(Carregando Índice)... (Carregando Índice)... |
Última revisão: 20/08/2015
Comentários de assinantes: 0
Lawrence L.K. Leung, MD
Artigo original: L.K. Leung L , MD. Hemostasis and its Regulation. ACP Medicine. 2012.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr.Lucas Santos Zambon
A hemostasia, processo de formação do coágulo sanguíneo, é uma série coordenada de respostas à lesão vascular. Requer interações complexas entre plaquetas, cascata de coagulação, fluxo sanguíneo e cisalhamento, células endoteliais e fibrinólise.
As plaquetas são ativadas no sítio de lesão vascular para formar um tampão que contenha sangramentos. Os estímulos fisiológicos plaquetários incluem o difosfato de adenosina (ADP), adrenalina, trombina e colágeno. ADP e adrenalina são relativamente fracos como estimuladores de plaquetas, enquanto a trombina e o colágeno são agonistas fortes. A ativação da trombina é mediada por receptores acoplados à proteína G ativados por protease (PARs),1 especificamente PAR-1 e PAR-4. A trombina cliva o domínio externo do PAR para iniciar a sinalização transmembrana.2 As respostas plaquetárias ao ADP requerem a ativação coordenada de dois receptores acoplados à proteína G, P2Y1 e P2Y12, com consequente ativação da fosfolipase C e supressão da formação de monofosfato de adenosina cíclico (cAMP), respectivamente. Os fármacos antiplaquetários, como ticlopidina e clopidogrel, bloqueiam a ativação de P2Y12.3 Há também receptores específicos para adrenalina, tromboxano A2 e colágeno.
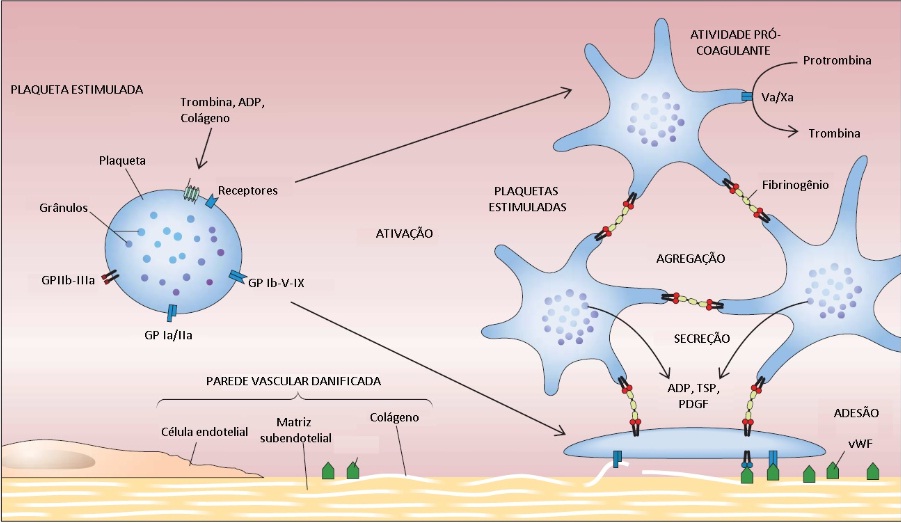
A ativação plaquetária envolve quatro processos distintos: adesão (deposição de plaquetas na matriz subendotelial) agregação (coesão plaquetária) secreção (liberação de proteínas dos grânulos plaquetários) e atividade pró-coagulante (intensificação da geração de trombina) [ver Figura 1].
A adesão plaquetária é mediada primariamente pela ligação do complexo receptor de superfície de plaqueta/glicoproteína (GP) Ib-IX-V à proteína de adesão chamada fator de von Willebrand (vWF), na matriz subendotelial.4 A deficiência do complexo GPIb-IX-V ou vWF acarreta dois distúrbios hemorrágicos congênitos, a doença de Bernard-Soulier e a doença de Von Willebrand, respectivamente [pesquise informação sobre distúrbios hemorrágicos no ACP Medicine]. Outras interações adesivas (p. ex., ligação do receptor de colágeno plaquetário GPIa-IIa às fibrilas de colágeno na matriz) também contribuem para a adesão plaquetária.5
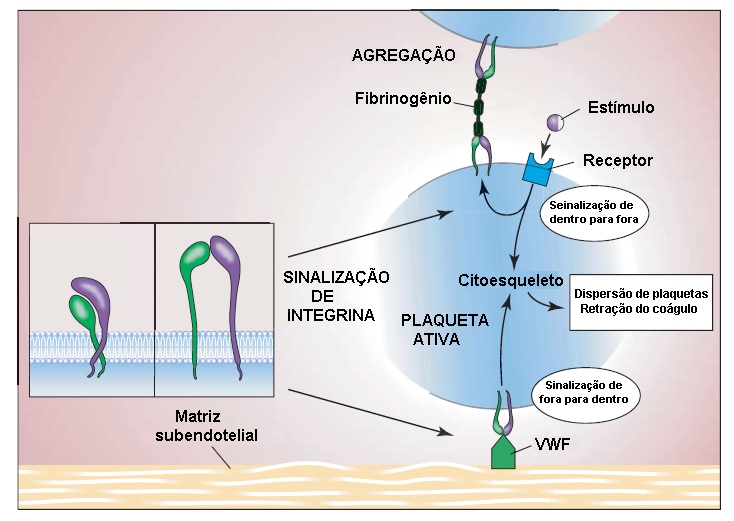
A agregação plaquetária envolve a ligação de fibrinogênio ao receptor plaquetário de fibrinogênio (i.e., complexo GPIIb-IIIa ). A GPIIb-IIIa (também chamada aIIbß3) é membro de uma superfamília de receptores de proteínas de adesão, chamadas integrinas, compostas por um complexo de subunidades a e ß, encontradas em diversos tipos celulares. É o receptor mais abundante na superfície das plaquetas. A GPIIb-IIIa não liga fibrinogênio nas plaquetas em repouso. Após a estimulação plaquetária, a GPIIb-IIIa sofre alteração conformacional e é convertida de receptor de fibrinogênio de baixa afinidade em receptor de alta afinidade, via ligação de proteínas intracelulares à porção citoplasmática de GPIIb-IIIa (sinalização de fora para dentro). O fibrinogênio, uma molécula bivalente, serve de ponte entre as plaquetas ativadas [ver Figura 2]. Por outro lado, o fibrinogênio (ou vWF) ligado à matriz subendotelial pode se ligar à porção extracelular de GPIIb-IIIa e ativá-la. A porção citosólica (ou citoplasmática) do complexo GPIIb-IIIa então ativado se liga ao citoesqueleto plaquetário e mede a dispersão das plaquetas e retração do coágulo (sinalização de fora para dentro).6 A deficiência congênita de GPIIb-IIIa ou de fibrinogênio leva ao desenvolvimento da trombastenia de Glanzmann e afibrinogenemia. A interação GPIIb-IIIa-fibrinogênio é a via final comum para agregação plaquetária. O bloqueio desta via é a base de uma classe importante de fármacos antiplaquetários potentes, os antagonistas de GPIIb-IIIa (p. ex., abciximabe, eptifibatide, tirofibana), que são úteis no tratamento da síndrome coronariana aguda.
A formação do agregado plaquetário estreitando o contato entre plaquetas adjacentes promove sinalização plaquetária adicional. As moléculas de adesão presentes na superfície de uma plaqueta podem ativar diretamente seus receptores em uma plaqueta adjacente (p. ex., efrinas (adrenalina) e receptores Eph; proteína semaforina sema4D (CD100) e seus receptores).7 Estas interações adicionais levam à estabilização e consolidação adicional do tampão plaquetário.
Após a estimulação, os grânulos plaquetários liberam ADP e serotonina, que estimulam e recrutam plaquetas adicionais; proteínas de adesão, como a fibronectina e a trombospondina, que reforçam e estabilizam os agregados plaquetários; fator V, um componente da cascata de coagulação; tromboxano, que estimula a vasoconstrição e agregação plaquetária e fatores de crescimento, como o fator de crescimento derivado de plaquetas (PDGF), que estimula a proliferação de células musculares lisas e mede o reparo tecidual. O PDGF também pode contribuir para o desenvolvimento de aterosclerose e reobstrução subsequentemente à angioplastia coronariana.
A atividade pró-coagulante plaquetária envolve a montagem de complexos enzimáticos da cascata de coagulação na superfície da plaqueta ativada (ver adiante).
As principais características da coagulação sanguínea são a rapidez (com a cessação do sangramento ocorrendo em questão de segundos), proporcionalidade (com o tamanho do trombo sendo apropriado para a magnitude da lesão vascular) e reversibilidade (com eventual dissolução do trombo e remodelamento vascular). A cascata de coagulação, com seu caráter embutido de amplificação, é responsável pela rapidez da resposta. A proporcionalidade indica que a cascata está sujeita a uma regulação rigorosa, enquanto a reversibilidade é resultado da fibrinólise.

Figura 1 - Depois de serem ativadas, as plaquetas sofrem alterações morfológicas significativas e produzem pseudópodos alongados. Também se tornam extremamente adesivas. A resposta funcional das plaquetas ativadas envolve quatro processos distintos: adesão (deposição de plaquetas na matriz subendotelial); agregação (coesão plaquetária); secreção (liberação de proteínas dos grânulos plaquetários); e atividade pró-coagulante (intensificação da geração de trombina). ADP = difosfato de adenosina; GP = glicoproteína; PDGF = fator de crescimento derivado de plaqueta; TSP = trombospondina; vWF = fator de Von Willebrand.
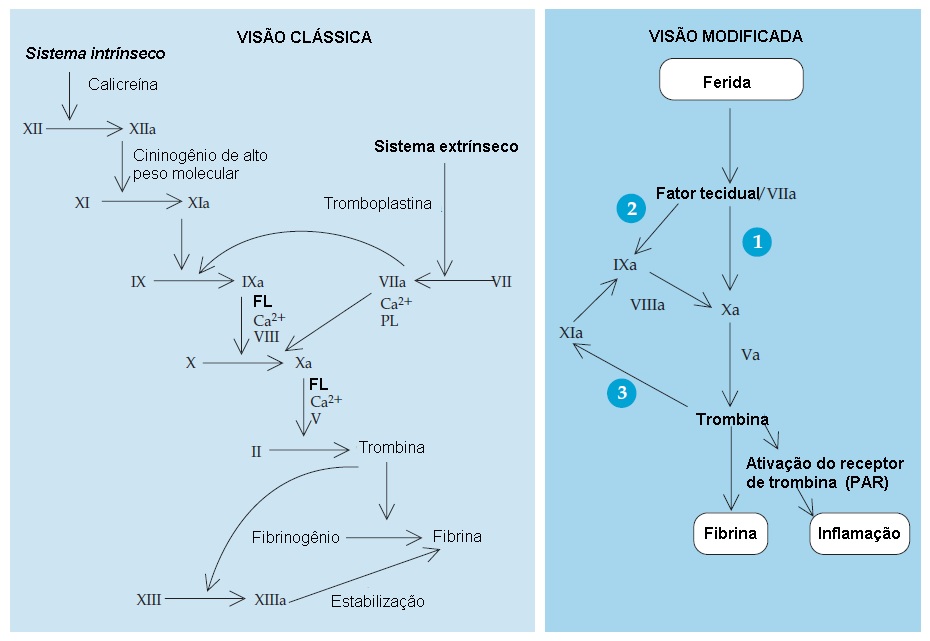
A cascata de coagulação consiste na ativação sequencial de uma série de pró-enzimas (zimogênios) em enzimas, gerando por fim a fibrina e reforçando o tampão plaquetário. Esta cascata é tradicionalmente descrita como sendo composta pelas vias intrínseca e extrínseca [ver Figura 3]. A via intrínseca é iniciada pela exposição do sangue a uma superfície negativamente carregada (p. ex., vidro), enquanto a via extrínseca é ativada por fator tecidual ou tromboplastina. Ambas as vias convergem na ativação do fator X, que então ativa a protrombina (fator II) em trombina - a enzima final da cascata de coagulação.
Embora esta perspectiva clássica da cascata de coagulação tenha sido útil na interpretação dos tempos de coagulação, não é totalmente precisa. Pacientes com deficiência grave de fator XII, bem como muitos pacientes deficientes de fator XI, não apresentam sangramento do ponto de vista clínico, indicando que a parte da iniciação da via intrínseca (a fase de contato) é irrelevante in vivo. Hoje, está estabelecido que a geração ou exposição de fator tecidual no sítio de uma ferida é o evento fisiológico primário que inicia a coagulação, a qual então prossegue ao longo de três vias [ver Figura 4].8 O fator tecidual atua como cofactor essencial para que o fator VII/fator VII ativado (VIIa) inicie a coagulação. O fator VIIa ativa o fator X diretamente e, indiretamente, via ativação do fator IX. Esta via dupla de ativação do fator X é necessária devido à quantidade limitada de fator tecidual gerada in vivo e à presença do inibidor da via do fator tecidual (TFPI) (ver adiante) que, ao se fixar ao fator Xa, inibe o complexo fator tecidual/fator VIIa. A trombina pode promover feedback de ativação do fator XI que, então, ativa o fator IX. Esta via de amplificação terciária gera trombina extra que se torna necessária no contexto de lesões vasculares significativas, e que provavelmente explica a observação de que os pacientes com deficiência de fator XI em geral sangram somente após sofrerem traumatismos graves ou se submeterem a cirurgias.
Esta interpretação também é consistente com o fenótipo observado em camundongos deficientes de fator XII ou fator XI. Estes camundongos não exibem nenhum problema de sangramento sob circunstâncias normais, similarmente aos pacientes com deficiência de fator XII ou fator XI, mas se mostram surpreendentemente protegidos contra o desenvolvimento de trombose arterial em modelos experimentais de trombose arterial.9,10 Neste sentido, é interessante o fato de um estudo recente ter mostrado que pacientes com deficiência grave de fator XI (< 15%) se mostraram protegidos contra o desenvolvimento de acidente vascular encefálico isquêmico e não contra infarto do miocárdio.11 Isto sugere que a trombina extra gerada pela via de amplificação terciária é importante na formação de trombos arteriais patológicos, levantando a possibilidade de que os inibidores dirigidos contra o fator XI possam ter perfil terapêutico superior, em comparação aos inibidores diretos de trombina ou fator Xa, por serem capazes de prevenir a trombose arterial patológica sem comprometer significativamente a hemostasia normal.12
Figura 2 - A agregação plaquetária envolve ligação da molécula bivalente de fibrinogênio ao receptor plaquetário de fibrinogênio (o complexo GPIIb-IIIa ). No estado de repouso, os domínios extracelulares de GPIIb-IIIa estão dobrados e o sítio de ligação de fibrinogênio está escondido. Após a estimulação plaquetária, os domínios extracelulares se tornam estendidos e expõem o sítio de ligação de fibrinogênio, enquanto GPIIb-IIIa é convertida da forma de receptor de fibrinogênio de baixa afinidade na forma de receptor de fibrinogênio de alta afinidade (sinalização de dentro para fora). A porção citosólica do complexo GPIIb-IIIa ativado pode mediar a dispersão plaquetária e a retração do coágulo (sinalização de fora para dentro) por meio de interações com duas proteínas ligadoras de integrina principais, talina e kindlina. VWF = fator de Von Willebrand.
Todos os pró-coagulantes são sintetizados no fígado, com exceção do VWF, que é sintetizado nos megacariócitos e nas células endoteliais. Vários pró-coagulantes, bem como anticoagulantes, requerem vitamina K. Os pró-coagulantes dependentes de vitamina K são protrombina, fator VII, fator IX e fator X; os anticoagulantes dependentes de vitamina K são proteína C e proteína S. Para estes fatores, a formação de resíduos de ácido C-carboxiglutâmico pela C-carboxilação de resíduos de ácido glutâmico dependente de vitamina K lhes confere propriedades ligadoras de cálcio e capacidade de interagir com fosfolipídios aniônicos na superfície de plaquetas ativadas.13
In vivo existe uma estreitíssima interação entre a cascata de coagulação e a superfície das plaquetas ativadas. Quando as plaquetas são ativadas, os lipídios aniônicos são expostos e o fator V (armazenado nos grânulos plaquetários) é liberado e expresso na superfície da plaqueta ativada. O fator V presente na superfície da plaqueta é ativado ao fator Va (pela trombina) e atua como sítio de montagem para ligação de fator Xa (enzima) e protrombina (substrato), o conhecido complexo protrombinase. A geração de trombina pelo complexo protrombinase é cerca de 300.000 vezes mais eficiente do que a geração de trombina por fator Xa de fase-fluída e protrombina isoladamente, sendo que o tampão plaquetário mantém a trombina localizada. O fator Xa ligado ao fator Va também está protegido contra a inibição por ação de inibidores circulantes, como a antitrombina (AT) (ver adiante). Uma montagem de complexo enzimático similar se aplica à ativação do fator X pelo fator VIIIa (cofator) e fator IXa (tenase intrínseca). O resultado destes processos é a amplificação eficiente e a localização da coagulação.
O curso temporal da geração de trombina, medido pela velocidade da formação do complexo trombina-AT no sangue total, indica uma fase de iniciação com apenas uma pequena quantidade de trombina sendo gerada, seguida por uma fase que consiste no grosso da formação de trombina e, por fim, na cessação da geração de trombina.14 É notável que o tempo de coagulação laboratorial (que detecta a formação do coágulo de fibrina inicial) mede primariamente a fase de iniciação e não a fase de propagação. A fase de iniciação é amplamente mediada pela ativação do fator X pelo fator tecidual/fator VIIa [ver Figura 3], dando origem a uma pequena quantidade de trombina que, por sua vez, ativa o fator V, fator VIII e plaquetas, expondo os fosfolipídios aniônicos para sustentar a montagem de complexos enzimáticos de componentes (tenase intrínseca e pró-trombinase). Assim, a pequena quantidade inicial de trombina gerada condiciona a cascata de coagulação e ativa as plaquetas que, então, levam à geração explosiva de trombina durante a fase de propagação [ver Figura 3].
Figura 3 - Na visão clássica da cascata de coagulação (esquerda), a via intrínseca é iniciada pela exposição do sangue a uma superfície de carga negativa (p. ex., vidro) e a via extrínseca é ativada pelo fator tecidual ou tromboplastina. Na perspectiva renovada (direita), a geração ou exposição de fator tecidual no sítio da ferida é o evento fisiológico primário que inicia a coagulação. O fator tecidual ativa o fator X diretamente, atuando como cofator com o fator VIIa (via 1), e indiretamente, ativando o fator IX (via 2). A trombina pode ativar o fator XI por meio de feedback e isto ativa o fator IX (via 3). Esta via de amplificação terciária gera trombina adicional, a qual é necessária no evento de uma lesão vascular significativa. PAR = receptor ativado por protease; FL = fosfolipídio.
Embora as plaquetas ativadas nitidamente exerçam papel essencial na intensificação da coagulação sanguínea, é comum observar que a trombocitopenia e a deficiência de fator de coagulação estão associadas a manifestações clínicas bastante distintas. A primeira em geral se manifesta com sangramento de mucosa, enquanto a segunda se manifesta na forma de hematomas e sangramento articular. Como as plaquetas são indispensáveis para a coagulação sanguínea eficiente, seria esperado que o sangramento decorrente de trombocitopenia grave também apresentasse características de deficiência de fator de coagulação. Este aparente paradoxo pode ser parcialmente explicado pela constatação recente de que apenas um pequeno número de plaquetas é requerido para formação adequada de fibrina. Desta forma, mesmo na presença de trombocitopenia grave (p. ex., contagem plaquetária de cerca de 10.000/mL), os pacientes geralmente apresentam apenas sangramento de mucosa, em vez de sangramento articular. Por outro lado, pacientes com trombocitopenia moderada não estão protegidos contra o desenvolvimento de tromboembolia venosa.
A coagulação é modulada por alguns mecanismos: diluição de pró-coagulantes no sangue em fluxo; remoção de fatores ativados por meio do sistema retículo endotelial, em especial no fígado; e controle por vias antitrombóticas naturais. Pelo menos sete sistemas de controle separados e distintos modulam cada fase da hemostasia e conferem proteção contra trombose, inflamação vascular e dano tecidual [ver Tabela 1]. AT, proteína C, proteína S e TFPI regulam coletivamente a cascata de coagulação; prostaciclina (PGI2) e óxido nítrico (NO) modulam a reatividade vascular e plaquetária; a ecto-ADPase inibe o recrutamento de plaquetas; e fibrinólise remove o coágulo de fibrina.
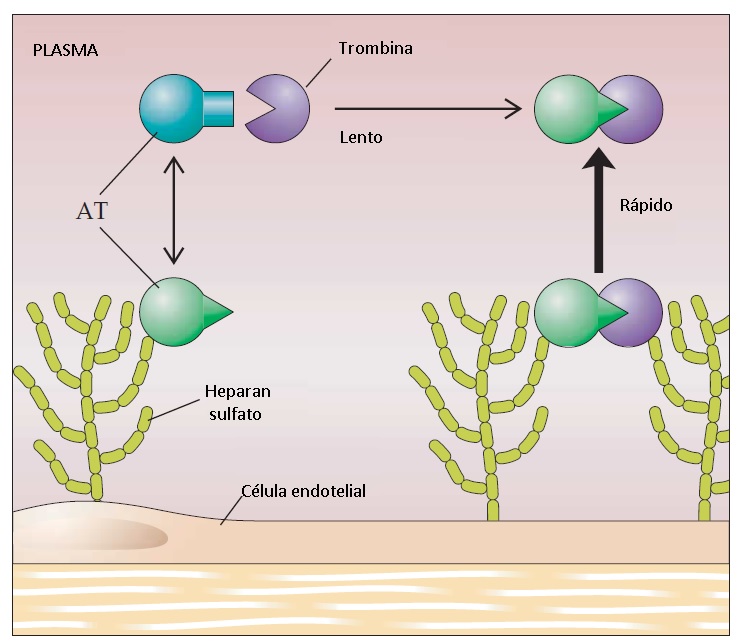
A AT é um inibidor de protease plasmático circulante. Inibe a trombina e o fator Xa, que são duas enzimas essenciais para a cascata de coagulação. A AT também inibe fator XII ativado e o fator XI. Na ausência da glicosaminoglicana heparina, a AT inibe a trombina e o fator Xa de forma relativamente lenta (a inibição complete requer pouco minutos). Quando presente, a heparina se liga a um sítio discreto na molécula de AT, a qual então sofre uma alteração conformacional que inibe a trombina de forma instantânea e irreversível. Este aumento da inibição de trombina e fator Xa é a base do uso terapêutico da heparina como anticoagulante.
Figura 4 - Na ausência de heparan sulfato (HS), a antitrombina (AT) inibe lentamente a trombina. Em presença de HS, este se liga a um sítio específico na molécula de AT, na qual produz uma alteração conformacional que permite que a molécula alcance o sítio ativo da trombina e iniba a enzima de maneira instantânea. HS também se liga a um sítio específico (exosítio ligador de ânion II) na trombina, posicionando-a para inibição ótima pela AT.
A ligação da heparina à AT é mediada por um pentassacarídeo presente na heparina. A heparina de baixo peso molecular é um fragmento de heparina regular que contém este pentassacarídeo específico. O fondaparinux é a versão sintética deste pentassacarídeo.
Os proteoglicanos do heparan sulfato presentes na superfície luminal das células endoteliais ativam a AT de modo similar à heparina [ver Figura 4].16 Portanto, a superfície endotelial normalmente é coberta com uma camada de AT já ativada pelo heparan sulfato endógeno. Como 1 mL de sangue pode ser exposto a até 5.000 cm2 de superfície endotelial, o sistema AT-heparan sulfato equilibrado para rapidamente inativar qualquer trombina presente na circulação geral.
|
Tabela 1 Mecanismos antitrombóticos naturais de células endoteliais |
|
Regulação da cascata de coagulação Fator tecidual via inibidor Antitrombina Proteína C/proteína S |
|
Modulação da reatividade de vasos e plaquetas Prostaciclina Óxido nítrico |
|
Inibição do recrutamento de plaquetas Ecto-ADPase (CD39) |
|
Remoção do coágulo de fibrina Fibrinólise |
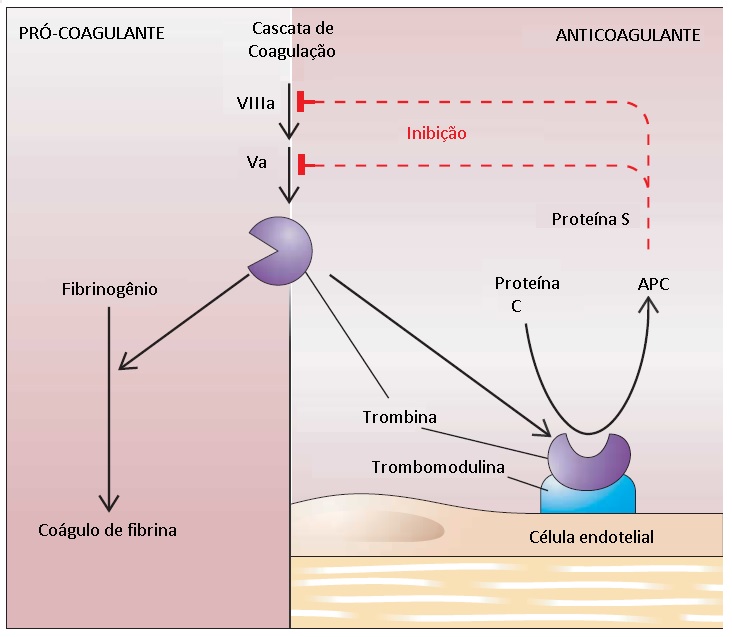
A trombomodulina é uma proteína integral de membrana encontrada na superfície luminal do endotélio vascular, na microcirculação. A ligação da trombina à trombomodulina resulta em mudança significativa das especificidades do substrato da trombina: esta deixa de formar coágulos de fibrinogênio ou de ativar plaquetas [ver Figura 5]. Por outro lado, a trombina adquire a capacidade de ativar a proteína C no plasma.17 Foi descoberto um receptor endotelial distinto para proteína C, que intensifica a ativação desta proteína pelo complexo trombina-trombomodulina.18 A proteína C ativada degrada o fator Va e fator VIIIa, que são os dois cofatores responsáveis pela montagem do complexo de protrombinase e tenase intrínseca na cascata de coagulação. A proteína S serve de cofator para a proteína C ativada. As deficiências de AT, proteína C e proteína S são causas importantes de um estado hipercoagulável.
A proteína C e a proteína S mostram, ambas, certo grau de similaridade estrutural com fatores de coagulação dependentes de vitamina K (protrombina, fator VII, fator IX e fator X). A proteína S circula em duas formas: uma forma livre, em que é ativa como anticoagulante; e uma forma ligada inativa, em que está complexada à proteína ligadora de C4b do sistema complemento. A proteína ligadora de C4b atua como reagente de fase aguda. A resultante intensificação dos estados inflamatórios diminui a atividade da proteína S livre, aumentando a probabilidade de trombose.
TFPI é um inibidor de protease plasmático circulante sintetizado pelo endotélio microvascular. Diferente de AT, a concentração plasmática de TFPI é baixíssima. O TFPI inibe o fator Xa. O complexo TFPI/fator Xa se torna um inibidor efetivo do fator tecidual/fator VIIa, mediando assim a inibição por feedback do fator tecidual/fator VIIa. Estudos realizados com animais têm mostrado que a depleção de TFPI endógeno sensibiliza os animais ao desenvolvimento de coagulação intravascular disseminada induzida por fator tecidual ou endotoxina.19
Figura 5 - A via da proteína C/proteína S é complementar à via da antitrombina (AT). Quando a trombina se liga à trombomodulina, a trombina sofre alteração conformacional e deixa de formar coágulos de fibrinogênio ou ativar plaquetas. Entretanto, adquire a capacidade de ativar a proteína C no plasma. A proteína S serve de cofator para a proteína C ativada (APC). A APC degrada os fatores V e VIII ativados, que são os dois cofatores na cascata de coagulação.
O TFPI é sintetizado primariamente pelo endotélio microvascular. A maioria do TFPI está associada à superfície endotelial, aparentemente ligada às glicosaminoglicanas da superfície celular. Os níveis plasmáticos de TFPI aumentam bastante após a administração intravenosa de heparina. Esta liberação de TFPI endotelial pode contribuir para a eficácia antitrombótica da heparina e da heparina de baixo peso molecular. Embora estudos realizados com animais tenham sugerido usos terapêuticos para o TFPI recombinante, os estudos clínicos fracassaram.
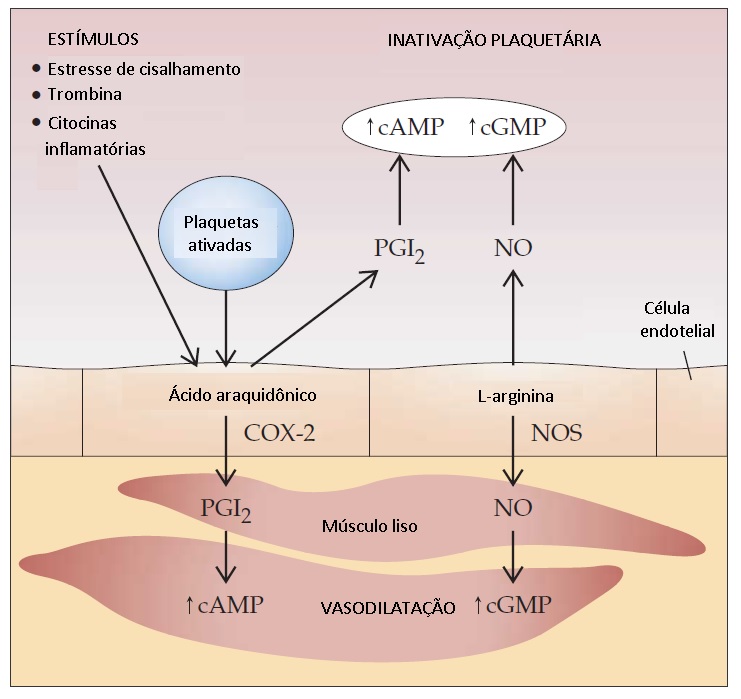
Diante de uma perturbação à célula, o ácido araquidônico, um ácido graxo, é liberado dos fosfolipídios da membrana celular pela fosfolipase A2. A enzima prostaglandina endoperóxido H sintase-1 (PGHS-1) converte o ácido araquidônico em prostaglandina endoperóxidos e, por fim, em tromboxano A2 (TXA2) nas plaquetas, bem como em PGI2 nas células endoteliais. TXA2 e PGI2 exercem funções opostas: TXA2 é um potente estimulador de agregação plaquetária e causa vasoconstrição, enquanto a PGI2 inibe a agregação plaquetária e induz vasodilatação. A PGI2 atua ativando a adenilato ciclase e isto leva ao aumento dos níveis intracelulares de cAMP [ver Figura 6].
Figura 6 - Existe um sinergismo significativo entre o óxido nítrico (NO) e a prostaciclina (PGI2), levando à inativação plaquetária e à vasodilatação. A enzima prostaglandina endoperóxido H sintase–1 (PGHS-1) converte o ácido araquidônico em PGI2 nas células endoteliais. A PGI2 ativa a adenilato ciclase e isto leva ao aumento da concentração intracelular de monofosfato de adenosina cíclico (cAMP), inibindo a agregação plaquetária e induzindo vasodilatação. O NO, formado a partir da L-arginina, estimula a produção de monofosfato de guanosina cíclico (cGMP). A ciclo-oxigenase-2 (COX-2) é a isoforma induzida da PGHS; sua formação provavelmente resulta do cisalhamento hemodinâmico na circulação. A formação de NO é catalisada pelas óxido nítrico sintases (NOSs).
A ciclo-oxigenase-1 (COX-1) é a isoforma constitutiva da PGHS. A ciclo-oxigenase-2 (COX-2) é uma isoforma induzível da PGHS. A COX-2 é indetectável na maioria dos tecidos. Entretanto, fatores de crescimento, endotoxinas e citocinas podem rapidamente induzir COX-2 nas células endoteliais e monócitos (mas não em plaquetas).20 Evidências recentes indicam que, sob condições fisiológicas, a COX-2 endotelial é uma fonte importante de PGI2 em seres humanos, resultante da indução contínua de COX-2 por cisalhamento hemodinâmico na circulação.21 A aspirina acetila e inibe de modo irreversível a COX-1 (e, fracamente, a COX-2). Como as plaquetas não podem fabricar COX-1 nova, a breve exposição das plaquetas à aspirina inibirá de modo permanente a produção de TXA2 ao longo de todo o tempo de expectativa de vida destas plaquetas. Os fármacos anti-inflamatórios não hormonais (AINHs) também inibem COX-1 e COX-2, ainda que de modo não permanente. Os efeitos colaterais cardiovasculares inesperados dos AINHs observados com o uso dos inibidores seletivos de COX-2 (p. ex., rofecoxibe) podem resultar de sua ação inibitória sobre a produção de PGI2 endotelial.
O NO é formado a partir da L-arginina nas células endoteliais. A formação de NO é catalisada pelas NO sintases, que existem em diferentes isoformas em diversos tecidos. O NO estimula a guanilato ciclase, levando ao aumento de monofosfato de guanosina ciclíco (cGMP) nas células-alvo, causa vasodilatação e inibe a adesão e agregação plaquetárias [ver Figura 6].22 O NO é rapidamente destruído pela hemoglobina e, assim, atua como hormônio local (i.e., parácrino). A infusão intravenosa de um análogo de arginina que bloqueia a produção de NO leva à elevação imediata e substancial da pressão arterial, sugerindo que o NO é liberado de forma continua e basal para regular o tônus vascular (em contraste com a produção de PGI2, que é liberada em resposta a um estímulo). Existe um sinergismo significativo entre NO e PGI2. Além de regular os eventos vasculares, o NO produz uma ampla gama de efeitos biológicos (p. ex., atua como neurotransmissor no sistema nervoso central; é secretado por macrófagos como parte de uma resposta imune inata; e é tóxico para bactérias).
O CD39 é uma proteína de membrana integral encontrada na superfície da célula endotelial. É uma enzima ativa que rapidamente hidrolisa ADP em adenosina monofosfato, atuando assim como uma ecto-ADPase ligada à célula. Limita o recrutamento de plaquetas adicionais para dentro do tampão plaquetário crescente, removendo o ADP liberado dos grânulos densos das plaquetas ativadas, bem como de eritrócitos danificados e células endoteliais.23
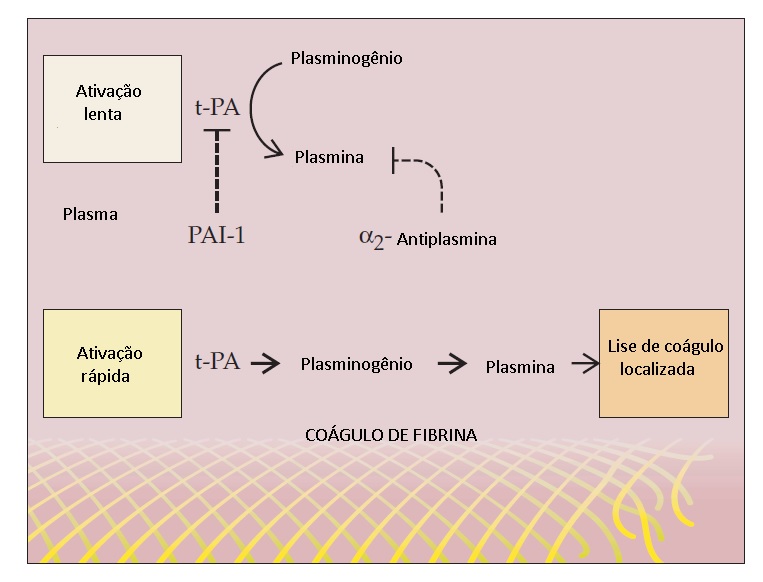
O ativador de plasminogênio tecidual (t-PA) é liberado por células endoteliais perturbadas nas proximidades do sítio de lesão vascular. O t-PA converte plasminogênio em plasmina. Assim como a interação de AT com a trombina, que é acelerada na presença de heparan sulfato na superfície da célula endotelial, a geração de plasmina é ideal em superfícies (neste caso, o coágulo de fibrina). Ambos, t-PA e plasminogênio, se ligam à fibrina (via reconhecimento dos resíduos de lisina), que facilita a geração de plasmina e a fibrinólise localizada [ver Figura 7].
Figura 7 - O ativador de plasmonigênio tecidual (t-PA), liberado por células endoteliais perturbadas nas proximidades de um vaso sanguíneo lesado, liga-se à fibrina. E o mesmo ocorre com o plasminogênio. Quando ambos estão ligados na superfície da fibrina, t-PA converte eficientemente o plasminogênio em plasmina, levando à fibrinólise localizada. A plasmina ligada ao coágulo de fibrina também fica protegida contra a inativação. A plasmina livre liberada na circulação é rapidamente inativada pela a2-antiplasmina plasmática, prevenindo assim a proteólise inespecífica generalizada pela plasmina.
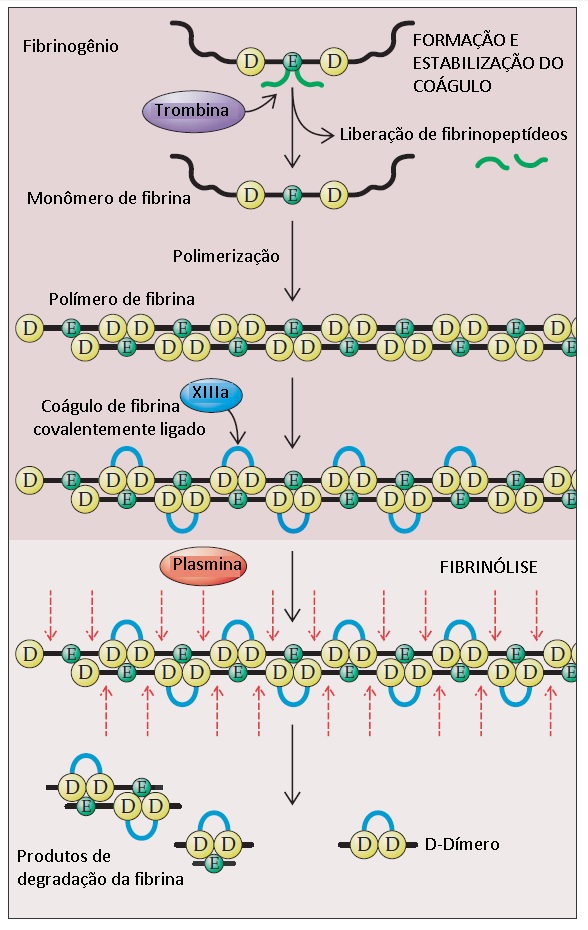
A plasmina cliva a fita de fibrina polimerizada em múltiplos sítios, liberando produtos da degradação da fibrina. Um dos principais produtos de degradação da fibrina é o D-dímero, que consiste em dois domínios D de monômeros de fibrina adjacentes que sofreram ligação cruzada por ação do fator XIII ativado [ver Figura 8]. A plasmina tem especificidade de substrato ampla e, além da fibrina, cliva fibrinogênio e várias proteínas plasmáticas e fatores de coagulação. A plasmina ligada ao coágulo de fibrina está protegida da inativação, enquanto a plasmina liberada na circulação é rapidamente inativada pela a2-antiplasmina plasmática. Assim, a fibrinólise localizada é alcançada, porém a degradação inespecífica de proteínas plasmáticas por ação da plasmina é evitada. Em casos raros, os pacientes apresentam problemas de sangramento causados por uma deficiência congênita em a2-antiplasmina.
A uroquinase é o segundo ativador de plasmonigênio fisiológico. Está presente em alta concentração na urina. Embora t-PA seja amplamente responsável pelo início da fibrinólise intravascular, a uroquinase é o principal ativador de fibrinólise no compartimento extravascular. A uroquinase é secretada por muitos tipos celulares na forma de pró-uroquinase, também denominada ativador de plasmonigênio tipo uroquinase de cadeia única (scu-PA). A pró-uroquinase é convertida em uroquinase pela plasmina. A uroquinase não tem especificidade para fibrina em termos de conversão de plasminogênio em plasmina, ao contrário da pró-uroquinase.
O principal inibidor fisiológico de t-PA e de ativador de plasmonigênio tipo uroquinase (u-PA) é o inibidor de ativador de plasmonigênio–1 (PAI-1).24 Quantidades substanciais de PAI-1 são encontradas nas plaquetas. PAI-1 também é liberado das células endoteliais. A deficiência congênita de PAI-1 é um distúrbio hemorrágico raro, geralmente associado ao sangramento relacionado a traumatismos ou cirurgia.25 Ao contrário, níveis elevados de PAI-1 têm sido encontrados em diversos estados patológicos, como obesidade e síndrome metabólica, podendo exercer algum papel na hipercoagulabilidade.
Figura 8 - A transformação do fibrinogênio em fibrina é iniciada com a trombina clivando os fibrinopeptídeos A e B dos domínios E do fibrinogênio para formar um monômero de fibrina. A clivagem muda a carga negativa geral do domínio E para uma carga positiva. Esta mudança de carga permite a polimerização espontânea dos monômeros de fibrina, porque o domínio E positivamente carregado é montado com os domínios D de carga negativa dos outros monômeros. O polímero é inicialmente unido por ligações de hidrogênio. A trombina ativa o fator XIII, que catalisa a formação de ligações covalentes entre domínios D adjacentes no polímero de fibrina. A plasmina cliva a fita de fibrina polimerizada em múltiplos sítios e libera produtos de degradação da fibrina, entre os quais o D-dímero.
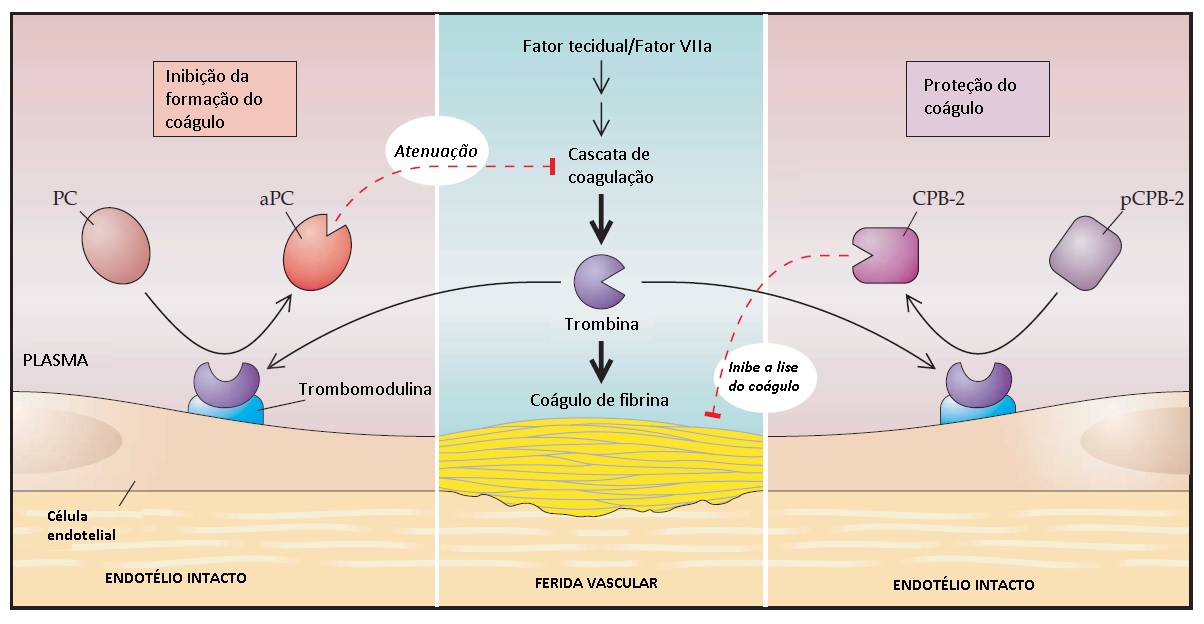
A carboxipeptidase B plasmática (que será referida como carboxipeptidase B-2 [CPB-2] para concordar com o nome de seu próprio gene) é um inibidor de fibrinólise trombina-ativável (TAFI)26,27 A CPB-2 é o segundo substrato fisiológico conhecido do complexo trombina-trombomodulina. A CPB-2 ativada remove as lisinas do terminal carboxil do coágulo de fibrina parcialmente digerido, impedindo que t-PA e plasminogênio adicional se liguem ao coágulo e, assim, inibam a fibrinólise. É possível prever que, após a formação do coágulo inicial de fibrina pela trombina no sítio de uma ferida vascular, a trombina se liga à trombomodulina em uma superfície endotelial intacta adjacente. A trombina ligada à trombomodulina leva à geração de proteína C ativa que inibe a cascata de coagulação e previne a geração excessiva de trombina, bem como de CPB-2 ativada, com consequente retardo da lise do coágulo formado [ver Figura 9]. Na hemofilia, a geração diminuída de trombina pode levar à ativação subótima de CPB-2 e resultar em lise precoce do coágulo, contribuindo para o sangramento tardio observado nestes pacientes.27 Níveis elevados de CPB-2 podem representar um fator de risco modesto de trombose.28,29 Há evidências crescentes indicando que CPB-2 exerce um amplo papel anti-inflamatório—exemplificando, inativa a bradicinina e o componente C5a do complemento—podendo assim operar com a proteína C ativada em um feedback negativo para inibir a função inflamatória da trombina.30
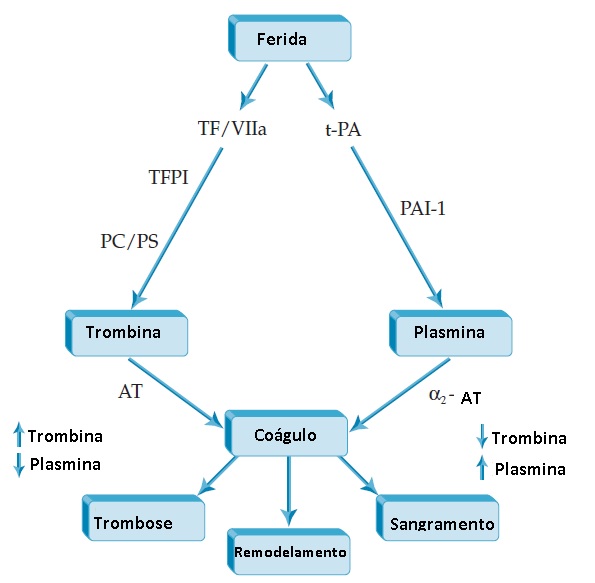
A cascata de coagulação é iniciada pela exposição do fator tecidual em uma ferida vascular, levando à geração de trombina e deposição de um coágulo de fibrina [ver Figura 10]. Ao mesmo tempo, o endotélio danificado libera t-PA, que converte plasminogênio em plasmina, e esta então lisa o coágulo. Ambas as vias são reguladas: o fator tecidual/fator VIIa é regulado pelo complexo TFPI/fator Xa, enquanto a geração de trombina é regulada pelas proteínas C e S. Similarmente, a atividade de t-PA é regulada por PAI-1. Trombina e plasmina estão sob controle de seus respectivos inibidores, AT e a2-antiplasmina. Quando estas duas vias atuam em simetria coordenada, a previsão é de que um coágulo pare de sangrar, seguindo-se a lise do coágulo e o remodelamento tecidual. A geração diminuída de trombina (como na deficiência de fator VIII) ou a produção aumentada de plasmina (como na deficiência de a2-antiplasmina) causa hemorragia [pesquise informação sobre distúrbios hemorrágicos no ACP Medicine]. Por outro lado, a produção excessiva de trombina (como na deficiência de AT ou de proteína C) acarreta trombose [pesquise informação sobre distúrbios trombóticos no ACP Medicine].
Figura 9 - A proteína C ativada (aPC) e a carboxipeptidase B-2 (CPB-2) (também chamada inibidor de fibrinólise trombina-ativável [TAFI]) têm papeis complementares nos sítios de lesão vascular. Depois que o coágulo de fibrina é formado pela trombina no sítio de ferida vascular, a trombina se liga à trombomodulina na superfície endotelial intacta próxima. A trombina ligada à trombomodulina leva à geração de aPC, que inibe a cascata de coagulação e previne a geração de trombina em excesso. A CPB-2 ativada (TAFIa) remove as lisinas carboxil terminais do coágulo de fibrina parcialmente digerido e impede a ligação de ativador de plasmonigênio tecidual e de plasminogênio ao coágulo, inibindo assim a fibrinólise. Em adição aos seus papéis na regulação da cascata de coagulação e na estabilização do coágulo, aPC e CPB-2 exercem funções anti-inflamatórias e citoprotetoras, representando assim um amplo mecanismo de feedback negativo na modulação da ação inflamatória da trombina no sítio de lesão tecidual. pCPB-2 = pró-carboxipeptidase B-2.
Embora o endotélio em geral seja considerado um Sistema orgânico homogêneo, existem diferenças significativas entre as células endoteliais arteriais, venosas e capilares, em termos de morfologia e suscetibilidade a doenças. Conjuntos distintos de proteínas marcam as células endoteliais arteriais e venosas desde os estágios iniciais da angiogênese. A efrina-B2, um ligante transmembrana membro da família Eph, marca somente as células endoteliais arteriais. Por outro lado, Eph-B4, um receptor tirosina quinase de efrina-B2, marca somente veias.31
É igualmente provável que os endotélios de diferentes leitos vasculares não sejam idênticos.32 Por exemplo, o endotélio alto nas vênulas pós-capilares dos linfonodos e placas de Peyer regula circulação de linfócitos do sangue para os linfáticos e tecidos periféricos. Receptores de proteínas de adesão específicos e proteínas da matriz são altamente expressos nestas vênulas de endotélio alto. O endotélio especializado representando a barreira hematoencefálica é outro exemplo.
Figura 10 - A exposição de fator tecidual em uma ferida vascular inicia a cascata de coagulação. A geração de trombina e deposição de um coágulo de fibrina ocorre de modo simultâneo com a liberação de ativador de plasmonigênio tecidual (t-PA) a partir do endotélio danificado e com a conversão de plasminogênio em plasmina. A plasmina então lisa o coágulo. Quando estas duas vias atuam em simetria coordenada, a previsão é de que o coágulo pare de sangrar e, em seguida, ocorra lise do coágulo e remodelamento. Por outro lado, a geração de trombina diminuída ou a geração aumentada de plasmina causa hemorragia. Em contraste, a geração excessiva de trombina resulta em trombose. a2-AP = a2-antiplasmina; AT = antitrombina; PAI-1 = inibidor de ativador de plasminogênio-1; PC/PS = proteína C/proteína S; TF = fator tecidual; TFPI = inibidor da via do fator tecidual.
Estas diferenças entre células endoteliais arteriais e venosas e o endotélio específico do leito vascular podem ser parcialmente responsáveis pelas diferentes suscetibilidades à trombose. Exemplificando, enquanto as deficiências de AT e proteína C em geral estão associadas à trombose venosa profunda de membros inferiores, a trombose das veias porta e hepática frequentemente está associada a doenças mieloproliferativas. Em ambas as condições, o defeito subjacente é um estado hipercoagulável, apesar da clara predisposição da trombose a leitos vasculares específicos. Assim, a trombose clínica é atribuível a um desequilíbrio entre estímulos pró-trombóticos sistêmicos e mecanismos antitrombóticos locais [pesquisar informação sobre distúrbios trombóticos no ACP Medicine].
As plaquetas são derivadas dos megacariócitos, que surgem de células-tronco hematopoiéticas pluripotentes. A produção de plaquetas é controlada por uma trombopoetina que está envolvida na maturação final do megacariócito. A trombopoetina exerce múltiplas ações no desenvolvimento do megacariócito,33 compartilha certo grau de homologia estrutural com a eritropoetina e é produzida principalmente pelo fígado. Aumenta o tamanho e o número dos megacariócitos, estimula a expressão de marcadores específicos de plaquetas, e um potente fator estimulador de colônia de megacariócito. Embora a trombopoetina seja nitidamente um fator essencial, o fator de célula-tronco (também chamado ligante kit), a interleucina-3 (IL-3), a IL-6 e a IL-11 contribuem para o controle da megacariocitopoiese.
Os megacariócitos sofrem endomitose, processo no qual ocorrem divisões nucleares sem divisão celular e acompanhadas de fusão nuclear, rendendo uma célula com conteúdo cromossômico de 8n, 16n ou 32n. Os megacariócitos maduros se aglomeram ao redor dos vasos sanguíneos na medula óssea e elaboram longos “processos pró-plaquetas” que se estendem para dentro do lúmen do vaso sanguíneo. O estresse de cisalhamento a partir do fluxo sanguíneo fragmenta estes processos, liberando plaquetas na circulação.34 O volume de megacariócito está correlacionado com a ploidia e a maturidade citoplasmática, sendo que os megacariócitos maiores produzem o maior número de plaquetas. As plaquetas maiores, chamadas megatrombócitos, são vistas no sangue periférico em estados trombocitopênicos, sobretudo na púrpura trombocitopênica. Estes megatrombócitos são provavelmente pró-plaquetas jovens e respondem pelo aumento do volume médio plaquetário que ocorre durante a resposta ou recuperação da trombocitopenia aguda.
As plaquetas que entram na circulação sobrevivem cerca de 8,5 a 10 dias e têm meia-vida aproximada de quatro dias. Cerca de 30-40% das plaquetas presentes estão presentes em um pool esplênico aberto a trocas livres com a circulação. Quando aumenta a necessidade de plaquetas, a produção pode aumentar 7-8 vezes. Como não há pool medular de plaquetas aguardando para ser liberado, pode demorar alguns dias para que os requisitos aumentados de plaquetas sejam atendidos. As plaquetas têm receptores para trombopoetina, a qual removem do plasma, sendo que a massa plaquetária atua como um dos principais reguladores de trombopoetina.35 Nos estados de hipoplasia de megacariócito e trombocitopenia (p. ex., anemia aplásica), pouca trombopoetina é metabolizada e os níveis plasmáticos de trombopoetina aumentam, levando à produção aumentada de megacariócitos e plaquetas. No contexto de trombocitose, há aumento da quebra de trombopoetina, diminuindo os níveis plasmáticos de trombopoetina e a produção de plaquetas.
A maioria dos ensaios de coagulação mede o tempo requerido para que o fibrinogênio do plasma forme faixas de fibrina. A formação de faixas pode ser detectada com dispositivos ópticos ou elétricos. O prolongamento pode representar um fator de concentração baixo, um ou mais fatores inativos, ou ainda a presença de inibidores.
O tempo de tromboplastina parcial (TTP), por vezes denominado tempo de tromboplastina parcial ativada (TTPa), testa o sistema de coagulação intrínseco. Uma superfície negativamente carregada (p. ex., caolin ou sílica), seguida de cefalina, é adicionada ao plasma para ativar os fatores XII e XI. O TTP é bastante sensível à ativação da heparina. É usado para monitorar e ajustar a terapia anticoagulante com heparina regular, mas não com heparinas de baixo peso molecular.
O tempo de protrombina (TP) é um teste do sistema extrínseco. O fator tecidual é adicionado ao plasma, levando à formação de fibrina normalmente em 9-12 segundos. Como uma ampla quantidade de fator tecidual é usada para iniciar a coagulação, o fator X é rapidamente ativado e desvia em grande parte a etapa de ativação do fator IX [ver Figura 4]. Assim, detecta as deficiências de fibrinogênio, fator II (protrombina), fator V, fator VII e fator X. Os resultados são relatados usando a razão normalizada internacional (INR). A INR é calculada usando a seguinte equação:
INR = (Log TPpaciente /Log TPcontrole)C
Onde C representa o índice de sensibilidade internacional (ISI). Neste sentido, a tromboplastina usada em um laboratório individual, com seu ISI específico, é calibrada contra um padrão de referência de tromboplastina, e um TP é relatado como uma INR.36 O TP é usado para monitorar a anticoagulação pela varfarina, sendo que uma INR de 2-3 é a faixa terapêutica padrão para varfarina. A presença de um anticoagulante lúpico pode interferir no TP.37
TTP e TP medem a atividade funcional geral da cascata de coagulação [ver Figura 3]. Assim, um TP prolongado com TTP normal indica deficiência de fator VII, enquanto um TTP prolongado com TP normal indica deficiência(s) envolvendo a fase de contato da cascata de coagulação (cininogênio de alto peso molecular, pré-calicreína, fator XII, fator XI, fator IX e fator VIII). O prolongamento de ambos, TP e TTP, indica a existência de anormalidades na via comum de coagulação, envolvendo fator X, fator V, protrombina, fibrinogênio ou polimerização da fibrina.
O veneno da víbora de Russell contém uma enzima que ativa o fator X; portanto o tempo do veneno diluído da víbora de Russell (DRVVT) mede a via comum da cascata de coagulação. É sensível à presença de um anticoagulante do tipo lúpico que inibe o complexo protrombinase fosfolipídio-dependente. Nestes casos, a adição de fosfolipídios (como lipídios hexagonais) encurtará significativamente (sem, no entanto, normalizar totalmente) o DRVVT prolongado.
O tempo de trombina (TT) é usado para testar anormalidades da conversão de fibrinogênio em fibrina. Pode estar prolongado em consequência de hipofibrinogenemia, fibrinogênio anormal (desfibrinogênio) ou presença de inibidores (p. ex., produtos de degradação da fibrina) que interferem na polimerização da fibrina. Os fatores clínicos comumente associados ao TT prolongado são a doença hepática grave, coagulação intravascular disseminada e terapia com heparina.
A reptilase é uma enzima análoga à trombina que converte fibrinogênio em fibrina. O tempo de reptilase (RT) é prolongado sob condições similares as que prolongam o TT, com apenas uma diferença significativa: a reptilase não é inibida pelo complexo AT-heparina. Desta forma, o RT não é prolongado pela heparina. Um tempo de trombina longo e um RT normal sugerem efeito da heparina.
O teste de atividade de antifator Xa é um ensaio cromogênico em que o fator Xa e substrato cromogênico específico do fator Xa são adicionados ao plasma. Em presença de heparina ou heparina de baixo peso molecular, AT é ativada e inibe a hidrólise do substrato cromogênico pelo fator Xa. Por comparação com uma curva padrão, a intensidade da atividade de antifator Xa é determinada e este valor fornece uma medida objetiva da atividade anticoagulante da heparina de baixo peso molecular. Uma atividade de antifator Xa igual a 0,5-1,2 unidades/mL em 4-6 horas após a última dose de heparina de baixo peso molecular é considerada terapêutica.
O teste de atividade de antifator Xa também pode ser usado para determinar os níveis plasmáticos de heparina quando a curva de calibração usada é baseada em concentrações conhecidas de heparina. Este teste é feito por um analisador automático e proporciona monitoramento mais preciso da atividade anticoagulante da heparina, em comparação ao TTPa. Os níveis terapêuticos de heparina geralmente estão na faixa de 0,3-0,7 unidades/mL.
Os níveis plasmáticos de fibrinogênio podem ser quantificados antigenicamente ou, de forma mais comum, por meio de ensaios de coagulação. Os resultados são relatados em miligramas por decilitro. O fibrinogênio é um reagente de fase aguda e seus níveis em geral aumentam nas doenças inflamatórias agudas. Desta forma, um nível de fibrinogênio relatado dentro da faixa normal em um paciente com condição inflamatória aguda, como a sepse, deve ser interpretado com cautela, pois pode sugerir um consumo substancial de fibrinogênio agudo.
Os produtos de degradação do fibrinogênio e os produtos de quebra de fibrina-fibrinogênio (PQFFs) resultam na degradação pela plasmina do fibrinogênio e do coágulo de fibrina [ver Figura 8]. O D-dímero é liberado pela degradação plasmina-mediada da fibrina totalmente polimerizada. A clivagem pela plasmina do fibrinogênio ou monômero solúvel de fibrina não rende D-dímero. Sendo assim, a concentração elevada de D-dímero é uma medida específica da deposição intravascular de fibrina e degradação de plasmina característica da coagulação intravascular disseminada. O teste de D-dímero é hoje um teste de laboratório padrão e substituiu o teste de PQFF.
O fator XIII é o único fator de coagulação cuja atividade não é avaliada por PT ou TTP, porque o ponto final destes dois testes é a formação de polímeros de fibrina, independentemente de estes polímeros estarem em ligação cruzada covalente por ação do fator XIII ativado. A suspeita de deficiência de fator XIII pode ser considerada em casos de bebês que apresentem sangramento significativo após a circuncisão ou, mais raramente, em casos de pacientes adultos com sangramento inexplicável. O fator XIII geralmente é ensaiado pelo teste de estabilidade de coágulo, em que o coágulo de fibrina derivado de plasma coagulado é colocado em solução de ureia a 5 M. O coágulo de fibrina sensibilizado pelo fator XIII ativado será resistente à dissolução. Considerando que níveis muito baixos de fator XIII (2-5%) são suficientes para promover estabilização normal do coágulo, este teste geralmente é adequado para detectar a deficiência de fator XIII clinicamente grave.
Não existe nenhum teste laboratorial eficiente para medir a atividade do sistema fibrinolítico. O tempo de lise do coágulo de euglobulina não é sensível nem específico. Durante a trombose e fibrinólise extensivas, tanto plasminogênio como a2-antiplasmina são consumidos. A quantificação direta dos níveis plasmáticos de plasminogênio e a2-antiplasmina por vezes é útil para avaliar a extensão da fibrinólise e a necessidade de reposição destas proteínas plasmáticas usando plasma fresco congelado.
Um tempo prolongado de coagulação (p. ex., TTPa de 60 segundos [normal = 28-30 segundos]) pode ser causado por deficiência de fator de coagulação ou por um inibidor. Um inibidor em geral é um anticorpo dirigido contra um fator de coagulação específico ou contra um complexo fosfolipídio-proteína, o conhecido anticoagulante lúpico [pesquise informação sobre distúrbios trombóticos no ACP Medicine]. Em um estudo de mistura, um volume de plasma de um paciente é misturado a um volume igual de plasma normal. A mistura resultante fornece pelo menos 50% de um fator deficiente e corrige a anormalidade. Se o problema for causado por um inibidor, a mistura de plasmas resultante ainda terá tempo de coagulação prolongado. Um ensaio de mistura deve ser feito sempre que um tempo de coagulação prolongado for detectado.
Bioensaios e imunoensaios são disponibilizados para avaliar a atividade de AT. Um ensaio funcional é preferível a um ensaio antigênico.
Existem métodos funcionais e imunológicos disponíveis pra avaliar as proteínas C e S. Como estas proteínas dependem da vitamina K, seus níveis estão diminuídos em pacientes que tomam varfarina. É melhor medir a proteína C ou a proteína S quando o paciente estiver sem tomar varfarina há 3-4 semanas.
Existem dois ensaios novos disponíveis: tromboelastografia (TEG) e ensaio de geração de trombina. No TEG, um pequeno volume de sangue total é colocado em uma cubeta de rotação lenta, que ativa a coagulação sanguínea. Uma haste sensora é inserida na amostra de sangue e diferentes parâmetros de coagulação sanguínea são medidos, incluindo o tempo que demora para detectar a formação do primeiro coágulo, a velocidade da formação de coágulo e a força do coágulo. Isto então fornece uma avaliação global do estado funcional da cascata de coagulação, plaquetas e sistema fibrinolítico.38 A geração de trombina é medida com auxílio de um trombinoscópio.39 Neste ensaio, que usa plasma ou plasma enriquecido com plaquetas, a coagulação é tipicamente iniciada com uma dose baixa de fator tecidual para similar a ativação fisiológica. A geração de trombina é quantificada pelo monitoramento contínuo da hidrólise de um substrato de trombina fluorescente. O ensaio relata cinco parâmetros de geração de trombina: o tempo que demora para fazer a primeira detecção de geração de trombina (tempo lag); a velocidade da geração de trombina; a concentração máxima de trombina gerada; a quantidade total de trombina gerada (trombina endógena em potencial) e a taxa de inibição de trombina por seus inibidores plasmáticos naturais. Ambos os ensaios fornecem muito mais informação do que os tempos de coagulação convencionais (p. ex., os tempos de coagulação geralmente estão correlacionados apenas com o tempo lag no processo de geração de trombina; assim o grosso da informação relacionada à geração de trombina não é medido). Em adição, novas gerações de dispositivos permitem que ambos os ensaios sejam realizados de modo automático, levando à maior reproducibilidade e menor variação entre os laboratórios. Atualmente, ambos os ensaios continuam sendo disponibilizados apenas em laboratórios clínicos especializados em coagulação, porém está previsto que virão a se tornar amplamente disponíveis em um futuro próximo. Ainda é preciso estabelecer se estes ensaios novos terão a sensibilidade e especificidade requeridas para terem utilidade diagnóstica superior, conforme o prometido.
A avaliação de esfregaço de sangue periférico fornece informação rápida e definitiva para confirmar ou questionar uma contagem plaquetária. Normalmente, existem 8-12 plaquetas por campo de maior aumento (aumento de 1.000×), correspondendo a uma contagem de plaquetas de 150.000-300.000/mL. O esfregaço também revela a granularidade plaquetária e se há megatrombócitos presentes.
Este ensaio mede primariamente a função plaquetária. Um dispositivo com mola é usado para fazer uma incisão padrão na pele do antebraço. Um tempo de sangramento prolongado em um paciente com contagem plaquetária acima de 100.000/mL sugere comprometimento da função plaquetária. É difícil padronizar o tempo de sangramento e um tempo de sangramento normal não prevê a segurança dos procedimentos cirúrgicos nem prediz precisamente uma hemorragia.40 Em consequência, este ensaio tem sido amplamente descartado.
O ensaio de função plaquetária-100 (PFA-100) é um teste automático para função de plaquetas. Sangue total tratado com citrato é aspirado através de um tubo capilar, sob forte tensão de cisalhamento, sobre uma membrana revestida com colágeno e adrenalina ou colágeno e ADP, na qual é criada uma abertura. O tempo que demora para o fluxo de sangue pela membrana parar é denotado como tempo de fechamento e constitui uma medida da função plaquetária. O tempo de fechamento é prolongado em pacientes com doença de Von Willebrand ou outros defeitos de função plaquetária.41 O PFA-100 geralmente é considerado o ensaio de primeira linha para os distúrbios de função plaquetária, embora sua sensibilidade e especificidade sejam pouco definidas.
Os agregômetros de plaqueta são dispositivos fotométricos para registro da transmissão de luz através de uma suspensão de plaquetas. Quando as plaquetas se agregam, a luz atravessa a suspensão mais rápido. Para testar a agregação, concentrações diluídas de agonistas de plaqueta (p. ex., ADP, adrenalina, colágeno e ristocetina) são adicionadas ao plasma rico em plaquetas tratadas com citrato. Com os agonistas fracos, como ADP e adrenalina, a onda primária de agregação é seguida por uma onda secundária. A onda secundária reflete a indução da reação de liberação das plaquetas, em que o conteúdo dos grânulos plaquetários é liberado para aumentar ainda mais a agregação plaquetária. Uma onda secundária subótima é vista quando há defeitos no pool de armazenamento de plaquetas, com o conteúdo dos grânulos diminuído ou com sua atividade de liberação comprometida. Este último defeito comumente está associado à ingesta de aspirina ou à trombocitopatia relacionada à uremia. Pacientes com doença de Von Willebrand apresentam resposta subótima de agregação plaquetária à ristocetina e, todavia, resposta normal a outros agonistas. O ensaio de agregação plaquetária é trabalhoso e caro, e deve ser realizado somente em laboratórios clínicos de coagulação, que fazem este teste regularmente. Embora não reproduza a ativação plaquetária fisiológica em um ambiente com alta tensão de cisalhamento nem preveja com segurança o sangramento cirúrgico, a agregometria plaquetária, quando combinada a uma história clínica detalhada, ainda tem utilidade diagnóstica na avaliação de pacientes com suspeita de defeito de função plaquetária.42
O autor não tem relações comerciais com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
1.Coughlin SR. Thrombin signaling and protease-activatedreceptors. Nature 2000; 4:258Ð64.
2.Kahn ML, Zheng YW, Huang W, et al. A dual thrombin receptor system for platelet activation. Nature 1998; 394:690.
3.Woulfe D, Yang J, Brass L. ADP and platelets: the end of the beginning (commentary). J Clin Invest 2001; 107:1503.
4.Clemetson KJ. Platelet GPIb-V-IX complex. Thromb Hae- most 1997; 78:266.
5.Sixma JJ, Zanten HV, Huizinga EG, et al. Platelet adhesion to collagen: an update. Thromb Haemost 1997; 78:434.
6.Banno A, Ginsberg MH. Integrin activation. Biochem Soc Trans 2008; 36:229.
7.Brass L. Understanding and evaluating platelet function. Hematology 2010; 2010:387Ð96.
8.Williams JC, Mackman N. Tissue factor in health and disease. Front Biosci 2012; 4:358.
9.Renne T, Pozgajova M, Gruner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med 2005; 202:271Ð81.
10.Rosen ED, Gailani D, Castellino FJ. FXI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost 2002; 87: 774Ð6.
11.Salomon O, Steinberg DM, Koren-Morag N, et al. Reduced incidence of ischemic stroke in patients with severe factor XI deÞciency. Blood 2008; 111:4113Ð7.
12.Lowenberg EC, Meijers JCM, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost 2010; 8:2349Ð57.
13.Ansell J, Hirsch J, Poller L, et al. The pharmacology and management of the vitamin K antagonists. The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004;126Ð204S.
14.Mann KG, Brummel-Ziedins K, Orfeo T, et al. Models of blood coagulation. Blood Cells Mol Dis 2006; 36:108.
15.Vandendries ER, Hamilton JR, Coughlin SR, et al. Par4 is required for platelet thrombus propagation but not Þbrin generation in a mouse model of thrombosis. Proc Natl Acad Sci U S A 2007; 104:288.
16.Adams TE, Huntington JA. Thrombin-cofactor interactions: structural insights into regulatory mechanisms. Arterioscler Thromb Vasc Biol 2006; 26:1738.
17.Esmon CT. The protein C pathway. Chest 2003; 124:265.
18.Gleeson EM, OÕDonnell JS, Preston RJ. The endothelial cell protein C receptor: cell surface conductor of cytoprotective coagulation factor signaling. Cell Mol Life Sci 2011 Oct 4. [Epub ahead of print]
19.Morten S, Bendz B. Tissue factor pathway inhibitor: clinical deÞciency states. Thromb Haemost 1997;78:467.
20.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endo- peroxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem 1996; 271:33157.
21.McAdam BF, Mardini IA, Kapoor S, et al. Systemic biosyn- thesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A 1999; 96:272.
22.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 1993; 329:2002.
23.Marcus AJ, Broekman JM, Drosopoulos J, et al. Thrombo- regulation by endothelial cells (brief reviews). Arterioscler Thromb Vasc Biol 2001; 21:178.
24.Van Meijer M, Pannekoek H. Structure of plasminogen activator inhibitor 1 (PAI-1) and its function in Þbrinolysis: an update. Fibrinolysis 1995; 9:263.
25.Fay WP, Parker AC, Condrey LR, et al. Human plasmino- gen activator inhibitor-1 (PAI-1) deÞciency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood 1997; 90:204.
26.Bajzar L, Morser J, Nesheim M. TAFI, or plasma procar- boxypeptidase B, couples coagulation and Þbrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem 1996; 271:16603.
27.Broze GJ, Higuchi DA. Coagulation-dependent inhibition of Þbrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood 1996;88: 3815.
28.de Bruijne EL, Murad SD, de Maat MP, et al. Genetic variation in thrombin-activatable Þbrinolysis inhibitor (TAFI) is associated with the risk of splanchnic vein thrombosis. Thromb Haemost 2007; 97:181.
29.Martini CH, Brandts A, de Bruijne EL, et al. The effect of genetic variants in the thrombin activatable Þbrinolysis inhibitor (TAFI) gene on TAFI-antigen levels, clot lysis time and the risk of venous thrombosis. Br J Haematol 2006; 134:92.
30.Nishimura T, Myles T, Piloponsky A, et al. Thrombin- activatable procarboxypeptidase B regulates complement C5a in vivo. Blood 2007; 109:1992Ð7.
31.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998; 93:741.
32.Atkins GB, Jain MK, Hamik A. Endothelial differentiation: molecular mechanism of speciÞcation and heterogeneity. Arterioscler Thromb Vasc Biol 2011; 31:1476.
33.de Graf CA, Metcalf D. Thrombopoietin and hematopoietic stem cells. Cell Cycle 2011; 10:1582.
34.Junt T, Schulze H, Chen Z, et al. Dynamic visualization of thrombopoiesis within the bone marrow. Science 2007; 317:1767.
35.Kuter DJ, Rosenberg RD. The reciprocal relationship of thrombopoietin (c-Mpl ligand) to changes in the platelet mass during busulfan-induced thrombocytopenia in the rabbit. Blood 1995; 85:2720.
36.Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1998; 114 Suppl:445S.
37.Moll S, Ortel TL. Monitoring warfarin therapy in patients with lupus anticoagulants. Ann Intern Med 1997; 127:177.
38.Whitten CW, Greilich PE. Thromboelastography: past, present, and future. Anesthesiology 2000; 92:1223Ð5.
39.Hemker HC, Dieri RA, De Smedt E, Beguin S. Thrombin generation, a function of the haemostatic-thrombotic system. Thromb Haemost 2006; 96:553Ð61.
40.The bleeding time [editorial]. Lancet 1991; 337:1447.
41.Fressinaud E, Veyradier A, Truchaud F, et al. Screening for von Willebrand disease with a new analyzer using high shear stress: a study of 60 cases. Blood 1998; 91:1325.
42.Hayward CP, Pai M, Liu Y, et al. Diagnostic utility of light transmission platelet aggregometry: results from a prospec- tive study of individuals referred from bleeding disorder assessments. J Thromb Haemost 2009; 7:676Ð84.
Figuras 1 e 4-9 – Seward Hung
Figura 2 – Christine Kenney
Figuras 3 e 10 – Marcia Kammerer
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.