(Carregando Índice)... (Carregando Índice)... |
Autores:
Patrícia Lima Junqueira
Médica Hematologista do Instituto do Câncer do Estado de São Paulo – Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Erica Okazaki
Médica Hematologista do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP) e da Fundação Pró-Sangue.
Paula Ribeiro Villaça
Médica Hematologista da Fundação Pró-Sangue e Professora Colaboradora da Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Elbio Antônio D’Amico
Médico Hematologista do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP) e Professor Colaborador da Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Última revisão: 13/08/2009
Comentários de assinantes: 0
O tromboembolismo venoso acomete cerca de 1 em cada 1.000 indivíduos ao ano, sendo que destes,
Alterações genéticas e adquiridas estão envolvidas na sua patogênese e, atualmente, um fator de risco pode ser reconhecido em mais de 80% dos pacientes com trombose, identificando-se pelo menos dois na maioria dos casos.
As bases fisiopatológicas surgiram em 1856, quando Virchow propôs que três fatores contribuíam para a formação do trombo: alterações no fluxo sanguíneo (estase), na parede vascular (lesão endotelial) e nos componentes do sangue. Desde então, o conhecimento das vias hemostáticas tem permitido a identificação de fatores que aumentam o risco de trombose, tornando possível instituir medidas terapêuticas e profiláticas.
O termo trombofilia tem sido utilizado para descrever anormalidades hereditárias e adquiridas do sistema hemostático, que predispõem a eventos trombóticos. Tais alterações interagem de forma complexa e, juntas, recebem a denominação genérica de fatores de risco (Tabela 1).
As apresentações clínicas mais comuns são a trombose venosa profunda (TVP) de membros inferiores e o tromboembolismo pulmonar (TEP), ainda que o evento vaso-oclusivo possa, mais raramente, ocorrer em outros locais (veias retinianas, mesentéricas, supra-hepáticas, cerebrais, de membros superiores e veias superficiais) ou, até mesmo, acometer o território arterial. As trombofilias podem também se manifestar por perdas gestacionais recorrentes e outras complicações durante a gravidez.
O reconhecimento dos fatores predisponentes é fundamental, particularmente quando há possibilidade de controle ou remoção deles, conseguindo-se, assim, reduzir o estado de hipercoagulabilidade
Tabela 1: Fatores de risco primários (hereditários) e secundários (adquiridos)
|
Hereditários |
Adquiridos |
|
Fator V Leiden |
Imobilização |
|
Mutação G20210A da protrombina |
Cirurgia / Trauma |
|
Deficiência de proteína C |
Infecções |
|
Deficiência de proteína S |
Cateter venoso central |
|
Deficiência de antitrombina |
Câncer / Quimioterápicos |
|
Resistência à proteína C ativada |
Gestação / Puerpério |
|
Deficiência de plasminogênio |
Terapia hormonal |
|
Desfibrinogenemia |
SAAF* |
|
Outros |
Síndrome nefrótica |
|
Hiper-homocisteinemia |
Doenças reumatológicas |
|
? Fator VIII |
Síndromes de hiperviscosidade |
|
? Fator IX |
Neoplasias mieloproliferativas |
|
? Fator XI |
Hemoglobinúria paroxística noturna |
|
? TAFI / ? TFPI |
Anemia falciforme |
*SAAF: síndrome do anticorpo antifosfolípide; TAFI: inibidor da fibrinólise ativado por trombina; TFPI: inibidor da via do fator tecidual.
As trombofilias hereditárias ocorrem quando mutações gênicas alteram propriedades de fatores envolvidos na coagulação do sangue, proporcionando seja um ganho de função aos pró-coagulantes (fator V, protrombina) ou perda de função de anticoagulantes naturais (antitrombina, proteína C, proteína S).
Em 1965, Egeberg reconheceu o primeiro fator de risco hereditário ao identificar que a deficiência de antitrombina estava associada a múltiplos eventos trombóticos em uma família norueguesa. Subsequentemente, foram descritas outras alterações genéticas, como deficiência da proteína C (1981), deficiência da proteína S (1984), fator V Leiden (1994) e a mutação G20210A no gene da protrombina (1996).
Os estados hipercoagulantes primários são causas pouco frequentes de trombose, sendo que as mutações Arg506Gln no gene do fator V (fator V Leiden) e a G20210A no gene da protrombina correspondem, juntas, a 50 a 60% dos casos. Deficiências de proteína C, S e antitrombina são menos comuns e as desfibrinogenemias são formas ainda mais raras de trombofilia hereditária. Níveis plasmáticos aumentados de homocisteína estão relacionados a tromboses venosas e arteriais, independentemente da condição ou polimorfismo genético subjacente.
A incidência de trombose em portadores de trombofilia hereditária é altamente variável e ter ou não evento trombótico depende do genótipo, da coexistência de outra alteração genética, além da associação com fatores adquiridos, como imobilidade e procedimentos cirúrgicos (Tabela 2).
Tabela 2: Prevalência estimada das principais trombofilias congênitas e risco de TEV
|
|
População geral (%) |
Pacientes (%) |
RR TEV* |
|
Fator V Leiden † |
4 a 4,8 |
15 a 25 |
5 a 8 |
|
Protrombina G20210A† |
2,7 |
6 |
2 a 4 |
|
Deficiência de proteína C |
0,2 a 0,4 |
3,7 |
4 a 5 |
|
Deficiência de proteína S |
0,7 a 2,3 |
2,3 |
4 a 5 |
|
Deficiência de antitrombina |
0,02 |
1,9 |
10 |
†Mutação heterozigótica. * Risco relativo de tromboembolismo venoso.
Síndromes familiares de tromboses repetidas e/ou tromboses idiopáticas/recorrentes em indivíduos jovens (menos de 50 anos de idade) sugerem fortemente uma causa genética subjacente.
O fator V é uma glicoproteína de 330 kDa que, quando ativada, potencializa a conversão da protrombina
O risco de trombose venosa aumenta significativamente em casos de mutações homozigóticas e quando há associação com outro fator de risco, como em mulheres usuárias de contraceptivos orais.
O diagnóstico laboratorial inclui teste funcional para proteína C ativada e pesquisa da mutação para o fator V Leiden.
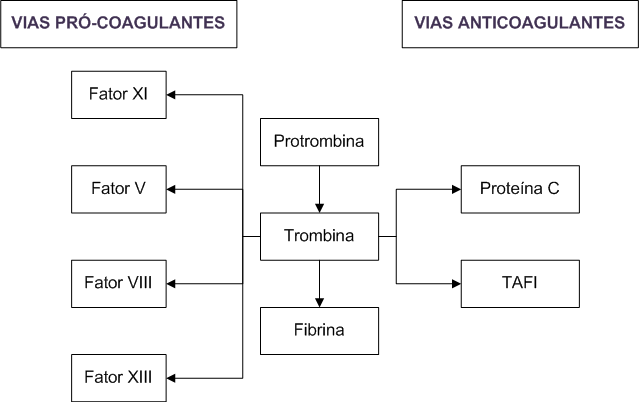
Quando ativada (Protrombina ? Trombina), a trombina apresenta atividades pró-coagulantes, anticoagulantes e antifibrinolíticas (Figura 1). Glicoproteína vitamina K-dependente, sua ativação é acelerada em torno de 300.000 vezes na presença de fator V ativado, cálcio e fosfolípide. A substituição da guanina por adenina na posição 20.210 do gene da protrombina induz a um ganho de função, elevando o risco de trombose em
Assim como acontece com o fator V Leiden, a chance de evento trombótico aumenta consideravelmente quando há associação com outros fatores e em casos de homozigose. A pesquisa da mutação confirma o diagnóstico.
Figura 1: Trombina e a ativação das vias pró-coagulantes e anticoagulantes.

TAFI: inibidor da fibrinólise ativada por trombina.
A proteína C é um zimógeno dependente de vitamina K que se ativa mediante ligação com seu receptor específico e pela ligação entre a trombina e a trombomodulina na superfície endotelial. Na presença do seu cofator (a proteína S, também dependente da vitamina K), a proteína C ativada inativa os fatores V e VIII, impedindo a geração de trombina. Ambas as deficiências são transmitidas por traços autossômicos dominantes de penetrância incompleta e induzem ao surgimento de um estado pró-trombótico.
São reconhecidos dois tipos de deficiência da proteína C: o tipo I, secundário à síntese diminuída, e o tipo II, que se define por níveis normais e funcionalidade comprometida. A mutação homozigótica da proteína C culmina com púrpura fulminante neonatal, podendo levar à morte.
Cerca de 60% da proteína S plasmática circula na forma inativa (ligada à proteína C4b), sendo que apenas 40% apresentam-se com atividade de cofator da proteína C (proteína S livre). Torna-se possível, então, identificar três tipos de deficiência: o tipo I, que corresponde a baixas concentrações de proteína S total e livre; o tipo II, com concentrações normais, porém com baixa atividade de cofator (molécula disfuncional) e o tipo III, que se caracteriza por concentração normal de proteína S total, porém com níveis baixos da fração não ligada ao C4b (proteína S livre).
A confirmação laboratorial da deficiência de proteína C costuma ser feita por avaliação direta da atividade plasmática da proteína C, enquanto que, até o momento, o teste imunológico é o melhor método para a determinação da proteína S. Resultados falso-positivos são encontrados na fase aguda da trombose, gestação, uso de anticoagulantes orais, hepatopatia e nas mais variadas causas de deficiência da vitamina K.
A antitrombina é uma serino-protease de 58 kDa, cujo efeito anticoagulante consiste em inativar a trombina e os fatores X e IX ativados. Esta ação é potencializada em até 1.000 vezes pela heparina e outros glicosaminoglicanos presentes na superfície endotelial. Condição rara, a deficiência é transmitida por traço autossômico dominante e correlaciona-se com risco trombótico muito alto. Portadores da mutação em homozigose, via de regra, morrem antes do nascimento.
Existem dois tipos de deficiência de antitrombina: o tipo I (quantitativo) e o tipo II (qualitativo), e testes cromogênicos permitem o diagnóstico diferencial entre eles.
A pesquisa da deficiência não deve ser realizada durante o uso da heparina, na fase aguda do evento trombótico ou se houver hepatopatia.
O aumento dos níveis plasmáticos da homocisteína pode ser secundário a alterações genéticas ou adquiridas. A principal alteração primária ocorre devido a uma mutação na enzima metilenotetra-hidrofolato-redutase. Causas adquiridas são mais comuns, como nas deficiências de vitamina B12, B6, folato e na insuficiência renal.
A hiper-homocisteinemia constitui fator de risco independente para aterosclerose e tem sido associada a maior risco de trombose venosa. Todavia, evidências sugerem que a correção dos níveis de homocisteína não diminuem o risco trombótico.
Diversos fatores adquiridos predispõem a eventos tromboembólicos e, assim como acontece nas trombofilias hereditárias, a chance de desencadearem trombose venosa ou arterial é extremamente variável.
Condições clínicas (imobilidade, gestação, período pós-operatório), entidades patológicas (malignidade, síndrome nefrótica, hemoglobinúria paroxística noturna), alterações laboratoriais (anticorpos antifosfolípides) e medicamentos (contraceptivos orais) perfazem o espectro de situações secundárias que elevam o risco trombótico. Na maioria das vezes, encontra-se uma sobreposição de vários destes fatores, tornando significativamente maior a possibilidade de episódios tromboembólicos.
As principais síndromes trombofílicas adquiridas encontram-se comentadas a seguir.
A estase venosa associada à imobilização prolongada (> 48 horas) constitui importante fator de risco, sendo que os pacientes hospitalizados e em cuidados domiciliares são responsáveis por até 60% de todos os casos de tromboembolismo venoso.
Indivíduos aparentemente sadios também podem estar suscetíveis (cerca de
O risco trombótico encontra-se elevado no pós-operatório, particularmente em cirurgias ortopédicas, oncológicas, neurológicas, abdominais e pélvicas maiores. Características do paciente, como idade avançada, doença cardiovascular, evento trombótico prévio, assim como o tipo de anestesia utilizada (anestesia geral) tornam o risco ainda maior. Traumatismos maiores (politrauma, cranioencefálico, fratura de fêmur) também se relacionam com maior incidência de tromboembolismo venoso.
Menor fluxo sanguíneo nas extremidades inferiores, fibrinólise diminuída, liberação/exposição ao fator tecidual e depleção de anticoagulantes endógenos parecem explicar a ativação do sistema de coagulação em tais situações.
Por ano,
Também há associação do risco ao tempo de uso, sendo maior no 1º ano, com redução em mais de 50% nos anos subsequentes e desaparecimento cerca de 3 meses após a interrupção da droga.
A terapia de reposição hormonal (TRH) está associada a um risco
O tromboembolismo venoso ocorre cerca de 6 vezes mais durante a gestação e o risco eleva-se em até 11 vezes no puerpério (primeiras 6 semanas após o parto).
A compressão de grandes veias pelo útero gravídico diminui o retorno venoso; além disso, surgem importantes alterações no sistema hemostático: elevam-se os níveis dos fatores I, II, VII, VIII, IX, X com progressiva redução da proteína S. Resistência à proteína C ativada pode ser observada no 2º e 3º trimestres, assim como maior produção de inibidores da fibrinólise pela placenta.
O parto é acompanhado por significativas alterações vasculares e lesão endotelial, induzindo a um estado pró-trombótico após a gestação.
A presença de neoplasia maligna eleva em 5 vezes a incidência anual do tromboembolismo venoso, associando-se com maior recorrência e pior prognóstico. O risco é maior em pacientes com tumores cerebrais e do pâncreas. A causa costuma ser multifatorial e inclui a produção de substâncias pró-coagulantes pela célula tumoral. Em algumas situações, o evento tromboembólico antecede ou propicia o diagnóstico do câncer.
O tratamento quimioterápico potencializa o estado pró-trombótico em pacientes oncológicos, principalmente se utilizada terapia hormonal e drogas inibidoras da angiogênese. A lesão endotelial secundária à quimioterapia, em associação com efeitos específicos sobre o sistema hemostático de determinados medicamentos, determinam o maior risco trombótico.
A trombose, principalmente venosa, constitui a principal causa de morbidade em portadores de síndrome nefrótica, que é clinicamente caracterizada por edema, proteinúria, hipoalbuminemia e dislipidemia. As veias renais são acometidas em torno de 35% dos casos e a incidência de trombose em outros locais chega a 20%. Diversas alterações da hemostasia têm sido descritas, como diminuição nos níveis plasmáticos de antitrombina, secundária à perda renal e elevação dos fatores X, VIII, VII e V. Evidências sugerem maior reatividade plaquetária associada.
A hemoglobinúria paroxística noturna (HPN) é uma rara alteração clonal de células progenitoras da medula óssea, caracterizada por hemólise intravascular, episódios de hemoglobinúria macroscópica, leucopenia e trombocitopenia. Apesar da plaquetopenia, episódios de trombose venosa, especialmente em vasos intra-abdominais e cerebrais, são mais frequentes do que intercorrências hemorrágicas. HPN deve ser considerada em pacientes com história familiar negativa de trombose e/ou evidência de hemólise ou pancitopenia.
Outras anemias hemolíticas, como a anemia falciforme e a talassemia, podem apresentar eventos tromboembólicos como complicações.
Alterações inerentes à hemácia falcêmica são capazes de criar um estado pró-coagulante mediante ativação endotelial, plaquetária e de todo o sistema hemostático.
A síndrome do anticorpo antifosfolípide pode ser primária ou secundária a uma doença reumatológica (lúpus eritematoso sistêmico, entre outras). Tromboses venosas e arteriais caracterizam esta síndrome.
O diagnóstico requer a presença do anticorpo contra proteínas plasmáticas ligadas a fosfolípides – antilúpico e/ou altos títulos da anticardiolipina IgG/IgM e/ou anti-beta-2-glicoproteína IgG ou IgM com título superior ao percentil 99 do teste realizado (duas dosagens em intervalo de 12 semanas e em menos de 5 anos da manifestação clínica) – em associação a um episódio de trombose venosa ou arterial ou mau passado gestacional (morte fetal com = 10 semanas inexplicada; 3 ou mais abortos com < 10 semanas sem causa obstétrica; um ou mais partos antes de 34 semanas por eclâmpsia, pré-eclâmpsia ou insuficiência placentária).
História clínica e exame físico minuciosos são fundamentais na avaliação inicial do paciente com trombose prévia ou atual e, muitas vezes, norteiam a investigação laboratorial.
A idade em que ocorreu o evento, fatores ou condições predisponentes (pós-operatório, viagem prolongada, gestação, infecção), comorbidades (obesidade, câncer, doenças reumatológicas) e medicações (contraceptivos orais) são dados de extrema relevância. Em mulheres, é necessário que o passado obstétrico sempre seja considerado.
A localização da trombose (proximal ou distal, sítio típico ou não), assim como episódios anteriores são detalhes de igual importância.
Tromboembolismo venoso em parentes de 1º grau sugere fortemente a presença de causa hereditária.
Um meticuloso exame físico deve ser realizado, com foco nas extremidades, sistema cardiovascular, abdome e pele. Toque retal e exame ginecológico devem ser considerados, uma vez que não é raro o evento trombótico ser a primeira manifestação de uma neoplasia.
Uma causa adquirida sempre deve ser considerada em indivíduos com trombose. Com base na avaliação clínica, exames laboratoriais e de imagem podem ser solicitados.
Em indivíduos jovens (< 50 anos de idade), com trombose em local atípico, recorrente e/ou com história familiar positiva, indica-se pesquisa de causas hereditárias. Todavia, devido a alterações nos níveis plasmáticos de antitrombina, proteína C, proteína S e mesmo da homocisteína durante a fase aguda da trombose, a avaliação laboratorial para trombofilia hereditária só deve ser realizada 3 meses após o evento vaso-oclusivo, exceto para as mutações no gene do fator V e da protrombina.
Gestantes e puérperas também não devem ser investigadas, devido às alterações hemostáticas que ocorrem durante o ciclo gravídico-puerperal.
Vale ainda ressaltar o efeito da heparina sobre a quantificação da antitrombina e dos anticoagulantes orais sobre as proteínas C e S, possibilitando a obtenção de resultados falso-positivos.
O tratamento do tromboembolismo venoso consiste em evitar a propagação/embolização do trombo por tempo suficiente até que ocorra a lise ou a organização do coágulo.
A presença de fatores de risco biológicos ou adquiridos não altera o manejo inicial do tromboembolismo venoso agudo. Entretanto, algumas considerações devem ser feitas quando se trata de pacientes com deficiência de antitrombina ou de proteína C.
Pacientes com deficiência de antitrombina podem apresentar resistência à heparina, necessitando de doses elevadas para obtenção do efeito anticoagulante adequado.
Quando não se consegue anticoagular satisfatoriamente os pacientes que apresentam recorrência durante o uso da heparina ou trombose em sítio atípico, recomenda-se a associação do concentrado de antitrombina. A infusão de 50 UI/kg é capaz de elevar o nível da antitrombina para aproximadamente 120% em um paciente com nível basal em torno de 50%. Os níveis plasmáticos devem ser mantidos por volta de 80%.
Embora o concentrado de antitrombina seja seguro e eficaz em pacientes com deficiência de antitrombina e trombose venosa aguda, deve ser restrito às indicações mencionadas, uma vez que seu uso como terapia adjuvante ou alternativa à heparina não foi avaliado em estudos controlados.
Em raras situações, a deficiência hereditária da proteína C associa-se à necrose de pele induzida pela varfarina devido a um estado de hipercoagulabilidade transitório nas primeiras 24 horas. Apesar do risco, a dosagem rotineira dos níveis da proteína C em pacientes com trombose, antes da terapia anticoagulante oral, não está indicada. Três observações sustentam esta contraindicação:
a necrose induzida por varfarina é um evento raro;
a frequência da deficiência de proteína C é baixa (1/200 na população em geral);
resultados falso-positivos são observados durante a fase aguda da trombose.
Recomenda-se iniciar o anticoagulante oral em doses baixas (varfarina 2 mg/dia nos primeiros 3 dias, seguido por aumento gradual da dose), ainda durante a heparinização, em indivíduos com deficiência conhecida.
Os pacientes com antecedente de necrose induzida por varfarina podem ser retratados com antagonistas da vitamina K, sendo que, para casos de necroses recorrentes, o uso é possível com reposição de proteína C exógena associada (plasma fresco congelado ou concentrado de proteína C).
A necrose induzida pela varfarina pode ocorrer em portadores de deficiência hereditária da proteína S, sendo ainda mais raro.
Após a abordagem inicial, tem-se por objetivo completar o tratamento do evento agudo, além de prevenir novas tromboses.
Preconiza-se anticoagulação oral (INR entre 2 e 3) por
Em casos de câncer, recomenda-se que o tratamento seja realizado preferencialmente com heparina de baixo peso molecular por um período mínimo de 6 meses.
Manter anticoagulação como medida profilática secundária depende da avaliação de risco do paciente tanto no que se refere à retrombose quanto à possibilidade de sangramento. Em geral, não se recomenda após o primeiro episódio de trombose, especialmente se associado a fator de risco transitório.
Os pacientes de alto risco para recorrência devem ser considerados para anticoagulação por tempo indeterminado (Tabela 3).
Tabela 3: Situações com maior risco de recorrência do tromboembolismo venoso
|
2 ou mais episódios de TVP idiopática |
|
1 evento espontâneo com risco de morte |
|
1 trombose idiopática em sítio atípico (cerebral, mesentérica) |
|
1 TVP idiopática + SAAF#, deficiência AT* ou câncer |
|
1 TVP idiopática + mais de 1 alteração genética ou em homozigose |
# SAAF: síndrome do anticorpo antifosfolípide. *AT: antitrombina.
Alterações fisiológicas do sistema hemostático durante a gestação e o puerpério elevam significativamente o potencial trombogênico das portadoras de trombofilias, sejam hereditárias ou adquiridas.
Para as pacientes de alto risco (deficiência de antitrombina, homozigose para fator V Leiden ou mutação da protrombina G20210A, presença de dupla mutação heterozigótica, SAAF) recomendam-se doses ajustadas de heparina não fracionada (HNF) ou heparina de baixo peso molecular (HBPM) durante toda a gestação e puerpério.
Mulheres de risco intermediário devem receber doses profiláticas de HNF ou HBPM a partir do 2º ou 3º trimestre da gestação, mantendo-se por todo o período puerperal.
Trombofilia corresponde a toda alteração hereditária e adquirida do sistema hemostático que aumenta o risco de trombose.
As síndromes trombofílicas hereditárias são menos frequentes e devem ser consideradas em indivíduos jovens (< 50 anos de idade) com evento trombótico idiopático/recorrente ou história familiar positiva.
Diversas condições clínicas, entidades patológicas e medicações induzem a um estado pró-trombótico, sendo denominadas de trombofilias adquiridas ou secundárias.
História clínica e exame físico minuciosos são ferramentas fundamentais e norteiam a investigação complementar.
Quando indicada, a pesquisa de trombofilia primária deve ser realizada 3 meses após o evento agudo e evitada durante o ciclo gravídico-puerperal (salvo a pesquisa das mutações).
O uso de anticoagulantes orais interfere nas dosagens de proteína C e S, assim como a heparina altera os níveis de antitrombina, obtendo-se resultados falso-positivos.
Em geral, o tratamento instituído para o tromboembolismo venoso independe da causa associada.
O pronto reconhecimento e, se possível, a remoção do fator desencadeante é de fundamental importância para a redução do estado de hipercoagulabilidade.
Identificar os pacientes de alto risco para recorrência associa-se a significativa diminuição de morbimortalidade.
1. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh. 1965;13:516-30.
2. Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353:1167-73.
3. Zoller B, et al. Thrombophilia as a multigenic disease. Haematologica. 1999;84:59-70.
4. Matei D, et al. Acquired thrombophilic syndromes. Blood Reviews. 2001;15:31-48.
5. Bertina RM. Genetic approach to thrombophilia. Thromb Haemost. 2001;86(1):92-103.
6. Bauer KA. The thrombophilias: well-defined risk factors with uncert therapeutic implications. Ann Intern Med. 2001;135:367-373.
7. Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001;344(16):1222-1231.
8. Chandler WL, Rodgers GM, et al. Elevated hemostatic factor levels as potential risk factors for thrombosis. Arch Pathol Lab Med. 2002;126(11):1405-1414.
9. Heit JA, O’Fallon WM, Petterson TM, et al. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: a population-based study. Arch Intern Med. 2002;162:1245-1248.
10. Bauer KA. Management of thrombophilia. J Thromb Haemost. 2003;1:1429-34.
11. Mannucci PM. Laboratory detection of inherited thrombophilia: a historical perspective. Semin Thromb Hemost. 2005;31(1):5-10.
12. Cushman M. Inherited risk factors for venous thrombosis. american society of hematology educ program. Hematology. 2005:452-457.
13. Bauuer KA. Hipercoagulable states. In: Hoffman R, Benz Jr. EJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, et al., eds. Hematology: basic principles and practice. 4. ed. Philadelphia: Elsevier; 2005. p.2197-2224.
14. Crowther MA, Ginsberg JS. Venous thromboembolism. In: Hoffman R, Benz Jr. EJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, et al., eds. Hematology: basic principles and practice. 4. ed. Philadelphia: Elsevier; 2005. p.2225-2240.
15. Bick RL. Hereditary and acquired thrombophilic disorders. Clin Appl Throm Hemost. 2006;12(2):125-135.
16. Bockenstedt PL. Management of hereditary hypercoagulable disorders. American Society of Hematology Educ Program. Hematology. 2006:444-449.
17. Emmerich J, Aiach M. Thrombophilia genetics. In: Colman RW, Clowes AW, Goldhaber SZ, Marder VJ, George JN, eds. Hemostasis and thrombosis. basic principles and clinical practice. 5. ed. Philadelphia: Lippincott; 2006.
18. Heit J. Thrombophilia: common questions on laboratory assessment and management. American Society of Hematology Educ Program. Hematology. 2007:127-135.
19. Battaglioli T, Martinelli I. Hormone therapy and thromboembolic disease. Current Opinion in Hematology. 2007;14:488-493.
20. Den Heijer M, et al. Homocysteine lowering by B vitamins and the secondary prevention of deep vein thrombosis and pulmonary embolism: a randomized, placebo-controlled, double-blind trial. Blood. 2007;109(1):139-44.
21. Bates SM, Greer IA, Pabinger I, Sofaer S, Hirsh J. Venous thromboembolism, thrombophilia, antithrombotic therapy, and pregnancy. American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). CHEST. 2008;133:844S-886S.
22. Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease. American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). CHEST. 2008;133:454S-545S.
23. Dahlback B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19-27.
24. Varga EA. Genetic counseling for inherited thrombophilias. J Thromb Thrombolysis. 2008;25:6-9.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.