(Carregando Índice)... (Carregando Índice)... |
Autor:
Rodrigo Antonio Brandão Neto
Médico Assistente da Disciplina de Emergências Clínicas do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 09/10/2015
Comentários de assinantes: 0
As Porfirias são doenças metabólicas resultantes de deficiências usualmente genéticas na atividade de enzimas específicas na via do heme. Neste caso, os intermediários desta via, que são o ácido alfa aminolevulanico e o porfirobilinogênio, são produzidos em excesso e se acumulam nos tecidos.
Apesar de o heme ser produzido em todos os tecidos, os mais importantes são o fígado e a medula óssea. Cerca de 80% da produção de heme diária é realizada na medula óssea, e quase todo o heme produzido na medula óssea é utilizada como substrato para produção de hemoglobina, com o fígado sendo responsável pela maior parte do restante e, portanto responsável pela produção dos outros produtos derivados do heme, como mioglobina, catalase, citocromos respiratórios, óxido nítrico sintase entre outros produtos. A primeira enzima envolvida nesta via é a delta-aminolulinato sintase, outras enzimas seguem e catalizam a formação do porfirobilinogênio, com exceção da protoporfirina todos os intermediários da porfirina estão em suas formas reduzidas.
As Porfirias podem ser dividas em eritropoiéticas ou hepáticas. O termo Porfiria foi utilizado pela primeira vez por Stekss em 1889, quando uma mulher com urina avermelhada evoluiu para óbito depois que dois irmãos foram afetados.
O heme é essencial para função das células aeróbicas, além da hemoglobina, funcionando também como substrato para produção de outras proteínas como mioglobina, citocromos mitocondriais, catalase, peroxidade, entre outros.
Estas proteínas são envolvidas no transporte de oxigênio e elétrons no metabolismo oxidativo.
O heme ou protoporfirina 9 é constituídoa de citocromo de ferro, coordenado com quatro anéis pirrólicos de protoporfirina. Na sua biossíntese, o primeiro passo é dependente da Alfa-aminolevulanico-sintase, que catalisa a condensação da ALA ou acido aminolevulínico que servirá como substrato para ação de enzimas no citosol hepático.
Nas mitocôndrias a primeira via enzimátic é a A DAla-sintase, que catalisa a condensação da glicina e da succinilcolina para formar o ácido aminolevulínico ou ALA. Este processo ocorre parcialmente no citosol, como já citado e parcialmente na mitocôndria. A segunda enzima da via do heme é a ALA-dehidrase, que é inibida quando ocorre intoxicação por chumbo. Outros passos na cadeia do heme incluem na ordem:
-Formação porfirobilinogênio;
-Formação uroporfirobilinogênio;
-Formação do Uroporfirogênio III;
-Formação coproporfirogênio;
-Formação protoporfirogênio IX (coproporfirogênio oxidase);
-Formação de protoporfirina IX;
-Formação do Heme.
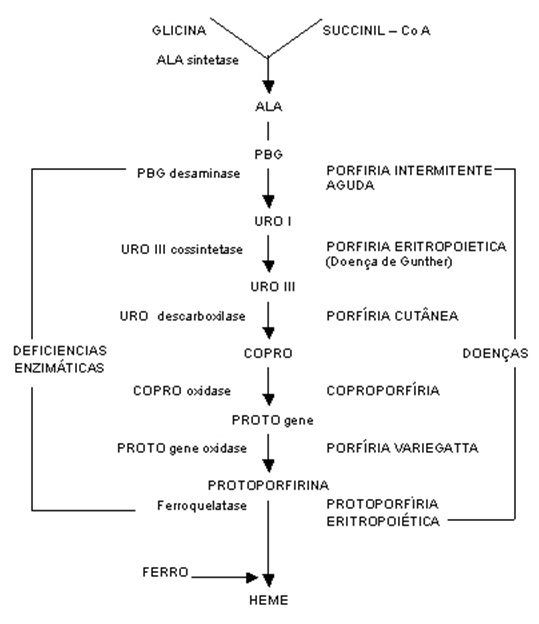
A figura abaixo resume o metabolismo do heme e as doenças associadas. A tabela 1 sumariza as diferentes alterações enzimáticas e o tipo de Porfirias resultantes.
Figura 1: cadeia metabólica do heme

Tabela 1: Classificação das Porfirias
|
Deficiência enzima |
Porfiria |
Sítio principal |
Transmissão |
|
ALA dehydrase |
Porfiria deficiente da ALA-dehidrase |
Fígado |
Recessiva |
|
PBG dease |
Porfiria Intermitente Aguda |
|
Dominante |
|
PBG dease |
Porfiria Intermitente Aguda 1 |
Fígado e medula óssea (M.O) |
Dominante |
|
PBG dease |
Porfiria Intermitente Aguda 2 |
MO |
Dominante |
|
PBG dease |
Porfiria Intermitente Aguda 3 |
Fígado e MO |
Dominante |
|
Uro cossintase |
Porfiria Eritropoética Congênita |
Fígado e MO |
Recessiva |
|
Uro decarboxilase |
Porfiria Cutânea Tarda |
Fígado |
Recessiva |
|
Uro decarboxilase |
Porfiria Cutânea Tarda P 1 |
Fígado |
Adquirido |
|
Uro gen decarboxilase |
Porfiria Cutânea Tarda 2 |
Fígado |
Dominante |
|
Uro gen decarboxilase |
Porfiria Cutânea Tarda 3 |
Fígado |
Dominante |
|
Copro oxidase |
Coproporfiria Hereditária |
Fígado |
Dominante |
|
Protogenoxidase Ferrohydrase |
Porfiria Variegatta |
Fígado |
Dominante |
|
Protogenoxidase Ferrohidrase |
Porfiria Eritropoiética |
Fígado |
Dominante |
As Porfirias podem ser divididas em:
-Porfirias agudas
- Porfiria deficiente da ALA-dehidrase
-Porfiria intermitente aguda
-Coproporfiria hereditária
-Porfiria Variegatta
-Porfirias cutâneas
As manifestações podem ser através de fotossensibilidade com erupções em geral vesiculares. Entre elas temos:
-Porfiria eritropoética congênita;
-Coproporfiria hereditária;
-Porfiria Hepatoeritropoética;
-Porfiria Cutânea Tarda;
-Porfiria Variegatta.
As lesões crônicas ocorrem principalmente em áreas expostas ao sol, ocorrendo principalmente no dorso das mãos, e se severa podem infectar e ser mutilantes. A fotossensibilidade nas Porfirias cutâneas ocorre pela ativação das Porfirinas na pele por raios ultravioleta.
Porfirias cutâneas agudas sem vesículas (blistering e fotossensibilidade):
-Protoporfiria eritropoética;
-Protoporfiria eritropoética;
-Protoporfiria ligada ao X.
Depende da mensuração dos precursores da Porfirina ALA e PBG e Porfirinas na urina, no plasma, nos eritrócitos e nas fezes. Deve-se considerar sua investigação em todos pacientes com sintomas neuroviscerais, como dor abdominal.
Nas Porfirias agudas o diagnóstico é realizado prontamente, se na vigência de um ataque temos aumento substancial do PBG urinário. O diagnóstico pode ser excluído sem esta elevação. O PBG pode ser mensurado no plasma em pacientes com disfunção renal, quando a excreção urinária pode estar prejudicada.
O ácido delta-aminolevulínico tem excreção urinária normal <7 mg/24 horas , em um ataque agudo de Porfiria intermitente aguda a excreção urinária é 10 vezes maior (25 a 100 mg/dia). O porfirobilinogênio, por sua vez, tem excreção urinária menor que 2 para 4 mg/24 horas, esta excreção aumenta 5-10 vezes nos ataques agudos. Nas Porfirias cutâneas com erupção vesicular, por sua vez, o diagnóstico usualmente é realizado com a mensuração das Porfirinas plasmáticas. Nas Porfirinas cutâneas agudas sem vesiculação, o exame diagnóstico mais sensível é a dosagem de protoporfirina eritrocitátia, que normalmente tem dosagens menores que 80 mcg/dl, mas em ataques agudos podem ter dosagens superiores acima de 500 mcg/dl.
Comentaremos brevemente sobre as principais manifestações dos mais importantes e prevalentes tipos de Porfirias:
Herança Autossômica Dominante. Os principais achados clínicos são lesões de fotossensibilidade de ocorrência precoce, após exposição à luz com desenvolvimento de lesões bolhosas subepidérmicas que evoluem para lesões crostosas. Os pacientes podem evoluir com hemólise e cálculos biliares com porfirinas. Nesta forma de Porfiria os pacientes apresentam aumento compensatório da medula óssea com ocorrência de fraturas patológicas, compressão vertebral com diminuição da altura e raramente lesões osteolíticas e escleróticas no esqueleto.
A doença pode ser reconhecida no útero por causa do líquido amniótico amarronzado, em crianças pode aparecer pigmento nas fraldas. A confirmação laboratorial pode ser realizada pela dosagem das Porfirinas urinárias que estão de 20 a 60 vezes com o limite da normalidade aumentado.
O tratamento envolve evitar o sol com uso de protetores solares, trauma e infecções. Pode ser necessária a transfusão de hemácias para evitar a hemólise, além de esplenectomia com pacientes com hemólise muito importante. A suplementação de ácido ascórbico e do tocoferol pode levar à melhora da anemia.
Ocorre por deficiência parcial de ferroquelase. A biópsia cutânea mostra paredes capilares espessadas circuladas por depósitos hialinos, imunoglobulinas, mucopolissacárides e substância PAS positiva.
Os sintomas pioram na primavera, com sensação de queimação na pele, seguida depois por eritema e edema com púrpura e vesículas. Uma série de 32 casos mostrou os seguintes sintomas:
-Queimação 97%;
-Edema 94%;
-Prurido 88%;
-Eritema 69%;
-Formação de cicatrizes 19%;
-Vesículas 3%;
-Anemia 27%;
-Colelitíase 12%;
-Alteração de resultados de exames hepáticos 4%.
O diagnóstico é confirmado por aumento de protoporfirinas nos eritrócitos, fezes e plasmas, embora em níveis menores do que em outras Porfirias e porfirinas normais na urina. Os pacientes podem apresentar anemia leve e hipertrigliceridemia.
O tratamento consiste em evitar o sol com uso de protetores solares, suplementação de beta-caróteno. Transfusões podem diminuir as lesões por fotossensibilidade, mas não são recomendadas usualmente. A colestirmamina diminui fotossensibilidade e diminui concentração das protoporfirnas hepáticas.
A doença é autossômica recessiva, sendo rara com poucos casos documentados. Os achados clínicos incluem vômitos, dores em membros inferiores e neuropatia. Assim como em outras Porfirias, estresse, ingesta de álcool e alimentar pode precipitar sintomas, inclusive dor abdominal como ocorre na Porfiria intermitente aguda. Os pacientes podem desenvolver paralisia de membros e hipotonia.
O diagnóstico é realizado por elevação do ácido aminolevulínico urinário e também protoporfirinas nas fezes e nos eritrócitos, mas o porfirobilinogênio urinário é normal.
Um diagnóstico diferencial é a intoxicação por chumbo, neste caso para realizar a diferenciação pela dosagem da plumbemia, que é aumentada na intoxicação por chumbo e baixa na Porfiria. A intoxicação por chumbo pode cursar com aumento de Porfirinas e diminuição da função e da atividade da enzima ALA-dehydrase. O tratamento é semelhante ao da Porfiria intermitente aguda. Outra situação que inibe a ALA-dehidrase é a tirosinemia tipo 1.
Doença autossômica dominante causada por deficiência da atividade da enzima PBG-deaminase. Dentre as Porfirias é a mais importante tanto em incidência como em sua severidade. Cerca de 90% dos pacientes têm deficiência da enzima em todos os tecidos, incluindo eritrócitos.
Mais de 90% dos pacientes apresentam anormalidades no gene da PBG-deaminase e quase 90% dos pacientes podem permanecer assintomáticos durante toda sua vida. As crises podem ser desencadeadas por diversos fatores exógenos ou endógenos incluindo:
-Indutores de ALA-sintase hepáticos;
-Fatores endócrinos: papel importante dos estrógenos ;
-Ingestão calórica: diminuição da ingesta calórica leva à exacerbação da Porfiria, mas dietas ricas em carboidratos diminuem a excreção da PBG e suprimem os ataques:
-Drogas e agentes químicos: muitos exacerbam por induzirem o citrocromo p450 demandando sintese do heme;
-Stress, álcool, cirurgias e outras patologias podem induzir ataques.
A doença apresenta curso variável, com ataques que duram de poucos dias a vários meses. Dor abdominal é quase sempre presente e é frequentemente o sintoma inicial, podendo ser generalizada ou localizada, mimetizando abdome agudo cirúrgico. Os sintomas incluem náuseas, vômitos e constipação, e em muitos casos o paciente é submetido à laparotomia exploradora inapropriada. Os pacientes podem ter também dor torácica, dor lombar e em membros. A neuropatia é principalmente motora e comum, mas qualquer tipo de neuropatia pode ocorrer e envolver nervos cranianos. Os ataques são frequentemente acompanhados de convulsões principalmente se hiponatremia associada. A tabela 2 sumariza os principais sintomas encontrados na Porfiria intermitente aguda.
Tabela 2: principais sintomas na porfiria intermitente aguda
|
Sintomas |
Prevalência |
|
Dor abdominal |
85-95% |
|
Vômito |
43-88% |
|
Diarreia |
5-12% |
|
Constipação |
48-84% |
|
Dores musculares |
50% |
|
Fraqueza muscular |
42-68% |
|
Perda sensorial |
9-38% |
|
Convulsões |
10-20% |
|
Paralisia respiratória |
9-14% |
|
Sintomas mentais |
40-58% |
|
Hipertensão |
36-54% |
|
Taquicardia |
28-80% |
|
Febre |
9-37% |
O diagnóstico pode ser realizado, mesmo fora da crise pela verificação da diminuição da atividade da PDG-deaminase em eritrócitos, alguns pacientes com a forma II da Porfiria intermitente aguda podem ter esta atividade normal.
Pacientes carreadores de genes silenciosos ou em latência clínica podem não ter aumento da PBG e da ALA.
O diagnóstico durante a crise pode ser realizado pela verificação do aumento importante dos precursores das porfirinas com produção de ALA de 25-100 mg ao dia e PBG de 50-200 mg/dia. As porfirinas urinárias e em fezes também são importantes.
Porfiria por deficiência da ala-hidrase, coproporfia hereditária e Porfiria variante agregatta são similares. Deve-se adequar a ingesta calórica e tratar rapidamente quaisquer infecções concomitantes.
Em pacientes não responsivos: deve-se administrar dextrose e manter uma ingesta mínima de 300 gramas de carboidrato ao dia.
A infusão de hematina endovenosa diminuirá excreção de ALA e PBG, com diminuição do ataque agudo e talvez da severidade da neuropatia.
Em mulheres que menstruam, o uso LHRH de longa ação inibe ovulação e pode diminuir os sintomas em mulheres.
A dor nestes pacientes deve ser tratada com analgésicos opioides, como o tramadol que são medicações com boa resposta nestes pacientes.
Em algumas séries é a Porfiria mais comum.
Há três subtipos principais: tipo I, que apresenta atividade do uroporfirinogênio descarboxilase normal em eritrócitos e casos esporádicos; tipo II, que apresenta atividade eritrocitária enzimática reduzida e casos familiares; e a tipo III, que apresenta casos familiares e atividade eritrocitária enzimática normal.
O quadro clínico é caracterizado por lesões bolhosas crônicas em áreas expostas ao sol. Cronicamente, o paciente pode apresentar atrofia cutânea ou áreas de pseudoescleroderma. Os pacientes apresentam frequentemente hemossiderose hepática.
Os pacientes laboratorialmente apresentam aumento do uroporfirogênio urinário e isocoporporfirina nas fezes.
O tratamento consiste em medidas para fotoproteção. Evitar álcool e traumas, flebotomias a cada 2-3 semanas pode ser necessário para hemossiderose hepática. É indicado uso de cloroquina em doses de 125 mg por duas vezes na semana até normalização das porfirinas e melhora das lesões cutâneas.
1-Sassa S. The hematologic aspects of porphyria. In: Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT, editors. Williams hematology. New York: McGraw-Hill; 2006. p. 803-22.
2-Dinardo C et al. Rev Med 2010; 89(2): 106-114.
3-Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135, 281-92.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.