(Carregando Índice)... (Carregando Índice)... |
Última revisão: 26/03/2015
Comentários de assinantes: 0
Alexandra Villa-Forte - MD, MPH - não possui relações comerciais com os fabricantes de produtos e/ou prestadores de serviços mencionados neste capítulo.
Brian F. Mandell - MD, PhD, MACP - recebe auxílio educacional da Abbott Laboratories, Aventis Pharmaceuticals, Inc. e TAP Pharmaceutical Products, Inc.; também recebe auxílio educacional e atuado como conselheiro ou consultor junto às empresas TAP, Sanofi-Aventis, Novartis, Pfizer e Savient Pharmaceuticals, Inc. (nada relacionado à terapia para vasculite).
Artigo original: Villa-Forte A, Mandell BF. Systemic vasculitis syndromes. ACP Medicine. 2012.
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.]
Agradecimentos: Figuras 1 e 2 – Seward Hung e Figura 8 – Gary S. Hoffman.
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Lucas Zambon.
Citoxano, corticosteroides, metotrexato, azatioprina, pentoxifilina, colchicina, aspirina e dapsona não têm aprovação do Food and Drug Administration para os usos descritos neste capítulo.
A vasculite é definida por evidência histológica de inflamação envolvendo os vasos sanguíneos. Pode ser classificada como primária, cuja causa é desconhecida, ou secundária, quando está associada a outras doenças ou à exposição a agentes deflagradores comprovados, como medicação, doença neoplásica ou agente infeccioso. O diagnóstico de um distúrbio vasculítico primário específico depende do padrão de envolvimento orgânico, da histopatologia, do tamanho dos vasos sanguíneos afetados e da exclusão de doenças que possam causar vasculite secundária. O diagnóstico deve ser estabelecido unicamente com base em exames laboratoriais (p. ex., achados de níveis séricos de anticorpos anticitoplasma de neutrófilo [ANCAs] ou crioglobulinas).
Os principais determinantes do prognóstico e da terapia apropriada incluem o distúrbio vasculítico específico, a gravidade e extensão do envolvimento de órgãos essenciais, a taxa de progressão da doença e a etiologia, quando identificável. O processo inflamatório, muitas vezes, está associado a sintomas inespecíficos e a anormalidades laboratoriais (p. ex., velocidade de hemossedimentação elevada, anemia e febres) que não distinguem as doenças vasculíticas de outras condições inflamatórias, infecciosas ou neoplásicas. A natureza tóxica das terapias para vasculite sistêmica determina a necessidade de um diagnóstico rápido.
Os exames invasivos, como a biópsia e a arteriografia, frequentemente, são necessários para a avaliação diagnóstica de pacientes com suspeita de vasculite sistêmica; contudo, a biópsia de tecido sem envolvimento clínico e o uso de exames menos específicos devem ser evitados.
Apesar de aparentemente evidente, as primeiras etapas do diagnóstico de uma possível síndrome vasculítica consistem em obter uma história detalhada do paciente e realizar um exame físico enfocado, com o intuito de documentar envolvimento de órgãos específicos. É preciso dar atenção especial à pele, aos olhos, às orelhas, às vias aéreas superiores, às articulações, aos linfonodos, aos nervos periféricos e aos grandes vasos. Os exames laboratoriais [ver Tabela 1] devem ser incluídos de modo seletivo na avaliação inicial. Exames especializados, inclusive as sorologias, devem ser obtidos e interpretados somente após a formulação de um diagnóstico diferencial. A glomerulonefrite costuma ser assintomática. Um teste de fita com urina que revele sangue, leucócitos ou proteína deve levar prontamente o médico a examinar sedimentos de urina fresca (Urina tipo 1). A urina que não é examinada dentro de algumas horas perde a utilidade para identificação de cilindros celulares, que degeneram rapidamente ex vivo. A presença de cilindros hemáticos é praticamente diagnóstica de glomerulonefrite e os cilindros leucocitários, também, podem ser encontrados na glomerulonefrite. Com base no padrão de envolvimento orgânico, um diagnóstico diferencial que inclua tipos específicos de vasculite sistêmica e outros distúrbios podem, então, ser gerados, levando a testes dirigidos adicionais.
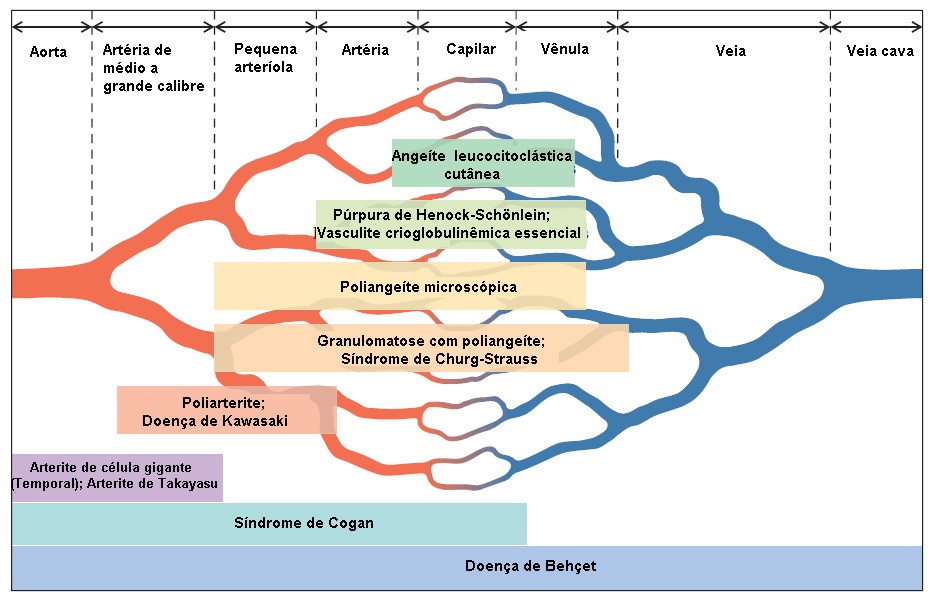
Vários esquemas de classificação têm sido propostos para a organização dos distúrbios vasculíticos sistêmicos em um paradigma consistente. As classificações são úteis para distinguir distúrbios clínicos que diferem em termos de prognóstico e resposta ao tratamento.1 Entretanto, nenhum esquema é universalmente aceito. Todos reiteram as características de doença clássica, enfatizando a especificidade do diagnóstico. Se a aderência a um esquema de classificação fosse estrita, um paciente que acabasse de adoecer e não manifestasse plenamente a condição tenderia a ficar sem receber um diagnóstico definitivo. Até as etiologias específicas das síndrome vasculíticas serem definidas, estas entidades diagnósticas permanecerão teóricas e a sobreposição entre as doenças não será incomum. Isto não deve ser impedimento à instituição da terapia para pacientes com risco de dano orgânico de progressão rápida. Mesmo assim, os sistemas de classificação fornecem constructos úteis para a comunicação, para a determinação da terapia inicial e para a inclusão de pacientes em protocolos de pesquisa [ver Figura 1]. Os esquemas de classificação mais amplamente usados são baseados no calibre dos vasos afetados, padrão de envolvimento de órgão, presença/ausência de inflamação granulomatosa e deposição significativa de imunocomplexos. Alguns autores propuseram a categoria de vasculite associada à ANCA com base na presença/ausência de ANCAs específicos no soro, em particular anticorpos contra proteinase 3 e mieloperoxidase. Atualmente, o papel apropriado para o teste de ANCA é dar suporte a um diagnóstico clínico desenvolvido de modo lógico. Em casos de pacientes que não se ajustam extremamente bem em um dado fenótipo clínico bem definido, estes testes sorológicos não devem suplantar as tentativas de obter diagnóstico tecidual. A presença de ANCA é insuficiente para estabelecer um diagnóstico de síndrome vasculítica primária. O teste de ANCA não deve ser usado para triagem de “vasculite.”
Quando os sintomas dominantes e achados (p. ex., neuropatia ou vasculite cutânea) não sugerem um único distúrbio vasculítico específico, os testes sorológicos podem ser úteis. A confirmação histológica é mais valiosa para estabelecer um diagnóstico. Não há valor reconhecível na realização indiscriminada de testes para anticorpos antinúcleo (FAN), ANCAs, fator reumatoide e enzima conversora de angiotensina. Em contraste, a infecção por hepatite B ou C pode estar associada a uma ampla gama de síndromes vasculíticas (secundárias). Estas infecções devem ser consideradas em casos de pacientes com vasculite envolvendo vasos pequenos e/ou médios.2
As vasculites sistêmicas são potencialmente fatais e podem requerer terapia anti-inflamatória e imunossupressora potente. Os diagnósticos devem ser estabelecidos com o máximo de certeza possível. Entretanto, as questões relacionadas com diagnósticos alternativos ou doenças coexistentes surgem com frequência. Até mesmo após o início da terapia, os médicos devem manter um alto grau de vigilância para detecção de problemas médicos não relacionados, complicações da terapia ou ambos. Os sinais e os sintomas de infecção não identificados podem ser confundidos com recaídas da síndrome vasculítica subjacente e é possível eliminá-los temporariamente à base de terapia com esteroides.3 Com a iniciação de uma terapia imunossupressora potente, há aumento da suscetibilidade a infecções oportunistas. Os maiores riscos ocorrem em pacientes com neutropenia acentuada ou naqueles sob tratamento com doses altas de corticosteroides. Os médicos devem ser, particularmente, cautelosos com relação à atribuição de novos problemas a “exacerbações” da doença subjacente, sem antes excluir a hipótese de infecção nova ou recrudescente. Os pacientes infectados pelo vírus varicela-zoster podem apresentar febre e dor antes do aparecimento das vesículas. Infecções por Pneumocystis jiroveci, citomegalovírus, infecções fúngicas sistêmicas e reativação de doença micobacteriana são observadas com maior frequência em pacientes submetidos à imunossupressão. A imunossupressão com esteroides e outras medicações frequentemente está associada à candidíase mucosa e, menos comumente, ao molusco contagioso e ao sarcoma de Kaposi.
|
Tabela 1 Exames laboratoriais selecionados para pacientes com doença multissistêmica e possível vasculite | |
|
Teste |
Comentários |
|
Contagem de plaquetas |
A trombocitose pode ocorrer em paralelo com a resposta de fase aguda. A trombocitopenia não é esperada em síndromes vasculíticas primárias; considerar LES, infiltração medular, leucemia de células pilosas ou outras malignidades com vasculite secundária, PTT, CIVD, hiperesplenismo, SAAF, HIV, crise renal do escleroderma e trombocitopenia induzida por heparina. |
|
Contagem de leucócitos |
A leucopenia não é esperada na vasculite primária; considerar LES, leucemias, hiperesplenismo, sepse, mielodisplasia e HIV. A eosinofilia é comum na síndrome de Churg-Strauss; pode ocorrer na GPA, artrite reumatoide, crise renal do escleroderma e embolia por colesterol. |
|
VHS |
Uma VHS relativamente baixa é observada na CIVD, insuficiência hepática e hiperviscosidade; a VHS frequentemente é normal na PHS, pode ser normal em 50% dos casos de arterite de Takayasu, e é normal em 20% dos casos de arterite de células gigantes. |
|
Aminotransferases |
Níveis de ALT ou AST elevados na doença hepática, miosite, rabdomiólise, hemólise ou necrose do miocárdio. |
|
Antimembrana basal glomerular |
Útil para avaliação de hemorragia alveolar, com ou sem glomerulonefrite; igualmente útil para avaliação de glomerulonefrite normocomplementêmica. Pode estar associada com um prognóstico ruim, diante da positividade para ANCA e anti-MBG na vasculite que se manifesta como doença grave. |
|
Anticorpo antinúcleo (FAN) |
Solicitar o teste quando houver suspeita clínica de LES (ou casos inexplicáveis de serosite, glomerulonefrite, hemorragia alveolar, leucopenia ou trombocitopenia) e não como teste de triagem geral para pacientes doentes. O resultado negativo torna a hipótese de LES improvável. |
|
ANCA |
Solicitar quando houver suspeita clínica de GPA ou PAM ou vasculite fármaco-induzida; o teste deve ser realizado por IF e ELISA. |
|
Triagem farmacológica |
Solicitar em caso de sintomas inexplicáveis do SNC, isquemia do miocárdio, espasmo vascular, ataques de pânico com achados sistêmicos, taquicardia, perfuração nasal. A triagem de urina deve ser feita. |
|
Hemoculturas |
Útil para qualquer paciente com doença febril, multissistêmica ou debilitante; infiltrados pulmonares; isquemia focal/infarto ou para aqueles que tenham sido previamente submetidos a procedimentos vasculares ou endovasculares. |
|
AAF/TTPA/TVVR |
Solicitar em casos inexplicáveis de trombose venosa ou arterial, ou trombocitopenia. |
|
Teste cutâneo com derivado proteico purificado (Panergy) |
Realizar em qualquer paciente que possa requerer terapia com esteroide ou com piúria estéril inexplicável, ou hematúria, inflamação granulomatosa, meningite crônica ou possível exposição à tuberculose. |
|
Exame de sedimento urinário fresco |
Realizar em todos os pacientes com febre inexplicável, hipertensivos ou com doença multissistêmica, ou ainda com níveis inexplicavelmente elevados de creatinina. |
|
Testes de sorologia viral: hepatite B e C, e possivelmente CMV e HIV |
Solicitar para avaliação de transaminases anormais ou fosfatase alcalina hepática elevada; hipertensão porta; PAN ou síndrome PAM; ou crioglobulinemia inexplicável, poliartrite ou vasculite cutânea. |
|
Complemento C3, C4 |
Não é um teste de triagem para vasculite; é útil no diagnóstico diferencial de glomerulonefrite. Níveis baixos na crioglobulinemia e no LES podem estar baixos na endocardite, glomerulonefrite relacionada à hepatite ou vasculite. |
|
Aldolase |
A aldolase não é órgão-específico; sua distribuição nos órgãos é similar a da lactato desidrogenase. |
ALT = alanina aminotransferase; ANCA = anticorpo anticitoplasma de neutrófilo; AAF = anticorpo antifosfolipídio; SAAF = síndrome do anticorpo antifosfolipídio; AST = aspartato aminotransferase; CMV = citomegalovírus; SNC = sistema nervoso central; CIVD = coagulação intravascular disseminada; ELISA = ensaio imunossorvente ligado à enzima; VHS = velocidade de hemossedimentação; MBG = membrana basal glomerular; GPA = granulomatose com poliangeíte; PHS = púrpura de Henoch-Schönlein; IF = imunofluorescência; PAM = poliangeíte microscópica; PAN = poliartrite nodosa; PR3 = proteinase 3; TTPA = tempo de tromboplastina parcial; TVVR = teste do veneno da víbora de Russell; LES = lúpus eritematoso sistêmico; PTT = púrpura trombocitopênica trombótica.
Metotrexato, azatioprina e ciclofosfamida podem causar leucopenia e, menos comumente, outras citopenias. Em pacientes com função renal diminuída, o metotrexato deve ser administrado com cautela, se usado. A dose de ciclofosfamida deve ser diminuída e cuidadosamente monitorada, uma vez que o medicamento é excretado pelos rins. A disfunção do esvaziamento da bexiga é uma contraindicação relativamente forte ao uso prolongado de ciclofosfamida, pois a exposição aumentada aos metabólitos tóxicos do fármaco predispõe à cistite hemorrágica e/ou câncer de bexiga. A abordagem padrão para tratamento de pacientes com certas síndrome vasculíticas sistêmicas potencialmente fatais tem sido a introdução da terapia com um curso de dose alta de corticosteroides aliado a um segundo agente imunossupressor para indução de remissão e, em seguida, dependendo da doença, afunilamento gradual dos corticosteroides e manutenção da terapia imunossupressora com o agente imunossupressor efetivo mais seguro, para manter a remissão. O segundo agente, que não é um corticosteroide, pode ser a ciclofosfamida, que parece ser o mais potente dos imunossupressores não corticosteroides. Após 3-6 meses, a ciclofosfamida é, então, substituída por um agente com perfil de segurança melhor (p. ex., metotrexato ou azatioprina). Este tipo de abordagem tem sido melhor avaliada em pacientes com granulomatose com poliangeíte (GPA; antigamente conhecida como granulomatose de Wegener), nos quais tem se mostrado efetiva. Recentemente, o rituximabe tem emergido como alternativa à ciclofosfamida para pacientes com GPA ou com poliangeíte microscópica (PAM).

Figura 1 - Classificação das síndromes de vasculite sistêmica.1
A vasculite que afeta primariamente os capilares, as vênulas e as arteríolas não musculares é o padrão mais comum de vasculite e quase sempre envolve a pele. Pode ocorrer em qualquer idade e afeta indivíduos de ambos os sexos com igual frequência.
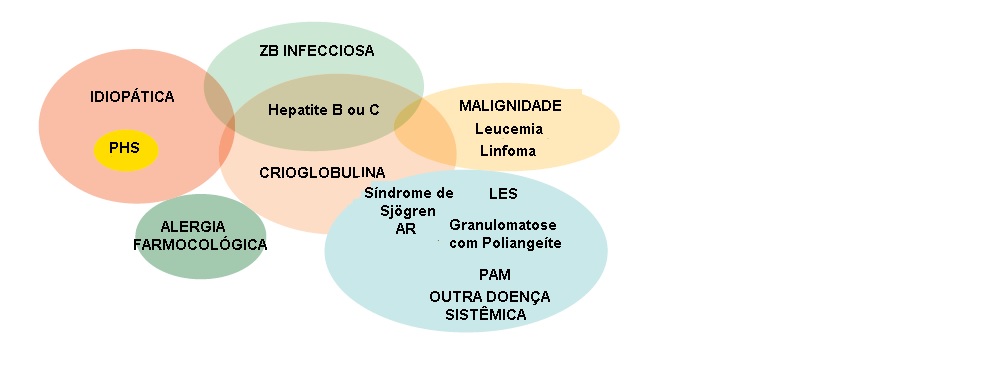
A vasculite de pequenos vasos pode ocorrer como distúrbio idiopático (primário), mas frequentemente é secundária a alergias farmacológicas, a infecções (endocardite bacteriana, a infecções virais como as causadas pelos vírus da hepatite B ou C, a infecção por Neisseria disseminada ou riquetsiose), aos distúrbios autoimunes e, menos frequentemente, às malignidades. Pode ser parte de um distúrbio autoimune sistêmico definido, como a síndrome de Sjögren, lúpus eritematoso sistêmico (LES) ou artrite reumatoide, ou pode ocorrer associada a malignidades hematológicas, linfoides e de órgão sólido [ver Figura 2].
O envolvimento cutâneo ocorre em muitas das síndromes vasculíticas primárias e secundárias. A obstrução ou estenose de vasos dérmicos profundos pode causar livedo, fenômeno de Raynaud ou necrose. Estas lesões cutâneas, também, podem ser causadas por vasculite envolvendo vasos de médio ou grande calibre, sendo que a identificação do tamanho do vaso muitas vezes é impossível. As lesões de púrpura palpáveis que descoloram parcialmente sob compressão são as manifestações mais comuns de vasculite de pequenos vasos. A vasculite de pequenos vasos, em particular quando associada a infecções, é tipicamente caracterizada pela deposição de imunocomplexos. Na literatura antiga, a vasculite com envolvimento primário das vênulas pós-capilares era denominada vasculite por hipersensibilidade.4 A vasculite primária de pequenos vasos pode ser limitada à pele ou estar associada ao envolvimento visceral, causando hemorragia alveolar, isquemia ou hemorragia intestinal e glomerulonefrite.
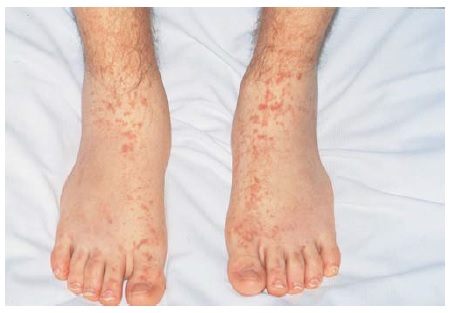
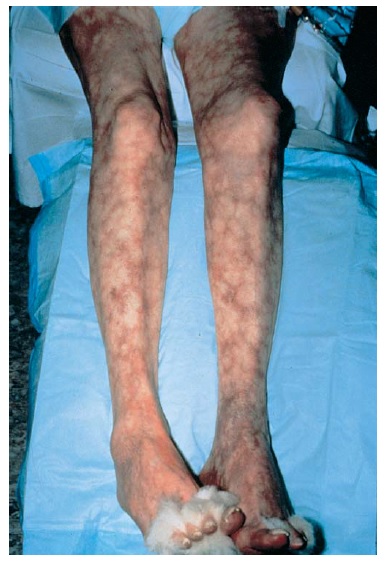
A púrpura tende a ocorrer em grupos de lesões de tempo de surgimento semelhante e é mais pronunciada em áreas dependentes de gravidade [ver Figura 3]. Quando a púrpura não está localizada primariamente em áreas dependentes de gravidade, podem ser consideradas as hipóteses de doença da aglutinina fria, crioglobulinemia (que pode estar associada a infecções como hepatite C ou ao linfoma), meningococemia, embolia, doenças infiltrantes e lesão autoinfligida. A vasculite cutânea, seja qual for a etiologia, pode estar associada a um edema marcante e dependente.
Figura 2 - Diagrama de Venn ilustrando as relações existentes entre as causas de vasculite de pequenos vasos (“hipersensibilidade”). EB = endocardite bacteriana; PHS = Púrpura de Henoch-Schönlein; PAM = poliangeíte microscópica; AR = artrite reumatoide; LES = lúpus eritematoso sistêmico.
Em uma série de casos de vasculite cutânea de pequenos vasos, cerca de 40% dos 172 pacientes com mais de 20 anos tinham um distúrbio sistêmico associado ou subjacente.4 A vasculite necrotizante sistêmica estava presente em 17 adultos; 4 tinham malignidade; 4 apresentavam infecção bacteriana causadora de vasculite; 11 estavam com crioglobulinemia e 59 tinham púrpura de Henoch-Schönlein. A prevalência da infecção pelo vírus da hepatite C, provavelmente a causa mais comum de crioglobulinemia mista,2 não foi relatada nesta série. Em adultos de idade mais avançada com púrpura recém-manifesta, a hipótese de malignidade (em especial, a doença mieloproliferativa e a linfoproliferativa) deve ser considerada.
A biópsia é mais útil para excluir as causas de púrpura não vasculítica, como amiloidose, leucemia cutânea, sarcoma de Kaposi, linfomas de célula T, traumatismo e êmbolos de colesterol ou mixomatosos. A imunofluorescência de tecidos é uma coloração útil para dar suporte ao diagnóstico de púrpura de Henoch-Schönlein (especificamente, a coloração para IgA), LES ou infecção (o percentual de pacientes com resultados positivos de imunofluorescência é desconhecido). As células que infiltram a parede vascular podem ser neutrófilos ou linfócitos, dependendo da doença subjacente. A patologia, na maioria dos casos de vasculite de pequenos vasos, é a angeíte leucocitoclástica. A hipótese de infecção pelo vírus da hepatite C deve ser excluída de modo rotineiro em casos de pacientes com púrpura inexplicável – um exemplo significativo do fato de a presença de angeíte leucocitoclástica não ser indicativa de que a doença de um paciente resulta de síndrome vasculítica primária. A histopatologia e presença (ou ausência) de imunoglobulina detectada depende da idade da lesão de origem da biópsia.
A angeíte leucocitoclástica cutânea é limitada à pele e, frequentemente, está associada a um fator precipitante, como as medicações. As lesões têm aproximadamente a mesma idade, uma vez que tendem a aparecer ao mesmo tempo. Pode haver artralgias, contudo sem envolvimento sistêmico. A causa nem sempre é identificada e as tentativas iniciais de tratamento incluem fármacos anti-inflamatórios não esteroidais, colchicina ou dapsona. Os casos refratários ou recidivantes podem requerer uso de corticosteroide. Outras medicações imunossupressoras, como azatioprina ou metotrexato, têm sido usadas em casos refratários ou recorrentes, com resultados variáveis.
A púrpura de Henoch-Schönlein é uma síndrome vasculítica de pequenos vasos clinicamente definida, caracterizada por quatro manifestações clínicas principais: púrpura palpável, artrite, doença gastrintestinal e glomerulonefrite. Os achados cutâneos usualmente são marcantes. A púrpura de Henoch-Schönlein, que é menos frequente em adultos do que em crianças, usualmente está associada com a deposição vascular e com a renal de imunocomplexos contendo IgA.5 Entre as manifestações comuns da púrpura de Henoch-Schönlein, estão a púrpura palpável; dor abdominal, vômitos, sangramento gastrintestinal ou intussuscepção (principalmente em crianças); artralgias ou artrite; hematúria; e proteinúria. Os sintomas viscerais raramente precedem as lesões cutâneas. A púrpura de Henoch-Schönlein, muitas vezes, parece ser precipitada por medicações ou infecções estreptocócicas ou virais. Trata-se de um distúrbio usualmente autolimitado, porém com casos raros (mais comumente em adultos) de progressão da glomerulonefrite associada para insuficiência renal. Na ausência de disfunção renal, a púrpura de Henoch-Schönlein frequentemente é uma síndrome autolimitada que pode requerer apenas terapia sintomática. A recorrência pode ser observada em até 1/3 dos pacientes. Como o envolvimento visceral pode ser significativo, o paciente deve ser monitorado periodicamente, até a resolução completa dos sintomas.
Figura 3 - A púrpura palpável da região distal dos membros é a manifestação mais comum da vasculite de pequenos vasos.
A vasculite urticariforme representa um subgrupo peculiar de vasculite de pequenos vasos.6 A manifestação clínica é a de pápulas ou pápulas sinuosas, por vezes com angioedema circundante ou geograficamente isolado. As lesões individuais apresentam resolução mais lenta do que a urticária clássica, muitas vezes com duração de vários dias. A doença segue um curso mais prolongado, em comparação à urticária típica. Com frequência, há uma sensação de desconforto disestésico com ardência decorrente das lesões. Assim como a púrpura, as lesões da vasculite urticariforme frequentemente estão localizadas em áreas dependentes de gravidade e, muitas vezes, cicatrizam com aparecimento de uma área de pele hiperpigmentada ou equimótica. A maioria dos casos é idiopática, embora tenha sido descrita uma associação com distúrbio autoimune sistêmico subjacente, como LES, paraproteinemia de IgM ou infecção viral. Em casos raros, a vasculite urticariforme tem sido associada a uma síndrome rara que inclui hipocomplementemia, doença pulmonar obstrutiva e glomerulonefrite (síndrome da vasculite urticariforme hipocomplementêmica). Esta síndrome é distinta do angioedema associado à deficiência de C1 esterase, que não causa urticária, glomerulonefrite nem neuropatia.
A terapia para vasculite cutânea é voltada, primeiramente, para reconhecer e eliminar qualquer precipitante subjacente. As etiologias infecciosas devem ser procuradas e tratadas. Os fármacos potencialmente agressores devem ser suspendidos. A associação com mielodisplasia e doença mieloproliferativa deve ser considerada, especialmente quando houver evidência de citopenias ou formas celulares anormais em esfregaços de sangue periférico. Na ausência de precipitantes evidentes, é possível tentar uma terapia de baixo risco com anti-inflamatórios não esteroides, colchicina, dapsona ou um curso de curta duração com dose baixa de corticosteroide (< 0,3 mg/kg/dia, para começar). Estas terapias não são uniformemente efetivas para diminuir a frequência ou gravidade dos ataques. A terapia prolongada com corticosteroide deve ser evitada, sempre que possível. O suporte com meias de compressão pode ser útil para limitar o edema significativo que, muitas vezes, acompanha a vasculite cutânea nas pernas.
O envolvimento visceral com disfunção de órgão pode requerer uma abordagem mais agressiva do que aquela usada na vasculite cutânea limitada. Doses moderadas de corticosteroides (0,5 mg/kg/dia) geralmente são efetivas. No contexto das potenciais complicações do uso crônico de corticosteroides ou no cenário de envolvimento visceral grave, o uso de metotrexato, azatioprina, ciclofosfamida ou outros agentes imunossupressores, ocasionalmente, pode ser necessário [ver Tabela 2]. A plasmaférese pode ser efetiva no tratamento da vasculite crioglobulinêmica. Ao tratar a doença de pequenos vasos crônica e refratária, na ausência de prejuízo para órgãos ou de potencial fatal, é preciso prestar bastante atenção à razão risco:benefício das terapias escolhidas.
A GPA é doença potencialmente fatal e relativamente incomum, caracterizada por inflamação granulomatosa necrotizante e vasculite de vasos de pequeno e médio calibre.7,8 Indivíduos de ambos os sexos e de todas as idades podem ser afetados.
A GPA é caracterizada por necrose parenquimal com um componente de vasculite variável. Há envolvimento de múltiplos órgãos, com predileção pelos tratos respiratórios superior e inferior, bem como pelos rins.
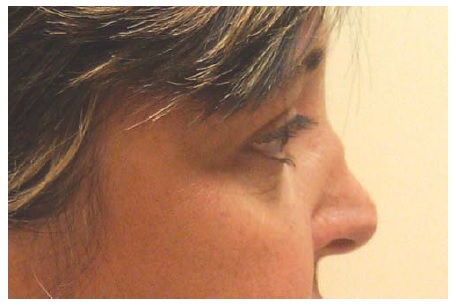
Envolvimento do trato respiratório superior. A doença do trato respiratório superior pode ser marcante, mas frequentemente é indolente e atribuída a meses ou anos de doença sinusal rotineira, até outras manifestações de GPA serem reconhecidas. Em cerca de 70% dos pacientes, há envolvimento da orelha, nariz e garganta no início da doença. Mesmo após o estabelecimento do diagnóstico e fornecimento de terapia imunossupressora, a doença sinusal pode ser recalcitrante ao tratamento. Esta cronicidade pode ser causada em parte por infecções recorrentes e/ou tecido danificado. O dano anatômico pode incluir perfurações de septo e deformação do nariz em sela [ver Figura 4]. O envolvimento laringotraqueal pode resultar em estenose subglótica, que é melhor tratada com injeção local de corticosteroide aliada à terapia de dilatação. O envolvimento da orelha é comum, em particular a otite média, que pode produzir perda da condução auditiva.
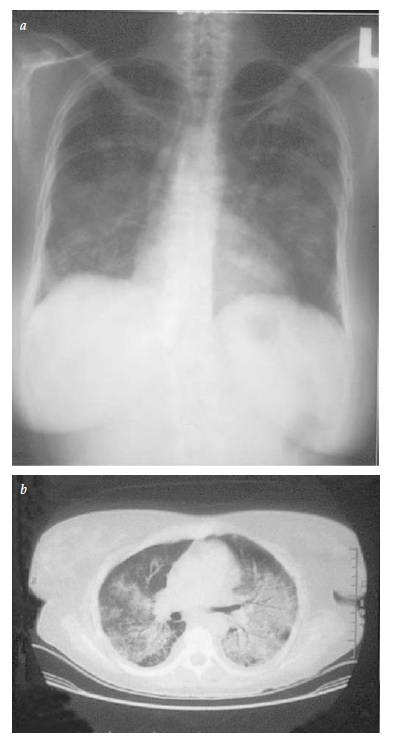
Envolvimento do trato respiratório inferior. É possível que não haja envolvimento pulmonar no início da doença, que esta seja assintomática ou que se manifeste como uma hemorragia alveolar drasticamente difusa. Um terço das lesões pulmonares observadas nos exames de imagem [ver Figura 5] são assintomáticas (a tomografia computadorizada é mais sensível do que a radiografia). Os nódulos podem sofrer necrose, levando à formação de cavidade. O broncoespasmo não é característico da GPA. Se houver suspeita de obstrução de vias respiratórias, o uso de broncoscopia deve ser considerado para exclusão da hipótese de estenose endobrônquica ou subglótica. Com frequência, é necessário excluir as causas infecciosas de infiltrados pulmonares e a broncoscopia com lavado é útil para esta finalidade. Entretanto, o tecido obtido por biópsia transbrônquica, em geral, é quantitativamente insuficiente para confirmar o diagnóstico patológico de GPA.
A biópsia pulmonar aberta ou toracoscópica costuma ser o melhor método para demonstrar os achados patológicos típicos da GPA, bem como para excluir malignidades e infecções atípicas. Os cortes típicos de biópsia aberta do pulmão9 podem conter áreas de necrose, frequentemente em padrão geográfico, inflamação granulomatosa e vasculite. É possível que nem todos os achados histopatológicos estejam presentes no mesmo corte de biópsia, e a vasculite pode não ser evidente. Dada a possibilidade de demonstrar patologias similares à GPA em infecções micobacterianas e fúngicas crônicas, a realização de colorações especiais e culturas para estes agentes é obrigatória.
|
Tabela 2 Terapias imunossupressoras para vasculite: comentários práticos | |||
|
Fármaco |
Dose |
Classificação da eficácia |
Comentários |
|
Corticosteroide |
No início, usado com frequência na dose de 1 mg/kg diária (doses partidas na doença grave); realizar desmame, com meta de parar em 9–12 meses ou, se possível, antes. A princípio, pode ser usado como doses pulsadas de 1 g/dia durante 3 dias, na doença grave. |
Terapia primária para todas as formas de vasculite prejudiciais a órgãos ou potencialmente fatais; é provavelmente a terapia de ação mais rápida. |
De modo ideal, checar o status basal de tuberculose latente; considerar a profilaxia contra Pneumocystis (ao usar doses altas) e osteoporose; monitorar o desenvolvimento de glaucoma em pacientes idosos. Doses altas podem aumentar a pressão arterial e a glicemia. |
|
Ciclofosfamida |
2 mg/kg VO, diariamente; evitar neutropenia; o nadir, geralmente, ocorre em 9–14 dias após a iniciação da terapia ou mudança de dose; diminuir a dose no contexto de insuficiência renal. A dosagem em “pulsos” mensal tem sido usada (0,5–1 g/m2), entretanto com uma probabilidade maior de neutropenia. A dose em pulso deve ser administrada após a diálise. |
Terapia imunossupressora não hormonal mais potente; início da ação indefinido; deve ser administrada quando do reconhecimento de doença grave, particularmente de glomerulonefrite de progressão rápida. |
Os principais efeitos colaterais limitam seu uso prolongado: leucopenia, doença mieloproliferativa, dano à bexiga e malignidade. A meta do uso corrente é induzir remissão, tanto na GPA como em outras formas graves de vasculite, com uso concomitante de prednisona, seguido de afunilamento desta com substituição por ciclofosfamida para obtenção de uma medicação menos tóxica após 3–6 meses (p. ex., azatioprina ou metotrexato). A gravidez deve ser evitada. |
|
Azatioprina |
2–3 mg/kg VO, diariamente. |
Útil para manter a remissão durante o afunilamento de corticosteroide. |
Não fornecida como terapia de indução primária; evitar leucopenia; pode causar uma reação de hipersensibilidade confusa, que inclui febre alta, com ou sem erupção, e eosinofilia. Evitar a gravidez. |
|
Metotrexato |
Administrado semanalmente (até cerca de 0,3 mg/kg/dose) com ácido fólico (1 mg) diário. |
É menos potente do que a ciclofosfamida para tratar doença grave; útil para manter a remissão durante o afunilamento da dosagem de corticosteroide; diminuir a dose para insuficiência renal leve; evitar em pacientes com creatinina > 2,0 mg/dL. |
Útil para manter a remissão; pode ser usado como terapia de indução primária com prednisona no tratamento de pacientes com GPA leve a moderada; monitorar níveis de LEU, creatinina e transaminase (causa hepatite e pode causar cirrose; evitar totalmente a ingesta de etanol); pode ser administrado por via oral ou injeção semanal; o ácido fólico diminui os efeitos colaterais “incômodos”. Evitar a gravidez. |
GPA = granulomatose com poliangeíte; PPD = derivado proteico purificado; LEU = contagem de leucócitos.
Glomerulonefrite. A glomerulonefrite é uma causa comum de morbidade e mortalidade na GPA. Na literatura, a GPA costuma ser referida como uma doença generalizada ou limitada. A presença de glomerulonefrite define a forma generalizada da doença. A glomerulonefrite, frequentemente, é agressiva, mas pode ser relativamente indolente. Do ponto de vista clínico e patológico, pode ser indistinguível da glomerulonefrite em crescente idiopática rapidamente progressiva, mas no início costuma ser clinicamente silenciosa. A evolução da doença renal subclínica para doença renal dependente de diálise pode se estender por várias semanas. A glomerulonefrite está presente no início da doença em 20% dos pacientes e se desenvolve tardiamente ao longo do curso da doença em 80% dos pacientes. A importância das urinálises frequentes na avaliação inicial e nas avaliações de seguimento dos pacientes com GPA não pode ser exagerada. Este monitoramento pode ser feito pelos próprios pacientes em casa, por meio da análise com fita de rotina para a detecção de hematúria oculta. Especialmente em pacientes idosos ou debilitados, informação valiosa pode ser obtida a partir de coletas ocasionais de urina de 24 horas, que podem estabelecer uma estimativa mais precisa da taxa de filtração glomerular do que aquela fornecida pela medida da creatinina sérica. A biópsia renal pode revelar a presença de glomerulonefrite focal e segmentar com alterações proliferativas glomerulares variáveis, formação de crescente e necrose, na ausência de deposição significativa de imunocomplexos (conhecida como glomerulonefrite pauci-imune).
Figura 4 - Deformidade do nariz em sela.
Embora sustentem o diagnóstico de GPA, estes achados não são diagnósticos da doença [ver Exames laboratoriais, adiante].
Figura 5 - Os infiltrados nodulares do pulmão na granulomatose com poliangeíte são mostrados de forma menos extensiva em uma radiografia padrão (a) do que em uma varredura de tomografia computadorizada (b).
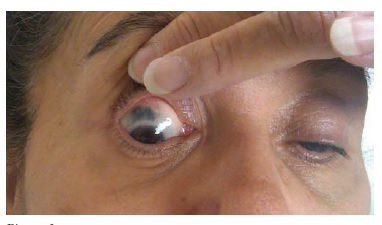
Manifestações clínicas adicionais. O envolvimento musculoesquelético ocorre em metade dos pacientes com GPA. Os sintomas podem incluir artralgias ou artrite, sendo que estes sintomas podem ser migratórios, aditivos ou de distribuição fixa. O fator reumatoide, frequentemente, está presente em pacientes com GPA e pode causar confusão diagnóstica com artrite reumatoide, ao produzir sintomas articulares significativos. A doença articular da GPA raramente produz erosões. Os sinais e sintomas neurológicos ocorrem em menos de 50% dos pacientes; a neuropatia periférica está presente em menos de 20% dos casos; e menos de 10% dos pacientes apresentam envolvimento do sistema nervoso central. Os pseudotumores orbitais podem causar proptose, oftalmoplegia, dor intratável e perda da visão. Estas massas inflamatórias e fibrosas podem ser refratárias à terapia anti-inflamatória, terapia imunossupressora e, até mesmo, à radioterapia. Pode haver defeitos oculomotores em consequência do empurramento por uma massa retrorbital ou doença sinusal inflamatória. A ocorrência de conjuntivite, de uveíte e de esclerite, de forma isolada ou combinada, é comum [ver Figura 6]. As ulcerações e isquemia gastrintestinal são infrequentes, mas podem ser confundidas com enteropatia inflamatória, em especial por esta última condição estar associada ao ANCA (usualmente, anticorpo anticitoplasma de neutrófilo perinuclear [p-ANCA]). Até 50% dos pacientes com GPA exibem envolvimento cutâneo com púrpura, paniculite ou ulcerações. Quando presente, a doença cutânea, geralmente, ocorre em paralelo com a atividade da doença sistêmica. Observações baseadas em estudos clínicos sugerem que os pacientes com GPA apresentam predisposição ao desenvolvimento de trombose venosa profunda.10
A inflamação crônica inexplicável do trato respiratório em presença de glomerulonefrite é bastante sugestiva de GPA. A probabilidade de GPA aumenta quando há envolvimento de múltiplos órgãos, doença de vias respiratórias destrutiva e demonstração de nódulos pulmonares (em especial com cavidades) por radiografia. Qualquer combinação de envolvimento de órgão é possível, porém mais de 90% dos pacientes apresentam envolvimento do trato respiratório superior e/ou inferior no momento do diagnóstico.
Se o quadro clínico como um todo for compatível com GPA e os diagnósticos alternativos apropriados tiverem sido excluídos, o achado de anticorpo anticitoplasma de neutrófilo citoplasmático (c-ANCA) circulante com especificidade antiproteinase 3 no ensaio imunossorvente ligado à enzima (ELISA) é suficiente para estabelecer um diagnóstico provisório e iniciar a terapia sem diagnóstico tecidual. Cerca de 20% dos pacientes com GPA podem apresentar p-ANCA com especificidade antimieloperoxidase. Havendo quaisquer achados atípicos ou preocupações especiais com relação à iniciação da terapia imunossupressora, ou se o paciente não responder apropriadamente à terapia, a confirmação histopatológica do diagnóstico deve ser agressivamente buscada. A presença de ANCA não equivale à presença de vasculite e o ANCA pode ser encontrado em outras doenças.
Os níveis de ANCA não são confiáveis como biomarcador da atividade da doença.11–13 Como a GPA requer terapia com corticosteroide acrescido de um segundo agente para induzir remissão e limitar a probabilidade de recaída, é preciso distinguí-la de outros distúrbios inflamatórios, inclusive de outras síndromes vasculíticas [ver Tabela 3], que podem ser tratados de modo efetivo com um regime menos tóxico.
Figura 6 - Esclerite necrotizante.
O tratamento da GPA justifica o uso combinado de corticosteroides com um segundo agente imunossupressivo. Os corticosteroides podem produzir envolvimento sintomático das vias respiratórias superiores, dos pulmões, da pele e do sistema musculoesquelético, porém o desmame usualmente resulta em exacerbação imediata da doença, a menos que um segundo agente seja administrado ao mesmo tempo. O tratamento da GPA deve ser determinado pela gravidade da doença. Pacientes com doença potencialmente fatal ou envolvimento de órgão grave devem ser tratados com ciclofosfamida combinada a doses altas de um corticosteroide (embora tenham proposto o uso de rituximabe neste contexto, em vez de ciclofosfamida). Uma vez alcançada a melhora ou remissão (em 3-6 meses), a terapia com ciclofosfamida deve ser substituída por metotrexato ou azatioprina para manutenção da remissão da doença [ver Tabela 2]. Esta abordagem atualmente é sustentada por vários estudos clínicos.14–16 Segundo um estudo randomizado que comparou a azatioprina com o metotrexato para uso na terapia de manutenção, não há diferença em termos de taxa de recidiva e toxicidade entre ambos os agentes.17 O micofenolato de mofetil tem sido uma alternativa para uso na terapia de manutenção da remissão em casos de pacientes que sofrem recaída durante o tratamento com metotrexato ou azatioprina, ou que tiveram contraindicação ao uso de um destes agentes, porém os resultados obtidos por um estudo randomizado mostraram que o micofenolato de mofetil é menos efetivo do que a azatioprina para fins de manutenção da remissão.18
Existem algumas contraindicações relativas fortes ao uso prolongado de ciclofosfamida, tais como a disfunção de bexiga (risco aumentado de câncer de bexiga e cistite induzida por metabólitos do fármaco) e leucopenia. Entretanto, mesmo na ausência dessas contraindicações, a morbidade associada à ciclofosfamida é significativa.7 Em pacientes com doença leve a moderada, as doses semanais de metotrexato com ácido fólico (ou leucovorina) podem ser substituídas por ciclofosfamida para induzir e manter a remissão.17 Pacientes sob tratamento com agentes imunossupressores devem ser continuamente monitorados quanto à ocorrência de exacerbação da doença, infecções oportunistas e efeitos colaterais da medicação. Como as exacerbações são frequentes após a suspensão do tratamento, a terapia de manutenção de longa duração tem sido recomendada.14,19 Os efeitos colaterais do metotrexato incluem as citopenias, hepatotoxicidade e pneumonite fármaco-induzida. Seu uso deve ser evitado no contexto de insuficiência renal ou consumo de álcool. A azatioprina pode causar reação de hipersensibilidade febril e leucopenia. Alguns autores têm sugerido o uso diário de trimetoprima-sulfametoxazol como terapia auxiliar para tratamento da GPA, com base em dados limitados que sugerem que esta terapia pode diminuir a frequência das exacerbações da doença do trato respiratório superior.20 Entretanto, esta abordagem ainda é controversa e recomenda-se cautela com o uso conjunto de doses integrais de metotrexato, uma vez que a combinação pode resultar em toxicidade antifolato aditiva. A administração de trimetoprima-sulfametoxazol com uma frequência de 3 vezes por semana, todavia, é útil para conferir proteção contra pneumonia por P. jiroveci (antigo P. carinii) durante a administração da terapia imunossupressiva intensiva. A higiene sinusal e nasal local, bem como a realização de avaliações otolaringoscópicas periódicas são parte rotineira do cuidado dos pacientes com doença de via respiratória superior.
Um estudo randomizado controlado demonstrou que a terapia antinecrose tumoral com etanercept é inefetiva como terapia auxiliar para tratamento da GPA. Essa terapia, aparentemente, também aumenta o risco de malignidade em pacientes com GPA previamente tratado com agentes citotóxicos.21,22 Apesar de sua alta eficácia para indução de remissão da doença e prolongamento da sobrevida, a ciclofosfamida está associada a uma toxicidade substancial. Dois estudos randomizados demonstraram que o rituximabe é tão efetivo quanto a ciclofosfamida para indução da remissão da doença em pacientes com GPA e PAM.23,24 Um estudo sugeriu que essa terapia pode ser superior para indução de remissão em casos de doença recidivante.23 Não foram observadas diferenças significativas em termos de eventos adversos entre estas duas terapias. Um estudo aberto, comparando a terapia à base de plasmaférese com pulsos intravenosos de metilprednisolona em pacientes sob tratamento com ciclofosfamida para doença renal grave, observou diminuição do risco de progressão para doença renal em estágio terminal. Entretanto, a mortalidade continuou sendo alta e não foi diminuída pela plasmaférese em um período de um ano.25
A adoção de medidas profiláticas para prevenção de osteoporose e a realização de varreduras ocasionais de absorciometria por raios x de dupla energia para determinação da densidade óptica devem ser consideradas com o uso prolongado de corticosteroides.
|
Tabela 3 Achados clínicos de vasculite | ||||
|
Distúrbio |
Órgãos-alvo comuns |
Achados patológicos especiais |
Exames laboratoriais especiais |
Comentários |
|
Poliangeíte microscópica |
Nervo periférico, glomérulo, pulmão (pequenos vasos), trato GI, pele. |
Vasculite, GN proliferativa (imunodepósitos* raros ou ausentes); sem células gigantes. |
p-ANCA (anti-MPO). |
Excluir a hipótese de hepatite C. |
|
Poliartrite nodosa |
Nervo periférico, trato GI; sem GN. |
Arterite de artérias musculares medianas, sem células gigantes. |
Sem ANCA. |
Ausência de envolvimento de pequenos vasos; excluir hipótese de hepatite B. |
|
Granulomatose com poliangeíte |
Vias aéreas superiores, pulmão (pequenos vasos), olho, glomérulo, nervo periférico, sistema musculoesquelético. |
Células gigantes, Necrose geográfica, vasculite, GN proliferativa (imunodepósitos raros ou ausentes). |
c-ANCA (anti-PR3). |
Doença da orelha ou sinusal crônica . |
|
Síndrome de Churg-Strauss
|
Asma, sinusite, rinite, pólipos nasais ou sinusais Nervo periférico, pulmão, coração, pele. |
Células gigantes, eosinofilia, vasculite, GN proliferativa (imunodepósitos raros ou ausentes) . |
Eosinofilia ANCA (30–50% de positividade). |
História positiva para atopia. |
|
Doença de Kawasaki |
Linfonodos, pele e mucosa oral, coronárias. |
Aneurismas inflamatórios da artéria coronária .
|
|
O tratamento inicial pode prevenir a formação de aneurisma.
|
|
PHS |
Pele, sistema musculoesquelético, trato GI, glomérulo. |
VLC cutânea Deposição tecidual de imunocomplexos de IgA . |
|
As recidivas ocorrem em cerca de 1/3 das crianças .
|
ANCA = anticorpo anticitoplasma de neutrófilo; c-ANCA = anticorpo anticitoplasma de neutrófilo citoplasmático; GI = gastrintestinal; GN = glomerulonefrite; PHS = púrpura de Henoch-Schönlein; VLC = vasculite leucocitoclástica; MPO = mieloperoxidase; p-ANCA = anticorpo anticitoplasma de neutrófilo perinuclear; PR3 = proteinase 3.
*A presença de imunodepósitos sugere uma possível infecção por vírus da hepatite B ou C.
A síndrome de Churg-Strauss (SCS) é uma forma rara de vasculite que afeta veias e artérias de tamanho pequeno a médio, associada à asma e à hipereosinofilia.
O componente inflamatório da SCS exibe similaridades clínicas com a GPA, em termos de envolvimento de órgãos e patologia, especialmente em pacientes com doença de vias aéreas inferiores ou superiores ou glomerulonefrite. A SCS difere mais notavelmente da GPA no sentido de que ocorre em pacientes com história de atopia, asma ou rinite alérgica, frequentemente em curso. A doença tem sido tipicamente descrita como tendo três fases: uma fase atópica inicial com rinite alérgica e asma; uma segunda fase de eosinofilia; e uma fase final de vasculite sistêmica. As três fases nem sempre estão presentes em todos os pacientes e, muitas vezes, não seguem uma sequência em particular. A eosinofilia é característica e frequentemente acentuada (> 1.000 eosinófilos/µL). Quando a GPA é acompanhada de eosinofilia, esta usualmente é mais modesta (500 eosinófilos/µL).
Os achados órgão-específicos da SCS incluem certa combinação de infiltrados pulmonares, miocardiopatia, arterite coronária, polineuropatia (simétrica ou mononeurite múltipla), enteropatia isquêmica, gastrenterite eosinofílica, inflamação ocular, perfurações nasais, glomerulonefrite, nódulos cutâneos e púrpura.26,27
Os infiltrados pulmonares irregulares da SCS costumam ser transitórios e podem estar associados à hemorragia alveolar. Os nódulos pulmonares são incomuns e, diferente da GPA, raramente cavitam. Os derrames pleurais frequentemente são ricos em eosinófilos. A distinção clínica da pneumonite por hipersensibilidade, aspergilose alérgica e linfoma pulmonar, às vezes, é difícil. Têm sido relatados vários casos de SCS em pacientes asmáticos ocorridos após a introdução dos inibidores de 5-lipoxigenase, durante o desmame dos corticosteroides.
Na SCS, a cardiopatia pode ser grave e é uma das principais causas de mortalidade. A infiltração cardíaca ou arterite coronária podem produzir insuficiência cardíaca e síndromes isquêmicas. Pode haver cardiopatia valvular, mas esta não é tão marcante nem tão comum na SCS quanto a síndrome hipereosinofílica idiopática. O envolvimento neurológico ocorre em mais de 60% dos pacientes com SCS. Este tipo de envolvimento pode ser grave e geralmente é atribuível à arterite. Pode haver púrpura cutânea, urticária, erupções eritematosas polimórficas e nódulos. O envolvimento gastrintestinal resultante de vasculite isquêmica, gastrenterite eosinofílica ou de ambas as condições pode causar dor, cólicas e diarreia.
A histopatologia tipicamente exibe inflamação granulomatosa extravascular, com um infiltrado eosinofílico proeminente, enquanto a presença de vasculite é variável. Granulomas podem ser encontrados no tecido em áreas separadas da região com vasculite demonstrável. Os infiltrados eosinofílicos na SCS são mais marcantes do que aqueles encontrados na GPA. Eosinófilos abundantes, granulomas e células gigantes não são encontradas na poliartrite nodosa (PAN) nem na PAM. A patologia dos nódulos em si não basta para estabelecer o diagnóstico de SCS, porque uma patologia similar pode ser vista no linfoma e na sarcoidose. A glomerulonefrite, na SCS, muitas vezes não é tão grave quando na GPA. Entretanto, quando presente na SCS, a glomerulonefrite costuma ser focal, segmentar e indistinguível de outras formas de glomerulonefrite pauci-imune (p.ex., glomerulonefrite com pouca ou nenhuma deposição tecidual de imunocomplexos). Embora os ANCAs circulantes sejam encontrados com menos frequência na SCS (30-50% dos pacientes), alguns autores incluem esta doença no grupo das vasculites ANCA-associadas.
Frequentemente, a SCS é responsiva à terapia com corticosteroide. A persistência da asma após a remissão da doença dificulta a descontinuação dos corticosteroides. A asma brônquica e a doença sinusal podem requerer terapia contínua, mesmo que o componente vasculítico da doença tenha sofrido remissão. Os pacientes com envolvimento orgânico visceral refratário ou grave são tratados empiricamente com agentes adicionais, como ciclofosfamida, metotrexato ou azatioprina, dependendo da gravidade do envolvimento orgânico. Os corticosteroides são desmamados depois que a remissão é alcançada [ver Tabela 2].
A PAM é uma vasculite de pequenos vasos predominantemente necrotizante, que tipicamente envolve os pulmões, rins e sistema nervoso periférico. Uma conferência internacional propôs que a distinção entre PAN e PAM fosse baseada na presença de vasculite afetando pequenos vasos (arteríolas, capilares, vênulas e capilares glomerulares) na PAM e não na PAN.1 O reconhecimento de que a PAM, e não a PAN, com frequência está associada à presença de ANCA (com os ANCAs quase sempre dirigidos contra a mieloperoxidase) enfatiza adicionalmente a distinção entre estas entidades. Em muitos aspectos, a PAM é similar à GPA, e os estudos terapêuticos,muitas vezes, incluem as duas condições.
A PAM, diferente da PAN, envolve vasos menores com tamanho que varia de capilares e vênulas a artérias de médio calibre [ver Figura 1].28 Devido ao envolvimento dos pequenos vasos, a PAM frequentemente se manifesta como glomerulonefrite ou hemorragia alveolar, muitas vezes acompanhada de neuropatia e púrpura cutânea. Clinicamente, a PAM pode mimetizar a GPA, embora alguns autores tenham arbitrariamente definido a PAM como uma condição que exclui o envolvimento das vias aéreas superiores. O diagnóstico de PAM na ausência de doença renal pode ser desafiador, levando ao atraso no início da terapia.
É preciso tentar confirmar o diagnóstico por patologia tecidual, mostrando a ocorrência de vasculite necrotizante na ausência de inflamação granulomatosa. Em oposição à GPA, a necrose e inflamação parenquimal observadas PAM não são marcantes (exceto nas áreas de dano isquêmico), e não há células gigantes na PAM. A biópsia de pulmão no contexto de hemorragia ou infiltrados pulmonares revela capilarite, um padrão histopatológico que também pode ser visto na GPA, LES e doença antimembrana basal glomerular. A biópsia de pulmão obtida de pacientes com suspeita de PAM também é útil para excluir diagnósticos pulmonares alternativos. As técnicas de pulmão aberto e toracoscopia fornecem rendimento maior em casos de vasculite, em comparação à biópsia transbrônquica. A histopatologia renal revela glomerulonefrite segmentar focal, com pouca ou nenhuma deposição de imunocomplexos, que é indistinguível da glomerulonefrite vista na GPA. A presença no soro de p-ANCA com especificidade antimieloperoxidase (detectada em cerca de 70-85% dos pacientes com PAM) sustenta o diagnóstico clínicos de PAM, mas não é específica para esta doença.
Como resultado da comprovada eficácia dos atuais regimes terapêuticos para GPA, uma abordagem de tratamento similar é usada para PAM. Não há estudos terapêuticos controlados incluindo apenas pacientes com PAM, entretanto dados retrospectivos mostraram que esta abordagem está associada a uma mortalidade menor.28 Pacientes com doença grave inicialmente devem ser tratados com uma combinação de ciclofosfamida e com uma dose alta de corticosteroide. A taxa de recidivas parece ser menor na PAM, em comparação com a GPA.
Os relatos iniciais sobre PAN e PAM não diferenciam corretamente as duas entidades. Hoje, em geral é aceito que a PAN é um distúrbio raro associado à arterite de artérias musculares de médio calibre; os pequenos vasos não são afetados e, deste modo, a púrpura deve estar ausente em pacientes com PAN clássica. Ainda mais importante, os pacientes com hepatite viral B ou C não foram excluídos dos estudos antigos. O reconhecimento da hepatite viral é essencial, porque a hepatite B crônica2,29 pode deflagrar uma síndrome vasculítica secundária que é indistinguível da PAN à apresentação, mas que difere quanto ao prognóstico e resposta à terapia.30
Glomerulonefrite e hemorragia alveolar estão ausentes, por definição, em pacientes com PAN.
A PAN afeta as artérias musculares de médio calibre e frequentemente está associada com neuropatia periférica e isquemia intestinal.31–33 Na PAN, pode haver azotemia e hipertensão em consequência da arterite das artérias renais e de nefropatia isquêmica, mas não por causa da glomerulonefrite. A formação de microaneurisma em artérias viscerais de médio calibre pode ser um achado marcante na PAN, sendo que as artérias ocasionalmente podem sofrer ruptura.
Os sintomas constitutivos, como febre, astenia e mialgias, são comuns. Níveis elevados de reagentes de fase aguda, trombocitose, leucocitose e a anemia da doença inflamatória são achados frequentes, embora não ocorram de modo uniforme e não diferenciem a PAN de outras síndromes vasculíticas.
Havendo suspeita de síndrome clínica de PAN, as hipóteses de infecção bacteriana crônica (p. ex., endocardite) e infecção viral (p. ex., hepatite B ou C) devem ser excluídas. A associação com hepatite B ou C pode não alterar drasticamente alguns aspectos das síndromes de PAN ou PAM, contudo a glomerulonefrite membranosa, crioglobulinemia, glomerulonefrite associada a imunocomplexos, insuficiência hepática e trombocitopenia tendem mais a ocorrer com a vasculite associada à hepatite viral. Uma deposição significativa de imunocomplexos não ocorre na PAN nem na PAM.
A síndrome do anticorpo antifosfolípide (SAAF) pode mimetizar a PAN manifestando-se como isquemia mesentérica ou insuficiência renal causada por obstrução trombótica de vasos mesentéricos e renais.34 O livedo reticular é um achado clínicos que pode ser encontrado na SAF e na vasculite com envolvimento de artérias musculares [ver Figura 7]. Crioglobulinemia, glomerulonefrite associada a imunocomplexos e à neuropatia periférica não têm associação com a SAF, a menos que o paciente também tenha LES. A trombocitopenia pode ocorrer com SAF, mas usualmente está ausente na PAN. A embolia por colesterol também deve ser considerada como causa de livedo, insuficiência renal e sintomas constitutivos,35 em particular se a história clínica incluir um procedimento vascular recente. A biópsia tecidual, muitas vezes, estabelecerá o diagnóstico.
De forma ideal, o diagnóstico deve ser estabelecido com base na demonstração histopatológica da arterite e no padrão clínico da doença. A obtenção de uma amostra de biópsia do tecido não necrótico com envolvimento clínico, que mostra a presença de arterite de artérias musculares é o achado de suporte ideal para o diagnóstico de arterite de vaso de médio calibre, contudo a obtenção deste tipo de amostra de biópsia nem sempre é possível. A PAN não está associada com ANCAs.
A PAN não causa glomerulonefrite, capilarite pulmonar nem doença parenquimal pulmonar.
Pode ser difícil demonstrar a arterite afetando artérias musculares na PAN, em especial no paciente com sintomas constitutivos dominantes e ausência de tecido afetado por doença de fácil acesso. As tentativas de obtenção de biópsia devem ser dirigidas para o tecido anormal, conforme demonstrado pelos sintomas ou por testes objetivos. A biópsia do nervo sural tem se tornado uma opção popular para tentar diagnosticar uma arterite que esteja afetando vasos musculares de médio calibre. O nervo sural é um nervo sensorial puro acessível e seus vasa nervorum contêm artérias musculares de pequeno e médio calibre. Estudos de condução nervosa podem identificar um nervo sural isquêmico adoecido antes do aparecimento de achados clínicos.36
Figura 7 - O livedo reticular é caracterizado pelo aparecimento de manchas vermelho-azuladas nos membros, causadas pela obstrução das arteríolas dérmicas profundas.
Entretanto, múltiplos relatos têm enfatizado o baixo rendimento diagnóstico da biópsia de um nervo assintomático e eletricamente normal. Até mesmo os nervos que exibem condução anormal têm mostrado declaradamente ausência de patologia diagnostica em 46% dos casos.37 Existe uma notável (ainda que subestimada) morbidade associada à biópsia do nervo sural. A cada 60 pacientes, 13 tiveram as feridas infeccionadas ou com cicatrização retardada, enquanto 3 pacientes sofreram com dor pós-procedimento no nervo submetido à biópsia.37 A biópsia de tecido sem envolvimento clínico (i.e., músculo assintomático) fornece um rendimento inferior a 30% para fins de diagnóstico de PAN.
A arteriografia abdominal é realizada com frequência na avaliação dos pacientes que podem ter arterite de vaso de médio calibre, quando a biópsia não é viável ou não é uma opção. As artérias afetadas pela PAN ou por outros distúrbios de artérias musculares de médio calibre podem desenvolver microaneurismas ou estenoses que podem ser visualizadas por arteriografia. Quando a arteriografia é usada para tentar diagnosticar vasculite necrotizante sistêmica na ausência de evidência patológica da doença, várias ressalvas devem ser observadas. A arteriografia apresenta resolução espacial limitada; os vasos menores não são bem visualizados.
Em pacientes com doença primariamente de pequenos vasos, é improvável que o arteriografia seja diagnóstica. Diferentes pesquisadores têm relatado aneurismas em 60-90% dos pacientes com PAN. Os aneurismas demoram certo tempo para se desenvolver e podem estar ausentes no início do curso da doença. Além de estar associada a aneurismas, a arterite pode estar associada a estenoses, as quais podem ser mais longas e regulares do que a obstrução ou as lesões ateroscleróticas típicas. Para maximizar o rendimento do procedimento, a arteriografia deve incluir os vasos celíacos, renais e mesentéricos. A ausência de envolvimento clinicamente evidente de um órgão (p. ex., ausência de isquemia intestinal) não anula a possibilidade de encontrar vasos anormais na arteriografia. Foi sugerido que a visualização de aneurismas na PAN denota uma doença mais grave e não está claro se sua presença pode estar alternativamente relacionada à duração da doença não tratada. Os aneurismas podem se resolver com o tratamento bem-sucedido da arterite associada à hepatite primária ou viral. A presença de microaneurismas viscerais não é diagnóstica de PAN e este achado também foi descrito de forma pouco confiável em pacientes com GPA e PAM, provavelmente representando o envolvimento de artéria muscular de médio calibre nestas doenças. Os microaneurismas também ocorrem em distúrbios não vasculíticos. Relatos de casos isolados têm descrito aneurismas em pacientes com mixoma atrial, endocardite bacteriana, carcinomatose peritoneal e hipertensão arterial grave, bem como após o uso abusivo de metanfetamina. Os dados disponíveis são inadequados para avaliar a sensibilidade e especificidade ou o valor preditivo da arteriografia abdominal no diagnóstico da arterite necrotizante. Como ocorre ao interpretar um resultado de biópsia de um caso com suspeita de vasculite, os exames de imagem devem ser considerados à luz do perfil clínico como um todo. A arteriografia, geralmente, é evitada no contexto de insuficiência renal progressiva ou significativa.
O tratamento da PAN é empírico38 [ver Tabela 2]. Doses altas de corticosteroides (1 mg/kg/dia de prednisona ou equivalente) continuam sendo a base inicial da terapia para o paciente com doença aguda. O uso apenas de corticosteroides pode ser suficiente para pacientes com PAN que não apresentam envolvimento de órgão significativo, definido como insuficiência renal (por isquemia renal), isquemia gastrintestinal grave, cardiopatia ou neuropatia periférica. Pacientes que necessitam de terapia à base de corticosteroide prolongada para controle da doença ou pacientes que apresentam marcadores clínicos da doença grave usualmente são tratados com glicocorticoides e um agente imunossupressor adicional, como a ciclofosfamida ou metotrexato. As indicações para a terapia combinada inicial ainda não estão suficientemente definidas.
Quando há infecção ativa por vírus da hepatite B ou C, deve ser considerada a instituição de um curso relativamente breve de esteroides, com base na gravidade da doença extra-hepática e nos órgãos que apresentam risco agudo de insuficiência, aliado a uma terapia antiviral agressiva. O afunilamento de esteroide tem sido associado à exacerbação da necrose hepática, quando a infecção viral não é tratada.
A doença de Kawasaki (DK) foi descrita pela primeira vez em 1967, como uma síndrome de linfonodo mucocutâneo.39 Afeta tipicamente bebês e crianças menores, causando manifestações dominantes na pele e mucosa oral, febre e arterite coronária. Em casos raros, pode afetar adultos.
A presença dos achados clínicos característicos tem permitido o estabelecimento de critérios diagnósticos para DK [ver Tabela 4]. A vasculite pode envolver vasos com tamanhos que variam de vênulas até a aorta. Inflamação proeminente é observada nas artérias coronárias maiores, resultando em formação de aneurisma em cerca de 25% dos pacientes não tratados. As complicações cardíacas imediatas e tardias potencialmente fatais da doença, aliadas a sua terapia exclusiva (aspirina e imunoglobulina intravenosa), obrigam o diagnóstico clínico imediato. A biópsia geralmente é desnecessária e tende a não fornecer um diagnóstico específico.
As febres altas podem persistir por 1-2 semanas, se não houver tratamento. A defervescência rápida usualmente é observada com a iniciação de terapia apropriada. A conjuntivite não exsudativa frequentemente aparece com a febre. É comum haver meningite asséptica (linfocítica). O envolvimento oral inclui eritema, ressecamento e rachadura dos lábios, faringite não exsudativa e eritema de língua com papilas bastante proeminentes. As ulcerações de mucosa não são características desta condição. O edema distal dos membros pode ocorrer dias após a febre, com eritema e sensibilidade não limitados às articulações. A descamação das mãos e pés, muitas vezes em lâminas, pode ter início em dias ou semanas após o aparecimento da febre. Quando a descamação ocorre no início da DK, pode surgir de modo concomitante com erupção truncal e alterações oculares e labiais. Pode mimetizar uma reação farmacológica ou a síndrome de Stevens-Johnson. A erupção usualmente é difusa e polimórfica, com componentes pruriginosos, morbiliformes, anulares ou de placa, mas não é vesicular. A adenopatia, que está presente em até 75% dos pacientes, é mais evidente na região cervical.
A morbidade e mortalidade (< 3%) da DK estão notavelmente associadas ao desenvolvimento de aneurismas inflamatórios de artéria coronária, a maioria dos quais é assintomática no momento de sua formação. Os aneurismas podem ser detectados por ecocardiografia. Pode haver trombose nos aneurismas, resultando em obstrução direta ou embólica da artéria coronária. Os eventos coronarianos agudos podem ocorrer em semanas ou até muitos anos após a doença febril. Um ecocardiograma basal deve ser obtido no momento da doença aguda e repetido após 2 e 6 semanas. O reconhecimento e tratamento antecipado da doença com administração de imunoglobulina intravenosa e aspirina têm diminuído significativamente a frequência de formação de aneurisma e de eventos coronarianos trombóticos.
|
Tabela 4 Critérios diagnósticos para doença de Kawasaki |
|
Febre persistente (> 5 dias) mais pelo menos quatro das seguintes condições: 1. Conjuntivite bilateral não purulenta; 2. Envolvimento da mucosa oral: faringe eritematosa, lábios avermelhados ou rachados, ou “língua de morango”; 3. Anomalias de tecido mole das mãos e pés: eritema, edema ou descamação; 4. Erupção não vesicular polimórfica; 5. Adenopatia cervical. |
O tratamento da DK deve ser iniciado com administração de imunoglobulina intravenosa (2 g/kg como dose única) e de aspirina (80-100 mg/kg/dia), tão logo a seja levantada a suspeita da condição.40 A aspirina é mais efetiva do que os corticosteroides na prevenção dos aneurismas. A terapia com corticosteroide usualmente é desnecessária, e alguns autores a consideram relativamente contraindicada. Os sintomas tendem a responder dentro de alguns dias após a instituição da aspirina e da imunoglobulina intravenosa. Por outro lado, nos casos resistentes, os corticosteroides frequentemente são adicionados às terapias supramencionadas. Crianças com aneurismas coronarianos amplos ou múltiplos podem precisar de anticoagulação por tempo prolongado, numa tentativa de prevenir o infarto do miocárdio tardio.
A arterite temporal ou arterite de célula gigante (ACG) e a arterite de Takayasu (AT) são as doenças inflamatórias mais comuns da aorta e de seus ramos. Um direcionamento vascular semelhante pode ocorrer na doença de Behçet, síndrome de Cogan e sarcoidose. Estas três condições são distinguidas pelo padrão de envolvimento de órgão extra-aórtico. Não está claro se AT e ACG são distúrbios diferentes ou se são o mesmo distúrbio com expressões modificadas em diferentes faixas etárias.
A ACG, geralmente, afeta indivíduos com mais de 50 anos de idade.41,42 A ACG pode se manifestar com quatro padrões clínicos principais: arterite cranial, vasculite de grandes vasos, polimialgia reumática (PMR) e uma doença inflamatória sistêmica inespecífica. Estes padrões clínicos podem ocorrer isolados, simultaneamente ou em momentos distintos ao longo do curso da doença, em um mesmo paciente. A PMR é caracterizada por dor muscular proximal, que piora à noite e de manhã cedo. Pode haver uma sensação subjetiva de fraqueza, sem que um enfraquecimento real seja detectado ao exame e na ausência de elevação dos níveis séricos de enzimas musculares. A sinovite pode estar presente, muitas vezes, dificultando a distinção entre ACG e artrite reumatoide senil, embora a ACG geralmente seja negativa para fator reumatoide. Os sintomas de arterite craniana incluem cefaleia, anormalidades ou perda da visão, sensibilidade no couro cabeludo e/ou temporal, e claudicação do masseter ou da língua. A ACG pode se manifestar com sinais inflamatórios sistêmicos, como febre e perda de peso. A vasculite de grandes vasos afeta a aorta e seus ramos principais, resultando em aneurismas aórticos inflamatórios ou estenose vascular. Os sinais e sintomas isquêmicos podem ser clinicamente indistinguíveis daqueles associados à doença ateroembólica ou arterioesclerótica obstrutiva.
O exame para detecção de pulsos assimétricos nos quatro membros e tomada de pressão arterial, aneurismas abdominais e ruídos no pescoço, abdome e membros deve fazer parte das consultas de seguimento de rotina para pacientes com ACG ou PMR. Os achados patológicos da ACG podem ser encontrados nas artérias temporais superficiais dos pacientes com PMR, mesmo na ausência de sintomas de ACG. Entretanto, a biópsia de rotina das artérias temporais superficiais de pacientes com PMR, na ausência de sintomas cranianos, não é justificada.
Os níveis de reagentes de fase aguda estão altos em mais de 80% dos pacientes com ACG, mas não são totalmente confiáveis como marcadores da atividade da doença durante e após a terapia. Um diagnóstico definitivo de ACG geralmente é estabelecido por meio de biópsia da artéria temporal superficial. Na ACG, a patologia usualmente revela infiltrados crônicos de células mononucleares, destruição da lâmina elástica interna e células gigantes. A presença de células gigantes não é requisito para o diagnóstico. A presença de achados clínicos característicos, como uma nova cefaleia e claudicação mandibular, especialmente com PMR concomitante, pode possibilitar um diagnóstico presuntivo sem biópsia ou quando a biópsia de artéria temporal superficial resulta negativa. Entretanto, como outras condições podem mimetizar a ACG, incluindo a aterosclerose e a amiloidose, uma tentativa de diagnosticar a ACG por biópsia é justificada para a maioria dos pacientes.43 A terapia com corticosteroide não afeta rapidamente os resultados da biópsia e não deve ser suspendida no caso de um paciente com forte suspeita de ACG que esteja aguardando uma biópsia, contudo a obtenção desta não pode ser adiada por mais de 1-2 semanas. A biópsia de artéria temporal superficial bilateral pode aumentar o rendimento diagnóstico, mas nem sempre resulta positiva.
Os diagnósticos diferenciais destas doenças inflamatórias obstrutivas de grandes vasos incluem aterosclerose, dissecção carotídea, glaucoma, dissecção aórtica, SAAF, displasia fibromuscular, sarcoidose, doença de Buerger e outras síndromes vasculíticas primárias menos comuns (p. ex., doença de Behçet).
A AT (doença sem pulsação) é uma doença inflamatória crônica que afeta a aorta e seus ramos principais.44 Usualmente diagnosticada em pacientes do sexo feminino jovens, em idade fértil, a AT também pode ocorrer em crianças menores e pacientes idosos de ambos os sexos. A condição é mais comumente associada com estenoses e aneurismas da aorta e vasos do ramo aórtico, em comparação à ACG.
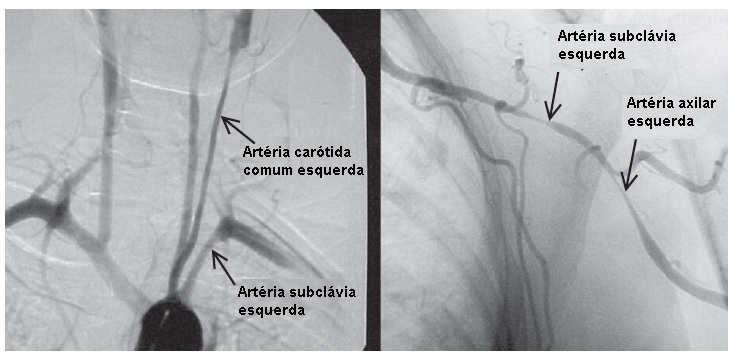
A síndrome clínica que se manifesta pode incluir uma doença semelhante à gripe, incluindo um padrão de dor muscular da PMR. Muitos pacientes inicialmente apresentam sintomas de isquemia de membro, cerebral ou cardíaca na ausência de quaisquer achados constitutivos. Os sintomas isquêmicos característicos refletem a localização e a gravidade dos vasos envolvidos, bem como a presença de fluxo sanguíneo colateral. A isquemia renal pode deflagrar hipertensão com níveis altos de renina. Os sítios de estenose predominantes são os vasos do arco aórtico, em particular as artérias subclávias [ver Figura 8]. A claudicação de membro com ruído supraclavicular ou axilar é comum. Dor e sensibilidade na artéria superficial (p. ex., carodinia) podem ser encontradas ao exame, mas não são diagnósticas de AT. A hipertensão central grave decorrente da estenose da artéria renal pode não ser reconhecida, devido à estenose coexistente da artéria do braço. Desta forma, as leituras de pressão arterial dos quatro membros devem ser inicialmente avaliadas e monitoradas com frequência. Ocasionalmente, são encontradas estenoses em todos os principais vasos dos membros, sendo que o monitoramento com manguito pode ser uma medida pouco confiável para medir as pressões aórticas centrais. O acidente vascular encefálico não é incomum e, frequentemente, está relacionado à hipertensão central não detectada. É extremamente difícil avaliar a atividade da AT. A presença ou ausência de achados constitutivos ou níveis elevados de reagentes de fase aguda são medidas precárias da atividade da doença. Esta impressão é sustentada pela histopatologia vascular obtida durante a cirurgia de reconstrução. Mais de 40% das amostras vasculares obtidas de pacientes considerados como estando em remissão revelaram a existência de inflamação ativa. Por este motivo, o monitoramento anatômico regular destes vasos por exame físico e exames de imagem (seja por imagem de ressonância magnética [IRM] ou arteriografia) é obrigatório.
O diagnóstico de AT usualmente é estabelecido por IRM ou evidência arteriográfica das lesões estenóticas. Os aneurismas são observados com menos frequência. A aorta inteira e seus principais ramos devem ser avaliados. É fundamentalmente importante que a pressão arterial central medida rotineiramente no momento da arteriografia e comparada com as pressões medidas simultaneamente com manguito no braço e na perna. O papel da IRM vascular sequencial e da tomografia por emissão de pósitrons na avaliação e seguimento dos pacientes com suspeita de AT está sendo investigada. É muito importante determinar se as propriedades de imagem da parede vascular podem contribuir para a avaliação da atividade da doença.45 Estas técnicas de imagem podem revelar alterações relacionadas com a terapia envolvendo a espessura da parede vascular, edema e alterações no tamanho do lúmen. Quando há necessidade de procedimentos cirúrgicos, é obrigatório realizar uma discussão pré-operatória com o cirurgião vascular para garantir que amostras de tecido apropriadas sejam obtidas, sempre que possível.
Os corticosteroides são o tratamento inicial para AT e ACG. A ACG geralmente é bastante responsiva à terapia com esteroide, embora a dose inicial mais apropriada ainda seja controversa. Doses iniciais diárias entre 20 mg e 1 mg/kg têm sido defendidas, com desmame ao longo de 9-12 meses. A maioria dos autores se sente confortável em relação a uma dose inicial de 40-60 mg/dia. Em geral, recomenda-se (ainda que sem dados de suporte de estudos controlados) que os pacientes com quaisquer sintomas de isquemia ocular sejam inicialmente tratados com doses altas de corticosteroides (no mínimo 1 mg/kg de prednisona ou equivalente; alguns autores, principalmente oftalmologistas, sugerem a administração intravenosa de metilprednisolona em doses de até 1 g/dia por vários dias). Uma proporção significativa dos pacientes com ACG requer vários anos de terapia, apresentando exacerbações com o afunilamento dos corticosteroides. A administração de prednisona em dias alternados não é suficientemente efetiva para o tratamento inicial da ACG.
A terapia com doses altas de corticosteroide, especialmente em idosos, está associada a uma morbidade significativa. É preciso considerar, especialmente, os pacientes sob terapia crônica com corticosteroide para prevenção de infecções oportunistas, osteoporose, glaucoma, hiperglicemia e hiperlipidemia. A medida dos reagentes de fase aguda fornece um índice imperfeito da atividade da doença e não deve ser tomada como único guia para ajuste da dosagem de esteroide. Se o esteroide produzir efeitos colaterais significativos ou se um paciente sofrer recaída durante o afunilamento, um agente de segunda linha (p. ex., metotrexato), às vezes, é fornecido empiricamente com a terapia à base de corticosteroide. Entretanto, o valor dos agentes poupadores de esteroide auxiliares na ACG ainda não está comprovado. Um amplo estudo prospectivo randomizado falhou em demonstrar um efeito positivo significativo da terapia com metotrexato,46 embora um segundo estudo tenha sugerido um pequeno benefício.47 Em outro estudo randomizado, a terapia antifator de necrose tumoral não conferiu nenhum benefício adicional ao tratamento com corticosteroide de pacientes com ACG.48 Tem sido demonstrado que a adição de doses baixas de aspirina à terapia com corticosteroide minimiza as complicações isquêmicas da arterite temporal.49 Tem havido apenas relatos pouco confiáveis acerca do sucesso do uso adjunto de azatioprina, metotrexato e micofenolato no tratamento da AT resistente. Os relatos iniciais sobre a terapia com infliximabe para AT resistente mostram resultados promissores, contudo estudos randomizados se fazem necessários para confirmar sua eficácia.50,51 Com base em evidências do papel da interleucina-6 na patogênese da ACG, o uso de um anticorpo antirrecetor de interleucina-6 no tratamento da ACG está sendo avaliado.
Figura 8 - Arteriografias de um paciente com arterite de Takayasu, mostrando lesões estenóticas longas e regulares na artéria subclávia esquerda, bem como envolvimento de outros ramos de vasos do arco aórtico.
Cirurgia reconstrutiva vascular, angioplastia e colocação de stent são alternativas terapêuticas auxiliares para alguns pacientes com AT. Entretanto, a experiência ainda bastante incipiente sugere um algo grau de falha dos stents, devido à ocorrência de estenose em alguns centros.52 O envolvimento frequente de vasos subclávios na AT deve ser considerado ao escolher o sítio de implantação de enxerto para procedimentos de desvio coronariano ou carotídeo, sendo que o enxerto a partir destes vasos deve ser evitado.
A doença de Behçet é uma doença inflamatória recidivante multissistêmica que tipicamente se manifesta com lesões orais e genitais recorrentes, além de diversas manifestações clínicas. A histopatologia é caracterizada por vasculite, que pode afetar artérias ou veias de qualquer tamanho.
A doença de Behçet pode afetar vários sistemas de órgãos: pele, mucosas oral e genital, ocular, musculoesquelético, nervoso central, gastrintestinal e pulmonar. A manifestação clínica mais frequente são as ulcerações orais dolorosas recorrentes, que ocorrem em quase todos os pacientes. As lesões genitais dolorosas estão presentes em pelo menos 75% dos pacientes e, diferente das úlceras orais, geralmente estão associadas à cicatrização. A doença ocular mais típica é a panuveíte, que pode ser difícil de tratar e pode estar associada com inflamação persistente entre episódios de exacerbações agudas. A doença retinal obstrutiva pode levar à cegueira. A manifestação cutânea mais comum é uma lesão acneiforme com pápulas e pústulas que podem ser indistinguíveis da acne vulgar. Outras lesões incluem eritema nodoso, nódulos e tromboflebite superficial. As manifestações neurológicas são incomuns e usualmente envolvem o sistema nervoso central, com lesões cerebrais, meningite asséptica e trombose arterial ou venosa. O envolvimento de artérias amplas pode resultar na formação de aneurisma. A glomerulonefrite raramente ocorre na doença de Behçet.
O diagnóstico é estabelecido com base nas manifestações clínicas e não há exames laboratoriais específicos. O diagnóstico pode ser bastante desafiador no caso de pacientes que não apresentam expressão clínica plena da doença. Um teste de patergia positivo pode ser obtido de pacientes com doença de Behçet, mas requer a adoção de técnica padronizada para evitar resultados falso-negativos. A patologia pode revelar presença de vasculite, que é considerada a principal lesão associada à maioria dos achados clínicos. Embora a trombose venosa profunda seja encontrada em até 25% dos pacientes, acredita-se que a tromboembolia pulmonar seja rara na doença de Behçet. A trombose pode envolver as veias intra-abdominais de grande calibre. O manejo dos eventos trombóticos nestes pacientes é controverso, com uma combinação de imunossupressão e anticoagulação sendo usada com frequência.53
O tratamento é determinado pelo envolvimento de sistema orgânico e gravidade das manifestações clínicas. A doença grave pode requerer uso de altas doses de glicocorticoides e ciclofosfamida. A azatioprina tem eficácia no controle da doença ocular inflamatória. Apesar da falta de estudos controlados, o infliximabe e o adalimumabe têm sido usados com certo sucesso no tratamento de pacientes com doença ocular e outras manifestações de doença grave.54,55 A colchicina e a pentoxifilina podem proporcionar algum benefício para pacientes com úlcera mucosa. A terapia à base de interferon e talidomida apresentam maior eficácia no tratamento da úlcera mucosa grave, mas exibem maior toxicidade associada ao tratamento.
Agradecimentos
Figuras 1 e 2 – Seward Hung
Figura 8 – Gary S. Hoffman
1.Jennette C, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994;37:187.
2.Vassilopoulos D, Calabrese L. Hepatitis C virus infection and vasculitis. Arthritis Rheum 2002;46:585.
3.Lawrence EC, Mills J. Bacterial endocarditis mimicking vasculitis with steroid-induced remission. West J Med 1976;124:333.
4.Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Cutaneous vasculitis in children and adults: associated disease and etiologic factors in 303 patients. Medicine (Baltimore) 1998; 77:403.
5.Gedalia A. Henoch-Schonlein purpura. Curr Rheumatol Rep 2004; 6:195.
6.Davis MD, Brewer JD. Urticarial vasculitis and hypocom- plementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am 2004;24:183.
7.Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488.
8.Rheinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long term outcome in 155 patients. Arthritis Rheum 2000; 43:1021.
9.Travis WD, Hoffman GS, Leavitt RY, et al. Surgical pathology of the lung in Wegener’s granulomatosis. Am J Surg 1991;15:315.
10.Merkel PA, Lo GH, Holbrook JT, et al. Brief communication: high incidence of venous thrombotic events among patients with Wegener’s granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis Study. Ann Intern Med 2005;142:620.
11.Hoffman GS. Classi?cation of the systemic vasculitides: antineutrophil cytoplasmic antibodies, consensus and controversy. Clin Exp Rheumatol 1998;16:111.
12.Hoffman GS, Specks U. Antineutrophil cytoplasmic anti- bodies: diagnostic value in systemic vasculitis. Arthritis Rheum 1998;41:1521.
13.Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007; 147:611–9.
14.Specks U. Methotrexate for Wegener’s granulomatosis: what is the evidence? Arthritis Rheum 2005; 52 :2237.
15.Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36.
16.Langford CA. Use of a cyclophosphamide-induction methotrexate-maintenance regimen for the treatment of Wegener’s granulomatosis: extended follow-up and rate of relapse. Am J Med2003;114 :463–9.
17.Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008; 359:2790–803.
18.Hiemstra TF, Walsh M, Mahr A, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in anti- neutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA 2010;304:2381–8.
19.De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induc- tion of remission in early systemic antineutrophil cytoplasmic antibody-associatedvasculitis. Arthritis Rheum 2005;52: 2461–9.
20.Stegeman CA, Tervaert JW, de Jong PE, et al. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N Engl J Med 1996; 335:16.
21.The GPAET Research Group. Etanercept in addition to standard therapy in patients with Wegener’s granulomato- sis. N Engl J Med2005;352:351–6.
22.Silva F, Seo P, Schroeder DR, et al. Solid malignancies amongetanercept-treated patients with granulomatosis with poly- angiitis (Wegener’s): long-term followup of a multicenter longitudinal cohort. Arthritis Rheum 2011;63:2495–503.
23.Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med2010;363:221–32.
24.Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010; 363:211–20.
25.Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol2007;18:2180–8.
26.Guillevin L, Cohen P, Gayraud M, et al. Churg-Strauss syn- drome: clinical study and long-term follow up of 96 patients. Medicine (Baltimore) 1999; 78:26.
27.Reid AC, Harrison BW, Watts RA, et al. Churg-Strauss syndrome in a district hospital. Q J Med 1998;91:219.
28.Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory ?ndings in 85 patients. Arthritis Rheum 1999; 42:421.
29.Hadziyannis SJ. The spectrum of extrahepatic manifestations in hepatitis C virus infection. J Viral Hepat 1997;4:9.
30.Guillevin L, Lhote F, Cohen P, et al. Polyarteritis nodosa related to hepatitis B virus: a prospective study with long- term observation of 41 patients. Medicine (Baltimore) 1995; 74:238.
31.Travers RL, Allison DJ, Brettle RP, et al. Polyarteritis nodosa: a clinical and angiographic analysis of 17 cases. Semin Arthritis Rheum 1979;8:184.
32.Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. Medicine (Baltimore) 1996;75:17.
33.Mandell BF, Hoffman GS. Differentiating the vasculitides. Rheum Dis Clin North Am 1994;20:409.
34.Franchini M, Veneri D. The antiphospholipid syndrome. Hematology 2005;10:265.
35.Om A, Ellahham S, DiScascio G. Cholesterol embolism: an underdiagnosed clinical entity. Am Heart J 1992;124:1321.
36.Wees SJ, Sunwoo IN, Oh SJ. Sural nerve biopsy in systemic necrotizing vasculitis. Am J Med 1981;71:525.
37.Rappaport WD, Valente J, Hunter GC, et al. Clinical utilization and complications of sural nerve biopsy. Am J Surg 1993;166:252.
38.Guillevin L, Lhote F. Treatment of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1998;41:2100.
39.Yeung RS. Pathogenesis and treatment of Kawasaki’s disease. Curr Opin Rheumatol 2005;17:617.
40.Leung DY, Schlievert PM, Meissner HC. The immuno- pathogenesis and management of Kawasaki syndrome. Arthritis Rheum 1998;41:1538.
41.Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med 2003;139:505.
42.Hoffman GS. Treatment of giant cell arteritis: where we have been and why we must move on (review). Cleve Clin J Med 2002;69 Suppl II:SII117.
43.Ponge T, Barrier JH, Grolleau JY, et al. The ef?cacy of selective unilateral temporal artery biopsy versus bilateral biop- sies for diagnosis of giant cell arteritis. J Rheumatol 1988; 15:997.
44.Kerr GS, Hallahan CW, Giordano J, et al. Takayasu’s arteritis. Ann Intern Med 1994;120:919.
45.Tso E, Flamm SD, White RD, et al. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum 2002;46:1634.
46.Hoffman GS, Cid MC, Hellmann DB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002;46:1309.
47.Jover JA, Hernandez-Garcia C, Morado IC, et al. Combined treatment of giant-cell arteritis with methotrexate and prednisone, a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001;134:106.
48.Hoffman GS, Cid MC, Rendt-Zagar KE, et al. In?iximab- GCA Study Group: In?iximab for maintenance of glucocor-ticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007;146:621–30.
49.Nesher G, Berkun Y, Mates M, et al. Risk factors for cranial ischemic complications in giant cell arteritis. Medicine (Baltimore) 2004;83:114.
50.Hoffman GS, Merkel PA, Brasington RD, et al. Anti-tumor necrosis factor therapy in patients with dif?cult to treat Takayasu arteritis. Arthritis Rheum 2004; 50:2296–304
51.Molloy ES, Langford CA, Clark TM, et al. Anti-tumour necrosis factor therapy in patients with refractory Takayasu arteritis:long-term follow-up. Ann Rheum Dis 2008; 67: 1567–9.
52.Liang P, Tan-Ong M, Hoffman GS. Takayasu’s arteritis: vascular interventions and outcomes. J Rheumatol 2004;31: 102.
53.Calamia KT, Schirmer M, Melikoglu M, et al. Major vessel involvement in Behcet disease. Curr Opin Rheumatol 2005;17:1–8.
54.Lindstedt EW, Baarsma GS, Kuijpers RW, et al. Anti-TNF- alpha therapy for sight threatening uveitis. Br J Ophthalmol2005;89:533–6.
55.van Laar J, Missotten T, van Daele P. Adalimumab: a new modality for Behçet’s disease? Ann Rheum Dis 2007; 66: 565–6.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.