–205.
42. Hely MA, Reid WGJ, Adena MA, et al. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord 2008;23:837–44.
43. Diederich NJ, Fénelon G, Stebbins G, Goetz CG. Hallucinations in Parkinson disease. Nat Rev Neurol 2009;5:331–42.
44. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. The Parkinson Study Group. N Engl J Med 1999;340:757–63.
45. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet 2014;383:533–40.
46. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord 2011;26 Suppl 3:S42–80.
47. Skapinakis P, Bakola E, Salanti G, et al. Efficacy and acceptability of selective serotonin reuptake inhibitors for the treatment of depression in Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. BMC Neurol 2010;10:49.
48. Pagonabarraga J, Kulisevsky J, Strafella AP, Krack P. Apathy in Parkinson’s disease: clinical features, neural substrates, diagnosis, and treatment. Lancet Neurol 2015;14:518–31.
49. Fasano A, Visanji NP, Liu LWC, et al. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 2015;14:625–39.
50. Sakakibara R, Panicker J, Finazzi-Agro E, et al, Parkinson’s Disease Subcommittee, The Neurourology Promotion Committee in The In-ternational Continence Society. A guideline for the management of bladder dysfunction in Parkinson’s disease and other gait disorders. Neurourol Urodyn 2015 Mar 25. DOI: 10.1002/nau.22764.
51. Slow EJ, Postuma RB, Lang AE. Implications of nocturnal symptoms towards the early diagnosis of Parkinson’s disease. J Neural Transm 2014 Feb 6. DOI: 10.1007/s00702-014-1168-4.
52. Oosterveld LP, Allen JC, Reinoso G, et al. Prognostic factors for early mortality in Parkinson’s disease. Parkinsonism Relat Disord 2015;21:226–30.
53. Reinoso G, Allen JC, Au W-L, et al. Clinical evolution of Parkinson’s disease and prognostic factors affecting motor progression: 9-year follow-up study. Eur J Neurol 2015;22:457–63.
54. Macleod AD, Taylor KSM, Counsell CE. Mortality in Parkinson’s disease: a systematic review and meta-analysis. Mov Disord 2014;29:1615–22.
55. Kalia LV, Kalia SK, Lang AE. Disease-modifying strategies for Parkinson’s disease. Mov Disord 2015 Jul 24. DOI: 10.1002/mds.26354. [Epub ahead of print]
56. Mayo MC, Bordelon Y. Dementia with Lewy bodies. Semin Neurol 2014;34:182–8.
57. McKeith I, Mintzer J, Aarsland D, et al. Dementia with Lewy bodies. Lancet Neurol 2004;3:19–28.
58. Savica R, Grossardt BR, Bower JH, et al. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol 2013;70:1396–402.
59. McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis 2006;9(3 Suppl):417–23.
60. Aarsland D, Perry R, Larsen JP, et al. Neuroleptic sensitivity in Parkinson’s disease and parkinsonian dementias. J Clin Psychiatry 2005;66:633–7.
61. Mori E, Ikeda M, Kosaka K, Donepezil-DLB Study Investigators. Donepezil for dementia with Lewy bodies: a randomized, placebo-controlled trial. Ann Neurol 2012;72:41–52.
62. Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med 2015;372:249–63.
63. Jecmenica-Lukic M, Poewe W, Tolosa E, Wenning GK. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol 2012;11:361–8.
64. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–6.
65. Wadia PM, Howard P, Ribeirro MQ, et al. The value of GRE, ADC and routine MRI in distinguishing Parkinsonian disorders. Can J Neurol Sci 2013;40:389–402.
66. Köllensperger M, Geser F, Ndayisaba J-P, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010;25:2604–12.
67. Golbe LI. Progressive supranuclear palsy. Semin Neurol 2014;34:151–9.
68. Pittman AM, Myers AJ, Abou-Sleiman P, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet 2005;42:837–46.
69. Colosimo C, Bak TH, Bologna M, Berardelli A. Fifty years of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2014;85:938–44.
70. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996;47:1–9.
71. Coakeley S, Strafella AP. Imaging pathological tau in atypical parkinsonian disorders. Curr Opin Neurol 2015;28:447–52.
72. Williams DR, Lees AJ, Wherrett JR, Steele JC. J. Clifford Richardson and 50 years of progressive supranuclear palsy. Neurology 2008;70:566–73.
73. Nieforth KA, Golbe LI. Retrospective study of drug response in 87 patients with progressive supranuclear palsy. Clin Neuropharmacol 1993;16:338–46.
74. Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord 2007;13 Suppl 3:S336–40.
75. Mahapatra RK, Edwards MJ, Schott JM, Bhatia KP. Corticobasal degeneration. Lancet Neurol 2004;3:736–43.
76. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503.
77. Korczyn AD. Vascular parkinsonism—characteristics, pathogenesis and treatment. Nat Rev Neurol 2015;11:319–26.
78. Kalra S, Grosset DG, Benamer HTS. Differentiating vascular parkinsonism from idiopathic Parkinson’s disease: a systematic review. Mov Disord 2010;25:149–56.
79. Vale TC, Caramelli P, Cardoso F. Clinicoradiological comparison between vascular parkinsonism and Parkinson’s disease. J Neurol Neurosurg Psychiatry 2015;86:547–53.
80. Vizcarra JA, Lang AE, Sethi KD, Espay AJ. Vascular parkinsonism: deconstructing a syndrome. Mov Disord 2015;30:886–94.
81. Jaraj D, Rabiei K, Marlow T, et al. Prevalence of idiopathic normal-pressure hydrocephalus. Neurology 2014;82:1449–54.
82. Toma AK, Stapleton S, Papadopoulos MC, et al. Natural history of idiopathic normal-pressure hydrocephalus. Neurosurg Rev 2011;34:433–9.
83. Gallia GL, Rigamonti D, Williams MA. The diagnosis and treatment of idiopathic normal pressure hydrocephalus. Nat Clin Pract Neurol 2006;2:375–81.
84. Chotai S, Medel R, Herial NA, Medhkour A. External lumbar drain: a pragmatic test for prediction of shunt outcomes in idiopathic normal pressure hydrocephalus. Surg Neurol Int 2014;5:12.
85. Williams MA, Relkin NR. Diagnosis and management of idiopathic normal-pressure hydrocephalus. Neurol Clin Pract 2013;3:375–85.
86. Curran T, Lang AE. Parkinsonian syndromes associated with hydrocephalus: case reports, a review of the literature, and pathophysiolog-ical hypotheses. Mov Disord 1994;9:508–20.
87. Muscettola G, Barbato G, Pampallona S, et al. Extrapyramidal syndromes in neuroleptic-treated patients: prevalence, risk factors, and association with tardive dyskinesia. J Clin Psychopharmacol 1999;19:203–8.
88. Lorberboym M, Treves TA, Melamed E, et al. [123I]-FP/CIT SPECT imaging for distinguishing drug-induced parkinsonism from Par-kinson’s disease. Mov Disord 2006;21:510–4.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.