(Carregando Índice)... (Carregando Índice)... |
Autores:
Amanda Veiga Cheuiche
Acadêmica da Faculdade de Medicina da UFRGS e aluna de iniciação científica na área de Endocrinologia da mesma instituição.
Eduardo Guimarães Camargo
Médico endocrinologista. Doutor em Ciências Médicas: Endocrinologia pela UFRGS.
Ariana Aguiar Soares
Farmacêutica. Mestre em Ciências Médicas: Endocrinologia pela UFRGS.
Sandra Pinho Silveiro
Médica endocrinologista. Professora associada do Departamento de Medicina Interna da UFRGS. Preceptora do Programa de Residência
Médica em Endocrinologia do HCPA. Professora
do Programa de Pós-graduação em Ciências Médicas: Endocrinologia da UFRGS. Doutora em Medicina: Ciências Médicas pela UFRGS.
Maria Júlia Rostirolla
Acadêmica da Faculdade de Medicina da PUCRS.
Última revisão: 12/05/2014
Comentários de assinantes: 0
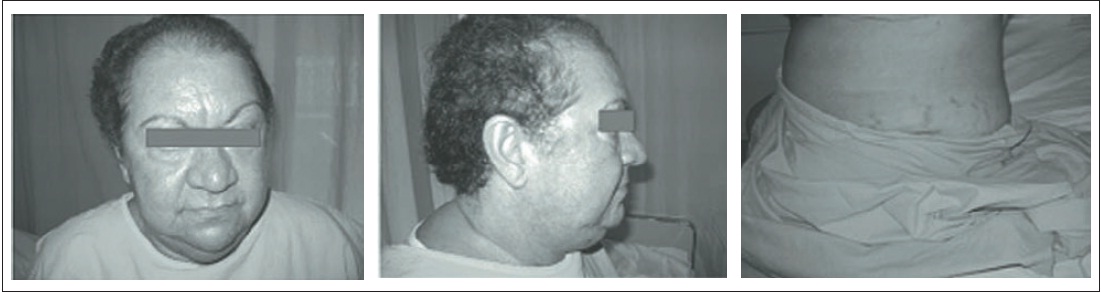
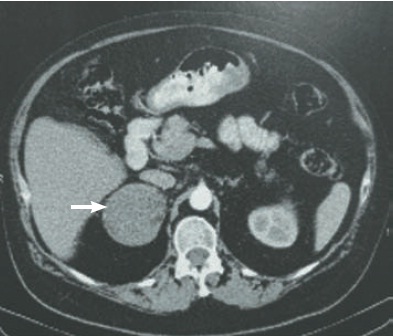
Uma paciente do sexo feminino, 53 anos, branca, apresentou aumento de peso e surgimento de pelos faciais há três meses. Ao realizar exame físico, constatou-se fácies de lua cheia, alopecia de padrão androgênico, pletora facial, hirsutismo, giba, obesidade centrípeta e hipertensão arterial sistêmica (Fig. 30.1). Com a suspeita de síndrome de Cushing, foram realizados exames e obtidos os seguintes resultados: cortisolúria de 191,73 g/24h (valor de referência [VR]: 36 a 137), cortisol pós-supressão com dexametasona 1 mg de 23,2 g/dL (VR 1,8), testosterona de 4,4 ng/mL (VR: 0,08 a 0,35); androstenediona de 20 ng/mL (VR: 0,5 a 3,7); sulfato de desidroepiandrosterona (SHDEA) de 791,8 g/ dL (VR: 35,4 a 256) e adrenocorticotrópico (ACTH) menor do que 10 pg/mL (VR: 10 a 52). A tomografia computadorizada de abdome evidenciou uma lesão expansiva na glândula suprarrenal direita de 6,0 cm por 6,4 cm, sem calcificações (Fig. 30.2).

Figura 30.1
Paciente com quadro clínico suspeito de síndrome de Cushing.
A síndrome de Cushing (SC) é uma doença decorrente da exposição prolongada a concentrações excessivas de glicocorticoides (GC) endógenos ou exógenos. O cortisol é um hormônio GC liberado pelo córtex da glândula suprarrenal, que tem importante papel sobre o metabolismo de carboidratos, proteínas e lipídeos. Além de modular o equilíbrio hidreletrolítico e as respostas inflamatória e imunológica, os GCs também regulam as funções cardiovascular, hematopoética e osteomuscular e estão envolvidos na resposta do organismo ao estresse. Devido às características anti-inflamatória e imunossupressora, os GCs são medicamentos comumente empregados na prática clínica.
Figura 30.2
Tomografia computadorizada de abdome: lesão expansiva de 6,0 por 6,4 cm na glândula suprarrenal direita (seta).
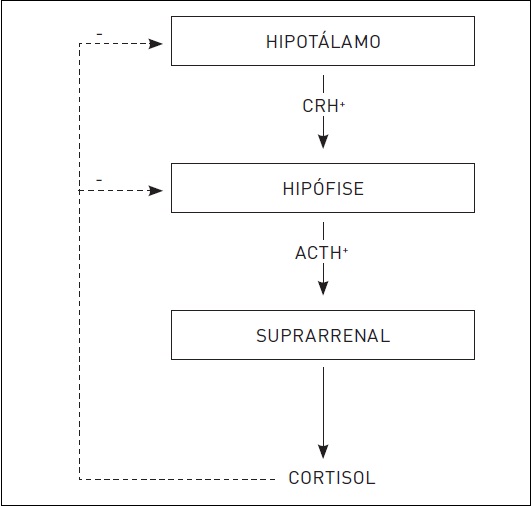
O hormônio adrenocorticotrófico (ACTH) é o principal estimulador da produção de cortisol (Fig. 30.3). A liberação do ACTH pela hipófise anterior é controlada principalmente pelo hormônio liberador de corticotrofina (CRH), produzido pelo hipotálamo. Em condições normais, o hipotálamo produz CRH, estimulando a liberação de ACTH pela hipófise. Esse hormônio, por sua vez, age na zona fasciculada do córtex suprarrenal, induzindo a secreção do cortisol. A secreção do cortisol e do ACTH apresentam um ritmo circadiano, sendo mais elevada pela manhã (entre 5 e 9 horas) e mais baixa a partir do final da tarde (entre 18 e 24 horas). Doenças que atingem o hipotálamo, a hipófise ou as suprarrenais e ocasionam aumento persistente de CRH, ACTH ou cortisol resultam em SC. De forma similar, a produção ectópica de ACTH ou de CRH causa hipercortisolismo, bem como a administração prolongada e excessiva de GCs exógenos. A SC cíclica consiste em um padrão de hipercortisolismo em que a produção de cortisol flutua ritmicamente.
Figura 30.3
Eixo hipotálamo-hipófise-suprarrenal.
(Sinal negativo (-), INIBIÇÃO; sinal positivo (+), ESTIMULAÇÃO.
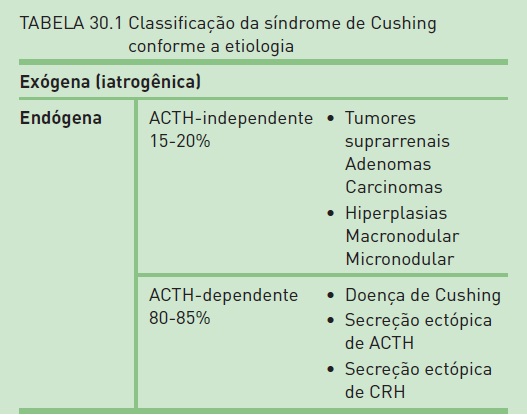
A causa mais comum de SC é o uso prolongado de doses excessivas de GC. As diferentes doenças que causam a SC podem ser classificadas em ACTH-dependentes e ACTH- independentes (Tab. 30.1).
•Doença de Cushing (DC): hipersecreção de ACTH pela hipófise decorrente da presença de um adenoma, em geral microadenoma (< 1 cm). Essa doença é a causa mais frequente de SC não iatrogênica, correspondendo a 60 a 80% dos casos. A incidência da DC é de 5 a 25/1.000.000 por ano. As mulheres apresentam uma tendência três a oito vezes maior de desenvolver DC em comparação aos homens. A DC afeta indivíduos geralmente entre 25 e 45 anos.
•Secreção ectópica de ACTH por tumores não pituitários: corresponde a 10 a 15% dos casos. Aproximadamente 1% dos pacientes com câncer de pequenas células do pulmão apresentam síndrome de secreção ectópica de ACTH, sendo essa neoplasia a causa de metade de todos os casos da síndrome ectópica. Outras causas são tumores carcinoides do pulmão, timo e pâncreas.
•Hiperplasia suprarrenal micronodular bilateral: corresponde a menos de 1% dos casos.
•Hiperplasia suprarrenal macronodular bilateral: corresponde a menos de 1% dos casos.
•Adenomas e carcinomas adrenocorticais: causam 18 a 20% de todos os casos de SC. A incidência de carcinoma suprarrenal é desconhecida, mas é estimada em 0,2 a 2/1.000.000 por ano. Em mulheres, há cerca de quatro vezes mais chance de manifestação da síndrome associada a tumor suprarrenal quando comparadas aos homens. Ao contrário dos adultos, em crianças, o carcinoma suprarrenal corresponde a 50% dos casos de SC da infância, e o adenoma, a um sexto. Tanto as neoplasias benignas quanto as malignas apresentam um pequeno pico de ocorrência aos 10 anos e picos mais significativos aos 40 anos, para o carcinoma, e aos 50 anos, para o adenoma.
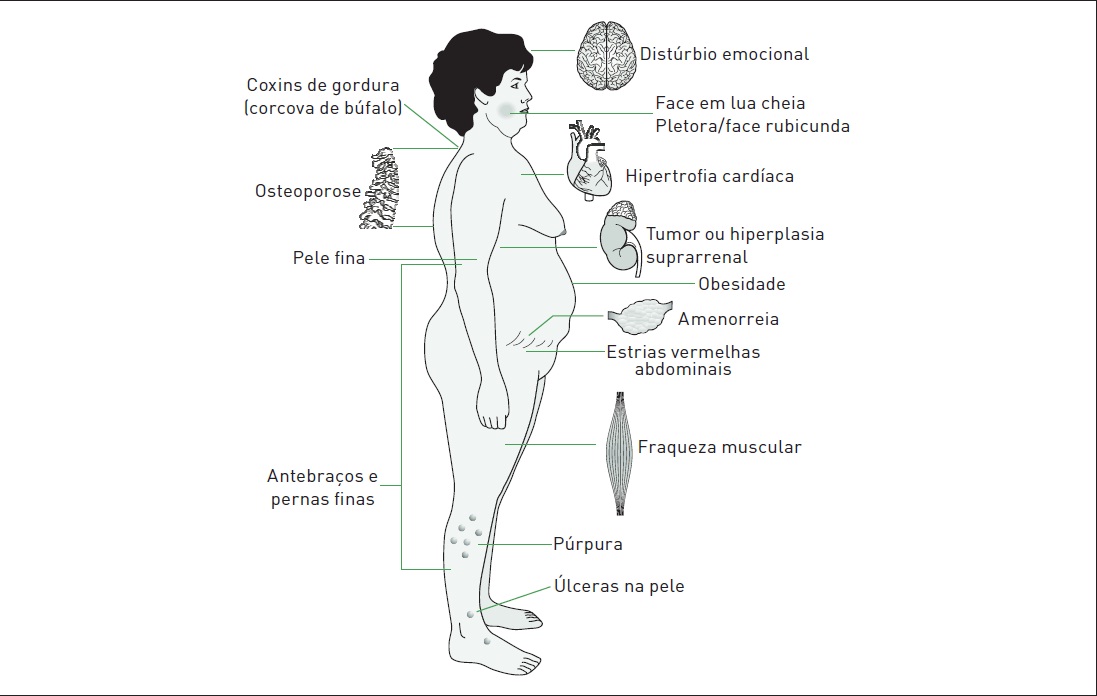
A manifestação mais comum é a obesidade centrípeta progressiva, envolvendo face, pescoço, tronco e abdome e geralmente poupando as extremidades. Pode haver as denominadas face em lua cheia e giba de búfalo (Fig. 30.4). A ocorrência de disfunção gonadal é frequente, causando irregularidade menstrual em mulheres e perda de libido em homens. Anormalidades psiquiátricas afetam metade dos pacientes com SC. Os sintomas mais comuns são labilidade emocional, depressão, irritabilidade, ansiedade, ataques de pânico e paranoia leve. A insônia é um sintoma precoce.
A osteoporose manifesta-se frequentemente em pacientes com SC, podendo ocorrer fratura de costelas e ossos longos, bem como fratura por compressão vertebral. O hipercortisolismo resulta em atrofia cutânea e perda da gordura subcutânea, manifestada por afinamento da pele, a qual se torna mais frágil. As estrias típicas de um paciente com SC são violáceas e com diâmetro maior do que 1 cm, frequentemente encontradas no abdome. Sintomas como fraqueza e perda de massa muscular proximal são comuns. O excesso de cortisol predispõe à hipertensão e à tolerância diminuída à glicose. Em casos de adenoma hipofisário produtor de ACTH, as mulheres podem apresentar sinais de excesso de androgênio devido ao estímulo da produção suprarrenal de androgênio pelo ACTH elevado. Os principais sintomas são hirsutismo leve, alopecia, pele oleosa e acne. Em casos de câncer de suprarrenal, pode haver envolvimento direto da camada produtora de andrógeno, com quadro de virilismo na mulher, ocasionando hirsutismo grave e clitoromegalia.
Figura 30.4
Quadro clínico da síndrome de Cushing.
A suspeita de SC ocorre quando há sinais e sintomas sugestivos de hipercortisolismo. Nenhuma das manifestações clínicas, no entanto, é fator patognomônico, e muitas são inespecíficas.Devem-se investigar os seguintes casos:
•Pacientes com osteoporose e hipertensão de ocorrência precoce.
•Pacientes com manifestações múltiplas e progressivas sugestivas.
•Crianças que apresentam percentil de altura decrescente e que estão ganhando peso.
•Pacientes com incidentaloma suprarrenal.
Não se deve esquecer de questionar o paciente a respeito de medicamentos utilizados no passado e no presente (incluindo cremes tópicos e medicamentos inalatórios para asma ou rinite), a fim de excluir causa iatrogênica por administração exógena de GC.
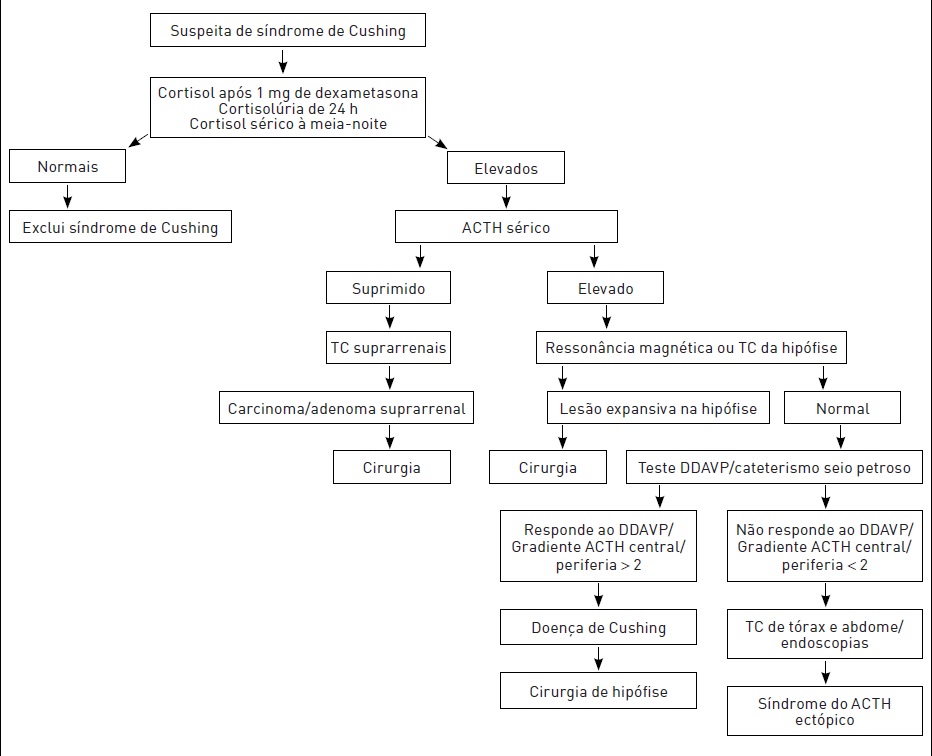
Tem-se o algoritmo diagnóstico para a SC na Figura 30.5. Na avaliação laboratorial, devem-se constatar os seguintes resultados dos exames:
•Cortisolúria de 24 horas basal elevada (coletar pelo menos duas amostras, com creatinina urinária para checar adequação da coleta).
•Não supressão do cortisol sérico após 1 mg de dexametasona ingerido às 23 horas (overnight).
•Elevação do cortisol sérico medido à meia-noite.
A dosagem do ACTH separa as causas dependentes do ACTH (nível elevado ou normal-alto) das não dependentes (ACTH baixo ou indetectável). Os testes com altas doses de dexametasona e do DDAVP são utilizados para diferenciar doença de Cushing da síndrome do ACTH ectópico.
A SC iatrogênica é indistinguível clinicamente da hiperfunção adrenocortical endógena. A distinção pode ser feita por meio da medida de cortisol sérico ou urinário, em que, no caso da síndrome iatrogênica, os níveis são baixos (secundário à supressão do eixo hipófise-suprarrenal).
Teste com 1 mg de dexametasona overnight. Administra-se 1 mg, via oral, de dexametasona (crianças: 0,015 mg/kg), às 23 horas, e dosa-se o cortisol sérico às 8 horas da manhã seguinte. A supressão dos níveis de cortisol para valores inferiores a 1,8 g/dL geralmente exclui síndrome de Cushing, e a não supressão é um indicativo desta, que deve ser confirmada com outros exames.
Dosagem de cortisol à meia-noite. Um cateter é colocado em uma veia periférica pelo menos duas horas antes da coleta. O paciente deve ficar em jejum e em repouso no leito a partir das 22 horas. Uma taxa de cortisol sérico superior a 7,5 g/dL apresenta alta especificidade para SC.
Teste com baixas doses de dexametasona. Administra-se 0,5 mg, via oral, de dexametasona, a cada 6 horas, por 48 horas. Coleta-se cortisolúria nas últimas 24 horas, e, seis horas após a última dose de dexametasona, dosa-se o cortisol sérico. O teste é considerado normal quando a cortisolúria for inferior a 10 g em 24 horas e o cortisol sérico, inferior a 1,8 g/dL. Na síndrome de Cushing, não ocorre supressão do cortisol sérico e/ou urinário. Esse teste, portanto, é utilizado para confirmar hipercortisolismo, mas em geral não é necessário. Porém, pacientes com doença de Cushing podem apresentar supressão do cortisol urinário com baixas doses de dexametasona. Além disso, 15 a 25% dos pacientes com pseudocushing (depressão, alcoolismo, obesidade) podem não apresentar supressão.
Teste com altas doses de dexametasona. Administram-se 2 mg, via oral, de dexametasona, a cada 6 horas, por 48 horas. Coleta-se cortisolúria nas últimas 24 horas, e, seis horas após a última dose de dexametasona,dosa-se o cortisol sérico. Redução de 90% da cortisolúria de 24 horas comparada à basal sugere a presença de doença de Cushing, em contraste com a não supressão nos casos de síndrome do ACTH ectópico. Esse teste em geral não é necessário e pode agravar o quadro de hipercortisolismo do paciente.
Teste do DDAVP. Coleta-se ACTH e cortisol nos tempos -15, 0, 15, 30, 45, 60, 90 e 120 minutos após a administração intravenosa de 10 g de DDAVP (va sopressina). O aumento do cortisol em 20 a 40% e do ACTH em 35 a 50% é compatível com doença de Cushing (adenoma de hipófise). Nos pacientes com síndrome de Cushing independente do ACTH, e na maioria dos com síndrome do ACTH ectópico, essa resposta não ocorre. No entanto, uma proporção expressiva de casos de ACTH ectópico pode mimetizar a resposta do adenoma de hipófise, confundindo o diagnóstico. Esse teste deve ser realizado antes daqueles de supressão com dexametasona.
Figura 30.5
Algoritmo diagnóstico para síndrome de Cushing.
Fonte: Adaptada de Rollin e colaboradores.¹
Cateterismo de seio petroso. É indicado em pacientes com síndrome de Cushing dependente de ACTH sem imagem diagnóstica na hipófise, para diagnóstico diferencial com a síndrome do ACTH ectópico. O procedimento deve ser realizado em centro especializado. Uma razão da medida de ACTH central-periférica maior do que 2 em condições basais e maior do que 3 pós-CRH (ou DDAVP) diagnostica um adenoma hipofisário secretor de ACTH. Razões inferiores a esses valores indicam produção ectópica de ACTH.
Conforme a sugestão de SC dependente ou independente de ACTH, deve-se realizar exame de imagem correspondente, respectivamente ressonância magnética de hipófise ou tomografia computadorizada (TC) de abdome superior com ênfase em suprarrenal. Caso haja suspeita de síndrome de produção ectópica de ACTH, deve-se realizar TC de tórax e abdome e/ou fibrobroncoscopia e/ ou esofagogastroduodenoscopia.
O tratamento de escolha para a doença de Cushing é a ressecção transesfenoidal seletiva. A taxa de remissão é de cerca de 80% para microadenomas e inferior a 50% para macroadenomas.
A maioria dos pacientes apresenta um período de deficiência sintomática de ACTH por até 12 meses após a cirurgia, necessitando de reposição com pequenas doses de GC. Em casos de síndromes de ACTH ectópico, a excisão cirúrgica do tumor causador, quando possível, pode promover a cura.
A irradiação hipofisária é geralmente um tratamento de segunda linha para a doença de Cushing, sendo mais eficaz em crianças e adultos jovens. Ele pode ser realizado também quando não há cura cirúrgica.
Inibidores esteroidogênicos (Tab. 30.2) podem ser utilizados em combinação com a irradiação para bloquear os efeitos suprarrenais de níveis persistentemente altos de ACTH.
Os adenomas suprarrenais devem ser tratados com adrenalectomia unilateral, com 100% de cura. Os carcinomas de suprarrenal são tumores agressivos, cujo prognóstico depende do grau de invasão local e da presença de metástases (ósseas, pulmonares e hepáticas).
O mitotano, medicamento adrenolítico, pode ser administrado como terapia adjuvante em pacientes com carcinoma de suprarrenal (Tab. 30.2).
A realização de suprarrenalectomia bilateral oferece uma cura definitiva para a síndrome de Cushing decorrente de hiperplasia.
Independentemente da causa da SC, deve-se fazer tratamento com tiabendazol (25 mg/kg/dia, por três dias consecutivos), oferecendo cobertura para a estrongiloidíase decorrente do estado de imunossupressão do paciente.
O quadro laboratorial confirmando a presença de síndrome de Cushing e a ocorrência de produção excessiva de mais de um hormônio suprarrenal (cortisol e SHDEA) associados à lesão suprarrenal com mais de 4 cm levantaram a hipótese de carcinoma suprarrenal.
Foi realizada suprarrenalectomia direita, e o exame anatomopatológico confirmou o diagnóstico de carcinoma adrenocortical com invasão vascular. Esse tumor causa cerca de 5% dos casos de síndrome de Cushing e tem como característica o rápido aparecimento das manifestações clínicas de hipercortisolismo. As lesões são em geral grandes, maiores do que 5 cm, podendo secretar também andrôgenio e mineralocorticoide.
Em mulheres, ele manifesta-se ocasionando virilização, com hirsutismo, clitoromegalia e acne grave. O prognóstico é reservado.
1. Rollin GA, Copstein E, Czepielewski MA. síndrome de Cushing. In: Gross JL, Silveiro SP, editores. Rotinas diagnósticas em endocrinologia. Porto Alegre: Artmed; 2004.
Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of ACTH-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93(7):2454-62.
Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab. 2009;94(9):3121-31.
Carroll TB, Findling JW. The diagnosis of Cushing’s syndrome. Rev Endocr Metab Disord. 2010;11(2):147-53.
233234
Elamin MB, Murad MH, Mullan R, Erickson D, Harris K, Nadeem S, et al. Accuracy of diagnostic tests for cushing’s syndrome: a systematic review and metaanalyses. J Clin Endocrinol Metab. 2008;93(5):1553-62.
Guignat L, Bertherat J. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline: commentary from a European perspective. Eur J Endocrinol. 2010;163(1):9-13.
Mancini T, Porcelli T, Giustina A. Treatment of Cushing disease: overview and recent findings. Ther Clin Risk Manag. 2010;6:505-16.
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367(9522):1605-17.
Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an endo- crine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93(5):1526-40.
Pivonello R, De Martino MC, De Leo M, Lombardi G, Colao A. Cushing’s syndrome. Endocrinol Metab Clin North Am. 2008;37(1):135-49.
Reimondo G, Pia A, Bovio S, Allasino B, Daffara F, Paccotti P, et al. Laboratory differentiation of Cushing’s syndrome. Clin Chim Acta.2008;388(1-2):5-14.
Rollin GA, Ferreira NP, Junges M, Gross JL, Czepielewski MA. Dynamics of serum cortisol levels after transsphenoidal surgery in a cohort of patients with Cushing’s disease. J Clin Endocrinol Metab. 2004;89(3):1131-9.
Sakihara S, Kageyama K, Oki Y, Doi M, Iwasaki Y, Takayasu S, et al. Evaluation of plasma, salivary, and urinary cortisol levels for diagnosis of Cushing’s syndrome. Endocr J. 2010;57(4):331-7.
Sonino N, Boscaro M, Fallo F. Pharmacological management of Cushing syndrome. Treat Endocrinol. 2005;4(2):87-94.
Tristos NA, Biller BMK, Swearingen B. Management of Cushing disease. Nat Rev Endocrinol. 2011;7(5):279-89.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.